the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 May 2019
| 16 May 2019
Effect of sea salt aerosol on tropospheric bromine chemistry
Daniel J. Jacob
Sebastian D. Eastham
Melissa P. Sulprizio
Xuan Wang
Tomás Sherwen
Mat J. Evans
Qianjie Chen
Becky Alexander
Theodore K. Koenig
Rainer Volkamer
L. Gregory Huey
Michael Le Breton
Thomas J. Bannan
Carl J. Percival
Bromine radicals influence global tropospheric chemistry by depleting ozone and by oxidizing elemental mercury and reduced sulfur species. Observations typically indicate a 50 % depletion of sea salt aerosol (SSA) bromide relative to seawater composition, implying that SSA debromination could be the dominant global source of tropospheric bromine. However, it has been difficult to reconcile this large source with the relatively low bromine monoxide (BrO) mixing ratios observed in the marine boundary layer (MBL). Here we present a new mechanistic description of SSA debromination in the GEOS-Chem global atmospheric chemistry model with a detailed representation of halogen (Cl, Br, and I) chemistry. We show that observed levels of SSA debromination can be reproduced in a manner consistent with observed BrO mixing ratios. Bromine radical sinks from the HOBr + S(IV) heterogeneous reactions and from ocean emission of acetaldehyde are critical in moderating tropospheric BrO levels. The resulting HBr is rapidly taken up by SSA and also deposited. Observations of SSA debromination at southern midlatitudes in summer suggest that model uptake of HBr by SSA may be too fast. The model provides a successful simulation of free-tropospheric BrO in the tropics and midlatitudes in summer, where the bromine radical sink from the HOBr + S(IV) reactions is compensated for by more efficient HOBr-driven recycling in clouds compared to previous GEOS-Chem versions. Simulated BrO in the MBL is generally much higher in winter than in summer due to a combination of greater SSA emission and slower conversion of bromine radicals to HBr. An outstanding issue in the model is the overestimate of free-tropospheric BrO in extratropical winter–spring, possibly reflecting an overestimate of the HOBr∕HBr ratio under these conditions where the dominant HOBr source is hydrolysis of BrNO3.
- Article
(5644 KB) - Full-text XML
-
Supplement
(2375 KB) - BibTeX
- EndNote





Bromine radicals (BrOx ≡ Br + BrO) influence global tropospheric chemistry by depleting ozone and thus OH, as well as by oxidizing species such as elemental mercury and dimethylsulfide (Saiz-Lopez and von Glasow, 2012; Simpson et al., 2015; Long et al., 2014). Tropospheric bromine radical chemistry is initiated by the production of reactive inorganic bromine (Bry) from sea salt aerosol (SSA) debromination, decomposition of organobromines primarily of marine origin (CHBr3, CH2Br2, and CH3Br), and transport from the stratosphere. Within the Bry family, bromine radicals cycle with non-radical reservoir species such as HBr, HOBr, BrNO2, BrNO3, Br2, BrCl, and IBr. Loss of Bry is by wet and dry deposition to the surface, mainly as HBr, which is highly soluble in water.
Sea salt aerosol (SSA) is thought to be the largest source of tropospheric bromine. Observations show extensive debromination of SSA relative to seawater composition (Sander et al., 2003; Newberg et al., 2005). Parrella et al. (2012) estimate a global Bry source of 1420 Gg Br a−1 from SSA debromination, as compared with 520 Gg a−1 from organobromines and 36 Gg a−1 from the stratosphere. Volatilization of bromide from SSA can take place by heterogeneous reactions with HOBr, HOCl, N2O5, ozone, and ClNO3 (Vogt et al., 1996; Hirokawa et al., 1998; Keene et al., 1998; Fickert et al., 1999; von Glasow et al., 2002a, b; Yang et al., 2005; Ordóñez et al., 2012; Saiz-Lopez et al., 2012; Long et al., 2014). A standing conundrum has been that observations of BrO in the marine boundary layer (MBL) do not show large enhancements relative to the free troposphere, where background mixing ratios are typically of the order of 1 ppt (ppt ≡ pmol mol−1, Leser et al., 2003; Sander et al., 2003; Theys et al., 2011; Volkamer et al., 2015; Wang et al., 2015; Le Breton et al., 2017). Ozone observations in the MBL similarly do not show depletion that would be expected from high mixing ratios of BrO (de Laat and Lelieveld, 2000; Sherwen et al., 2016). This has led recent global models not to include SSA debromination as a source of Bry (Schmidt et al., 2016; Sherwen et al., 2016).
Here we present a new mechanistic description of sea salt debromination in the GEOS-Chem global 3-D model of tropospheric chemistry, including detailed representation of halogens (Cl, Br, and I) (Sherwen et al., 2016; Wang et al., 2019). We find that we can reproduce the observed levels of SSA debromination while also being consistent with the relatively low BrO mixing ratios observed in the MBL. This is because the previously recognized fast production of Bry from SSA debromination is offset by fast removal of Br atoms by acetaldehyde emitted from the ocean (Toyota et al., 2004; Millet et al., 2010; Badia et al., 2019) and by fast removal of HOBr by dissolved SO2 (S(IV)) in cloud (Long et al., 2014; Chen et al., 2017). We examine the implications for the global budget of tropospheric bromine and tropospheric oxidants.
We use GEOS-Chem 12.3.0 (https://doi.org/10.5281/zenodo.2620535, GEOS-Chem 12.3.0, 2019), which includes a detailed representation of ozone–NOx–volatile-organic-compound–aerosol–halogen tropospheric chemistry (Sherwen et al., 2016), to which we have added the comprehensive tropospheric chlorine chemistry of Wang et al. (2019) with some additional modifications as described below. The model is driven by assimilated meteorological data for 2011–2012 from the Modern-Era Retrospective analysis for Research and Applications, Version 2 (MERRA2), produced by the NASA Global Modeling and Assimilation Office (Gelaro et al., 2017). The horizontal resolution of MERRA2 is 0.5∘ × 0.625∘ and is degraded here to 4∘ × 5∘ for input to GEOS-Chem. Dynamic and chemical time steps are 30 and 60 min, respectively. GEOS-Chem stratospheric chemistry (Eastham et al., 2014) is linearized following Murray et al. (2012) to serve as boundary conditions for stratospheric input to the troposphere. The model is spun up for 1 yr and we use simulation results for 2012.
Tropospheric bromine chemistry in GEOS-Chem was first introduced by Parrella et al. (2012). Loss of Bry from the troposphere is mainly by deposition of HBr, which is highly water-soluble unlike HOBr or BrO. Parrella et al. (2012) found that the acid-catalyzed HOBr + Br− reaction taking place in liquid-water clouds, ice crystal quasi-liquid surfaces, and aqueous aerosols was critical for recycling bromide and maintaining background tropospheric BrO at observed ∼1 ppt levels:
The current standard version of the model (GEOS-Chem 12.3.0) includes more extensive heterogeneous bromine chemistry (Schmidt et al., 2016), coupling to other halogens (Sherwen et al., 2016), HOBr + S(IV) reactions in clouds (Chen et al., 2017), and oceanic emission of acetaldehyde which reacts rapidly with Br atoms (Toyota et al., 2004; Millet et al., 2010; Badia et al., 2019). Wang et al. (2019) more recently added a comprehensive treatment of tropospheric chlorine chemistry in GEOS-Chem including explicit accounting of SSA chloride volatilization and aerosol pH. The model does not attempt to simulate the fast but localized bromine chemistry taking place in the Arctic MBL in spring due to volatilization of bromine deposited on sea ice (Simpson et al., 2015).
Here we added several updates to the computation of heterogeneous chemistry recycling bromine radicals. Uptake of HOBr(aq) by clouds involves competition between reactions with Br−, Cl−, , and , as given by Reactions (R1), (R4), and (R5):
Reactions (R1) and (R4) recycle bromine radicals, while Reaction (R5) is effectively a terminal sink because of HBr deposition. The standard GEOS-Chem code computes the reactive uptake coefficient (γ) independently for each pathway, which incorrectly assumes that other pathways do not affect mass transfer. Here we express the first-order aqueous-phase loss of HOBr as a sum of the four pathways to compute γ, and we then distribute the loss by pathways on the basis of the relative rates (see Supplement Sect. S1). When calculating HOBr uptake by ice crystals, we assume a radius of 38.5 µm based on cloud observations (Fu, 1996) rather than 75 µm in the standard GEOS-Chem code and further increase the effective surface area of ice crystals by a factor of 10 to account for their irregular shape (Schmitt and Heymsfield, 2005). Finally, we correct a registration error for SSA alkalinity in the standard GEOS-Chem code that caused an underestimate of alkalinity titration (see Sect. S2). The overall result of our updates is to have more efficient heterogeneous recycling of bromine radicals, both in the MBL and in the free troposphere.
The GEOS-Chem SSA simulation is from Jaeglé et al. (2011), who showed that it could reproduce successfully SSA observations over the oceans from ship cruises and coastal/island stations, as well as observations of aerosol optical depth (AOD) from the Aerosol Robotic Network (AERONET) and the MODIS satellite instrument. The model separates fine (≤0.5 µm radius) and coarse SSA as two separate transported species. The global dry SSA source in our simulation is 3140 Tg a−1. We emit bromide as part of fine and coarse SSA with a seawater ratio of kg Br per kilogram of dry SSA (Sander et al., 2003; Lewis and Schwartz, 2004), since observations show that fresh SSA has a bromide content equal to that of seawater (Duce and Woodcock, 1971; Duce and Hoffman, 1976; Turekian et al., 2003). SSA alkalinity is emitted at a ratio of 0.07 equivalents per kilogram of dry SSA and is depleted by strong acids following Alexander et al. (2005). The cloud pH is calculated as described in Alexander et al. (2012) and is 3.5–6.5 for clean marine conditions, consistent with the observed range (3.8–6.1) reported in the literature (Gioda et al., 2009; Hegg et al., 1984; Lenschow et al., 1988; Vong et al., 1997; Watanabe et al., 2001).
Activation of SSA bromide takes place by heterogeneous reactions with HOBr, ozone, and ClNO3 once alkalinity has been titrated and SSA is acidified (Hirokawa et al., 1998; Keene et al., 1998; Fickert et al., 1999):
We also consider parameterized SSA debromination by HOI(aq) following McFiggans et al. (2002), where HOI(aq) may be taken up from the gas phase or produced by hydrolysis of INO2 and INO3:
Inorganic oceanic iodine (HOI and I2) emissions are from Carpenter et al. (2013). Unlike for chloride, SSA debromination does not take place by acid displacement because of the much stronger acidity of HBr than HCl or HNO3 (Sander et al., 2003). On the contrary, uptake of gas-phase HBr can lead to bromine enrichment in SSA.
Sander et al. (2003) introduced the dimensionless enrichment factor (EF) as a measure of SSA debromination. EF is computed from aerosol measurements as
where aerosol [Na+] is assumed to be mainly from sea salt, a reliable assumption in marine air. In GEOS-Chem we treat SSA as a chemically inert tracer and account for sea salt bromide as a separate species; therefore EF is computed as
We sum [Br−] and [SSA] from both fine and coarse SSA to calculate EF. We will also present EF values for fine and coarse SSA separately.
Figure 1 (top panel) shows the annual mean SSA bromine enhancement factors (EFs) in surface air computed by GEOS-Chem and compares to annual mean observations compiled by Sander et al. (2003) and from Newberg et al. (2005). We consider six island and four coastal sites with bulk aerosol EF measurements available for more than 1 year. The observations are for different years than the GEOS-Chem simulation, but we assume that interannual variability is a minor source of error. The mean GEOS-Chem EF averaged over the sites is 0.75±0.23 (±1 standard deviation), compared with the observed value of 0.66±0.32. SSA bromide over the Southern Ocean in the model is less depleted (EF∼0.9) than over the northern midlatitudes (EF∼0.6), because SSA tends to retain its alkalinity over the Southern Ocean (Alexander et al., 2005; Schmidt et al., 2016; Fig. S1). Similarly in the observations, mean EF is 0.78±0.08 over the Southern Ocean compared with 0.42±0.11 at northern midlatitudes. Most of the SSA debromination in the model is from Reaction (R1).
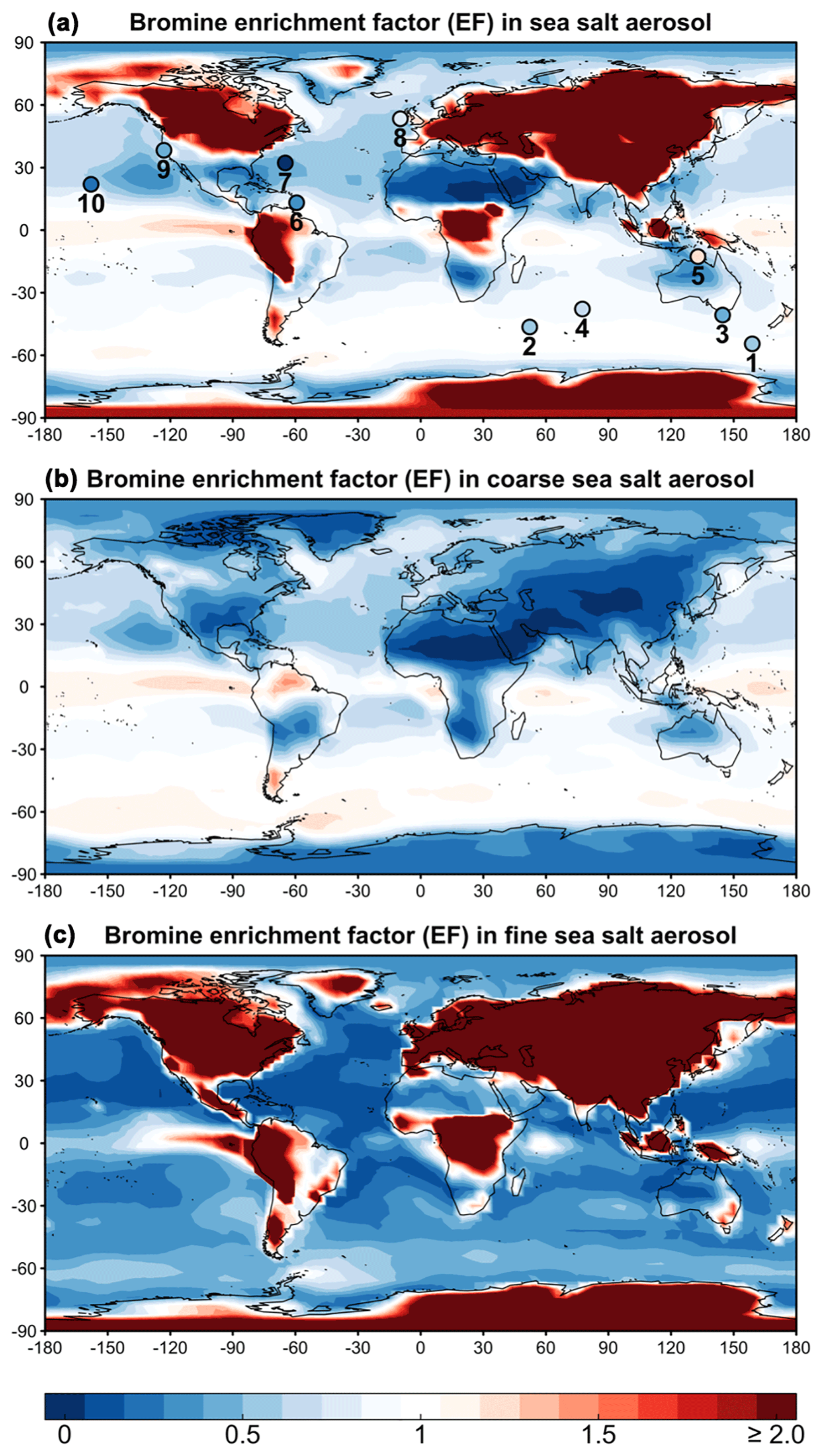
Figure 1Annual mean bromine enrichment factor (EF) of sea salt aerosol (SSA) in surface air. GEOS-Chem model results (contours) are compared to observations (circles in a). (a) shows results for total SSA (fine + coarse), (b) is for coarse SSA (>0.5 µm radius), and (c) is for fine SSA (≤0.5 µm radius). Observations at 10 sites compiled by Sander et al. (2003) and from Newberg et al. (2005) are superimposed as circles and labeled in order 1–10: Macquarie Island, Crozet Islands, Cape Grim, Amsterdam Island, Jabiru, Barbados, Bermuda, Mace Head, Bodega Bay, and Hawaii, respectively. The simulation is for 2012 and the observations are for different years. Color bar saturates at 2.0. Maximum modeled EF is 75.0.
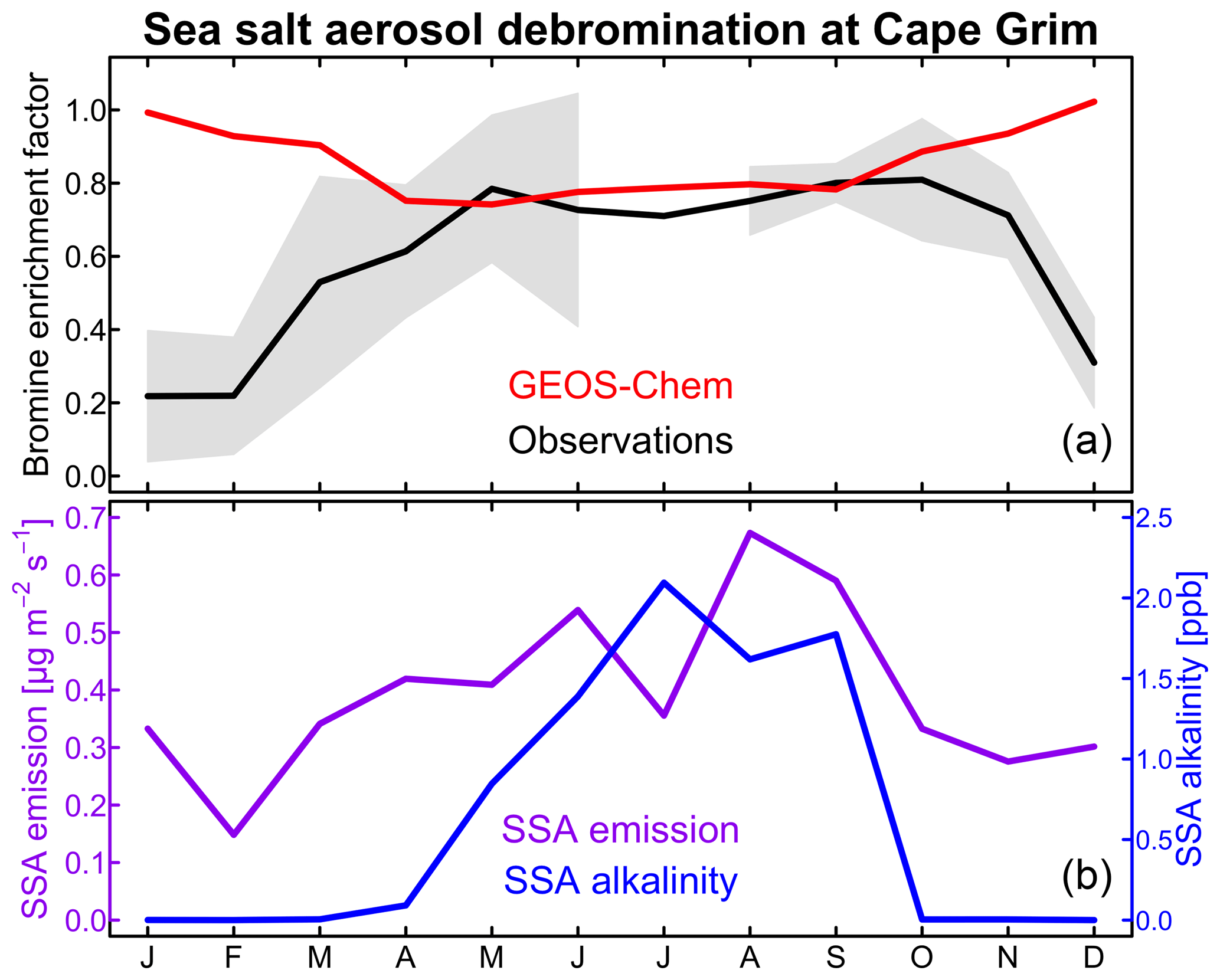
Figure 2Seasonal variation of sea salt aerosol (SSA) debromination at Cape Grim, Tasmania (40.7∘ S, 144.7∘ E). (a) shows the bromine enrichment factors (EFs) of monthly mean SSA in surface air. Observations from Ayers et al. (1999) and Sander et al. (2003) for 1996–1998 are compared to GEOS-Chem model values for 2012. Shading gives the interannual standard deviation in the observations. (b) shows the GEOS-Chem monthly SSA emission flux at Cape Grim (site 3 in Fig. 1) and the SSA alkalinity. The SSA emission flux is for the oceanic fraction of the Cape Grim grid square.
SSA mass in GEOS-Chem is dominated by the coarse component over the oceans and by the fine component over land (due to fast dry deposition of the coarse component). Thus the EF values for bulk SSA over the ocean are dominated by the coarse component. SSA debromination is more extensive for the fine component of SSA because the initial supply of bromide is less and loss of alkalinity is more extensive. Some areas of the ocean have weak SSA bromide enrichment (EF>1) because of uptake of HBr. Over land the EF values are dominated by the fine SSA component because the coarse component deposits close to the coast. These EF values can be very large because Bry volatilized from the coarse SSA component is then taken up as HBr by the fine SSA after the coarse component has deposited.
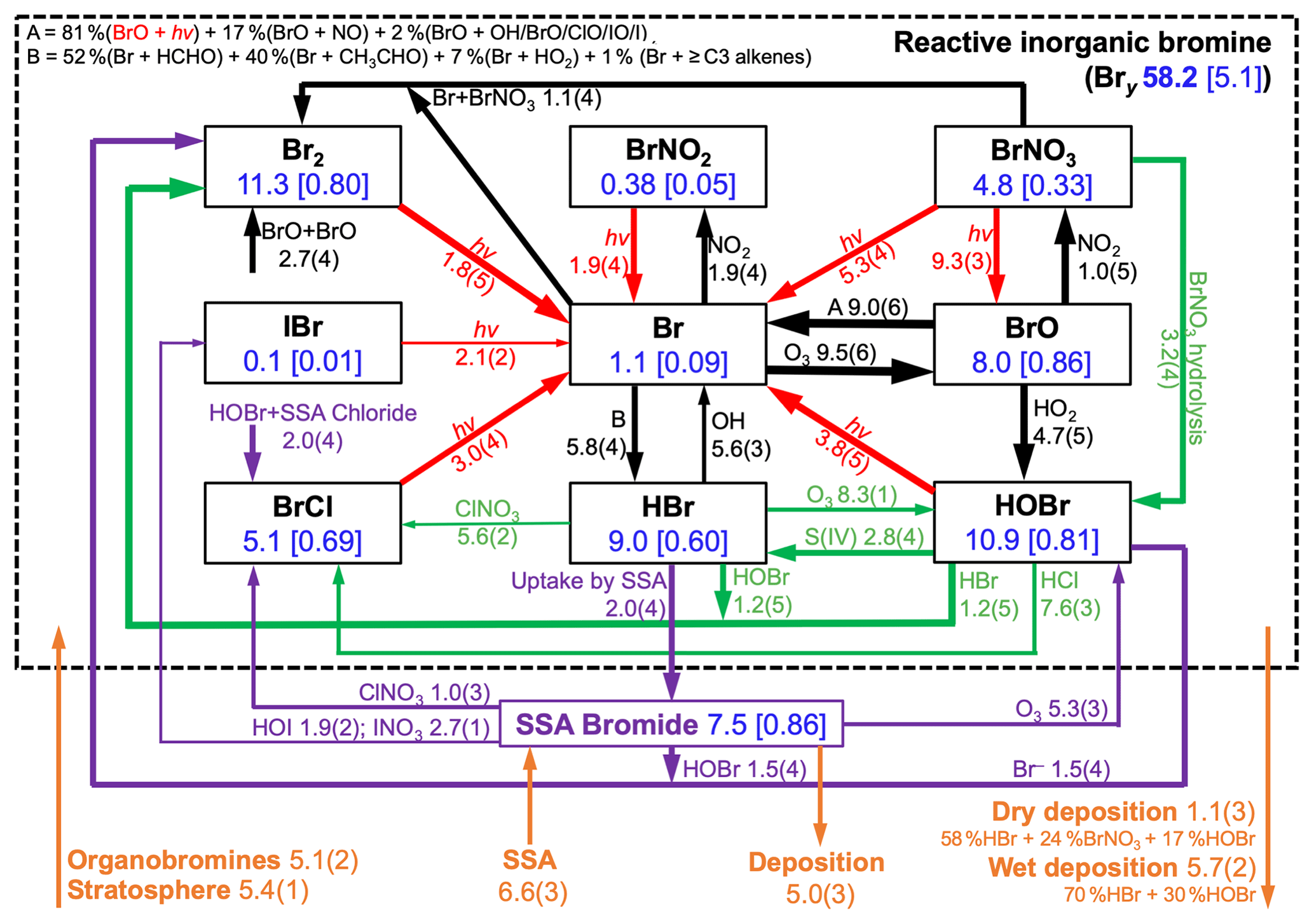
Figure 3Global annual mean tropospheric budget and cycling of reactive inorganic bromine (Bry) and sea salt aerosol (SSA) bromide. Results are from our GEOS-Chem simulation for 2012 including SSA debromination. Bry is defined as the ensemble of species inside the dashed box. Rates are in gigagrams of Br per year (Gg Br a−1), masses in the boxes are in Gg Br, and numbers in brackets are mean mixing ratios (ppt). Read 5.8(4) as 5.8×104 Gg Br a−1. Arrows in black are for gaseous reactions, red for photolysis, purple for heterogeneous reactions in SSA, and green for other heterogeneous reactions taking place in cloud and sulfate aerosol. Sources and sinks of total inorganic bromine (Bry + SSA bromide) are in orange. Arrow thickness scales with its corresponding rate.
The model overestimates the observed EF over the Southern Ocean. This appears to reflect a seasonal bias in the model. Figure 2 compares the simulated and observed seasonality at Cape Grim, Tasmania (Ayers et al., 1999; Sander et al., 2003). To our knowledge, this is the only site for which seasonal information is available in the observations. The observed EF is 0.6–0.8 for most of the year, consistent with the model, but decreases to below 0.4 in summer while the model does not. The summer minimum in the observations has been attributed to increased SSA acidity (Ayers et al., 1999; Sander et al., 2003). Indeed, Fig. 2 shows that SSA alkalinity in the model is titrated in summer due to the combination of weaker SSA emission (lower winds) and larger photochemical production of strong acids (H2SO4 and HNO3). This drives volatilization of Bry from SSA, but we find in the model that the resulting HBr mainly returns to SSA rather than deposits to the surface because SSA emission is still relatively high. Uptake of HBr by SSA proceeds in the model with a reactive uptake coefficient exp(4290 K/T) as recommended by IUPAC (Amman et al., 2013) but with large uncertainty, ranging from −90 % to +860 % at 278 K.
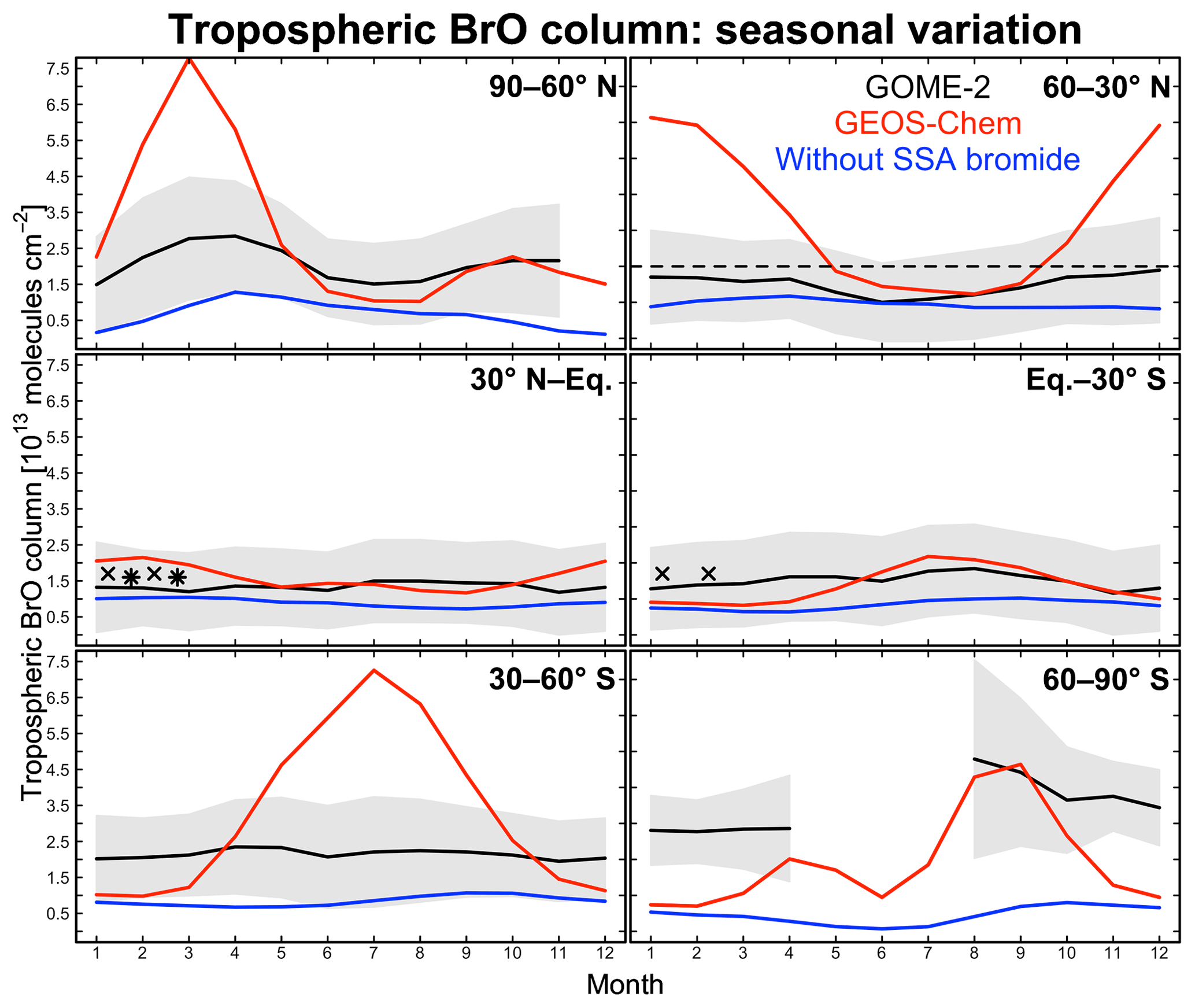
Figure 4Seasonal variation of zonal mean tropospheric BrO columns in different latitudinal bands. Monthly GOME-2 BrO observations are for 2007 and taken from Theys et al. (2011); shading represents 1 standard deviation about the monthly mean GOME-2 BrO columns. GEOS-Chem BrO columns are sampled at the GOME-2 local overpass time (09:00–10:00). Red lines are from our standard simulation including sea salt aerosol (SSA) debromination; blue lines are from a sensitivity simulation without SSA debromination. The dashed black line indicates observations for 2009–2011 reported by Coburn et al. (2011) in Florida, USA, without seasonality information. Black crosses and stars represent average BrO columns measured during aircraft campaigns over the eastern tropical Pacific (Volkamer et al., 2015; Wang et al., 2015; Dix et al., 2016) and western tropical Pacific (Koenig et al., 2017), respectively.
Figure 3 shows the global budget and speciation of tropospheric Bry in our simulation. This updates a similar figure by Schmidt et al. (2016) to include SSA debromination, the HOBr + S(IV) reactions in clouds, oceanic emission of acetaldehyde, and full coupling with the other halogens. SSA debromination is the largest global source, mainly from Reactions (R1) and (R7) producing Br2 and HOBr, respectively. The dominant sink of Bry is uptake of HBr by SSA, rather than deposition, emphasizing the importance of competition between these two processes in determining the extent of SSA debromination. Bromine radical (Br) is converted to HBr by formaldehyde, acetaldehyde, HO2 radical, and ≥ C3 alkenes with relative contributions of 52 %, 40 %, 7 %, and 1 %, respectively. In the Schmidt et al. (2016) budget, acetaldehyde contributed only 17 % of this bromine radical sink; the larger contribution in our simulation reflects its oceanic emission. Observations from the recent ATom aircraft campaign (Wofsy et al., 2018) over the remote Pacific and Atlantic show mean MBL acetaldehyde mixing ratios within 10 % of those simulated by GEOS-Chem including the Millet et al. (2010) ocean source (Bates et al., 2018).

Figure 5Vertical profiles of BrO mixing ratios over the tropical Pacific. Observations from the TORERO (Volkamer et al., 2015; Wang et al., 2015; Dix et al., 2016), CONTRAST (Chen et al., 2016; Koenig et al., 2017), and CAST (Le Breton et al., 2017) aircraft campaigns are compared to model values. Solid black lines indicate mean observed values in 1 km vertical bins and with standard deviations (shading). We use two independent CONTRAST BrO data sets. The black line with dots shows the median values (dashed lines are 25 % and 75 % quantiles) as reported in Koenig et al. (2017). The solid line denotes the mean values from Chen et al. (2016). GEOS-Chem is sampled along the flight tracks at the time of the measurements. Model results are shown from our (i) standard simulation including sea salt aerosol debromination (red lines) and (ii) sensitivity simulations not including SSA debromination (blue lines) and HOBr + S(IV) reactions (purple lines). Solid lines are mean values, dotted lines are median values.
The global tropospheric loading of BrO in our simulation is 8.0 Gg Br, corresponding to a mean tropospheric mixing ratio of 0.86 ppt (1.7 ppt as daytime average). The BrO loading is higher than in previous GEOS-Chem versions starting with 3.8 Gg in Parrella et al. (2012), 5.7 Gg in Schmidt et al. (2016), 6.4 Gg in Sherwen et al. (2016), 3.6 Gg in Chen et al. (2017), and 4.2 Gg in Wang et al. (2019). Wang et al. (2019) described this evolution between versions. Our high BrO loading reflects our updates to HOBr uptake and correction of SSA alkalinity.
Figure 4 compares simulated tropospheric BrO columns with Global Ozone Monitoring Experiment (GOME)-2 satellite observations from Theys et al. (2011) as a function of season and for different latitudinal bands. Also shown are tropospheric columns from ground-based measurements in Florida, USA (Coburn et al., 2011), and derived from mean aircraft vertical profiles over the tropical Pacific from the TORERO (Volkamer et al., 2015; Wang et al., 2015; Dix et al., 2016) and CONTRAST (Koenig et al., 2017) campaigns. There is general consistency between these observations. Model results are from our standard simulation and from a sensitivity simulation without SSA debromination. The standard simulation provides a good fit to the observations in the tropics but is much too high at extratropical latitudes in winter and spring. High model mixing ratios under these conditions are due to high SSA emissions and fast bromide recycling via Reaction (R1), with the latter due to a large HOBr source from BrNO3 hydrolysis. BrNO3 hydrolysis mainly takes place in cloud (droplets and ice crystals) rather than in aerosols. The dominant global sink for BrNO3 is photolysis (Fig. 3), but under extratropical winter–spring conditions we find that hydrolysis is more important because of weak radiation. Another reason for the high modeled BrO in extratropical winter–spring is that removal of Bry via deposition of HBr becomes slower under these conditions, as described below.
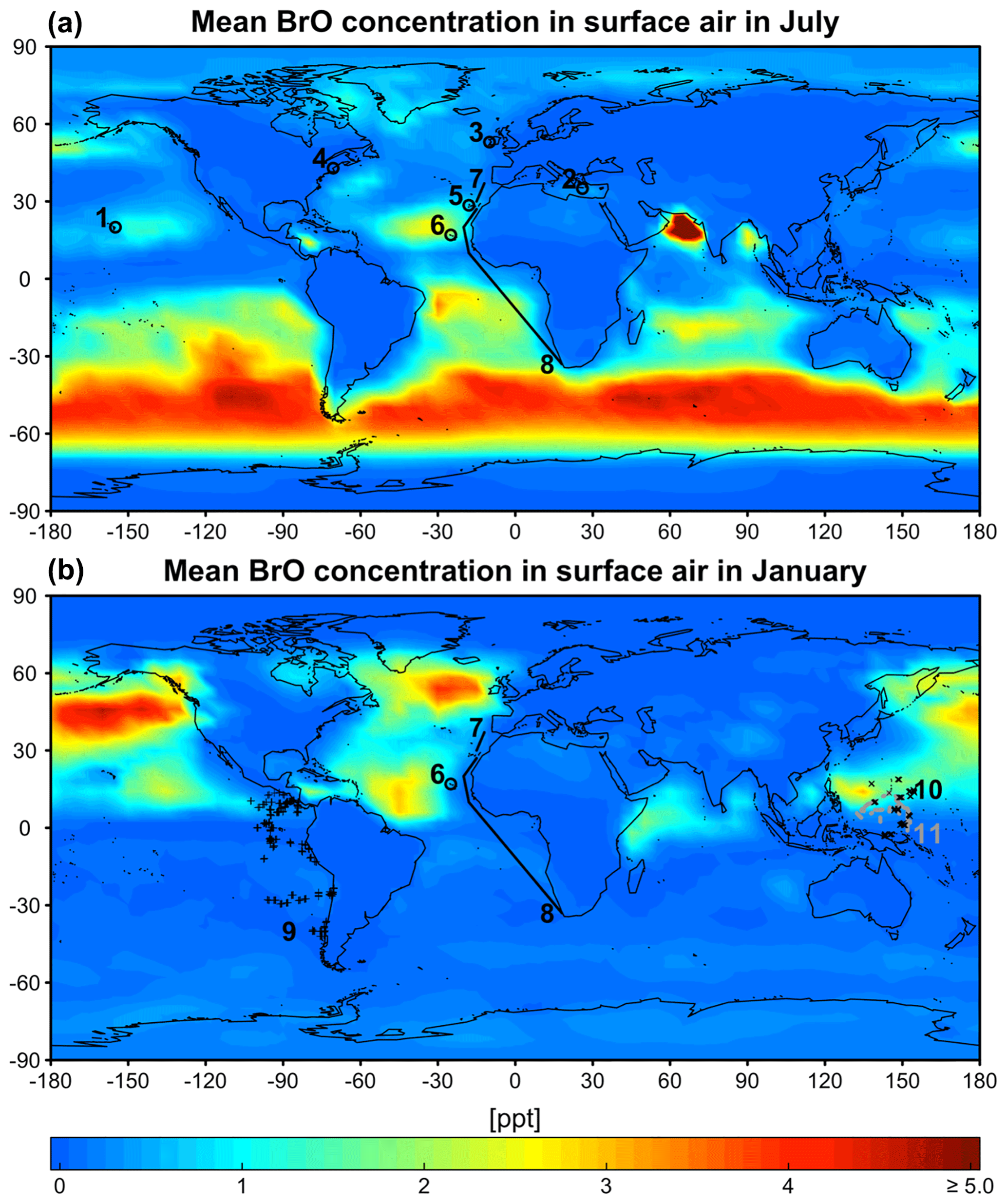
Figure 6GEOS-Chem mean BrO mixing ratios in surface air in July (a) and January (b). Locations of BrO observations in Table 1 are shown as symbols for the closest season. Open circles are ground sites and solid lines are ship tracks. Flight tracks in the marine boundary layer (<2 km) during the TORERO (Volkamer et al., 2015; Wang et al., 2015; Dix et al., 2016), CONTRAST (Chen et al., 2016), and CAST (Le Breton et al., 2017) aircraft campaigns are shown as pluses, crosses, and dots (gray), respectively. Daytime BrO mixing ratios are about double the values shown here since BrO drops to near zero at night. Color bar saturates at 13.3 ppt near India.
Table 1Daytime mixing ratios (ppt) of BrO in the marine boundary layera.
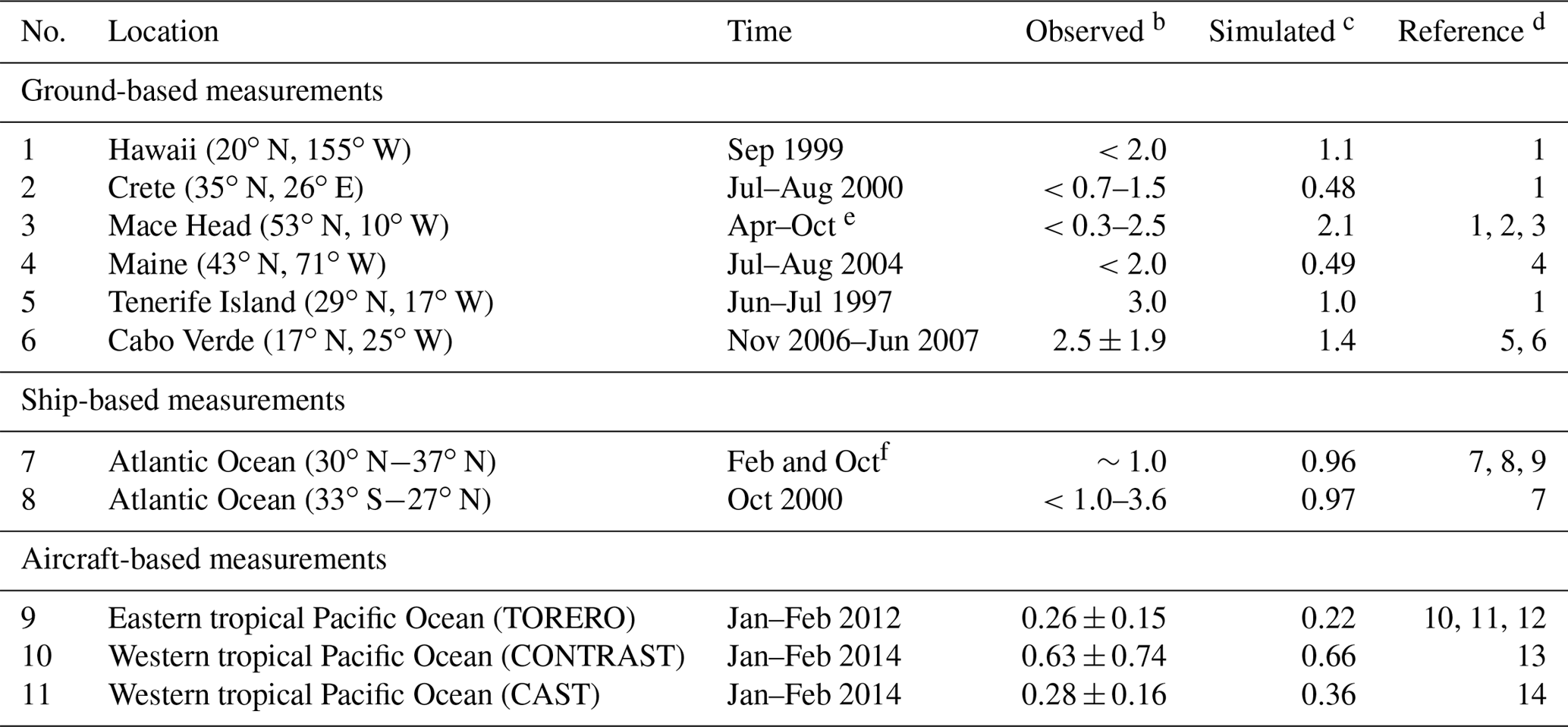
a Locations of measurements are shown in Fig. 6. b Values reported as ranges, means, and means ± standard deviations depending on availability. The symbol “<” indicates that BrO is below the corresponding detection limit. c Mean values for the simulated model year of 2012. The observations are for different years. Model values are sampled at the location and time of year of the observations. d (1) Sander et al. (2003), (2) Saiz-Lopez et al. (2004), (3) Saiz-Lopez et al. (2006), (4) Keene et al. (2007), (5) Read et al. (2008), (6) Mahajan et al. (2010), (7) Leser et al. (2003), (8) Martin et al. (2009), (9) Saiz-Lopez et al. (2012), (10) Volkamer et al. (2015), (11) Wang et al. (2015), (12) Dix et al. (2016), (13) Chen et al. (2016), and (14) Le Breton et al. (2017) e From 1996, 1997, and 2002. f From 2000 and 2007.
Figure 5 compares simulated vertical profiles with aircraft BrO observations over the tropics in January and February. Schmidt et al. (2016) and Shermen et al. (2016) reported negative biases of 0.6–1.0 ppt in modeled BrO compared with TORERO observations in the upper free troposphere. Our standard simulation shows a much smaller bias (∼0.3 ppt) because of the impact of faster HOBr uptake on cloud ice (Sect. 2). Figure 5 also shows BrO results from two sensitivity simulations not including SSA debromination or the HOBr + S(IV) reactions. Model–observation agreement in those two cases is generally not as good as in our standard simulation. As shown in Fig. 5, the impact of SSA debromination extends through the depth of the troposphere by increasing BrO by 0.1–0.5 ppt with larger impact near the surface. This is generally seen in other seasons over the tropical latitudes as well (Fig. S2). For the extratropical latitudes, the impact of SSA debromination on BrO is much larger (up to 3 ppt) especially in winter–spring, which is also reflected in BrO columns (Fig. 4). Besides high SSA emissions and fast bromide recycling as described above, we also find that only ∼15 % of Bry there is present as HBr (Figs. S3 and S4), reflecting the relatively lower Br∕BrO ratio (∼0.02 at winter while ∼0.08 at summer) when radiation is weak. Bry has then a longer lifetime because non-HBr species are much less water-soluble (Parrella et al., 2012) and can be effectively transported to the free troposphere.
Figure 6 shows the simulated global distribution of BrO mixing ratios in surface air in January and July. We attribute the elevated marine surface BrO in winter time to a combination of greater SSA emission and slower removal of Bry via deposition of HBr. However, we fail to find enough information in previous studies to evaluate the modeled seasonality in marine surface BrO. Only Cabo Verde has seasonal information (Read et al., 2008; Mahajan et al., 2010) and shows insignificant seasonal variation consistent with the model, but this is for the tropics. Observations are compiled in Table 1 with corresponding model values. The model is generally consistent with observations in showing daytime mixing ratios in the range 0.5–2 ppt, including low BrO (0.3–0.6) in the MBL measured from aircraft campaigns. The low level of BrO (∼0.3 ppt) over the eastern tropical Pacific Ocean (Volkamer et al., 2015; Wang et al., 2015; Dix et al., 2016) observed during the TORERO flight campaign is driven by weak SSA emission in the austral summer. The higher BrO in CONTRAST than in CAST is reproduced by the model where it is due to regional variations in SSA emission. The tropical North Atlantic is enhanced with BrO (1–3 ppt) relative to other tropical oceans, both in the model and observations (Tenerife, Cabo Verde). This is due in the model to slow removal of Bry via dry deposition of BrNO3, resulting in elevated Bry (and BrO through Bry cycling). We find the production rate of BrNO3 by BrO + NO2 in this region is ∼76 % slower than over the surrounding Atlantic. Isolated hotspots near India (July) and the Caribbean (January and July) in the model correspond to localized hotspots of SSA emissions.
Observations of sea salt aerosol (SSA) debromination over oceans worldwide imply a large source of bromine radicals in the marine boundary layer (MBL), yet measured BrO mixing ratios in the MBL are relatively low. Here we attempted to reconcile these observations with a global simulation of tropospheric bromine chemistry in the GEOS-Chem model, including detailed representation of processes.
We find that we can successfully simulate the observations of SSA debromination in the literature as measured by the SSA bromine enrichment factor (EF). Most of the debromination in the model is by the HOBr(aq) + Br− + H+ and O3(aq) + Br− + H+ reactions taking place in acidified SSA. Debromination is more extensive at northern latitudes than at southern latitudes because of higher acidity. Observations at southern midlatitudes show extensive debromination in summer that is not captured by the model, and we attribute this to model competition for HBr between uptake by SSA and deposition, where HBr uptake by SSA (taken from the IUPAC recommendation) may be too fast. The model predicts large bromide enrichments for fine SSA over land (EF>1) as bromine lost from the coarse SSA transfers as HBr to the fine SSA that is transported inland.
Our model simulation improves over previous GEOS-Chem versions in the simulation of surface, aircraft, and satellite observations of BrO mixing ratios in the tropics and summertime midlatitudes. Previous model versions including SSA debromination overestimated BrO in the MBL and underestimated it in the free troposphere. Our lower BrO in the MBL reflects the inclusion of radical sinks to HBr from the HOBr + S(IV) and CH3CHO + Br reactions. Our higher BrO in the free troposphere reflects more efficient recycling of bromine radicals by HOBr reactions in clouds. However, the model appears to generate excessive free-tropospheric BrO in the extratropics in winter–spring. A distinctive feature of these conditions in the model is a HOBr∕HBr ratio in excess of unity, reflecting a large source of HOBr from BrNO3 hydrolysis and the inefficient production of HBr in the MBL, allowing MBL bromine to be transported to the free troposphere. Further investigation into the chemistry mechanism and uncertainty may be needed, including uptake of HBr by SSA, uptake of HOBr by aerosols (Roberts et al., 2014), BrNO3 hydrolysis, unexplained observations of oxygenated volatile organic compounds in the free troposphere (Volkamer et al., 2015; Anderson et al., 2017; Badia et al., 2019), and oxidation of bromide by ozone involving the BrOOO− ozonide (Artiglia et al., 2017).
The GEOS-Chem model is available at http://acmg.seas.harvard.edu/geos/ (last access: 9 May 2019). GEOS-Chem chemistry mechanism and monthly bromine simulation output used in this study are available at https://doi.org/10.7910/DVN/BADJDE (Zhu, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-6497-2019-supplement.
LZ and DJJ designed the research and wrote the paper; LZ, DJJ, SDE, MPS, XW, TS, MJE, QC, and BA led the model development; TKK, RV, LGH, ML, TJB, and CJP provided BrO observations.
The authors declare that they have no conflict of interest.
This work was supported by the NSF Atmospheric Chemistry Program. We acknowledge contributions from the TORERO, CONTRAST, and CAST science team. We thank Dexian Chen for the CONTRAST BrO observations. Qianjie Chen and Becky Alexander acknowledge support from NSF AGS 1343077. Rainer Volkamer and Theodore K. Koenig acknowledge funding from NSF AGS-1620530.
This research has been supported by the NSF (Atmospheric Chemistry Program and grant nos. NSF AGS 1343077 and AGS-1620530).
This paper was edited by Markus Ammann and reviewed by Rolf Sander and one anonymous referee.
Alexander, B., Park, R. J., Jacob, D. J., Li, Q. B., Yantosca, R. M., Savarino, J., Lee, C. C. W., and Thiemens, M. H.: Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes, J. Geophys. Res., 110, D10307, https://doi.org/10.1029/2004JD005659, 2005.
Alexander, B., Allman, D. J., Amos, H. M., Fairlie, T. D., Dachs, J, Hegg, D. A., and Sletten, R. S.: Isotopicconstraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean, J. Geophys. Res., 117, D06304, https://doi.org/10.1029/2011JD016773, 2012.
Ammann, M., Cox, R. A., Crowley, J. N., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates, Atmos. Chem. Phys., 13, 8045–8228, https://doi.org/10.5194/acp-13-8045-2013, 2013.
Anderson, D. C., Nicely, J. M., Wolfe, G. M., Hanisco, T. F., Salawitch, R. J., Canty, T. P., Dickerson, R. R., Apel, E. C., Baidar, S., Bannan, T. J., Blake, N. J., Chen, D., Dix, B., Fer- nandez, R. P., Hall, S. R., Hornbrook, R. S., Gregory Huey, L., Josse, B., Jöckel, P., Kinnison, D. E., Koenig, T. K., Le Breton, M., Marécal, V., Morgenstern, O., Oman, L. D., Pan, L. L., Percival, C., Plummer, D., Revell, L. E., Rozanov, E., Saiz-Lopez, A., Stenke, A., Sudo, K., Tilmes, S., Ullmann, K., Volkamer, R., Weinheimer, A. J., and Zeng, G.: Formaldehyde in the Tropical Western Pacific: Chemical Sources and Sinks, Convective Transport, and Representation in CAM-Chem and the CCMI Models, J. Geophys. Res.-Atmos., 122, 11201–11226, https://doi.org/10.1002/2016JD026121, 2017.
Artiglia, L., Edebeli, J., Orlando, F., Chen, S., Lee, M.-T., Corral Arroyo, P., Gilgen, A., Bartels-Rausch, T., Kleibert, A., Vaz-dar, M., Andres Carignano, M., Francisco, J. S., Shepson, P. B., Gladich, I., and Ammann, M.: A surface-stabilized ozonide trig- gers bromide oxidation at the aqueous solution-vapour interface, Nat. Commun., 8, 700, https://doi.org/10.1038/s41467-017-00823-x, 2017.
Ayers, G. P., Gillett, R. W., Cainey, J. M., and Dick, A. L.: Chloride and bromide loss from sea-salt particles in southern ocean air, J. Atmos. Chem., 33, 299–319, 1999.
Badia, A., Reeves, C. E., Baker, A. R., Saiz-Lopez, A., Volkamer, R., Koenig, T. K., Apel, E. C., Hornbrook, R. S., Carpenter, L. J., Andrews, S. J., Sherwen, T., and von Glasow, R.: Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem, Atmos. Chem. Phys., 19, 3161–3189, https://doi.org/10.5194/acp-19-3161-2019, 2019.
Bates, K. H., Kim, M., and Jacob, D. J.: New Constraints on Remote Tropospheric Budgets of Oxidized VOCs, 15th IGAC Science Conference, Takamatsu, Kagawa, Japan, 2018.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Chen, D., Huey, L. G., Tanner, D. J., Salawitch, R. J., Anderson, D. C., Wales, P. A., Pan, L. L., Atlas, E. L., Hornbrook, R. S., Apel, E. C., Blake, N. J., Campos, T. L., Donets, V., Flocke, F. M., Hall, S. R., Hanisco, T. F., Hills, A. J., Honomichl, S. B., Jensen, J. B., Kaser, L., Montzka, D. D., Nicely, J. M., Reeves, J. M., Riemer, D. D., Schauffler, S. M., Ullmann, K., Weinheimer, A. J., and Wolfe, G. M.: Airborne measurements of BrO and the sum of HOBr and Br2 over the Tropical West Pacific from 1 to 15 km during the CONvective TRansport of Active Species in the Tropics (CONTRAST) experiment, J. Geophys. Res.-Atmos., 121, 12560–12578, https://doi.org/10.1002/2016JD025561, 2016.
Chen, Q., Schmidt, J. A., Shah, V., Jaegleì, L., Sherwen, T., and Alexander, B.: Sulfate production by reactive bromine: Implications for the global sulfur and reactive bromine budgets, Geophys. Res. Lett., 44, 7069–7078, https://doi.org/10.1002/2017GL073812, 2017.
Coburn, S., Dix, B., Sinreich, R., and Volkamer, R.: The CU ground MAX-DOAS instrument: characterization of RMS noise limitations and first measurements near Pensacola, FL of BrO, IO, and CHOCHO, Atmos. Meas. Tech., 4, 2421–2439, https://doi.org/10.5194/amt-4-2421-2011, 2011.
de Laat, A. T. J. and Lelieveld, J.: The diurnal O3 cycle in the tropical and subtropical marine boundary layer, J. Geophys. Res., 105, 11547–11559, 2000.
Dix, B., Koenig, T. K., and Volkamer, R.: Parameterization retrieval of trace gas volume mixing ratios from Airborne MAX-DOAS, Atmos. Meas. Tech., 9, 5655–5675, https://doi.org/10.5194/amt-9-5655-2016, 2016.
Duce, R. A. and Hoffman, E.: Chemical fractionation at the air/sea interface, Annu. Rev. Earth Planet. Sci., 4, 187–228, 1976.
Duce, R. A. and Woodcock, A. H.: Difference in chemical composition of atmospheric sea salt particles produced in the surf zone and on the open sea in Hawaii, Tellus, 23, 427–435, 1971.
Eastham, S. D., Weisenstein, D. K., and Barrett, S. R. H.: Development and evaluation of the unified tropospheric–stratospheric chemistry extension (UCX) for the global chemistry-transport model GEOS-Chem, Atmos. Environ., 89, 52–63, https://doi.org/10.1016/j.atmosenv.2014.02.001, 2014.
Fickert, S., Adams, J. W., and Crowley, J. N.: Activation of Br2 and BrCl via uptake of HOBr onto aqueous salt solutions, J. Geophys. Res., 104D, 23719–23727, 1999.
Fu, Q.: An accurate parameterization of the solar radiative properties of cirrus clouds for climate models, J. Climate, 9, 2058–2082, 1996.
Gelaro, R., McCarty, W., Suárez, M. J., Todling, R., Molod, A., Takacs, L., Randles, C. A., Darmenov, A., Bosilovich, M. G., Re- ichle, R., Wargan, K., Coy, L., Cullather, R., Draper, C., Akella, S., Buchard, V., Conaty, A., da Silva, A. M., Gu, W., Kim, G.- K., Koster, R., Lucchesi, R., Merkova, D., Nielsen, J. E., Partyka, G., Pawson, S., Putman, W., Rienecker, M., Schubert, S. D., Sienkiewicz, M., and Zhao, B.: The Modern-Era Retrospective Analysis for Research and Applications, Version 2 (MERRA-2), J. Climate, 30, 5419–5454, https://doi.org/10.1175/JCLI-D-16-0758.1, 2017.
GEOS-Chem 12.3.0: The International GEOS-Chem User Community, Zenodo, https://doi.org/10.5281/zenodo.2620535, 2019.
Gioda, A., Mayol-Bracer, O. L., Morales-García, F., Collett, J., Decesari, S., Emblico, L., Facchini, M. C., Morales-De Jesús, R. J., Mertes, S., Borrmann, S., Walter, S., and Schneider, J.: Chemical Composition of Cloud Water in the Puerto Rican Tropical Trade Wind Cumuli, Water Air Soil Pollut., 200, 3–14, https://doi.org/10.1007/s11270-008-9888-4, 2009.
Hegg, D. A., Radke, L. F., and Hobbs, P. V. : Measurements of Transformations in the Physical and Chemical Properties of Clouds Associated with Onshore Flow in Washington State, J. Appl. Clim. Meteorol., 23, 979–984, https://doi.org/10.1175/1520-0450(1984)023<0979:MOTITP>2.0.CO;2, 1984.
Hirokawa, J., Onaka, K., Kajii, Y., and Akimoto, H.: Heterogeneous processes involving sodium halide particles and ozone: Molecular bromine release in the marine boundary layer in the absence of nitrogen oxides, Geophys. Res. Lett., 25, 2449–2452, 1998.
Jaeglé, L., Quinn, P. K., Bates, T. S., Alexander, B., and Lin, J.-T.: Global distribution of sea salt aerosols: new constraints from in situ and remote sensing observations, Atmos. Chem. Phys., 11, 3137–3157, https://doi.org/10.5194/acp-11-3137-2011, 2011.
Keene, W. C., Sander, R., Pszenny, A. A. P., Vogt, R., Crutzen, P. J., and Galloway, J. N.: Aerosol pH in the marine boundary layer: A review and model evaluation, J. Aerosol Sci., 29, 339–356, 1998.
Keene, W. C., Stutz, J., Pszenny, A. A. P., Maben, J. R., Fischer, E. V., Smith, A. M., von Glasow, R., Pechtl, S., Sive, B. C., and Varner, R. K.: Inorganic chlorine and bromine in coastal New England air during summer, J. Geophys. Res., 112, D10S12, https://doi.org/10.1029/2006JD007689, 2007.
Koenig, T. K., Volkamer, R., Baidar, S., Dix, B., Wang, S., Anderson, D. C., Salawitch, R. J., Wales, P. A., Cuevas, C. A., Fernandez, R. P., Saiz-Lopez, A., Evans, M. J., Sherwen, T., Jacob, D. J., Schmidt, J., Kinnison, D., Lamarque, J.-F., Apel, E. C., Bresch, J. C., Campos, T., Flocke, F. M., Hall, S. R., Honomichl, S. B., Hornbrook, R., Jensen, J. B., Lueb, R., Montzka, D. D., Pan, L. L., Reeves, J. M., Schauffler, S. M., Ullmann, K., Weinheimer, A. J., Atlas, E. L., Donets, V., Navarro, M. A., Riemer, D., Blake, N. J., Chen, D., Huey, L. G., Tanner, D. J., Hanisco, T. F., and Wolfe, G. M.: BrO and inferred Bry profiles over the western Pacific: relevance of inorganic bromine sources and a Bry minimum in the aged tropical tropopause layer, Atmos. Chem. Phys., 17, 15245–15270, https://doi.org/10.5194/acp-17-15245-2017, 2017.
Le Breton, M., Bannan, T. J., Shallcross, D. E., Khan, M. A., Evans, M. J., Lee, J., Lidster, R., Andrews, S., Carpenter, L., Schmidt, J., Jacob, D., Harris, N. R. P., Bauguitte, S.-J., Gallagher, M., Bacak, A., Leather, K. E., and Percival, C. J.: Enhanced ozone loss by active inorganic bromine chemistry in the tropical troposphere, Atmos. Environ., 155, 21–28, 2017.
Lenschow, D. H., Paluch, I. R., Bandy, A. R., Pearson Jr., R., Kawa, S. R., Weaver, C. J., Huebert, B. J., Kay, J. G., Thornton, D. C., and Driedger III, A. R.: Dynamics and Chemistry of MarineStratocumulus (DYCOMS) experiment, Bull. Am. Meteorol. Soc., 69, 1058–1067, https://doi.org/10.1175/1520-0477(1988)069<1058:DACOMS>2.0.CO;2, 1988.
Leser, H., Hönninger, G., and Platt, U.: MAX-DOAS Measure-ments of BrO and NO2 in the Marine Boundary Layer, Geophys. Res. Lett., 30, 1537, https://doi.org/10.1029/2002GL015811, 2003.
Lewis, E. R. and Schwartz, S. E.: Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models – A Critical Review, AGU, 413 pp., Washington, D. C., 2004.
Long, M. S., Keene, W. C., Easter, R. C., Sander, R., Liu, X., Kerkweg, A., and Erickson, D.: Sensitivity of tropospheric chemical composition to halogen-radical chemistry using a fully coupled size-resolved multiphase chemistry-global climate system: halogen distributions, aerosol composition, and sensitivity of climate-relevant gases, Atmos. Chem. Phys., 14, 3397–3425, https://doi.org/10.5194/acp-14-3397-2014, 2014.
Mahajan, A. S., Plane, J. M. C., Oetjen, H., Mendes, L., Saunders, R. W., Saiz-Lopez, A., Jones, C. E., Carpenter, L. J., and McFiggans, G. B.: Measurement and modelling of tropospheric reactive halogen species over the tropical Atlantic Ocean, Atmos. Chem. Phys., 10, 4611–4624, https://doi.org/10.5194/acp-10-4611-2010, 2010.
Martin, M., Pöhler, D., Seitz, K., Sinreich, R., and Platt, U.: BrO measurements over the Eastern North-Atlantic, Atmos. Chem. Phys., 9, 9545–9554, https://doi.org/10.5194/acp-9-9545-2009, 2009.
McFiggans, G., Cox, R. A., Mossinger, J. C., Allan, B. J., and Plane, J. M. C.: Active chlorine release from marine aerosols: Roles for reactive iodine and nitrogen species, J. Geophys. Res.-Atmos., 107, ACH10-1–ACH10-13, https://doi.org/10.1029/2001jd000383, 2002.
Millet, D. B., Guenther, A., Siegel, D. A., Nelson, N. B., Singh, H. B., de Gouw, J. A., Warneke, C., Williams, J., Eerdekens, G., Sinha, V., Karl, T., Flocke, F., Apel, E., Riemer, D. D., Palmer, P. I., and Barkley, M.: Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys., 10, 3405–3425, https://doi.org/10.5194/acp-10-3405-2010, 2010.
Murray, L. T., Jacob, D. J., Logan, J. A., Hudman, R. C., and Koshak, W. J.: Optimized regional and interannual variability of lightning in a global chemical transport model constrained by LIS/OTD satellite data, J. Geophys. Res.-Atmos., 117, D20307, https://doi.org/10.1029/2012JD017934, 2012.
Newberg, J. T., Matthew, B. M., and Anastasio, C.: Chloride and bromide depletions in sea-salt particles over the northeastern Pacific Ocean, J. Geophys. Res., 110, D06209, https://doi.org/10.1029/2004JD005446, 2005.
Ordóñez, C., Lamarque, J.-F., Tilmes, S., Kinnison, D. E., Atlas, E. L., Blake, D. R., Sousa Santos, G., Brasseur, G., and Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 12, 1423–1447, https://doi.org/10.5194/acp-12-1423-2012, 2012.
Parrella, J. P., Jacob, D. J., Liang, Q., Zhang, Y., Mickley, L. J., Miller, B., Evans, M. J., Yang, X., Pyle, J. A., Theys, N., and Van Roozendael, M.: Tropospheric bromine chemistry: implications for present and pre-industrial ozone and mercury, Atmos. Chem. Phys., 12, 6723–6740, https://doi.org/10.5194/acp-12-6723-2012, 2012.
Read, K. A., Mahajan, A. S., Carpenter, L. J., Evans, M. J., Faria, B. V. E., Heard, D. E., Hopkins, J. R., Lee, J. D., Moller, S. J., Lewis, A. C., Mendes, L., McQuaid, J. B., Oetjen, H., Saiz-Lopez, A., Pilling, M. J., and Plane, J. M. C.: Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 453, 1232–1235, https://doi.org/10.1038/nature07035, 2008.
Roberts, T. J., Jourdain, L., Griffiths, P. T., and Pirre, M.: Re-evaluating the reactive uptake of HOBr in the troposphere with implications for the marine boundary layer and volcanic plumes, Atmos. Chem. Phys., 14, 11185–11199, https://doi.org/10.5194/acp- 14-11185-2014, 2014.
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chem- istry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/c2cs35208g, 2012.
Saiz-Lopez, A., Plane, J. M. C., and Shillito, J. A.: Bromine oxide in the mid-latitude marine boundary layer, Geophys. Res. Lett., 31, L03111, https://doi.org/10.1029/2003GL018956, 2004.
Saiz-Lopez, A., Shillito, J. A., Coe, H., and Plane, J. M. C.: Measurements and modelling of I2, IO, OIO, BrO and NO3 in the mid-latitude marine boundary layer, Atmos. Chem. Phys., 6, 1513–1528, https://doi.org/10.5194/acp-6-1513-2006, 2006.
Saiz-Lopez, A., Lamarque, J.-F., Kinnison, D. E., Tilmes, S., Ordóñez, C., Orlando, J. J., Conley, A. J., Plane, J. M. C., Mahajan, A. S., Sousa Santos, G., Atlas, E. L., Blake, D. R., Sander, S. P., Schauffler, S., Thompson, A. M., and Brasseur, G.: Estimating the climate significance of halogen-driven ozone loss in the tropical marine troposphere, Atmos. Chem. Phys., 12, 3939–3949, https://doi.org/10.5194/acp-12-3939-2012, 2012.
Sander, R., Keene, W. C., Pszenny, A. A. P., Arimoto, R., Ayers, G. P., Baboukas, E., Cainey, J. M., Crutzen, P. J., Duce, R. A., Hönninger, G., Huebert, B. J., Maenhaut, W., Mihalopoulos, N., Turekian, V. C., and Van Dingenen, R.: Inorganic bromine in the marine boundary layer: a critical review, Atmos. Chem. Phys., 3, 1301–1336, https://doi.org/10.5194/acp-3-1301-2003, 2003.
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Suleiman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res.-Atmos., 121, 11819–11835, https://doi.org/10.1002/2015JD024229, 2016.
Schmitt, C. G. and Heymsfield, A. J.: Total Surface Area Estimates for Individual Ice Particles and Particle Populations, J. Appl. Meteor., 44, 467–474, 2005.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and von Glasow, R.: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Theys, N., Van Roozendael, M., Hendrick, F., Yang, X., De Smedt, I., Richter, A., Begoin, M., Errera, Q., Johnston, P. V., Kreher, K., and De Mazière, M.: Global observations of tropospheric BrO columns using GOME-2 satellite data, Atmos. Chem. Phys., 11, 1791–1811, https://doi.org/10.5194/acp-11-1791-2011, 2011.
Toyota, K., Kanaya, Y., Takahashi, M., and Akimoto, H.: A box model study on photochemical interactions between VOCs and reactive halogen species in the marine boundary layer, Atmos. Chem. Phys., 4, 1961–1987, https://doi.org/10.5194/acp-4-1961-2004, 2004.
Turekian, V. C., Macko, S. A., and Keene, W. C.: Concentrations, isotopic compositions, and sources of size-resolved, particulate organic carbon and oxalate in near-surface marine air at Bermuda during spring, J. Geophys. Res., 108D, 4157, https://doi.org/10.1029/2002JD002053, 2003.
Vogt, R., Crutzen, P. J., and Sander, R.: A mechanism for halogen release from sea-salt aerosol in the remote marine boundary layer, Nature, 383, 327–330, 1996.
Volkamer, R., Baidar, S., Campos, T. L., Coburn, S., DiGangi, J. P., Dix, B., Eloranta, E. W., Koenig, T. K., Morley, B., Ortega, I., Pierce, B. R., Reeves, M., Sinreich, R., Wang, S., Zondlo, M. A., and Romashkin, P. A.: Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2−O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements, Atmos. Meas. Tech., 8, 2121–2148, https://doi.org/10.5194/amt-8-2121-2015, 2015.
Vong, R. J., Baker, B. M., Brechtel, F. J., Collier, R. T., Harris, J. M., Kowalski, A. S., McDonald, N. C., and Mcinnes, L. M. : Ionic and trace element composition of cloudwater collected on the Olympic peninsula of Washington State, Atmos. Environ., 31, 1991–2001, https://doi.org/10.1016/S1352-2310(96)00337-8, 1997.
von Glasow, R., Sander, R., Bott, A., and Crutzen, P. J.: Modeling halogen chemistry in the marine boundary layer. 1. Cloud-free MBL, J. Geophys. Res., 107D, 4341, https://doi.org/10.1029/2001JD000942, 2002a.
von Glasow, R., Sander, R., Bott, A., and Crutzen, P. J.: Modeling halogen chemistry in the marine boundary layer. 2. Interactions with sulfur and the cloud-covered MBL, J. Geophys. Res., 107D, 4323, https://doi.org/10.1029/2001JD000943, 2002b.
Wang, S.-Y., Schmidt, J., Baidar, S., Coburn, S., Dix, B., Koenig, T., Apel, E., Bowdalo, D., Campos, T., Eloranta, E., Evans, M., DiGangii, J., Zondlo, M., Gao, R.-S., Haggerty, J., Hall, S., Hornbrook, R., Jacob, D., Morley, B., Pierce, B., Reeves, M., Romashkin, P., ter Schure, A., and Volkamer, R.: Active and widespread halogen chemistry in the tropical and subtropical free troposphere, P. Natl. Acad. Sci. USA, 112, 9281–9286, https://doi.org/10.1073/pnas.1505142112, 2015.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Watanabe, K., Ishizaka, I., and Takenaka, C. : Chemical characteristics of cloud water overthe Japan Sea and the northwestern Pacific Ocean near the central partof Japan: Airborne measurements, Atmos. Environ., 35, 645–655, https://doi.org/10.1016/S1352-2310(00)00358-7, 2001.
Wofsy, S. C., Apel, E., Blake, D. R., Brock, C. A., Brune, W. H., Bui, T. P., Daube, B. C., Dibb, J. E., Diskin, G. S., Elki- ins, J. W., Froyd, K., Hall, S. R., Hanisco, T. F., Huey, L. G., Jimenez, J. L., McKain, K., Montzka, S. A., Ryerson, T. B., Schwarz, J. P., Stephens, B. B., Weinzierl, B., and Wennberg, P.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, ORNL DAAC, Oak Ridge, Tennessee, USA, https://doi.org/10.3334/ORNLDAAC/1581, 2018.
Yang, X., Cox, R., Warwick, N., Pyle, J., Carver, G., O'Connor, F., and Savage, N.: Tropospheric bromine chemistry and its impacts on ozone: A model study, J. Geophys. Res., 110, D23311, https://doi.org/10.1029/2005JD006244, 2005.
Zhu, L.: GEOS-Chem chemistry mechanism and monthly bromine simulation output, Harvard Dataverse, V2, https://doi.org/10.7910/DVN/BADJDE, 2019.