the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Jan 2019
| 03 Jan 2019
Aircraft-based measurements of High Arctic springtime aerosol show evidence for vertically varying sources, transport and composition
Megan D. Willis
Heiko Bozem
Daniel Kunkel
Alex K. Y. Lee
Hannes Schulz
Julia Burkart
Amir A. Aliabadi
Andreas B. Herber
W. Richard Leaitch
Jonathan P. D. Abbatt
The sources, chemical transformations and removal mechanisms of aerosol transported to the Arctic are key factors that control Arctic aerosol–climate interactions. Our understanding of sources and processes is limited by a lack of vertically resolved observations in remote Arctic regions. We present vertically resolved observations of trace gases and aerosol composition in High Arctic springtime, made largely north of 80∘ N, during the NETCARE campaign. Trace gas gradients observed on these flights defined the polar dome as north of 66–68∘ 30′ N and below potential temperatures of 283.5–287.5 K. In the polar dome, we observe evidence for vertically varying source regions and chemical processing. These vertical changes in sources and chemistry lead to systematic variation in aerosol composition as a function of potential temperature. We show evidence for sources of aerosol with higher organic aerosol (OA), ammonium and refractory black carbon (rBC) content in the upper polar dome. Based on FLEXPART-ECMWF calculations, air masses sampled at all levels inside the polar dome (i.e., potential temperature <280.5 K, altitude km) subsided during transport over transport times of at least 10 days. Air masses at the lowest potential temperatures, in the lower polar dome, had spent long periods (>10 days) in the Arctic, while air masses in the upper polar dome had entered the Arctic more recently. Variations in aerosol composition were closely related to transport history. In the lower polar dome, the measured sub-micron aerosol mass was dominated by sulfate (mean 74 %), with lower contributions from rBC (1 %), ammonium (4 %) and OA (20 %). At higher altitudes and higher potential temperatures, OA, ammonium and rBC contributed 42 %, 8 % and 2 % of aerosol mass, respectively. A qualitative indication for the presence of sea salt showed that sodium chloride contributed to sub-micron aerosol in the lower polar dome, but was not detectable in the upper polar dome. Our observations highlight the differences in Arctic aerosol chemistry observed at surface-based sites and the aerosol transported throughout the depth of the Arctic troposphere in spring.
- Article
(5457 KB) - Full-text XML
-
Supplement
(4334 KB) - BibTeX
- EndNote





Arctic regions are warming faster than the global average, with significant impacts on local ecosystems and local people (e.g, Bindoff et al., 2013; Hinzman et al., 2013). While Arctic warming is driven largely by increasing concentrations of anthropogenic greenhouse gases and local feedback mechanisms, short-lived climate forcing agents also impact Arctic climate. In particular, short-lived species such as aerosol, tropospheric ozone and methane are important climate forcers (e.g., Law and Stohl, 2007; Quinn et al., 2008). The impact of pollution aerosol, transported northward over long distances, on Arctic climate has been significant. For example, a large fraction of greenhouse-gas-induced warming (∼60 %) has been offset by anthropogenic aerosol over the past century, such that reductions in sulfur emissions in Europe since 1980 can explain a large amount of Arctic warming since that time (∼0.5 K) (Fyfe et al., 2013; Najafi et al., 2015; Navarro et al., 2016). These estimates are compelling, and at the same time global models that form the basis of our predictive capability often struggle to reproduce key characteristics of Arctic aerosol, such as the seasonal cycle and vertical distribution (Shindell et al., 2008; Emmons et al., 2015; Monks et al., 2015; Eckhardt et al., 2015; Arnold et al., 2016). Our incomplete understanding of Arctic aerosol processes results in diverse and frequently poor model skill in simulating Arctic aerosol both at the surface and through the troposphere, and therefore also in accurately simulating aerosol–climate interactions (Arnold et al., 2016). This challenge arises in part due to a lack of vertically resolved observations in Arctic regions.
Particle composition drives aerosol optical properties (e.g., Boucher and Anderson, 1995; Jacobson, 2001; Wang et al., 2008), ice nucleation efficiency (e.g., Abbatt et al., 2006; Hoose and Möhler, 2012), and heterogeneous chemistry that impacts both gas and particle composition (e.g., Fan and Jacob, 1992; Mao et al., 2010; Abbatt et al., 2012). The vertical distribution of aerosol and its chemical and physical properties can impact Arctic regional climate in a number of ways. First, absorption of incoming solar radiation by aerosol (e.g., black carbon) can lead to warming in the lower troposphere when present near the surface. In contrast, absorbing aerosol present at higher altitudes causes cooling at the surface and impacts atmospheric stratification (Rinke et al., 2004; Ritter et al., 2005; Treffeisen et al., 2005; Shindell and Faluvegi, 2009; Engvall et al., 2009). Further, the location in the troposphere impacts deposition to high albedo surfaces, depending on the mechanism of removal (e.g., Macdonald et al., 2017). Absorbing aerosol deposited to the surface has a strong impact on the albedo of ice and snow, efficiently leading to warming (Clarke and Noone, 1985; Hansen and Nazarenko, 2004; Flanner et al., 2009; Flanner, 2013). Second, neutralization of acidic sulfate impacts aerosol water content and aerosol phase, with implications for the magnitude of aerosol–radiation interactions (Boucher and Anderson, 1995; Wang et al., 2008). Third, sulfuric acid coatings on particles can decrease their ability to act as ice-nucleating particles (INPs), leading to larger, more readily precipitating ice crystals (Blanchet and Girard, 1994, 1995). This process can lead to enhanced atmospheric dehydration, leading to diminished long-wave forcing (Curry and Herman, 1985; Blanchet and Girard, 1994, 1995). Further, particles containing mineral dust, organic species, sea salt or neutralized sulfate can act as ice nuclei and increase ice crystal number, also leading to impacts on long-wave and short-wave cloud forcing (Sassen et al., 2003; Abbatt et al., 2006; Baustian et al., 2010; Wagner et al., 2018). Finally, Arctic pollution aerosol can impact the micro-physical properties of liquid-containing clouds, by increasing liquid water path and decreasing droplet radius. Such micro-physical changes can result in enhanced long-wave warming effects during winter and spring (Garrett and Zhao, 2006; Lubin and Vogelmann, 2006; Zhao and Garrett, 2015).
Observations at Arctic ground-based monitoring stations form the basis of our current knowledge about Arctic aerosol seasonality, chemical composition and sources. These long-term observations have demonstrated a pronounced seasonal cycle in Arctic aerosol mass concentrations, particle size distribution and composition, driven by seasonal variations in northward long-range transport and aerosol wet removal (e.g., Quinn et al., 2007; Sharma et al., 2013; Tunved et al., 2013; Croft et al., 2016; Asmi et al., 2016; Nguyen et al., 2016; Freud et al., 2017; Leaitch et al., 2018). Aerosol mass concentrations peak in winter to early spring when long-range-transported accumulation mode particles (200–400 nm mode diameter), referred to broadly as “Arctic haze”, dominate the particle size distribution (e.g., Croft et al., 2016; Freud et al., 2017). A mixture of natural and anthropogenic aerosol is transported to Arctic regions by near-isentropic transport along surfaces of constant potential temperature that slope upwards toward the Arctic (Klonecki et al., 2003; Stohl, 2006). The sloping isentropic surfaces form a closed “dome” over the polar region; this polar dome is zonally asymmetric, extends further south in winter and contracts northward in spring to summer (Shaw, 1995; Law and Stohl, 2007; Stohl, 2006; Law et al., 2014).
Arctic haze observed near the surface is largely acidic sulfate, with fewer contributions from organic aerosol (OA), dust, nitrate, ammonium and sea salt (Li and Winchester, 1989; Quinn et al., 2002, 2007; Shaw et al., 2010; Leaitch et al., 2018). Aerosol acidity increases during winter and reaches a peak in late spring (Sirois and Barrie, 1999; Toom-Sauntry and Barrie, 2002), before the return of wet removal brings the Arctic toward near-pristine conditions with more neutralized aerosol (Engvall et al., 2008; Browse et al., 2012; Breider et al., 2014; Wentworth et al., 2016; Croft et al., 2016). Sea salt is thought to be an important contributor to Arctic haze in winter to early spring owing to stronger wind speeds over nearby oceans, potential wind-driven sources in ice and snow-covered regions, and open leads (Leck et al., 2002; Shaw et al., 2010; May et al., 2016; Huang and Jaeglé, 2017; Kirpes et al., 2018). The major source region of near-surface Arctic haze in winter and early spring is northern Europe and northern Asia/Siberia, but the magnitudes of sources in this region have been decreasing in recent decades (Barrie and Hoff, 1984; Sharma et al., 2004; Koch and Hansen, 2005; Sharma et al., 2006; Hirdman et al., 2010; Huang et al., 2010b; Gong et al., 2010; Bourgeois and Bey, 2011; Stohl et al., 2013; Sharma et al., 2013; Monks et al., 2015; Qi et al., 2017). Surface-based observations have provided substantial insight into Arctic aerosol processes, but owing to the stability of the troposphere the surface can be decoupled from the atmosphere above. Therefore, surface-based observations may not represent the overall composition of aerosol transported to the Arctic troposphere (e.g., Stohl, 2006; McNaughton et al., 2011). How transported aerosol present throughout the troposphere is related to Arctic haze observed near the surface remains an unresolved question (Law et al., 2014; Arnold et al., 2016).
Vertically resolved observations of the Arctic atmosphere, in the last 20 years, have furthered our understanding of the properties, processes, and impacts of Arctic aerosol. Some of the only seasonal airborne observations of aerosol sulfate suggested that the aerosol seasonal cycle may differ aloft compared to near the surface (Klonecki et al., 2003; Scheuer et al., 2003). Clean-out may begin to take place near the surface in late April to May, before significant changes occur aloft. Intensive observations were made during the International Polar Year (IPY) in 2007–2008. During IPY, high concentrations of aerosol and trace gases from biomass and fossil fuel burning were observed in discrete layers that did not appear related to Arctic haze observed near the surface (e.g., Warneke et al., 2009; Schmale et al., 2011; Brock et al., 2011; Law et al., 2014). Also during IPY, aerosol ammonium content increased from near the surface toward the middle to upper troposphere (Fisher et al., 2011). The largest fraction of sulfate was observed in the lower ∼2 km, in general agreement with long-term monitoring observations. In years with high burned area in the Northern Hemisphere, such as 2008, biomass burning sources contribute a significant fraction of black carbon and organic aerosol in the Arctic troposphere (Warneke et al., 2009; Hecobian et al., 2011; McNaughton et al., 2011; Bian et al., 2013). In years with moderate burned area consistent with decadal mean conditions, anthropogenic sources can still lead to enhanced absorbing aerosol in the Arctic mid-troposphere (Liu et al., 2015). IPY observations in the Alaskan Arctic demonstrated that background pollution aerosol (i.e., in air masses with CO<170 ppbv) and aerosol in the near-surface layer (i.e., in air masses with depleted O3) contained a larger fraction of sulfate compared to aerosol attributed to biomass or fossil fuel burning (Brock et al., 2011). The properties of Arctic background air masses were generally consistent with median observations at a nearby ground station, Utqiaġvik (Barrow), Alaska (Brock et al., 2011). This background aerosol often has diffuse source regions that are difficult to diagnose precisely using 10-day backward trajectories (e.g, Brock et al., 2011; Qi et al., 2017; Leaitch et al., 2018).
Our knowledge of the vertical distribution of Arctic aerosol source regions has also been extended by recent airborne observations. Results from modelling efforts generally agree that Arctic pollution aerosol is a result of a combination of anthropogenic and natural sources from mid-latitudes in the Northern Hemisphere; particularly a combination of European, north and south Asian, and North American source regions (e.g., Law et al., 2014; Arnold et al., 2016). However, modelling efforts provide less quantitative agreement on the magnitude of the contributions of each region near the surface and as a function of altitude. Our emerging understanding is of northern Eurasian sources dominating near the surface in winter, while North America and southern and eastern Asia can be important in the middle to upper troposphere (e.g., Koch and Hansen, 2005; Shindell et al., 2008; Huang et al., 2010a; Law et al., 2014; Liu et al., 2015; Arnold et al., 2016; Qi et al., 2017). In spring, as the polar dome recedes northward, North American and Asian sources become more important at all altitudes (Koch and Hansen, 2005; Fisher et al., 2011; Xu et al., 2017). Overall, more southerly source regions become more important at higher altitudes (e.g., Stohl, 2006; Fisher et al., 2011; Harrigan et al., 2011), and the importance of Asian sources above the Arctic surface is being increasingly recognized (e.g., Koch and Hansen, 2005; Fisher et al., 2011; Xu et al., 2017). The magnitude of Asian influence on the lower troposphere inferred from models in spring varies significantly and depends on emissions estimates and assumptions about aerosol removal efficiency during transport (e.g., Matsui et al., 2011a, b). Source apportionment of recent vertically resolved Arctic black carbon observations demonstrated that eastern and southern Asia make important contributions throughout the troposphere in spring, with a more significant contribution at higher altitudes (Xu et al., 2017). Northern Asia was a more important source region near the surface (Xu et al., 2017). Changes in source strengths at mid-latitudes and within the Arctic strongly impact the dominant source regions for different aerosol species (Arnold et al., 2016).
Previous vertically resolved observations of Arctic pollution aerosol frequently focused on episodic events of high pollutant concentrations, largely owing to their potential radiative impact (e.g., Rahn et al., 1977; Engvall et al., 2009; Warneke et al., 2009; Law et al., 2014). We know less about the vertical distribution of Arctic aerosol properties within the High Arctic polar dome and under conditions consistent with Arctic background conditions (e.g., CO<170 ppbv; Brock et al., 2011). Improved understanding of different anthropogenic and natural contributions to Arctic aerosol will provide a scientific basis for sustainable climate mitigation and adaptation strategies. Within the framework of the NETCARE project, airborne observations of Arctic haze aerosol were made across the North American and European Arctic in April 2015. Observations of trace gas gradients during this campaign were used by Bozem et al. (2018) to define the boundaries of the polar dome. The location of the maximum trace gas gradient defined the polar dome as north of 66–68∘ 30′ N and below potential temperatures of 283.5–287.5 K. Based on Bozem et al. (2018) we use a conservative definition of the polar dome area based on the interquartile range of the location of maximum trace gas gradient: north of 69∘ 30′ N and below 280.5 K. In this work, we quantify vertical changes in sub-micron aerosol composition in the Canadian High Arctic within the boundaries of the polar dome and in the absence of episodic transport events of high pollutant concentrations. Using the Lagrangian particle dispersion model FLEXPART, we explore the source regions that drive observed sub-micron aerosol in the springtime polar dome. Finally, we examine the depth over which aerosol consistent with surface monitoring observations extends vertically in the polar dome, and assess the representativeness of ground-based observations for aerosol transported to the polar dome in spring.
2.1 High Arctic measurements
2.1.1 Measurement platform and inlets
Measurements of aerosol, trace gases and meteorological parameters were made in High Arctic spring aboard the Alfred Wegener Institute (AWI) Polar 6 aircraft, an unpressurized DC-3 aircraft converted to a Basler BT-67 (Herber et al., 2008), as part of the Network on Climate and Aerosols: Addressing Key Uncertainties in Remote Canadian Environments project (NETCARE, http://www.netcare-project.ca, last access: 20 December 2018), and in partnership with the Polar Airborne Measurements and Arctic Regional Climate Model Simulation Project (PAMARCMiP; Herber et al., 2012). Measurements on a total of 10 flights took place from 4 to 22 April 2015, based at four stations along the PAMARCMiP track: Longyearbyen, Svalbard (78.2 ∘ N, 15.6 ∘ E); Alert, Nunavut, Canada (82.5 ∘ N, 62.3 ∘ W); Eureka, Nunavut, Canada (80.0 ∘ N, 85.9 ∘ W); and Inuvik, Northwest Territories, Canada (68.4 ∘ N, 133.7 ∘ W). To focus our analysis on aerosol within the polar dome, a subset of six flights in the High Arctic during 7–13 April 2015 are considered in this analysis (Fig. 1). The vertical extent of these flights is shown in Fig. S1 in the Supplement. During measurement flights aircraft speed was maintained at ∼75 m s−1 (∼270 km h−1), with ascent and descent rates of ∼150 m min−1.
Aerosol and trace gas inlets were identical to those used aboard Polar 6 during the NETCARE 2014 summer campaign and are described in Leaitch et al. (2016) and Willis et al. (2016). Briefly, aerosol was sampled approximately isokinetically through a stainless steel shrouded diffuser inlet, with near-unity transmission of particles 20 nm to ∼1 µm in diameter at typical survey airspeeds and a total flow rate of approximately 55 L min−1. Bypass lines off the main inlet, at angles of 45∘, carried aerosol to various instruments. Performance of the aerosol inlet used here was characterized by Leaitch et al. (2016). Aerosol was not actively dried prior to sampling; however, the temperature in the inlet line within the aircraft cabin was at least 15 ∘C warmer than the ambient temperature so that the relative humidity (RH) decreased significantly.
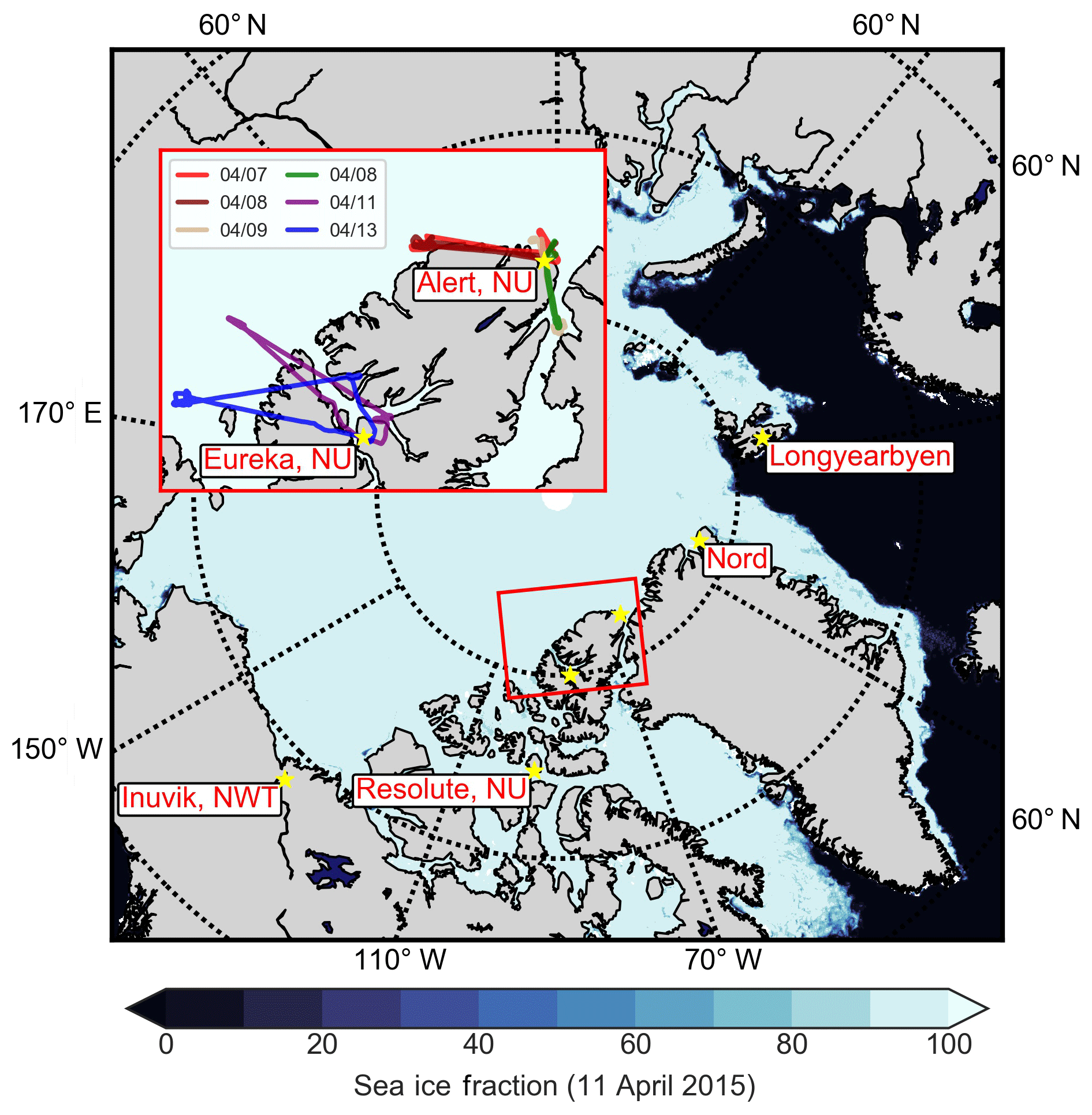
Figure 1Map of the NETCARE 2015 campaign study area, showing sea ice concentrations on 11 April 2015 (Spreen et al., 2008). All stations along the NETCARE/PAMARCMiP 2015 track are shown with yellow stars (Longyearbyen, Svalbard; Alert, Nunavut; Eureka, Nunavut; Resolute Bay, Nunavut; and Inuvik, Northwest Territories). Parallels are shown in dashed circles at 60, 70 and 80∘ N. Inset: flight tracks from six flights during 7–13 April 2015 based in Alert and Eureka, Nunavut, which are the focus of this work.
2.1.2 State parameters
State parameters and meteorological conditions were measured with an AIMMS-20, manufactured by Aventech Research Inc. (Barrie, ON, Canada; https://aventech.com/products/aimms20.html, last access: 20 December 2018). The AIMMS-20 consists of three modules: (1) an Air Data Probe, which measures temperature and the three-dimensional aircraft-relative flow vector (total air speed TAS, angle of attack, and side slip) with a three-dimensional accelerometer for measurement of turbulence; (2) an Inertial Measurement Unit, which provides the aircraft angular rate and acceleration; and (3) a Global Positioning System for aircraft three-dimensional position and inertial velocity. Vertical and horizontal wind speeds are measured with accuracies of 0.75 and 0.50 m s−1 respectively. Accuracy and precision of the temperature measurement are 0.30 and 0.10 ∘C respectively. Potential temperature was calculated using temperature and pressure measured by the AIMMS-20.
2.1.3 Trace gases
Carbon monoxide. CO concentrations were measured at 1 Hz with an Aerolaser ultra-fast carbon monoxide monitor (model AL 5002), based on VUV fluorimetry using excitation of CO at 150 nm. The instrument was modified such that in situ calibrations could be conducted in flight. Measured concentrations were significantly higher than the instrument detection limit. The measurement precision is ±1.5 ppbv, with an instrument stability based on in-flight calibrations of 1.7 %.
Water vapour and carbon dioxide. H2O and CO2 measurements were made at 1 Hz using non-dispersive infrared absorption with a LI-7200 enclosed CO2∕H2O analyzer from LI-COR Biosciences. In situ calibrations were performed during flight at regular intervals (15–30 min) using a NIST traceable CO2 standard with zero water vapour concentration. Measured concentrations were significantly higher than the instrument detection limit. The measurement precision for CO2 is ±0.05 ppmv, with an instrument stability based on in-flight calibrations of 0.5 %. The measurement precision for H2O is ±18.5 ppmv, with an instrument stability based on in-flight calibrations of 2.5 %.
Ozone. O3 concentrations were measured, with a time resolution of 10 s, using UV absorption at 254 nm with a Thermo Scientific ozone analyzer (model 49i). The measurement uncertainty is ±0.2 ppbv.
2.1.4 Particle concentrations
Aerosol number size distributions from 100 nm to 1 µ m were acquired with two instruments: (1) a Droplet Measurement Technology (DMT) Ultra-High Sensitivity Aerosol Spectrometer (UHSAS) with a flow rate of 55 cm3 min−1 from a bypass flow off the main aerosol inlet, and (2) a GRIMM sky optical particle counter (Sky-OPC, model 1.129) with a flow rate of 1200 cm3 min−1 from a bypass flow off the main aerosol inlet (Cai et al., 2008). In their overlapping size range, comparison of UHSAS and OPC particle number concentrations suggested that the UHSAS underestimated the concentration of larger particles (>500 nm). This comparison is presented in Fig. S2 and discussed further in Supplement Sect. S1. We therefore present these observations as the number of particles between 100 and 500 nm (N100–500) derived from UHSAS observations and the number greater than 500 nm (N>500) from the OPC. Recent work has highlighted the impact of rapid pressure changes, during aircraft ascent and descent, on reported UHSAS particle concentrations (Brock et al., 2011; Kupc et al., 2018). However, comparison between particle measurements during NETCARE 2015 suggests that these effects are not significant, likely owing to the relatively slow vertical speed of the Polar 6 (Schulz et al., 2018). Owing to these instrumental discrepancies present at low particle number concentrations, we emphasize that absolute particle number concentrations should be treated with caution.
2.1.5 Particle composition
Refractory black carbon. Concentrations of particles containing refractory black carbon (rBC) were measured with a DMT single-particle soot photometer (SP2) (Schwarz et al., 2006; Gao et al., 2007). The SP2 uses a continuous intra-cavity Nd:YAG laser (1064 nm) to classify particles as either incandescent (rBC) or scattering (non-rBC), based on the individual particle's interaction with the laser beam. The peak incandescence signal is linearly related to the rBC mass. The SP2 was calibrated with Fullerene Soot (Alfa Aesar) standard by selecting a narrow size distribution of particles with a differential mobility analyzer upstream of the SP2 (Laborde et al., 2012). The SP2 efficiently detected particles with rBC mass of 0.6 to 328.8 fg, which corresponds to 85–704 nm mass equivalent diameter (assuming a void free bulk material density of 1.8 g cm−3). rBC mass concentrations were not corrected for particles outside the instrument size range, and the measurement uncertainty is ±15 % (Laborde et al., 2012). Measurements of rBC during NETCARE 2015 are discussed in detail by Schulz et al. (2018).
Non-refractory aerosol composition. Non-refractory aerosol composition was measured with an Aerodyne time-of-flight aerosol mass spectrometer (ToF-MS) (DeCarlo et al., 2006). Operation of the ToF-AMS aboard Polar 6 and characterization of the pressure-controlled inlet system is described in Willis et al. (2016, 2017). The ToF-AMS deployed here was equipped with an infrared laser vaporization module similar to that of the DMT SP2 (SP laser) (Onasch et al., 2012); however, rBC concentrations during the flights discussed here were generally below ToF-AMS detection limits (∼0.1 µg m−3 for rBC) so SP2 measurements of rBC are used in this work. The instrument was operated up to an altitude of ∼3.5 km, and the temperature of the ToF-AMS was passively maintained using a modular foil-lined insulating cover. The ToF-AMS was operated in “V-mode” with a mass range of m∕z 3–290, alternating between ensemble mass spectrum (MS) mode for 10 s (two cycles of 5 s MS open and 5 s MS closed) with the SP laser on, MS mode with the SP laser off for 10 s, and efficient particle time-of-flight (epToF) mode with the SP laser on for 10 s (Supplement Table S1) (DeCarlo et al., 2006; Onasch et al., 2012). Single-particle observations were made on two flights; this ToF-AMS operation mode is described below. Only observations made with the SP laser off are used to quantify non-refractory aerosol composition. Filtered ambient air was sampled with the ToF-AMS at least 3 times per flight, for a duration of at least 5 min, to account for contributions from air signals.
Species comprising non-refractory particulate matter are quantified by the ToF-AMS, including sulfate (SO4), nitrate (NO3), ammonium (NH4), and the sum of organic species (OA). The ToF-AMS is also capable of detecting sea salt (Ovadnevaite et al., 2012). The detection efficiency of sea-salt-containing particles is dependent on not only the ambient RH but also the temperature of the tungsten vaporizer (Ovadnevaite et al., 2012). A quantitative estimate of sea salt mass is not possible with these measurements and this species is not included in the calculation of aerosol chemical mass fractions, such that the mass fractions presented represent non-refractory aerosol species and rBC measured by the SP2. The vaporizer temperature was calibrated with sodium nitrate particles and was operated at a temperature of ∼650 ∘C. ToF-AMS signals for sea salt, in particular NaCl+ (m∕z 57.96), can be used to quantify sea salt (Ovadnevaite et al., 2012); however, here we use the NaCl+ signal only as a qualitative indication for the presence of sea salt owing to uncertainties in sea salt collection efficiency as a function of RH and the lack of RH measurement in the sampling line. Ammonium nitrate calibrations (Jimenez et al., 2003) were carried out twice during the campaign as well as before and after, owing to restricted access to calibration instruments during the campaign. Air-beam corrections were referenced to the appropriate calibration in order to account for differences in instrument sensitivity between flights. The relative ionization efficiencies for sulfate and ammonium (RIE and RIE) were 0.9±0.1 and 3.4±0.3. The default relative ionization efficiency for organic species (i.e., RIEOrg=1.4) was used, which is appropriate for oxygenated organic aerosol (Jimenez et al., 2003, 2016). Elemental composition was calculated using the method presented in Canagaratna et al. (2015). Data were analyzed using the Igor Pro-based analysis tool PIKA (v.1.16H) and SQUIRREL (v.1.57l) (DeCarlo et al., 2006; Sueper, 2010). Detection limits and propagated uncertainties (i.e., ±(detection limit + total uncertainty)) for sulfate, nitrate, ammonium, and organics at a 10 s time resolution were ±(0.009 µg m−3 + 35 %), ±(0.001 µg m−3 + 33 %), ±(0.003 µg m−3 + 33 %), and ±(0.08 µg m−3 + 37 %), respectively. We note that ion ratios commonly reported from ToF-AMS measurements of ammonium and sulfate are not appropriate for estimating aerosol neutralization (Hennigan et al., 2015), so we do not report these here. A composition-dependent collection efficiency (CDCE) was applied to correct ToF-AMS mass loadings for non-unity particle detection due to particle bounce on the tungsten vaporizer (Middlebrook et al., 2012), which resulted in a median (quartile range) collection efficiency correction of 18 % (12 %–28 %) applied uniformly to non-refractory aerosol species.
ToF-AMS total non-refractory aerosol mass correlated well with estimated aerosol mass from the UHSAS and OPC, but was generally higher by approximately a factor of 2 (Fig. S4, assuming a mean density of 1.5 g cm−3). An important exception to this observation occurred when the ToF-AMS measured significant NaCl+; at these times, the ToF-AMS total aerosol mass was relatively constant while the estimated mass increased, indicating that sea salt was an important contributor to aerosol mass. These discrepancies are discussed further in Sect. S1 of the Supplement. Owing to the discrepancies between measured and estimated particle mass, we emphasize that absolute mass concentrations presented in this work should be treated with caution; however, these discrepancies do not prevent a useful interpretation of the ToF-AMS data based upon relative changes in particle composition.
ToF-AMS single-particle measurements. The ToF-AMS was operated in Event Trigger Single Particle (ETSP) mode on two flights (Table S1). ETSP is run in the single-slit particle-time-of-flight (pToF) mode. A particle event is defined as a single mass spectrum (MS) extraction or set of consecutive MS extractions associated with a single particle being vaporized and producing MS signals. The number of MS extractions obtained during a particle event is determined by the pulser frequency, and thus the mass range, set during acquisition; in this case 30.9 kHz, corresponding to a pulser period of 32.4 µs (m∕z 3–290). Under these conditions, at least a single mass spectrum is collected per particle event. Saving mass spectra associated with a particle event is triggered in real time based on the signals present in up to three continuous ranges of mass-to-charge ratios, called regions of interest (ROIs). Three ROIs were used in this work such that a signal above a specified ion threshold in any ROI would trigger saving a mass spectrum (Table S2). Ion thresholds were purposely set low to collect a large number of false positives that are subsequently removed based on the relationship between total aerosol ion signal (i.e., excluding air) and particle size (Fig. S5), similar to the approach described in Lee et al. (2015). Two background regions in the particle size distribution (10–50 and 2000–4000 nm) were selected to determine the average background ion signal excluding air peaks, and particle events considered “real” must be between 80 and 1000 nm with ion signals above the mean background plus 3 times its standard deviation (Fig. S5). A simplified fragmentation table, described in Lee et al. (2015), was applied to particle mass spectra identified as “real” and fragmentation corrections were based on higher mass resolution ensemble MS spectra collected concurrently. A total of 1677 “real” particle spectra were collected over two flights (8 and 13 April 2015). A k-means cluster analysis was applied to particle spectra to explore different particle mixing states, following Lee et al. (2015). A two-cluster solution was selected to describe the 1677 total “real” particle spectra. Owing to the small number of particle spectra and the lack of specificity in organic aerosol peaks from highly oxygenated aerosol, increasing the number of clusters did not yield physically meaningful information. Mean mass spectra and mass spectral histograms for each particle class are shown in Fig. S6. ETSP data were analyzed using the Igor Pro-based analysis tools Tofware version 2.5.3.b (developed by TOFWERK and Aerodyne Research, Inc.), clustering input preparation panel (CIPP) version ETv2.1b and cluster analysis panel (CAP) version ETv2.1 (developed by Alex K. Y. Lee and Megan D. Willis).
2.2 Air mass history from particle dispersion modelling
The Lagrangian particle dispersion model FLEXible PARTicle (FLEXPART) (Brioude et al., 2013) driven by meteorological analysis data from the European Centre for Medium-Range Weather Forecasts (ECMWF) was used to study the history of air masses prior to sampling during NETCARE flights. The ECMWF data had a horizontal grid spacing of 0.25∘ and 137 vertical levels. Here, we use FLEXPART-ECMWF run in backward mode to study the origin of air influencing aircraft-based aerosol and trace gas measurements. Individual FLEXPART parcels were initialized along the flight track every 3 min and then traced back in time for 10 days, providing time-resolved information on source regions of trace species measured along the flight track. FLEXPART-ECMWF output was provided every 3 h over the 10-day period, with horizontal grid spacing of 0.25∘ and 10 vertical levels (50, 100, 200, 500, 1000, 2000, 4000, 6000, 8000 and 10 000 m). In backward mode, the model provides an emission sensitivity function called the potential emission sensitivity (PES). The PES in a particular grid cell, or air volume, is the response function of a source–receptor relationship, and is proportional to the particle residence time in that grid cell (e.g., Hirdman et al., 2010). PES values can be combined with emission distributions to calculate receptor concentrations, assuming the species is inert; however, we use the PES directly and show maps of PES with units of seconds (i.e., proportional to air mass residence time). Absolute residence times depend on the model output time step and the extent of spatial averaging. Maps of PES represent integration of model output over a period of time prior to sampling (i.e., 10 days), also referred to as the “time before measurement”, and over a vertical range. We show maps of both the total column PES (i.e., 0–20 km) and partial column PES (i.e., 0–200 m), as emissions near the surface are of particular interest.
By integrating model output at each model release over specific pressure levels and/or latitude ranges we used FLEXPART-ECMWF to calculate the residence time of air in the middle-to-lower polar dome. The horizontal extent of the polar dome was defined based on Bozem et al. (2018) as north of 69∘ 30′ N. The vertical extent of the middle-to-lower polar dome was defined based on trace gas profiles as below 265 K (∼1550 m). Calculation of this quantity is analogous to calculating the PES (i.e., by integrating in time and space), with constraints on altitude and location. This residence time is reported as a relative residence time over the 10-day FLEXPART-ECMWF backward integration time. Aircraft observations were sub-sampled to the model time resolution by taking a 1 min average of measurements around the model release time, when the aircraft altitude was within ±100 m of the model release altitude.

Figure 2Mean potential temperature profiles of trace gases (CO, CO2, O3 and H2O) and particle concentrations (N100–500 and N>500) in the polar dome observed during 7–13 April 2015. Coloured lines indicate the mean profile for each flight, the black line represents the mean profile over all flights, and gray shading shows the range of observations in each potential temperature bin. Horizontal dashed blue lines separate the lower, middle and upper polar dome defined as 245–252, 252–265 and 265–280 K.
3.1 Transport regimes in the polar dome
We focus on observations made on six flights in the High Arctic during NETCARE 2015 over the period 7–13 April 2015. Figure 1 illustrates flight tracks during this period on a map of the sea ice concentration from 11 April 2015. Observations of trace gas gradients during this campaign defined the region inside the polar dome as north of 69∘ 30′ N and below 280.5 K (∼3.5 km) (Bozem et al., 2018). Zonal mean potential temperature cross sections from ECMWF for the period 7–13 April 2015 generally agree with this definition of the polar dome, and this demonstrates that our observations were made in the coldest air masses present in the Arctic region during this time (Fig. S7). CO concentrations observed in the polar dome were consistent with “Arctic background” air masses identified in previous airborne observations and with monthly mean CO concentrations at Alert, Nunavut, Canada (Fig. S8). This suggests that our observations during April 2015 in the polar dome were not strongly impacted by episodic transport events of high pollutant concentrations (Brock et al., 2011). We restrict our analysis to those air masses residing in the polar dome, to determine the sources and processes contributing to aerosol composition within this region during spring. When discussing observations and model predictions, we use potential temperature instead of height or pressure for two reasons. First, the location of the polar dome and transport northward are dictated by potential temperature rather than absolute height. Second, trace gases and aerosol observed in the polar dome varied systematically with potential temperature, but showed less systematic variability with pressure (Fig. S9). Altitude profiles of absolute and potential temperature are shown in Fig. S10. In this section, we discuss transport patterns inferred from trace gas observations and FLEXPART-ECMWF air mass history, and in Sect. 3.2 we discuss observed aerosol composition in the context of these transport patterns.

Figure 3FLEXPART-ECMWF potential emission sensitivity (PES) and plume centroid altitude averaged over three potential temperature ranges in the polar dome. (a–c) Mean total column PES, (d–f) mean partial column (<200 m) PES, (g–i) mean plume centroid altitudes for 245–252 K (a, d, g), 252–265 K (b, e, h) and 265–280 K (c, f, i). Fire locations during 28 March to 13 April 2015 from MODIS are purple points, gas flaring locations associated with oil and gas extraction from the ECLIPSE emission inventory (V5) for 2015 are light blue points. Parallels are shown in dashed circles at 45, 60 and 80∘ N.
Trends in trace gas concentrations with potential temperature illustrate different transport regimes within the polar dome (Fig. 2). Based on the mean vertical profiles of trace gases, we divided observed vertical profiles into three ranges of potential temperature (Fig. 2: 245–252, 252–265 and 265–280 K) to guide interpretation of air mass history, transport characteristics and aerosol composition in the polar dome. We refer to these three ranges of potential temperature as the lower, middle and upper polar dome, respectively (dashed horizontal lines in Fig. 2), and discuss the characteristics of each region in turn. First, in the coldest and driest air masses (245–252 K), we consistently observed temperature inversion conditions, with potential temperature increasing by 37 K km−1 compared to 11 K km−1 above the lower polar dome (Fig. S10). Temperature inversions are frequent in the High Arctic spring, with median inversion strengths of ∼5–10 K occurring frequently in March, April and May (Bradley et al., 1992; Tjernström and Graversen, 2009; Zhang et al., 2011; Devasthale et al., 2016). Owing to the static stability of the lower polar dome under these conditions, these air masses may be isolated from the air aloft and may be sensitive to different sources and transport history (Stohl, 2006). Under these stable conditions, CO and CO2 were relatively constant (mean (quartile range), 144.5 (144.2–146.5) ppbv and 405.8 (405.4–406.2) ppmv, respectively) in the lower polar dome and O3 was depleted to 11.4 (3.1–23.4) ppbv. Active halogen production and resulting O3 depletion may occur largely at the surface (e.g., Spackman et al., 2010; Oltmans et al., 2012; Abbatt et al., 2012; Pratt et al., 2013). It follows that the observed O3 profile could be interpreted as an indication of mixing of O3-depleted air from the surface up to ∼252 K (∼400 m). Particle number concentrations between 100 and 500 nm (N100–500) were relatively constant in the lower polar dome (∼150 cm−3), while larger accumulation mode particles (N>500) were most abundant in the lower polar dome compared to higher potential temperatures (∼4 cm−3 compared to <1 cm−3). Second, in the middle polar dome (252–265 K), O3 increased toward ∼50 ppbv and CO and CO2 remained relatively constant while water vapour showed more variability. Finally, at the highest potential temperatures we observed more variability in CO, CO2 and H2O, while O3 concentrations were relatively constant at 49.6 (45.7–54.1) ppbv. N>500 was near zero in the upper polar dome, while N100–500 showed more variability compared to colder potential temperatures.
The importance of lower latitude source regions increases as potential temperature increases in the polar dome. The distribution of FLEXPART-ECMWF potential emission sensitivities (Fig. 3) indicates that most air masses in the lower and middle polar dome had resided there for at least 10 days, with significant sensitivity to the surface north of 80∘ N and some sensitivity to high-latitude Eurasia. The fraction of the previous 10 days spent in the polar dome is highest in the middle and lower polar dome, while above ∼265 K this quantity decreases significantly (Fig. 4, S8). This observation indicates a clear separation in air mass history between the middle-to-lower polar dome and the upper polar dome. Sensitivity to lower latitude regions increases as potential temperature increases in the polar dome, particularly in high-latitude Eurasia and North America (Fig. 3). Locations of active fires during 28 March 2015–13 April 2015 and of oil and gas extraction emissions (Fig. 3) indicate that biomass burning emissions likely had a stronger influence on the upper polar dome, while oil and gas extraction emissions may be more important in the lower polar dome. Total March–May 2015 fire counts in the Northern Hemisphere were comparable to previous years (Fig. S12), but were significantly lower than 2008. This suggests that biomass burning sources are often less important sources of Arctic aerosol than has been suggested by previous observations from the year 2008 (e.g., Warneke et al., 2009; Brock et al., 2011; Hecobian et al., 2011; McNaughton et al., 2011; Liu et al., 2015).
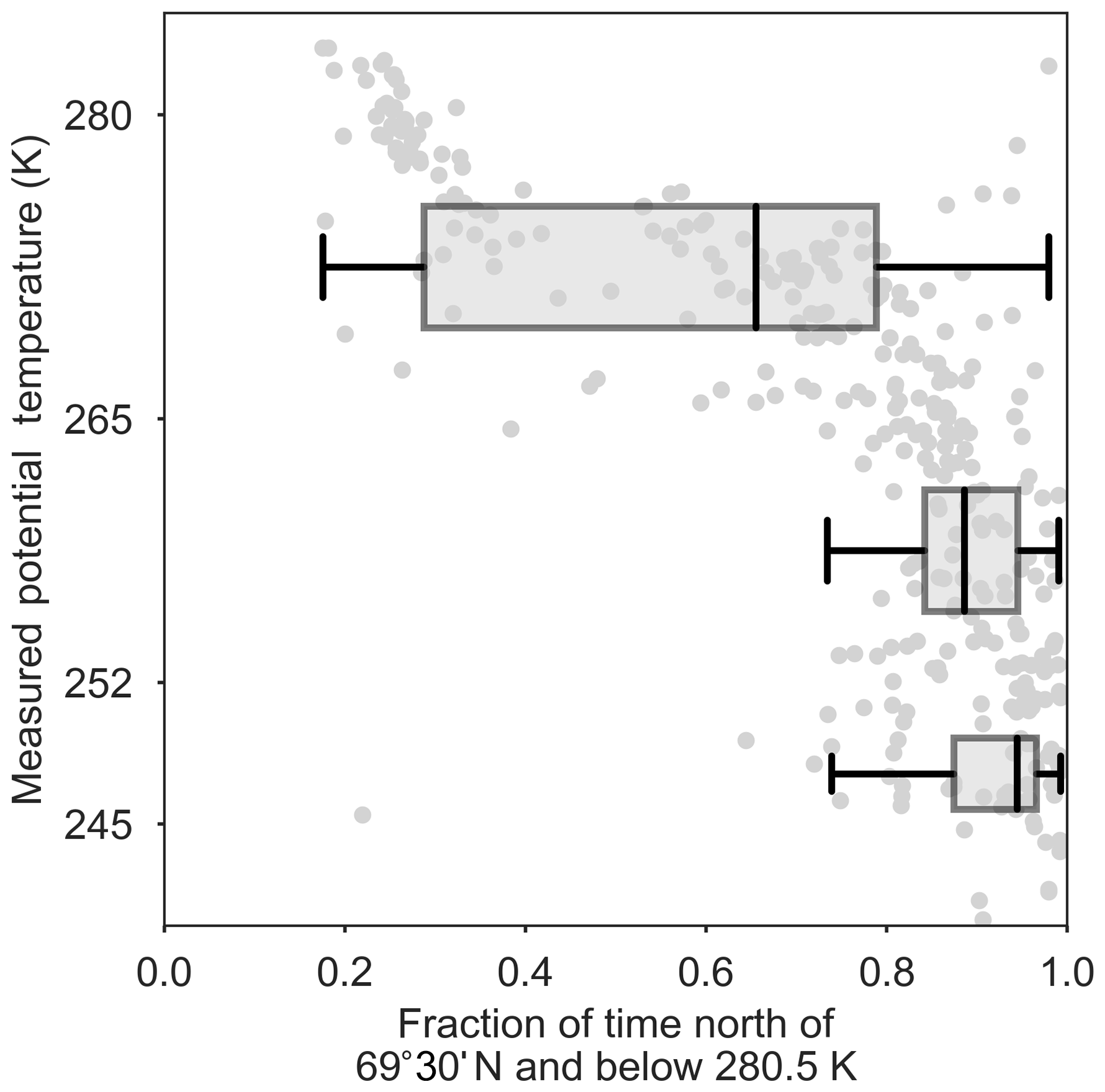
Figure 4Observed potential temperature (K) versus FLEXPART-ECMWF-predicted fraction of the past 10 days in the polar dome (i.e., below 280.5 K and north of 69∘ 30′ N). The FLEXPART-ECMWF relative residence time is binned in the lower (245–252 K), middle (252–265 K) and upper (265–280 K) polar dome.
A prevalent feature of air mass histories in the lower and middle polar dome is descent from aloft over at least 10 days prior to our measurements (Fig. 3g, h). The FLEXPART-ECMWF-predicted plume centroid also shows some evidence for descent in the upper polar dome (Fig. 3i), though we note that descent from aloft in the plume centroid does not preclude some sensitivity to the surface. Air mass descent in the polar dome is likely caused by a combination of both radiative cooling (on the order of 1 K day−1; Klonecki et al., 2003) and orographic effects over nearby elevated terrain on Ellesmere Island and Greenland. With long aerosol lifetimes under cold and relatively dry conditions in the polar dome, this suggests that aerosol in the upper polar dome can influence the lower and middle polar dome on the timescale of 10 days and longer. Transport times to the Arctic lower troposphere are likely longer than 10 days (e.g., Brock et al., 2011; Qi et al., 2017; Leaitch et al., 2018), suggesting that a major springtime transport mechanism may be lofting near source regions, followed by northward transport and descent into the polar dome (Stohl, 2006). In the next section, we discuss observed aerosol composition in the context of these transport patterns.
3.2 Aerosol composition in the polar dome
Vertical variability in aerosol composition was systematic across flights in the polar dome during April 2015. Sub-micron aerosol present in the coldest air masses of the lower polar dome contained the highest fraction of sulfate (74 % by mass, Fig. 5). This trend in the sulfate mass fraction (mf) was driven by both decreasing sulfate and increasing organic aerosol concentrations as potential temperature increased (Figs. 6, S5). This observation is broadly consistent with previous vertically resolved measurements of aerosol sulfate in both the Canadian Arctic and Alaskan Arctic during spring that have indicated increasing sulfate concentrations toward lower altitudes (Scheuer et al., 2003; Bourgeois and Bey, 2011). Large contributions of sulfate to near-surface Arctic spring aerosol is also consistent with ground-based observations at long-term monitoring stations including Zeppelin, Svalbard; Alert, Nunavut; and Utqiaġvik (Barrow), Alaska (e.g., Barrie and Hoff, 1985; Quinn et al., 2007; Breider et al., 2017; Leaitch et al., 2018). The mass fraction of ammonium (mf) increases with increasing potential temperature. This trend is driven by both decreasing sulfate concentration and increasing ammonium concentration as potential temperature increases (Fig. 6). This observation is broadly consistent with previous vertically resolved measurements in the North American Arctic from April 2008 that demonstrated increased ammonium relative to sulfate toward higher altitudes (Fisher et al., 2011). However, Fisher et al. (2011) observed significantly higher ammonium relative to sulfate compared to our measurements. These differences may arise from the larger altitude range in Fisher et al. (2011) (up to ∼10 km) and differences in source regions or source strengths between 2008 and 2015.

Figure 5Mean potential temperature profiles of relative aerosol composition, including mass fractions of sulfate (mf), organic aerosol (mfOA), refractory black carbon (mfrBC), and ammonium (mf), in the polar dome observed during 7–13 April 2015. Coloured lines indicate the mean profile for each flight, the black line represents the mean profile over all six flights, and gray shading shows the range of observations in each potential temperature bin.
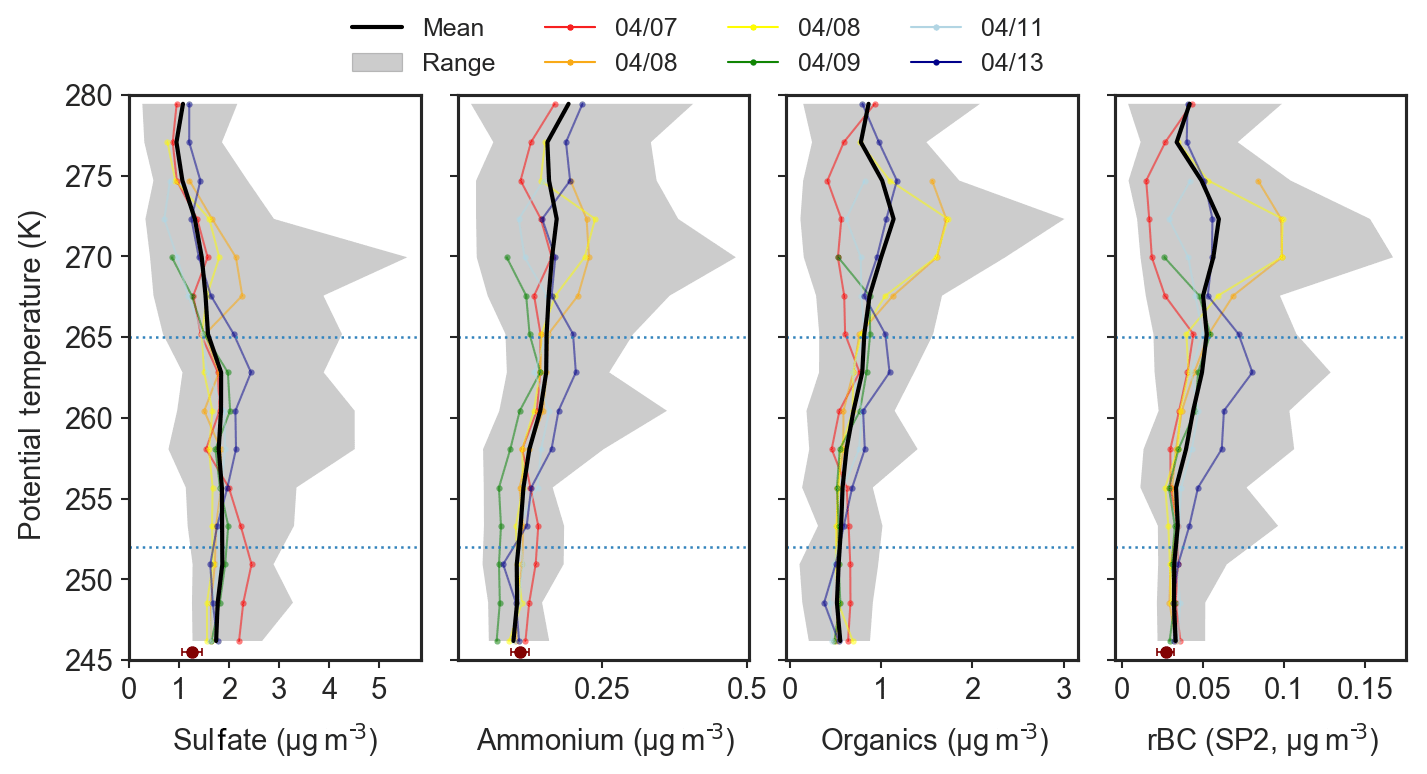
Figure 6Mean potential temperature profiles of absolute (STP) sub-micron aerosol composition in the polar dome observed during 7–13 April 2015, including sulfate, organics and ammonium from the ToF-AMS and refractory black carbon (rBC) from the SP2. Nitrate concentrations were negligible, and largely below detection limits. Coloured lines indicate the mean profile for each flight, the black line represents the mean profile over all six flights, and gray shading shows the range of observations in each potential temperature bin. Single points at the lowest potential temperature represent concentrations of sulfate, ammonium and rBC measured at Alert, NU, during 6–13 April 2015 from Macdonald et al. (2017). Points represent the mean concentration and error bars represent measurement uncertainty.
Organic aerosol and refractory black carbon were more abundant in the upper polar dome, while sulfate was less abundant. On average, OA and rBC contributed 42 % and 2 % to aerosol mass, respectively, in the upper polar dome. OA was highly oxygenated throughout the polar dome, with oxygen-to-carbon (O ∕ C) ratios above 0.5 in the majority of measurements (Fig. S13). High O ∕ C ratios are consistent with an abundance of highly functionalized organic acids observed in Arctic haze aerosol at Alert, Nunavut, during spring (Kawamura et al., 1996, 2005, 2010; Narukawa et al., 2008; Fu et al., 2009; Leaitch et al., 2018). Owing to the lack of unique mass spectral fragments from this highly oxygenated OA, our ToF-AMS spectra cannot distinguish differences in OA composition in the polar dome. Overall, our observations suggest that surface-based measurements may underestimate the contribution of OA, rBC and ammonium to aerosol transported to the Arctic troposphere in spring.
Air masses spent the longest times in the middle to lower polar dome (Fig. 4), and aerosol composition varied systematically with time spent in this portion of the polar dome. The mass fractions of OA and rBC decrease with the FLEXPART-ECMWF-predicted fraction of the previous 10 days spent north of 69∘ 30′ N and below 265 K (Fig. 7). OA and rBC were well-correlated in the middle and upper polar dome (Fig. S14), suggesting that these species have a similar source region and/or have undergone similar processing. A dominance of anthropogenic (fossil fuel) sources of black carbon to the High Arctic during April 2015 may explain this relationship between rBC and OA. The importance of anthropogenic emissions of black carbon from eastern and southern Asia to measured Arctic black carbon in spring was recently demonstrated using a chemical transport model constrained by our measurements of black carbon in combination with surface sites and previous aircraft-based campaigns (Xu et al., 2017). European and north Asian anthropogenic emissions contributed significantly to Arctic black carbon in the lowest kilometre, with eastern and southern Asian sources increasing in importance toward higher altitudes (Xu et al., 2017). Southern Asian regions are not well-represented in 10-day FLEXPART-ECMWF backward simulations, which likely do not capture transport back to all source regions (Qi et al., 2017; Leaitch et al., 2018). OA and rBC are largely uncorrelated in the lower polar dome, suggesting shifting source regions and/or chemical processing of OA toward lower potential temperatures. This observation is consistent with multi-year observations from Alert, Nunavut, showing that black carbon and organic matter are correlated during winter, but become uncorrelated during spring (Leaitch et al., 2018).
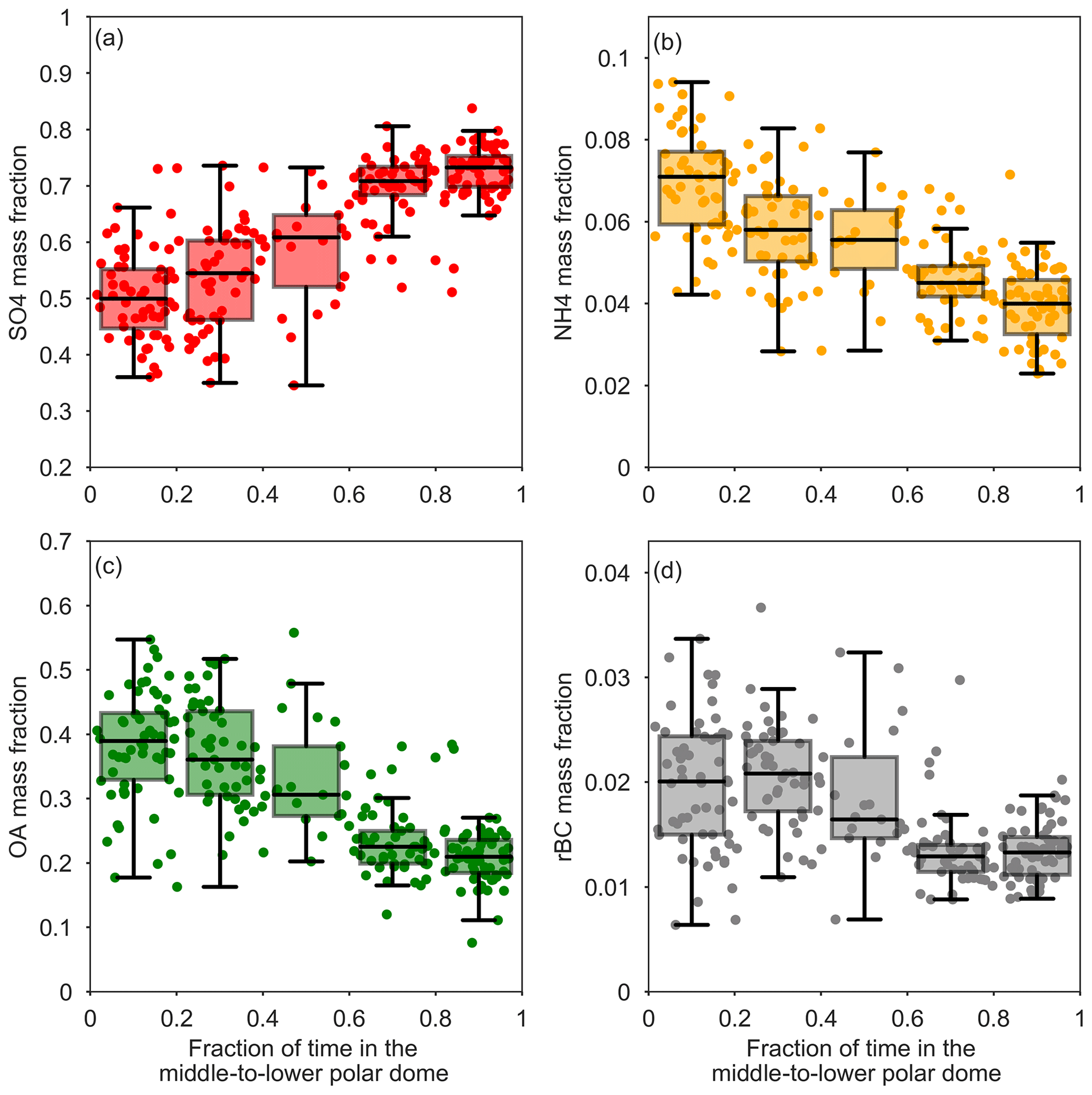
Figure 7Sub-micron aerosol mass fractions versus FLEXPART-ECMWF-predicted fraction of the previous 10 days prior to measurement spent in the middle-to-lower polar dome (north of 69∘ 30′ N, Bozem et al., 2018, and, based on trace gas profiles, below 265 K, ∼1600 m). Data points corresponding to individual FLEXPART-ECMWF releases are shown as circles, and summary statistics are shown as boxes (25th, 50th, 75th percentiles) and whiskers (5th, 95th percentiles) for data binned by time spent in the middle and lower polar dome.
In contrast to OA and rBC, the mass fraction of sulfate increases with increasing time spent in the middle-to-lower polar dome (Fig. 7). In the upper polar dome the ammonium-to-sulfate molar ratio is at times consistent with ammonium bisulfate, while more sulfuric acid is likely present at lower potential temperatures. The enhanced fraction of sulfate in the lower polar dome compared to higher potential temperatures could arise from a combination of possible mechanisms. First, the stability of the polar dome may cause systematic vertical variability in source regions throughout the polar dome (e.g., Stohl, 2006). The observed middle-to-lower polar dome aerosol composition could arise from high-latitude, sulfur-rich emissions in the absence of significant ammonia and organic aerosol sources. A complex mixture of natural and anthropogenic sources has previously been shown to contribute to observed variations in sulfate and ammonium with altitude in Arctic spring (Fisher et al., 2011). In the lowest 2 km, emissions from non-Arctic Russia and Kazakhstan (included as part of eastern and southern Asia in Xu et al., 2017), along with North American emissions, were the dominant sources of sulfate in April 2008 (Fisher et al., 2011). At higher altitudes, model results suggested eastern Asian sources of sulfate became more important and, along with European sources, were the main contributor of sulfate aerosol in Arctic spring. Second, and possibly in addition to shifting source regions, aerosol composition could be changing as a result of chemical processing over the long aerosol lifetime. The fraction of sulfate could be increasing with decreasing potential temperature as a result of oxidation of transported sulfur dioxide and subsequent condensation of sulfuric acid onto existing particles as air masses slowly descend (Fig. 3). In addition, oxidation of existing OA, resulting in fragmentation and loss of aerosol mass to the gas phase, could contribute to a decrease in OA concentrations toward lower altitudes (e.g., Kroll et al., 2009); however, this process may be less important at low temperatures. Descent from aloft appears to be an important transport mechanism influencing the lower polar dome in our flight area, lending some support to this second set of processes. Finally, wet removal or cloud processing of aerosol over long transport times likely impacts the aerosol composition we observe, though we cannot distinguish this influence with our measurements. In the next section, we examine the characteristics of lower polar dome aerosol in detail and compare it to aerosol present in the middle and upper polar dome.

Figure 8Potential temperature profiles of the ToF-AMS NaCl+ signal (top axis), as a qualitative indication of the presence of sea salt aerosol, and N>500 (bottom axis). The solid lines represent the mean profile for 7–13 April 2015, and shading represents the range of measurements in each potential temperature bin.
3.3 Characteristics of lower polar dome aerosol
Lower polar dome air masses had resided for the longest times within the polar dome (Figs. 3, 4 and S11), suggesting that this aerosol likely had a lifetime of 10 days or longer. This aerosol was comprised largely of sulfate, with smaller amounts of OA, rBC and ammonium compared to aerosol present in the middle and upper polar dome (Sect. 3.2). In addition, ToF-AMS spectra provide qualitative evidence for the presence of sea salt aerosol in the lower polar dome, which decreases to negligible concentrations through the middle polar dome (Fig. 8). The ToF-AMS NaCl+ signal and N>500 have a similar profile, suggesting that sea salt may be associated with the increase in larger accumulation mode particles observed in the lower polar dome. This observation is consistent with previous airborne measurements in the Alaskan Arctic during spring that showed the largest fraction of sea salt particles were present in air masses identified as associated with the “Arctic boundary layer” (i.e., identified by depleted O3 concentrations) (Brock et al., 2011). Sea salt contributes significantly to aerosol observed at ground-based long-term monitoring stations (e.g., Quinn et al., 2002; Leaitch et al., 2013; Huang and Jaeglé, 2017; Leaitch et al., 2018), and peaks in concentration during winter to early spring. Sources of sea salt at high northern latitudes in spring include transport of sea salt from northern oceans, production of sea salt aerosol from open leads in sea ice (e.g., Leck et al., 2002; Held et al., 2011; May et al., 2016), and production of saline aerosol through wind driven processes over ice and snow (Yang et al., 2008; Shaw et al., 2010; Xu et al., 2016; Huang and Jaeglé, 2017). The strong decrease in NaCl+ signal and N>500 above the lower polar dome is suggestive of a near-surface source of sea salt in the High Arctic; open leads or wind-driven ice and snow processes may contribute to lower polar dome aerosol. Recent observations at Utqiaġvik (Barrow), Alaska, have demonstrated the prevalence of sea salt aerosol in Arctic winter and significant mixing with sulfate (Kirpes et al., 2018). Sulfate may be internally mixed with sea salt in the lower polar dome; however, owing to low particle concentrations we are unable to obtain an NaCl+ signal from size-resolved mass spectra.
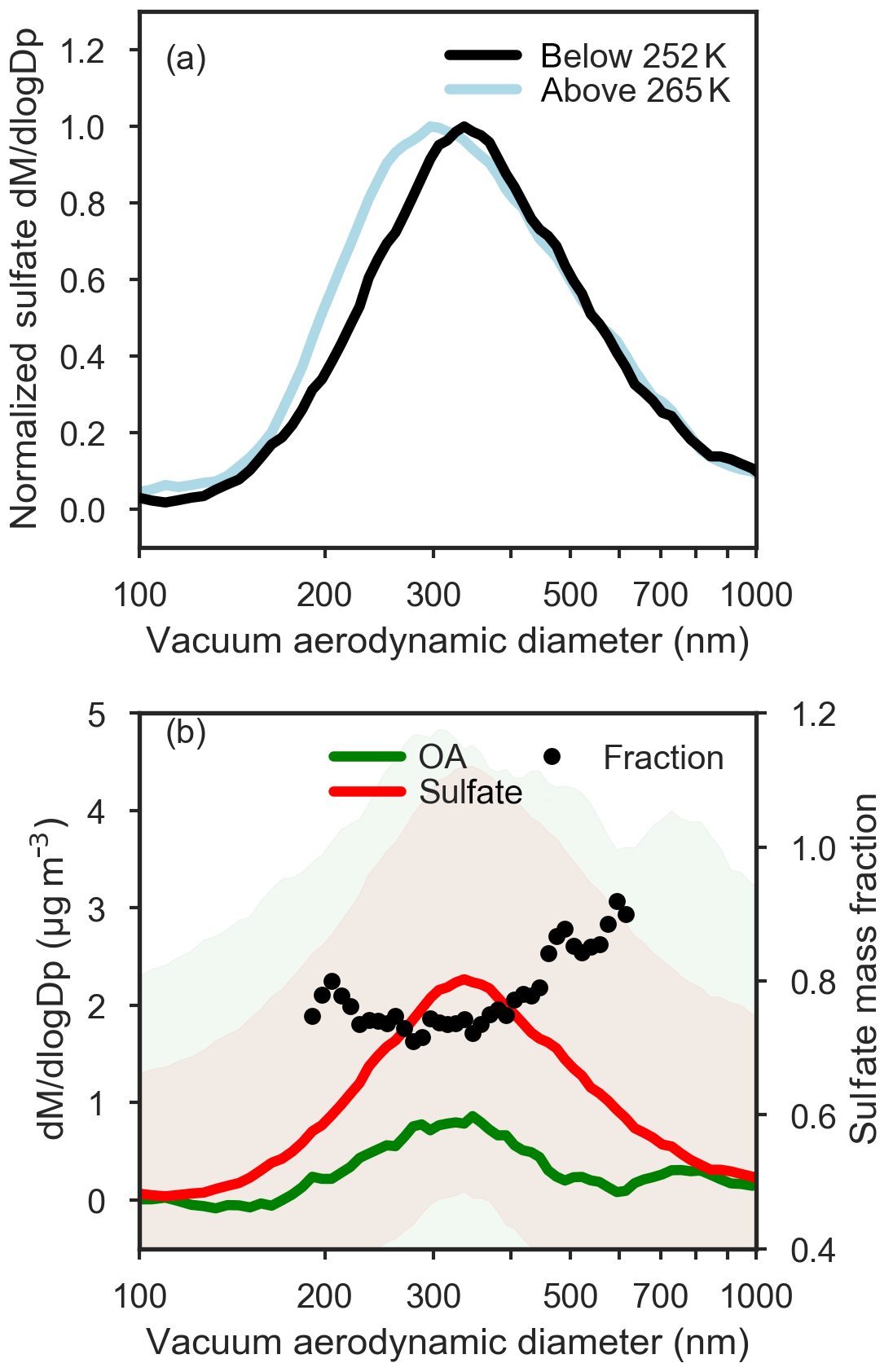
Figure 9(a) Normalized mean ToF-AMS size distributions of sulfate subset by observed potential temperature: below 252 K (black), above 265 K (light blue). (b) ToF-AMS size distributions of sulfate (red) and total organic aerosol (green) below 252 K. The mass fraction of sulfate calculated from ToF-AMS size distributions is shown on the right axis in black circles, and is calculated only between 200 and 600 nm owing to low OA signals at smaller and larger sizes. Shading corresponds to ±1 standard deviation for sulfate and organic aerosol size distributions, and the relatively large variation in size-resolved composition indicates that the derived mass fraction of sulfate as a function of size is uncertain.
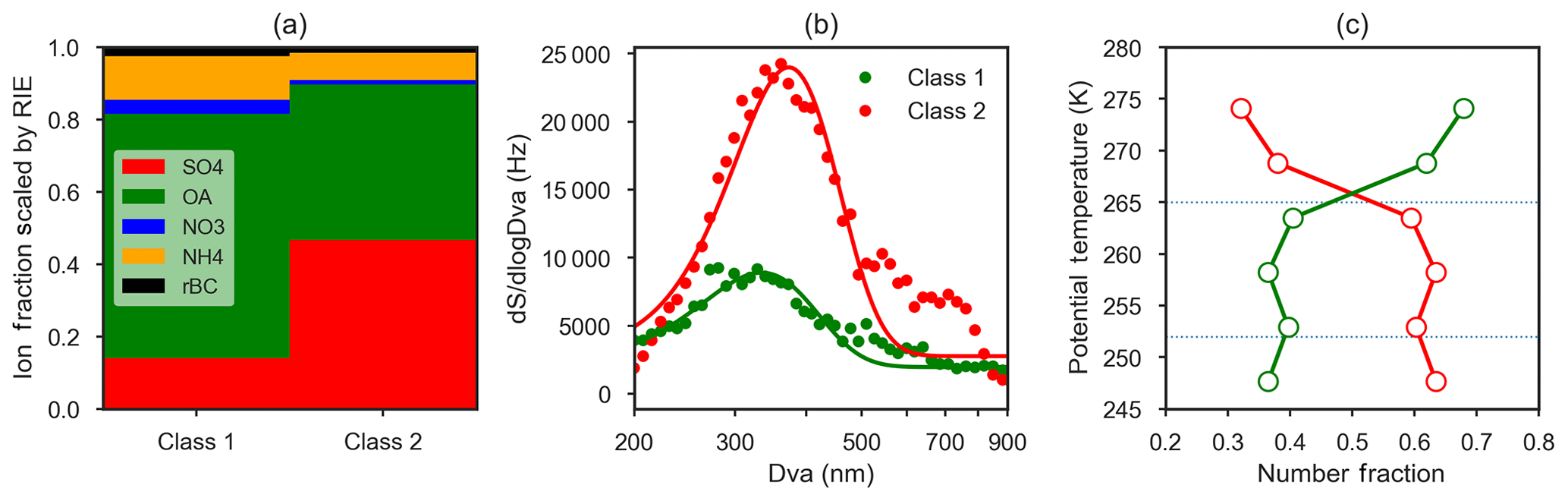
Figure 10Summary of k-means cluster analysis of 1677 single-particle (ETSP) spectra obtained on two flights (8 and 13 April 2015). (a) Bar chart of relative ion fraction scaled by the relative ionization efficiency (RIE) of each species, for two particle classes obtained by k-means cluster analysis. Particle class 1 is referred to as “organic-rich” and class 2 is referred to as “sulfate-rich”. (b) Mean size distributions (expressed as dSignal∕dlog Dva, Hz) of the two particle classes (points) and Gaussian fits to the observations (lines). (c) Mean relative abundance of class 1 (green, organic-rich) and class 2 (red, sulfate-rich) binned by potential temperature. Horizontal lines represent the divisions between the lower, middle and upper polar dome.
Size-resolved observations of non-refractory aerosol composition provide evidence for different particle mixing states across the size distribution. On average, sulfate was present in larger particle sizes in the lower polar dome compared to the middle and upper polar dome (Fig. 9). In contrast, OA size distributions were very similar in the lower and upper polar dome (Fig. S15). In the lower polar dome, the fraction of sulfate increases with particle size (Fig. 9), implying the presence of different particle mixing states, and different particle sources or chemical processing in the polar dome. Single-particle observations from two flights (Figs. 10, S16) are consistent with these bulk size-resolved observations. Accumulation mode particles were highly internally mixed, consistent with very aged particles, but the presence of sulfate-rich and organic-rich particles was discernible from cluster analysis of ToF-AMS spectra. Sulfate-rich particles were dominant in the coldest air masses and were larger in size compared to organic-rich particles (Figs. 10 and S16). Increasing sulfate fraction toward larger particle sizes (Fig. 9) suggests that sulfuric acid may have condensed on existing particles, growing them to larger sizes. An increase in aerosol sulfate toward lower altitudes and a simultaneous decrease in gas phase SO2 has been observed previously in Arctic spring; this could be consistent with oxidation of SO2 and condensation on pre-existing particles in the lowest 1–2 km (Bourgeois and Bey, 2011). Barrie and Hoff (1984) estimated a mean oxidation rate of SO2 to sulfuric acid in April of 2.4 % day−1–4.8 % day−1, which could explain the enhanced concentrations of sulfate toward lower potential temperatures in our observations. While the smaller OA size in the lower polar dome could be consistent with loss of OA mass through fragmentation processes, similar OA size distributions in the lower and upper polar dome appear to negate this possibility (Fig. S15). Our observations do not distinguish unambiguously between vertical variability in source composition, chemical processing during descent in the polar dome, and wet removal or cloud processing during transport. All processes likely contribute to the systematic vertical variability in High Arctic aerosol composition that we observe.
In the Arctic spring polar dome, aerosol composition and trace gas concentrations varied systematically with potential temperature. We defined the lower (245–252 K), middle (252–265 K) and upper (265–280 K) polar dome based on vertical profiles of trace gases. The contribution of sulfate increased from the upper to lower polar dome (mean mass fractions 48 % and 74 %, respectively), while organic aerosol, refractory black carbon and ammonium were more abundant in the upper polar dome (mean mass fractions 42 %, 2 % and 8 %, respectively). At the lowest potential temperatures, in the lower polar dome, sulfate-rich particles were present at larger accumulation mode sizes compared to the upper polar dome.
While observations at long-term monitoring stations provide the majority of our knowledge about Arctic aerosol, decoupling of air masses near the surface from the rest of the polar dome means that surface-based observations may not represent the altitude-dependent composition of aerosol transported to the Arctic troposphere. Our observations indicate that long-term, surface-based measurements may underestimate the contribution of organic aerosol, refractory black carbon and ammonium to aerosol transported to the High Arctic troposphere in spring, while overestimating the contribution of sulfate. In addition, our observations of sea salt signals in the lower polar dome suggest that the significant sea salt concentrations observed at long-term monitoring stations in spring may not occur throughout the depth of the polar dome.
Systematic differences in aerosol composition with potential temperature likely arise through a combination of mechanisms. First, aerosol from different source regions, with differing composition, arrives at a range of potential temperatures in a stable atmosphere. Second, aerosol composition can be altered by chemical processing of transported aerosol and sulfur dioxide during descent into the polar dome over periods of 10 days or longer. Third, wet removal and cloud processing near emission and along the transport path may impact the composition of aerosol arriving in the polar dome, though this influence is difficult to distinguish with our observations. Modelled air mass history from FLEXPART-ECMWF demonstrates that this systematic variation in aerosol composition is in part related to differing transport regimes as a function of potential temperature. In the lower polar dome, air masses had resided in the High Arctic region for at least 10 days prior to measurement and had largely descended from higher altitudes. Some sensitivity to the High Arctic surface could explain the observed sea salt in the lower polar dome. Lower latitude source regions in Europe, Asia and North America became more important toward higher potential temperatures in the upper polar dome. Long transport times make source diagnosis difficult using 10-day backward trajectories, and chemical processing during long Arctic residence times contributes to challenges in identifying source regions of lower polar dome aerosol. Using our observations, we cannot quantitatively distinguish the relative importance of vertical variability in source composition, chemical processing during descent in the polar dome, and removal or cloud processing during transport. Our observations present a challenge to chemical transport models for their representation of the processes impacting High Arctic aerosol in spring.
NETCARE data are available on the Government of Canada Open Data Portal (https://open.canada.ca/data/en/dataset). Data from NETCARE 2015 are available at https://open.canada.ca/data/dataset/efe0e41c-890d-404d-bb1b-421456022d51 (Abbatt et al., 2018). Sea ice data shown in Fig. 1 are available at https://seaice.uni-bremen.de/sea-ice-concentration/ (Spreen et al., 2008). Global MODIS active fire locations shown in Fig. 3 are available at https://earthdata.nasa.gov/earth-observation-data/near-real-time/firms/active-fire-data (NASA, 2015). Gas-flaring locations from the ECLIPSE inventory V5 shown in Fig. 3 are available at http://www.iiasa.ac.at/web/home/research/researchPrograms/air/ECLIPSEv5.html (International Institute for Applied Systems Analysis, 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-57-2019-supplement.
MDW wrote the paper, with significant conceptual input from DK, HB, WRL and JPDA, and critical feedback from all co-authors. MDW, HB, JB, AKYL, HS and WRL operated instruments in the field and analyzed resulting data. AAA analyzed flight data. WRL, JPDA and ABH designed the field experiment. DK ran FLEXPART simulations, and MDW analyzed the resulting data with input from DK and HB.
The authors declare that they have no conflict of interest.
This article is part of the special issue “NETCARE (Network on Aerosols and Climate: Addressing Key Uncertainties in Remote Canadian Environments) (ACP/AMT/BG inter-journal SI)”. It is not associated with a conference.
Funding for this work was provided by the Natural Sciences and Engineering
Research Council of Canada (NSERC) through the NETCARE project
(https://www.netcare-project.ca/) of the Climate Change and Atmospheric Research
Program, the Alfred Wegener Institute (AWI) and Environment and Climate
Change Canada (ECCC). We gratefully acknowledge Kenn Borek Air Ltd, in
particular our pilots and crew Garry Murtsell, Neil Traverse and Doug Mackenzie, for their support of our measurements. Logistical and technical
support before and during the campaign was provided by a number of
contributors, in particular by Desiree Toom-Sauntry (ECCC), Ralf Staebler
(ECCC), Katherine Hayden (ECCC), Andrew Elford (ECCC), Anne Marie MacDonald
(ECCC), Maurice Watt (ECCC), Mohammed Wasey (ECCC), Jason Iwachow (ECCC),
Alina Chivulescu (ECCC), Ka Sung (ECCC), Dan Veber (ECCC), Julia Binder
(AWI), Lukas Kandora (AWI), Jens Herrmann (AWI) and Manuel Sellmann (AWI).
Extensive logistical and technical support was provided by Andrew Platt
(ECCC), Mike Harwood (ECCC) and Martin Gerhmann (AWI). We are grateful to CFS
Alert and Eureka Weather Station for supporting the measurements presented in
this work. We gratefully acknowledge funding by the Deutsche
Forschungsgemeinschaft (DFG, German Research Foundation) – project number
268020496 – TRR 172, within the Transregional Collaborative Research Center
“ArctiC Amplification: Climate Relevant Atmospheric and SurfaCe Processes,
and Feedback Mechanisms (AC)3”. Data were analyzed in Igor Pro 6.37
(https://www.wavemetrics.com/, last access: 20 December 2018) and Python 3.5.2
(https://www.python.org/downloads/release/python-352/, last access: 20 December 2018). We acknowledge Charles Brock,
Jennifer Murphy and Dylan Jones for their comments on an early version of
this paper.
Edited by: Barbara Ervens
Reviewed by: four anonymous referees
Abbatt, J. P. D., Benz, S., Cziczo, D. J., Kanji, Z., Lohmann, U., and Moehler, O.: Solid ammonium sulfate aerosols as ice nuclei: A pathway for cirrus cloud formation, Science, 313, 1770–1773, https://doi.org/10.1126/science.1129726, 2006. a, b
Abbatt, J. P. D., Thomas, J. L., Abrahamsson, K., Boxe, C., Granfors, A., Jones, A. E., King, M. D., Saiz-Lopez, A., Shepson, P. B., Sodeau, J., Toohey, D. W., Toubin, C., von Glasow, R., Wren, S. N., and Yang, X.: Halogen activation via interactions with environmental ice and snow in the polar lower troposphere and other regions, Atmos. Chem. Phys., 12, 6237–6271, https://doi.org/10.5194/acp-12-6237-2012, 2012. a, b
Abbatt, J. P. D., Leaitch, W. R., Herber, A. B., Bertram, A. K., Blanchet, J. P., Korolev, A., Burkart, J., Bozem, H., Willis, M. D., Lee, A. K. Y., Schulz, H., Hanna, S., Aliabadi, A. A., and Staebler, R.: NETCARE 2015 POLAR6 aircraft campaign, available at: https://open.canada.ca/data/dataset/efe0e41c-890d-404d-bb1b-421456022d51, last access: 20 December 2018. a
Arnold, S. R., Law, K. S., Brock, C. A., Thomas, J. L., Starkweather, S. M., von Salzen, K., Stohl, A., Sharma, S., Lund, M. T., Flanner, M. G., Petäjä, T., Tanimoto, H., Gamble, J., Dibb, J. E., Melamed, M., Johnson, N., Fidel, M., Tynkkynen, V. P., Baklanov, A., Eckhardt, S., Monks, S. A., Browse, J., and Bozem, H.: Arctic air pollution: Challenges and opportunities for the next decade, Elementa, 4, 000104, https://doi.org/10.12952/journal.elementa.000104, 2016. a, b, c, d, e, f
Asmi, E., Kondratyev, V., Brus, D., Laurila, T., Lihavainen, H., Backman, J., Vakkari, V., Aurela, M., Hatakka, J., Viisanen, Y., Uttal, T., Ivakhov, V., and Makshtas, A.: Aerosol size distribution seasonal characteristics measured in Tiksi, Russian Arctic, Atmos. Chem. Phys., 16, 1271–1287, https://doi.org/10.5194/acp-16-1271-2016, 2016. a
Barrie, L. A. and Hoff, R. M.: The Oxidation Rate and Residence Time of Sulfur-Dioxide in the Arctic Atmosphere, Atmos. Environ., 18, 2711–2722, https://doi.org/10.1016/0004-6981(84)90337-8, 1984. a, b
Barrie, L. A. and Hoff, R. M.: Five Years of Air Chemistry Observations in the Canadian Arctic, Atmos. Environ., 19, 1995–2010, https://doi.org/10.1016/0004-6981(85)90108-8, 1985. a
Baustian, K. J., Wise, M. E., and Tolbert, M. A.: Depositional ice nucleation on solid ammonium sulfate and glutaric acid particles, Atmos. Chem. Phys., 10, 2307–2317, https://doi.org/10.5194/acp-10-2307-2010, 2010. a
Bian, H., Colarco, P. R., Chin, M., Chen, G., Rodriguez, J. M., Liang, Q., Blake, D., Chu, D. A., da Silva, A., Darmenov, A. S., Diskin, G., Fuelberg, H. E., Huey, G., Kondo, Y., Nielsen, J. E., Pan, X., and Wisthaler, A.: Source attributions of pollution to the Western Arctic during the NASA ARCTAS field campaign, Atmos. Chem. Phys., 13, 4707–4721, https://doi.org/10.5194/acp-13-4707-2013, 2013. a
Bindoff, N., Stott, P., AchutaRao, K., Allen, M., Gillett, N., Gutzler, D., Hansingo, K., Hegerl, G., Hu, Y., Jain, S., Mokhov, I., Overland, J., Perlwitz, J., Sebbari, R., and Zhang, X.: Detection and Attribution of Climate Change: from Global to Regional, chap. 10, 867–952, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2013. a
Blanchet, J.-P. and Girard, E.: Arctic “greenhouse effect”, Nature, 371, p. 383, https://doi.org/10.1038/371383a0, 1994. a, b
Blanchet, J.-P. and Girard, E.: Water vapor-temperature feedback in the formation of continental Arctic air: its implication for climate, Sci. Total Environ., 160–161, 793–802, https://doi.org/10.1016/0048-9697(95)04412-T, 1995. a, b
Boucher, O. and Anderson, T. L.: General circulation model assessment of the sensitivity of direct climate forcing by anthropogenic sulfate aerosols to aerosol size and chemistry, J. Geophys. Res.-Atmos., 100, 26117–26134, https://doi.org/10.1029/95JD02531, 1995. a, b
Bourgeois, Q. and Bey, I.: Pollution transport efficiency toward the Arctic: Sensitivity to aerosol scavenging and source regions, J. Geophys. Res.-Atmos., 116, D08213, https://doi.org/10.1029/2010jd015096, 2011. a, b, c
Bozem, H., Hoor, P., Kunkel, D., Köllner, F., Schneider, J., Herber, A., Schulz, H., Leaitch, W., Willis, M., Burkart, J., and Abbatt, J.: Characterization of Transport Regimes and the polar dome during both Arctic Spring and Summer using in-situ Aircraft Measurements, in preparation for Atmospheric Chemistry and Physics, 2018. a, b, c, d, e
Bradley, R. S., Keimig, F. T., and Diaz, H. F.: Climatology of Surface-Based Inversions in the North-American Arctic, J. Geophys. Res.-Atmos., 97, 15699–15712, 1992. a
Breider, T. J., Mickley, L. J., Jacob, D. J., Wang, Q. Q., Fisher, J. A., Chang, R. Y. W., and Alexander, B.: Annual distributions and sources of Arctic aerosol components, aerosol optical depth, and aerosol absorption, J. Geophys. Res.-Atmos., 119, 4107–4124, https://doi.org/10.1002/2013jd020996, 2014. a
Breider, T. J., Mickley, L. J., Jacob, D. J., Ge, C., Wang, J., Sulprizio, M. P., Croft, B., Ridley, D. A., McConnell, J. R., Sharma, S., Husain, L., Dutkiewicz, V. A., Eleftheriadis, K., Skov, H., and Hopke, P. K.: Multidecadal trends in aerosol radiative forcing over the Arctic: Contribution of changes in anthropogenic aerosol to Arctic warming since 1980, J. Geophys. Res.-Atmos., 122, 3573–3594, https://doi.org/10.1002/2016jd025321, 2017. a
Brioude, J., Arnold, D., Stohl, A., Cassiani, M., Morton, D., Seibert, P., Angevine, W., Evan, S., Dingwell, A., Fast, J. D., Easter, R. C., Pisso, I., Burkhart, J., and Wotawa, G.: The Lagrangian particle dispersion model FLEXPART-WRF version 3.1, Geosci. Model Dev., 6, 1889–1904, https://doi.org/10.5194/gmd-6-1889-2013, 2013. a
Brock, C. A., Cozic, J., Bahreini, R., Froyd, K. D., Middlebrook, A. M., McComiskey, A., Brioude, J., Cooper, O. R., Stohl, A., Aikin, K. C., de Gouw, J. A., Fahey, D. W., Ferrare, R. A., Gao, R.-S., Gore, W., Holloway, J. S., Hübler, G., Jefferson, A., Lack, D. A., Lance, S., Moore, R. H., Murphy, D. M., Nenes, A., Novelli, P. C., Nowak, J. B., Ogren, J. A., Peischl, J., Pierce, R. B., Pilewskie, P., Quinn, P. K., Ryerson, T. B., Schmidt, K. S., Schwarz, J. P., Sodemann, H., Spackman, J. R., Stark, H., Thomson, D. S., Thornberry, T., Veres, P., Watts, L. A., Warneke, C., and Wollny, A. G.: Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project, Atmos. Chem. Phys., 11, 2423–2453, https://doi.org/10.5194/acp-11-2423-2011, 2011. a, b, c, d, e, f, g, h, i, j
Browse, J., Carslaw, K. S., Arnold, S. R., Pringle, K., and Boucher, O.: The scavenging processes controlling the seasonal cycle in Arctic sulphate and black carbon aerosol, Atmos. Chem. Phys., 12, 6775–6798, https://doi.org/10.5194/acp-12-6775-2012, 2012. a
Cai, Y., D. C. Montague, D., Mooiweer-Bryan, W., and Deshler, T.: Performance characteristics of the ultra-high sensitivity aerosol spectrometer for particles between 55 and 800 nm: Laboratory and field studies, J. Aerosol Sci., 39, 759–769, 2008. a
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015. a
Clarke, A. D. and Noone, K. J.: Soot in the Arctic Snowpack - A Cause for Perturbations in Radiative-Transfer, Atmos. Environ., 19, 2045–2053, https://doi.org/10.1016/0004-6981(85)90113-1, 1985. a
Croft, B., Martin, R. V., Leaitch, W. R., Tunved, P., Breider, T. J., D'Andrea, S. D., and Pierce, J. R.: Processes controlling the annual cycle of Arctic aerosol number and size distributions, Atmos. Chem. Phys., 16, 3665–3682, https://doi.org/10.5194/acp-16-3665-2016, 2016. a, b, c
Curry, J. A. and Herman, G. F.: Infrared Radiative Properties of Summertime Arctic Stratus Clouds, J. Clim. Appl. Meteorol., 24, 525–538, https://doi.org/10.1175/1520-0450(1985)024<0525:IRPOSA>2.0.CO;2, 1985. a
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006. a, b, c
Devasthale, A., Sedlar, J., Kahn, B. H., Tjernström, M., Fetzer, E. J., Tian, B., Teixeira, J., and Pagano, T. S.: A Decade of Spaceborne Observations of the Arctic Atmopshere Novel Insights from NASA's AIRS Instrument, B. Am. Meteorol. Soc., 97, 2163–2176, https://doi.org/10.1175/bams-d-14-00202.1, 2016. a
Eckhardt, S., Quennehen, B., Olivié, D. J. L., Berntsen, T. K., Cherian, R., Christensen, J. H., Collins, W., Crepinsek, S., Daskalakis, N., Flanner, M., Herber, A., Heyes, C., Hodnebrog, Ø., Huang, L., Kanakidou, M., Klimont, Z., Langner, J., Law, K. S., Lund, M. T., Mahmood, R., Massling, A., Myriokefalitakis, S., Nielsen, I. E., Nøjgaard, J. K., Quaas, J., Quinn, P. K., Raut, J.-C., Rumbold, S. T., Schulz, M., Sharma, S., Skeie, R. B., Skov, H., Uttal, T., von Salzen, K., and Stohl, A.: Current model capabilities for simulating black carbon and sulfate concentrations in the Arctic atmosphere: a multi-model evaluation using a comprehensive measurement data set, Atmos. Chem. Phys., 15, 9413–9433, https://doi.org/10.5194/acp-15-9413-2015, 2015. a
Emmons, L. K., Arnold, S. R., Monks, S. A., Huijnen, V., Tilmes, S., Law, K. S., Thomas, J. L., Raut, J.-C., Bouarar, I., Turquety, S., Long, Y., Duncan, B., Steenrod, S., Strode, S., Flemming, J., Mao, J., Langner, J., Thompson, A. M., Tarasick, D., Apel, E. C., Blake, D. R., Cohen, R. C., Dibb, J., Diskin, G. S., Fried, A., Hall, S. R., Huey, L. G., Weinheimer, A. J., Wisthaler, A., Mikoviny, T., Nowak, J., Peischl, J., Roberts, J. M., Ryerson, T., Warneke, C., and Helmig, D.: The POLARCAT Model Intercomparison Project (POLMIP): overview and evaluation with observations, Atmos. Chem. Phys., 15, 6721–6744, https://doi.org/10.5194/acp-15-6721-2015, 2015. a
Engvall, A.-C., Krejci, R., Ström, J., Treffeisen, R., Scheele, R., Hermansen, O., and Paatero, J.: Changes in aerosol properties during spring-summer period in the Arctic troposphere, Atmos. Chem. Phys., 8, 445–462, https://doi.org/10.5194/acp-8-445-2008, 2008. a
Engvall, A. C., Ström, J., Tunved, P., Krejci, R., Schlager, H., and Minikin, A.: The radiative effect of an aged, internally mixed Arctic aerosol originating from lower-latitude biomass burning, Tellus B, 61, 677–684, https://doi.org/10.1111/j.1600-0889.2009.00431.x, 2009. a, b
Fan, S.-M. and Jacob, D. J.: Surface ozone depletion in Arctic spring sustained by bromine reactions on aerosols, Nature, 359, 522–524, https://doi.org/10.1038/359522a0, 1992. a
Fisher, J. A., Jacob, D. J., Wang, Q. Q., Bahreini, R., Carouge, C. C., Cubison, M. J., Dibb, J. E., Diehl, T., Jimenez, J. L., Leibensperger, E. M., Lu, Z. F., Meinders, M. B. J., Pye, H. O. T., Quinn, P. K., Sharma, S., Streets, D. G., van Donkelaar, A., and Yantosca, R. M.: Sources, distribution, and acidity of sulfate-ammonium aerosol in the Arctic in winter-spring, Atmos. Environ., 45, 7301–7318, https://doi.org/10.1016/j.atmosenv.2011.08.030, 2011. a, b, c, d, e, f, g, h, i
Flanner, M. G.: Arctic climate sensitivity to local black carbon, J. Geophys. Res.-Atmos., 118, 1840–1851, https://doi.org/10.1002/jgrd.50176, 2013. a
Flanner, M. G., Zender, C. S., Hess, P. G., Mahowald, N. M., Painter, T. H., Ramanathan, V., and Rasch, P. J.: Springtime warming and reduced snow cover from carbonaceous particles, Atmos. Chem. Phys., 9, 2481–2497, https://doi.org/10.5194/acp-9-2481-2009, 2009. a
Freud, E., Krejci, R., Tunved, P., Leaitch, R., Nguyen, Q. T., Massling, A., Skov, H., and Barrie, L.: Pan-Arctic aerosol number size distributions: seasonality and transport patterns, Atmos. Chem. Phys., 17, 8101–8128, https://doi.org/10.5194/acp-17-8101-2017, 2017. a, b
Fu, P. Q., Kawamura, K., and Barrie, L. A.: Photochemical and Other Sources of Organic Compounds in the Canadian High Arctic Aerosol Pollution during Winter-Spring, Environ. Sci. Technol., 43, 286–292, https://doi.org/10.1021/es803046q, 2009. a
Fyfe, J. C., von Salzen, K., Gillett, N. P., Arora, V. K., Flato, G. M., and McConnell, J. R.: One hundred years of Arctic surface temperature variation due to anthropogenic influence, Sci. Rep., 3, 2645, https://doi.org/10.1038/srep02645, 2013. a
Gao, R. S., Schwarz, J. P., Kelly, K. K., Fahey, D. W., Watts, L. A., Thompson, T. L., Spackman, J. R., Slowik, J. G., Cross, E. S., Han, J.-H., Davidovits, P., Onasch, T. B., and Worsnop, D. R.: A Novel Method for Estimating Light-Scattering Properties of Soot Aerosols Using a Modified Single-Particle Soot Photometer, Aerosol Sci. Tech., 41, 125–135, https://doi.org/10.1080/02786820601118398, 2007. a
Garrett, T. J. and Zhao, C. F.: Increased Arctic cloud longwave emissivity associated with pollution from mid-latitudes, Nature, 440, 787–789, https://doi.org/10.1038/nature04636, 2006. a
Gong, S. L., Zhao, T. L., Sharma, S., Toom-Sauntry, D., Lavoue, D., Zhang, X. B., Leaitch, W. R., and Barrie, L. A.: Identification of trends and interannual variability of sulfate and black carbon in the Canadian High Arctic: 1981–2007, J. Geophys. Res.-Atmos., 115, D07305, https://doi.org/10.1029/2009jd012943, 2010. a
Hansen, J. and Nazarenko, L.: Soot climate forcing via snow and ice albedos, P. Natl. Acad. Sci. USA, 101, 423–428, https://doi.org/10.1073/pnas.2237157100, 2004. a
Harrigan, D. L., Fuelberg, H. E., Simpson, I. J., Blake, D. R., Carmichael, G. R., and Diskin, G. S.: Anthropogenic emissions during Arctas-A: mean transport characteristics and regional case studies, Atmos. Chem. Phys., 11, 8677–8701, https://doi.org/10.5194/acp-11-8677-2011, 2011. a
Hecobian, A., Liu, Z., Hennigan, C. J., Huey, L. G., Jimenez, J. L., Cubison, M. J., Vay, S., Diskin, G. S., Sachse, G. W., Wisthaler, A., Mikoviny, T., Weinheimer, A. J., Liao, J., Knapp, D. J., Wennberg, P. O., Kürten, A., Crounse, J. D., Clair, J. St., Wang, Y., and Weber, R. J.: Comparison of chemical characteristics of 495 biomass burning plumes intercepted by the NASA DC-8 aircraft during the ARCTAS/CARB-2008 field campaign, Atmos. Chem. Phys., 11, 13325–13337, https://doi.org/10.5194/acp-11-13325-2011, 2011. a, b
Held, A., Brooks, I. M., Leck, C., and Tjernström, M.: On the potential contribution of open lead particle emissions to the central Arctic aerosol concentration, Atmos. Chem. Phys., 11, 3093–3105, https://doi.org/10.5194/acp-11-3093-2011, 2011. a
Hennigan, C. J., Izumi, J., Sullivan, A. P., Weber, R. J., and Nenes, A.: A critical evaluation of proxy methods used to estimate the acidity of atmospheric particles, Atmos. Chem. Phys., 15, 2775–2790, https://doi.org/10.5194/acp-15-2775-2015, 2015. a
Herber, A., Dethloff, K., Haas, C., Steinhage, D., Strapp, J. W., Bottenheim, J., McElroy, T., and Yamanouchi, T.: Polar 5 – A new research aircraft for improved access to the Arctic, ISAR-1, Drastic Change under Global Warming, Extended Abstract, Alfred Wegener Institute, Bremen, Germany, 54–57, 2008. a
Herber, A., Haas, C., Bottenheim, J., Liu, P., Shao-Meng, L., Staebler, R., Strapp, J. W., and Dethloff, K.: Regular Airborne Surveys of Arctic Sea Ice and Atmosphere, EOS, 93, 41–48, https://doi.org/10.1029/2010gl042652, 2012. a
Hinzman, L. D., Deal, C. J., McGuire, A. D., Mernild, S. H., Polyakov, I. V., and Walsh, J. E.: Trajectory of the Arctic as an integrated system, Ecol. Appl., 23, 1837–1868, https://doi.org/10.1890/11-1498.1, 2013. a
Hirdman, D., Sodemann, H., Eckhardt, S., Burkhart, J. F., Jefferson, A., Mefford, T., Quinn, P. K., Sharma, S., Ström, J., and Stohl, A.: Source identification of short-lived air pollutants in the Arctic using statistical analysis of measurement data and particle dispersion model output, Atmos. Chem. Phys., 10, 669–693, https://doi.org/10.5194/acp-10-669-2010, 2010. a, b
Hoose, C. and Möhler, O.: Heterogeneous ice nucleation on atmospheric aerosols: a review of results from laboratory experiments, Atmos. Chem. Phys., 12, 9817–9854, https://doi.org/10.5194/acp-12-9817-2012, 2012. a
Huang, J. and Jaeglé, L.: Wintertime enhancements of sea salt aerosol in polar regions consistent with a sea ice source from blowing snow, Atmos. Chem. Phys., 17, 3699–3712, https://doi.org/10.5194/acp-17-3699-2017, 2017. a, b, c
Huang, L., Gong, S. L., Jia, C. Q., and Lavoue, D.: Relative contributions of anthropogenic emissions to black carbon aerosol in the Arctic, J. Geophys. Res.-Atmos., 115, D19208, https://doi.org/10.1029/2009jd013592, 2010a. a
Huang, L., Gong, S. L., Sharma, S., Lavoué, D., and Jia, C. Q.: A trajectory analysis of atmospheric transport of black carbon aerosols to Canadian high Arctic in winter and spring (1990–2005), Atmos. Chem. Phys., 10, 5065–5073, https://doi.org/10.5194/acp-10-5065-2010, 2010b. a
International Institute for Applied Systems Analysis: ECLIPSE V5 global emission fields, available at: http://www.iiasa.ac.at/web/home/research/researchPrograms/air/ECLIPSEv5.html, last access: 30 April 2018. a
Jacobson, M. Z.: Global direct radiative forcing due to multicomponent anthropogenic and natural aerosols, J. Geophys. Res.-Atmos., 106, 1551–1568, https://doi.org/10.1029/2000JD900514, 2001. a
Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X. F., Smith, K. A., Morris, J. W., and Davidovits, P.: Ambient aerosol sampling using the Aerodyne Aerosol Mass Spectrometer, J. Geophys. Res.-Atmos., 108, 8425, https://doi.org/10.1029/2001jd001213, 2003. a, b
Jimenez, J. L., Canagaratna, M. R., Drewnick, F., Allan, J. D., Alfarra, M. R., Middlebrook, A. M., Slowik, J. G., Zhang, Q., Coe, H., Jayne, J. T., and Worsnop, D. R.: Comment on “The effects of molecular weight and thermal decomposition on the sensitivity of a thermal desorption aerosol mass spectrometer”, Aerosol Sci. Tech., 50, i–xv, https://doi.org/10.1080/02786826.2016.1205728, 2016. a
Kawamura, K., Kasukabe, H., and Barrie, L. A.: Source and reaction pathways of dicarboxylic acids, ketoacids and dicarbonyls in Arctic aerosols: One year of observations, Atmos. Environ., 30, 1709–1722, https://doi.org/10.1016/1352-2310(95)00395-9, 1996. a
Kawamura, K., Imai, Y., and Barrie, L. A.: Photochemical production and loss of organic acids in high Arctic aerosols during long-range transport and polar sunrise ozone depletion events, Atmos. Environ., 39, 599–614, https://doi.org/10.1016/j.atmosenv.2004.10.020, 2005. a
Kawamura, K., Kasukabe, H., and Barrie, L. A.: Secondary formation of water-soluble organic acids and alpha-dicarbonyls and their contributions to total carbon and water-soluble organic carbon: Photochemical aging of organic aerosols in the Arctic spring, J. Geophys. Res.-Atmos., 115, D21306, https://doi.org/10.1029/2010jd014299, 2010. a
Kirpes, R. M., Bondy, A. L., Bonanno, D., Moffet, R. C., Wang, B., Laskin, A., Ault, A. P., and Pratt, K. A.: Secondary sulfate is internally mixed with sea spray aerosol and organic aerosol in the winter Arctic, Atmos. Chem. Phys., 18, 3937–3949, https://doi.org/10.5194/acp-18-3937-2018, 2018. a, b
Klonecki, A., Hess, P., Emmons, L., Smith, L., Orlando, J., and Blake, D.: Seasonal changes in the transport of pollutants into the Arctic troposphere-model study, J. Geophys. Res.-Atmos., 108, 8367, https://doi.org/10.1029/2002jd002199, 2003. a, b, c
Koch, D. and Hansen, J.: Distant origins of Arctic black carbon: A Goddard Institute for Space Studies ModelE experiment, J. Geophys. Res.-Atmos., 110, D04204, https://doi.org/10.1029/2004jd005296, 2005. a, b, c, d
Kroll, J. H., Smith, J. D., Che, D. L., Kessler, S. H., Worsnop, D. R., and Wilson, K. R.: Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol, Phys. Chem. Chem. Phys., 11, 8005–8014, https://doi.org/10.1039/B905289E, 2009. a
Kupc, A., Williamson, C., Wagner, N. L., Richardson, M., and Brock, C. A.: Modification, calibration, and performance of the Ultra-High Sensitivity Aerosol Spectrometer for particle size distribution and volatility measurements during the Atmospheric Tomography Mission (ATom) airborne campaign, Atmos. Meas. Tech., 11, 369–383, https://doi.org/10.5194/amt-11-369-2018, 2018. a
Laborde, M., Schnaiter, M., Linke, C., Saathoff, H., Naumann, K.-H., Möhler, O., Berlenz, S., Wagner, U., Taylor, J. W., Liu, D., Flynn, M., Allan, J. D., Coe, H., Heimerl, K., Dahlkötter, F., Weinzierl, B., Wollny, A. G., Zanatta, M., Cozic, J., Laj, P., Hitzenberger, R., Schwarz, J. P., and Gysel, M.: Single Particle Soot Photometer intercomparison at the AIDA chamber, Atmos. Meas. Tech., 5, 3077–3097, https://doi.org/10.5194/amt-5-3077-2012, 2012. a, b
Law, K. S. and Stohl, A.: Arctic air pollution: Origins and impacts, Science, 315, 1537–1540, https://doi.org/10.1126/science.1137695, 2007. a, b
Law, K. S., Stohl, A., Quinn, P. K., Brock, C. A., Burkhart, J. F., Paris, J.-D., Ancellet, G., Singh, H. B., Roiger, A., Schlager, H., Dibb, J., Jacob, D. J., Arnold, S. R., Pelon, J., and Thomas, J. L.: Arctic Air Pollution: New Insights from POLARCAT-IPY, B. Am. Meteorol. Soc., 95, 1873–+, https://doi.org/10.1175/bams-d-13-00017.1, 2014. a, b, c, d, e, f
Leaitch, W. R., Sharma, S., Huang, L., Toom-Sauntry, D., Chivulescu, A., Macdonald, A. M., von Salzen, K., Pierce, J. R., Bertram, A. K., Schroder, J. C., Shantz, N. C., Chang, R. Y. W., and Norman, A.-L.: Dimethyl sulfide control of the clean summertime Arctic aerosol and cloud, Elementa, 1, 000017, https://doi.org/10.12952/journal.elementa.000017, 2013. a
Leaitch, W. R., Korolev, A., Aliabadi, A. A., Burkart, J., Willis, M. D., Abbatt, J. P. D., Bozem, H., Hoor, P., Köllner, F., Schneider, J., Herber, A., Konrad, C., and Brauner, R.: Effects of 20–100 nm particles on liquid clouds in the clean summertime Arctic, Atmos. Chem. Phys., 16, 11107–11124, https://doi.org/10.5194/acp-16-11107-2016, 2016. a, b
Leaitch, W. R., Russell, L. M., Liu, J., Kolonjari, F., Toom, D., Huang, L., Sharma, S., Chivulescu, A., Veber, D., and Zhang, W.: Organic functional groups in the submicron aerosol at 82.5∘ N, 62.5∘ W from 2012 to 2014, Atmos. Chem. Phys., 18, 3269–3287, https://doi.org/10.5194/acp-18-3269-2018, 2018. a, b, c, d, e, f, g, h, i
Leck, C., Norman, M., Bigg, E. K., and Hillamo, R.: Chemical composition and sources of the high Arctic aerosol relevant for cloud formation, J. Geophys. Res.-Atmos., 107, 4135, https://doi.org/10.1029/2001JD001463, 2002. a, b
Lee, A. K. Y., Willis, M. D., Healy, R. M., Onasch, T. B., and Abbatt, J. P. D.: Mixing state of carbonaceous aerosol in an urban environment: single particle characterization using the soot particle aerosol mass spectrometer (SP-AMS), Atmos. Chem. Phys., 15, 1823–1841, https://doi.org/10.5194/acp-15-1823-2015, 2015. a, b, c
Li, S. M. and Winchester, J. W.: Geochemistry of Orangic and Inorganic ions of Late Winter Arctic Aerosol, Atmos. Environ., 23, 2401–2415, 1989. a
Liu, D., Quennehen, B., Darbyshire, E., Allan, J. D., Williams, P. I., Taylor, J. W., Bauguitte, S. J.-B., Flynn, M. J., Lowe, D., Gallagher, M. W., Bower, K. N., Choularton, T. W., and Coe, H.: The importance of Asia as a source of black carbon to the European Arctic during springtime 2013, Atmos. Chem. Phys., 15, 11537–11555, https://doi.org/10.5194/acp-15-11537-2015, 2015. a, b, c
Lubin, D. and Vogelmann, A. M.: A climatologically significant aerosol longwave indirect effect in the Arctic, Nature, 439, 453–456, https://doi.org/10.1038/nature04449, 2006. a
Macdonald, K. M., Sharma, S., Toom, D., Chivulescu, A., Hanna, S., Bertram, A. K., Platt, A., Elsasser, M., Huang, L., Tarasick, D., Chellman, N., McConnell, J. R., Bozem, H., Kunkel, D., Lei, Y. D., Evans, G. J., and Abbatt, J. P. D.: Observations of atmospheric chemical deposition to high Arctic snow, Atmos. Chem. Phys., 17, 5775–5788, https://doi.org/10.5194/acp-17-5775-2017, 2017. a, b
Mao, J., Jacob, D. J., Evans, M. J., Olson, J. R., Ren, X., Brune, W. H., Clair, J. M. St., Crounse, J. D., Spencer, K. M., Beaver, M. R., Wennberg, P. O., Cubison, M. J., Jimenez, J. L., Fried, A., Weibring, P., Walega, J. G., Hall, S. R., Weinheimer, A. J., Cohen, R. C., Chen, G., Crawford, J. H., McNaughton, C., Clarke, A. D., Jaeglé, L., Fisher, J. A., Yantosca, R. M., Le Sager, P., and Carouge, C.: Chemistry of hydrogen oxide radicals (HOx) in the Arctic troposphere in spring, Atmos. Chem. Phys., 10, 5823–5838, https://doi.org/10.5194/acp-10-5823-2010, 2010. a
Matsui, H., Kondo, Y., Moteki, N., Takegawa, N., Sahu, L. K., Koike, M., Zhao, Y., Fuelberg, H. E., Sessions, W. R., Diskin, G., Anderson, B. E., Blake, D. R., Wisthaler, A., Cubison, M. J., and Jimenez, J. L.: Accumulation-mode aerosol number concentrations in the Arctic during the ARCTAS aircraft campaign: Long-range transport of polluted and clean air from the Asian continent, J. Geophys. Res.-Atmos., 116, D20217, https://doi.org/10.1029/2011jd016189, 2011a. a
Matsui, H., Kondo, Y., Moteki, N., Takegawa, N., Sahu, L. K., Zhao, Y., Fuelberg, H. E., Sessions, W. R., Diskin, G., Blake, D. R., Wisthaler, A., and Koike, M.: Seasonal variation of the transport of black carbon aerosol from the Asian continent to the Arctic during the ARCTAS aircraft campaign, J. Geophys. Res.-Atmos., 116, D05202, https://doi.org/10.1029/2010jd015067, 2011b. a
May, N. W., Quinn, P. K., McNamara, S. M., and Pratt, K. A.: Multiyear study of the dependence of sea salt aerosol on wind speed and sea ice conditions in the coastal Arctic, J. Geophys. Res.-Atmos., 121, 9208–9219, https://doi.org/10.1002/2016jd025273, 2016. a, b
McNaughton, C. S., Clarke, A. D., Freitag, S., Kapustin, V. N., Kondo, Y., Moteki, N., Sahu, L., Takegawa, N., Schwarz, J. P., Spackman, J. R., Watts, L., Diskin, G., Podolske, J., Holloway, J. S., Wisthaler, A., Mikoviny, T., de Gouw, J., Warneke, C., Jimenez, J., Cubison, M., Howell, S. G., Middlebrook, A., Bahreini, R., Anderson, B. E., Winstead, E., Thornhill, K. L., Lack, D., Cozic, J., and Brock, C. A.: Absorbing aerosol in the troposphere of the Western Arctic during the 2008 ARCTAS/ARCPAC airborne field campaigns, Atmos. Chem. Phys., 11, 7561–7582, https://doi.org/10.5194/acp-11-7561-2011, 2011. a, b, c
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012. a
Monks, S. A., Arnold, S. R., Emmons, L. K., Law, K. S., Turquety, S., Duncan, B. N., Flemming, J., Huijnen, V., Tilmes, S., Langner, J., Mao, J., Long, Y., Thomas, J. L., Steenrod, S. D., Raut, J. C., Wilson, C., Chipperfield, M. P., Diskin, G. S., Weinheimer, A., Schlager, H., and Ancellet, G.: Multi-model study of chemical and physical controls on transport of anthropogenic and biomass burning pollution to the Arctic, Atmos. Chem. Phys., 15, 3575–3603, https://doi.org/10.5194/acp-15-3575-2015, 2015. a, b
Najafi, M. R., Zwiers, F. W., and Gillett, N. P.: Attribution of Arctic temperature change to greenhouse-gas and aerosol influences, Nat. Clim. Change, 5, 246–249, https://doi.org/10.1038/nclimate2524, 2015. a
Narukawa, M., Kawamura, K., Li, S. M., and Bottenheim, J. W.: Stable carbon isotopic ratios and ionic composition of the high-Arctic aerosols: An increase in delta-13C values from winter to spring, J. Geophys. Res., 113, D02312, https://doi.org/10.1029/2007jd008755, 2008. a
NASA: MODIS Collection 6 NRT Hotspot/Active Fire Detections MCD14DL, https://doi.org/10.5067/FIRMS/MODIS/MCD14DL.NRT.006, 2015. a
Navarro, J. C. A., Varma, V., Riipinen, I., Seland, O., Kirkevag, A., Struthers, H., Iversen, T., Hansson, H. C., and Ekman, A. M. L.: Amplification of Arctic warming by past air pollution reductions in Europe, Nat. Geosci., 9, 277–281, https://doi.org/10.1038/ngeo2673, 2016. a
Nguyen, Q. T., Glasius, M., Sørensen, L. L., Jensen, B., Skov, H., Birmili, W., Wiedensohler, A., Kristensson, A., Nøjgaard, J. K., and Massling, A.: Seasonal variation of atmospheric particle number concentrations, new particle formation and atmospheric oxidation capacity at the high Arctic site Villum Research Station, Station Nord, Atmos. Chem. Phys., 16, 11319–11336, https://doi.org/10.5194/acp-16-11319-2016, 2016. a
Oltmans, S. J., Johnson, B. J., and Harris, J. M.: Springtime boundary layer ozone depletion at Barrow, Alaska: Meteorological influence, year-to-year variation, and long-term change, J. Geophys. Res.-Atmos., 117, D00R18, https://doi.org/10.1029/2011JD016889, 2012. a
Onasch, T. B., Trimborn, A., Fortner, E. C., Jayne, J. T., Kok, G. L., Williams, L. R., Davidovits, P., and Worsnop, D. R.: Soot Particle Aerosol Mass Spectrometer: Development, Validation, and Initial Application, Aerosol Sci. Tech., 46, 804–817, https://doi.org/10.1080/02786826.2012.663948, 2012. a, b
Ovadnevaite, J., Ceburnis, D., Canagaratna, M., Berresheim, H., Bialek, J., Martucci, G., Worsnop, D. R., and O'Dowd, C.: On the effect of wind speed on submicron sea salt mass concentrations and source fluxes, J. Geophys. Res.-Atmos., 117, D16201, https://doi.org/10.1029/2011jd017379, 2012. a, b, c
Pratt, K. A., Custard, K. D., Shepson, P. B., Douglas, T. A., Poehler, D., General, S., Zielcke, J., Simpson, W. R., Platt, U., Tanner, D. J., Huey, L. G., Carlsen, M., and Stirm, B. H.: Photochemical production of molecular bromine in Arctic surface snowpacks, Nat. Geosci., 6, 351–356, https://doi.org/10.1038/ngeo1779, 2013. a
Qi, L., Li, Q., Henze, D. K., Tseng, H.-L., and He, C.: Sources of springtime surface black carbon in the Arctic: an adjoint analysis for April 2008, Atmos. Chem. Phys., 17, 9697–9716, https://doi.org/10.5194/acp-17-9697-2017, 2017. a, b, c, d, e
Quinn, P. K., Miller, T. L., Bates, T. S., Ogren, J. A., Andrews, E., and Shaw, G. E.: A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska, J. Geophys. Res.-Atmos., 107, 4130, https://doi.org/10.1029/2001jd001248, 2002. a, b
Quinn, P. K., Shaw, G., Andrews, E., Dutton, E. G., Ruoho-Airola, T., and Gong, S. L.: Arctic haze: current trends and knowledge gaps, Tellus B, 59, 99–114, https://doi.org/10.1111/j.1600-0889.2006.00238.x, 2007. a, b, c
Quinn, P. K., Bates, T. S., Baum, E., Doubleday, N., Fiore, A. M., Flanner, M., Fridlind, A., Garrett, T. J., Koch, D., Menon, S., Shindell, D., Stohl, A., and Warren, S. G.: Short-lived pollutants in the Arctic: their climate impact and possible mitigation strategies, Atmos. Chem. Phys., 8, 1723–1735, https://doi.org/10.5194/acp-8-1723-2008, 2008. a
Rahn, K. A., Borys, R. D., and Shaw, G. E.: Asian Source of Arctic Haze Bands, Nature, 268, 713–715, https://doi.org/10.1038/268713a0, 1977. a
Rinke, A., Dethloff, K., and Fortmann, M.: Regional climate effects of Arctic Haze, Geophys. Res. Lett., 31, L16202, https://doi.org/10.1029/2004gl020318, 2004. a
Ritter, C., Notholt, J., Fischer, J., and Rathke, C.: Direct thermal radiative forcing of tropospheric aerosol in the Arctic measured by ground based infrared spectrometry, Geophys. Res. Lett., 32, L23816, https://doi.org/10.1029/2005gl024331, 2005. a
Sassen, K., DeMott, P. J., Prospero, J. M., and Poellot, M. R.: Saharan dust storms and indirect aerosol effects on clouds CRYSTAL–FACE results, Geophys. Res. Lett., 30, 1633, https://doi.org/10.1029/2003GL017371, 2003. a
Scheuer, E., Talbot, R. W., Dibb, J. E., Seid, G. K., DeBell, L., and Lefer, B.: Seasonal distributions of fine aerosol sulfate in the North American Arctic basin during TOPSE, J. Geophys. Res.-Atmos., 108, 8370, https://doi.org/10.1029/2001jd001364, 2003. a, b
Schmale, J., Schneider, J., Ancellet, G., Quennehen, B., Stohl, A., Sodemann, H., Burkhart, J. F., Hamburger, T., Arnold, S. R., Schwarzenboeck, A., Borrmann, S., and Law, K. S.: Source identification and airborne chemical characterisation of aerosol pollution from long-range transport over Greenland during POLARCAT summer campaign 2008, Atmos. Chem. Phys., 11, 10097–10123, https://doi.org/10.5194/acp-11-10097-2011, 2011. a
Schulz, H., Bozem, H., Zanatta, M., Leaitch, W. R., Herber, A. B., Burkart, J., Willis, M. D., Hoor, P. M., Abbatt, J. P. D., and Gerdes, R.: High-Arctic aircraft measurements characterising black carbon vertical variability in spring and summer, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2018-587, in review, 2018. a, b
Schwarz, J. P., Gao, R. S., Fahey, D. W., Thomson, D. S., Watts, L. A., Wilson, J. C., Reeves, J. M., Darbeheshti, M., Baumgardner, D. G., Kok, G. L., Chung, S. H., Schulz, M., Hendricks, J., Lauer, A., Kärcher, B., Slowik, J. G., Rosenlof, K. H., Thompson, T. L., Langford, A. O., Loewenstein, M., and Aikin, K. C.: Single-particle measurements of midlatitude black carbon and light-scattering aerosols from the boundary layer to the lower stratosphere, J. Geophys. Res.-Atmos., 111, D16207, https://doi.org/10.1029/2006JD007076, 2006. a
Sharma, S., Lavoue, D., Cachier, H., Barrie, L. A., and Gong, S. L.: Long-term trends of the black carbon concentrations in the Canadian Arctic, J. Geophys. Res.-Atmos., 109, D15203, https://doi.org/10.1029/2003jd004331, 2004. a
Sharma, S., Andrews, E., Barrie, L. A., Ogren, J. A., and Lavoue, D.: Variations and sources of the equivalent black carbon in the high Arctic revealed by long-term observations at Alert and Barrow: 1989-2003, J. Geophys. Res.-Atmos., 111, D14208, https://doi.org/10.1029/2005jd006581, 2006. a
Sharma, S., Ishizawa, M., Chan, D., Lavoue, D., Andrews, E., Eleftheriadis, K., and Maksyutov, S.: 16-year simulation of Arctic black carbon: Transport, source contribution, and sensitivity analysis on deposition, J. Geophys. Res.-Atmos., 118, 943–964, https://doi.org/10.1029/2012jd017774, 2013. a, b
Shaw, G. E.: The Arctic haze phenomenon, B. Am. Meteorol. Soc., 76, 2403–2413, https://doi.org/10.1175/1520-0477(1995)076<2403:tahp>2.0.co;2, 1995. a
Shaw, P. M., Russell, L. M., Jefferson, A., and Quinn, P. K.: Arctic organic aerosol measurements show particles from mixed combustion in spring haze and from frost flowers in winter, Geophys. Res. Lett., 37, L10803, https://doi.org/10.1029/2010gl042831, 2010. a, b, c
Shindell, D. and Faluvegi, G.: Climate response to regional radiative forcing during the twentieth century, Nat. Geosci., 2, 294–300, https://doi.org/10.1038/ngeo473, 2009. a
Shindell, D. T., Chin, M., Dentener, F., Doherty, R. M., Faluvegi, G., Fiore, A. M., Hess, P., Koch, D. M., MacKenzie, I. A., Sanderson, M. G., Schultz, M. G., Schulz, M., Stevenson, D. S., Teich, H., Textor, C., Wild, O., Bergmann, D. J., Bey, I., Bian, H., Cuvelier, C., Duncan, B. N., Folberth, G., Horowitz, L. W., Jonson, J., Kaminski, J. W., Marmer, E., Park, R., Pringle, K. J., Schroeder, S., Szopa, S., Takemura, T., Zeng, G., Keating, T. J., and Zuber, A.: A multi-model assessment of pollution transport to the Arctic, Atmos. Chem. Phys., 8, 5353–5372, https://doi.org/10.5194/acp-8-5353-2008, 2008. a, b
Sirois, A. and Barrie, L. A.: Arctic lower tropospheric aerosol trends and composition at Alert, Canada: 1980–1995, J. Geophys. Res.-Atmos., 104, 11599–11618, https://doi.org/10.1029/1999jd900077, 1999. a
Spackman, J. R., Gao, R. S., Neff, W. D., Schwarz, J. P., Watts, L. A., Fahey, D. W., Holloway, J. S., Ryerson, T. B., Peischl, J., and Brock, C. A.: Aircraft observations of enhancement and depletion of black carbon mass in the springtime Arctic, Atmos. Chem. Phys., 10, 9667–9680, https://doi.org/10.5194/acp-10-9667-2010, 2010. a
Spreen, G., Kaleschke, L., and Heygster, G.: Sea ice remote sensing using AMSR-E 89-GHz channels, J. Geophys. Res., 113, C02S03, https://doi.org/10.1029/2005jc003384, 2008 (data available at: https://seaice.uni-bremen.de/sea-ice-concentration/, last access: 30 April 2018). a, b
Stohl, A.: Characteristics of atmospheric transport into the Arctic troposphere, J. Geophys. Res.-Atmos., 111, D11306, https://doi.org/10.1029/2005jd006888, 2006. a, b, c, d, e, f, g
Stohl, A., Klimont, Z., Eckhardt, S., Kupiainen, K., Shevchenko, V. P., Kopeikin, V. M., and Novigatsky, A. N.: Black carbon in the Arctic: the underestimated role of gas flaring and residential combustion emissions, Atmos. Chem. Phys., 13, 8833–8855, https://doi.org/10.5194/acp-13-8833-2013, 2013. a
Sueper, D. T.: ToF-AMS analysis software downloads, available at: http://cires.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/index.html (last access: 15 December 2018), 2010. a
Tjernström, M. and Graversen, R. G.: The vertical structure of the lower Arctic troposphere analysed from observations and the ERA-40 reanalysis, Q. J. Roy. Meteor. Soc., 135, 431–443, https://doi.org/10.1002/qj.380, 2009. a
Toom-Sauntry, D. and Barrie, L. A.: Chemical composition of snowfall in the high Arctic: 1990-1994, Atmos. Environ., 36, 2683–2693, https://doi.org/10.1016/s1352-2310(02)00115-2, 2002. a
Treffeisen, R., Rinke, A., Fortmann, M., Dethloff, K., Herber, A., and Yamanouchi, T.: A case study of the radiative effects of Arctic aerosols in March 2000, Atmos. Environ., 39, 899–911, https://doi.org/10.1016/j.atmosenv.2004.09.066, 2005. a
Tunved, P., Ström, J., and Krejci, R.: Arctic aerosol life cycle: linking aerosol size distributions observed between 2000 and 2010 with air mass transport and precipitation at Zeppelin station, Ny-Ålesund, Svalbard, Atmos. Chem. Phys., 13, 3643–3660, https://doi.org/10.5194/acp-13-3643-2013, 2013. a
Wagner, R., Kaufmann, J., Möhler, O., Saathoff, H., Schnaiter, M., Ullrich, R., and Leisner, T.: Heterogeneous Ice Nucleation Ability of NaCl and Sea Salt Aerosol Particles at Cirrus Temperatures, J. Geophys. Res.-Atmos., 123, 2841–2860, https://doi.org/10.1002/2017JD027864, 2018. a
Wang, J., Jacob, D. J., and Martin, S. T.: Sensitivity of sulfate direct climate forcing to the hysteresis of particle phase transitions, J. Geophys. Res.-Atmos., 113, D11207, https://doi.org/10.1029/2007JD009368, 2008. a, b
Warneke, C., Bahreini, R., Brioude, J., Brock, C. A., de Gouw, J. A., Fahey, D. W., Froyd, K. D., Holloway, J. S., Middlebrook, A., Miller, L., Montzka, S., Murphy, D. M., Peischl, J., Ryerson, T. B., Schwarz, J. P., Spackman, J. R., and Veres, P.: Biomass burning in Siberia and Kazakhstan as an important source for haze over the Alaskan Arctic in April 2008, Geophys. Res. Lett., 36, L02813, https://doi.org/10.1029/2008gl036194, 2009. a, b, c, d
Wentworth, G. R., Murphy, J. G., Croft, B., Martin, R. V., Pierce, J. R., Côté, J.-S., Courchesne, I., Tremblay, J.-É., Gagnon, J., Thomas, J. L., Sharma, S., Toom-Sauntry, D., Chivulescu, A., Levasseur, M., and Abbatt, J. P. D.: Ammonia in the summertime Arctic marine boundary layer: sources, sinks, and implications, Atmos. Chem. Phys., 16, 1937–1953, https://doi.org/10.5194/acp-16-1937-2016, 2016. a
Willis, M. D., Burkart, J., Thomas, J. L., Köllner, F., Schneider, J., Bozem, H., Hoor, P. M., Aliabadi, A. A., Schulz, H., Herber, A. B., Leaitch, W. R., and Abbatt, J. P. D.: Growth of nucleation mode particles in the summertime Arctic: a case study, Atmos. Chem. Phys., 16, 7663–7679, https://doi.org/10.5194/acp-16-7663-2016, 2016. a, b
Willis, M. D., Köllner, F., Burkart, J., Bozem, H., Thomas, J. L., Schneider, J., Aliabadi, A. A., Hoor, P. M., Schulz, H., Herber, A. B., Leaitch, W. R., and Abbatt, J. P. D.: Evidence for marine biogenic influence on summertime Arctic aerosol, Geophys. Res. Lett., 44, 6460–6470, https://doi.org/10.1002/2017gl073359, 2017. a
Xu, J.-W., Martin, R. V., Morrow, A., Sharma, S., Huang, L., Leaitch, W. R., Burkart, J., Schulz, H., Zanatta, M., Willis, M. D., Henze, D. K., Lee, C. J., Herber, A. B., and Abbatt, J. P. D.: Source attribution of Arctic black carbon constrained by aircraft and surface measurements, Atmos. Chem. Phys., 17, 11971–11989, https://doi.org/10.5194/acp-17-11971-2017, 2017. a, b, c, d, e, f, g
Xu, L., Russell, L. M., and Burrows, S. M.: Potential sea salt aerosol sources from frost flowers in the pan-Arctic region, J. Geophys. Res.-Atmos., 121, 10840–10856, https://doi.org/10.1002/2015jd024713, 2016. a
Yang, X., Pyle, J. A., and Cox, R. A.: Sea salt aerosol production and bromine release: Role of snow on sea ice, Geophys. Res. Lett., 35, L16815, https://doi.org/10.1029/2008gl034536, 2008. a
Zhang, Y., Seidel, D. J., Golaz, J.-C., Deser, C., and Tomas, R. A.: Climatological Characteristics of Arctic and Antarctic Surface-Based Inversions, J. Climate, 24, 5167–5186, https://doi.org/10.1175/2011JCLI4004.1, 2011. a
Zhao, C. F. and Garrett, T. J.: Effects of Arctic haze on surface cloud radiative forcing, Geophys. Res. Lett., 42, 557–564, https://doi.org/10.1002/2014gl062015, 2015. a