the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 Mar 2019
| 25 Mar 2019
Seasonality in the Δ33S measured in urban aerosols highlights an additional oxidation pathway for atmospheric SO2
David Au Yang
Pierre Cartigny
Karine Desboeufs
David Widory
Sulfates present in urban aerosols collected worldwide usually exhibit significant non-zero Δ33S signatures (from −0.6 ‰ to 0.5 ‰) whose origin still remains unclear. To better address this issue, we recorded the seasonal variations of the multiple sulfur isotope compositions of PM10 aerosols collected over the year 2013 at five stations within the Montreal Island (Canada), each characterized by distinct types and levels of pollution. The δ34S-values (n= 155) vary from 2.0 ‰ to 11.3 ‰ (±0.2 ‰, 2σ), the Δ33S-values from −0.080 ‰ to 0.341 ‰ (±0.01 ‰, 2σ) and the Δ36S-values from −1.082 ‰ to 1.751 ‰ (±0.2 ‰, 2σ). Our study evidences a seasonality for both the δ34S and Δ33S, which can be observed either when considering all monitoring stations or, to a lesser degree, when considering them individually. Among them, the monitoring station located at the most western end of the island, upstream of local emissions, yields the lowest mean δ34S coupled to the highest mean Δ33S-values. The Δ33S-values are higher during both summer and winter, and are < 0.1 ‰ during both spring and autumn. As these higher Δ33S-values are measured in “upstream” aerosols, we conclude that the mechanism responsible for these highly positive S-MIF also occurs outside and not within the city, at odds with common assumptions. While the origin of such variability in the Δ33S-values of urban aerosols (i.e. −0.6 ‰ to 0.5 ‰) is still subject to debate, we suggest that oxidation by Criegee radicals and/or photooxidation of atmospheric SO2 in the presence of mineral dust may play a role in generating such large ranges of S-MIF.
- Article
(4259 KB) - Full-text XML
-
Supplement
(327 KB) - BibTeX
- EndNote





Sulfur (S) is an element of environmental interest due to its key role in climate change and air pollution (Seinfeld and Pandis, 2012). Indeed, gaseous sulfur-bearing compounds are ubiquitous in both the marine and urban environments (Bardouki et al., 2003; Wall et al., 1988). The SO2 which could come from direct emissions or which result from the oxidation of H2S, dimethylsulfide (DMS) is the most common S-bearing gas in the atmosphere of which about 60 % is deposited and eliminated from the atmosphere (Berglen et al., 2004; Harris et al., 2013b). The remaining 40 % are ultimately oxidized into sulfate () following two major chemical pathways (gaseous and aqueous) that will actually condensate, forming secondary sulfate aerosols (Seinfeld and Pandis, 2012; Tomasi and Lupi, 2017). The sulfate particles have chemical compositions ranging from sulfuric acid droplets to ammonium sulfates (Sinha et al., 2008), depending on the availability of gaseous ammonia to neutralize the sulfuric acid. Sulfate aerosols affect both human health and climate (Albrecht, 1989; Lelieveld et al., 2015; Levy et al., 2013; Myhre et al., 2013; Penner et al., 1992, 2006; Ramanathan et al., 2001, 2005). The chemical pathway converting precursors to sulfates is important because it changes radiative effects in the atmosphere. The gaseous phase oxidation, which occurs predominantly via OH, permits the formation of new sulfate particles by homogeneous nucleation process (Benson, 2008; Kulmala et al., 2004; Tanaka et al., 1994). This nucleation via gas-to-particle conversion is the largest source of atmospheric aerosol particles in the free troposphere (Kulmala et al., 2004), providing up to half of the global cloud condensation nuclei (Merikanto et al., 2009; Yu and Luo, 2009). The SO2 aqueous phase oxidation which occurs via several oxidants, the major ones being O2+TMI (Transition Metal Ion), H2O2, O3 and NO2 (Alexander et al., 2009, 2012; Cheng et al., 2016; Harris et al., 2013a, b; Herrmann, 2003; Sarwar et al., 2013; Seinfeld and Pandis, 2012; Lee and Schwartz, 1983), produces sulfates which will be released during the evaporation of cloud water. These sulfates will condense on pre-existing particles present in the cloud droplets (Mertes et al., 2005a, b). By causing some to condense onto larger particles with lower scattering efficiencies and shorter atmospheric lifetimes, the heterogeneous reactions of SO2 on mineral aerosols (Andreae and Crutzen, 1997) and the oxidation of SO2 into sulfate in sea-salt-containing cloud droplets and deliquesced sea-salt aerosols reduce the radiative impact of sulfate aerosols.
Sulfur has four stable isotopes, 32S, 33S, 34S and 36S whose natural abundances are approximately 95 %, 0.75 %, 4.2 % and 0.015 %, respectively (Ding et al., 2001). The S-isotope compositions are expressed using the δ-notation defined as (Coplen, 2011):
where 3xS is one of the S heavy isotopes (33S, 34S or 36S) and CDT is the Vienna Canyon Diablo Troilite 34S∕32S international standard. There is no international standard for the 33S∕32S and 36S∕32S. Accuracy of the measured values is established by direct comparison with data measured by other laboratories.
Stable isotopes fractionate during unidirectional (kinetic) and/or exchange (equilibrium) reactions, resulting in the product and reactant having distinct isotope compositions. Isotope fractionation factors are expressed using the α-notation, which relates the two isotope compositions as follows (expressed here for the SO2 oxidation into SO4). (Coplen, 2011):
At equilibrium, the three sulfur isotope ratios are usually scaled to each other according to their mass ((1∕ m m2) ∕ (1∕ m m3)), following a “mass-dependent fractionation” model (Farquhar et al., 2000). The isotope fractionation of 33S over 32S (1 amu difference) has approximately half the magnitude of the fractionation of the 34S over 32S (2 amu difference). More rigorously, mass-dependent fractionation is expressed by Young et al. (2002) and Dauphas and Schauble (2016):
where 3yα is either 33α or 36α and 3yβ is either 33β or 36β. The 3yβ-exponent describes the relative fractionation of 3yS ∕ 32S and 34S∕32S. The β-exponent is usually expressed as θ to refer to isotope equilibrium. We are using β-instead as the processes describing the SO2-oxidation are actually not at the isotope equilibrium. Its value depends on the reaction considered (Farquhar et al., 2001; Harris et al., 2013a; Ono et al., 2013; Watanabe et al., 2009). At high temperature (> 500 ∘C, i.e. under equilibrium), 33θ and 36θ-values are, respectively, 0.515 and 1.889 (Eldridge et al., 2016; Otake et al., 2008). Deviation of the 3yβ-value from these high temperature values usually leads to non-zero Δ33S and Δ36S values typically in the range of ±0.1 ‰ and ±1 ‰, respectively. Δ33S and Δ36S are expressed as follows (Farquhar and Wing, 2003):
Non-zero Δ33S-values can result from non-equilibrium (kinetic) processes and their combination through Rayleigh fractionation and other mass conservation effects (Farquhar et al., 2007; Ono et al., 2006a; Harris et al., 2013a).
Previous studies showed that sulfates in urban aerosols display δ34S-values from 0 ‰ to 20 ‰ and Δ33S-values from −0.6 ‰ to 0.5 ‰ (Guo et al., 2010; Romero and Thiemens, 2003; Shaheen et al., 2014; Lin et al., 2018b; Han et al., 2017). Two main lines of reasoning are usually evoked to explain such S-isotope compositions. The first one neglects the role of S-isotope fractionation and uses S-isotopes as a direct tracer of emission sources that have been shown to be characterized by large and distinct δ34S-values (Becker and Hirner, 1998; Calhoun et al., 1991; Gaffney et al., 1980; Guo et al., 2016; Newman and Forrest, 1991; Nielsen, 1974; Norman et al., 2006; Premuzic et al., 1986; Smith and Batts, 1974; Wadleigh et al., 1996; Wasiuta et al., 2015). For example, sea-salt sulfate is characterized by a δ34S-value of 21 ‰ (Rees et al., 1978), marine biogenic non-sea-salt sulfate has a δ34S ranging from 12 ‰ to 19 ‰ (Calhoun et al., 1991; Sanusi et al., 2006; Oduro et al., 2012), while anthropogenic sulfur emissions are often lighter although there are significant variations between sources ranging from −40 ‰ to 30 ‰ (Nielsen, 1974; Norman et al., 2006; Wasiuta et al., 2015; Krouse and Grinenko, 1991). The alternative interpretation relies on a constant SO2 isotope composition. The variations observed in the sulfur multiple isotope compositions (δ34S and Δ33S) of rural sulfate aerosols reflect changes in the atmospheric concentrations of SO2 oxidants, each having distinct fractionation factors (Harris et al., 2012b, c, 2013a, b). In this case, high (up to ∼7 ‰) and low (down to ∼1 ‰) δ34S-values are predicted during winter and summer, respectively (Harris et al., 2013a).
However to date, these −0.6 ‰ to 0.5 ‰ Δ33S-values reported in urban aerosols cannot be fully explained by a source effect, given that corresponding isotope compositions for emission sources vary from −0.2 ‰ to 0.2 ‰ (Lee et al., 2002). These also cannot be explained by the experimentally determined 34α and 33α-values currently available in the literature (implicating O2+TMI, H2O2 and OH; Harris et al., 2013a, and NO2; Au Yang et al., 2018) or their potential combinations that predict Δ33S-values centered around 0 ‰; i.e. at odds with available data for urban aerosols (Guo et al., 2010; Romero and Thiemens, 2003; Shaheen et al., 2014; Lin et al., 2018b; Han et al., 2017).
Non-zero Δ33S-values may thus ultimately be attributed to urban-specific chemical reactions linked to the polluted urban environment. With that in mind and using Montreal as our study site, our objectives were (i) to identify where the most positive Δ33S-values are produced: outside or inside the city, and (ii) to characterize the Δ33S urban seasonality and decipher whether local emissions tend to increase/decrease the Δ33S-values.
2.1 Sampling site
PM10 aerosols (particles with an aerodynamical diameter < 10 µm) were sampled over a 1-year period in 2013 by the RSQA (Réseau de Surveillance de la Qualité de l'Air) in the city of Montreal (Canada; 45∘ N 73∘ W) and its vicinity. Montreal is considered as a relatively lowly polluted city with an average annual PM10 concentration of 16 µg m−3 (World Health Organization, 2016). Montreal therefore respects the 20 µg m−3 guidelines set by the World Health Organization (WHO), while also exhibiting local variations with several stations recording concentrations punctually exceeding the mean 50 µg m−3 24 h guidelines (Boulet and Melançon, 2012, 2013).
Five monitoring stations (03, 06, 13, 50, and 98) disseminated onto the Montreal island were selected: (i) station 03, “Saint-Jean-Baptiste”, is located at the north-eastern end of the island and due to the dominant wind directions is likely more influenced by local petro-chemistry industries. (ii) Station 06, “Anjou”, is located close to a two high-traffic highways interchange (highways 40 and 25). (iii) Downtown station 13, “Drummond”, represents the urban background. (iv) Station 50, “Hochelaga-Maisonneuve”, located at the Old Port of Montreal, is expected to be influenced by maritime activities. (v) Station 98, “Sainte-Anne de Bellevue”, is at the western end of the island in a semi-rural environment about 35 km upstream of Montreal downtown and is thus less likely impacted by the global city anthropogenic emissions (Boulet and Melançon, 2012). The four stations 03, 06, 13 and 50 are thus usually considered to be representative of the different polluted environments in the city, while station 98 is more akin to sample aerosols generated elsewhere upstream and transported to Montreal.
PM10 samples were weekly collected on pre-combusted quartz filters using a high volume PM10 size selective inlet, with an average flow of 1.13 m3 min−1 for a period of 24 h, i.e. a sampled air volume around 1627 m3 by filters. Three to four filters were selected and analyzed for each month trying to select the same sampling periods for all stations. The atmospheric PM10 concentrations during the sampling periods in the five stations range from 2 to 138 µg m−3, covering typical values of both background and particulate pollution periods. Blank filters were analyzed by RSQA and showed no significant amount of S (below the detection limit of 0.2 mg L−1) and generated no sulfate when we followed the chemistry method that we are describing below.
2.2 Sulfur multi-isotope analysis
Before analyzing the S multi-isotope compositions, speciation of S in the aerosol samples needed to be addressed to understand the form(s) under which S was present (i.e. sulfates, sulfur and/or sulfide; Longo et al., 2016). Neither elemental sulfur nor sulfide was detected in our selected representative samples (n= 5) indicating that S probably only occurs as sulfates. For each filter sample, a ∼5 cm × 5 cm piece was cut and inserted into a reaction vessel heated at 180 ∘C with 20 mL of Thode solution, a mixture of hydrochloric, hydroiodic and hypophosphorous acids (Thode et al., 1961), for 1.3 h to quantitatively reduce sulfate into H2S. The formed gases were purged from the vessel using nitrogen gas, bubbled through deionized water and subsequently passed through a 0.3 M silver nitrate (AgNO3) solution to form silver sulfide (Ag2S). This solid Ag2S was then rinsed twice with Millipore water and dried at 70 ∘C overnight. Ag2S was then loaded into an aluminum foil, weighted and degassed under vacuum.
Ag2S was subsequently converted to SF6 by reacting with approximately 200 Torr of excess fluorine in a nickel bomb at 250 ∘C. The produced SF6 was purified using both cryogenic techniques and gas chromatography, quantified and subsequently analyzed by dual inlet isotope ratio mass spectrometry (Thermo-Fisher MAT-253) where , 128, 129 and 131 ion beams were monitored. For S amounts down to 0.1 µmol, samples were analyzed using a micro-volume device (Au Yang et al., 2016). For S amounts smaller than 0.1 µmol, only the Δ33S-values are reported, as the intensity of the 36S is then too small to be properly analyzed.
The δ34S-values were measured against our in-house SF6 tank that had been previously calibrated with respect to the IAEA-S1, IAEA-S2 and IAEA-S3 international standards and expressed vs. V-CDT assuming a δ34S ‰ vs. V-CDT isotope composition. To express our Δ33S and Δ36S data with respect to V-CDT, we anchored our data using CDT data measured previously in our laboratory following Defouilloy et al. (2016). No further corrections were carried out, other than normalization of the data to CDT. Δ33S and Δ36S IAEA-standards were within values reported elsewhere (Labidi et al., 2012; Defouilloy et al., 2016; Au Yang et al., 2016). Our analysis (n=8) of the IAEA-S1 standard yielded ‰, ‰ and ‰ vs. CDT. Analysis of IAEA-S2 standard (n=8) gave ‰, ‰ and ‰ vs. CDT. Analysis of IAEA-S3 standard (n=8) gave ‰, ‰ and ‰ vs. CDT. Analyses of the international sulfate standard NBS-127 was also performed and gave a δ34S of 20.8±0.4 ‰ (2σ; n=12), consistent with the 20.3±0.4 ‰ value reported by the IAEA.
2.3 Chemical analysis
Concentrations of selected soluble inorganic species (Na2+, , K+, Ca2+, Mg2+, , , Cl−, F−, ) were measured by ion chromatography (Professional IC 850 by Metrohm®) after extraction of another 3 cm × 3 cm filter piece in 30 mL Milli-Q water (Paris et al., 2010). Detection limits for ionic species are usually in the order of 5 µg L−1, i.e. 0.1 ng m−3 considering our sampling and extraction protocol.
3.1 Multiple sulfur isotopic compositions
3.1.1 Description
Sulfur multi-isotope compositions in aerosol sulfates from stations 03, 06, 13, 50 and 98 are reported in Tables S1, S2, S3, S4, and S5 in the Supplement, respectively. For station 03, δ34S-values vary from 2 ‰ to 8 ‰, Δ33S-values from −0.006 ‰ to 0.208 ‰ and Δ36S-values from 0.9 ‰ to −0.7 ‰. For station 06, δ34S-values vary from 2 ‰ to 11 ‰, Δ33S-values from −0.075 ‰ to 0.319 ‰ and Δ36S-values from −0.8 ‰ to 0.7 ‰. For station 13, δ34S-values vary from 2 ‰ to 11 ‰, Δ33S-values from −0.080 ‰ to 0.209 ‰ and Δ36S-values from −0.5 ‰ to 0.8 ‰. For station 50, δ34S-values vary from 3 ‰ to 11 ‰, Δ33S-values from −0.018 ‰ to 0.316 ‰ and Δ36S-values from −0.5 ‰ to 0.8 ‰. For station 98, δ34S-values vary from 2 ‰ to 8 ‰, Δ33S-values from −0.022 ‰ to 0.341 ‰ and Δ36S-values from −1 ‰ to 1.7 ‰.
Station 98 (i.e. the westernmost station likely less influenced by local anthropogenic emissions) presents δ34S-values ranging from 2 ‰ to 8 ‰ while other stations, likely impacted by local anthropogenic sources (stations 03, 06, 13 and 50), have δ34S ranging from 2 ‰ to 12 ‰. Stations 06, 13 and 50, i.e. more influenced by highways, the downtown and maritime traffic, respectively, typically present the highest δ34S-values and display a similar general trend with the seasonality. All the stations (03, 06, 13, 50 and 98) present similar range of Δ33S-values ranging from −0.075 ‰ to 0.341 ‰. By contrast, the Δ36S-values for stations around Montreal downtown (from −0.8 ‰ to 0.8 ‰) are in the isotope range found for station 98 (from −1 ‰ to 1.7 ‰).
3.1.2 Evidence of seasonality
In order to reveal the seasonal variations of the S multi-isotope compositions for each station, a locally weighted scatter plot smoothing (LOWESS) was applied (Fig. 1). All stations present seasonal δ34S variations that follow a similar pattern (Fig. 1): low δ34S-values from early winter on, followed by significant 34S enrichment during spring. Then, the isotope compositions decrease from the summer to autumn, back to the early winter values. However, isotope differences in the δ34S range are observed between sites.
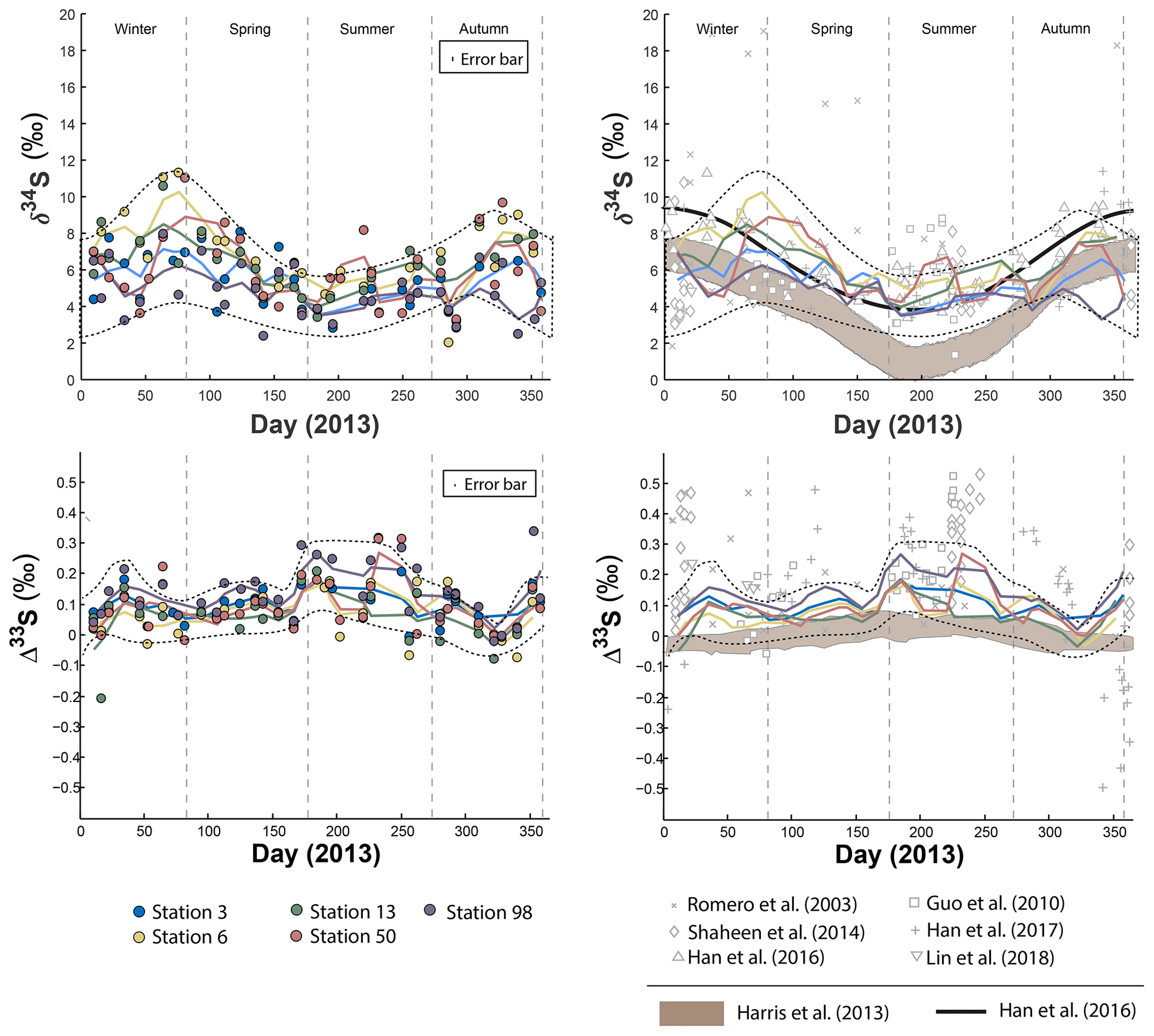
Figure 1Sulfate δ34S and Δ33S variations over time in Montreal PM10 aerosols. Grey bands represent the modelled sulfate isotope characteristics if SO4 is formed following the oxidation pathways proposed by Harris et al. (2013a).
During spring, all stations display a similar δ34S pattern characterized by an enrichment in 34S, with station 06 yielding the highest δ34S (up to 10 ‰), followed by stations 03, 50 and 13 with isotope compositions up to 8 ‰. Station 98, in contrast, presents the lowest δ34S-values during that period, down to 5 ‰. However, the difference in δ34S-values between the stations 03, 06, 13 and 50 becomes less significant the rest of the year, yielding a near constant δ34S-value of 8 ‰ during both summer and winter. During winter, station 98 still shows lower δ34S-values (around 5 ‰).
The seasonal variations proposed by Harris et al. (2013a) for rural aerosols from continental northern mid-latitudes (45∘ N) is also represented (brown field) in Fig. 1. This model (i.e. second model described in the introduction section) relies upon the isotope seasonal variations induced by three major oxidation pathways (OH, H2O2, O2+TMI) involved in the oxidation of 43 % of the atmospheric SO2, the other 57 % being dry-deposited. As Montreal latitude (45∘ N 73∘W) is similar to the one considered by Harris et al. (2013a) both datasets may be compared. The seasonality trend documented in Beijing by Han et al. (2016) (thick black line in Fig. 1) highlights high δ34S-values up to 10 ‰ during spring and winter while summer is characterized by low δ34S-values down to 4 ‰. According to Han et al. (2016), following the first model in the introduction section (i.e. S isotope systematic as a direct tracer of emission sources), this seasonality would reflect changes in the respective contributions of sources of atmospheric sulfate during different times of the year rather than changes in the SO2 oxidation chemical pathways. While our data are consistent at the first order with the seasonality highlighted for urban aerosols (Han et al., 2016) and the seasonality modelled for rural aerosols (Harris et al., 2013a) for the period bridging from the end of spring to the end of autumn, they show a significant deviation with a δ34S decrease between early winter and early spring in Montreal which is neither predicted by Harris et al. (2013a) model nor observed in Beijing.
The Δ33S measured at the five stations also show seasonal variations displaying 33S enrichment up to 0.3 ‰ during early winter and summer and Δ33S∼0 ‰ in spring and autumn (Fig. 1; dotted lines). While stations 03, 06, 13 and 50 present a similar Δ33S-range than station 98, mean Δ33S-values from the LOWESS show that station 98 is characterized by the highest Δ33S-value (0.143 ‰) compared to the others (ranging from 0.064 ‰ to 0.101 ‰). The lowest Δ33S-values are recorded along the year by stations 03, 06 and 13. In contrast, most of the stations present similar Δ36S-values, except for station 98 that yields the largest range of Δ36S from −1 ‰ to 1.7 ‰ (Table S5) so no seasonal Δ36S-variations are highlighted (Fig. S1 in the Supplement). The model proposed by Harris et al. (2013a) also suggested a seasonal variation (brown area in Fig. 1) for rural aerosols with a maximum Δ33S-value of 0.05 ‰ during summer and a minimum of −0.05 ‰ in winter. Clearly, this model cannot explain the larger range of S isotope compositions observed in Montreal.
3.1.3 Comparison with the literature
S-isotope data have been previously obtained on aerosols collected in the United States of America (Romero and Thiemens, 2003; Shaheen et al., 2014): La Jolla (32∘ N and 117∘ W, rural environment), Bakersfield (35∘ N and 119∘ W, urban environment), White Mountain (37∘ N, 118∘ W, rural environment) and in China (Guo et al., 2010; Lin et al., 2018b; Han et al., 2017): Beijing (39∘ N and 116∘ E, urban environment), Guangzhou (23∘ N and 113∘ E, urban environment) and Xianghe (39∘ N and 116∘ E, urban environment).
The Δ33S-values in Montreal aerosols share common characteristics with most of the available data (Guo et al., 2010; Romero and Thiemens, 2003; Shaheen et al., 2014; Lin et al., 2018b) with high Δ33ST-values (i.e. the Δ33S measured and non-corrected for sea salt) occurring during both summer and winter sampling periods (open grey marks in Fig. 1). Collectively these data differ from the study by Han et al. (2017) who recently reported the first negative Δ33S-values, down to −0.6 ‰, in Beijing.
Neglecting seasonality, it can be noted that worldwide urban aerosols present a range of δ34S-values varying mostly from 0 ‰ to 20 ‰ with a mean value of 6±5 ‰ (2σ) and Δ33S-values varying from −0.6 ‰ to 0.7 ‰ with a mean value of 0.17±0.4 ‰ (2σ) (Fig. 2). The model presented by Harris et al. (2013a) cannot explain these δ34S-values as it predicts isotope variations ranging between 1 ‰ and 7 ‰. Also, Montreal samples present a wider range of Δ36S-values (from −1 ‰ to 2 ‰) compared to the one (−2 ‰ to 0 ‰) reported in the literature with significant positive isotope compositions for half of our samples (Fig. 3). The observation of positive Δ36S-values is surprising but do not come from analytical issue as these are observed in several samples, some of them having been duplicated. This represents, to our knowledge, the first positive Δ36S-values reported for urban aerosols. To date, Baroni et al. (2008) is the only other study reporting both positive Δ33S and slightly positive Δ36S-values for one volcanic sample that the authors considered to result from mass-dependent fractionation. Recently, the Δ36S-values have been suggested to be decoupled from the Δ33S-values, the Δ36S-values in aerosols being explained by combustion while the Δ33S-values would reflect another atmospheric process, e.g. input of stratospheric sulfates (Lin et al., 2018b). Both combustion process and stratospheric inputs could explain the background tropospheric sulfates in China, which is characterized by positive Δ33S and negative Δ36S-values (Lin et al., 2018b), but they cannot account for both the positive Δ33S and Δ36S-values measured in Montreal. Up to now, only the experimental photooxidation has been shown to produce both positive Δ33S and Δ36S-values (slope ∼0.64) (Whitehill and Ono, 2012), which could highlight the implication of this process in the samples in Montreal.
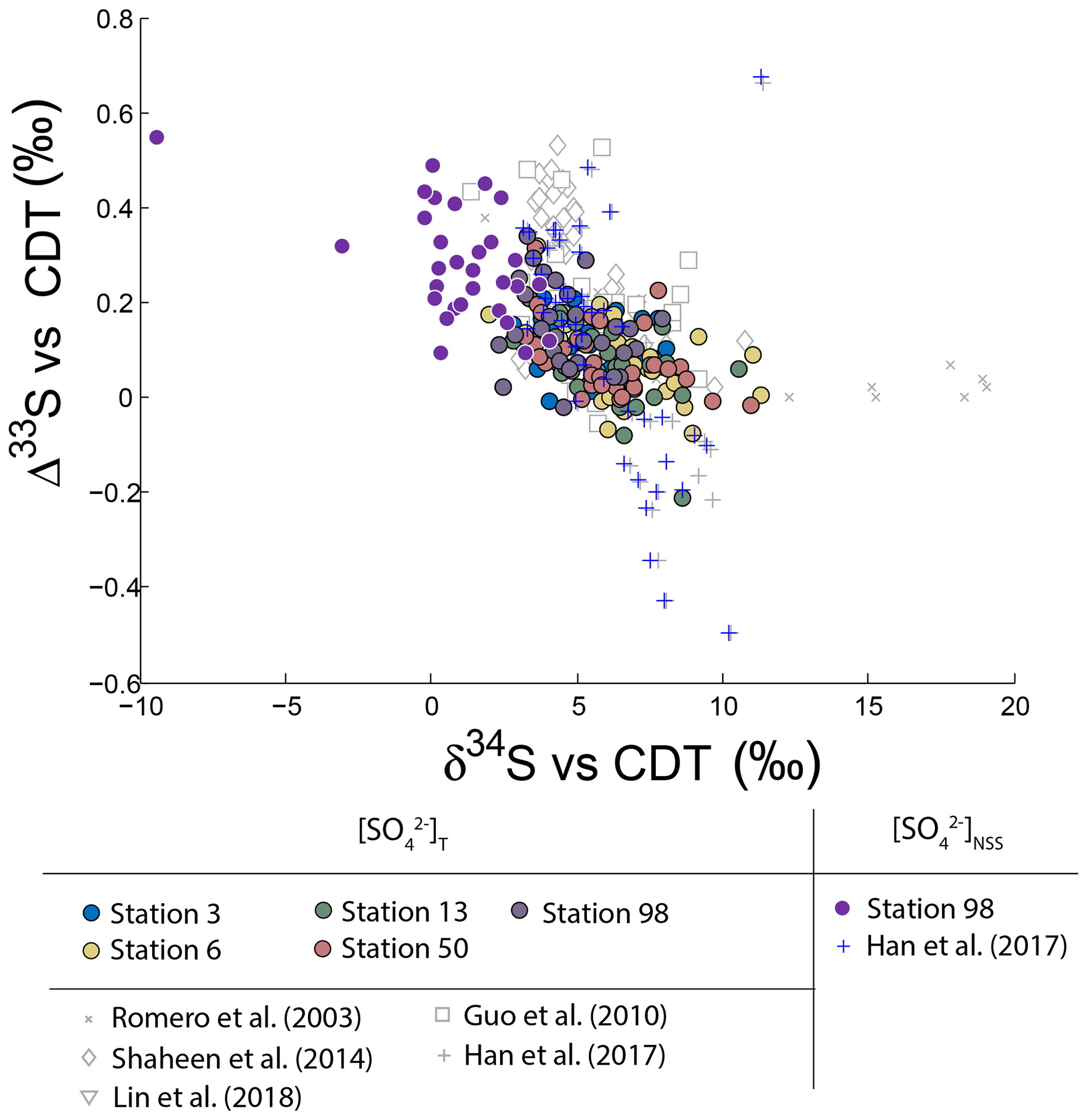
Figure 2Variations of Δ33S as a function of δ34S in sulfates from PM10 aerosols collected in Montreal. Light grey dots are compiled from Guo et al. (2010), Romero and Thiemens (2003), Shaheen et al. (2014) and Han et al. (2017). Dark grey dots represent the NSS-s-corrected multiple sulfur isotope compositions.
3.1.4 Potential sea-salt contribution
The concentrations of Na+ in aerosols from station 98 ranged from 0.08 to 11.85 µg m−3 and those of from 0.04 to 2.42 µg m−3 (Table S5). The presence of detectable Na+ concentrations in all samples from this station may either reflect the local use of NaCl deicing road salts during winter (Zinger and Delisle, 1988) and/or the contribution of sea-salt aerosols. If deicing NaCl was the source of aerosol Na, we would expect higher Na+ amounts in aerosols during winter. Yet, Na+ concentrations do not show any significant variations along the year, precluding the input from deicing salts highlighting the contribution of sea salt in the observed Na+ concentrations.
It is worth mentioning that among the aerosols collected at station 98, two of them show a Na∕Cl ratio (∼1) similar to sea-salt sulfates (∼0.6). The other samples show very high Na∕Cl ratios (> 10). This shows a depletion of Cl in most of our samples, which could be explained by a reaction between NaCl and sulfate ∕ nitrate that forms NaSO4 or NaNO3, a process also known as the “aging of sea-salt particles” (Sinha et al., 2008; Song and Carmichael, 1999). The aging process involves the exposure of secondary aerosols precursors, which would form sulfates in a marine environment. In this context sulfates could either result from the oxidation of SO2 formed by the oxidation of DMS or from the oxidation of anthropogenic SO2 (Harris et al., 2012d). DMS is characterized by a mean δ34S-value of 18 ‰ and a mean Δ33S-value of 0.03 ‰ (Oduro et al., 2012) and anthropogenic SO2 by variable δ34S-values and a mean Δ33S-value of −0.018 ‰ (Lin et al., 2018b).
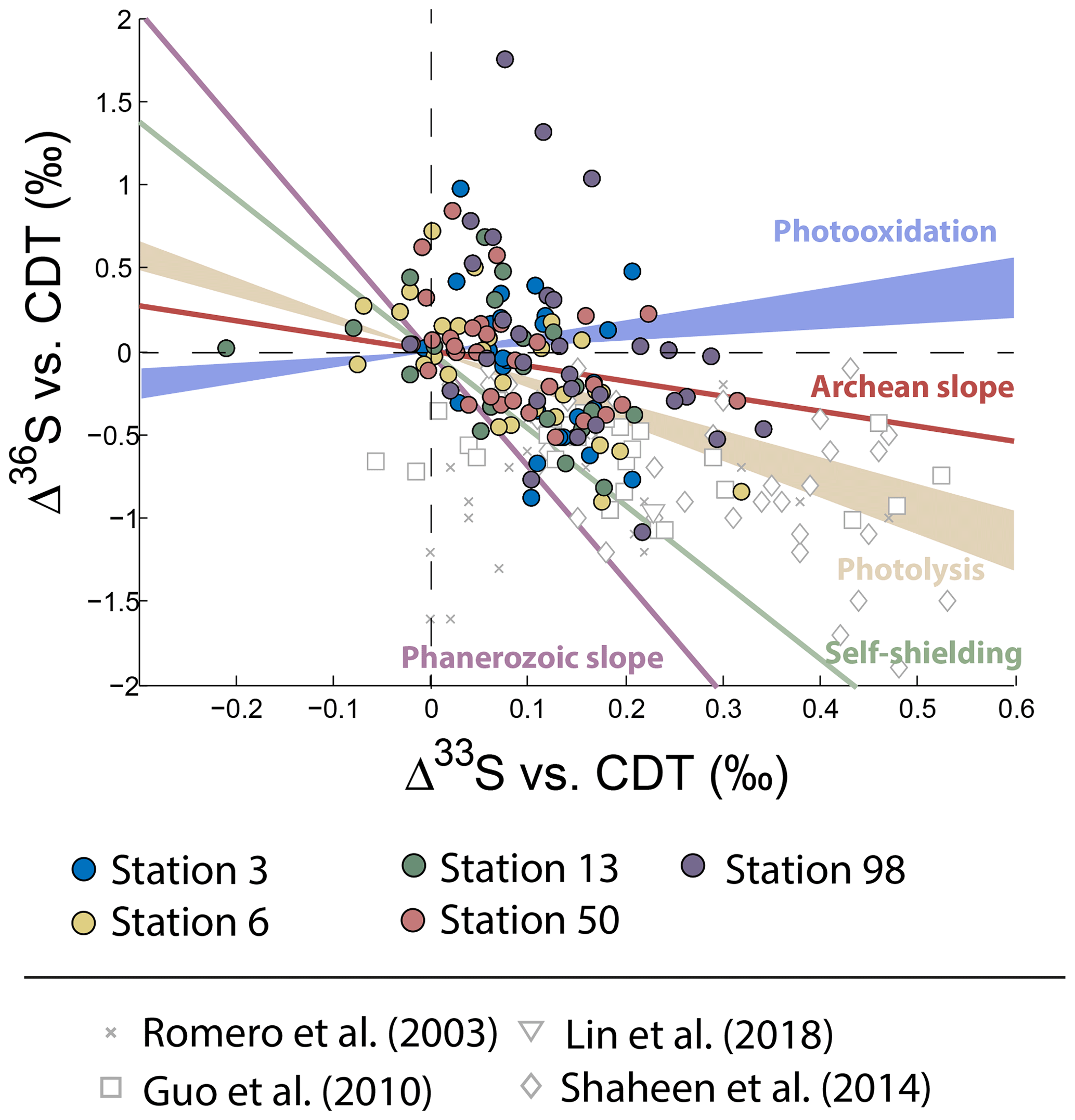
Figure 3Variations of Δ33S as a function of δ34S in sulfates from PM10 aerosols collected in Montreal. Archean and Phanerozoic slopes taken from the literature are also reported.
Thus, any contribution from aged sea salt to the sulfates budget would decrease the Δ33S-values measured in aerosols. It is thus essential to correct for these contributions. Considering that the different major oxidants in the marine environment (H2O2∕O3, O2+TMI, OH) would form sulfates with similar Δ33S-anomalies than the initial SO2 and that the Δ33S-values of sea-salt sulfates, DMS and anthropogenic SO2 are very similar (∼0.05 ‰), we corrected our Δ33S-values using the 0.05 % Δ33SSS-values reported by previous studies (Labidi et al., 2012; Ono et al., 2006a, b). Both DMS and sea-salt sulfate are characterized by similar δ34S-values. The δ34S of anthropogenic SO2 is more variable but the corresponding correction for the δ34S-value is of little importance and will thus not change the conclusion we are making following the study of the Δ33SNSS-values. Ultimately, we thus applied an isotope balance equation to calculate the δ34SNSS and Δ33SNSS isotope compositions corresponding to the non-sea-salt (NSS) sulfate fraction for each aerosol sample using average δ34SSS (=22 ‰) and Δ33SSS values (0.05 %) for sea-salt sulfate. A detailed description and results (δ34S, Δ33S and percentages) are reported in Table S6. A consequence of this correction is that it both decreases the δ34S-values of the aerosol samples and increases their corresponding Δ33S-values. Results show that in average sulfates sampled at station 98 include % of NSS sulfates that are characterized by Δ33SNSS values varying from 0.010 ‰ to 0.550±0.1 ‰ with a mean value of 0.280±0.118 ‰. Two Δ33SNSS values from station 98, calculated at 2.197 ‰ and −0.723 ‰ (01/22 and 12/06, respectively; Table S6), were not considered as one is characterized by a low ∼30 % NSS sulfate concentration making it very sensitive to the SS correction and the other one present a too high concentration of Na which induces a negative percentage (−153 %) of NSS (Table S6). As the SS contribution appears constant along the year, this correction does not affect the seasonality pattern highlighted in Sect. 3.1.2. Other stations (03, 06, 13, 50) show similar NSS contributions (∼80 %; Table S7) and Δ33SNSS-values that also range between 0 ‰ and 0.5±0.1 ‰, but station 98 still presents the highest mean Δ33SNSS-values.
It is worth noting that the highest Δ33ST-value measured in Montreal (0.35 ‰) is lower than the one reported in Xianghe and Beijing (China; 0.5 ‰; Guo et al., 2010; Han et al., 2017). This difference may be attributed to significantly different sea-salt contributions: 20 % in Montreal compared to 1 % in Beijing (Han et al., 2017). Due to this low 1 % sea-salt contribution in Beijing, the corresponding corrected isotope compositions NSS-s (blue marker in Fig. 2) are not significantly affected. For Montreal, the corrected Δ33SNSS reach values as high as ‰, therefore similar in magnitude to other studies. The fact that sulfate aerosols from Beijing and Montreal display the same highest Δ33S may highlight a common reaction scheme. Thus, our discussion will be based on the assumption of a common process explaining the high Δ33S-values observed in the two cities.
4.1 Anthropogenic emissions, Δ33S-values and seasonality
The large majority of coal and oil used worldwide as an energy source is extracted from Proterozoic sediments (< 2.3 Gy) and, as such, does not have significant non-zero Δ33S (typically within ±0.1 ‰, e.g. Farquhar and Wing, 2003). The complete conversion of sulfur (as organic S, sulfate and/or pyrite) to SO2 implies that SO2 has the same isotope composition than that of its starting material, i.e. no isotope fractionation or ‰. Only if part of the SO2 is scavenged and the fractionation process is strongly non-mass dependent (β≠0.515) would the emitted SO2 have a non-zero Δ33S.
Iron extraction from Archean banded-iron formation (BIF) is another source of atmospheric S that therefore produces non-zero Δ33S. However, the 5800 tons of SO2 emitted each year in the Quebec/Ontario region by mining activities (Environnement Canada, 2013) only represents 1.5 % of the annual 370 000 tons of national SO2 emissions (Environnement Canada, 2013). If we consider a high average Δ33S of 2 ‰ (Thomassot et al., 2015) and a proportion of 1.5 % of SO2 resulting from iron processing, this would lead to an average Δ33S-anomaly of the final SO2 of up to 0.02 ‰. Thus, iron processing can hardy account for the origin of the non-zero Δ33S-values observed in most aerosols. More specifically, the Canadian iron ore production is split between Quebec (50 %), Labrador (45 %) and Nunavut (5 %). With respect to Quebec, iron production is mainly operated from the Algoma BIFs (∼2.8 Ga) typified by the Temagami deposits for which Diekrup et al. (2018) give an average Δ33S of 0.467±0.707 (samples including oxidic facies, cherts, BIF sulfides and sulfidic clays, sulfide veins), which makes the emitted SO2 having even smaller Δ33S. In the following discussion we will therefore consider that sources of SO2 have Δ33S∼0 ‰ and that only specific chemical reactions (photochemical or not) can produce non-zero Δ33S.
Aerosols collected at stations likely impacted by local emission sources (i.e. stations 03, 06, 13 and 50) present the lowest Δ33S-values ( ‰) and the highest δ34S-values (up to ∼12 ‰) compared to station 98 (less influenced by anthropogenic emissions). This suggests that local emissions in Montreal are characterized by δ34S-values up to 12 ‰ and mean Δ33S-values close to 0 ‰, which implies that the high Δ33S-anomalies with lower δ34S are transported to rather than produced in Montreal. Local anthropogenic sources could then isotopically impact these imported aerosol sulfates by decreasing their Δ33S-values towards 0 ‰. This is consistent with Lee et al. (2002) who showed the ability of these primary aerosols, resulting from the combustion process, to decrease Δ33S-values towards 0 ‰, as they are characterized by zero to slightly negative Δ33S-values down to −0.2 ‰. Secondary sulfates formed by SO2 oxidation within cities by the main oxidation pathways (OH, O2+TMI, NO2, H2O2, O3) would not generate significant MIF and would also lead to decrease the Δ33S-value of imported aerosols (Harris et al., 2013a; Au Yang et al., 2018).
This contrasts with the interpretation where the negative Δ33S-values (down to −0.6 ‰) measured during winter in Beijing would relate to anthropogenic sources, in particular those generating incomplete, i.e. low-temperature, coal or wood combustion (Han et al., 2017). Still, this model cannot explain the total range of isotope compositions observed. The authors mostly rely on data showing that primary aerosols are characterized by negative Δ33S-values but only down to −0.2 ‰ (Lee et al., 2002). Also the complementary positive Δ33S still need to be addressed. Furthermore, Han et al. (2017) interpretation would predict (i) a seasonality with negative Δ33S-values down to −0.6 ‰ during winter as a result from increased coal and wood burning and (ii) a gradient in the Δ33S-values from the outer towards the inner city with isotope shifting from ∼0 ‰ to negative Δ33S-values. This would contradict our observations, since our data in Montreal show the opposite to what was observed in Beijing. It comes that based on the available data of S anthropogenic emissions, the combustion of coal or wood at low temperature can neither explain the Δ33S seasonality nor the highest Δ33S-values up to 0.5 ‰ measured in urban aerosols.
This conclusion is reinforced by the fact that coal is not the major source of energy in Montreal, oil representing 50 % of the fuel energy in Quebec (Ville de Montréal, 2015). Oil would thus display Δ33S-values close to 0 ‰ (Lee et al., 2002). Taken together, our observations suggest that anthropogenic activities (both coal and oil combustion) are unlikely responsible for the Δ33S seasonality nor the highest Δ33S-values up to 0.5 ‰ measured in Montreal urban aerosols. This implies that non-zero Δ33S-values are produced in rural rather than in urban environments. Thus, the following discussion mostly focuses on data from station 98, located on the western part of the island, upstream of the main blowing winds and supposedly less affected by emissions from local anthropogenic activities.
4.2 Input of stratospheric sulfates, Δ33S-values and seasonality
To date, sulfates samples trapped in the Antarctica ice are showing the most extreme non-zero Δ33S-values with negative and positive Δ33S-values down to −2 ‰ and up to 1 ‰, respectively (Baroni et al., 2007, 2008; Bindeman et al., 2007; Savarino et al., 2003; Shaheen et al., 2014; Gautier et al., 2018). Their formation results from the photochemical oxidation of atmospheric SO2. This is because SO2 possesses two dominant absorption bands in the ultraviolet region: one at 190–220 nm (photolysis) and the other one at 250–330 nm (photooxidation), which are able to create high Δ33S-values up to 15 ‰ (Farquhar et al., 2000, 2001; Whitehill et al., 2013, 2015; Whitehill and Ono, 2012). While produced under distinct O2 levels, this process has been suggested to account for sulfur multiple isotope signatures of both Archean sediments (Whitehill and Ono, 2012) and modern aerosols in Antarctica (Savarino et al., 2003; Hattori et al., 2013; Baroni et al., 2007; Gautier et al., 2018).
Given the similar Δ33S-values between urban (≦0.5 ‰) and Antarctica aerosols (≦2 ‰), the Δ33S-values of urban aerosols could result from inputs of stratospheric sulfate aerosols, supposedly carrying significant Δ33S anomalies into the troposphere (Guo et al., 2010; Romero and Thiemens, 2003; Lin et al., 2018a, b). However, according to the HYSPLIT 3-day back-trajectory analysis carried out for each of our samples in Montreal (Fig. 4), the probability of injecting stratospheric air masses into the troposphere is relatively low, the HYSPLIT model predicting that only ∼10 % of the aerosols were coming from altitudes higher than 500 m. Furthermore the stratosphere troposphere exchange (STE) in the Northern Hemisphere is preferentially located in the northern Pacific and northern Atlantic (Boothe and Homeyer, 2017; Gettelman et al., 2011; Sprenger and Wernli, 2003) and occurs during a period less than 80 days year−1 (Boothe and Homeyer, 2017). This again is not favoring the hypothesis that injection of stratospheric sulfate into Montreal urban atmosphere can explain the occurrence of non-zero Δ33S-values. This is consistent with the study of Lin et al. (2016) who also estimated a very low (∼1 %) input of stratospheric SO4 in their study. Considering 1 % associated with a maximal Δ33S anomaly of 10 ‰ for stratospheric sulfates (Ono et al., 2013) this would result in a Δ33S-value of only 0.1 ‰ in tropospheric sulfates. Finally, based on the Antarctica sulfate isotope record, the Δ33S anomalies of the stratospheric sulfates produced by photochemical processes would only occur when high SO2 concentrations are reached, typically following stratospheric volcanic eruptions (Ono et al., 2013; Baroni et al., 2007, 2008; Savarino et al., 2003; Martin, 2018). Ultimately, inputs of stratospheric aerosol sulfates can thus hardly explain the sustainable urban aerosol Δ33S anomalies that are observed worldwide.

Figure 4Three-day back-trajectories modelled using the HYSPLIT software for the sampling stations in Montreal. Back trajectories are calculated using an initial height of 50 m a.s.l. (above the sea level). American deserts are also shown in brown and include the Great Basin, the Mohave desert, the Chihuahua desert and the Sonoran Desert.
4.3 Formation of secondary sulfates, Δ33S-values and seasonality
Romero and Thiemens (2003) highlighted a negative dependence between Δ33S-values and the aerosol size, down to typical secondary aerosol sub-micron sizes (Seinfeld and Pandis, 2012) associated with a Δ33S increase with values up to 0.5 ‰, similar to values measured among aerosols. The fact that the main oxidants OH, H2O2, O2+TMI, O3 and NO2 cannot produce Δ33S-values higher than ±0.2 ‰ (Au Yang et al., 2018; Harris et al., 2013a) arises the need for another oxidation. It comes that the only process that could preserve the non-zero Δ33S signatures in urban aerosols after mixing with aerosols that underwent the main near mass-dependent oxidation reactions highlighted above would be an oxidation associated with high Δ33S-values. We explore in the following a non-exhaustive series of reactions which we believe require special attention and discussion.
4.3.1 Oxidation by the Stabilized Criegee Intermediate (SCI)
Previous studies suggested that SO2 oxidation by the Stabilized Criegee Intermediate (SCI) may represent a reliable possibility (Mauldin III et al., 2012; Sarwar et al., 2014; Boy et al., 2013; Ye et al., 2018; Sipilä et al., 2014). The SCI oxidation pathway is important in rural environments (characterized by higher VOC∕NOx ratios compared to the urban atmosphere) and would mitigate the discrepancy between modelled and measured SO4 concentrations (Sarwar et al., 2013, 2014). Boy et al. (2013) showed, using a one-dimensional model that SCI reaction with SO2 may represent 40 % of the total sulfuric acid production during winter in the atmospheric boundary layer above the forest canopy, while this contribution varies between 15 % and 20 % for the other seasons. Thus, although the contribution of the SCI oxidation pathway might be low (representing 0.69 % of the NO2 oxidation pathway; Cheng et al., 2016), it might represent a viable alternative to be considered in the future, notably in winter. If this SCI oxidation pathway generates high Δ33S-anomalies, it would account for the observation that Δ33S-values in winter is more important at station 98 (i.e. background) than at stations located downtown. In this case, the high anomalies found during summer for station 98 could be related to another process.
4.3.2 SO2 photooxidation
Another possibility, based on the study by Whitehill and Ono (2012) and Ono et al. (2013) could be the still unexplored SO2 photooxidation in the troposphere. The wavelength range of the actual actinic flux at sea level (in the troposphere) varies from 300 to 2500 nm (Eltbaakh et al., 2011) because ozone (O3) absorbs the bulk of the solar UV radiation in the 290–320 nm region (Molina and Molina, 1986). The overlapping region shows that tropospheric SO2 absorption can only occur within a narrow wavelength range (typically 320 to 330 nm; Calvert et al., 1978) that still leave room for S-MIF to be produced. Although the cross section in this overlapping region is small ( cm2; Blackie et al., 2011), experimental studies need to be performed to estimate the S multiple isotope compositions of sulfates formed under these particular conditions. To our knowledge, there is no data available yet within this narrow wavelength range (Farquhar et al., 2001; Whitehill et al., 2013, 2015; Whitehill and Ono, 2012; Danielache et al., 2012). This process would likely be more significant during summer, but would hardly explain the high Δ33S-values observed in winter. Due to the narrow wavelength range where photooxidation could occur the rate of this mechanism would unlikely be a few orders of magnitude larger than that in the gas phase.
4.3.3 Aging of sea-salt sulfate
Chemical analyses of our samples show a correlation between sulfate and Na concentrations, suggesting sodium sulfate is probably the main form of sulfate in our samples. The formation of sodium sulfate result from the SO2 oxidation coming either from direct emission or DMS oxidation on sea-salt aerosols surface (Harris et al., 2012c; Sinha et al., 2008). Thus, the aging process, which induces a Cl depletion, might explain such Δ33S-values. In this perspective and according to the hypotheses in Sect. 3.1.4, as for urban aerosols sulfates in Montreal, a Cl depletion should also be observed in the aerosols in Beijing. However, the contribution of the sea-salt sulfates in those samples represents only 1 % of the total aerosols without showing a Cl depletion which rules out this hypothesis and thus indicate the occurrence of another oxidation process.
4.3.4 Photooxidation of SO2 in the presence of mineral dust
Finally, we suggest that the photooxidation of SO2 in the presence of mineral dust may represent an alternative way to generate these non-zero Δ33S-values in sulfate aerosols. To date, the mechanisms behind the in-particle chemistry remain little studied and several SO2 heterogeneous oxidation reactions may have been overlooked. Several studies showed that mineral dusts promote sulfate formation by either heterogeneous SO2 oxidation (i.e. oxidation of the SO2 adsorbed onto the mineral dust by several possible oxidants including O3, H2O2, NO2) or by gaseous oxidation by radicals OH•. Heterogeneous SO2 oxidation should a priori not lead to distinct Δ33S-values whether it occurs on mineral dust or not (Harris et al., 2012a). Given that the in-particle chemistry remains elusive, many other reactions may occur such as the oxidation by the superoxide radical anion O2• formed on semi-conducting metal oxides like Al2O3, Fe2O3 and TiO2 by UV radiation (Yu et al., 2017; He et al., 2014; Dupart et al., 2012; George et al., 2015; Usher et al., 2003; Zhao et al., 2018; Ma et al., 2018). Although, S-isotope fractionations have not been reported for this oxidation pathway yet. Furthermore, others radicals/oxidant might be involved, leading the SO2 oxidation by mineral dust to have specific S-isotope fractionations compared to the oxidation by OH.
Thus, depending on the oxidation pathway, mineral dust could promote either the increase in the aerosol size or the formation of new particles, respectively. This oxidation pathway would also mitigate the discrepancy between modelled and measured SO4 concentrations (Fu et al., 2016).
Our chemical analyses do not indicate any significant contributions of dust particles in Montreal as (i) samples present low Fe∕Al ratios (typically 0.05), distinct from the ones characterizing desert dusts, which vary from 0.48 to 1.74 (Formenti et al., 2011) and (ii) modelled back trajectories (in Fig. 4) indicate that the air masses reaching the city are unlikely influenced by the western deserts. Still, we suggest that oxidation of SO2 in the presence of mineral dust could still occur.
Asian deserts (Cottle et al., 2013; McKendry et al., 2001) and to a lesser extent the Sahara (Chin et al., 2007) have been shown to represent the main sources of mineral dust affecting the North American continent, with events mainly recorded during spring and summer (Zhao et al., 2006; Prospero, 1996). The seasonality of long-ranged transport of mineral dust over the North Atlantic is concomitant with the period when the lowest δ34S-values and highest Δ33S-values are measured. This highlights a potential link between the S isotope variations and mechanisms involving mineral dust. It is particularly interesting to note that the transport of SO2 from the East Asian major sources to North America is typically observed (Clarisse et al., 2011) with a mixing of sulfates on mineral dust reported over the Northern Hemisphere continents (Bauer and Koch, 2005). This suggests that the transported mineral dust is typically coupled with sulfates or in mixing with SO2. However, models show a decline in the dust/total sulfate ratio during trans-Pacific transport due to an enhanced settling of super-micron dust particles compared to the fine ammonium sulfate (Fairlie et al., 2010), precluding the observation of mixed dust over North America. Thus, the current knowledge of the transport and reactivity of mineral dust and SO2 over North America is consistent with the photooxidation of SO2 in the presence of mineral dust.
It is also worth noting that the seasonality reported in dust particles fluxes by Félix-Bermúdez et al. (2017) in southern California displays a strong similarity with the δ34S seasonality observed in our Montreal aerosols (Fig. 5) with high δ34S-values associated with high dust particles fluxes. Importantly, Fig. 5 also highlights an anti-correlation trend between dust particle fluxes and Δ33S-values with low dust particle fluxes (∼14 mg m−2 d−1) associated with the high Δ33S-values (0.3 ‰). Although we are comparing two locations separated by several thousand kilometers (i.e. California and Quebec), Félix-Bermúdez et al. (2017) suggest that the seasonality they are observing is extendable to a global scale as the dust deposition rate is mainly driven by the contrasting atmospheric air mass transport taking place during the warm and cool seasons.
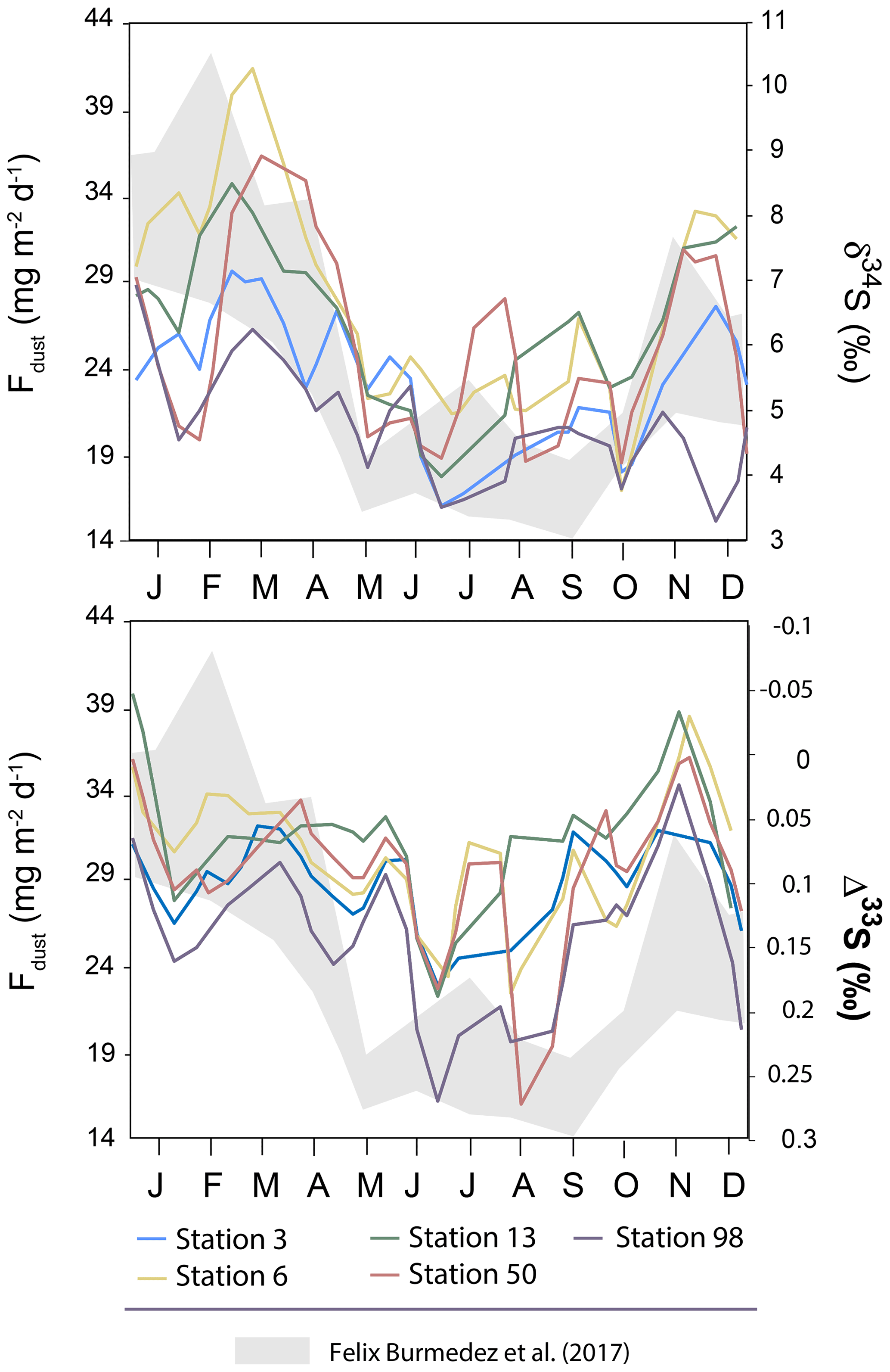
Figure 5Variations of the mean δ34S and Δ33S-values over time in sulfate aerosols collected in Montreal. The grey band represents the seasonality of the dust deposition rate determined by Félix-Bermúdez et al. (2017).
In order to confirm that the Δ33S seasonality we are observing in Montreal aerosols does not reflect the signatures of primary mineral dust particles transported to the city, we analyzed desert dust samples from China, Morocco, Tunisia and Jordan. Results show δ34S-values varying from 5 ‰ to 13 ‰ but importantly no significant Δ33S-anomaly (Table S8).
Thus, in order to explain our data (i.e. most positive Δ33S-values at station 98, seasonality of the S-isotope compositions, no dust particles detected in Montreal aerosols), we suggest that the SO2 photooxidation reaction may occur at the dust surface and, by oxidizing the surrounding SO2 into sulfates, it would deplete the resulting SO4 in 33S and by mass balance, leave the residual SO2 enriched in 33S (Fig. 6). Sulfates associated with dust would be characterized by negative Δ33S-values and will be deposited while the residual atmospheric SO2 (i.e. characterized by positive Δ33S-values) would be transported to Montreal. The transported SO2 enriched in 33S would then be oxidized into sulfates in the vicinity of Montreal through the major oxidation pathways (O2+TMI, H2O2, O3, OH, NO2). We suggest the presence of two different types of sulfates: (i) the first type would be formed by photooxidation and would be associated with coarse particles (dust particles), while (ii) the second type would be formed by the oxidation of the remaining SO2 and thus likely be associated with finer particles. These sulfates supposedly characterized by positive Δ33S up to 0.5 ‰ would be mixed with both primary and secondary sulfates emitted and formed within the city and supposedly characterized by Δ33S-values close to 0 ‰ (i.e. oxidation by O2+TMI, H2O2, O3, OH, NO2; Fig. 6).
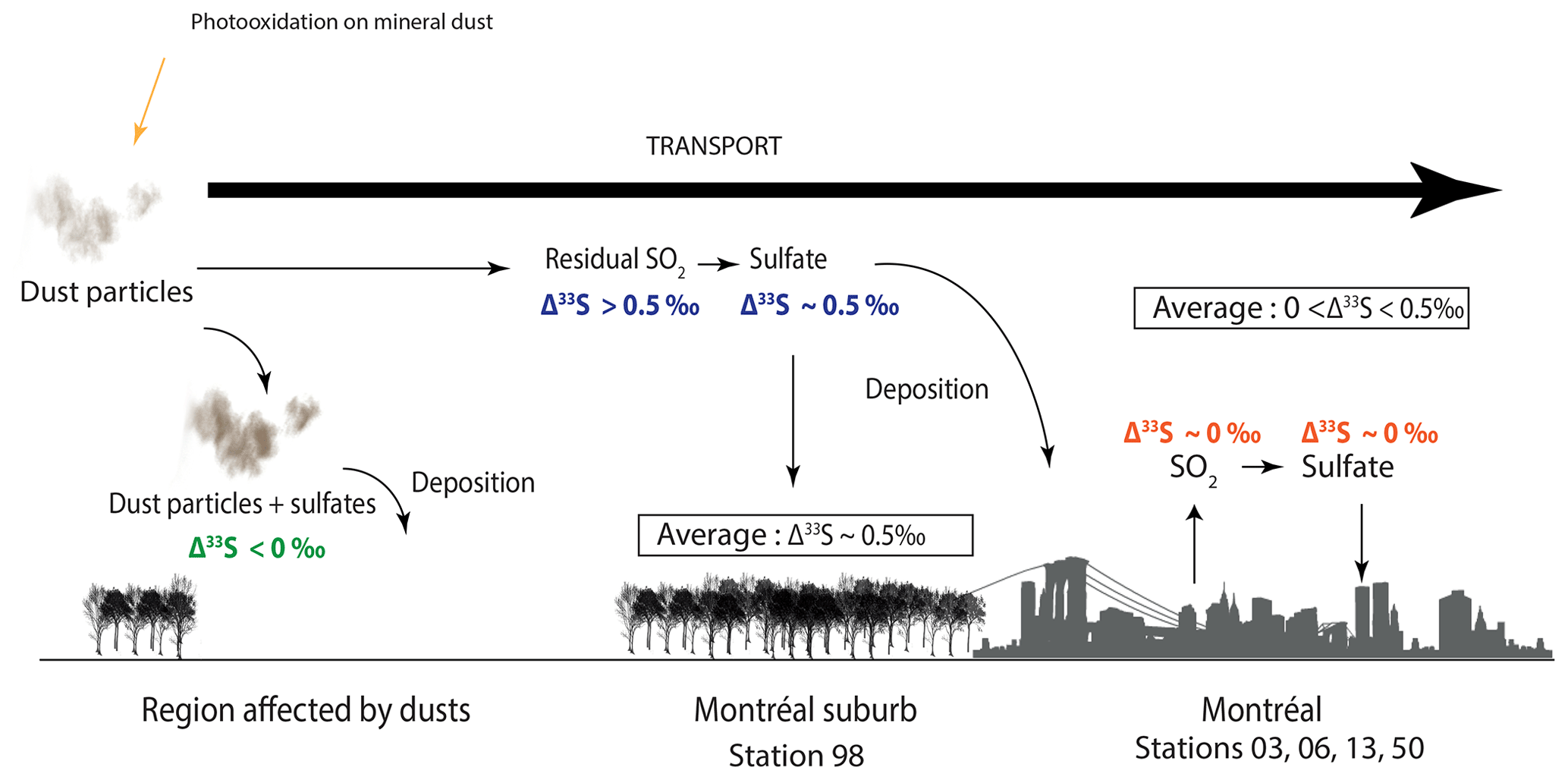
Figure 6Scheme of the reaction mechanisms leading to the formation of sulfates driven by airborne mineral dust. The 33S-enriched residual SO2 is supposedly transported to cities, while the 33S-depleted sulfates are deposited along with dust particles in the rural environment.
It is worth mentioning that our model would thus generate a different temporal pattern from the one recorded in sulfates from the Antarctica snowpack which are first characterized by positive Δ33S-values that then shift to negative Δ33S-values, reflecting a depletion in 33S in the residual SO2 pool (Baroni et al., 2008; Gautier et al., 2018). Although the origin of the Δ33S-values in snowpack remains unclear, a combination of different oxidation pathways with similar contributions of S-MDF (high or lower contribution of OH oxidation pathway) and S-MIF processes (photoexcitation and photolysis) has been recently suggested to explain such Δ33S-values (Gautier et al., 2018). The OH oxidation pathway is occurring in both the troposphere and the stratosphere. However, in the troposphere as (i) photolysis cannot occur because of the ozone layer and (ii) photooxidation would only occur in a narrow range of UV (see Sect. 4.3.2), but would unlikely display a seasonal variation, we suggest that the reactions responsible for S-MIF in the stratosphere and in the troposphere are different. Thus, the contrasting patterns observed in sulfates in Antarctica and in Montreal could be explained by the implication of different combinations of oxidation pathways where a S-MIF process other than photolysis and photooxidation is involved.
It is worth noting that the exact photochemical mechanism which would be responsible for that relation remains speculative but some reactions can be highlighted and discussed. The recently proposed oxidation of SO2 by NO2 on mineral dust (Ma et al., 2018) is unlikely because NO2 is predominant in the urban environment, i.e. at odds with the present evidence. The oxidation implicating heterogeneous oxidation and OH radicals should a priori not show significant differences from the one that occurs in gaseous and aqueous phase; i.e. heterogeneous oxidation of SO2 is likely to induce a mass-dependent fractionation of S-isotopes (Harris et al., 2012a) while the gas phase by OH would induce negative Δ33S-values < −0.15 ‰ (Harris et al., 2013a). Among other reactions, SO2 oxidation by the O2• superoxide radical anion is another oxidation reaction that has not yet been isotopically characterized (Dupart et al., 2014; Usher et al., 2003). If this latest oxidation pathway could promote the formation of sulfates characterized by high Δ33S-values (hypothetically 10 ‰), then a small contribution (hypothetically ∼10 %) from this oxidation pathway would produce a significant signal on the sulfur isotope composition of tropospheric sulfate aerosols (i.e. Δ33S∼1 ‰ based on these hypotheses). In this case, even a small proportion of those sulfates mixed with sulfates formed by the major oxidation pathways locally produced (i.e. Δ33S∼0 ‰) could explain the Δ33S-values observed in the troposphere (Δ33S < 0.5 ‰). This hypothesis needs to be further tested in the future.
Our hypothesis could explain sulfates with positive Δ33S-values transported to Montreal but implies that negative Δ33S-values also need to be found in dust particles. This hypothesis could leave a new room to explain negative Δ33S-values measured in Beijing aerosols (Han et al., 2017). Indeed this oxidation pathway may occur at a larger scale and may also be involved in the formation of urban aerosols reported in the literature. Indeed, urban aerosol sulfates previously studied in La Jolla, Bakersfield and White Mountain in the United States, and in Xianghe and Beijing may also be influenced by Asian mineral dust.
Intuitively, dust particles may be transported during discrete storm episodes (Marticorena and Bergametti, 1995; Kok et al., 2012) which have been reported mostly during spring in China (Zhao et al., 2006; An et al., 2018). Following this hypothesis, negative Δ33S-values would be found in spring, which is not the case in both PM2.5 and PM10 collected in 2016 and 2005, respectively (Guo et al., 2010; Han et al., 2017). In fact, five dust episodes were identified in China in 2016 (An et al., 2018) with one (4 March) happening close to the sampling period (15 March to 26 April; Han et al., 2017). However, the images recorded by the NASA satellite show that the dust storm in the Gobi would unlikely reach Beijing that day (https://modis.gsfc.nasa.gov/gallery/individual.php?db_date=2016-03-11, last access: 14 March 2019), possibly explaining why such negative values have not been measured (Han et al., 2017). Negative Δ33S values have also not been measured in PM10 during spring. Guo et al. (2010) data show positive Δ33S-values, similar to ours and to other studies but different from Han et al. (2017). However, Guo et al. (2010) did not report major elements in their aerosol samples, making it difficult to detect any significant dust contribution. Nevertheless, while Guo et al. (2010) measured sulfate S isotope compositions until 11 April, Cao et al. (2014) reported a significant dust event on 27 April of the same year. In that respect this does not contradict the hypothesis: SO2 photooxidation on mineral dust could lead to positive Δ33S of the residual SO2 transported to Beijing. Moreover, for our model to be consistent with the data of Han et al. (2017), their aerosol fine fraction would need to be dominated by dust which is consistent with the observation that Asian dust storms contribute to the PM2.5 budget in Beijing (Han et al., 2015).
Weak wind conditions can also be responsible for a high contribution of fine (0.15–15 µm) mineral dust fraction in the total aerosol content (Golitsyn et al., 1997, 2003; Chkhetiani et al., 2012), being also observed in Beijing during the winter haze episodes (Yang et al., 2017, 2018) where negative Δ33S-values primarily occur (Han et al., 2017). Although the contribution of sulfates from terrigenous sources has been estimated to a maximal value of 3.84±4.40 to 5.62±6.52 % deduced from Ca2+ concentration (Han et al., 2017). However, Asian dusts present a high variability of Ca2+ concentration (the Ca∕Al ratio varying from < 0.1 % to 35 %; Formenti et al., 2011) reflecting that Ca2+ is mainly a tracer for carbonate mineral (Formenti et al., 2011). Using conventional crustal references (Fe∕Al and K∕Al) (Guieu et al., 2002; Wagener et al., 2008; Paris et al., 2010; Formenti et al., 2011) may help to better discuss the contribution of dust particles.
In that perspective, we predict that locations characterized by low or no mineral dust inputs but still emitting sulfates (e.g. South Africa or Brazil; observable at https://svs.gsfc.nasa.gov/30017, last access: 14 March 2019) would be characterized by low or zero-Δ33S-values. Identically, we predict that regions highly affected by dust could be characterized by negative Δ33S-values. Our study also suggests that the aerosol sulfate-coating could be characterized by negative Δ33S-values. This further needs to be tested by isotopically characterizing the formation of sulfate coating in aerosols through SO2 photooxidation in the presence of mineral dust.
In this study, we determined for the first time the multiple sulfur isotope compositions of PM10 sampled during a 1-year period in Montreal at several monitoring stations disseminated within the island, each characterized by a specific environment (high-traffic highways interchange, urban background, maritime and festive activities, rural background). We demonstrated that stations impacted by local anthropogenic emissions are characterized by higher δ34S (from 2 ‰ to 12 ‰) and lower Δ33S-values that tend towards 0 ‰. The rural background station, which likely collects aerosols that are transported by upstream winds to Montreal, yielded lower δ34S-values (2 ‰) and higher Δ33S-values, up to 0.35 ‰. Our results suggest that aerosols collected within the city have their Δ33S-values lowered by mixing with emissions from local sources compared to aerosols sampled in the vicinity of the city. We conclude that the non-zero Δ33S-values we measured were rather generated upstream the city than produced locally.
We also identified an urban seasonality for both δ34S and Δ33S in PM10, with higher δ34S-values during early spring and autumn and higher Δ33S-values in summer and winter. Our results indicate that these seasonal trends cannot be explained by corresponding seasonal variations in the atmospheric concentrations of the OH, H2O2, O2+TMI and O3 oxidants. In turn, we suggest that this seasonality may be better explained by either SO2 oxidation by the Criegee radicals and/or SO2 photooxidation in the presence of mineral dust. Still, further studies are required to isotopically characterize these latest oxidation pathways (i.e. Criegee radicals and SO2 photooxidation in the presence of mineral dust), which are still neglected in most current atmospheric models.
All data needed to draw the conclusions in the present study are shown in this paper and/or the Supplement. For additional data related to this study, please contact the corresponding author (auyang@mail.gyig.ac.cn).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-3779-2019-supplement.
DAY conducted sulfur isotope measurements under the supervision of PC and conducted chemical composition measurements under the supervision of KD. DW provided the samples. DAY, PC, KD and DW interpreted the data. DAY wrote the paper with contributions from all the coauthors.
The authors declare that they have no conflict of interest.
This study was supported by the Fond France Canada pour la Recherche (Grant 12/51) to David Au Yang. This project was supported by a grant from the Agence Nationale de la Recherche (ANR) via contract 14-CE33-0009-02-FOFAMIFS. We warmly thank the RSQA members for providing the samples, especially Diane Boulet and Véronique Chalut. We also thank Stefan Lalonde for discussions on the S-isotopes of Archean BIF, Marjorie Bagur for the logistic help and thank Hao Thi Bui and Boswell Wing for their analyses assistance performed in Montreal and Nelly Assayag and Guillaume Landais for analysis assistance performed at IPGP. IPGP contribution number 4020.
This paper was edited by Eliza Harris and reviewed by Huiming Bao and one anonymous referee.
Albrecht, B. A.: Aerosols, cloud microphysics, and fractional cloudiness, Science, 245, 1227–1230, 1989.
Andreae, M. O. and Crutzen, P. J.: Atmospheric Aerosols: Biogeochemical Sources and Role in Atmospheric Chemistry, Science, 276, 1052–1058, https://doi.org/10.1126/science.276.5315.1052, 1997.
Alexander, B., Park, R., Jacob, D., and Gong, S.: Transition metal-catalyzed oxidation of atmospheric sulfur: Global implications for the sulfur budget, J. Geophys. Res.-Atmos., 114, D02309, https://doi.org/10.1029/2008JD010486, 2009.
Alexander, B., Allman, D., Amos, H., Fairlie, T., Dachs, J., Hegg, D. A., and Sletten, R. S.: Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean, J. Geophys. Res.-Atmos., 117, D06304, https://doi.org/10.1029/2011JD016773, 2012.
An, L., Che, H., Xue, M., Zhang, T., Wang, H., Wang, Y., Zhou, C., Zhao, H., Gui, K., and Zheng, Y.: Temporal and spatial variations in sand and dust storm events in East Asia from 2007 to 2016: Relationships with surface conditions and climate change, Sci. Total Environ., 633, 452–462, 2018.
Au Yang, D., Landais, G., Assayag, N., Widory, D., and Cartigny, P.: Improved analysis of micro-and nanomole-scale sulfur multi-isotope compositions by gas source isotope ratio mass spectrometry, Rapid Commun. Mass Sp., 30, 897–907, 2016.
Au Yang, D., Bardoux, G., Assayag, N., Laskar, C., Widory, D., and Cartigny, P.: Atmospheric SO2 oxidation by NO2 plays no role in the mass independent sulfur isotope fractionation of urban aerosols, Atmos. Environ., 193, 109–117, https://doi.org/10.1016/j.atmosenv.2018.09.007, 2018.
Bardouki, H., Liakakou, H., Economou, C., Sciare, J., Smolık, J., Ždìmal, V., Eleftheriadis, K., Lazaridis, M., Dye, C., and Mihalopoulos, N.: Chemical composition of size-resolved atmospheric aerosols in the eastern Mediterranean during summer and winter, Atmos. Environ., 37, 195–208, 2003.
Baroni, M., Thiemens, M. H., Delmas, R. J., and Savarino, J.: Mass-independent sulfur isotopic compositions in stratospheric volcanic eruptions, Science, 315, 84–87, 2007.
Baroni, M., Savarino, J., Cole-Dai, J., Rai, V. K., and Thiemens, M. H.: Anomalous sulfur isotope compositions of volcanic sulfate over the last millennium in Antarctic ice cores, J. Geophys. Res.-Atmos., 113, D20112, https://doi.org/10.1029/2008JD010185, 2008.
Bauer, S. E. and Koch, D.: Impact of heterogeneous sulfate formation at mineral dust surfaces on aerosol loads and radiative forcing in the Goddard Institute for Space Studies general circulation model, J. Geophys. Res.-Atmos., 110, D17202, https://doi.org/10.1029/2005JD005870, 2005.
Becker, S. and Hirner, A.: Characterisation of Crude Oils by Carbon and Sulphur Isotope Ratio Measurements as a Tool for Pollution Control, Isot. Environ. Healt. S, 34, 255–264, 1998.
Benson, D. R., Young, L.-H., Kameel, F. R., and Lee, S.-H.: Laboratory-measured nucleation rates of sulfuric acid and water binary homogeneous nucleation from the SO2+OH reaction, Geophys. Res. Lett., 35, L11801, https://doi.org/10.1029/2008GL033387, 2008.
Berglen, T. F., Berntsen, T. K., Isaksen, I. S., and Sundet, J. K.: A global model of the coupled sulfur/oxidant chemistry in the troposphere: The sulfur cycle, J. Geophys. Res.-Atmos., 109, D19310, https://doi.org/10.1029/2003JD003948, 2004.
Bindeman, I., Eiler, J., Wing, B., and Farquhar, J.: Rare sulfur and triple oxygen isotope geochemistry of volcanogenic sulfate aerosols, Geochim. Cosmochim. Ac., 71, 2326–2343, 2007.
Blackie, D., Blackwell-Whitehead, R., Stark, G., Pickering, J., Smith, P., Rufus, J., and Thorne, A.: High-resolution photoabsorption cross-section measurements of SO2 at 198 K from 213 to 325 nm, J. Geophys. Res.-Planet., 116, E03006, https://doi.org/10.1029/2010JE003707, 2011.
Boothe, A. C. and Homeyer, C. R.: Global large-scale stratosphere–troposphere exchange in modern reanalyses, Atmos. Chem. Phys., 17, 5537–5559, https://doi.org/10.5194/acp-17-5537-2017, 2017.
Boulet, D. and Melançon, S.: Bilan environnemental, Qualité de l'air à Montréal, Rapport Annuel 2011, Ville de Montréal, Service des infrastructures, du transport et de l'environnement, Direction de l'environnement et du développement durable, Division de la planification et du suivi environnemental, RSQA, 8, 2012.
Boulet, D. and Melançon, S.: Bilan environnemental, Qualité de l'air à Montréal, Rapport Annuel 2013, Ville de Montréal, Service de l'environnement Division de la planification et du suivi environnemental, RSQA, 8, 2013.
Boy, M., Mogensen, D., Smolander, S., Zhou, L., Nieminen, T., Paasonen, P., Plass-Dülmer, C., Sipilä, M., Petäjä, T., Mauldin, L., Berresheim, H., and Kulmala, M.: Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations, Atmos. Chem. Phys., 13, 3865–3879, https://doi.org/10.5194/acp-13-3865-2013, 2013.
Calhoun, J. A., Bates, T. S., and Charlson, R. J.: Sulfur isotope measurements of submicrometer sulfate aerosol particles over the Pacific Ocean, Geophys. Res. Lett., 18, 1877–1880, 1991.
Calvert, J. G., Su, F., Bottenheim, J. W., and Strausz, O. P.: Mechanism of the homogeneous oxidation of sulfur dioxide in the troposphere, Atmos. Environ., 12, 197–226, 1978.
Cao, C., Zheng, S., and Singh, R. P.: Characteristics of aerosol optical properties and meteorological parameters during three major dust events (2005–2010) over Beijing, China, Atmos. Res., 150, 129–142, https://doi.org/10.1016/j.atmosres.2014.07.022, 2014.
Cheng, Y., Zheng, G., Wei, C., Mu, Q., Zheng, B., Wang, Z., Gao, M., Zhang, Q., He, K., and Carmichael, G.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Science Advances, 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Chin, M., Diehl, T., Ginoux, P., and Malm, W.: Intercontinental transport of pollution and dust aerosols: implications for regional air quality, Atmos. Chem. Phys., 7, 5501–5517, https://doi.org/10.5194/acp-7-5501-2007, 2007.
Chkhetiani, O. G., Gledzer, E. B., Artamonova, M. S., and Iordanskii, M. A.: Dust resuspension under weak wind conditions: direct observations and model, Atmos. Chem. Phys., 12, 5147–5162, https://doi.org/10.5194/acp-12-5147-2012, 2012.
Clarisse, L., Fromm, M., Ngadi, Y., Emmons, L., Clerbaux, C., Hurtmans, D., and Coheur, P.-F.: Intercontinental transport of anthropogenic sulfur dioxide and other pollutants: An infrared remote sensing case study, Geophys. Res. Lett., 38, L19806, https://doi.org/10.1029/2011GL048976, 2011.
Coplen, T. B.: Guidelines and recommended terms for expression of stable-isotope-ratio and gas-ratio measurement results, Rapid Commun. Mass Sp., 25, 2538–2560, 2011.
Cottle, P., Strawbridge, K., McKendry, I., O'Neill, N., and Saha, A.: A pervasive and persistent Asian dust event over North America during spring 2010: lidar and sunphotometer observations, Atmos. Chem. Phys., 13, 4515–4527, https://doi.org/10.5194/acp-13-4515-2013, 2013.
Danielache, S. O., Hattori, S., Johnson, M. S., Ueno, Y., Nanbu, S., and Yoshida, N.: Photoabsorption cross-section measurements of 32S, 33S, 34S, and 36S sulfur dioxide for the B1B1-X1A1 absorption band, J. Geophys. Res.-Atmos., 117, D24301, https://doi.org/10.1029/2012JD017464, 2012.
Dauphas, N. and Schauble, E. A.: Mass fractionation laws, mass-independent effects, and isotopic anomalies, Annu. Rev. Earth Pl. Sc., 44, 709–783, 2016.
Defouilloy, C., Cartigny, P., Assayag, N., Moynier, F., and Barrat, J.-A.: High-precision sulfur isotope composition of enstatite meteorites and implications of the formation and evolution of their parent bodies, Geochim. Cosmochim. Ac., 172, 393–409, 2016.
Diekrup, D., Hannington, M. D., Strauss, H., and Ginley, S. J.: Decoupling of Neoarchean sulfur sources recorded in Algoma-type banded iron formation, Earth Planet. Sc. Lett., 489, 1–7, 2018.
Ding, T., Valkiers, S., Kipphardt, H., De Bievre, P., Taylor, P., Gonfiantini, R., and Krouse, R.: Calibrated sulfur isotope abundance ratios of three IAEA sulfur isotope reference materials and V-CDT with a reassessment of the atomic weight of sulfur, Geochim. Cosmochim. Ac., 65, 2433–2437, 2001.
Dupart, Y., King, S. M., Nekat, B., Nowak, A., Wiedensohler, A., Herrmann, H., David, G., Thomas, B., Miffre, A., and Rairoux, P.: Mineral dust photochemistry induces nucleation events in the presence of SO2, P. Natl. Acad. Sci. USA, 109, 20842–20847, 2012.
Dupart, Y., Fine, L., D'Anna, B., and George, C.: Heterogeneous uptake of NO2 on Arizona Test Dust under UV-A irradiation: An aerosol flow tube study, Aeolian Res., 15, 45–51, 2014.
Eldridge, D., Guo, W., and Farquhar, J.: Theoretical estimates of equilibrium sulfur isotope effects in aqueous sulfur systems: Highlighting the role of isomers in the sulfite and sulfoxylate systems, Geochim. Cosmochim. Ac., 195, 171–200, 2016.
Eltbaakh, Y. A., Ruslan, M. H., Alghoul, M., Othman, M. Y., Sopian, K., and Fadhel, M.: Measurement of total and spectral solar irradiance: Overview of existing research, Renew. Sust. Energ. Rev., 15, 1403–1426, 2011.
Environnement Canada: National Pollutant Release Inventory, available at: http://donnees.ec.gc.ca/data/substances/plansreports/national-pollutant-release (last access: 21 March 2019), 2013.
Fairlie, T. D., Jacob, D. J., Dibb, J. E., Alexander, B., Avery, M. A., van Donkelaar, A., and Zhang, L.: Impact of mineral dust on nitrate, sulfate, and ozone in transpacific Asian pollution plumes, Atmos. Chem. Phys., 10, 3999–4012, https://doi.org/10.5194/acp-10-3999-2010, 2010.
Farquhar, J., Bao, H., and Thiemens, M.: Atmospheric influence of Earth's earliest sulfur cycle, Science, 289, 756–758, 2000.
Farquhar, J. and Wing, B. A.: Multiple sulfur isotopes and the evolution of the atmosphere, Earth Planet. Sc. Lett., 213, 1–13, 2003.
Farquhar, J., Savarino, J., Airieau, S., and Thiemens, M. H.: Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere, J. Geophys. Res.-Planet., 106, 32829–32839, 2001.
Farquhar, J., Johnston, D. T., and Wing, B. A.: Implications of conservation of mass effects on mass-dependent isotope fractionations: influence of network structure on sulfur isotope phase space of dissimilatory sulfate reduction, Geochim. Cosmochim. Ac., 71, 5862–5875, 2007.
Félix-Bermúdez, A., Delgadillo-Hinojosa, F., Huerta-Diaz, M., Camacho-Ibar, V., and Torres-Delgado, E.: Atmospheric Inputs of Iron and Manganese to Coastal Waters of the Southern California Current System: Seasonality, Santa Ana Winds, and Biogeochemical Implications, J. Geophys. Res.-Oceans, 122, 9230–9254, https://doi.org/10.1002/2017JC013224, 2017.
Formenti, P., Schütz, L., Balkanski, Y., Desboeufs, K., Ebert, M., Kandler, K., Petzold, A., Scheuvens, D., Weinbruch, S., and Zhang, D.: Recent progress in understanding physical and chemical properties of African and Asian mineral dust, Atmos. Chem. Phys., 11, 8231–8256, https://doi.org/10.5194/acp-11-8231-2011, 2011.
Fu, X., Wang, S., Chang, X., Cai, S., Xing, J., and Hao, J.: Modeling analysis of secondary inorganic aerosols over China: pollution characteristics, and meteorological and dust impacts, Sci. Rep. UK, 6, 35992, https://doi.org/10.1038/srep35992, 2016.
Gaffney, J., Premuzic, E., and Manowitz, B.: On the usefulness of sulfur isotope ratios in crude oil correlations, Geochim. Cosmochim. Ac., 44, 135–139, 1980.
Gautier, E., Savarino, J., Erbland, J., and Farquhar, J.: SO2 oxidation kinetics leave a consistent isotopic imprint on volcanic ice core sulfate, J. Geophys. Res.-Atmos., 123, https://doi.org/10.1029/2018JD028456, 2018.
George, C., Ammann, M., D'Anna, B., Donaldson, D., and Nizkorodov, S. A.: Heterogeneous photochemistry in the atmosphere, Chem. Rev., 115, 4218–4258, 2015.
Gettelman, A., Hoor, P., Pan, L., Randel, W., Hegglin, M. I., and Birner, T.: The extratropical upper troposphere and lower stratosphere, Rev. Geophys., 49, RG3003, https://doi.org/10.1029/2011RG000355, 2011.
Golitsyn, G., Granberg, I., Aloyan, A., Andronova, A., Gorchakov, G., Ponomarev, V., and Shishkov, P.: Study of emissions and transport of dust aerosol in Kalmykia Black Lands, J. Aerosol Sci., 1001, S725–S726, 1997.
Golitsyn, G., Granberg, I., Andronova, A., Ponomarev, V., Zilitinkevich, S., Smirnov, V., and Yablokov, M. Y.: Investigation of boundary layer fine structure in arid regions: Injection of fine dust into the atmosphere, Water Air Soil Poll., 3, 245–257, 2003.
Guieu, C., Loÿe-Pilot, M. D., Ridame, C., and Thomas, C.: Chemical characterization of the Saharan dust end-member: Some biogeochemical implications for the western Mediterranean Sea, J. Geophys. Res.-Atmos., 107, https://doi.org/10.1029/2001JD000582, 2002.
Guo, Q., Zhu, G., Strauss, H., Peters, M., Chen, T., Yang, J., Wei, R., Tian, L., and Han, X.: Tracing the sources of sulfur in Beijing soils with stable sulfur isotopes, J. Geochem. Explor., 161, 112–118, 2016.
Guo, Z., Li, Z., Farquhar, J., Kaufman, A. J., Wu, N., Li, C., Dickerson, R. R., and Wang, P.: Identification of sources and formation processes of atmospheric sulfate by sulfur isotope and scanning electron microscope measurements, J. Geophys. Res.-Atmos., 115, D00K07, https://doi.org/10.1029/2009JD012893, 2010.
Han, L., Cheng, S., Zhuang, G., Ning, H., Wang, H., Wei, W., and Zhao, X.: The changes and long-range transport of PM2.5 in Beijing in the past decade, Atmos. Environ., 110, 186–195, 2015.
Han, X., Guo, Q., Liu, C., Fu, P., Strauss, H., Yang, J., Hu, J., Wei, L., Ren, H., and Peters, M.: Using stable isotopes to trace sources and formation processes of sulfate aerosols from Beijing, China, Sci. Rep. UK, 6, 29958, https://doi.org/10.1038/srep29958, 2016.
Han, X., Guo, Q., Strauss, H., Liu, C., Hu, J., Guo, Z., Wei, R., Peters, M., Tian, L., and Kong, J.: Multiple sulfur isotope constraints on sources and formation processes of sulfate in Beijing PM2.5 aerosol, Environ. Sci. Technol., 51, 7794–7803, 2017.
Harris, E., Sinha, B., Foley, S., Crowley, J. N., Borrmann, S., and Hoppe, P.: Sulfur isotope fractionation during heterogeneous oxidation of SO2 on mineral dust, Atmos. Chem. Phys., 12, 4867–4884, https://doi.org/10.5194/acp-12-4867-2012, 2012a.
Harris, E., Sinha, B., Hoppe, P., Crowley, J. N., Ono, S., and Foley, S.: Sulfur isotope fractionation during oxidation of sulfur dioxide: gas-phase oxidation by OH radicals and aqueous oxidation by H2O2, O3 and iron catalysis, Atmos. Chem. Phys., 12, 407–423, https://doi.org/10.5194/acp-12-407-2012, 2012b.
Harris, E., Sinha, B., Hoppe, P., Foley, S., and Borrmann, S.: Fractionation of sulfur isotopes during heterogeneous oxidation of SO2 on sea salt aerosol: a new tool to investigate non-sea salt sulfate production in the marine boundary layer, Atmos. Chem. Phys., 12, 4619–4631, https://doi.org/10.5194/acp-12-4619-2012, 2012c.
Harris, E., Sinha, B., Hoppe, P., Foley, S., and Borrmann, S.: Fractionation of sulfur isotopes during heterogeneous oxidation of SO2 on sea salt aerosol: a new tool to investigate non-sea salt sulfate production in the marine boundary layer, Atmos. Chem. Phys., 12, 4619–4631, https://doi.org/10.5194/acp-12-4619-2012, 2012d.
Harris, E., Sinha, B., Hoppe, P., and Ono, S.: High-precision measurements of 33S and 34S fractionation during SO2 oxidation reveal causes of seasonality in SO2 and sulfate isotopic composition, Environ. Sci. Technol., 47, 12174–12183, 2013a.
Harris, E., Sinha, B., Van Pinxteren, D., Tilgner, A., Fomba, K. W., Schneider, J., Roth, A., Gnauk, T., Fahlbusch, B., and Mertes, S.: Enhanced role of transition metal ion catalysis during in-cloud oxidation of SO2, Science, 340, 727–730, 2013b.
Hattori, S., Schmidt, J. A., Johnson, M. S., Danielache, S. O., Yamada, A., Ueno, Y., and Yoshida, N.: SO2 photoexcitation mechanism links mass-independent sulfur isotopic fractionation in cryospheric sulfate to climate impacting volcanism, P. Natl. Acad. Sci. USA, 110, 17656–17661, 2013.
He, H., Wang, Y., Ma, Q., Ma, J., Chu, B., Ji, D., Tang, G., Liu, C., Zhang, H., and Hao, J.: Mineral dust and NOx promote the conversion of SO2 to sulfate in heavy pollution days, Sci. Rep. UK, 4, 4172, https://doi.org/10.1038/srep04172, 2014.
Herrmann, H.: Kinetics of aqueous phase reactions relevant for atmospheric chemistry, Chem. Rev., 103, 4691–4716, 2003.
Kok, J. F., Parteli, E. J., Michaels, T. I., and Karam, D. B.: The physics of wind-blown sand and dust, Rep. Prog. Phys., 75, 106901, 2012.
Krouse, H. R. and Grinenko, V. A.: Stable isotopes: natural and anthropogenic sulphur in the environment, published on behalf of the Scientifc Committee on Problems of the Environment (SCOPE) of the International Council of Scientific Unions (ICSU) in collaboration with the United Nations Environment Programme by Wiley, Chichester, New York, 1991.
Kulmala, M., Vehkamäki, H., Petäjä, T., Dal Maso, M., Lauri, A., Kerminen, V.-M., Birmili, W., and McMurry, P. H.: Formation and growth rates of ultrafine atmospheric particles: a review of observations, J. Aerosol Sci., 35, 143–176, 2004.
Labidi, J., Cartigny, P., Birck, J., Assayag, N., and Bourrand, J.: Determination of multiple sulfur isotopes in glasses: A reappraisal of the MORB δ34S, Chem. Geol., 334, 189–198, 2012.
Lee, C. W., Savarino, J., Cachier, H., and Thiemens, M.: Sulfur (32S, 33S, 34S, 36S) and oxygen (16O, 17O, 18O) isotopic ratios of primary sulfate produced from combustion processes, Tellus B, 54, 193–200, 2002.
Lee, Y. N. and Schwartz, S. E.: Kinetics of oxidation of aqueous sulfur (IV) by nitrogen dioxide, Precipitation Scavenging, Dry Deposition, and Resuspension, edited by: Pruppacher, H. R., Semonin, R. G., and Slinn, W. G. N., Elsevier, New York, Vol. 1, 453–466, 1983.
Lelieveld, J., Evans, J., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, 2015.
Levy, H., Horowitz, L. W., Schwarzkopf, M. D., Ming, Y., Golaz, J. C., Naik, V., and Ramaswamy, V.: The roles of aerosol direct and indirect effects in past and future climate change, J. Geophys. Res.-Atmos., 118, 4521–4532, 2013.
Lin, M., Zhang, Z., Su, L., Hill-Falkenthal, J., Priyadarshi, A., Zhang, Q., Zhang, G., Kang, S., Chan, C. Y., and Thiemens, M. H.: Resolving the impact of stratosphere-to-troposphere transport on the sulfur cycle and surface ozone over the Tibetan Plateau using a cosmogenic 35S tracer, J. Geophys. Res.-Atmos., 121, 439–456, 2016.
Lin, M., Kang, S., Shaheen, R., Li, C., Hsu, S.-C., and Thiemens, M. H.: Atmospheric sulfur isotopic anomalies recorded at Mt. Everest across the Anthropocene, P. Natl. Acad. Sci. USA, 115, 6964–6969, https://doi.org/10.1073/pnas.1801935115, 2018a.
Lin, M., Zhang, X., Li, M., Xu, Y., Zhang, Z., Tao, J., Su, B., Liu, L., Shen, Y., and Thiemens, M. H.: Five-S-isotope evidence of two distinct mass-independent sulfur isotope effects and implications for the modern and Archean atmospheres, P. Natl. Acad. Sci. USA, 115, 8541–8546, 2018b.
Longo, A. F., Vine, D. J., King, L. E., Oakes, M., Weber, R. J., Huey, L. G., Russell, A. G., and Ingall, E. D.: Composition and oxidation state of sulfur in atmospheric particulate matter, Atmos. Chem. Phys., 16, 13389–13398, https://doi.org/10.5194/acp-16-13389-2016, 2016.
Ma, J., Chu, B., Liu, J., Liu, Y., Zhang, H., and He, H.: NOx promotion of SO2 conversion to sulfate: An important mechanism for the occurrence of heavy haze during winter in Beijing, Environ. Pollut., 233, 662–669, 2018.
Marticorena, B. and Bergametti, G.: Modeling the atmospheric dust cycle: 1. Design of a soil-derived dust emission scheme, J. Geophys. Res.-Atmos., 100, 16415–16430, 1995.
Martin, E.: Volcanic Plume Impact on the Atmosphere and Climate: O-and S-Isotope Insight into Sulfate Aerosol Formation, Geosciences, 8, 198, https://doi.org/10.3390/geosciences8060198, 2018.
Mauldin III, R., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V.-M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, 2012.
McKendry, I. G., Hacker, J. P., Stull, R., Sakiyama, S., Mignacca, D., and Reid, K.: Long-range transport of Asian dust to the Lower Fraser Valley, British Columbia, Canada, J. Geophys. Res.-Atmos., 106, 18361–18370, https://doi.org/10.1029/2000JD900359, 2001.
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S.: Impact of nucleation on global CCN, Atmos. Chem. Phys., 9, 8601–8616, https://doi.org/10.5194/acp-9-8601-2009, 2009.
Mertes, S., Galgon, D., Schwirn, K., Nowak, A., Lehmann, K., Massling, A., Wiedensohler, A., and Wieprecht, W.: Evolution of particle concentration and size distribution observed upwind, inside and downwind hill cap clouds at connected flow conditions during FEBUKO, Atmos. Environ., 39, 4233–4245, 2005a.
Mertes, S., Lehmann, K., Nowak, A., Massling, A., and Wiedensohler, A.: Link between aerosol hygroscopic growth and droplet activation observed for hill-capped clouds at connected flow conditions during FEBUKO, Atmos. Environ., 39, 4247–4256, 2005b.
Molina, L. and Molina, M.: Absolute absorption cross sections of ozone in the 185-to 350-nm wavelength range, J. Geophys. Res.-Atmos., 91, 14501–14508, 1986.
Myhre, G., Shindell, D., Bréon, F.-M., Collins, W., Fuglestvedt, J., Huang, J., Koch, D., Lamarque, J.-F., Lee, D., Mendoza, B., Nakajima, T., Robock, A., Stephens, G., Takemura, T., and Zhang, H.: Anthropogenic and Natural Radiative Forcing. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013.
Newman, L. and Forrest, J.: Sulphur isotope measurements relevant to power plant emissions in the northeastern United States, in: Stable isotopes: Natural and Anthropogenic sulphur in the environment, edited by: Krouse, H. R. and Grinenko, V. A., John Wiley and Sons, 133–176, 1991.
Nielsen, H.: Isotopic composition of the major contributors to atmospheric sulfur, Tellus, 26, 213–221, 1974.
Norman, A.-L., Anlauf, K., Hayden, K., Thompson, B., Brook, J. R., Li, S.-M., and Bottenheim, J.: Aerosol sulphate and its oxidation on the Pacific NW coast: S and O isotopes in PM2.5, Atmos. Environ., 40, 2676–2689, 2006.
Oduro, H., Van Alstyne, K. L., and Farquhar, J.: Sulfur isotope variability of oceanic DMSP generation and its contributions to marine biogenic sulfur emissions, P. Natl. Acad. Sci. USA, 109, 9012–9016, 2012.
Ono, S., Wing, B., Johnston, D., Farquhar, J., and Rumble, D.: Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles, Geochim. Cosmochim. Ac., 70, 2238–2252, 2006a.
Ono, S., Wing, B., Rumble, D., and Farquhar, J.: High precision analysis of all four stable isotopes of sulfur (32S, 33S, 34S and 36S) at nanomole levels using a laser fluorination isotope-ratio-monitoring gas chromatography – mass spectrometry, Chem. Geol., 225, 30–39, 2006b.
Ono, S., Whitehill, A., and Lyons, J.: Contribution of isotopologue self-shielding to sulfur mass-independent fractionation during sulfur dioxide photolysis, J. Geophys. Res.-Atmos., 118, 2444–2454, 2013.
Otake, T., Lasaga, A. C., and Ohmoto, H.: Ab initio calculations for equilibrium fractionations in multiple sulfur isotope systems, Chem. Geol., 249, 357–376, 2008.
Paris, R., Desboeufs, K. V., Formenti, P., Nava, S., and Chou, C.: Chemical characterisation of iron in dust and biomass burning aerosols during AMMA-SOP0/DABEX: implication for iron solubility, Atmos. Chem. Phys., 10, 4273–4282, https://doi.org/10.5194/acp-10-4273-2010, 2010.
Penner, J. E., Dickinson, R. E., and O'Neill, C. A.: Effects of aerosol from biomass burning on the global radiation budget, Science, 256, 1432–1434, 1992.
Penner, J. E., Quaas, J., Storelvmo, T., Takemura, T., Boucher, O., Guo, H., Kirkevåg, A., Kristjánsson, J. E., and Seland, Ø.: Model intercomparison of indirect aerosol effects, Atmos. Chem. Phys., 6, 3391–3405, https://doi.org/10.5194/acp-6-3391-2006, 2006.
Premuzic, E., Gaffney, J., and Manowitz, B.: The importance of sulfur isotope ratios in the differentiation of Prudhoe Bay crude oils, J. Geochem. Explor., 26, 151–159, 1986.
Prospero, J. M.: Saharan Dust Transport Over the North Atlantic Ocean and Mediterranean: An Overview, in: The Impact of Desert Dust Across the Mediterranean, edited by: Guerzoni, S. and Chester, R., Springer Netherlands, Dordrecht, 133–151, 1996.
Ramanathan, V., Crutzen, P. J., Kiehl, T. J., and Rosenfeld, D.: Aerosols, Climate, and the Hydrological Cycle, Science, 294, 2119–2124, 2001.
Ramanathan, V., Chung, C., Kim, D., Bettge, T., Buja, L., Kiehl, J., Washington, W., Fu, Q., Sikka, D., and Wild, M.: Atmospheric brown clouds: Impacts on South Asian climate and hydrological cycle, P. Natl. Acad. Sci. USA, 102, 5326–5333, 2005.
Rees, C., Jenkins, W., and Monster, J.: The sulphur isotopic composition of ocean water sulphate, Geochim. Cosmochim. Ac., 42, 377–381, 1978.
Romero, A. B. and Thiemens, M. H.: Mass-independent sulfur isotopic compositions in present-day sulfate aerosols, J. Geophys. Res.-Atmos., 108, 4524, https://doi.org/10.1029/2003JD003660, 2003.
Sanusi, A. A., Norman, A.-L., Burridge, C., Wadleigh, M., and Tang, W.-W.: Determination of the S isotope composition of methanesulfonic acid, Anal. Chem., 78, 4964–4968, 2006.
Sarwar, G., Fahey, K., Kwok, R., Gilliam, R. C., Roselle, S. J., Mathur, R., Xue, J., Yu, J., and Carter, W. P.: Potential impacts of two SO2 oxidation pathways on regional sulfate concentrations: aqueous-phase oxidation by NO2 and gas-phase oxidation by Stabilized Criegee Intermediates, Atmos. Environ., 68, 186–197, 2013.
Sarwar, G., Simon, H., Fahey, K., Mathur, R., Goliff, W. S., and Stockwell, W. R.: Impact of sulfur dioxide oxidation by Stabilized Criegee Intermediate on sulfate, Atmos. Environ., 85, 204–214, 2014.
Savarino, J., Romero, A., Cole-Dai, J., Bekki, S., and Thiemens, M.: UV induced mass-independent sulfur isotope fractionation in stratospheric volcanic sulfate, Geophys. Res. Lett., 30, 2131, https://doi.org/10.1029/2003GL018134, 2003.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, 3. Edn, John Wiley & Sons, Inc., Hoboken, New Jersey, 2012.
Shaheen, R., Abaunza, M. M., Jackson, T. L., McCabe, J., Savarino, J., and Thiemens, M. H.: Large sulfur-isotope anomaly in nonvolcanic sulfate aerosol and its implications for the Archean atmosphere, P. Natl. Acad. Sci. USA, 111, 11979–11983, 2014.
Sinha, B. W., Hoppe, P., Huth, J., Foley, S., and Andreae, M. O.: Sulfur isotope analyses of individual aerosol particles in the urban aerosol at a central European site (Mainz, Germany), Atmos. Chem. Phys., 8, 7217–7238, https://doi.org/10.5194/acp-8-7217-2008, 2008.
Sipilä, M., Jokinen, T., Berndt, T., Richters, S., Makkonen, R., Donahue, N. M., Mauldin III, R. L., Kurtén, T., Paasonen, P., Sarnela, N., Ehn, M., Junninen, H., Rissanen, M. P., Thornton, J., Stratmann, F., Herrmann, H., Worsnop, D. R., Kulmala, M., Kerminen, V.-M., and Petäjä, T.: Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids, Atmos. Chem. Phys., 14, 12143–12153, https://doi.org/10.5194/acp-14-12143-2014, 2014.
Smith, J. and Batts, B.: The distribution and isotopic composition of sulfur in coal, Geochim. Cosmochim. Ac., 38, 121–133, 1974.
Song, C. H. and Carmichael, G. R.: The aging process of naturally emitted aerosol (sea-salt and mineral aerosol) during long range transport, Atmos. Environ., 33, 2203–2218, 1999.
Sprenger, M. and Wernli, H.: A Northern Hemispheric climatology of cross-tropopause exchange for the ERA15 time period (1979–1993), J. Geophys. Res.-Atmos., 108, 8521, https://doi.org/10.1029/2002JD002636, 2003.
Tanaka, N., Rye, D. M., Xiao, Y., and Lasaga, A. C.: Use of stable sulfur isotope systematics for evaluating oxidation reaction pathways and in-cloud-scavenging of sulfur dioxide in the atmosphere, Geophys. Res. Lett., 21, 1519–1522, 1994.
Thode, H., Monster, J., and Dunford, H.: Sulphur isotope geochemistry, Geochim. Cosmochim. Ac., 25, 159–174, 1961.
Thomassot, E., O'Neil, J., Francis, D., Cartigny, P., and Wing, B. A.: Atmospheric record in the Hadean Eon from multiple sulfur isotope measurements in Nuvvuagittuq Greenstone Belt (Nunavik, Quebec), P. Natl. Acad. Sci. USA, 112, 707–712, 2015.
Tomasi, C. and Lupi, A.: Primary and Secondary Sources of Atmospheric Aerosol, Atmospheric Aerosols: Life Cycles and Effects on Air Quality and Climate, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 1–86, 2017.
Usher, C. R., Michel, A. E., and Grassian, V. H.: Reactions on mineral dust, Chem. Rev., 103, 4883–4940, 2003.
Ville de Montréal: Reduced dependance on fossil fuels in Montréal, Ville de Montréal, Reduced dependance on fossil fuels in Montréal, Office de consultation publique de Montréal, 32, 2015.
Wadleigh, M., Schwarcz, H., and Kramer, J.: Isotopic evidence for the origin of sulphate in coastal rain, Tellus B, 48, 44–59, 1996.
Wagener, T., Guieu, C., Losno, R., Bonnet, S., and Mahowald, N.: Revisiting atmospheric dust export to the Southern Hemisphere ocean: Biogeochemical implications, Global Biogeochem. Cy., 22, GB2006, https://doi.org/10.1029/2007GB002984, 2008.
Wall, S. M., John, W., and Ondo, J. L.: Measurement of aerosol size distributions for nitrate and major ionic species, Atmos. Environ., 22, 1649–1656, 1988.
Wasiuta, V., Lafrenière, M. J., Norman, A.-L., and Hastings, M. G.: Summer deposition of sulfate and reactive nitrogen to two alpine valleys in the Canadian Rocky Mountains, Atmos. Environ., 101, 270–285, 2015.
Watanabe, Y., Farquhar, J., and Ohmoto, H.: Anomalous fractionations of sulfur isotopes during thermochemical sulfate reduction, Science, 324, 370–373, 2009.
Whitehill, A. R. and Ono, S.: Excitation band dependence of sulfur isotope mass-independent fractionation during photochemistry of sulfur dioxide using broadband light sources, Geochim. Cosmochim. Ac., 94, 238–253, 2012.
Whitehill, A. R., Xie, C., Hu, X., Xie, D., Guo, H., and Ono, S.: Vibronic origin of sulfur mass-independent isotope effect in photoexcitation of SO2 and the implications to the early earth's atmosphere, P. Natl. Acad. Sci. USA, 110, 17697–17702, 2013.
Whitehill, A. R., Jiang, B., Guo, H., and Ono, S.: SO2 photolysis as a source for sulfur mass-independent isotope signatures in stratospehric aerosols, Atmos. Chem. Phys., 15, 1843–1864, https://doi.org/10.5194/acp-15-1843-2015, 2015.
World Health Organization: Ambient (outdoor) air quality and health, 2016, available at: http://www.who.int/mediacentre/factsheets/fs313/en/ (last access: 29 November 2017), 2016.
Yang, Y., Russell, L. M., Lou, S., Liao, H., Guo, J., Liu, Y., Singh, B., and Ghan, S. J.: Dust-wind interactions can intensify aerosol pollution over eastern China, Nat. Commun., 8, 15333, https://doi.org/10.1038/ncomms15333, 2017.
Yang, Y., Wang, H., Smith, S. J., Zhang, R., Lou, S., Qian, Y., Ma, P.-L., and Rasch, P. J.: Recent intensification of winter haze in China linked to foreign emissions and meteorology, Sci. Rep. UK, 8, 2107, https://doi.org/10.1038/s41598-018-20437-7, 2018.
Ye, J., Abbatt, J. P. D., and Chan, A. W. H.: Novel pathway of SO2 oxidation in the atmosphere: reactions with monoterpene ozonolysis intermediates and secondary organic aerosol, Atmos. Chem. Phys., 18, 5549–5565, https://doi.org/10.5194/acp-18-5549-2018, 2018.
Young, E. D., Galy, A., and Nagahara, H.: Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance, Geochim. Cosmochim. Ac., 66, 1095–1104, 2002.
Yu, F. and Luo, G.: Simulation of particle size distribution with a global aerosol model: contribution of nucleation to aerosol and CCN number concentrations, Atmos. Chem. Phys., 9, 7691–7710, https://doi.org/10.5194/acp-9-7691-2009, 2009.
Yu, Z., Jang, M., and Park, J.: Modeling atmospheric mineral aerosol chemistry to predict heterogeneous photooxidation of SO2, Atmos. Chem. Phys., 17, 10001–10017, https://doi.org/10.5194/acp-17-10001-2017, 2017.
Zhao, D., Song, X., Zhu, T., Zhang, Z., Liu, Y., and Shang, J.: Multiphase oxidation of SO2 by NO2 on CaCO3 particles, Atmos. Chem. Phys., 18, 2481–2493, https://doi.org/10.5194/acp-18-2481-2018, 2018.
Zhao, T., Gong, S., Zhang, X., Blanchet, J.-P., McKendry, I., and Zhou, Z.: A simulated climatology of Asian dust aerosol and its trans-Pacific transport. Part I: Mean climate and validation, J. Climate, 19, 88–103, 2006.
Zinger, I. and Delisle, C.: Quality of used-snow discharged in the ST-Lawrence river, in the region of the Montreal Harbor, Water Air Soil Poll., 39, 47–57, 1988.