the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 01 Mar 2019
| 01 Mar 2019
Enhancement of secondary organic aerosol formation and its oxidation state by SO2 during photooxidation of 2-methoxyphenol
Changgeng Liu
Tianzeng Chen
Jun Liu
Peng Zhang
2-Methoxyphenol (guaiacol) is derived from the lignin pyrolysis and taken as a potential tracer for wood smoke emissions. In this work, the effect of SO2 at atmospheric levels (0–56 ppbv) on secondary organic aerosol (SOA) formation and its oxidation state during guaiacol photooxidation was investigated in the presence of various inorganic seed particles (i.e., NaCl and (NH4)2SO4). Without SO2 and seed particles, SOA yields ranged from (9.46±1.71) % to (26.37±2.83) % and could be well expressed by a one-product model. According to the ratio of the average gas-particle partitioning timescale ( over the course of the experiment to the vapor wall deposition timescale (τg−w), the determined SOA yields were underestimated by a factor of ∼2. The presence of SO2 resulted in enhancing SOA yield by 14.04 %–23.65 %. With (NH4)2SO4 and NaCl seed particles, SOA yield was enhanced by 23.07 % and 29.57 %, respectively, which further increased significantly to 29.78 %–53.43 % in the presence of SO2, suggesting that SO2 and seed particles have a synergetic contribution to SOA formation. The decreasing trend of the ratio in the presence of seed particles and SO2 suggested that more SOA-forming vapors partitioned into the particle phase, consequently increasing SOA yields. It should be noted that SO2 was found to be in favor of increasing the carbon oxidation state (OSC) of SOA, indicating that the functionalization or the partitioning of highly oxidized products into particles should be more dominant than the oligomerization. In addition, the average N∕C ratio of SOA was 0.037, which revealed that NOx participated in the photooxidation process, consequently leading to the formation of organic N-containing compounds. The experimental results demonstrate the importance of SO2 on the formation processes of SOA and organic S-containing compounds and are also helpful to further understand SOA formation from the atmospheric photooxidation of guaiacol and its subsequent impacts on air quality and climate.
- Article
(1014 KB) - Full-text XML
-
Supplement
(2862 KB) - BibTeX
- EndNote





Biomass burning is considered as one of the major sources of gas and particulate pollutants in the atmosphere (Lauraguais et al., 2014b; Yang et al., 2016). Therefore, it has significant adverse impacts on regional and global air quality (Bari and Kindzierski, 2016; Lelieveld et al., 2001), climate (Chen and Bond, 2010), and human health (Naeher et al., 2007). The chemical species emitted by biomass burning is mainly dependent on fuel source and combustion conditions (O'Neill et al., 2014). Natural wood is composed of cellulose (40 wt %–50 wt %), hemicellulose (25 wt %–35 wt %), and lignin (18 wt %–35 wt %) (Nolte et al., 2001). During the burning process, lignin pyrolysis could result in the formation of methoxyphenols, mainly including guaiacol (2-methoxyphenol), syringol (2,6-dimethoxyphenol), and their derivatives (Nolte et al., 2001; Schauer et al., 2001). Due to the high emission rate of methoxyphenols (900–4200 mg kg−1 wood), methoxyphenols are considered as the potential tracers for wood burning (Hawthorne et al., 1989, 1992; Simoneit et al., 1993).
As a representative type of methoxyphenols, guaiacol mainly exists in the gas phase and is widely found in the atmosphere (Schauer et al., 2001). Its emission factor of wood burning is in the range of 172–279 mg kg−1 wood (Schauer et al., 2001). In recent years, the reactivity of gas-phase guaiacol toward OH radicals (Coeur-Tourneur et al., 2010a), NO3 radicals (Lauraguais et al., 2016; Yang et al., 2016), Cl atoms (Lauraguais et al., 2014a), and O3 (El Zein et al., 2015) has been investigated, suggesting that its degradation by OH and NO3 radicals might be predominant in the atmosphere. Meanwhile, several studies have reported the significant secondary organic aerosol (SOA) formation from guaiacol oxidation by OH radicals, produced from the photolysis of the OH precursors (i.e., H2O2 and CH3ONO) (Ahmad et al., 2017; Lauraguais et al., 2014b; Yee et al., 2013). However, SOA formation from the photooxidation of guaiacol in the presence of NOx has not been investigated without adding a direct OH precursor, even though it has been recently reported that the atmospheric level of NOx could reach up to 200 ppbv in severely polluted atmospheres of China (Li et al., 2017).
Although many studies concentrated on the SOA production from the oxidation of volatile organic compounds (VOCs), the reported SOA yields showed high variability for a given precursor (Chu et al., 2016, 2017; Ge et al., 2017a; Lauraguais et al., 2012, 2014b; Ng et al., 2007; Sarrafzadeh et al., 2016; Yee et al., 2013). This variability is mainly dependent on the numerous factors, e.g., preexisting seed particles, SO2 level, NOx level, humidity, and temperature. Two of the critical factors are the impacts of preexisting seed particles and SO2 level on SOA formation (Chu et al., 2016, 2017; Ge et al., 2017a). In addition, the atmospheric concentration of SO2 could be close to 200 ppbv in severely polluted atmospheres of China, and SOA from biomass burning and sulfate formation could significantly contribute to severe haze pollution (Li et al., 2017). During the transport process, smoke plumes from biomass burning would be inevitably mixed with suspended particles (e.g., (NH4)2SO4 particles), SO2, and NOx in the atmosphere. However, the influences of these co-existing pollutants on the transformation of guaiacol and its SOA formation are still unclear. For these reasons, the aim of this work was to investigate the SOA formation from guaiacol photooxidation in the presence of NOx in a 30 m3 indoor smog chamber, as well as the effect of SO2 on SOA formation with various inorganic seed particles.
2.1 Smog chamber
The photooxidation experiments were performed in a 30 m3 indoor smog chamber (4 m height × 2.5 m width × 3 m length), which was built in a temperature-controlled room located at the Research Center for Eco-Environment Sciences, Chinese Academy of Sciences (RCEES-CAS). The details have been described elsewhere (Chen et al., 2019) and are shown in Fig. S1 in the Supplement. Briefly, 120 UV lamps (365 nm, Philips TL 60/10R) were taken as the light source with a NO2 photolysis rate of 0.55 min−1, which was comparable to the irradiation intensity at noon in Beijing (Chou et al., 2011). A maglev fan installed at the bottom center of the smog chamber was used to mix sufficiently the introduced gas species and seed particles. Temperature (T) and relatively humidity (RH) in the chamber were (302±1) K and (39±1) %, respectively. Before each experiment, the chamber was flushed by purified dry zero air for ∼36 h with a flow rate of 100 L min−1 until the particle number concentration in the chamber was lower than 20 cm−3.
2.2 Experimental procedures
Gas-phase guaiacol was firstly introduced into the chamber by purified dry zero air flowing through the gently heated injector with a known volume of pure liquid guaiacol until guaiacol fully vaporized. Its concentration in the chamber was measured in real time by a high-resolution proton-transfer reaction time-of-flight mass spectrometer (HR-ToF-PTRMS) (Ionicon Analytik GmbH) and was calibrated by a commercial permeation tube (VICI AG International Valco Instruments Co., Inc.). When guaiacol concentration was stable, NO and SO2 were introduced into the chamber by a mass flow meter using purified dry zero air as the carrier gas. Their concentrations were controlled by the injection time preset through the electromagnetic valve and were measured by a NOx analyzer (model 42i-TL, Thermo Fisher Scientific, Inc.) and a SO2 analyzer (model 43i, Thermo Fisher Scientific Inc.), respectively. In this work, the initial ratio (V∕V) of guaiacol concentration to NOx concentration in the chamber was similar in all experiments (∼1.2) (Tables 1 and 2). In addition, sodium chloride (NaCl) and ammonium sulfate ((NH4)2SO4) were used as the inorganic seeds. The seed aerosols in the chamber were generated by the atomization of a 0.02 M aqueous solution. Through atomization, the size distribution of seed particles peaked at 51–58 nm was achieved, with a number concentration of 10 100–11 400 cm−3 (Table 2). After gas species and seed particles in the chamber were mixed well, the photooxidation experiment was carried out with the fan turned off. In this work, the OH concentrations in the chamber were (1.3–2.2) × 106 molecules cm−3, calculated based on the degradation rate ( cm3 molecule−1 s−1) of guaiacol with OH radicals (Coeur-Tourneur et al., 2010a). The chemicals and gas samples used in this work were described in Supplement.
2.3 Data analysis
The HR-ToF-PTRMS with a time resolution of 1 min was used online to measure the gas-phase concentration of guaiacol, and its m∕z range was 10–500 in the process of data acquisition. Before data collection, the peaks of the protonated water () and protonated acetone ([C3H7O]+) ions at and 59.0491 were used for mass calibration, with the aim of obtaining accurate mass determination during the experimental process. All data obtained by the HR-ToF-PTRMS were analyzed with the PTR-MS Viewer software (version 3.1.0, IONICON Analytik).
An Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) was applied to online measure the chemical composition of particles and the non-refractory submicron aerosol mass (DeCarlo et al., 2006). For all experiments, the acquisition time of the HR-ToF-AMS was 2 min. The inlet flow rate, ionization efficiency, and particle sizing of the HR-ToF-AMS were calibrated at regular intervals, according to the standard protocols using the size-selected pure ammonium nitrate particles (Drewnick et al., 2005; Jimenez et al., 2003). All data obtained by the HR-ToF-AMS were analyzed by the ToF-AMS analysis toolkit SQUIRREL 1.57I/PIKA 1.16I version, in Igor Pro version 6.37. The size distribution and concentration of particles were monitored by a scanning mobility particle sizer (SMPS), which is composed of a differential mobility analyzer (DMA) (model 3082, TSI Inc.) and a condensation particle counter (CPC) (model 3776, TSI Inc.). Assuming that particles are spherical and non-porous, the average particle density could be calculated to be 1.4 g cm−3 using the equation (DeCarlo et al., 2004), where dva is the mean vacuum aerodynamic diameter measured by the HR-ToF-AMS and dm is the mean volume-weighted mobility diameter measured by the SMPS. The mass concentration of particles measured by the HR-ToF-AMS was corrected by the SMPS data in this work using the same method as Gordon et al. (2014). In this work, the wall loss rate (kdep) of (NH4)2SO4 particles could be expressed as (Dp is the particle diameter, nm), which was measured according to the literature method (Takekawa et al., 2003) and was used to correct the wall loss of SOA. In addition, its wall loss rate was determined at predetermined time intervals, which only had a slight change among different experiments.
2.4 Vapor wall-loss correction
Previous studies have indicated that the losses of SOA-forming vapors to chamber walls can result in the substantial and systematic underestimation of SOA yield (Zhang et al., 2014, 2015). Therefore, SOA yields obtained in this work were also corrected by vapor wall loss. The effect of vapor wall deposition on SOA yields mainly depends on the competition between the uptake of organic vapors by aerosol particles and the chamber wall (Zhang et al., 2015). Thus, the ratio of the average gas-particle partitioning timescale () over the course of the experiment to the vapor wall deposition timescale (τg−w) could be reasonably used to evaluate the underestimation of SOA yields. The detailed calculation of and τg−w was shown in the Supplement.
3.1 SOA yields
A series of experiments were conducted at different guaiacol∕NOx concentrations under atmospheric pressure. The experimental conditions and results are shown in Table 1. SOA yield was calculated to be the ratio of SOA mass concentration (Mo, µg m−3) to the consumed guaiacol concentration (Δ[guaiacol], µg m−3) at the end of each experiment (Kang et al., 2007). The results showed that SOA yield was dependent on the initial guaiacol concentration ([Guaiacol]0). Higher precursor concentration would result in a higher amount of condensable products, subsequently enhancing SOA formation (Lauraguais et al., 2012). In addition, it should be noted that SOA mass could directly affect the gas-particle partitioning via acting as the adsorption medium of oxidation products; thus, higher SOA mass generally leads to a higher SOA yield (Lauraguais et al., 2014b).
Table 1Experimental conditions and results for guaiacol photooxidation in the presence NOx.
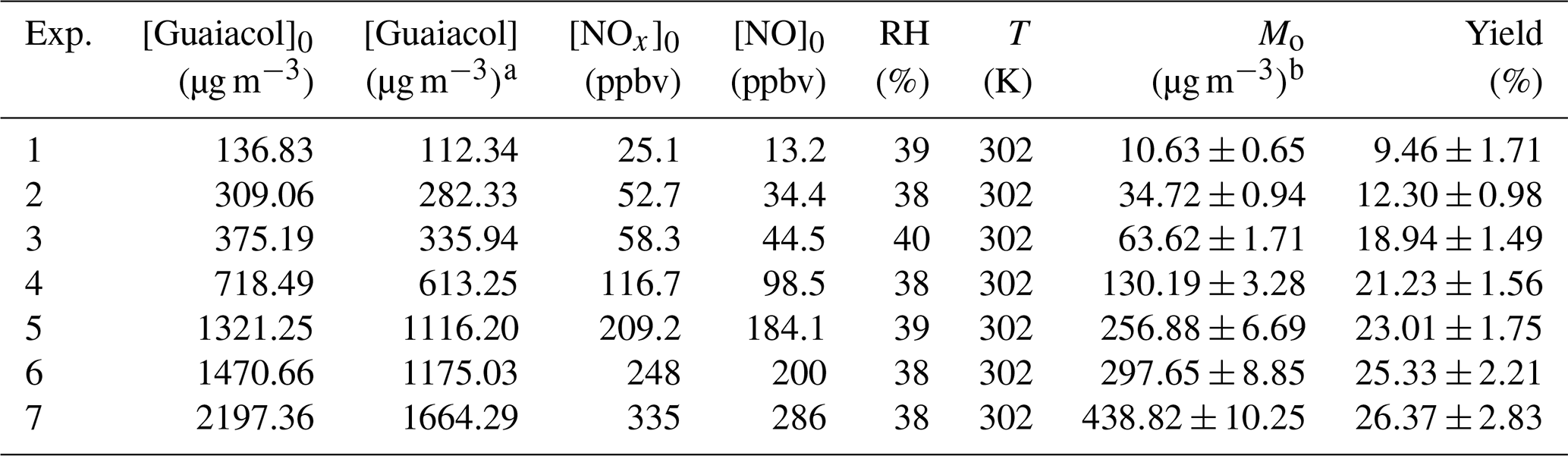
a The consumed guaiacol concentration at the end of each
experiment.
b Mo is the mass concentration of SOA.
SOA yield (Y) could be represented by a widely used semi-empirical model based on the absorptive gas-particle partitioning of semi-volatile products, typically calculated using the following equation (Odum et al., 1996):
where αi is the mass-based stoichiometric coefficient for the reaction producing the semi-volatile product i, Kom,i is the gas-particle partitioning equilibrium constant, and Mo is the total aerosol mass concentration.
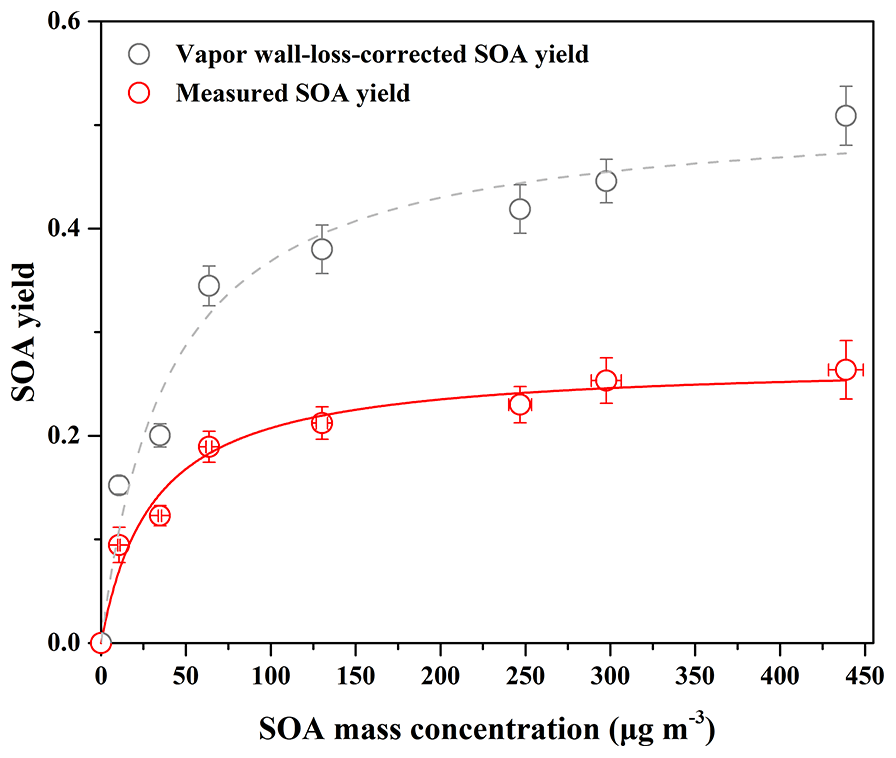
Figure 1SOA yield as a function of SOA mass concentration (Mo) for guaiacol photooxidation in the presence of NOx at different guaiacol concentrations. The lines were fit to the experimental data using a one-product model. Values of α and Kom,i used to generate the solid line were (0.27±0.01) and (0.033±0.008), and their values for the dashed line were (0.52±0.03) and (0.025±0.006), respectively.
The yield curve for guaiacol photooxidation is shown in Fig. 1, obtained by plotting the SOA yield data in Table 1 according to Eq. (1). The yield data were accurately reproduced by a one-product model (R2=0.97), while two or more products used in the model did not significantly improve the fitting quality. The obtained values of αi and Kom,i for one-product model were (0.27±0.01) and (0.033±0.008) m3 µg−1, respectively. In previous studies, the one-product model was widely applied to describe SOA yields from the oxidation of aromatic compounds including methoxyphenols (Coeur-Tourneur et al., 2010b; Lauraguais et al., 2012, 2014b). In this work, this simulation suggests that the products in SOA have similar values of αi and Kom,i, i.e., the obtained αi and Kom,i are the average values. The plot shown in Fig. S2 is the relationship between Mo versus Δ[guaiacol], of which slope (0.28) is slightly higher than αi value (0.27). This suggests that the formed low-volatile products almost completely partitioned into the particle-phase according to the theoretical partition model (Lauraguais et al., 2012, 2014b).
In the previous studies, the significant SOA formation from the OH-initiated reaction of guaiacol has been reported (Lauraguais et al., 2014b; Yee et al., 2013). In this work, SOA yields for guaiacol photooxidation range from (9.46±1.71) % to (26.37±2.83) %, shown in Table 1. According to the ratios of (0.61–0.93), the determined SOA yields were underestimated by a factor of ∼2, suggesting that vapor wall loss in the chamber could significantly affect SOA formation. The similar results were reported previously by Zhang et al. (2014), who observed that SOA yields for toluene photooxidation were substantially underestimated by factors of as much as 4, caused by vapor wall loss. As shown in Fig. 1, the vapor wall-loss-corrected SOA yields were in the range of (15.24±0.85) % to (50.89±2.87) %, and could also be reproduced by a one-product model (R2=0.96). This range overlaps SOA yields of 0.6 %–87 % for guaiacol oxidation under high NOx condition (∼10 ppmv NO), reported by Lauraguais et al. (2014b), using CH3ONO as the OH source. Under low NOx conditions (<5 ppbv NO), SOA yields for guaiacol oxidation were in the range of 44 %–50 %, reported by Yee et al. (2013) using H2O2 as the OH source and (NH4)2SO4 as seed particles; they also indicated that high NOx concentration (>200 ppbv NO) played an opposite role in SOA formation. Overall, the vapor wall-loss-corrected SOA yields in this work are well in agreement with those reported previously (Lauraguais et al., 2014b; Yee et al., 2013), but the determined SOA yields are much lower. Therefore, the effect of vapor wall loss on SOA formation should be seriously taken into account.
In addition, the average N∕C ratio of SOA for guaiacol photooxidation in the presence of NOx is 0.037, calculated according to the element analysis by the HR-ToF-AMS. This indicates that NOx participates in SOA formation and growth. This phenomenon is well supported by the previous studies, which have reported that the nitro-substituted products are the main products of the OH-initiated reaction of guaiacol in the presence of NOx (Ahmad et al., 2017; Lauraguais et al., 2014b). The relative low volatility of these products could reasonably contribute to SOA formation (Duporté et al., 2016; Liu et al., 2016a). The average ratio of SOA from guaiacol photooxidation is 4.08, which is higher than that (2.06–2.54) for ammonium nitrate, determined by the HR-ToF-AMS in this work. The possible explanation might be that nitro-organics and organonitrates both exist in SOA (Farmer et al., 2010; Sato et al., 2010). The relative abundance of organic N-containing compounds could be estimated from the average N∕C ratio. Assuming that the oxidation products in the SOA retain seven carbon atoms, the yield of organic N-containing compounds is 25.9 %, which is the upper limit due to the possible C–C bond scission during photooxidation process.
3.2 Effect of SO2 on SOA formation
3.2.1 SOA yields
In China, atmospheric SO2 concentration is always in the range of several to dozens of parts per billion by volume, while in the severely polluted atmosphere it could be up to close 200 ppbv (Han et al., 2015; Li et al., 2017). In addition, a recent field measurement study has reported that the decrease in biogenic SOA mass concentration in the atmosphere has a positive correlation with SO2 emission controls (Marais et al., 2017). Therefore, the effect of SO2 at atmospheric levels on SOA formation from guaiacol photooxidation under atmospheric NOx conditions was investigated. The experimental conditions and results are shown in Table 2. The formation of SOA, sulfate, and nitrate as a function of SO2 concentration for guaiacol photooxidation is shown in Fig. S3, and the time-series variations in the concentrations of sulfate and nitrate are shown in Fig. S4. The decays of guaiacol, NOx, and SO2 are shown in Figs. S5a, S6a, and S7, respectively, which have similar changing trends for different experiments. As illustrated in Fig. 2, the induction period became shorter with the increase in SO2 concentration. The similar results caused by SO2 have also been reported previously (Chu et al., 2016; Liu et al., 2016b). Meanwhile, Mo for the experiment without SO2 (Exp. 1 in Table 2) increased from (63.62±1.71) to (71.88±1.43) and (78.59±2.06) µg m−3, enhanced by 12.98 % and 23.53 %, respectively, when SO2 concentration raised from 0 to 33 and 56 ppbv. The corresponding SOA yields were (21.60±1.27) % and (23.42±1.80) %, respectively. The similar results were reported by previous studies (Kleindienst et al., 2006; Lin et al., 2013; Liu et al., 2016b), which observed the significant enhancement of SOA yields for VOC oxidation and the photochemical aging of gasoline vehicle exhaust in the presence of SO2.
Table 2Experimental conditions and results for guaiacol photooxidation in the presence of seed particles and SO2.
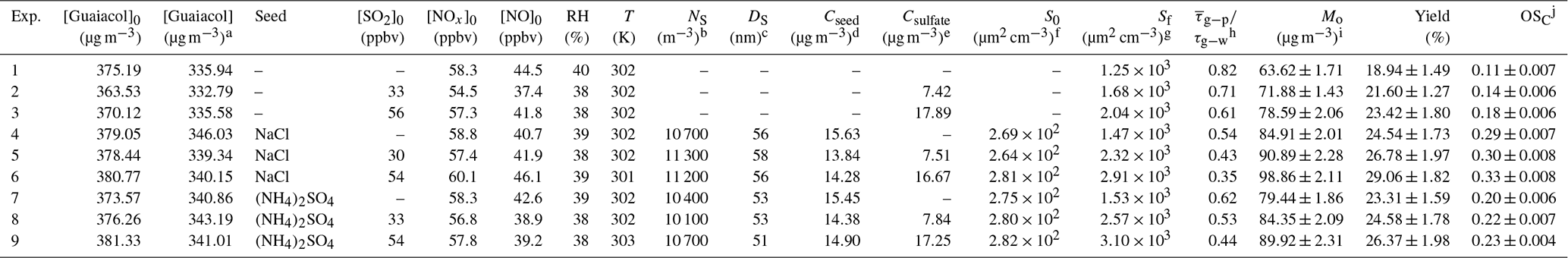
a The consumed guaiacol concentration at the end of each experiment.
b NS is the initial seed number.
c DS is the average
diameter of seed particles.
d Cseed is the initial concentration of seed.
e Csulfate is the sulfate concentration formed by SO2
oxidation.
f The initial surface area of seed particles.
g The final surface
area of aerosol particles (seed + organic aerosol), measured by the
SMPS.
h The ratio of the average gas-particle partitioning timescale
( over the course of the experiment to the
vapor
wall deposition timescale (τg−w).
i Mo is the
mass concentration of SOA.
j OSC is the average oxidation state of
carbon of SOA.
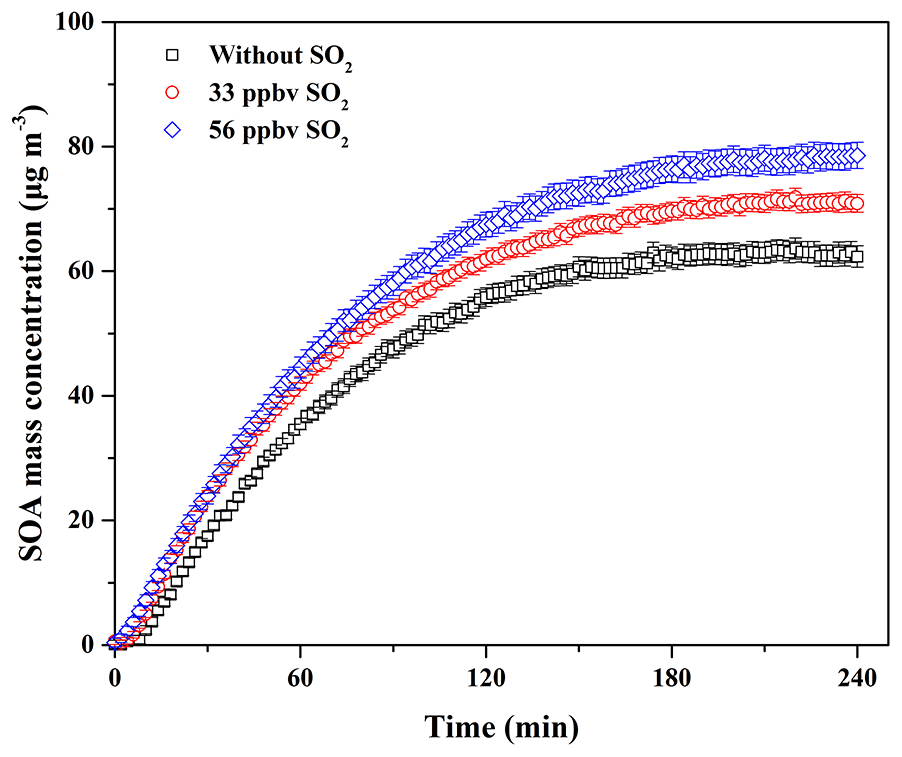
Figure 2Time-dependent growth curves of SOA mass concentration for guaiacol photooxidation at different SO2 levels (Exps. 1–3 in Table 2).
As shown in Fig. 3, the ratio decreased from 0.82 to 0.71 and 0.61 when the SO2 concentration increased from 0 to 33 and 56 ppbv. It suggests that the sulfate formed via SO2 oxidation could serve as seed particles (Jaoui et al., 2008) and increase the surface areas of particles (Xu et al., 2016). These roles are favorable to partitioning more SOA-forming vapors into the particle phase (Zhang et al., 2014), consequently enhancing SOA yields. At the same time, as shown in Fig. S4 and Table 2, the sulfate concentration increased significantly from 7.42 to 17.89 µg m−3 when SO2 concentration increased from 33 to 56 ppbv. Nevertheless, the particle peak attributed to sulfate formed via SO2 oxidation was not observed by the SMPS during the experimental process due to the quick particle growth in the presence of organic vapors. In this work, it is difficult to completely remove traces of NH3 from zero air; thus, the formed sulfate should be the mixture of H2SO4 and (NH4)2SO4. The time-series changes in the concentration of ammonium salt at different SO2 concentrations are shown in Fig. S8. Its concentration increased obviously with increasing SO2 concentration, suggesting that more (NH4)2SO4 was produced. Similar results have also been reported recently by Chu et al. (2016). In addition, the surface area concentration of aerosol particles at the end time was calculated. As shown in Table 2, the final surface area of aerosol particles formed via guaiacol photooxidation increased from 1.25×103 to 1.68×103 and 2.04×103 µm2 cm−3 when SO2 concentration increased from 0 to 33 and 56 ppbv. The increased surface area could be in favor of outcompeting the wall loss for low-volatility vapors produced from guaiacol photooxidation; i.e., more low-volatility vapors would be diverted from wall loss to the particles, consequently increasing SOA yields (Kroll et al., 2007). This is well supported by the decrease in the ratio with increasing SO2 concentration, shown in Fig. 3.
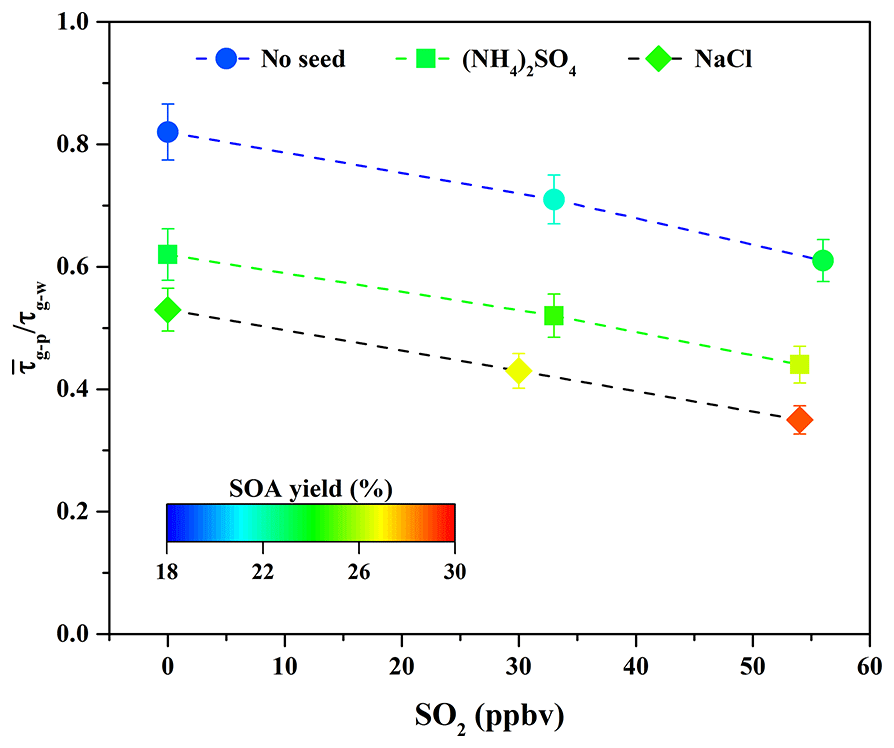
Figure 3Variations in the ratio in the presence of various seed particles as a function of SO2 concentration.
The time-series changes in the mass concentrations of NO+ and are shown in Fig. S9a. The mass concentration of NO+ increased more quickly than that of and had a positive correlation with SO2 concentration. But compared to the experiment without SO2, the presence of SO2 had little impact on and N∕C ratios obtained at the end time, shown in Figs. S9b and S10b, respectively. These ratios indicated that organic N-containing compounds were also produced in this system (Farmer et al., 2010; Sato et al., 2010).
3.2.2 Oxidation state of SOA
The average carbon oxidation state () of OA is widely used to represent the oxidation degree of atmospheric OA, because it takes into account the saturation level of carbon atoms in the OA (Kroll et al., 2011). As shown in Table 2, increasing SO2 concentration (0–56 ppbv, Exps. 1–3) leads to the increase in OSC (0.11–0.18). The variations in H∕C, O∕C, and N∕C ratios as a function of irradiation time are shown in Fig. S10. In order to further identify the effect of SO2 on the chemical properties of SOA, positive matrix factorization (PMF) analysis for all AMS data obtained at different SO2 concentrations over the course of the experiments was carried out. Two factors were obtained from the PMF analysis, and their mass spectra are shown in Fig. 4. The organic mass fraction of (), named f44, was 0.122 for Factor 2, which is higher than that (0.094) for Factor 1. Therefore, Factor 2 was tentatively assigned to the more-oxidized SOA, while Factor 1 was the less-oxidized SOA (Ulbrich et al., 2009). During the photooxidation process, these two factors had different variations as a function of irradiation time. As shown in Fig. S11, Factor 1 increased along with the reaction and then decreased, while Factor 2 had an increasing trend. Compared to Exps. 1 and 2 in Table 2, the higher fraction of Factor 2 mass obtained at 56 ppbv SO2 (Exp. 3 in Table 2) suggests that the formed SOA mainly consists of more-oxidized products with relatively low volatility. This is well supported by the time-series variations in the fraction of organic ion groups (CH+, CHO+, and – containing more than one oxygen atom) (Fig. S12a), which shows the higher fraction of and lower fraction of CH+ obtained at higher SO2 concentration, consequently resulting in a higher OSC of SOA.
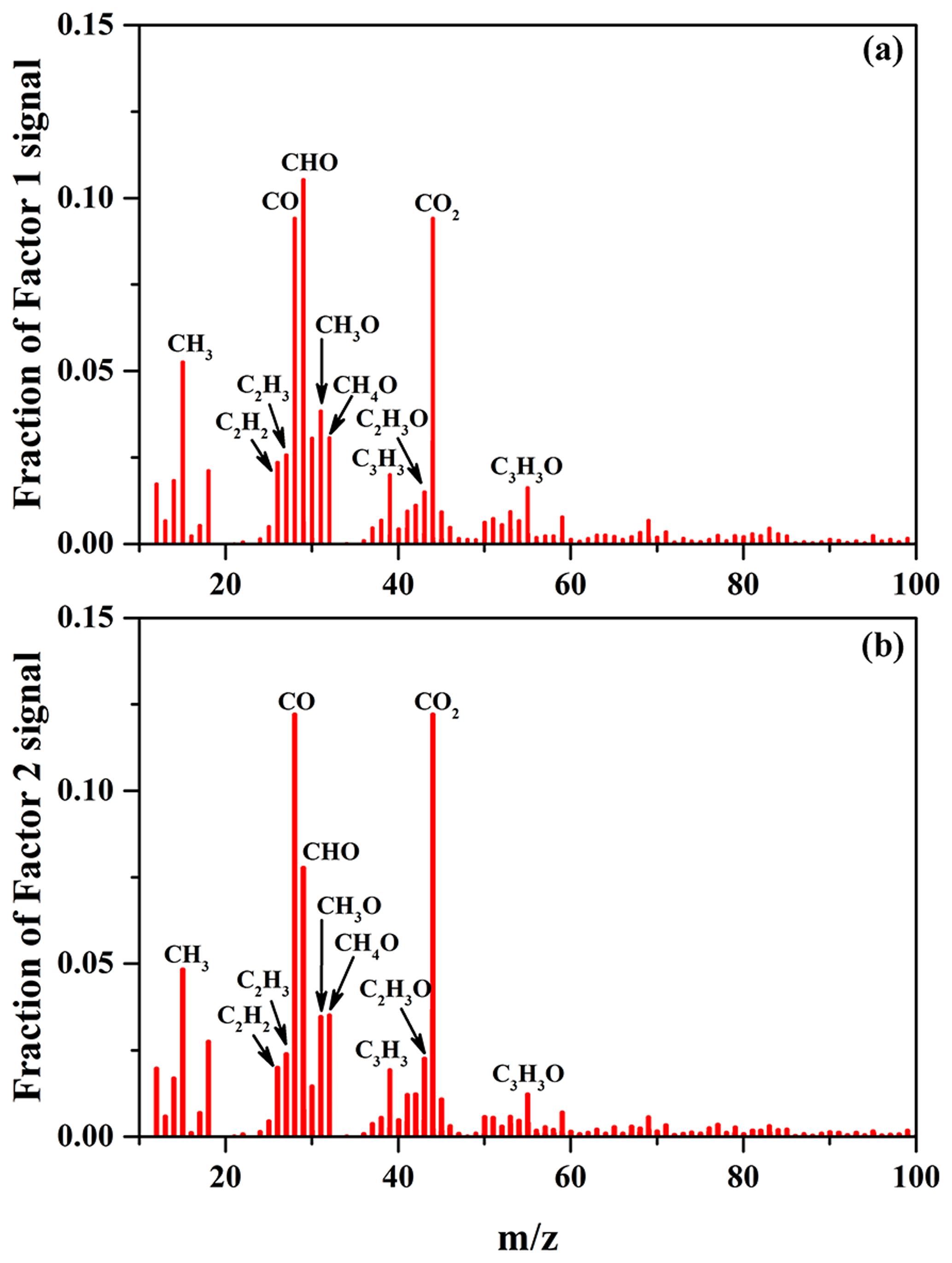
Figure 4Mass spectra of Factor 1 (a) and Factor 2 (b) for the formed SOA identified by applying PMF analysis to the AMS data, obtained at different SO2 concentrations over the course of the experiments.
Previous studies mostly reported that the enhancement of SOA yield in the presence of SO2 was ascribed to the functionalization and oligomerization reactions (Cao and Jang, 2007; Jaoui et al., 2008; Liu et al., 2016b; Xu et al., 2016). If the oligomerization reaction plays a predominant role in the presence of SO2 which will lead to particle-phase H2SO4, the carbon number of oligomers will increase but their net O∕C or H∕C values will show little change, consequently resulting in little change in the oxidation state of SOA (Chen et al., 2011). Nevertheless, we observed that SO2 not only enhanced SOA yields, but also resulted in higher OSC (Table 2 and Fig. 5). This suggests that the functionalization reaction might be predominant with SO2, which leads to higher OSC of products with low molecular weight (MW) (Ye et al., 2018), consequently resulting in an overall increase in OSC and SOA yields. More recently, Ye et al. (2018) also found the similar results in the ozonolysis of limonene. Figure S13 shows the differences among the normalized mass spectra of SOA formed at different SO2 concentrations. As shown in Fig. S13a, the signal fractions from the low-MW species were enhanced significantly in the presence of SO2 and were much higher than those from the high-MW species (m∕z>300). The similar results were also observed in Fig. S13b when increasing SO2 concentration. In other words, SO2 played a more important role in the formation of organic S-containing compounds and the formation or uptake of low-MW species, compared to the formation of high-MW species (i.e., oligomers) that should be reasonably produced via the acid-catalyzed heterogeneous reactions (Cao and Jang, 2007; Jaoui et al., 2008; Liu et al., 2016b; Xu et al., 2016).
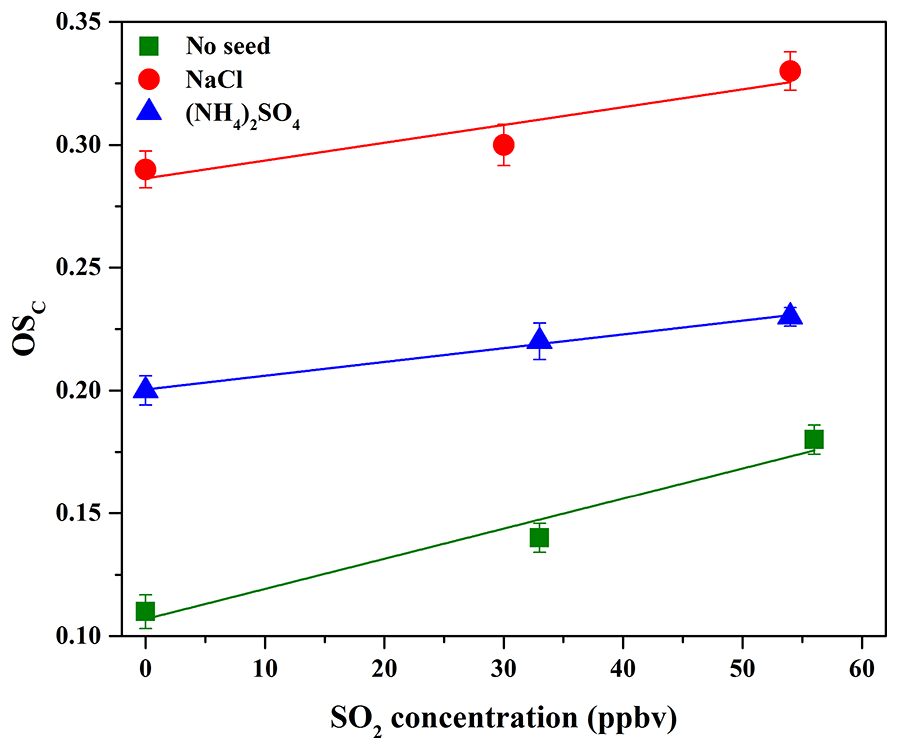
Figure 5OSC of SOA formed in the presence of various seed particles as a function of SO2 concentration.
In this work, assuming that all organic S-containing compounds are organosulfates and have the same response factor and fragmentation as methyl sulfate, the conservative lower-bound concentration of organosulfates was calculated to be in the range of (2.1±0.8) to (4.3±1.7) ng m−3 using the method described by Huang et al. (2015) shown in the Supplement and increased with the increase in SO2 concentration. This concentration range is close to those derived from the atmospheric oxidation of polycyclic aromatic hydrocarbons and alkane (Meade et al., 2016; Riva et al., 2015). Figure S14 is the examples of the ions (i.e., CSO+, , and ) of methyl sulfate obtained at 56 ppbv SO2 (Exp. 3 in Table 2). On the other hand, sulfuric acid formed from SO2 may be favorable for the uptake of water-soluble low-MW species (e.g., small carboxylic acids and aldehydes) and also be helpful for retaining them in the aerosol phase, which would result in the increase in OSC. This is well supported by the time-series variations in the concentrations of acetic acid at different SO2 concentrations measured by the HR-ToF-PTRMS (Fig. S15a), which shows that acetic acid concentration decreased with the increase in SO2 concentration (0–56 ppbv). These results were in good agreement with those reported by Liggio et al. (2005) and Liu et al. (2010), who observed that the uptake of organic compounds under acidic conditions would be enhanced significantly. Recently, Huang et al. (2016) have also reported that acetic acid is present in SOA formed via α-pinene ozonolysis and its uptake would increase in the presence of seed particles. In addition, Krapf et al. (2016) have indicated that peroxides in SOA are unstable and liable to decompose into volatile compounds, consequently leading to decreases in SOA yield and OSC. But Ye et al. (2018) found that the reactions of SO2 with organic peroxides were the dominant sink of SO2, initiated by the heterogeneous uptake of SO2 under humidity conditions. These reactions would result in the formation of organic S-containing compounds, consequently increasing SOA yields and OSC.
3.3 Effect of inorganic seed particles on SOA formation
Seed particles are one of the critical factors influencing SOA formation (Ge et al., 2017a); thus, the effects of inorganic seeds (i.e., NaCl and (NH4)2SO4) on SOA formation from guaiacol photooxidation were investigated. As shown in Fig. 6, the presence of inorganic seed particles could accelerate the SOA growth rate at the initial stage of photooxidation (i.e., it would shorten the induction period), followed by the decrease in the growth rate along with the reaction, because the presence of inorganic seeds could promote the condensation of SOA-forming organic products and consequently increase SOA formation (Yee et al., 2013). The results showed that Mo for the experiment without seed particles (Exp. 1 in Table 2) increased from (63.62±1.71) to (79.44±1.86) and (84.91±2.01) µg m−3 (Table 2), enhanced by 24.87 % and 33.46 %, respectively, with (NH4)2SO4 and NaCl seed particles. The corresponding SOA yields were (23.31±1.59) % and (24.54±1.73) %, respectively. In previous work, the similar results on the enhancements of SOA formation by NaCl and (NH4)2SO4 seed particles were reported in the oxidation of VOCs (Ge et al., 2017a, b; Huang et al., 2013, 2017). As shown in Fig. 3, ratios with (NH4)2SO4 and NaCl seed particles were 0.62 and 0.54, respectively, which suggested that more SOA-forming vapors partitioned into the particle phase in the presence of NaCl seed particles (Zhang et al., 2014), consequently resulting in a relatively higher SOA yield.

Figure 6Time-dependent growth curves of SOA mass concentration for guaiacol photooxidation in the presence of inorganic seed particles (Exps. 1, 4, and 7 in Table 2).
As shown in Table 2 and Fig. 6, the SOA mass concentration in the presence of NaCl seed particles was higher than that in the presence of (NH4)2SO4 seed particles. In addition, the OSC of SOA in the presence of NaCl seed particles is 0.29, slightly higher than that (0.20) in the presence of (NH4)2SO4 seed particles. Recently, it has been also reported that the presence of (NH4)2SO4 and NaNO3 seed particles could significantly enhance the oxidation state of SOA, compared to without seed particles (Huang et al., 2016). In this work, the experimental conditions for seed experiments are almost the same (Table 2), including reactant concentration, temperature, RH, and the number and diameter of seed particles. Therefore, the differences in the yield and oxidation state of SOA resulted from the different chemical compositions of SOA in the presence of different inorganic seeds. As shown in Fig. S12b and c, compared to (NH4)2SO4 seed particles, the higher fraction of and lower fraction of CH+ were obtained with NaCl seed particles, consequently resulting in higher OSC of SOA. The time-series evolution of O∕C, H∕C, and N∕C ratios is shown in Figs. S16 and S17, which indicate that O∕C ratios (0.94–0.99) with NaCl seed particles at the end of experiments are higher than those (0.90–0.93) with (NH4)2SO4 seed particles. Figure 7 shows the mass spectra of SOA in the presence of NaCl and (NH4)2SO4 seed particles obtained by the HR-ToF-AMS, as well as their difference mass spectrum. As shown in Fig. 7, f44 for SOA in the presence of NaCl seed particles was higher than that obtained in the presence of (NH4)2SO4 seed particles, while the mass fractions of (CH3) and 29 (CHO) fragments were both lower. The ion () is mainly contributed from acids or acid-derived species, such as esters (Ng et al., 2011). The higher f44 of SOA with NaCl than (NH4)2SO4 seed particles suggests that the distribution of highly oxidized small carboxylic acids onto seed particles plays an important role in SOA formation, consequently resulting in a higher oxidation state of SOA (Huang et al., 2016; Ng et al., 2011). Compared to (NH4)2SO4, the hygroscopicity of NaCl is stronger (Ge et al., 2017a; Gysel et al., 2002). The molar ratio of H2O to NaCl is about 0.1 at 40 % RH, and water is mainly adsorbed on NaCl particles (Weis and Ewing, 1999). Thus, the greater water content on the particle surface could facilitate the uptake of highly oxidized small carboxylic acids onto NaCl particles, which might explain the higher SOA oxidation state observed in the presence of NaCl seed particles (Huang et al., 2016). As shown in Fig. S15, the concentration of acetic acid in the gas phase with NaCl seed particles was lower than that with (NH4)2SO4 seed particles. It suggests that the uptake of acetic acid on NaCl seed particles might be higher than that on (NH4)2SO4 seed particles under similar experimental conditions (i.e., NOx and guaiacol concentrations, temperature, and RH). Moreover, the adsorbed acid products would also generate H+ ions, which could catalyze heterogeneous reactions to produce more-oxidized products or oligomers with relatively low volatility (Fig. S18), consequently resulting in the enhancement of SOA formation (Huang et al., 2013, 2017; Cao and Jang, 2007; Jaoui et al., 2008; Liu et al., 2016b; Xu et al., 2016).
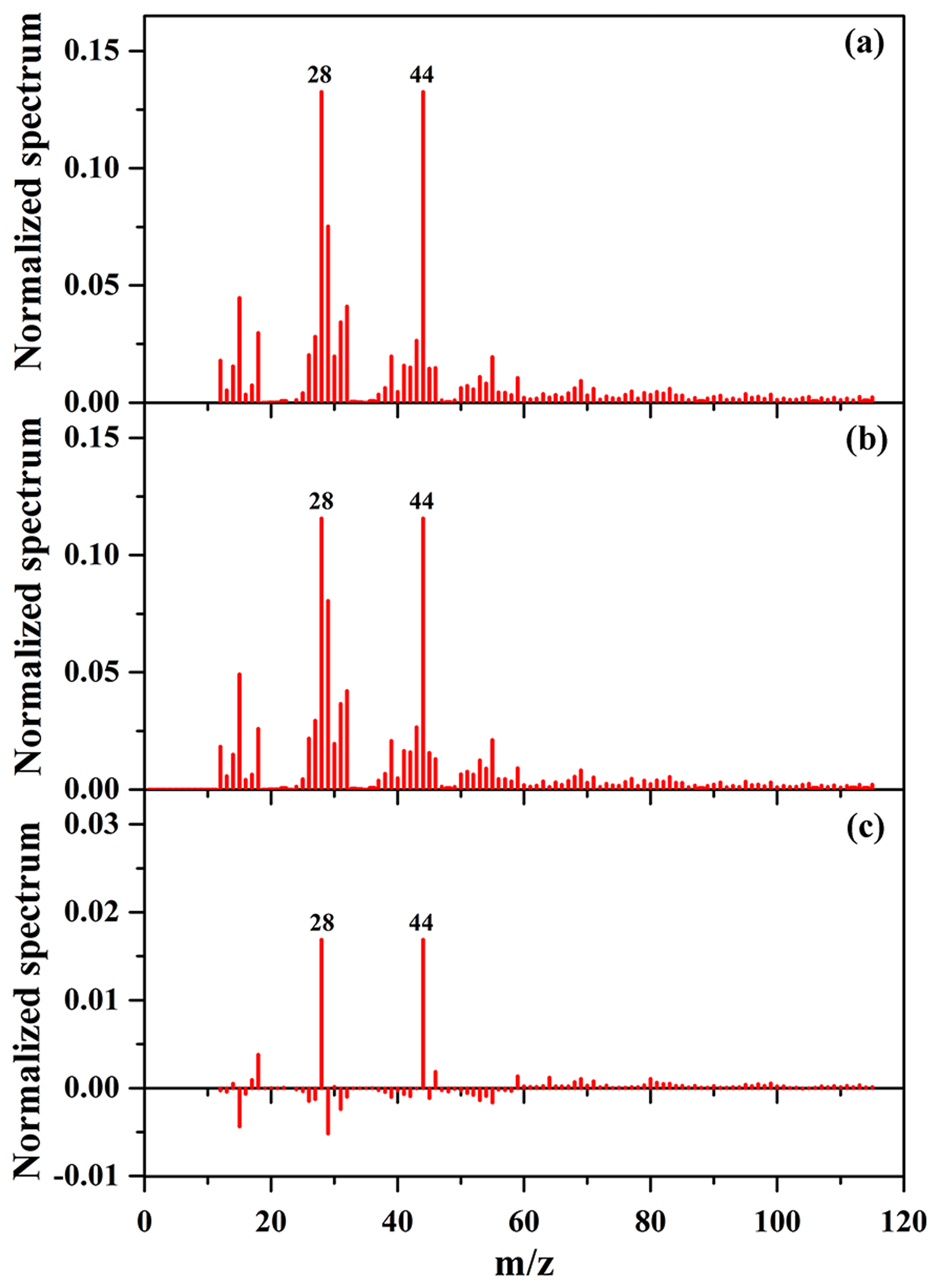
Figure 7Mass spectra of SOA with NaCl (a) and (NH4)2SO4 (b) as seed particles obtained by the HR-ToF-AMS, as well as their difference mass spectrum (c ).
In addition, the possible formation of Cl atoms from the photolysis of nitryl chloride (, s−1) (Mielke et al., 2011) and the reaction of OH radicals with Cl− (, M−1 s−1) (Fang et al., 2014) would also initiate a series of reactions to oxidize SOA composition, which might be another reason for higher OSC observed with NaCl seed particles. According to the rate constant (109 M−1 s−1) (Fang et al., 2014), the uptake coefficient () of OH radicals on NaCl particles (Park et al., 2008), and the concentrations of OH radicals and Cl−, the concentration of Cl atoms produced from the reaction of OH radical with Cl− was estimated to be less than 38 molecules cm−3, which was much higher than that from the photolysis of ClNO2 due to the slow photolysis rate constant of s−1 (Mielke et al., 2011). Compared to OH concentration in the chamber, the oxidation of SOA composition by Cl atoms should be insignificant.
3.4 Synergetic effect of SO2 and inorganic seed particles on SOA formation
According to the former results obtained in this work, it is known that SO2 and inorganic seed particles both have a positive role in enhancing SOA formation. Therefore, their possible synergetic effects on SOA formation were investigated. Considering the experiments performed under the comparable conditions (Table 2), the results should be reasonably reliable. The decays of guaiacol, NOx, and SO2 are shown in Figs. S5, S6, and S7, respectively, which have the similar changing trends for different experiments. Figure S19 shows the time-series evolution in the sulfate concentration in the presence of different SO2 concentrations and seed particles, which indicates that sulfate concentration is dependent on SO2 concentration. As shown in Fig. 8, the addition of SO2 into the chamber in the presence of inorganic seed particles significantly promoted SOA formation from guaiacol photooxidation but had an ignorable impact on the induction period. When SO2 concentration raised from 0 to 30 and 54 ppbv in the presence of NaCl seed particles, Mo increased from (84.91±2.01) to (90.89±2.28) and (98.86±2.11) µg m−3, enhanced by 7.04 % and 16.43 %, respectively, and the corresponding SOA yields were (26.78±1.97) % and (29.06±1.82) %. For (NH4)2SO4 seed particles, Mo increased from (79.44±1.86) to (84.35±2.09) for 33 ppbv SO2 and (89.92±2.31) µg m−3 for 54 ppbv SO2, enhanced by 6.18 % and 13.19 %, respectively, and the corresponding SOA yields were (24.58±1.78) % and (26.37±1.98) %. As shown in Fig. 3, the ratio had a decreasing trend when increasing SO2 concentration in the presence of seed particles, suggesting that the underestimation of SOA yields caused by vapor wall loss was weakened significantly because of the additional sulfate formed from SO2 oxidation. Thus, inorganic seed particles and SO2 showed a synergetic effect on SOA formation.
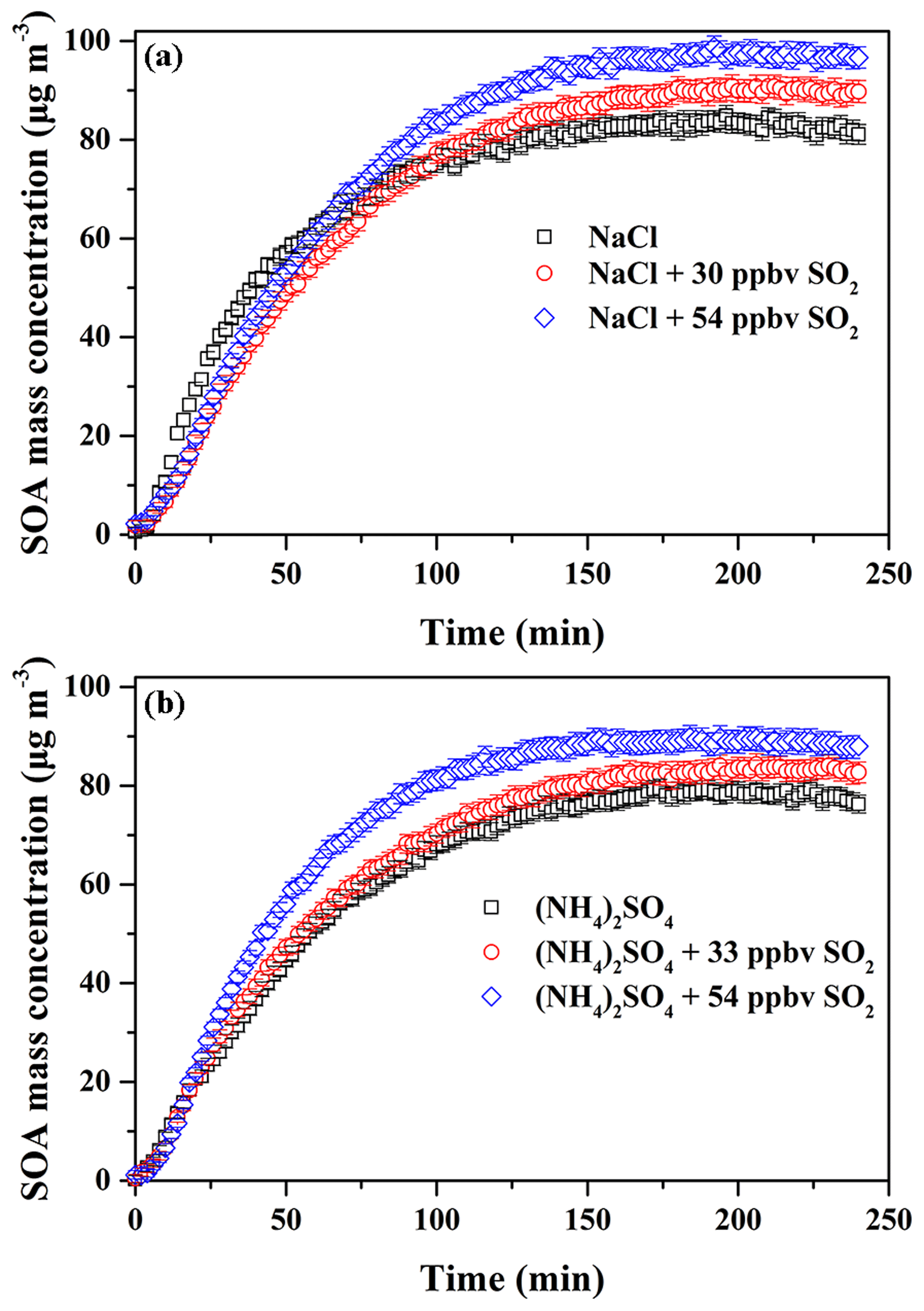
Figure 8Time-dependent growth curves of SOA mass concentration for guaiacol photooxidation in the presence of SO2 and inorganic seed particles (a NaCl; b (NH4)2SO4) (Exps. 4–9 in Table 2).
As shown in Table 2 and Fig. 5, it should be noted that OSC of SOA increased in the presence of SO2, which was well supported by the time-series variations in H∕C, O∕C, and N∕C ratios at different SO2 concentrations with NaCl and (NH4)2SO4 as seed particles, shown in Figs. S16 and S17. In addition, as shown in Fig. S12b and c, the higher fraction of and lower fraction of CH+ were obtained at higher SO2 concentration, consequently resulting in higher OSC of SOA. Figure S20 shows the mass spectra of SOA with NaCl and (NH4)2SO4 as seed particles at different SO2 concentrations obtained by the HR-ToF-AMS. As illustrated in Fig. S20, SO2 addition was in favor of increasing the value of f44, suggesting that more products with higher OSC are produced by the functionalization reaction (Ye et al., 2018). Meanwhile, Table 2 shows that the final surface area of aerosol particles increased in the presence of SO2, which played a positive role in diverting more low-volatility vapors from wall loss to the particles, consequently enhancing SOA yields (Kroll et al., 2007). In addition, the presence of inorganic seeds could promote the condensation of SOA-forming organic products and the heterogeneous uptake of SO2 (Yee et al., 2013), providing favorable conditions for the following reactions. Meanwhile, the higher hygroscopicity of NaCl than (NH4)2SO4 might be helpful to dissolve more acid substances on NaCl particle surface (e.g., H2SO4 and organic acid), especially in the presence of SO2. This hypothesis could be supported by the variations in acetic acid concentration in the presence of different seed particles and SO2 concentrations (Fig. S15), which shows that acetic acid concentration decreased with the increase in SO2 concentration (0–54 ppbv). The dissolved acid compounds might be helpful to catalyze heterogeneous reactions (Cao and Jang, 2007; Huang et al., 2013, 2017; Jaoui et al., 2008; Liu et al., 2016b; Xu et al., 2016). Figures S21 and S22 show the differences among the normalized mass spectra of SOA formed at different SO2 concentrations with various seed particles. The results indicated that the signal fractions from the low-MW species increased significantly in the presence of SO2 and were much higher than those from the high-MW species (m∕z>300). Compared to Exps. 2 and 3 in Table 2 with no seed particles, the conservative lower-bound concentrations of organosulfates formed with seed particles were similar and in the range of (2.2±0.7) to (4.6±1.8) ng m−3, which might be caused by the similar SO2 concentrations applied for experiments. With NaCl and (NH4)2SO4 as seed particles, SOA yields and OSC both increased with the increase in SO2, suggesting that the functionalization reaction should be more dominant than the oligomerization reaction during photooxidation process.
In this work, SOA formation from guaiacol photooxidation in the presence of NOx was investigated in a 30 m3 smog chamber. SOA yields for guaiacol photooxidation were in the range of (9.46±1.71) % to (26.37±2.83) % and could be expressed well by a one-product model. These yields were underestimated by a factor of ∼2 according to ratios. The presence of SO2 could increase SOA yield and OSC, indicating that the functionalization reaction should be more dominant than the oligomerization reaction. Meanwhile, the similar effect of SO2 was also observed with NaCl and (NH4)2SO4 seed particles. But SOA yield and OSC in the presence of NaCl seed particles were both slightly higher than those in the presence of (NH4)2SO4 seed particles. In addition, the results indicated the synergetic contribution of SO2 and inorganic seed particles to SOA formation. The decreasing trend of ratio in the presence of seed particles and SO2 suggested that more SOA-forming vapors partitioned into the particle phase, consequently increasing SOA yields. The average N∕C ratio (0.037) of SOA suggested that NOx participated in the process of guaiacol photooxidation, resulting in the formation of organic N-containing compounds.
The significant SOA formation from guaiacol photooxidation at the atmospheric levels of SO2 and NOx in this work suggests that more attention needs to be given to the SOA formation from biomass burning and its subsequent effects on haze evolution, especially in China with nationwide biomass burning, because recent studies have indicated that SOA formed from biomass burning plays an important role in haze pollution in China (Ding et al., 2017; Li et al., 2017). In addition, the results imply that the oxidation of SO2 and VOCs are tightly coupled, and SO2 has a direct impact on the physics and chemistry of SOA formation. Although guaiacol concentrations in the chamber study are higher than those in the ambient atmosphere, the results obtained in this work could provide new information for SOA formation from the photooxidation of methoxyphenols and might be useful for SOA modeling, especially for air quality simulation modeling of the specific regions experiencing serious pollution caused by fine particulate matter. In addition, the results would help to further understand the photochemical aging process of smoke plumes from biomass burning in the atmosphere.
The experimental data are available upon request to the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-2687-2019-supplement.
CL and TC contributed equally to this work and should be considered as co-first authors. CL, TC, YL, and HH designed the research and wrote the paper. CL, TC, and JL performed the experiments. CL, TC, YL, JL, HH, and PZ carried out the data analysis. All authors contributed to the final paper.
The authors declare that they have no conflict of interest.
This work was financially supported by the National Key R&D Program of
China (2016YFC0202700), National Natural Science Foundation of China
(21607088 and 41877306), China Postdoctoral Science Foundation funded
project (2017M620071), Applied Basic Research Project of Science and
Technology Department of Sichuan Province (2018JY0303), and Key Research
Program of Frontier Sciences, CAS (QYZDB-SSW-DQC018). Yongchun Liu would like to
thank Beijing University of Chemical Technology for financial support.
The authors would also like to acknowledge the experimental help provided by Xiaolei Bao from Hebei Provincial Academy of Environmental Sciences, Shijiazhuang,
China.
Edited by: Jason Surratt
Reviewed by: Sophie Tomaz and two anonymous referees
Ahmad, W., Coeur, C., Tomas, A., Fagniez, T., Brubach, J.-B., and Cuisset, A.: Infrared spectroscopy of secondary organic aerosol precursors and investigation of the hygroscopicity of SOA formed from the OH reaction with guaiacol and syringol, Appl. Opt., 56, E116–E122, https://doi.org/10.1364/ao.56.00e116, 2017.
Bari, M. A. and Kindzierski, W. B.: Fine particulate matter (PM2.5) in Edmonton, Canada: Source apportionment and potential risk for human health, Environ. Pollut., 218, 219–229, https://doi.org/10.1016/j.envpol.2016.06.014, 2016.
Cao, G. and Jang, M.: Effects of particle acidity and UV light on secondary organic aerosol formation from oxidation of aromatics in the absence of NOx, Atmos. Environ., 41, 7603–7613, https://doi.org/10.1016/j.atmosenv.2007.05.034, 2007.
Chen, Q., Liu, Y., Donahue, N. M., Shilling, J. E., and Martin S. T.: Particle-phase chemistry of secondary organic material: Modeled compared to measured O:C and H:C elemental ratios provide constraints, Environ. Sci. Technol., 45, 4763–4770, https://doi.org/10.1021/es104398s, 2011.
Chen, T., Liu, Y., Chu, B., Liu, C., Liu, J., Ge, Y., Ma, Q., Ma, J., and He, H.: Differences of the oxidation process and secondary organic aerosol formation at low and high precursor concentrations, J. Environ. Sci., 79, 256–263, https://doi.org/10.1016/j.jes.2018.11.011, 2019.
Chen, Y. and Bond, T. C.: Light absorption by organic carbon from wood combustion, Atmos. Chem. Phys., 10, 1773–1787, https://doi.org/10.5194/acp-10-1773-2010, 2010.
Chou, C. C.-K., Tsai, C.-Y., Chang, C.-C., Lin, P.-H., Liu, S. C., and Zhu, T.: Photochemical production of ozone in Beijing during the 2008 Olympic Games, Atmos. Chem. Phys., 11, 9825–9837, https://doi.org/10.5194/acp-11-9825-2011, 2011.
Chu, B., Zhang, X., Liu, Y., He, H., Sun, Y., Jiang, J., Li, J., and Hao, J.: Synergetic formation of secondary inorganic and organic aerosol: effect of SO2 and NH3 on particle formation and growth, Atmos. Chem. Phys., 16, 14219–14230, https://doi.org/10.5194/acp-16-14219-2016, 2016.
Chu, B., Liggio, J., Liu, Y., He, H., Takekawa, H., Li, S.-M., and Hao, J.: Influence of metal-mediated aerosol-phase oxidation on secondary organic aerosol formation from the ozonolysis and OH-oxidation of α-pinene, Sci. Rep., 7, 40311, https://doi.org/10.1038/srep40311, 2017.
Coeur-Tourneur, C., Cassez, A., and Wenger, J. C.: Rate coefficients for the gas-phase reaction of hydroxyl radicals with 2-methoxyphenol (guaiacol) and related compounds, J. Phys. Chem. A, 114, 11645–11650, https://doi.org/10.1021/jp1071023, 2010a.
Coeur-Tourneur, C., Foulon, V., and Lareal, M.: Determination of aerosol yields from 3-methylcatechol and 4-methylcatechol ozonolysis in a simulation chamber, Atmos. Environ., 44, 852–857, https://doi.org/10.1016/j.atmosenv.2009.11.027, 2010b.
DeCarlo, P. F., Slowik, J. G., Worsnop, D. R., Davidovits, P., and Jimenez, J. L.: Particle morphology and density characterization by combined mobility and aerodynamic diameter measurements. Part 1: Theory, Aerosol Sci. Technol., 38, 1185–1205, https://doi.org/10.1080/027868290903907, 2004.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Ding, X., Zhang, Y.-Q., He, Q.-F., Yu, Q.-Q., Wang, J.-Q., Shen, R.-Q., Song, W., Wang, Y.-S., and Wang, X.-M.: Significant increase of aromatics-derived secondary organic aerosol during fall to winter in China, Environ. Sci. Technol., 51, 7432–7441, https://doi.org/10.1021/acs.est.6b06408, 2017.
Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R.: A new time-of-flight aerosol mass spectrometer (TOF-AMS)-instrument description and first field deployment, Aerosol Sci. Technol., 39, 637–658, https://doi.org/10.1080/02786820500182040, 2005.
Duporté, G., Parshintsev, J., Barreira, L. M. F., Hartonen, K., Kulmala, M., and Riekkola, M.-L.: Nitrogen-containing low volatile compounds from pinonaldehyde-dimethylamine reaction in the atmosphere: A laboratory and field study, Environ. Sci. Technol., 50, 4693–4700, https://doi.org/10.1021/acs.est.6b00270, 2016.
El Zein, A., Coeur, C., Obeid, E., Lauraguais, A., and Fagniez, T.: Reaction kinetics of catechol (1,2-benzenediol) and guaiacol (2-methoxyphenol) with ozone, J. Phys. Chem. A, 119, 6759–6765, https://doi.org/10.1021/acs.jpca.5b00174, 2015.
Fang, J., Fu, Y., and Shang, C.: The roles of reactive species in micropollutant degradation in the UV/free chlorine system, Environ. Sci. Technol., 48, 1859–1868, https://doi.org/10.1021/es4036094, 2014.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Ge, S., Xu, Y., and Jia, L.: Effects of inorganic seeds on secondary organic aerosol formation from photochemical oxidation of acetone in a chamber, Atmos. Environ., 170, 205–215, https://doi.org/10.1016/j.atmosenv.2017.09.036, 2017a.
Ge, S., Xu, Y., and Jia, L.: Secondary organic aerosol formation from propylene irradiations in a chamber study, Atmos. Environ., 157, 146–155, https://doi.org/10.1016/j.atmosenv.2017.03.019, 2017b.
Gordon, T. D., Presto, A. A., Nguyen, N. T., Robertson, W. H., Na, K., Sahay, K. N., Zhang, M., Maddox, C., Rieger, P., Chattopadhyay, S., Maldonado, H., Maricq, M. M., and Robinson, A. L.: Secondary organic aerosol production from diesel vehicle exhaust: impact of aftertreatment, fuel chemistry and driving cycle, Atmos. Chem. Phys., 14, 4643–4659, https://doi.org/10.5194/acp-14-4643-2014, 2014.
Gysel, M., Weingartner, E., and Baltensperger, U.: Hygroscopicity of aerosol particles at low temperatures. 2. Theoretical and experimental hygroscopic properties of laboratory generated aerosols, Environ. Sci. Technol., 36, 63–68, https://doi.org/10.1021/es010055g, 2002.
Han, T., Liu, X., Zhang, Y., Qu, Y., Zeng, L., Hu, M., and Zhu, T.: Role of secondary aerosols in haze formation in summer in the Megacity Beijing, J. Environ. Sci., 31, 51–60, https://doi.org/10.1016/j.jes.2014.08.026, 2015.
Hawthorne, S. B., Krieger, M. S., Miller, D. J., and Mathiason, M. B.: Collection and quantitation of methoxylated phenol tracers for atmospheric pollution from residential wood stoves, Environ. Sci. Technol., 23, 470–475, https://doi.org/10.1021/es00181a013, 1989.
Hawthorne, S. B., Miller, D. J., Langenfeld, J. J., and Krieger, M. S.: PM10 high-volume collection and quantitation of semivolatile and nonvolatile phenols, methoxylated phenols, alkanes, and polycyclic aromatic hydrocarbons from winter urban air and their relationship to wood smoke emissions, Environ. Sci. Technol., 26, 2251–2262, https://doi.org/10.1021/es00035a026, 1992.
Huang, D. D., Li, Y. J., Lee, B. P., and Chan, C. K.: Analysis of organic sulfur compounds in atmospheric aerosols at the HKUST supersite in Hong Kong using HR-ToF-AMS, Environ. Sci. Technol., 49, 3672–3679, https://doi.org/10.1021/es5056269, 2015.
Huang, D. D., Zhang, X., Dalleska, N. F., Lignell, H., Coggon, M. M., Chan, C.-M., Flagan, R. C., Seinfeld, J. H., and Chan, C. K.: A note on the effects of inorganic seed aerosol on the oxidation state of secondary organic aerosol-alpha-Pinene ozonolysis, J. Geophys. Res.-Atmos., 121, 12476–12483, https://doi.org/10.1002/2016jd025999, 2016.
Huang, M., Hao, L., Gu, X., Hu, C., Zhao, W., Wang, Z., Fang, L., and Zhang, W.: Effects of inorganic seed aerosols on the growth and chemical composition of secondary organic aerosol formed from OH-initiated oxidation of toluene, J. Atmos. Chem., 70, 151–164, https://doi.org/10.1007/s10874-013-9262-9, 2013.
Huang, M., Hao, L., Cai, S., Gu, X., Zhang, W., Hu, C., Wang, Z., Fang, L., and Zhang, W.: Effects of inorganic seed aerosols on the particulate products of aged 1,3,5-trimethylbenzene secondary organic aerosol, Atmos. Environ., 152, 490–502, https://doi.org/10.1016/j.atmosenv.2017.01.010, 2017.
Jaoui, M., Edney, E. O., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Surratt, J. D., and Seinfeld, J. H.: Formation of secondary organic aerosol from irradiated α-pinene/toluene/NOx mixtures and the effect of isoprene and sulfur dioxide, J. Geophys. Res.-Atmos., 113, D09303, https://doi.org/10.1029/2007jd009426, 2008.
Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X., Smith, K. A., Morris, J. W., and Davidovits, P.: Ambient aerosol sampling using the Aerodyne Aerosol Mass Spectrometer, J. Geophys. Res.-Atmos., 108, 8425, https://doi.org/10.1029/2001JD001213, 2003.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Kleindienst, T. E., Edney, E. O., Lewandowski, M., Offenberg, J. H., and Jaoui, M.: Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2, Environ. Sci. Technol., 40, 3807–3812, https://doi.org/10.1021/es052446r, 2006.
Krapf, M., El Haddad, I., Bruns, E. A., Molteni, U., Daellenbach, K. R., Prevot, A. S. H., Baltensperger, U., and Dommen, J.: Labile peroxides in secondary organic aerosol, Chem, 1, 603–616, https://doi.org/10.1016/j.chempr.2016.09.007, 2016.
Kroll, J. H., Chan, A. W. H., Ng, N. L., Flagan, R. C., and Seinfeld, J. H.: Reactions of semivolatile organics and their effects on secondary organic aerosol formation, Environ. Sci. Technol., 41, 3545–3550, https://doi.org/10.1021/es062059x, 2007.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nature Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Lauraguais, A., Coeur-Tourneur, C., Cassez, A., and Seydi, A.: Rate constant and secondary organic aerosol yields for the gas-phase reaction of hydroxyl radicals with syringol (2,6-dimethoxyphenol), Atmos. Environ., 55, 43–48, https://doi.org/10.1016/j.atmosenv.2012.02.027, 2012.
Lauraguais, A., Bejan, I., Barnes, I., Wiesen, P., Coeur-Tourneur, C., and Cassez, A.: Rate coefficients for the gas-phase reaction of chlorine atoms with a series of methoxylated aromatic compounds, J. Phys. Chem. A, 118, 1777–1784, https://doi.org/10.1021/jp4114877, 2014a.
Lauraguais, A., Coeur-Tourneur, C., Cassez, A., Deboudt, K., Fourmentin, M., and Choel, M.: Atmospheric reactivity of hydroxyl radicals with guaiacol (2-methoxyphenol), a biomass burning emitted compound: Secondary organic aerosol formation and gas-phase oxidation products, Atmos. Environ., 86, 155–163, https://doi.org/10.1016/j.atmosenv.2013.11.074, 2014b.
Lauraguais, A., El Zein, A., Coeur, C., Obeid, E., Cassez, A., Rayez, M.-T., and Rayez, J.-C.: Kinetic study of the gas-phase reactions of nitrate radicals with methoxyphenol compounds: Experimental and theoretical approaches, J. Phys. Chem. A, 120, 2691–2699, https://doi.org/10.1021/acs.jpca.6b02729, 2016.
Lelieveld, J., Crutzen, P. J., Ramanathan, V., Andreae, M. O., Brenninkmeijer, C. A. M., Campos, T., Cass, G. R., Dickerson, R. R., Fischer, H., de Gouw, J. A., Hansel, A., Jefferson, A., Kley, D., de Laat, A. T. J., Lal, S., Lawrence, M. G., Lobert, J. M., Mayol-Bracero, O. L., Mitra, A. P., Novakov, T., Oltmans, S. J., Prather, K. A., Reiner, T., Rodhe, H., Scheeren, H. A., Sikka, D., and Williams, J.: The Indian Ocean Experiment: Widespread air pollution from South and Southeast Asia, Science, 291, 1031–1036, https://doi.org/10.1126/science.1057103, 2001.
Li, H., Zhang, Q., Zhang, Q., Chen, C., Wang, L., Wei, Z., Zhou, S., Parworth, C., Zheng, B., Canonaco, F., Prévôt, A. S. H., Chen, P., Zhang, H., Wallington, T. J., and He, K.: Wintertime aerosol chemistry and haze evolution in an extremely polluted city of the North China Plain: significant contribution from coal and biomass combustion, Atmos. Chem. Phys., 17, 4751–4768, https://doi.org/10.5194/acp-17-4751-2017, 2017.
Liggio, J., Li, S.-M., and McLaren, R.: Reactive uptake of glyoxal by particulate matter, J. Geophys. Res.-Atmos., 110, D10304, https://doi.org/10.1029/2004JD005113, 2005.
Lin, Y.-H., Knipping, E. M., Edgerton, E. S., Shaw, S. L., and Surratt, J. D.: Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches, Atmos. Chem. Phys., 13, 8457–8470, https://doi.org/10.5194/acp-13-8457-2013, 2013.
Liu, J., Lin, P., Laskin, A., Laskin, J., Kathmann, S. M., Wise, M., Caylor, R., Imholt, F., Selimovic, V., and Shilling, J. E.: Optical properties and aging of light-absorbing secondary organic aerosol, Atmos. Chem. Phys., 16, 12815–12827, https://doi.org/10.5194/acp-16-12815-2016, 2016a.
Liu, T., Wang, X., Hu, Q., Deng, W., Zhang, Y., Ding, X., Fu, X., Bernard, F., Zhang, Z., Lü, S., He, Q., Bi, X., Chen, J., Sun, Y., Yu, J., Peng, P., Sheng, G., and Fu, J.: Formation of secondary aerosols from gasoline vehicle exhaust when mixing with SO2, Atmos. Chem. Phys., 16, 675–689, https://doi.org/10.5194/acp-16-675-2016, 2016b.
Liu, Z., Ge, M., Yin, S., and Wang, W.: Uptake and reaction kinetics of α-pinene and β-pinene with sulfuric acid solutions, Chem. Phys. Lett., 491, 146–150, https://doi.org/10.1016/j.cplett.2010.04.004, 2010.
Marais, E. A., Jacob, D. J., Turner, J. R., and Mickley, L. J.: Evidence of 1991-2013 decrease of biogenic secondary organic aerosol in response to SO2 emission controls, Environ. Res. Lett., 12, 054018, https://doi.org/10.1088/1748-9326/aa69c8, 2017.
Meade, L. E., Riva, M., Blomberg, M. Z., Brock, A. K., Qualters, E. M., Siejack, R. A., Ramakrishnan, K., Surratt, J. D., and Kautzman, K. E.: Seasonal variations of fine particulate organosulfates derived from biogenic and anthropogenic hydrocarbons in the mid-Atlantic United States, Atmos. Environ., 145, 405–414, https://doi.org/10.1016/j.atmosenv.2016.09.028, 2016.
Mielke, L. H., Furgeson, A., and Osthoff, H. D.: Observation of CINO2 in a mid-continental urban environment, Environ. Sci. Technol., 45, 8889–8896, https://doi.org/10.1021/es201955u, 2011.
Naeher, L. P., Brauer, M., Lipsett, M., Zelikoff, J. T., Simpson, C. D., Koenig, J. Q., and Smith, K. R.: Woodsmoke health effects: A review, Inhal. Toxicol., 19, 67–106, https://doi.org/10.1080/08958370600985875, 2007.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011.
Nolte, C. G., Schauer, J. J., Cass, G. R., and Simoneit, B. R. T.: Highly polar organic compounds present in wood smoke and in the ambient atmosphere, Environ. Sci. Technol., 35, 1912–1919, https://doi.org/10.1021/es001420r, 2001.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
O'Neill, E. M., Kawam, A. Z., Van Ry, D. A., and Hinrichs, R. Z.: Ozonolysis of surface-adsorbed methoxyphenols: kinetics of aromatic ring cleavage vs. alkene side-chain oxidation, Atmos. Chem. Phys., 14, 47–60, https://doi.org/10.5194/acp-14-47-2014, 2014.
Park, J.-H., Ivanov, A. V., and Molina, M. J.: Effect of relative humidity on OH uptake by surfaces of atmospheric importance, J. Phys. Chem. A, 112, 6968–6977, https://doi.org/10.1021/jp8012317, 2008.
Riva, M., Tomaz, So, Cui, T., Lin, Y. H., Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an unrecognized secondary anthropogenic source of organosulfates and sulfonates: Gas-phase oxidation of polycyclic aromatic hydrocarbons in the presence of sulfate aerosol, Environ. Sci. Technol., 49, 6654–6664, https://doi.org/10.1021/acs.est.5b00836, 2015.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Sato, K., Takami, A., Isozaki, T., Hikida, T., Shimono, A., and Imamura, T.: Mass spectrometric study of secondary organic aerosol formed from the photo-oxidation of aromatic hydrocarbons, Atmos. Environ., 44, 1080–1087, https://doi.org/10.1016/j.atmosenv.2009.12.013, 2010.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources. 3. C-1-C-29 organic compounds from fireplace combustion of wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Simoneit, B. R. T., Rogge, W. F., Mazurek, M. A., Standley, L. J., Hildemann, L. M., and Cass, G. R.: Lignin pyrolysis products, lignans, and resin acid as specific tracers of plant classes in emissions from biomass combustion, Environ. Sci. Technol., 27, 2533–2541, https://doi.org/10.1021/es00048a034, 1993.
Takekawa, H., Minoura, H., and Yamazaki, S.: Temperature dependence of secondary organic aerosol formation by photo-oxidation of hydrocarbons, Atmos. Environ., 37, 3413–3424, https://doi.org/10.1016/s1352-2310(03)00359-5, 2003.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Weis, D. D. and Ewing, G. E.: Water content and morphology of sodium chloride aerosol particles, J. Geophys. Res.-Atmos., 104, 21275–21285, https://doi.org/10.1029/1999jd900286, 1999.
Xu, L., Middlebrook, A. M., Liao, J., de Gouw, J. A., Guo, H., Weber, R. J., Nenes, A., Lopez-Hilfiker, F. D., Lee, B. H., Thornton, J. A., Brock, C. A., Neuman, J. A., Nowak, J. B., Pollack, I. B., Welti, A., Graus, M., Warneke, C., and Ng, N. L.: Enhanced formation of isoprene-derived organic aerosol in sulfur-rich power plant plumes during Southeast Nexus, J. Geophys. Res.-Atmos., 121, 11137–11153, https://doi.org/10.1002/2016jd025156, 2016.
Yang, B., Zhang, H., Wang, Y., Zhang, P., Shu, J., Sun, W., and Ma, P.: Experimental and theoretical studies on gas-phase reactions of NO3 radicals with three methoxyphenols: Guaiacol, creosol, and syringol, Atmos. Environ., 125, 243–251, https://doi.org/10.1016/j.atmosenv.2015.11.028, 2016.
Ye, J., Abbatt, J. P. D., and Chan, A. W. H.: Novel pathway of SO2 oxidation in the atmosphere: reactions with monoterpene ozonolysis intermediates and secondary organic aerosol, Atmos. Chem. Phys., 18, 5549–5565, https://doi.org/10.5194/acp-18-5549-2018, 2018.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015.