the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Nov 2019
| 27 Nov 2019
New particle formation and its effect on cloud condensation nuclei abundance in the summer Arctic: a case study in the Fram Strait and Barents Sea
Simonas Kecorius
Teresa Vogl
Pauli Paasonen
Janne Lampilahti
Daniel Rothenberg
Heike Wex
Sebastian Zeppenfeld
Manuela van Pinxteren
Markus Hartmann
Silvia Henning
Xianda Gong
Andre Welti
Markku Kulmala
Frank Stratmann
Hartmut Herrmann
Alfred Wiedensohler
In a warming Arctic the increased occurrence of new particle formation (NPF) is believed to originate from the declining ice coverage during summertime. Understanding the physico-chemical properties of newly formed particles, as well as mechanisms that control both particle formation and growth in this pristine environment, is important for interpreting aerosol–cloud interactions, to which the Arctic climate can be highly sensitive. In this investigation, we present the analysis of NPF and growth in the high summer Arctic. The measurements were made on-board research vessel Polarstern during the PS106 Arctic expedition. Four distinctive NPF and subsequent particle growth events were observed, during which particle (diameter in a range 10–50 nm) number concentrations increased from background values of approx. 40 up to 4000 cm−3. Based on particle formation and growth rates, as well as hygroscopicity of nucleation and the Aitken mode particles, we distinguished two different types of NPF events. First, some NPF events were favored by negative ions, resulting in more-hygroscopic nucleation mode particles and suggesting sulfuric acid as a precursor gas. Second, other NPF events resulted in less-hygroscopic particles, indicating the influence of organic vapors on particle formation and growth. To test the climatic relevance of NPF and its influence on the cloud condensation nuclei (CCN) budget in the Arctic, we applied a zero-dimensional, adiabatic cloud parcel model. At an updraft velocity of 0.1 m s−1, the particle number size distribution (PNSD) generated during nucleation processes resulted in an increase in the CCN number concentration by a factor of 2 to 5 compared to the background CCN concentrations. This result was confirmed by the directly measured CCN number concentrations. Although particles did not grow beyond 50 nm in diameter and the activated fraction of 15–50 nm particles was on average below 10 %, it could be shown that the sheer number of particles produced by the nucleation process is enough to significantly influence the background CCN number concentration. This implies that NPF can be an important source of CCN in the Arctic. However, more studies should be conducted in the future to understand mechanisms of NPF, sources of precursor gases and condensable vapors, as well as the role of the aged nucleation mode particles in Arctic cloud formation.
- Article
(7422 KB) - Full-text XML
-
Supplement
(2555 KB) - BibTeX
- EndNote




Atmospheric new particle formation (NPF), during which particles with diameters from 1 to 2 nm are formed, is a phenomenon observed in many different environments around the world (Kerminen et al., 2018). Initial steps involved in particle formation and subsequent growth are usually clustering and condensation of both organic and inorganic vapors (Schobesberger et al., 2013). Ions are also known to be involved in the nucleation process (e.g., Jokinen et al., 2018). If newly formed particles are not lost due to coagulation (Lehtinen, et al., 2007), and manage to grow to sizes >50 nm, they can act as cloud condensation nuclei (CCN, Kerminen et al., 2012). Under the presence of sufficient water vapor, CCN activate to form cloud droplets (Köhler, 1936). Atmospheric NPF is estimated to be a substantial source of the world's CCN budget (Merikanto et al., 2009). Thus, in a highly sensitive atmosphere such as the Arctic, where CCN number concentration is usually low (<100 cm−3, Mauritsen et al., 2011), NPF may be an important phenomenon controlling the radiative forcing (Allan et al., 2015; Croft et al., 2016).
During the last decade, Arctic regions have experienced remarkable changes. Here, the near-surface temperature has increased at least 2-fold compared to the Northern Hemisphere (a phenomenon known as Arctic amplification, Overland et al., 2011; Jeffries and Richter-Menge, 2012). In parallel, a substantial decline in multiyear sea-ice cover (e.g., Bi et al., 2018), an increase in sea-ice mean speed and deformation (Rampal et al., 2009), and development of melt ponds (Polashenski et al., 2017) were also observed. Such changes are not only reflected in the dynamics of the Arctic ecosystem (Meier et al., 2014), but are also predicted to impact the mid-latitude climate (Serreze and Barry, 2011; Cohen et al., 2014; Walsh, 2014).
Recent studies suggest that the amplified warming in the Arctic and related changes are a result of a complex interaction between different feedback mechanisms including parameters such as temperature (Pithan and Mauritsen, 2014), surface albedo (e.g., Screen and Simmonds, 2010; Taylor et al., 2013), water vapor (Graversen and Wang, 2009), cloud (Vavrus, 2004), and the lapse rate (Bintanja et al., 2012). Additionally, variations in atmospheric and oceanic heat transport were also identified as active players in the changing Arctic climate (Spielhagen et al., 2011; Alexeev and Jackson, 2013). Increase in latent heat and moisture transport towards the poles may drive the low-cloud formation, and thus, Arctic surface warming (Praetorius et al., 2018). And while the mechanisms of lapse rate, surface albedo, temperature and water vapor feedbacks are rather well understood, the net cloud feedback still has one of the largest uncertainties (Zhang et al., 2018).
The multi-year analysis of particle number size distributions from the sites around the Arctic Ocean revealed frequent new particle formation, occurring either locally or at higher elevations and prevailing mostly during spring and summer months (Freud et al., 2017; Nguyen et al., 2016; Dall'Osto et al., 2019). In the near future, the frequency of atmospheric NPF occurrences is expected to increase due to Arctic sea-ice melt (Dall'Osto, et al., 2017, 2018a, b). This makes measurements of the ultrafine particle physico-chemical properties in the Arctic increasingly valuable if aerosol–cloud–climate interactions need to be understood (Willis et al., 2018; Abbatt et al., 2019). Contrary to scientific interest, such studies in this remote environment still remain limited, mainly because of logistic challenges in the region (e.g., Willis et al., 2017; Wendisch et al., 2019). The following are studies which focus on nucleation mode particles in the Arctic. Wiedensohler et al. (1996) reported the occurrence of ultrafine particles in the Arctic as a result of NPF. However, no correlation with potential precursor gases has been found. Karl et al. (2012) found that a sulfuric acid nucleation mechanism best explains the observed growth of nucleation mode particles over the central Arctic Ocean. In another study by Karl et al. (2013), marine granular nanogels were proposed as a novel route to atmospheric nanoparticles in the high Arctic. Furthermore, NPF in the Arctic region was associated with marine biological processes, such as the seasonal cycle of the gel-forming phytoplankton by Heintzenberg et al. (2017). From the results of volatility measurements, Giamarelou et al. (2016) have proposed that particles during NPF events in the high Arctic exist in the form of partly or fully neutralized ammoniated sulfates. Iodine from coastal macro algae was detected in the growing particles (Allan et al., 2015; Sipilä et al., 2016), suggesting the iodine as a nucleation precursor. A large body of studies comes from the Canadian Arctic region. For example, Croft et al. (2016) showed that ammonia from seabird-colony guano is a key factor contributing to bursts of newly formed particles at Alert, Nunavut, Canada. Aerosol particle growth in the Canadian Arctic Archipelago during summer was correlated with organic species, trimethylamine, and methanesulfonic acid (MSA), suggesting an important marine influence (Leaitch et al., 2013; Willis et al., 2016, Abbatt et al., 2019). Park et al. (2017) provided compelling evidence of the contribution of marine biogenic dimethyl sulfide (DMS) to the formation of aerosol particles. Collins et al. (2017) also reported frequent ultrafine particle formation and growth in Canadian Arctic marine and coastal environments. The authors emphasized that the low condensation sink, high solar radiation, low sea-ice concentration, and marine microbial processes all contribute to a higher frequency of particle formation and growth. Most recently, Tremblay et al. (2019) correlated particle formation and growth events with the melting of the sea ice. The authors indicated that besides oxidation of DMS to produce particle-phase sulfate, other gas-phase organic compounds are important for particle growth.
Compared to NPF research in Arctic environments, studies on whether nucleation mode particles (diameter of 20 nm) can act as CCN are even scarcer. Leaitch et al. (2016) investigated effects of 20–100 nm particles on liquid clouds in the clean summertime Arctic and found that particles as small as 20–50 nm can activate to cloud droplets. This was also confirmed by Burkart et al. (2017b), who found that in the Canadian high Arctic marine boundary layer, newly formed particles (approx. 30 nm in diameter) are capable of being involved in cloud activation, suggesting that in the pristine environment, where cloud radiative forcing is limited by the CCN available (Mauritsen et al., 2011), information about aerosol sources is crucial in understanding the link between sea-ice melt and low-altitude clouds.
In this investigation, we analyzed four cases of NPF and a subsequent growth from a perspective of particle physical (number concentration, number size distribution, and formation and growth rates) and indirect chemical (hygroscopicity) properties. Our main goal here is to test the hypothesis that NPF and secondary aerosol production can influence the CCN budget in the summertime Arctic. The study is structured as follows. After a short description of materials and methods in Sect. 2, we proceed by describing each NPF event separately (Sect. 3). This includes specification of the meteorological conditions during which NPF occurred, characterization of particle formation and growth rates, followed by the observed hygroscopicity of newly formed particles, and the measured CCN concentrations during NPF events. We start the discussion of the results (Sect. 4) with a general overview of our observations, putting the results into perspective of other studies. This leads to Sect. 4.1, where we discuss the indirect evidence of the composition of newly formed particles. Here, we reflect on our observational data as well as various techniques to gain information on particle formation mechanisms, possible sources of precursor gases, etc. The discussion section is closed by investigating the implication of NPF for cloud formation. This is done by using zero-dimensional parcel model to examine, whether newly formed and slightly grown particles can become CCN. Model results are compared to measured number concentration of CCN during the NPF events. The main results are summarized at the end of the work; general conclusions are also provided.
2.1 Description of observations
The data used in this study were obtained during two legs of an expedition of the German Research Vessel Polarstern (PS 106/1 and PS 106/2): the “Physical feedbacks of Arctic boundary layer, Sea ice, Cloud and AerosoL (PASCAL, PS 106/1)” and “Survival of Polar Cod in a Changing Arctic Ocean (SiPCA, PS 106/2)” (Macke and Flores, 2018; Wendisch et al., 2019). Both expeditions took place in the vicinity of Svalbard (Norway) from May to July 2017. PASCAL was performed in the framework of the ArctiC Amplification: Climate Relevant Atmospheric and SurfaCe Processes, and Feedback Mechanisms (AC)3 project and was designed to explore cloud properties, aerosol impact on clouds, atmospheric radiation and turbulent-dynamical processes. During the first leg of the trip (PS 106/1, PASCAL), RV Polarstern reached approx. 82∘ north, where an ice-floe camp was established (5–14 June). The first leg of the expedition ended at Longyearbyen, Svalbard, by 21 June. On 22 June, RV Polarstern left Svalbard for the SiPCA expedition. On the second expedition leg aerosol particle measurements were performed until 16 July. The cruise track and the ice drift are shown in Fig. 1.
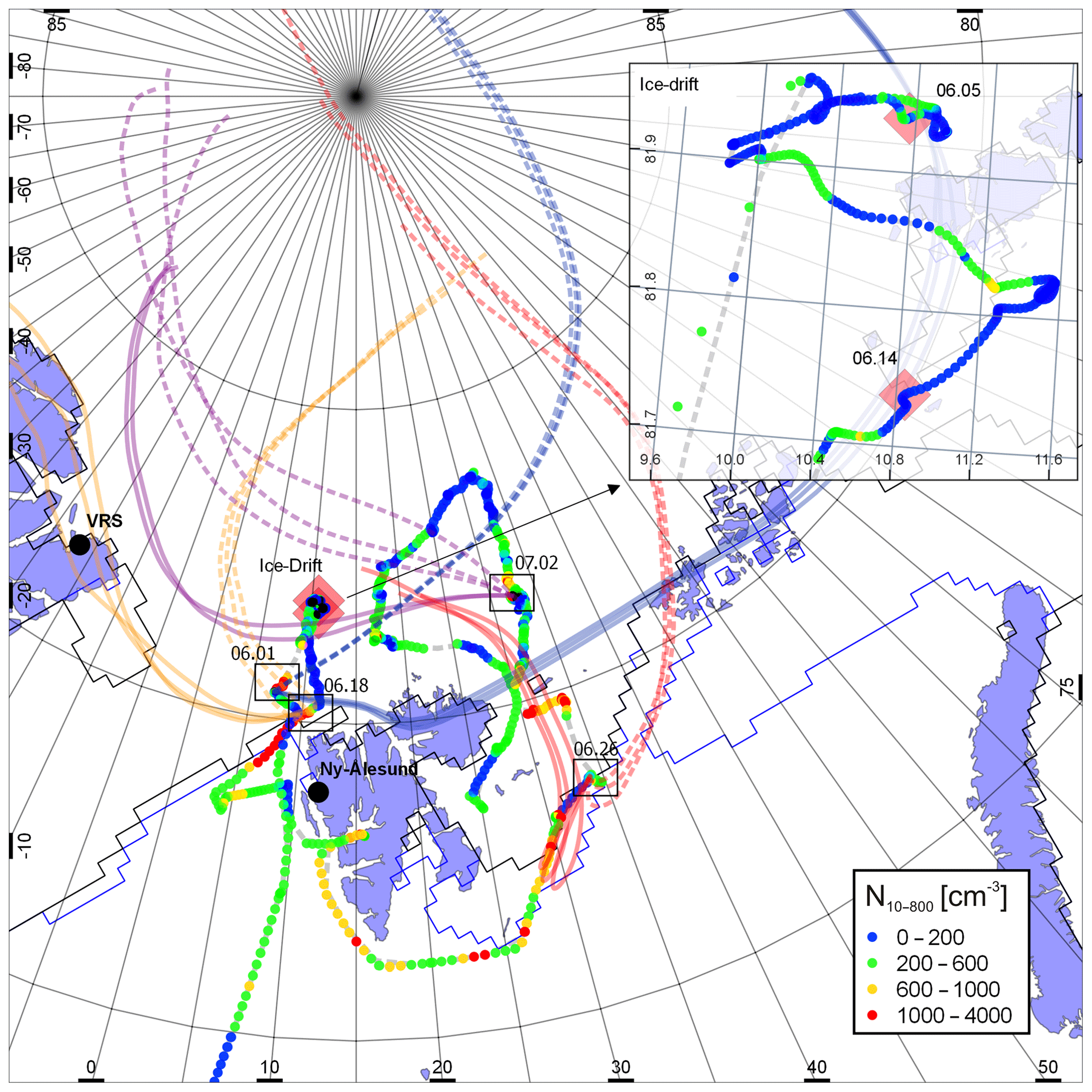
Figure 1Cruise track and particle number concentration (integrated in a size range from 10 to 800 nm) during PASCAL and SiPCA expeditions. The days which were picked to analyze NPF events and subsequent particle growth are indicated with square boxes. Backward air mass trajectories (72 h) were calculated using HYSPLIT (Draxler and Rolph, 2012) and are shown by solid (200 m a.s.l) and dotted (2000 m a.s.l) lines corresponding to each NPF event. Ice drift is shown in the insert. Thin blue and black lines are the observed ice edge for June and July 2017, respectively (Fetterer et al., 2002).
2.2 Measurement setup and equipment
To measure aerosol particle physico-chemical properties, a temperature controlled measurement container, prepared and operated by the Leibniz Institute for Tropospheric Research, Leipzig, Germany, was installed on the observation deck of RV Polarstern. The aerosol container was air-conditioned to 24 ∘C and the aerosol inlet head was heated to 30 ∘C to ensure the stability of aerosol instrumentation and prevent icing, respectively. The aerosol inlet was made of 6 m length stainless steel tubing, with an inner tube diameter of 40 mm. It was placed on top of the measurement container with an angle of 45 ∘, pointing away from the ship. The aerosol flow in the 6 m long inlet was set to 40 L min−1 (Reynolds number <2000, laminar flow) to minimize particle losses. Inside the container, an isokinetic splitter was used together with short and vertical conductive tubes to feed the measurement instrumentation with an aerosol sample. Aerosol instrumentation (relevant to this study) included a neutral cluster and air ion spectrometer (NAIS), a mobility particle size spectrometer (MPSS), the Volatility/Hygroscopicity-Tandem Differential Mobility Analyzer (VH-TDMA), and the Cloud Condensation Nuclei Counter (CCNC) to measure aerosol particle number size distribution, volatility/hygroscopicity properties of aerosol particles, and the number concentration of CCN, respectively.
2.2.1 Neutral cluster and air ion spectrometer (NAIS)
A neutral cluster and air ion spectrometer (NAIS, Mirme and Mirme 2013) and guidelines by Kulmala et al. (2012) were used to study early stages of NPF and subsequent growth (including NPF event classification, formation (J), and growth rate (GR) calculation). The NAIS measures the number size distribution of neutral particles in the diameter range of approx. 2–40 nm and charged particles and clusters in the size range of approx. 0.8–40 nm. The instrument is an extended version of the air ion spectrometer (Mirme et al., 2007) and utilizes a sample preconditioning section to enable measurements of neutrally charged particles. Unipolar corona chargers are used for both charging and charge neutralization. Charged particle classification is carried out in the multichannel differential mobility analyzer (DMA) where 21 individual electrometers are used to record electric current carried by the charged particles. Due to high total flow of NAIS (60 L min−1) a dedicated 1.3 m long copper inlet (3.5 cm in diameter) was installed to sample ambient air. Measurement data were inverted using the v14-lrnd inversion algorithm (Wagner et al., 2016). Particle losses due to diffusion were corrected before data processing.
2.2.2 Mobility particle size spectrometer (MPSS)
Particle number size distributions (PNSD), in a mobility size range from 10 to 800 nm, were measured with a TROPOS-type mobility particle size spectrometer (MPSS, Wiedensohler et al., 2012). The MPSS consisted of a Hauke-type DMA (effective length of 28 cm), a condensation particle counter (CPC, model 3772, TSI Inc., USA, flow rate 1 L min−1), a closed-loop sheath flow arrangement, and a bipolar diffusion charger, ensuring the bipolar charge equilibrium as described in Wiedensohler (1988). The sample flow rate was controlled by a CPC (1 L min−1) and the sheath flow rate was 5 L min−1. The time resolution of an up-and-down scan was 5 min. Electrical particle mobility distributions were inverted to PNSDs using the inversion algorithm presented by Pfeifer et al. (2014). The final PNSDs were corrected for transmission losses in the sampling lines using the method of equivalent length and CPC counting efficiencies (Wiedensohler et al., 1997). Sizing accuracy of MPSS was controlled using nebulized polystyrene latex spheres (PSL, Thermo Scientific™, Duke Standards™) of 203 nm (Wiedensohler et al., 2018). High-voltage supply offset calibration, instrument flows, and tests for leakage were performed on a regular basis (once per week).
2.2.3 Volatility/Hygroscopicity-Tandem Differential Mobility Analyzer (VH-TDMA)
Aerosol particle affinity with water and volatility properties (not discussed here) was measured using the TROPOS-type Volatility/Hygroscopicity-Tandem Differential Mobility Analyzer (VH-TDMA, Augustin-Bauditz et al., 2016). The instrument consists of a DMA-1 that selects chosen quasi-monodisperse particles, a thermodenuder (not used in this study), an aerosol humidification section that conditions the particles selected by the DMA-1, and an MPSS-equivalent closed-loop sheath flow unit inside the temperature-controlled box, which is used to obtain the hygroscopic growth factor (HGF). The HGF is defined as the ratio between the measured particle electrical mobility diameter at a given RH as measured by the second DMA and the initially selected dry diameter.
During the whole expedition, two constant aerosol particle sizes, 50 and 150 nm, were selected for the measurement of HGF at a target RH of 90 %. Additionally, HGF of 15, 20 and 30 nm size particles were measured during NPF and growth events. The system RH, measured by a humidity sensor, was periodically calibrated by an automatic calibration unit, using pure ammonium sulfate. Scans with RH ±2 % from target RH were excluded from data analysis. Sizing accuracy, high-voltage supply offset calibration, flow rates, and zero tests were performed regularly (once per week). In general, recommendations have been followed as described in Massling et al. (2011).
The VH-TDMA data were inverted using a TDMAinv routine (Gysel et al., 2009) to retrieve the probability density functions of GF (GF-PDF). Scans with RH <20 % were used to calibrate size offset in the system, as well as to define the width of the transfer function (Gysel et al., 2009). The particle hygroscopicity parameter kappa (κ) was derived from VH-TDMA data following the κ-Köhler theory by Petters and Kreidenweis (2007):
where S is the saturation ratio; σs is the surface tension of the solution; MW is the molecular weight of water; R is the universal gas constant; T is the temperature; ρW is the density of water; and Dd is the particle dry diameter.
2.2.4 Cloud condensation particle counter (CCNC)
The CCNC (model CCN-100 from Droplet Measurement Technologies, Roberts and Nenes, 2005) measured CCN number concentrations, subsequently at six different supersaturations (0.1 %, 0.15 %, 0.2 %, 0.3 %, 0.5 % and 1 %), where each supersaturation was sampled for 10 min. Hence an hourly average concentration at each supersaturation is available. The instrument was calibrated before and directly following the campaign using pure ammonium sulfate particles of known sizes, based on the ACTRIS protocol (Gysel and Stratmann, 2013). Only poly-disperse aerosol was sampled by CCNC.
2.2.5 Offline chemical analysis
The sampling of aerosol particles was conducted using five-stage low-pressure Berner impactors (Hauke, Austria) with a flow rate of 75 L min−1, which was installed on the top of the observation deck facing the ocean at a height of ca. 25 m. Particles were collected in the size ranges 0.05–0.14 µm (stage 1), 0.14–0.42 µm (stage 2), 0.42–1.2 µm (stage 3), 1.2–3.5 µm (stage 4), and 3.5–10 µm (stage 5) aerodynamic particle diameter (50 % cut-off) on aluminum foils as impaction substrates, which had been heated at 350 ∘C for at least 2 h to reduce blank levels prior to sampling. To avoid condensation of atmospheric water on the surface of these aluminum foils, a conditioning unit was mounted between the impactor inlet and the sampling unit consisting of a 3 m tube. By heating the sampled air, high relative humidity of the ambient air was reduced to 75 %–80 % before the collection of the aerosol particles. The temperature difference between the ambient air at the impactor inlet and the sampled air after the conditioning unit did not exceed 9 K. Thus, the losses due to evaporation of semi-volatile compounds are expected to be minimal.
After sampling, the aluminum foils were stored in aluminum boxes at −20 ∘C and transported in dry ice to the TROPOS laboratories in Leipzig, Germany. Field blanks were collected by loading the Berner impactor with the aluminum foils at the sampling site with no air drawn through it. Please note that the sampling time was set to 72 or 144 h (to accumulate enough particle mass on the filters), thus, it does not exclusively comprise the discussed NPF events. For example, during NPF Event 1, chemical particle composition was determined from samples that were collected between 29 May (midday) and 1 June (approx. 08:00). During NPF Event 3, sampling was done between 25 June (11:00) and 28 June (09:00).
Particle mass determination was performed by weighing clean (blank) and particle-loaded filters using a microbalance UMT-2 (Mettler-Toledo, Switzerland). The concentrations of water-soluble methanesulfonic acid (MSA) and inorganic compounds relevant to this study (, , Na+) in filtered (0.45 µm syringe) aqueous extracts (50 % of the filter in 2 mL) were determined using ion chromatography (ICS3000, Dionex, Sunnyvale, CA,USA), as described in Müller et al. (2010). Assuming that the ocean is the major source of the measured atmospheric sodium, sea salt sulfate (ss-sulfate) was calculated from the constant mass ratio () in bulk seawater (Galloway et al., 1993; Fomba et al., 2014). Non-sea salt sulfate (nss-sulfate) was calculated by subtracting ss-sulfate from the total sulfate concentration. The determination of total carbon (TC) as organic carbon (OC) and elemental carbon (EC) was carried out by a two-step thermographic method (C-mat 5500, Ströhlein, Germany) with nondispersive infrared sensor (NDIR) detection as described in Müller et al. (2010). Organic matter (OM) was calculated by considering OM as twice OC () for remote aerosols (Turpin and Lim, 2001).
2.3 Analysis of PNSD measurements
Before NPF event classification, inverted and loss-corrected NAIS and MPSS PNSDs were merged together. For the smallest particle diameter, from 2 to 10 nm, exclusively NAIS data were chosen. This is because the MPSS used in this study was optimized to operate in a diameter range from 10 to 800 nm. The diffusional losses of sub-10 nm particles were too great to accurately recover the PNSD at initial steps of nucleation. Contrarily, uncertainties in the NAIS measured particle number concentration increases for particle diameters larger than 10 nm (Wagner et al., 2016). For these reasons, PNSDs from both NAIS and MPSS were merged at 10 nm diameter. No additional treatment (e.g., spline fit to smooth merging distributions) was performed on merged PNSDs.
Following the protocol by Kulmala et al. (2012), NPF events were visually identified from the merged PNSDs. Although different types of NPFs were recorded (e.g., short bursts in the smallest particle number as, e.g., described for the Arctic region by Heintzenberg et al., 2017 and Dall'Osto et al., 2017), in this work we will only focus on NPF events with subsequent particle growth. This type of event includes not only particle formation, but also includes later particle growth lasting for several hours, thus representing a more regional phenomenon (Ström et al., 2009). It also allows us to calculate the GR of the particles, which would not be possible in the case of short nucleation mode particle bursts.
Different methods exist to determine the GR based on the measured PNSD. For example, maximum-concentration and log-normal distribution function methods were proposed by Kulmala et al. (2012). Tracking regions of PNSD and interpreting the change rate of the size-integrated general dynamic equation methods was suggested by Pichelstorfer et al. (2018). In this work, we used a trial-and-error approach to find the best fit to determine the GR by selectively applying all the mentioned methods for certain NPF cases. The formation rate of particles of a certain size (J) was calculated as described by Kulmala et al. (2012), based on the observed changes in particle concentrations, GR determined, and particle losses characterized by a coagulation sink (CoagS).
2.4 Adiabatic cloud parcel model
To study the climatic relevance of NPF in the Arctic, we have used a zero-dimensional, adiabatic cloud parcel model. Thorough formulation of the model is given by Rothenberg and Wang (2016) and will not be discussed here. Model code is also freely available at https://pyrcel.readthedocs.io (last access: 21 August 2019). Shortly, at the initial step, the model calculates an equilibrium wet-size distribution from the set of given parameters. This includes the description of the aerosol population and environmental specifications of temperature, pressure, relative humidity, parcel ascending velocity, and the height of the planetary boundary layer. The aerosol particle population, consisting of two modes, is described by the total number concentration, the geometric mean diameter, and the geometric standard deviation of the log-normal distribution. The hygroscopicity parameter κ following Petters and Kreidenweis (2007) is used to describe particle chemical composition. The evolutions of the parcel supersaturation, temperature, pressure, and liquid/vapor water content are then integrated forward in time to describe the thermodynamic evolution of an adiabatically lifted, non-entraining parcel. In the model, the evolution of supersaturation S is
where α and γ are functions depending on temperature and pressure (Leaitch et al., 1986) and wc is the liquid cloud water mass mixing ratio. Change in temperature is described as
V is the updraft velocity, g is gravitational acceleration, cp is the specific heat of dry air at constant pressure, L is the latent heat of water, and wv is the water vapor mass mixing ratio. Water mass conservation is ensured as vapor condenses into cloud water. Pressure change within the ascending parcel can be written as
where Tv is temperature, Rd – gas constant for dry air. The change in cloud water is
Here ρa and ρw is the density of air and water, respectively. Ni is a number concentration and ri is radius in a size bin, S is environmental saturation, Seq is the predicted equilibrium supersaturation under framework described by Petters and Kreidenweis (2007). G is a growth coefficient, which is a function of both the chemical and physical properties of particles.
2.5 Contamination from ship exhaust
During the cruise, ship exhaust occasionally disturbed measurements on-board RV Polarstern. This was mostly pronounced during the periods when the ship was breaking the ice (rapid forward–backward direction change) and/or was drifting during sea experiments. Ship exhaust contamination can be seen in Fig. 2 contour plots as a sharp increase in particle number concentrations over the whole particle diameter range. The contamination from online measurements was removed manually. For this, we referred to total particle number concentration observed by separate CPC with 2 s time resolution. The comparison between the total particle number concentration and the signal from the single-particle soot photometer (results are not shown here) confirmed that the total CPC indeed is able to observe sharp increase in particle number, which is related to ship exhaust (black carbon particles).

Figure 2The NPF events observed during RV Polarstern cruise PS106. The PNSDs from NAIS (negative polarity) and MPSS are shown as contour plots. The color scale represents particle number concentration as dN∕dlog Dp. Inside the contour plots, particle number concentration, integrated between two size ranges (10 to 50 and 100 to 800 nm), is shown with dashed and dotted black lines. The presence of corona charger ions (<2 nm, Manninen et al., 2011) can also be seen in NAIS data. This artifact was excluded from data analysis. Coagulation and condensation sinks, meteorological parameters (wind speed and direction, global radiation, temperature, and relative humidity), and formation rates (J) for each NPF event are shown in the panels below the contour plots. Note: sample contamination by ship exhaust was removed from data analysis; however, for better representation of particle growth, the contour plots include all the data (contamination not removed).
For offline measurements, an automatic system (to measure relative wind direction) was installed together with a high-volume sampler to stop the pumps when the wind direction was associated with the pollution sector. A similar approach was used by Huang et al. (2018). The Berner impactor, on the other hand, did not have such a system to prevent samples from contamination. To avoid measurement artifacts, only samples with the same order of organic carbon as from high-volume samplers were used for data analysis and discussion.
During the PS106 cruise, a number of instances were recorded whereby a total particle number concentration (integrated from MPSS between 10 and 800 nm) steadily increased from the background concentrations of several hundred to several thousand particles per cm3 (Fig. 1). After eliminating the contribution from the ship exhaust (by filtering abrupt and short increases in particle number concentration recorded by a total CPC with 2 s time resolution), these cases were associated with new particle formation (NPF) events. For further discussion, we have selected four NPF events with a subsequent particle growth which represent the phenomenon on a regional scale (Ström et al., 2009). To gain information about the scale of NPF, additional data of PNSD information from the Villum Research Station and Zeppelin Mountain Observatory were also taken into account (data for visual inspection were taken from http://ebas.nilu.no/, last access: 18 March 2019).
The geographic location of the observed NPF events can be seen in Fig. 1 (indicated with black rectangles and date of occurrence), and took place between 78.55 to 81.66∘ N and 7.28 to 33.96∘ E. The most intense event (NPF 1) occurred on 1 June 2017, with the total particle number concentration increasing from 100 to more than 4000 particles cm−3. During the NPF event, the lateral distance between RV Polarstern and the nearest coast of Svalbard archipelago was 150 km. The least intensive NPF event (NPF 3) was recorded on 26 June, during which the total particle number concentration increased from 160 to 700 particles cm−3. Nevertheless, the subsequent particle growth from 3 to approx. 50 nm lasted for 3 d. All the events that were recorded during June (1, 18, and 26 June) took place in the vicinity of the marginal ice zone. The most northern event (NPF 4, 81.6 ∘ north) was observed on 2 July 2017. At this time, RV Polarstern was further away from the marginal ice zone. The average total particle number concentration before the NPF event was approx. 100 particles cm−3, which increased to 1400 particles cm−3 during the event.
3.1 Overview of the NPF events
In this paragraph, a detailed overview of the events is presented with the focus on environmental conditions during which NPF occurred, as well as the formation and growth rates of newly formed particles.
3.1.1 NPF 1: 1 June
The first NPF event with a subsequent particle growth was observed from around 06:00 onwards on 1 June 2017. RV Polarstern reached the marginal ice zone at 11:00 and entered the pack ice at around 15:00 on 31 May 2017 (note that all times in this study are given in UTC). This can be seen from the air and water temperature profiles (Fig. 2). The temperature of air and water decreased from approx. +5 to −5 ∘C (air) and −2 ∘C (water). In this area, the ice was broken up by leads, which facilitated the passage of the vessel towards the north. Around 20:00 a region with more densely packed ice was reached, which obstructed the movement of the ship (Nicolaus, 2018). On these occasions, due to frequent reverse–forward ship movement, pollution highly affected the measurements on-board (see PNSDs in Fig. 1). On 1 June, the vessel could once again pass through open leads in the pack ice, allowing for contamination-free scans for the time period from 04:00 to 20:00. During this time, RV Polarstern moved 26 km (from 80.39∘ N 7.58∘ E to 80.62∘ N 7.94∘ E) in mostly cloud-free conditions. From 18:00 to 20:00, a thin ice cloud was present at over 8 km altitude. Also, over a short period from 14:00 to 15:00, intermittent low-level liquid clouds were present, which however did not decrease the global radiation significantly. For a more detailed description of local and associated large-scale weather patterns during PS106, please refer to Knudsen et al. (2018).
Before the NPF event, the average particle number concentration in a size range from 10 to 50 nm (PNC10–50) was 50 particles cm−3. The particle number concentration in the size range from 100 to 800 nm (PNC100–800) before the event decreased from 150 to as low as 2 particles cm−3. This resulted in a sharp decrease in the coagulation sink for 3 nm particles from to s−1. The condensation sink also decreased by 1 order of magnitude from to s−1, creating favorable conditions for particles to form. The NPF event occurred when the RH was approx. 90 % and the particle formation rate peaked when the global radiation approached the maximum (600 W m−2). The wind speed gradually decreased from an average of 8 m s−1 on 31 May 2017 to 5 m s−1 during the NPF event. As a result of the NPF, the number of ultrafine particles increased by almost 2 orders of magnitude.
The backward air mass trajectories (calculated for 200 and 2000 m above sea level, Draxler and Rolph, 2012) showed possible intrusion of air from higher altitudes and also that air was arriving at the ship following the 80 ∘ north latitude, passing over the Prince George Land and northeastern Svalbard archipelago (Fig. 1). This can be confirmed by the increase in ozone concentration at Zeppelin Observatory (Aas et al., 2018; data available from http://ebas.nilu.no/). Following the NPF event on 1 June, the wind direction gradually changed from northeast to southwest and brought in a sudden fog (at 19:00, evident from a steep increase in ambient RH to 100 % and a simultaneous decrease in visibility measured by the vessel's meteorology station). This can be seen as a sharp increase in both air temperature and RH (to over 100 %) causing disruption in the PNSD (onwards from approx. 20:00, 1 June). At the same time, further observations of the event were corrupted by the local pollution from ship exhaust.
Table 1Calculated parameters for observed NPF events during RV Polarstern cruise 106. The GR is obtained from the NAIS size spectrum using the methods proposed by Kulmala et al. (2012) and Pichelstorfer et al. (2018). J is the formation rate of 1.6 nm sized positive/negative ion clusters and 3 nm sized particles. Please note that in some instances the size range for GR and J calculations is different (due to measured PNSD). Nevertheless, we calculated both parameters from the smallest possible particle/ion size range. The value after “±” shows the standard deviation. Date format: mm/dd.
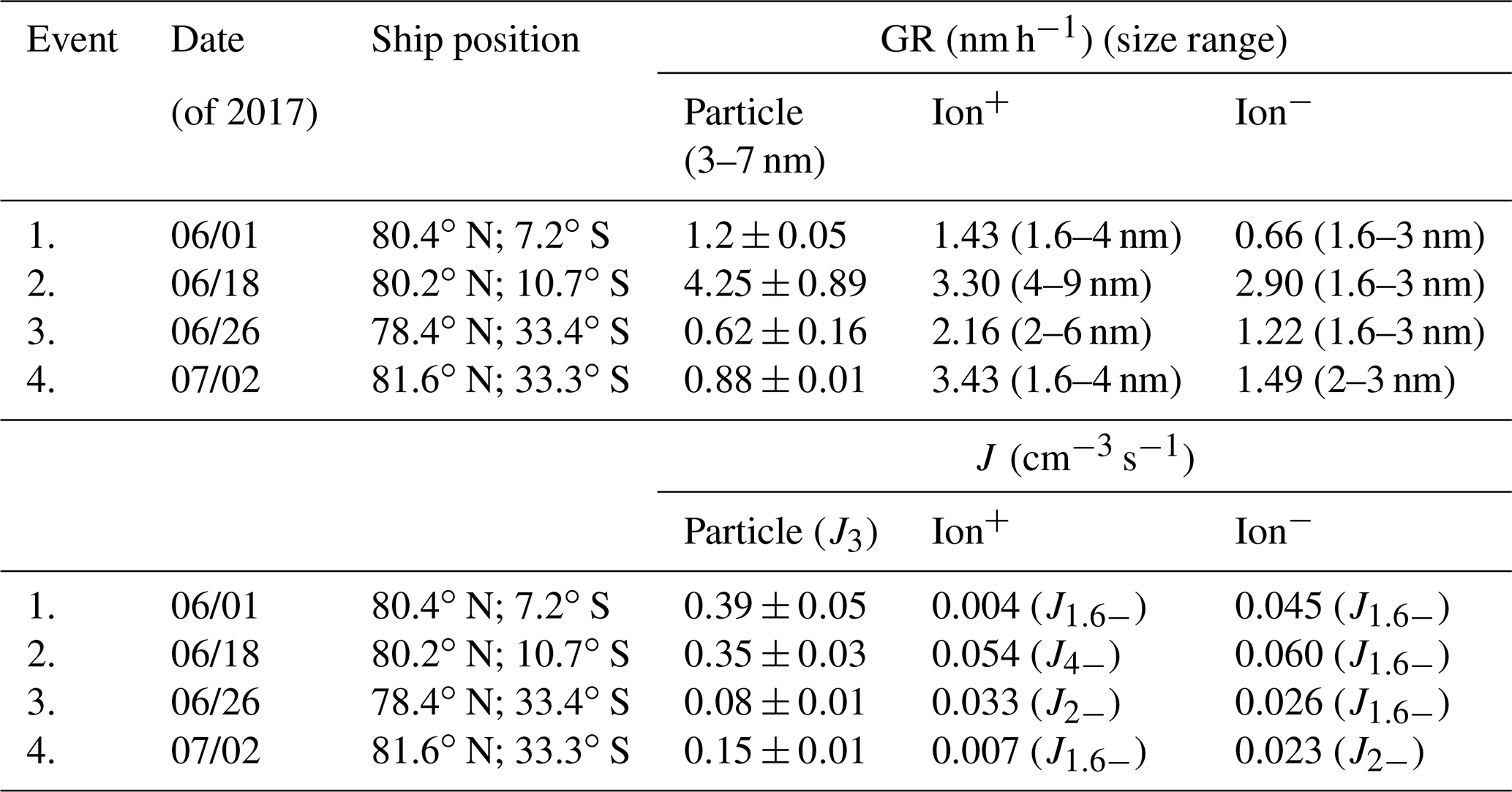
Some parameters describing newly formed particles and ions are shown in Table 1. The particle GR in a size range from 3 to 7 nm was 1.2 nm h−1. After the NPF event, subsequent particle growth lasted for about 12 h, during which the particles were able to grow to approx. 30 nm in diameter (geometric mean diameter). The GR for 1.6 to 3 nm ions was somewhat more variable – 0.7 for negative and 1.4 nm h−1 for positive ions. Please note that we were not able to calculate the positive ion GR in a size range from 1.6 to 3 nm. Instead, the GR for a particle size range 1.6 to 4 nm was calculated. The formation rate of 3 nm (J3−) sized neutral particles and negative ions (1.6 nm, J1.6−) was approx. 0.4 and 0.045 cm−3 s−1, respectively.
3.1.2 NPF 2: 18 June
On 17 June, the ship was moving southward through packed ice area, breaking floes and navigating through polynyas (Nicolaus, 2018). Over the complete day of 17 June, low-level stratocumulus clouds were present, which were broken up occasionally between 07:00 and 13:00 and between 04:00 and 22:00. Between 23:00 on 17 June and 01:00 on 18 June, measured visibility decreased, accompanied by an increase in relative humidity (RH), indicating fog. This low-level cloud layer was present until approx. 08:00 on 18 June, when RV Polarstern left the packed ice, entering the marginal ice zone. This resulted in water and air temperature increases from −1.9 ∘C to approx. 2 and 0.5 ∘C above zero, respectively. At the same time, local wind speed decreased from 5 to 2 m s−1. During the following hours, until 18:00, no clouds were present except for a very thin, high ice cloud at 8 km from approx. 11:30 to 12:00. This period of high incident radiation was only briefly interrupted by a short fog event from 15:00 to 15:30. During this whole time, RV Polarstern moved through open water, but was always surrounded by floating ice. Starting at 18:00, a thin low-level cloud layer was present above the ship, which decreased the global radiation significantly. This cloud layer was present until the next day, 19 June, at approx. 12:00. During 19 June, RV Polarstern moved through open water and ice along the western coast of Spitsbergen (Fig. 1). From approx. 12:30 to 15:00 another short cloud-free period led to high global radiation. At 16:00 at approx. 3 km altitude a cloud moved in, decreasing the global radiation once again.
The PNC10–50 and PNC100–800 from 17 June prior to the NPF event were rather stable, with an average value of approx. 30 cm−3. The corresponding coagulation (for 3 nm particles) and condensation sink was and s−1, respectively. Analysis of backward trajectories showed that since midnight of 17 June, air masses were passing over the Arctic Ocean and Greenland Sea. From the beginning of 18 June and onwards, air masses were already passing over the northeastern coastal area of Greenland (Fig. 1). The NPF event occurred when the global radiation reached its maximum at 570 W m−2 and the RH decreased to 85 %. During the event, the PNC10–50 increased to 3200 cm−3. Particle growth was slightly disturbed by a fog episode (this can be seen in PNSD and as RH increase to 100 % in Fig. 2) at around 15:00 and drizzle at 23:00. Nevertheless, the particle growth remained observable until the evening of 19 June. During this time (after a period of 32 h), newly formed particles grew to approx. 50 nm (geometric mean diameter).
The GRs for particles in the size range from 3 to 7 nm were in a range from 3.6 to 4.9 nm h−1. The GR for 1.6 to 3 nm negative ions was 2.9 nm h−1, and 4 to 9 nm positive ions 3.3 nm h−1. The J3− of particles was approx. 0.35 cm−3 s−1. Formation rate for positive (J1.6−) and negative (J4−) ions were 0.05 and 0.06 cm−3 s−1, respectively. If compared to Event 1, it can be seen that despite similar intensity of NPF, particle growth during the second event was approx. 2 times faster, and particles were able to grow to larger diameters (30 nm during event 1 versus 50 nm during Event 2).
3.1.3 NPF 3: 26 June
The third, least intensive NPF event occurred during the second leg of the expedition, 26 June, when RV Polarstern was at the marginal ice zone, around 200 km east of Svalbard, moving towards the north. Areas dominated by open water were passed by the vessel, as well as ice-covered water (Nicolaus, 2018). However, the ice was never very densely packed and the transit of the ship did not require the ice to be broken. Low-level clouds and fog were present during all of 25 to 27 June; on 28 June a short period of cloud-free conditions was observed from around 04:00 to 06:00. There were two short floe stations, one on 25 June from around 17:00 until midnight and the other on 27 June from around midnight to 03:00.
The formation and growth of particles were already observed on both 24 and 25 June during less pronounced NPF events (not shown), when the ship was approx. 100 km south of the Svalbard coast. New particle formation along the eastern coast of Svalbard can be seen in Fig. 1 as an increase in total particle number concentrations, which were measured from 24 to 28 June, along a distance of more than 600 km. The daily average of PNC10–50 and PNC100–800 from 24 June up to the NPF event (26 June) were approx. 600 and 50 cm−3. As a result of NPFs on 24 and 25 June, an interesting pattern emerged in 26 June PNSD (Fig. 2). At the beginning of 26 June (midnight to 03:00), three distinctive modes with geometric mean diameters of 15, 40, and 150 nm can be seen. The smallest mode at 15 nm is a result of the NPF, which occurred on 25 June. These newly formed particles slowly grew in size and, by 08:00 on 26 June, the mode at 40 nm emerged, which was in turn a result of NPF and subsequent particle growth observed on 24 June. Larger size particles (150 nm in diameter) seem to exist independently of the NPF events and were present before, during, and after the NPF on 26 June. However, because we were not able to identify particle growth, the NPF events on 24 and 25 June were excluded from the result and discussion sections.
The event on 26 June started with relatively calm winds (2 m s−1), which gradually increased to 10 m s−1 over a 3 d period (26 to 28 June) with a constant rate of 0.3 m s−1 h−1. The direction of the wind remained stable during the event, with prevailing winds from the south-southwesterly (190 to 200∘) direction and stagnant air masses coming from the marginal ice zone. At the beginning of the event, relative humidity was at around 87 %, and remained below 95 % during the whole 3 d period. Air and water temperature during the event was approx. −1.5 ∘C. During the described 3 d period, water temperature remained the same (with some short episodes of warmer water), while air temperature steadily increased to 0 ∘C. The NPF event occurred with a global radiation being at its maximum (200 W m−2); however, this time solar radiation was at least 2-fold lower than observed during previous cases. This is due to the presence of a low-level cloud layer topped at 2 km during the whole day of 26 June. The corresponding coagulation and condensation sink just before the event was and s−1, respectively.
The GR of 3 to 7 nm particles was in a range from 0.5 to 0.7 nm h−1. The GRs of negative (1.6 to 3 nm) and positive ions (2 to 6 nm) were accordingly 1.2 and 2.2 nm h−1. Despite the noticeable pollution from ship exhaust, particle growth after the NPF event was observed over the period of 3 d (Fig. 2). During this time period, particles grew from several nanometers up to sizes of 50 nm (geometric mean diameter). The formation rate of positive (J2−) and negative (J1.6−) ions were 0.03 cm−3 s−1, and the J3− for particles was approx. 0.08 cm−3 s−1.
3.1.4 NPF 4: 2 July
From midnight of 1 to 4 July, RV Polarstern was moving northwards from 81.64∘ N 32.62∘ E to 82.16∘ N 32.87∘ E. This region was mostly ice-covered with some open leads, through which the vessel could pass without having to break the ice. At this time of the expedition, melt ponds were observed frequently on the ice floes. On 1 July, there was a thick (up to 3 km altitude) low-level cloud layer present until 14:00 associated with some snowfall. After 13:00, the cloud bottom height increased steadily; however, some intermittent fog was still present at sea level. A single fogbow was observed between 18:20 and 19:00. The fog dissolved at midnight on 2 July. Almost throughout the entire day of 2 July, no clouds were present except for optically thin cirrus clouds, allowing for high solar irradiation.
On 2 July, RV Polarstern ventured further into the Arctic ice, more than 300 km from the coasts of Svalbard and Prince George Land (81.51∘ N, 32.97∘ E). The prevailing westerly winds were rather stable during a 3 d period (from 1 to 4 June) at 6 m s−1. The same was true for water temperature, which remained approx. 2 ∘C below zero during the whole event period. The air temperature, on the other hand, varied between −1 and −5 ∘C. The calculated backward air mass trajectories indicated that before the midday of 1 July, air was coming from the direction of Prince George Land. The average PNC10–50 and PNC100–800 during this time was 60 and 70 cm−3, respectively (Fig. 2). From 1 July onwards, air masses arriving at RV Polarstern passed closer and closer to the northeastern coast of Greenland, but did not pass over the land, as was the case for Event 2 (Fig. 1). Effective wet removal of particles by fog could be observed during the afternoon hours of 1 July, leading to extremely low particle number concentrations prior to the NPF event. The PNC10–50 and PNC100–800, respectively, decreased to 40 and 10 cm−3. The resulting coagulation and condensation sink became and s−1, respectively. The NPF event started at 08:00 on 2 July at an ambient RH of approx. 90 % and a maximum global radiation of 500 W m−2. In parallel to RV Polarstern measurements, the formation of new particles was also observed at both Villum Research Station and Zeppelin Observatory, indicating a regional phenomenon.
The particle GR, in a size range from 3 to 7 nm, was 0.9 nm h−1. After 40 h of growth, the geometric mean diameter of the particles reached 30 nm. The GR of negative ions was 1.5 nm h−1 (in a size range from 2 to 3 nm). Once again, it has to be noted that, for ions, the GR in the 1.6 to 3 nm size range was difficult to obtain. Particle formation rate J3− was approx. 0.15 cm−3 s−1. The formation rate of negative ions (J2−) was 0.02 cm−3 s−1. As in the case of Event 1, negative ions seemed to be more prominent than positive ones.
3.2 Particle hygroscopicity during NPF events
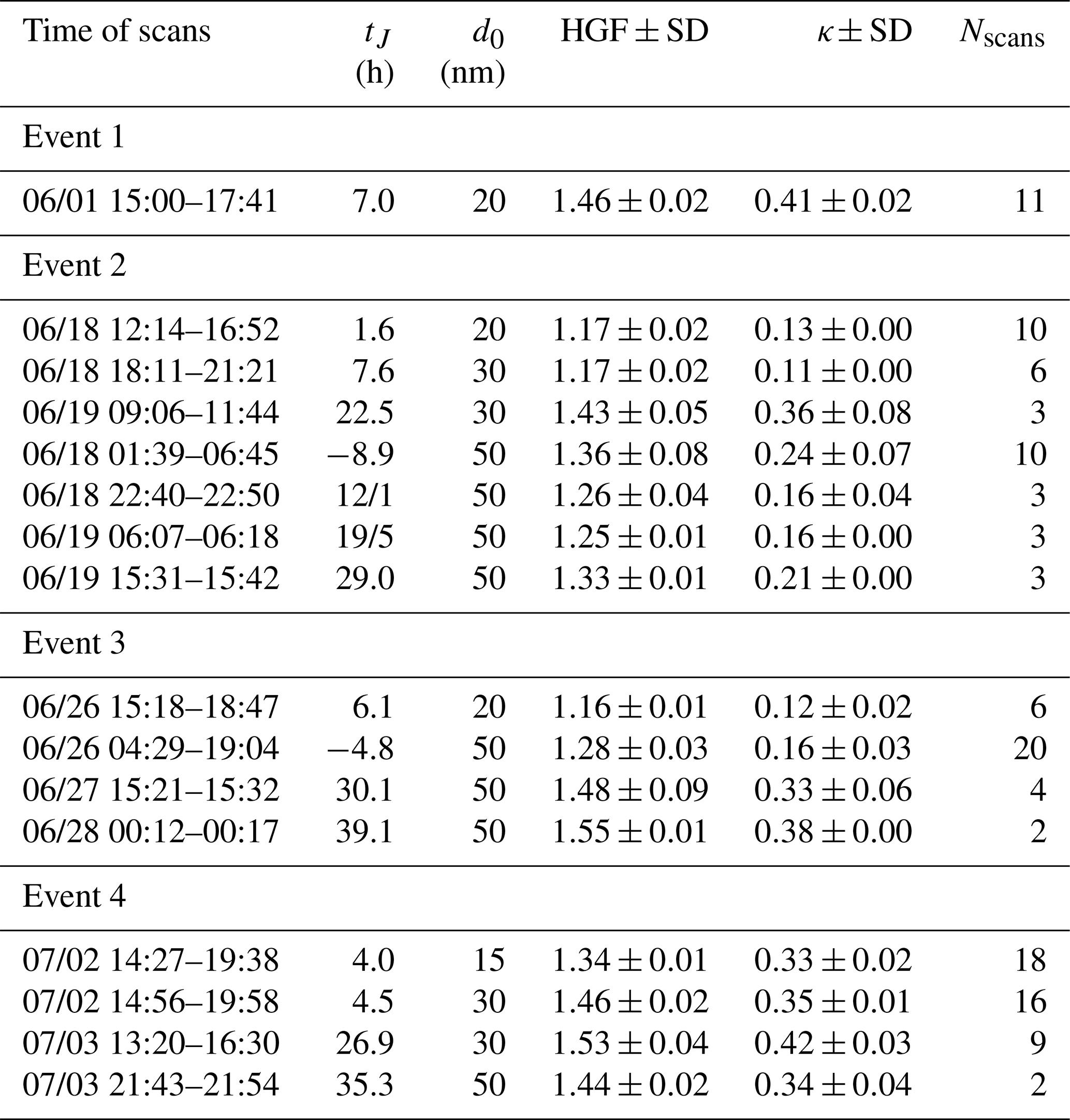
The size segregated HGF and hygroscopicity parameter κ during NPF events is presented in Table 2. Diameters and scan times of dry particles that were selected for HGF measurements are also indicated in Fig. 2. The HGF scans were performed following the growth of freshly formed particles from MPSS PNSD measurements. In most of the instances newly formed particles grew rather slowly and took between 2 and 7 h to grow to diameters of 20–30 nm, when its HGF was measured. The HGF of 30–50 nm particles was measured between 20 and 40 h after the initial NPF event. Despite the size of selected dry particles, the measured HGF distributions were exclusively mono-modal, indicating internal mixture of the aerosol particle. The highest HGF of nucleation mode particles (15–20 nm) was observed during Event 1 and Event 4. The HGF of 20 nm particles during Event 1 was measured 7 h after the beginning of the NPF and was 1.46±0.02 (± standard deviation, SD; ). At the time of Event 4, HGF of the 15 nm particles was 1.34±0.01 (). The lowest HGF of 20 nm particles was observed throughout both Event 2 and Event 3 and was 1.17 () and 1.16 (), respectively. Hygroscopicity of slightly grown Aitken mode particle (30 to 50 nm) varied from 1.17±0.02 () to 1.55±0.01 (). In general, the longer the particles aged, the more hygroscopic they became. For example, 8 h after the new particles were formed during Event 2, the HGF of 20 nm particles was 1.17±0.02. After another 15 h, these particles grew to sizes of approx. 30 nm, which HGF increased to 1.43±0.05 (). Interestingly, the HGF of 50 nm particles was somewhat lower, 1.25±0.01 (). Nevertheless, it followed the same pattern and with time increased to the values recorded before the NPF event.
Table 2Hygroscopic growth factor (at 90 % RH) and hygroscopicity parameter κ during NPF events. Here: time of scans – a time window during which hygroscopicity distributions were measured; tJ – approx. time between the observed formation rate maximum and the measurements of HGF. In other words, tJ indicates how long before/after the NPF events the HGF was measured. For example, if tJ=7, the HGF was measured 7 h after the maximum in J. Negative tJ indicates the measurements of HGF prior NPF event; d0 – selected diameter of dry particles; Nscans – number of scans; SD – standard deviation. Date format: mm/dd.
3.3 Measured CCN concentrations during NPF events
Concentrations of CCN (NCCN) measured during the four NPF events can be seen in Fig. 3. An increase in NCCN during these events can be seen across all supersaturations. To determine the increase, measured data were fitted, visible as lines in Fig. 3. Data included in the fitting were taken from times on when formation rates of particles were noticeably increased (10 % of the maximum signal) and go up to the time when the NPF event was interrupted by a change in air mass or fog formation. These periods span 10, 39.5, 44.5, and 29 h for NPF events 1 to 4, respectively. Independent of the duration of the event, the observed increases in NCCN during these periods were mostly roughly a factor of 2 for supersaturations from 0.1 % to 0.5 % and roughly a factor of 3 to 6 at 1 %. This larger increase at the highest supersaturation is related to the fact that the number concentrations of smaller particles, which are only activated at higher supersaturations, increased the strongest. During NPF event 2, the increase was somewhat lower, mostly below a factor of 2. These measurements clearly show that during NPF events not only new particles are generated, but also that particulate mass is gained on particles of all sizes, increasing their size and hence their ability to act as CCN at a given supersaturation. A similar observation was made in Antarctica (Herenz et al., 2019), where NPF events with increases in total particle number concentrations from a few hundred to thousands of particles per cm3 were also accompanied by an increase in NCCN of at least a factor of 2 at all examined supersaturations. Burkart et al. (2017b) came to similar conclusion. This is in agreement with modeling results by Merikanto et al. (2009), where CCN in Arctic regions were found to almost exclusively originate from NPF.
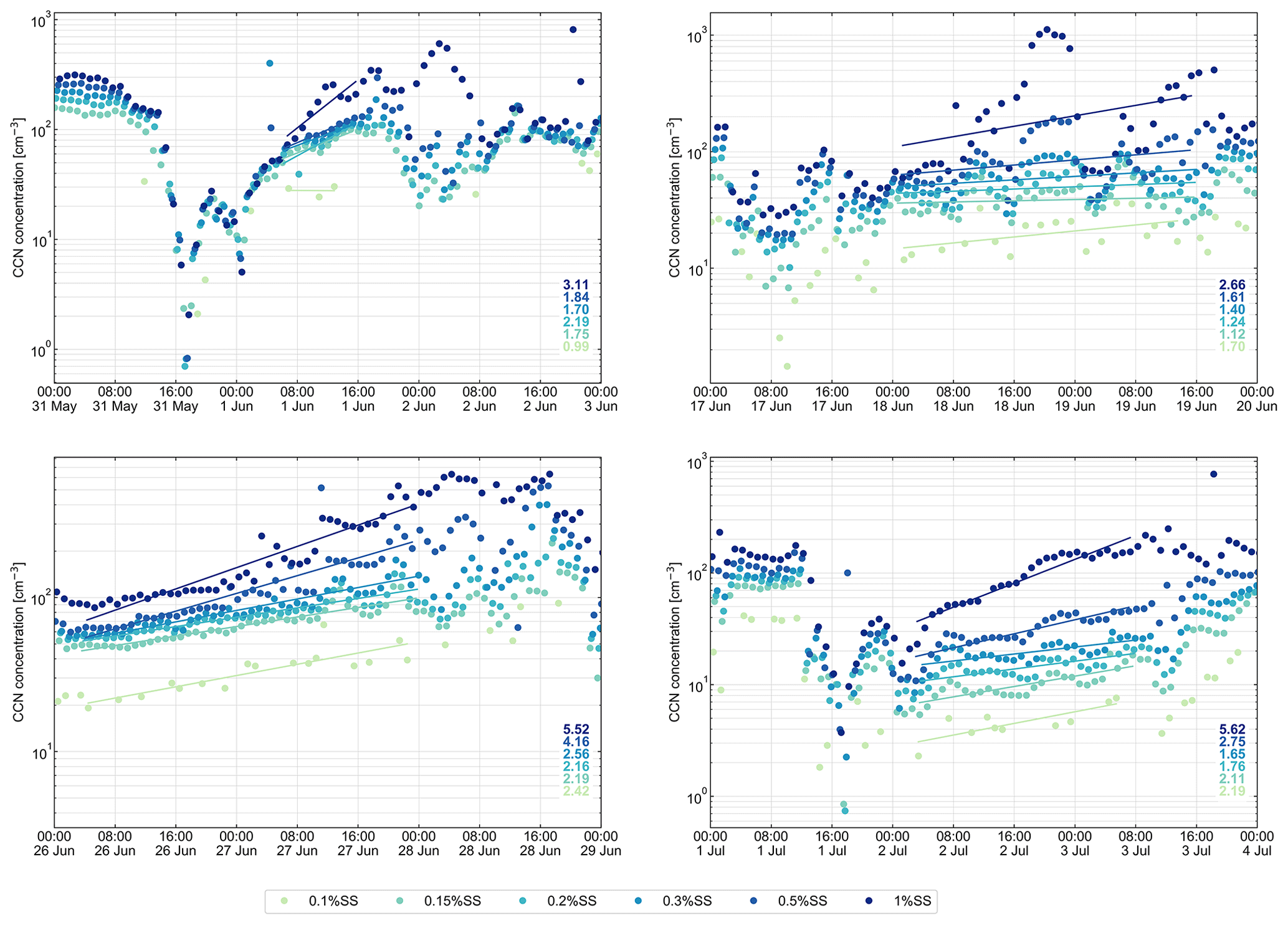
Figure 3The CCN number concentration measured during NPF events (1 to 4). The lines and corresponding values show the increase in CCN concentrations (prior to NPF versus particles that have grown to the Aitken mode).
3.4 Chemical composition of size-resolved particles
The size-resolved absolute atmospheric concentrations of ammonium, MSA, nss-sulfate, and sea salt (sodium) for the selected periods versus campaign average are shown in Fig. 4. On average, the highest concentrations of nss-sulfate (81 and 70 ng m−3), MSA (18 and 10 ng m−3), and ammonium (16 and 8.7 ng m−3) were found in a size range of 0.14–0.42 µm (impactor stage 2) and 0.42–1.2 µm (impactor stage 3), respectively. While the concentrations of nss-sulfate and ammonium on the impactor samples from 25 to 28 June were comparable to the average values, the impactor samples from 29 May to 1 June stood out with much higher values, especially in the accumulation mode (nss-sulfate: 251 and 295 ng m−3 and ammonium: 34 and 17 ng m−3 in size range of 0.14–0.42 and 0.42–1.2 µm, respectively). Also for smaller particles (size range of 0.05–0.14 µm, stage 1), nss-sulfate was found at a much higher concentration (35 ng m−3) than the average (8.3 ng m−3).

Figure 4Size-resolved atmospheric concentrations for ammonium, MSA, nss-sulfate, sodium, and OM for two sampling periods and the whole campaign average. Stages 1, 2, 3, 4, and 5 correspond to aerodynamic particle diameter ranges of 0.05–0.14, 0.14–0.42, 0.42–1.2, 1.2–3.5, and 3.5–10 µm, respectively.
It must also be noted that no action was taken (e.g., sampling interrupt-dependent on the specific wind sector) to reduce ship contamination for the size-segregated aerosol particle measurements. Thus, the contamination from the ship exhaust cannot be ruled out completely. However, the high concentrations of biogenic compounds like MSA and the presence of sodium on the aerosol particles suggested a strong marine influence on the particle composition.
The highest organic matter (OM) mass concentrations were found at stage 2 (106 ng m−3) and the lowest at stage 5 (39 ng m−3). OM mass concentration for the period from 25 to 28 June strongly exceeded the average concentration, especially in the accumulation mode (218 and 147 ng m−3 for stages 2 and 3, respectively). For a time period from 29 May to 1 June the OM mass concentration ranged close to the average values.
Sodium was mainly found at Berner stages 3–5. The sodium values for the sampling period from 25 to 28 June (Berner stage 4: 49 ng m−3) were quite similar to the average values, while the impactor samples from 29 May to 1 June showed much higher atmospheric concentrations (Berner stage 4: 386 ng m−3). This agrees well with previous studies, which show that atmospheric sea salt is mostly present in super-micron particles, while OM contributes strongly to the submicron particle composition (e.g., Müller et al., 2010). Previous works also suggest that OM is strongly enriched during the bubble bursting process (compared to sea salt), and therefore OM and sea salt are not transferred to the same extent from seawater to the aerosol particles (Keene et al., 2007; Quinn et al., 2015; Van Pinxteren et al., 2017). It is possible that increased sodium and OM, observed during NPF 1, is a result of sea spray; however, due to the low sampling time resolution of the Berner cascade impactor, we do not allow ourselves such a conclusion. Moreover, please note that the increased values of sodium during this time period may be related to the ship's proximity to open water (RV Polarstern reached the marginal ice zone only on 31 May), while the increase in OM could have happened later (e.g., 1 June) but been included in the same sample. In chemical sample analysis, we did not find any positive correlation between OM∕OC and sodium concerning the different aerosol size classes. A more detailed chemical characterization of the aerosol particles during the PS 106 cruise will be addressed in a separate publication.
4.1 General overview
Although NPF events in the high Arctic were reported by several studies, there are no observations using the same or equivalent measurement equipment as in this study which are able to observe the dynamic changes in the smallest particles (formation and growth of >1.6 nm clusters). Because of this, we have also calculated the rate at which new particles appear at larger diameter (10 nm, J10−). The values of so-called apparent nucleation rates are more frequently reported in the literature. For example, in several studies from the Svalbard region, GRs for 5 to 25 nm particles were reported to be from 0.1 to 0.6 nm h−1, but in general ≤1.0 nm h−1 (Ström et al., 2009; Giamarelou et al., 2016; Heintzenberg et al., 2017). The corresponding J10− values were in a range from 0.1 to 1.4 cm−3 s−1. Nieminen et al. (2018), on the other hand, reviewed NPF events based on long-term measurements and reported GRs for the Arctic region to be 1.1–1.2 nm h−1 (for the June–August time period). The reported formation rates were somewhat lower, 0.008–0.032 cm−3 s−1. In the case of this study, the GRs for 5–25 nm and J10− values varied correspondingly from 0.7 to 5.4 nm h−1 and from 0.04 to 0.4 cm−3 s−1, respectively. The GR of 5–25 nm size particles in this study was on average 0.9 nm h−1. The GR of 5–25 nm particles on 18 June, however, exceeds other NPF events, with the GR being significantly higher, 5.4 nm h−1. During the same event, the J10− was also higher, 0.4 cm−3 s−1. Nevertheless, on average, the observed GR and J10− values were on the same order as reported in other studies from the Arctic region (e.g., Asmi et al., 2016; Nieminen et al., 2018). Some studies for similar environmental conditions also exist. Jokinen et al. (2018) provided a comprehensive study on the particle formation in coastal Antarctica. The growth and formation rates for 3 nm particles were found to be between 0.3 and 1.3 nm h−1 and between 0.03 and 0.14 cm−3 s−1. It was concluded that ion-induced nucleation of sulfuric acid and ammonia is a major source of secondary aerosol particles in the pristine Antarctic environment. Kyrö et al. (2013) reported formation rates of negative clusters (J1.6−, 0.01 to 0.4 cm−3 s−1) measured at the Finnish Antarctic Research Station, Aboa, in Dronning Maud Land. In addition, apparent nucleation rates of 10 nm particles at Aboa ranged from 0.003 to 0.3 cm−3 s−1. In yet another Antarctic study, Weller et al. (2015) reported the average growth and formation rates (in a size range from 3 to 25 nm) to be 0.9 nm h−1 and 0.06 cm−3 s−1, respectively. These authors also concluded that due to an insufficient concentration of low-volatility organic compounds, the particle growth was restricted to the nucleation mode. All of these studies showed some resemblance to the results observed in our study.
The question is which mechanism drives the nucleation and which are the condensable vapors responsible for the observed particle growth in the pristine high-altitude environments. Most recent studies indicate the importance of semi-volatile organics (Willis et al., 2016; Burkart et al., 2017a). The subsequent growth of newly formed particles was associated with organic precursors from meltwater ponds (Kyrö et al., 2013), while Weller et al. (2015) speculated that low-volatility organic compounds of marine origin govern the growth of newly formed particles in Antarctica. It was also shown that in a clean environment, sufficiently high sulfuric acid concentrations (107 molecules cm−3) can fully explain particle growth (Jokinen et al., 2018). The GRs observed in our study are somewhat similar to those from similar environments; however, they remain difficult to compare because of case-to-case variability.
Insights on the chemical composition of nucleation mode particles and the climatic relevance of NPF can be drawn from the hygroscopicity measurements either at water vapor sub-saturation (measurements of HGF) or supersaturation (measurements of the number of CCN). While κ is a parameter that is independent of experimental conditions, HGF still depends on the dry particle size and RH for which it was determined. Still, for the Arctic more data are available for HGF, so that we will use this parameter for comparison with the literature in the following. Zhou et al. (2001) measured the HGF during the Arctic Ocean Expedition 1996. The HGF of nucleation mode particles (just after a NPF event, dry diameters of 15) was 1.38. The HGF of 35 nm particles was 1.56. After some time, the particles that grew to sizes of 50 nm were found to be less hygroscopic (HGF of 1.05). It was suggested that these particles were produced at the sea surface and not in the free troposphere. However, the authors could not derive the composition of those nucleation mode particles. Park et al. (2014) reported HGF values of 50 nm particles during enhanced number concentration of the Aitken mode to be 1.46. Sulfate and biogenic volatile organic species were identified to contribute to the Aitken mode particle formation. Compared to our measured HGF of 15 and 20 nm particles, we can see that during events 1 and 4 values agree reasonably well with previously measured particle hygroscopicity. The HGF values of nucleation mode particles during events 2 and 3, on the other hand, are significantly lower. The hygroscopicity of the Aitken mode particles, measured during Event 4 was almost identical to that noted by Park et al. (2014). On the other instances, for example Event 2, the HGF of the Aitken mode particles was lower (1.33 versus 1.46) than previously reported values. It clearly indicates that different condensable vapors were driving the growth of newly formed particles into sizes of 30 to 50 nm.
Based on particle hygroscopicity, formation and growth rates of positive/negative ions and neutral clusters, and offline chemical analysis, our observed NPF events represent two different cases: (1) more hygroscopic particle formation favored by negative ions, events 1 and 4 (1 June and 2 July, respectively); and (2) relatively low hygroscopicity particle formation during events 2 and 3 (18 and 26 June, respectively), suggesting the presence of condensable organics in particle growth. Further, we would like to discuss event-specific particle growth/formation rates and hygroscopicity with respect to formation mechanism and condensable vapors.
4.2 Indirect evidence of the composition of newly formed particles
4.2.1 NPF 1 and 4
Occurrences of nucleation mode particles in the summer Arctic were associated with intrusion from higher altitudes and new particle production in upper layers of the marine boundary layer (MBL, e.g., Wiedensohler et al., 1996). It is possible that the NPF precursors can be brought from either open ocean or anthropogenic continental sources by air masses. Coupled with low condensation and a coagulation sink and with plentiful global radiation, it creates favorable conditions for new particles to be formed. However, in all of our observed NPF cases the particle formation started from nucleation of 1–2 nm clusters, suggesting that the NPF took place right at the sea level, rather than in upper layers of the MBL. In this study, unfortunately neither the high-resolution online chemical composition of aerosol particles nor relevant gases (e.g., SO2, O3) were directly measured on-board RV Polarstern. To gain some insights into the chemical composition of newly formed and slightly grown particles, as well as precursor gases, we used measured particle physico-chemical properties (e.g., hygroscopicity, growth rate) as well as satellite imagery.

Figure 5Sea-ice concentration (white – 100 %, dark blue – 1 %; from NASA Worldview; Maslanik and Stroeve, 1999) and chlorophyll-a surface concentration (taken from http://marine.copernicus.eu, last access: 29 April 2019) during NPF events (a, b, d, e). On the right – the ice alga and diatom Melosira arctica (c, f) observed from the ship deck during NPF Event 1. RV Polarstern track and location during the NPF event are indicated by the red line and black circle, respectively. Orange lines mark the 72 h backward air mass trajectory at 200 m a.s.l.
It is known that Arctic phytoplankton contributes to the production of dimethyl sulfide (DMS), which is the main source of biogenic sulfur (Stefels et al., 2007; Levasseur, 2013, and reference therein). Released into the atmosphere, DMS can be involved in NPF through oxidation and creation of sulfuric acid (H2SO4) (Kulmala et al., 2001; Park et al., 2017). In a study by Nguyen et al. (2016), NPF and particle growth at Station Nord, Greenland, were found to be linked to O3, most likely through creation of a hydroxyl (OH) radical and oxidation of sulfur dioxide (SO2) and volatile organic compounds. The satellite-derived chlorophyll-a mass concentration in surface seawater, as an indicator of phytoplankton biomass (Becagli et al., 2016), can be seen in Fig. 5 (left). It is evident that during all NPF events RV Polarstern was in close proximity to an area of increased biological activity in the Arctic Ocean. During Event 1, we also observed large ice-attached mats of the Melosira arctica (Fig. 5), which suggests the presence of DMS (Levasseur, 2013). It can also be seen from Fig. 5 that sea-ice retreat is somewhat linked to increase in chlorophyll-a mass concentration in surface seawater. For example, on 26 June some ice coverage in the southern part of Prince George Land can still be visible, while on 2 July it is all gone, replaced by biological activity. This is most likely because the ice edge provides increased stability from the meltwater, which facilitates the seasonal production of phytoplankton (Conover and Huntley, 1991).
An interesting feature was observed with respect to formation rates and the number size distributions of positive and negative ions during events 1 and 4 (Fig. 2; see also Fig. S1 in the Supplement). Firstly, it seems that the formation of ions occurred before that of neutral particles. The peak ion formation rate was observed approx. half an hour prior to the formation of neutral particles. Although not in the polar regions, similar behavior was noticed in several other studies, suggesting the importance of ions in NPF events (Manninen et al., 2010; Jayaratne et al., 2016). The role of ions in NPF was investigated in both laboratory and field studies (e.g., Wagner et al., 2017; Jokinen et al., 2018). It was shown that ions enhance the nucleation and condensation of the vapor molecules by stabilizing the molecular clusters and/or are involved in charged cluster neutralization via recombination with oppositely charged clusters. The second interesting feature that was observed only during events 1 and 4 was the absence of the smallest (<1.6 nm) positive ions. Negatively charged ions seemed to be involved in the particle formation more favorably than the positive ones. This was also observed in previous studies (e.g., Hirsikko et al., 2007; Asmi et al., 2010; Jokinen et al., 2018) and was associated with sulfuric-acid nucleation. Although H2SO4 concentrations were not determined directly, the presence of negative clusters suggests that in the case of events 1 and 4, sulfuric acid was somewhat involved in observed NPF, too.
From previous studies, it was shown that H2SO4 concentration of 107 molecules cm−3 are sufficient to explain the observed new particle GRs in coastal Antarctica (Jokinen et al., 2018). In our case, the hypothesis was tested that H2SO4 was involved in NPF events 1 and 4 by using the look-up tables from an ion-mediated nucleation model for the H2SO4–H2O binary system (Yu, 2010). At a given temperature (TEvent1=268.8 K; TEvent4=268.4 K), relative humidity (RHEvent1=96.3 %; RHEvent4=96.23 %), and surface area concentration of pre-existing particles (SEvent1=2.9 µm2 cm−3; SEvent4=0.5 µm2 cm−3), and assumed ionization rate (Q=2 ion-pairs cm−3 s−1) – the corresponding H2SO4 concentration was calculated to be approx. 106 molecules cm−3. If compared to a study from Antarctica (Jokinen et al., 2018) or laboratory studies by Dunne et al. (2016) from the CERN CLOUD (Cosmics Leaving Outdoor Droplets) chamber, our calculated H2SO4 concentration is 10 to 30 times lower than that from previous studies. On the other hand, the results of this study are in agreement with a study by Ehn et al. (2007), who studied the relationship between particle hygroscopicity and sulfuric acid concentration in boreal forest. The authors reported that the concentration of H2SO4, corresponding to 15 and 20 nm particle HGFs of 1.34 and 1.46, was in a range of 107 molecules cm−3. Nevertheless, there were numerous instances when the same hygroscopic growth was also observed at lower H2SO4 concentrations (<107). Moreover, both in the Arctic and Antarctica, H2SO4 concentrations of 106 molecules cm−3 were associated with NPF by Croft et al. (2016) and Kyrö et al. (2013), respectively.
The fraction of the particle growth that can be explained by sulfuric acid can be found from the comparison of observed versus predicted particle growths. From Vakkari et al. (2015), the particle growth due to sulfuric acid can be found from the relation
where coefficient A is equal to 1.58×107, 1.99×107, and 2.28×107 for particle growth in the size range from 1.5 to 3, 3 to 7, and 7 to 20 nm, respectively. Using our estimated concentration of H2SO4 we found that growth (in a size range from 1.5 to 3 nm) due to sulfuric acid alone accounts only from 4 % to 10 % of the observed growth during Event 4 and 1, respectively. The contribution to particle growth in a size range from 3 to 20 nm gets even lower, 4 %–5 %. Our values are somewhat comparable to those observed in Antarctica (Kyrö et al., 2013). It suggests that besides sulfuric acid, other vapors have to be present to reach the observed particle growth. From offline chemical analysis, we see that during Event 1, ammonium and nss-sulfate in accumulation and the Aitken mode particles were somewhat higher than campaign average (Fig. 4). Some studies (e.g., Croft et al., 2016; Köllner et al., 2017) identified that certain nitrogen-containing species such as ammonia and amines are linked to particle growth in the Arctic region. To test this, we investigate the formation rate of critical clusters using a parametrization of the ternary H2SO4–NH3–H2O system, presented by Napari et al. (2002). That is, we adjust the concentrations of H2SO4 and NH3 until we get the formation rate close to that of observed value. The estimated concentrations of H2SO4 and NH3 varied from 1×104 to 5×106 cm−3 and 0.1 to 100 ppt, respectively (see Supplement). According to Wentworth et al. (2016), such concentrations of NH3 can be indeed found in the Arctic region. There is evidence that H2SO4–NH3–H2O clusters are only partly neutralized under atmospheric conditions (e.g., Kurtén et al., 2007; Schobesberger et al., 2015). On the other hand, Asmi et al. (2010) reported that Aitken mode particles are somewhat more neutralized. Now, if we assume that the newly formed particles were partly neutralized by ammonia (as suggested by Giamarelou et al., 2016), we would expect particle hygroscopicity to be close to that of ammoniated sulfates. However, our observed HGFs of 20 and 30 nm particles during both events were somewhat lower (e.g., 1.46 versus 1.64, Asmi et al., 2010). Similar hygroscopicity of ultrafine particles (HGF = 1.38 for 15 nm particles) in the Arctic was observed by Zhou et al. (2001). However, the authors excluded the water–sulfuric acid nucleation as a source of such particles because <50 nm particles did not appear to be composed of either sulfuric acid or ammonium sulfate. Kim et al. (2016) measured the hygroscopicity of nanoparticles produced from homogeneous nucleation in the CLOUD experiment. If compared to CLOUD experiment results, the measured hygroscopicity of 20 nm particles during Event 1 was closest to the results of experiment, during which sulfuric acid and dimethylamine (DMA) concentrations were 7.6×106 molecules cm−3 and 23.8 ppt, respectively. With that being said, experiments with sulfuric acid (15.1×106 molecules cm−3) and organics produced from α-pinene ozonolysis (420 ppt) resulted in 15 nm particles with HGF = 1.33, which is identical to those observed during Event 4.
To conclude, one can only assume that during events 1 and 4, the NPF was initiated by sulfuric acid. Although the involvement of ammonia in new particle formation is possible, it cannot be proven by this work. The organics of marine origin could have been involved in particle growth to some extent. However, low (compared to the campaign average) organic matter concentrations, observed by offline chemical analysis, contradict the aforesaid conclusion. The hypothesis that NPF is driven by sulfuric acid can be supported by the results of neutral cluster and ion number size distribution and hygroscopicity measurements of nucleation mode particles.
4.2.2 NPF 2 and 3
Following the same line of thought as in the previous section, we investigate to what extent sulfuric acid may have been involved in the NPF and growth during events 2 and 3. From satellite imagery of chlorophyll a (Fig. 5), we can see that RV Polarstern remained in close proximity to somewhat decreased but still present biological activity in the Arctic Ocean. In addition to that, some depletion in sea-ice cover close to Greenland as well as an increase in biological activity south of Svalbard were also observed (Fig. 5). Thus, it is safe to say that air masses arriving at RV Polarstern were passing over regions which are a potential source of both DMS and organics of marine origin. Assuming a H2SO4–H2O binary system, the H2SO4 concentrations corresponding to formation rates of those observed for events 2 and 3 were 15 % to 50 % higher, compared to events 1 and 4. This is mainly because during events 2 and 3 both the condensation sink and temperature were higher too. Only between 1 % and 3 % of observed particle growth during Event 2 can be explained by H2SO4 alone. This fraction is somewhat higher on Event 3 (6 %–9 %). At the initial states of nucleation mode particle growth, particle hygroscopicity on both events was rather low (HGF between 1.16 and 1.18). Such low hygroscopic particle growth, coupled with a rather rapid increase in size (Event 2, from 3 to 20 nm, GR = 4.2 nm h−1), suggests that in these events the organics must have played a much bigger role during initial particle growth than during events 1 and 4. The observed particle hygroscopicity agrees rather well with less-hygroscopic particle values reported by Zhou et al. (2001). During Event 2, particle hygroscopicity did not change much when particles from nucleation mode grew into the Aitken mode, with HGF remaining between 1.16 and 1.18. Only after approx. 30 h after the new particles were created did they grow to a diameter of 50 nm with slightly increased hygroscopicity, HGF = 1.33. Contrarily, in Event 3 the HGF of 50 nm particles (after approx. 40 h after the nucleation) reached a value of 1.55. It is expected that with time newly formed particle hygroscopicity will increase due to the process known as aging. From smog chamber experiments, Tritscher et al. (2011) showed that organic aerosol photochemical aging increases the particle hygroscopicity mainly due to O3 induced condensation of organic molecules onto particles. The rate at which particle hygroscopicity parameter κ increases can be calculated from the change in κ over the time period (Δκ∕Δt). We found that during events 2 and 3 κ changed with the rates of 0.0027 and 0.0067 h−1, respectively. These values are surprisingly close to those observed by Tritscher et al. (2011), further supporting the evidence of organics participating in our observed particle growth.
Using our calculated formation rates (0.06 and 0.026 cm−3 s−1 during events 2 and 3, respectively) and sulfuric acid values from previous studies (5×106 molecules cm−3, Croft et al., 2016) as a guideline, we calculate the extremely low-volatility organic compound concentration from the parameterization of particle formation rate as a function of sulfuric acid and EL-VOC concentration (Riccobono et al., 2014):
The resulting EL-VOC concentration for Event 2 was found to be approx. 8.0×106 molecules cm−3. This is 40 times higher than what is expected from monoterpene air–sea flux in the Arctic Ocean (Croft et al., 2016). On the other hand, during Event 3, the estimated concentration of EL-VOC was in pair with results published by the same authors. The question is where the EL-VOC comes from. Kyrö et al. (2013) showed that NPF can be a result of precursor vapor emission from meltwater ponds. In Fig. 5, we can see that air masses during Event 2 are arriving from the coast of Greenland, with a pronounced sea-ice index change, indicating ice retreat. Moreover, measurements of PNSD at Villum Research Station also indicated the occurrence of NPF. However, it remains unclear whether the ice and biological activity development at the coast of Greenland could have produced the organic vapors that participated in NPF observed at RV Polarstern. Yet another source of condensable organic vapor could be the aged phytoplankton blooms, presented as irregularities in chlorophyll-a spatial distribution, at the marginal ice zone, close to the research vessel.
Atmospheric particulate methanesulfonic acid (MSA) and non-sea salt sulfate (nss-sulfate) are considered to be oxidation end products of DMS, which is released as a gas during biogenic processes and indicates the formation of secondary aerosol with biogenic origin (Leck et al., 2002; Miyazaki et al., 2010). MSA was shown to be involved in nucleation mode particle growth in the Arctic by Willis et al. (2016). The authors found that MSA and condensable organic species, originating from marine-derived biogenic volatile organic compounds, drive particle growth in a shallow marine inversion layer. Organic matter in Arctic submicron particles was found to be of both continental and biogenic marine origins (Kerminen et al., 1997; Chang et al., 2011). Orellana et al. (2011) showed that submicron OM can be composed of phytoplankton exudates in form of marine hydrogels. If we look at offline chemical analysis of aerosol samples, OM was found on all impactor stages, especially on the submicron particles between 0.14 and 1.2 µm. While submicron particles of the impactor samples for Episode 1 were mainly dominated by ammonium and nss-sulfate (see Fig. 4), higher concentrations of OM (together with MSA) were found for the sampling period between 25 and 28 June. These results corresponded to the observed differences between particle hygroscopicity during events 1 and 3.
To summarize, the rapid particle growth (Event 2) and the low but steadily increasing hygroscopicity (events 2 and 3) suggest that organics must have been involved in both NPF and subsequent particle growth. Although our observed results agree with previously made conclusions that particle growth in the Arctic is largely via organic condensation (Burkart et al., 2017a), due to a lack of measurements, we cannot specify which organic species may/or may not have been involved in these processes. We also cannot exclude either the role of iodine (Allan et al., 2015) in the initial steps of NPF or other pathways for initial particle growth (e.g., aminium salts; Smith et al., 2010). In the future, measurements of the chemical composition of naturally charged air ions and ion clusters and low-volatility aerosol precursor gases would greatly improve our understanding of NPF processes and particle growth in the Arctic.
4.3 Implication for CCN abundance
In the last section of this work, the climatic relevance of the newly formed particles in the Arctic is discussed. In several studies (e.g., Allan et al., 2015; Willis et al., 2016; Burkart et al., 2017b) it was reported that nucleated particles in the Arctic atmosphere rarely grow beyond the Aitken mode. It is the result of low organic vapor/precursor gas concentrations involved in NPF and subsequent growth, as well as particle lifetime (particles being scavenged by fog or precipitation, Karl et al., 2012). These findings are also comparable to those from Antarctica. Weller et al. (2015) reported that particle growth is governed by the deficit or availability of low-volatility organic compounds of marine origin and drew the conclusion that particles do not grow to a diameter range relevant for action as CCN. On the other hand, some studies both from Arctic and Antarctica proved that particles do not have to grow beyond 50–60 nm in diameter to be able to act as CCN (Kyrö et al., 2013; Croft et al., 2016; Leaitch et al., 2016). This is because in the pristine Arctic environment the absence of larger particles may lower water uptake, which will increase supersaturation, enabling cloud water to condense on smaller particles (Leaitch et al., 2016).
Table 3Input parameters and the results from parcel model (Rothenberg and Wang, 2016). Here: P – pressure (Pascal), T – temperature (Kelvin), RH – relative humidity (%), GMD – geometric mean diameter of two modes fitted to PNSD (in nanometers); N – number concentration of particles in the mode (in particles per cubic centimeter), κ – hygroscopicity parameter kappa (derived for particle sizes indicated in bracket), σ – the shape parameter (standard deviation of the log of the distribution), NCCN,0.1, and NCCN,3.2 – the number concentration of CCN at two different vertical wind velocities, 0.1 and 3.2 m s−1. Note: κ for specific GMDs was adopted from the nearest value of measured 15, 20, 30, 50, and 150 nm particle hygroscopicity. For example, hygroscopicity of 20 nm particles was used as an input value for the GMD of 16 nm mode particles. Date format: yyyy-mm-dd.
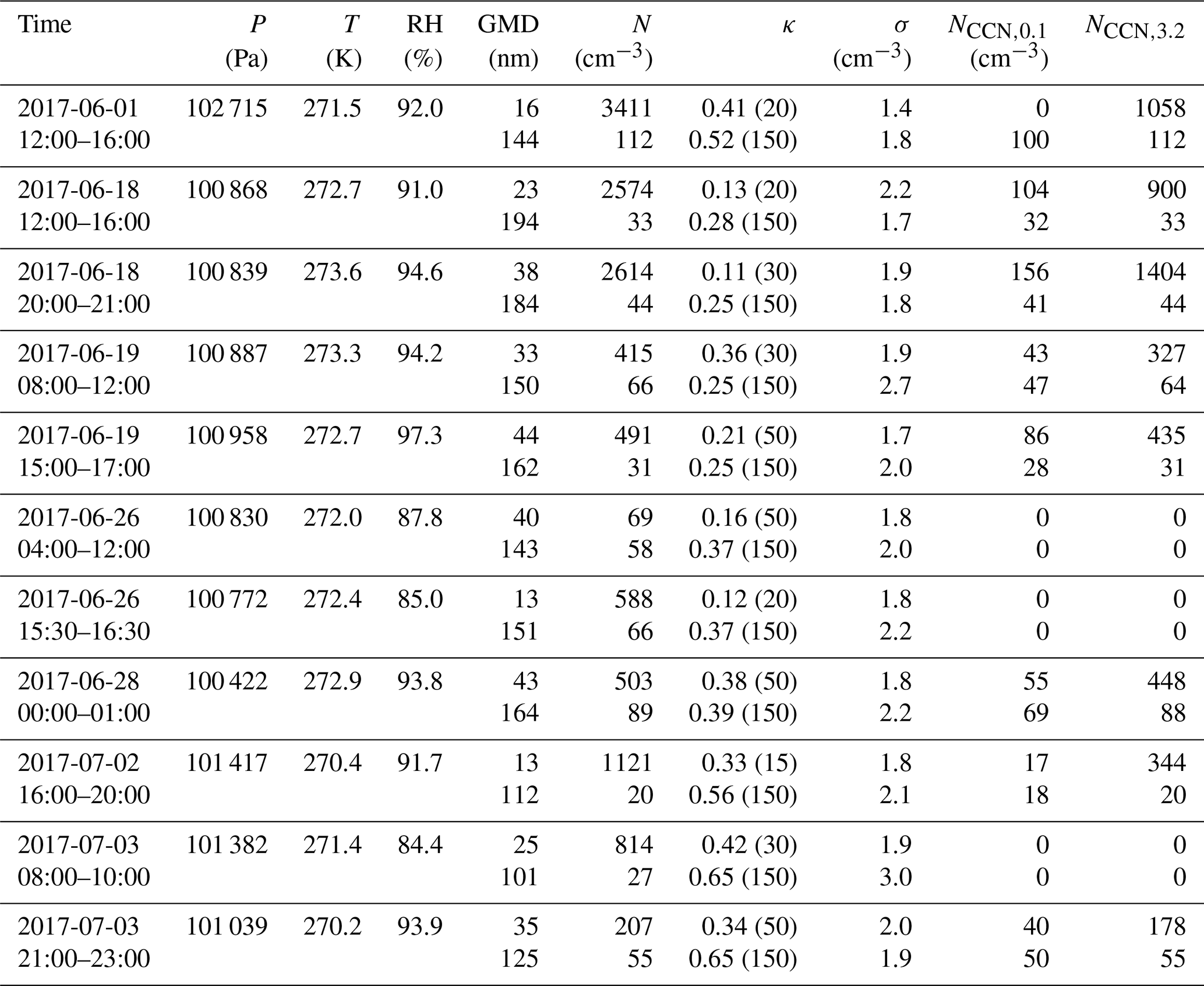
To examine to which degree NPF may influence the CCN nuclei budget in the Arctic, we used an adiabatic non-entraining cloud parcel model (described in Sect. 2.4). All the initial parameters and simulation results can be found in Table 3. The change in CCN number was calculated for two different updraft wind velocities, 0.1 and 3.2 m s−1, representing the 75th percentile and maximum value, respectively. The measurements of vertical wind velocity was performed during the ice-drift station, as described by Egerer et al. (2019). We define the CCN number concentration (NCCN) increase due to particles created in the nucleation process as
where ) is the number concentration of CCN resulting from the particles created in NPF event (calculated from bi-modal PNSD using parcel model; see Table 3 for simulation parameters) and NCCN,bp is the CCN number concentration resulting entirely from accumulation mode particles present during the NPF event (the newly formed particle mode is suppressed in parcel simulation). For a more detailed discussion about the CCN increase calculation, please refer to the Supplement. It can be seen that for most of the cases (when RH >90 %), the CCN number concentrations increased by a factor of 2 to 5 (at upward wind velocities of 0.1 m s−1) and 4 to 32 (at upward wind velocities of 3.2 m s−1). Although the activated number fraction in a size range from 15 to 20 nm was rather low (1.5 %–4 %), the high number of nucleation mode particles resulted in a noticeable increase in total CCN. The CCN fraction was higher (30 %–50 %) when 3.2 m s−1 updraft wind speed was assumed. For the Aitken mode particles, CCN fraction was approx. 12 % and 80 % for updraft wind speeds of 0.1 and 3.2 m s−1, respectively. In some cases, the particles did not activate to CCN. This is because activation supersaturation was not reached during the parcel updraft. The maximum supersaturation achieved with an updraft velocity of 0.1 m s−1 was 0.17 %. The updraft velocity of 3.2 m s−1 would represent, although rare, however, a not unlikely situation when supersaturations of 0.9 % can be reached. It can be anticipated that an even higher fraction of CCN may result from nucleation mode particles when higher supersaturation values are reached. Measurements of CCN number concentration on-board RV Polarstern corroborate the results obtained by our modeling efforts, which all are in good agreement with previous works. For example, Croft et al. (2016) reported maximum supersaturation in the Arctic region of 0.15 %–0.25 % for the updraft speed of 0.1 m s−1. From a comprehensive study on the ultrafine particle effects on liquid clouds in the clean summertime Arctic, Leaitch et al. (2016) determined the supersaturation for low- and high-altitude clouds to be approx. 0.3 % and 0.6 %, respectively. In the Arctic environment with the lack of aerosol particles upon which clouds may form, even a small increase in aerosol loading can lead to cloud formation and thus influence the ice-covered Arctic surface (Mauritsen et al., 2011). From our results, we conclude that NPF in the Arctic can play a significant role in determining the future changes in this pristine and remote environment.
Aerosol particle physico-chemical properties were determined in the summer Arctic on-board research vessel (RV) Polarstern from 26 May to 16 July 2017 as a part of the PASCAL/SiPCA campaign. Here, regional NPF events are analyzed and put into prospective of producing the CCN. From the measurements of neutral cluster and air ion number size distributions, it can be concluded that new particles were formed within the marine boundary layer and not mixed down from aloft. Therefore, the majority of particles in a size range up to 50 nm in diameter can be related to secondary aerosol production rather than primary emissions. Two different types of NPF were distinguished: (a) NPF favored by negative ions, and more-hygroscopic nucleation mode particles; and (b) NPF with subsequent rapid growth (Event 2), resulting in less-hygroscopic particles. From analysis of particle formation and growth rates, as well as the hygroscopicity of slightly grown particles, it seems that sulfuric acid–water ion-mediated nucleation is an acceptable mechanism explaining the observed NPF during events 1 and 4. Meanwhile, low particle hygroscopicity and rapid growth suggest that condensable organics were somewhat involved in particle growth during events 2 and 3. Although the imagery from satellites confirms the biological activity as a possible source of marine sulfur and organics, due to the lack of appropriate measurements, we cannot provide quantitative information about the extent to which these precursor gases may have been involved in the observed particle formation and growth. For the same matter, we also cannot exclude other species (e.g., iodine) participating in NPF. To answer these questions, high temporal resolution measurements of nucleation and the Aitken mode particle chemical composition after the NPF are necessary, which remain a topic for future research.
After the nucleation, in 12 to 56 h newly formed particles grew to the Aitken mode sizes (approx. 30–50 nm). We have traced particle growth and measured particle hygroscopicity for dry diameters of 15, 20, 30, and 50 nm. Here, one of our main objectives was to test whether particles created in the Arctic marine boundary layer can act as CCN. To accomplish this task we have used a zero-dimensional, adiabatic cloud parcel model. Measured particle physico-chemical properties and ambient information (relative humidity, pressure, temperature) were used to simulate particle population activation to cloud droplets at two different updraft velocities of 0.1 and 3.2 m s−1. Simulation results showed that although the activated fractions of nucleation mode particles were below 5 % at an updraft wind velocity of 0.1 m s−1, background CCN number concentration increased by up to a factor of 5. The Aitken mode particle activation was somewhat higher, approx. 12 %. Such an increase in CCN number concentrations was also confirmed by direct measurements for supersaturations from 0.1 % to 1 % on-board RV Polarstern. Our findings support previous observations suggesting that in a pristine Arctic environment particles do not have to grow to sizes above 50 nm to act as CCN. We conclude that in a changing Arctic, NPF can be an important source of CCN. New particle formation and the Aitken mode particles' ability to become CCN require more in-depth studies with the focus on mechanisms of NPF, chemical composition of the precursor gases and condensable vapors, as well as the identification of their sources and impact on Arctic clouds.
Processed and raw data are available on request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-14339-2019-supplement.
SK operated aerosol instrumentation on-board RV Polarstern, evaluated data, and wrote the manuscript. TV operated aerosol instrumentation on-board RV Polarstern and contributed to the manuscript writing. PP, JL, and MK contributed to the NAIS data evaluation, discussion, and manuscript writing. DR contributed to the simulation of CCN. HW contributed to the writing of the manuscript. SZ and MvP collected samples for chemical analysis and contributed to the writing of the manuscript. MH operated the CCNC on-board RV Polarstern and evaluated CCNC data. XG and AWe operated CCNC on-board RV Polarstern. SH calibrated the CCNC prior measurement campaign. FS, HH, and AWi participated in fund raising for the measurement campaign.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Arctic mixed-phase clouds as studied during the ACLOUD/PASCAL campaigns in the framework of (AC)3 (ACP/AMT inter-journal SI)”. It is not associated with a conference.
We gratefully acknowledge the funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 268020496 – TRR 172, within the Transregional Collaborative Research Center “ArctiC Amplification: Climate Relevant Atmospheric and SurfaCe Processes, and Feedback Mechanisms (AC)3, as well as funding of RV Polarstern cruise PS106 (expedition grant number AWI-PS-106-00) by AWI. The authors would also like to acknowledge a number of people who were involved in this work. We acknowledge the discussions and support (H2SO4–H2O nucleation look-up tables) by Fangqun Yu (UAlbany). We also thank Sebastian Ehrhart (MPIC), Joachim Curtius (IAU), Steffen Münch (ETHZ), and Andreas Kürten (IAU) for the discussions concerning sulfuric acid–water nucleation, Ella-Maria Duplissy, Veli-Matti Kerminen, Jenni Kontkanen, Stephany N. Buenrostro Mazon from Helsinki University for their time, valuable suggestions, and discussions, Ulrike Egerer for providing the updraft wind velocities during the ice-drift station, Hannes Griesche, Ronny Engelmann, and Martin Radenz for providing ship-based remote sensing data to characterize the cloud situations during the selected events, Peter Gege (DLR), Svenja Kohnemann (UniTrier), and Marcel Nicolaus (AWI) for sharing the ship-deck photos, Andreas Macke (TROPOS) and Hauke Flores (AWI), Chief Scientists of PS106 cruise, for the attitude and phenomenal attention to all our requests regarding scientific activities on-board RV Polarstern and on the ice, and finally, the RV Polarstern crew, staff members, numerous scientists, and polar bear guards and watchers, who made the expedition not only exceptional, but also a safe experience. Villum Research Station, Robert Lange, Andreas Massling, Henrik Skov, and Niels Bohse Hendriksen are acknowledged for providing PNSD data. We acknowledge Hartmut and Andrea Haudek for building the conditioning system for both the aerosol inlet and the Berner impactor for these Arctic environmental conditions. Maik Merkel and Rene Rabe were a huge technical support for setting up the measurement container and Berner impactors. Susanne Fuchs performed the ion chromatography analysis and Anke Rödger the OC∕EC thermographic analysis. We also acknowledge the use of imagery from the NASA Worldview application (https://worldview.earthdata.nasa.gov, last access: 3 June 2019), part of the NASA Earth Observing System Data and Information System (EOSDIS). Also, this study has been conducted using E.U. Copernicus Marine Service Information (Arctic Chlorophyll Concentration from Satellite observations (daily average) Reprocessed L3 (ESA-CCI). We acknowledge two anonymous referees for their time, comments, and suggestions, which improved the final version of the manuscript.
This research has been supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (grant no. 268020496 – TRR 172) and the Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research (grant no. AWI-PS-106-00).
This paper was edited by Amy Solomon and reviewed by two anonymous referees.
Aas, W., Fiebig, M., Solberg, S., and Yttri, K. E.: Monitoring of long-range transported air pollutants in Norway, annual Report 2017, NILU rapport, Norwegian Environment Agency, available at: https://www.miljodirektoratet.no/globalassets/publikasjoner/m1395/m1395.pdf (last access: 12 March 2019), 2018.
Abbatt, J. P. D., Leaitch, W. R., Aliabadi, A. A., Bertram, A. K., Blanchet, J.-P., Boivin-Rioux, A., Bozem, H., Burkart, J., Chang, R. Y. W., Charette, J., Chaubey, J. P., Christensen, R. J., Cirisan, A., Collins, D. B., Croft, B., Dionne, J., Evans, G. J., Fletcher, C. G., Galí, M., Ghahremaninezhad, R., Girard, E., Gong, W., Gosselin, M., Gourdal, M., Hanna, S. J., Hayashida, H., Herber, A. B., Hesaraki, S., Hoor, P., Huang, L., Hussherr, R., Irish, V. E., Keita, S. A., Kodros, J. K., Köllner, F., Kolonjari, F., Kunkel, D., Ladino, L. A., Law, K., Levasseur, M., Libois, Q., Liggio, J., Lizotte, M., Macdonald, K. M., Mahmood, R., Martin, R. V., Mason, R. H., Miller, L. A., Moravek, A., Mortenson, E., Mungall, E. L., Murphy, J. G., Namazi, M., Norman, A.-L., O'Neill, N. T., Pierce, J. R., Russell, L. M., Schneider, J., Schulz, H., Sharma, S., Si, M., Staebler, R. M., Steiner, N. S., Thomas, J. L., von Salzen, K., Wentzell, J. J. B., Willis, M. D., Wentworth, G. R., Xu, J.-W., and Yakobi-Hancock, J. D.: Overview paper: New insights into aerosol and climate in the Arctic, Atmos. Chem. Phys., 19, 2527–2560, https://doi.org/10.5194/acp-19-2527-2019, 2019.
Alexeev, V. A. and Jackson, C. H.: Polar amplification: is atmospheric heat transport important?, Clim. Dynam., 41, 533–547, 2013.
Allan, J. D., Williams, P. I., Najera, J., Whitehead, J. D., Flynn, M. J., Taylor, J. W., Liu, D., Darbyshire, E., Carpenter, L. J., Chance, R., Andrews, S. J., Hackenberg, S. C., and McFiggans, G.: Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA, Atmos. Chem. Phys., 15, 5599–5609, https://doi.org/10.5194/acp-15-5599-2015, 2015.
Asmi, E., Frey, A., Virkkula, A., Ehn, M., Manninen, H. E., Timonen, H., Tolonen-Kivimäki, O., Aurela, M., Hillamo, R., and Kulmala, M.: Hygroscopicity and chemical composition of Antarctic sub-micrometre aerosol particles and observations of new particle formation, Atmos. Chem. Phys., 10, 4253–4271, https://doi.org/10.5194/acp-10-4253-2010, 2010.
Asmi, E., Kondratyev, V., Brus, D., Laurila, T., Lihavainen, H., Backman, J., Vakkari, V., Aurela, M., Hatakka, J., Viisanen, Y., Uttal, T., Ivakhov, V., and Makshtas, A.: Aerosol size distribution seasonal characteristics measured in Tiksi, Russian Arctic, Atmos. Chem. Phys., 16, 1271–1287, https://doi.org/10.5194/acp-16-1271-2016, 2016.
Augustin-Bauditz, S., Wex, H., Denjean, C., Hartmann, S., Schneider, J., Schmidt, S., Ebert, M., and Stratmann, F.: Laboratory-generated mixtures of mineral dust particles with biological substances: characterization of the particle mixing state and immersion freezing behavior, Atmos. Chem. Phys., 16, 5531–5543, https://doi.org/10.5194/acp-16-5531-2016, 2016.
Becagli, S., Lazzara, L., Marchese, C., Dayan, U., Ascanius, S. E., Cacciani, M., Caiazzo, L., Di Biagio, C., Di Iorio, T., Di Sarra, A., and Eriksen, P.: Relationships linking primary production, sea ice melting, and biogenic aerosol in the Arctic, Atmos. Environ., 136, 1–15, 2016.
Bi, H., Zhang, J., Wang, Y., Zhang, Z., Zhang, Y., Fu, M., Huang, H., and Xu, X.: Arctic Sea Ice Volume Changes in Terms of Age as Revealed From Satellite Observations, IEEE J. Sel. Top. Appl., 11, 2223–2237, 2018.
Bintanja, R., Van der Linden, E. C., and Hazeleger, W.: Boundary layer stability and Arctic climate change: A feedback study using EC-Earth, Clim. Dynam., 39, 2659–2673, 2012.
Burkart, J., Hodshire, A. L., Mungall, E. L., Pierce, J. R., Collins, D. B., Ladino, L. A., Lee, A. K., Irish, V., Wentzell, J. J., Liggio, J., and Papakyriakou, T.: Organic condensation and particle growth to CCN sizes in the summertime marine Arctic is driven by materials more semivolatile than at continental sites, Geophys. Res. Lett., 44, 10725–10734, https://doi.org/10.1002/2017GL075671, 2017a.
Burkart, J., Willis, M. D., Bozem, H., Thomas, J. L., Law, K., Hoor, P., Aliabadi, A. A., Köllner, F., Schneider, J., Herber, A., Abbatt, J. P. D., and Leaitch, W. R.: Summertime observations of elevated levels of ultrafine particles in the high Arctic marine boundary layer, Atmos. Chem. Phys., 17, 5515–5535, https://doi.org/10.5194/acp-17-5515-2017, 2017b.
Chang, R. Y.-W., Leck, C., Graus, M., Müller, M., Paatero, J., Burkhart, J. F., Stohl, A., Orr, L. H., Hayden, K., Li, S.-M., Hansel, A., Tjernström, M., Leaitch, W. R., and Abbatt, J. P. D.: Aerosol composition and sources in the central Arctic Ocean during ASCOS, Atmos. Chem. Phys., 11, 10619–10636, https://doi.org/10.5194/acp-11-10619-2011, 2011.
Cohen, J., Screen, J. A., Furtado, J. C., Barlow, M., Whittleston, D., Coumou, D., Francis, J., Dethloff, K., Entekhabi, D., Overland, J., and Jones, J.: Recent Arctic amplification and extreme mid-latitude weather, Nat. Geosci., 7, 627–637, https://doi.org/10.1038/ngeo2234, 2014.
Collins, D. B., Burkart, J., Chang, R. Y.-W., Lizotte, M., Boivin-Rioux, A., Blais, M., Mungall, E. L., Boyer, M., Irish, V. E., Massé, G., Kunkel, D., Tremblay, J.-É., Papakyriakou, T., Bertram, A. K., Bozem, H., Gosselin, M., Levasseur, M., and Abbatt, J. P. D.: Frequent ultrafine particle formation and growth in Canadian Arctic marine and coastal environments, Atmos. Chem. Phys., 17, 13119–13138, https://doi.org/10.5194/acp-17-13119-2017, 2017.
Conover, R. J. and Huntley, M.: Copepods in ice-covered seas – distribution, adaptations to seasonally limited food, metabolism, growth patterns and life cycle strategies in polar seas, J. Marine Syst., 2, 1–41, 1991.
Croft, B., Wentworth, G. R., Martin, R. V., Leaitch, W. R., Murphy, J. G., Murphy, B. N., Kodros, J. K., Abbatt, J. P., and Pierce, J. R.: Contribution of Arctic seabird-colony ammonia to atmospheric particles and cloud-albedo radiative effect, Nat. Commun., 7, 13444, https://doi.org/10.1038/ncomms13444, 2016.
Dall'Osto, M., Beddows, D. C. S., Tunved, P., Krejci, R., Ström, J., Hansson, H. C., Yoon, Y. J., Park, K. T., Becagli, S., Udisti, R., and Onasch, T.: Arctic sea ice melt leads to atmospheric new particle 70 formation, Sci. Rep., 7, 3318, https://doi.org/10.1038/s41598-017-03328-1, 2017.
Dall'Osto, M., Geels, C., Beddows, D. C. S., Boertmann, D., Lange, R., Nøjgaard, J. K., Harrison, R. M., Simo, R., Skov, H., and Massling, A.: Regions of open water and melting sea ice drive new particle formation in North East Greenland, Sci. Rep., 8, 6109, https://doi.org/10.1038/s41598-018-24426-8, 2018a.
Dall'Osto, M., Beddows, D. C. S., Asmi, A., Poulain, L., Hao, L., Freney, E., Allan, J. D., Canagaratna, M., Crippa, M., Bianchi, F., and De Leeuw, G.: Novel insights on new particle formation derived from a pan-european observing system, Sci. Rep., 8, 1482, https://doi.org/10.1038/s41598-017-17343-9, 2018b.
Dall'Osto, M., Beddows, D. C. S., Tunved, P., Harrison, R. M., Lupi, A., Vitale, V., Becagli, S., Traversi, R., Park, K.-T., Yoon, Y. J., Massling, A., Skov, H., Lange, R., Strom, J., and Krejci, R.: Simultaneous measurements of aerosol size distributions at three sites in the European high Arctic, Atmos. Chem. Phys., 19, 7377–7395, https://doi.org/10.5194/acp-19-7377-2019, 2019.
Draxler, R. R. and Rolph, G. D.: HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory), NOAA Air Resources Laboratory, Silver Spring, USA, 2012.
Dunne, E. M., Gordon, H., Kürten, A., Almeida, J., Duplissy, J., Williamson, C., Ortega, I. K., Pringle, K. J., Adamov, A., Baltensperger, U., and Barmet, P.: Global atmospheric particle formation from CERN CLOUD measurements, Science, 354, 1119–1124, 2016.
Egerer, U., Gottschalk, M., Siebert, H., Ehrlich, A., and Wendisch, M.: The new BELUGA setup for collocated turbulence and radiation measurements using a tethered balloon: first applications in the cloudy Arctic boundary layer, Atmos. Meas. Tech., 12, 4019–4038, https://doi.org/10.5194/amt-12-4019-2019, 2019.
Ehn, M., Petäjä, T., Aufmhoff, H., Aalto, P., Hämeri, K., Arnold, F., Laaksonen, A., and Kulmala, M.: Hygroscopic properties of ultrafine aerosol particles in the boreal forest: diurnal variation, solubility and the influence of sulfuric acid, Atmos. Chem. Phys., 7, 211–222, https://doi.org/10.5194/acp-7-211-2007, 2007.
Fetterer, F., Knowles, K., Meier, W., and Savoie, M.: Sea ice index. National Snow and Ice Data Center, Boulder, CO, digital media, available at: http://nsidc.org/data/g02135.html (last access: 25 May 2019), 2002.
Fomba, K. W., Müller, K., van Pinxteren, D., Poulain, L., van Pinxteren, M., and Herrmann, H.: Long-term chemical characterization of tropical and marine aerosols at the Cape Verde Atmospheric Observatory (CVAO) from 2007 to 2011, Atmos. Chem. Phys., 14, 8883–8904, https://doi.org/10.5194/acp-14-8883-2014, 2014.
Freud, E., Krejci, R., Tunved, P., Leaitch, R., Nguyen, Q. T., Massling, A., Skov, H., and Barrie, L.: Pan-Arctic aerosol number size distributions: seasonality and transport patterns, Atmos. Chem. Phys., 17, 8101–8128, https://doi.org/10.5194/acp-17-8101-2017, 2017.
Galloway, J. N., Savoie, D. L., Keene, W. C., and Prospero, J. M.: The temporal and spatial variability of scavenging ratios for NSS sulfate, nitrate, methanesulfonate and sodium in the atmosphere over the North Atalantic Ocean, Atmos. Environ. A-Gen., 27, 235–250, 1993.
Giamarelou, M., Eleftheriadis, K., Nyeki, S., Tunved, P., Torseth, K., and Biskos, G.: Indirect evidence of the composition of nucleation mode atmospheric particles in the high Arctic, J. Geophys. Res.-Atmos., 121, 965–975, 2016.
Graversen, R. G. and Wang, M.: Polar amplification in a coupled climate model with locked albedo, Clim. Dynam., 33, 629–643, 2009.
Gysel, M. and Stratmann, F.: WP3 – NA3: In-situ chemical, physical and optical properties of aerosols, Deliverable D3.11: Standardized protocol for CCN measurements, Tech. rep., available at: http://www.actris.net (last access: 25 May 2019), 2013.
Gysel, M., McFiggans, G. B., and Coe, H.: Inversion of tandem differential mobility analyser (TDMA) measurements, J. Aerosol Sci., 40, 134–151, 2009.
Heintzenberg, J., Tunved, P., Galí, M., and Leck, C.: New particle formation in the Svalbard region 2006–2015, Atmos. Chem. Phys., 17, 6153–6175, https://doi.org/10.5194/acp-17-6153-2017, 2017.
Herenz, P., Wex, H., Mangold, A., Laffineur, Q., Gorodetskaya, I. V., Fleming, Z. L., Panagi, M., and Stratmann, F.: CCN measurements at the Princess Elisabeth Antarctica research station during three austral summers, Atmos. Chem. Phys., 19, 275–294, https://doi.org/10.5194/acp-19-275-2019, 2019.
Hirsikko, A., Bergman, T., Laakso, L., Dal Maso, M., Riipinen, I., Hõrrak, U., and Kulmala, M.: Identification and classification of the formation of intermediate ions measured in boreal forest, Atmos. Chem. Phys., 7, 201–210, https://doi.org/10.5194/acp-7-201-2007, 2007.
Huang, S., Wu, Z., Poulain, L., van Pinxteren, M., Merkel, M., Assmann, D., Herrmann, H., and Wiedensohler, A.: Source apportionment of the organic aerosol over the Atlantic Ocean from 53∘ N to 53∘ S: significant contributions from marine emissions and long-range transport, Atmos. Chem. Phys., 18, 18043–18062, https://doi.org/10.5194/acp-18-18043-2018, 2018.
Jayaratne, E. R., Pushpawela, B., and Morawska, L.: Temporal evolution of charged and neutral nanoparticle concentrations during atmospheric new particle formation events and its implications for ion-induced nucleation, Front. Env. Sci. Eng., 10, 13, https://doi.org/10.1007/s11783-016-0862-x, 2016.
Jeffries, M. O. and Richter-Menge, J.: State of the climate in 2011: The Arctic, B. Am. Meteorol. Soc., 93, 127–148, 2012.
Jokinen, T., Sipilä, M., Kontkanen, J., Vakkari, V., Tisler, P., Duplissy, E. M., Junninen, H., Kangasluoma, J., Manninen, H. E., Petäjä, T., and Kulmala, M.: Ion-induced sulfuric acid–ammonia nucleation drives particle formation in coastal Antarctica, Sci. Adv., 4, 9744, https://doi.org/10.1126/sciadv.aat9744, 2018.
Karl, M., Leck, C., Gross, A., and Pirjola, L.: A study of new particle formation in the marine boundary layer over the central Arctic Ocean using a flexible multicomponent aerosol dynamic model, Tellus B, 64, 17158, https://doi.org/10.3402/tellusb.v64i0.17158, 2012.
Karl, M., Leck, C., Coz, E., and Heintzenberg, J.: Marine nanogels as a source of atmospheric nanoparticles in the high Arctic, Geophys. Res. Lett., 40, 3738–3743, 2013.
Keene, W. C., Maring, H., Maben, J. R., Kieber, D. J., Pszenny, A. A. P., Dahl, E. E., Izaguirre, M. A., Davis, A. J., Long, M. S., Zhou, X., Smoydzin, L., and Sander, R.: Chemical and Physical Characteristics of Nascent Aerosols Produced by Bursting Bubbles at a Model Air-Sea Interface, J. Geophys. Res.-Atmos., 112, D21202, https://doi.org/10.1029/2007JD008464, 2007.
Kerminen, V. M., Aurela, M., Hillamo, R. E., and Virkkula, A.: Formation of particulate MSA: deductions from size distribution measurements in the Finnish Arctic, Tellus B, 49, 159–171, 1997.
Kerminen, V.-M., Paramonov, M., Anttila, T., Riipinen, I., Fountoukis, C., Korhonen, H., Asmi, E., Laakso, L., Lihavainen, H., Swietlicki, E., Svenningsson, B., Asmi, A., Pandis, S. N., Kulmala, M., and Petäjä, T.: Cloud condensation nuclei production associated with atmospheric nucleation: a synthesis based on existing literature and new results, Atmos. Chem. Phys., 12, 12037–12059, https://doi.org/10.5194/acp-12-12037-2012, 2012.
Kerminen, V. M., Chen, X., Vakkari, V., Petäjä, T., Kulmala, M., and Bianchi, F.: Atmospheric new particle formation and growth: review of field observations, Environ. Res. Lett., 13, 103003, https://doi.org/10.1088/1748-9326/aadf3c, 2018.
Kim, J., Ahlm, L., Yli-Juuti, T., Lawler, M., Keskinen, H., Tröstl, J., Schobesberger, S., Duplissy, J., Amorim, A., Bianchi, F., Donahue, N. M., Flagan, R. C., Hakala, J., Heinritzi, M., Jokinen, T., Kürten, A., Laaksonen, A., Lehtipalo, K., Miettinen, P., Petäjä, T., Rissanen, M. P., Rondo, L., Sengupta, K., Simon, M., Tomé, A., Williamson, C., Wimmer, D., Winkler, P. M., Ehrhart, S., Ye, P., Kirkby, J., Curtius, J., Baltensperger, U., Kulmala, M., Lehtinen, K. E. J., Smith, J. N., Riipinen, I., and Virtanen, A.: Hygroscopicity of nanoparticles produced from homogeneous nucleation in the CLOUD experiments, Atmos. Chem. Phys., 16, 293–304, https://doi.org/10.5194/acp-16-293-2016, 2016.
Knudsen, E. M., Heinold, B., Dahlke, S., Bozem, H., Crewell, S., Gorodetskaya, I. V., Heygster, G., Kunkel, D., Maturilli, M., Mech, M., Viceto, C., Rinke, A., Schmithüsen, H., Ehrlich, A., Macke, A., Lüpkes, C., and Wendisch, M.: Meteorological conditions during the ACLOUD/PASCAL field campaign near Svalbard in early summer 2017, Atmos. Chem. Phys., 18, 17995–18022, https://doi.org/10.5194/acp-18-17995-2018, 2018.
Köhler, H.: The nucleus in and the growth of hygroscopic droplets, T. Faraday Soc., 32, 1152–1161, 1936.
Köllner, F., Schneider, J., Willis, M. D., Klimach, T., Helleis, F., Bozem, H., Kunkel, D., Hoor, P., Burkart, J., Leaitch, W. R., Aliabadi, A. A., Abbatt, J. P. D., Herber, A. B., and Borrmann, S.: Particulate trimethylamine in the summertime Canadian high Arctic lower troposphere, Atmos. Chem. Phys., 17, 13747–13766, https://doi.org/10.5194/acp-17-13747-2017, 2017.
Kulmala, M., Maso, M. D., Mäkelä, J. M., Pirjola, L., Väkevä, M., Aalto, P., Miikkulainen, P., Hämeri, K., and O'dowd, C. D.: On the formation, growth and composition of nucleation mode particles, Tellus B, 53, 479–490, 2001.
Kulmala, M., Petäjä, T., Nieminen, T., Sipilä, M., Manninen, H. E., Lehtipalo, K., Dal Maso, M., Aalto, P. P., Junninen, H., Paasonen, P., and Riipinen, I.: Measurement of the nucleation of atmospheric aerosol particles, Nat. Protoc., 7, 1651, https://doi.org/10.1038/nprot.2012.091, 2012.
Kurtén, T., Torpo, L., Sundberg, M. R., Kerminen, V.-M., Vehkamäki, H., and Kulmala, M.: Estimating the NH3:H2SO4 ratio of nucleating clusters in atmospheric conditions using quantum chemical methods, Atmos. Chem. Phys., 7, 2765–2773, https://doi.org/10.5194/acp-7-2765-2007, 2007.
Kyrö, E.-M., Kerminen, V.-M., Virkkula, A., Dal Maso, M., Parshintsev, J., Ruíz-Jimenez, J., Forsström, L., Manninen, H. E., Riekkola, M.-L., Heinonen, P., and Kulmala, M.: Antarctic new particle formation from continental biogenic precursors, Atmos. Chem. Phys., 13, 3527–3546, https://doi.org/10.5194/acp-13-3527-2013, 2013.
Leaitch, W. R., Strapp, J. W., Isaac, G. A., and Hudson, J. G.: Cloud droplet nucleation and cloud scavenging of aerosol sulphate in polluted atmospheres, Tellus B, 38, 328–344, 1986.
Leaitch, W. R., Sharma, S., Huang, L., Toom-Sauntry, D., Chivulescu, A., Macdonald, A. M., von Salzen, K., Pierce, J. R., Bertram, A. K., Schroder, J. C., Shantz, N. C., Chang, R. Y.-W., and Norman, A.-L.: Dimethyl sulfide control of the clean summertime Arctic aerosol and cloud, Elem. Sci. Anth., 1, 00017, https://doi.org/10.12952/journal.elementa.000017, 2013.
Leaitch, W. R., Korolev, A., Aliabadi, A. A., Burkart, J., Willis, M. D., Abbatt, J. P. D., Bozem, H., Hoor, P., Köllner, F., Schneider, J., Herber, A., Konrad, C., and Brauner, R.: Effects of 20–100 nm particles on liquid clouds in the clean summertime Arctic, Atmos. Chem. Phys., 16, 11107–11124, https://doi.org/10.5194/acp-16-11107-2016, 2016.
Leck, C., Norman, M., Bigg, E. K., and Hillamo, R.: Chemical composition and sources of the high Arctic aerosol relevant for cloud formation, J. Geophys. Res.-Atmos., 107, 4135, https://doi.org/10.1029/2001JD001463, 2002.
Lehtinen, K. E., Dal Maso, M., Kulmala, M., and Kerminen, V. M.: Estimating nucleation rates from apparent particle formation rates and vice versa: Revised formulation of the Kerminen–Kulmala equation, J. Aerosol Sci., 38, 988–994, 2007.
Levasseur, M.: Impact of Arctic meltdown on the microbial cycling of sulphur, Nat. Geosci., 6, 691–700, https://doi.org/10.1038/ngeo1910, 2013.
Macke, A. and Flores, H.: The Expeditions PS106/1 and 2 of the Research Vessel POLARSTERN to the Arctic Ocean in 2017. Berichte zur Polar- und Meeresforschung, Reports on polar and marine research, Bremerhaven, Alfred Wegener Institute for Polar and Marine Research, 719, 171, https://doi.org/10.2312/BzPM_0719_2018, 2018.
Manninen, H. E., Nieminen, T., Asmi, E., Gagné, S., Häkkinen, S., Lehtipalo, K., Aalto, P., Vana, M., Mirme, A., Mirme, S., Hõrrak, U., Plass-Dülmer, C., Stange, G., Kiss, G., Hoffer, A., Törő, N., Moerman, M., Henzing, B., de Leeuw, G., Brinkenberg, M., Kouvarakis, G. N., Bougiatioti, A., Mihalopoulos, N., O'Dowd, C., Ceburnis, D., Arneth, A., Svenningsson, B., Swietlicki, E., Tarozzi, L., Decesari, S., Facchini, M. C., Birmili, W., Sonntag, A., Wiedensohler, A., Boulon, J., Sellegri, K., Laj, P., Gysel, M., Bukowiecki, N., Weingartner, E., Wehrle, G., Laaksonen, A., Hamed, A., Joutsensaari, J., Petäjä, T., Kerminen, V.-M., and Kulmala, M.: EUCAARI ion spectrometer measurements at 12 European sites – analysis of new particle formation events, Atmos. Chem. Phys., 10, 7907–7927, https://doi.org/10.5194/acp-10-7907-2010, 2010.
Manninen, H. E., Franchin, A., Schobesberger, S., Hirsikko, A., Hakala, J., Skromulis, A., Kangasluoma, J., Ehn, M., Junninen, H., Mirme, A., Mirme, S., Sipilä, M., Petäjä, T., Worsnop, D. R., and Kulmala, M.: Characterisation of corona-generated ions used in a Neutral cluster and Air Ion Spectrometer (NAIS), Atmos. Meas. Tech., 4, 2767–2776, https://doi.org/10.5194/amt-4-2767-2011, 2011.
Maslanik, J. and Stroeve, J.: Near-Real-Time DMSP SSMIS Daily Polar Gridded Sea Ice Concentrations, 1 Boulder, Colorado USA. NASA National Snow and Ice Data Center Distributed Active Archive Center, https://doi.org/10.5067/U8C09DWVX9LM, 1999.
Massling, A., Niedermeier, N., Hennig, T., Fors, E. O., Swietlicki, E., Ehn, M., Hämeri, K., Villani, P., Laj, P., Good, N., McFiggans, G., and Wiedensohler, A.: Results and recommendations from an intercomparison of six Hygroscopicity-TDMA systems, Atmos. Meas. Tech., 4, 485–497, https://doi.org/10.5194/amt-4-485-2011, 2011.
Mauritsen, T., Sedlar, J., Tjernström, M., Leck, C., Martin, M., Shupe, M., Sjogren, S., Sierau, B., Persson, P. O. G., Brooks, I. M., and Swietlicki, E.: An Arctic CCN-limited cloud-aerosol regime, Atmos. Chem. Phys., 11, 165–173, https://doi.org/10.5194/acp-11-165-2011, 2011.
Meier, W. N., Hovelsrud, G. K., van Oort, B. E., Key, J. R., Kovacs, K. M., Michel, C., Haas, C., Granskog, M. A., Gerland, S., Perovich, D. K., and Makshtas, A.: Arctic sea ice in transformation: A review of recent observed changes and impacts on biology and human activity, Rev. Geophys., 52, 185–217, 2014.
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S.: Impact of nucleation on global CCN, Atmos. Chem. Phys., 9, 8601–8616, https://doi.org/10.5194/acp-9-8601-2009, 2009.
Mirme, A., Tamm, E., Mordas, G., Vana, M., Uin, J., Mirme, S., Bernotas, T., Laakso, L., Hirsikko, A., and Kulmala, M.: A wide-range multi-channel Air Ion Spectrometer, Boreal Environ. Res.,12, 247–264, 2007.
Mirme, S. and Mirme, A.: The mathematical principles and design of the NAIS – a spectrometer for the measurement of cluster ion and nanometer aerosol size distributions, Atmos. Meas. Tech., 6, 1061–1071, https://doi.org/10.5194/amt-6-1061-2013, 2013.
Miyazaki, Y., Kawamura, K., and Sawano, M.: Size distributions of organic nitrogen and carbon in remote marine aerosols: Evidence of marine biological origin based on their isotopic ratios, Geophys. Res. Lett., 37, L06803, https://doi.org/10.1029/2010GL042483, 2010.
Müller, K., Lehmann, S., van Pinxteren, D., Gnauk, T., Niedermeier, N., Wiedensohler, A., and Herrmann, H.: Particle characterization at the Cape Verde atmospheric observatory during the 2007 RHaMBLe intensive, Atmos. Chem. Phys., 10, 2709–2721, https://doi.org/10.5194/acp-10-2709-2010, 2010.
Napari, I., Noppel, M., Vehkamäki, H., and Kulmala, M.: Parametrization of ternary nucleation rates for H2SO4-NH3-H2O vapors, J. Geophys. Res.-Atmos., 107, AAC 6-1–AAC 6-6, https://doi.org/10.1029/2002JD002132, 2002.
Nguyen, Q. T., Glasius, M., Sørensen, L. L., Jensen, B., Skov, H., Birmili, W., Wiedensohler, A., Kristensson, A., Nøjgaard, J. K., and Massling, A.: Seasonal variation of atmospheric particle number concentrations, new particle formation and atmospheric oxidation capacity at the high Arctic site Villum Research Station, Station Nord, Atmos. Chem. Phys., 16, 11319–11336, https://doi.org/10.5194/acp-16-11319-2016, 2016.
Nicolaus, M.: Shipborne visual observations of Arctic sea ice during POLARSTERN cruise PS106, PANGAEA, https://doi.org/10.1594/PANGAEA.889264, 2018.
Nieminen, T., Kerminen, V.-M., Petäjä, T., Aalto, P. P., Arshinov, M., Asmi, E., Baltensperger, U., Beddows, D. C. S., Beukes, J. P., Collins, D., Ding, A., Harrison, R. M., Henzing, B., Hooda, R., Hu, M., Hõrrak, U., Kivekäs, N., Komsaare, K., Krejci, R., Kristensson, A., Laakso, L., Laaksonen, A., Leaitch, W. R., Lihavainen, H., Mihalopoulos, N., Németh, Z., Nie, W., O'Dowd, C., Salma, I., Sellegri, K., Svenningsson, B., Swietlicki, E., Tunved, P., Ulevicius, V., Vakkari, V., Vana, M., Wiedensohler, A., Wu, Z., Virtanen, A., and Kulmala, M.: Global analysis of continental boundary layer new particle formation based on long-term measurements, Atmos. Chem. Phys., 18, 14737–14756, https://doi.org/10.5194/acp-18-14737-2018, 2018.
Orellana, M. V., Matrai, P. A., Leck, C., Rauschenberg, C. D., Lee, A. M., and Coz, E.: Marine microgels as a source of cloud condensation nuclei in the high Arctic, P. Natl. Acad. Sci. USA, 108, 13612–13617, 2011.
Overland, J. E., Wood, K. R., and Wang, M.: Warm Arctic – cold continents: climate impacts of the newly open Arctic Sea, Polar Res., 30, 15787, https://doi.org/10.3402/polar.v30i0.15787, 2011.
Park, K., Kim, G., Kim, J. S., Yoon, Y. J., Cho, H. J., and Ström, J.: Mixing state of size-selected submicrometer particles in the Arctic in May and September 2012, Environ. Sci. Technol., 48, 909–919, 2014.
Park, K.-T., Jang, S., Lee, K., Yoon, Y. J., Kim, M.-S., Park, K., Cho, H.-J., Kang, J.-H., Udisti, R., Lee, B.-Y., and Shin, K.-H.: Observational evidence for the formation of DMS-derived aerosols during Arctic phytoplankton blooms, Atmos. Chem. Phys., 17, 9665–9675, https://doi.org/10.5194/acp-17-9665-2017, 2017.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 7, 1961–1971, https://doi.org/10.5194/acp-7-1961-2007, 2007.
Pfeifer, S., Birmili, W., Schladitz, A., Müller, T., Nowak, A., and Wiedensohler, A.: A fast and easy-to-implement inversion algorithm for mobility particle size spectrometers considering particle number size distribution information outside of the detection range, Atmos. Meas. Tech., 7, 95–105, https://doi.org/10.5194/amt-7-95-2014, 2014.
Pichelstorfer, L., Stolzenburg, D., Ortega, J., Karl, T., Kokkola, H., Laakso, A., Lehtinen, K. E. J., Smith, J. N., McMurry, P. H., and Winkler, P. M.: Resolving nanoparticle growth mechanisms from size- and time-dependent growth rate analysis, Atmos. Chem. Phys., 18, 1307–1323, https://doi.org/10.5194/acp-18-1307-2018, 2018.
Pithan, F. and Mauritsen, T.: Arctic amplification dominated by temperature feedbacks in contemporary climate models, Nat. Geosci., 7, 181–184, https://doi.org/10.1038/ngeo2071, 2014.
Polashenski, C., Golden, K. M., Perovich, D. K., Skyllingstad, E., Arnsten, A., Stwertka, C., and Wright, N.: Percolation blockage: A process that enables melt pond formation on first year Arctic sea ice, J. Geophys. Res.-Oceans, 122, 413–440, 2017.
Praetorius, S., Rugenstein, M., Persad, G., and Caldeira, K.: Global and Arctic climate sensitivity enhanced by changes in North Pacific heat flux, Nat. Commun., 9, 3124, https://doi.org/10.1038/s41467-018-05337-8, 2018.
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A., and Bates, T. S.: Chemistry and Related Properties of Freshly Emitted Sea Spray Aerosol, Chem. Rev., 115, 4383–4399, 2015.
Rampal, P., Weiss, J., and Marsan, D.: Positive trend in the mean speed and deformation rate of Arctic sea ice, 1979–2007, J. Geophys. Res.-Oceans, 114, C05013, https://doi.org/10.1029/2008JC005066, 2009.
Riccobono, F., Schobesberger, S., Scott, C. E., Dommen, J., Ortega, I. K., Rondo, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., and David, A.: Oxidation products of biogenic emissions contribute to nucleation of atmospheric particles, Science, 344, 717–721, 2014.
Roberts, G. C. and Nenes, A.: A continuous-flow streamwise thermal-gradient CCN chamber for atmospheric measurements, Aerosol Sci. Tech., 39, 206–221, 2005.
Rothenberg, D. and Wang, C.: Metamodeling of droplet activation for global climate models, J. Atmos. Sci., 73, 1255–1272, 2016.
Schobesberger, S., Junninen, H., Bianchi, F., Lönn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., and Nieminen, T.: Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules, P. Natl. Acad. Sci. USA, 110, 17223–17228, 2013.
Schobesberger, S., Franchin, A., Bianchi, F., Rondo, L., Duplissy, J., Kürten, A., Ortega, I. K., Metzger, A., Schnitzhofer, R., Almeida, J., Amorim, A., Dommen, J., Dunne, E. M., Ehn, M., Gagné, S., Ickes, L., Junninen, H., Hansel, A., Kerminen, V.-M., Kirkby, J., Kupc, A., Laaksonen, A., Lehtipalo, K., Mathot, S., Onnela, A., Petäjä, T., Riccobono, F., Santos, F. D., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Viisanen, Y., Wagner, P. E., Wimmer, D., Curtius, J., Donahue, N. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: On the composition of ammonia–sulfuric-acid ion clusters during aerosol particle formation, Atmos. Chem. Phys., 15, 55–78, https://doi.org/10.5194/acp-15-55-2015, 2015.
Screen, J. A. and Simmonds, I.: The central role of diminishing sea ice in recent Arctic temperature amplification, Nature, 464, 1334, https://doi.org/10.1038/nature09051, 2010.
Serreze, M. C. and Barry, R. G.: Processes and impacts of Arctic amplification: A research synthesis, Global Planet. Change, 77, 85–96, 2011.
Sipilä, M., Sarnela, N., Jokinen, T., Henschel, H., Junninen, H., Kontkanen, J., Richters, S., Kangasluoma, J., Franchin, A., Peräkylä, O., and Rissanen, M. P.: Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3, Nature, 537, 532–534, https://doi.org/10.1038/nature19314, 2016.
Smith, J. N., Barsanti, K. C., Friedli, H. R., Ehn, M., Kulmala, M., Collins, D. R., Scheckman, J. H., Williams, B. J., and McMurry, P. H.: Observations of aminium salts in atmospheric nanoparticles and possible climatic implications, P. Natl. Acad. Sci. USA, 107, 6634–6639, 2010.
Spielhagen, R. F., Werner, K., Sørensen, S. A., Zamelczyk, K., Kandiano, E., Budeus, G., Husum, K., Marchitto, T. M., and Hald, M.: Enhanced modern heat transfer to the Arctic by warm Atlantic water, Science, 331, 450–453, 2011.
Stefels, J., Steinke, M., Turner, S., Malin, G., and Belviso, S.: Environmental constraints on the production and removal of the climatically active gas dimethylsulphide (DMS) and implications for ecosystem modelling, Biogeochemistry, 83, 245–275, 2007.
Ström, J., Engvall, A. C., Delbart, F., Krejci, R., and Treffeisen, R.: On small particles in the Arctic summer boundary layer: observations at two different heights near Ny-Ålesund, Svalbard, Tellus B, 61, 473–482, 2009.
Taylor, P. C., Cai, M., Hu, A., Meehl, J., Washington, W., and Zhang, G. J.: A decomposition of feedback contributions to polar warming amplification, J. Climate, 26, 7023–7043, 2013.
Tremblay, S., Picard, J.-C., Bachelder, J. O., Lutsch, E., Strong, K., Fogal, P., Leaitch, W. R., Sharma, S., Kolonjari, F., Cox, C. J., Chang, R. Y.-W., and Hayes, P. L.: Characterization of aerosol growth events over Ellesmere Island during the summers of 2015 and 2016, Atmos. Chem. Phys., 19, 5589–5604, https://doi.org/10.5194/acp-19-5589-2019, 2019.
Tritscher, T., Dommen, J., DeCarlo, P. F., Gysel, M., Barmet, P. B., Praplan, A. P., Weingartner, E., Prévôt, A. S. H., Riipinen, I., Donahue, N. M., and Baltensperger, U.: Volatility and hygroscopicity of aging secondary organic aerosol in a smog chamber, Atmos. Chem. Phys., 11, 11477–11496, https://doi.org/10.5194/acp-11-11477-2011, 2011.
Turpin, B. J. and Lim, H. J.: Species contributions to PM2.5 mass concentrations: Revisiting common assumptions for estimating organic mass, Aerosol Sci. Tech., 35, 602–610, 2001.
Vakkari, V., Tiitta, P., Jaars, K., Croteau, P., Beukes, J. P., Josipovic, M., Kerminen, V. M., Kulmala, M., Venter, A. D., van Zyl, P. G., and Worsnop, D. R.: Reevaluating the contribution of sulfuric acid and the origin of organic compounds in atmospheric nanoparticle growth, Geophys. Res. Lett., 42, 10–486, 2015.
Van Pinxteren, M., Barthel, S., Fomba, K. W., Müller, K., Von Tümpling, W., and Herrmann, H.: The Influence of Environmental Drivers on the Enrichment of Organic Carbon in the Sea Surface Microlayer and in Submicron Aerosol Particles – Measurements from the Atlantic Ocean, Elem. Sci. Anth., 5, 35, https://doi.org/10.1525/elementa.225, 2017.
Vavrus, S.: The impact of cloud feedbacks on Arctic climate under greenhouse forcing, J. Climate, 17, 603–615, 2004.
Wagner, R., Manninen, H. E., Franchin, A., Lehtipalo, K., Mirme, S., Steiner, G., Petäjä, T., and Kulmala, M.: On the accuracy of ion measurements using a Neutral cluster and Air Ion Spectrometer, Boreal Environ. Res., 21, 230–241, 2016.
Wagner, R., Yan, C., Lehtipalo, K., Duplissy, J., Nieminen, T., Kangasluoma, J., Ahonen, L. R., Dada, L., Kontkanen, J., Manninen, H. E., Dias, A., Amorim, A., Bauer, P. S., Bergen, A., Bernhammer, A.-K., Bianchi, F., Brilke, S., Mazon, S. B., Chen, X., Draper, D. C., Fischer, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., Heikkinen, L., Heinritzi, M., Hofbauer, V., Hoyle, C. R., Kirkby, J., Kürten, A., Kvashnin, A. N., Laurila, T., Lawler, M. J., Mai, H., Makhmutov, V., Mauldin III, R. L., Molteni, U., Nichman, L., Nie, W., Ojdanic, A., Onnela, A., Piel, F., Quéléver, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Sengupta, K., Simon, M., Stolzenburg, D., Stozhkov, Y., Tröstl, J., Viisanen, Y., Vogel, A. L., Wagner, A. C., Xiao, M., Ye, P., Baltensperger, U., Curtius, J., Donahue, N. M., Flagan, R. C., Gallagher, M., Hansel, A., Smith, J. N., Tomé, A., Winkler, P. M., Worsnop, D., Ehn, M., Sipilä, M., Kerminen, V.-M., Petäjä, T., and Kulmala, M.: The role of ions in new particle formation in the CLOUD chamber, Atmos. Chem. Phys., 17, 15181–15197, https://doi.org/10.5194/acp-17-15181-2017, 2017.
Walsh, J. E.: Intensified warming of the Arctic: Causes and impacts on middle latitudes, Global Planet. Change, 117, 52–63, 2014.
Weller, R., Schmidt, K., Teinilä, K., and Hillamo, R.: Natural new particle formation at the coastal Antarctic site Neumayer, Atmos. Chem. Phys., 15, 11399–11410, https://doi.org/10.5194/acp-15-11399-2015, 2015.
Wendisch, M., Macke, A., Ehrlich, A., Lüpkes, C., Mech, M., Chechin, D., Dethloff, K., Barientos, C., Bozem, H., Brückner, M., and Clemen, H. C.: The Arctic Cloud Puzzle: Using ACLOUD/PASCAL Multi-Platform Observations to Unravel the Role of Clouds and Aerosol Particles in Arctic Amplification, B. Am. Meteorol. Soc., 100, 841–871, https://doi.org/10.1175/BAMS-D-18-0072.1, 2019.
Wentworth, G. R., Murphy, J. G., Croft, B., Martin, R. V., Pierce, J. R., Côté, J.-S., Courchesne, I., Tremblay, J.-É., Gagnon, J., Thomas, J. L., Sharma, S., Toom-Sauntry, D., Chivulescu, A., Levasseur, M., and Abbatt, J. P. D.: Ammonia in the summertime Arctic marine boundary layer: sources, sinks, and implications, Atmos. Chem. Phys., 16, 1937–1953, https://doi.org/10.5194/acp-16-1937-2016, 2016.
Wiedensohler, A.: An approximation of the bipolar charge distribution for particles in the submicron size range, J. Aerosol Sci., 19, 387–389, 1988.
Wiedensohler, A., Covert, D. S., Swietlicki, E., Aalto, P., Heintzenberg, J., and Leck, C.: Occurrence of an ultrafine particle mode less than 20 nm in diameter in the marine boundary layer during Arctic summer and autumn, Tellus B, 48, 213–222, 1996.
Wiedensohler, A., Orsini, D., Covert, D. S., Coffmann, D., Cantrell, W., Havlicek, M., Brechtel, F. J., Russell, L. M., Weber, R. J., Gras, J., and Hudson, J. G.: Intercomparison study of the size-dependent counting efficiency of 26 condensation particle counters, Aerosol Sci. Tech., 27, 224–242, 1997.
Wiedensohler, A., Birmili, W., Nowak, A., Sonntag, A., Weinhold, K., Merkel, M., Wehner, B., Tuch, T., Pfeifer, S., Fiebig, M., Fjäraa, A. M., Asmi, E., Sellegri, K., Depuy, R., Venzac, H., Villani, P., Laj, P., Aalto, P., Ogren, J. A., Swietlicki, E., Williams, P., Roldin, P., Quincey, P., Hüglin, C., Fierz-Schmidhauser, R., Gysel, M., Weingartner, E., Riccobono, F., Santos, S., Grüning, C., Faloon, K., Beddows, D., Harrison, R., Monahan, C., Jennings, S. G., O'Dowd, C. D., Marinoni, A., Horn, H.-G., Keck, L., Jiang, J., Scheckman, J., McMurry, P. H., Deng, Z., Zhao, C. S., Moerman, M., Henzing, B., de Leeuw, G., Löschau, G., and Bastian, S.: Mobility particle size spectrometers: harmonization of technical standards and data structure to facilitate high quality long-term observations of atmospheric particle number size distributions, Atmos. Meas. Tech., 5, 657–685, https://doi.org/10.5194/amt-5-657-2012, 2012.
Wiedensohler, A., Wiesner, A., Weinhold, K., Birmili, W., Hermann, M., Merkel, M., Müller, T., Pfeifer, S., Schmidt, A., Tuch, T., and Velarde, F.: Mobility particle size spectrometers: Calibration procedures and measurement uncertainties, Aerosol Sci. Tech., 52, 146–164, 2018.
Willis, M. D., Burkart, J., Thomas, J. L., Köllner, F., Schneider, J., Bozem, H., Hoor, P. M., Aliabadi, A. A., Schulz, H., Herber, A. B., Leaitch, W. R., and Abbatt, J. P. D.: Growth of nucleation mode particles in the summertime Arctic: a case study, Atmos. Chem. Phys., 16, 7663–7679, https://doi.org/10.5194/acp-16-7663-2016, 2016.
Willis, M. D., Köllner, F., Burkart, J., Bozem, H., Thomas, J. L., Schneider, J., Aliabadi, A. A., Hoor, P. M., Schulz, H., Herber, A. B., and Leaitch, W. R.: Evidence for marine biogenic influence on summertime Arctic aerosol, Geophys. Res. Lett., 44, 6460–6470, 2017.
Willis, M. D., Leaitch, W. R., and Abbatt, J. P.: Processes controlling the composition and abundance of Arctic aerosol, Rev. Geophys., 56, 621–671, 2018.
Yu, F.: Ion-mediated nucleation in the atmosphere: Key controlling parameters, implications, and look-up table, J. Geophys. Res.-Atmos., 115, D17205, https://doi.org/10.1029/2009JD012630, 2010.
Zhang, W., Miller, P. A., Jansson, C., Samuelsson, P., Mao, J., and Smith, B.: Self-Amplifying Feedbacks Accelerate Greening and Warming of the Arctic, Geophys. Res. Lett., 45, 7102–7111, 2018.
Zhou, J., Swietlicki, E., Berg, O. H., Aalto, P. P., Hämeri, K., Nilsson, E. D., and Leck, C.: Hygroscopic properties of aerosol particles over the central Arctic Ocean during summer, J. Geophys. Res.-Atmos., 106, 32111–32123, 2001.