the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Oct 2019
| 22 Oct 2019
Carboxylic acids from limonene oxidation by ozone and hydroxyl radicals: insights into mechanisms derived using a FIGAERO-CIMS
Julia Hammes
Anna Lutz
Thomas Mentel
Cameron Faxon
This work presents the results from a flow reactor study on the formation of carboxylic acids from limonene oxidation in the presence of ozone under NOx-free conditions in the dark. A High-Resolution Time-of-Flight acetate Chemical Ionisation Mass Spectrometer (HR-ToF-CIMS) was used in combination with a Filter Inlet for Gases and AEROsols (FIGAERO) to measure the carboxylic acids in the gas and particle phases. The results revealed that limonene oxidation produced large amounts of carboxylic acids which are important contributors to secondary organic aerosol (SOA) formation. The highest 10 acids contributed 56 %–91 % to the total gas-phase signal, and the dominant gas-phase species in most experiments were C8H12O4, C9H14O4, C7H10O4 and C10H16O3. The particle-phase composition was generally more complex than the gas-phase composition, and the highest 10 acids contributed 47 %–92 % to the total signal. The dominant species in the particle phase were C8H12O5, C9H14O5, C9H12O5 and C10H16O4. The measured concentration of dimers bearing at least one carboxylic acid function in the particle phase was very low, indicating that acidic dimers play a minor role in SOA formation via ozone (O3)/hydroxyl (OH) oxidation of limonene. Based on the various experimental conditions, the acidic compositions for all experiments were modelled using descriptions from the Master Chemical Mechanism (MCM). The experiment and model provided a yield of large (C7–C10) carboxylic acid of the order of 10 % (2 %–23 % and 10 %–15 %, respectively). Significant concentrations of 11 acids, from a total of 16 acids, included in the MCM were measured with the CIMS. However, the model predictions were, in some cases, inconsistent with the measurement results, especially regarding the OH dependence. Reaction mechanisms are suggested to fill-in the knowledge gaps. Using the additional mechanisms proposed in this work, nearly 75 % of the observed gas-phase signal in our lowest concentration experiment (8.4 ppb converted, ca. 23 % acid yield) carried out under humid conditions can be understood.
- Article
(1315 KB) - Full-text XML
-
Supplement
(1480 KB) - BibTeX
- EndNote





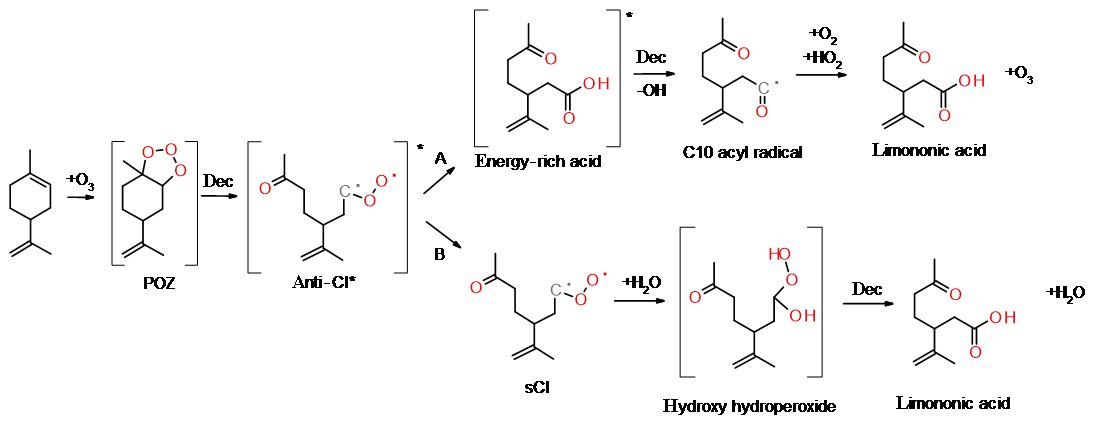
Figure 1(a) Example of the initial reactions of limonene with ozone to form limononic acid from the anti-CI* via the hot acid channel and (b) the collisional stabilisation channel (Vereecken and Francisco, 2012).
Atmospheric aerosol particles have an impact on climate and human health, and their respective effects depend on particle properties determined by the particle size and chemical composition. Among the many constituents of atmospheric aerosol particles, organic aerosol particles are the least understood (Glasius and Goldstein, 2016). Secondary organic aerosol (SOA) is the major component of organic aerosols. Identifying the chemical pathways of condensable products is essential for predicting SOA formation (Hallquist et al., 2009; Ziemann and Atkinson, 2012; Ehn et al., 2014; Shrivastava et al., 2017; McFiggans et al., 2019). However, this identification is inherently difficult as such products often reside in both the gas and particulate phases and continuous partitioning occurs between these two phases. Low vapour pressure products from radical-initiated (i.e. OH-initiated) oxidation or ozonolysis of volatile organic compounds (VOCs), such as monoterpenes (C10H16), contribute significantly to atmospheric aerosol particle formation and growth (Hallquist et al., 2009). Limonene, the main constituent of the essential oil from citrus plants, is a widely used chemical in personal care and household-related consumer products (owing to its pleasant smell); therefore, elevated indoor concentrations of limonene can be expected (Brown et al., 1994; Langer et al., 2008) as well as subsequent SOA formation (Youssefi and Waring, 2014). The total global forest emission of limonene has been estimated at 11.4 Tg yr−1, placing it in the top four among monoterpenes (Guenther et al., 2012). A high aerosol yield and the two chemically different double bonds, an endocyclic and an exocyclic double bond, make limonene ozonolysis of specific interest (Koch et al., 2000; Saathoff et al., 2009; Chen and Hopke, 2010; Gong et al., 2018). The initial reaction predominantly occurs at the endocyclic double bond. However, the first-generation products may be unsaturated and exhibit high reactivity for further oxidation. The oxidation of limonene eventually leads to the formation of SOA in both the atmosphere and in indoor environments. The oxidation of monoterpenes and, specifically, limonene has been previously reported (Leungsakul et al., 2005a, b; Walser et al., 2008; Maksymiuk et al., 2009) and reaction mechanisms that describe first- and second-generation oxidation products have been proposed (Carslaw, 2013; Chen and Griffin, 2005). Due to their low vapour pressure, carboxylic acids, a major class of limonene-oxidation products, can play an important role in SOA formation (Salo et al., 2010). The relative contribution of carboxylic acids from limonene oxidation to SOA formation was assessed via a model by Pathak et al. (2012). According to that study, limonene ozonolysis produces significant amounts of carboxylic acids, and the distribution of these acids is affected by the OH and ozone concentrations.
During ozonolysis, limonene is attacked by ozone and forms an unstable and energy-rich primary ozonide, POZ (see Fig. 1). The POZ undergoes decomposition during which the oxygen atoms contribute to the formation of a carbonyl and a carbonyl oxide group, the so-called excited Criegee intermediate (CI*). The 10-carbon skeleton is retained during this process if an endocyclic double bond is attacked. The CI* has a planar structure, and the orientation of the outer oxygen determines its chemical fate. The dominant reaction pathway (86 %; Atkinson et al., 1992) for limonene syn-CI* is the vinyl hydroperoxide channel (VHP) that generates an alkyl radical under the loss of an OH radical. This pathway provides a source for night-time OH in the atmosphere. The VHP requires an alkyl group in the syn position and is, hence, inaccessible to anti-CI*. The dominant fate of the anti-CI* is decomposition via the ester or the “hot acid” channel where an energy-rich ester or acid formed will undergo decomposition, resulting in various products. A possibility is formation of OH and an acyl radical, which can subsequently react with O2 and HO2 to form a carboxylic acid and ozone. Furthermore, the CI* can, to some extent, become collisionally stabilised (sCI); exocyclic CI* are stabilised more efficiently than endocyclic CI*. The sCI formed undergos further reactions and the sCI + water reaction produces a carbonyl, an alkyl or an alkoxy radical. If the sCI contains an α-hydrogen, a carboxylic acid can be produced directly from the water reaction. Although the sCI + water reaction is likely the most dominant in the atmosphere, sCI may also react with carboxylic acids and form stable adducts that have been identified as dimer esters (Kristensen et al., 2016). The decomposition of CI* can lead to the formation of alkyl radicals. These radicals rapidly react with oxygen to form alkyl peroxy radicals (RO2), which are an important intermediate in the gas-phase oxidation of organic compounds.
The atmospheric fate of RO2 radicals in the absence of NOx includes a self-reaction (Reactions R1–R3), isomerisation via an internal H-shift (Reaction R4) and a reaction with HO2 (Reaction 5) (Ehn et al., 2014). If RO2 is an acyl peroxy radical, a carboxylic acid can be formed.
These reactions lead to further functionalisation, e.g. the formation of acids, alcohols, carbonyls or peroxides, and may also produce alkoxy radicals. Subsequently, alkoxy radicals can be converted by oxygen to a carbonyl if an α-hydrogen is present. Alkoxy radicals that lack this hydrogen will undergo isomerisation or decomposition via β-scission.
During ozonolysis experiments, OH radicals are produced and react with the precursor as well as the reaction products. This process occurs in the laboratory as well as in the actual atmosphere and increases the complexity of the degradation mechanisms. In the laboratory, one can scavenge the OH radicals produced by adding a compound, e.g. 2-butanol, that reacts rapidly with OH, thereby reducing OH. The OH scavenger reduces the OH concentration but leads to an increase in the HO2 and RO2 concentrations. This yields changes in the distribution of radicals and, subsequently, the radical-dependent chemistry (Keywood et al., 2004; Jonsson et al., 2008b). For example, the reaction of 2-butanol with OH produces HO2 radicals with a yield of 64 % (MCM v3.3.1), thereby increasing the HO2∕RO2 ratio. In laboratory experiments, these features can be employed in investigating the importance of various radicals/pathways for product distribution and subsequent SOA formation.
The Gothenburg Flow Reactor for Oxidation Studies at Low Temperatures (G-FROST) has been used in previous studies (Jonsson et al., 2006, 2008a, b; Faxon et al., 2018; Kristensen et al., 2014) to investigate the dependence of aerosol properties on different parameters (e.g. humidity and radical conditions). The G-FROST set-up has now been extended with a High-Resolution Time-of-Flight Chemical Ionisation Mass Spectrometer (HR-ToF-CIMS) that will provide insight into the chemical composition of the gas and particle phase via the connection to the Filter Inlet for Gases and AEROsols (FIGAERO). These new techniques allow for sensitive simultaneous detection in the gas and particle phases. Herein, an ionisation using acetate allows for the investigation of carboxylic acid formation. In the following, we analyse the carboxylic acid product spectrum of limonene. The goal is to detect major pathways and to compare the results with a model using the existing master chemical mechanism (MCM) that was primarily developed for gas-phase chemistry related to the impact on tropospheric ozone formation, but now is frequently used as a link to particle formation. This work considers (i) ozonolysis under dark conditions and NOx-free conditions (for various limonene concentrations) as well as the effect of humidity, OH scavenging and ozone level on carboxylic acid formation, and (ii) provides an outlook and suggestions for mechanistic gaps with the aim of eventually describing major acidic products found in the gas and particle phases under realistic atmospheric conditions, i.e. ozonolysis is performed in the absence of an OH scavenger under low concentration and humidity conditions.
Table 1Summary of experimental conditions and an overview of the selected results. OH, HO2 and RO2 are integrated concentrations (ppb s) as calculated with the model using the MCM. Water in the system increases the SOA mass and yield in the system, whereas the presence of 2-butanol decreases the SOA mass and yield. The contribution of the highest 10 compounds is linked to the complexity of the product distribution, i.e. if 10 compounds dominate then any minor product is less important and the composition appears less complex. The rate-normalised HO2∕RO2 ratio assumed rate coefficients of 8.8 and 9.1 for the RO2 and HO2 reactions, respectively (MCM v3.3.1).

* Some data for experiments 4-6, 10–12 and 28–29 are unavailable due to no/low particle concentrations or malfunctioning of the FIGAERO unit.
Oxidation studies of limonene in the presence of ozone have been performed under a variety of experimental conditions (Table 1). The experimental matrix was chosen to help understand the ozonolysis of limonene under dark, NOx-free conditions. The laminar-flow reactor approach is well-suited to investigating changes in experimental condition, e.g. dry vs. humid or high vs. low concentration. Due to the short residence time, the absolute concentration is higher than the ambient concentration even if the amount of limonene converted (a few ppb) and higher) is similar to larger simulation chamber studies. The conversion of any RO2 radicals is biased towards self-reaction, which is of importance in VOC-dominated rural forest conditions and in the indoor environment. The G-FROST system employed has been described in detail elsewhere (Jonsson et al., 2008b, a) and will only be presented briefly here. G-FROST consists of a laminar-flow reactor (a vertical Pyrex glass cylinder with a length of 191 cm, an inner diameter 10 cm and a halocarbon wax coating) in a temperature-controlled housing (see Fig. S1). The total inflow into the system was 1.6 L min−1 and the sample outflow was 0.94 L min−1, yielding an average residence time of 240 s. The aerosol was sampled with a funnel system from the centre part of the laminar flow, to minimise wall effects. Limonene (Alfa Aesar, (R)-(+)-limonene, 97 %) was added by passing synthetic air (laboratory zero air generator, N-GC-6000, Linde Gas) through a characteristic diffusion source. Limonene was then premixed with a dry or humidified bulk flow, with or without 2-butanol (Merck, p.a. > 99 %) as an OH scavenger. During each experiment, limonene concentrations were increased stepwise (15, 40, 150 ppb), while the temperature inside G-FROST was kept constant at 20 ∘C for either dry, relative humidity (RH) < 2 %, or 40 % RH conditions. Ozone (400, 1000, 5000 ppb) was generated by passing oxygen gas through a set of Pen-Ray® mercury lamps (UVP, λ 254 nm) and was added to G-FROST through a separate 6 mm Teflon line. The ozone level was kept constant during each experimental run.
A summary of the experimental conditions is provided in Table 1. The product distribution in the gas and particle phases was analysed with an acetate HR-ToF-CIMS (Aerodyne) (Bertram et al., 2011) coupled to the FIGAERO inlet (Lopez-Hilfiker et al., 2014). The reagent ion acetate is especially susceptible to acidic organic compounds such as carboxylic acids (Bertram et al., 2011). One may note that proxy acids also have high sensitivities (Lopez-Hilfiker et al., 2015) and that acetate ionisation has previously been used to detect nitro phenols (Mohr et al., 2013; Le Breton et al., 2019) and organic sulfates (Le Breton et al., 2019). However, here we assume carboxylic acids and peroxy acids to be the primarily compounds observed with the current set-up of the CIMS. The sensitivity used for larger carboxylic acids was 5.5 Hz ppt−1 (Le Breton et al., 2019). This sensitivity was used to estimate molar yields; even if one is cautious to provide absolute yields from this type of study, it provides indications on the product contribution. The sample flow from G-FROST was diluted with ultra-high purity (UHP) nitrogen gas and pumped at 2×4 L min−1 by two diaphragm pumps (KNF, N816.3KN.18) through the FIGAERO inlet. The dilution was necessary for analytical reasons and might evaporate some of the most volatile compounds from the condensed phase. However, the time between the dilution and the analysis was short (< 1 s), exactly the same for all conditions and a possible effect would somewhat mimic atmospheric conditions. Perfluoroheptanoic acid (Sigma Aldrich, 99 %) was used as the internal mass calibration standard. The gas-phase composition was determined via 60 min measurements, and particles were collected simultaneously on a 1 µm 24 mm Zefluor® PTFE filter (Pall Corporation). During desorption, the temperature was increased from 25 to 200 ∘C over a 50 min period (3.5 ∘C min−1) and kept constant at 200 ∘C for 10 min. Subsequently, UHP nitrogen gas was bubbled (flow rate: 0.02 L min−1) through acetic anhydride (Sigma Aldrich, Puriss p.a. ≥ 99 %) and diluted with a bulk flow of UHP nitrogen to 2.2 L min−1. This flow was reduced to 2 L min−1 using a critical orifice (O'Keefe Controls Co) and passed through a commercial 210Po alpha emitter (NDR, P-2021) to produce acetate reagent ions. A scanning mobility particle sizer (SMPS; CPC 3775 and DMA 3081, TSI Inc.) was used to measure the particle size distribution. The mass of the aerosol produced was determined, assuming a particle density of 1.4 g cm−3 (Hallquist et al., 2009). The CIMS data were analysed using the Tofware package (Tofwerk/Aerodyne) for Igor Pro (WaveMetrics). The data were acquired at 1 Hz and pre-averaged to 0.0167 Hz (1 min) for further analysis. To account for thermal decomposition (double or triple peaks in the desorption profile), the average (four desorption cycles per reaction condition) FIGAERO desorption profiles (thermograms) were analysed in Python 3.6.0 using the NumPy (v1.11.3), SciPy library (v0.18.1) and pandas (v0.19.2) packages. The exponentially modified Gaussian function (Foley and Dorsey, 1984) was used as a peak shape function for peak fitting of the thermograms (Fig. S2). The area of the fitted peaks was calculated by integrating along the given axis using the composite trapezoidal rule. A Spearman correlation analysis was carried out based on major products, experimental conditions and calculated radical concentrations. Compared with a standard correlation, the Spearman correlation is more robust to outliers and independent of any assumptions about the distribution of the data. Therefore, it was preferred to assess the degree of association between each dominant acid and the experimental parameters. The evaluation using a Spearman correlation is similar to other correlation methods giving 0, −1 and 1 for no correlation, a perfect negative correlation and a perfect positive correlation, respectively. All experiments have been modelled utilising an open access mechanism (MCM v3.3.1; see Table 1 for the initial conditions). The initial concentration of 2-butanol was set to 3×105 µg m−3 for the OH scavenger experiments (Pathak et al., 2012). Based on the calculations, the amount of reacted limonene was derived. The OH, HO2 and RO2 levels that enabled the calculation of the corresponding values integrated over a reaction time of 240 s were used in the Spearman correlation analysis.
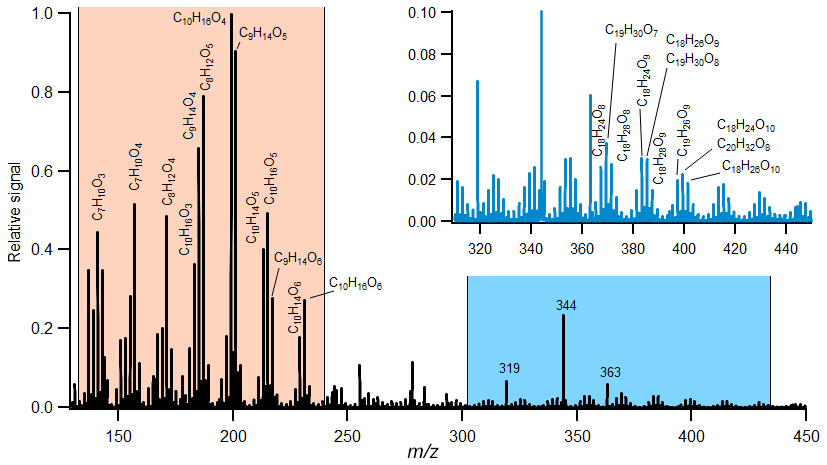
Figure 2Example of derived mass spectra of the condensed phase taken from the experiment (Experiment no. 21) with medium-ozone and high-limonene conditions with added OH scavenger. The regions with identified monomers (orange region) and selected dimers (blue region) are indicated. The peaks at 319, 344 and 363 are associated with the HPFA used for mass calibration. The complexity of this mass spectra is described using the fraction of 10 dominant product ions, and the spectra are compared to other experiments with similar conditions in Fig. 3.
A total of 33 different experiments were performed under various reaction conditions (Table 1). In the following, we will characterise the distribution of gas- and particle-phase organic acid. Figure 2 shows an example of a mass spectrum from one of the experiments. This experiment was carried out at medium-ozone and high-limonene concentrations with an estimated 8 % molar yield of large carboxylic acids. Over 100 different molecular formulas for acids were still identified, far exceeding the number of acids reported in previous studies (Leungsakul et al., 2005a; Jaoui et al., 2005, 2006; Rossignol et al., 2012, 2013; Walser et al., 2008; Glasius et al., 2000). Here, we focus on the analysis of acids with carbon numbers ranging from 7 to 10 (and the dimers formed from these acids). Typically, these acids represent 2 %–23 % of the reacted limonene on a molar basis, assuming an average reaction time and CIMS sensitivity (Table 1). The distribution between the gas and particle phase varied between compounds where the average particle fraction was between 5 % and 80 % depending on experimental conditions. The contribution of each acid to the total signal is calculated, and the highest 10 ion signals are selected from each experiment. This gave a total of 32 different molecular compositions, representing the greatest fraction (47 %–91 %) of the total signal.
The fraction corresponding to the sum of the 10 highest ions to the total signal can reveal the diversity of the product distribution for each condition. A low coverage indicates an experiment where several compounds with the same intensities are generated. Figure 3 shows the fraction of the 10 most prevalent ions for the comparable data to Fig. 2 (medium ozone, high limonene). This was the most complete sub-step (dry–humid, with and without scavenger) of the total matrix and the illustrated pairwise features (e.g. dry vs. humid) that were also common among other concentrations (e.g. for other condition we had missing particle-phase data, see Table 1). For these four selected experiments the estimated total yields of larger carboxylic acids (C7–C10) were very similar: 6 % (dry, scavenger), 8 % (humid, scavenger), 6 % (dry, no scavenger) and 7 % (humid, no scavenger).
In general, the particle-phase composition is more diverse (less dominated by the 10 top compounds) than the gas-phase composition (Fig. 3). The presence of water in the system also increases the diversity of the product distribution in both the gas and the particle phases, i.e. a small effect in Fig. 3 but consistent for all pairwise dry/humid experiments as shown in Table 1. Compared with lower ozone concentrations, higher concentrations generally give larger product diversity, owing to the greater possibility for exocyclic double-bond oxidation or unsaturated-acid oxidation that yields a wider variety of products. The OH reaction pathways are suppressed in experiments with an OH scavenger and the oxidation can then only occur via ozonolysis. This apparently reduces the oxidation product diversity of the particle phase, which is consistent with the findings of Watne et al. (2017). In that study, the volatility of limonene SOA produced via ozonolysis only was found to be more homogeneous than that of limonene produced via other/additional processes.
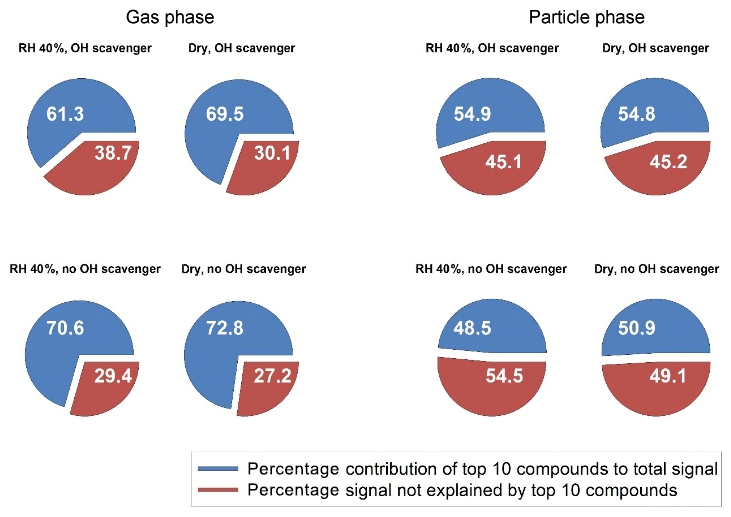
Figure 3Contribution of the highest 10 compounds (blue) to the total signal in the gas and particle phase. The red fraction shows the signal not explained by the 10 highest compounds. Thus, a larger red fraction indicates a more complex composition. Data are shown for selected experiments with 1000 ppb ozone and 150 ppb limonene under different conditions. The estimated total yield of larger carboxylic acids (C7–C10) for these four experiments were very similar: 6 % (dry, scavenger), 8 % (humid, scavenger), 6 % (dry, no scavenger) and 7 % (humid, no scavenger).
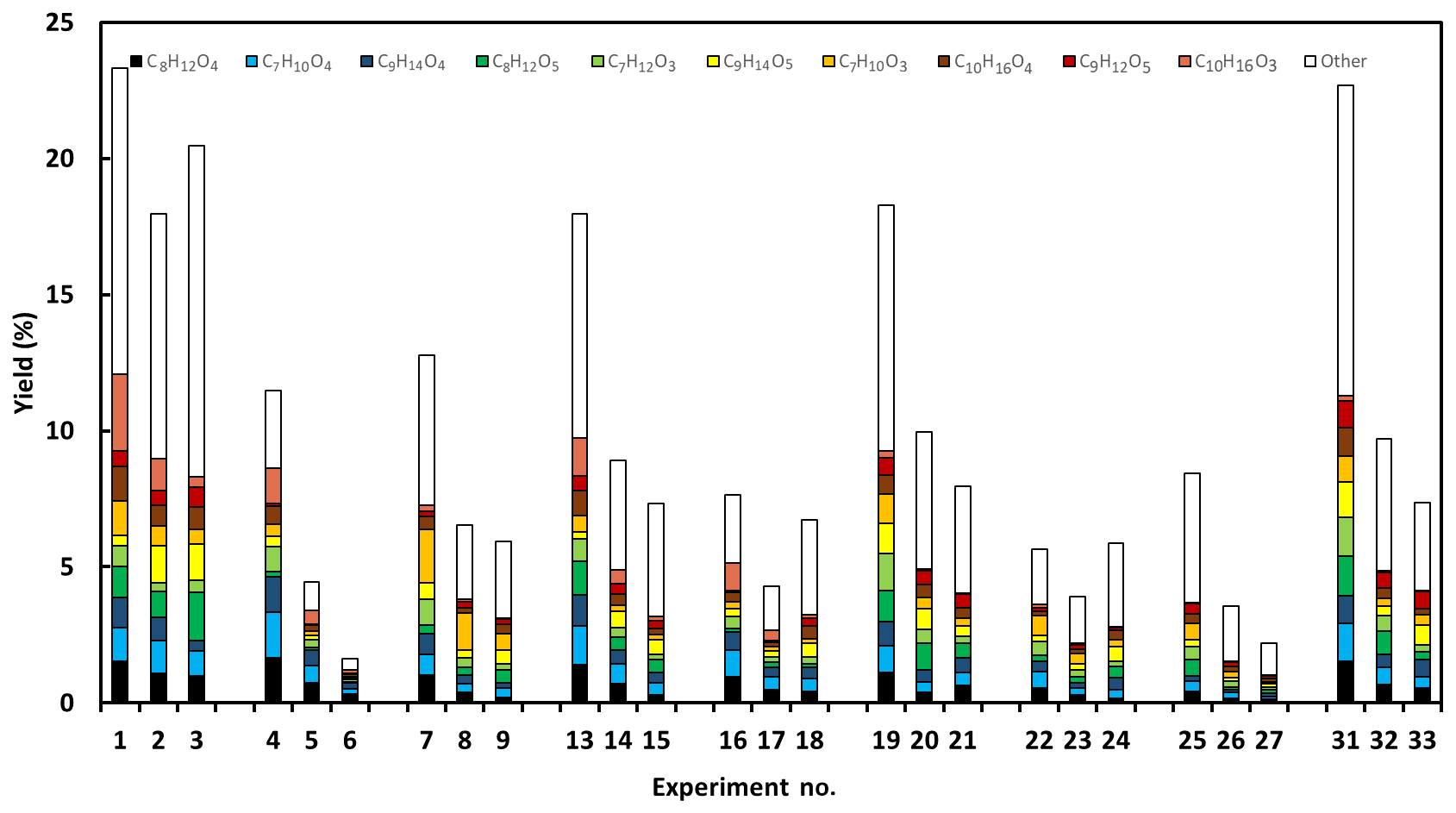
Figure 4The molar yields of identified C7–C10 acids. The most important acids (averaged over all experiments) are C7H10O3, C7H10O4, C8H12O4, C8H12O5, C9H14O4, C9H14O5, C10H16O3 and C10H16O4 and are illustrated using individual colours (see legend). Experiments 10–12 and 28–30 were removed due to uncompleted particle-phase characterisation.
Figure 4 shows the total molar yield of the acids identified in this study. One may note that the absolute yield presented here has significant uncertainties, although the relative importance of an acid can guide us in our mechanistic interpretation. Generally, the most important acids (averaged over all experiments) are C7H10O3, C7H10O4, C8H12O4, C8H12O5, C9H14O4, C9H14O5, C10H16O3 and C10H16O4 with yields at or below 1 %, which is in line with the yield of major acids in the study of Glasius et al. (2000). The most important acids in the present study are compared with an overview (Table S1) of previously reported carboxylic acids (C7–C10) resulting from limonene ozonolysis. Table S1 also illustrates the proposed structures of these acids based on the current literature. A total of 10 of the previously reported acid formulas are found in this study, while 3 acids, C7H10O6, C8H12O6 and C8H14O4, lie outside the 10 highest corresponding ions identified in any of our 33 experiments. Leungsakul et al. (2005a) and Walser et al. (2008) reported that C9H14O4 and C10H16O3 were the most and second-most dominant particle-phase compounds, respectively. However, in our study, the more oxidised (compared with C9H14O4 and C10H16O3) compounds, C9H14O5 and C10H16O4, are the dominant in the particle-phase. From Fig. 4 it is evident that the composition of acids is very complex and many compounds contribute to the total signal. Furthermore, among the most prominent acids, there is no acid that clearly dominates in any of the experiments. We should also remember that, in addition to the acids, there are many other product categories contributing to the product distribution, e.g. recently, Gong et al. (2018) carried out a similar study focusing on peroxides and carbonyls. Utilising the complexity to further understand the mechanism requires some strategy. Here we decide to start with the 10 most prominent ions for each experiment. Thus, we created a correlation matrix of 32 different molecular compositions, where each composition might include several isomers. The intensities measured for each compound are presented in the Supplement (Tables S2 and S3). In Figs. S3–S4, we show the corresponding correlations, using Spearman ranking, for each of the 32 different molecular compositions representing the majority of the ion signals both with and without OH scavenger (Fig. S3) and for both humid and dry conditions (Fig. S4). The results for the eight most important acid formulas, i.e. C7H10O3, C7H10O4, C8H12O4, C8H12O5, C9H14O4, C9H14O5, C10H16O3 and C10H16O4, are presented and discussed. These are all oxidation products with a mass ranging from 130 m∕z to 250 m∕z and are identified as carboxylic acids with carbon numbers ≤ 10. Based on other studies (Kristensen et al., 2013, 2014, 2016; Mohr et al., 2017; Witkowski and Gierczak, 2014), dimer formation is expected. These dimers are expected to contribute significantly to the particle phase. For the particle-phase data, compounds with mass above 300 m∕z are detected and are classified as dimer species if they have a carbon numbers > 10. These compounds only occur in the particle phase; however, the relative signals are significantly lower than those reported for dimer formation in a study on limonene with nitrate radicals (Faxon et al., 2018) or the ozonolysis of other terpenes such as α-pinene (Kristensen et al., 2016). In the present study the identified products must be acids, as we apply CI using the acetate ion. We conclude that, although dimer formation may occur (in general), no important acidic dimers are formed in the system. Consequently, we will focus on the formation of the monomer acids.
3.1 Water effect
Generally, most of the 32 top ions have higher signals in humidity experiments than in other environments (see Tables S2 and S3 and the correlation matrix in Fig. S4). The opposite is true for the 400 ppb ozonolysis-only (OH-scavenged) cases (gas and particle phases). For experiments with OH scavenger, the importance of water is evidenced by the prominent formation of gas-phase C10H16O3, C9H16O3, C9H14O3 and C8H14O3. The water dependence of these acids is less pronounced in the mixed oxidation cases (except for C8H14O3), than in other cases, but water seems to be favourable for the formation of other acids, such as C8H10O4−5 and C7H10O2−3. In general, water enhances the formation of the particle-phase acids. This concurs with the findings of Jonsson et al. (2006), who reported an increase in the SOA number and mass under humid conditions. The authors attributed this to (i) an increase in the number of low-volatility products with increasing water concentration during the ozonolysis of limonene, and (ii) the water effect on SOA formation to C10H16O3 formation. For the humidity experiments considered in the present study, we observe a considerable increase and a slight increase in the formation of gas-phase C10H16O3 and particle-phase C10H16O3, respectively. Assuming that the humidity effect on C10H16O3 production is responsible for the SOA dependence on humidity, the subsequent transformation of condensed material is required as the particulate phase is deficient in C10H16O3.
3.2 Radical effect
Consistent with previously reported results on the SOA mass (Jonsson et al., 2008b; Pathak et al., 2012), the intensities of most acids in the low- and medium-ozone cases are higher for experiments employing mixed oxidation than for experiments employing an OH scavenger. For low-ozone and low-VOC experiments, the scavenger-provided SOA mass decreases with 2-butanol addition, as previously reported by Jonsson et al. (2008a), although the effect observed here is weaker than the effect reported in that work. However, for relatively high concentrations of limonene, the opposite effect is observed, i.e. the SOA mass increases with the use of a scavenger. Notably, this effect occurs independently of the acid-intensity behaviour, and may have resulted from the fact that (i) the SOAs associated with mixed oxidation are quite volatile and (ii) increased oxidation in the presence of OH, rather than converting semi-volatiles to low/extremely low volatiles, converts volatiles/intermediate volatiles to semi-volatile species, as suggested by Pathak et al. (2012); these species are then lost during the dilution process. Another possibility is that changes in the chemistry affect nucleation, as indicated by a size-distribution shift to smaller sizes which (compared with larger sizes) are more susceptible to evaporative losses in the dilution step. Separation of these effects during the experiments is impossible; hence, the SOA formation potential associated with mixed oxidation may have been underestimated in this study. Owing to the sufficiently low ozone levels employed in the low and medium experiments, OH has an influence on the reaction pathways. At the highest ozone level, however, the intensities of acids associated with mixed oxidation are lower than those resulting from the use of an OH scavenger. To investigate the effect of radical chemistry on the reaction pathways leading to the observed carboxylic acids, the OH, HO2 and RO2 concentrations are calculated and integrated using the model for each experiment (Table 1). Regarding correlation (Fig. S3), a comparison of the mixed oxidation cases reveals that the formation of most gas-phase acids (e.g. C10H16O3, C9H14O4 and C7H10O3) decreases with an increase in the amount of OH radicals in the system. The HO2∕RO2 ratio only has a small influence on the mixed oxidation. However, when an OH scavenger is used, the amount of gas-phase products (C10H16O3, C10H16O4, C9H14O4 and C8H14O3) decreases considerably with increasing HO2∕RO2. The general influence of OH on acid formation is most pronounced for experiments performed under dry conditions. Under these conditions, OH and HO2∕RO2 have a significant effect on the formation of C10H16O3, C9H14O3 and C7H10O4. For example, C10H16O3 and C9H14O3 formation increases with increasing OH and decreasing HO2∕RO2. The opposite is true for C7H10O4 formation, which decreases with increasing OH and decreasing HO2∕RO2.
3.3 Effect of excess ozone
Experiments with high ozone levels are performed to assess the effect of excess ozone on acidic oxidation products. The aim is to oxidise (with ozone) the remaining double bond of the unsaturated carboxylic acids produced. The results show that ozone has a distinct negative impact on C7H10O2−3 in the pure ozonolysis cases (see correlation matrix in Fig. S3); hence, we conclude that those compounds are unsaturated. Furthermore, the levels of gas-phase C10H16O3, C9H16O3, C9H14O3−4 and C8H14O3 are positively correlated with ozone in the absence of OH. For C10H16O3, this is surprising as this compound is assumed to be limononic acid, which is an unsaturated compound. This positive correlation may have resulted from the fact that the production of C10H16O3 dominates over the removal (via ozonolysis) of the remaining double bond. The correlation with ozone is negative for most acids in the presence of OH and is most pronounced for gas-phase C10H16O3, C9H14O3 and C8H12O3. The negative ozone correlation observed for mixed oxidation cases considering C10H16O3 and C9H14O3 concurs with the modelling results of a previous study that assessed the influence of ozone on limonene oxidation (Pathak et al., 2012). A positive (albeit slightly positive) correlation with ozone is only observed for particle-phase C8H14O3. The acid–ozone correlation obtained for humid conditions differs significantly from that obtained for dry conditions. The negative acid–ozone correlation is quite pronounced in the dry experiment cases and becomes increasingly negative (in general) for acids with relatively low carbon numbers, which is a trend that is unique to these experiments. The level of C10H16O3 (especially the particle-phase) is positively correlated with ozone levels in the dry experiments. Generally, the amount of gas-phase acids has a stronger positive correlation with the limonene consumption (Δlimonene) under humid conditions compared with dry experiments. In the dry experiments, C10H16O5 and C9H14O4 are the only acids with a strong positive correlation to Δ limonene. Compared with the occurrence of gas-phase acids, the occurrence of particle-phase acids is (in general) more strongly correlated with Δ limonene.
3.4 Model results and comparison with experiments
Model calculations using the scheme presented by the master chemical mechanism (MCM v3.3.1; Saunders et al., 2003) were performed for all 33 experimental conditions, in order to calculate Δ limonene and radical concentrations as well as product distributions, based on the experimental conditions. The model only considered the gas-phase scheme of the MCM. Most of the previously reported carboxylic acid molecular formulas (see Table S1) are included in the MCM, which was originally developed to provide accurate, robust and current information regarding the role of specific organic compounds in ground-level ozone formation, in relation to air-quality policy development in Europe. Over the years, the MCM has been employed for models in studies linked to SOA formation (Jenkin, 2004), although this mechanism is still under development with respect to capturing descriptions on the fraction of low-volatility and often very oxygenated organic compounds (Barley et al., 2011).
Generally, the model provides a small variation in the total molar yield for the large carboxylic acids (C7–C10) of 10 %–15 %, whereas the experiments show larger variability (2 %–23 % molar yield); this is plausible due to the complication of aerosol formation not covered by the model. A key to understanding the chemical mechanism leading to various products is the radical distribution. The experimental set-up requiring the product distribution to be measured at the end of the flow reactor restricts dynamic information. However, the variation of the radical distribution between experiments is illustrated in Table 1. Here the values of radicals are given as the integral concentration over the reaction time (unit ppb s). Furthermore, the integral HO2∕RO2 ratio is presented along with a rate-normalised ratio of these reactions, i.e. the HO2+RO2 reaction is rapid and the typical rate constant is 1 order of magnitude larger than that of the RO2+RO2 self-reaction (Orlando and Tyndall, 2012).
Regarding oxidant/radical variation, the modelled OH levels decrease with the initial limonene concentration, except for the highest ozone cases. High-ozone experiments yield the highest OH dose. The model results show that the HO2∕RO2 ratio in experiments employing the OH scavenger 2-butanol is approximately 1 order of magnitude higher than that of the mixed oxidant experiments. This higher ratio results from the HO2 radicals generated by the reaction of 2-butanol with OH and have more influence over the HO2+RO2 reaction in the experiment with a scavenger. However, the RO2 self-reaction is still the major pathway in these experiments, with twice the normalised rate of the HO2 reaction. One may note that the RO2 reaction rates are very much structure dependent and might be faster or slower than the assumed rates (see Jenkin et al. (2019) for a recent review on RO2 chemistry).
In the MCMv3.3.1, 25 closed-shell carboxylic acids with 16 different chemical formulas are included for limonene. We identify 11 of the 16 acids (Tables S4 and S5; all MCM species used in the model are presented in Table S6). C9H14O3 and C9H14O4 are the most dominant and second-most dominant acids in the 33 modelled experiments, respectively. C10H16O3 (LIMONONIC), formed by the reaction of sCI + water, is the only acid that exhibits an overall positive water dependence. The model calculations predict that water should also have a positive influence on KLIMONONIC and CO25C6CO2H. However, this influence is undetectable in our experiments, owing to the extremely low concentrations of these compounds and the stronger influence exerted on other compounds with the same molecular mass.
The model predicts that the presence of OH radicals has a greater influence on the product distribution, compared with the presence of water. Most individual species from the MCM exert a strong positive OH-chemistry effect in the model, except for LIMONONIC (C10H16O3), C823CO3H (C9H14O5), C823OOH (C8H14O4) and C825OOH (C8H12O5). In all cases, the concentrations of the last three compounds estimated with the model are highest when the OH chemistry is “turned off” (2-butanol added in model). C82CO2H (C9H14O3) is produced to a lesser extent under humid and high-ozone conditions and for the highest OH conditions. It was produced to a higher extent under medium and low ozone and for the medium and lowest OH conditions. In the presence of OH chemistry, the LIMONONIC concentration is lower under humid conditions than under dry conditions. The presence of OH is essential for the formation of numerous compounds and only yields a significant concentration in the absence of 2-butanol, i.e. the modelled concentrations are close to zero in the presence of 2-butanol. For example, C731CO2H, KLIMONIC and KLIMONONIC are formed by ozone attack on the limona ketone which, in the model, is formed by the initial OH attack on the exocyclic double bond of limonene. Owing to the presence of 2-butanol, this attack on the double bond is reduced, minimising the amount of products generated. The correlation results for the humid and dry cases show that C823CO3H, C823OOH and C825OOH are negatively correlated with OH levels in the model. The reaction with OH represents the only destruction pathway for the acids produced in the model (even if unsaturated). This negative correlation indicates that the OH-induced destruction of the respective acid dominates over acid production as the OH levels increase. However, the reactions of unsaturated acids with ozone are not included in the MCM.
The experimental results reveal that the four dominant compounds are C8H12O4, C8H12O5, C9H14O4 and C9H14O5. However, C9H14O3, which plays only a minor role in the experiments, represents the dominant compound in the modelling results. C8H12O4, which contributes significantly to the experimental results, is characterised by a medium-level contribution to the model. C8H12O4 and C8H12O5 exhibit a positive OH-dependence in the model that is consistent with the gas-phase results obtained for C8H12O4 under humid low-ozone and all medium-ozone experiments. The estimated concentration of C8H12O5 is lower in the presence of OH chemistry for most conditions except for humid low-ozone experiments. The model reveals a positive OH dependence and a negative OH dependence for C9H14O4 and C9H14O5, respectively. The behaviour of the C9H14O4 gas phase concurs with the model results for low- and medium-ozone experiments. For the highest ozone-level experiments, the levels observed for mixed oxidation are lower than those observed for oxidation in the presence of an OH scavenger. The OH dependence of C9H14O5 in the experimental results differs from the overall negative OH dependence of modelled C9H14O5. In contrast to the model predictions, the C7 acids C7H10O4 and C7H12O3 contribute significantly to the gas-phase results and exhibit only a weak OH dependency. The model predicts a weak OH dependence for C10H16O3 which is in stark contrast to the strong dependence revealed by the experimental results. Overall, most acids exhibit a positive RH dependence in the medium-ozone and humid low-ozone cases, which is a behaviour that is lacking from the modelling results. However, consistent with the modelling results, water in the system increases the concentration of C10H16O3 by a factor of 2. This hold true for all cases, except for the highest ozone cases where the concentrations observed in the experiments are higher than the values predicted for dry conditions. In conclusion, significant concentrations of 11 acids (from a total of 16) included in the MCM are measured with the CIMS. The model predictions are, in some cases, inconsistent with the measurement results, with the most notable inconsistencies occurring for the OH dependency.

Figure 5Proposed reaction mechanisms for secondary ozone chemistry of limononic (R1), limonic (R2) and limonalic (R3) acid. The dotted bond shows where R1-3 is connected to the 2-methyl-ethenyl group (i.e. ) in the parent compounds.
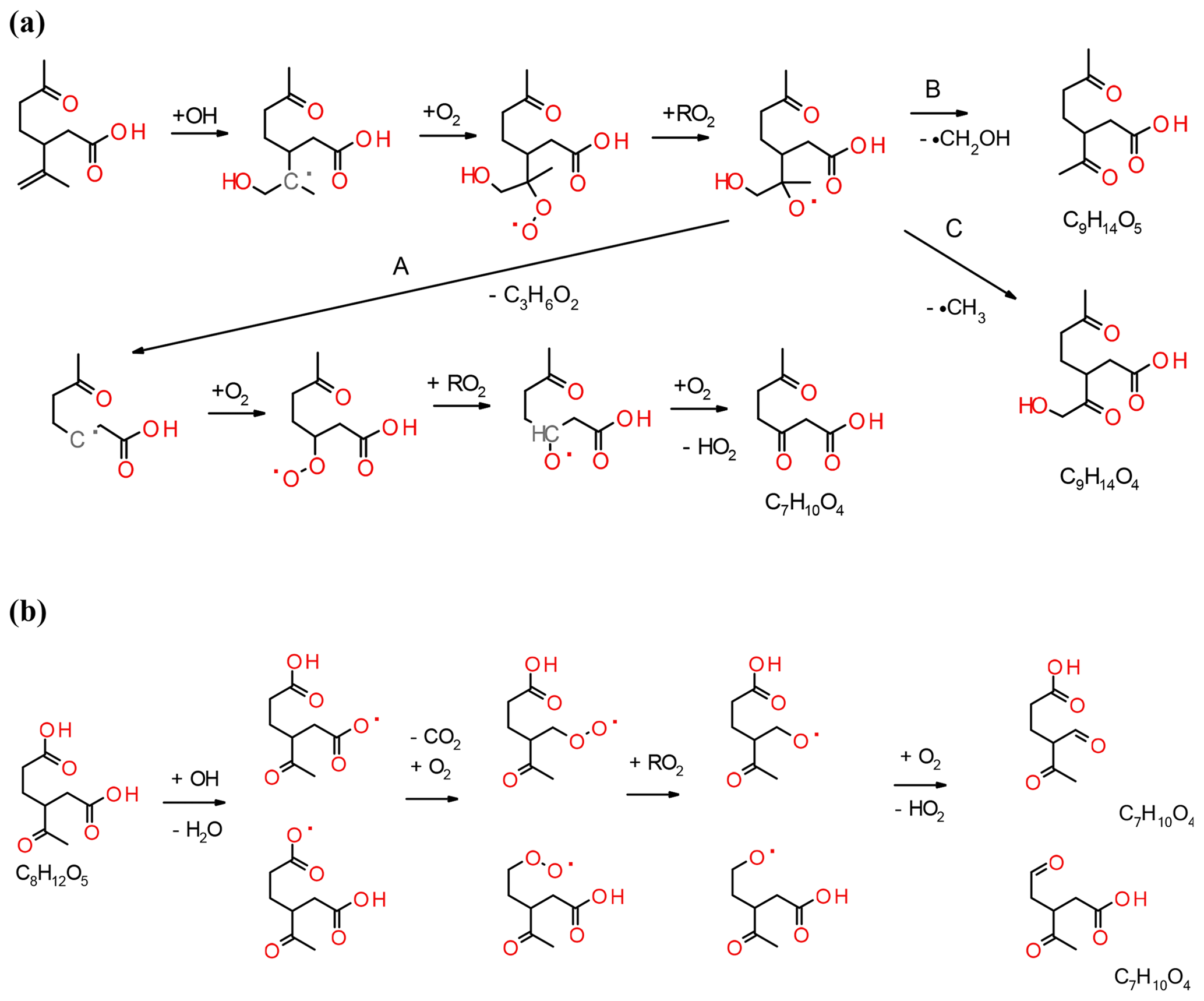
Figure 6(a) Secondary chemistry of the addition of OH to the unsaturated double bond of the primary product limononic acid and the subsequent formation of C7H10O4, C9H14O5 and C9H14O4. (b) Secondary chemistry of a selected saturated carboxylic acid product (C8H12O5) giving the observed C7 products.
The formation and dependence of the eight most prominent ions in the experiments are only partly explained by the model and e.g. the reaction pathways that form compounds with the molecular formulas C7H10O3 and C10H16O4 are absent. In the following, we propose reaction pathways for explaining the formation of some ions not accounted for in the model and propose additional pathways for compounds already included in the MCM. Examples include the reactions of unsaturated acid products with ozone or the formation of C10 acyl radicals via the hot acid channel (see pathway A in Fig. 1). The largest discrepancy between model and experimental results is observed for the formation of compounds, such as the group of C7 acids or ketolimononic, -limonic or -limonalic acid which are OH-dependent in the model but are OH-independent in the observations. The dominance of C9H14O4 and the positive correlation with ozone can be explained by the additional formation of ketolimononic acid via reaction pathways, as seen in Fig. 5. Here, ozone attacks the double bond of the primary product limononic acid, forming a CI. In the case of exocyclic CI, sCI can be formed directly and the remaining CI* are usually more effectively stabilised than endocyclic CI*; therefore, a larger yield of sCI can be expected. The sCI produced can produce ketolimononic acid via the water channel (see Fig. 5). Ketolimonalic (C8H12O4) and ketolimonic (C8H12O5) acid may be formed via the reaction of limonalic (R3 in Fig. 5, C9H14O3) and limonic acid (R2 in Fig. 5, C9H14O4), respectively, with ozone. The formation of a vinyl hydro peroxide (VHP) and its subsequent decomposition via OH elimination and oxygen addition to the alkyl radical yields an alkyl peroxy radical. The bimolecular reaction of the alkyl peroxy with other RO2 can lead to an alkoxy radical which can then form a carbonyl and HO2 upon reacting with oxygen. This reaction chain may explain the formation of C9H12O5 and C8H10O5−6.
The model predictions for cases with and without the scavenger only differ slightly, but the reaction pathway involving OH is an important contributor to C10H16O3 formation in the experiments. This becomes especially clear when dry experiments with/without OH (with no possibility for the water pathway) are compared. The pathway leading to the formation of C10H16O3 via the hot acid channel from the anti-CI* (see Fig. 1) is also neglected by the model. Figure 6 illustrates how the remaining double bond can also be attacked by OH which would lead to the formation of an alkyl radical and the subsequent addition of O2. The reaction pathways shown in Fig. 6 lead to the observed acid formation and may explain the formation of C7H10O4, C9H14O5 and C9H14O4. The alkoxy radical produced will probably follow pathway A which produces the most stable radical and, subsequently, C7H10O4. This pathway involves two bimolecular steps and is positively correlated with RO2 levels in the system. Saturated compounds, although non-reactive with ozone, are susceptible to secondary chemical reactions induced by OH. Figure 6b illustrates how the fate of the saturated compounds depends on the relative reactivity of different sites to OH, and may include the abstraction of the acidic hydrogen followed by the splitting off of CO2; the subsequent bimolecular reactions will produce C7H10O4. This reaction competes with the abstraction of the tertiary hydrogen, but will lead to products that are inconsequential to the present experimental results. The formation of C10H16O4 results from processes other than ozonolysis or OH attack on the exocyclic double bond of an acid product due to the fragmentation of the POZ produced and excessively high resulting oxygen numbers. C10H16O4 may have resulted from the reaction of an acyl peroxy radical with HO2 (see Fig. 1) and the formation of a peroxy acid. However, the pathway for C7H10O3 formation remains unclear. For the particle phase, C8H12O5 and C9H14O5 are the dominant compounds in most of the experiments performed in this study, whereas C10H16O3 (a major gas-phase compound) only represents a minor contributor to this phase. C10H16O4 is excluded in the model, but plays a role in the particle-phase results. The formation of C10H16O4 is positively correlated with the presence of RO2 and HO2. C9H14O3 and C9H14O4 are the dominant acids in the model calculations, but are only minor compounds in the particle phase. C9H14O4 formation only seems to occur in experiments with the highest limonene content. Rapid autoxidation for the formation of highly oxidised molecules (HOMs) has recently gained significant attention (Ehn et al., 2014). This autoxidation proceeds via intramolecular H abstraction of RO2 and the subsequent formation of hydroperoxide groups. RO2 lifetimes in low-NOx environments are usually sufficient for the occurrence of this process (Orlando and Tyndall, 2012). During this process, large amounts of oxygen are rapidly introduced into the molecules, leading to a decrease in their vapour pressure. Most of the RO2 will originate from the VHP channel, in the case of limonene ozonolysis, and products will probably be non-identifiable unless the radical termination reaction yields a carboxylic acid. Even if the compounds formed contain one or more carboxylic acid group, the corresponding low vapour pressure may be undetectable by the FIGAERO inlet used. Jokinen et al. (2014) investigated the formation of HOM from limonene and found that highly oxygenated monomers (C10) and dimers (C20) with oxygen numbers ranging from 5 to 11 and 7 to 18, respectively, play a crucial role in this formation. Only one compound with the same chemical formula (C9H14O5) has been found in this study, but it is unclear if the chemical structure is the same.

Figure 7Proposed formation of observed dimers (C18H28O8, C19H20O7 and C19H30O8) from monomer–Criegee reactions.
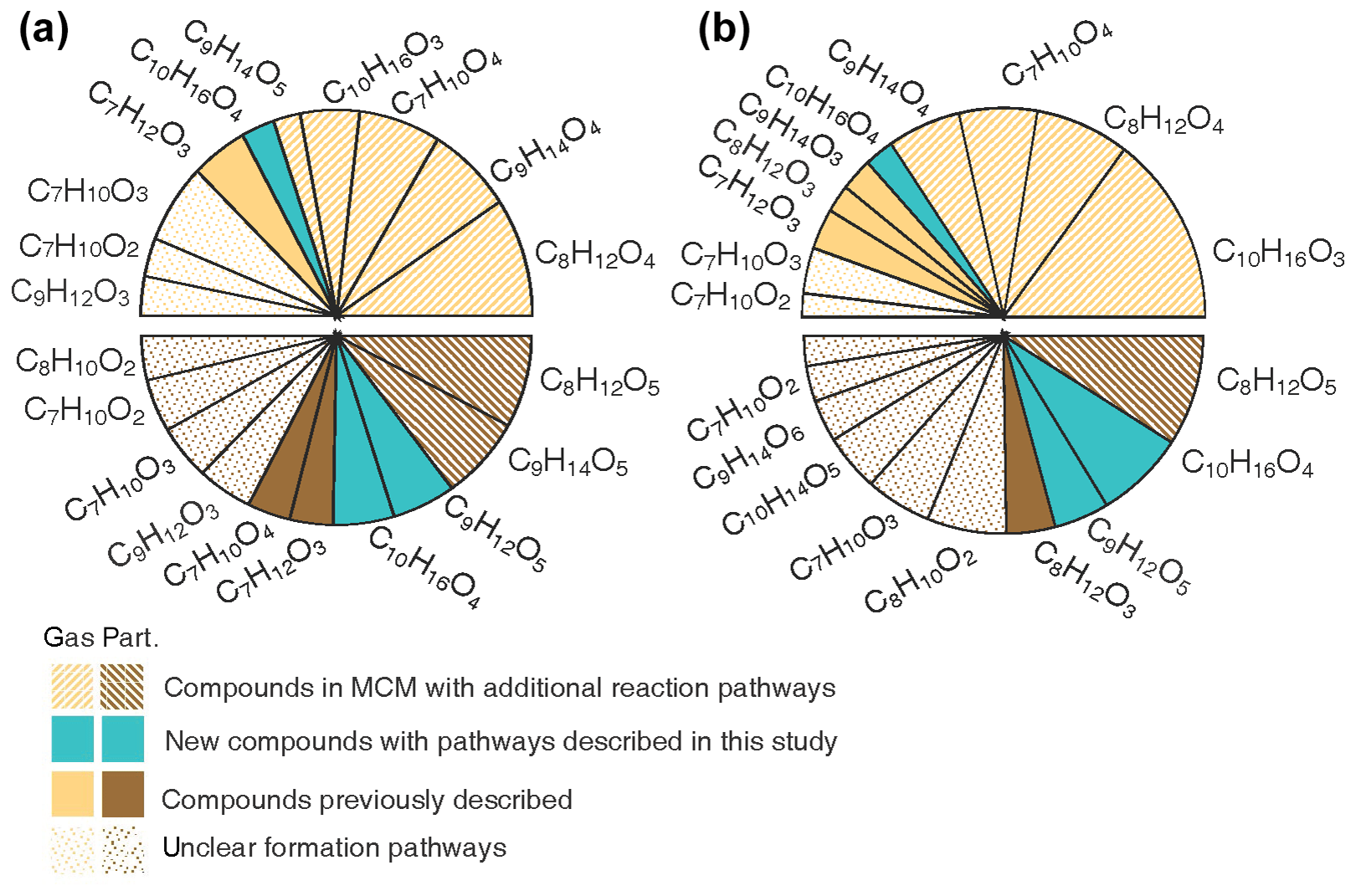
Figure 8Pie charts showing the percentage contribution of each compound for the 10 major compounds observed. The top half of the pies shows the gas-phase data, and the bottom half of the pies shows the particle-phase data. The compounds are classified according to current knowledge, i.e. previously described reaction mechanism, mechanism suggested in this study and unclear formation pathways. (a) The averaged contribution for all experiments. (b) The specific contributions for Experiment 1, which had the lowest concentrations of reactants with mixed oxidants and humid conditions.
In this study, dimers have exclusively been detected in the particle phase and are absent from the gas phase, owing to their potentially low vapour pressure. The formation of dimer esters from α-pinene ozonolysis has recently been investigated by Kristensen et al. (2016). In that work, the reaction of sCI with carboxylic acids, suggested as the formation pathway in the gas phase, was followed by partitioning into the particle phase. Consequently, the carboxylic acid group is lost in the esterification process which may explain the relatively low signals observed for acidic dimers in the present study. A potential acidic dimer ester will only be detectable if the dimer has a carboxylic acid group, as in the case of a di- or tricarboxylic acid, or if the sCI carries a carboxylic acid group. Unsaturated dimers may react with ozone. C18H28O8 and C19H30O7 can form via the reaction of the endocyclic limonene sCI with ketolimonic (C8H12O5) or limonic (C9H14O4) acid, respectively (see Fig. 7). In addition, C19H30O8 may be formed from the dimerisation reaction of limononic acid and the limononic-sCI. Gas-phase dimerisation reactions of dominating C7–C10 acids with sCI account for only some of the dimer formulas. Reactions of acids with relatively small carbon numbers (< C7), RO2 dimerisation reactions or condensed-phase reactions may account for the other formulas.
Figure 8a provides an overview of the most important acidic compounds found in this study. These are identified by comparing the average contribution of each compound to all 33 experiments. Explicit formation pathways for the C10H16O4 and C9H12O5 compounds implemented in the model and additional reaction pathways for C7H10O4, C8H12O4−5, C9H14O4−5 and C10H16O3 are proposed. Structures for C10H14O5 and C10H16O4 have been proposed in previous studies (Jaoui et al., 2006; Rossignol et al., 2012; Leungsakul et al., 2005a; Walser et al., 2008; Glasius et al., 2000), but the current mechanistic understanding is inadequate to explain the formation of compounds with the proposed structures. In Fig. 8a, we show that the mechanisms proposed in this work can improve the qualitative understanding of the formation characterising (on average) 65 % of the dominant gas-phase compounds and 50 % of the particle-phase compounds. Notably, the particle-phase data correspond partly to compounds with a low oxygen content (2–3 oxygen), and their formation and negative correlation with ozone remain unclear and require further study. However, only a few acidic dimers are detected. This may have resulted from the lack of evaporation of these dimers (i.e. as acidic dimers) or the loss of the acid functional group from potential acid monomer precursors during the dimer formation, as suggested in previous studies (Kristensen et al., 2016; Witkowski and Gierczak, 2014; Wang et al., 2016). Experiment 1, performed at low concentrations, for mixed oxidants and under humid conditions, should best represent atmospheric conditions. The summarised signal of the highest 10 acids in Experiment 1 can be qualitatively attributed to 89 % of the gas phase, and the proposed mechanisms in this study account for 74 % of the total signal (see Fig. 8b). The particle-phase composition can be qualitatively explained (by up to 42 %) by the mechanisms proposed in this work. The relatively large percentage of unexplained signal in the atmospheric case will result in large uncertainties when the acidic-particle phase composition of limonene SOA is modelled based on existing mechanisms (e.g. MCM) and partitioning theory.
For a more quantitative mechanism (compared with the mechanism considered), the inclusion of non-acidic products is required for a complete picture of the oxidation products. Furthermore, secondary and tertiary chemistry must be considered when the oxidation of compounds is modelled. Subsequent aerosol formation as well as dimerisation and condensed-phase reactions must also be evaluated.
The data used in this study are available upon request from the authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-13037-2019-supplement.
The experiments were designed by JH with support from CF. Data collection and interpretation were carried out by JH with support from AL. MH designed the project. The paper was written by JH with support from MH and TM. All co-authors provided article feedback and comments.
The authors declare that they have no conflict of interest.
This research has been supported by the Vetenskapsrådet (grant nos. 2013-06917 and 2014-05332) and the Svenska Forskningsrådet Formas (grant no. 2015-1537).
This paper was edited by Alexander Laskin and reviewed by two anonymous referees.
Atkinson, R., Aschmann, S. M., Arey, J., and Shorees, B.: Formation of OH radicals in the gas phase reactions of O3 with a series of terpenes, J. Geophys. Res., 97, 6065–6073, https://doi.org/10.1029/92jd00062, 1992.
Barley, M. H., Topping, D., Lowe, D., Utembe, S., and McFiggans, G.: The sensitivity of secondary organic aerosol (SOA) component partitioning to the predictions of component properties – Part 3: Investigation of condensed compounds generated by a near-explicit model of VOC oxidation, Atmos. Chem. Phys., 11, 13145–13159, https://doi.org/10.5194/acp-11-13145-2011, 2011.
Bertram, T. H., Kimmel, J. R., Crisp, T. A., Ryder, O. S., Yatavelli, R. L. N., Thornton, J. A., Cubison, M. J., Gonin, M., and Worsnop, D. R.: A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 4, 1471–1479, https://doi.org/10.5194/amt-4-1471-2011, 2011.
Brown, S. K., Sim, M. R., Abramson, M. J., and Gray, C. N.: Concentrations of Volatile Organic-Compounds in Indoor Air – a Review, Indoor Air, 4, 123–134, https://doi.org/10.1111/j.1600-0668.1994.t01-2-00007.x, 1994.
Carslaw, N.: A mechanistic study of limonene oxidation products and pathways following cleaning activities, Atmos. Environ., 80, 507–513, https://doi.org/10.1016/j.atmosenv.2013.08.034, 2013.
Chen, J. J. and Griffin, R. J.: Modeling secondary organic aerosol formation from oxidation of alpha-pinene, beta-pinene, and d-limonene, Atmos. Environ., 39, 7731–7744, https://doi.org/10.1016/j.atmosenv.2005.05.049, 2005.
Chen, X. and Hopke, P. K.: A chamber study of secondary organic aerosol formation by limonene ozonolysis, Indoor Air, 20, 320–328, https://doi.org/10.1111/j.1600-0668.2010.00656.x, 2010.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018.
Foley, J. P., and Dorsey, J. G.: A Review of the Exponentially Modified Gaussian (Emg) Function – Evaluation and Subsequent Calculation of Universal Data, J. Chromatogr. Sci., 22, 40–46, https://doi.org/10.1093/chromsci/22.1.40, 1984.
Glasius, M., Lahaniati, M., Calogirou, A., Di Bella, D., Jensen, N. R., Hjorth, J., Kotzias, D., and Larsen, B. R.: Carboxylic Acids in Secondary Aerosols from Oxidation of Cyclic Monoterpenes by Ozone, Environ. Sci. Technol., 34, 1001–1010, 2000.
Glasius, M. and Goldstein, A. H.: Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry, Environ. Sci. Technol., 50, 2754–2764, https://doi.org/10.1021/acs.est.5b05105, 2016.
Gong, Y., Chen, Z., and Li, H.: The oxidation regime and SOA composition in limonene ozonolysis: roles of different double bonds, radicals, and water, Atmos. Chem. Phys., 18, 15105–15123, https://doi.org/10.5194/acp-18-15105-2018, 2018.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., and Edney, E. O.: Identification and quantification of aerosol polar oxygenated compounds bearing carboxylic or hydroxyl groups. 2. Organic tracer compounds from monoterpenes, Environ. Sci. Technol., 39, 5661–5673, 2005.
Jaoui, M., Corse, E., Kleindienst, T. E., Offenberg, J. H., Lewandowski, M., and Edney, E. O.: Analysis of Secondary Organic Aerosol Compounds from the Photooxidation ofd-Limonene in the Presence of NOx and their Detection in Ambient PM2.5, Environ. Sci. Technol., 40, 3819–3828, https://doi.org/10.1021/es052566z, 2006.
Jenkin, M. E.: Modelling the formation and composition of secondary organic aerosol from α- and β-pinene ozonolysis using MCM v3, Atmos. Chem. Phys., 4, 1741–1757, https://doi.org/10.5194/acp-4-1741-2004, 2004.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jokinen, T., Sipila, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: Impact of Humidity on the Ozone Initiated Oxidation of Limonene, Δ3-Carene, and α-Pinene, Environ. Sci. Technol., 40, 188–194, https://doi.org/10.1021/es051163w, 2006.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: The effect of temperature and water on secondary organic aerosol formation from ozonolysis of limonene, Δ3-carene and α-pinene, Atmos. Chem. Phys., 8, 6541–6549, https://doi.org/10.5194/acp-8-6541-2008, 2008a.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: Influence of OH Scavenger on the Water Effect on Secondary Organic Aerosol Formation from Ozonolysis of Limonene, Δ3-Carene, and α-Pinene, Environ. Sci. Technol., 42, 5938–5944, https://doi.org/10.1021/es702508y, 2008b.
Keywood, M. D., Kroll, J. H., Varutbangkul, V., Bahreini, R., Flagan, R. C., and Seinfeld, J. H.: Secondary Organic Aerosol Formation from Cyclohexene Ozonolysis: Effect of OH Scavenger and the Role of Radical Chemistry, Environ. Sci. Technol., 38, 3343–3350, https://doi.org/10.1021/es049725j, 2004.
Koch, S., Winterhalter, R., Uherek, E., Kolloff, A., Neeb, P., and Moortgat, G. K.: Formation of new particles in the gas-phase ozonolysis of monoterpenes, Atmos. Environ., 34, 4031–4042, 2000.
Kristensen, K., Enggrob, K. L., King, S. M., Worton, D. R., Platt, S. M., Mortensen, R., Rosenoern, T., Surratt, J. D., Bilde, M., Goldstein, A. H., and Glasius, M.: Formation and occurrence of dimer esters of pinene oxidation products in atmospheric aerosols, Atmos. Chem. Phys., 13, 3763–3776, https://doi.org/10.5194/acp-13-3763-2013, 2013.
Kristensen, K., Cui, T., Zhang, H., Gold, A., Glasius, M., and Surratt, J. D.: Dimers in α-pinene secondary organic aerosol: effect of hydroxyl radical, ozone, relative humidity and aerosol acidity, Atmos. Chem. Phys., 14, 4201–4218, https://doi.org/10.5194/acp-14-4201-2014, 2014.
Kristensen, K., Watne, Å. K., Hammes, J., Lutz, A., Petäjä, T., Hallquist, M., Bilde, M., and Glasius, M.: High-Molecular Weight Dimer Esters Are Major Products in Aerosols from α-Pinene Ozonolysis and the Boreal Forest, Environ. Sci. Technol. Lett., 3, 280–285, https://doi.org/10.1021/acs.estlett.6b00152, 2016.
Langer, S., Moldanova, J., Arrhenius, K., Ljungstrom, E., and Ekberg, L.: Ultrafine particles produced by ozone/limonene reactions in indoor air under low/closed ventilation conditions, Atmos. Environ., 42, 4149–4159, https://doi.org/10.1016/j.atmosenv.2008.01.034, 2008.
Le Breton, M., Psichoudaki, M., Hallquist, M., Watne, A. K., Lutz, A., and Hallquist, A. M.: Application of a FIGAERO ToF CIMS for on-line characterization of real-world fresh and aged particle emissions from buses, Aerosol Sci. Technol., 53, 244–259, https://doi.org/10.1080/02786826.2019.1566592, 2019.
Leungsakul, S., Jaoui, M., and Kamens, R. M.: Kinetic mechanism for predicting secondary organic aerosol formation from the reaction of d-limonene with ozone, Environ. Sci. Technol., 39, 9583–9594, 2005a.
Leungsakul, S., Jeffries, H. E., and Kamens, R. M.: A kinetic mechanism for predicting secondary aerosol formation from the reactions of d-limonene in the presence of oxides of nitrogen and natural sunlight, Atmos. Environ., 39, 7063–7082, 2005b.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Carrasquillo, A. J., Daumit, K. E., Hunter, J. F., Kroll, J. H., Worsnop, D. R., and Thornton, J. A.: Phase partitioning and volatility of secondary organic aerosol components formed from α-pinene ozonolysis and OH oxidation: the importance of accretion products and other low volatility compounds, Atmos. Chem. Phys., 15, 7765–7776, https://doi.org/10.5194/acp-15-7765-2015, 2015.
Maksymiuk, C. S., Gayahtri, C., Gil, R. R., and Donahue, N. M.: Secondary organic aerosol formation from multiphase oxidation of limonene by ozone: mechanistic constraints via two-dimensional heteronuclear NMR spectroscopy, Phys. Chem. Chem. Phys., 11, 7810–7818, https://doi.org/10.1039/b820005j, 2009.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mohr, C., Lopez-Hilfiker, F. D., Zotter, P., Prévôt, A. S. H., Xu, L., Ng, N. L., Herndon, S. C., Williams, L. R., Franklin, J. P., Zahniser, M. S., Worsnop, D. R., Knighton, W. B., Aiken, A. C., Gorkowski, K. J., Dubey, M. K., Allan, J. D., and Thornton, J. A.: Contribution of Nitrated Phenols to Wood Burning Brown Carbon Light Absorption in Detling, United Kingdom during Winter Time, Environ. Sci. Technol., 47, 6316–6324, https://doi.org/10.1021/es400683v, 2013.
Mohr, C., Lopez-Hilfiker, F. D., Yli-Juuti, T., Heitto, A., Lutz, A., Hallquist, M., D'Ambro, E. L., Rissanen, M. P., Hao, L. Q., Schobesberger, S., Kulmala, M., Mauldin, R. L., Makkonen, U., Sipila, M., Petaja, T., and Thornton, J. A.: Ambient observations of dimers from terpene oxidation in the gas phase: Implications for new particle formation and growth, Geophys. Res. Lett., 44, 2958–2966, https://doi.org/10.1002/2017gl072718, 2017.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Pathak, R. K., Salo, K., Emanuelsson, E. U., Cai, C., Lutz, A., Hallquist, A. M., and Hallquist, M.: Influence of ozone and radical chemistry on limonene organic aerosol production and thermal characteristics, Environ. Sci. Technol., 46, 11660–11669, https://doi.org/10.1021/es301750r, 2012.
Rossignol, S., Chiappini, L., Perraudin, E., Rio, C., Fable, S., Valorso, R., and Doussin, J. F.: Development of a parallel sampling and analysis method for the elucidation of gas/particle partitioning of oxygenated semi-volatile organics: a limonene ozonolysis study, Atmos. Meas. Tech., 5, 1459–1489, https://doi.org/10.5194/amt-5-1459-2012, 2012.
Rossignol, S., Rio, C., Ustache, A., Fable, S., Nicolle, J., Meme, A., D'Anna, B., Nicolas, M., Leoz, E., and Chiappini, L.: The use of a housecleaning product in an indoor environment leading to oxygenated polar compounds and SOA formation: Gas and particulate phase chemical characterization, Atmos. Environ., 75, 196–205, https://doi.org/10.1016/j.atmosenv.2013.03.045, 2013.
Saathoff, H., Naumann, K.-H., Möhler, O., Jonsson, Å. M., Hallquist, M., Kiendler-Scharr, A., Mentel, Th. F., Tillmann, R., and Schurath, U.: Temperature dependence of yields of secondary organic aerosols from the ozonolysis of α-pinene and limonene, Atmos. Chem. Phys., 9, 1551–1577, https://doi.org/10.5194/acp-9-1551-2009, 2009.
Salo, K., Jonsson, A. M., Andersson, P. U., and Hallquist, M.: Aerosol volatility and enthalpy of sublimation of carboxylic acids, J. Phys. Chem. A, 114, 4586–4594, https://doi.org/10.1021/jp910105h, 2010.
Shrivastava, M., Cappa, C. D., Fan, J. W., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 41, 6259–6293, https://doi.org/10.1039/c2cs35070j, 2012.
Walser, M. L., Desyaterik, Y., Laskin, J., Laskin, A., and Nizkorodov, S. A.: High-resolution mass spectrometric analysis of secondary organic aerosol produced by ozonation of limonene, Phys. Chem. Chem. Phys. 10, 1009–1022, https://doi.org/10.1039/b712620d, 2008.
Wang, M., Yao, L., Zheng, J., Wang, X., Chen, J., Yang, X., Worsnop, D. R., Donahue, N. M., and Wang, L.: Reactions of Atmospheric Particulate Stabilized Criegee Intermediates Lead to High-Molecular-Weight Aerosol Components, Environ. Sci. Technol., 50, 5702–5710, https://doi.org/10.1021/acs.est.6b02114, 2016.
Watne, A. K., Westerlund, J., Hallquist, A. M., Brune, W. H., and Hallquist, M.: Ozone and OH-induced oxidation of monoterpenes: Changes in the thermal properties of secondary organic aerosol (SOA), J. Aerosol Sci., 114, 31–41, https://doi.org/10.1016/j.jaerosci.2017.08.011, 2017.
Witkowski, B. and Gierczak, T.: Early stage composition of SOA produced by α-pinene/ozone reaction: α-Acyloxyhydroperoxy aldehydes and acidic dimers, Atmos. Environ., 95, 59–70, https://doi.org/10.1016/j.atmosenv.2014.06.018, 2014.
Youssefi, S. and Waring, M. S.: Transient Secondary Organic Aerosol Formation from Limonene Ozonolysis in Indoor Environments: Impacts of Air Exchange Rates and Initial Concentration Ratios, Environ. Sci. Technol., 48, 7899–7908, https://doi.org/10.1021/es5009906, 2014.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, 2012.