the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Sep 2019
| 02 Sep 2019
High time-resolved measurement of stable carbon isotope composition in water-soluble organic aerosols: method optimization and a case study during winter haze in eastern China
Wenqi Zhang
Fang Cao
Yankun Xiang
Yuanyuan Zhang
Mengying Bao
Xiaoyan Liu
Yu-Chi Lin
Water-soluble organic carbon (WSOC) is a significant fraction of organic carbon (OC) in atmospheric aerosols. WSOC is of great interest due to its significant effects on atmospheric chemistry, the Earth's climate and human health. The stable carbon isotope (δ13C) can be used to track the potential sources and investigate atmospheric processes of organic aerosols. However, the previous methods measuring the δ13C values of WSOC in ambient aerosols require a large amount of carbon content, are time-consuming and require labor-intensive preprocessing. In this study, a method of simultaneously measuring the mass concentration and the δ13C values of WSOC from aerosol samples is established by coupling the GasBench II preparation device with isotopic ratio mass spectrometry. The precision and accuracy of isotope determination is better than 0.17 ‰ and 0.5 ‰, respectively, for samples containing WSOC amounts larger than 5 µg. This method is then applied for the aerosol samples collected every 3 h during a severe wintertime haze period in Nanjing, eastern China. The WSOC values vary between 3 and 32 µg m−3, whereas δ13C−WSOC ranges from −26.24 ‰ to −23.35 ‰. Three different episodes (Episode 1, Episode 2 and Episode 3) are identified in the sampling period, showing a different tendency of δ13C−WSOC with the accumulation process of WSOC aerosols. The increases in both the WSOC mass concentrations and the δ13C−WSOC values in Episode 1 indicate that WSOC is subject to a substantial photochemical aging during the air mass transport. In Episode 2, the decline of the δ13C−WSOC is accompanied by the increase in the WSOC mass concentrations, which is associated with regional-transported biomass burning emissions. In Episode 3, heavier isotope (13C) is exclusively enriched in total carbon (TC) in comparison to WSOC aerosols. This suggests that the non-WSOC fraction in total carbon may contain 13C-enriched components such as dust carbonate, which is supported by the enhanced Ca2+ concentrations and air mass trajectory analysis. The present study provides a novel method to determine the stable carbon isotope composition of WSOC, and it offers a great potential to better understand the source emission, the atmospheric aging and the secondary production of water-soluble organic aerosols.
- Article
(2942 KB) - Full-text XML
-
Supplement
(551 KB) - BibTeX
- EndNote





Water-soluble organic carbon (WSOC) contributes to a large fraction (9 %–75 %) of organic carbon (OC) (Sullivan et al., 2004; Decesari et al., 2007; Anderson, et al., 2008) and affects substantially the global climate change and human health (Ramanathan et al., 2001; Myhre, 2009). Due to its hydrophilic nature, WSOC has a great impact on the hygroscopic properties of aerosols and promotes the increase of cloud condensation nuclei (CCN) activity (Asa-Awuku et al., 2011). WSOC is a contributor to cardiovascular and respiratory problems because it is easy to be incorporated in biological systems such as human blood and lungs (Mills et al., 2009).
WSOC can be emitted as primary organic carbon (POC) and secondary organic carbon (SOC) produced from atmospheric oxidation of volatile organic compounds (VOCs) (Sannigrahi et al., 2006; Weber et al., 2007; Zhang et al., 2018). Due to the hygroscopic property of the WSOC, the origins of POC may be from biomass burning or marine emissions. However, the SOC may stem from various sources including coal combustion, vehicle emissions, biogenic emissions, marine emissions and biomass burning (Decesari et al., 2007; Jimenez et al., 2009; Kirillova et al., 2010, 2013; Bozzetti et al., 2017a, b).
Stable carbon isotopic composition (δ13C) can provide valuable information to track both potential sources and atmospheric processes of carbonaceous aerosols (Rudolph, 2007; Pavuluri and Kawamura, 2012; Kirillova et al., 2013, 2014). Carbonaceous aerosols from coal combustion have an isotope signature from −24.9 ‰ to −21 ‰ (Cao et al., 2011). Particulate matter emitted from motor vehicles exhibits with isotopes from −26 ‰ to −28 ‰ (Widory, 2006). Due to the different pathways of metabolism, C3 and C4 plants exhibit significant differences of δ13C (approximately −27 ‰ for C3 and −13 ‰ for C4; Martinelli et al., 2002; Sousa Moura et al., 2008). Laboratory studies demonstrate that there is no significant isotope fractionation (±0.5 ‰) between the produced aerosols and the C3 plant material (Turekian et al., 1998; Currie et al., 1999; Das et al., 2010). The burning of C4 plants results in 13C depletion (< 0.5 % to 7.2 %) of the produced aerosols (Turekian et al., 1998; Das et al., 2010). Marine organic aerosol sources have a carbon isotope signature of −22 ‰ to −18 ‰ (Miyazaki et al., 2011) and play an important role in the aerosols at coastal sites. In contrast, carbonate carbon exhibits with a pretty high isotopic ratio of −0.3 ‰ (Kawamura et al., 2004) and generally shows a large proportion in dust aerosols. Thus, the isotope signatures of particulate matter emitted from these various sources may have different effects on the characteristics of δ13C in ambient WSOC.
In addition, atmospheric processes like secondary formation and photochemical aging may change the constitution and properties of WSOC, as well as the stable carbon isotope of WSOC (δ13C−WSOC). According to the kinetic isotope effect (KIE), the reaction rate of molecules containing heavier isotopes is usually lower than the molecules containing lighter isotopes (Atkinson, 1986; Fisseha et al., 2009; Kirillova et al., 2013). The change in reaction rate primarily results from the greater energetic need for molecules containing heavier isotopes to reach the transition state (Brüniche-Olsen and Ulstrup, 1979). Consequently, the oxidants preferentially react with molecules with lighter isotopes (inverse kinetic isotope effect), which would result in an enrichment of 13C in the residual materials and a depletion in 13C of the particulate oxidation products (Rudolph et al., 2002). Therefore, organic compounds formed via secondary formation are generally depleted in 13C compared with their precursors (Sakugawa and Kaplan, 1995; Fisseha et al., 2009), and this isotope depletion has been proved by both field measurements and laboratory studies (Pavuluri and Kawamura, 2012). For example, the studies of KIE clearly indicate that the compounds formed via the oxidation are depleted in the 13C compared with their precursors during the reaction of VOCs with OH and ozone (dominant atmospheric oxidants) (Rudolph et al., 2000; Iannone et al., 2003; Anderson et al., 2004; Fisseha et al., 2009). An enrichment of 13C in the particulate organic aerosol may occur in the atmospheric aging processes, such as interactions with photochemical oxidants (e.g., hydroxyl radical and ozone), during the long-range transport. For instance, studies have demonstrated that the substantial enrichment of 13C in the residual, aged aerosols (e.g., isoprene, a precursor of oxalic acid; Rudolph et al., 2003) occurred after a long-range transport. In that case, the stable carbon isotope can be used to study the sources and the atmospheric processes that contribute to the carbonaceous aerosols.
Several studies report the temporal and spatial variation, complex chemical species, light absorption and thermal characteristics of WSOC, as well as its relationship with other compounds in fine particles (Wang et al., 2006; Wozniak et al., 2008; Martinez et al., 2016; Zhang et al., 2018). However, only few studies focus on the analysis of δ13C−WSOC (Fisseha et al., 2006; Kirillova et al., 2010; Lang et al., 2012; Zhou et al., 2015; Suto and Kawashima, 2018). This is partially due to the limited techniques to analyze the δ13C signatures of WSOC in ambient aerosols, as their concentrations are usually very small. In the recent years, some efforts have been made to measure the δ13C values of WSOC. Bauer et al. (1991) used potassium persulfate to convert organic carbon in natural waters into CO2 for δ13C measurements. This wet oxidation method requires more than 0.5 mM C and 1 h for the pretreatment (from sample injection to the isolation of purified CO2). Fisseha et al. (2006) boiled the oxidizing solution for 45 min to remove the organic matter, and the total time required for the pretreatment (for 15 samples) is 1.5 h. Kirillova et al. (2010) developed a combustion method that applies the aerosol extract without filtration for isotope measurement and involves complicated processes such as freeze-drying of the aerosol extract under vacuum for 16 h. This combustion method is the most widely used for the δ13C measurements in WSOC aerosols (Kirillova et al., 2010, 2013, 2014; Miyazaki et al., 2012; Pavuluri Kawamura, 2017). Although these methods are able to provide the δ13C values of WSOC in natural waters and/or ambient aerosols, the analytical methods require either a large amount of WSOC (from 100 µg C to 0.5 mM C) or time-consuming preprocessing. And some of the methods oxidize the WSOC extract without filtration and/or decarbonation in the pretreatment, which would result in higher uncertainty of the δ13C results. The high detection limit of the previous methods is difficult to determine the δ13C−WSOC in aerosol samples with low carbon concentrations. In that case, an easily operated method detecting the δ13C−WSOC values in aerosol samples with a low detection limit and high precision is urgently needed. The objectives of this study are as follows: (1) to provide an accurate, precise and easily operated method to measure the WSOC and δ13C−WSOC in ambient aerosol samples and (2) to apply this method for analyzing the high time-resolution aerosol samples during a severe haze and discuss the potential sources and the atmospheric processes of WSOC. In addition, the concentrations of inorganic ions and air mass back trajectories coupled with MODIS fire maps are also analyzed to substantiate the results obtained from the δ13C analysis.
2.1 Standards
Four working standards were used in this study: potassium hydrogen phthalate (KHP), benzoic acid (BA), sucrose (CH6) and sodium oxalate (C2). KHP and BA are widely used as the standards of WSOC measurements (Kirillova et al., 2010), and they were used here as the WSOC test substances. Also, their isotope signatures are close to the δ13C values of aerosol samples (Fisseha et al., 2009; Miyazaki et al., 2012; Suto and Kawashima, 2018). Sucrose and oxalic are taken as the standards to represent the characteristics of the components in atmospheric WSOC (Pathak et al., 2011; Pavuluri and Kawamura, 2012; Liang et al., 2015; Fowler et al., 2018). The carbon isotope composition of these four standards is as follows: −12.20 ‰ (CH6), −13.84 ‰ (C2), −27.17 ‰ (BA) and −30.40 ‰ (KHP), respectively. The wide range of the delta values of the working standards is able to cover the majority of the δ13C−WSOC values in ambient aerosol samples. Standards were resolved in Milli-Q water (18.2 MΩ quality) to make standard solutions of 0.25, 0.75, 1.5, 3, 6, 12 and 24 µg mL−1, which means a carbon content of 1, 3, 6, 12, 24, 48 and 96 µg in 4 mL standard solution to test the procedures during the pretreatment.
2.2 Aerosol samples
The aerosol samples were collected during a severe haze in January (from 14 to 28 January) of 2015 in a suburb of Nanjing, a megacity in eastern China. The sampling site is located at the agrometeorological station in the campus of the Nanjing University of Information Science and Technology. It is close to a busy traffic road and surrounded by a large number of industrial factories. The PM2.5 samples were collected on pre-combusted quartz-fiber filters (180×230 mm) every 3 h with a high-volume aerosol sampler (KC100, Qingdao, China) at a flow rate of 1 m3 min−1. After sampling, all the filters were wrapped in the aluminum foil, sealed in air-tight polyethylene bags and stored at −26 ∘C for later analysis. A field blank was obtained by placing the blank filter in the filter holder for 10 min without sampling.
2.3 Chemical analysis
The PM2.5 concentrations were observed at Pukou environmental supervising station. Concentrations of total carbon (TC) and δ13C−TC values were analyzed with and elemental analyzer combined with an isotope ratio mass spectrometer (EA-IRMS,Thermo Fisher Scientific, Bremen, Germany). The WSOC mass concentrations were measured with the total organic carbon (TOC) analyzer (Shimadzu). Ion concentrations were obtained from the ion chromatography system (IC, Thermo Fisher Scientific, Bremen, Germany). The meteorological data were observed close to the sampling site (Envis automatic meteorological station).
2.4 Sample pretreatment
The wet oxidation method was used to covert the WSOC to CO2 (Sharp, 1973), and the resulting CO2 levels were measured by an isotope ratio mass spectrometer (IRMS). The overview of the optimized method for measuring WSOC and δ13C−WSOC in the aerosols is shown in Fig. 1. The process of the pretreatment consisted of six steps: WSOC on a 20 mm diameter disk was extracted with 6 mL Milli-Q water in an ultrasonic water bath for 30 min (step 1–2). The WSOC extract was filtered with a 0.22 µm syringe filter to remove the particles in step 3. A total of 2.0 g potassium persulfate (K2S2O8, Aladdin Industrial Corporation, Shanghai) and 100 µL phosphoric acid (85 % H3PO4, AR, ANPEL Laboratory Technologies Inc., Shanghai) were dissolved in 50 mL Milli-Q water to make the oxidizing solution. The oxidizing solution made within 24 h was added into the filtered WSOC extract as shown in step 4 of Fig. 1. The phosphoric acid was added to remove the inorganic carbon resolved in the solution, and the persulfate was added for the preparation to convert the organic compounds to CO2. The vials were sealed tightly with the caps as soon as the oxidizing solution was added to the WSOC extract.
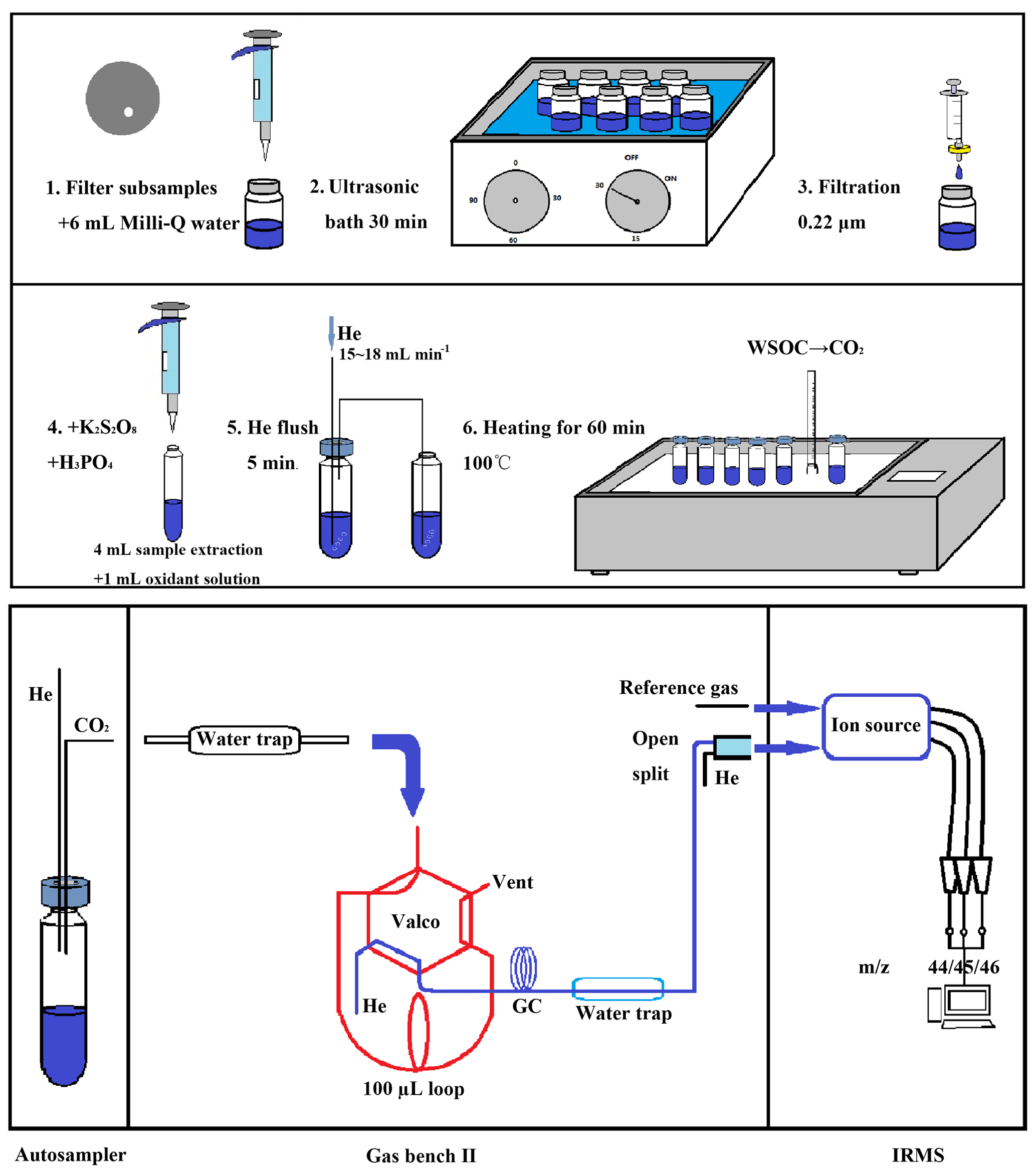
Figure 1Schematic of the optimized method for the measurement of WSOC mass concentrations and the δ13C−WSOC values. A filter disk is dissolved with 6 mL Milli-Q water in a 20 mL pre-combusted glass bottle in the first step. After 30 min ultrasonic bath, the WSOC extract is filtered with 0.22 µm syringe filter and transferred to another 20 mL pre-combusted glass bottle in step 3. A total of 4 mL of filtrate is transferred to a 12 mL pre-combusted glass vial, which contains 1 mL oxidant solution (2.0 g K2S2O8 and 100 µL 85 % H3PO4 dissolved in 50 mL Milli-Q water) in the vial in step 4. Next, the mixed solution of WSOC extract and the oxidant solution is flushed with helium at a flow rate of 15–18 mL min−1 as shown in step 5. Finally, the vials are heated for 60 min under 100 ∘C in the sand bath pot (step 6).
To remove the ambient CO2 dissolved in the mixture (mixed solution of the oxidizing solution and the WSOC extract) and the atmospheric CO2 in the headspace of the sealed sample vials, high-purity helium (Grade 5.0, 99.999 % purity) was flushed into the vials for 5 min in step 5. The aim of this step was to exclude the possible contamination from the atmospheric CO2, and it had to be finished within 12 h after the mixing of the WSOC extract and the oxidizing solution to avoid the loss of CO2 produced under room temperature. High-purity helium (15–18 mL min−1) is flushed under the water surface, and a stainless steel tube was set for the output gas stream. The open end of this tube was submerged in Milli-Q water to prevent any backflow of atmospheric CO2 (Fig. 1, step 5). After flushing, the vials were heated at 100 ∘C for 60 min in the sand bath pot (quartz sand, Y-2, Guoyu, China) to start the oxidation of WSOC in step 6. The heated vials were stored overnight at room temperature to condense the moisture before the analysis on the IRMS in order to prevent damage to the measuring equipment.
2.5 Determination of the carbon content and stable carbon isotopic ratios
CO2 gas produced in the headspace of the prepared sample was extracted and purified by the GasBench II system (Thermo Fisher Scientific, Bremen, Germany) and was introduced into an isotope ratio mass spectrometer (IRMS) (MAT 253, Thermo Fisher Scientific, Bremen, Germany) for analysis. The extracted gas was purified with a Nafion water trap to remove the water vapor, and then the gas was loaded into a 100 µL sample loop through an eight-port Valco valve. After 120 s loading time (the duration time from the beginning of the analysis to the first rotation of the eight port in the GasBench II), the eight-port Valco valve rotated every 70 s to inject the sample gas from the loop into a GC column (PoraPLOT Q fused-silica cap, 25 m, 0.32 mm; Agilent Technologies). The GC column was set at 40 ∘C for the CO2 separation from the matrix gases. The separated CO2 was introduced into another Nafion water trap and subsequently entered into the IRMS through an open split. The CO2 gas in each vial was detected 10 times in 15 min, showing 10 sample peaks after five reference peaks. The peak areas and the isotope compositions of the 10 sample peaks were given correspondingly, and the results of the first two sample peaks were abandoned considering the possible effect on the memory of the system. The average peak area and the isotope composition of the last eight peaks were taken as the result of a certain sample determined by the GasBench II–IRMS.
The wet oxidation method was adapted from the stable isotope analysis of organic matter in ground water (Lang et al., 2012; Zhou et al., 2015). Several tests were performed to adjust the optimal conditions for measuring WSOC aerosols with relative low carbon amounts.
3.1 The carbon content in the procedural blank
In order to quantify the low concentration of WSOC in aerosols, it is critical to reduce the carbon content in the procedural blank for minimizing the detection limit of the method. To achieve this goal, the procedural blanks were analyzed to test for contamination that the reagents could have introduced to the results (shown in Table 1). The average carbon content in the procedural blank was about 0.5 µg C (corresponding with a peak area of 0.23 Vs) with a δ13C value of ‰ (n=15). The carbon content and the isotope compositions of Milli-Q water and the agents dissolved in the oxidizing solution were also determined in order to identify the source of contamination in the procedural blank. The peak area of Milli-Q water was not detected (Table 1) after going through all the processes in the pretreatment without adding any other materials, suggesting that no contamination had been introduced from the Milli-Q water. Furthermore, the contamination from 85 % H3PO4 with different purities (acid-1: analytical reagent, AR; acid-2: high performance liquid chromatography, HPLC) was compared. The carbon content in the 85 % H3PO4 dissolved in Milli-Q water was 0.03–0.04 µg C and showed no significant discrepancy between different purities.
Table 1Various blank preparations and results.
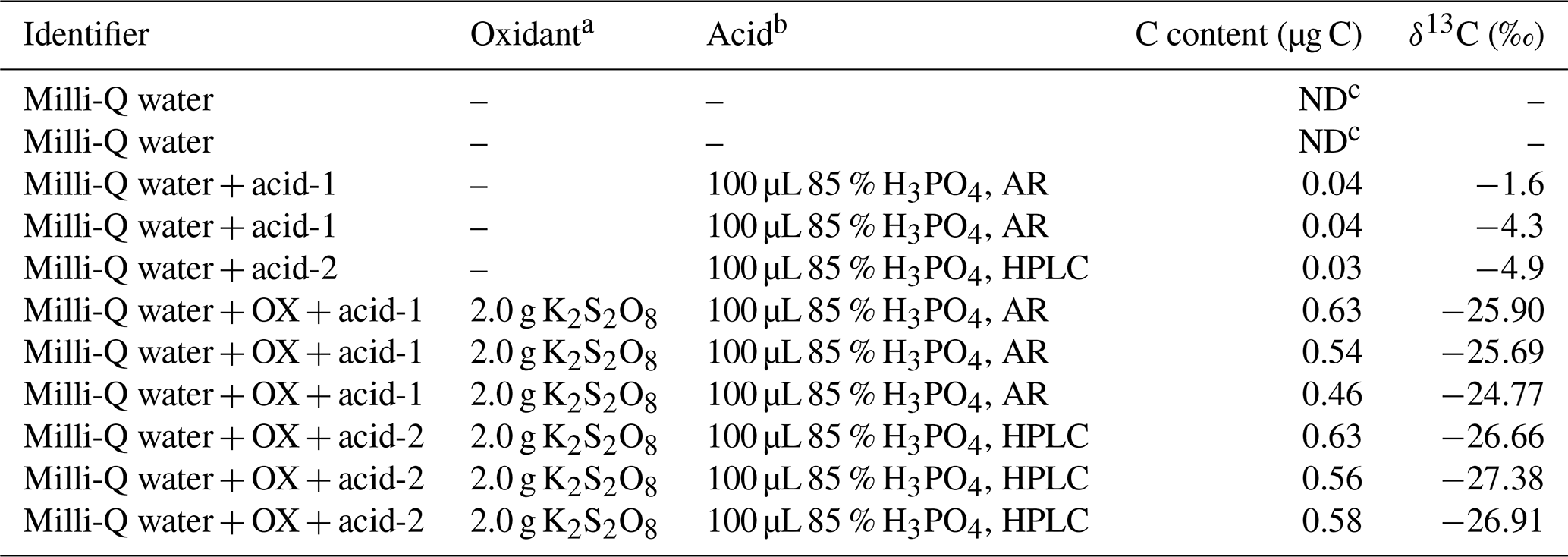
a, b Oxidant and acid added to 50 mL Milli-Q water. c Not detected.
Interestingly, the carbon content increased to 0.5–0.6 µg C after the persulfate was added, implicating that the CO2 in the procedural blank was mainly produced from the oxidation of organic substance in the persulfate. The carbon content in HPLC grade of 85 % H3PO4 mixed with the persulfate (0.56–0.63 µg C) was close to that of the AR grade (0.46–0.63 µg C; see Table 1). Thus, AR grade with purity of 85 % H3PO4 was utilized to prepare the oxidizing solution in this method. The average carbon content of the procedural blank was estimated to be 0.5±0.06 µg C, and the detection limit was expected to be 10 times the procedural blank (i.e., 5 µg C). The carbon content in the procedural blank of this method is much lower than that of the methods analyzing isotopes of WSOC in aquatic environment or soil (Werner et al., 1999; Atekwana and Krishnamurthy, 2004; Polissar et al., 2009). The smaller carbon content of the procedural blank suggests the possibility of correctly measuring the WSOC and δ13C−WSOC of samples containing low carbon content.
3.2 Flushing methods
To avoid any contamination, the headspace of the sample vial had to be flushed with the high-purity helium to remove the CO2 (both dissolved and gas phase). Two different flushing methods (F1 and F2) were compared here. The F1 method is a one-step flushing: helium is bubbled under the water surface for 5 min in a sealed vial, and the gas in the headspace is released through a stainless steel tube to the atmosphere. The open end of this tube is submerged in Milli-Q water to balance the air pressure and to prevent any backflow of the atmospheric CO2. The F2 method requires two steps: the helium is first bubbled under the water surface for 5 min in an open vial to remove the dissolved CO2 in the solution. After the vial is sealed, the helium is flushed again into the headspace for 5 min by piercing the septum with a two-hole sample needle. The two holes act as the inlet of the helium and the exit of the outflow, respectively. Since the flow rate of the inlet helium is larger than that of the outflow, the headspace pressure is considered to be greater than 1 atm. In that case, the most noticeable difference between F1 and F2 is the air pressure of the headspace.
Different concentrations of the working standard (KHP) were tested to compare the flushing methods. The results obtained from F1 and F2 show no significant difference regardless of the concentration of KHP. This indicates that F1 and F2 are both able to completely remove the CO2 in the vials. But it should be noted that F2 produces excessive air pressure in the headspace, and the heating step may increase the risk of gas leak. Gas leaking during the preparation usually results in the loss of carbon content and the isotope fractionation. Furthermore, the flushing step in F2 takes 5 more minutes for each sample compared with F1. Consequently, the F1 method is considered as the suitable flushing method to remove CO2 dissolved in the solution and the headspace.
3.3 Heating time
In order to assure the complete oxidation of WSOC, the duration time for heating the samples was tested with KHP, a widely used WSOC standard, which is difficult to oxidize. Figure 2 shows the carbon content and the δ13C values of KHP solutions heated from 15 to 120 min at 100 ∘C. Some caps of the sample vials were out of shape after heating for a longer time (more than 60 min), suggesting a gas leak from the vials. High pressure can be built up in the headspace with the increase in temperature during the longer heating time, especially for the vials with a higher carbon content. The CO2 gas produced in the headspace may leak through the minor holes on the septum pierced by the stainless tube during the helium flushing step (step 5 in Fig. 1). According to the kinetic isotope effect (KIE), isotope fractionation occurs during the gas leaking. The light carbon isotopes (12C) are easier to escape from the vials than the heavy ones (13C); thus the remaining CO2 would be more enriched with heavy isotopes (13C). In that case, lower carbon content and higher δ13C values were expected to be observed in the results of leaking vials. In the results of the KHP standards, some of the vials containing a larger amount of organic carbon were detected to have extremely low carbon content, which corresponds to very high isotopic ratios. For example, one of the 10 µg C KHP standards was measured to be 1.2 µg C and δ13C ‰ after 120 min of heating; one of the 30 µg C KHP standards was measured to be 2.4 µg C and δ13C ‰ after 90 min of heating (Fig. 2). The stable results (both carbon content and the isotopes, Fig. 2) of 4 µg C standards are probably due to the less CO2 gas and lower pressure produced in the headspace during the heating. Accordingly, a heating time longer than 60 min increased the probability of gas leak in the measurement.
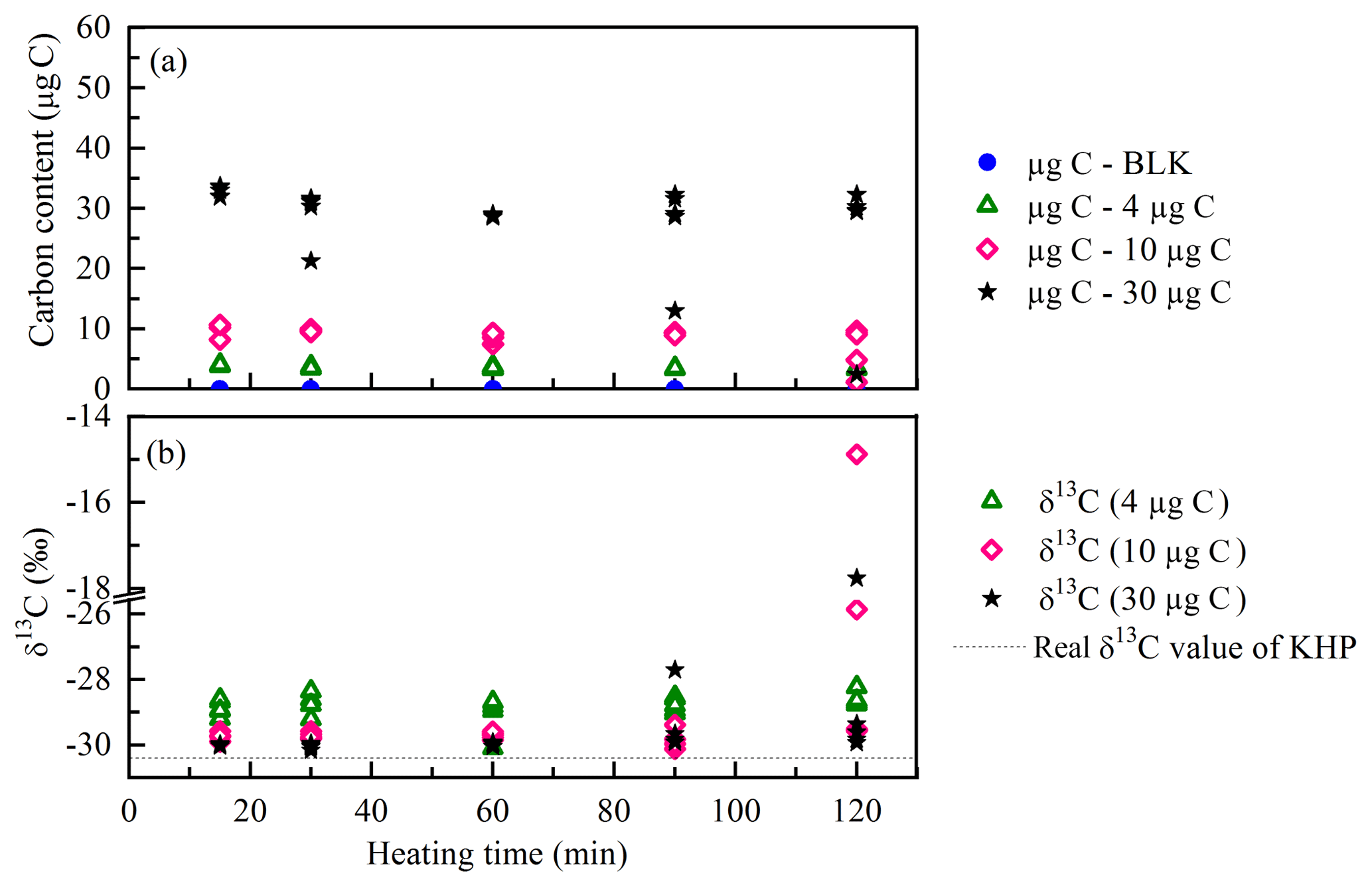
With respect to isotope composition, KHP standards heated for 15, 30 and 60 min all show stable results with similar standard deviations (from 0.51 to 0.57; see Table S1). The heating times of 15 and 30 min are not long enough for the complete oxidation, which is shown by the presence of a lower carbon content (Fig. 2). Therefore, heating for 60 min at 100 ∘C is found to be the most suitable duration to produce constant results without gas leak and isotope fractionation.
3.4 Waiting time and instrument settings
The waiting time of the mixture (the aerosol extract and the oxidizing solution) between steps 4 and 5 in Fig. 1 was tested to prevent the CO2 loss during the flushing. Some of the compounds in aerosol samples could be oxidized at room temperature. The CO2 generated from the mixture before heating could be lost during the flushing step (Sharp, 1973). The ambient sample was tested to detect the room-temperature-oxidized CO2 (Fig. S1 in the Supplement). Replicates of the ambient aerosol extract (from one filter) were mixed with the oxidizing solution, and the mixtures of the aerosol extract and the oxidizing solution were flushed with He to exclude the effect of CO2 (both in the headspace and in the mixture) as soon as possible. After flushing, the mixtures were stored at room temperature from 1 to 31 h before analysis without heating. The carbon content produced in the mixtures that were stored less than 12 h before analysis was smaller than 0.02 µg, which contributes to ∼7 % to the carbon content in the procedural blank (0.5 µg C). But when the waiting time was extended to 31 h, up to 2.3 µg C (about 5 times more than the procedural blank) was oxidized into CO2. The room-temperature-oxidized CO2 produced during the waiting time could be flushed out by the He in the later procedure and could result in significant isotope fractionation in the delta results. Therefore, the mixture should be flushed with He within 12 h to avoid the CO2 loss and isotope fractionation.
In addition, various combinations of shorter loading times (30–90 s) and/or fewer sample peaks (i.e., five sample peaks) were tested with reference gas (CO2 mixed with He) to shorten the analysis in the system. However, the amount of CO2 in the reference gas detected by the mass spectrometry was about 2 µg C lower compared the results obtained with longer loading times and more sample peaks. And there was a decrease in isotope value (∼0.4 ‰) when the loading time was shorter or the sample peaks were less than 10. Thus, a 120 s loading time and 10 sample peaks are necessary for the precise results, and the standard deviation is < 0.03 ‰ for the 10 sample peaks within a run.
3.5 Calibration of the results
3.5.1 Quantification of the carbon content
The sample peak area is proportional to the carbon content in the vial and is used to quantify the amount of CO2 in the inflow of IRMS. The average value of the peak areas for the last eight sample peaks is taken as the peak area of a certain sample. The first two sample peaks are excluded to avoid the effect of the residual CO2 of the former vial. We established a carbon content standard curve (linear equation) by measuring the peak areas of CO2 gas samples containing 1–24 µg C (Fig. S2). It has to be noted that the gas samples with a larger carbon content are not tested for the difficulty of injecting a too large volume of mixed CO2 and He gas. Then the amount of CO2 oxidized from the unknown samples can be quantified with the following linear equation: carbon content (µg) = peak area (Vs) ), R2=0.98. The standard curve (linear equation) of the peak areas against the carbon content in the WSOC solution (KHP solution containing 1–100 µg C) is also established (Fig. S2). And a linear equation similar with the peak areas against CO2 gas is obtained: carbon content (µg) = peak area (Vs) ), R2=1.00.
Then the conversion efficiency of the WSOC extract containing 1–100 µg C can be roughly calculated as 104±3 %. A high conversion efficiency demonstrates a complete conversion and the negligible isotope fractionation during the oxidation. In that case, the carbon content in the WSOC extract of unknown samples can be calculated based on the standard curve of peak areas against the carbon content in the WSOC extract. And the standard curve quantifying the carbon content has to be established with every batch of unknown samples to assure the complete conversion.
3.5.2 Blank correction
The blank contribution to the WSOC mass concentrations and the δ13C−WSOC values are evaluated with the peak area and the isotope value of the procedural blank. The peak area (average value of the last eight peaks) from the measurement is proportional to the carbon content in the vial and then is taken to represent the CO2 amounts in the inflow of IRMS. The procedural blank can be corrected according to the mass balance as follows:
where δ13Ccorr, δ13Cmeas and δ13Cblk are the blank-corrected δ13C, the measured δ13C of the samples and the δ13C of the procedural blank, respectively. Ameas and Ablk denote the peak areas of the samples and the blank, correspondingly.
In order to calibrate the contribution of the procedural blank to the isotope results, Ablk and δ13Cblk were calculated with an indirect method (Polissar et al., 2009). KHP (δ13C ‰) and CH6 (δ13C ‰) with various concentrations were measured to calculate Ablk and δ13Cblk. The wide range of their isotopes can basically cover the δ13C−WSOC values in most ambient aerosol samples. According to Eq. (1), δ13Cmeas can be written as the following:
According to Eq. (2), there is a linear relationship of the δ13Cmeas values and the reciprocal of peak areas (1/Ameas). Based on the Keeling plot theory, linear equations of the δ13Cmeas values and 1/Ameas for the two standards can be set up separately (e.g., δ13C and 1/Ameas values obtained from the measurement of CH6 and their linear relationship are shown in Fig. S3). The slopes (k1 and k2) and the intercepts (b1 and b2) of this linear relationship can be expressed with δ13Cblk, Ablk and δ13Ccorr as follows.
δ13Ccorr−std1 and δ13Ccorr−std2 are the blank-corrected δ13C values of two standard materials, for example, CH6 and KHP. Thus, Ab and δ13Cb can be calculated as follows.
Thus, the blank contribution is able to be calibrated with the equation below:
For example, δ13Cblk and Ablk are calculated to be −27.43 ‰ and 0.3 Vs (∼0.5 µg C) based on the results of KHP and CH6 (shown in Fig. 3). The carbon content in the procedural blank contributed to 1 %–10 % of the carbon content of an ambient aerosol sample. Although the δ13Cblk and Ablk are not strongly varied values, they need to be measured before every batch of the ambient samples to assure the stable status of the system (IRMS) and the proper processes during the pretreatment.
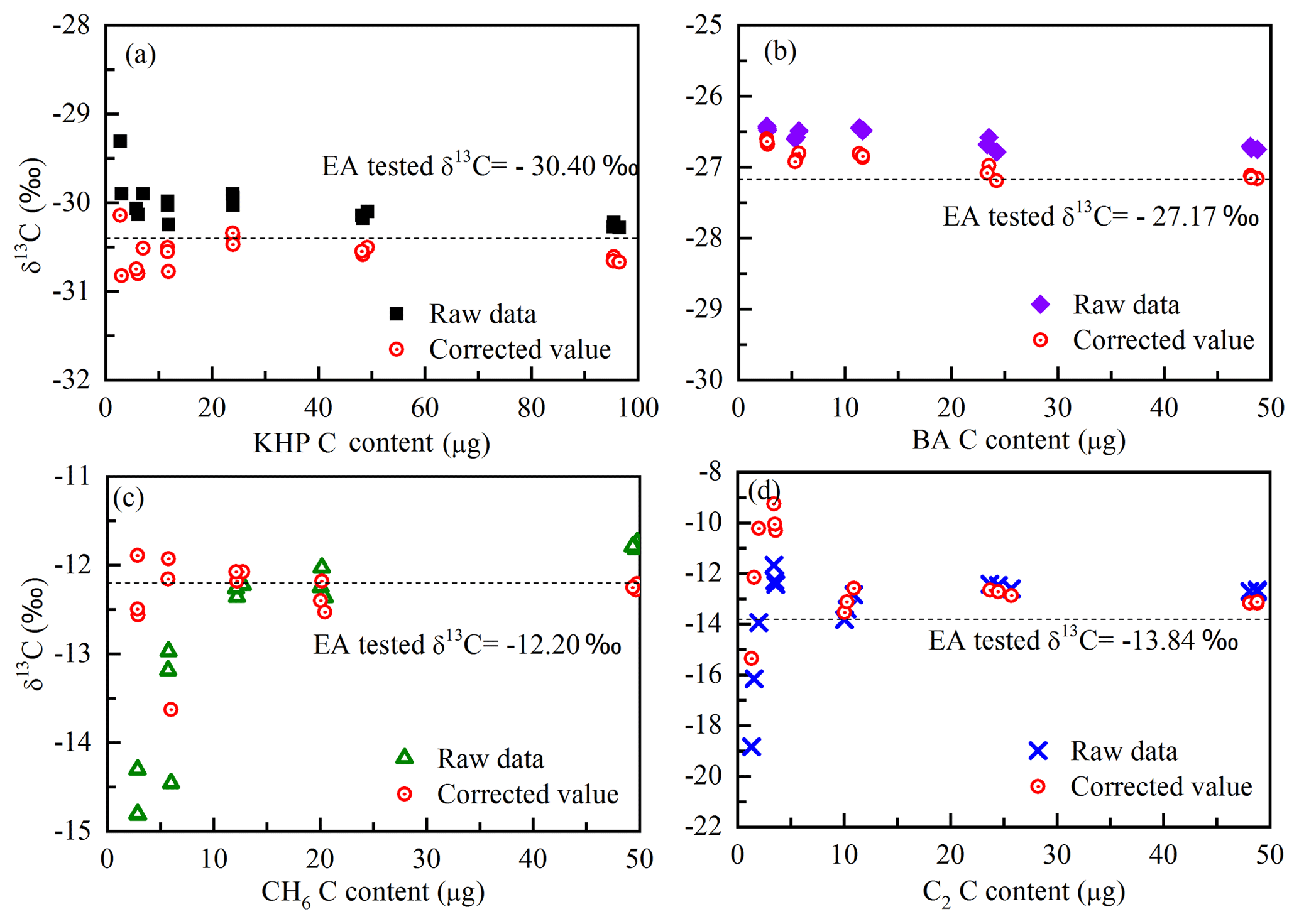
Figure 3Isotope results before and after the two-step correction of the four standards (a KHP, b BA, c CH6, d C2). Red circles with a spot represent the two-step corrected isotopic ratios; the other symbols represent the raw data from the GasBench II; the dotted line represents the blank-corrected δ13C values tested by EA.
3.5.3 Calibration of isotope results
In order to calibrate the isotope results, four working standards (KHP, BA, CH6 and C2) containing different carbon contents were measured with EA-IRMS and GasBench II–IRMS. The standards measured with the elemental analyzer (EA) were combusted at 1000 ∘C to convert the organic materials into CO2 for the measurement in IRMS without pretreatment. More than 10 repetitions of each standard were measured in this way, and the average delta values (after blank correction) of each standard are defined as correct values here. On the other hand, the average isotope compositions (after blank correction) of 10 repetitions obtained from the wet oxidation method (determined with GasBench II) are defined as measured values. Thus a calibration curve can be established on the basis of the measured values and the correct values (Fig. S4). For instance, the isotope results can be calibrated as follows:
where δ13Ccali is the isotope composition after the isotope calibration, δ13Cblk−corr is the blank corrected isotope composition determined with GasBench II, k and b are the slope and the intercept obtained from the calibration curve. Similar with the blank correction, the isotope calibration curve needs to be established with each batch of the ambient samples to assure the stable status of the IRMS and the proper processes during the pretreatment.
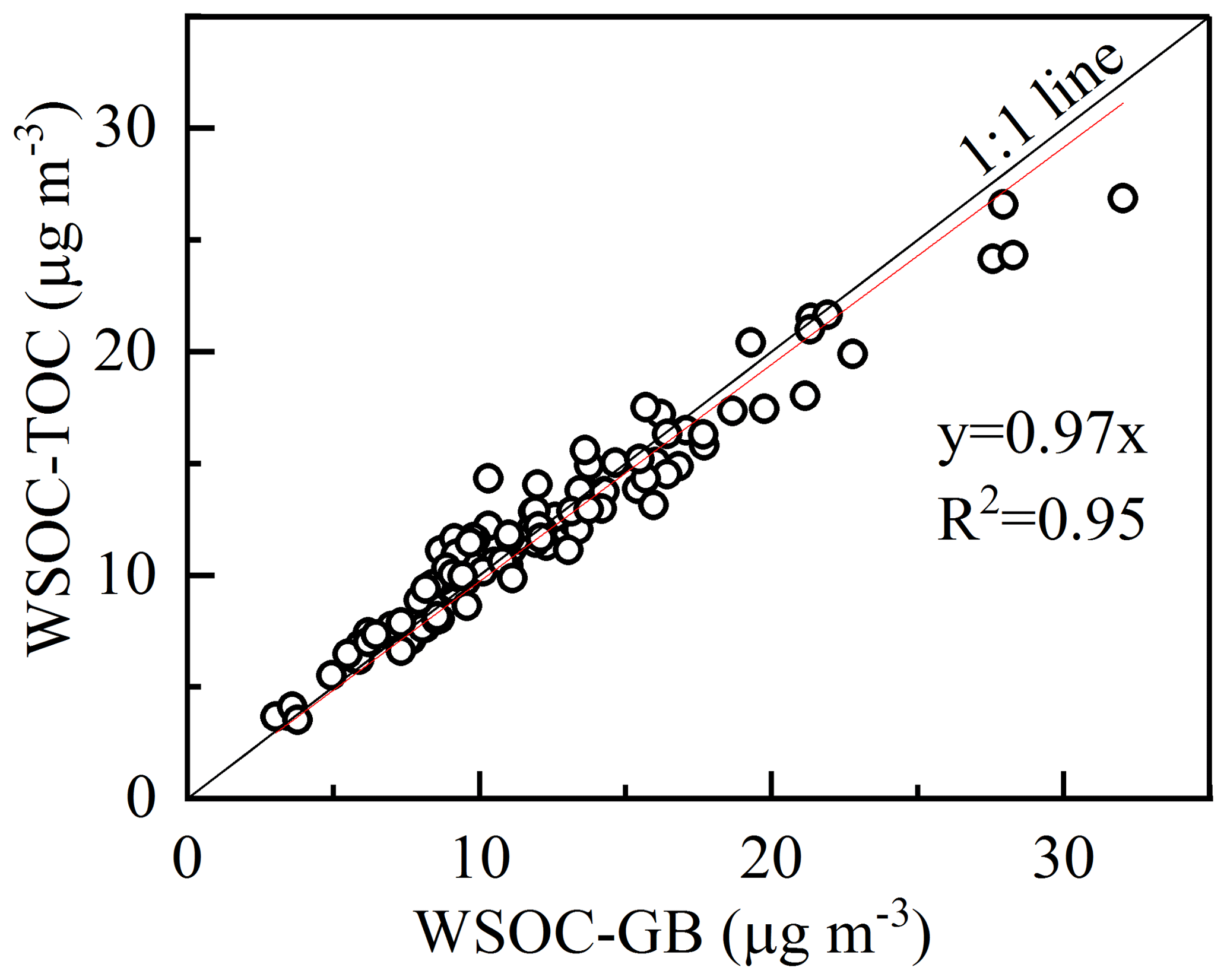
Figure 4Correlation of WSOC mass concentrations measured with the GasBench II–IRMS and TOC analyzer.
In this way, the isotope results can be calibrated. The raw data, the isotope composition after the blank correction and the isotope calibration determined with GasBench II are compared in Fig. 3. The correct values of standard carbon isotopes are plotted in Fig. 3. The isotope results after two steps of correction (the blank correction and the calibration of isotope results) are closer to the correct values (isotopes measured with EA), and the blank contribution is drastically eliminated. But for the standards containing carbon content smaller than 5 µg C, the contribution of the procedural blank (with an isotope ratio about −27.43 ‰) is still significant. According to the isotope variation of the ambient aerosols, the analysis of isotope compositions is not reliable if the repetitions of the standards show a difference larger than 1 ‰ (SD > 0.5 ‰). After correction, the standard deviations of isotope results of each standard are better than 0.17 ‰ (regardless of the carbon content of a certain standard) when the carbon content is larger than 5 µg C. In that case, the detection limit of this method is 5 µg C and the results (both carbon content and the isotopic ratios) of WSOC lower than 5 µg C are not reliable.
3.6 Quality control and quality assurance procedures
A batch of working standards with different carbon content were measured to evaluate the optimized method in this study (data shown in Fig. 3). The qualities of the unknown samples were assured with a standard curve established with the peak areas and the corresponding input carbon content of WSOC extract (e.g., in Fig. S2). The conversion efficiency of the WSOC oxidation was found to be 104±3 %. The average recovery of the working standards and the ambient samples were tested to be 97±6 % and 99±10 %, respectively. The conversion efficiency and the recoveries suggest complete oxidation of WSOC extract without significant isotope fractionation in the pretreatment. The blank contribution was evaluated with the peak area and the isotopic ratio, and these values are calculated with the indirect method introduced in Sect. 3.5.2. According to the carbon content (0.3–0.5 µg C) and the isotope composition ( ‰) of the procedural blank, the WSOC detection limit of this method is 5 µg C, 10 times more than the carbon content in the procedural blank. The blank-corrected isotope compositions should be calibrated again with the calibration curve as described in Sect. 3.5.3 to obtain the isotopic ratios of the unknown samples.
In order to obtain the carbon content and the corrected isotope compositions of the unknown samples, at least two kinds of standards need to be measured before every batch of the unknown samples. The range of the carbon content and the isotope compositions of the standards are required to cover the range of WSOC and δ13C−WSOC in the ambient samples, e.g., KHP, BA and CH6. Hence, the concentration standard curve, the linear equations for the blank correction and the isotope calibration curve are able to be established according to the results of the standards. Besides, one standard should be measured after every 10 unknown samples to assure the stable status of the equipment.
As for the isotope measurement, the precision of the last eight sample peaks is < 0.15 ‰ within a run for standards containing more than 1 µg C; between runs, the deviation of the standards with different carbon content (> 5 µg C, n≥10) was < 0.17 ‰. The accuracy is estimated to be better than 0.5 ‰ by comparing the calibrated δ13C results from the GasBench II and the blank-corrected isotopic ratios from EA. Isotope results tested by GasBench II are slightly lower compared to the results of EA. The ambient aerosol filters were tested repeatedly to evaluate the reproducibility of the ambient samples. The standard deviation of the WSOC concentrations and the isotope results of the repeated ambient samples are 0.25±0.04 µg C (n≥3) and 0.14±0.07 ‰ (n≥3), respectively. To conclude, the method presented is considered to be precise and accurate in the detection of low abundance WSOC and isotopes in aerosol samples.
To test the applicability of this method in measuring the atmospheric WSOC, the ambient aerosol samples collected in Nanjing were analyzed. And the WSOC concentrations were measured with a TOC analyzer (Shimadzu) for comparison. Figure 4 shows the scatter plot of WSOC concentrations measured with the two peripherals (TOC analyzer and GasBench II–IRMS). The strong correlation (R2=0.95, p<0.01) and the slope (0.97) demonstrate the reliability of measuring WSOC with the presented method. It suggests complete oxidation of WSOC in aerosol samples, which means no significant carbon isotope fractionation happens during the preparation. Moreover, the δ13C−WSOC values (between −26.24 ‰ to −23.35 ‰) of ambient aerosols are close to the published data (from −26.5 ‰ to −17.5 ‰) (Kirillova et al., 2013, 2014). In that case, the δ13C values determined from this method are considered to be effective for ambient WSOC.
4.1 Temporal variation
Time series of PM2.5, δ13C values, chemical tracers and meteorological data observed at the sampling site during the studied period are illustrated in Fig. 5. WSOC ranges from 3.0 to 32.0 µg m−3, occupying 49±10 % of total carbon in PM2.5. The stable carbon isotopes of WSOC and TC vary between −26.24 ‰ to −23.35 ‰ and −26.83 ‰ to −22.25 ‰, respectively. δ13C values shift over 2 ‰ in 24 h and over 1 ‰ in 3 h, which was not able to be captured in lower time-resolution samples (e.g., 12 or 24 h). In that case, this data set can be interpreted with more detailed information about the WSOC sources and the atmospheric processes. Biomass burning tracer (nss-K+), dust tracer (Ca2+), MODIS fire spots and air mass trajectories were analyzed to investigate the potential sources of WSOC. Nss-K+ is used as a proxy of biomass burning (Zhang et al., 2013). Nss-K+ concentrations are evaluated from Na+ concentrations in the samples according to their respective ratios ( =0.037 w∕w) in seawater (Osada et al., 2007):
where [K+] and [Na+] are the total mass concentrations of K+ and Na+ of the aerosol samples. The concentration of nss-K+ ranges from 0.16 to 6.70 µg m−3 with an average of 1.31 µg m−3. The high concentrations and the intense increase on 24 January indicate a significant biomass burning event and will be discussed later.
As shown in Fig. 5, δ13C−TC and δ13C−WSOC show similar patterns during the sampling period. In general, δ13C−TC is slightly lower than δ13C−WSOC, and the trend is also observed elsewhere (Fisseha et al., 2009). The difference is related to the sources and the atmospheric processes during the formation and transformation of carbonaceous particles in the atmosphere. The C4 plant biomass burning and the marine organic materials are the sources with relatively enriched 13C. Smith and Epstein (1971) suggest that C4 plants have a mean δ13C isotope signature of −13 ‰. And the isotope composition of carbon emitted from phytoplankton, an example of primary marine aerosol, is about −22 ‰ to −18 ‰ (Miyazaki et al., 2011). However, January is not a specific time period for the growing or combustion of C4 plants in eastern China, indicating a small possibility of C4 plant biomass burning as a major source of WSOC aerosols. In addition, both WSOC and non-WSOC components can be emitted from biomass burning; thus the C4 plant combustion would generally result in the enrichment of 13C in both TC and WSOC. The air parcel transported from marine areas normally has little effect on the aerosols during winter in Nanjing (Qin et al., 2016), suggesting the negligible contribution of marine emissions to WSOC during the sampling period. Therefore, the WSOC sources with higher isotope signatures (compared with non-WSOC sources) are not able to explain the higher values of δ13C−WSOC over δ13C−TC.

Figure 5Time series of PM2.5, WSOC, precipitation, δ13C values, nss-K+, Ca2+, TC, wind speed and wind direction at the sampling site during the studied period. The time period framed with the rectangles is defined as Episode 1 (green), Episode 2 (red) and Episode 3 (orange). The dotted line in 5e is the average value of nss-K+ during the studied period. The high concentration and intense increase in nss-K+ in Episode 2 indicate a significant biomass burning (BB) event and is marked with “BB event” in (e). The similar trends of Ca2+ and TC suggest a dust event in Episode 3.
Apart from the sources, the secondary formation (Saarikoski et al., 2008; Jimenez et al., 2009; Hecobian et al., 2010) of WSOC is reported to affect the isotope compositions. Precursors like VOCs can be oxidized with the hydroxyl radicals and ozone to produce WSOC in the atmosphere (Pathak et al., 2011). Laboratory and field studies demonstrate that the lighter isotopes have the priority to be oxidized and produce particulates with lower isotopic ratios. For example, the oxidation of VOCs in the atmosphere would result in the 13C depletion in the products and the 13C enrichment in the residual VOCs (Rudolph et al., 2002). In other words, the secondary formation tends to lower the δ13C value of ambient WSOC; thus the secondary formation could not explain the 13C enrichment in WSOC compared to TC.
Studies demonstrate that the photochemical aging process during the long-range transport causes significant enrichment in 13C (Aggarwal and Kawamura, 2008; Wang et al., 2010). The isotope fractionation is up to 3 ‰–7 ‰ of the residual during the photolysis of oxalic acid, a dominant species in WSOC aerosols (Pathak et al., 2011). Due to the hydrophilic property, WSOC is associated with the aerosol aging processes. The WSOC ∕ OC ratio is normally considered to represent the aging status of aerosol samples (Agarwal et al., 2010; Pathak et al., 2011), and it increases with the photochemical aging process. The ratio of WSOC ∕ OC is 0.67±0.12 (Fig. S5) in this study, which is higher than the aged aerosols with WSOC ∕ OC =0.41 reported elsewhere (Huang et al., 2012). The high ratio of WSOC ∕ OC indicates aged aerosols during the sampling period. Thus the photochemical aging process could partially explain the reason of higher values of δ13C−WSOC (compared with δ13C−TC).
According to the principle of mass balance, 13C-depleted sources of non-WSOC can also result in the depletion of 13C in TC. TC consists of OC, EC and carbonate carbon (CC) (Huang et al., 2006), and OC can be divided into WSOC and water-insoluble OC (WIOC) according to the hydrophilic character (Eq. 10). In most circumstances, CC is negligible to the amount of TC in PM2.5 (Ten Brink et al., 2004; Huang et al., 2006). Thus non-WSOC component can be presented as Eq. (11).
WIOC and EC generally originate from primary emissions (Park et al., 2013; Y. L. Zhang et al., 2014), and the δ13C values are better in representing their sources. In that case, the 13C-depleted source, which only contributes to non-WSOC components, such as WIOC emitted from the vegetation, is likely to be another reason of δ13C−TC depletion during the sampling period.
4.2 Three episodes
During the sampling period, three significant haze events (Episode 1, Episode 2 and Episode 3) are observed in Nanjing. These three episodes show different tendencies of δ13C−WSOC variation during the accumulation of WSOC aerosols (see Fig. 5). Episode 1 and Episode 2 are compared here due to the distinct δ13C−WSOC trends with WSOC accumulation. In Episode 3, 13C is found to be enriched in TC compared to WSOC (δ13C−WSOC < δ13C−TC, p<0.01), in contrast to the trend of isotope compositions during other periods (δ13C−WSOC > δ13C−TC, p<0.01).
4.2.1 Episode 1
As for Episode 1, the δ13C−WSOC values increase with the mass concentrations of WSOC (r=0.84, p<0.001; see Fig. 6d), indicating that the sampling site is impacted by 13C-enriched WSOC sources and/or photochemical-aged aerosols. As shown in Fig. 6a, air mass trajectories of WSOC with higher δ13C−WSOC values (> 24 ‰) originate mainly from northern China, and the northerly wind prevails at this site (Fig. 5g). During the long-range transport, the studied WSOC mass concentration increases with the 13C enrichment of WSOC due to the isotope fractionation in the photochemical aging process. This is supported by the increasing ratio of WSOC ∕ OC (from 0.73 to 0.91) in Episode 1 (Fig. S5).
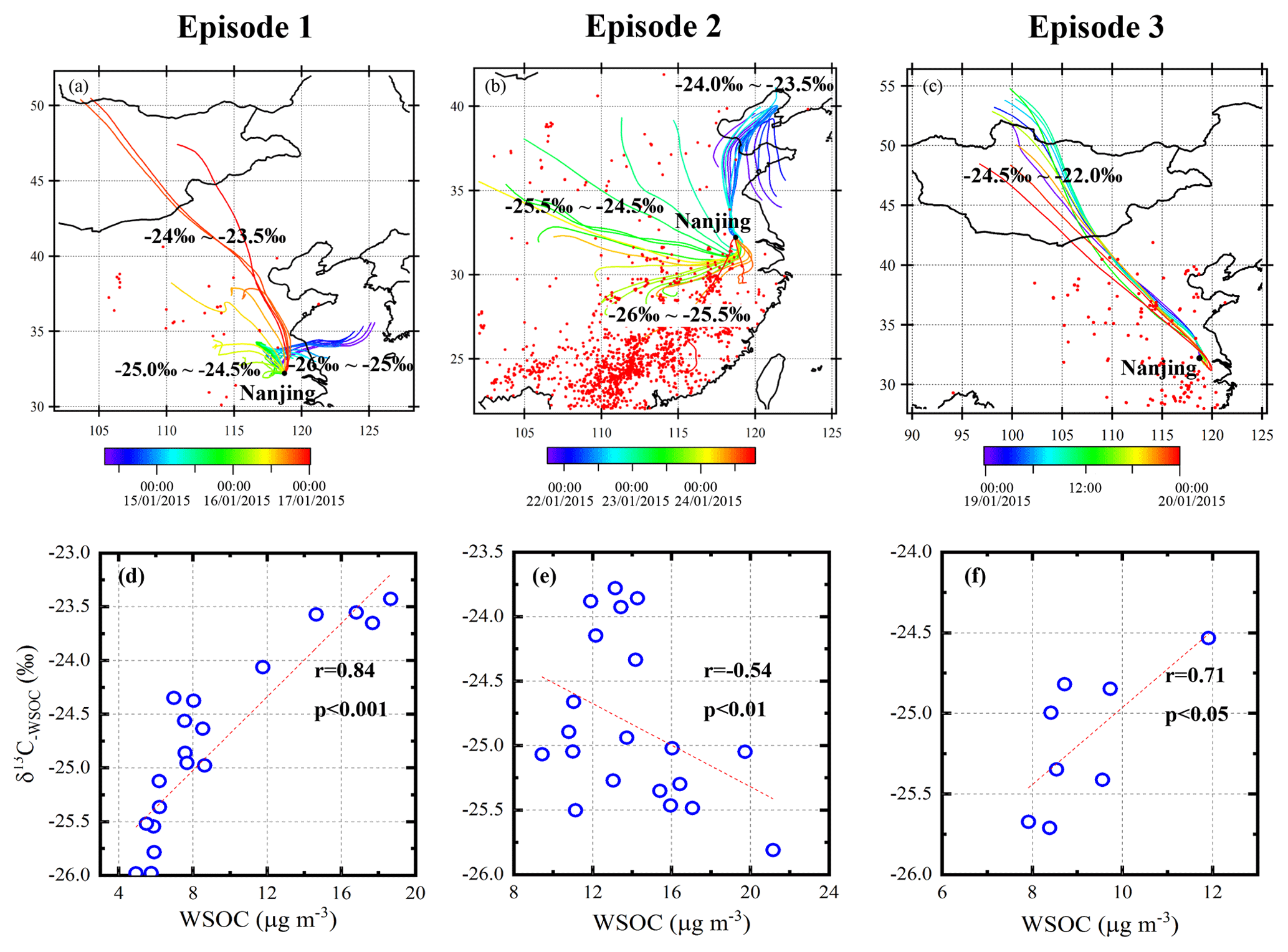
Figure 6The 48 h air mass back trajectories at 500 m and MODIS fire maps for the three episodes and the corresponding relationship between WSOC and δ13C−WSOC. Panels (a–c) represent the back trajectories and the fire maps of Episode 1, Episode 2 and Episode 3, separately. The colors of the back trajectories are marked according to the time of the specific trajectory. Red points represent the fire spots in each episode obtained from the Fire Information for Resource Management System (FIRMS) derived from the Moderate Resolution Imaging Spectroradiometer. The ranges of the δ13C values of the back trajectories are labeled: the marked isotopic ratios are the δ13C−WSOC values (for a and b) and the δ13C−TC values (for c). Panels (d–f) are the correlation between WSOC and δ13C−WSOC in each episode.
According to the higher isotopes (δ13C−WSOC > −24 ‰ ) and the corresponding trajectories (Fig. 6a), C4 plant biomass burning (δ13C ‰; Martinelli et al., 2002; Sousa Moura et al., 2008) and coal combustion (δ13C ‰ to −21 ‰; Cao et al., 2011) are considered to be possible sources of WSOC. Nss-K+ mostly originates from plant combustion (Zhang et al., 2013) and is analyzed as a proxy of biomass burning. However, during this period the nss-K+ level (0.56±0.41 µg m−3) was not significantly increased and was generally lower than the average value (1.31 µg m−3, Fig. 5e), indicating that the C4 plant biomass burning is not a major source of WSOC. The main crops growing in northern China are mainly C3 plants such as wheat and rice instead of C4 plants during the sampling period (Chen et al., 2004). And the biomass burning contribution of C3 plants would even lower the δ13C values of WSOC. Furthermore, there are only few MODIS fire spots along with the trajectories from northern China (Fig. 6a). In that case, open-field biomass burning is not considered as a major source of WSOC at the sampling site during Episode 1.
Furthermore, the WSOC mass concentrations and the δ13C−WSOC values decrease synchronously with the change of the wind direction (from north to southeast) after Episode 1. The southeast wind breaks the continuous transport of WSOC from northern China. And the relatively lower δ13C−WSOC values are then observed, suggesting a regional isotope signal of WSOC without the substantial aging. Besides, the WSOC ∕ OC ratio declines obviously with the isotope after Episode 1 (Fig. S5), indicating less contribution of aged aerosols to the sampling site. Therefore, the elevated δ13C−WSOC values with the increased WSOC mass concentrations in Episode 1 are mainly affected by the aged aerosols transported from northern China.
4.2.2 Episode 2
The δ13C−WSOC values show an opposite trend with WSOC mass concentrations (, p<0.01; see Fig. 6e) in Episode 2. At the beginning of Episode 2 (22 January), the sampling site was mainly affected by the air mass from the north of Nanjing, and the WSOC displayed relatively higher δ13C−WSOC values at the same time (Fig. 6b). After 22 January, the shift of the wind direction and the air mass trajectories correspond well to the decline of the δ13C−WSOC values (Fig. 6b). The large amount of fire spots in the potential source regions suggests the significant impact of open-field biomass burning. It should be noted that the stable carbon isotope composition of C3 plant combustion is relatively low (i.e., δ13C ‰; Martinelli et al., 2002; Sousa Moura et al., 2008). The δ13C−WSOC values decrease and the WSOC mass concentrations peak to the maximum when the air mass travels throughout the regions with a great many hot spots. The concentration of nss-K+ has a positive correlation with WSOC concentration (r=0.82, p<0.001) and a negative correlation with δ13C−WSOC (, p<0.05) during Episode 2. And the concentration of nss-K+ increases up to 6.70 µg m−3, about 5 times the average value, indicating a significant biomass burning contribution (Fig. 5e). The decrease in the δ13C−WSOC values and the increase in the biomass burning tracers (i.e., nss-K+) suggest that the biomass burning emission is a major contributing factor to WSOC aerosols. Also, the WSOC ∕ OC ratio declines from 0.88 to 0.53 (Fig. S5), indicating that the increased WSOC is rather from fresh biomass-burning aerosols without a substantial aging process.
4.2.3 Episode 3
The 13C is clearly enriched (p<0.01) in TC ( ‰) compared to WSOC ( ‰) during Episode 3 (see Fig. 6f). This might be related to a 13C-enriched source and/or the aging process of the non-WSOC fraction in TC. The non-WSOC fraction consists mainly of WIOC, EC and carbonate carbon (CC). Among these carbonaceous species, carbonate carbon (CC) exhibits much higher δ13C values than EC and OC (Kawamura et al., 2004). CC could be a significant fraction of dust aerosols, even though it is a very small part of TC in PM2.5 in most cases.
To study the dust contribution in Episode 3, Ca2+ is determined as an indicator of dust (Jankowski et al., 2008; Huang et al., 2010). Ca2+ and TC show similar patterns (R2=0.84, p<0.01), indicating dust origins in this period. The argument is also supported by the 48 h backward trajectory analysis (Fig. 6c). It shows that the air mass mainly originates from a semi-arid region, Mongolia. The photochemical aging of dust aerosols during the long-range transport from Mongolia to Nanjing could possibly promote the 13C enrichment. Briefly speaking, the enrichment of 13C in TC over WSOC is due to a dust event transported to the studied site.
According to the mass balance, the isotopic ratio of TC affected by CC in the dust aerosols can be expressed as follows:
where the δ13C−TC, δ13C−CC and δ13C−NC are the measured stable carbon isotope of TC, the isotopic ratio of CC in dust aerosols and the isotope composition of non-CC fractions. The fcc represents the CC contribution to TC. The CC contribution during Episode 3 is roughly estimated based on a few assumptions: (1) the increase in TC and δ13C−TC is only affected by the dust origin, (2) the average value of δ13C−TC (−25 ‰) during the studied period (except Episode 3) is taken as the value of δ13C−NC, and (3) δ13C−CC =0.3 ‰ in dust sources (Kawamura et al., 2004). With these considerations, CC is estimated to contribute up to 10 % of TC according to the Eq. (12).
An optimized method for the determination of WSOC mass concentrations and δ13C−WSOC values in aerosol samples with GasBench II–IRMS is presented. A two-step correction is applied to correct the blank contribution and to calibrate the isotope results. The procedural blank is estimated to be 0.5 µg C with isotope composition of −27.43 ‰. The detection limit is demonstrated to be 5 µg C according to the measurement of working standards with various carbon contents. The method yields a high recovery of the standards (97±6 %) and ambient samples (99±10 %). According to the high recoveries, the isotope fractionation during the pretreatment tended to be negligible. The precision and the accuracy is better than 0.17 ‰ and 0.5 ‰, respectively. WSOC concentrations determined with this optimized method are consistent (R2=0.95) with the results of the TOC analyzer. Compared with the previous methods, the optimized method presented in this study is more precise and accurate and requires less time-consuming pretreatment.
The method presented was applied to analyze the δ13C−WSOC of the high time-resolution aerosol samples collected during a severe winter haze in eastern China. WSOC ranged from 3.0 to 32.0 µg m−3, and δ13C−WSOC varied between −26.24 ‰ and −23.35 ‰. 13C was more enriched in WSOC than TC in the majority of the sampling period, indicating aged aerosols and/or 13C-depleted primary sources of non-WSOC component. Three haze events (Episode 1, Episode 2 and Episode 3) are identified with different tendencies of δ13C−WSOC during the accumulation of WSOC aerosols. Similar patterns of the WSOC concentrations and the δ13C−WSOC values in Episode 1 are demonstrated to be affected by the air mass transported from northern China. The increase in δ13C−WSOC indicates that the WSOC aerosols from the studied site are subject to a substantial photochemical aging process during the long-range transport. The contrasting trend of the WSOC and δ13C−WSOC values in Episode 2 is interpreted as the contribution of regional C3 plant biomass burning sources. In Episode 3, the heavier isotope (13C) is clearly enriched in total carbon (TC) compared to WSOC fraction due to the dust contribution.
The optimized method is demonstrated to be accurate and precise in the detection of the WSOC mass concentration and its isotope compositions (δ13C−WSOC) in aerosols. Our results indicate that the high time-resolved measurement of δ13C−WSOC can be used to distinguish different atmospheric processes such as photochemical aging and aerosol sources (e.g., biomass burning and dust). However, a quantitative understanding of sources and formation processes of WSOC aerosols is still a great challenge. To reduce the knowledge gaps, a combination of multiple methodologies is needed in future studies, such as high time-resolved measurement of radiocarbon (14C) and stable carbon isotope compositions (δ13C), and the real-time measurement of chemical compositions (e.g., aerosol mass spectrometers, AMS or thermal desorption aerosol gas chromatograph-AMS).
Data are available from the corresponding author on request (dryanlinzhang@outlook.com).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-11071-2019-supplement.
YLZ conceived and designed the study; YLZ, FC and WZ designed the experimental strategy; WZ, YZ and YX performed the sampling and isotope measurements; YLZ and WZ analyzed the experimental data; YLZ and WZ proposed the hypotheses; WZ wrote the paper with YCL, and all other co-authors contributed to writing.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Multiphase chemistry of secondary aerosol formation under severe haze”. It is not associated with a conference.
This study is supported by the National Natural Science Foundation of China (grant nos. 91644103, 41977305, 41761144056, and 41603104), the National Key Research and Development Program of China (grant no. 2017YFC0212704) and the Provincial Natural Science Foundation of Jiangsu (grant no. BK20180040).
This paper was edited by Jingkun Jiang and reviewed by two anonymous referees.
Agarwal, S., Aggarwal, S. G., Okuzawa, K., and Kawamura, K.: Size distributions of dicarboxylic acids, ketoacids, α-dicarbonyls, sugars, WSOC, OC, EC and inorganic ions in atmospheric particles over Northern Japan: implication for long-range transport of Siberian biomass burning and East Asian polluted aerosols, Atmos. Chem. Phys., 10, 5839–5858, https://doi.org/10.5194/acp-10-5839-2010, 2010.
Aggarwal, S. G. and Kawamura, K.: Molecular distributions and stable carbon isotopic compositions of dicarboxylic acids and related compounds in aerosols from Sapporo, Japan: Implications for photochemical aging during long-range atmospheric transport, J. Geophys. Res.-Atmos., 113, 1–13, https://doi.org/10.1029/2007JD009365, 2008.
Anderson, R. S., Iannone, R., Thompson, A. E., Rudolph, J., and Huang, L.: Carbon kinetic isotope effects in the gas-phase reactions of aromatic hydrocarbons with the OH radical at 296±4 K, Geophys. Res. Lett., 31, L15108, https://doi.org/10.1029/2004GL020089, 2004.
Anderson, C., Dibb, J. E., Griffin, R. J., and Bergin, M. H.: Simultaneous measurements of particulate and gas-phase water-soluble organic carbon concentrations at remote and urban-influenced locations, Geophys. Res. Lett., 35, 2–5, https://doi.org/10.1029/2008GL033966, 2008.
Asa-Awuku, A., Moore, R. H., Nenes, A., Bahreini, R., Holloway, J. S., Brock, C. A., Middlebrook, A. M., Ryerson, T. B., Jimenez, J. L., DeCarlo, P. F., Hecobian, A., Weber R. J., Stickel, R., Tanner D. J., and Huey, L. G.: Airborne Cloud Condensation Nuclei Measurements during the 2006 Texas Air Quality Study, J. Geophys. Res.-Atmos., 116, 1–18. https://doi.org/10.1029/2010JD014874, 2011.
Atekwana, E. A. and Krishnamurthy, R. V.: Chapter 10 – Extraction of Dissolved Inorganic Carbon (DIC) in Natural Waters for Isotopic Analyses, edited by: Pier, A. B. T., Handbook of Stable Isotope Analytical Techniques, de Groot, Elsevier, Amsterdam, 203–28, https://doi.org/10.1016/B978-044451114-0/50012-0, 2004.
Atkinson, R.: Kinetics and Mechanisms of the Gas-Phase Reactions of the Hydroxyl Radical with Organic Compounds under Atmospheric Conditions, Chem. Rev., 86, 69–201, https://doi.org/10.1021/cr00071a004, 1986.
Bauer, J. E., Haddad, R. I., and Des Marais, D. J.: Method for determining stable isotope ratios of dissolved organic carbon in interstitial and other natural marine waters, Mar. Chem., 33, 335–351, https://doi.org/10.1016/0304-4203(91)90076-9, 1991.
Bozzetti, C., El Haddad, I., Salameh, D., Daellenbach, K. R., Fermo, P., Gonzalez, R., Minguillón, M. C., Iinuma, Y., Poulain, L., Elser, M., Müller, E., Slowik, J. G., Jaffrezo, J.-L., Baltensperger, U., Marchand, N., and Prévôt, A. S. H.: Organic aerosol source apportionment by offline-AMS over a full year in Marseille, Atmos. Chem. Phys., 17, 8247–8268, https://doi.org/10.5194/acp-17-8247-2017, 2017a.
Bozzetti, C., Sosedova, Y., Xiao, M., Daellenbach, K. R., Ulevicius, V., Dudoitis, V., Mordas, G., Byčenkienė, S., Plauškaitė, K., Vlachou, A., Golly, B., Chazeau, B., Besombes, J.-L., Baltensperger, U., Jaffrezo, J.-L., Slowik, J. G., El Haddad, I., and Prévôt, A. S. H.: Argon offline-AMS source apportionment of organic aerosol over yearly cycles for an urban, rural, and marine site in northern Europe, Atmos. Chem. Phys., 17, 117–141, https://doi.org/10.5194/acp-17-117-2017, 2017b.
Brüniche-Olsen, N. and Ulstrup, J.: Quantum theory of kinetic isotope effects in proton transfer reactions, Journal of the Chemical Society Faraday Transactions Physical Chemistry in Condensed Phases, 75, 205–226, 1979.
Cao, J. J., Chow, J. C., Tao, J., Lee, S. C., Watson, J. G., Ho, K. F., Wang, G. H., Zhu, C. S., and Han, Y. M.: Stable carbon isotopes in aerosols from Chinese cities: Influence of fossil fuels, Atmos. Environ., 45, 1359–1363, https://doi.org/10.1016/j.atmosenv.2010.10.056, 2011.
Chen, X. P., Zhou, J. C., Wang, X. R., Blackmer, A. M., and Zhang, F.: Optimal rates of nitrogen fertilization for a winter wheat-corn cropping system in northern china, Commun. Soil Sci. Plan, 35, 583–597, https://doi.org/10.1081/CSS-120029734, 2004.
Currie, L. A., Klouda, G. A., Benner, B. A., Garrity, K., and Eglinton, T. I.: Isotopic and molecular fractionation in combustion; three routes to molecular marker validation, including direct molecular “dating” (GC/AMS), Atmos. Environ., 33, 2789–2806, http://https://doi.org/10.1016/S1352-2310(98)00325-2, 1999.
Das, O., Wang, Y., and Hsieh, Y. P.: Chemical and carbon isotopic characteristics of ash and smoke derived from burning of C and C grasses, Org. Geochem., 41, 263–269, http://https://doi.org/10.1016/j.orggeochem.2009.11.001, 2010.
Decesari, S., Mircea, M., Cavalli, F., Fuzzi, S., Moretti, F., Tagliavini, E., and Facchini, M. C.: Source attribution of water-soluble organic aerosol by nuclear magnetic resonance spectroscopy, Environ. Sci. Technol., 41, 2479–2484, https://doi.org/10.1021/Es061711l, 2007.
Fisseha, R., Saurer, M., Jäggi, M., Szidat, S., Siegwolf, R. T. W., and Baltensperger, U.: Determination of stable carbon isotopes of organic acids and carbonaceous aerosols in the atmosphere, Rapid Commun. Mass Sp., 20, 2343–2347, 2006.
Fisseha, R., Saurer, M., Jäggi, M., Siegwolf, R. T., Dommen, J., Szidat, S., Samburova, V., and Baltensperger, U.: Determination of primary and secondary sources of organic acids and carbonaceous aerosols using stable carbon isotopes, Atmos. Environ., 43, 431–437, https://doi.org/10.1016/j.atmosenv.2008.08.041, 2009.
Fowler, K., Connolly, P. J., Topping, D. O., and O'Meara, S.: Maxwell–Stefan diffusion: a framework for predicting condensed phase diffusion and phase separation in atmospheric aerosol, Atmos. Chem. Phys., 18, 1629–1642, https://doi.org/10.5194/acp-18-1629-2018, 2018.
Hecobian, A., Zhang, X., Zheng, M., Frank, N., Edgerton, E. S., and Weber, R. J.: Water-Soluble Organic Aerosol material and the light-absorption characteristics of aqueous extracts measured over the Southeastern United States, Atmos. Chem. Phys., 10, 5965–5977, https://doi.org/10.5194/acp-10-5965-2010, 2010.
Huang, H., Ho, K. F., Lee, S. C., Tsang, P. K., Ho, S. S. H., Zou, C. W., Zou, S. C., Cao, J. J., and Xu, H. M.: Characteristics of carbonaceous aerosol in PM2.5: Pearl Delta River region, China, Atmos. Res., 104, 227–236, https://doi.org/10.1016/j.atmosres.2011.10.016, 2012.
Huang, K., Zhuang, G., Li, J., Wang, Q., Sun, Y., Lin, Y., and Fu, J. S.: Mixing of Asian dust with pollution aerosol and the transformation of aerosol components during the dust storm over China in spring 2007, J. Geophys. Res., 115, D00K13, https://doi.org/10.1029/2009JD013145, 2010.
Huang, L., Brook, J. R., Zhang, W., Li, S. M., Graham, L., Ernst, D., Chivulescu, A., and Lu, G.: Stable isotope measurements of carbon fractions (OC/EC) in airborne particulate: A new dimension for source characterization and apportionment, Atmos. Environ., 40, 2690–2705, https://doi.org/10.1016/j.atmosenv.2005.11.062, 2006.
Iannone, R., Anderson, R. S., Rudolph, J., Huang, L., and Ernst, D.: The carbon kinetic isotope effects of ozone-alkene reactions in the gas-phase and the impact of ozone reactions on the stable carbon isotope ratios of alkenes in the atmosphere, Geophys. Res. Lett., 30, 1684, https://doi.org/10.1029/2003GL017221, 2003.
Jankowski, N., Schmidl, C., Marr, I. L., Bauer, H., and Puxbaum, H.: Comparison of methods for the quantification of carbonate carbon in atmospheric PM10 aerosol samples, Atmos. Environ., 42, 8055–8064, https://doi.org/10.1016/j.atmosenv.2008.06.012, 2008.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., and Aiken, A. C.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kawamura, K., Kobayashi, M., Tsubonuma, N., Mochida, M., Watanabe, T., and Lee, M.: Organic and inorganic compositions of marine aerosols from East Asia: Seasonal variations of water-soluble dicarboxylic acids, major ions, total carbon and nitrogen, and stable C and N isotopic composition, Geo. Soc. S. P., 9, 243–265, https://doi.org/10.1016/S1873-9881(04)80019-1, 2004.
Kirillova, E. N., Sheesley, R. J., Andersson, A., and Gustafsson, Ö. : Natural Abundance 13C and 14C Analysis of Water-Soluble Organic Carbon in Atmospheric, Anal. Chem., 82, 7973–7978, https://doi.org/10.1029/2006GL028325, 2010.
Kirillova, E. N., Andersson, A., Sheesley, R. J., Kruså, M., Praveen, P. S., Budhavant, K., Safai, P. D., Rao, P. S. P., and Gustafsson, Ö.: 13C- and 14C-based study of sources and atmospheric processing of water-soluble organic carbon (WSOC) in South Asian aerosols, J. Geophys. Res.-Atmos., 118, 614–626, https://doi.org/10.1002/jgrd.50130, 2013.
Kirillova, E. N., Andersson, A., Tiwari, S., Kumar Srivastava, A., Singh Bisht, D., and Gustafsson, Ö.: Water-soluble organic carbon aerosols during a full New Delhi winter: Isotope-base source apportionment and optical properties, J. Geophys. Res.-Atmos., 119, 3476–3485, https://doi.org/10.1002/2013JD021272, 2014.
Lang, S. Q., Bernasconi, S. M., and Früh-Green, G. L.: Stable isotope analysis of organic carbon in small (µg C) samples and dissolved organic matter using a GasBench preparation device, Rapid Commun. Mass Sp., 26, 9–16, https://doi.org/10.1002/rcm.5287, 2012.
Liang, L. L., Guenter, E., Duan, F. K., Ma, Y. L., Cheng, Y., Du, Z. Y., and He, K. B.: Composition and Source Apportionments of Saccharides in Atmospheric Particulate Matter in Beijing, Huanjing Kexue/Environmental Science, 36, 3935–3942, https://doi.org/10.13227/j.hjkx.2015.11.001, 2015.
Martinelli, L. A., Camargo, P. B., Lara, L. B. L. S., Victoria, R. L., and Artaxo, P.: Stable carbon and nitrogen isotopic composition of bulk aerosol particles in a C4 plant landscape of southeast Brazil, Atmos. Environ., 36, 2427–2432. https://doi.org/10.1016/S1352-2310(01)00454-X, 2002.
Martinez, R. E., Williams, B. J., Zhang, Y., Hagan, D., Walker, M., Kreisberg, N. M., Hering S. V., Hohaus T., Jayne J. T., and Worsnop D. R.: Development of a volatility and polarity separator (VAPS) for volatility- and polarity-resolved organic aerosol measurement, Aerosol Sci. Technol., 50, 255–271, https://doi.org/10.1080/02786826.2016.1147645, 2016.
Mills, N. L., Donaldson, K., Hadoke, P. W., Boon, N. A., MacNee, W., Cassee, F. R., Sandström, T., Blomberg, A., and Newby, D. E.: Adverse cardiovascular effects of air pollution, Nat. Clin. Pract. Card., 6, 36–44, https://doi.org/10.1038/ncpcardio1399, 2009.
Miyazaki, Y., Kawamura, K., Jung, J., Furutani, H., and Uematsu, M.: Latitudinal distributions of organic nitrogen and organic carbon in marine aerosols over the western North Pacific, Atmos. Chem. Phys., 11, 3037–3049, https://doi.org/10.5194/acp-11-3037-2011, 2011.
Miyazaki, Y., Fu, P. Q., Kawamura, K., Mizoguchi, Y., and Yamanoi, K.: Seasonal variations of stable carbon isotopic composition and biogenic tracer compounds of water-soluble organic aerosols in a deciduous forest, Atmos. Chem. Phys., 12, 1367–1376, https://doi.org/10.5194/acp-12-1367-2012, 2012.
Myhre, G.: Consistency between satellite-derived and modeled estimates of the direct aerosol effect, Science, 325, 187–190, https://doi.org/10.1126/science.1174461, 2009.
Osada, K., Kido, M., Nishita, C., Matsunaga, K., Iwasaka, Y., Nagatani, M., and Nakada H.: Temporal variation of water-soluble ions of free tropospheric aerosol particles over central japan, Tellus B, 59, 742–754, https://10.1111/j.1600-0889.2007.00296.x, 2007.
Park, S. S., Schauer, J. J., and Cho, S. Y.: Sources and their contribution to two water-soluble organic carbon fractions at a roadway site, Atmos. Environ., 77, 348–357, https://doi.org/10.1016/j.atmosenv.2013.05.032, 2013.
Pathak, R. K., Wang, T., Ho, K. F., and Lee, S. C.: Characteristics of summertime PM2.5 organic and elemental carbon in four major Chinese cities: Implications of high acidity for water-soluble organic carbon (WSOC), Atmos. Environ., 45, 318–325, https://doi.org/10.1016/j.atmosenv.2010.10.021, 2011.
Pavuluri, C. M. and Kawamura, K.: Evidence for 13-carbon enrichment in oxalic acid via iron catalyzed photolysis in aqueous phase, Geophys. Res. Lett., 39, 1–6, https://doi.org/10.1029/2011GL050398, 2012.
Pavuluri C. M. and Kawamura K.: Seasonal changes in TC and WSOC and their 13C isotope ratios in Northeast Asian aerosols: land surface–biosphere–atmosphere interactions, Acta Geochimica, 36, 355–358, https://doi.org/10.1007/s11631-017-0157-3, 2017.
Polissar, P. J., Fulton, J. M., Junium, C. K., Turich, C. C., and Freeman, K. H.: Measurement of 13C and 15N Isotopic Composition on Nanomolar Quantities of C and N, Anal. Chem., 81, 755–763. https://doi.org/10.1021/ac801370c, 2009.
Qin, X., Zhang, Z. F., Li, Y. W., Shen, Y., and Zhao, S. H.: Sources analysis of heavy metal aerosol particles in north suburb of nanjing, Environ. Sci., 37, 4467–4474, https://doi.org/10.13227/j.hjkx.201605237, 2016.
Ramanathan, V., Crutzen, P. J., Kiehl, J. T., and Rosenfeld, D.: Aerosols, climate, and the hydrological cycle, Science, 294, 2119–2124, https://doi.org/10.1126/science.1064034, 2001.
Rudolph, J.: Gas Chromatography-Isotope Ratio Mass Spectrometry, Volatile Organic Compounds in the Atmosphere, Blackwell Publishing, Oxford, UK, 388–466, 2007.
Rudolph, J., Czuba, E., and Huang, L.: The Stable Carbon Isotope Fractionation for Reactions of Selected Hydrocarbons with OH-Radicals and Its Relevance for Atmospheric Chemistry, J. Geophys. Res.-Atmos., 105, 29329–29346, https://doi.org/10.1029/2000JD900447, 2000.
Rudolph, J., Czuba, E., Norman, A. L., Huang, L., and Ernst, D.: Stable carbon isotope composition of nonmethane hydrocarbons in emissions from transportation related sources and atmospheric observations in an urban atmosphere, Atmos. Environ., 36, 1173–1181, https://doi.org/10.1016/S1352-2310(01)00537-4, 2002.
Rudolph, J., Anderson R. S., Czapiewski K. V., Czuba E., Ernst D., Gillespie T., Huang L., Rigby C., and Thompson A. E.: The stable carbon isotope ratio of biogenic emissions of isoprene and the potential use of stable isotope ratio measurements to study photochemical processing of isoprene in the atmosphere, J. Atmos. Chem., 44, 39–55, https://doi.org/10.1023/A:1022116304550, 2003.
Saarikoski, S., Timonen, H., Saarnio, K., Aurela, M., Järvi, L., Keronen, P., Kerminen, V.-M., and Hillamo, R.: Sources of organic carbon in fine particulate matter in northern European urban air, Atmos. Chem. Phys., 8, 6281–6295, https://doi.org/10.5194/acp-8-6281-2008, 2008.
Sakugawa, H. and Kaplan, I. R.: Stable carbon isotope measurements of atmospheric organic acids in Los Angeles, California, Geophys. Res. Lett., 22, 1509–1512, https://doi.org/10.1029/95GL01359, 1995.
Sannigrahi, P., Sullivan, A. P., Weber, R. J., and Ingall, E. D.: Characterization of water-soluble organic carbon in urban atmospheric aerosols using solid-state C-13 NMR spectroscopy, Environ. Sci. Technol., 40, 666–672, 2006.
Sharp, J. H.: Total organic carbon in seawater – comparison of measurements using persulfate oxidation and high temperature combustion, Mar. Chem., 1, 211–229, https://doi.org/10.1016/0304-4203(73)90005-4, 1973.
Smith, B. N. and Epstein, S.: Two Categories of 13C∕12C Ratios for Higher Plants, Plant Physiol., 47, 380–384, https://doi.org/10.1104/pp.47.3.380, 1971.
Sousa Moura, J. M., Martens, C. S., Moreira, M. Z., Lima, R. L., Sampaio, I. C. G., Mendlovitz, H. P., and Menton, M. C.: Spatial and seasonal variations in the stable carbon isotopic composition of methane in stream sediments of eastern Amazonia, Tellus B, 60, 21–31, https://doi.org/10.1111/j.1600-0889.2007.00322.x, 2008.
Sullivan, A. P., Weber, R. J., Clements, A. L., Turner, J. R., Bae, M. S., and Schauer, J. J.: A method for on-line measurement of water-soluble organic carbon in ambient aerosol particles: Results from an urban site, Geophys. Res. Lett., 31, 14–17, https://doi.org/10.1029/2004GL019681, 2004.
Suto, N. and Kawashima, H.: Online wet oxidation/isotope ratio mass spectrometry method for determination of stable carbon isotope ratios of water-soluble organic carbon in particulate matter, Rapid Commun. Mass Sp., 32, 1668–1674, https://doi.org/10.1002/rcm.8240, 2018.
Ten Brink, H., Maenhaut, W., Hitzenberger, R., Gnauk, T., Spindler, G., Even, A., Chi, X., Bauer, H., Puxbaum, H., Putaud, J. P., and Tursic, J: INTERCOMP2000: The comparability of methods in use in Europe for measuring the carbon content of aerosol, Atmos. Environ., 38, 6507–6519, https://doi.org/10.1016/j.atmosenv.2004.08.027, 2004.
Turekian, V. C., Macko, S., Ballentine, D., Swap, R. J., and Garstang, M.: Causes of bulk carbon and nitrogen isotopic fractionations in the products of vegetation burns: Laboratory studies, Chem. Geol., 152, 181–192, https://doi.org/10.1016/S0009-2541(98)00105-3, 1998.
Wang, G., Xie, M., Hu, S., Gao, S., Tachibana, E., and Kawamura, K.: Dicarboxylic acids, metals and isotopic compositions of C and N in atmospheric aerosols from inland China: implications for dust and coal burning emission and secondary aerosol formation, Atmos. Chem. Phys., 10, 6087–6096, https://doi.org/10.5194/acp-10-6087-2010, 2010.
Wang, H., Kawamura, K., and Shooter, D.: Wintertime organic aerosols in Christchurch and Auckland, New Zealand: Contributions of residential wood and coal burning and petroleum utilization, Environ. Sci. Technol., 40, 5257–5262, https://doi.org/10.1021/es052523i, 2006.
Weber, R. J., Sullivan, A. P., Peltier, R. E., Russell, A., Yan, B., Zheng, M., de Gouw, J., Warneke, C., Brock, C., Holloway, J. S., Atlas, E. L., and Edgerton, E.: A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States, J. Geophys. Res., 112, D13302, https://doi.org/10.1029/2007JD008408, 2007.
Werner, R. A., Bruch, B. A., and Brand, W. A.: ConFlo III – an interface for high precision δ13C and δ15N analysis with an extended dynamic range, Rapid Commun. Mass Sp., 13, 1237–1241, https://doi.org/10.1002/(SICI)1097-0231(19990715)13:13<1237::AID-RCM633>3.0.CO;2-C, 1999.
Widory, D.: Combustibles, fuels and their combustion products: A view through carbon isotopes, Combust. Theor. Model., 10, 831–841, https://doi.org/10.1080/13647830600720264, 2006.
Wozniak, A. S., Bauer, J. E., Sleighter, R. L., Dickhut, R. M., and Hatcher, P. G.: Technical Note: Molecular characterization of aerosol-derived water soluble organic carbon using ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry, Atmos. Chem. Phys., 8, 5099–5111, https://doi.org/10.5194/acp-8-5099-2008, 2008.
Zhang, R., Jing, J., Tao, J., Hsu, S.-C., Wang, G., Cao, J., Lee, C. S. L., Zhu, L., Chen, Z., Zhao, Y., and Shen, Z.: Chemical characterization and source apportionment of PM2.5 in Beijing: seasonal perspective, Atmos. Chem. Phys., 13, 7053–7074, https://doi.org/10.5194/acp-13-7053-2013, 2013.
Zhang, Y. L., Li, J., Zhang, G., Zotter, P., Huang, R. J., Tang, J. H., Wacker, L., Prévôt, A. S., and Szidat, S.: Radiocarbon-based source apportionment of carbonaceous aerosols at a regional background site on Hainan Island, South China, Environ. Sci. Technol., 48, 2651–2659, https://doi.org/10.1021/es4050852, 2014.
Zhang, Y.-L., El-Haddad, I., Huang, R.-J., Ho, K.-F., Cao, J.-J., Han, Y., Zotter, P., Bozzetti, C., Daellenbach, K. R., Slowik, J. G., Salazar, G., Prévôt, A. S. H., and Szidat, S.: Large contribution of fossil fuel derived secondary organic carbon to water soluble organic aerosols in winter haze in China, Atmos. Chem. Phys., 18, 4005–4017, https://doi.org/10.5194/acp-18-4005-2018, 2018.
Zhou, Y., Guo, H., Lu, H., Mao, R., Zheng, H., and Wang, J.: Analytical methods and application of stable isotopes in dissolved organic carbon and inorganic carbon in groundwater, Rapid Commun. Mass Sp., 29, 1827–1835, https://doi.org/10.1002/rcm.7280, 2015.