the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 May 2018
| 25 May 2018
Cloud condensation nuclei activity of CaCO3 particles with oleic acid and malonic acid coatings
Mingjin Wang
Defeng Zhao
Florian Rubach
Andreas Wahner
Astrid Kiendler-Scharr
Condensation of carboxylic acids on mineral particles leads to coatings and impacts the particles' potential to act as cloud condensation nuclei (CCN). To determine how the CCN activity of mineral particles is impacted by carboxylic acid coatings, the CCN activities of CaCO3 particles and CaCO3 particles with oleic acid and malonic acid coatings were compared in this study. The results revealed that small amounts of oleic acid coating (volume fraction (vf) ≤4.3 %) decreased the CCN activity of CaCO3 particles, while more oleic acid coating (vf ≥16 %) increased the CCN activity of CaCO3 particles. This phenomenon has not been reported before. In contrast, the CCN activity of CaCO3 particles coated with malonic acid increased with the thickness of the malonic acid coating (vf =0.4–40 %). Even the smallest amounts of malonic acid coating (vf =0.4 %) significantly enhanced the CCN activity of CaCO3 particles from to . This indicates that a small amount of water-soluble organic acid coating may significantly enhance the CCN activity of mineral particles. The presence of water vapor during the coating process with malonic acid additionally increased the CCN activity of the coated CaCO3 particles, probably because more CaCO3 reacts with malonic acid when sufficient water is available.
- Article
(1077 KB) - Full-text XML
-
Supplement
(458 KB) - BibTeX
- EndNote





Atmospheric aerosols serve as cloud condensation nuclei (CCN) and change the radiative properties (cloud albedo effect) and lifetime (cloud lifetime effect) of clouds, thus affecting the Earth's climate indirectly (Liu and Wang, 2010; Gantt et al., 2012; Penner et al., 2004; Haywood and Boucher, 2000). Mineral aerosol is one of the most abundant components of the atmospheric aerosol. It is estimated that 1500–2600 Tg of mineral aerosol particles with radii between 0.1 and 8 µm are emitted annually into the atmosphere on a global scale (Cakmur et al., 2006). Mineral aerosol particles are mainly composed of substances that are slightly soluble or insoluble in water. CCN activity measurements show that the hygroscopicity parameter κ (Petters and Kreidenweis, 2007) varies between 0.001 and 0.08 for mineral aerosols, including CaCO3 aerosol, clay aerosols, and mineral dust aerosols generated in the laboratory or sampled from various locations worldwide (Garimella et al., 2014; Yamashita et al., 2011; Zhao et al., 2010; Koehler et al., 2009; Sullivan et al., 2010; Herich et al., 2009). The low κ indicates that the CCN activity of mineral aerosol is much lower than that of water-soluble salts like (NH4)2SO4 (κ=0.61) and NaCl (κ=1.28), which are also common in atmospheric aerosols (Petters and Kreidenweis, 2007). Tang et al. (2016) reviewed recently the interaction of mineral dust particles with water.
Mineral aerosol particles can be coated by organic vapors during their residence and transport in the atmosphere. Many individual particle measurements have shown that mineral components and organic matter can coexist in the same individual aerosol particle in the real atmosphere (Falkovich et al., 2004, 2001; Russell et al., 2002; Li and Shao, 2010). Carboxylic acids (R(C=O)OH) are abundant species among the organic matter that coexists with mineral particles. Russell et al. (2002) found that carbonyls (R(C=O)R), alkanes, and R(C=O)OH are present in individual mineral (and sea salt) aerosol particles, with enhanced concentration of R(C=O)OH. They also found that Ca2+, , R(C=O)OH, and R(C=O)R coexisted in some individual mineral aerosol particles with a strong correlation between and R(C=O)OH. These particles could be formed by CaCO3 particles (partly) coated with organic film. Falkovich et al. (2004) also found that organic and inorganic components coexisted in individual mineral aerosol particles with the organic component consisting of various short-chain (C1–C10) mono- and dicarboxylic acids (MCA and DCA). The concentration of short-chain carboxylic acids in mineral aerosol particles increased with the increase of the ambient relative humidity (RH). A possible explanation for such observations could be that when more water is condensed onto mineral particles at higher ambient RH, the adsorbed carboxylic acids are ionized in the aqueous environment and react with mineral particles forming organic acid salts. Of the major components of mineral aerosol particles (clay, calcite (CaCO3), quartz, mica, feldspar, etc.), only CaCO3 with alkaline character can react with carboxylic acids in this way. Thus CaCO3 may play a key role in the uptake of carboxylic acids by mineral aerosol particles.
Carboxylic acid coatings on mineral aerosol particles change their chemical composition and thus may have an impact on their CCN activity. Many previous studies have investigated the CCN activity of pure mineral aerosol (Garimella et al., 2014; Yamashita et al., 2011; Zhao et al., 2010; Koehler et al., 2009; Sullivan et al., 2010; Herich et al., 2009) and pure carboxylic acid aerosol (Kumar et al., 2003; Hori et al., 2003; Cruz and Pandis, 1997; Hartz et al., 2006), but only a few studies have investigated the CCN activity of mineral aerosol particles with carboxylic acid coatings (Tang et al., 2015; Hatch et al., 2008; Gierlus et al., 2012).
In this study we used malonic acid (MA) and oleic acid (OA) as coating materials and CaCO3 particles as cores and investigated the CCN activity of the coated CaCO3 particles. Herein we varied the coating thickness and the RH during the coating process. MA is a representative of the class of DCA acids and OA is an example of surfactant-like compounds. DCA acids are ubiquitous in the atmosphere (Kawamura et al., 1996; Kawamura and Ikushima, 1993; Ho et al., 2007; Mkoma and Kawamura, 2013; Kawamura and Bikkina, 2016) and formed by photochemical reactions and ozonolysis (Chebbi and Carlier, 1996; Charbouillot et al. 2012; Kawamura and Bikkina, 2016; Kawamura et al., 1996; Khare et al., 1999; Mellouki et al., 2015). It has been reported that DCA acids (C2–C10) account for 0.06–1.1 % of the total aerosol mass, with higher values in the summer, and 1.8 % of the total aerosol carbon in urban aerosol, in which oxalic acid, MA, and succinic acid are the most abundant species (Kawamura et al., 1996; Kawamura and Ikushima, 1993; Ho et al., 2007; Mkoma and Kawamura, 2013). OA, which is emitted into the atmosphere by the cooking of meat, wood burning, and automobile sources (Schauer et al., 1999; Rogge et al., 1998, 1993), is present in atmospheric aerosols of urban, rural, and forest areas (Cheng et al., 2004; Ho et al., 2010). The water solubility of the two organic acids is complementary; it is high for MA while it is very low for OA. Coatings of MA and OA could thus have different effects on CCN activity of mineral particles.
As general procedure, CaCO3 aerosol was generated according to Zhao et al. (2010), and then poly- or monodisperse CaCO3 aerosol particles were coated by malonic or oleic acid in a coating device. A flow tube was optionally applied to extend the residence time. The particle size, chemical composition, and CCN activity of the CaCO3 particles were measured before and after coating. Figure 1 shows the schematic of the experimental setup.
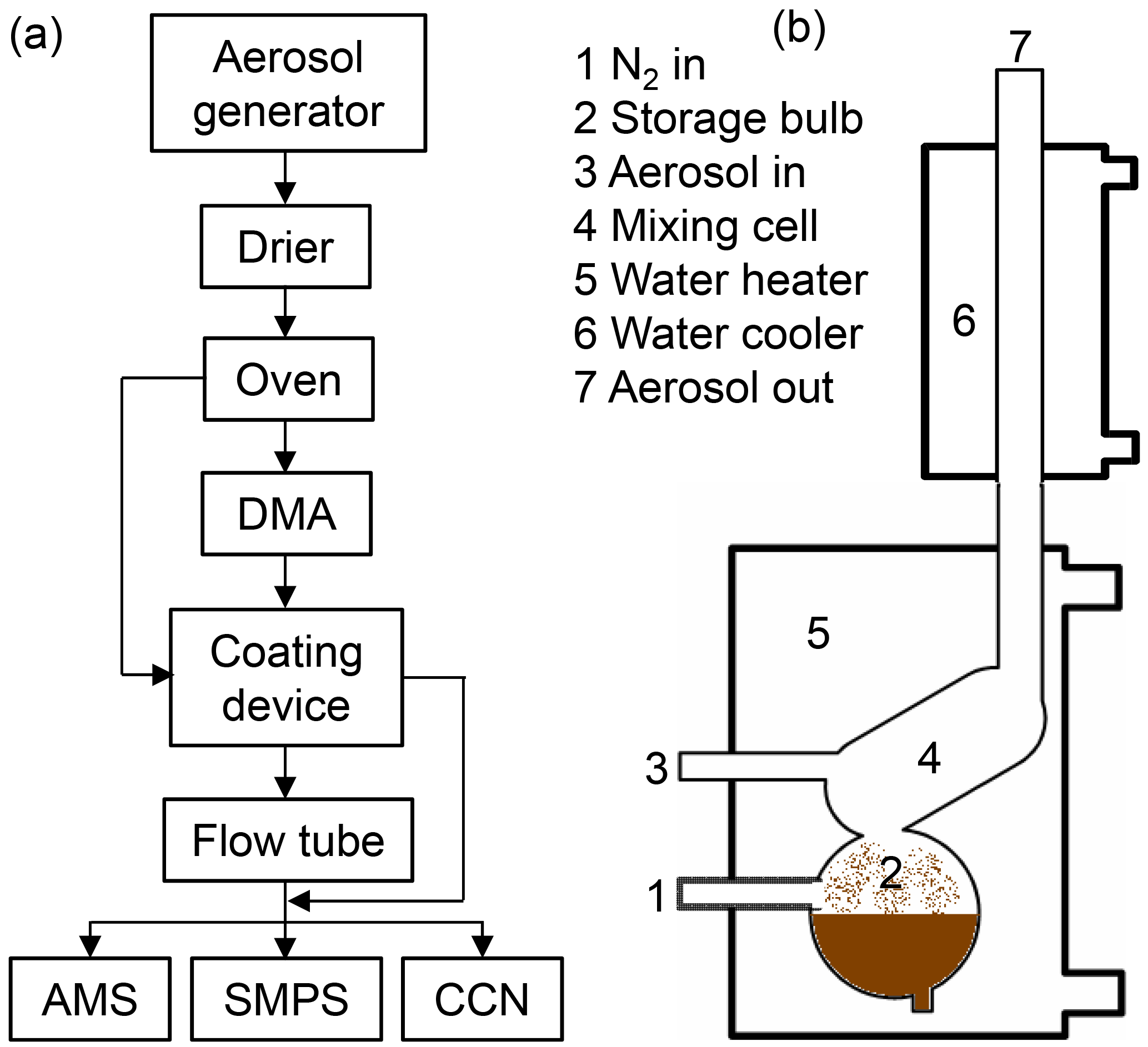
Figure 1Schematics of the experimental setup (a). CaCO3 aerosol is generated by spray-drying saturated Ca(HCO3)2 solutions and tempering the aerosol passing through an oven at 300 ∘C. The polydisperse CaCO3 aerosol is either led directly to the coating device (b, after Roselli, 2006) or led to a differential mobility analyzer (DMA) for size selection first. An optional flow tube can be switched into the pass to enhance the reaction time of the coated particles. The stream of coated particles is finally split to the analytical instruments, namely aerosol mass spectrometer (AMS), scanning mobility particle sizer (SMPS) and CCN counter.
2.1 Generation of CaCO3 aerosol
CaCO3 aerosol was generated by spraying a saturated Ca(HCO3)2 solution. A sample of CaCO3 powder (2 g, pro analysis, ≥99 %, Merck, Darmstadt, Germany) was suspended in 1 L Milli-Q water (18.2 MΩ cm, TOC <5 ppb). Then about 1.5 L min−1 CO2 (purity ≥99.995 %, Praxair Industriegase GmbH & Co. KG, Magdeburg, Germany) was bubbled into the suspension at room temperature (RT) for 3 h, while the suspension was stirred using a magnetic stirrer. During bubbling, CO2 reacted with CaCO3 to produce Ca(HCO3)2. After bubbling, the suspension was allowed to settle for 10 min, the supernatant clear Ca(HCO3)2 solution was decanted and used for spraying by a constant output atomizer (model 3076, TSI, Shoreview, MN, USA) using 1.75 L min−1 high-purity N2 (Linde LiPur 6.0, purity 99.9999 %, Linde AG, Munich, Germany).
The major portion (0.9 L min−1) of the aerosol flow generated by spraying was dried in a diffusion drier filled with silica gel. The RH was below 10 % after drying. The remainder of the aerosol flow was drawn off by a pump and discarded. The dry aerosol was passed through a tube furnace (model RS 120/1000/12, Nabertherm GmbH, Lilienthal, Germany) set at 300 ∘C. The residence time of the aerosol in the furnace was 5.2 s. Zhao et al. (2010) described this method for generating CaCO3 aerosol in detail. At room temperature, dry Ca(HCO3)2 is thermodynamically unstable and decays into CaCO3, CO2, and H2O (Keiser and Leavitt, 1908). With this method the aerosol still contained some Ca(HCO3)2 after drying, but after heating at 300 ∘C it was completely converted into CaCO3 (Zhao et al., 2010). The CaCO3 aerosol generated was either first size selected by a differential mobility analyzer (DMA, TSI 3081) with a neutralizer on the inlet or entered the coating device directly as polydisperse aerosol.
Figure 2a shows the total number concentration and mean size of the bare CaCO3 aerosol particles generated at different spraying time, which were measured with the scanning mobility particle sizer (SMPS) described below. The mean size stabilized after about 50 min in the range 49.8–55.5 nm. Over the 232 min spraying time, the total number concentration varied in the range 1.8×106–4.5×106 cm−3. The total number concentration decreased by about one-third in the initial 70 min. The decrease became slower after 70 min and the total number concentration tended to stabilize after 155 min. After 70 min the total number concentration varied in a smaller range of 1.8×106–2.9×106 cm−3; therefore, the measurements in this study typically started after 70 min spraying. The typical size distribution of the CaCO3 aerosol particles after 70 min spraying is shown in Fig. 2b. The CaCO3 particles showed a single mode distribution with a mode diameter at 32.2 nm. The number concentration was more than 10 000 cm−3 for particles between 12.6 and 151.2 nm.
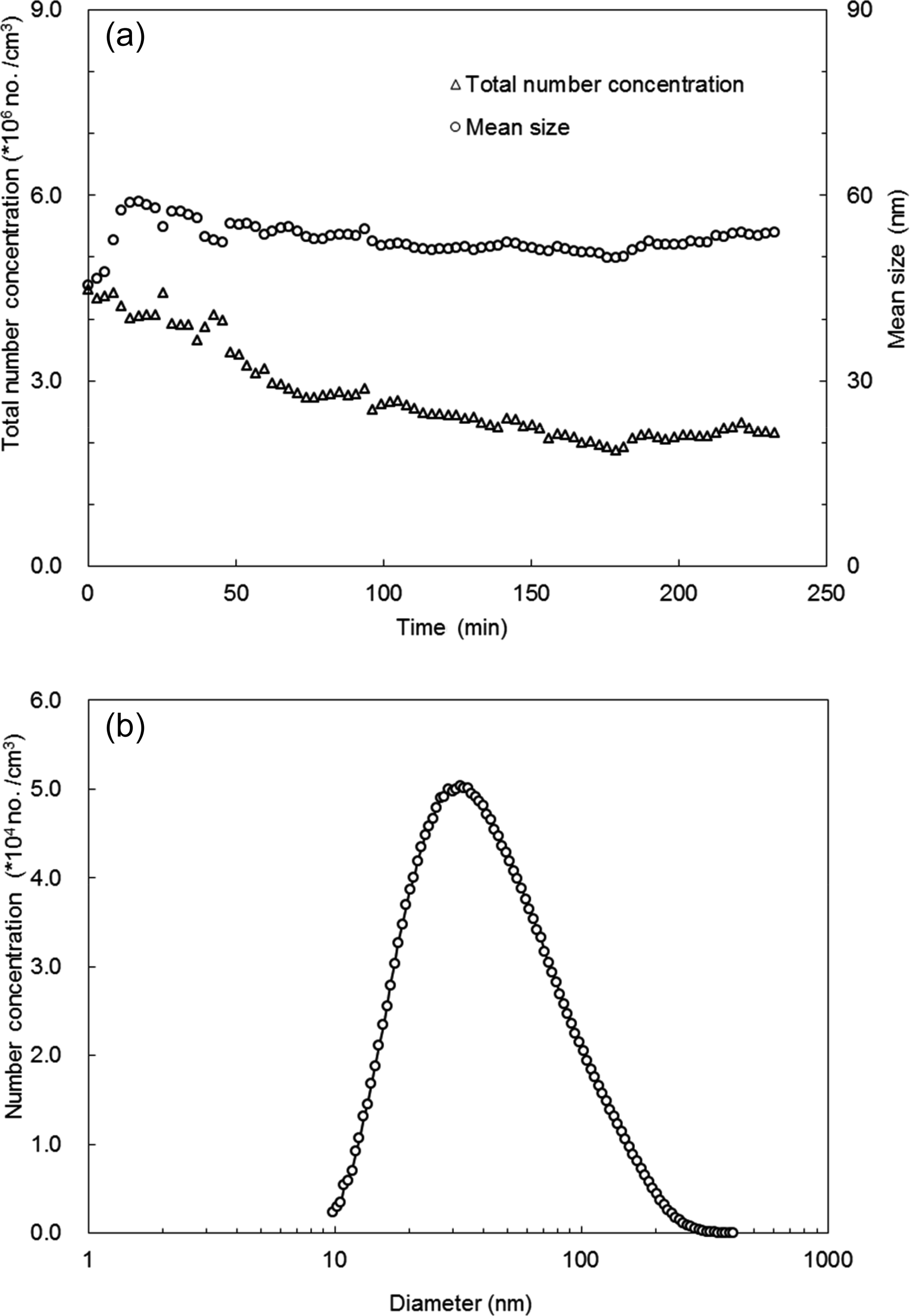
Figure 2Total number concentration and mean diameter of CaCO3 aerosol particles generated as a function of the spraying time (a). Typical size distribution of the CaCO3 aerosol after 70 min spraying (b).
2.2 Organic acid coating
The coating device (Fig. 1b) used in this study was designed by Roselli (2006) and showed good reproducibility, controllability, and stability. The glass apparatus consisted of a small storage bulb (100 mL) holding the organic coating substances which was directly connected to a mixing cell (about 35 mL). The storage bulb and mixing cell were fully immersed in a flow-through water heater connected to a thermostatic bath (F25, Julabo GmbH, Seelbach, Germany). The temperature range of the thermostatic bath used in this study was 30–80 ∘C. An extra N2 stream could be passed through the storage bulb in order to enhance the organic vapors flowing into the mixing cell. The outflow of the coating device was connected to a Liebig type water cooler. The water cooler was controlled by another thermostatic bath (F25, Julabo GmbH, Seelbach, Germany) operated at 25 ∘C throughout all of the experiments.
The bottom of the storage bulb was filled with either 5.0 g MA powder (assay ≥98 % (T), Fluka Chemika, Sigma-Aldrich, St Louis, MO, USA) or 10.0 mL OA (chemical purity (GC) 99.5 %, Alfa Aesar, Ward Hill, MA, USA). A flow of 0.9 L min−1 high-purity N2 was used to carry the organic acid vapor up into the mixing cell. The flow of 0.9 L min−1 CaCO3 aerosol was passed through the mixing cell and mixed with the 0.9 L min−1 N2 flow carrying the organic acid vapor. The mixed flow then entered the water cooler. The organic acid vapor was condensed on CaCO3 aerosol particles in both the mixing cell and the water cooler. The residence time of the aerosol in the coating device including the cooler was about 6 s. Three identical coating devices, with the same heating and cooling thermostatic bath, were used: one for MA coating, one for OA coating, and a blank one without organic acid for assessing the impact caused by heating the CaCO3 aerosol in the coating device without organic acid (Roselli, 2006).
The aerosol could enter the measuring instruments directly or after passing through a flow tube to increase its residence time. The flow tube was made of a straight circular glass tube with a 2.5 cm internal diameter. The aerosol flow in the flow tube was laminar flow. The average residence time of the aerosol in the flow tube was 23.7 s.
For the coating process we mixed flows of 0.9 L min−1 of dry N2 and of aerosol dried to <10 % RH at RT. As a consequence RH at the outlet of the coating device was <5 % at RT. To investigate the impact of water on the coating process and CCN activity, organic coating at a higher RH was also performed. For that a bubbling device filled with Milli-Q water was utilized to saturate the N2 stream with water vapor before it entered the storage bulb (RH >90 % at RT). After mixing with the aerosol stream at RH <10 %, the water concentration in the mixing cell corresponded to RH ≈50 % at RT or a partial pressure of ≈1500 Pa. The RH of the aerosol at the outlet of the coating device at RT was indeed ∼47 % when humidification was applied. For the partial water vapor pressure of 1500 Pa we calculated RH >7 % at 60 ∘C (for MA) and RH >3 % at 80 ∘C (for OA), which is about an order of magnitude higher than RH in the dry cases. In fact RH will be somewhat higher as the gas phase may not reach the bath temperature, which primarily serves to warm up the coating agent and control its vapor pressure.
2.3 Size and chemical composition measurements
The number size distribution of the aerosol particles was measured using a SMPS (TSI 3080 Electrostatic Classifier with TSI 3081 DMA, TSI 3786 UWCPC). The sample flow was set to 0.6 L min−1 and the sheath flow was set to 6.0 L min−1. The size range measured was 9.82–414.2 nm with a size resolution of 64 channels per decade and the time resolution was 3 min for a complete scan. Despite the maximum resolution of the SMPS the size bin width was still substantial compared to the observed growth by coating. We therefore derived the diameter of the coated (and the respective bare CaCO3 particles) by interpolating in between the size bins. For that we considered 5–9 size bins around the size bin of nominal mode and fitted a lognormal distribution to these data. The fitted mode positions are listed in Table 1. The error bars in x direction in Fig. S1 in the Supplement show the shifts of the fitted mode position relative to the nominal size bin.
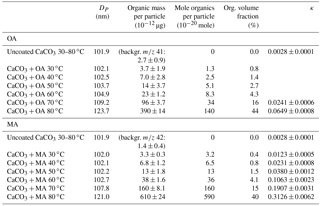
Table 1Mode diameters, chemical compositions, and κ values of CaCO3 aerosol particles (size selected by DMA at 101.8 nm) before (uncoated) and after coating with oleic acid (OA) or malonic acid (MA) at 30–80 ∘C.
The chemical composition and the vacuum aerodynamic diameter of the aerosol particles were measured using a High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-ToF-AMS, Aerodyne Research Inc., Billerica, MA, USA; DeCarlo et al., 2006). The aerosol particles were vaporized at 600 ∘C and ionized by electron impact ionization at 70 eV; i.e., we focused on the measurements of the organic coatings and sacrificed a direct CaCO3 determination by AMS (compare Zhao et al., 2010). The AMS was routinely operated in V mode in two alternating modes: 1 min MS mode to measure the chemical composition and 2 min PToF mode. Only MS mode data were analyzed. AMS measurements and SMPS measurements were synchronous and both were repeated at least four times for each sample. Size information for bare CaCO3 was taken from SMPS data in the blank coating device.
We used specific marker m∕z to derive the amount of organic coating. For pure oleic acid the signal at m∕z 41 () was reported to be the strongest signal in the mass spectrum measured by Q-AMS at EI energy of 70 eV and vaporizer temperature of 600 ∘C (Sage et al., 2009). The signal at m∕z 41 was also strongest for OA coatings in our HR mass spectra. In order to get a high signal-to-noise ratio we choose the signal at m∕z 41 in the MS mode of the AMS measurement as a marker for OA in the coated CaCO3 particles. There was no significant signal at m∕z 41 for the uncoated CaCO3 particles. The average background signal at m∕z 41 per single aerosol particle corresponded to µg for bare CaCO3. The average value presented the baseline of the mass spectra and the standard deviation (SD) was derived from the noise of the mass spectra at m∕z 41. Similarly, the signal at m∕z 42 (C2H2O+) was one of the strongest signals in the mass spectrum of pure malonic particles measured by Q-AMS with EI energy of 70 eV and vaporizer temperature of 600 ∘C (Takegawa et al., 2007). That signal was also observed for MA coatings in our HR mass spectra and used as marker for MA coatings. The average background signal per aerosol particle at m∕z 42 for bare CaCO3 particles was µg. The average value represented the baseline of the mass spectra at m∕z 42 and the SD was derived from the noise in the mass spectra.
The coating amount for both organic compounds was derived as follows. The observed signal at the respective marker m∕z was corrected for the background signal from bare CaCO3 and then scaled to the volume increase (per particle) calculated from the shift of the particle diameter DP for the largest coating amount achieved at 80 ∘C coating temperature. Because of the relative large bin width compared to the growth by coating we used the DP's, interpolated between the nominal size bins of the SMPS (see above). This assumed spherical core shell morphology, based on Zhao et al. (2010), where it was shown that the CaCO3 particles generated by the spray-drying method were spherical. The relation between AMS-derived organic mass (baseline corrected marker signals at m∕z 41 or m∕z 42 particle−1) and SMPS-derived organic mass () is linear within the limits of the method (see Fig. S1). For discussion we will refer to the AMS results, as we are able to detect amounts of organic coatings as small as several 10−12 µg per particle with the AMS, while these could be not be detected by the SMPS.
2.4 CCN activity measurement
The aerosol was dried to RH <3 % by another diffusion drier before the CCN activity was measured. To determine the CCN activity of the aerosol, the number concentration of the CCN of the aerosol was measured with a continuous flow CCN counter (CCNC, DMT-100, Droplet Measurement Technologies, Boulder, CO, USA). The total number concentration (CN) of the aerosol particles was synchronously measured using an ultrafine water-based condensation particle counter (UWCPC, TSI 3786; see Zhao et al., 2010). The ratio of CCN to CN (CCN ∕ CN) is called the activated fraction (af). In cases where a polydisperse aerosol was coated, the coated aerosol particles were size selected by scanning a DMA between 10.6 and 478.3 nm, and the CCN and CN concentrations were determined for each size bin while the supersaturation (SS) kept constant (known as Scanning Mobility CCN Analysis (SMCA); Moore et al., 2010). The activated fraction was calculated after the CCN and CN concentrations were corrected for the multiple charged particles.
The activated fraction as a function of the particle size was fitted with a cumulative Gaussian distribution function (Rose et al., 2008). The turning point of the function is the critical dry diameter (Dcrit or D50) at the set SS. The activation efficiency (i.e., the activated fraction when aerosol particles are completely activated) was 83 % for the CCN instrument, determined using 150 nm (NH4)2SO4 particles at SS=0.85 %. Besides CaCO3 and coated CaCO3 particles, the CCN activities of MA particles, OA particles, and mixed particles of CaCO3 and MA were also measured. The MA particles were generated by spray-drying a MA solution. The OA particles were generated by heating 10.0 mL OA to 97 ∘C in the storage bulb and then cooling the vapor to 2 ∘C in the water cooler in a clean coating device. Then, 1.75 L min−1 high-purity N2 was used as carrying gas and went into the storage bulb through “1 N2 in” entrance in Fig. 1; the “3 Aerosol in” entrance in Fig. 1 was closed. This way, pure OA particles with diameters up to 333 nm were generated. Mixed CaCO3–MA particles were generated by spraying the supernatant clear solutions which were prepared by settling suspensions containing CaCO3 and MA in molar ratios of about 1:1 and 3:1. The suspensions were prepared with 0.020 g MA and 0.021 g CaCO3 and 0.025 g MA and 0.076 g CaCO3 in 1000 mL Milli-Q water, respectively. The suspensions were allowed to stand for 24 h.
For aerosols where monodisperse aerosol particles with a dry diameter DP were coated, the CCN concentration was measured at different SS and the CN concentration was measured synchronously. Similarly, the activated fraction as a function of SS was fitted with a sigmoidal function. The turning point of the function is the critical supersaturation (SScrit) and the corresponding dry diameter DP is called the critical diameter, Dcrit. The hygroscopicity parameter κ (Petters and Kreidenweis, 2007) was then calculated from the DP(Dcrit)−SScrit or SS(SScrit)−Dcrit data set. The SS settings of the CCN counter were calibrated weekly using (NH4)2SO4 aerosol based on the theoretic values in the literature (summarized by Rose et al., 2008).
3.1 CCN activity of CaCO3 aerosol
Before the coating experiments we determined the CCN activity of the bare CaCO3 aerosol particles. It was measured by the SMCA method using polydisperse CaCO3 aerosol particles. The value of the hygroscopicity parameter κ of the CaCO3 aerosol was 0.0028±0.0001 derived by the least-square fitting of Dcrit as a function of SS(SScrit). This κ value is quite small, indicating that the CCN activity of the CaCO3 aerosol is low. Our κ is well within the range of κ's of 0.0011 ± 0.0004 to 0.0070 ± 0.0017 found in previous studies for wet-generated CaCO3 particles (Zhao et al., 2010; Sullivan et al., 2009; Gierlus et al., 2012; Tang et al., 2016), but larger than κ for dry-generated CaCO3 aerosols (0.0008–0.0018, Sullivan et al., 2009).
The CCN activity for CaCO3 aerosol passed through the blank coating device exposed to temperatures of 60 and 80 ∘C was determined using the same method. The κ value remained at 0.0028±0.0001 up to 60 ∘C and increased to 0.0036±0.0001 at 80 ∘C. The increase in κ by 0.0008 at 80 ∘C was smaller than the differences of reported κ values for CaCO3 aerosol in various studies and much smaller than the changes of κ values measured in this study when the CaCO3 aerosol particles were coated by MA or OA. So the effect of heating the CaCO3 aerosol during the coating process on the CCN activity of the CaCO3 aerosol was neglected. The Dcrit at different supersaturations (SScrit) for the CaCO3 aerosol and for the CaCO3 aerosol passed through a blank coating device at heating temperatures of 60 and 80 ∘C are shown in Fig. 5 (red, yellow, and green circles).
As the solubility of CaCO3 in water is very low, droplet activation of CaCO3 (and other mineral dust components) is often described by a water adsorption approach, wherein the solute term B in the Köhler equation (Köhler, 1936; Seinfeld and Pandis, 2006; see Eqs. S1–S3 in the Supplement) is replaced by a water adsorption term. Equations (1) and (2) show application of the Frenkel–Halsey–Hill (FHH) adsorption isotherm as proposed by Sorjamaa and Laaksonen (2007) and Kumar et al. (2009):
Therein the water coverage θ (Sorjamaa and Laaksonen, 2007) is given as
where Dw and du are the diameter of the wet particles and the insoluble core. We applied the FHH parameter for CaCO3 (AFHH=0.25 and BFHH=1.19; Kumar et al., 2009) and derived a critical supersaturation of 1.52 % for CaCO3 particles with du=101.9 nm (Fig. 7, blue line). In comparison κ-Koehler theory predicts SScrit=1.49 % for κ=0.0028. A SScrit of1.49 % would also be achieved by mole solute per particle (Fig. 7, black line). Figure 7 also shows the SScrit for the bare CaCO3 particles processed at 80 ∘C temperature and the range of SScrit for 101.9 nm particles calculated from the range of κ's given in the literature (Tang et al., 2016, and references therein) for wet-generated CaCO3 particles.
We conclude that the surface of our CaCO3 particles is a little more wettable than the dry-generated particles studied by Kumar et al. (2009). We presume formation of Ca(OH) (HCO3) structures on the surface during the spray-drying generation process as commonly observed whenever the CaCO3 surface has been exposed to gaseous water or liquid water (Stipp, 1999; Stipp and Hochella, 1991; Neagle and Rochester, 1990). In case of soluble components causing the lower SScrit, their amount must be of the order of mole particles−1.
3.2 CCN activity of CaCO3 particles with oleic acid coating
For the coating with OA, we selected monodisperse CaCO3 aerosol particles of 101.8 nm diameter using the DMA and measured the size and chemical composition of the particles before (uncoated) and after (coated) coating with OA. The results are listed in the upper part of Table 1.
The mode diameters of number size distribution for the uncoated CaCO3 particles at 30–80 ∘C remained in the 101.8 nm size bin, identical to that selected by the DMA. Interpolation in between the size bins as described in the experimental section led to an average dry diameters of bare CaCO3 of du=101.9 nm. The mode diameters of the CaCO3 particles after coating with OA in the range of 30–50 ∘C stayed in the pre-selected size bin at 101.8 nm, which means that the layers were too thin to effectively grow the particles to the next size bin; the mode diameters increased distinctively in the temperature range of 60–80 ∘C (Fig. 3a). However, the bin-interpolated diameters DP which are shown in Table 1 increased monotonically over the whole temperature range.
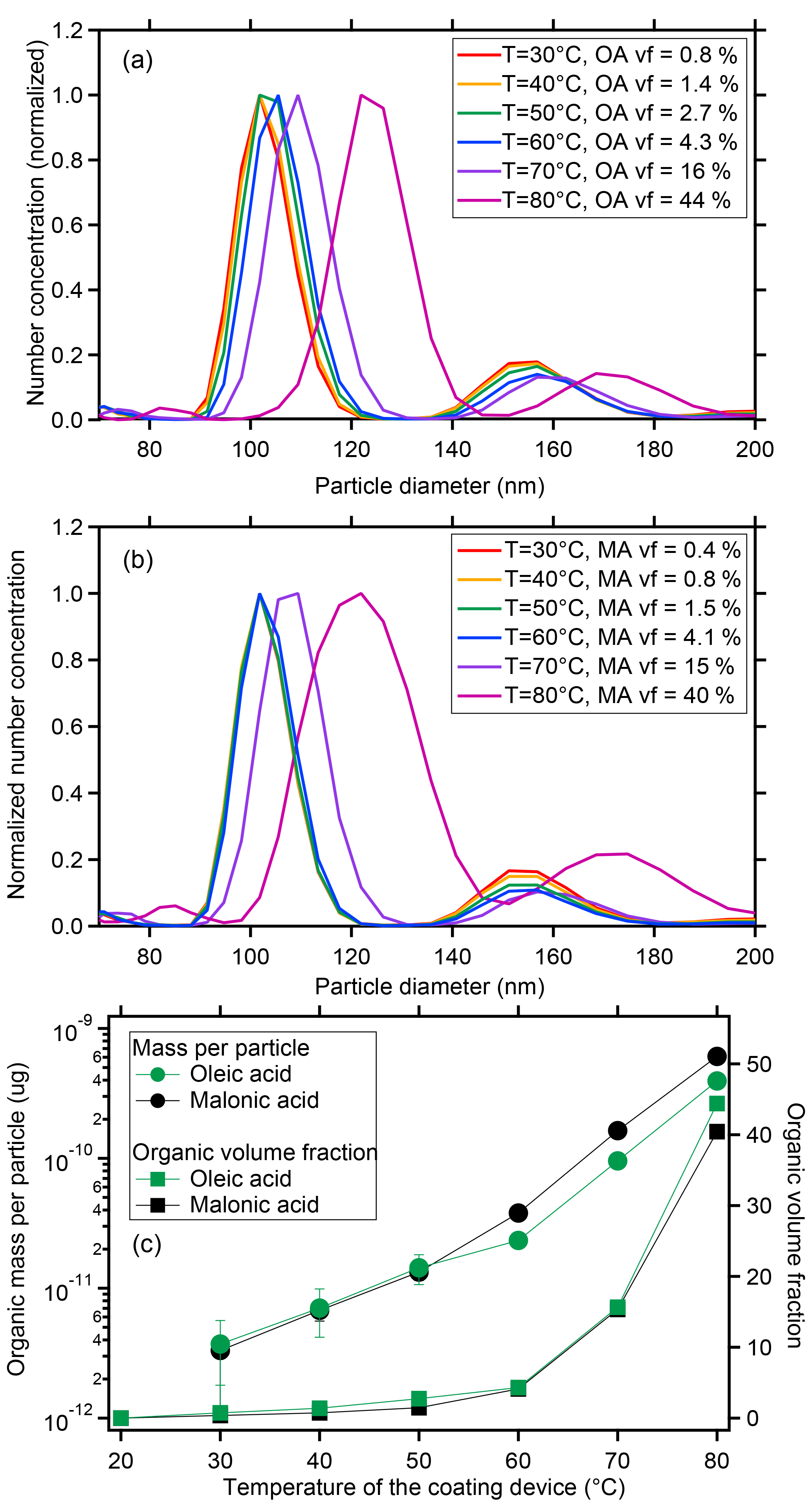
Figure 3Size distribution of monodisperse CaCO3 aerosol particles after coating with oleic acid (a) or malonic acid (b). Coating amount and organic volume fraction for oleic and malonic acid as a function of the coating temperature for the same experiments (c, data in Table 1).
The values of m∕z 41 (µg per particle) originating from the OA coating for the coated CaCO3 particles at 30–80 ∘C were at all temperatures significantly larger than for the bare CaCO3 particles and increased with the increasing coating temperature (– µg per particle; compare Table 1 and Fig. 3c, red circles). The AMS detected increase at m∕z 41 showed that the CaCO3 particles already contained small amounts of OA after coating with OA at temperatures below 60 ∘C, although the DP shifted less than a size bin.
The organic volume fraction (vf) in the aerosol particles, VOA∕Vpar (%), was calculated. Herein ), where VOA is the OA volume derived by AMS and is the volume of the bare CaCO3 before coating (101.9 nm). VOA∕Vpar for the uncoated CaCO3 particles is by definition zero. The vf for the coated CaCO3 particles at 30–80 ∘C increased with the increase in the coating temperature from 0.8 % at 30 ∘C to 44 % at 80 ∘C (Fig. 3c, green square and Table 1). The CaCO3 particles were indeed coated with a significant amount of OA and the amount of OA coating increased with the increase in the coating temperature. The experiments were repeated at least four times. The according SDs for the OA mass per particle in Table 1 demonstrate that the reproducibility of the experiments was good and the performance of the coating device was stable.
The activated fractions at different SS for monodisperse CaCO3 particles with du=101.9 nm before and after OA coating at 30–80 ∘C are shown in Fig. 4. The top panel in Fig. 4 shows the results at 30–60 ∘C with up to µg of coating material deposited on the CaCO3 particles (vf up to 4.3 %). At the lowest SS of 0.17 and 0.35 %, the activated fractions were very low and independent of the presence of the coating material within the errors. When the SS increased to 0.52, 0.70, and 0.87 %, the activated fractions for the coated CaCO3 particles were lower than those for the uncoated particles. Notably the activated fractions for the coated CaCO3 particles decreased with the increase in the coating material in the range of vf 0.8–2.7 %. The activated fractions for the CaCO3 particles with different amounts of coating spread with larger SS applied. However, this trend reversed at the coating temperature of 60 ∘C and an OA vf of 4.3 %, and the activated fractions at vf =4.3 % became higher than those at 2.7 % at the three largest SS. In summary, we found that the CCN activity of the coated CaCO3 particles with vf of OA in a range 0.8–4.3 % was lower than that of the uncoated CaCO3 particles. The CCN activity of the coated CaCO3 particles decreased with the increasing vf in between 0.8 and 2.7 %; i.e., the CCN activity became lower when more coating material deposited on the CaCO3 particles. This trend turned at a vf somewhere between 2.7 and 4.3 %. As the DP also increased at 60 ∘C we cannot differentiate whether the increase in the activated fractions is due to increasing size or increasing wettability.
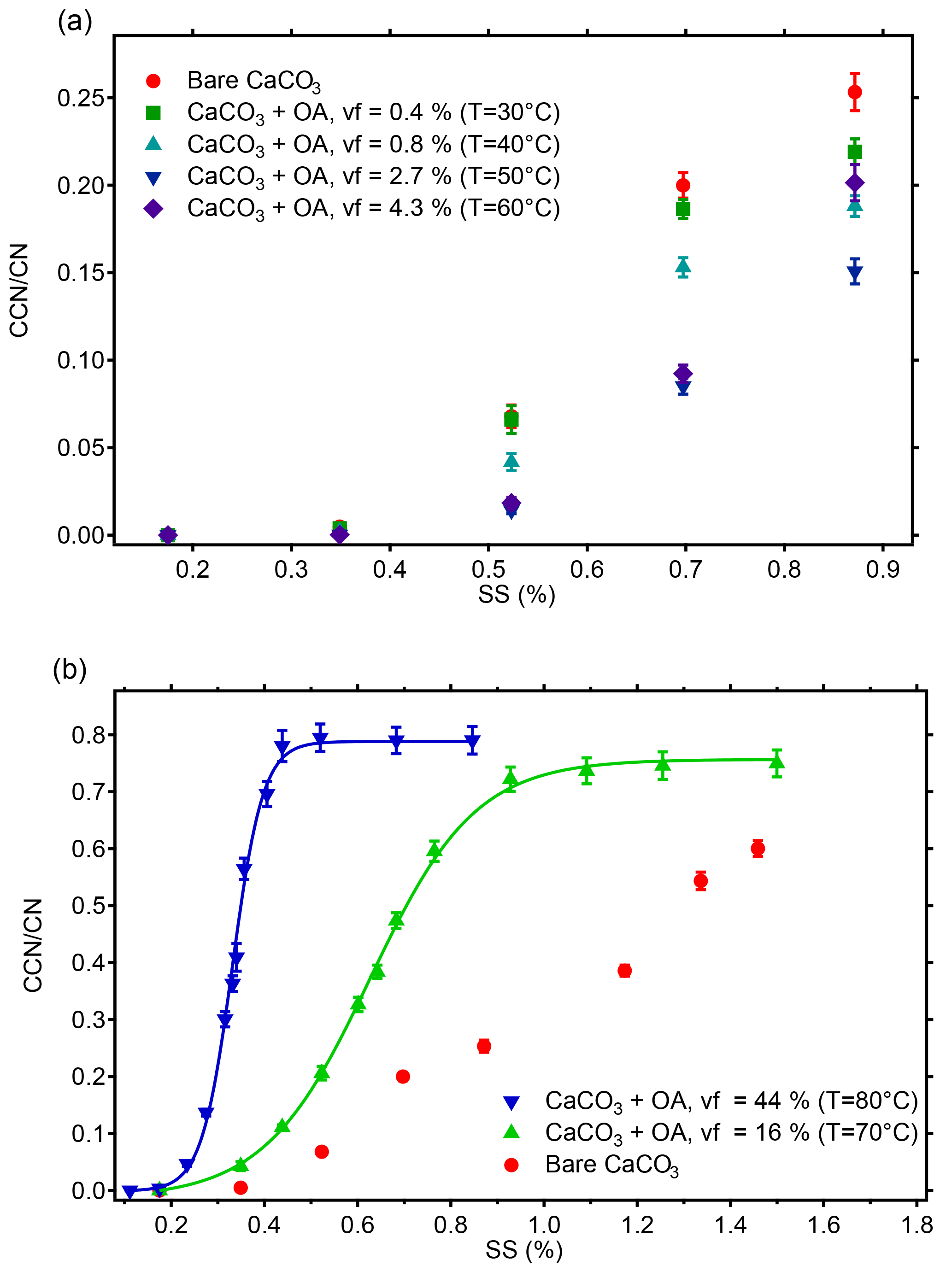
Figure 4Activated fractions (CCN ∕ CN) of monodisperse CaCO3 aerosol particles (diameter du=101.9 nm) at different supersaturations before and after OA coating. With increasing coating temperatures of 30–50 ∘C the activated fraction decreases despite the increase of organic volume fraction (vf) from 0.8 to 2.7 %. At a vf of 4.3 % at 60 ∘C this trend turns. Considering the increased particle diameter at 30–60 ∘C and the reduced activated fraction simultaneously, the CCN activity of the coated CaCO3 particles at 30–60 ∘C was lower than that of the uncoated CaCO3 particles (a). At coating vf of 16 and 44 % (T=70–80 ∘C) the activated fractions, and thus CCN activities, are higher than for bare CaCO3 and increase with coating vf. In these two cases all particles are activated at the highest SS and SScrit and κ can be determined from the turning point of the sigmoidal fit (b, compare Table 1).
The activated fractions of CaCO3 particles after coating with OA with vf of 16 and 44 % (coating temperatures of 70 and 80 ∘C, respectively) were considerably higher than that before coating, as shown in Fig. 4b. The increased activated fractions resulted from both the increase in particle size (Fig. 3) and the increase of the OA vf of particles. At vf of 16 and 44 %, the activated fractions of the CaCO3 particles after coating increased with the increase of SS and reached complete activation. (Note that because the activation efficiency is 83 %, the activated fractions appear at values less than 100 % at the points of full activation.) For vf of 16 and 44 %, SScrit was determined by fitting a sigmoidal function to the activated fraction as a function of SS. The particle dry diameter DP, which is Dcrit in these cases, is given in Table 1. The hygroscopicity parameter κ was determined from DP (Dcrit) and the corresponding SScrit. The κ values of the CaCO3 particles coated with vf of OA of 16 and 44 % were 0.0241±0.0006 and 0.0649±0.0008, respectively. The respective κ values for the CaCO3 particles with a diameter of 101.9 nm without coating and after coating with OA at 30–60 ∘C (OA vf ≤4.3 %) could not be determined by this method because these particles could not be fully activated at the highest SS reachable by the CCN counter. Therefore we give for the uncoated CaCO3 particles determined by scanning the size of the polydisperse CaCO3 aerosol particles as described above (see Fig. 5).
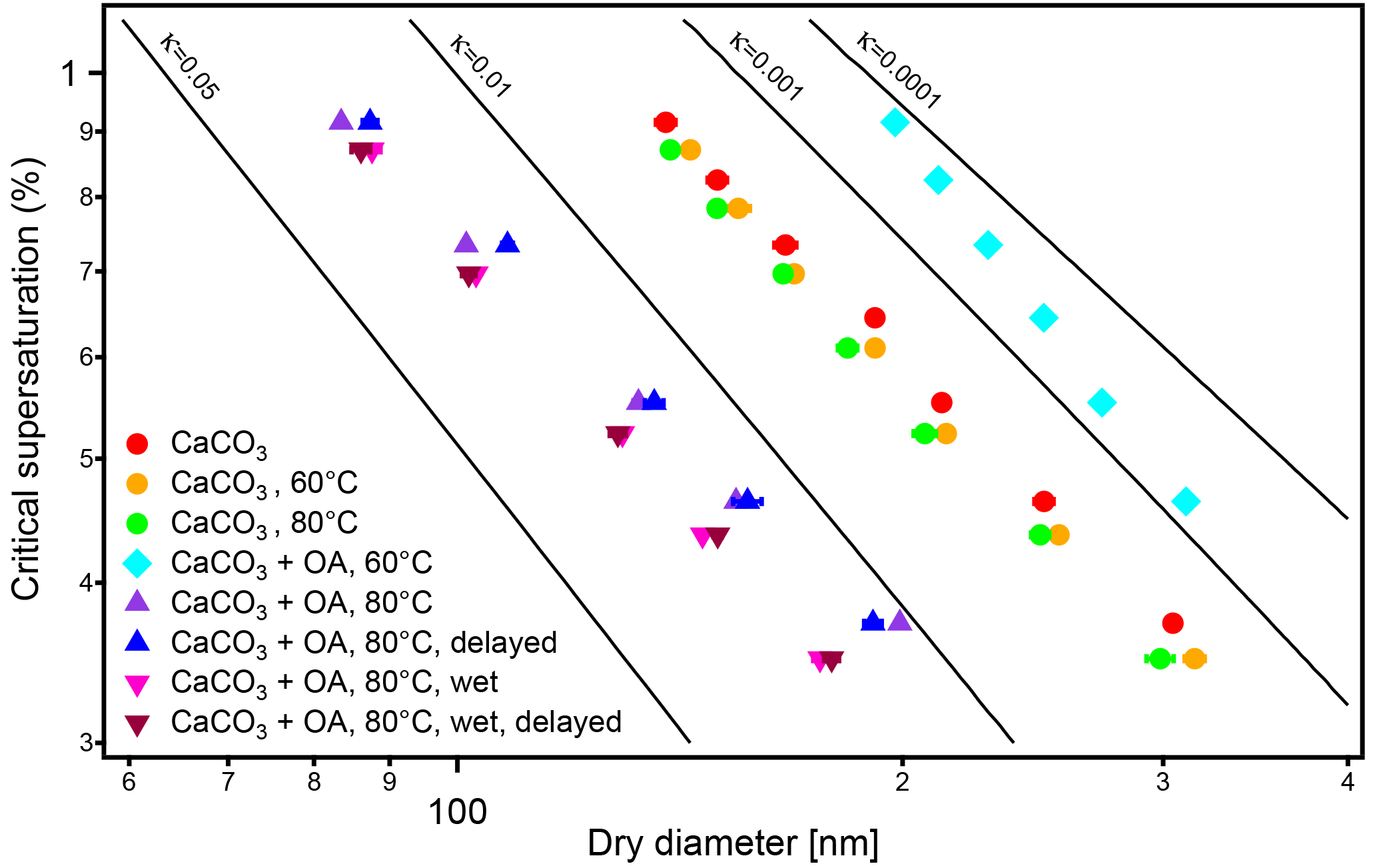
Figure 5Critical dry diameters at different supersaturations (SS) of polydisperse CaCO3 aerosol before (circles) and after oleic acid (OA) coating. Experiments were performed at 60 ∘C (turquoise diamond) and 80 ∘C (triangles) coating temperatures. The flow tube experiments at 80 ∘C were performed (indicated by “delayed”) at dry conditions (normal, blue triangles) and at enhanced water vapor (“wet”, brown triangles). The effect of the temperature in the coating device on the CaCO3 core is negligible (red, green, and orange circles). As for the monodisperse case in Fig. 4, at 60 ∘C coating temperature the particles are less CCN active than bare CaCO3 while at 80 ∘C the coated particles are more CCN active. The presence of water vapor (1500 Pa) in the coating process enhances CCN activity. The increasing of residence time (23.7 s) of the coated aerosol in the flow tube had no significant effect on CCN activity in both experiments.
So we conclude that for vf of OA of 0.8–2.7 % the CCN activity of CaCO3 particles after coating is lower than that of uncoated CaCO3 particles and decreases with the fraction of OA. The trend turns at a vf between 2.7 and 4.3 %. CCN activity was higher than that of the bare CaCO3 particles at vf of OA of 16 % (70 ∘C) and 44 % (80 ∘C) with CCN activity and κ=0.0649 ± 0.0008, respectively. The enhanced and reduced CCN activity of CaCO3 particles coated with OA at 80 and 60 ∘C, respectively, was also evident from the CCN activity measurement using polydisperse aerosols (Fig. 5).
A possible explanation for our observation can be based on the amphiphilic character of OA, namely that one end of the OA molecule is hydrophobic (the hydrocarbon chain), while the other is hydrophilic (the carboxyl group).
We refer to Ca(OH) (HCO3) structures at the surface which offer polar surface sites to bind the hydrophilic ends (the carboxyl groups) of the OA molecules. The hydrophobic ends of OA molecules (the hydrocarbon chains) that are then exposed on the particle surface thus increase the hydrophobicity of the particle surface. Such a formation of a hydrophobic layer should be occurring until all polar sites are occupied or monolayer coverage is reached, maybe in the form of a self-assembled layer. This can hinder the uptake of water. Activation of CaCO3 particles can be described by the Kelvin term and a water absorption term, e.g., FHH isotherm (Sorjamaa and Laaksonen, 2007; Kumar et al., 2009). In terms of Kelvin–FHH theory the hydrophobic OA coating will lower AFHH and/or likely increase BFHH. The formation of a monolayer of OA on black carbon particles with the polar groups pointing outwards was postulated by Dalirian et al. (2017), which leads to increased activation of the black carbon particles. Thus, they observed a similar effect of layer formation, but with switched polarity.
Garland et al. (2008) suggested that OA at sub-monolayer coverage forms self-associated islands rather than uniformly covering the surfaces, and OA molecules are oriented vertically, with polar heads facing to the surface. This is in support of our working hypothesis: the formation of a hydrophobic surface film. We conclude that all hygroscopic sides on the CaCO3 surface are covered at OA vf somewhere between 2.7 and 4.3 %, as here the trend turns and droplet activation starts to increase again. This would place the monolayer coverage above 3 % organic volume fraction. According to the measurements and calculations of the length of OA molecule, the thickness of OA sub-monolayer on solid surfaces, and the thickness of deuterated OA monolayers at the air–water interface (Garland et al., 2008; King et at., 2009; Iwahashi et al., 2000), we estimate 2.3 nm as the likely thickness of OA monolayer on CaCO3 particles; accordingly a monolayer would be achieved at about 12–13 % organic vf. As a consequence the re-increase of hygroscopicity starts at sub-monolayer coverage and we propose that a fraction of OA binds to already adsorbed OA tail by tail such that carboxylic groups are facing outwards.
For CaCO3 particles coated with more than an OA monolayer (vf =16 and 44 % at 70 and 80 ∘C coating temperatures), OA in the first layer should still combine with the CaCO3 surface, the heads pointing downwards. We suppose that now a portion of the carboxyl groups of the OA molecules, which are not in the first layer, will be exposed to the particle surface, similar to the formation of lipid bilayers in cells. However, We have no experimental proof for that. The particle surface then would become more hydrophilic.
When carboxylic groups of OA are exposed at the surface, the interaction of water with the OA layer becomes stronger, and the surface becomes wettable. In terms of the Kelvin–FHH approach, the surface water interaction becomes stronger and AFHH increases and likely also the interaction between the higher water layers (BFHH decreases). From this point of view water adsorption by the “OA bilayer” should become similar to thin MA layers (compare next section). In addition, when droplets form, OA will transfer to the surface of the droplets and lower the surface tension of the solution (the surface tension of OA is 0.033 J m−2 (Chumpitaz et al., 1999), which is much lower than that of pure water of 0.072 J m−2). Thus, the activation of OA-coated particles is probably a complex interaction between formation of a specific hydrophobic layers and more hydrophilic multilayers, surface tension effects, and, for the largest coating amounts, simple size effects. As shown in Fig. 7, SScrit for OA is lower than for thin MA coatings, probably because of the surface tension effect, but higher than for thick MA coatings, because of the missing solute effect.
The CCN activity of all oleic-coated particles is higher than the CCN activity of pure OA. Our CCN activity measurement showed that pure OA particles up to 333 nm did not activate at 0.87 % SS; this sets an upper limit for CCN activity of OA particles (κ<0.0005), in agreement with Kumar et al. (2003) and Broekhuizen et al. (2004). In liquid state OA forms micelle-like structures; the hydrophilic ends (the carboxyl groups) of OA molecules tend to combine together by hydrogen bonds and the hydrophobic tails (the hydrocarbon chains) are exposed at the outside (Iwahashi et al., 2000; Garland et al., 2008). The arrangement of OA molecules in pure OA particles should be similar. Hydrophobic tails facing outwards can explain the hydrophobicity of the particle surface and the hindrance of the uptake of water, making the CCN activity of pure OA particles very low. For sub-monolayer coatings of OA of vf 0.8–2.7 % the CCN activity seems to approach that of pure OA. However, the arrangement of OA molecules in these thin coatings will be influenced by the CaCO3 core with its polar, hydrophilic sides differing from pure OA particles and can thus be less hydrophobic.
Even at the largest coating with an organic vf of 44 %, the coating thickness is about 10 nm, which corresponds to about only four monolayers of OA (assuming the thickness of OA monolayer on CaCO3 particles is about 2.3 nm). Additionally, the arrangement of OA molecules will still be likely influenced by the CaCO3 core. Water can probably adsorb at the carboxylic groups facing outward (“bilayer” type structure) and diffuse through the thin OA coatings. It may form an adsorbed water phase near the CaCO3 surface. This could push the OA out to act as surfactant which lowers the Kelvin term. Such processes should also happen in pure OA particles. Because of the presence of CaCO3 core the SS to achieve this is lower than for pure OA.
The phenomenon described above is reported for the first time in the studies on the CCN activity of multicomponent aerosols. This phenomenon also shows a limitation of the otherwise very useful mixing rule (Petters and Kreidenweis, 2007) for multicomponent aerosols with specific morphologies.
In Fig. 5 we additionally show the influence of water vapor on CCN activity of CaCO3 particles coated with OA for the highest coating temperature (80 ∘C) and thus largest OA amount. Herein we determined Dcrit at different supersaturations (SScrit) for polydisperse CaCO3 aerosol particles (by SMCA). The experiments were performed at RH 0.3 % and at RH 3 % at the coating temperature of 80 ∘C on cooling to RT the RH increased to 47 %. The presence of more water vapor (1500 Pa) in the coating process increased κ somewhat and enhanced the CCN activity. This is of importance since RH will often be larger than 0.3 % when coating appears in the atmosphere. This will be discussed further in context of MA coatings at enhanced water vapor.
3.3 CCN activity of CaCO3 particles with malonic acid coating
For the study with MA coatings, the CaCO3 particles were also size selected with a diameter of 101.8 nm. The size DP and chemical composition of CaCO3 aerosol particles are listed in Table 1 before and after coating with MA at temperatures in a range of 30–80 ∘C. The mode diameter did not shift after coating in a temperature range of 30–60 ∘C, but it increased for coatings at 70 and 80 ∘C with increasing coating temperature. The size bin interpolated particle diameter DP of the MA-coated particles increased monotonically with the coating temperature. The average of the interpolated diameter of bare CaCO3 particles in the temperature range 30–80 ∘C was du=101.9 nm.
Values of the MA marker m∕z 42 per particle were significantly larger for CaCO3 particles after coating at 30–80 ∘C and the MA mass increased from to µg per particle with the coating temperature (Table 1, Fig. 3c). The organic vf of MA (, %) was calculated as in the case of the OA and ranged from 0.4 to 40 %. As for OA, the MA experiments were repeated at least four times and the reproducibility and stability were good (see SDs in Table 1).
The activated fractions at different SS for 101.9 nm CaCO3 particles before and after coating with MA at 30–80 ∘C are shown in Fig. 6. SScrit was determined by fitting a sigmoidal function to the data and the κ value was calculated from the DP (Dcrit) and the corresponding SScrit. The results are listed in Table 1. In this procedure we had to neglect the contribution of double-charged particles because the step in the CN∕CCN vs. SS data in Fig. 6 is not sufficiently expressed to separate a plateau for multiply charged particles (e.g., Sullivan et al., 2009). The exception is the MA coating with vf =0.4 %. For this case we compared a sigmoidal fitting both from the beginning (the first point) and from the multiply charged plateau (the third point) to the “completely activated plateau” (Fig. S3). We yield % for fitting from the beginning and % for fitting from the multiply charged plateau, which is a difference of 0.013 %. The underestimate in SScrit is the largest (0.013 %) when the MA mass is the smallest (vf =0.4 %) and the underestimate will be reduced with increasing vf of MA. At the largest two MA vf it can be neglected. We have to concede a systematic error in SScrit, but it is distinctively less than 0.02 %.
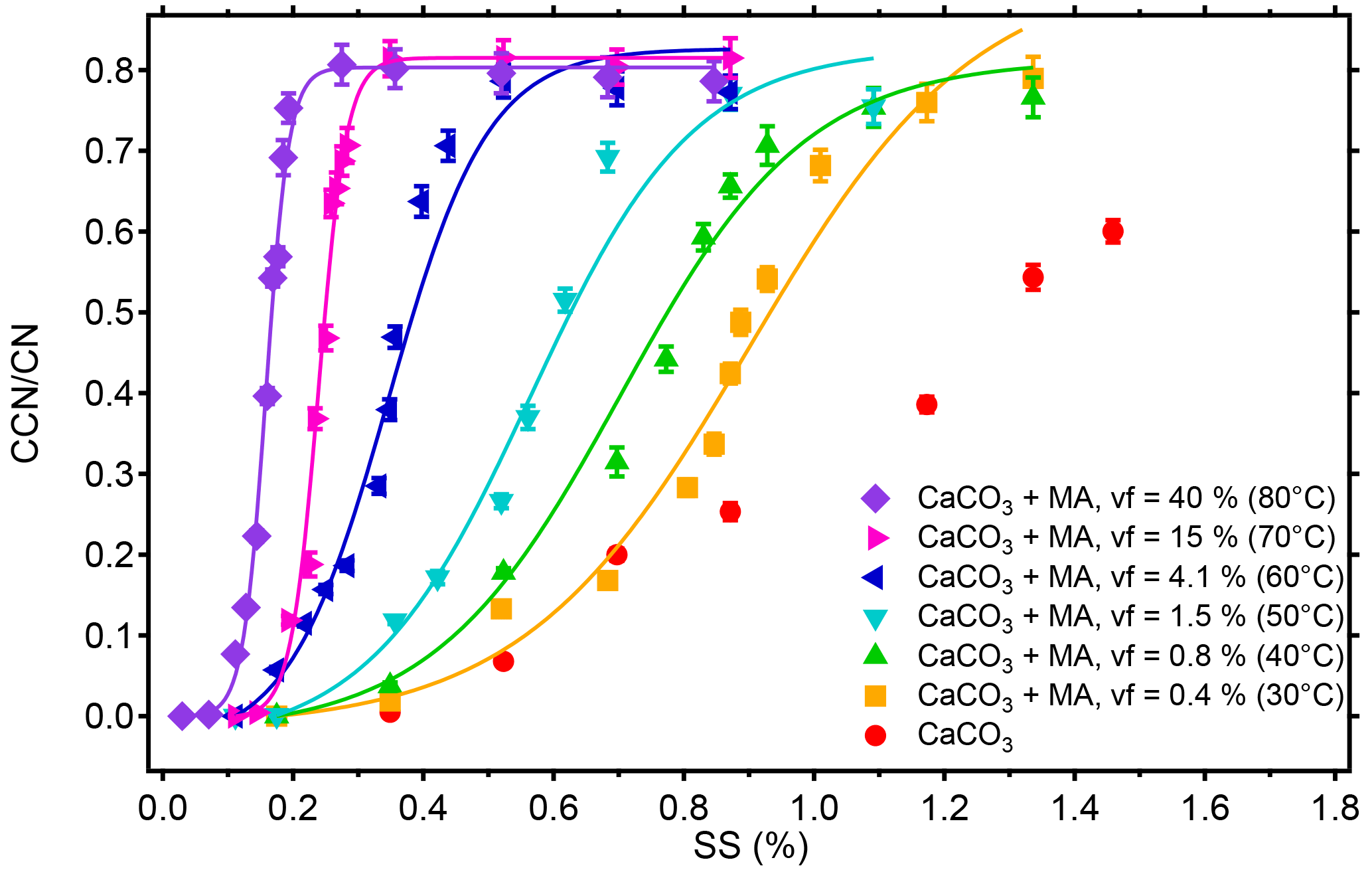
Figure 6Activated fractions (CCN ∕CN) of monodisperse CaCO3 aerosol particles (with CaCO3 core, du=101.9 nm) at different supersaturations before (red circles) and after malonic acid (MA) coating. With increasing coating, i.e., MA volume fraction (vf), the activated fraction, and thus CCN activity, increases compared to bare CaCO3 particles. All coated particles can be activated at sufficiently high SS and SScrit and κ was determined (see Table 1).
The κ values of the CaCO3 particles after coating with MA at 30–80 ∘C were higher than the κ value of the uncoated CaCO3 particles () and increased with the increasing coating MA mass per particle and increasing MA vf. The CCN activity of the CaCO3 particles increased monotonically after coating with increasing MA mass. This result differs from that of OA, which is not surprising since MA is easily soluble in water.
The κ value for the CaCO3 particles after coating with a mass of MA as small as µg per particle and vf of MA of only 0.4 % was 0.0123±0.0005, thus considerably larger than the κ value for the uncoated CaCO3 particles (). This suggests already that a small amount of MA can significantly enhance the CCN activity of CaCO3 particles. The phenomenon that traces of water-soluble substances can strongly affect droplet activation has been reported before (Bilde and Svenningsson, 2004).
We applied Koehler theory to CaCO3 particles coated with MA, assuming that the MA coating will fully dissolve in water when droplets form (see Eqs. S1–S3 in the Supplement). Increasing MA solute decreases the activity of water in solution and lowers the critical supersaturation SScrit for droplet activation.
The resulting Koehler curves, i.e., equilibrium SS over the solution droplet as a function of the wet diameter Dw, are shown in Fig. S2. Therein the maximum of each SS curve is the critical supersaturation (theory SScrit). In Table S1 and Fig. 7 we compare the SScrit predicted by the Köhler approach (red) with the observed SScrit (black). Koehler theory overpredicts SScrit for thin coatings substantially, meaning it underestimates the hygroscopicity of the thinly coated particles. But with increasing coating Koehler theory approaches the observed SScrit and SScrit for a particle of 121 nm diameter composed of pure MA (red circle).
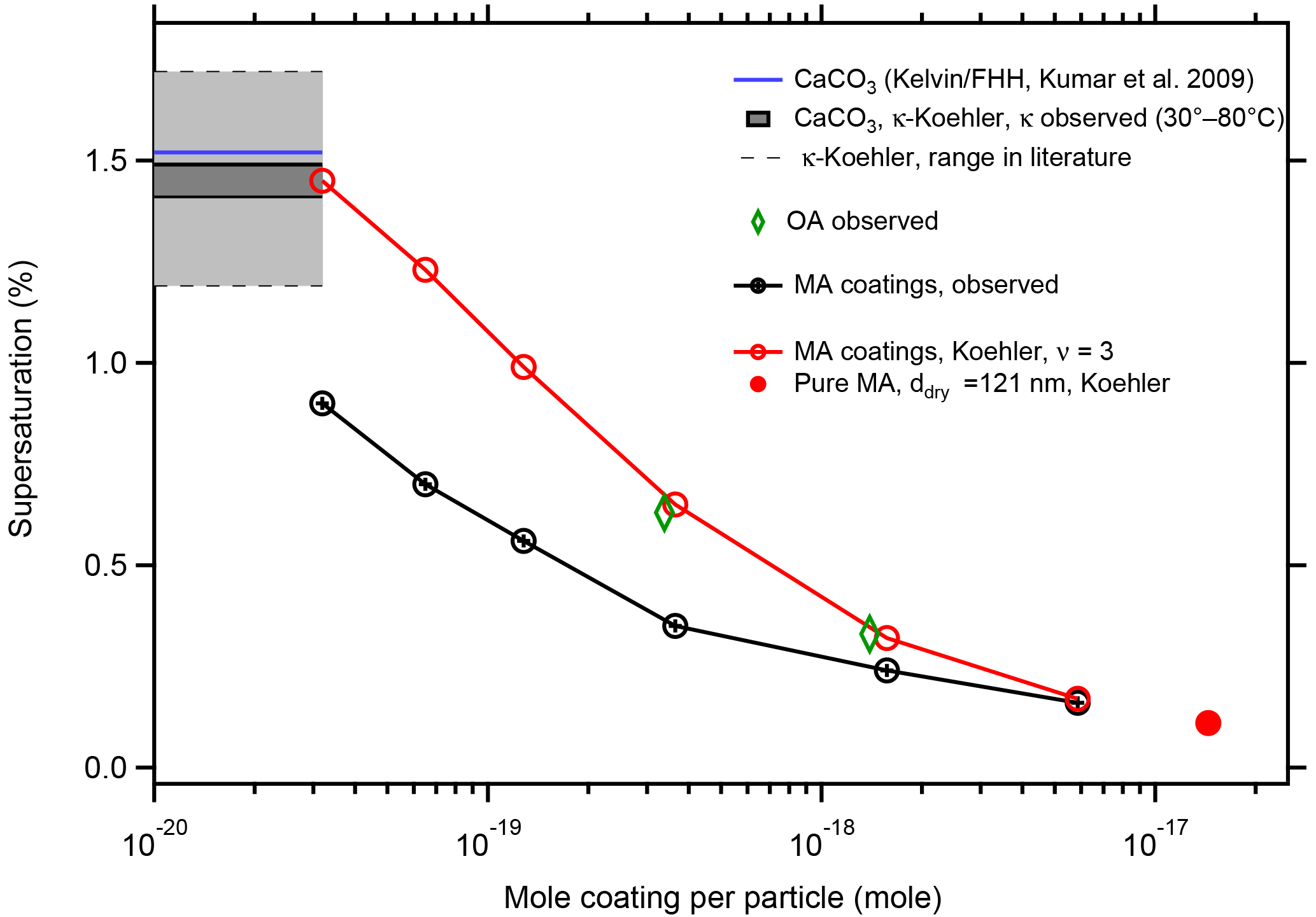
Figure 7Comparison of SScrit predicted by Koehler theory with observations. Koehler theory for aqueous MA solutions assuming full dissociation (ν=3) overpredicts SScrit (red circles) compared to the observation (black circles). With increasing coating amount Koehler theory approaches the observation, with the limiting SScrit for 121.0 nm particles made of pure malonic acid. For comparison we also show observed SScrit for the two thickest OA coatings (green diamonds). The horizontal lines indicate SScrit of the bare CaCO3 particles as calculated from our observed κ (black) and predicted by Kelvin–FHH theory (blue). Light grey area between the thin dashed grey lines shows the range of SScrit for 101.9 nm particles calculated from the range of κ in literature for wet-generated CaCO3 particles (compare Tang et al., 2016).
From the Koehler results we derived the water content of the particles at SScrit and we calculated molality and mass fraction of the solute in the solution at the point of activation. The molality at minimum and maximum MA load of and µg particle−1 were 0.006 and 0.0015 mol kg−1, respectively. We used these values in the AIOMFAC model (Zuend et al., 2011, http://www.aiomfac.caltech.edu, last access: 29 April 2014) to calculate the deviation from ideality for the solution at point of activation for a flat solution. The both solutions are highly non-ideal with respect to the MA (ax=0.4), wherein MA was treated as solute with reference state infinite dilution (mole fraction xsolute→0). However, this did not much affect the activity coefficient of water, which is essentially 1, i.e., water treated as solvent with reference state pure liquid (mole fraction xwater=1). Moreover, in this concentration range, the surface tension of aqueous MA solutions is about 0.070 J m−2 and thus the nearly same as for water (Table S2 in the Supplement). One should expect that Koehler theory would predict SS quite well under such conditions.
To bring Köhler theory in agreement with the observation for the thinnest coating, more solute entities would be required. Thus, disagreement cannot be caused by an ill-determined van't Hoff factor as we used already the maximum ν of 3 and reducing ν would increase the deviation. Note that recent observations point to the importance of the surface effect by organic surface films over the solute effect for water-soluble inorganics in presence of organics, including MA (Ruehl et al., 2016). A lower surface tension will bring Koehler prediction and observation punctually in better agreement and still allow for smaller van't Hoff factors (Varga et al., 2007). As an example, a surface tension of 55 % of σw and a van't Hoff factor of 1 will bring SScrit predicted by Koehler theory and observation in agreement for the thinnest coating. However, a surface tension of 55 % of σw will cause disagreement for the thickest coating, because the solute term gains in importance. The findings for the mixed solutions of MA and water-soluble ammonium sulfate are likely not directly transferable to our systems with insoluble inorganic core, where we expect dilute aqueous solutions of 0.006 mol kg−1 of MA at the activation point. At such concentrations MA does not reduce σw; moreover, in the study of Ruehl et al. (2016) MA was one of the more Koehler κ behaving organics.
In Fig. 7 we show the prediction of SScrit=1.52 % for activating CaCO3 by the Kelvin–FHH theory with the CaCO3 parameters taken from Kumar et al. (2009). SScrit for our bare CaCO3 particles is 1.49 % and the lower SScrit should be due to a more adsorptive surface, e.g., the presence of Ca(OH)(HCO3) structures. According to classical Koehler theory the equivalent of moles of dissolvable entities would be needed to explain a κ of 0.0028 and SScrit of 1.49 %, which is only about one-quarter of the moles MA in the thinnest MA coating. Therefore, whatever makes our CaCO3 particles wettable is not sufficient to explain the low SScrit of 0.9 % at the thinnest MA coating in terms of Koehler theory.
We estimate monolayer coverage by MA at 2–3 % vf; this would be achieved in between MA mass loads of and µg per particle. Thus a sub-monolayer coating of µg MA per particle caused a drop of SScrit from 1.49 to 0.9 and increased κ from 0.0028 to 0.012. Therefore we conclude that CaCO3–MA coatings show a non-Koehler behavior at thin coatings, but approach Koehler behavior with increasing MA load.
This means there must be specific interactions between MA and the CaCO3 surface which eases water adsorption and CCN activation. We refer to the Ca(OH)(HCO3) structures that likely exist on the particle surface. When CaCO3 particles are coated by MA (or OA) the hydrophilic sides can serve as polar surface active sites for accommodation of the acids. In the case of MA there is no long hydrophobic organic chain, but there is a second carboxylic group which could still support the adsorption of water films.
In terms of Kelvin–FHH theory one could explain the observed low SScrit for thin MA coatings by net stronger interaction with water (higher AFHH) and/or stronger interaction between the adsorbed water layers (lower BFHH) compared to bare CaCO3. If coatings become thicker the Koehler solute effect starts increasingly to contribute and eventually controls the CCN activation. Our data are not sufficient to determine AFHH and BFHH. (The only system in the literature which comes close – in a far sense – is calcium oxalate monohydrate, with AFHH=0.57 of and BFHH=0.88 (Kumar et al., 2009). Using these FHH parameters will lead to SScrit=0.53 %, comparable with our observed value of 0.56 % for µg MA coating, which represents an organic vf of 1.5 % and thus is close to monolayer.)
Dcrit at different supersaturations (SScrit) for polydisperse CaCO3 aerosol particles before and after coating with MA is shown in Fig. 8. Our observation of for pure malonic is consistent with the κ derived from the data of Kumar et al. (2003) (κ=0.20–0.25) and Prenni et al. (2001) (κ=0.24), but significantly lower than the κ derived from the data of Giebl et al. (2002) (κ=0.41–1.04). The behavior of polydisperse coated aerosol was similar to the result obtained from the monodisperse CaCO3 aerosol particles.
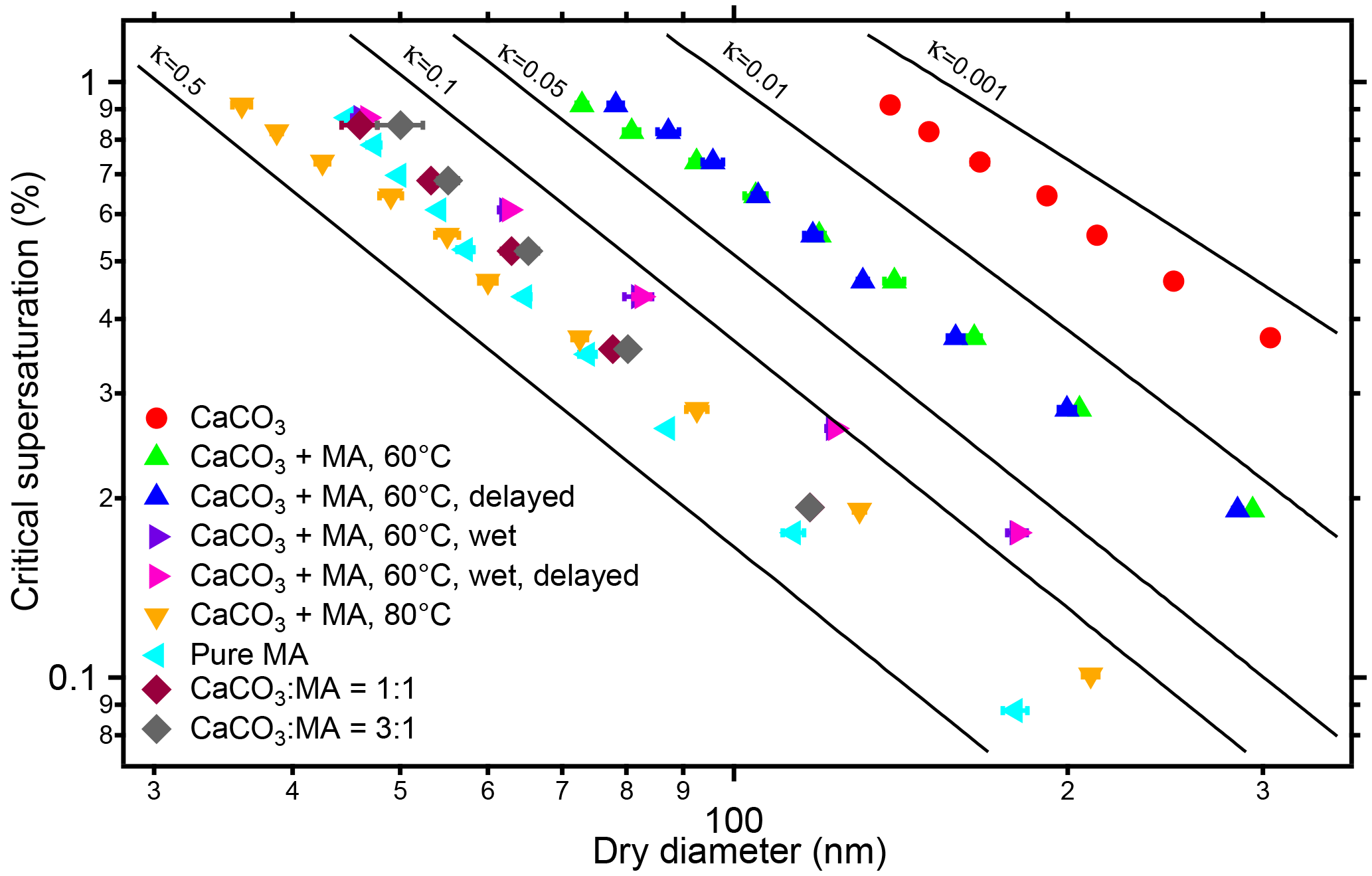
Figure 8Critical dry diameters at different supersaturations (SS) of polydisperse CaCO3 aerosol before (circles) and after malonic acid coating (triangles). Experiments were performed at 60 and 80 ∘C coating temperatures. The results are similar to the monodisperse case in Fig. 6. Critical dry diameters as a function of SS are also shown for malonic acid particles and particles that were generated by spraying mixed solution with molar ratios of CaCO3 ∕ malonic acid of 1 : 1 and 3 : 1. The CCN activity decreases with increasing CaCO3 content. The flow tube experiments at 60 ∘C were performed (indicated by “delayed”) at dry condition (blue triangles) and in presence of 1500 Pa water vapor (magenta triangles). The presence of water in the coating process substantially enhanced κ and CCN activity. The increasing of residence time (23.7 s) of the coated aerosol in the flow tube had no significant effect on CCN activity in both experiments.
In Fig. 8 we added results for coating in presence of enhanced water vapor (1500 Pa) and aerosols generated by spraying mixtures of MA and CaCO3. At the coating temperature of 60 ∘C, when the RH increased from 0.7 to 7 % and eventually to 47 % at RT, the CCN activity of the coated CaCO3 particles increased substantially (compare “dry” (green triangles) and “wet” (lilac triangles) in Fig. 8). The effect is more distinct than for the OA coating shown in Fig. 5, and κ increases by about an order of magnitude. In wet conditions, the reaction between CaCO3 and MA may be more efficient and formation of calcium malonate will reduce du, i.e., the diameter of the insoluble core; according to Eq. (S1) this may be the reason for the higher CCN activity at the higher RH. The hypothesis of malonate formation is supported by the CCN activity of “calcium malonate” aerosols, generated by spraying solutions containing CaCO3 and MA with molar ratios of about 1:1 and 3:1. Here the CCN activity is similar to that arising in the coating process in the presence of water vapor. The change of the Ca ∕ malonate ratio from 3 : 1 to 1 : 1 had no large effects. But, taking the data of pure MA particles also into account, there is a trend to lower κ with increasing Ca in the initial solution.
The increase of residence time (by 23.7 s) had no significant impact on CCN activity for both OA coating and MA coating at both dry and enhanced water vapor conditions, probably because the coating process was already completed in the coating device and no further reactions occurred in the flow tube.
Our findings may be important for aging processes of mineral particles in the atmosphere. The dependence of CCN activity of the coated particles on RH during the coating process will help to enhance the increase of the CCN activity by the coating process as water will be abundant in many instances. The effect probably will be relatively small for OA and similar organics, which are hardly water-soluble, but strong for MA and similar organic acids, which are highly water-soluble.
The CCN activity of CaCO3 particles with oleic acid and malonic acid coatings was investigated in this study. The results show that oleic acid coating and malonic acid coating have different impacts on the CCN activity of CaCO3 particles. This can be attributed to the amphiphilic property of oleic acid in contrast to the high water solubility of malonic acid. Small amounts of oleic acid coating (vf ≤4.3 %) decreased the CCN activity of the CaCO3 particles, while more oleic acid coating (vf ≥16 %) increased it. This phenomenon was reported here for the first time and was attributed to stepwise passivation of the active sites of CaCO3 by oleic acid. Once all active sites are occupied we suggest the formation of a lipid-like bilayer with the carboxylic groups facing outwards.
In contrast, malonic acid coating (0.4–40 %) increased the CCN activity of CaCO3 particles regardless of the amount of the coating. The CCN activity of CaCO3 particles with malonic acid coating increased with the amount of the coating. Even a small amount of malonic acid coating (vf =0.4 %) significantly enhanced the CCN activity of CaCO3 particles from to Increasing the RH during the coating increased the CCN activity of the CaCO3 particles with malonic acid coating, probably because more CaCO3 reacted with malonic acid to soluble calcium malonate. This process will help to increase the CCN activity.
Although malonic acid is soluble in water, SScrit for MA-coated particles was overpredicted by Köhler theory. Our results indicate that thin MA coatings provide a wettable particle surface, which favors adsorption of water. For thicker coatings the coated particles approached Köhler behavior because of increasing importance of the solute effect.
Mineral aerosol is one of the most abundant components of the atmospheric aerosol, but its low water solubility limits its CCN activity. This study showed that water-soluble organic acid coating might significantly enhance the CCN activity of mineral aerosol particles. This could lead to mineral aerosol playing a more important role in cloud formation.
Access to data can be requested from the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-7345-2018-supplement.
The authors declare that they have no conflict of interest.
This study was supported by Forschungszentrum Jülich, the National
Natural Science Foundation Committee of China (41421064, 21190051, 41121004),
and the China Scholarship Council.
The article processing charges for this open-access
publication were covered by a Research
Centre of the Helmholtz Association.
Edited by:
Frank Keutsch
Reviewed by: two anonymous referees
Bilde, M. and Svenningsson, B.: CCN activation of slightly soluble organics: the importance of small amounts of inorganic salt and particle phase, Tellus B, 56, 128–134, https://doi.org/10.1111/j.1600-0889.2004.00090.x, 2004.
Broekhuizen, K. E., Thornberry, T., Kumar, P. P., and Abbatt, J. P. D.: Formation of cloud condensation nuclei by oxidative processing: Unsaturated fatty acids, J. Geophys. Res.-Atmos., 109, D24206, https://doi.org/10.1029/2004jd005298, 2004.
Cakmur, R. V., Miller, R. L., Perlwitz, J., Geogdzhayev, I. V., Ginoux, P., Koch, D., Kohfeld, K. E., Tegen, I., and Zender, C. S.: Constraining the magnitude of the global dust cycle by minimizing the difference between a model and observations, J. Geophys. Res.-Atmos., 111, D06207, https://doi.org/10.1029/2005jd005791, 2006.
Charbouillot, T., Gorini, S., Voyard, G., Parazols, M., Brigante, M., Deguillaume, L., Delort, A. M., and Mailhot, G.: Mechanism of carboxylic acid photooxidation in atmospheric aqueous phase: Formation, fate and reactivity, Atmos. Environ., 56, 1–8, https://doi.org/10.1016/j.atmosenv.2012.03.079, 2012.
Chebbi, A. and Carlier, P.: Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review, Atmos. Environ., 30, 4233–4249, 1996.
Cheng, Y., Li, S. M., Leithead, A., Brickell, P. C., and Leaitch, W. R.: Characterizations of cis-pinonic acid and n-fatty acids on fine aerosols in the Lower Fraser Valley during Pacific 2001 Air Quality Study, Atmos. Environ., 38, 5789–5800, https://doi.org/10.1016/j.atmosenv.2004.01.051, 2004.
Chumpitaz, L. D. A., Coutinho, L. F., and Meirelles, A. J. A.: Surface tension of fatty acids and triglycerides, J. Am. Oil Chem. Soc., 76, 379–382, https://doi.org/10.1007/s11746-999-0245-6, 1999.
Cruz, C. N. and Pandis, S. N.: A study of the ability of pure secondary organic aerosol to act as cloud condensation nuclei, Atmos. Environ., 31, 2205–2214, https://doi.org/10.1016/s1352-2310(97)00054-x, 1997.
Dalirian, M., Ylisirniö, A., Buchholz, A., Schlesinger, D., Ström, J., Virtanen, A., and Riipinen, I.: Cloud droplet activation of black carbon particles coated with organic compounds of varying solubility, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2017-1084, in review, 2017.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Falkovich, A. H., Ganor, E., Levin, Z., Formenti, P., and Rudich, Y.: Chemical and mineralogical analysis of individual mineral dust particles, J. Geophys. Res.-Atmos., 106, 18029–18036, https://doi.org/10.1029/2000jd900430, 2001.
Falkovich, A. H., Schkolnik, G., Ganor, E., and Rudich, Y.: Adsorption of organic compounds pertinent to urban environments onto mineral dust particles, J. Geophys. Res.-Atmos., 109, D02208, https://doi.org/10.1029/2003jd003919, 2004.
Gantt, B., Xu, J., Meskhidze, N., Zhang, Y., Nenes, A., Ghan, S. J., Liu, X., Easter, R., and Zaveri, R.: Global distribution and climate forcing of marine organic aerosol – Part 2: Effects on cloud properties and radiative forcing, Atmos. Chem. Phys., 12, 6555–6563, https://doi.org/10.5194/acp-12-6555-2012, 2012.
Garimella, S., Huang, Y.-W., Seewald, J. S., and Cziczo, D. J.: Cloud condensation nucleus activity comparison of dry- and wet-generated mineral dust aerosol: the significance of soluble material, Atmos. Chem. Phys., 14, 6003–6019, https://doi.org/10.5194/acp-14-6003-2014, 2014.
Garland, E. R., Rosen, E. P., Clarke, L. I., and Baer, T.: Structure of submonolayer oleic acid coverages on inorganic aerosol particles: evidence of island formation, Phys. Chem. Chem. Phys., 10, 3156–3161, https://doi.org/10.1039/b718013f, 2008.
Giebl, H., Berner, A., Reischl, G., Puxbaum, H., Kasper-Giebl, A., and Hitzenberger, R.: CCN activation of oxalic and malonic acid test aerosols with the University of Vienna cloud condensation nuclei counter, J. Aerosol Sci., 33, 1623–1634, https://doi.org/10.1016/s0021-8502(02)00115-5, 2002.
Gierlus, K. M., Laskina, O., Abernathy, T. L., and Grassian, V. H.: Laboratory study of the effect of oxalic acid on the cloud condensation nuclei activity of mineral dust aerosol, Atmos. Environ., 46, 125–130, https://doi.org/10.1016/j.atmosenv.2011.10.027, 2012.
Hartz, K. E. H., Tischuk, J. E., Chan, M. N., Chan, C. K., Donahue, N. M., and Pandis, S. N.: Cloud condensation nuclei activation of limited solubility organic aerosol, Atmos. Environ., 40, 605–617, https://doi.org/10.1016/j.atmosenv.2005.09.076, 2006.
Hatch, C. D., Gierlus, K. M., Schuttlefield, J. D., and Grassian, V. H.: Water adsorption and cloud condensation nuclei activity of calcite and calcite coated with model humic and fulvic acids, Atmos. Environ., 42, 5672–5684, https://doi.org/10.1016/j.atmosenv.2008.03.005, 2008.
Haywood, J. and Boucher, O.: Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review, Rev. Geophys., 38, 513–543, https://doi.org/10.1029/1999rg000078, 2000.
Herich, H., Tritscher, T., Wiacek, A., Gysel, M., Weingartner, E., Lohmann, U., Baltensperger, U., and Cziczo, D. J.: Water uptake of clay and desert dust aerosol particles at sub- and supersaturated water vapor conditions, Phys. Chem. Chem. Phys., 11, 7804–7809, https://doi.org/10.1039/b901585j, 2009.
Ho, K. F., Cao, J. J., Lee, S. C., Kawamura, K., Zhang, R. J., Chow, J. C., and Watson, J. G.: Dicarboxylic acids, ketocarboxylic acids, and dicarbonyls in the urban atmosphere of China, J. Geophys. Res.-Atmos., 112, D22s27, https://doi.org/10.1029/2006jd008011, 2007.
Ho, K. F., Lee, S. C., Ho, S. S. H., Kawamura, K., Tachibana, E., Cheng, Y., and Zhu, T.: Dicarboxylic acids, ketocarboxylic acids, alpha-dicarbonyls, fatty acids, and benzoic acid in urban aerosols collected during the 2006 Campaign of Air Quality Research in Beijing (CAREBeijing-2006), J. Geophys. Res.-Atmos., 115, D19312, https://doi.org/10.1029/2009jd013304, 2010.
Hori, M., Ohta, S., Murao, N., and Yamagata, S.: Activation capability of water soluble organic substances as CCN, J. Aerosol Sci., 34, 419–448, https://doi.org/10.1016/s0021-8502(02)00190-8, 2003.
Iwahashi, M., Kasahara, Y., Matsuzawa, H., Yagi, K., Nomura, K., Terauchi, H., Ozaki, Y., and Suzuki, M.: Self-diffusion, dynamical molecular conformation, and liquid structures of n-saturated and unsaturated fatty acids, J. Phys. Chem. B, 104, 6186–6194, https://doi.org/10.1021/jp000610l, 2000.
Kawamura, K. and Bikkina, S.: A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation, Atmos. Res., 170, 140–160, https://doi.org/10.1016/j.atmosres.2015.11.018, 2016.
Kawamura, K. and Ikushima, K.: Seasonal-changes in the distribution of dicarboxylic-acids in the urban atmosphere, Environ. Sci. Technol., 27, 2227–2235, https://doi.org/10.1021/es00047a033, 1993.
Kawamura, K., Kasukabe, H., and Barrie, L. A.: Source and reaction pathways of dicarboxylic acids, ketoacids and dicarbonyls in arctic aerosols: One year of observations, Atmos. Environ., 30, 1709–1722, https://doi.org/10.1016/1352-2310(95)00395-9, 1996.
Keiser, E. H. and Leavitt, S.: On the preparation and the composition of the acid carbonates of calcium and barium, J. Am. Chem. Soc., 30, 1711–1714, https://doi.org/10.1021/ja01953a008, 1908.
Khare, P., Kumar, N., Kumari, K. M., and Srivastava, S. S.: Atmospheric formic and acetic acids: An overview, Rev. Geophys., 37, 227–248, 1999.
King, M. D., Rennie, A. R., Thompson, K. C., Fisher, F. N., Dong, C. C., Thomas, R. K., Pfrang, C., and Hughes, A. V.: Oxidation of oleic acid at the air–water interface and its potential effects on cloud critical supersaturations, Phys. Chem. Chem. Phys., 11, 7699–7707, https://doi.org/10.1039/b906517b, 2009.
Köhler, H.: The nucleus in and the growth of hygroscopic droplets, T. Faraday Soc., 32, 1152–1161, https://doi.org/10.1039/tf9363201152, 1936.
Koehler, K. A., Kreidenweis, S. M., DeMott, P. J., Petters, M. D., Prenni, A. J., and Carrico, C. M.: Hygroscopicity and cloud droplet activation of mineral dust aerosol, Geophys. Res. Lett., 36, L08805, https://doi.org/10.1029/2009gl037348, 2009.
Kumar, P., Nenes, A., and Sokolik, I. N.: Importance of adsorption for CCN activity and hygroscopic properties of mineral dust aerosol, Geophys. Res. Lett., 36, L24804, https://doi.org/10.1029/2009gl040827, 2009.
Li, W. J. and Shao, L. Y.: Mixing and water-soluble characteristics of particulate organic compounds in individual urban aerosol particles, J. Geophys. Res.-Atmos., 115, D02301, https://doi.org/10.1029/2009jd012575, 2010.
Liu, X. H. and Wang, J. A.: How important is organic aerosol hygroscopicity to aerosol indirect forcing?, Environ. Res. Lett., 5, 044010, https://doi.org/10.1088/1748-9326/5/4/044010, 2010.
Mellouki, A., Wallington, T. J., and Chen, J.: Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate, Chem. Rev., 115, 3984–4014, https://doi.org/10.1021/cr500549n, 2015.
Mkoma, S. L. and Kawamura, K.: Molecular composition of dicarboxylic acids, ketocarboxylic acids, α-dicarbonyls and fatty acids in atmospheric aerosols from Tanzania, East Africa during wet and dry seasons, Atmos. Chem. Phys., 13, 2235–2251, https://doi.org/10.5194/acp-13-2235-2013, 2013.
Moore, R. H., Nenes, A., and Medina, J.: Scanning Mobility CCN Analysis – A Method for Fast Measurements of Size-Resolved CCN Distributions and Activation Kinetics, Aerosol Sci. Tech., 44, 861–871, https://doi.org/10.1080/02786826.2010.498715, 2010.
Neagle, W. and Rochester, C. H.: Infrared study of the adsorption of water and ammonia on calcium carbonate, J. Chem. Soc. Faraday T., 86, 181–183, 1990.
Penner, J. E., Dong, X. Q., and Chen, Y.: Observational evidence of a change in radiative forcing due to the indirect aerosol effect, Nature, 427, 231–234, https://doi.org/10.1038/nature02234, 2004.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 7, 1961–1971, https://doi.org/10.5194/acp-7-1961-2007, 2007.
Pradeep Kumar, P., Broekhuizen, K., and Abbatt, J. P. D.: Organic acids as cloud condensation nuclei: Laboratory studies of highly soluble and insoluble species, Atmos. Chem. Phys., 3, 509–520, https://doi.org/10.5194/acp-3-509-2003, 2003.
Prenni, A. J., DeMott, P. J., Kreidenweis, S. M., Sherman, D. E., Russell, L. M., and Ming, Y.: The effects of low molecular weight dicarboxylic acids on cloud formation, J. Phys. Chem. A, 105, 11240–11248, https://doi.org/10.1021/jp012427d, 2001.
Rogge, W. F., Hildemann, L. M., Mazurek, M. A., Cass, G. R., and Simoneit, B. R. T.: Sources of fine organic aerosol. 2. Noncatalyst and catalyst-equipped automobiles and heavy-duty diesel trucks, Environ. Sci. Technol., 27, 636–651, https://doi.org/10.1021/es00041a007, 1993.
Rogge, W. F., Hildemann, L. M., Mazurek, M. A., Cass, G. R., and Simoneit, B. R. T.: Sources of fine organic aerosol. 9. Pine, oak and synthetic log combustion in residential fireplaces, Environ. Sci. Technol., 32, 13–22, https://doi.org/10.1021/es960930b, 1998.
Rose, D., Gunthe, S. S., Mikhailov, E., Frank, G. P., Dusek, U., Andreae, M. O., and Pöschl, U.: Calibration and measurement uncertainties of a continuous-flow cloud condensation nuclei counter (DMT-CCNC): CCN activation of ammonium sulfate and sodium chloride aerosol particles in theory and experiment, Atmos. Chem. Phys., 8, 1153–1179, https://doi.org/10.5194/acp-8-1153-2008, 2008.
Roselli, D.: Development and test of a system for simultaneous measurement of hygroscopic properties and chemical composition of multi-component aerosols, Univerista degli Studi “Carlo Bo” of Urbino, Urbino, Italy, 2006.
Ruehl, C. R., Davies, J. F., and Wilson, K. R.: An interfacial mechanism for cloud droplet formation on organic aerosols, Science, 351, 1447–1450, https://doi.org/10.1126/science.aad4889, 2016.
Russell, L. M., Maria, S. F., and Myneni, S. C. B.: Mapping organic coatings on atmospheric particles, Geophys. Res. Lett., 29, 1779, https://doi.org/10.1029/2002gl014874, 2002.
Sage, A. M., Weitkamp, E. A., Robinson, A. L., and Donahue, N. M.: Reactivity of oleic acid in organic particles: changes in oxidant uptake and reaction stoichiometry with particle oxidation, Phys. Chem. Chem. Phys., 11, 7951–7962, https://doi.org/10.1039/b904285g, 2009.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources. 1. C1 through C29 organic compounds from meat charbroiling, Environ. Sci. Technol., 33, 1566–1577, https://doi.org/10.1021/es980076j, 1999.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics-From Air Pollution to Climate Change, 2nd edn., John Wiley & Sons, Inc., Hoboken, New Jersey, 773–776, 2006.
Sorjamaa, R. and Laaksonen, A.: The effect of H2O adsorption on cloud drop activation of insoluble particles: a theoretical framework, Atmos. Chem. Phys., 7, 6175–6180, https://doi.org/10.5194/acp-7-6175-2007, 2007.
Stipp, S. L. S.: Toward a conceptual model of the calcite surface: hydration, hydrolysis, and surface potential, Geochim. Cosmochim. Ac., 63, 3121–3131, 1999.
Stipp, S. L. and Hochella Jr., M. F.: Structure and bonding environments at the calcite surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy electron diffraction (LEED), Geochim. Cosmochim. Ac., 55, 1723–1736, 1991.
Sullivan, R. C., Moore, M. J. K., Petters, M. D., Kreidenweis, S. M., Roberts, G. C., and Prather, K. A.: Effect of chemical mixing state on the hygroscopicity and cloud nucleation properties of calcium mineral dust particles, Atmos. Chem. Phys., 9, 3303–3316, https://doi.org/10.5194/acp-9-3303-2009, 2009.
Sullivan, R. C., Moore, M. J. K., Petters, M. D., Kreidenweis, S. M., Qafoku, O., Laskin, A., Roberts, G. C., and Prather, K. A.: Impact of Particle Generation Method on the Apparent Hygroscopicity of Insoluble Mineral Particles, Aerosol Sci. Tech., 44, 830–846, https://doi.org/10.1080/02786826.2010.497514, 2010.
Takegawa, N., Miyakawa, T., Kawamura, K., and Kondo, Y.: Contribution of selected dicarboxylic and omega-oxocarboxylic acids in ambient aerosol to the m∕z 44 signal of an aerodyne aerosol mass spectrometer, Aerosol Sci. Tech., 41, 418–437, https://doi.org/10.1080/02786820701203215, 2007.
Tang, M. J., Whitehead, J., Davidson, N. M., Pope, F. D., Alfarra, M. R., McFiggans, G., and Kalberer, M.: Cloud condensation nucleation activities of calcium carbonate and its atmospheric ageing products, Phys. Chem. Chem. Phys., 17, 32194–32203, https://doi.org/10.1039/c5cp03795f, 2015.
Tang, M. J., Cziczo, D. J., and Grassian, V. H.: Interactions of Water with Mineral Dust Aerosol: Water Adsorption, Hygroscopicity, Cloud Condensation, and Ice Nucleation, Chem. Rev., 116, 4205–4259, https://doi.org/10.1021/acs.chemrev.5b00529, 2016.
Varga, Z., Kiss, G., and Hansson, H.-C.: Modelling the cloud condensation nucleus activity of organic acids on the basis of surface tension and osmolality measurements, Atmos. Chem. Phys., 7, 4601–4611, https://doi.org/10.5194/acp-7-4601-2007, 2007.
Yamashita, K., Murakami, M., Hashimoto, A., and Tajiri, T.: CCN Ability of Asian Mineral Dust Particles and Their Effects on Cloud Droplet Formation, J. Meteorol. Soc. Jpn., 89, 581–587, https://doi.org/10.2151/jmsj.2011-512, 2011.
Zhao, D. F., Buchholz, A., Mentel, Th. F., Müller, K.-P., Borchardt, J., Kiendler-Scharr, A., Spindler, C., Tillmann, R., Trimborn, A., Zhu, T., and Wahner, A.: Novel method of generation of Ca(HCO3)2 and CaCO3 aerosols and first determination of hygroscopic and cloud condensation nuclei activation properties, Atmos. Chem. Phys., 10, 8601–8616, https://doi.org/10.5194/acp-10-8601-2010, 2010.
Zuend, A., Marcolli, C., Booth, A. M., Lienhard, D. M., Soonsin, V., Krieger, U. K., Topping, D. O., McFiggans, G., Peter, T., and Seinfeld, J. H.: New and extended parameterization of the thermodynamic model AIOMFAC: calculation of activity coefficients for organic-inorganic mixtures containing carboxyl, hydroxyl, carbonyl, ether, ester, alkenyl, alkyl, and aromatic functional groups, Atmos. Chem. Phys., 11, 9155–9206, https://doi.org/10.5194/acp-11-9155-2011, 2011.