the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Oct 2018
| 26 Oct 2018
A comprehensive organic nitrate chemistry: insights into the lifetime of atmospheric organic nitrates
Azimeh Zare
Paul S. Romer
Tran Nguyen
Frank N. Keutsch
Kate Skog
Organic nitrate chemistry is the primary control over the lifetime of nitrogen oxides (NOx) in rural and remote continental locations. As NOx emissions decrease, organic nitrate chemistry becomes increasingly important to urban air quality. However, the lifetime of individual organic nitrates and the reactions that lead to their production and removal remain relatively poorly constrained, causing organic nitrates to be poorly represented by models. Guided by recent laboratory and field studies, we developed a detailed gas-phase chemical mechanism representing most of the important individual organic nitrates. We use this mechanism within the Weather Research and Forecasting (WRF) model coupled with Chemistry (WRF-Chem) to describe the role of organic nitrates in nitrogen oxide chemistry and in comparisons to observations. We find the daytime lifetime of total organic nitrates with respect to all loss mechanisms to be 2.6 h in the model. This is consistent with analyses of observations at a rural site in central Alabama during the Southern Oxidant and Aerosol Study (SOAS) in summer 2013. The lifetime of the first-generation organic nitrates is ∼2 h versus the 3.2 h lifetime of secondary nitrates produced by oxidation of the first-generation nitrates. The different generations are subject to different losses, with dry deposition to the surface being the dominant loss process for the second-generation organic nitrates and chemical loss being dominant for the first-generation organic nitrates. Removal by hydrolysis is found to be responsible for the loss of ∼30 % of the total organic nitrate pool.
- Article
(6296 KB) - Full-text XML
-
Supplement
(7520 KB) - BibTeX
- EndNote





In remote continental regions, biogenic volatile organic compounds (BVOCs), including isoprene and terpenes, are the most reactive organic compounds in the atmosphere (Guenther, 2013). The oxidative chemistry of BVOCs affects the distribution of oxidants (OH, O3, NO3) and the lifetime of NOx ), creating a feedback loop that affects oxidant concentrations, the lifetime of BVOCs and secondary organic aerosol (SOA) formation. Along the pathway to complete oxidation of BVOCs, reactions with the nitrogen oxide family radicals (NO, NO2 and NO3) to form organic nitrate products (e.g., Perring et al., 2013) are an important branch point that sets the importance of this feedback (Carlton et al., 2018; Mao et al., 2018).
During the day, BVOCs react with the hydroxyl radical (HO) and peroxy radicals (RO2) are formed. At high and even modest concentrations of NOx, the peroxy radicals react primarily with NO. The major products of that reaction are NO2 and an alkoxy radical (RO). There is also a minor channel (with a branching fraction α) that results in addition of the NO to the peroxy radical, resulting in an organic nitrate (RONO2) product. During the night, nitrate radicals (NO3), the product of the oxidation of NO2 by O3, are also a major source of RONO2. BVOCs react with NO3, resulting in the formation of nitrooxy peroxy radicals in high yields. The radicals subsequently react to form closed shell RONO2, with branching ratio β.
In the last decade, there have been major updates to our understanding of the chemical reactions that occur during isoprene oxidation (Paulot et al., 2009a, b; Crounse et al., 2011; Liu et al., 2013; Peeters et al., 2014; Nguyen et al., 2014; Wolfe et al., 2016; Mills et al., 2016; Teng et al., 2017). This understanding includes recognition that the yield of RONO2 from reaction of isoprene peroxy radicals with NO is 11 %–15 %, which is at the high end of the range reported in earlier laboratory experiments (Wennberg et al., 2018). The yield of nitrates from monoterpene oxidation is less clear as laboratory data indicate a very wide range (e.g., from greater than 1 %, Aschmann et al., 2002, to 26 %, Rindelaub et al., 2015). For NO3 oxidation of isoprene experimental data show that the yield, β, is high and varies in the range of 65 %–80 % (Perring et al., 2009a; Rollins et al., 2009; Kwan et al., 2012).
Once formed, RONO2 can be photolyzed or oxidized to produce NOx or HNO3 along with an organic partner, or they can serve as reservoirs of NOx that can be transported or deposited to the surface. An additional pathway for gas-phase RONO2 loss is partitioning into aerosol in either an organic phase where vapor pressure would describe partitioning or an aqueous phase where a Henry's law constant would describe solubility. In the aerosol, the RONO2 can undergo liquid-phase reaction. Some RONO2 are rapidly hydrolyzed with timescales on the order of hours to minutes under environmentally relevant pH conditions (Jacobs et al., 2014; Boyd et al., 2015; Rindelaub et al., 2016), while other nitrates are thought to be relatively stable against hydrolysis in neutral conditions (Hu et al., 2011). The main nitrogen-containing product of organic nitrate hydrolysis is nitric acid (Darer et al., 2011). Using measurements of organic nitrates and nitric acid over the Canadian boreal forest and southeast United States, Browne et al. (2013) and Romer et al. (2016) provide evidence that hydrolysis of monoterpene and isoprene nitrates is likely a significant loss process and contributes to HNO3 production. The short lifetime of HNO3 to deposition in the boundary layer means that organic nitrate loss through hydrolysis in the boundary layer is a permanent sink of NOx.
For any organic nitrate, its structure determines the rate of its oxidation and photolysis as well as the rate of hydrolysis and deposition. Multifunctional nitrates containing hydroxyl or peroxide groups are likely to have deposition rates much faster than the rates for monofunctional nitrates (Shepson et al., 1996). The dry deposition of organic nitrates has been discussed in the studies by Farmer and Cohen (2008) and Nguyen et al. (2015). Nguyen et al. (2015) directly measured deposition rates of organic nitrates from BVOCs and the first-generation isoprene nitrates were observed to have a daytime dry deposition velocity of ∼2 cm s−1, which is higher than the values currently used in most models (Ito et al., 2009; Mao et al., 2013; Browne et al., 2014).
Unlike hydrolysis of organic nitrates in aerosol and deposition of organic nitrates to the surface, which is considered a sink of nitrogen oxides from the atmosphere, oxidation and photolysis of RONO2 may recycle NOx. Different assumptions regarding NOx recycling during organic nitrate oxidation result in large variations in simulating NOx and O3 (von Kuhlmann et al., 2004; Fiore et al., 2005; Wu et al., 2007; Horowitz et al., 2007; Paulot et al., 2012). For example, Xie et al. (2013) showed that the uncertainty in the fraction of NOx returned to the atmosphere during isoprene nitrate oxidation had a larger impact than uncertainty in isoprene nitrate yield on O3 production. This affirms the need for characterization of the fate and lifetime of RONO2 in the atmosphere. New clarity is available for the chemical fate of the first-generation isoprene nitrates (e.g., Lee et al., 2014; Xiong et al., 2015, 2016), while much less is known about the fate of organic nitrates formed from monoterpenes. Because few of these loss processes have been measured, especially for highly oxidized or monoterpene nitrates, there is large uncertainty associated with any description of the lifetime of organic nitrates. Several modeling studies (Paulot et al., 2012; Xie et al., 2013; Mao et al., 2013) have focused specifically on the fate of isoprene nitrates and have found that how their chemistry is represented has major consequences for NOx and O3. Recently, Browne et al. (2014) extended the representation of organic nitrate chemistry by including in detail the gas-phase chemistry of monoterpenes and discussed different scenarios for uncertain loss processes of monoterpene nitrates. Their improved mechanism for BVOC chemistry has been used as a skeleton for several subsequent modeling studies (e.g., Fisher et al., 2016 and this work).
However, none of these models has yet combined detailed molecular representations of individual RONO2 derived from anthropogenic, isoprene and monoterpene VOC precursors. Here we describe the development of a gas-phase mechanism along those lines. In a forthcoming paper we couple the mechanism described here to aerosol and cloud properties. Here we approximate the effects of aerosols and clouds with simpler parameters representing the effects of the condensed-phase chemistry. The model calculations are compared to observations from the SOAS (the Southern Oxidant and Aerosol Study) campaign in the rural southeastern United States in summer 2013. We explore the relative contributions of OH and NO3 chemistry to the production of organic nitrates from BVOCs and investigate the importance of different organic nitrate loss processes. Then we explore the lifetime of organic nitrates and consequences of organic nitrate chemistry for atmospheric NOx. This information helps to understand the role of RONO2 in the NOx and ozone budgets in the moderate NOx, BVOC-dominated terrestrial environments that represent the most common chemical regime on the continents during summer.
We use the Weather Research and Forecasting (WRF) model coupled with Chemistry (WRF-Chem) version 3.5.1 (Grell et al., 2005) with a horizontal resolution of 12 km over the eastern United States. Our simulation domain is defined on the Lambert projection, which is centered at 35∘ N, 87∘ W and has 290 and 200 grid points in the west–east and south–north directions, respectively (see Fig. 3 for the horizontal domain). The vertical coordinate is hybrid sigma-pressure that covers 30 levels from the surface to 100 hPa. Near-surface levels follow terrain and gradually transition to constant pressure at higher levels. Vertical grid spacing varies with height such that finer spacing is assigned to the lower atmosphere while coarser vertical spacing is applied at higher levels. In this analysis, the model predictions are averaged over two lowest model levels (∼25 m) used for comparison with ground-based measurements taken from a 20 m walk-up tower. The predicted concentrations in the boundary layer are described as an average over eight vertical model levels with a height (∼1000 m) that is comparable with the planetary boundary layer depth at midday in the southeastern United States in June 2013. Meteorological data for initial and boundary conditions are driven by the North American Regional Reanalysis (NARR) and data for the chemical initial and boundary conditions are taken from MOZART (Emmons et al., 2010). The model simulation period is from 27 May to 30 June 2013, with the first 5 days as spin-up, similar to Browne et al. (2014), to remove the impact of initial conditions.
Anthropogenic emissions are based on the US EPA 2011 National Emission Inventory (NEI) and scaled to 2013 based on the changes in the annual average emissions from 2011 to 2013. The appropriate scale factors have been derived from the NEI Air Pollutant Emissions Trend Data. We also adjust NOx emissions (uniformly reduced by 50 %) following Travis et al. (2016), who suggest that reduced NOx emissions can better reproduce the SEAC4RS aircraft measurements for the southeastern United States. Lightning emissions of NOx are not included in the model. Lightning NOx is mainly released at the top of convective updrafts (Ott et al., 2010) and does not strongly impact the distribution of NO2 in the boundary layer (e.g., Laughner and Cohen, 2017). Biogenic emissions of isoprene, monoterpenes, other BVOCs, oxygenated VOCs (OVOCs) and nitrogen gas emissions from the soil are parameterized using the Model of Emissions of Gases and Aerosol from Nature (MEGAN) (Guenther et al., 2006).
Gas-phase reactions are simulated using the second-generation Regional Atmospheric Chemistry Mechanism (RACM2) (Goliff et al., 2013), as updated by Browne et al. (2014) and with additions to the mechanisms as described below, which is implemented with the kinetic preprocessor (KPP) software (Damian et al., 2002). The Modal Aerosol Dynamics Model for Europe–Secondary Organic Aerosol Model (MADE/SORGAM) aerosol module (Ackermann et al., 1998; Schell et al., 2001) is used to treat organic and inorganic aerosols. This two-product aerosol scheme does not treat the partitioning of individual chemical species such as organic nitrates. Therefore, we focus here only on investigating the impacts of the gas-phase representation of the chemistry and a full consideration of the gas and aerosol in a coupled framework is a subject of continuing research.
2.1 Chemical mechanism
We base our core chemistry on a modified version of RACM2 (Goliff et al., 2013). The base we begin with is described by Browne and Cohen (2012) and Browne et al. (2014) and is referred to here as the RACM2_Berkeley scheme. Full details of the RACM2_Berkeley mechanism and a complete list of the compounds can be found in Browne et al. (2014). RACM2_Berkeley includes updates to the isoprene oxidation mechanism (Paulot et al., 2009a, b; Peeters and Müller, 2010; Lockwood et al., 2010; Stavrakou et al., 2010; Crounse et al., 2011), an extended mechanism for anthropogenic-originated organic nitrates (Carter and Atkinson, 1989; Middleton et al., 1990; Arey et al., 2001), and updates for monoterpene chemistry. Browne et al. (2014) evaluated the RACM2_Berkeley mechanism using aircraft observations over the Canadian boreal forest.
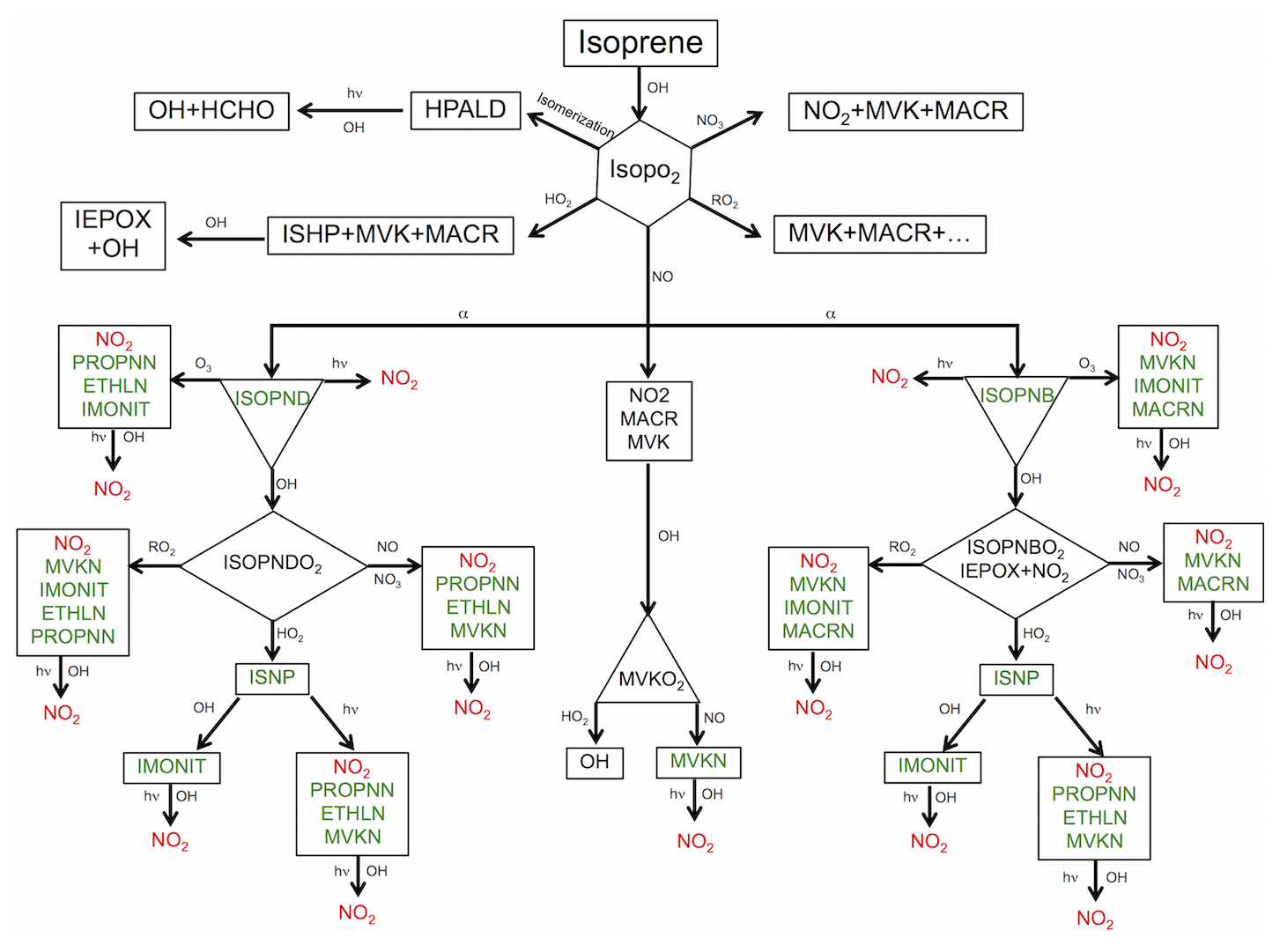
Figure 1Schematic representation of the formation of isoprene nitrates (in green) initiated by OH oxidation. Rerelease of the consumed NOx to the atmosphere by chemical loss processes of oxidation, ozonolysis and photolysis of organic nitrate is shown in red. See Table S2 in the Supplement for species descriptions.
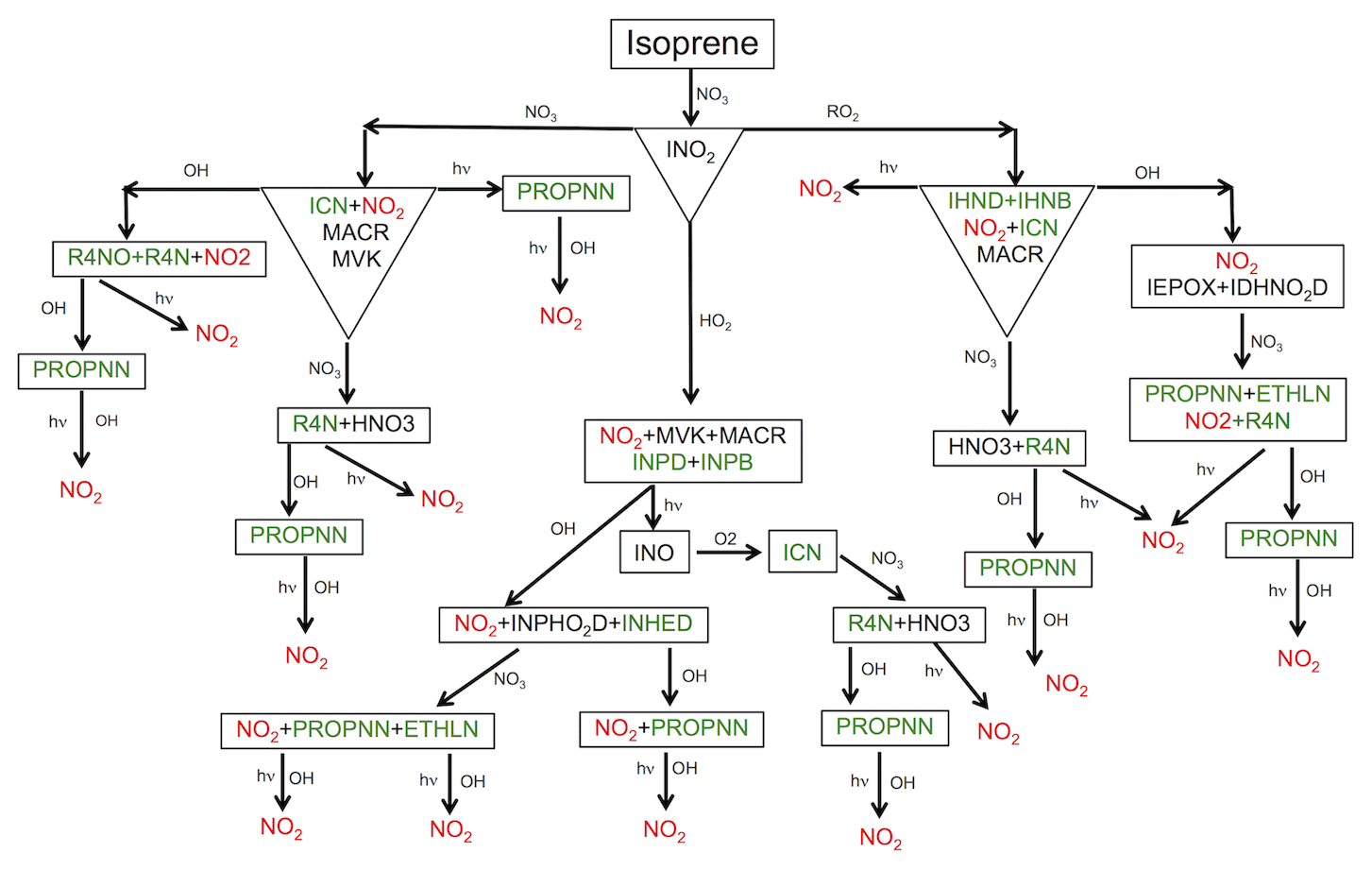
Figure 2Schematic representation of the formation of isoprene nitrates (in green) initiated by NO3 oxidation. For simplification, fates of only one isomer of hydroxy nitrates (IHNB and IHND) and nitrooxy hydroperoxide (INPB and INPD) are shown. See Table S2 in the Supplement for species descriptions.
In this study, the RACM2_Berkeley scheme is further updated with recent advances in the representation of OH- and NO3-initiated BVOC oxidation under both low- and high-NOx conditions, as well as with improved deposition rates, and is denoted RACM2_Berkeley2 (see Tables S1–S3 in the Supplement). We begin with a more complete description of recent advances in our understanding of isoprene chemistry (Fig. 1). The hydroxy peroxy radical (ISOPO2) that is the product of isoprene oxidation by OH has multiple potential fates. ISOPO2 can undergo unimolecular isomerization, leading to the production of hydroperoxy aldehydes (HPALD), among other products. It can react with HO2 to produce isoprene hydroxy hydroperoxide (ISHP), methyl vinyl ketone (MVK), methacrolein (MACR) and CH2O. The latter three species can also be formed from the reactions of ISOPO2 with other (acetyl or methyl) peroxy radicals. ISHP reacts with OH to form isoprene epoxydiols (IEPOX) and regenerate OH. St. Clair et al. (2015) found that the reaction rate of ISHP+OH is approximately 10 % faster than the rate given by Paulot et al. (2009b) and indicate the relative role of the different isomers of ISHP. Here we use kinetics and products of the reactions of three different isomers of ISHP with OH based on St. Clair et al. (2015). We also increase the molar yield of total ISHP from the ISOPO2+HO2 reaction to 93.7 % (Liu et al., 2013), with a decrease in the yields of MVK, MACR and HOx to maintain mass balance. We use rates from Bates et al. (2016) for reactions of three different isomers of IEPOX with OH.
We maintain the overall branching ratio of isoprene nitrates at 11.7 % as in Browne et al. (2014) while changing the mix of isomers. Browne et al. (2014) implemented a scheme for the reaction of ISOPO2 with NO, based on experiments conducted by Paulot et al. (2009b), including β- and δ-hydroxy isoprene nitrates (ISOPNB and ISOPND) with yields of 4.7 % and 7.0 %, respectively. Here we update the yield of β versus δ isomers to 10.5 % and 1.2 %, respectively. A theoretical study by Peeters et al. (2014) showed that the peroxy radical redissociations are fast and peroxy isomers may interconvert, so that β isomers comprise ∼95 % of the radical pool. The experimental findings of Teng et al. (2017) are also consistent with that idea. Simulations by Fisher et al. (2016) showed that an isoprene hydroxy radical distribution leading to 90 % ISOPNB and 10 % ISOPND is consistent with SOAS observations.
ISOPND and ISOPNB then photolyze or react with O3 or OH to yield either NOx or second-generation organic nitrates. We update reaction rates of isoprene hydroxy nitrate oxidation based on Lee et al. (2014). Compared to the RACM2_Berkeley mechanism (based on Paulot et al., 2009b and Lockwood et al., 2010), the reaction rates for ISOPNs+OH are increased and the rate coefficients of ISOPNs+O3 are decreased. The model represents the products of the reactions of ISOPND and ISOPNB with OH as ISOPNDO2 and ISOPNBO2. We update the reaction of ISOPNB+OH to include a small yield of IEPOX and NO2 (12 %) as found by Jacobs et al. (2014). We also update the rate constants for reaction of ISOPNDO2 and ISOPNBO2 with NO, producing second-generation isoprene nitrates following Lee et al. (2014). Second-generation isoprene nitrates from the OH-initiated pathway include ethanal nitrate (ETHLN), propanone nitrate (PROPNN), multifunctional isoprene nitrate (IMONIT), methacrolein nitrate (MACRN) and methyl vinyl ketone nitrate (MVKN). MACRN and MVKN can also be formed directly from photooxidation of MVK and MACR under high-NOx conditions. Here, we follow Praske et al. (2015) to update MVK chemistry under both low- and high-NOx conditions, resulting in greater recycling of OH and NO2 and decreased formation of organic nitrates from reactions with NO or HO2.
In addition to OH chemistry, isoprene is oxidized by NO3. In RACM2_Berkeley, the isoprene + NO3 chemistry was parameterized with one generic organic nitrate as the product. Recently, Schwantes et al. (2015) developed a kinetic mechanism for NO3-initiated oxidation of isoprene in which products, branching ratios and rate constants are estimated based on recent experimental results. Their suggested products from NO3 oxidation of isoprene are consistent with organic nitrates detected in the ambient atmosphere during SOAS. RACM2_Berkeley2 treats the NO3-initiated oxidation of isoprene in some detail with formation and subsequent oxidation of isoprene nitrates largely based on the work of Schwantes et al. (2015) and Rollins et al. (2009) (Fig. 2). In the first step, NO3 addition to isoprene forms a nitrooxy peroxy radical (INO2) and then, depending on the radical the INO2 reacts with, first-generation isoprene nitrates are formed, namely C5 carbonyl nitrate (ICN), β- and δ-hydroxy nitrate (IHNB and IHND) and β and δ-nitrooxy hydroperoxide (INPB and INPD).
We set the ICN yield at 54 % and 72 % for the INO2+NO3 and INO2+INO2 reactions, respectively. ICN is a first-generation isoprene nitrate that is reactive towards NO3 (Rollins et al., 2009). Subsequent oxidation of ICN by NO3 forms second-generation isoprene nitrates as well as nitric acid at rates and yields based on the Master Chemical Mechanism (MCM v3.2) (Jenkin et al., 1997; Saunders et al., 2003). MCM v3.2 uses a reaction rate coefficient of cm3 molec−1 s−1 at 298 K, which is 5 times slower than the rate given by Rollins et al. (2009) and an order of magnitude faster than the rate given by Schwantes et al. (2015). Given the differences in the experimental data, splitting the difference by using the MCM rate seems a reasonable choice.
IHNB and IHND are also identified in the chamber experiments as products of the INO2+INO2 reaction. We follow Schwantes et al. (2015) and use rate constants and products, respectively, from Lee et al. (2014) and Jacob et al. (2014), for the subsequent fate of IHNB and IHND upon reaction with OH. We note that these rate constants and products correspond to the rate constants and products for the reactions of ISOPND and ISOPNB (OH-initiated isoprene hydroxy nitrates) with OH. In Fig. 2, we only show reactions for one of the isomers.
Nitrooxy hydroperoxides (INPD and INPB) are the dominant products of the INO2+HO2 reaction with a combined 77 % yield, most of which is δ isomers (INPD). Schwantes et al. (2015) found that the total molar yield of INPD and INPB per reacted isoprene is higher than the yield found in previous studies (Ng et al., 2008; Kwan et al., 2012). In these previous studies, the carbonyl nitrate (ICN) was the main contributor to the family of isoprene nitrates produced by NO3-initiated chemistry and nitrooxy hydroperoxides (INPD and INPB) were a minor fraction. The difference is likely caused by variation in the fate of the nitrooxy peroxy radical (INO2+HO2 vs. INO2+NO3) under different experimental conditions. The fate of this radical (INO2) in the nighttime atmosphere is still highly uncertain (Brown and Stutz, 2012; Boyd et al., 2015).
Chamber experiments by Schwantes et al. (2015) suggested that OH reacts with INPD and INPB to form second-generation isoprene nitrates and nitrooxy hydroxy epoxides (INHE), a newly identified product that undergoes a similar heterogeneous chemistry as IEPOX and can impact secondary organic aerosol formation. We set the yield of the INHE δ isomer from INPD at 37 % and yield of the INHE β isomer from INPB at 78 %. Those are lower than the IEPOX yield formed from ISHP. INHE isomers will be further oxidized by OH, leading to recycling of NOx or forming a later generation of organic nitrates. We use the same rates that we use for OH oxidation of IEPOX isomers (Schwantes et al., 2015; Bates et al., 2016). In this scheme, we also assume that INPD and INPB undergo hydrogen abstraction from the hydroperoxide group with the same rate constants as those we use for hydrogen abstraction from the hydroperoxide group of ISHP as suggested by St. Clair et al. (2015).
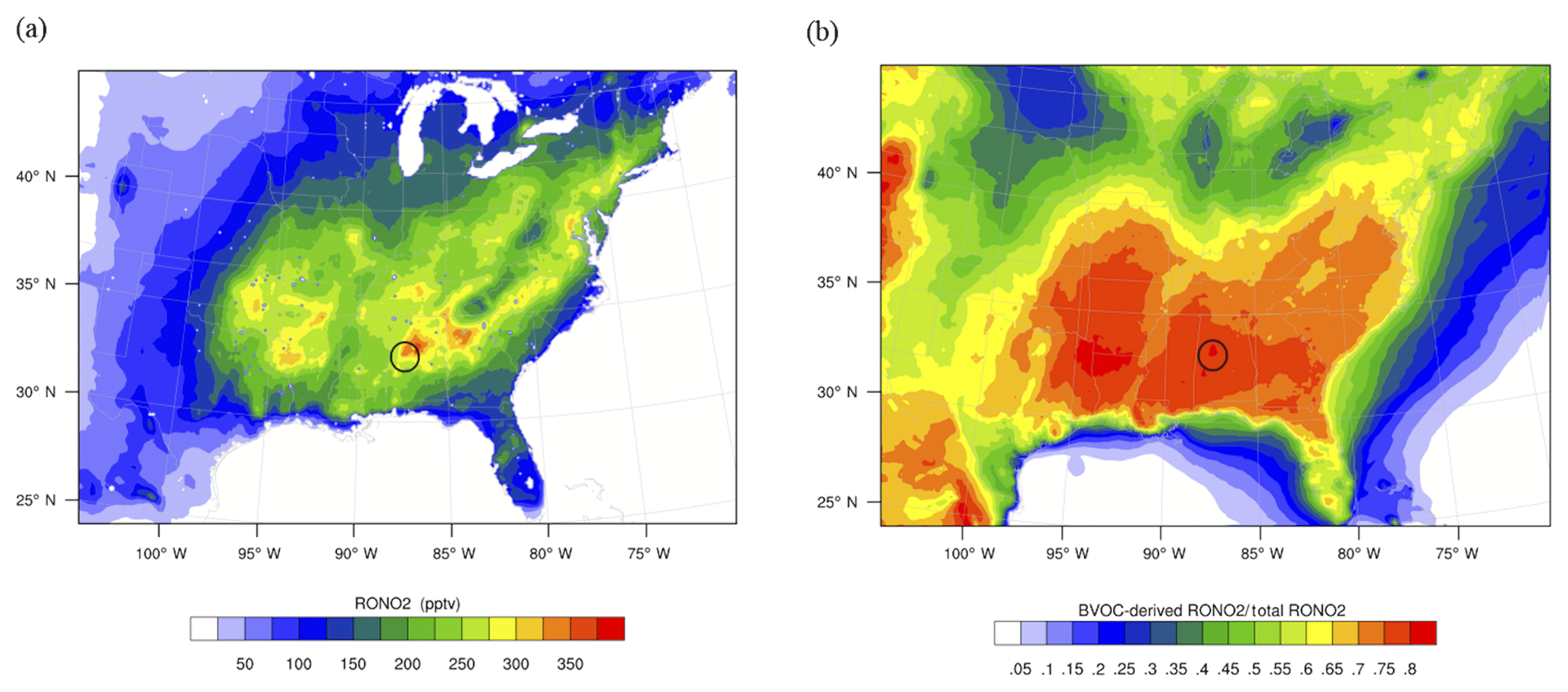
Figure 3(a) Concentrations of total organic nitrates for the average of the model simulation period (June 2013). (b) Fractional contribution of BVOC-derived organic nitrates to total organic nitrates. The location of the SOAS CTR ground site is circled in the figure.
C4 carbonyl hydroperoxy nitrates (R4NO) and C4 carbonyl hydroxy nitrates (R4N) form from oxidation of the first-generation isoprene nitrates. In reaction with OH, they produce propanone nitrate. The subsequent fate of PROPNN is dominated by oxidation by OH and photolysis, resulting in the return of NOx to the atmosphere. Here, we include a photolysis rate for PROPNN and other carbonyl nitrates (ICN, ETHLN), MVKN and MACRN that is faster than that used in some other recent mechanisms following the recommendations in Müller et al. (2014). These rates are 10 times larger than the rates given by Paulot et al. (2009a). We use the enhanced photolysis rate of PROPNN for carbonyl nitrate ICN formed from NO3-initiated isoprene oxidation and also include photolysis reactions for other new isoprene nitrate species, following Schwantes et al. (2015) and MCM v3.2. The fast photolysis reactions of organic nitrates implemented in this work lead to an increase in NOx concentrations.
2.2 Organic nitrate deposition to surfaces
We use the resistance-based approach based on the original formulation of Wesely (1989) to calculate dry deposition velocities. Dry deposition rates of RONO2 in RACM2_Berkeley follow Ito et al. (2007). In RACM2_Berkeley2, using the same formalism, we update the dry deposition parameters (effective Henry's Law coefficients, H*; and reactivity factors, f0) for isoprene nitrates as recommended by Nguyen et al. (2015), which results in more rapid removal than in RACM2_Berkeley (Table S3 in the Supplement). We also update the dry deposition parameters for monoterpene-derived nitrates that were previously assumed to deposit at a rate similar to the deposition rate of isoprene hydroxy nitrates (Browne et al., 2014; Ito et al., 2007). Dry deposition velocities of the nitrooxy hydroxy epoxides (INHE) are assumed to be similar to the updated depositional loss rate of IEPOX, given by Nguyen et al. (2015).
2.3 Hydrolysis of tertiary nitrates
In addition to oxidation, photolysis and deposition to the surface, another possible fate of organic nitrates is uptake to the aerosol phase followed by hydrolysis. A rapid hydrolysis (Hu et al., 2011; Jacobs et al., 2014) is recognized for tertiary nitrates, while non-tertiary nitrates under atmospheric conditions are considered unreactive (Darer et al., 2011; Boyd et al., 2015). Due to the limitations of the model representation of organic nitrate aerosol from either aqueous (Marais et al., 2016) or vapor-pressure-dependent pathways (Pye et al., 2015), we represent this process for gas-phase organic nitrates by applying a timescale of 3 h for tertiary nitrates based on the laboratory chamber study by Boyd et al. (2015). The fraction of tertiary (Ftertiary) vs. non-tertiary nitrates is estimated, depending upon the molecular structure of the nitrate, from MCM v3.2.
We apply Ftertiary at 41 % for β isomers of isoprene nitrates from OH oxidation while we assume that all δ isomers are non-tertiary. Most of the nitrates formed by NO3-initiated chemistry of isoprene are not tertiary nitrates. The fraction of tertiary nitrates for monoterpene-derived nitrates is also different for species formed by OH oxidation than from NO3 oxidation. In RACM2_Berkeley2, we introduce TONIH (C10 nitrooxy hydroperoxide), TONIN (saturated) and UTONIN (unsaturated) monoterpene-derived nitrates from NO3 oxidation, which differ from the unsaturated (UTONIT) and saturated (TONIT) monoterpene-derived nitrates from OH oxidation. Since contributions of tertiary limonene, α-pinene and β-pinene nitrates from NO3 reaction are 35 %, 15 % and 50 % (MCM v3.2), respectively, we define Ftertiary at 35 % as an average for TONIH, UTONIN and TONIN. Ftertiary is defined at 77 % for UTONIT and TONIT. The value is the average of the 62 % for α-pinene nitrates and 92 % for β-pinene nitrates and is equal to the 77 % for limonene nitrates from OH chemistry.
Further changes in the RACM2_Berkeley2 mechanism for monoterpene nitrate chemistry consist of a revised reaction rate for API + NO3. The rate constant is calculated as an average of the rates given in MCM v3.2 for α-pinene and β-pinene, as API in the mechanism indicates a 50–50 mixture of α-pinene and β-pinene. In our mechanism, following Browne et al. (2014), first-generation monoterpene nitrates react with O3 and OH and form second-generation nitrates. Here, we also add reaction of first-generation monoterpene nitrates with NO3 with the rate constant K exp(), following Fisher et al. (2016). We assume the second-generation monoterpene nitrate can oxidize, photolyze and deposit identically to nitric acid (Browne et al., 2014). In summary, we have described a detailed chemical mechanism tracking individual organic nitrates in some detail through second-generation products of isoprene and monoterpene oxidation.
We evaluate our mechanism by comparison to SOAS observations in Bibb County, Alabama (32.90∘ N latitude, 87.25∘ W longitude), in summer 2013 (Carlton et al., 2018; Mao et al., 2018). These observations together with field campaign data from the long-term monitoring site in the Southeastern Aerosol Research and Characterization (SEARCH) Network (Hansen et al., 2003) (at the same location) provide unique resources for evaluation of our model of organic nitrate chemistry. The measurements include total and speciated organic nitrates, gas-phase and aerosol organic nitrates, HOx radicals, a wide range of VOCs and ozone (Pye et al., 2015; Romer et al., 2016; Lee et al., 2016).
3.1 Organic nitrate concentrations
Figure 3a shows the spatial distribution of total organic nitrates for the 24 h average of the model simulation period at the surface. The location of the SOAS ground site (at Centreville, CTR) is circled in the figure. The campaign area is in a location with among the highest modeled organic nitrate concentrations in the region, up to 350 ppt. Figure 3b highlights that the modeled RONO2 originating from biogenic VOCs dominate over the organic nitrates with anthropogenic VOC precursors over most of the domain. In the southeast, up to 80 % of organic nitrates are biogenic. Biogenic nitrates are 40 %–50 % in the northern portion.
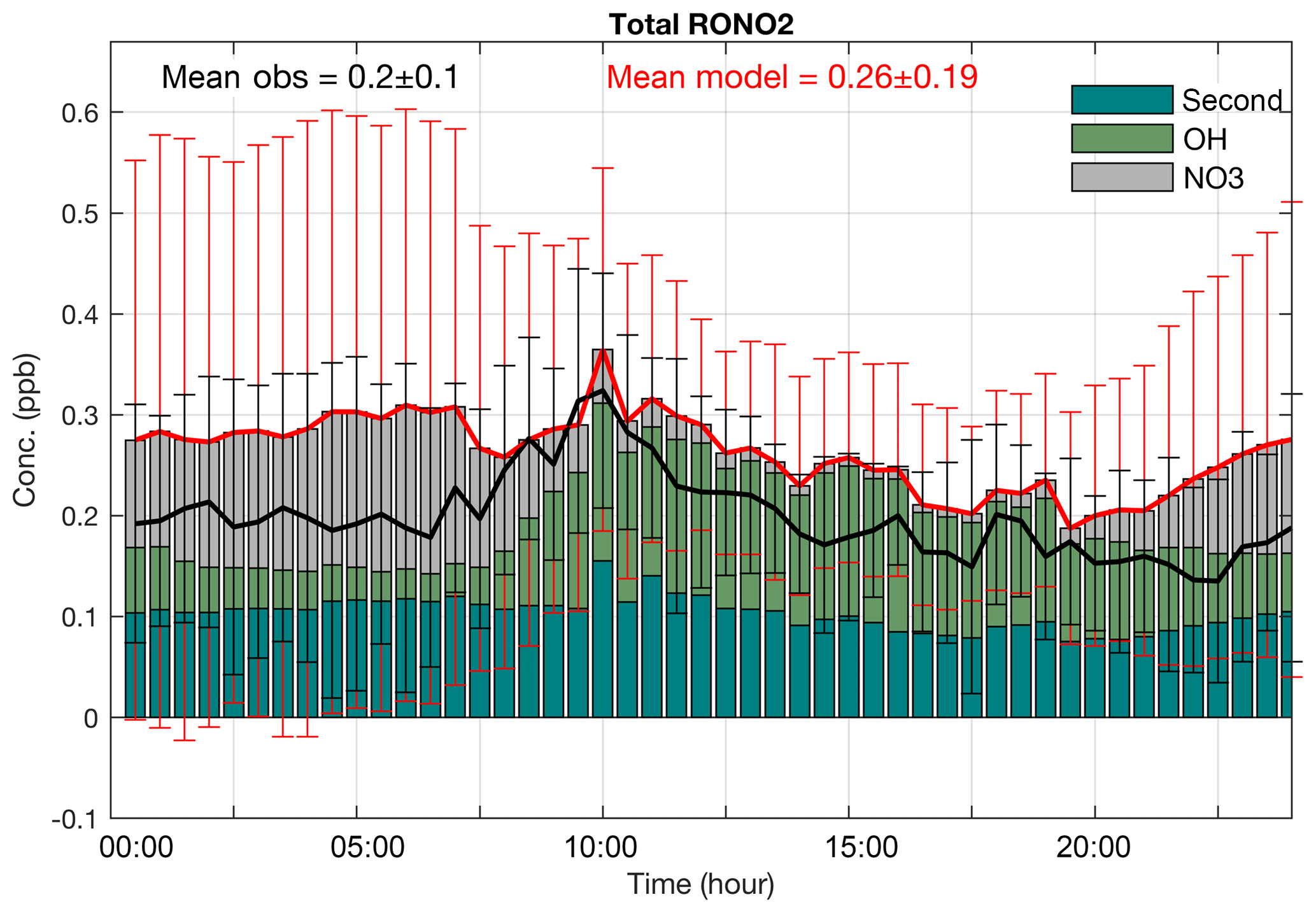
Figure 4Median diurnal cycles of observed (black) and simulated (red) total organic nitrates at Centreville during the 2013 SOAS campaign. The vertical bars show the interquartile range of the hourly data. The panel includes the mean of the simulated and observed organic nitrates. Diurnal cycles of the OH-initiated, NO3-initiated and second-generation organic nitrate concentrations are shown as the stacked bars.
Figure 4 compares the median diurnal cycle of observed total organic nitrates from the SOAS campaign to the model simulation during the simulation period. Total organic nitrates in both the gas and particle phase were measured by TD-LIF (thermal dissociation laser-induced fluorescence, Day et al., 2002). Temporal variability in the total organic nitrates for the entire time series is reproduced with little bias (r2=0.8 and normalized mean bias (NMB) = 32 %). Although the mean of the simulated organic nitrates (0.26±0.19) slightly overestimates the mean of the observations (0.20±0.1), the medians are found to be within variability of the observations. The simulated mean of total RONO2 in this study is in the range of two other recent modeling studies over the southeastern United States in summer 2013 that simulated 200 ppt (Fisher et al., 2016) and 270 ppt (Li et al., 2018). However, in both of these studies RONO2 derived from anthropogenic VOC precursors were not included. In our simulation, these organic nitrates represent ∼20 % of total RONO2. Specific sources of the differences include the slightly smaller yield of 10 % for isoprene nitrates and application of a 3 h hydrolysis lifetime only for ISOPNB in Li et al. (2018). Fisher et al. (2016) apply a faster hydrolysis rate (1 h) for all organic nitrates and a lower yield (9 % for isoprene nitrates).
The highest bias in the model median values and variability is observed after sunset to sunrise, which is likely caused by mismatch in vertical turbulent mixing in the simulated and actual boundary layers. Inclusion of hydrolysis as a possible fate for tertiary organic nitrates results in significant improvement of the simulations compared to the observations (not shown here). Tertiary nitrates have shorter lifetime against hydrolysis under atmospheric conditions, compared to the lifetime against deposition (Fig. S1 in the Supplement), making them the most important sink of nitrates. Li et al. (2018) also showed, by introducing the hydrolysis of ISOPNB, the model relative bias of total RONO2 was reduced by 18 % during ICARTT (International Consortium for Atmospheric Research on Transport and Transformation, summer 2004) over the southeastern United States.
Diurnal cycles of measured and simulated RONO2 have maximum values at 320 and 370 ppt around 10:00 CDT, respectively, with a slow decline through the rest of the day. Throughout the night, the mixing ratios were observed and modeled to remain nearly constant. Around 10:00, when the highest total organic nitrates are observed during SOAS, the simulated OH-initiated and second-generation organic nitrate concentrations both reach their maximum. Second-generation nitrates do not show sharp variability over day and night because of their longer lifetime but they do slightly increase after sunrise (around 07:00). OH-initiated organic nitrates that can remain in the residual layer overnight contribute to the total organic nitrate during the morning. At sunrise, when OH and NO begin to increase (Fig. S2 in the Supplement), OH-initiated organic nitrates increase until they reach their maximum at around 10:00. In contrast, NO3-initiated organic nitrates reach their peak mixing ratio before sunrise, and, immediately after sunrise, they decline sharply to a minimum concentration during the day. As the sun sets, NO drops to near zero and NO3 production initiates the formation of organic nitrates. OH- and NO3-initiated reactions occur out of phase in the diurnal cycle, resulting in the relatively flat diurnal profile for total organic nitrate throughout the night. Observations of individual molecules are predicted to have more strongly varying diurnal cycles, consistent with observations (Xiong et al., 2015).
3.2 Organic nitrate composition
The composition of the simulated organic nitrates by our model during SOAS at the Centreville site is shown in Fig. 5a. Monoterpene nitrates are calculated to be one-third of total organic nitrates, which is comparable to the Browne et al. (2014) calculation for boreal regions of North America. We define total isoprene-derived nitrates in WRF-Chem as the sum of isoprene hydroxy nitrates, isoprene carbonyl nitrates, MVK and MACR nitrates, isoprene nitrooxy hydroperoxides, ethanal nitrate, propanone nitrate and multifunctional isoprene nitrates. We find the contribution of the total isoprene-derived nitrates to total organic nitrates to be 44 %. This is consistent with the range of 25 %–50 % observed from SEAC4RS airborne measurements taken onboard the NASA DC-8 in August–September 2013 over the southeastern United States (Fisher et al., 2016). However, it is in contrast to other recent modeling studies over the southeastern United States by Mao et al. (2013) and Xie et al. (2013) that suggested, respectively, more than 90 % and 60 % of total organic nitrates are from isoprene oxidation. This discrepancy is likely due to the simulated longer lifetime of these nitrates as well as omission of organic nitrates produced from monoterpenes and anthropogenic VOCs in those models.
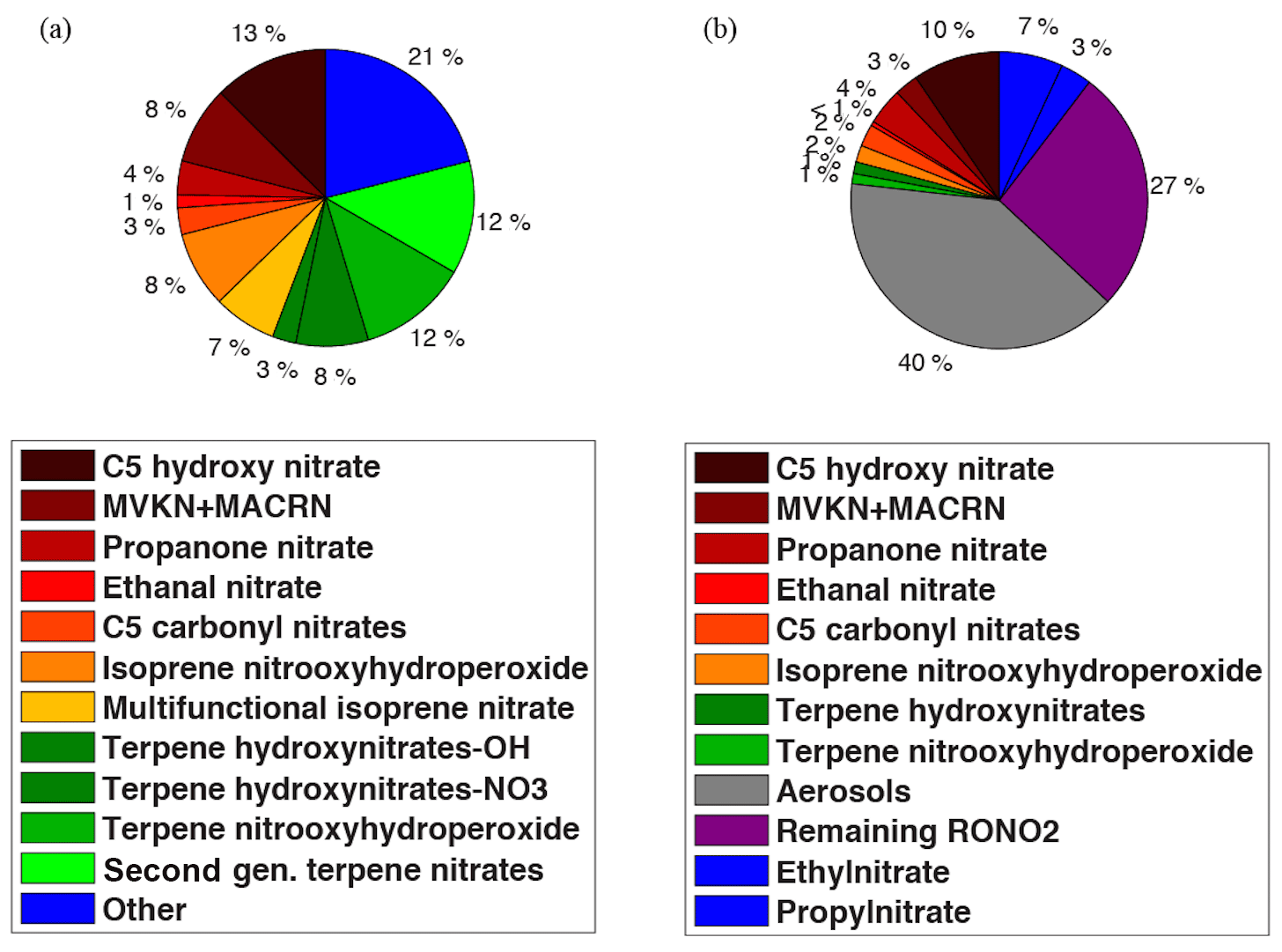
Figure 5The composition of the (a) simulated organic nitrates by WRF-Chem using RACM2_Berkeley2 and (b) observed organic nitrates during SOAS at CTR.
The observed RONO2 composition during SOAS is shown in Fig. 5b. The sums of the individual isomers of isoprene nitrates, terpene hydroxy nitrates and terpene nitrooxy hydroperoxides were measured in the gas phase by chemical ionization time-of-flight mass spectrometry using CF3O− reagent ion (Crounse et al., 2006; Schwantes et al., 2015; Teng et al., 2015), and ethyl and isopropyl nitrates were measured by gas chromatography–mass spectrometry (de Gouw et al., 2003). Similar to the model results, the largest contributions to the total organic nitrates in the observations are isoprene oxidation products, which represent 22 % against 44 % in the model. Carbonyl isoprene nitrates including ICN, ETHLN and PROPNN as a fraction of total RONO2 (8 % in the model and <7 % in the observations) and their concentrations (Fig. S3a and b in the Supplement) are reproduced well by the model. However, the model overestimates the fraction of RONO2 that is isoprene hydroxy nitrates and MVKN + MACRN (21 % modeled vs. 13 % observed). Isoprene hydroxy nitrates from NO3-initiated chemistry are a small portion of the total simulated isoprene hydroxy nitrates (∼15 %). The difference between the modeled and observed contribution of isoprene hydroxy nitrates to total organic nitrates is thus more likely a result of differences between the modeled and observed nitrates that are products of OH-initiated chemistry. Insights from very recent studies by Teng et al. (2017) and Wennberg et al. (2018) suggest a larger Ftertiary for OH-initiated isoprene hydroxy nitrates than the value we calculated from MCM. That suggests a larger fraction of these nitrates is subject to hydrolysis and thus it perhaps explains part of the discrepancy between the model simulations and observations.
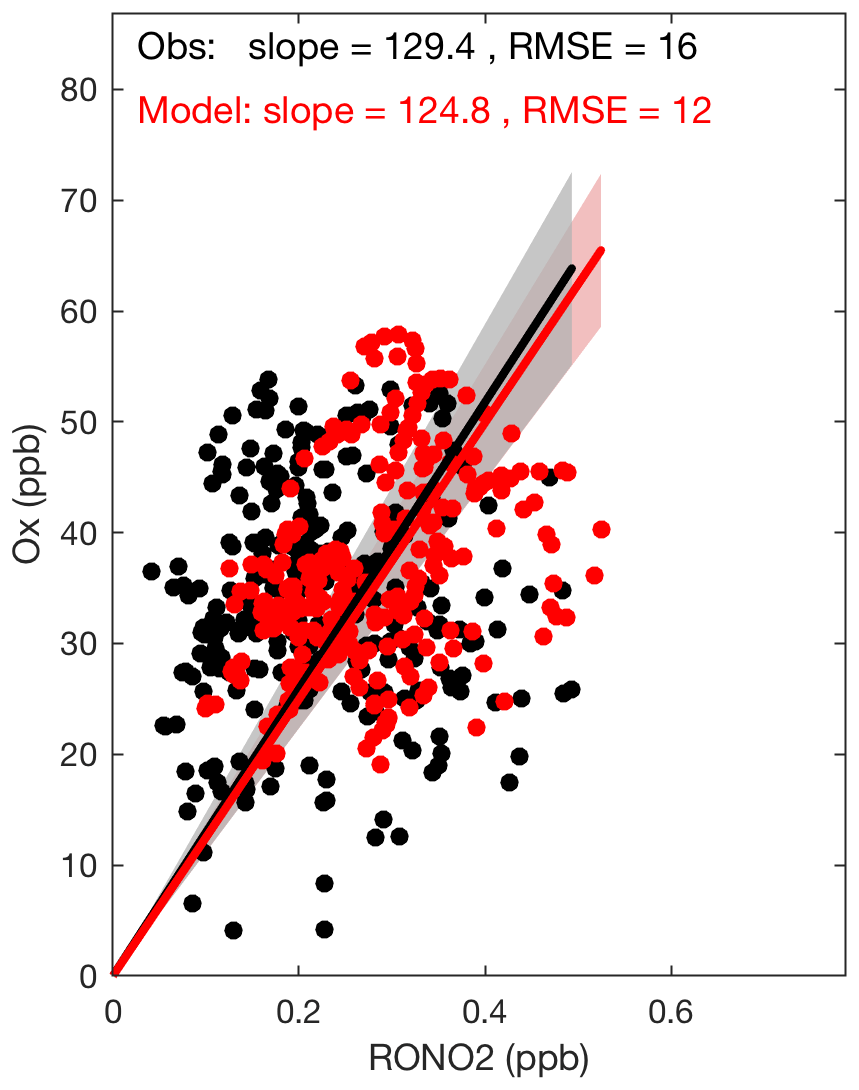
Figure 6The modeled and observed correlations between Ox () and organic nitrate concentrations during daytime at SOAS. The lines indicate linear regression (intercept fixed at 0) and confidence intervals. The panel includes slopes of the lines and root mean square errors (RMSE).
The largest difference between the modeled and observed contribution of isoprene nitrates to total organic nitrates is due to the modeled gas-phase multifunctional isoprene nitrates and isoprene nitrooxy hydroperoxides. Aerosol- and gas-phase second-generation multifunctional isoprene nitrates and aerosol-phase isoprene nitrooxy hydroperoxides were not individually measured during SOAS. Instead, total aerosol-phase organic nitrates were measured by TD-LIF, using an activated charcoal denuder to remove gas-phase organic nitrates and found to contribute around 40 % of total organic nitrates at the SOAS CTR site (Fig. 5b). Ng et al. (2008) and Rollins et al. (2009) found isoprene oxidation can form 4 %–23 % nitrate aerosol yields and showed multifunctional nitrates to be a dominant nitrate aerosol. If the isoprene nitrooxy hydroperoxides are favored to partition to aerosol this would explain the model-measurement discrepancy for the calculated contribution of multifunctional isoprene nitrates and isoprene nitrooxy hydroperoxide. They are simulated in the gas phase using RACM2_Berkeley2 but we might interpret them as contributing to particle-phase organic nitrate. That is consistent with the Lee et al. (2016) finding from observations of speciated particle organic nitrates during the SOAS campaign. They showed particle isoprene nitrates have a dominant contribution from highly functionalized isoprene nitrates containing between six and eight oxygen atoms.
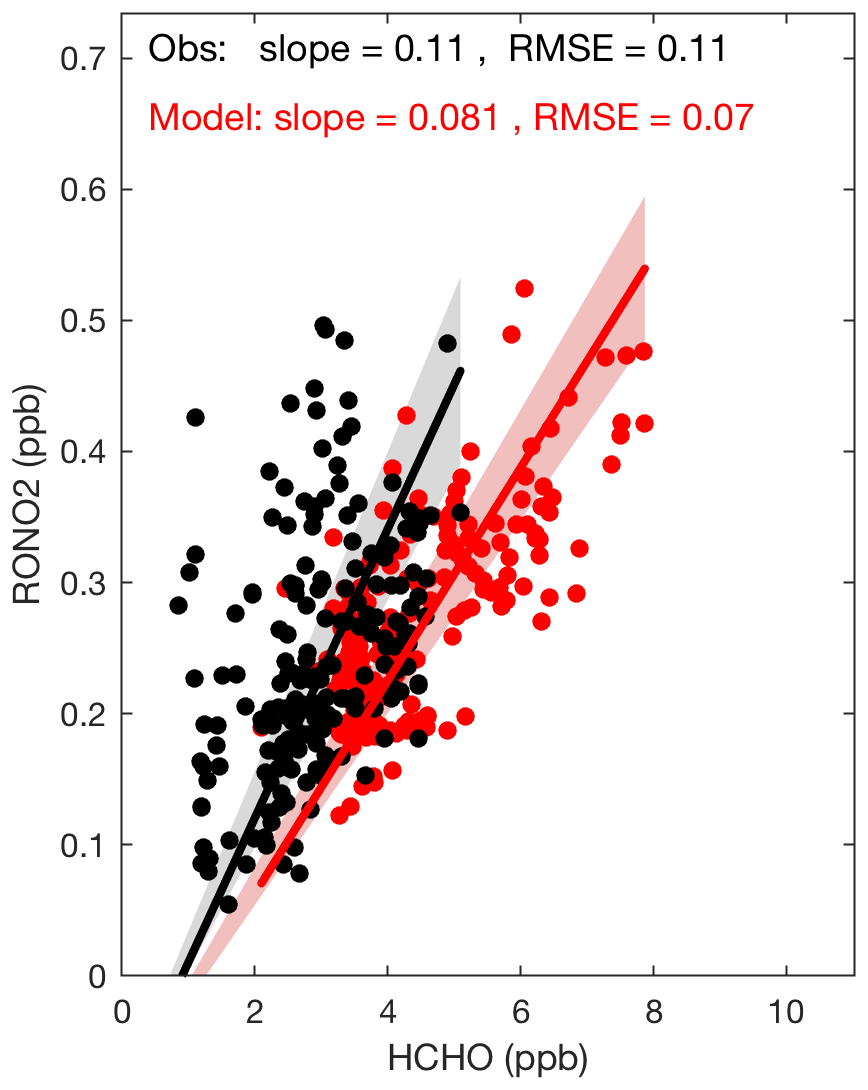
Figure 7The modeled and observed correlations between CH2O and organic nitrates during daytime at SOAS. The slope shows the best-fit line, with an intercept allowed to differ from zero to consider the possibility of background CH2O. The panel includes slopes of the lines and root mean square errors (RMSE).
Nitrate aerosol yields for monoterpene oxidation reactions from different laboratory chamber experiments, field measurements and modeling studies have been reported to be very high (up to 100 %) (Russell and Allen, 2005; Fry et al., 2009, 2011, 2013; Pye et al., 2015; Boyd et al., 2015). Among monoterpene nitrates, NO3-initiated nitrates (Ayres et al., 2015) and functionalized nitrates (Lee et al., 2016) have been shown to be an especially significant fraction of the total particle organic nitrate source at SOAS site. These findings imply that the remainder of the measured particle organic nitrates can be attributed to mono- or sesquiterpene-derived RONO2 including NO3-initiated terpene hydroxy nitrates, terpene nitrooxy hydroperoxides and multifunctional terpene nitrates, which are simulated and present in the gas phase in our mechanism. If we interpret the aerosol nitrates to be these compounds, then we find a rough correspondence between the model and observations (see Fig. 5a and b). However, Li et al. (2018) estimated a smaller contribution of gas-phase NO3-initiated monoterpene nitrates to total RONO2 due to a lower molar yield (10 % vs. 70 % for high-reactivity monoterpenes and 10 % for low-reactivity monoterpenes in this study). In contrast, due to other differences in the mechanism they found a larger contribution of OH-initiated monoterpene nitrates to total RONO2 than our finding in this study.
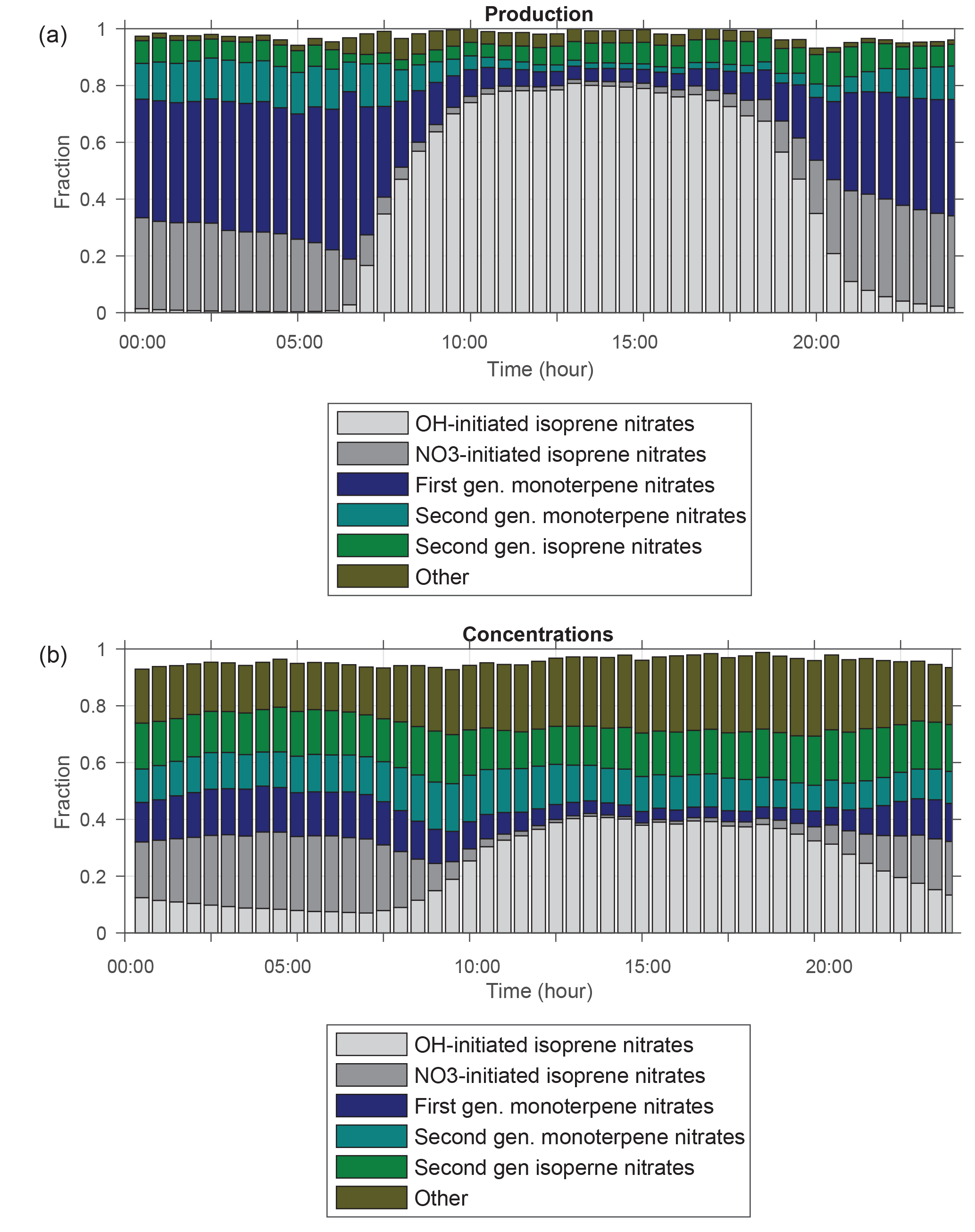
Figure 8Diurnal cycle of fractional organic nitrate (a) production and (b) concentrations simulated by WRF-Chem averaged over the boundary layer at the CTR site during SOAS.
In RACM2_Berkeley2, the contribution from organic nitrates of anthropogenic origin is simulated to be 21 % of total RONO2 (referred as Other at Fig. 5a). That is higher than the 10 % inferred from the observations of the measured anthropogenic organic nitrates at SOAS. Some caution should be taken in the interpretation of such a comparison, as the observations at SOAS do not represent the same species as the modeled ones and include only ethyl and propyl nitrates (Fig. 5b). In RACM2_Berkeley2, a wide range of the organic nitrates of anthropogenic origin (C1–C5 nitrates) is categorized into four groups including monofunctional saturated, multifunctional unsaturated, multifunctional saturated and aromatic-derived nitrates that are partitioned from the lumped precursors including alkanes, aromatics, alcohols and alkenes. However, the remaining unspeciated measured RONO2 contributes 27 % of the total organic nitrates observed by TD-LIF. One hypothesis to explain this difference is that the rest of the simulated organic nitrates of anthropogenic origin might be a portion of the unspecified measured RONO2.
3.3 Relationships between RONO2, Ox and CH2O
During daytime, ozone and organic nitrates are produced in a common reaction with branches that yield one or the other. Therefore, their observed and modeled correlation provides an additional constraint on our understanding of organic nitrates. The sum of O3 and NO2 is conserved on longer timescales than O3 alone; accordingly we use in this analysis. As shown in Fig. 6, during the daytime (from 08:00 to 18:00 local time) the modeled and observed correlations between Ox and RONO2 are nearly identical. A linear fit to the observations yields a line with slope of 129±4 ppbv(Ox) ppbv(RONO2)−1 and a fit to the model output yields 125±4 ppbv(Ox) ppbv(RONO2)−1.
This slope has typically been used to estimate the approximate branching ratio of the entire VOC mixture (αeff) with an assumption that photochemical production is rapid as compared to removal processes; αeff is inversely proportional to the Ox vs. RONO2 slope (Perring et al., 2013). The quantified α's in the laboratory for BVOCs are much higher than typical α's for anthropogenic VOCs (Perring et al., 2013, and references therein). Therefore, for regions like the southeastern United States where BVOCs dominate the VOC mixture, a much lower slope than our calculated value is expected. We conclude that the observed slope is reflecting the short lifetime of organic nitrates at SOAS.
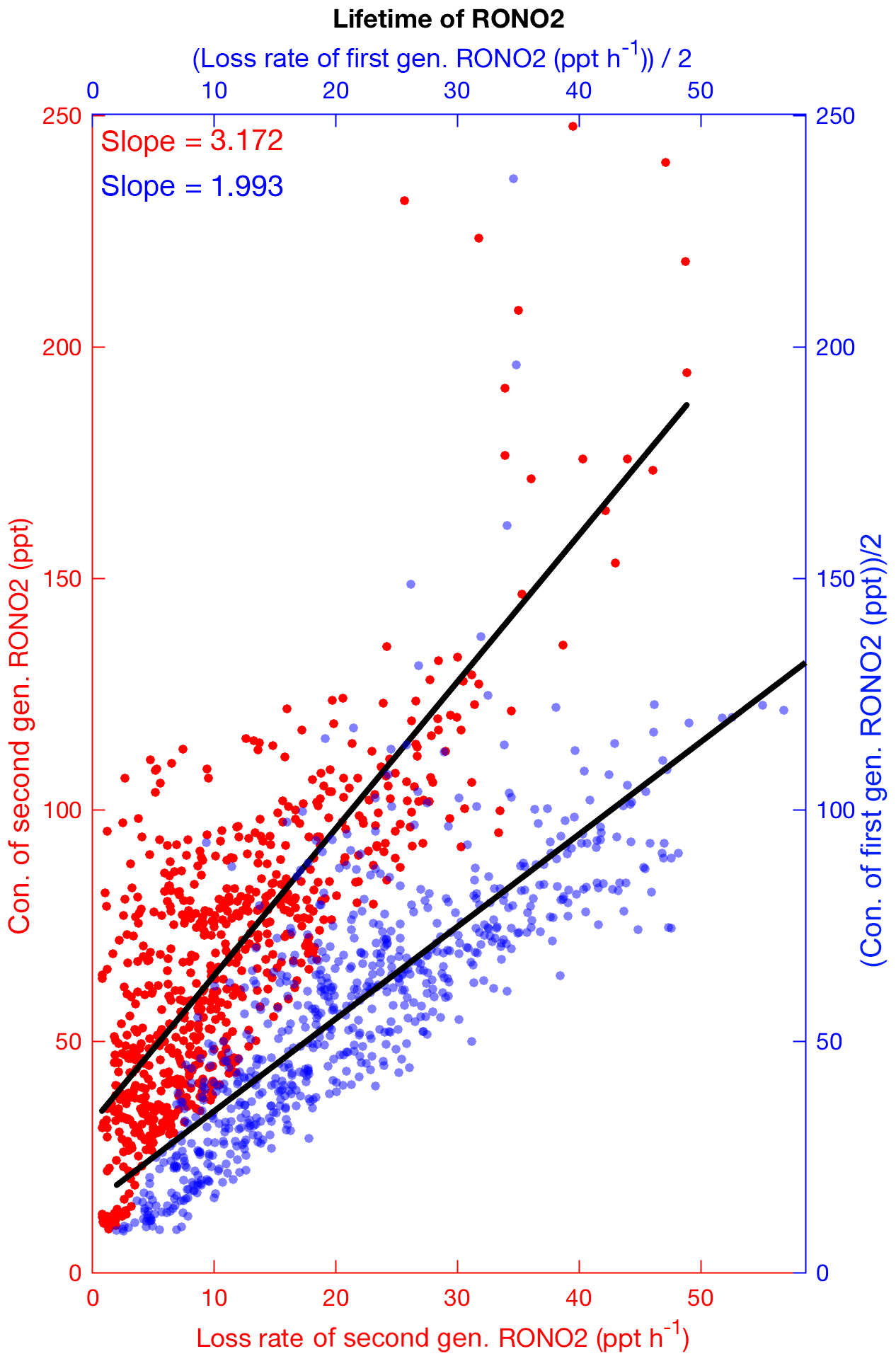
Figure 9The simulated concentrations of first (blue) and second (red) generation organic nitrates versus their loss rates during daytime at SOAS. Slopes of the linear fit give their lifetimes. The concentrations and loss rates of first-generation nitrates are divided by 2.

Figure 10Contribution of different fates to (a) the first and (b) second generation of isoprene and monoterpene nitrates loss.
Formaldehyde (CH2O) is another co-product to RONO2 and, as Perring et al. (2009b) discussed, the slope of the RONO2∕CH2O correlation is related to the ratio of the production of both species, as both have similar lifetimes (Perring et al., 2009b). We would expect the slope could provide a constraint on the yield of isoprene nitrates, especially since in much of the domain isoprene is the dominant source of both RONO2 and CH2O. Figure 7 shows the correlation between observed CH2O and RONO2 during SOAS. The slope of the best-fit line, with an intercept allowed to differ from zero to consider the possibility of background CH2O that mixes in from the free troposphere, is found to be 0.116, consistent with previous estimates by Perring et al. (2009b), who observed a slope of 0.119 during INTEX-NA (Intercontinental Chemical Transport Experiment) in 2004. The slope would imply an OH-initiated isoprene nitrate yield of 12 % (Perring et al., 2009b) if we use a lifetime of 1.7 h at SOAS for RONO2 as reported by Romer et al. (2016). This is nearly identical to the yield used in the mechanism described in this manuscript. However, the correlation of modeled CH2O and modeled total RONO2 has a smaller slope of 0.085. The discrepancy between the slopes from the simulated and observed data can be attributed to model overestimation of CH2O (Fig. S4 in the Supplement). In Fig. S5, we also provide additional model evaluation for isoprene and monoterpene concentrations.
3.4 Organic nitrate formation
Figure 8a shows the diurnal cycle of fractional RONO2 production simulated using RACM2_Berkeley2 averaged over the boundary layer at the CTR site during SOAS. During the day, production of organic nitrates is dominated by reaction of isoprene with OH. This is consistent with the OH reactivity (OHR) of individually measured compounds at SOAS which was dominated by reaction with isoprene (Kaiser et al., 2016; Romer et al., 2016). In contrast, the vast majority of RONO2 production at night is of monoterpene nitrates, which are formed as a result of NO3 oxidation of nighttime monoterpene emissions. NO3 chemistry of isoprene leading to isoprene nitrate formation is also found to be significant during the nighttime. This fraction from isoprene that accumulates in the boundary layer in late afternoons can reach 35 % of the total organic nitrates formed at night. In addition to investigating the relative importance of instantaneous organic nitrate production based on the VOC precursors, we calculate the fraction of isoprene nitrates produced from NO3 chemistry to the total isoprene nitrate production (∼44 %, Fig. S5 in the Supplement) over day and night of the entire modeling period confirming the relative importance of this pathway for producing isoprene nitrates versus OH oxidation of isoprene. This finding is consistent with the modeling result (∼40 %) from Xie et al. (2013).
The fraction of anthropogenic VOC-derived organic nitrates to total simulated production of organic nitrates is estimated to be negligible; however their contribution to the simulated concentrations of organic nitrates is much higher and reached up to 0.25 (Fig. 8b). This is due to their relatively long lifetime (>100 h lifetime to oxidation by 1×106 molecules cm−3 of OH at 298 K and a similarly long lifetime to deposition – Henry's law constant of ∼1 Matm−1; Browne et al., 2014, and references therein) that causes them to persist longer in the atmosphere. Similarly, the fraction of second-generation nitrates formed from oxidation of first-generation isoprene and monoterpene nitrates (Fig. 8a) is predicted to be ∼0.04, which is much less than the calculated contribution of their concentrations to total organic nitrate concentrations (∼0.3) (Fig. 8b). In the next subsection we will discuss more about the loss processes and lifetime of these organic nitrates.
3.5 Organic nitrate lifetime
We define the lifetime of organic nitrates as the concentration of RONO2 divided by the combined loss rate via all proposed loss mechanisms. The loss mechanisms include chemical loss processes (oxidation, photolysis and hydrolysis of RONO2) and deposition. The nighttime lifetime of organic nitrates might be longer than the daytime value (and might be similar to or longer than the length of a single night). Because of uncertainties associated with simulation of the boundary layer height and organic nitrate concentrations at nighttime, we focus on the daytime lifetime as a guide for thinking about the organic nitrate fate. Figure 9 shows the estimated daytime lifetime of 2 h for first-generation biogenic organic nitrates and a longer lifetime for the second-generation organic nitrates (3.2 h). Including organic nitrates from anthropogenic sources we estimate a fairly short overall lifetime of 2.6 h for total RONO2. This short lifetime results in less efficient transport of organic nitrates to the free troposphere and over large distances from sources. Using the SOAS field observations, Romer et al. (2016) suggested ∼1.7 h for the atmospheric lifetime of RONO2. They calculated the lifetime by the assumption that RONO2 are near steady state in the afternoon. If we constrain our calculation to 12:00–16:00 and give an intercept of 40 ppt as Romer et al. (2016) did, the overall estimated lifetime in the model is estimated to be 2.9 h, but using the production rates of organic nitrates instead of the loss rates (by assumption of the atmospheric steady state condition applied in Romer et al., 2016) our result remarkably shows very good agreement with their finding (identical value, Fig. S6 in the Supplement). GEOS-Chem simulations by Fisher et al. (2016) reported a similar short lifetime by assuming a hydrolysis lifetime of 1 h for all tertiary and non-tertiary nitrates and not including the longer-lived small alkyl nitrates. However, Li et al. (2018) estimated much longer lifetimes for individual nitrates except ISOPNB, which they assumed to be hydrolyzed.
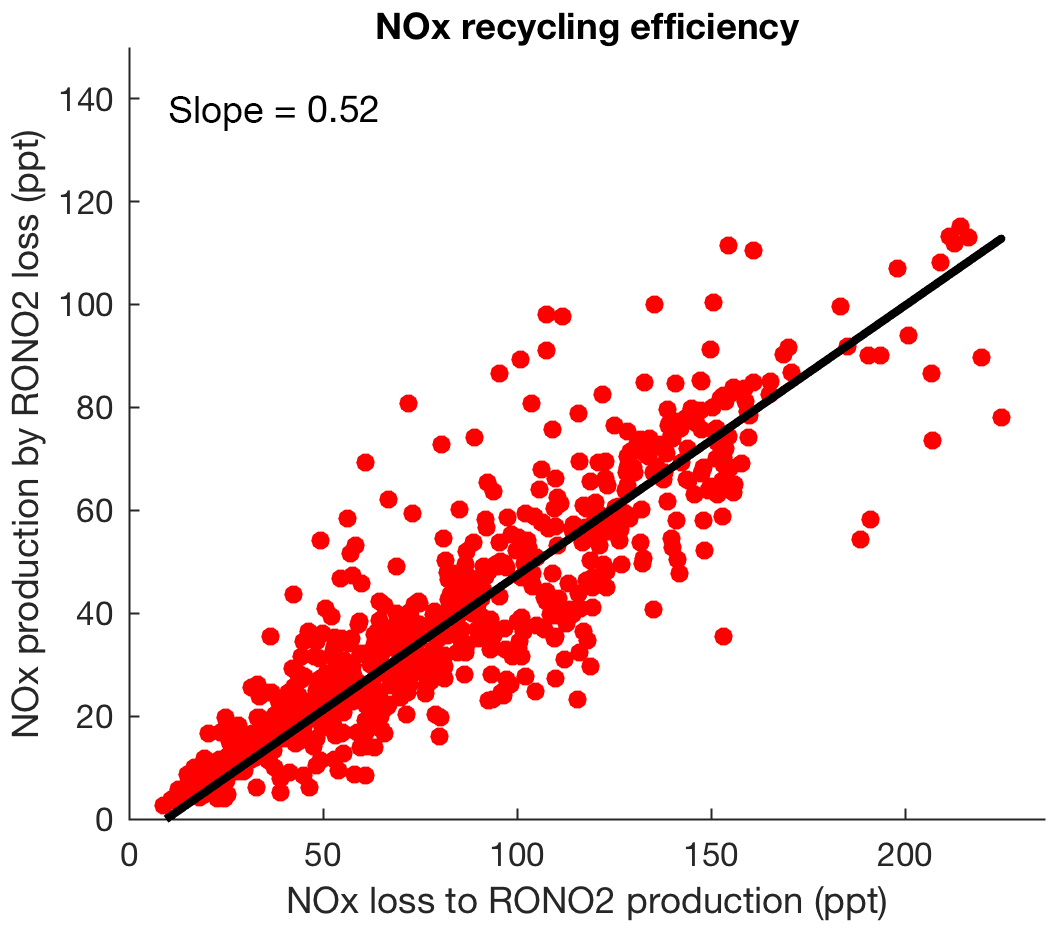
Figure 11The simulated instantaneous production of NOx from loss of organic nitrates versus the instantaneous loss of NOx to production of organic nitrates. The slope shows the NOx recycling efficiency.
Accurate determination of the lifetime of organic nitrates is a major challenge for assessing the influence of organic nitrates on atmospheric chemistry. However, the estimated lifetime of ∼3 h for organic nitrates found here, as well as in other studies over the southeastern United States (Perring et al., 2009b; Romer et al., 2016; Fisher et al., 2016), is less than the range of NOx lifetimes (5.5–11 h) calculated by observational studies (e.g., Valin et al., 2013; Romer et al., 2016). Organic nitrates should therefore generally be categorized as short-lived NOx reservoirs, which remove NOx in a plume, but act as a source of NOx in remote regions.
3.6 NOx recycling efficiency
To determine the fraction of NOx converted to RONO2 and then released back to the gas phase as NOx, the relative importance of different loss pathways of organic nitrates must be known. Oxidation and photolysis of organic nitrates recycle NOx but hydrolysis and deposition cause permanent removal of NOx from the atmosphere. Recent field studies suggested that isoprene nitrates are removed quickly by dry deposition (Nguyen et al., 2015) and some have concluded that deposition is the primary sink of nitrates (Rosen et al., 2004; Horii et al., 2006; Horowitz et al., 2007), while others estimated that oxidation or ozonolysis is the dominant loss mechanism of isoprene nitrates (Shepson et al., 1996; Ito et al., 2007; Perring et al., 2009a; Browne et al., 2013). Similar uncertainty for the fate and dominant loss processes of monoterpene nitrates was found by Browne et al. (2014). Here, we update possible fates of organic nitrates in WRF-Chem from recent findings including photolysis (Müller et al., 2014), oxidation and ozonolysis (Lee et al., 2014), deposition (Nguyen et al., 2015) and hydrolysis (Boyd et al., 2015) and then estimate the contribution of different fates to first- and second-generation isoprene and monoterpene nitrates (Fig. 10a and b). We note that our calculation represents the loss of the nitrate functionality and does not include the fraction of loss processes of first-generation organic nitrates by oxidation and ozonolysis that retain the nitrate functionality by forming second-generation organic nitrates.
Figure 10b shows that the loss of second-generation organic nitrates is dominated by deposition (60 %), causing permanent loss of NOx. That is due to the assumed rapid depositional loss of second-generation monoterpene nitrates (deposited as fast as HNO3) in this study following Browne et al. (2014). Fractional contributions of photolysis (∼25 %) and oxidation (∼15 %) are not negligible and are much larger than those estimated by Browne et al. (2014), which is a consequence of using the rapid photolysis rates of PROPNN and ETHLN as second-generation isoprene nitrates (Müller et al., 2014). In contrast, the loss of first-generation nitrates occurs largely by the sum of chemical mechanisms that recycle NOx to the atmosphere: reaction with ozone (24 %), OH (11 %) and photolysis (20 %), with additional loss by deposition (15 %) and hydrolysis (∼29 %). Fisher et al. (2016) predicted much larger losses of RONO2 by aerosol hydrolysis (∼60 % of total nitrate losses), reflecting the short lifetime of nitrates with respect to hydrolysis applied in their study for all nitrates. However, with our representation of hydrolysis of nitrates in aqueous solutions (slower rate and only tertiary nitrates), hydrolysis is still important (the calculated loss rate for nitrates is ∼3 to 11 pptv h−1), accounting for one-third of the organic nitrate loss and leading to a large increase in HNO3 production in the atmosphere. Assessing the impact of hydrolysis of nitrates on the budget of nitric acid is beyond the scope of this work. We note that the relative contribution of nitrate hydrolysis in aqueous solutions differs widely for individual RONO2 species due to their different structures.
Including loss of RONO2 from anthropogenic sources, we find that loss of overall RONO2 via hydrolysis with an additional contribution from deposition becomes comparable to loss via other processes that return NOx to the atmosphere. Figure 11 shows the NOx recycling efficiency, defined as the ratio between the instantaneous production of NOx from loss of organic nitrates and the instantaneous loss of NOx to production of organic nitrates. Since chemical degradation of nitric acid is much slower than deposition, the slope of 0.52 is interpreted as the fraction of the sequestered NOx that can be exported away from its emission region and released downwind through organic nitrate chemistry. Our finding is consistent with a recycling of about half of isoprene nitrates to NOx calculated by Paulot et al. (2009a) and Horowitz et al. (2007). Of this total, we calculated ∼38 % of NOx cycled back relatively quickly while first-generation nitrates are oxidizing and producing second-generation nitrates.
The lifetimes of organic nitrates with respect to hydrolysis, oxidation and deposition play an important role in the NOx budget and formation of O3 and secondary organic aerosols. Analyses from recent field studies in the southeastern United States found a lifetime of ∼2–3 h for organic nitrates. By incorporating new findings from recent laboratory, field and modeling studies into a gas-phase mechanism we provide a state-of-the-science representation of expanded organic nitrates in the WRF-Chem model. Using the updated model we are able to reproduce a short organic nitrate lifetime (2.6 h), similar to that observed during SOAS.
After adding hydrolysis as a possible fate of tertiary gas-phase biogenic organic nitrates in our mechanism and in combination with all other loss mechanisms, we find that the lifetime of second-generation organic nitrates is longer that the lifetime of first-generation nitrates. We find dry deposition is the dominant loss process for second-generation organic nitrates and chemical mechanisms of ozonolysis, photolysis and oxidation that can recycle NOx to the atmosphere have a more important role in loss of first-generation organic nitrates from the atmosphere. The contribution of tertiary nitrate hydrolysis to total organic nitrate removal from the atmosphere is found to be 30 %. We find, therefore, that 52 % of the NOx sequestered by production of organic nitrates can be cycled back to the atmosphere.
To accurately estimate organic nitrate lifetime, the production, loss and fate of these compounds must be well constrained. Evaluation of our updated mechanism using SOAS observations in summer 2013 indicates the model represents much of the important chemistry governing organic nitrates. We show that the simulated concentrations of total organic nitrates, correlations with CH2O and ozone, and the contribution of individual RONO2 to total organic nitrates are in fairly good agreement with observations at the SOAS CTR ground site. We find the largest difference between the modeled and observed contributions of individual organic nitrate compounds to total RONO2 is for highly functionalized isoprene nitrates and monoterpene nitrates. We attribute this difference to possible high aerosol yields of these organic nitrate species, which are represented in the gas phase in our mechanism. Future analysis for developing a complete representation of organic nitrate chemistry including an organic nitrate aerosol formation mechanism, either from aqueous-phase uptake or vapor-pressure partitioning onto preexisting organic aerosol, in addition to the detailed gas-phase mechanism described here will benefit the model approximation.
Measurements from the SOAS campaign are available at https://esrl.noaa.gov/csd/groups/csd7/measurements/2013senex/Ground/DataDownload (SOAS Science Team, 2013).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-15419-2018-supplement.
AZ and RC designed the study. AZ carried out the simulations and performed the data analysis. PSR, TN, KS and FNK contributed their insights on the field measurements. AZ wrote the manuscript with feedback from all co-authors. RC supervised the project.
The authors declare that they have no conflict of interest.
We gratefully acknowledge support from NSF grants AGS-1352972, AGS-1247421
and AGS-1628530; NOAA Office of Global Programs grant NA13OAR4310067; and NASA
grant NNX 15AE37G. We thank Allen Goldstein and Pawel Konrad Misztal for
PTRTOFMS VOC data and William Brune for OH data. We also thank the SOAS field
campaign team including Ann Marie Carlton.
Edited by: James Roberts
Reviewed by: two anonymous referees
Ackermann, I. J., Hass, H., Memmesheimer, M., Ebel, A., Binkowski, F. S., and Shankar, U.: Modal Aerosol Dynamics model for Europe: Development and first applications, Atmos. Environ., 32, 2981–2999, https://doi.org/10.1016/S1352-2310(98)00006-5, 1998.
Arey, J., Aschmann, S. M., Kwok, E. S. C., and Atkinson, R.: Alkyl Nitrate, Hydroxyalkyl Nitrate, and Hydroxycarbonyl Formation from the NOx-Air Photooxidations of C5-C8 n-Alkanes, J. Phys. Chem. A, 105, 1020–1027, https://doi.org/10.1021/jp003292z, 2001.
Aschmann, S. M., Atkinson, R., and Arey, J.: Products of reaction of OH radicals with α-pinene, J. Geophys. Res.-Atmos., 107, 4191, https://doi.org/10.1029/2001JD001098, 2002.
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015.
Bates, K. H., Nguyen, T. B., Teng, A. P., Crounse, J. D., Kjaergaard, H. G., Stoltz, B. M., Seinfeld, J. H., and Wennberg, P. O.: Production and Fate of C4 Dihydroxycarbonyl Compounds from Isoprene Oxidation, J. Phys. Chem. A, 120, 106–117, https://doi.org/10.1021/acs.jpca.5b10335, 2016.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/C2CS35181A, 2012.
Browne, E. C. and Cohen, R. C.: Effects of biogenic nitrate chemistry on the NOx lifetime in remote continental regions, Atmos. Chem. Phys., 12, 11917–11932, https://doi.org/10.5194/acp-12-11917-2012, 2012.
Browne, E. C., Min, K.-E., Wooldridge, P. J., Apel, E., Blake, D. R., Brune, W. H., Cantrell, C. A., Cubison, M. J., Diskin, G. S., Jimenez, J. L., Weinheimer, A. J., Wennberg, P. O., Wisthaler, A., and Cohen, R. C.: Observations of total RONO2 over the boreal forest: NOx sinks and HNO3 sources, Atmos. Chem. Phys., 13, 4543–4562, https://doi.org/10.5194/acp-13-4543-2013, 2013.
Browne, E. C., Wooldridge, P. J., Min, K.-E., and Cohen, R. C.: On the role of monoterpene chemistry in the remote continental boundary layer, Atmos. Chem. Phys., 14, 1225–1238, https://doi.org/10.5194/acp-14-1225-2014, 2014.
Carlton, A. G., de Gouw, J., Jimenez, J. L., Ambrose, J. L., Attwood, A. R., Brown, S., Baker, K. R., Brock, C., Cohen, R. C., Edgerton, S., Farkas, C. M., Farmer, D., Goldstein, A. H., Gratz, L., Guenther, A., Hunt, S., Jaeglé, L., Jaffe, D. A., Mak, J., McClure, C., Nenes, A., Nguyen, T. K., Pierce, J. R., de Sa, S., Selin, N. E., Shah, V., Shaw, S., Shepson, P. B., Song, S., Stutz, J., Surratt, J. D., Turpin, B. J., Warneke, C., Washenfelder, R. A., Wennberg, P. O., and Zhou, X.: Synthesis of the Southeast Atmosphere Studies: Investigating Fundamental Atmospheric Chemistry Questions, B. Am. Meteorol. Soc., 99, 547–567, https://doi.org/10.1175/BAMS-D-16-0048.1, 2018.
Carter, W. P. L. and Atkinson, R.: Alkyl nitrate formation from the atmospheric photoxidation of alkanes; a revised estimation method, J. Atmos. Chem., 8, 165–173, https://doi.org/10.1007/BF00053721, 1989.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/C1CP21330J, 2011.
Damian, V., Sandu, A., Damian, M., Potra, F., and Carmichael, G. R.: The kinetic preprocessor KPP-a software environment for solving chemical kinetics, Comput. Chem. Eng., 26, 1567–1579, https://doi.org/10.1016/S0098-1354(02)00128-X, 2002.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
Day, D. A., Wooldridge, P. J., Dillon, M. B., Thornton, J. A., and Cohen, R. C.: A thermal dissociation laser-induced fluorescence instrument for in situ detection of NO2, peroxy nitrates, alkyl nitrates, and HNO3, J. Geophys. Res.-Atmos., 107, 4046, https://doi.org/10.1029/2001JD000779, 2002.
de Gouw, J. A., Goldan, P. D., Warneke, C., Kuster, W. C., Roberts, J. M., Marchewka, M., Bertman, S. B., Pszenny, A. a. P., and Keene, W. C.: Validation of proton transfer reaction-mass spectrometry (PTR-MS) measurements of gas-phase organic compounds in the atmosphere during the New England Air Quality Study (NEAQS) in 2002, J. Geophys. Res.-Atmos., 108, 4682, https://doi.org/10.1029/2003JD003863, 2003.
Emmons, L. K., Walters, S., Hess, P. G., Lamarque, J.-F., Pfister, G. G., Fillmore, D., Granier, C., Guenther, A., Kinnison, D., Laepple, T., Orlando, J., Tie, X., Tyndall, G., Wiedinmyer, C., Baughcum, S. L., and Kloster, S.: Description and evaluation of the Model for Ozone and Related chemical Tracers, version 4 (MOZART-4), Geosci. Model Dev., 3, 43–67, https://doi.org/10.5194/gmd-3-43-2010, 2010.
Farmer, D. K. and Cohen, R. C.: Observations of HNO3, ΣAN, ΣPN and NO2 fluxes: evidence for rapid HOx chemistry within a pine forest canopy, Atmos. Chem. Phys., 8, 3899–3917, https://doi.org/10.5194/acp-8-3899-2008, 2008.
Fiore, A. M., Horowitz, L. W., Purves, D. W., Levy, H., Evans, M. J., Wang, Y., Li, Q., and Yantosca, R. M.: Evaluating the contribution of changes in isoprene emissions to surface ozone trends over the eastern United States, J. Geophys. Res.-Atmos., 110, D12303, https://doi.org/10.1029/2004JD005485, 2005.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Rohrer, F., Tillmann, R., Wahner, A., Wooldridge, P. J., and Cohen, R. C.: SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 11, 3879–3894, https://doi.org/10.5194/acp-11-3879-2011, 2011.
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013.
Grell, G. A., Peckham, S. E., Schmitz, R., McKeen, S. A., Frost, G., Skamarock, W. C., and Eder, B.: Fully coupled “online” chemistry within the WRF model, Atmos. Environ., 39, 6957–6975, https://doi.org/10.1016/j.atmosenv.2005.04.027, 2005.
Guenther, A.: Biological and Chemical Diversity of Biogenic Volatile Organic Emissions into the Atmosphere, Int. Sch. Res. Not., 2013, e786290, https://doi.org/10.1155/2013/786290, 2013.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Hansen, D. A., Edgerton, E. S., Hartsell, B. E., Jansen, J. J., Kandasamy, N., Hidy, G. M., and Blanchard, C. L.: The Southeastern Aerosol Research and Characterization Study: Part 1 – Overview, J. Air Waste Manage. Assoc., 53, 1460–1471, https://doi.org/10.1080/10473289.2003.10466318, 2003.
Horii, C. V., William Munger, J., Wofsy, S. C., Zahniser, M., Nelson, D., and Barry McManus, J.: Atmospheric reactive nitrogen concentration and flux budgets at a Northeastern U.S. forest site, Agric. For. Meteorol., 136, 159–174, https://doi.org/10.1016/j.agrformet.2006.03.005, 2006.
Horowitz, L. W., Fiore, A. M., Milly, G. P., Cohen, R. C., Perring, A., Wooldridge, P. J., Hess, P. G., Emmons, L. K., and Lamarque, J.-F.: Observational constraints on the chemistry of isoprene nitrates over the eastern United States, J. Geophys. Res.-Atmos., 112, D12S08, https://doi.org/10.1029/2006JD007747, 2007.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Ito, A., Sillman, S., and Penner, J. E.: Effects of additional nonmethane volatile organic compounds, organic nitrates, and direct emissions of oxygenated organic species on global tropospheric chemistry, J. Geophys. Res.-Atmos., 112, D06309, https://doi.org/10.1029/2005JD006556, 2007.
Ito, A., Sillman, S., and Penner, J. E.: Global chemical transport model study of ozone response to changes in chemical kinetics and biogenic volatile organic compounds emissions due to increasing temperatures: Sensitivities to isoprene nitrate chemistry and grid resolution, J. Geophys. Res.-Atmos., 114, D09301, https://doi.org/10.1029/2008JD011254, 2009.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Kaiser, J., Skog, K. M., Baumann, K., Bertman, S. B., Brown, S. B., Brune, W. H., Crounse, J. D., de Gouw, J. A., Edgerton, E. S., Feiner, P. A., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K. F., St. Clair, J. M., Teng, A. P., Toma, S., Wennberg, P. O., Wild, R. J., Zhang, L., and Keutsch, F. N.: Speciation of OH reactivity above the canopy of an isoprene-dominated forest, Atmos. Chem. Phys., 16, 9349–9359, https://doi.org/10.5194/acp-16-9349-2016, 2016.
Kwan, A. J., Chan, A. W. H., Ng, N. L., Kjaergaard, H. G., Seinfeld, J. H., and Wennberg, P. O.: Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 12, 7499–7515, https://doi.org/10.5194/acp-12-7499-2012, 2012.
Laughner, J. L. and Cohen, R. C.: Quantification of the effect of modeled lightning NO2 on UV-visible air mass factors, Atmos. Meas. Tech., 10, 4403–4419, https://doi.org/10.5194/amt-10-4403-2017, 2017.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., Gouw, J. de, Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Lee, L., Teng, A. P., Wennberg, P. O., Crounse, J. D., and Cohen, R. C.: On Rates and Mechanisms of OH and O3 Reactions with Isoprene-Derived Hydroxy Nitrates, J. Phys. Chem. A, 118, 1622–1637, https://doi.org/10.1021/jp4107603, 2014.
Li, J., Mao, J., Fiore, A. M., Cohen, R. C., Crounse, J. D., Teng, A. P., Wennberg, P. O., Lee, B. H., Lopez-Hilfiker, F. D., Thornton, J. A., Peischl, J., Pollack, I. B., Ryerson, T. B., Veres, P., Roberts, J. M., Neuman, J. A., Nowak, J. B., Wolfe, G. M., Hanisco, T. F., Fried, A., Singh, H. B., Dibb, J., Paulot, F., and Horowitz, L. W.: Decadal changes in summertime reactive oxidized nitrogen and surface ozone over the Southeast United States, Atmos. Chem. Phys., 18, 2341–2361, https://doi.org/10.5194/acp-18-2341-2018, 2018.
Liu, Y. J., Herdlinger-Blatt, I., McKinney, K. A., and Martin, S. T.: Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation, Atmos. Chem. Phys., 13, 5715–5730, https://doi.org/10.5194/acp-13-5715-2013, 2013.
Lockwood, A. L., Shepson, P. B., Fiddler, M. N., and Alaghmand, M.: Isoprene nitrates: preparation, separation, identification, yields, and atmospheric chemistry, Atmos. Chem. Phys., 10, 6169–6178, https://doi.org/10.5194/acp-10-6169-2010, 2010.
Mao, J., Paulot, F., Jacob, D. J., Cohen, R. C., Crounse, J. D., Wennberg, P. O., Keller, C. A., Hudman, R. C., Barkley, M. P., and Horowitz, L. W.: Ozone and organic nitrates over the eastern United States: Sensitivity to isoprene chemistry, J. Geophys. Res.-Atmos., 118, 2013JD020231, https://doi.org/10.1002/jgrd.50817, 2013.
Mao, J., Carlton, A., Cohen, R. C., Brune, W. H., Brown, S. S., Wolfe, G. M., Jimenez, J. L., Pye, H. O. T., Lee Ng, N., Xu, L., McNeill, V. F., Tsigaridis, K., McDonald, B. C., Warneke, C., Guenther, A., Alvarado, M. J., de Gouw, J., Mickley, L. J., Leibensperger, E. M., Mathur, R., Nolte, C. G., Portmann, R. W., Unger, N., Tosca, M., and Horowitz, L. W.: Southeast Atmosphere Studies: learning from model-observation syntheses, Atmos. Chem. Phys., 18, 2615–2651, https://doi.org/10.5194/acp-18-2615-2018, 2018.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
Middleton, P., Stockwell, W. R., and Carter, W. P. L.: Aggregation and analysis of volatile organic compound emissions for regional modeling, Atmospheric Environ. Part Gen. Top., 24, 1107–1133, https://doi.org/10.1016/0960-1686(90)90077-Z, 1990.
Mills, G. P., Hiatt-Gipson, G. D., Bew, S. P., and Reeves, C. E.: Measurement of isoprene nitrates by GCMS, Atmos. Meas. Tech., 9, 4533–4545, https://doi.org/10.5194/amt-9-4533-2016, 2016.
Müller, J.-F., Peeters, J., and Stavrakou, T.: Fast photolysis of carbonyl nitrates from isoprene, Atmos. Chem. Phys., 14, 2497–2508, https://doi.org/10.5194/acp-14-2497-2014, 2014.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014.
Nguyen, T. B., Crounse, J. D., Teng, A. P., Clair, J. M. S., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, 392–401, https://doi.org/10.1073/pnas.1418702112, 2015.
Ott, L. E., Pickering, K. E., Stenchikov, G. L., Allen, D. J., DeCaria, A. J., Ridley, B., Lin, R.-F., Lang, S., and Tao, W.-K.: Production of lightning NOx and its vertical distribution calculated from three-dimensional cloud-scale chemical transport model simulations, J. Geophys. Res.-Atmos., 115, D04301, https://doi.org/10.1029/2009JD011880, 2010.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kroll, J. H., Seinfeld, J. H., and Wennberg, P. O.: Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 9, 1479–1501, https://doi.org/10.5194/acp-9-1479-2009, 2009a.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., Clair, J. M. S., Seinfeld, J. H., and Wennberg, P. O.: Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009b.
Paulot, F., Henze, D. K., and Wennberg, P. O.: Impact of the isoprene photochemical cascade on tropical ozone, Atmos. Chem. Phys., 12, 1307–1325, https://doi.org/10.5194/acp-12-1307-2012, 2012.
Peeters, J. and Müller, J.-F.: HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: experimental evidence and global impact, Phys. Chem. Chem. Phys., 12, 14227–14235, https://doi.org/10.1039/C0CP00811G, 2010.
Peeters, J., Müller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl Radical Recycling in Isoprene Oxidation Driven by Hydrogen Bonding and Hydrogen Tunneling: The Upgraded LIM1 Mechanism, J. Phys. Chem. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014.
Perring, A. E., Wisthaler, A., Graus, M., Wooldridge, P. J., Lockwood, A. L., Mielke, L. H., Shepson, P. B., Hansel, A., and Cohen, R. C.: A product study of the isoprene+NO3 reaction, Atmos. Chem. Phys., 9, 4945–4956, https://doi.org/10.5194/acp-9-4945-2009, 2009a.
Perring, A. E., Bertram, T. H., Wooldridge, P. J., Fried, A., Heikes, B. G., Dibb, J., Crounse, J. D., Wennberg, P. O., Blake, N. J., Blake, D. R., Brune, W. H., Singh, H. B., and Cohen, R. C.: Airborne observations of total RONO2: new constraints on the yield and lifetime of isoprene nitrates, Atmos. Chem. Phys., 9, 1451–1463, https://doi.org/10.5194/acp-9-1451-2009, 2009b.
Perring, A. E., Pusede, S. E., and Cohen, R. C.: An Observational Perspective on the Atmospheric Impacts of Alkyl and Multifunctional Nitrates on Ozone and Secondary Organic Aerosol, Chem. Rev., 113, 5848–5870, https://doi.org/10.1021/cr300520x, 2013.
Praske, E., Crounse, J. D., Bates, K. H., Kurtén, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 49, 14195–14203, https://doi.org/10.1021/acs.est.5b03738, 2015.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015.
Rindelaub, J. D., Borca, C. H., Hostetler, M. A., Slade, J. H., Lipton, M. A., Slipchenko, L. V., and Shepson, P. B.: The acid-catalyzed hydrolysis of an a-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact, Atmos. Chem. Phys., 16, 15425–15432, https://doi.org/10.5194/acp-16-15425-2016, 2016.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Allen, H. M., Ayres, B. R., Brown, S. S., Brune, W. H., Crounse, J. D., de Gouw, J., Draper, D. C., Feiner, P. A., Fry, J. L., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K., Teng, A. P., Wennberg, P. O., Wild, R. J., Zhang, L., and Cohen, R. C.: The lifetime of nitrogen oxides in an isoprene-dominated forest, Atmos. Chem. Phys., 16, 7623–7637, https://doi.org/10.5194/acp-16-7623-2016, 2016.
Rosen, R. S., Wood, E. C., Wooldridge, P. J., Thornton, J. A., Day, D. A., Kuster, W., Williams, E. J., Jobson, B. T., and Cohen, R. C.: Observations of total alkyl nitrates during Texas Air Quality Study 2000: Implications for O3 and alkyl nitrate photochemistry, J. Geophys. Res.-Atmos., 109, D07303, https://doi.org/10.1029/2003JD004227, 2004.
Russell, M. and Allen, D. T.: Predicting secondary organic aerosol formation rates in southeast Texas, J. Geophys. Res.-Atmos., 110, D07S17, https://doi.org/10.1029/2004JD004722, 2005.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schell, B., Ackermann, I. J., Hass, H., Binkowski, F. S., and Ebel, A.: Modeling the formation of secondary organic aerosol within a comprehensive air quality model system, J. Geophys. Res.-Atmos., 106, 28275–28293, https://doi.org/10.1029/2001JD000384, 2001.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St. Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2+HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Shepson, P. B., Mackay, E., and Muthuramu, K.: Henry's Law Constants and Removal Processes for Several Atmospheric β-Hydroxy Alkyl Nitrates, Environ. Sci. Technol., 30, 3618–3623, https://doi.org/10.1021/es960538y, 1996.
SOAS Science Team: SOAS 2013 Centreville Site Data, NOAA, available at: https://esrl.noaa.gov/csd/groups/csd7/measurements/2013senex/Ground/DataDownload/ (24 October 2018), 2013.
St. Clair, J. M., Rivera-Rios, J. C., Crounse, J. D., Knap, H. C., Bates, K. H., Teng, A. P., Jørgensen, S., Kjaergaard, H. G., Keutsch, F. N., and Wennberg, P. O.: Kinetics and Products of the Reaction of the First-Generation Isoprene Hydroxy Hydroperoxide (ISOPOOH) with OH, J. Phys. Chem. A, 120, 1441–1451, https://doi.org/10.1021/acs.jpca.5b06532, 2015.
Stavrakou, T., Peeters, J., and Müller, J.-F.: Improved global modelling of HOx recycling in isoprene oxidation: evaluation against the GABRIEL and INTEX-A aircraft campaign measurements, Atmos. Chem. Phys., 10, 9863–9878, https://doi.org/10.5194/acp-10-9863-2010, 2010.
Teng, A. P., Crounse, J. D., Lee, L., St. Clair, J. M., Cohen, R. C., and Wennberg, P. O.: Hydroxy nitrate production in the OH-initiated oxidation of alkenes, Atmos. Chem. Phys., 15, 4297–4316, https://doi.org/10.5194/acp-15-4297-2015, 2015.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
Valin, L. C., Russell, A. R., and Cohen, R. C.: Variations of OH radical in an urban plume inferred from NO2 column measurements, Geophys. Res. Lett., 40, 1856–1860, https://doi.org/10.1002/grl.50267, 2013.
von Kuhlmann, R., Lawrence, M. G., Pöschl, U., and Crutzen, P. J.: Sensitivities in global scale modeling of isoprene, Atmos. Chem. Phys., 4, 1–17, https://doi.org/10.5194/acp-4-1-2004, 2004.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Wesely, M. L.: Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ., 23, 1293–1304, https://doi.org/10.1016/0004-6981(89)90153-4, 1989.
Wolfe, G. M., Kaiser, J., Hanisco, T. F., Keutsch, F. N., de Gouw, J. A., Gilman, J. B., Graus, M., Hatch, C. D., Holloway, J., Horowitz, L. W., Lee, B. H., Lerner, B. M., Lopez-Hilifiker, F., Mao, J., Marvin, M. R., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., Thornton, J. A., Veres, P. R., and Warneke, C.: Formaldehyde production from isoprene oxidation across NOx regimes, Atmos. Chem. Phys., 16, 2597–2610, https://doi.org/10.5194/acp-16-2597-2016, 2016.
Wu, S., Mickley, L. J., Jacob, D. J., Logan, J. A., Yantosca, R. M., and Rind, D.: Why are there large differences between models in global budgets of tropospheric ozone?, J. Geophys. Res.-Atmos., 112, D05302, https://doi.org/10.1029/2006JD007801, 2007.
Xie, Y., Paulot, F., Carter, W. P. L., Nolte, C. G., Luecken, D. J., Hutzell, W. T., Wennberg, P. O., Cohen, R. C., and Pinder, R. W.: Understanding the impact of recent advances in isoprene photooxidation on simulations of regional air quality, Atmos. Chem. Phys., 13, 8439–8455, https://doi.org/10.5194/acp-13-8439-2013, 2013.
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015.
Xiong, F., Borca, C. H., Slipchenko, L. V., and Shepson, P. B.: Photochemical degradation of isoprene-derived 4,1-nitrooxy enal, Atmos. Chem. Phys., 16, 5595–5610, https://doi.org/10.5194/acp-16-5595-2016, 2016.