the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 May 2026
| 22 May 2026
Reactions of carbonyl oxide with aldehydes: accurate electronic structure methods, kinetic insights, and atmospheric implications
Chaolu Xie
Carbonyl oxide (CH2OO) is paramount in atmospheric oxidation chemistry, yet quantitative kinetics data for its bimolecular reactions are very limited and even unknown. Here we establish a computational framework to obtain quantitative kinetics from small to large reaction systems. For CH2OO + HCHO, we develop electronic structure methods to reach CCSDTQ/CBS accuracy for its activation enthalpies at 0 K. For CH2OO + aldehydes (RCHO; R= CH3–C5H11, CH2F, CHF2, CF3), we introduce two strategies that recover CCSDTQ/CBS-quality activation enthalpies at 0 K. A dual-level strategy has been used to calculate their kinetics. The calculated rate constants show excellent agreement with available experimental data for CH2OO + RCHO (R = CH3–C3H7), which validates the designed computational framework. We find that fluorination leads to exceptional rate enhancement, with reactions of CHF2CHO and CF3CHO exceeding 10−10 cm3 molecule−1 s−1 over 200–320 K, approaching the collision limit. We also find that fluorination-driven reactivity enhancement originates predominantly from lower-level electronic effects than that of post-CCSD(T). Incorporation of the kinetics into a global chemical transport model uncovers previously unrecognized atmospheric impacts, with CH2OO + HCHO reducing nighttime CH2OO and gas-phase sulfate concentrations by 25.3 % in Antarctica and 12.2 % over Canada, respectively. The present findings address a long-term challenge in how to obtain quantitative kinetics for large molecular systems, where post-CCSD(T) calculations are prohibitive and provide new insights into the chemical transformation of CH2OO and fluorinated aldehydes in the atmosphere.
- Article
(1784 KB) - Full-text XML
-
Supplement
(2070 KB) - BibTeX
- EndNote




Aldehydes are a major class of oxygenated volatile organic compounds (OVOCs) that substantially influence atmospheric oxidative capacity, secondary organic aerosol (SOA) formation, and air quality (Lary and Shallcross, 2000; Liu et al., 2022; Zhao et al., 2024; Li et al., 2024; Mellouki et al., 2015; Bao et al., 2025; Zhang et al., 2012; Bari and Kindzierski, 2018; Edwards et al., 2014; Yang et al., 2018). They originate from both direct emissions – including biomass and fossil-fuel combustion, biogenic sources, and vehicle exhaust – and secondary production via VOC oxidation (Zhao et al., 2024; Knote et al., 2014; Parrish et al., 2012; Chen et al., 2014; Luecken et al., 2012; Grosjean et al., 1983). Their atmospheric removal is governed primarily by photolysis and OH reactions during daytime, whereas fluorinated aldehydes exhibit notably reduced OH reactivity (Wenger, 2006; Jiménez et al., 2007; Atkinson and Pitts, 1978; Lily et al., 2021; Sellevåg et al., 2005; Scollard et al., 1993; Thévenet et al., 2000; D'anna et al., 2001). NO3 reactions constitute a nighttime sink but proceed extremely slow, highlighting the need to identify alternative nocturnal loss pathways (Cabañas et al., 2001; Bossmeyer et al., 2006; Papagni et al., 2000).
Stabilized Criegee intermediates (sCIs), key intermediate species of O3-initiated alkene ozonolysis (Criegee, 1975; Criegee and Wenner, 1949), play critical roles in atmospheric oxidation and SOA formation (Khan et al., 2018; Novelli et al., 2014; Percival et al., 2013; Chhantyal-Pun et al., 2020) and react rapidly with acids (Cabezas and Endo, 2019; Chung et al., 2019; Peltola et al., 2020; Foreman et al., 2016; Raghunath et al., 2017), amides (Wei et al., 2022; Long et al., 2025), and SO2 (Berndt et al., 2014; Boy et al., 2013; Manonmani et al., 2023; Kukui et al., 2021). Accurate kinetics for their bimolecular reactions are therefore essential for constraining their atmospheric fate.
Despite numerous studies on CH2OO + aldehydes, important gaps remain (Table 1 and Fig. 1) for CH2OO + HCHO, where theoretical and experimental rate constants differ by an order of magnitude (Luo et al., 2023; Enders et al., 2024; Long et al., 2021; Zhang et al., 2023); prior work on CH2OO + CH3CHO/C2H5CHO/C3H7CHO (Tables 2–3) relied primarily on CCSD(T) despite evidence that higher-level excitations are required (Taatjes et al., 2012; Elsamra et al., 2016; Stone et al., 2014; Berndt et al., 2015; Jiang et al., 2024; Kaipara and Rajakumar, 2018; Liu et al., 2020, 2023; Cornwell et al., 2023; Debnath and Rajakumar, 2024); and key effects such as anharmonicity, torsional anharmonicity, and recrossing were generally neglected (Luo et al., 2023; Enders et al., 2024; Kaipara and Rajakumar, 2018; Debnath and Rajakumar, 2024; Jalan et al., 2013).
Table 1Rate constants of CH2OO + HCHO by previous investigation at different temperatures and pressures.
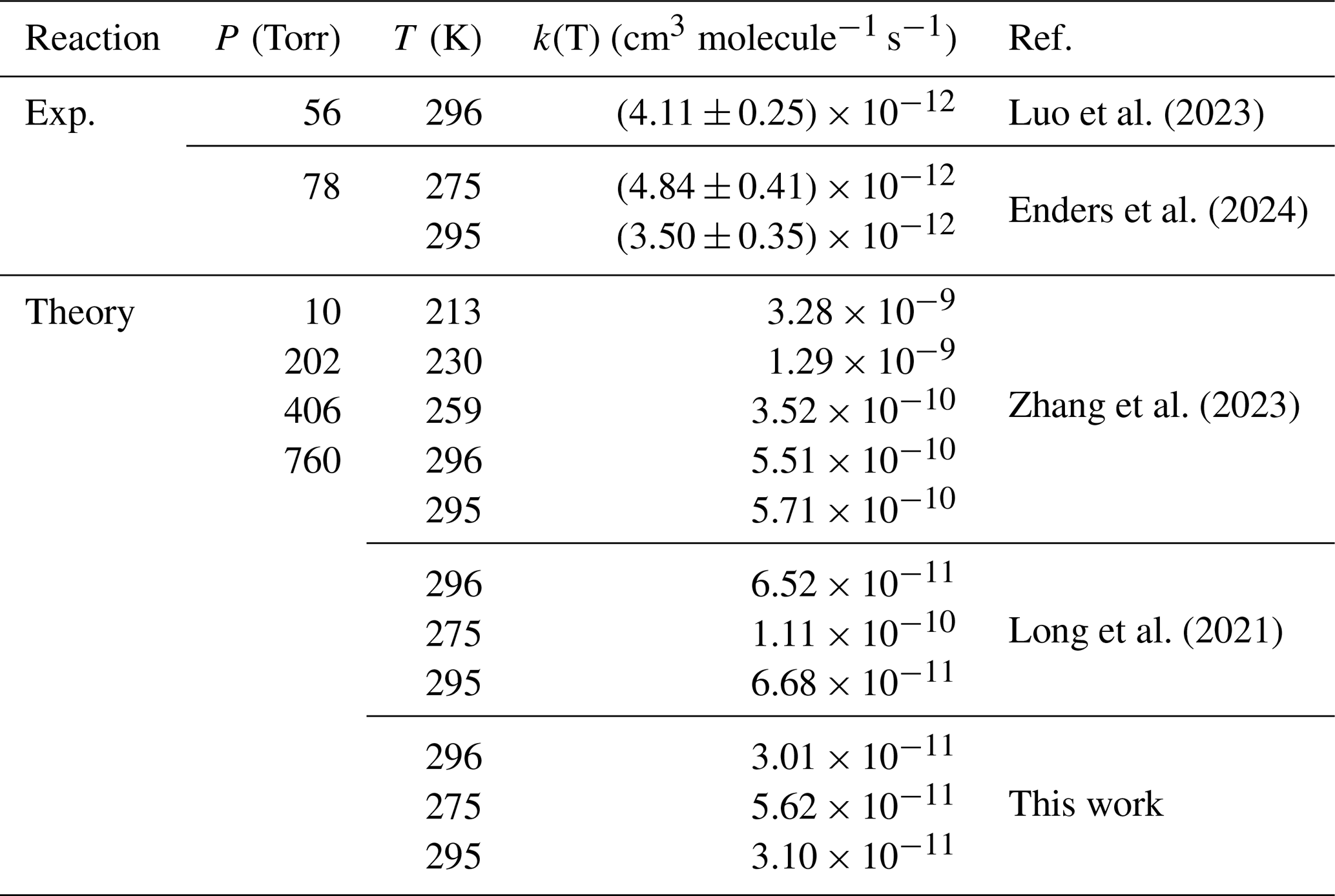
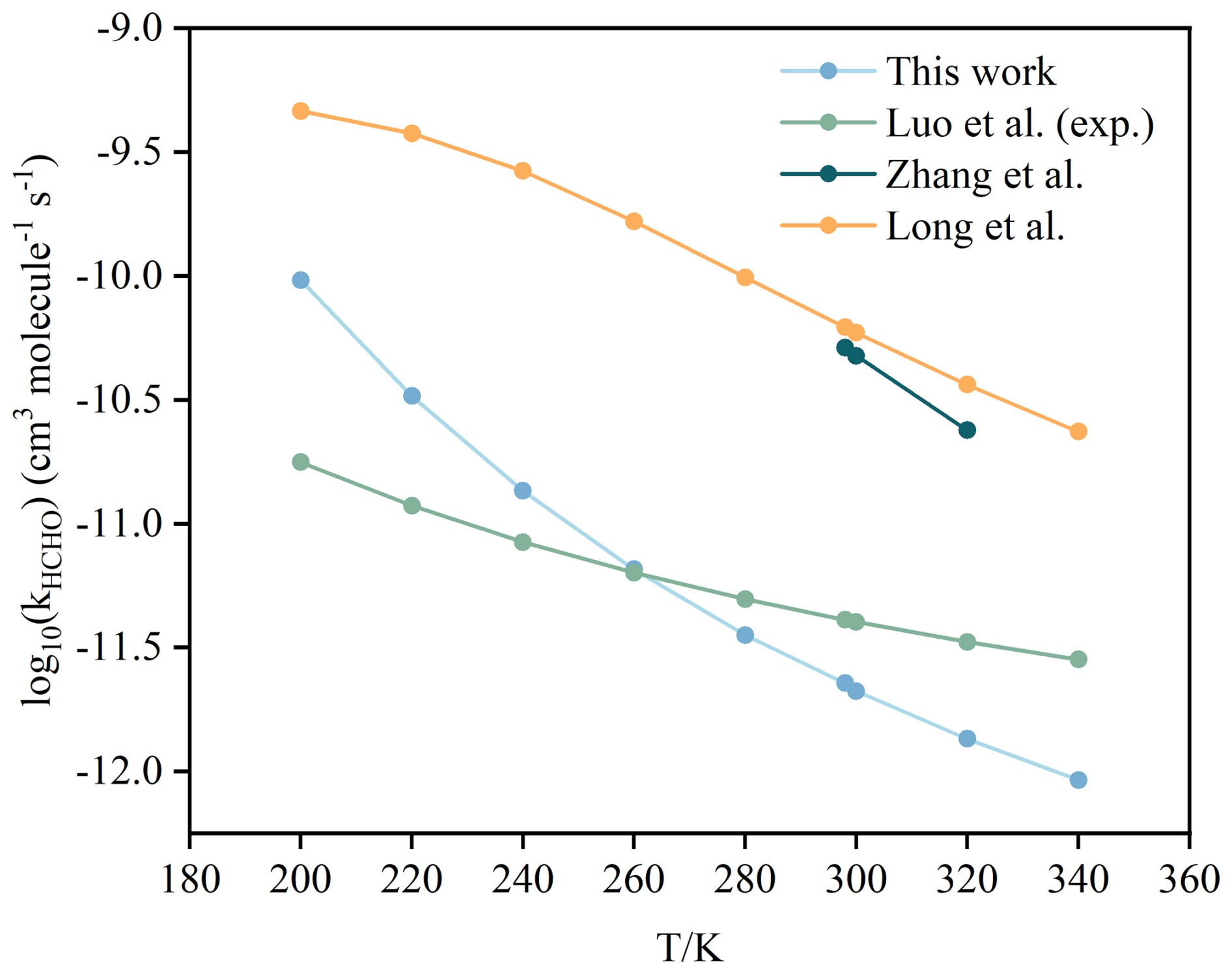
Figure 1A comparison of reported rate constants for the CH2OO + HCHO reaction from previous studies at different temperatures and high-pressure limit.
Table 2Rate constants of CH2OO + CH3CHO by previous investigation at different temperatures and pressures.
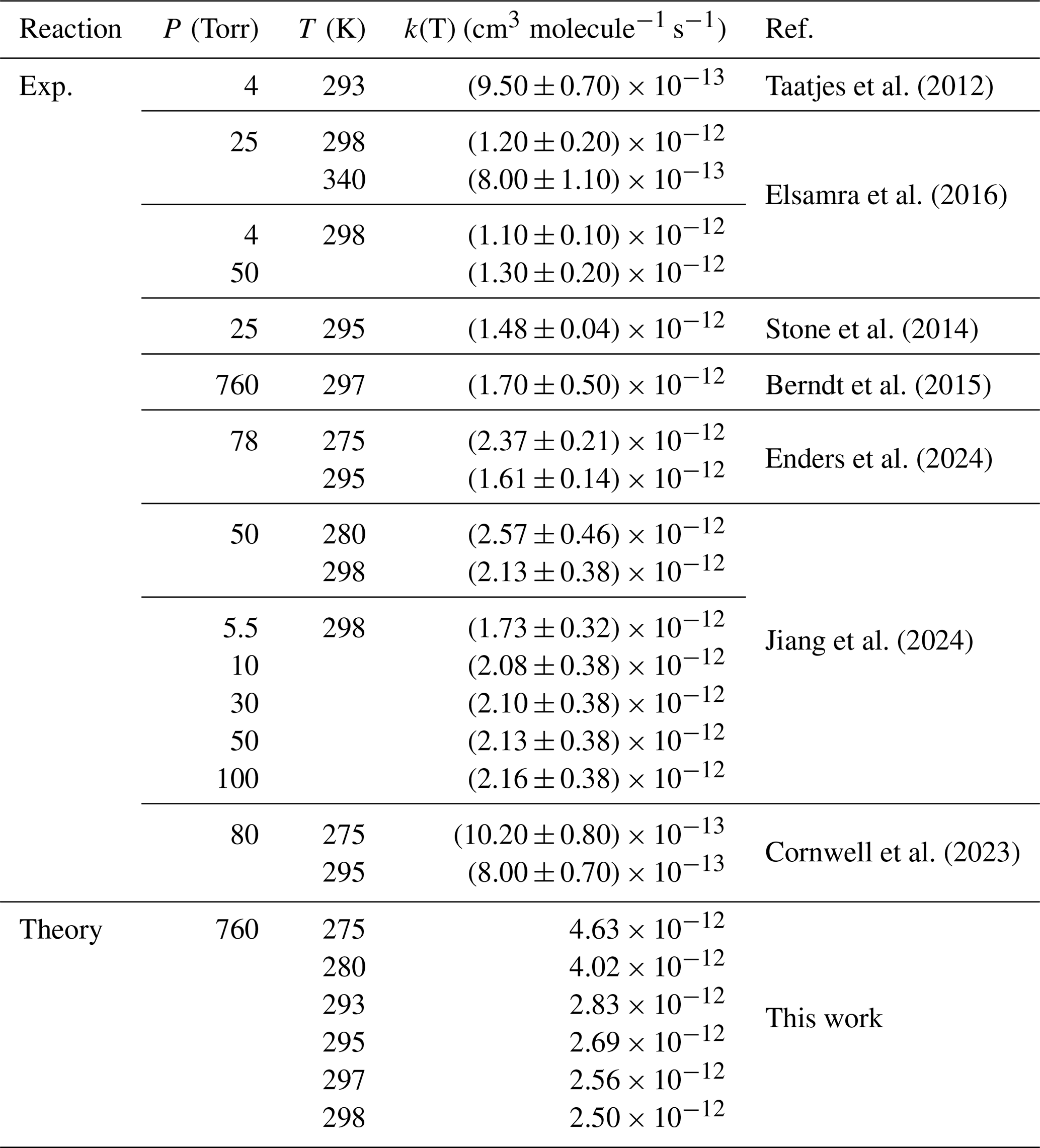
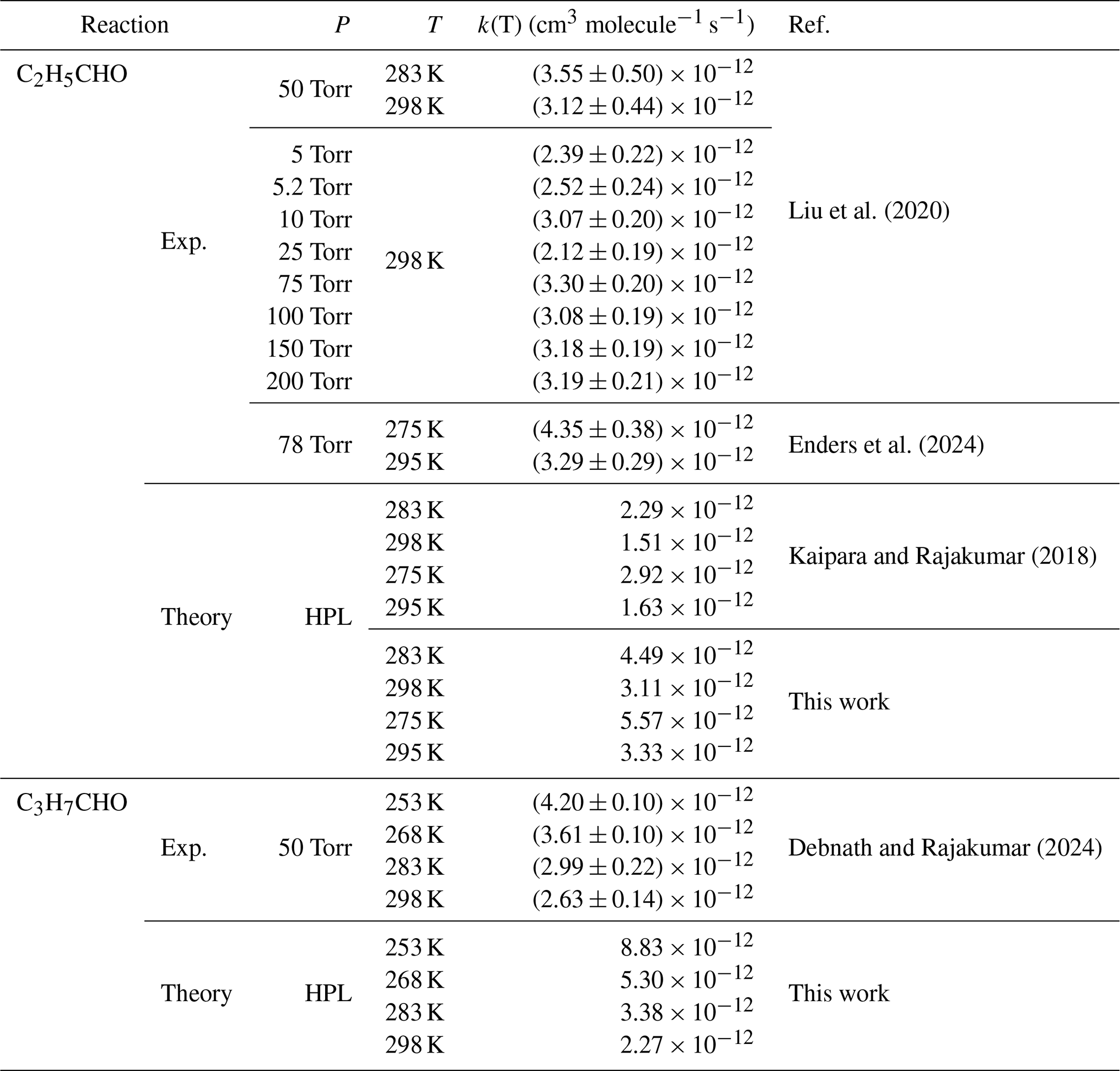
Table 3Rate constants of CH2OO + RCHO (R= C2H5/C3H7) by previous investigation at different temperatures and pressures.
Moreover, no kinetic data exist for reactions with larger or fluorinated aldehydes, including pentanal, hexanal, CH2FCHO, CHF2CHO, and CF3CHO. To address these gaps, atmospheric models have effectively utilized rate constants derived from empirical structure–reactivity relationships (SRRs) – such as those proposed by Jenkin et al. (2018) – which provide a practical and robust framework for large-scale modeling. Given the inherent complexity of computing atmospheric kinetics, these empirical methods remain a primary tool for estimation.
Here, we investigate CH2OO reactions with nine aldehydes (RCHO; R= H, CH3, C2H5, C3H7, C4H9, C5H11, CH2F, CHF2, CF3) to obtain quantitative rate constants and to establish a general high-accuracy computational protocol applicable from small benchmark systems to large atmospheric molecules. For the prototypical CH2OO + HCHO reaction, we develop the GMM(Q).L4 composite scheme that approaches full-CI accuracy, and for the broader reaction suite we devise a scalable strategy capable of delivering near–full-CI activation energies. Dual-level strategy calculations accounting for all major anharmonic and dynamical effects yield benchmark-quality rate constants, which are subsequently implemented in GEOS-Chem to quantify their atmospheric impacts. This work provides a broadly extensible computational framework and significantly advances the understanding of CH2OO–aldehyde chemistry.
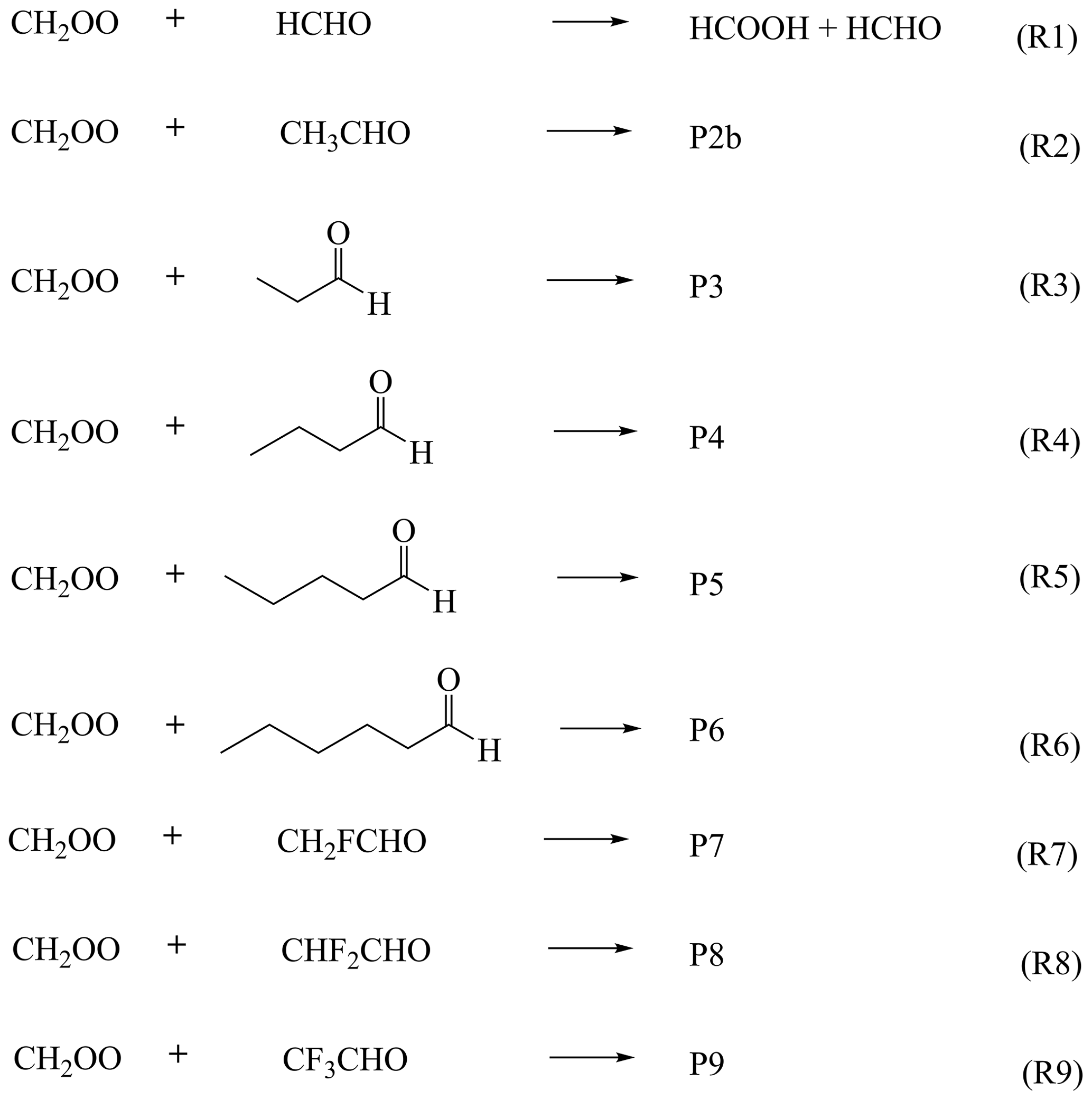
2.1 Electronic structure best estimates for the CH2OO + HCHO reaction
Accurate electronic-structure data are essential for quantitative kinetics. All geometries and harmonic frequencies were optimized at the CCSD(T)-F12a/cc-pVTZ-F12 level (Adler et al., 2007; Knizia et al., 2009; Bischoff et al., 2009). To approach the full-CI limit for single-point energies, we developed a composite protocol, GMMQ.L4, which effectively reproduces CCSDTQ/CBS quality:
Here, is obtained from the previously validated MW2-F12.L scheme which detailed in Table S7 (Long et al., 2021). ΔET−(T) (CCSDT–CCSD(T)) and ΔE(Q)−T (CCSDT(Q)–CCSDT) are extrapolated to the CBS limit (cc-pVDZ → cc-pVTZ and cc-pVDZ → VTZ(d)) using
with L=2 for cc-pVDZ and 3 for cc-pVTZ and VTZ(d). The final correction, ΔEQ−(Q), is evaluated at the CCSDTQ–CCSDT(Q) level using the VDZ(NP) basis set. VTZ(d) employs H(s) and heavy-atom(spd), while VDZ(NP) uses H(s) and heavy-atom(sp) functions (Chan and Radom, 2015).
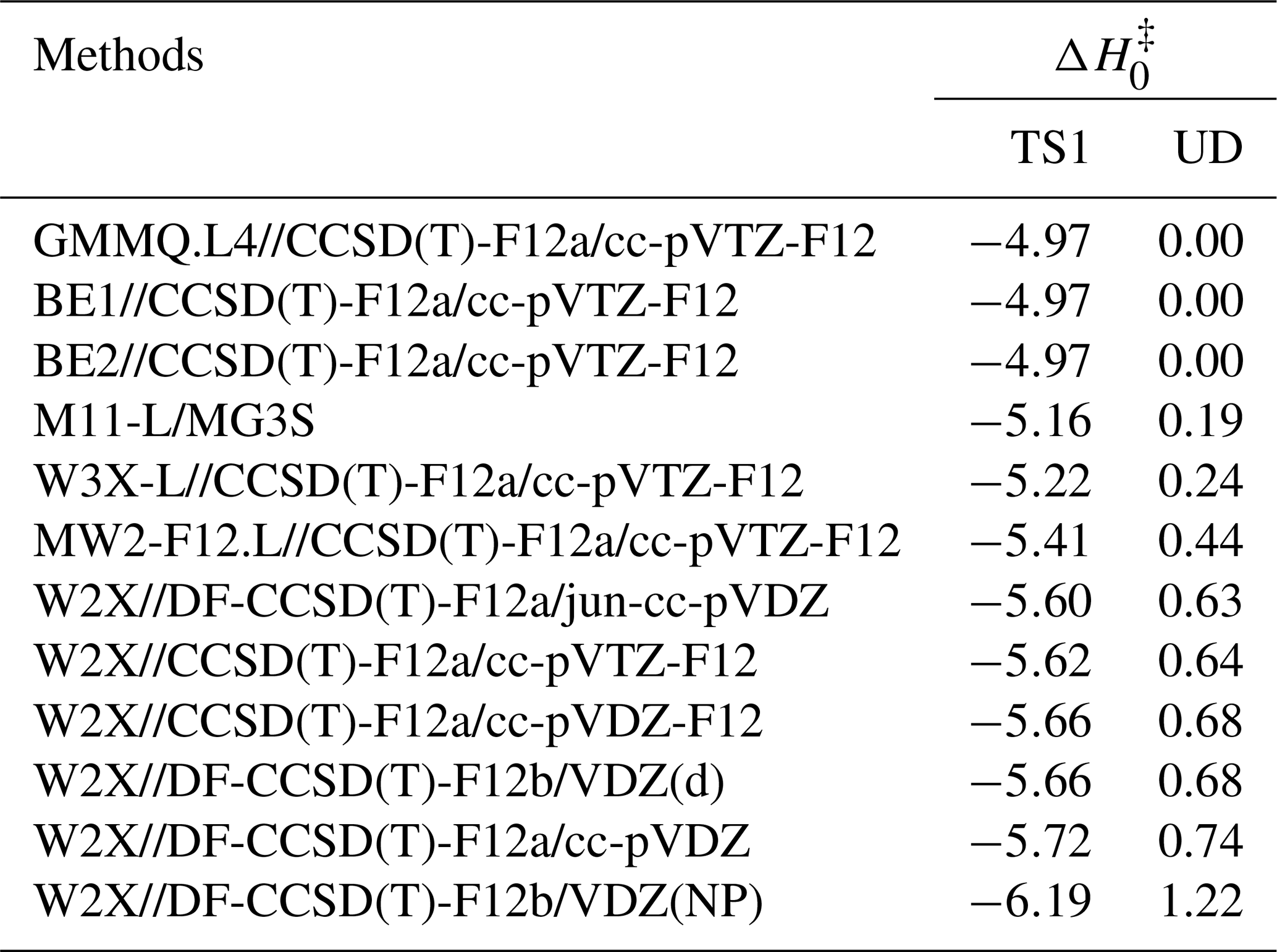
Coupled-cluster theory converges systematically toward Full configuration interaction (Full-CI), but the steep scaling necessitates truncation. Previous studies have established rapid basis-set convergence for both CCSDT(Q)–CCSDT and CCSDT–CCSD(T) (Long et al., 2021, 2019; Xia et al., 2025). Consistently, the CCSDTQ–CCSDT(Q) contribution in our system is only 0.096 kcal mol−1, indicating that excitations beyond quadruples contribute <0.10 kcal mol−1 in Table S1. Thus, GMMQ.L4//CCSD(T)-F12a/cc-pVTZ-F12 serves as the benchmark level in our dual-level kinetics framework.
We further compared GMMQ.L4 with the W3X-L composite method (Chan and Radom, 2015) for Reaction (R1). Although both protocols include identical post-CCSD(T) contributions, GMMQ.L4 employs the MW2-F12.L component, whereas W3X-L is based on W2X. Detailed comparisons are provided in Tables S1, S7, and S8. The observed deviation of 0.24 kcal mol−1 indicates that W3X-L does not achieve quantitatively reliable barrier heights for this system. Our analysis shows that this discrepancy primarily originates from the difference between MW2-F12.L and W2X. Specifically, MW2-F12.L includes HF energies, ΔCCSD and Δ(T) correlation contributions, core–valence (Δ(C+V)) corrections, and scalar relativistic (Δ(C+R)) effects, all evaluated with larger basis sets. In contrast, W2X comprises analogous HF, ΔCCSD, Δ(T), and Δ(C+R) terms, but these are computed using smaller basis sets. The calculated results showed the difference of 0.24 kcal mol−1 comes from the Δ(C+V) and Δ(C+R) terms, which differ by 0.19 and 0.12 kcal mol−1, respectively. Additionally, CCSD(T)-F12 convergence was verified by comparing W2X energies computed with cc-pVTZ-F12 and cc-pVDZ-F12 geometries; the difference of only 0.04 kcal mol−1 confirms near-CBS performance of CCSD(T)-F12 for structural and vibrational data (see Table 4).
Table 4Calculated enthalpies of activation at 0 K ( in kcal mol−1, relative to the bimolecular reactants) and unsigned deviation (MUD) (in kcal mol−1).
2.2 Electronic structure best estimates for Reactions (R2)–(R9)
2.2.1 Geometrical optimization and frequency calculations
Reliable optimized geometries and harmonic frequencies are essential for obtaining quantitative 0 K activation enthalpies. For Reaction (R1), we verified that CCSD(T)-F12a/cc-pVDZ-F12 delivers results essentially identical to CCSD(T)-F12a/cc-pVTZ-F12, allowing us to employ the lower-cost cc-pVDZ-F12 basis for Reaction (R2). However, for larger CH2OO + aldehyde systems, CCSD(T)-F12a/cc-pVDZ-F12 remains computationally prohibitive. To overcome this limitation, we systematically benchmarked density-fitted F12 coupled-cluster methods (DF-CCSD(T)-F12b) (Győrffy and Werner, 2018) across a range of compact basis sets (Table 4). Remarkably, DF-CCSD(T)-F12b/jun-cc-pVDZ (Parker et al., 2014) and DF-CCSD(T)-F12b/VDZ(d) exhibit exceptionally small mean unsigned deviations of only 0.03 and 0.04 kcal mol−1, respectively, relative to the best estimate for W2X reference (Table S2).This identifies a new, computationally efficient F12 protocol capable of retaining sub-0.05 kcal mol−1 accuracy for CH2OO–aldehyde reactions, representing a key methodological advance enabling routine treatment of larger Criegee intermediate–carbonyl systems. Accordingly, we employed DF-CCSD(T)-F12b/jun-cc-pVDZ for Reactions (R3)–(R5) and (R7)–(R8), and DF-CCSD(T)-F12b/VDZ(d) for Reactions (R3)–(R6) to obtain geometries and vibrational frequencies with near-CBS accuracy at greatly reduced cost.
2.2.2 Single point energy calculations
To further reduce the cost of CCSDTQ/CBS-quality calculations, we developed a new composite scheme, denoted BE1, which achieves near-GMMQ.L4 accuracy. The BE1 single-point energy is defined as
where ΔE(Q)−(T) is the CCSDT(Q) – CCSD(T) correction evaluated with the VDZ(NP) basis set for Reactions (R1)–(R8).
The term ΔESC1 introduces a structure-specific correction and is given by
This formulation anchors the composite energy to a single high-level reference transition state (TS1), ensuring the transferability of the correction across the reaction series. The value of ΔESC1 is 0.04 kcal mol−1.
For comparison, we also employed our previously reported strategy, BE2 (Sun et al., 2024), which augments the W2X energy with a constant post-CCSD(T) correction:
where ΔESC2 is the GMMQ.L4 − W2X difference for TS1 (0.64 kcal mol−1 in Table 4). Both BE1 and BE2 offer computationally inexpensive routes to emulate CCSDTQ/CBS performance by incorporating systematic, physically motivated corrections. In the present work, the BE1 protocol served as the high-level (HL) energy in our dual-level kinetics strategy, with the underlying structures obtained from
-
BE1//CCSD(T)-F12a/cc-pVDZ-F12 for Reaction (R2),
-
BE1//DF-CCSD(T)-F12b/jun-cc-pVDZ for Reactions (R3)–(R5) and Reactions (R7)–(R8), and
-
BE1//DF-CCSD(T)-F12b/VDZ(d) for Reaction (R6).
This composite strategy enables sub-kcal mol−1 accuracy at a fraction of the cost of full GMMQ.L4 or CCSDTQ/CBS calculations.
2.3 Electronic structure density functional methods
To enable efficient direct kinetics calculations for the full aldehyde series, we systematically evaluated a range of density functional methods against the BE1 benchmark. Among all tested functionals, M11-L (Peverati and Truhlar, 2012)/MG3S (Lynch et al., 2003) exhibits the best performance, yielding a remarkably small mean unsigned deviation (MUD) of 0.32 kcal mol−1 cross Reactions (R1)–(R8) (Fig. 2). This accuracy – well within sub-kcal mol−1 agreement with the BE1 high-level reference – identifies M11-L/MG3S as a reliable and computationally economical low-level (LL) method for the dual-level kinetics framework. Accordingly, M11-L/MG3S was used for all direct kinetics calculations involving CH2OO + aldehyde reactions. Standard vibrational scaling factors were applied as listed in Table S3.
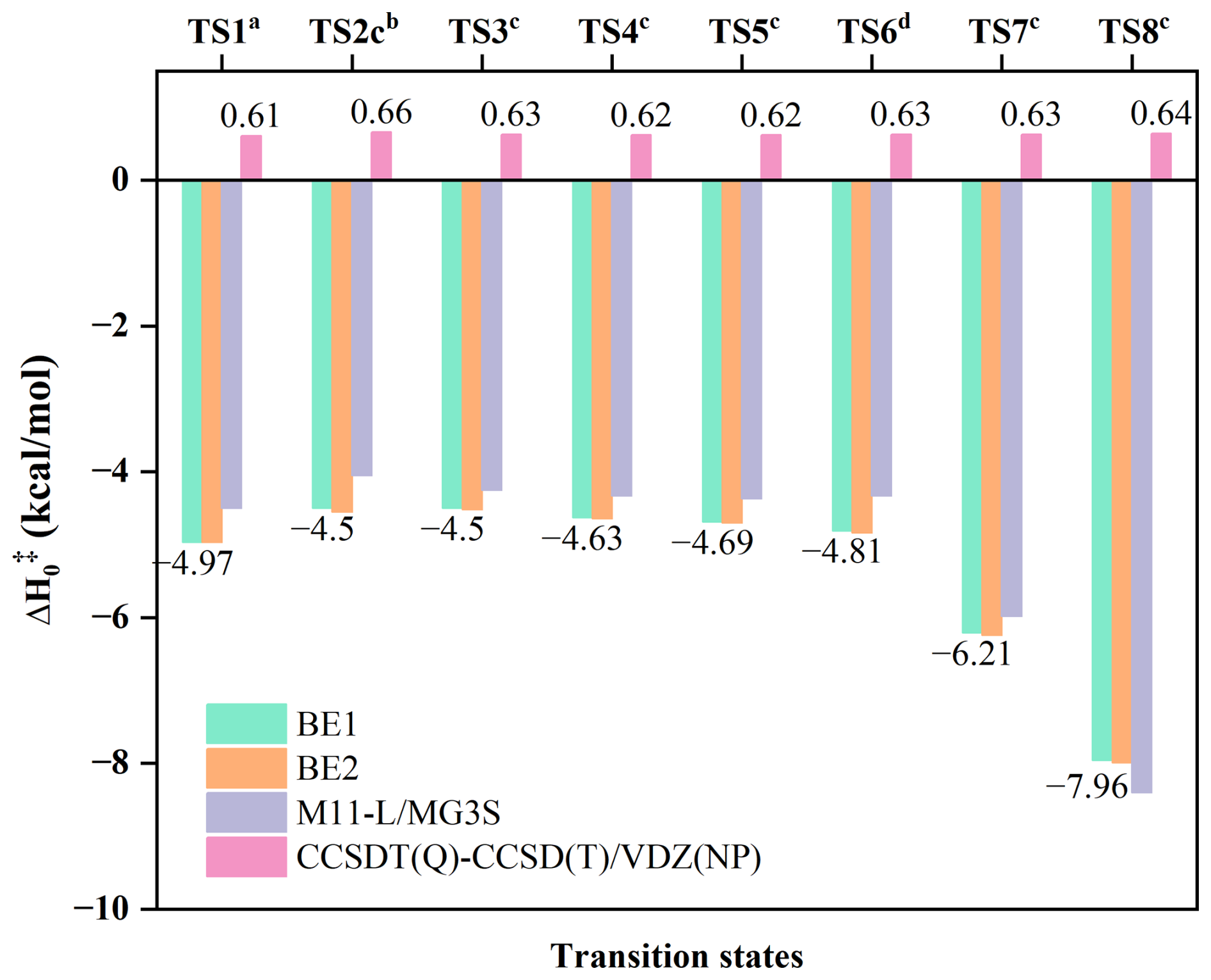
Figure 2Best estimate for Reactions (R1)–(R8) at different level. aThe best estimate results by BE1//CCSD(T)-F12a/cc-pVTZ-F12 in the CH2OO + HCHO reaction. bThe best estimate results by BE1//CCSD(T)-F12a/cc-pVDZ-F12 in the CH2OO + CH3CHO reaction. cThe best estimate results by BE1//DF-CCSD(T)-F12b/jun-cc-pVDZ in the CH2OO + XCHO (X=C2H5/C3H7/C4H9/CH2F/CHF2) reaction. dThe best estimate results by BE1//DF-CCSD(T)-F12b/VDZ(d) in the CH2OO + C5H11CHO reaction.
Previous studies have suggested that standard scaling factors may be unsuitable for certain transition states, we explicitly investigated the impact of anharmonicity. Using the method described by Long et al. (2023), we calculated specific scaling factors (see Tables S4 and S5). However, we found that anharmonicity corrections to the zero-point energy (ZPE) were negligible. Consequently, standard scaling factors are employed throughout this work. Full methodological details are provided in the Supplement.
2.4 Kinetics Methods
2.4.1 High-pressure limited rate constants for Reactions (R2)–(R6)
Dual-level strategy (Long et al., 2019, 2016; Sun et al., 2024) was employed, in which high-level (HL) conventional transition state theory (TST) provides the baseline rate constants, whereas canonical variational transition state theory with small-curvature tunneling (CVT/SCT) at the low-level (LL) supplies kinetic corrections. The high-pressure-limit rate constants were obtained according to Eq. (6):
where is the rate constants calculated at HL. κLL(T) and ΓLL(T) is tunneling and recrossing transmission coefficients calculated at the LL level. is referred to multi-structural anharmonic factor calculated by Eq. (7) at the M11-L/MG3S level
2.4.2 High-pressure limited rate constants for Reactions (R1) and (R7)–(R9)
The rate constants of Reactions (R1) and (R7)–(R9) were calculated by simultaneously considering both the loose transition state between reactants and the van der Waals complex, and the tight TS between reactants and products. The rate constant for the loose TS (kloose) was calculated using variable-reaction-coordinate variational transition-state theory (VRC-VTST) (Georgievskii and Klippenstein, 2003; Zheng et al., 2008; Bao et al., 2016b) with 500 configurations for Monte Carlo sampling. A single-faceted dividing surface was constructed with two pivot points, following procedures validated in previous work (Long et al., 2021). One pivot point was placed along a vector at a distance d from the center of mass (COM) of CH2OO, oriented perpendicular to the CH2OO plane, while the other was placed similarly with respect to CH2F/CHF2/CF3CHO. The pivot distance was set to d=0.05 Å. The reaction coordinate s was defined as the separation between the two pivot points, ranging from 3.5 to 10 Å for Reaction (R7), 3.9 to 10 Å for Reaction (R8), 4.4 to 10 Å for Reaction (R9) with increments of 0.1 Å. The rate constant for the tight TS (ktight) was calculated by using dual-level strategy presented above. The overall rate constant was then obtained using the steady-state approximation (Garrett and Truhlar, 1982; Zhang et al., 2020; Long et al., 2024) in Eq. (7).
2.4.3 Pressure-dependent rate constant
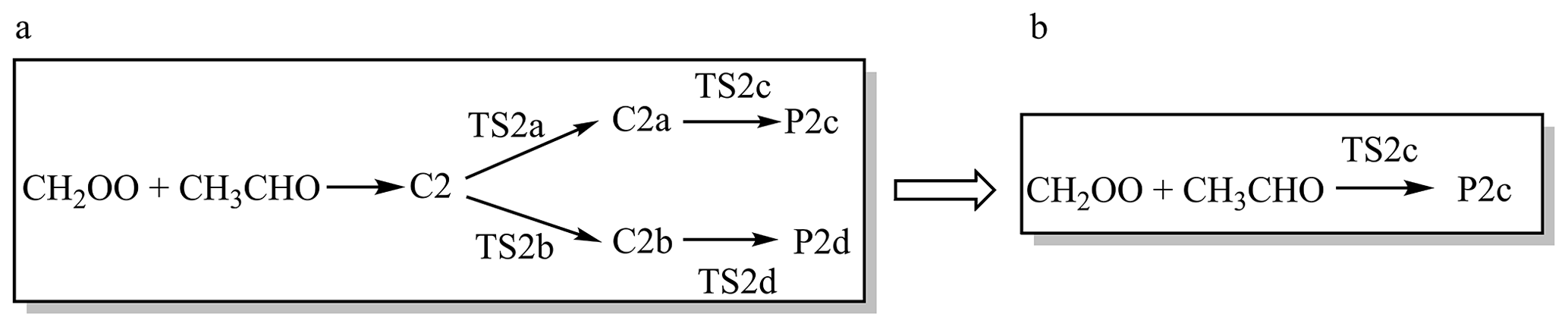
Master equation method with Rice−Ramsperger−Kassel−Marcus theory (ME/RRKM) (Kenneth A. Holbrook, 1996; Fernández-Ramos et al., 2006; Georgievskii et al., 2013; Klippenstein, 2003) was used to calculate pressure dependence of rate constants for the reactions of CH2OO with HCHO and CH3CHO. The calculation utilized parameters from W3X-L//CCSD(T)-F12a/cc-pVTZ-F12 for Reaction (R1) and W2X//DF-CCSD(T)-F12b/jun-cc-pVDZ for Reaction (R2). Both reactions were modeled with N2 as the bath gas, employing Lennard-Jones parameters from Table S6 and an average energy transfer parameter of down=200 cm−1. Within this framework, the pressure effect was approximated as the quotient of the high-pressure limit and a pressure ratio. This ratio is defined as the value at 7.5 × 103 Torr relative to its value at different pressures. We further inspect the simplification of Reaction (R2) in Scheme 2. The kinetic results for Schemes 2a and 2b demonstrate remarkable robustness, with the simplification introducing no statistically significant perturbations to the calculated rate constants.
2.5 Atmospheric modeling
We performed two atmospheric simulations included Reactions (R1) and (R2) to investigate the significance of these reactions by observing the change of concentration globally in GEOS-Chem. This included: (1) a “base” model using default setting (2) a “update1” model adding a new sink of HCHO in the base model, (3) a “update2” model adding a new sink of CH3CHO in the base model. These models include the meteorological data observations assimilated from the NASA Modern-Era Retrospective Analysis for Research and Applications (MERRA-2) (Gelaro et al., 2017) and Emissions data from the default Harmonized Emission Component (HEMCO) (Lin et al., 2021). For anthropogenic emissions, we used the Community Emissions Data System (CEDS) (Hoesly et al., 2018). For biogenic emissions, we used offline VOC emissions computed from the Model of Emissions of Gases and Aerosols from Nature (MEGAN) (Guenther et al., 2012). The simulation was carried out with 2° × 2.5° horizontal resolution at 47 vertical layers. The annual changes displayed are obtained from simulations that employed meteorological data from 1 February 2018, to 31 January 2019, following a six-month model spin-up.
2.6 Software
Density functional calculations were performed by using the Gaussian 16 (Frisch et al., 2016). The coupled cluster calculations were performed by using the Molpro 2019 (Werner, et al., 2019) and MRCC codes (Kállay et al., 2020). Multi-structural anharmonic calculations were performed in MSTor codes (Zheng et al., 2012). Rate constants were calculated using the Polyrate 2017-C (Zheng et al., 2017), Gaussrate 2017-B codes (Zheng et al., 2018), and KiSThlP 2021 (Canneaux et al., 2014). The master equation calculations were performed by utilizing the TUMME program (Zhang et al., 2022). Atmospheric modeling was performed by using GEOS-Chem 14.4.2 (Bey et al., 2001, http://www.geos-chem.org, last access: 4 November 2025).
The enthalpy of activation at 0 K () is referred to the relative energies with zero-point energy between transition states and reactants.
3.1 The electronic structure of CH2OO + HCHO
The reaction mechanism examined here is consistent with that established in earlier studies (Luo et al., 2023; Long et al., 2021; Jalan et al., 2013; Wang et al., 2022). The relative enthalpy profile for the CH2OO + HCHO reaction is depicted in Fig. 3, and the key data are summarized in Table 4. Notably, the activation enthalpy at 0 K obtained at the GMMQ.L4//CCSD(T)-F12a/cc-pVTZ-F12 level (−4.97 kcal mol−1) differs from that predicted by W3X-L//CCSD(T)-F12a/cc-pVTZ-F12 (−5.21 kcal mol−1 in Table 4) and deviates even more substantially from the RCCSD(T)-F12a/VTZ-F12//B3LYP/MG3S value (−6.30 kcal mol−1) (Jalan et al., 2013). These differences demonstrate the strong sensitivity of to the underlying electronic-structure treatment, thereby directly influencing predicted rate constants.
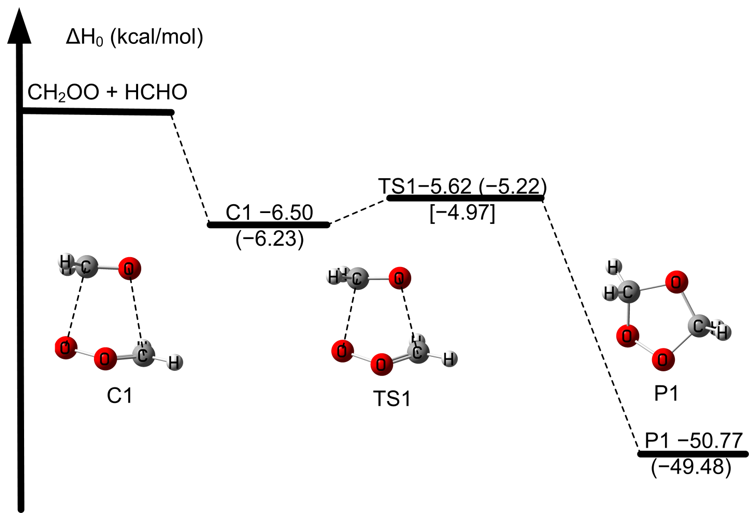
Figure 3The relative enthalpies at 0 K for the reaction of CH2OO + HCHO. Values are given for all species as calculated by W2X//CCSD(T)-F12a/cc-pVTZ-F12, and in parentheses and bracket, values are given for the transition state TS1 as calculated by W3X-L//CCSD(T)-F12a/cc-pVTZ-F12 and GMMQ.L4//CCSD(T)-F12a/cc-pVTZ-F12.
Previous studies have shown that post-CCSD(T) correlation is essential for quantitative barriers in Criegee chemistry (Long et al., 2021, 2016; Xia et al., 2022). For TS1, the unsigned deviation between GMMQ.L4 and MW2-F12.L is 0.40 kcal mol−1 – slightly different with the ∼ 0.50 kcal mol−1 benchmark established for post-CCSD(T) effects (Long et al., 2021) – reaffirming the need for high-level correlation to achieve quantitative accuracy. We further find that the post-CCSD(T) contribution through CCSDT(Q), quantified by the W3X-L–W2X difference, is 0.44 kcal mol−1, in excellent agreement with the 0.40 kcal mol−1 value. This concordance highlights the robustness of W3X-L in capturing post-CCSD(T) contributions (Table 4). The remaining 0.24 kcal mol−1 discrepancy between GMMQ.L4 and W3X-L primarily reflects differences between the MW2-F12.L and W2X components of TS1 (Tables S7 and S8). The 0.21 kcal mol−1 deviation between MW2-F12.L and W2X further illustrates that larger basis sets are required for fully quantitative predictions.
This present work provides a rigorously benchmarked assessment of for the CH2OO + HCHO reaction, explicitly quantifying post-CCSD(T) contributions and revealing their decisive role in achieving sub-kcal mol−1 accuracy. The systematic comparison among GMMQ.L4, MW2-F12.L, and W3X-L underscores the reliability of our calculated results.
3.2 The electronic structure of CH2OO + CH3CHO
We aim to demonstrate the feasibility of simplifying the reaction mechanism of larger aldehydes with CH2OO in Scheme 2. A partial reaction mechanism CH2OO + CH3CHO has been reported in our previous work (Wang et al., 2022). We first consider the seven-membered ring pre-reaction complex C2 formation in Fig. 4, which is consistent with our previous results (Wang et al., 2022). However, due to two distinct orientations of the methyl group in CH3CHO toward CH2OO, there are two rotation transition states TS2a and TS2b connecting C2 to the five-membered ring complexes C2a and C2b, respectively. Therefore, the process is only the transformation of complex in the reaction processes. Then, C2a and C2b undergo the corresponding transition state TS2c and TS2d responsible for the formation of P2a and P2b. The mechanism was depicted in Scheme 2a. However, the enthalpies of activation at 0 K for TS2a and TS2b are lower than those of TS2c and TS2d by 0.64 and 0.37 kcal mol−1 at W3X-L//CCSD(T)-F12a/cc-pVDZ-F12 in Fig. 4, respectively. Therefore, TS2a and TS2b could be neglected from energetic point of view. We will also discuss it from the kinetics point of view.
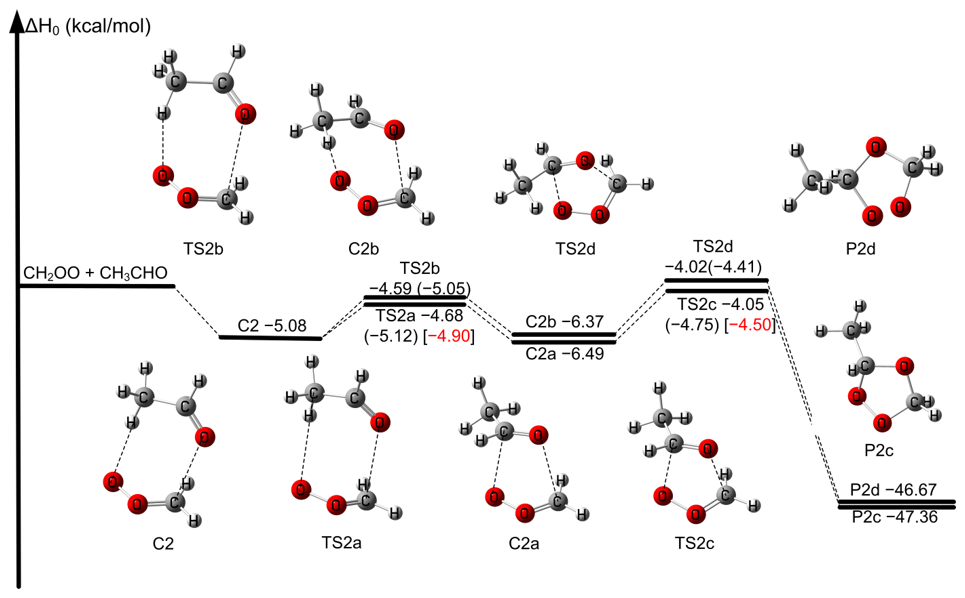
Figure 4The relative enthalpies at 0 K for the reaction of CH2OO + CH3CHO. Values are given for all species as calculated by M11-L/MG3S, and in parentheses and bracket, values are given for the transition states as calculated by W3X-L//CCSD(T)-F12a/cc-pVDZ-F12 and BE1//CCSD(T)-F12a/cc-pVDZ-F12.
The five-membered ring complexes C2a and C2b can interconvert via TS2ISO with C = O bond rotation, which lies 2.51 kcal mol−1 above C2a at the M11-L/MG3S level (Fig. S4), similar to the reaction between CH2OO and FCHO (Xia et al., 2024). For aldehydes with longer chains, the corresponding isomerization transition states of the five-membered ring complexes (Figs. S5–S6) exhibit similarly low barriers, indicating facile interconversion, which also verified from kinetics perspective. Consequently, the complex mechanism can be effectively reduced to the straightforward reaction pathway b depicted in Scheme 2. Accordingly, the mechanism for CH2OO with larger aldehydes was simplified to consider only the lowest-energy pathway corrected by torsional anharmonicity in kinetics calculations.
The for TS2c is −4.50 kcal mol−1 at the BE1//CCSD(T)-F12a/cc-pVDZ-F12 level (see Table S9), which is 0.8 kcal mol−1 higher than the result reported by Jalan et al. at the RCCSD(T)-F12a/VTZ-F12//B3LYP/MG3S level and 0.19 kcal mol−1 higher than that of Wang et al. (2022) at the WMS//M11-L/MG3S level (Wang et al., 2022; Jalan et al., 2013). BE1 and BE2 for TS2c agree well with each other in Fig. 2 and Table S9, not only demonstrating the reliability of the computational protocol, but also capturing the essential physical origin underlying the quantitative description of . The M11-L/MG3S has been chosen for direct dynamics calculations due to the MUD of 0.81 kcal mol−1 in Table S9.
The validity of the DF-CCSD(T)-F12/jun-cc-pVDZ and DF-CCSD(T)-F12b/VDZ(d) methods was also confirmed for Reaction (R2). As shown in Table S9, these methods yielded mean unsigned deviations (MUD) of 0.05 and 0.02 kcal mol−1, respectively, relative to the CCSD(T)-F12a/cc-pVDZ-F12 benchmark.
3.3 Electronic structure of CH2OO + RCHO (R = C2H5/C3H7/C4H9/C5H11)
The complexity of Reactions (R3)–(R6) increases with reactant system size owing to the presence of multiple conformers of both reactants and transition states (Table S10). Conformers for each reactant and transition state were obtained by rotating the dihedral angles listed in Table S10. Specifically, two conformers were identified for C2H5CHO, four for C3H7CHO, twelve for C4H9CHO, and thirty-five for C5H11CHO, arising from C–C bond rotations. In contrast, conformational diversity is even more pronounced for the transition states, with three conformers for TS3, eighteen for TS4, twenty-four for TS5, and seventy-nine for TS6, primarily due to internal C = O and C-C bond rotations.
As the carbon chain prolongs, the change in for Reactions (R1)–(R6) is not obvious, but it presents a trend. We find a slight decrease in with the elongation of carbon chain for Reactions (R2)–(R6) with the exception of Reaction (R1). The calculated by best estimate are −4.50, −4.50, −4.63, −4.70, and −4.80 kcal mol−1 for Reactions (R2)–(R6) (see Fig. 2 and Table S11), which are about 3 kcal mol−1 below the reaction of the corresponding reactants with HO2 (Gao et al., 2024; Long et al., 2022; Ding and Long, 2022). Moreover, the influence of carbon chain length on enthalpy of activation for Reactions (R2)–(R6) is analogue to the reaction of HO2 and aldehydes (Gao et al., 2024). Also, BE1 and BE2 for TS2c–TS6 (Fig. 2 and Table S11) exhibit excellent mutual consistency. This behavior can be attributed to the nearly invariant term (∼ 0.6 kcal mol−1) among these transition states, demonstrating that the post-CCSD(T) contributions are almost uniform across this reaction series. These observations provide compelling evidence that both alkyl substitution and carbon-chain elongation negligibly modulate the magnitude of post-CCSD(T) corrections, implying that such higher-order correlation effects are intrinsically insensitive to substituent-induced electronic and conformational changes.
3.4 Electronic structure of CH2OO + RCHO (R = CH2F/CHF2/CF3)
The electronic structure information was depicted in Fig. 2 and Table S12. The activation enthalpies at 0 K decrease significantly with the increasing number of fluorine substitutions in the methyl group of the aldehyde.
The for CH2OO + CH2FCHO (TS7) is −6.21 kcal mol−1 by our best estimate, which is 1.24 and 1.71 kcal mol−1 lower than the Reactions (R1) and (R2), respectively. Consequently, Reaction (R7) is expected to exhibit a significantly larger rate constant compared to the CH2OO + HCHO/CH3CHO reactions. This reduction in indicates that fluorine substitution enhances the reactivity of the aldehyde toward CH2OO, which is similar to HO2 + CF3CHO (Long et al., 2022). For the reaction of CH2OO + CHF2CHO (Reaction R8), the is −7.96 kcal mol−1, which is 1.75 kcal mol−1 lower than that of the corresponding transition state, TS7. This value is close to that of CH2OO + HCl (Foreman et al., 2016), which approaches the bimolecular collision limit, suggesting that the Reaction (R8) through the tight transition state is not the rate-determining step. Although fluorine substitution on the methyl group of the aldehyde leads to substantially enhanced reactivity toward CH2OO, the post-CCSD(T) contributions from the term (∼ 0.6 kcal mol−1) remain nearly identical across the transition states as shown in Fig. 2, revealing that the higher-order correlation effects are largely insensitive to fluorination and establishing that the fluorination-driven reactivity enhancement originates primarily from lower-level electronic effects than that of post-CCSD(T).
Given the demonstrated accuracy of the M11-L/MG3S method for Reactions (R7) and (R8), this method was subsequently applied to Reaction (R9), as depicted in Fig. S3. Regarding CF3CHO + CH2OO (Reaction R9), the further decreases to −9.74 kcal mol−1 at M11-L/MG3S level. However, this value is slightly higher than the activation enthalpies observed for the universal mechanism of Criegee intermediates reacting with amides (Long et al., 2025), which are significantly submerged below the reactants by approximately 9 to 11 kcal mol−1. This shows that this tight transition state is not the rate-determining step for Reaction (R9).
We further compare the calculated of the CH2OO + RCHO (R = CH2F, CHF2, CF3) reactions with those of the corresponding OH reactions. The for OH + CH2FCHO is −1.15 kcal mol−1 at the CCSD(T)//M06-2X/aug-cc-pVTZ level, which is 5.06 kcal mol−1 higher than that of Reaction (R7). We also find that the for Reaction (R8) by our best estimate is 8.19 kcal mol−1 lower than that of OH + CHF2CHO, calculated at the CCSD(T)/aug-cc-pVDZ//MP2(FC)/aug-cc-pVDZ level. The for Reaction (R9) calculated by M11-L/MG3S is 11.94 kcal mol−1 lower than that of OH + CF3CHO at QCISD(T)/6-311G(d,p) level (Chandra et al., 2001). The present findings reveal that the much lower for Reactions (R7)–(R9) leads to a much faster rate constant, indicating that oxidation by CH2OO contributes significantly to the atmospheric loss of fluorinated aldehydes relative to the OH-initiated pathway from energetic point of view.
3.5 Kinetics
3.5.1 Pressure-dependent rate constants
The pressure dependence of the rate constants for Reactions (R1) and (R2) was evaluated using the ME/RRKM framework, with the results summarized in Tables S13–S15. As shown in Table S13, Reaction (R1) exhibits no appreciable pressure dependence over the conditions examined, indicating that pressure effects can be safely neglected for this channel. This conclusion is fully consistent with the findings reported by Luo et al. (2023). For example, the falloff factor calculated for the CH2OO + HCHO reaction at 298 K and 0.0316 bar is 1.34 (Table S13). This factor, defined as the ratio of the rate constant at 1000 bar to that at 0.0316 bar, indicates only a weak pressure dependence for this system. We observed that at 295 K and 78 Torr, the pressure-dependent rate constant was 2.71 × 10−11 cm3 molecule−1 s−1 in Table S13, which is 7.74 times higher than the reported value ((3.50 ± 0.35) × 10−12 cm3 molecule−1 s−1) in Table 1 (Enders et al., 2024).
We assessed the validity of the simplified pathway by contrasting the full mechanism (Scheme 2a) with the model (Scheme 2b) from a kinetic perspective as listed in Tables S13 and S14. The pressure-dependent rate constants obtained from both models exhibit negligible deviations, thereby validating the simplified scheme as a computationally efficient strategy for larger aldehydes. The calculated pressure-dependent rate constant for Reaction (R2) is 1.84 × 10−12 cm3 molecule−1 s−1 at 293 K and 4 Torr in Table S14, in good agreement with the value of (9.50 ± 0.70) cm3 molecule−1 s−1 reported by Taatjes et al. (2012). Our pressure-dependent rate constant at 298 K and 25 Torr corroborates the experimental value of (1.20 ± 0.20) cm3 molecule−1 s−1 reported by Elsamra et al. (2016) (1.65 × 10−12 cm3 molecule−1 s−1 in Table S14) (Elsamra et al., 2016). We found that the fall-off factor is only 1.36 (Table S14) for the Reaction (R2) at 298 K and 4 Torr, which also shown that the rate constant of Reaction (R2) is negligibly pressure-dependent, which confirms the experimental results qualitatively (Enders et al., 2024; Stone et al., 2014; Berndt et al., 2015; Jiang et al., 2024). In addition, there is experimental evidence that the pressure effect is also insignificant for propionaldehyde and butyraldehyde (Liu et al., 2020; Debnath and Rajakumar, 2024).
3.5.2 High pressure limit rate constants
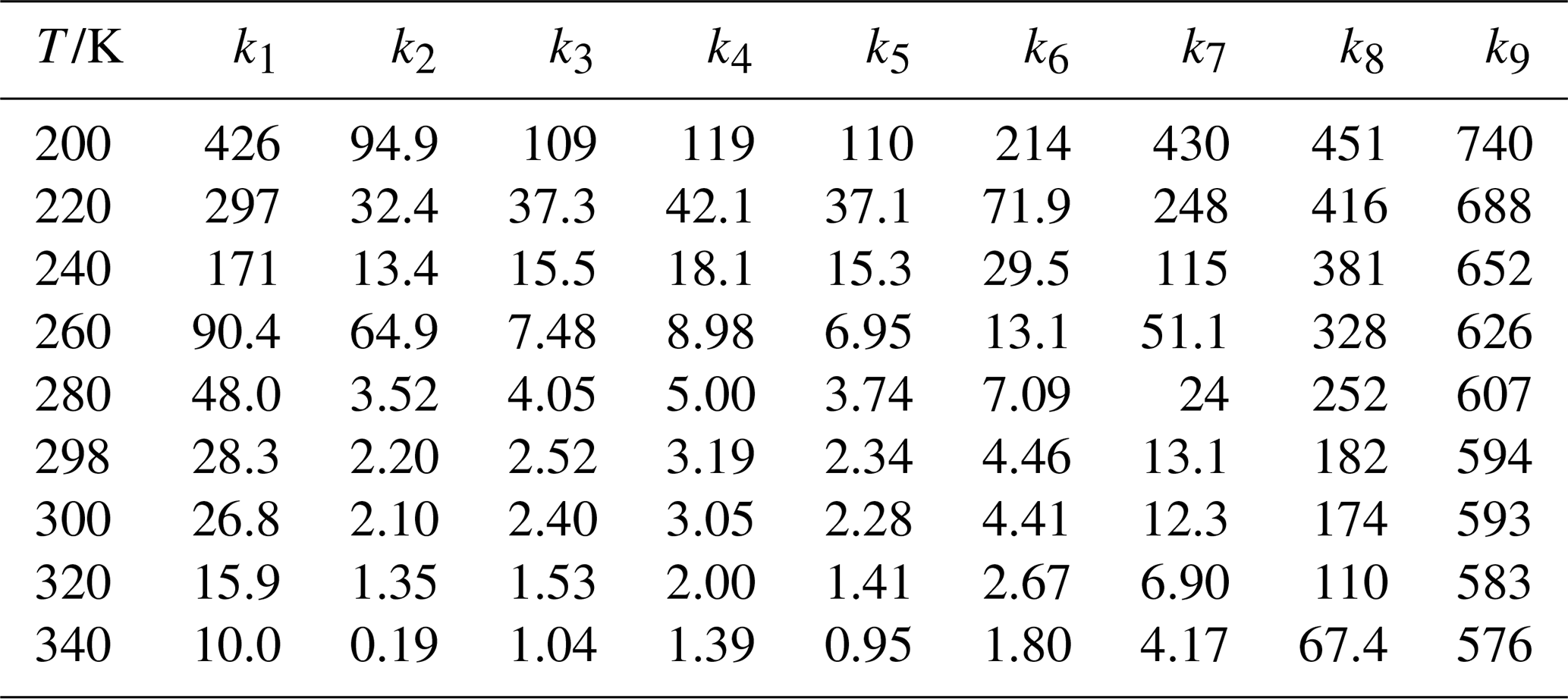
High-pressure limit rate constants for all reactions are summarized in Table 5, with additional details provided in Tables S16–S24. The rate constants in the temperature range of 190–350 K were fitted using the four-parameter expression (Zheng and Truhlar, 2012; Bao et al., 2016a):
Where R is the gas constant, T is temperature in K, the fitting parameters were listed in Table S25. The temperature dependence of the Arrhenius activation energies was further calculated using the following expression:
Table 5The high-pressure limiting rate constants (×10−12 cm3 molecule−1 s−1) of the CH2OO + RCHO (R = H/CH3/C2H5/C3H7/C4H9 /C5H12/CH2F/CHF2/CF3) reaction.
The reaction of CH2OO + HCHO
As summarized in Table 1 and Fig. 1, a long-standing order-of-magnitude discrepancy exists between previously reported experimental and theoretical rate constants for Reaction (R1). At 296 K, the rate constant obtained in this work is 3.01 × 10−11 cm3 molecule−1 s−1 in Table 1, which is 7.31 times larger than the experimental value reported by Luo et al. (2023), but 1.83 and 2.17 times smaller than the theoretical predictions of Zhang et al. (2023) and Long et al. (2021), respectively. We therefore consider two plausible explanations: The experimental determination of CH2OO kinetics may introduce systematic uncertainties. Alternatively, subtle dynamic effects beyond conventional transition state theory (e.g., non-statistical dynamics or complex-forming behavior) may play a role and require further investigation. Although the present value does not fully reconcile the experimental and theoretical results, it substantially narrows the gap between the two, providing a quantitatively improved estimate for this key reaction.
Notably, the derived rate constant for Reaction (R1) is approximately 8 times larger than that for the corresponding OH-initiated reaction and more than two orders of magnitude larger than that for the HO2-initiated pathway (Long et al., 2022; Sivakumaran et al., 2003), highlighting the unexpectedly high reactivity of CH2OO in this system. These findings underscore the need for further high-precision experimental measurements and establish the present computational protocol as a robust framework for resolving persistent discrepancies in atmospheric reaction kinetics.
The reaction of CH2OO + CH3CHO
To date, no theoretical kinetic studies have been reported for the CH2OO + CH3CHO reaction in Table 2. The earliest experimental determination yielded a rate constant of (9.50 ± 0.25) × 10−13 cm3 molecule−1 s−1 at 293 K and 4 Torr, as measured by Taatjes et al. (2012), which is a factor of 2.9 smaller than the present theoretical prediction in Table 2. At 298 K, the calculated rate constant for Reaction (R2) is 2.20 × 10−12 cm3 molecule−1 s−1 in Table 2, in excellent agreement with the experimental values reported by Elsamra et al. (2016) and Jiang et al. (2024) In addition, the value measured by Berndt et al. (2015) at 297 K, (1.7 ± 0.50) × 10−12 cm3 molecule−1 s−1, is fully consistent with our calculated result of 2.27 × 10−12 cm3 molecule−1 s−1 in Table 2. Overall, the rate constants obtained in this work are in good agreement with the available experimental data (Elsamra et al., 2016; Stone et al., 2014; Berndt et al., 2015; Jiang et al., 2024; Cornwell et al., 2023), providing the first reliable theoretical benchmark for the kinetics of the CH2OO + CH3CHO reaction. Notably, the rate constant for Reaction (R2) is approximately 5.6 times smaller than that for the corresponding OH-initiated reaction, yet nearly two orders of magnitude larger than that for the HO2-initiated pathway, highlighting the distinct and non-negligible role of CH2OO in aldehyde oxidation chemistry (Long et al., 2022; Zhu et al., 2008). The five-membered ring species C2a and C2b readily interconvert, as the rate constant for the isomerization process is approximately two orders of magnitude larger than that of the addition reaction (Table S26).
The reaction of CH2OO + (RCHO R = C2 H5/C3 H7/C4 H9/C5 H11.)
Rate constants for the reactions of CH2OO with C2H5CHO have been reported previously from both experimental and theoretical studies (see Table 3) (Enders et al., 2024; Kaipara and Rajakumar, 2018; Liu et al., 2020), whereas the reaction with C3H7CHO has been examined only experimentally. At 298 K, the calculated rate constant for CH2OO + C2H5CHO is 2.52 × 10−12 cm3 molecule−1 s−1 (Table 3), in excellent agreement with the experimental value reported by Liu et al. (2020).
For CH2OO + C3H7CHO, the calculated rate constant of cm3 molecule−1 s−1 (Table 3) closely reproduces the experimental value of (2.63 ± 0.14) × 10−12 cm3 molecule−1 s−1 (Debnath and Rajakumar, 2024), further validating the reliability of the present computational protocol. To the best of our knowledge, no prior experimental or theoretical studies have reported rate constants for the reactions of CH2OO with pentanal or hexanal. Our calculations indicate that the rate constant for CH2OO + C4H9CHO is comparable to that for CH3CHO, whereas the rate constant for CH2OO + C5H11CHO is approximately twice as large, yet remains within the same order of magnitude (Table 5). These results demonstrate that increasing alkyl chain length exerts only a minor influence on the reaction kinetics of CH2OO with aldehydes, revealing a weak and nonmonotonic size dependence across the C1–C5 series. This behavior is fully consistent with the computed activation enthalpies (see Fig. 2) and establishes a transferable structure–reactivity relationship for CH2OO reactions with larger aldehydes. Overall, aside from formaldehyde, the rate constants for CH2OO reactions with alkyl-substituted aldehydes vary only modestly, underscoring the limited role of substituent size in governing CH2OO reactivity.
The reaction of CH2OO + RCHO (R = CH2 F/CHF2 /CHF3)
A striking fluorination-induced reactivity enhancement emerges upon substitution of hydrogen atoms on the methyl group. Introduction of fluorine leads to a pronounced increase in the rate constants for CH2OO + CH3CHO reactions, revealing an unexpected structure–reactivity trend. At 298 K, the rate constant for Reaction (R7) is 1.31 × 10−11 cm3 molecule−1 s−1 (Table 5), which is about 6 times larger than that of Reaction (R2) and about 5 times larger than the corresponding OH + CH2FCHO reaction (Lily et al., 2021).
Even more dramatic behavior is observed for Reactions (R8) and (R9). For Reaction (R8), the calculated rate constants approach the collision limit, decreasing slightly from 4.51 × 10−10 cm3 molecule−1 s−1 at 200 K to 6.74 × 10−11 cm3 molecule−1 s−1 at 340 K in Table 5, indicating of a weak negative temperature dependence characteristic of barrierless processes. Notably, at 298 K the reaction of CHF2CHO with CH2OO is more than two orders of magnitude faster than its reactions with OH [(1.8 ± 0.4) × 10−12 cm3 molecule−1 s−1] (Sellevåg et al., 2005), underscoring the unusually high reactivity of CH2OO toward fluorinated aldehydes.
The most pronounced effect is found for Reaction (R9), for which the rate constant ranges from 7.40 × 10−10 cm3 molecule−1 s−1 at 200 K to 5.76 × 10−10 cm3 molecule−1 s−1 at 340 K in Table 5, fully approaching the collision limit and exceeding the corresponding OH-initiated reaction rates by orders of magnitude. These results demonstrate that fluorination fundamentally alters the reaction landscape of CH2OO with aldehydes, transforming otherwise moderately fast bimolecular reactions into near-collision-controlled processes.
3.6 Atmospheric Implications
The reaction of aldehydes with OH have been investigated extensively experimentally and theoretically. Here, we considered the competition for aldehydes relative to CH2OO and OH. The ratio of reaction rate was calculated by Eq. (9):
where the ki is the rate constants for the Reactions (R2)–(R9), kOH,i is the rate constant of OH + RCHO (R= CH3, C2H5, C3H7, C4H9, C5H11, CH2F, CHF2, CF3), and i is referred to is equal to 2–9. The concentrations of CH2OO and OH exhibit pronounced geographical and spatial distributions. The concentration of OH is varied from 104–106 molecules cm−3 (Khan et al., 2018; Ren et al., 2003; Stone et al., 2012), and the estimated concentration for CH2OO is range from 104 to 105 molecules cm−3 (peaking at 6 × 105 molecules cm−3) (Lelieveld et al., 2016; Novelli et al., 2017) In contrast, the base-version model simulations yield CH2OO concentrations approximately one order of magnitude lower than the estimated value. This discrepancy likely originates from (i) the adoption of relatively fast rate constants for CH2OO loss via reactions with H2O and (H2O)2, and (ii) an incomplete representation of CH2OO sources in the model framework. Consequently, the use of model-derived concentrations probably leads to an underestimation of the contribution of CH2OO to aldehyde removal.
Our results demonstrate that for aliphatic aldehydes, reactions with CH2OO constitute a negligible sink compared with OH oxidation, owing to both modest rate constants and low ambient CH2OO concentration (see Tables S27–S29). Although fluorine substitution generally enhances reactivity, the increase in the rate constant for CH2FCHO remains insufficient to meaningfully compete with the OH pathway. Effective competition is predicted only under highly specific conditions – namely, nighttime at ∼ 10 km altitude over the Malaysian region (Table S29). In stark contrast, the reactions of highly fluorinated aldehydes with CH2OO proceed at near-collision-limit rates. As a result, CH2OO constitutes a major atmospheric sink for CHF2CHO and CF3CHO. As summarized in Table 6, CH2OO competes effectively with OH for CHF2CHO at night near the surface over Russia and the Arctic, and influences its removal at 5 km over Russia and Indonesia, and contributes significantly at 10 km over Indonesia. Notably, because the reaction of CF3CHO with OH is intrinsically slow, CH2OO dominates its atmospheric removal over Indonesia at all altitudes considered, while in the Russian region its influence is confined to 0 and 5 km.
Table 6rate concentration ratios CH2OO to OH and the rate ratio at different heights from different region.
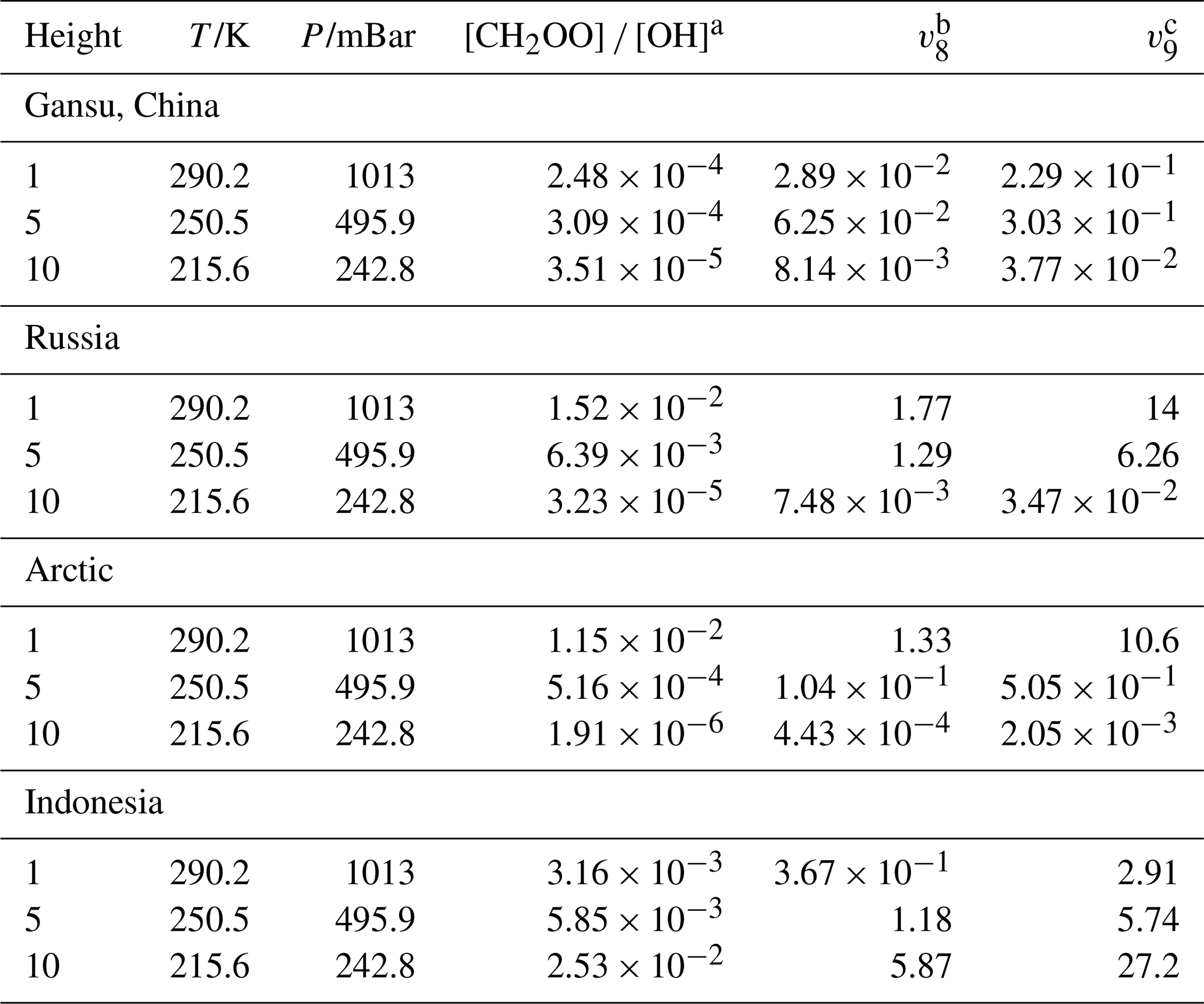
a The concentration ratio between CH2OO and OH from GEOS-Chem. b The rate ratio between CH2OO + CHF2CHO and CHF2CHO + OH. c The rate ration between CH2OO + CF3CHO and CF3CHO + OH.
Overall, these findings reveal a qualitative shift in aldehyde oxidation pathways upon heavy fluorination, identifying CH2OO as a previously underappreciated but potentially dominant oxidant for highly fluorinated aldehydes under specific atmospheric regimes – an effect with important implications for the atmospheric lifetimes of emerging fluorinated oxygenated VOCs.
3.7 Atmospheric modelling
Model simulations were further performed to assess the atmospheric significance of nighttime reactions between CH2OO and aldehydes. The Criegee intermediate (CI) chemistry implemented in the base model has been described in our previous work (Long et al., 2024). In this study, two targeted updates were introduced to isolate and quantify the impacts of newly identified CI–aldehyde reaction pathways. The first update incorporates the CH2OO + HCHO reaction into the base mechanism, reflecting an improved understanding of CI removal under aldehyde-rich nighttime conditions. The second update further expands the CI sink by including the reaction between CH2OO and CH3CHO, thereby providing a more comprehensive representation of acetaldehyde-driven CI loss. The aldehyde chemistry employed in the model is summarized in Table S30. We do not consider the impact of CH2OO on fluorinated aldehyde sinks by using GEOS-Chem, as fluorinated aldehydes are not involved in the current default GEOS-Chem version.
The simulated aldehyde concentrations exhibit pronounced spatial and vertical distributions. Surface-level HCHO concentrations reach up to 1.46×1011 molecules cm−3, while CH3CHO attains maxima of 8.06 × 1010 molecules cm−3, with the highest abundances over Malaysia and Indonesia. These values are consistent with field observations, which report peak HCHO concentrations of up to 3.63×1011 molecules cm−3 (Hu et al., 2025), lending confidence to the model performance. The simulated global mean surface concentration of CH3CHO (5.89 × 109 molecules cm−3, corresponding to ∼ 200 ppt) is in reasonable agreement with observational constraints and remains lower than values reported by Komazaki et al. (1999) and Tereszchuk and Bernath (2011).
The contribution of HCHO to the reduction of CH2OO has been assessed in our prior work and is once again validated by model simulations (Long et al., 2021). Figure 5 shows the relative changes in annual mean surface-layer CH2OO concentrations resulting from the inclusion of the CH2OO + HCHO (Reaction R1) and CH2OO + CH3CHO (Reaction R2) reactions. Incorporation of the updated rate constant for Reaction (R1) leads to a pronounced reduction in CH2OO, with a maximum decrease of 25.3 % over the Antarctic region (Fig. 5), highlighting the previously unrecognized importance of HCHO as a nighttime CI sink. In contrast, Reaction (R2) produces a more modest effect, with a maximum CH2OO reduction of 3.39 % over Russia in Fig. 5.
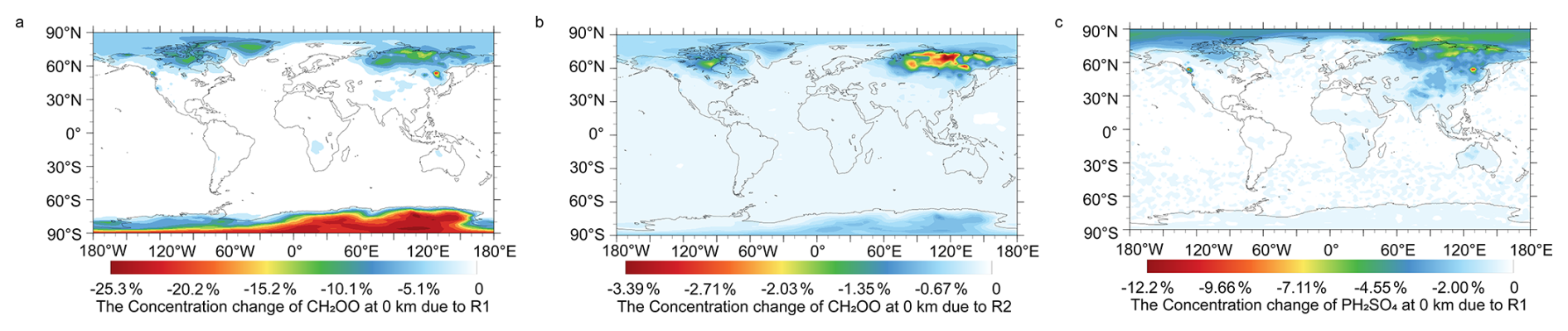
Figure 5Changes in global CH2OO concentrations due to Reactions (R1) and (R2) (a) Reaction (R1), (b) Reaction (R2), and (c) changes in global sulfate concentrations due to Reaction (R1).
Despite the substantial impact on CI abundances, the direct effects on aldehyde concentrations remain small. As shown in Fig. S8, surface acetaldehyde decreases by only 0.12 % in the Arctic. However, the influence on secondary oxygenated products is more pronounced. As illustrated in Fig. S9, inclusion of Reaction (R1) enhances formic acid concentrations by up to 5.44 % over Canada and Russia, while acetic acid increases by as much as 0.69 % in the Arctic. These results demonstrate that CI–aldehyde reactions, while exerting limited feedback on aldehydes themselves, can make significant contribution to the sinks of CH2OO and the formation of atmospheric acids.
The potential implications of Reaction (R1) for regional air quality were also assessed, particularly regarding the mitigation of gas-phase sulfate formation. We found that the concentration of gas-phase sulfate can reach 108 molecules cm−3 in Mexico region in Fig. S10. The inclusion of this reaction pathway effectively lowers the concentration of CH2OO, thereby diminishing its capacity to oxidize SO2 into sulfuric acid precursors. This depletion of oxidative capacity leads to a marked decrease in gas-phase sulfate concentration. The effect is geographical, with the reduction in gas-phase sulfate concentrations estimated to be 12.2 % in Canada and 6.01 % in Russia during the nighttime in Fig. 5c. While the relative changes might initially imply a substantial regional sink for atmospheric sulfate aerosols, a detailed comparison of Figs. 5c and S10 reveals that the largest percentage changes in gas-phase sulfate predominantly occur in regions with low baseline concentrations. Specifically, although peak concentrations over Canada and Russia reach ∼ 107 molecules cm−3, their regional averages remain on the order of 105 molecules cm−3. In contrast, regions with much higher absolute concentrations (e.g., ∼ 1 × 108 molecules cm−3 over Mexico) exhibit only minimal relative changes. This indicates that modest absolute variations can produce large percentage changes under low-background conditions, whereas comparable or even larger absolute changes appear insignificant in high-concentration environments. Consequently, this reaction has a negligible impact on the global atmospheric sulfate burden.
The present work establishes a transferable and systematically improvable theoretical framework for predicting quantitative atmospheric reaction kinetics across molecular complexity, using the reactions of CH2OO with a series of aldehydes as a definitive test case. By explicitly approaching the full configuration interaction (CI) limit for the benchmark CH2OO + HCHO system, we delineate the accuracy requirements necessary for reliable kinetic predictions and provide a rigorous reference against which lower-cost methods can be assessed. Energetic and kinetic analyses validate a simplified reaction mechanism, attributed to the facile interconversion between complexes and the energetic preference for rotational transition states over addition pathways.
Guided by the detailed electronic-structure insights obtained for CH2OO + HCHO, we develop a computational protocol that integrates optimized geometries, vibrational frequencies, and high-level single-point energies, enabling accurate kinetics for larger systems at feasible computational cost. We find that DF-CCSD(T)-F12b/VDZ(d) and DF-CCSD(T)-F12b/jun-cc-pVDZ can be used to reliably describe the optimized geometries and calculated frequencies. two generalizable strategies (BE1 and BE2) have been used to recover the CCSDTQ/CBS level single point energies, which provide new insight into how to obtain the quantitative enthalpy of activation.
In kinetics calculations, for reactions with appreciable barriers (Reactions R2–R6), this dual-level strategy yields robust rate constants, whereas for reactions characterized by exceptionally low or submerged barriers (Reactions R1 and R7–R9), the explicit application of VRC-VTST proves essential for capturing the correct dynamical behavior. This demonstrates a practical pathway for extending benchmark-level kinetics from small to chemically diverse, larger molecules.
The resulting kinetic trends reveal that alkyl-chain elongation exerts only a minor influence on reactivity, whereas fluorine substitution dramatically enhances reaction rates, driving the CH2OO + CHF2CHO and CH2OO + CF3CHO reactions toward the collision limit. All reactions exhibit negligible pressure dependence, underscoring their relevance under atmospheric conditions. These high-precision rate constants provide a mechanistically grounded explanation for the increasingly important role of Criegee intermediates in the oxidation of fluorinated aldehydes. We find that fluorine substitution on aldehydes dramatically enhances their reactivity toward CH2OO; however, the post-CCSD(T) contributions remain almost equal across the reaction series. This behavior indicates that fluorination-driven rate acceleration is governed primarily by lower-level electronic effects rather than by higher-order electron correlation than CCSD(T). This observation also provides a fundamental basis for the development of high-accuracy semiempirical correction schemes.
Beyond molecular-scale kinetics, global and regional modeling demonstrates that while reactions of CH2OO with HCHO and CH3CHO contribute negligibly to aldehyde removal, HCHO constitutes a major global sink for Criegee intermediates, accounting for a 25.3 % reduction in the global CH2OO burden during the night. In contrast, fluorination fundamentally alters atmospheric fate: for CH2FCHO, CH2OO reactions become regionally significant (e.g., near 10 km altitude over Malaysia), and for more heavily fluorinated aldehydes such as CHF2CHO, CH2OO overwhelmingly dominates over OH-initiated loss pathways. The associated enhancement in acid formation, although modest, further highlights the chemical implications of these processes. The inclusion of Reaction (R1) results in a reduction of gas-phase sulfate levels by 12.2 % over Canada and 6.01 % over Russia. These present findings deliver a generalizable, benchmark-anchored strategy for quantitative kinetic prediction, bridges electronic-structure theory with atmospheric modeling, and reveals how fluorination reshapes the atmospheric relevance of Criegee intermediates – insights that are critical for atmospheric chemical mechanisms.
Electronic structure calculations were performed using commercially available software (Gaussian 16, Revision A.03 and Molpro 2019). Access to the software is subject to licensing terms. The MRCC and MSTor codes can be accessed at https://www.mrcc.hu (last access: 4 November 2025) and https://comp.chem.umn.edu/mstor (last access: 4 November 2025), respectively. Polyrate 2017-C and Gaussrate 2017-B are available at https://comp.chem.umn.edu/polyrate and https://comp.chem.umn.edu/gaussrate (last access: 4 November 2025). KiSThlP 2021 is accessible at http://kisthelp.univ-reims.fr (last access: 4 November 2025), and the TUMME program can be found at https://comp.chem.umn.edu/tumme (last access: 4 November 2025). The GEOS-Chem 14.4.2 is available at http://www.geos-chem.org (last access: 4 November 2025). Optimized geometries, and calculated energies are available in Supplement. Other data are available from the corresponding author upon reasonable request.
The following information is provided in the Supplement: Details of Reaction (R9), enthalpies of binding and activation and barrier height; vibrational frequency scale factors; Lennard-Jone parameters; Rate constants and rate constant fits; Rate ratio; Absolute energies and the Cartesian coordinates and absolute energies; relative enthalpies for reaction of Reactions (R3)–(R9); Enthalpy profile for the conversion of pre-reaction complex; Changes in global CH3CHO, HCOOH, and CH3COOH concentrations. The supplement related to this article is available online at https://doi.org/10.5194/acp-26-6973-2026-supplement.
Chaolu Xie carried out the calculations, analysed and interpretation of data, and wrote the manuscript draft. Bo Long designed the project, analyzed and interpretation of data, and reviewed and edited the manuscript.
The contact author has declared that neither of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We also thank the Minnesota Supercomputing Institute for computational resources.
This work was supported in part by the National Natural Science Foundation of China (grant nos. 42120104007 and 41775125), by the Guizhou Provincial Science and Technology Projects, China (grant nos. CXTD [2022]001 and GCC [2023]026), and by the U.S. Department of Energy under (award no. DE-SC0015997), Guizhou Graduate Research Fund Project (grant no. 2024YJSK YJJ224).
This paper was edited by Ivan Kourtchev and reviewed by two anonymous referees.
Adler, T. B., Knizia, G., and Werner, H.-J.: A simple and efficient CCSD(T)-F12 approximation, J. Chem. Phys., 127, 221106, https://doi.org/10.1063/1.2817618, 2007.
Atkinson, R. and Pitts Jr., J. N.: Kinetics of the reactions of the OH radical with HCHO and CH3CHO over the temperature range 299–426 K, J. Chem. Phys., 68, 3581–3584, https://doi.org/10.1063/1.436215, 1978.
Bao, J., Zhang, X., Wu, Z., Zhou, L., Qian, J., Tan, Q., Yang, F., Chen, J., Li, Y., Liu, H., Deng, L., and Li, H.: Atmospheric carbonyl compounds are crucial in regional ozone heavy pollution: insights from the Chengdu Plain Urban Agglomeration, China, Atmos. Chem. Phys., 25, 1899–1916, https://doi.org/10.5194/acp-25-1899-2025, 2025.
Bao, J. L., Zhang, X., and Truhlar, D. G.: Predicting pressure-dependent unimolecular rate constants using variational transition state theory with multidimensional tunneling combined with system-specific quantum RRK theory: a definitive test for fluoroform dissociation, Phys. Chem. Chem. Phys., 18, 16659–16670, https://doi.org/10.1039/C6CP02765B, 2016a.
Bao, J. L., Zhang, X., and Truhlar, D. G.: Barrierless association of CF2 and dissociation of C2F4 by variational transition-state theory and system-specific quantum Rice–Ramsperger–Kassel theory, Proc. Natl. Acad. Sci., 113, 13606–13611, https://doi.org/10.1073/pnas.1616208113, 2016b.
Bari, M. A. and Kindzierski, W. B.: Ambient volatile organic compounds (VOCs) in Calgary, Alberta: Sources and screening health risk assessment, Sci. Total Environ., 627–640, https://doi.org/10.1016/j.scitotenv.2018.03.023, 2018.
Berndt, T., Jokinen, T., Sipilä, M., Mauldin, R. L., Herrmann, H., Stratmann, F., Junninen, H., and Kulmala, M.: H2SO4 formation from the gas-phase reaction of stabilized Criegee Intermediates with SO2: Influence of water vapour content and temperature, Atmos. Environ., 89, 603–612, https://doi.org/10.1016/j.atmosenv.2014.02.062, 2014.
Berndt, T., Kaethner, R., Voigtländer, J., Stratmann, F., Pfeifle, M., Reichle, P., Sipilä, M., Kulmala, M., and Olzmann, M.: Kinetics of the unimolecular reaction of CH2OO and the bimolecular reactions with the water monomer, acetaldehyde and acetone under atmospheric conditions, Phys. Chem. Chem. Phys., 17, 19862–19873, https://doi.org/10.1039/C5CP02224J, 2015.
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q., Liu, H. Y., Mickley, L. J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res.-Atmos., 106, 23073–23095, https://doi.org/10.1029/2001JD000807, 2001.
Bischoff, F. A., Wolfsegger, S., Tew, D. P., and Klopper, W.: Assessment of basis sets for F12 explicitly-correlated molecular electronic-structure methods, Mol. Phys., 107, 963–975, https://doi.org/10.1080/00268970802708942, 2009.
Bossmeyer, J., Brauers, T., Richter, C., Rohrer, F., Wegener, R., and Wahner, A.: Simulation chamber studies on the NO3 chemistry of atmospheric aldehydes, Geophys. Res. Lett., 33, https://doi.org/10.1029/2006GL026778, 2006.
Boy, M., Mogensen, D., Smolander, S., Zhou, L., Nieminen, T., Paasonen, P., Plass-Dülmer, C., Sipilä, M., Petäjä, T., Mauldin, L., Berresheim, H., and Kulmala, M.: Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations, Atmos. Chem. Phys., 13, 3865–3879, https://doi.org/10.5194/acp-13-3865-2013, 2013.
Cabañas, B., Martín, P., Salgado, S., Ballesteros, B., and Martínez, E.: An experimental study on the temperature dependence for the gas-phase reactions of NO3 radical with a series of aliphatic aldehydes, J. Atmos. Chem., 40, 23–39, https://doi.org/10.1023/A:1010797424283, 2001.
Cabezas, C. and Endo, Y.: The Criegee intermediate-formic acid reaction explored by rotational spectroscopy, Phys. Chem. Chem. Phys., 21, 18059–18064, https://doi.org/10.1039/C9CP03001H, 2019.
Canneaux, S., Bohr, F., and Henon, E.: KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 35, 82–93, https://doi.org/10.1002/jcc.23470, 2014.
Chan, B. and Radom, L.: W2X and W3X-L: cost-effective approximations to W2 and W4 with kJ mol−1 accuracy, J. Chem. Theory Comput., 11, 2109–2119, https://doi.org/10.1021/acs.jctc.5b00135, 2015.
Chandra, A. K., Uchimaru, T., and Sugie, M.: Kinetics of hydrogen abstraction reactions of CF3CHO, CF2ClCHO, CFCl2CHO and CCl3CHO with OH Radicals: An ab initio study, Phys. Chem. Chem. Phys., 3, 3961–3966, https://doi.org/10.1039/B104904F, 2001.
Chen, W. T., Shao, M., Lu, S. H., Wang, M., Zeng, L. M., Yuan, B., and Liu, Y.: Understanding primary and secondary sources of ambient carbonyl compounds in Beijing using the PMF model, Atmos. Chem. Phys., 14, 3047–3062, https://doi.org/10.5194/acp-14-3047-2014, 2014.
Chhantyal-Pun, R., Khan, M. A. H., Zachhuber, N., Percival, C. J., Shallcross, D. E., and Orr-Ewing, A. J.: Impact of Criegee intermediate reactions with peroxy radicals on tropospheric organic aerosol, ACS Earth Space Chem., 4, 1743–1755, https://doi.org/10.1021/acsearthspacechem.0c00147, 2020.
Chung, C.-A., Su, J. W., and Lee, Y.-P.: Detailed mechanism and kinetics of the reaction of Criegee intermediate CH2OO with HCOOH investigated via infrared identification of conformers of hydroperoxymethyl formate and formic acid anhydride, Phys. Chem. Chem. Phys., 21, 21445–21455, https://doi.org/10.1039/C9CP04168K, 2019.
Cornwell, Z. A., Enders, J. J., Harrison, A. W., and Murray, C.: Temperature-dependent kinetics of the reactions of CH2OO with acetone, biacetyl, and acetylacetone, Int. J. Chem. Kinet., 55, 154–166, https://doi.org/10.1002/kin.21625, 2023.
Criegee, R.: Mechanism of Ozonolysis, Angew. Chem. Int. Ed., 14, 745–752, https://doi.org/10.1002/anie.197507451, 1975.
Criegee, R. and Wenner, G.: Die Ozonisierung des 9,10-Oktalins, Justus Liebigs Ann. Chem., 564, 9–15, https://doi.org/10.1002/jlac.19495640103, 1949.
D'Anna, B., Andresen, Ø., Gefen, Z., and Nielsen, C. J.: Kinetic study of OH and NO3 radical reactions with 14 aliphatic aldehydes, Phys. Chem. Chem. Phys., 3, 3057–3063, https://doi.org/10.1039/B103623H, 2001.
Debnath, A. and Rajakumar, B.: Experimental and theoretical study of Criegee intermediate (CH2OO) reactions with n-butyraldehyde and isobutyraldehyde: kinetics, implications and atmospheric fate, Phys. Chem. Chem. Phys., 26, 6872–6884, https://doi.org/10.1039/D3CP05482A, 2024.
Ding, D.-P. and Long, B.: Reaction between propionaldehyde and hydroxyperoxy radical in the atmosphere: A reaction route for the sink of propionaldehyde and the formation of formic acid, Atmos. Environ., 284, 119202, https://doi.org/10.1016/j.atmosenv.2022.119202, 2022.
Edwards, P. M., Brown, S. S., Roberts, J. M., Ahmadov, R., Banta, R. M., deGouw, J. A., Dubé, W. P., Field, R. A., Flynn, J. H., Gilman, J. B., Graus, M., Helmig, D., Koss, A., Langford, A. O., Lefer, B. L., Lerner, B. M., Li, R., Li, S.-M., McKeen, S. A., Murphy, S. M., Parrish, D. D., Senff, C. J., Soltis, J., Stutz, J., Sweeney, C., Thompson, C. R., Trainer, M. K., Tsai, C., Veres, P. R., Washenfelder, R. A., Warneke, C., Wild, R. J., Young, C. J., Yuan, B., and Zamora, R.: High winter ozone pollution from carbonyl photolysis in an oil and gas basin, Nature, 514, 351–354, https://doi.org/10.1038/nature13767, 2014.
Elsamra, R. M. I., Jalan, A., Buras, Z. J., Middaugh, J. E., and Green, W. H.: Temperature- and Pressure-Dependent Kinetics of CH2OO + CH3COCH3 and CH2OO + CH3CHO: Direct Measurements and Theoretical Analysis, Int. J. Chem. Kinet., 48, 474–488, https://doi.org/10.1002/kin.21007, 2016.
Enders, J. J., Cornwell, Z. A., Harrison, A. W., and Murray, C.: Temperature-Dependent Kinetics of the Reactions of the Criegee Intermediate CH2OO with Aliphatic Aldehydes, J. Phys. Chem. A, 128, 7879–7888, https://doi.org/10.1021/acs.jpca.4c04990, 2024.
Fernández-Ramos, A., Miller, J. A., Klippenstein, S. J., and Truhlar, D. G.: Modeling the Kinetics of Bimolecular Reactions, Chem. Rev., 106, 4518–4584, https://doi.org/10.1021/cr050205w, 2006.
Foreman, E. S., Kapnas, K. M., and Murray, C.: Reactions between Criegee Intermediates and the Inorganic Acids HCl and HNO3: Kinetics and Atmospheric Implications, Angew. Chem. Int. Ed., 55, 10419–10422, https://doi.org/10.1002/anie.201604662, 2016.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams, Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian Inc, Wallingford CT, https://gaussian.com/gaussian16/ (last access: 4 November 2025), 2016.
Gao, Q., Shen, C., Zhang, H., Long, B., and Truhlar, D. G.: Quantitative kinetics reveal that reactions of HO2 are a significant sink for aldehydes in the atmosphere and may initiate the formation of highly oxygenated molecules via autoxidation, Phys. Chem. Chem. Phys., 26, 16160–16174, https://doi.org/10.1039/D4CP00693C, 2024.
Garrett, B. C. and Truhlar, D. G.: Canonical unified statistical model. Classical mechanical theory and applications to collinear reactions, J. Chem. Phys., 76, 1853–1858, https://doi.org/10.1063/1.443157, 1982.
Gelaro, R., McCarty, W., Suárez, M. J., Todling, R., Molod, A., Takacs, L., Randles, C. A., Darmenov, A., Bosilovich, M. G., Reichle, R., Wargan, K., Coy, L., Cullather, R., Draper, C., Akella, S., Buchard, V., Conaty, A., da Silva, A. M., Gu, W., Kim, G.-K., Koster, R., Lucchesi, R., Merkova, D., Nielsen, J. E., Partyka, G., Pawson, S., Putman, W., Rienecker, M., Schubert, S. D., Sienkiewicz, M., and Zhao, B.: The Modern-Era Retrospective Analysis for Research and Applications, Version 2 (MERRA-2), J. Clim., 30, 5419–5454, https://doi.org/10.1175/JCLI-D-16-0758.1, 2017.
Georgievskii, Y. and Klippenstein, S. J.: Variable reaction coordinate transition state theory: Analytic results and application to the C2H3+H→C2H4 reaction, J. Chem. Phys., 118, 5442–5455, https://doi.org/10.1063/1.1539035, 2003.
Georgievskii, Y., Miller, J. A., Burke, M. P., and Klippenstein, S. J.: Reformulation and Solution of the Master Equation for Multiple-Well Chemical Reactions, J. Phys. Chem. A, 117, 12146–12154, https://doi.org/10.1021/jp4060704, 2013.
Grosjean, D., Swanson, R. D., and Ellis, C.: Carbonyls in Los Angeles air: Contribution of direct emissions and photochemistry, Sci. Total Environ., 29, 65–85, https://doi.org/10.1016/0048-9697(83)90034-7, 1983.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Győrffy, W. and Werner, H.-J.: Analytical energy gradients for explicitly correlated wave functions. II. Explicitly correlated coupled cluster singles and doubles with perturbative triples corrections: CCSD(T)-F12, J. Chem. Phys., 148, 114104, https://doi.org/10.1063/1.5020436, 2018.
Hoesly, R. M., Smith, S. J., Feng, L., Klimont, Z., Janssens-Maenhout, G., Pitkanen, T., Seibert, J. J., Vu, L., Andres, R. J., Bolt, R. M., Bond, T. C., Dawidowski, L., Kholod, N., Kurokawa, J.-I., Li, M., Liu, L., Lu, Z., Moura, M. C. P., O'Rourke, P. R., and Zhang, Q.: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 11, 369–408, https://doi.org/10.5194/gmd-11-369-2018, 2018.
Hu, R., Zhang, G., Cai, H., Guo, J., Lu, K., Li, X., Lou, S., Tan, Z., Hu, C., Xie, P., and Liu, W.: Accurate elucidation of oxidation under heavy ozone pollution: a full suite of radical measurements in the chemically complex atmosphere, Atmos. Chem. Phys., 25, 3011–3028, https://doi.org/10.5194/acp-25-3011-2025, 2025.
Jalan, A., Allen, J. W., and Green, W. H.: Chemically activated formation of organic acids in reactions of the Criegee intermediate with aldehydes and ketones, Phys. Chem. Chem. Phys., 15, 16841–16852, https://doi.org/10.1039/C3CP52598H, 2013.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9329–9349, https://doi.org/10.5194/acp-18-9329-2018, 2018.
Jiang, H., Liu, Y., Xiao, C., Yang, X., and Dong, W.: Reaction Kinetics of CH2OO and syn-CH3CHOO Criegee Intermediates with Acetaldehyde, J. Phys. Chem. A, 128, 4956–4965, https://doi.org/10.1021/acs.jpca.4c01374, 2024.
Jiménez, E., Lanza, B., Martínez, E., and Albaladejo, J.: Daytime tropospheric loss of hexanal and trans-2-hexenal: OH kinetics and UV photolysis, Atmos. Chem. Phys., 7, 1565–1574, https://doi.org/10.5194/acp-7-1565-2007, 2007.
Kaipara, R. and Rajakumar, B.: Temperature-dependent kinetics of the reaction of a Criegee intermediate with propionaldehyde: A computational investigation, J. Phys. Chem. A, 122, 8433–8445, https://doi.org/10.1021/acs.jpca.8b06603, 2018.
Kállay, M., Nagy, P. R., Mester, D., Rolik, Z., Samu, G., Csontos, J., Csóka, J., Szabó, P. B., Gyevi-Nagy, L., Hégely, B., Ladjánszki, I., Szegedy, L., Ladóczki, B., Petrov, K., Farkas, M., Mezei, P. D., and Ganyecz, Á.: The MRCC program system: Accurate quantum chemistry from water to proteins, J. Chem. Phys., 152, 074107, https://doi.org/10.1063/1.5142048, 2020.
Kenneth A. Holbrook, M. J. P., Struan H. Robertson Unimolecular Reactions, 2nd ed., John Wiley & Sons, Chichester, 177–214, ISBN 978-0-471-92268-1, 1996.
Khan, M. A. H., Percival, C. J., Caravan, R. L., Taatjes, C. A., and Shallcross, D. E.: Criegee intermediates and their impacts on the troposphere, Environ. Sci. Processes Impacts, 20, 437–453, https://doi.org/10.1039/C7EM00585G, 2018.
Klippenstein, S. J.: RRKM theory and its implementation, in: Comprehensive Chemical Kinetics, Elsevier, 55–103, https://doi.org/10.1016/S0069-8040(03)80004-3, 2003.
Knizia, G., Adler, T. B., and Werner, H.-J.: Simplified CCSD(T)-F12 methods: Theory and benchmarks, J. Chem. Phys., 130, 054104, https://doi.org/10.1063/1.3054300, 2009.
Knote, C., Hodzic, A., Jimenez, J. L., Volkamer, R., Orlando, J. J., Baidar, S., Brioude, J., Fast, J., Gentner, D. R., Goldstein, A. H., Hayes, P. L., Knighton, W. B., Oetjen, H., Setyan, A., Stark, H., Thalman, R., Tyndall, G., Washenfelder, R., Waxman, E., and Zhang, Q.: Simulation of semi-explicit mechanisms of SOA formation from glyoxal in aerosol in a 3-D model, Atmos. Chem. Phys., 14, 6213–6239, https://doi.org/10.5194/acp-14-6213-2014, 2014.
Komazaki, Y., Hiratsuka, M., Narita, Y., Tanaka, S., and Fujita, T.: The development of an automated continuous measurement system for the monitoring of HCHO and CH3CHO in the atmosphere by using an annular diffusion scrubber coupled to HPLC, Fresen. J. Anal. Chem., 363, 686–695, https://doi.org/10.1007/s002160051272, 1999.
Kukui, A., Chartier, M., Wang, J., Chen, H., Dusanter, S., Sauvage, S., Michoud, V., Locoge, N., Gros, V., Bourrianne, T., Sellegri, K., and Pichon, J.-M.: Role of Criegee intermediates in the formation of sulfuric acid at a Mediterranean (Cape Corsica) site under influence of biogenic emissions, Atmos. Chem. Phys., 21, 13333–13351, https://doi.org/10.5194/acp-21-13333-2021, 2021.
Lary, D. J. and Shallcross, D. E.: Central role of carbonyl compounds in atmospheric chemistry, J. Geophys. Res.-Atmos., 105, 19771–19778, https://doi.org/10.1029/1999JD901184, 2000.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Li, F., Tang, S., Lv, J., Yu, S., Sun, X., Cao, D., Wang, Y., and Jiang, G.: Critical contribution of chemically diverse carbonyl molecules to the oxidative potential of atmospheric aerosols, Atmos. Chem. Phys., 24, 8397–8411, https://doi.org/10.5194/acp-24-8397-2024, 2024.
Lily, M., Hynniewta, S., Muthiah, B., Wang, W., Chandra, A. K., and Liu, F.: Quantum chemical insights into the atmospheric reactions of CH2FCH2OH with OH radical, fate of CH2FC•HOH radical and ozone formation potential, Atmos. Environ., 249, 118247, https://doi.org/10.1016/j.atmosenv.2021.118247, 2021.
Lin, H., Jacob, D. J., Lundgren, E. W., Sulprizio, M. P., Keller, C. A., Fritz, T. M., Eastham, S. D., Emmons, L. K., Campbell, P. C., Baker, B., Saylor, R. D., and Montuoro, R.: Harmonized Emissions Component (HEMCO) 3.0 as a versatile emissions component for atmospheric models: application in the GEOS-Chem, NASA GEOS, WRF-GC, CESM2, NOAA GEFS-Aerosol, and NOAA UFS models, Geosci. Model Dev., 14, 5487–5506, https://doi.org/10.5194/gmd-14-5487-2021, 2021.
Liu, Q., Gao, Y., Huang, W., Ling, Z., Wang, Z., and Wang, X.: Carbonyl compounds in the atmosphere: A review of abundance, source and their contributions to O3 and SOA formation, Atmos. Res., 274, 106184, https://doi.org/10.1016/j.atmosres.2022.106184, 2022.
Liu, S., Chen, Y., Jiang, H., Shi, J., Ding, H., Yang, X., and Dong, W.: Reaction between Criegee Intermediate CH2OO and Isobutyraldehyde: Kinetics and Atmospheric Implications, Chem. Select, 8, e202303129, https://doi.org/10.1002/slct.202303129, 2023.
Liu, Y., Zhou, X., Chen, Y., Chen, M., Xiao, C., Dong, W., and Yang, X.: Temperature- and pressure-dependent rate coefficient measurement for the reaction of CH2OO with CH3CH2CHO, Phys. Chem. Chem. Phys., 22, 25869–25875, https://doi.org/10.1039/D0CP04316H, 2020.
Long, B., Bao, J. L., and Truhlar, D. G.: Atmospheric chemistry of Criegee intermediates: unimolecular reactions and reactions with water, J. Am. Chem. Soc., 138, 14409–14422, https://doi.org/10.1021/jacs.6b08655, 2016.
Long, B., Bao, J. L., and Truhlar, D. G.: Kinetics of the strongly correlated CH3O + O2 reaction: The importance of quadruple excitations in atmospheric and combustion chemistry, J. Am. Chem. Soc., 141, 611–617, https://doi.org/10.1021/jacs.8b11766, 2019.
Long, B., Wang, Y., Xia, Y., He, X., Bao, J. L., and Truhlar, D. G.: Atmospheric kinetics: Bimolecular reactions of carbonyl oxide by a triple-level strategy, J. Am. Chem. Soc., 143, 8402–8413, https://doi.org/10.1021/jacs.1c02029, 2021.
Long, B., Xia, Y., and Truhlar, D. G.: Quantitative kinetics of HO2 reactions with aldehydes in the atmosphere: High-order dynamic correlation, anharmonicity, and falloff effects are all important, J. Am. Chem. Soc., 144, 19910–19920, https://doi.org/10.1021/jacs.2c07994, 2022.
Long, B., Xia, Y., Zhang, Y.-Q., and Truhlar, D. G.: Kinetics of sulfur trioxide reaction with water vapor to form atmospheric sulfuric acid, J. Am. Chem. Soc., 145, 19866–19876, https://doi.org/10.1021/jacs.3c06032, 2023.
Long, B., Zhang, Y.-Q., Xie, C.-L., Tan, X.-F., and Truhlar, D. G.: Reaction of carbonyl oxide with hydroperoxymethyl thioformate: Quantitative kinetics and atmospheric implications, Research, 7, 0525, https://doi.org/10.34133/research.0525, 2024.
Long, B., Xie, C., and Truhlar, D. G.: Criegee intermediates compete well with OH as a cleaning agent for atmospheric amides, J. Am. Chem. Soc., 147, 22237–22244, https://doi.org/10.1021/jacs.5c07439, 2025.
Luecken, D. J., Hutzell, W. T., Strum, M. L., and Pouliot, G. A.: Regional sources of atmospheric formaldehyde and acetaldehyde, and implications for atmospheric modeling, Atmos. Environ., 47, 477–490, https://doi.org/10.1016/j.atmosenv.2011.10.005, 2012.
Luo, P.-L., Chen, I. Y., Khan, M. A. H., and Shallcross, D. E.: Direct gas-phase formation of formic acid through reaction of Criegee intermediates with formaldehyde, Commun. Chem., 6, 130, https://doi.org/10.1038/s42004-023-00933-2, 2023.
Lynch, B. J., Zhao, Y., and Truhlar, D. G.: Effectiveness of Diffuse Basis Functions for Calculating Relative Energies by Density Functional Theory, J. Phys. Chem. A, 107, 1384–1388, https://doi.org/10.1021/jp021590l, 2003.
Manonmani, G., Sandhiya, L., and Senthilkumar, K.: Reaction of Criegee Intermediates with SO2-A Possible Route for Sulfurous Acid Formation in the Atmosphere, ACS Earth Space Chem., 7, 1890–1904, https://doi.org/10.1021/acsearthspacechem.3c00058, 2023.
Mellouki, A., Wallington, T. J., and Chen, J.: Atmospheric chemistry of oxygenated volatile organic compounds: Impacts on air quality and climate, Chem. Rev., 115, 3984–4014, https://doi.org/10.1021/cr500549n, 2015.
Novelli, A., Vereecken, L., Lelieveld, J., and Harder, H.: Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 16, 19941–19951, https://doi.org/10.1039/C4CP02719A, 2014.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Papagni, C., Arey, J., and Atkinson, R.: Rate constants for the gas-phase reactions of a series of C3-C6 aldehydes with OH and NO3 radicals, Int. J. Chem. Kinet., 32, 79–84, https://doi.org/10.1002/(SICI)1097-4601(2000)32:2<79::AID-KIN2>3.0.CO;2-A, 2000.
Parker, T. M., Burns, L. A., Parrish, R. M., Ryno, A. G., and Sherrill, C. D.: Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies, J. Chem. Phys., 140, 094106, https://doi.org/10.1063/1.4867135, 2014.
Parrish, D. D., Ryerson, T. B., Mellqvist, J., Johansson, J., Fried, A., Richter, D., Walega, J. G., Washenfelder, R. A., de Gouw, J. A., Peischl, J., Aikin, K. C., McKeen, S. A., Frost, G. J., Fehsenfeld, F. C., and Herndon, S. C.: Primary and secondary sources of formaldehyde in urban atmospheres: Houston Texas region, Atmos. Chem. Phys., 12, 3273–3288, https://doi.org/10.5194/acp-12-3273-2012, 2012.
Peltola, J., Seal, P., Inkilä, A., and Eskola, A.: Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: unimolecular decomposition of CH2OO and its reaction with formic acid, Phys. Chem. Chem. Phys., 22, 11797–11808, https://doi.org/10.1039/D0CP00302F, 2020.
Percival, C. J., Welz, O., Eskola, A. J., Savee, J. D., Osborn, D. L., Topping, D. O., Lowe, D., Utembe, S. R., Bacak, A., McFiggans, G., Cooke, M. C., Xiao, P., Archibald, A. T., Jenkin, M. E., Derwent, R. G., Riipinen, I., Mok, D. W. K., Lee, E. P. F., Dyke, J. M., Taatjes, C. A., and Shallcross, D. E.: Regional and global impacts of Criegee intermediates on atmospheric sulphuric acid concentrations and first steps of aerosol formation, Faraday Discuss., 165, 45–73, https://doi.org/10.1039/C3FD00048F, 2013.
Peverati, R. and Truhlar, D. G.: M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics, J. Phys. Chem. Lett., 3, 117–124, https://doi.org/10.1021/jz201525m, 2012.
Raghunath, P., Lee, Y.-P., and Lin, M. C.: Computational Chemical Kinetics for the Reaction of Criegee Intermediate CH2OO with HNO3 and Its Catalytic Conversion to OH and HCO, J. Phys. Chem. A, 121, 3871–3878, https://doi.org/10.1021/acs.jpca.7b02196, 2017.
Ren, X., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Shirley, T., Adams, J., Simpas, J. B., and Brune, W. H.: HOx concentrations and OH reactivity observations in New York City during PMTACS-NY2001, Atmos. Environ., 37, 3627–3637, https://doi.org/10.1016/S1352-2310(03)00460-6, 2003.
Scollard, D. J., Treacy, J. J., Sidebottom, H. W., Balestra-Garcia, C., Laverdet, G., LeBras, G., MacLeod, H., and Teton, S.: Rate constants for the reactions of hydroxyl radicals and chlorine atoms with halogenated aldehydes, J. Phys. Chem., 97, 4683–4688, https://doi.org/10.1021/j100120a021, 1993.
Sellevåg, S. R., Stenstrøm, Y., Helgaker, T., and Nielsen, C. J.: Atmospheric chemistry of CHF2CHO: Study of the IR and UV−Vis absorption cross sections, photolysis, and OH-, Cl-, and NO3-Initiated oxidation, J. Phys. Chem. A, 109, 3652–3662, https://doi.org/10.1021/jp050313m, 2005.
Sivakumaran, V., Hölscher, D., Dillon, T. J., and Crowley, J. N.: Reaction between OH and HCHO: temperature dependent rate coefficients (202–399 K) and product pathways (298 K), Phys. Chem. Chem. Phys., 5, 4821–4827, https://doi.org/10.1039/B306859E, 2003.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/C2CS35140D, 2012.
Stone, D., Blitz, M., Daubney, L., Howes, N. U. M., and Seakins, P.: Kinetics of CH2OO reactions with SO2, NO2, NO, H2O and CH3CHO as a function of pressure, Phys. Chem. Chem. Phys., 16, 1139–1149, https://doi.org/10.1039/C3CP54391A, 2014.
Sun, Y., Long, B., and Truhlar, D. G.: Unimolecular Reactions of E-Glycolaldehyde Oxide and Its Reactions with One and Two Water Molecules, Research, 6, 0143, https://doi.org/10.34133/research.0143, 2024.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Osborn, D. L., Lee, E. P. F., Dyke, J. M., Mok, D. W. K., Shallcross, D. E., and Percival, C. J.: Direct measurement of Criegee intermediate (CH2OO) reactions with acetone, acetaldehyde, and hexafluoroacetone, Phys. Chem. Chem. Phys., 14, 10391–10400, https://doi.org/10.1039/C2CP40294G, 2012.
Tereszchuk, K. A. and Bernath, P. F.: Infrared absorption cross-sections for acetaldehyde (CH3CHO) in the 3µm region, J. Quant. Spectrosc. Radiat. Transfer, 112, 990–993, https://doi.org/10.1016/j.jqsrt.2010.12.003, 2011.
Thévenet, R., Mellouki, A., and Le Bras, G.: Kinetics of OH and Cl reactions with a series of aldehydes, Int. J. Chem. Kinet., 32, 676–685, https://doi.org/10.1002/1097-4601(2000)32:11<676::AID-KIN3>3.0.CO;2-V, 2000.
Wang, P.-B., Truhlar, D. G., Xia, Y., and Long, B.: Temperature-dependent kinetics of the atmospheric reaction between CH2OO and acetone, Phys. Chem. Chem. Phys., 24, 13066–13073, https://doi.org/10.1039/D2CP01118B, 2022.
Wei, Y., Zhang, Q., Huo, X., Wang, W., and Wang, Q.: The reaction of Criegee intermediates with formamide and its implication to atmospheric aerosols, Chemosphere, 296, 133717, https://doi.org/10.1016/j.chemosphere.2022.133717, 2022.
Wenger, J. C.: Chamber Studies on the Photolysis of Aldehydes Environmental, Environmental Simulation Chambers: Application to Atmospheric Chemical Processes, Dordrecht, 111–119, https://doi.org/10.1007/1-4020-4232-9_8,, 2006.
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., Schütz, M., Celani, P., Györffy, W., Kats, D., Korona, T., Lindh, R., Mitrushenkov, A., Rauhut, G., Shamasundar, K. R., Adler, T. B., Amos, R. D., Bennie, S. J., Bernhardsson, A., Berning, A., Cooper, D. L., Deegan, M. J. O., Dobbyn, A. J., Eckert, F., Goll, E., Hampel, C., Hesselmann, A., Hetzer, G., Hrenar, T., Jansen, G., Köppl, C., Lee, S. J. R., Liu, Y., Lloyd, A. W., Ma, Q., Mata, R. A., May, A. J., McNicholas, S. J., Meyer, W., Miller III, T. F., Mura, M. E., Nicklass, A., O'Neill, D. P., Palmieri, P., Peng, D., Pflüger, K., Pitzer, R., Reiher, M., Shiozaki, T., Stoll, H., Stone, A. J., Tarroni, R., Thorsteinsson, T., Wang, M., and Welborn, M.: MOLPRO, version 2019.2, a package of ab initio programs, https://www.molpro.net/ (last access: 4 November 2025), 2019.
Xia, Y., Long, B., Lin, S., Teng, C., Bao, J. L., and Truhlar, D. G.: Large pressure effects caused by internal rotation in the s-cis-syn-Acrolein stabilized Criegee intermediate at tropospheric temperature and pressure, J. Am. Chem. Soc., 144, 4828–4838, https://doi.org/10.1021/jacs.1c12324, 2022.
Xia, Y., Long, B., Liu, A., and Truhlar, D. G.: Reactions with Criegee intermediates are the dominant gas-phase sink for formyl fluoride in the atmosphere, Fundam. Res., 4, 1216–1224, https://doi.org/10.1016/j.fmre.2023.02.012, 2024.
Xia, Y., Zhang, W., Tang, X., and Long, B.: Quantitative kinetics of the hydrogen shift reaction of methylthiomethyl peroxy radical (CH3SCH2OO) in the atmosphere, J. Phys. Chem. A, 129, 2275–2285, https://doi.org/10.1021/acs.jpca.4c06818, 2025.
Yang, X., Xue, L., Wang, T., Wang, X., Gao, J., Lee, S., Blake, D. R., Chai, F., and Wang, W.: Observations and explicit modeling of summertime carbonyl formation in Beijing: Identification of key precursor species, J. Geophys. Res.-Atmos., 123, 1426–1440, https://doi.org/10.1002/2017JD027403, 2018.
Zhang, L., Truhlar, D. G., and Sun, S.: Association of Cl with C2H2 by unified variable-reaction-coordinate and reaction-path variational transition-state theory, Proc. Natl. Acad. Sci., 117, 5610–5616, https://doi.org/10.1073/pnas.1920018117, 2020.
Zhang, R. M., Xu, X., and Truhlar, D. G.: TUMME: Tsinghua University Minnesota Master Equation program, Comput. Phys. Commun., 270, 108140, https://doi.org/10.1016/j.cpc.2021.108140, 2022.
Zhang, T., Wen, M., Ding, C., Zhang, Y., Ma, X., Wang, Z., Lily, M., Liu, J., and Wang, R.: Multiple evaluations of atmospheric behavior between Criegee intermediates and HCHO: Gas-phase and air-water interface reaction, J. Environ. Sci., 127, 308–319, https://doi.org/10.1016/j.jes.2022.06.004, 2023.
Zhang, Y., Mu, Y., Liu, J., and Mellouki, A.: Levels, sources and health risks of carbonyls and BTEX in the ambient air of Beijing, China, J. Environ. Sci., 24, 124–130, https://doi.org/10.1016/S1001-0742(11)60735-3, 2012.
Zhao, M., Shen, H., Zhang, J., Liu, Y., Sun, Y., Wang, X., Dong, C., Zhu, Y., Li, H., Shan, Y., Mu, J., Zhong, X., Tang, J., Guo, M., Wang, W., and Xue, L.: Carbonyl Compounds Regulate Atmospheric Oxidation Capacity and Particulate Sulfur Chemistry in the Coastal Atmosphere, Environ. Sci. Technol., 58, 17334–17343, https://doi.org/10.1021/acs.est.4c03947, 2024.
Zheng, J. and Truhlar, D. G.: Multi-path variational transition state theory for chemical reaction rates of complex polyatomic species: ethanol + OH reactions, Faraday Discuss., 157, 59–88, https://doi.org/10.1039/C2FD20012K, 2012.
Zheng, J., Zhang, S., and Truhlar, D. G.: Density Functional Study of Methyl Radical Association Kinetics, J. Phys. Chem. A, 112, 11509–11513, https://doi.org/10.1021/jp806617m, 2008.
Zheng, J., Mielke, S. L., Clarkson, K. L., and Truhlar, D. G.: MSTor: A program for calculating partition functions, free energies, enthalpies, entropies, and heat capacities of complex molecules including torsional anharmonicity, Comput. Phys. Commun., 183, 1803–1812, https://doi.org/10.1016/j.cpc.2012.03.007, 2012.
Zheng, J., Bao, J. L., Meana-Pañeda, R., Zhang, S., J.Lynch, B., Corchado, J. C., Chuang, Y., Fast, P. L., Hu, W.-P., Liu, Y.-P., Lynch, G. C., Nguyen, K. A., Jackels, C. F., Ramos, A. F., Ellingson, B. A., Melissas, V. S., Villà, J., Rossi, I., Coitiño, E. L., Pu, J., Albu, T. V., Ratkiewicz, A., Steckler, R., Garrett, B. C., Isaacson, A. D., and Truhlar, D. G.: Polyrate-version 2017-C; University of Minnesota: Minneapolis, https://comp.chem.umn.edu/polyrate/ (last access: 4 November 2025), 2017.
Zheng, J., Bao, J. L., Zhang, S., Corchado, J. C., Chuang, Y., Ellingson, B. A., and Truhlar, D. G.: Gaussrate, version 2017-B; University of Minnesota: Minneapolis, MN, https://comp.chem.umn.edu/polyrate/ (last access: 4 November 2025), 2018.
Zhu, L., Talukdar, R. K., Burkholder, J. B., and Ravishankara, A. R.: Rate coefficients for the OH + acetaldehyde (CH3CHO) reaction between 204 and 373 K, Int. J. Chem. Kinet., 40, 635–646, https://doi.org/10.1002/kin.20346, 2008.