the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Apr 2026
| 21 Apr 2026
Impacts of summertime photochemical aging on the physicochemical properties of aerosols in a Paris suburban forest region
Paola Formenti
Joel F. de Brito
Astrid Bauville
Antonin Bergé
Hichem Bouzidi
Mathieu Cazaunau
Manuela Cirtog
Claudia Di Biagio
Ludovico Di Antonio
Cécile Gaimoz
Franck Maisonneuve
Pascal Zapf
Tobias Seubert
Simone T. Andersen
Patrick Dewald
Gunther N. T. E. Türk
John N. Crowley
Alexandre Kukui
Chaoyang Xue
Cyrielle Denjean
Olivier Garrouste
Jean-Claude Etienne
Huihui Wu
James D. Allan
Dantong Liu
Yangzhou Wu
Christopher Cantrell
Vincent Michoud
Organic aerosols (OA) play a significant role in influencing both climate and human health. However, in source–receptor modelling, a large fraction of OA is typically attributed to highly aged, atmospherically processed species collectively referred to as oxygenated organic aerosol (OOA). Nevertheless, the formation pathways and evolution of OOA as well as their impacts on aerosol optical properties, remain poorly understood. To address this knowledge gap, an experiment was conducted in a suburban site in the Paris region to study the evolution of OOA and their optical properties. Our results show that in regionally transported air masses with mixed biogenic and anthropogenic emissions, the formation of OOA through photochemical processes explains most of the increase in submicron particle mass. Meteorological conditions played a critical role: under dry and strong solar radiation conditions, enhanced formation of more-oxidized OOA (MO-OOA) was observed. BrC absorption increased concurrently, with short-wavelength absorption rising by ∼ 35 % over relatively ∼ 24 h of photochemical aging. Conversely, under humid, low-radiation conditions, the OA composition shifted toward less-oxidized OOA (LO-OOA). Suppressed photochemistry limited MO-OOA production, resulting in a lower overall OA oxidation state. These findings highlight the role of photochemistry in shaping both the chemical evolution and resultant optical properties of OA, underscoring the need to consider meteorological dynamics when evaluating aerosol–climate interactions in suburban forest environments.
- Article
(3394 KB) - Full-text XML
-
Supplement
(3669 KB) - BibTeX
- EndNote




Atmospheric aerosols are complex combinations of different chemical species and have an important impact on the climate system by scattering and absorbing atmospheric radiation (Ravishankara et al., 2015) and by interacting with clouds (Farmer et al., 2015). They also negatively affect air quality by reducing visibility, causing respiratory problems, and damaging ecosystems (Manisalidis et al., 2020). Urban areas, with their intensive resource use, are major contributors to anthropogenic aerosol emissions globally (Zhang et al., 2020; Papachristopoulou et al., 2022; Franklin et al., 2025). Therefore, characterizing the physicochemical properties of aerosols is crucial for understanding their impact on climate, air quality, and human health, ultimately informing strategies for mitigation and environmental protection.
Aerosols contain various components varying from volatile to refractory materials. Globally, Organic Aerosols (OA) often comprise the dominated mass fraction of submicron aerosols, while other components include soot (also known as Black Carbon (BC)), inorganic salts (such as sulfates, nitrates, and ammonium), metals, and other elements (Seinfeld and Pandis, 2016). OA can be classified into primary OA (POA) and secondary OA (SOA) compounds. POA compounds are hydrocarbon components emitted from primary sources such as biomass burning, cooking, and transport (e.g. Allan et al., 2010; Lu et al., 2011), while SOA compounds are formed through atmospheric chemical reactions involving the oxidation of gas precursors such as volatile organic compounds (VOCs) (Kroll and Seinfeld, 2008). Source apportionment analysis on OA based on mass spectra from widely used instruments such as the Aerodyne Aerosol Mass Spectrometer (AMS) (Jayne et al., 2000) or Aerosol Chemical Speciation Monitor (ACSM) (Ng et al., 2011) can provide insight into its primary and secondary components. The Positive Matrix Factorization (PMF) analysis of AMS/ACSM (Ulbrich et al., 2009; Zhang et al., 2011) derived POA typically includes hydrocarbon-like OA (HOA), biomass burning OA (BBOA), and cooking-related OA (COA), while the SOA (also referred as oxygenated OA (OOA), often used as a proxy for SOA) is often typically separated into Less-Oxidized OOA (LO-OOA) and More-Oxidized OOA (MO-OOA). At some measurement sites, specific components like peat-emitted OA, shipping-related OA, or sea spray-related OA can also be identified (Lin et al., 2020; Fossum et al., 2024; Schmale et al., 2013). Previous AMS/ACSM studies revealed that the OOA dominated the total submicron OA mass in the European region with a yearly average fraction of more than 70 %, while the contribution from solid-fuel related POA is about 16 % yearly on average and mostly during the winter months (Chen et al., 2022). The chemical characteristics and evolution of OA have significant impacts on aerosol optical properties. Particulate Organic Nitrate (pON), an important part of OA formed either through primary emissions such as combustion-related sources (e.g. Reyes-Villegas et al., 2018), or through the oxidation of VOCs in the presence of nitrogen oxide radicals and atmospheric oxidants (e.g. Ng et al., 2017), can substantially contribute to the total light-absorbing brown carbon (BrC) loadings (Laskin et al., 2015). Studies show that the contribution of light absorption by BrC may be enhanced due to the increase of nitrogenous organic aerosols during photochemical aging (Yang et al., 2021; Jiang et al., 2022; Cappa et al., 2020). However, bleaching of BrC through intense photooxidation processes has also been documented, with significant temporal variability depending on factors such as oxidant concentrations and ambient relative humidity (RH) (Liu et al., 2021; Zhao et al., 2015). The variability of OOA also influences the aerosol hygroscopicity, and the complex aging process of OOA introduces significant uncertainty in representing the cloud condensation nuclei (CCN) activity of atmospheric aerosols (Mei et al., 2013; Wang et al., 2022). Given all these factors, understanding the aging process of OA is crucial to better constrain their role in the atmosphere.
Paris is one of the most developed and densely populated megacities in Europe, facing significant air pollution challenges. In the past decades, extensive research has been conducted to identify and characterize the properties of particulate pollutants in the Paris region (e.g. Beekmann et al., 2015). Previous aerosol mass spectrometer studies (Stirnberg et al., 2021; Bressi et al., 2013; Petit et al., 2015; Healy et al., 2013) indicate that Paris is often affected by the mid-range to long-range transport pollutants attribute to the flat orography of the city. Previous AMS-PMF analysis during summertime have shown that, in addition to the significant contributions from the continental polluted air mass to the total particulate matter with aerodynamic diameter less than 1 µm (PM1), Paris peri-urban area is also influenced by emissions from the Atlantic Ocean (Crippa et al., 2013). Previous investigations (Healy et al., 2013; Crippa et al., 2013; Petit et al., 2015) in the Paris region were primarily carried out in urban and peri-urban environments, where strong local anthropogenic emissions may obscure regional transport signals and introduce biases in characterising aerosol aging processes. However, most of these studies were conducted more than a decade ago. In recent years, ozone (O3) has emerged as a major pollutant in European urban areas, and reductions in nitrogen oxides (NOx) have been reported to contribute to increasing urban O3 concentrations (Grange et al., 2021). These changes imply a shift in atmospheric oxidation capacity, highlighting the need to reassess the evolution of aerosol physicochemical properties during transport under present-day chemical regimes. To address these issues, the Atmospheric Chemistry of the Suburban Forest (ACROSS) project (Cantrell and Michoud, 2022) included a ground-based field campaign in a forested suburban site to capture regionally transported air masses arriving in the Paris region. In this study, we analyse those observations to examine the evolution of aerosol chemical composition in relation to photochemical processing, with particular emphasis on its implications for aerosol optical properties in ambient conditions.
2.1 Atmospheric Chemistry of the Suburban Forest (ACROSS) project
A ground-based experiment was conducted in the suburban forested area of Rambouillet, (48.7° N, 1.7° E) ∼ 50 km southwest from the centre of Paris in the framework of the ACROSS project (Cantrell and Michoud, 2022). The forest is composed of approximately 70 % oak, 20 % pine, and 10 % beech and chestnut trees. The canopy height of the trees is on the order of ∼ 20–25 m. During the campaign, multiple containers were placed side by side in a clear area (with a size of 26.5 m × 26.3 m, ∼ 697 m2) within the forest. The broader experiment, encompassing both near-ground (∼ 5 m) and above-canopy (on a ∼ 40 m-high tower) measurements, was conducted from 13 June to 25 July 2022. As it will be discussed in the following sections, in this study we analyse near-ground measurements in the period 29 June to 20 July, ensuring instrument availability.
2.2 Particulate phase measurements
2.2.1 High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-AMS)
The Non-Refractory PM1 (NR-PM1) compositions, including organic (OA), sulfate (SO4), nitrate (NO3), ammonium (NH4), and chloride (Chl), was characterised through an Aerodyne HR-AMS (DeCarlo et al., 2006). Briefly, particles enter the AMS through a critical orifice and an aerodynamic lens, forming a narrow particle beam. The particle size (vacuum aerodynamic diameter) is determined in a Particle Time-of-Flight (PToF) region through the flight time of particles. NR components are subsequently vaporized on a metallic surface, ionized by electron impact ionization (EI, 70 eV), and analysed by Time-of-Flight Mass Spectrometry (ToF-MS) under high vacuum. During the campaign, the HR-AMS conducted measurements with a flow rate of ∼ 0.1 L min−1, drawn from the main 3 L min−1 flow from a PM2.5 cyclone and through a Nafion dryer. The HR-AMS sampled intermittently, alternating between near-ground and above-canopy measurements every 5 min. This study focuses exclusively on data collected at near-ground level. The HR-AMS was operated alternatively among two modes: V-mode and PToF mode. The Ionization Efficiency (IE) and the Relative Ionization Efficiency (RIE) of the HR-AMS were calibrated using mono-disperse (300 nm), nebulized ammonium nitrate and ammonium sulfate particles, and the IE and RIE results were presented in Table S1. A Composition-Dependent Collection Efficiency (CDCE) was applied to the final results (Middlebrook et al., 2012). Background CO2 in the HR-AMS was corrected by calibrating the measured CO2 in filtered air against external measurements obtained with a cavity ringdown spectrometer (CRD, Picarro G2302).
The mass concentrations of different NR-PM1 species and high-resolution mass spectra analysis were processed from the V-mode results. Elemental ratios of OA, including oxygen-to-carbon (O : C), hydrogen-to-carbon (H : C), and nitrogen-to-carbon (N : C), as well as the organic mass to organic carbon (OM : OC), were derived from high-resolution mass spectra using the “Improved-Ambient (I-A)” method (Canagaratna et al., 2015). The contribution of pON to the total HR-AMS measured NO3 is estimated following the method described by Day et al. (2022), as shown in Eq. (1):
Where fpON is the estimated fraction between the pON mass concentration and the total HR-AMS measured NO3, and Rambient, , and RpON are the ratios between NO and NO+ for ambient, pure NH4NO3, and pure pON (determined as 2.75), respectively.
The source apportionment analysis of OA was performed on the high-resolution OA mass spectral matrix for 12–120 in V mode from the HR-AMS using the PMF Evaluation Tool (PET) (Ulbrich et al., 2009). Though a Biomass Burning (BB) event was observed on the evening of 19 July (presented in the time-series results in Fig. 2), it only lasted for few hours and the unconstrained PMF analysis is unable to derive a distinct BB factor for this event. To avoid biasing the PMF solution, results from this period were excluded from the PMF analysis in this study. A six-factor solution with fPeak = 0 was chosen as the optimal solution. Although the mass spectra of the OOA factors were highly similar (Pearson correlation coefficient r > 0.8), their time series remained distinct, with r < 0.6 in the six-factor solution. The spectra of the six factors were comparable to those from previous summer AMS PMF study in Paris urban area (Crippa et al., 2013) documented in the AMS spectral database (Jeon et al., 2023), and the comparison is presented in Table S2 in the Supplement.
2.2.2 Aethalometer
The PM1 aerosol optical absorption coefficient (units of Mm−1 = 10−6 m−1) at seven different wavelengths (370, 470, 520, 590, 660, and 880 and 950 nm) was measured by a dual-spot aethalometer (AE33, Magee Sci.) (Drinovec et al., 2015) at the PortablE Gas and Aerosol Sampling UnitS (PEGASUS) mobile platform (Formenti et al., 2025) which was positioned close to the HR-AMS container. The AE33 was operated at a flow rate of 5 L min−1 and connected to a PM1 cyclone and a Nafion drier. The whole inlet system has a total flow rate of around 38 L min−1. The AE33 measures the light attenuation coefficient (σatn) through a filter tape, which was sampled at 1 min intervals. The spectral aerosol absorption coefficient is determined as:
Where Cref is the multiple scattering correction factor. While the filter-based AE33 quantifies σabs via transmittance attenuation from deposited particles, its accuracy is compromised by multi-scattering interactions within the filter matrix, including variable aerosol loading and filter leakage (Yus-Díez et al., 2021). A wavelength-independent Cref of 3.38 has been applied to correct the final absorption coefficient results, and more details about the corrections of AE33 results during the ACROSS campaign are presented in Di Antonio et al. (2025a, b). The light absorbing equivalent Black Carbon (eBC) mass concentration is derived from the multiple scattering corrected 880 nm channel results using a mass absorption cross section (MAC) of 7.77 m2 g−1 (Petzold et al., 2013; Bond et al., 2013). Due to anomalous peaks at 370 nm were observed when fresh filter tape was brought into the analytical airflow during the experiments, the results from 370 nm were excluded from this study, while other wavelengths remained unaffected.
To determine the absorption contributed by the BrC, firstly, the Absorption Ångström exponent (AAE) is calculated through:
Where and represent the absorption coefficient measured at wavelengths λ1 (470 nm) and λ2 (880 nm), respectively. The BrC absorption coefficient is determined in the assumption that absorption at 880 nm is contributed by BC only and the AAE for BC is 1. Henceforth, the σabs contributed by BC at 470 nm can be derived as:
Then the σabs of BrC at 470 nm can be estimated as:
A Single Particle Soot Photometer (SP2, DMT) was employed to characterise the BC mixing state but was only operational for a limited period during the campaign due to a technical issue, with data available partially between 11 and 19 July. The coating thickness of BC particles was derived from the SP2 results using leading-edge only (LEO) method (Gao et al., 2007), and the mass ratio between the non-BC coating material and BC core material (MR) was 1.65 (± 1.38), assuming densities as 1.8 g cm−3 for BC and 1.6 g cm−3 for non-BC coating. According to Liu et al. (2017), when MR is less than 3, the lensing effect becomes less pronounced. Therefore, the lensing effect of BC can be considered negligible in this study.
2.2.3 Total PM1 mass concentration calculation
The total PM1 mass concentration was determined as the sum of the NR-PM1 and eBC mass concentration measured by the HR-AMS and the AE-33, respectively. Although the HR-AMS was operated downstream of a PM2.5 cyclone, its aerodynamic lens transmits only submicron particles. The two sampling inlet systems showed comparable line loss rates for PM1, with maximum losses estimated at approximately 6 % for PM1 in both inlet systems following the method described in von der Weiden et al. (2009).
2.3 Gas phase measurements
The nitrogen oxides (NOx, NOx = NO + NO2) and total gas-phase nitrogen species (NOy) concentrations were measured by the MPIC thermal dissociation cavity ringdown spectroscopy (TD-CRD) instrument at 405 nm (Friedrich et al., 2020), and more details about the operations of the MPIC TD-CRD during ACROSS project are described in Andersen et al. (2024). NO2 concentration was measured by a chemiluminescence analyzer (HORIBA APNA 370). The O3 and CO concentrations were measured using a commercial monitor (HORIBA APOA-370) and a CRD (Picarro G2302) at PEGASUS platform, respectively. VOCs were characterised through the Proton Transfer Reaction Time-of-Flight Mass Spectrometer (PTR-MS, KORE Inc.). The instrument operated in the PEGASUS mobile lab in H3O+ mode with a drift tube pressure of 1.30 mbar and a temperature of 40 °C, along with a drift tube voltage of 397 V, resulting in an ratio of 134.2 Td. Air samples were drawn at a flow rate of approximately 300 mL min−1 through a 3 m-long Silcosteel® coated stainless steel tube (2.1 mm inner diameter). The PTR-MS conducted full mass scan mode analysis (10–270 amu). The PTR-MS was calibrated during the campaign using a NPL (National Physical laboratory) VOC standards for PTR-MS every three days. Water dependencies of various compounds were determined by performing calibration at various relative humidity before and after the campaign using the same NPL standard cylinder. Non-calibrated compounds were retrieved from reactor conditions, rate constants, fragmentations, and ion transmissions determined using the same NPL standard cylinder. An automated zero procedure was performed every hour for 10 min by sampling humid zero air generated by passing ambient air through a catalytic converter. OH radicals were measured through the Chemical Ionisation Mass Spectrometry (CIMS, LPC2E), and more details about the instrument was described in Kukui et al. (2008). Briefly, the CIMS determines OH radical concentrations by chemically converting sampled OH into H2SO4 by addition of SO2 in a chemical conversion reactor in the presence of water vapour and oxygen (Eisele and Tanner, 1991). H2SO4 is detected by the mass spectrometer as HSO ion. To distinguish this signal from ambient H2SO4, the conversion process uses isotopically labelled 34SO4 leading to the formation of HSO4.
2.4 Other supporting measurements
The surface downwelling shortwave radiation and wind parameters were measured by a net radiometer (Kipp & Zonen, CNR4: 192119) and a wind monitor (Young, 05103: 4655) mounted on the tower. As the measurements were conducted in a clear area with no trees, the tower-top observations were considered representative of near-surface atmospheric conditions relevant for radiation and wind.
2.5 Relative anthropogenic photochemical age estimation
Photochemical processing of the mixed anthropogenic–biogenic air masses arriving at the measurement site was evaluated using the ratio between NOx and NOy as proposed by Kleinman et al. (2003, 2007), as done in previous ground-based measurement studies (Hayes et al., 2013; Ensberg et al., 2014). The relative anthropogenic photochemical age (tanthrop-photo) in this study is calculated following the method described by Nault et al. (2018):
Where [NOx] and [NOy] are the measured concentrations of NOx and NOy, respectively, [OH]avg is the average measured OH concentration during the campaign period, which is 1.6 (± 1.3) × 106 molec. cm−3. is the rate coefficient of OH with NO2 and is assumed to be 9.2 × 10−12 cm3 molec.−1 s−1 (Mollner et al., 2010). The estimated tanthrop-photo was constrained to the period of 06:00–20:00 European Summer Time (EST) according to the diurnal trend of the radiation intensity (presented in Fig. 3a and e).
The results are expressed as “relative anthropogenic photochemical age” since the method mainly captures the aging of anthropogenic pollutants in the mixed anthropogenic–biogenic air masses arriving at the site. In addition, the potential short lifetime of NOy in the forest (Andersen et al., 2024) may compromise the accuracy of absolute photochemical age calculation. Only the calculated tanthrop-photo within 1 d (24 h) was used here to reduce the uncertainty of the estimated tanthrop-photo due to the potential short lifetime of HNO3 and other oxidised reservoirs via deposition in the forest (Nguyen et al., 2015; Romer et al., 2016).
2.6 UK Met Office NAME dispersion model
2.6.1 Air mass source region classification
Potential source regions were identified using the UK Met Office Numerical Atmospheric-dispersion Modelling Environment (NAME) dispersion model (Jones et al., 2007). The model calculates the integrated historical air mass contribution over 24 h by back-tracking tracer particles (released at 1 g s−1 from the sampling site) on a 0.25° × 0.25° horizontal grid within the boundary layer every 4 h. The model distinguishes between air masses primarily influenced by local sources and those transported regionally from broader areas. The potential air mass sources are classified into five main regions (presented in Fig. 1a) according to the CAMS EAC4 (ECMWF Atmospheric Composition Reanalysis 4) global reanalysis PM1 spatial distribution results (presented in Fig. 1b) (Inness et al., 2019) during the campaign period: (1) the Ile-de-France region (IDF (local), 48–50° N, 1–3° E) represents the local emission from the Paris region; (2) the Northeast (NE) region (50–54° N, 1–10° E) is identified as a source of anthropogenic polluted continental air masses, evidenced by significantly elevated PM1 concentrations in this area; (3) the Northwest (NW) region (50–54° N, −5–1° E) represents the source of polluted maritime air masses originating from the English Channel and the United Kingdom (UK); (4) the Southwest (SW) region (44–50° N, −5–3° E, exclude the IDF region) is characterized as a source of low-pollution maritime air masses, as evidenced by persistently low PM1 concentrations over Southwest France despite several wildfire activities (the spatial distribution of fire points information during the experiment period is presented in Fig. S2); and (5) the Southeast (SE) region (44–48° N, 3–10° E) represents another source of polluted continental air mass due to the high PM1 concentration over Southeast France and North Italy. The air mass examples are presented in Fig. S1 in the Supplement.
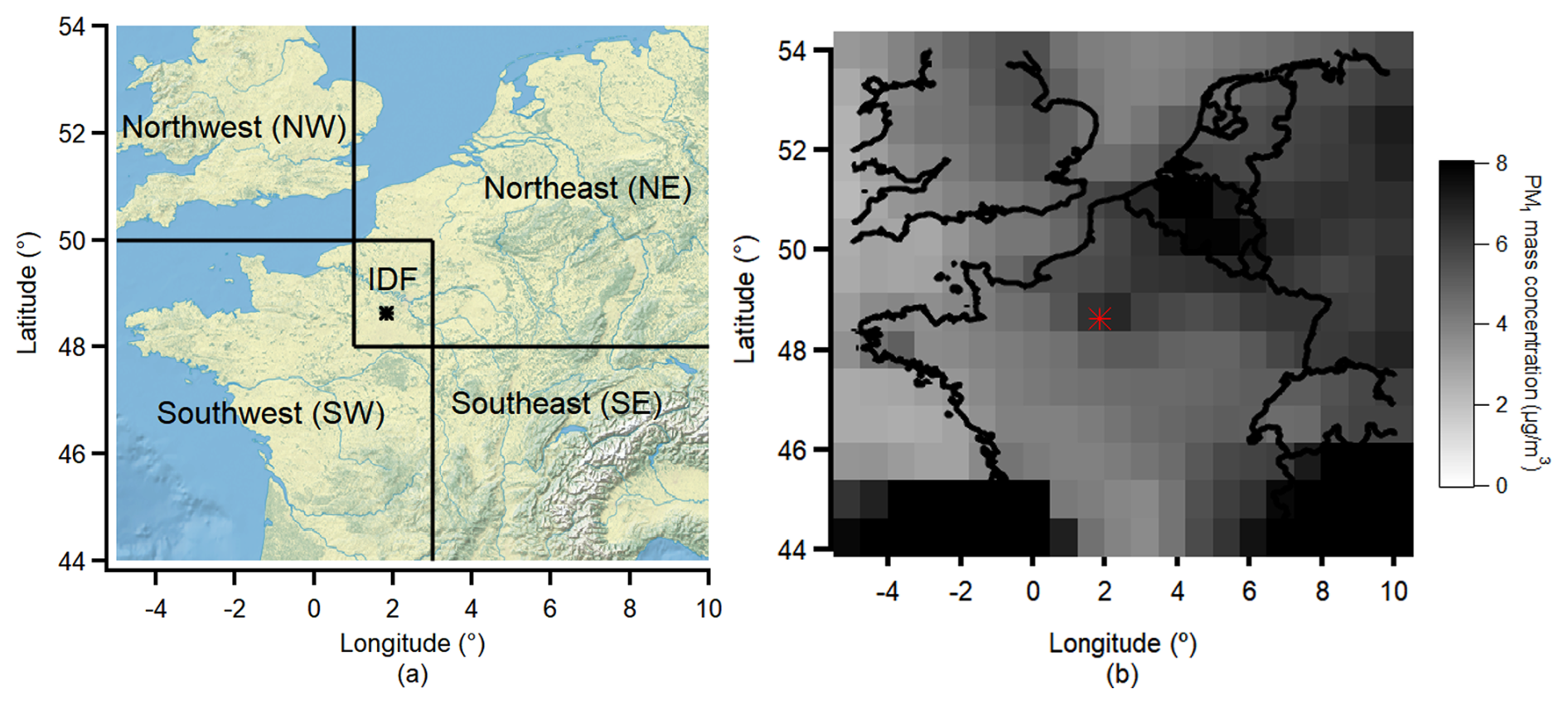
Figure 1(a) Classification of the potential air mass source regions; (b) ECMWF CAMS EAC4 (ECMWF Atmospheric Composition Reanalysis 4) reanalysis PM1 spatial distribution during the ACROSS campaign period. The dot in the figure represents the location of the Rambouillet measurement site. Maps were created from public domain GIS data on Natural Earth (http://www.naturalearthdata.com, last access: 1 April 2025).
2.6.2 Estimation of trajectory-based plume transport age
Plume transport age was derived from backward air-mass trajectories to provide an independent validation of tanthrop-photo inferred from NOx NOy ratios. Backward air-mass trajectories were simulated using the NAME model with the same model domain as applied in the backward dispersion analysis. The model generated 24 h backward trajectories by releasing ensembles of Lagrangian particles at hourly intervals and tracking their transport on a 0.25° × 0.25° grid, with particle positions recorded every 15 min. For each backward trajectory, the dominant potential air-mass source region was first identified following the method described in Sect. 2.6.1. The EDGAR anthropogenic PM2.5 emission inventory (0.01° × 0.01° resolution, reported as total PM2.5 emission in Tg) (Crippa et al., 2024) for the campaign period (June–July 2022) was used to determine the dominant anthropogenic emission areas associated with each identified source region (Fig. S12). The emission inventory indicates that no major anthropogenic emission hotspots are present between these source regions and the measurement site, supporting the interpretation that the observed air masses reflect regional-scale transport without substantial intermediate source contributions. The plume transport age was defined as the time elapsed between the observation time at the measurement site and the first backward-trajectory intersection with the corresponding anthropogenic emission region. The trajectory-derived transport age serves as a relative indicator for validating the temporal variability of tanthrop-photo based on the assumption that air masses with longer transport times generally experience longer cumulative photochemical processing. Detailed comparison between tanthrop-photo and transport age is presented in Sect. S10 in the Supplement.
3.1 Overview of meteorology and campaign conditions
Figure 2a presents the time series of temperature and RH, PM1 composition, and the fractional contributions of different air masses throughout the whole campaign period. The average mass concentrations of PM1 species and the normalised OA under different air mass dominated periods are displayed in Fig. 2b and c. While the contribution from the local emissions (IDF air mass) was significant, external air masses also had a substantial impact on PM1 mass concentrations. The SE air mass-dominated period was excluded from further analysis due to its short duration during the campaign. The NE air masses brought higher average aerosol mass loadings compared to the other sectors, in agreement with the previous summertime studies in Paris, indicating that the continental air mass is generally more polluted (Crippa et al., 2013). The continental air masses also typically resulted in the highest temperatures and the lowest RH. In contrast, air masses originating from oceanic influence, whether from the SW or NW sectors, resulted in lower aerosol loadings in the Paris region, while higher RH and lower temperatures prevailed. The ECMWF CAMS reanalysis shows higher average PM1 concentrations over the Southwest France due to the forest fires burning during the summer of 2022 (Menut et al., 2023). Pronounced increases in PM1 mass concentration and acetonitrile were observed on the evening of 19 July during periods dominated by SW and SE air masses, indicating a BB event. However, acetonitrile concentrations exhibited only limited variability during the rest of the measurement period. In addition, molecular analysis over the campaign period identified a significant enhancement of the BB tracer (nitrocatechols, C6H5NO4) only on 19 July (Pereira et al., 2025). These results indicate that BB emissions made a limited contribution during the campaign, apart from the event on 19 July.
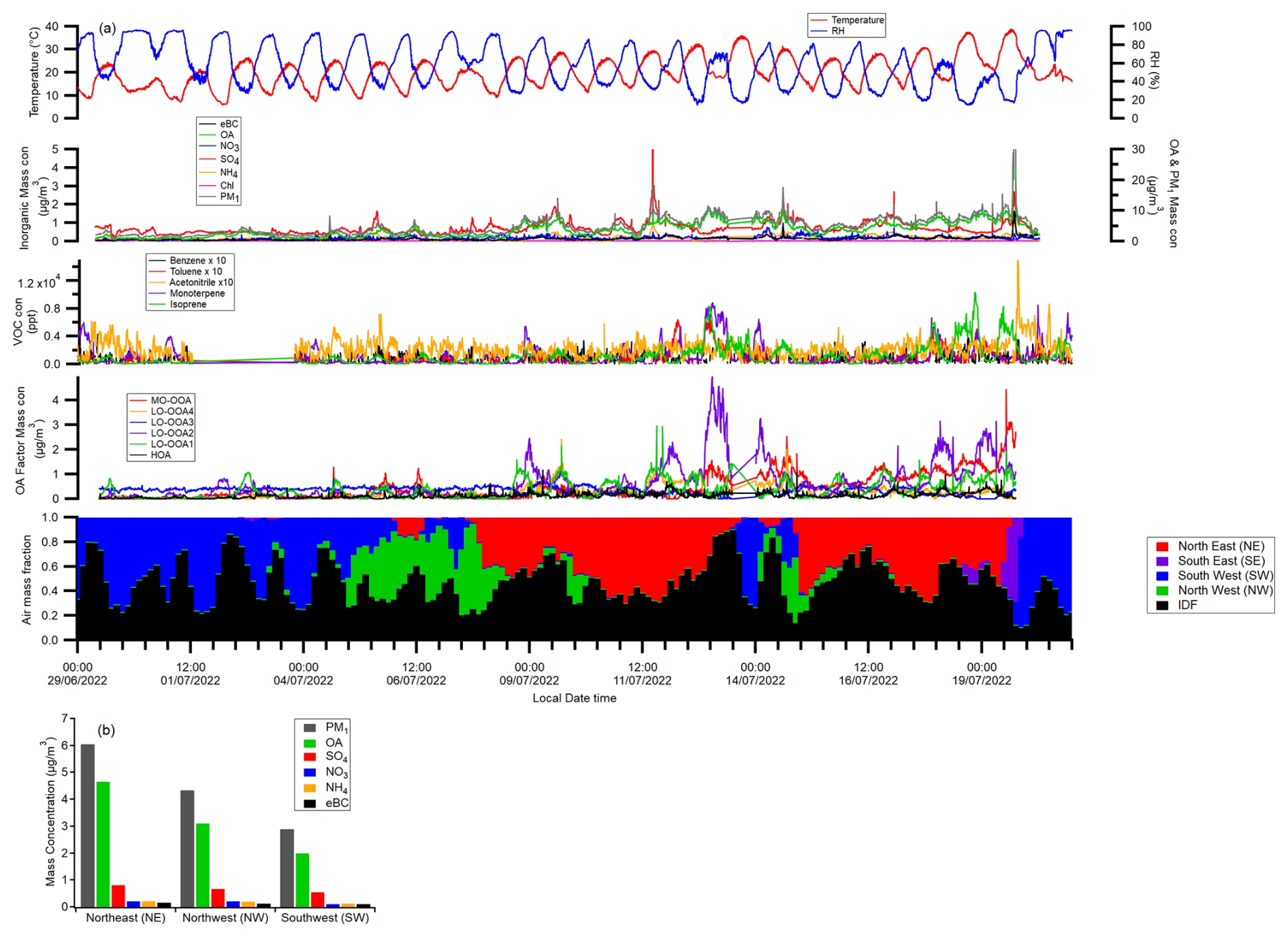
Figure 2(a) Time series of temperature, RH, PM1, VOC, PMF OA factors and historic air mass fractions; (b) the average PM1 composition mass concentrations under different external air mass dominated periods.
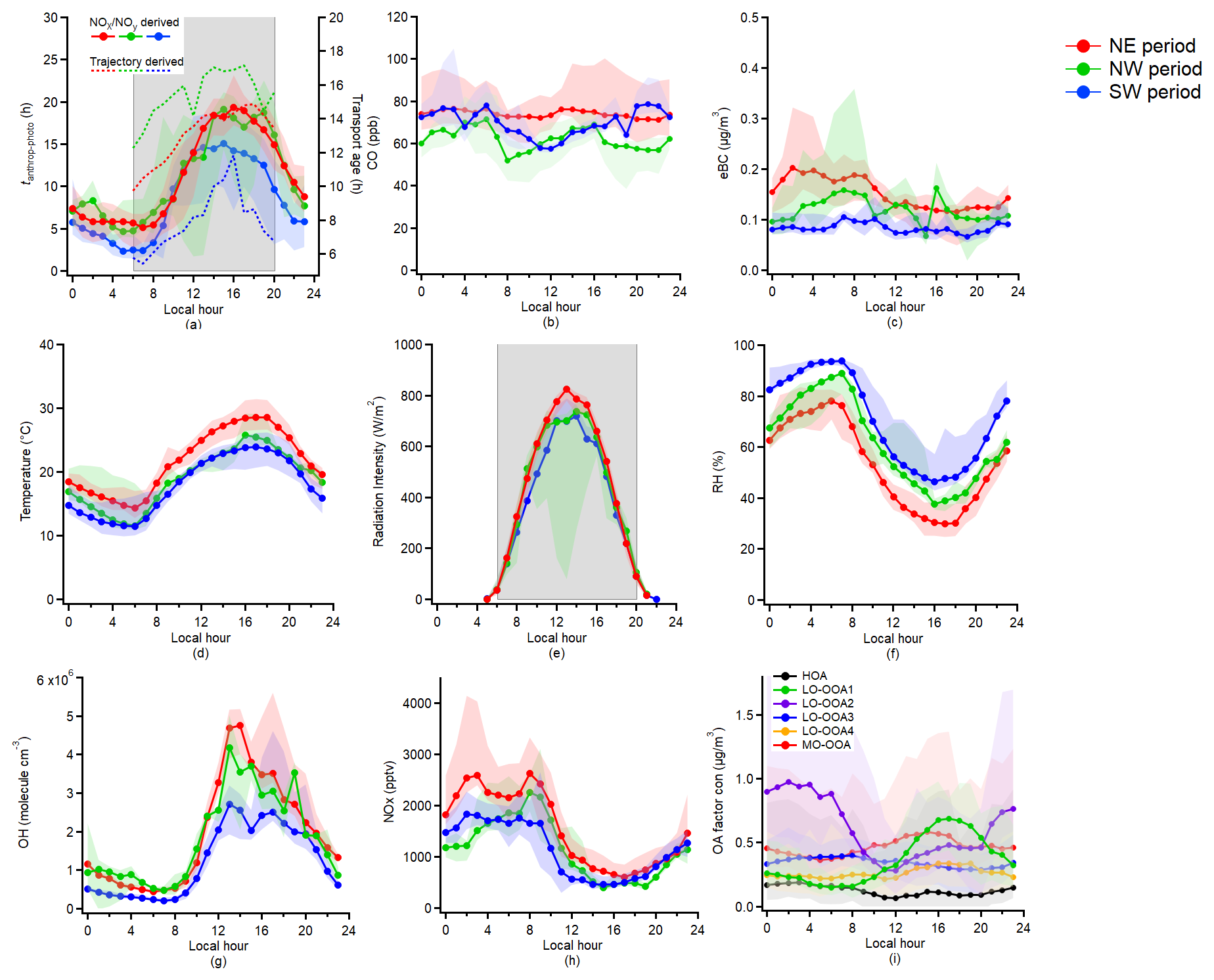
Figure 3Diurnal variation of the (a) Relative anthropogenic photochemical age (tanthrop-photo) derived from NOx NOy ratio and trajectory-derived transport age; (b) CO mixing ratio; (c) eBC mass concentration; (d) Temperature; (e) Radiation Intensity; (f) RH; (g) OH concentration; (h) NOx concentration; (i) PMF OA factors. The grey shades in panels (a) and (e) indicate the periods for the photochemical aging analysis. The lower and upper whiskers in the figure are the 25th and 75th percentiles, respectively.
Figure 3 presents the diurnal variations of the tanthrop-photo, CO concentration, eBC mass concentration, temperature, downward shortwave radiation, RH, OH and NOx concentrations under the different external air masses dominated periods. The BB event on the evening of 19 July was excluded from the analysis. Based on the diurnal trends in meteorological parameters, increases in radiation intensity during the day were inversely correlated with decreases in RH, which reached its lowest level in the late afternoon. As shown in Fig. 3a, the diurnal variation of tanthrop-photo followed that of the trajectory-based transport age derived from the NAME dispersion model. As presented in Sect. S10 in the Supplement, these two independent indicators exhibited comparable temporal trends, supporting the use of tanthrop-photo as a proxy for relative photochemical aging during the campaign. In addition, as illustrated in Fig. 3a and g, the diurnal pattern of tanthrop-photo was consistent with the observed diurnal cycles of OH concentration and radiation intensity, further supporting its robustness as a relative indicator of photochemical aging. Due to the weaker radiation intensity or the potential shorter transport distance, the average tanthrop-photo and OH concentration of the SW air mass dominated period were lower compared to the other air masses dominated periods. Since the measurements were conducted away from urban emission sources, the diurnal average eBC mass concentration was with no discernible peak during rush hours, and there was only limited increase in NOx concentrations during the morning rush hours during NE and NW periods. The daytime decrease of CO concentration during NW and SW periods was due to boundary layer dilution, with lowest CO concentrations at midday. In contrast, the NE period showed no diurnal variation of CO, suggesting the accumulation of regionally transported pollutants from surrounding areas at the suburban forest site.
3.2 Source apportionment of organic aerosols
Figure 4 presented the average mass spectra of the resolved six PMF OA factors: one Hydrocarbon-like OA (HOA) and five OOA factors. The Pearson correlation coefficient (r) between the HR-AMS PMF factors and the external tracers is presented in Fig. 5. The time series of the PMF-derived OA factors between 29 June and 20 July 2022, is presented in Fig. 2a. Figure 3i shows the diurnal variations of the OA factors, and Fig. 6 presents the mass fractions of the OA factors according to the dominated air mass periods. The comparison between below- and above-canopy PMF results is provided in the Supplement, showing that the trends of NR-PM1 and PMF for below- and above-canopy were consistent during the daytime throughout the campaign. Thus, the influence of localized fresh plumes and nearby human activities was minimal since the boundary layer was well mixed during the daytime during the campaign period, supporting our subsequent findings on regional pollution within the boundary layer during the daytime.
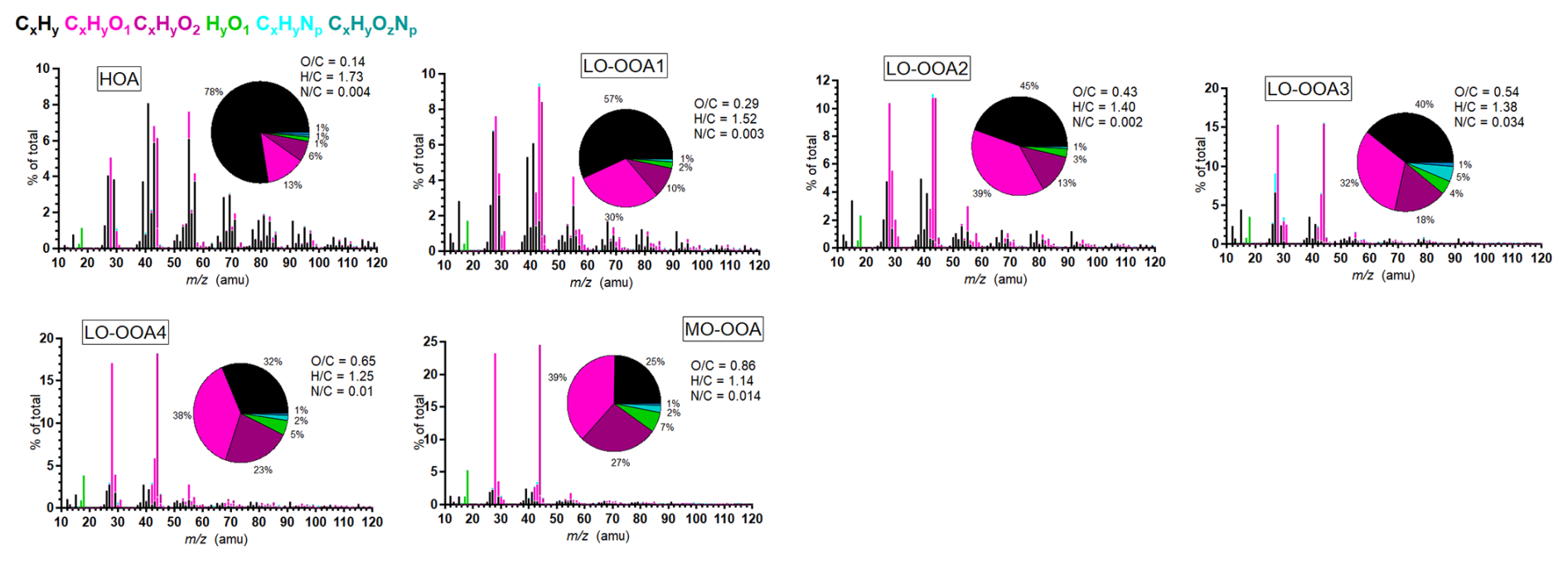
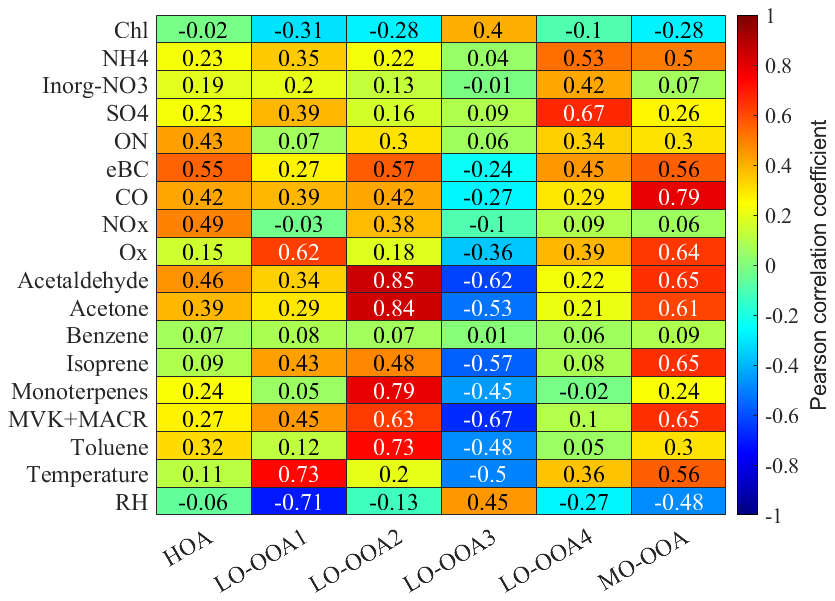
Figure 5Pearson correlation coefficient (r) between the PMF OA factors and the external tracers.
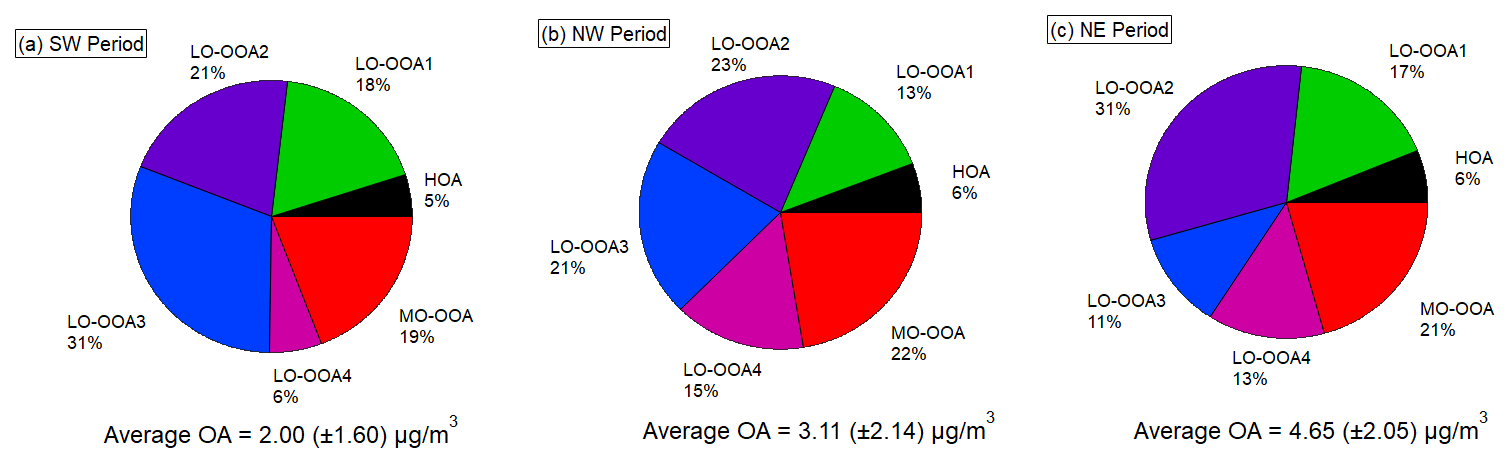
Figure 6Pie charts for the mass fractions of different PMF OA factors during the three different airmass dominated periods. Values are reported as mean ± standard deviation.
The OOA factors accounted for over 90 % of total OA during the campaign, contributing a larger mass fraction than in previous studies conducted in the Paris urban area or its surroundings (Crippa et al., 2013; Petit et al., 2015; Zhang et al., 2019). As expected, the HOA factor has the lowest O : C ratio (0.15) and the highest fraction of CxHy fragments, notably at 41, 43, 55, and 57. Temporal variability shows sporadic enhancements associated with occasional vehicular activity in the vicinity of the site. However, HOA only exhibited moderate correlation with eBC (r = 0.55) and with NOx (r = 0.49). This may be due to the remote location of the measurement site, primary anthropogenic emission sources were limited near the site.
LO-OOA1 and LO-OOA2 exhibited very similar mass spectral profiles but distinct temporal patterns, with LO-OOA1 peaking during the daytime and LO-OOA2 during the nighttime. The correlation between LO-OOA2 and toluene (r = 0.73) may be due to two reasons. Firstly, it may arise from instrumental interference, as a fraction of monoterpenes or biogenic compounds related to monoterpenes is known to fragment under PTR-MS ionization conditions and produce ions at 93 (C7H) (Tani et al., 2003; Ambrose et al., 2010), which overlap with the ion signal of toluene. This effect can be particularly pronounced at this remote measurement site, where ambient toluene concentrations were low. Secondly, toluene may also originate from biogenic sources. Previous studies (Misztal et al., 2015; White et al., 2009) have shown that certain vegetation types can also emit toluene, indicating that not all observed toluene necessarily derives from anthropogenic emissions. Given these two reasons, toluene may not serve as a reliable tracer of anthropogenic primary sources for our study, as only limited temporal variability relative to CO was observed during the campaign (presented in Sect. S7 in the Supplement). The moderate correlation between LO-OOA1 and isoprene was to be expected in such a temperate forest, given the typical mixtures of BSOA precursors and the differing lifetimes of aerosol- and gas-phase compounds. LO-OOA2 exhibited a pronounced nocturnal maximum and showed the strongest correlation with monoterpenes among the measured VOCs, indicating a close link to monoterpene oxidation. As shown in Fig. S10, LO-OOA2 concentrations were decoupled between the above- and below-canopy layers at night, with enhanced levels observed below the canopy. This vertical gradient suggests that LO-OOA2 formation was strongly influenced by locally emitted monoterpenes from surrounding vegetation. Dewald et al. (2024) reported that nighttime monoterpene oxidation was predominantly driven by O3 during the experimental period. Good correlation (r = 0.63) was observed between LO-OOA2 and the sum of methyl vinyl ketone (MVK) and methacrolein (MACR), while a fair correlation (r = 0.45) was found between LO-OOA1 and MVK + MACR. Isoprene epoxydiols (IEPOX)-OOA was not identified through the HR-AMS results during the experiment period, as the mass spectral profiles of both LO-OOA1 and LO-OOA2 lacked the characteristic enhancement at 82, a known tracer signal for IEPOX-OOA (Hu et al., 2015). However, both OOAs exhibited pronounced contributions at 91.05, attributed to C7H fragments likely from thermal decomposition of dimers and oligomers on the vaporizer (Riva et al., 2016). These two biogenic-related LO-OOA factors contributed approximately 40 % to 50 % of the total OA mass concentration throughout the campaign period.
The diurnal profile of LO-OOA3 showed a clear nocturnal enhancement, with concentrations increasing after approximately 20:00 and reaching a maximum between 06:00 and 08:00 EST. This accumulation is consistent with shallow nocturnal boundary layer conditions and limited vertical mixing, which favour the build-up of regionally transported and aged background OOA. The moderate positive correlation with RH (r = 0.45) further suggests that humid conditions may have facilitated its partitioning or accumulation. During daytime, LO-OOA3 concentrations declined and exhibited strong inverse correlations with photochemically related OVOCs (e.g., MVK + MACR, r = −0.69). The growth of the PBL after sunrise likely enhanced vertical mixing and dilution, further contributing to the daytime decrease. Together, these observations suggest that LO-OOA3 accumulated under stable nocturnal conditions and was progressively diluted and/or transformed during daytime photochemical aging. In terms of MS characteristics, LO-OOA3 exhibited the highest N : C ratio among the identified OOA factors. The dominant nitrogen-containing fragments were CHN+ ( 27.01) and CH4N+ ( 30.03), together accounting for approximately 6 % of the total LO-OOA3 signal. However, its weak correlation with pON (r = 0.06) suggests that the elevated N : C ratio did not correspond to a major contribution to pON variability. Overall, these features are consistent with LO-OOA3 representing a relatively less-oxidized regional background OOA component containing embedded nitrogen functionality.
LO-OOA4 exhibited a good correlation with sulfate (r = 0.67) and moderate correlation with inorganic nitrate (r = 0.42). The wind rose analysis (shown in Fig. S8 in the Supplement) reveals that LO-OOA4 predominantly originated from anthropogenic polluted air masses advected from the NE and NW regions. Consequently, during the SW period, LO-OOA4 contributed only 6 % to the total OA mass concentration. In contrast, its contribution increased to 15 % and 13 % with the increasing pollution level during the NW and NE periods, respectively.
The MO-OOA was with the highest average O : C ratio (0.86) among all the factors, and dominant peaks were presented at 28 (CO+) and 44 (CO). The good correlation between MO-OOA and isoprene was attributed to under daytime conditions, both elevated isoprene emissions and the oxidation of various precursors leading to MO-OOA formation occurred simultaneously. The significant increase in MO-OOA observed during the afternoon, despite boundary layer dilution, indicates that summertime MO-OOA formation was strongly driven by active photochemical processes. Due to the reduction of photochemical activity during the SW period, the mass fraction of MO-OOA during the SW period was smaller than the fraction during the other periods. The wind rose analysis shows that the MO-OOA was contributed significantly from the anthropogenic polluted NE regions.
3.3 Evolution of aerosol compositions due to photochemical aging
Figure 7 illustrates the evolution of Ox (O3 + NO2) mixing ratio, O : C ratio, and NR-PM1 components concentrations as a function of tanthrop-photo. The correlation between tanthrop-photo and Ox concentrations shown in Fig. 7a indicate that the photochemical process in summer has a significant impact on the formation of Ox throughout the experiment period. This agrees with the previous study in Beijing during summertime (Chen et al., 2021) and suggests that increased photochemical aging leads to greater consumption of VOCs, which in turn accelerates NOx cycling and promotes the accumulation of tropospheric O3. The NE period showed higher average Ox concentrations compared to the NW and SW periods during the relative ∼ 24 h photochemical aging period.
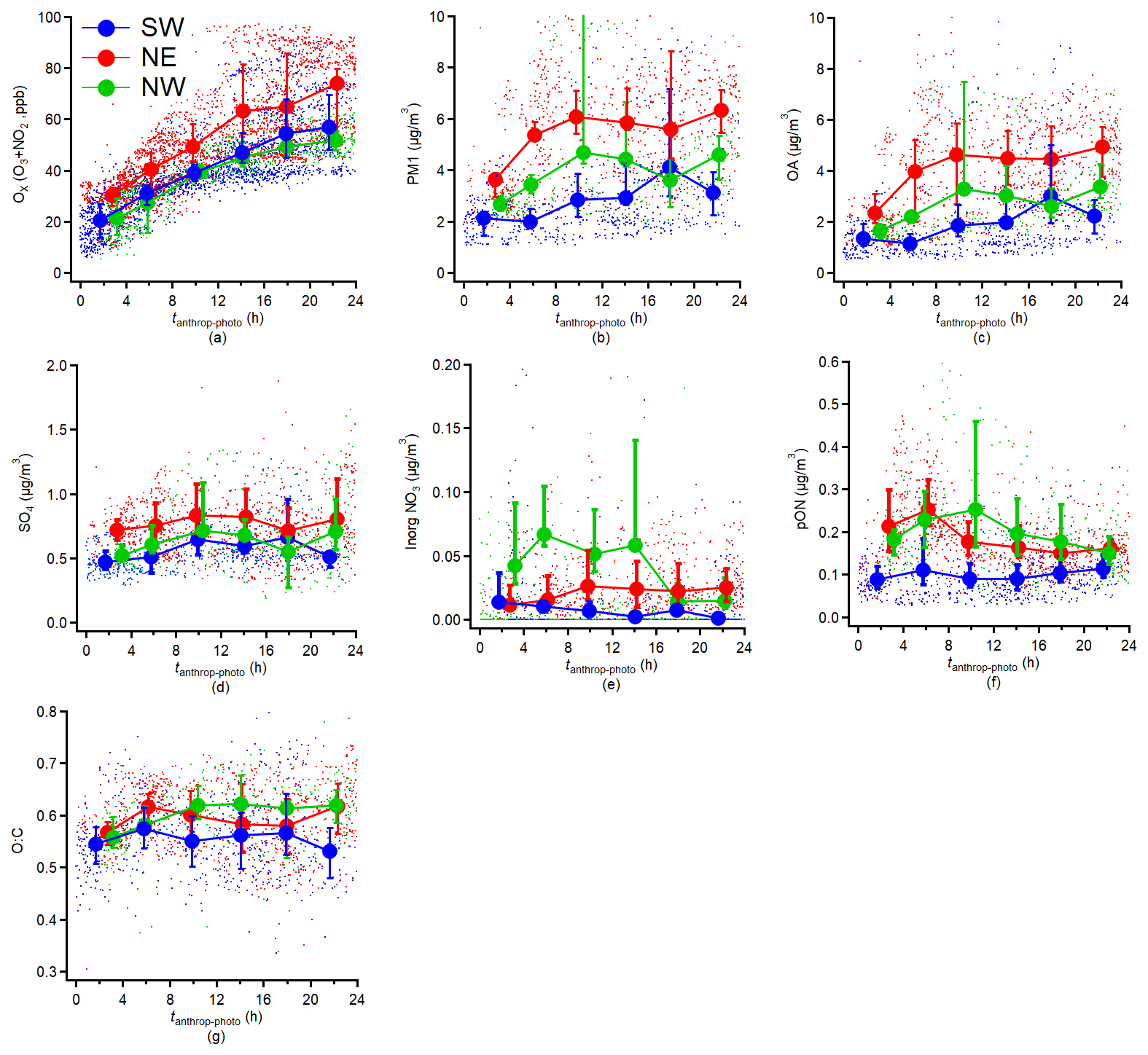
Figure 7Variations in aerosol compositions concentrations: (a) Ox (O3 + NO2), (b) PM1, (c) OA, (d) Sulfate, (e) Inorganic nitrate, (f) particulate Organic Nitrate (pON), (g) O : C ratio measured by HR-AMS as a function of tanthrop-photo. The results are averaged into 4 h intervals along tanthrop-photo and the error bar indicates the 25th and 75th percentiles, respectively.
The PM1 concentration, shown in Fig. 7b as a function of tanthrop-photo, exhibited an increase across all three periods with higher tanthrop-photo, and the OA mass concentration doubled following a relative ∼ 24 h photochemical aging process as presented in Fig. 7c. This high OA increase combined with no significant relative increase of other aerosol components (SO4, organic and inorganic NO3; Fig. 7d–f), indicates that photochemical OA formation played a dominant role in the observed increase in total PM1 mass after approximately 24 h of tanthrop-photo. This finding aligns with previous studies in Europe, which have shown that OOA formation substantially contributes to summer PM1 levels (Chen et al., 2022; Zhang et al., 2019). The increasing trend of OA also agreed with the results previously observed in urban plumes from Paris, Mexico City, and Seoul (Freney et al., 2014; DeCarlo et al., 2010; Nault et al., 2018) during photochemical process. However, there was a slight increase in the average bulk O : C ratio as a function of tanthrop-photo only during the NW period. Indeed, the average bulk O : C ratio did not show a corresponding increase with tanthrop-photo during the SW and NE period; instead, a slight decrease was observed at higher tanthrop-photo during the NE period. The average bulk O : C ratio at tanthrop-photo = 0 was approximately 0.55 across all periods, which was higher than the O : C ratios of most LO-OOA factors (LO-OOA1–3). This suggests that the regional background OA was already substantially oxidized prior to further photochemical processing. Consequently, additional aging may not necessarily lead to large increases in the average bulk O : C ratio. Other field studies in remote areas also report oxidation indicators reached a steady state rather than continuing to rise with photochemical age (Irei et al., 2015). During the NE period, the temporal evolution of bulk O : C exhibited a non-monotonic behaviour. Between 0 and 8 h of tanthrop-photo, the increase in MO-OOA contributed to a rise in average bulk O : C ratio. However, after ∼ 8 h, the more rapid formation of LO-OOA1 led to a greater relative contribution of less-oxidized components, thereby lowering the overall bulk oxidation level. As a result, the average bulk O : C decreased as tanthrop-photo increased from 8 to 20 h. Measurements in American urban environments report that LO-OOA increase more rapidly than MO-OOA during periods of active photochemical processing (Guo et al., 2024; Rogers et al., 2025).
The sulfate concentrations exhibited minimal variation with tanthrop-photo here, regardless of the period, likely due to the limited availability of SO2 precursors in the Rambouillet remote measurement site. Previous studies have demonstrated rapid conversion of SO2 into sulfate via aging processes in urban areas (Wu et al., 2022; Chen et al., 2021) or near emission sources (Yu et al., 2020). Concerning particulate nitrate, which was over 80 % attributed to pON on average during the experiment period, has shown distinct behaviours between its inorganic and organic fractions. For inorganic nitrate compounds, concentrations in the NW and SW period slightly decreased with increasing tanthrop-photo, whereas those in the NE period exhibited a minor increasing trend. It is likely that this variation was due to the inhomogeneous availability of NOx concentrations during different periods. The SW period showed a slight increase in the pON concentration with tanthrop-photo increases, while the NE and NW periods displayed a decreasing trend. This difference was due to depletion of primary pON and concurrent formation of secondary pON during photochemical aging, and this relationship will be further explored through linking to the trend of PMF-derived factors.
3.4 Evolution of PMF-derived OA factors due to photochemical aging
Figure 8 shows the evolution of the concentrations of HR-AMS PMF-derived factors as a function of tanthrop-photo. For the primary OA compounds, the HOA shown a decreasing trend with the increase of tanthrop-photo during the NE and NW periods. During the SW period, HOA concentrations exhibited a slight increase. Previous studies in the polluted urban areas have shown that strong photochemical processes lead to the oxidation of the HOA and traffic related primary VOCs (Wu et al., 2022; Chen et al., 2021). However, reduced radiation levels decreased the photochemical oxidation rate of primary traffic-related pollutants, potentially leading to their accumulation in suburban forested areas. As HOA has shown some correlation with the pON (r = 0.35), this may explain that the decline in pON concentration with tanthrop-photo was driven by photodepletion of primary pON during intense photochemical activity, while the photochemical loss of primary pON during the SW period was slower than the other periods.
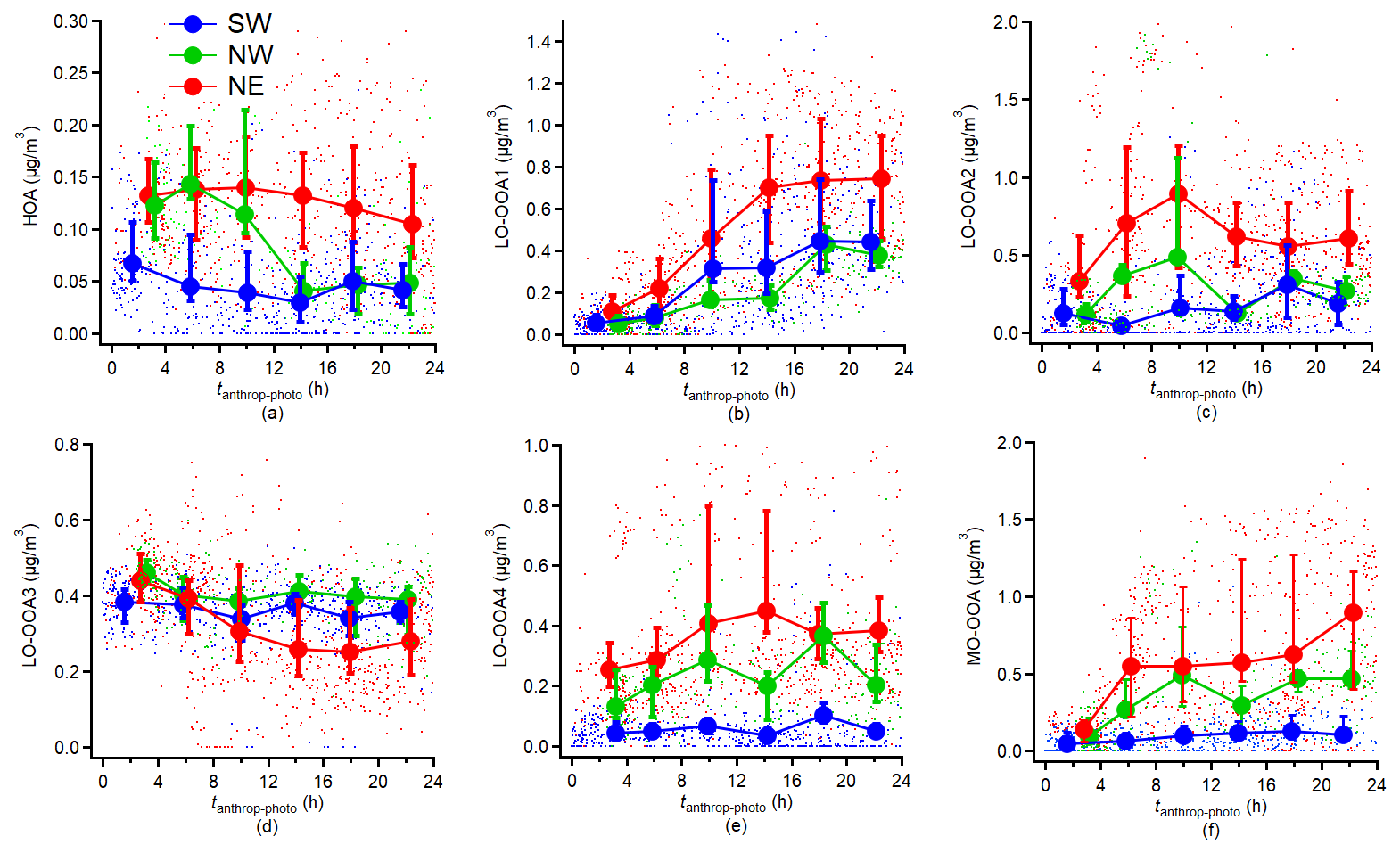
Figure 8Variations in (a–f) HR-AMS PMF-derived factors normalised or non-normalised concentrations. The results are averaged into 4 h intervals along tanthrop-photo and the error bar indicates the 25th and 75th percentiles, respectively.
LO-OOA1 showed an increasing trend during all three periods. However, LO-OOA2 showed no clear correlation with tanthropo−photo. As discussed in Sect. 3.2, LO-OOA2 can likely be attributed to SOA formation from locally emitted monoterpenes. As the Boundary Layer Height (BLH) increased during the daytime, LO-OOA2 was progressively diluted. Therefore, the variation of LO-OOA2 did not correspond to the evolution of tanthropo−photo.
During the NE period, LO-OOA3 exhibited a clear decreasing trend with increasing tanthrop-photo, whereas no distinct trend was observed in the other periods. Elevated OH radical and secondary oxidant concentrations during the NE period likely enhanced oxidative depletion of nitrogen-enriched LO-OOA3 relative to other periods. While continental pollution-related LO-OOA4 demonstrated a strong dependence on photochemical processes, its variability may also be influenced by dominant air masses, as the SW period was with lower average normalised mass concentrations compared to other periods. The MO-OOA showed strong correlation with the photochemical process. The normalised mass concentrations of MO-OOA during the NE and NW period were significantly higher than the concentration during the SW period after a relative ∼ 24 h photochemical aging. Limited formation of MO-OOA during the SW period led to a lower average bulk O : C ratio compared to other periods. Moreover, our findings indicate that highly oxidized particulate species were not favoured under conditions of low radiation and high RH. In addition, LO-OOA4 and MO-OOA also exhibited a correlation with pON (r = 0.34 and 0.3, respectively), suggesting that LO-OOA4 and MO-OOA importantly contributed to secondary pON formation during photochemical aging. Thereby, the concurrent depletion of HOA and formation of LO-OOA4 and MO-OOA likely govern the observed variations in total pON concentrations throughout the photochemical aging process.
3.5 The impact of photochemical aging on aerosol optical properties
Figure 9 illustrates the increasing contribution of BrC to the total aerosol light absorption coefficient at 470 nm as a function of tanthrop-photo, particularly during the NE and NW periods. As tanthrop-photo increased from around 2 h to nearly 24 h, the average BrC absorption fraction rose from about 17 % to 23 % in the NE period and from roughly 18 % to 26 % in the NW period. For the SW period, the average BrC absorption fraction remained relatively constant during the first 20 h of tanthrop-photo and increased to approximately 25 % after 24 h of tanthrop-photo. To identify and quantify the contribution of different OA factors to the total σabs BrC, 470, a Multiple Linear Regression (MLR) analysis was applied using the OA factors from the PMF analysis during the whole experiment period (with BB period removed). The p-test (p < 0.01) was also applied to assess the statistical significance of each parameter. The MLR relationship between σabs BrC, 470 and PMF-derived factors are formulated as:
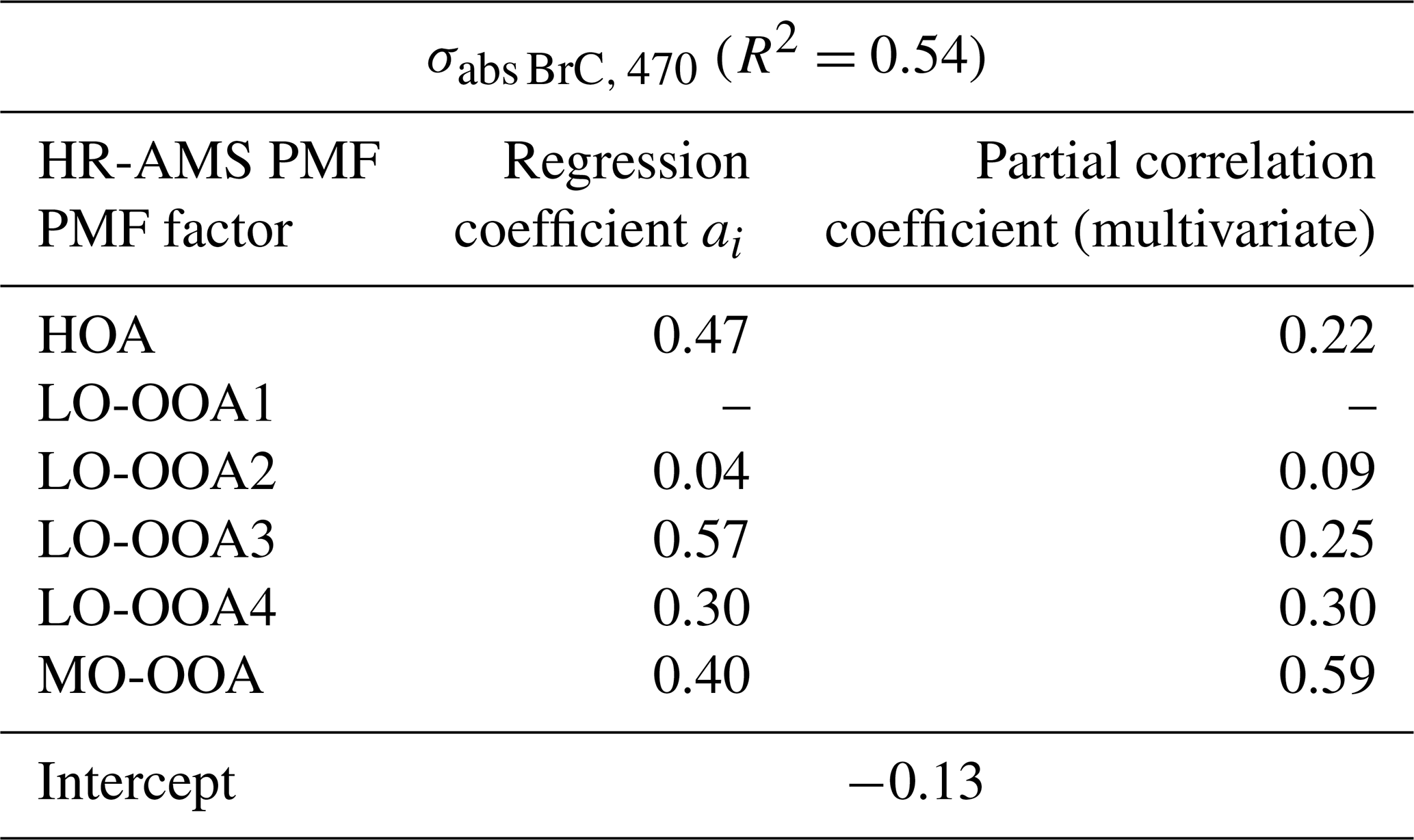
Where a0 represents the intercept, and ai represents the regression coefficient for each PMF-derived factor Ci. These regression coefficients can be associated with the Mass Absorption Coefficients (MAC) of each PMF-derived factor at 470 nm. The Variance Inflation Factors (VIF) for all factors were below 5, indicating that multicollinearity was not a concern. The partial correlation remained unchanged after variable standardization.
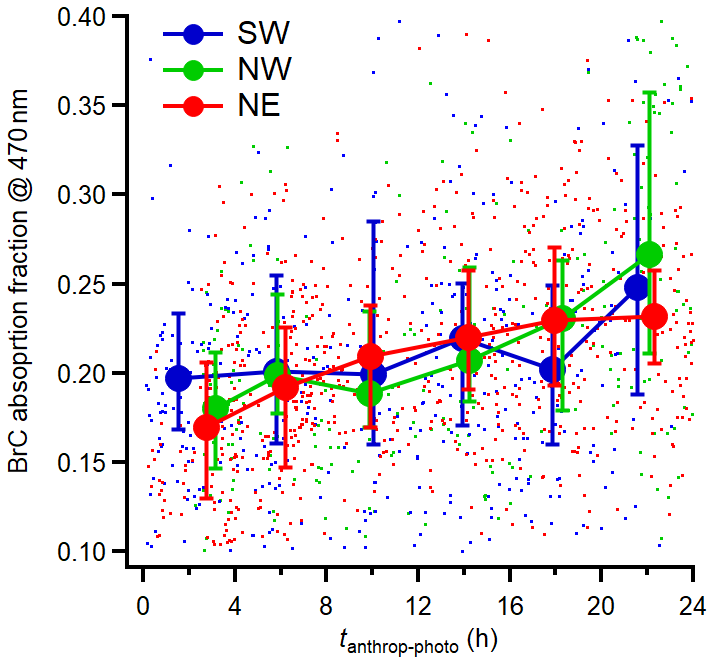
Figure 9Variations of absorption fraction of BrC at 470 nm as a function of tanthrop-photo. The results are averaged into 4 h intervals along tphoto and the error bar indicates the 25th and 75th percentiles, respectively.
Table 1 presents the results of the regression factors of the MLR analysis between σabs BrC, 470 and PMF-derived factors. The multivariate correlation coefficient is also presented to illustrate the contribution of each PMF-derived factor to the total BrC absorption during the whole experiment period. Interestingly, the LO-OOA3 and MO-OOA, which have the highest nitrogen content among all factors, also depict high MAC (regression coefficients). However, their multivariate associations with BrC differed substantially. MO-OOA showed the highest partial correlation with BrC among all the factors. In contrast, although LO-OOA3 displayed a relatively high MAC, its partial correlation with BrC was limited, suggesting a weaker contribution to the overall increase of BrC absorption during the daytime. One possible explanation is that LO-OOA3 represented a relatively labile nitrogen-containing precursor pool that underwent further transformation during daytime photochemical processing, with part of its chromophoric contribution being transferred to the more oxidized MO-OOA component. While this scenario is possible, it cannot be explicitly confirmed due to MO-OOA comprised contributions from multiple sources and oxidation pathways. Although bleaching of BrC after intensive photooxidation was observed in previous studies (Zhao et al., 2015), the available evidence did not indicate that bleaching was the dominant process during photochemical aging in the present observations. Under the oxidative conditions encountered here, MO-OOA likely retained net light-absorbing properties and continued to contribute to BrC absorption. The HOA and LO-OOA4 also showed high MAC which were 0.47 and 0.30 m2 g−1, respectively, and they exhibited fair correlation with BrC. The MAC reported here was close to the results from other suburban areas, for example, Jiang et al. (2022) observed MAC values of 0.4–0.8 m2 g−1 at 375 nm for OA with O : C ratios between 0.4 and 0.6. In contrast, our MAC values were lower than the MAC observed near emission source, for example, Washenfelder et al. (2015) reported a MAC of 1.35 m2 g−1 at 365 nm for BB emitted OA. It is interesting to note that HOA, LO-OOA4 and MO-OOA also exhibited correlation with the pON, typically associated as an important component of BrC (Laskin et al., 2015).
Table 1Results of multiple linear regression (MLR) between σabs BrC, 470 and individual PMF-derived factors. The final regression excludes the LO-OOA1 due to its negative regression coefficient value (−0.06). The Variance Inflation Factors (VIF) for HOA, LO-OOA2, LO-OOA3, LO-OOA4, and MO-OOA were 1.48, 1.16, 1.71, 2.27 and 1.31, respectively. Results include all the PMF-derived factors are presented in Table S2 in the Supplement.
Combined with the photochemical aging process discussed in Sect. 3.3, the decline in HOA concentration with photochemical aging during the NE and NW period demonstrated a reduced contribution of primary BrC following a relative ∼ 24 h photochemical aging process. In contrast, the BrC absorption fraction increased with tanthrop-photo, consistent with enhanced formation of secondary BrC. The covariance between MO-OOA and the BrC absorption fraction suggests that MO-OOA formation was associated with increased short-wavelength BrC absorption, especially under high solar radiation conditions during the NE and NW periods. By comparison, during the SW period, MO-OOA formation was less pronounced, and the corresponding increase in BrC absorption fraction with photochemical aging was also limited. Previous studies also demonstrated that the photochemical process promoted the enhancement of the secondary BrC absorption in urban (Li et al., 2023) and remote areas (Wu et al., 2024). The persistence of secondary BrC in rather remote regions demonstrates their enhanced summertime production efficiency, thus amplifying their direct radiative effects through solar radiation interactions. The weak absorption capacity of LO-OOA1 and LO-OOA2 suggested that these PMF-derived factors exhibited negligible short-wavelength absorption. While LO-OOA1 and LO-OOA2 were predominantly associated with Biogenic SOA (BSOA) derived from isoprene and monoterpene oxidation, they typically displayed limited light absorption properties due to the prevalence of non-chromophoric oxygenated products (Flores et al., 2014; Laskin et al., 2015). Previous study in a suburban forest area in the US also finds that BSOA was not correlated with the BrC absorption at short wavelengths (Washenfelder et al., 2015).
In this paper, we present a study in a suburban forest region of a European megacity in summer to demonstrate the impacts of photochemical processes on aerosol properties in regionally transported air masses containing a mixture of anthropogenic and biogenic emitted compounds. Based on the detailed mass spectra of the HR-AMS, we resolved primary and secondary OA sources via PMF analysis, enabling quantitative tracking of the evolution of OA during photochemical aging. The evolution of individual PMF-derived OA factors was further examined in relation to aerosol optical properties to determine their respective contributions to BrC. Throughout the entire experiment, OA dominated the total submicron aerosol mass. An increase in OA with relative anthropogenic photochemical age was primarily driven by the production of OOA. The production rates of different OOA types varied with meteorological conditions: dry air masses from NE continental regions coincided with strong solar radiation, while SW marine air masses were associated with humid, low-radiation conditions. OOA formed under humid, low-radiation conditions exhibited a lower oxidation state, reflected in reduced average bulk O : C ratios, with LO-OOA accounting for a substantially larger fraction of total OA. By contrast, under dry, high-radiation conditions, enhanced photochemical processing was associated with increased formation of MO-OOA. During NE period, primary anthropogenic OA and nitrogen-enriched LO-OOA decreased, while MO-OOA increased concurrently. The contribution of short-wavelength BrC absorption increased with tanthrop-photo and coincided with enhanced MO-OOA formation. Consequently, the fraction of total BrC associated with short-wavelength absorption increased with relative anthropogenic photochemical age. This highlights the critical need to incorporate BrC formation mechanisms into models to accurately simulate direct radiative effect (Drugé et al., 2022).
The below-canopy measurement data for Ramouillet site measurements during ACROSS project is available from the AERIES website (https://across.aeris-data.fr/, last access: 1 April 2025): which includes NR-PM1 data from HR-AMS (https://doi.org/10.25326/492, Brito and Riffault, 2023), NOx, NOy and O3 data (https://doi.org/10.25326/687, Crowley, 2024). OH data from CIMS (https://doi.org/10.25326/510, Kukui, 2023), eBC and multi-wavelength absorption data from AE33 (https://doi.org/10.25326/669, Di Antonio and Di Biagio, 2024), VOC data from LISA PTR-MS data (https://doi.org/10.25326/685, Michoud and Bouzidi, 2024), and meteorological data (https://doi.org/10.25326/437, Denjean, 2023).
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-5313-2026-supplement.
VM and CC designed the ACROSS Rambouillet experiments; CY performed the data analysis and wrote the paper; PF and VM provided guidance with the analysis and writing; CY, PF, JB, CDB, LDA, TS, STA, PD, GNTET, JNC, AK, CX, CD, MCi, MCa, CC and VM performed experiments; JB performed the operation and data analysis of the HR-AMS, and guidance on interpretation of results; CY and JB performed the HR-AMS PMF analysis; CDB and LDA performed the operation and data analysis of AE33; TS, STA, PD, GNTET and JNC performed the operation and data analysis of TD-CRD; AK and CX performed the operation and data analysis of CIMS; CD, OG and JCE performed the operation and data analysis of the meteorological data measurements; VM, HB, and AB performed the operation and data analysis of PTR-MS; CY and HW performed the back dispersion analysis of UK Met Office NAME dispersion model; CG coordinate the technical aspect of the Rambouillet experiments; FM, MCa, ABe and PZ contributed to the design and preparation of measurement infrastructures at the Rambouillet site; JDA, DL and YW contributed to the data analysis and discussions.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
This article is part of the special issue “Atmospheric Chemistry of the Suburban Forest – multiplatform observational campaign of the chemistry and physics of mixed urban and biogenic emissions”. It is not associated with a conference.
Authors are grateful to the ACROSS consortium for their work prior and during the field campaign. The authors wish to thank AERIS (https://www.aeris-data.fr/, last access: 1 April 2025), the French centre for atmospheric data and service, for providing the campaign website and organizing the curation and open distribution of the ACROSS data. Chenjie Yu would like to acknowledge the European Union Marie Skłodowska-Curie COFUND Postdoctoral Fellowship program supported by the Paris Region. Special thanks to Benjamin A. Nault and Peter F. DeCarlo (Johns Hopkins University) for their valuable discussions and suggestions contributing to this study.
The ACROSS project has received funding from the French National Research Agency (ANR), with the reference ANR-20-CE01-0010, and under the investment program integrated into France 2030, with the reference ANR-17-MPGA-0002, and it was supported by the French National program LEFE (Les Enveloppes Fluides et l'Environnement) of the CNRS/INSU (Centre National de la Recherche Scientifique/Institut National des Sciences de L'Univers). This research also receives funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 945298. The PEGASUS facility receives funding as a national facility (instrument national) of the CNRS INSU. The French State under the France-2030 programme and the Initiative of Excellence of the University of Lille are acknowledged for the funding and support granted to the R-CDP-24-003-AREA project.
This paper was edited by Roya Bahreini and reviewed by three anonymous referees.
Allan, J. D., Williams, P. I., Morgan, W. T., Martin, C. L., Flynn, M. J., Lee, J., Nemitz, E., Phillips, G. J., Gallagher, M. W., and Coe, H.: Contributions from transport, solid fuel burning and cooking to primary organic aerosols in two UK cities, Atmos. Chem. Phys., 10, 647–668, https://doi.org/10.5194/acp-10-647-2010, 2010.
Ambrose, J. L., Haase, K., Russo, R. S., Zhou, Y., White, M. L., Frinak, E. K., Jordan, C., Mayne, H. R., Talbot, R., and Sive, B. C.: A comparison of GC-FID and PTR-MS toluene measurements in ambient air under conditions of enhanced monoterpene loading, Atmos. Meas. Tech., 3, 959–980, https://doi.org/10.5194/amt-3-959-2010, 2010.
Andersen, S. T., McGillen, M. R., Xue, C., Seubert, T., Dewald, P., Türk, G. N. T. E., Schuladen, J., Denjean, C., Etienne, J.-C., Garrouste, O., Jamar, M., Harb, S., Cirtog, M., Michoud, V., Cazaunau, M., Bergé, A., Cantrell, C., Dusanter, S., Picquet-Varrault, B., Kukui, A., Mellouki, A., Carpenter, L. J., Lelieveld, J., and Crowley, J. N.: Measurement report: Sources, sinks, and lifetime of NOx in a suburban temperate forest at night, Atmos. Chem. Phys., 24, 11603–11618, https://doi.org/10.5194/acp-24-11603-2024, 2024.
Beekmann, M., Prévôt, A. S. H., Drewnick, F., Sciare, J., Pandis, S. N., Denier van der Gon, H. A. C., Crippa, M., Freutel, F., Poulain, L., Ghersi, V., Rodriguez, E., Beirle, S., Zotter, P., von der Weiden-Reinmüller, S.-L., Bressi, M., Fountoukis, C., Petetin, H., Szidat, S., Schneider, J., Rosso, A., El Haddad, I., Megaritis, A., Zhang, Q. J., Michoud, V., Slowik, J. G., Moukhtar, S., Kolmonen, P., Stohl, A., Eckhardt, S., Borbon, A., Gros, V., Marchand, N., Jaffrezo, J. L., Schwarzenboeck, A., Colomb, A., Wiedensohler, A., Borrmann, S., Lawrence, M., Baklanov, A., and Baltensperger, U.: In situ, satellite measurement and model evidence on the dominant regional contribution to fine particulate matter levels in the Paris megacity, Atmos. Chem. Phys., 15, 9577–9591, https://doi.org/10.5194/acp-15-9577-2015, 2015.
Bond, T. C., Doherty, S. J., Fahey, D. W., Forster, P. M., Berntsen, T., DeAngelo, B. J., Flanner, M. G., Ghan, S., Kärcher, B., Koch, D., Kinne, S., Kondo, Y., Quinn, P. K., Sarofim, M. C., Schultz, M. G., Schulz, M., Venkataraman, C., Zhang, H., Zhang, S., Bellouin, N., Guttikunda, S. K., Hopke, P. K., Jacobson, M. Z., Kaiser, J. W., Klimont, Z., Lohmann, U., Schwarz, J. P., Shindell, D., Storelvmo, T., Warren, S. G., and Zender, C. S.: Bounding the role of black carbon in the climate system: A scientific assessment, J. Geophys. Res.-Atmos., 118, 5380–5552, https://doi.org/10.1002/jgrd.50171, 2013.
Bressi, M., Sciare, J., Ghersi, V., Bonnaire, N., Nicolas, J. B., Petit, J.-E., Moukhtar, S., Rosso, A., Mihalopoulos, N., and Féron, A.: A one-year comprehensive chemical characterisation of fine aerosol (PM2.5) at urban, suburban and rural background sites in the region of Paris (France), Atmos. Chem. Phys., 13, 7825–7844, https://doi.org/10.5194/acp-13-7825-2013, 2013.
Brito, J. and Riffault, V.: ACROSS_IMTNE_RambForest_AMS_BelowCanopy_L2, ACROSS – AERIS-SEDOO [data set], https://doi.org/10.25326/492, 2023.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Cantrell, C. and Michoud, V.: An Experiment to Study Atmospheric Oxidation Chemistry and Physics of Mixed Anthropogenic–Biogenic Air Masses in the Greater Paris Area, B. Am. Meteorol. Soc., 103, 599–603, https://doi.org/10.1175/BAMS-D-21-0115.1, 2022.
Cappa, C. D., Lim, C. Y., Hagan, D. H., Coggon, M., Koss, A., Sekimoto, K., de Gouw, J., Onasch, T. B., Warneke, C., and Kroll, J. H.: Biomass-burning-derived particles from a wide variety of fuels – Part 2: Effects of photochemical aging on particle optical and chemical properties, Atmos. Chem. Phys., 20, 8511–8532, https://doi.org/10.5194/acp-20-8511-2020, 2020.
Chen, G., Canonaco, F., Tobler, A., Aas, W., Alastuey, A., Allan, J., Atabakhsh, S., Aurela, M., Baltensperger, U., Bougiatioti, A., De Brito, J. F., Ceburnis, D., Chazeau, B., Chebaicheb, H., Daellenbach, K. R., Ehn, M., El Haddad, I., Eleftheriadis, K., Favez, O., Flentje, H., Font, A., Fossum, K., Freney, E., Gini, M., Green, D. C., Heikkinen, L., Herrmann, H., Kalogridis, A.-C., Keernik, H., Lhotka, R., Lin, C., Lunder, C., Maasikmets, M., Manousakas, M. I., Marchand, N., Marin, C., Marmureanu, L., Mihalopoulos, N., Močnik, G., Nęcki, J., O'Dowd, C., Ovadnevaite, J., Peter, T., Petit, J.-E., Pikridas, M., Matthew Platt, S., Pokorná, P., Poulain, L., Priestman, M., Riffault, V., Rinaldi, M., Różański, K., Schwarz, J., Sciare, J., Simon, L., Skiba, A., Slowik, J. G., Sosedova, Y., Stavroulas, I., Styszko, K., Teinemaa, E., Timonen, H., Tremper, A., Vasilescu, J., Via, M., Vodička, P., Wiedensohler, A., Zografou, O., Cruz Minguillón, M., and Prévôt, A. S. H.: European aerosol phenomenology – 8: Harmonised source apportionment of organic aerosol using 22 Year-long ACSM/AMS datasets, Environ. Int., 166, 107325, https://doi.org/10.1016/j.envint.2022.107325, 2022.
Chen, T., Liu, J., Ma, Q., Chu, B., Zhang, P., Ma, J., Liu, Y., Zhong, C., Liu, P., Wang, Y., Mu, Y., and He, H.: Measurement report: Effects of photochemical aging on the formation and evolution of summertime secondary aerosol in Beijing, Atmos. Chem. Phys., 21, 1341–1356, https://doi.org/10.5194/acp-21-1341-2021, 2021.
Crippa, M., El Haddad, I., Slowik, J. G., DeCarlo, P. F., Mohr, C., Heringa, M. F., Chirico, R., Marchand, N., Sciare, J., Baltensperger, U., and Prévôt, A. S. H.: Identification of marine and continental aerosol sources in Paris using high resolution aerosol mass spectrometry, J. Geophys. Res.-Atmos., 118, 1950–1963, https://doi.org/10.1002/jgrd.50151, 2013.
Crippa, M., Guizzardi, D., Pagani, F., Schiavina, M., Melchiorri, M., Pisoni, E., Graziosi, F., Muntean, M., Maes, J., Dijkstra, L., Van Damme, M., Clarisse, L., and Coheur, P.: Insights into the spatial distribution of global, national, and subnational greenhouse gas emissions in the Emissions Database for Global Atmospheric Research (EDGAR v8.0), Earth Syst. Sci. Data, 16, 2811–2830, https://doi.org/10.5194/essd-16-2811-2024, 2024.
Crowley, J.: ACROSS_MPIC_RambForest_NOx_1min_L2, ACROSS – AERIS-SEDOO [data set], https://doi.org/10.25326/687, 2024.
Day, D. A., Campuzano-Jost, P., Nault, B. A., Palm, B. B., Hu, W., Guo, H., Wooldridge, P. J., Cohen, R. C., Docherty, K. S., Huffman, J. A., de Sá, S. S., Martin, S. T., and Jimenez, J. L.: A systematic re-evaluation of methods for quantification of bulk particle-phase organic nitrates using real-time aerosol mass spectrometry, Atmos. Meas. Tech., 15, 459–483, https://doi.org/10.5194/amt-15-459-2022, 2022.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
DeCarlo, P. F., Ulbrich, I. M., Crounse, J., de Foy, B., Dunlea, E. J., Aiken, A. C., Knapp, D., Weinheimer, A. J., Campos, T., Wennberg, P. O., and Jimenez, J. L.: Investigation of the sources and processing of organic aerosol over the Central Mexican Plateau from aircraft measurements during MILAGRO, Atmos. Chem. Phys., 10, 5257–5280, https://doi.org/10.5194/acp-10-5257-2010, 2010.
Denjean, C.: ACROSS_CNRM_RambForest_MTO-1MIN_L2, ACROSS – AERIS-SEDOO [data set], https://doi.org/10.25326/437, 2023.
Dewald, P., Seubert, T., Andersen, S. T., Türk, G. N. T. E., Schuladen, J., McGillen, M. R., Denjean, C., Etienne, J.-C., Garrouste, O., Jamar, M., Harb, S., Cirtog, M., Michoud, V., Cazaunau, M., Bergé, A., Cantrell, C., Dusanter, S., Picquet-Varrault, B., Kukui, A., Xue, C., Mellouki, A., Lelieveld, J., and Crowley, J. N.: NO3 reactivity during a summer period in a temperate forest below and above the canopy, Atmos. Chem. Phys., 24, 8983–8997, https://doi.org/10.5194/acp-24-8983-2024, 2024.
Di Antonio, L. and Di Biagio, C.: ACROSS_LISA_RambForest_AETH-Abs_PM1_1-Min_L2-station_processed, ACROSS – AERIS-SEDOO [data set], https://doi.org/10.25326/669, 2024.
Di Antonio, L., Beekmann, M., Siour, G., Michoud, V., Cantrell, C., Bauville, A., Bergé, A., Cazaunau, M., Chevaillier, S., Cirtog, M., de Brito, J. F., Formenti, P., Gaimoz, C., Garret, O., Gratien, A., Gros, V., Haeffelin, M., Hawkins, L. N., Kotthaus, S., Noyalet, G., Pereira, D. L., Petit, J.-E., Pronovost, E. D., Riffault, V., Yu, C., Foret, G., Doussin, J.-F., and Di Biagio, C.: Modelling of atmospheric variability in gas and aerosols during the ACROSS campaign 2022 of the greater Paris area: evaluation of the meteorology, dynamics and chemistry, Atmos. Chem. Phys., 25, 4803–4831, https://doi.org/10.5194/acp-25-4803-2025, 2025a.
Di Antonio, L., Di Biagio, C., Formenti, P., Gratien, A., Michoud, V., Cantrell, C., Bauville, A., Bergé, A., Cazaunau, M., Chevaillier, S., Cirtog, M., Coll, P., D'Anna, B., de Brito, J. F., De Haan, D. O., Dignum, J. R., Deshmukh, S., Favez, O., Flaud, P.-M., Gaimoz, C., Hawkins, L. N., Kammer, J., Language, B., Maisonneuve, F., Močnik, G., Perraudin, E., Petit, J.-E., Acharja, P., Poulain, L., Pouyes, P., Pronovost, E. D., Riffault, V., Roundtree, K. I., Shahin, M., Siour, G., Villenave, E., Zapf, P., Foret, G., Doussin, J.-F., and Beekmann, M.: Aerosol spectral optical properties in the Paris urban area and its peri-urban and forested surroundings during summer 2022 from ACROSS surface observations, Atmos. Chem. Phys., 25, 3161–3189, https://doi.org/10.5194/acp-25-3161-2025, 2025b.
Drinovec, L., Močnik, G., Zotter, P., Prévôt, A. S. H., Ruckstuhl, C., Coz, E., Rupakheti, M., Sciare, J., Müller, T., Wiedensohler, A., and Hansen, A. D. A.: The “dual-spot” Aethalometer: an improved measurement of aerosol black carbon with real-time loading compensation, Atmos. Meas. Tech., 8, 1965–1979, https://doi.org/10.5194/amt-8-1965-2015, 2015.
Drugé, T., Nabat, P., Mallet, M., Michou, M., Rémy, S., and Dubovik, O.: Modeling radiative and climatic effects of brown carbon aerosols with the ARPEGE-Climat global climate model, Atmos. Chem. Phys., 22, 12167–12205, https://doi.org/10.5194/acp-22-12167-2022, 2022.
Eisele, F. L. and Tanner, D. J.: Ion-assisted tropospheric OH measurements, J. Geophys. Res.-Atmos., 96, 9295–9308, https://doi.org/10.1029/91JD00198, 1991.
Ensberg, J. J., Hayes, P. L., Jimenez, J. L., Gilman, J. B., Kuster, W. C., de Gouw, J. A., Holloway, J. S., Gordon, T. D., Jathar, S., Robinson, A. L., and Seinfeld, J. H.: Emission factor ratios, SOA mass yields, and the impact of vehicular emissions on SOA formation, Atmos. Chem. Phys., 14, 2383–2397, https://doi.org/10.5194/acp-14-2383-2014, 2014.
Farmer, D. K., Cappa, C. D., and Kreidenweis, S. M.: Atmospheric Processes and Their Controlling Influence on Cloud Condensation Nuclei Activity, Chem. Rev., 115, 4199–4217, https://doi.org/10.1021/cr5006292, 2015.
Flores, J. M., Washenfelder, R. A., Adler, G., Lee, H. J., Segev, L., Laskin, J., Laskin, A., Nizkorodov, S. A., Brown, S. S., and Rudich, Y.: Complex refractive indices in the near-ultraviolet spectral region of biogenic secondary organic aerosol aged with ammonia, Phys. Chem. Chem. Phys., 16, 10629–10642, https://doi.org/10.1039/C4CP01009D, 2014.
Formenti, P., Giorio, C., Desboeufs, K., Zherebker, A., Gaetani, M., Baldo, C., Landrot, G., Montebello, S., Chevaillier, S., Triquet, S., Siour, G., Di Biagio, C., Battaglia, F., Doussin, J.-F., Feron, A., Namwoonde, A., and Piketh, S. J.: Elemental composition, iron mineralogy, and solubility of anthropogenic and natural mineral dust aerosols in Namibia: a case study analysis from the AEROCLO-sA campaign – Part 2, Atmos. Chem. Phys., 25, 16127–16145, https://doi.org/10.5194/acp-25-16127-2025, 2025.
Fossum, K. N., Lin, C., O'Sullivan, N., Lei, L., Hellebust, S., Ceburnis, D., Afzal, A., Tremper, A., Green, D., Jain, S., Byčenkienė, S., O'Dowd, C., Wenger, J., and Ovadnevaite, J.: Two distinct ship emission profiles for organic-sulfate source apportionment of PM in sulfur emission control areas, Atmos. Chem. Phys., 24, 10815–10831, https://doi.org/10.5194/acp-24-10815-2024, 2024.
Franklin, E. B., Rossell, R. K., Vermeuel, M. P., De Groodt, A., Richard, K., Yee, L. D., Marcantonio, J., Maddaleno, T., Osburn, C., O'Brien, R. E., Commane, R., Mak, J. E., Goldstein, A. H., Millet, D. B., and Farmer, D. K.: Emerging drivers of urban aerosol increase global change vulnerability in a US megacity, npj Clim. Atmos. Sci., 8, 333, https://doi.org/10.1038/s41612-025-01202-w, 2025.
Freney, E. J., Sellegri, K., Canonaco, F., Colomb, A., Borbon, A., Michoud, V., Doussin, J.-F., Crumeyrolle, S., Amarouche, N., Pichon, J.-M., Bourianne, T., Gomes, L., Prevot, A. S. H., Beekmann, M., and Schwarzenböeck, A.: Characterizing the impact of urban emissions on regional aerosol particles: airborne measurements during the MEGAPOLI experiment, Atmos. Chem. Phys., 14, 1397–1412, https://doi.org/10.5194/acp-14-1397-2014, 2014.
Friedrich, N., Tadic, I., Schuladen, J., Brooks, J., Darbyshire, E., Drewnick, F., Fischer, H., Lelieveld, J., and Crowley, J. N.: Measurement of NOx and NOy with a thermal dissociation cavity ring-down spectrometer (TD-CRDS): instrument characterisation and first deployment, Atmos. Meas. Tech., 13, 5739–5761, https://doi.org/10.5194/amt-13-5739-2020, 2020.
Gao, R. S., Schwarz, J. P., Kelly, K. K., Fahey, D. W., Watts, L. A., Thompson, T. L., Spackman, J. R., Slowik, J. G., Cross, E. S., Han, J. H., Davidovits, P., Onasch, T. B., and Worsnop, D. R.: A Novel Method for Estimating Light-Scattering Properties of Soot Aerosols Using a Modified Single-Particle Soot Photometer, Aerosol Sci.d Tech., 41, 125–135, https://doi.org/10.1080/02786820601118398, 2007.
Grange, S. K., Lee, J. D., Drysdale, W. S., Lewis, A. C., Hueglin, C., Emmenegger, L., and Carslaw, D. C.: COVID-19 lockdowns highlight a risk of increasing ozone pollution in European urban areas, Atmos. Chem. Phys., 21, 4169–4185, https://doi.org/10.5194/acp-21-4169-2021, 2021.
Guo, F., Bui, A. A. T., Schulze, B. C., Dai, Q., Yoon, S., Shrestha, S., Wallace, H. W., Sanchez, N. P., Alvarez, S., Erickson, M. H., Sheesley, R. J., Usenko, S., Flynn, J., and Griffin, R. J.: Airmass history, night-time particulate organonitrates, and meteorology impact urban SOA formation rate, Atmos. Environ., 322, 120362, https://doi.org/10.1016/j.atmosenv.2024.120362, 2024.
Hayes, P. L., Ortega, A. M., Cubison, M. J., Froyd, K. D., Zhao, Y., Cliff, S. S., Hu, W. W., Toohey, D. W., Flynn, J. H., Lefer, B. L., Grossberg, N., Alvarez, S., Rappenglück, B., Taylor, J. W., Allan, J. D., Holloway, J. S., Gilman, J. B., Kuster, W. C., de Gouw, J. A., Massoli, P., Zhang, X., Liu, J., Weber, R. J., Corrigan, A. L., Russell, L. M., Isaacman, G., Worton, D. R., Kreisberg, N. M., Goldstein, A. H., Thalman, R., Waxman, E. M., Volkamer, R., Lin, Y. H., Surratt, J. D., Kleindienst, T. E., Offenberg, J. H., Dusanter, S., Griffith, S., Stevens, P. S., Brioude, J., Angevine, W. M., and Jimenez, J. L.: Organic aerosol composition and sources in Pasadena, California, during the 2010 CalNex campaign, J. Geophys. Res.-Atmos., 118, 9233–9257, https://doi.org/10.1002/jgrd.50530, 2013.
Healy, R. M., Sciare, J., Poulain, L., Crippa, M., Wiedensohler, A., Prévôt, A. S. H., Baltensperger, U., Sarda-Estève, R., McGuire, M. L., Jeong, C.-H., McGillicuddy, E., O'Connor, I. P., Sodeau, J. R., Evans, G. J., and Wenger, J. C.: Quantitative determination of carbonaceous particle mixing state in Paris using single-particle mass spectrometer and aerosol mass spectrometer measurements, Atmos. Chem. Phys., 13, 9479–9496, https://doi.org/10.5194/acp-13-9479-2013, 2013.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
Inness, A., Ades, M., Agustí-Panareda, A., Barré, J., Benedictow, A., Blechschmidt, A.-M., Dominguez, J. J., Engelen, R., Eskes, H., Flemming, J., Huijnen, V., Jones, L., Kipling, Z., Massart, S., Parrington, M., Peuch, V.-H., Razinger, M., Remy, S., Schulz, M., and Suttie, M.: The CAMS reanalysis of atmospheric composition, Atmos. Chem. Phys., 19, 3515–3556, https://doi.org/10.5194/acp-19-3515-2019, 2019.
Irei, S., Takami, A., Sadanaga, Y., Miyoshi, T., Arakaki, T., Sato, K., Kaneyasu, N., Bandow, H., and Hatakeyama, S.: Transboundary secondary organic aerosol in western Japan: An observed limitation of the f44 oxidation indicator, Atmos. Environ., 120, 71–75, https://doi.org/10.1016/j.atmosenv.2015.08.070, 2015.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles, Aerosol Sci. Tech., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Jeon, S., Walker, M. J., Sueper, D. T., Day, D. A., Handschy, A. V., Jimenez, J. L., and Williams, B. J.: A searchable database and mass spectral comparison tool for the Aerosol Mass Spectrometer (AMS) and the Aerosol Chemical Speciation Monitor (ACSM), Atmos. Meas. Tech., 16, 6075–6095, https://doi.org/10.5194/amt-16-6075-2023, 2023.
Jiang, X., Liu, D., Li, Q., Tian, P., Wu, Y., Li, S., Hu, K., Ding, S., Bi, K., Li, R., Huang, M., Ding, D., Chen, Q., Kong, S., Li, W., Pang, Y., and He, D.: Connecting the Light Absorption of Atmospheric Organic Aerosols with Oxidation State and Polarity, Environ. Sci. Technol., 56, 12873–12885, https://doi.org/10.1021/acs.est.2c02202, 2022.
Jones, A., Thomson, D., Hort, M., and Devenish, B.: The U.K. Met Office's Next-Generation Atmospheric Dispersion Model, NAME III, Air Pollution Modeling and Its Application XVII, Boston, MA, 580–589, https://doi.org/10.1007/978-0-387-68854-1, 2007.
Kleinman, L. I., Daum, P. H., Lee, Y. N., Nunnermacker, L. J., Springston, S. R., Weinstein-Lloyd, J., Hyde, P., Doskey, P., Rudolph, J., Fast, J., and Berkowitz, C.: Photochemical age determinations in the Phoenix metropolitan area, J. Geophys. Res.-Atmos., 108, https://doi.org/10.1029/2002JD002621, 2003.
Kleinman, L. I., Daum, P. H., Lee, Y.-N., Senum, G. I., Springston, S. R., Wang, J., Berkowitz, C., Hubbe, J., Zaveri, R. A., Brechtel, F. J., Jayne, J., Onasch, T. B., and Worsnop, D.: Aircraft observations of aerosol composition and ageing in New England and Mid-Atlantic States during the summer 2002 New England Air Quality Study field campaign, J. Geophys. Res.-Atmos., 112, https://doi.org/10.1029/2006JD007786, 2007.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kukui, A.: ACROSS_LPC2E_Rambforest_OH_L2, ACROSS – AERIS-SEDOO [data set], https://doi.org/10.25326/510, 2023.
Kukui, A., Ancellet, G., and Le Bras, G.: Chemical ionisation mass spectrometer for measurements of OH and Peroxy radical concentrations in moderately polluted atmospheres, J. Atmos. Chem., 61, 133–154, https://doi.org/10.1007/s10874-009-9130-9, 2008.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of Atmospheric Brown Carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Li, Q., Liu, D., Jiang, X., Tian, P., Wu, Y., Li, S., Hu, K., Liu, Q., Huang, M., Li, R., Bi, K., Kong, S., Ding, D., and Yu, C.: Concurrent photochemical whitening and darkening of ambient brown carbon, Atmos. Chem. Phys., 23, 9439–9453, https://doi.org/10.5194/acp-23-9439-2023, 2023.
Lin, C., Ceburnis, D., Xu, W., Heffernan, E., Hellebust, S., Gallagher, J., Huang, R.-J., O'Dowd, C., and Ovadnevaite, J.: The impact of traffic on air quality in Ireland: insights from the simultaneous kerbside and suburban monitoring of submicron aerosols, Atmos. Chem. Phys., 20, 10513–10529, https://doi.org/10.5194/acp-20-10513-2020, 2020.
Liu, D., Whitehead, J., Alfarra, M. R., Reyes-Villegas, E., Spracklen, D. V., Reddington, C. L., Kong, S., Williams, P. I., Ting, Y.-C., Haslett, S., Taylor, J. W., Flynn, M. J., Morgan, W. T., McFiggans, G., Coe, H., and Allan, J. D.: Black-carbon absorption enhancement in the atmosphere determined by particle mixing state, Nat. Geosci., 10, 184–188, https://doi.org/10.1038/ngeo2901, 2017.
Liu, D., Li, S., Hu, D., Kong, S., Cheng, Y., Wu, Y., Ding, S., Hu, K., Zheng, S., Yan, Q., Zheng, H., Zhao, D., Tian, P., Ye, J., Huang, M., and Ding, D.: Evolution of Aerosol Optical Properties from Wood Smoke in Real Atmosphere Influenced by Burning Phase and Solar Radiation, Environ. Sci. Technol., 55, 5677–5688, https://doi.org/10.1021/acs.est.0c07569, 2021.
Lu, Z., Zhang, Q., and Streets, D. G.: Sulfur dioxide and primary carbonaceous aerosol emissions in China and India, 1996–2010, Atmos. Chem. Phys., 11, 9839–9864, https://doi.org/10.5194/acp-11-9839-2011, 2011.
Manisalidis, I., Stavropoulou, E., Stavropoulos, A., and Bezirtzoglou, E.: Environmental and Health Impacts of Air Pollution: A Review, Front. Public Health, 8, https://doi.org/10.3389/fpubh.2020.00014, 2020.
Mei, F., Hayes, P. L., Ortega, A., Taylor, J. W., Allan, J. D., Gilman, J., Kuster, W., de Gouw, J., Jimenez, J. L., and Wang, J.: Droplet activation properties of organic aerosols observed at an urban site during CalNex-LA, J. Geophys. Res.-Atmos., 118, 2903–2917, https://doi.org/10.1002/jgrd.50285, 2013.
Menut, L., Cholakian, A., Siour, G., Lapere, R., Pennel, R., Mailler, S., and Bessagnet, B.: Impact of Landes forest fires on air quality in France during the 2022 summer, Atmos. Chem. Phys., 23, 7281–7296, https://doi.org/10.5194/acp-23-7281-2023, 2023.
Michoud, V. and Bouzidi, H.: ACROSS_2022_RambForest_LISA_PTRMS_VOCs_Belowcanopy_10min_20220617 – 20220723, ACROSS – AERIS-SEDOO [data set], https://doi.org/10.25326/685, 2024.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Misztal, P. K., Hewitt, C. N., Wildt, J., Blande, J. D., Eller, A. S. D., Fares, S., Gentner, D. R., Gilman, J. B., Graus, M., Greenberg, J., Guenther, A. B., Hansel, A., Harley, P., Huang, M., Jardine, K., Karl, T., Kaser, L., Keutsch, F. N., Kiendler-Scharr, A., Kleist, E., Lerner, B. M., Li, T., Mak, J., Nölscher, A. C., Schnitzhofer, R., Sinha, V., Thornton, B., Warneke, C., Wegener, F., Werner, C., Williams, J., Worton, D. R., Yassaa, N., and Goldstein, A. H.: Atmospheric benzenoid emissions from plants rival those from fossil fuels, Sci. Rep., 5, 12064, https://doi.org/10.1038/srep12064, 2015.
Mollner, A. K., Valluvadasan, S., Feng, L., Sprague, M. K., Okumura, M., Milligan, D. B., Bloss, W. J., Sander, S. P., Martien, P. T., Harley, R. A., McCoy, A. B., and Carter, W. P. L.: Rate of Gas Phase Association of Hydroxyl Radical and Nitrogen Dioxide, Science, 330, 646–649, https://doi.org/10.1126/science.1193030, 2010.
Nault, B. A., Campuzano-Jost, P., Day, D. A., Schroder, J. C., Anderson, B., Beyersdorf, A. J., Blake, D. R., Brune, W. H., Choi, Y., Corr, C. A., de Gouw, J. A., Dibb, J., DiGangi, J. P., Diskin, G. S., Fried, A., Huey, L. G., Kim, M. J., Knote, C. J., Lamb, K. D., Lee, T., Park, T., Pusede, S. E., Scheuer, E., Thornhill, K. L., Woo, J.-H., and Jimenez, J. L.: Secondary organic aerosol production from local emissions dominates the organic aerosol budget over Seoul, South Korea, during KORUS-AQ, Atmos. Chem. Phys., 18, 17769–17800, https://doi.org/10.5194/acp-18-17769-2018, 2018.
Ng, N. L., Herndon, S. C., Trimborn, A., Canagaratna, M. R., Croteau, P. L., Onasch, T. B., Sueper, D., Worsnop, D. R., Zhang, Q., Sun, Y. L., and Jayne, J. T.: An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol, Aerosol Sci. Tech., 45, 780–794, https://doi.org/10.1080/02786826.2011.560211, 2011.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
Papachristopoulou, K., Raptis, I.-P., Gkikas, A., Fountoulakis, I., Masoom, A., and Kazadzis, S.: Aerosol optical depth regime over megacities of the world, Atmos. Chem. Phys., 22, 15703–15727, https://doi.org/10.5194/acp-22-15703-2022, 2022.
Pereira, D. L., Giorio, C., Gratien, A., Zherebker, A., Noyalet, G., Chevaillier, S., Alage, S., Almarj, E., Bergé, A., Bertin, T., Cazaunau, M., Coll, P., Di Antonio, L., Harb, S., Heuser, J., Gaimoz, C., Guillemant, O., Language, B., Lauret, O., Macias, C., Maisonneuve, F., Picquet-Varrault, B., Torres, R., Triquet, S., Zapf, P., Hawkins, L., Pronovost, D., Riley, S., Flaud, P.-M., Perraudin, E., Pouyes, P., Villenave, E., Albinet, A., Favez, O., Aujay-Plouzeau, R., Michoud, V., Cantrell, C., Cirtog, M., Di Biagio, C., Doussin, J.-F., and Formenti, P.: Molecular characterization of organic aerosols in urban and forested areas of Paris using high-resolution mass spectrometry, Atmos. Chem. Phys., 25, 4885–4905, https://doi.org/10.5194/acp-25-4885-2025, 2025.
Petit, J.-E., Favez, O., Sciare, J., Crenn, V., Sarda-Estève, R., Bonnaire, N., Močnik, G., Dupont, J.-C., Haeffelin, M., and Leoz-Garziandia, E.: Two years of near real-time chemical composition of submicron aerosols in the region of Paris using an Aerosol Chemical Speciation Monitor (ACSM) and a multi-wavelength Aethalometer, Atmos. Chem. Phys., 15, 2985–3005, https://doi.org/10.5194/acp-15-2985-2015, 2015.
Petzold, A., Ogren, J. A., Fiebig, M., Laj, P., Li, S.-M., Baltensperger, U., Holzer-Popp, T., Kinne, S., Pappalardo, G., Sugimoto, N., Wehrli, C., Wiedensohler, A., and Zhang, X.-Y.: Recommendations for reporting “black carbon” measurements, Atmos. Chem. Phys., 13, 8365–8379, https://doi.org/10.5194/acp-13-8365-2013, 2013.
Ravishankara, A. R., Rudich, Y., and Wuebbles, D. J.: Physical Chemistry of Climate Metrics, Chem. Rev., 115, 3682–3703, https://doi.org/10.1021/acs.chemrev.5b00010, 2015.
Reyes-Villegas, E., Priestley, M., Ting, Y.-C., Haslett, S., Bannan, T., Le Breton, M., Williams, P. I., Bacak, A., Flynn, M. J., Coe, H., Percival, C., and Allan, J. D.: Simultaneous aerosol mass spectrometry and chemical ionisation mass spectrometry measurements during a biomass burning event in the UK: insights into nitrate chemistry, Atmos. Chem. Phys., 18, 4093–4111, https://doi.org/10.5194/acp-18-4093-2018, 2018.
Riva, M., Budisulistiorini, S. H., Chen, Y., Zhang, Z., D'Ambro, E. L., Zhang, X., Gold, A., Turpin, B. J., Thornton, J. A., Canagaratna, M. R., and Surratt, J. D.: Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxyhydroperoxides, Environ. Sci. Technol., 50, 9889–9899, https://doi.org/10.1021/acs.est.6b02511, 2016.
Rogers, M. J., Joo, T., Hass-Mitchell, T., Canagaratna, M. R., Campuzano-Jost, P., Sueper, D., Tran, M. N., Machesky, J. E., Roscioli, J. R., Jimenez, J. L., Krechmer, J. E., Lambe, A. T., Nault, B. A., and Gentner, D. R.: Humid Summers Promote Urban Aqueous-Phase Production of Oxygenated Organic Aerosol in the Northeastern United States, Geophys. Res. Lett., 52, e2024GL112005, https://doi.org/10.1029/2024GL112005, 2025.
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Allen, H. M., Ayres, B. R., Brown, S. S., Brune, W. H., Crounse, J. D., de Gouw, J., Draper, D. C., Feiner, P. A., Fry, J. L., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K., Teng, A. P., Wennberg, P. O., Wild, R. J., Zhang, L., and Cohen, R. C.: The lifetime of nitrogen oxides in an isoprene-dominated forest, Atmos. Chem. Phys., 16, 7623–7637, https://doi.org/10.5194/acp-16-7623-2016, 2016.
Schmale, J., Schneider, J., Nemitz, E., Tang, Y. S., Dragosits, U., Blackall, T. D., Trathan, P. N., Phillips, G. J., Sutton, M., and Braban, C. F.: Sub-Antarctic marine aerosol: dominant contributions from biogenic sources, Atmos. Chem. Phys., 13, 8669–8694, https://doi.org/10.5194/acp-13-8669-2013, 2013.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, Wiley, ISBN 978-1-118-94740-1, 2016.
Stirnberg, R., Cermak, J., Kotthaus, S., Haeffelin, M., Andersen, H., Fuchs, J., Kim, M., Petit, J.-E., and Favez, O.: Meteorology-driven variability of air pollution (PM1) revealed with explainable machine learning, Atmos. Chem. Phys., 21, 3919–3948, https://doi.org/10.5194/acp-21-3919-2021, 2021.
Tani, A., Hayward, S., and Hewitt, C. N.: Measurement of monoterpenes and related compounds by proton transfer reaction-mass spectrometry (PTR-MS), Int. J. Mass Spectrom., 223–224, 561–578, https://doi.org/10.1016/S1387-3806(02)00880-1, 2003.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
von der Weiden, S.-L., Drewnick, F., and Borrmann, S.: Particle Loss Calculator – a new software tool for the assessment of the performance of aerosol inlet systems, Atmos. Meas. Tech., 2, 479–494, https://doi.org/10.5194/amt-2-479-2009, 2009.
Wang, Y., Voliotis, A., Hu, D., Shao, Y., Du, M., Chen, Y., Kleinheins, J., Marcolli, C., Alfarra, M. R., and McFiggans, G.: On the evolution of sub- and super-saturated water uptake of secondary organic aerosol in chamber experiments from mixed precursors, Atmos. Chem. Phys., 22, 4149–4166, https://doi.org/10.5194/acp-22-4149-2022, 2022.
Washenfelder, R. A., Attwood, A. R., Brock, C. A., Guo, H., Xu, L., Weber, R. J., Ng, N. L., Allen, H. M., Ayres, B. R., Baumann, K., Cohen, R. C., Draper, D. C., Duffey, K. C., Edgerton, E., Fry, J. L., Hu, W. W., Jimenez, J. L., Palm, B. B., Romer, P., Stone, E. A., Wooldridge, P. J., and Brown, S. S.: Biomass burning dominates brown carbon absorption in the rural southeastern United States, Geophys. Res. Lett., 42, 653–664, https://doi.org/10.1002/2014GL062444, 2015.
White, M. L., Russo, R. S., Zhou, Y., Ambrose, J. L., Haase, K., Frinak, E. K., Varner, R. K., Wingenter, O. W., Mao, H., Talbot, R., and Sive, B. C.: Are biogenic emissions a significant source of summertime atmospheric toluene in the rural Northeastern United States?, Atmos. Chem. Phys., 9, 81–92, https://doi.org/10.5194/acp-9-81-2009, 2009.
Wu, Y., Liu, D., Tian, P., Sheng, J., Liu, Q., Li, R., Hu, K., Jiang, X., Li, S., Bi, K., Zhao, D., Huang, M., Ding, D., and Wang, J.: Tracing the Formation of Secondary Aerosols Influenced by Solar Radiation and Relative Humidity in Suburban Environment, J. Geophys. Res.-Atmos., 127, e2022JD036913, https://doi.org/10.1029/2022JD036913, 2022.
Wu, Y., Liu, Q., Liu, D., Tian, P., Xu, W., Wang, J., Hu, K., Li, S., Jiang, X., Wang, F., Huang, M., Ding, D., Yu, C., and Hu, D.: Enhanced formation of nitrogenous organic aerosols and brown carbon after aging in the planetary boundary layer, npj Clim. Atmos. Sci., 7, 179, https://doi.org/10.1038/s41612-024-00726-x, 2024.
Yang, J., Au, W. C., Law, H., Lam, C. H., and Nah, T.: Formation and evolution of brown carbon during aqueous-phase nitrate-mediated photooxidation of guaiacol and 5-nitroguaiacol, Atmos. Environ., 254, 118401, https://doi.org/10.1016/j.atmosenv.2021.118401, 2021.
Yu, C., Pasternak, D., Lee, J., Yang, M., Bell, T., Bower, K., Wu, H., Liu, D., Reed, C., Bauguitte, S., Cliff, S., Trembath, J., Coe, H., and Allan, J. D.: Characterizing the Particle Composition and Cloud Condensation Nuclei from Shipping Emission in Western Europe, Environ. Sci. Technol., 54, 15604–15612, https://doi.org/10.1021/acs.est.0c04039, 2020.
Yus-Díez, J., Bernardoni, V., Močnik, G., Alastuey, A., Ciniglia, D., Ivančič, M., Querol, X., Perez, N., Reche, C., Rigler, M., Vecchi, R., Valentini, S., and Pandolfi, M.: Determination of the multiple-scattering correction factor and its cross-sensitivity to scattering and wavelength dependence for different AE33 Aethalometer filter tapes: a multi-instrumental approach, Atmos. Meas. Tech., 14, 6335–6355, https://doi.org/10.5194/amt-14-6335-2021, 2021.
Zhang, L., Wilson, J. P., MacDonald, B., Zhang, W., and Yu, T.: The changing PM2.5 dynamics of global megacities based on long-term remotely sensed observations, Environ. Int., 142, 105862, https://doi.org/10.1016/j.envint.2020.105862, 2020.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhang, Y., Favez, O., Petit, J.-E., Canonaco, F., Truong, F., Bonnaire, N., Crenn, V., Amodeo, T., Prévôt, A. S. H., Sciare, J., Gros, V., and Albinet, A.: Six-year source apportionment of submicron organic aerosols from near-continuous highly time-resolved measurements at SIRTA (Paris area, France), Atmos. Chem. Phys., 19, 14755–14776, https://doi.org/10.5194/acp-19-14755-2019, 2019.
Zhao, R., Lee, A. K. Y., Huang, L., Li, X., Yang, F., and Abbatt, J. P. D.: Photochemical processing of aqueous atmospheric brown carbon, Atmos. Chem. Phys., 15, 6087–6100, https://doi.org/10.5194/acp-15-6087-2015, 2015.