the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Feb 2026
| 16 Feb 2026
Aerosol iodine recycling is a major control on tropospheric reactive iodine abundance
Leyang Liu
Xuan Wang
Yuk-Chun Chan
Alyson Fritzmann
Ryan Pound
Amy Lees
Lewis Marden
Mat Evans
Lucy J. Carpenter
Jochen Stutz
Joel A. Thornton
Gordon Novak
Andrew Rollins
Gregory P. Schill
Xu-Cheng He
Henning Finkenzeller
Mago Reza
Rainer Volkamer
Kelvin H. Bates
Alfonso Saiz-Lopez
Anoop S. Mahajan
Tropospheric reactive iodine influences the oxidizing capacity of the atmosphere and serves as an important source of ultra-fine particles. However, the paucity of observations of gas-phase and aerosol iodine, combined with incomplete understanding and representation of iodine chemistry in models, leads to substantial uncertainties in understanding iodine abundance, speciation, and impacts. Motivated by known gaps in previous modeling studies, we introduced speciated aerosol iodine and aerosol iodide recycling to the global chemical transport model, GEOS-Chem. Modeled aerosol iodine is speciated into fine and coarse mode soluble organic iodine (SOI), iodate, and iodide. Aerosol iodide is recycled into the gas phase via heterogeneous chemistry involving halogen nitrates and hypohalous acids to form I2, ICl, and IBr, which represents an additional source of gas-phase iodine to the atmosphere. Iodide dehalogenation doubles the tropospheric burden of reactive iodine (Iy) while reducing model-measurement bias for IO and aerosol iodine. The rate of aerosol iodine conversion to Iy is more than twice as fast as the combined rates of inorganic ocean emissions and the photolysis of organic iodine gases, suggesting that aerosols are important in mediating the abundance and lifetime of tropospheric Iy. The incorporation of SOI and iodate into the model prevents iodide dehalogenation by partitioning iodide into less reactive reservoirs, which has a stabilizing effect for reactive iodine chemistry. These findings have implications for reactive halogen abundances and global oxidant budgets in the troposphere.
- Article
(9330 KB) - Full-text XML
- BibTeX
- EndNote




Gas-phase reactive halogens (chlorine, bromine, and iodine-containing compounds) affect the oxidation capacity of the atmosphere and global climate. Halogen chemistry is estimated to reduce tropospheric ozone and OH burdens by 10 %–20 % and 4 %–10 %, respectively (Badia et al., 2019, 2021; Saiz-Lopez et al., 2014; Sherwen et al., 2016a; Wang et al., 2021). This reduction in O3 and OH increases the lifetime of methane by 6 %–11 %, thus indirectly enhancing the warming potential of methane (Li et al., 2022a; Sherwen et al., 2016a; Wang et al., 2021).
Despite the low abundance of atmospheric iodine, iodine-induced tropospheric ozone loss is believed to be 2–5 times greater than chlorine and bromine-induced ozone depletion combined (Saiz-Lopez et al., 2014; Sherwen et al., 2016a; Wang et al., 2021). Besides its impacts on the oxidation capacity of the atmosphere, laboratory experiments have demonstrated that iodine oxoacids (iodic acid, HIO3 and iodous acid, HIO2) enhance new particle formation (NPF) by factors of 10–10 000 in marine and polar regions, which ultimately affects cloud condensation nuclei (CCN) formation (Baccarini et al., 2020; He, 2023; He et al., 2021a; Hoffmann et al., 2001; O'Dowd et al., 2002; Saiz-Lopez et al., 2012; Xavier et al., 2024). A global three dimensional modeling study by Zhao et al. (2024) found that iodine oxoacids are the dominant NPF source in the marine boundary layer, even with modeled HIO3 concentrations 80 %–100 % lower than observed values (Zhao et al., 2024). However, they were unable to reproduce the iodine levels observed in the free troposphere during aircraft campaigns, suggesting that the effective lifetime of iodine in their model is not sufficient for long-range transport to the upper troposphere (Koenig et al., 2020; Schill et al., 2025; Zhao et al., 2024).
Ocean emissions are the initial source of atmospheric iodine species, including molecular iodine (I2), hypoiodous acid (HOI), methyl iodide (CH3I), and other iodocarbons (Carpenter et al., 2013, 2021; Saiz-Lopez et al., 2012; Stemmler et al., 2014). The largest single source of iodine in the atmosphere is thought to be from reactions between O3 and aqueous iodide on the ocean surface, which releases both I2 and HOI into the atmosphere (Carpenter et al., 2013, 2021; MacDonald et al., 2014; Tinel et al., 2020). The emission of I2 and HOI is likely also sensitive to the chemical composition of organics and surfactants within the sea surface microlayer, complicating estimates of emissions (Carpenter et al., 2021; Tinel et al., 2020). Continental sources of iodine to the atmosphere include dust, biomass burning, and anthropogenic emissions, though the contribution of these sources to global iodine budgets is uncertain since they are not typically included in global models (Koenig et al., 2021; Schill et al., 2025; Shi et al., 2021; Zhang et al., 2024). Aerosol iodine is mostly formed by the uptake of gas-phase iodine species onto existing aerosol, which is dictated by aerosol surface area and alkalinity (Baker and Yodle, 2021; Gómez Martín et al., 2022b; Pechtl et al., 2007; Saiz-Lopez et al., 2012; Vogt et al., 1999). There are three main types of aerosol iodine: soluble organic iodine (SOI), iodide (I−), and iodate (), which are all globally ubiquitous in the marine boundary layer (Baker et al., 2001; Droste et al., 2021; Gilfedder et al., 2008; Gómez Martín et al., 2022b; Lai et al., 2008; Yu et al., 2019). Formation of aerosol iodine is usually regarded as a depositional sink for reactive iodine in chemical transport models. However, aerosol iodine is not inert and its reactions can be a potential source of gas-phase reactive iodine. For example, aerosol iodide undergoes heterogeneous reactions involving hypohalous acids and halogen nitrates with aerosol halides to yield IBr, ICl, and I2, a process we refer to as iodide dehalogenation (Pechtl et al., 2007; Tham et al., 2021; Vogt et al., 1999).
Aerosol halide dehalogenation refers to the recycling of chloride, bromide, and iodide to yield gas-phase reactive halogen species Cly, Bry, and Iy (Eqs. 1–3).
Iodide dehalogenation has not been explicitly modeled on a global scale prior to this work. Both GEOS-Chem and CAM-Chem have chloride and bromide dehalogenation but do not partition aerosol iodide back to the gas phase. For example, both models had the reaction of HOI with chloride and bromide to yield IBr and ICl. This contributes a new source of Bry and Cly to the atmosphere and repartitions HOI to dihalogen species. This chemistry is not comprehensive, however, since it did not include the reactions between HOBr, HOCl, HOI, BrNO3, ClNO3, or INO3 with iodide (Li et al., 2022b; Saiz-Lopez et al., 2014; Sherwen et al., 2016a; Wang et al., 2021). This can lead to underestimates in the importance of reactive iodine chemistry, since iodide that could be recycled back to the gas phase is only lost to deposition, reducing its effective lifetime and impact on the oxidation capacity of the atmosphere.
This study examines the role of iodine aerosol speciation and dehalogenation in controlling global gas-phase reactive iodine. Our results indicate that incorporating the formation and interconversion of soluble organic iodine, iodate, and iodide aerosol is crucial for accurately reproducing surface observations of speciated aerosol iodine. Including aerosol iodine recycling chemistry also improves model bias for IO, especially in the upper troposphere. Additionally, we show that aerosol iodide dehalogenation has a larger control on reactive iodine abundance than ocean emissions and photolysis of organic iodine gases. Therefore, reducing uncertainties in iodine aerosol chemical composition, species interconversion, and cycling is essential for understanding and modeling halogen impacts on global oxidant abundances. We explore the impact of incorporating speciated aerosol iodine and aerosol iodine recycling chemistry on oxidants in a follow-up paper.
2.1 Model configuration
We used GEOS-Chem version 14.4.0, a state-of-the-art global chemical transport model that includes detailed oxidant-aerosol chemistry in the troposphere and stratosphere (Bey et al., 2001). Aerosol thermodynamic calculations are performed using the HETerogeneous vectorized or Parallel (HETP) module for estimating NH3-, HNO3-, and HCl-Cl−, along with aerosol properties such as pH and liquid water content (Miller et al., 2024). HETP does not calculate the thermodynamic partitioning of sulfate as it does for semi-volatile species. Sulfate is formed kinetically via chemical oxidation reactions and is assumed to reside entirely in the aerosol phase.
Global anthropogenic emissions are from the Community Emissions Data System (CEDS v2) with aircraft emissions from the Aircraft Emissions Inventory Code (AEIC) 2019 (Simone et al., 2013). Shipping emissions of NOx (NO + NO2) are calculated in the PARANOx module (Holmes et al., 2014; Vinken et al., 2011). Marine emissions of dimethyl sulfide (DMS) are from Breider et al. (2017) based on Lana et al. (2011) (Breider et al., 2017; Lana et al., 2011). Wet and dry deposition (including gravitational settling) of aerosols and gases are from Liu et al. (2001), Emerson et al. (2020), and Li et al. (2023) (Emerson et al., 2020; Li et al., 2023; Liu et al., 2001). Photolysis rates are computed in Cloud-J (Prather, 2015). GEOS-Chem Classic simulations in this study were conducted at 4°×5° resolution with 72 vertical levels driven by MERRA-2 meteorology. Model runs were conducted for the year 2022 with 1 year spin-up (see Table 1 for configuration details).
Online sea salt aerosol emissions from the sea surface and blowing snow are from Jaeglé at al. (2011) and Huang and Jaeglé (2017). The current halogen chemistry in GEOS-Chem already includes sea salt debromination, anthropogenic HCl and aerosol chloride emissions, Iy uptake on alkaline sea salt aerosol, and stratospheric halogen chemistry (Eastham et al., 2014; Sherwen et al., 2016a, b; Wang et al., 2019, 2021; Zhang et al., 2022). Modeled sea salt aerosols are assumed to have an initial pH of 8 upon emission, after which their alkalinity begins to be titrated by uptake of gas-phase acidic species (HNO3, SO2, HCl). Fine mode SSA pH is calculated in HETP while coarse mode sea salt aerosols are assumed to have a pH of 5 after all of the initial alkalinity is depleted.
The continental chlorine emission inventory is described in Zhang et al. (2022). HCl emission factors from open fires are from Andreae (2019) utilizing Global Fire Emissions Database version 4 (GFED4) (Andreae, 2019). Organic halogen gases, bromocarbons, and iodocarbons are from Meinhausen et al. (2017), Bell et al. (2002), Liang et al. (2010), and Ordóñez et al. (2012) (Bell et al., 2002; Liang et al., 2010; Meinshausen et al., 2017; Ordóñez et al., 2012). These gases are photolyzed in the model to form reactive inorganic Iy. Surface emissions of inorganic iodine in the model are driven by O3 deposition on the sea surface, which reacts with iodide in seawater to produce I2 and HOI. Sea surface iodide concentrations in version 14.4.0 are from MacDonald et al. (2014), though this scheme has a known low bias to underestimate sea surface iodide by more than a factor of two globally with especially poor performance in polar regions (Sherwen et al., 2019). More recently, Chance et al. (2014) and Sherwen et al. (2019) used sea surface iodide parameterizations that show better agreement with observations of sea surface iodide concentrations than the scheme of MacDonald et al. (2014) (Chance et al., 2014; Pound et al., 2024; Sherwen et al., 2019). However, these new schemes have not been included in the GEOS-Chem base model yet and are not considered in this work.
Here we expand upon the heterogeneous and gas-phase iodine chemistry in GEOS-Chem. Additions include size-resolved, speciated iodine aerosol and heterogeneous dehalogenation of aerosol iodide to the gas-phase by reaction with hypohalous acids (HOCl, HOBr, and HOI, referred to collectively as HOX) and halogen nitrates (ClNO3, BrNO3, and INO3, referred to as XNO3). Additions to the model chemical mechanism are described in detail below.
2.2 Sources and sinks of speciated iodine aerosol
2.2.1 Primary emissions of aerosol iodine
We introduce primary emissions of SOI, iodate, and iodide in the model from the ocean surface (Table A1). On average, iodide and iodate constitute 42 % ± 19 % and 48 % ± 22 % of total dissolved iodine in seawater (Jones et al., 2024; Wong and Cheng, 1998). The concentration of SOI in seawater is more uncertain since it depends on marine biogenic activity, which can vary based on nutrient availability, sea surface temperature (SST), and latitude. SOI can be abundant in bulk seawater, with observed fractional contributions ranging between 7 % to 45 % of total iodine (Gong and Zhang, 2013; Jones et al., 2024; Schwehr and Santschi, 2003; Wong and Cheng, 1998). SOI may also be enriched in the sea surface microlayer (SSM) relative to bulk seawater based on studies that have found that SSM enrichment of organics and pollutants could be a factor of 0.8 to 5 (García-Flor et al., 2005; Mustaffa et al., 2018; Tinel et al., 2020; Wurl and Obbard, 2004).
We calculate the emission of primary SOI as a fraction of primary marine organic aerosol, assuming they share the same size distribution as sea salt upon emission (Gantt et al., 2015; Jaeglé et al., 2011). Gantt et al. (2015) parameterized emissions of fine-mode primary marine organic aerosol using a top-down interpolated MODIS/Aqua-derived [chl a] observations at horizontal resolution (Gantt et al., 2012, 2015). We added a coarse-mode primary marine organic aerosol tracer to GEOS-Chem, following Gantt et al. (2015). We assume that the emission of coarse-mode primary marine organic aerosol follows the same size distribution as sea salt (0.1–0.5 µm dry radius for the fine mode and 0.5–4 µm dry radius for the coarse mode). The primary emission of SOI is calculated using the observed iodine-to-carbon ratios in seawater from Satoh et al. (2023), assuming 0.01 % of primary marine organic aerosol is SOI by mass (Satoh et al., 2023). This estimate could be improved by better understanding the zonal and regional dependencies of soluble organic iodine in seawater and the role of the sea surface microlayer in mediating its emission to the atmosphere. Primary SOI contributes 0.5 % of the total aerosol SOI global production rate, making this a minor source of total SOI in the model.
Primary aerosol iodide emissions utilized the GEOS-Chem SST-based sea surface iodide concentration parameterization from MacDonald et al. (2014), assuming the same size distribution as sea salt aerosol upon emission (Carpenter et al., 2013; MacDonald et al., 2014). Consistent with the fine and coarse-mode sea salt emissions in the model, primary aerosol iodide and iodate have a dry radius of 0.1–0.5 and 0.5–4 µm in the fine and coarse mode, respectively. Primary aerosol iodate emission is calculated using the average ratio of iodate to iodide in bulk seawater from Wong and Cheng (1998) and Jones et al. (2024), which was 2.2 ± 1.7 (Jones et al., 2024; Wong and Cheng, 1998). Speciated iodine observations in bulk seawater are sparse, with a relatively wide range of measured iodate: iodide ratios (0.27–5.00) (Jones et al., 2024; Wong and Cheng, 1998). However, primary iodide and iodate only contribute 0.02 % and 0.01 % of their total production rates, respectively, suggesting that this is not important for the budgets of aerosol iodide and iodate.
2.2.2 HIO3 chemistry
We added HIO3 to the model based on the formation mechanism described in Finkenzeller et al. (2023), which is discussed in detail in Liu et al. (2024) (Finkenzeller et al., 2023; Liu, 2024) (Reactions R1 and R2).
HIO3 can undergo uptake to existing fine- and coarse-mode aerosol or new particle formation (NPF) to form aerosol iodate (Table A2). New particle formation of HIO3 to form fine mode is calculated according to the rate constants shown in Eqs. (4) and (5) based on He et al. (2021a, b), described in Liu (2024) (He et al., 2021a; Liu, 2024). The nucleation rate is effectively a temperature-dependent HIO3 loss function to form fine-mode iodate (Liu, 2024).
where is the HIO3 loss rate in , k0 is the nucleation rate in , Nmol/nucleus=40.7 and refers to the number of molecules of HIO3 per 1.7 nm nucleus, [HIO3] is in molecules cm−3, and T refers to temperature in K (Liu, 2024).
HIO3 also undergoes wet and dry deposition to the surface. HIO3 photolysis rate constants and absorption cross sections have not been measured, though it is likely slow compared to other loss processes. Given that the rates and products of HIO3 photolysis are unknown, this is currently not included in the model, though should be revisited once they become available. Another potential reaction that could affect the formation of HIO3 is the photolysis of I2O5. We can calculate the lifetime of I2O5 () against reaction with water by dividing the modeled reaction rate () by the modeled concentration of I2O5 (molec cm−3). In the marine boundary layer, the lifetime is between 0.001 and 10 s across all latitudes. At 200 hPa, is between 10 and 100 s across all latitudes. Given that I2O5 rapidly reacts with water to form HIO3 (Reaction R2) the photolysis rate for I2O5 would need to be very fast to outcompete the availability of water vapor and aerosol uptake of I2O5.
2.2.3 Secondary inorganic aerosol iodine sources
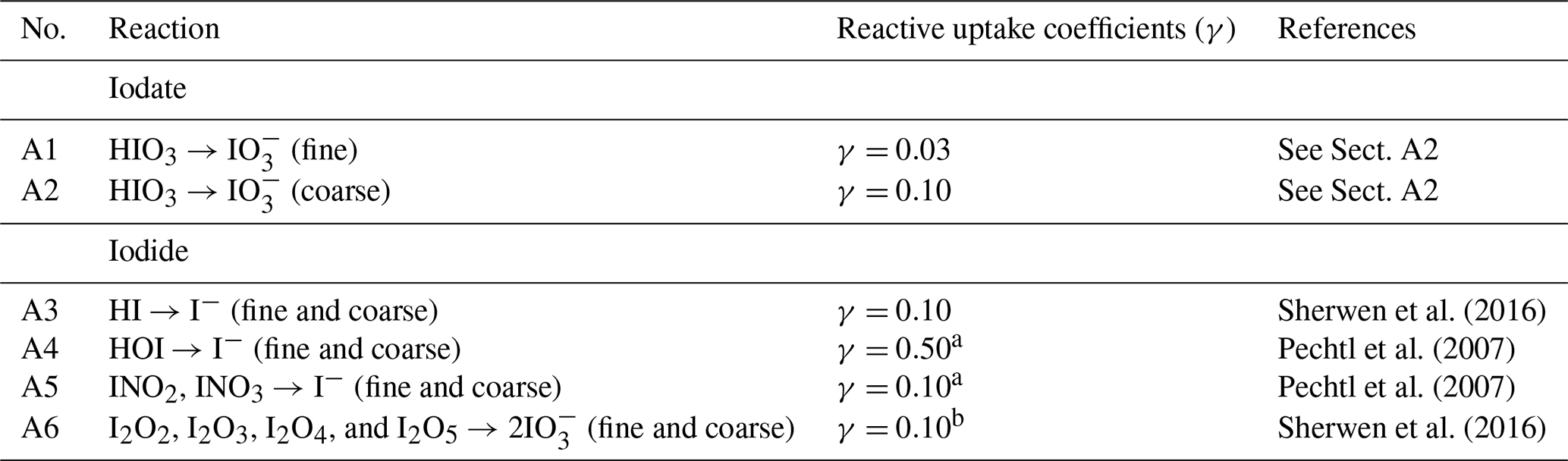
Aerosol iodide (I−) in GEOS-Chem forms through the uptake of gas-phase HI, HOI, INO2, INO3 onto fine and coarse-mode aerosol (Table A2). Aerosol iodate () forms from the uptake of gas-phase iodic acid (HIO3) and other iodine oxides (I2Ox=I2O2, I2O3, I2O4, and I2O5) (Table A2). Surface observations indicate that aerosol is more abundant in the coarse mode, as shown in Gómez Martín et al. (2022b). This size distribution is partly driven by the preferential uptake of acidic HIO3 on coarse mode aerosols, which have higher alkalinity, though the higher pH may also have a stabilizing effect for iodate. However, iodate is not necessarily an inert sink. Previous studies have indicated that iodate is reduced to iodide in aerosol (Baker and Yodle, 2021; Pechtl et al., 2007; Reza et al., 2024; Saunders et al., 2012). Iodate reduction to iodide is influenced by aerosol composition, particularly the presence of organics such as humic acid and photoactive chromophore-rich material like dust, which converts iodate to iodide (Baker and Yodle, 2021; Pechtl et al., 2007; Reza et al., 2024; Saunders et al., 2012).
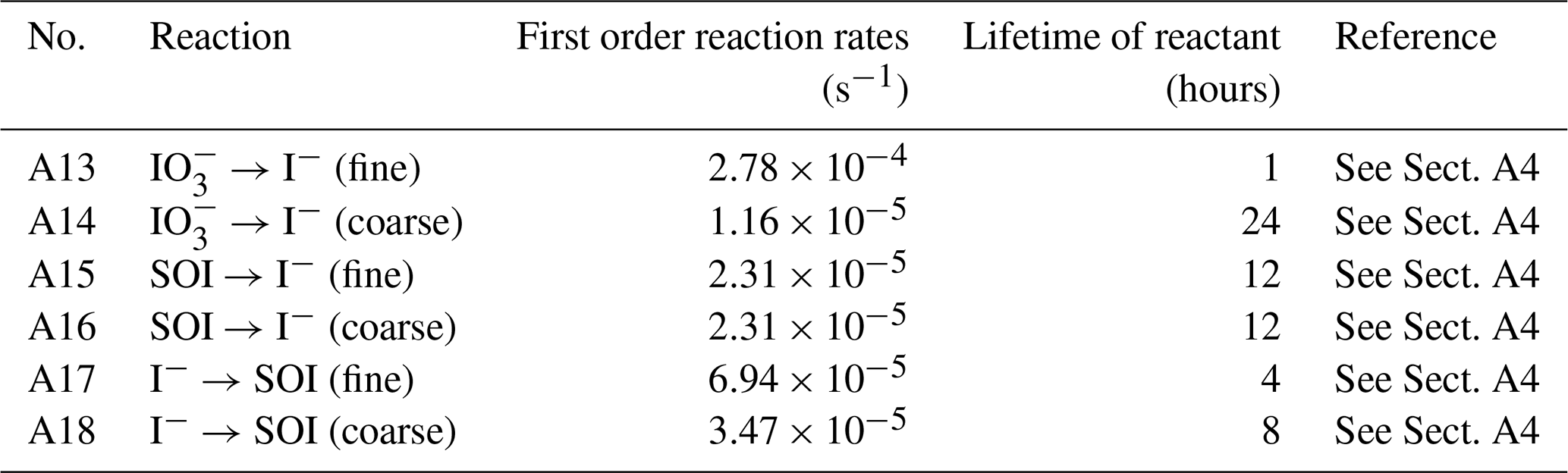
In this new version of GEOS-Chem, we implement the reduction of aerosol iodate to form aerosol iodide as a first-order reaction with a rate constant selected to achieve a size-resolved distribution of iodide and iodate consistent with global observations (Gómez Martín et al., 2022b) (Fig. 1). The lifetime of iodate against conversion to iodide is estimated to be 1 h in the fine mode and 24 h in the coarse mode in the model. This is consistent with the finding that 85 % of aerosol iodate resides in the coarse mode in observations and previous theories that the reduction of iodate to iodide is likely faster for fine mode aerosol due to its increased acidity and organic content (Baker and Yodle, 2021; Gómez Martín et al., 2022b; Saunders et al., 2012). Additionally, to represent the enhancing effect of aerosol alkalinity on HIO3 uptake, the reactive uptake coefficient for HIO3 onto coarse aerosol is assumed to be around 3 times higher than in the fine mode (Table A2). Li et al. (2024) used observations of HIO3 and particulate iodine to estimate that condensed-phase HIO3 could be recycled back to the gas-phase on a time scale of 1 to 3 h, which is consistent with the rapid fine-mode iodate parameterization in the model (Li et al., 2024). Fine mode iodate in the model contributes 24 % of total iodate, suggesting that the conversion rate of fine iodate to iodide may need to be even faster to reproduce ambient observations.
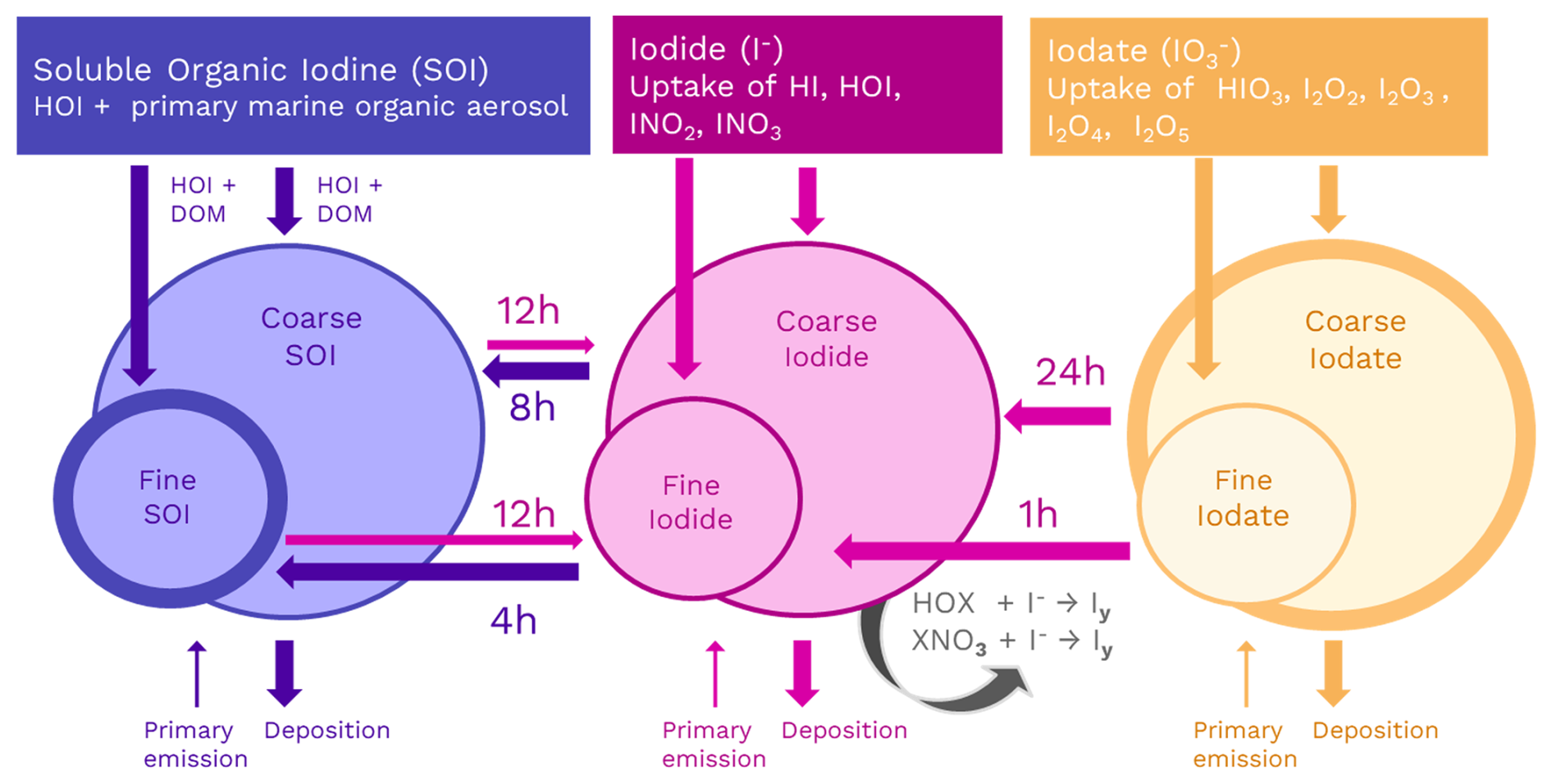
Figure 1Schematic representing heterogeneous iodine chemistry in GEOS-Chem. Soluble organic iodine (SOI) is in blue, iodide is in pink, and iodate is in orange. Each aerosol iodine species has two size bins. Arrows moving toward each species represent a source while arrows moving away represent a sink. Primary emission and deposition occur for both size bins. The iodide dehalogenation reactions are indicated by the black arrow. Interconversion between the aerosol iodine species (SOI → iodide, iodide → SOI, and iodate → iodide) is also represented, where the numbers above show the lifetime of each species in hours before conversion.
2.2.4 Secondary organic aerosol iodine sources
Soluble organic iodine (SOI) is the predominant iodine species in fine-mode aerosols, constituting 50 % of PM1 aerosol iodine mass on average (Gómez Martín et al., 2022b). While global observations have demonstrated that SOI is ubiquitous and abundant, its dominant formation mechanisms have yet to be fully elucidated. For modeling SOI, we add primary SOI emissions from the sea surface, as described previously, and two secondary sources to the model: (1) SOI formation from HOI reaction with primary marine organic aerosol, and (2) SOI formation from iodide to form iodide-organic adducts (Gómez Martín et al., 2022b; Yu et al., 2019).
One source of secondary SOI likely comes from the reaction between HOI and dissolved organic matter (DOM) (Baker, 2005; Gómez Martín et al., 2022b; Shi et al., 2021; Yu et al., 2019). Several secondary SOI species have been detected in aerosol and rainwater samples and were found to be abundant and relatively stable such as iodoacetic acid and iodopropenoic acid (Yu et al., 2019). Shi et al. (2021) identified 37 organic iodine species during their study in Beijing while Yu et al. (2019) detected 45 compounds (Shi et al., 2021; Yu et al., 2019). We have incorporated the reaction of HOI with primary marine organic aerosol to form secondary SOI, though other sources of organic aerosol (i.e. pollution, biomass burning, non-marine biogenic emissions) may also contribute to SOI formation (Shi et al., 2021; Yu et al., 2019) (Table A3).
Concentrations of SOI tend to be higher under acidic conditions (i.e. in fine mode aerosol and in more polluted air) (Gómez Martín et al., 2022b). To represent this feature in the model, we increase the reaction rate for the HOI + DOM reaction as a function of H+ concentration in solution (see Table A3 for details). This approach is similar to the parameterization of other acid-catalysed reactions between HOX and halides, where the reaction rates scale linearly as a function of H+ concentration between pH 2 and 6 for bromine (Roberts et al., 2014). This pH-dependency in secondary SOI formation makes the reaction rate faster in the fine mode relative to the coarse mode, which is also consistent with the larger abundance of fine mode SOI compared to the coarse mode in observations (Gómez Martín et al., 2022b). The modeled pH-dependency also allows this reaction to compete with reactions between HOI and halides (Cl−, Br−, I−), which are very fast (Roberts et al., 2014). Laboratory experiments that characterize the reaction rate of HOI + organic aerosol would better constrain the relative importance of these reactions.
Secondary SOI may be produced by the formation of iodide-organic adducts. Iodide-organic adducts are formed from dissolved iodide in aerosols, which can bind with hydroxyl, acid, or keto groups (Lee et al., 2014; Yu et al., 2019). While formation rates for iodide organic adducts have not been measured, we use global speciated iodine observations from Gómez Martín et al. (2022b) to tune the rates of iodide → SOI conversion in the model. In Lee et al. (2014), iodide organic adducts are formed within a chemical ionization mass spectrometer (CIMS). While it was demonstrated that organics can efficiently and quantitatively attach to iodide in Lee et al. (2014), it's unclear if this chemistry translates to form condensed-phase iodide organic adducts under ambient conditions. Based on the work in Yu et al. (2019), iodide-organic adducts are thought to be abundant at both the inland and coastal sites in ambient samples.
The first-order reaction rates for the interconversion of all of the aerosol iodine species are found in Table A4 (and depicted in Fig. 1). The use of the first-order rate constant for aerosol iodine interconversion makes this reaction easy to implement into 1D, box, or chemical transport models. We represent the formation of organic-iodide adducts with a first-order rate constant equivalent to a lifetime of 4 and 8 h for the fine and coarse mode, respectively (Table A4). The faster formation rate for iodide-organic adducts in the fine mode is supported by the higher abundance of SOI in the fine mode in global observations (Gómez Martín et al., 2022b). This also allows for the formation of secondary SOI over the continents, which is consistent with observations in Yu et al. (2019), who found that 64 % ± 8 % of total aerosol iodine at their inland site was in the form of iodide-organic adducts among the 45 organic iodine compounds they measured (Yu et al., 2019). Aerosol iodine interconversion rates for SOI → iodide and iodide → SOI were tuned to size-resolved and speciated iodide and SOI observations from Gómez Martín et al. (2022b). We explore model sensitivity to aerosol iodine interconversion rates on the order of minutes, hours, and days in a follow-up paper.
The C-I bond in many SOI compounds is likely relatively weak due to the large size of iodine atoms and diffuse orbital arrangement, leaving the possibility of dissociation into iodide in atmospheric aerosol or during sample extraction (Baker et al., 2000; Yodle and Baker, 2019; Yu et al., 2019). To represent the relative instability of iodide-organic adducts, we parameterize the dissociation of SOI to yield iodide using a first-order rate constant (Table A4). Further studies that quantify how the abundance and composition of organic aerosol impact SOI formation rates would be valuable for global modeling of SOI.
2.2.5 Aerosol dehalogenation as a source of Iy
Halogen nitrates (ClNO3, BrNO3, and INO3) and hypohalous acids (HOCl, HOBr, and HOI) react with aerosol halides to form gas-phase dihalogen species, collectively referred to in this work as aerosol dehalogenation (Reactions R3 and R4).
where HOX refers to HOCl, HOBr, and HOI, XNO3 refers to ClNO3, BrNO3, and INO3, halides are chloride, bromide, and iodide, and dihalogens are Cl2, Br2, I2, BrCl, ICl, and IBr.
The resulting dihalogen species photolyze readily and participate in other reactive halogen chemistry. While GEOS-Chem previously included dechlorination and debromination onto aerosol via HOBr, HOCl, ClNO3, and BrNO3 to form Br2, BrCl, and Cl2, iodide dehalogenation was absent (Sherwen et al., 2016a; Wang et al., 2019, 2021). As a result, aerosol iodine was effectively treated as a depositional sink for iodine species (Iy) before this work. To our knowledge, this is the first time explicit iodide conversion back to Iy has been represented in a global chemical transport model.
Appendix B details the parameters used for HOX and XNO3 dehalogenation, which use the same rate constants for iodide as bromide (Tables B1 and B2). Some of the rate constants for HOX and XNO3 reaction with iodide are not available; however, this is likely not a large source of uncertainty since the overall reaction rates are limited by the diffusion of the gases onto aerosol. We show this in Sect. B2 of the Appendix.
2.2.6 Sinks of aerosol iodine
The permanent sink for total aerosol iodine is wet and dry deposition. Iodide dehalogenation also serves as a temporary sink since the liberated Iy can either be deposited in the gas-phase or undergo aerosol uptake. The interconversion of aerosol iodine species: SOI → iodide, iodide → SOI, and iodate → iodide represent sinks for the individual aerosol species but not total aerosol iodine.
2.3 Model simulations
Table 1 details the assumptions for the three model simulations reported in this work. The base model is out-of-the-box GEOS-Chem 14.4.0 under the GEOS-Chem Classic configuration. The new iodine chemistry simulation incorporates speciated aerosol iodine, iodide dehalogenation chemistry, aerosol uptake of HIO3, and HIO3 new particle formation. New iodine chemistry (no HIO3 NPF) is the same as new iodine chemistry without NPF (Eqs. 4 and 5).

2.4 Observations used for measurement and model comparison
Global observations of speciated iodine aerosol are compiled from Gómez Martín (2022b), who synthesized all prior literature, including surface measurements from both site-based and shipborne campaigns. Surface HIO3 observations are from He et al. (2021), another synthesis of all prior measurements (Beck et al., 2021; Finkenzeller et al., 2023; He et al., 2021a, b; Jokinen et al., 2018; Sipilä et al., 2016; Thakur et al., 2022; Zhang et al., 2024). We compiled surface IO observations including site-based and cruise measurements (Allan et al., 2000; Butz et al., 2009; Carpenter et al., 2001; Gómez Martín et al., 2013; Grilli et al., 2012, 2013; Großmann et al., 2013; Huang et al., 2010; Inamdar et al., 2020; Mahajan et al., 2010b, a, 2012, 2021; Oetjen, 2009; Peters et al., 2005; Prados-Roman et al., 2015a; Read et al., 2008; Saiz-Lopez et al., 2008; Saiz-Lopez and Plane, 2004; Stutz et al., 2007). Vertical profiles of non-sea-salt iodine aerosol are from the NASA Atmospheric Tomography Mission (ATom) (Schill et al., 2025). Vertical profiles of IO are from the Tropical Ocean Troposphere Exchange of Reactive Halogen Species and Oxygenated VOC (TORERO) and Convective Transport of Active Species in the Tropics (CONTRAST) campaigns (Koenig et al., 2020; Pan et al., 2017; Volkamer et al., 2015, 2020; Volkamer and Dix, 2017). We use over two decades of iodine observations to ensure adequate spatial coverage for model and measurement comparison, even though the model was run for the year 2022. This may introduce uncertainties in model and measurement comparison in regions with high variability in iodine emissions (e.g., with strong interannual variability in surface ozone concentrations or marine biogenic production). The spatial coverage of the observations used for model comparison may be viewed in Fig. 2. See the data availability section for links to access these datasets.

Figure 2Modeled annual mean bulk (fine + coarse mode) surface iodine concentrations for total aerosol iodine (SOI + iodide + iodate) (a), bulk soluble organic iodine (b), bulk iodate (c), bulk iodide (d), and gas-phase HIO3 (e) and IO (f). Aerosol concentrations and gas-phase mixing ratios are reported in ppt.
3.1 Comparison between model and surface observations
Figure 2 compares modeled annual-average, surface bulk aerosol iodine (a–d), HIO3 (e), and IO (f) from the new iodine chemistry model simulation with surface observations (Gómez Martín et al., 2022b; Großmann et al., 2013; He et al., 2021a; Prados-Roman et al., 2015b). Modeled and observed total aerosol iodine concentrations peak in the tropics, with the highest concentrations in the equatorial Northern Hemisphere. Modeled SOI (Fig. 2b), iodate (Fig. 2c), and iodide (Fig. 2d) aerosols exhibit different spatial patterns despite similarities in their zonal distribution. SOI concentrations tend to be higher in biogenically productive marine environments (e.g. the equatorial Pacific and regions with coastal upwelling) and where iodide is also abundant. Iodate concentrations are enhanced in the Mediterranean and off the West coasts of the United States and Africa due to higher HIO3 abundance in these regions (Fig. 2c and e). Because iodide abundances result from gas-phase diffusion of a myriad of Iy species onto aerosol as well as the decomposition of SOI and iodate, it has the most diffuse spatial pattern (Fig. 2d). The model predicts that all three aerosol iodine species have the highest concentrations over the North Indian Ocean, a region that currently does not have speciated or bulk aerosol iodine observations.
Figures 2e, f and 3a, b show modeled annual-mean HIO3 (e) and IO (f) compared to surface observations. The model predicts surface HIO3 between 0.001–1.1 ppt. The normalized mean bias for modeled HIO3 in the new iodine chemistry simulation is −61.8 % for mean observed HIO3 concentrations in 3a and −17.7 % for median observed HIO3. Modeled HIO3 compares well with observations where available, though it's worth noting that the regions with the highest surface HIO3 in the model do not have observations available for model evaluation (Figs. 2e and 3a) (Beck et al., 2021; Finkenzeller et al., 2023; He et al., 2021a, b; Jokinen et al., 2018; Sipilä et al., 2016; Thakur et al., 2022; Zhang et al., 2024). HIO3 mixing ratios are enhanced near the West Coast of the US, the Mediterranean, and the Indian Ocean (Fig. 2e). The spatial pattern of HIO3 concentrations follow IO in the model since O3 deposition enhances the emission of Iy from the sea surface in these regions.

Figure 3Surface observation versus model comparisons for HIO3 (a) and IO (b). The boxplots in (a) compare GEOS-Chem and observed HIO3, organized by latitude. The light blue stars represent the monthly-mean concentration in the new iodine chemistry simulation, which coincide with the month of year the observations were made. The base model does not have HIO3 chemistry. The grey squares represent mean observations for each site. The boxplots show the median and interquartile ranges (IQR) for the observations (25th–75th percentiles). The whiskers represent 1.5 times the IQR. The black diamonds represent outliers, which exceed 1.5 times the IQR. (b) The pink circles show surface IO observations as a function of latitude. Zonal mean IO observations and model output from the new iodine chemistry simulation and base model are plotted in the pink solid, blue solid, and black dotted lines, respectively. The shading along the modeled zonal mean IO concentrations represents 1 standard deviation of the values at a given latitude.
Figure 2f shows that GEOS-Chem performs fairly well in reproducing surface IO over the open ocean (Allan et al., 2000; Butz et al., 2009; Carpenter et al., 2001; Gómez Martín et al., 2013; Grilli et al., 2012, 2013; Großmann et al., 2013; Huang et al., 2010; Inamdar et al., 2020; Mahajan et al., 2010b, a, 2012, 2021; Oetjen, 2009; Peters et al., 2005; Prados-Roman et al., 2015a; Read et al., 2008; Saiz-Lopez et al., 2008; Saiz-Lopez and Plane, 2004; Stutz et al., 2007). Figure 3b compares surface IO with the zonal-mean IO in the base and new iodine chemistry simulations, where, on average, the new iodine chemistry version underestimates surface IO by −0.5 ± 1.6 ppt. The normalized mean bias for IO in the new iodine chemistry simulation is −62.7 %, a slight improvement from the base model which had a normalized mean bias of −68.9 %. The largest differences in measured and modeled IO are at Mace Head in Ireland, the Isles of Shoals in Maine, Roscoff, France, and Halley Station, Antarctica, demonstrating that the coarse model resolution is not able to capture concentrated emissions from coastal iodine hot spots (Fig. 2f) (Alicke et al., 1999; Furneaux et al., 2010; Huang et al., 2010; Mahajan et al., 2009; Saiz-Lopez et al., 2007; Saiz-Lopez and Plane, 2004; Thurlow et al., 2014; Wada et al., 2007; Whalley et al., 2007). It is also possible that uncertainties in current chemical mechanisms and emissions of iodine contribute to the model biases in these coastal hot spots.
The overall bias for modeled HIO3 and IO may be improved further by changing the sea surface iodine emission scheme from MacDonald et al. (2014) to Sherwen et al. (2019), which had 80 % higher sea surface iodide concentrations on average with the largest increases in emissions in polar regions (Sherwen et al., 2019). When the Sherwen et al. (2019) scheme was incorporated into GEOS-Chem v14.1.1 by Pound et al. (2024), the relative mean bias of IO shifted from −0.43 ppt with the MacDonald et al. (2014) scheme to +0.43 ppt, with the largest increases occurring in the polar regions. However, v14.1.1 had sea-salt debromination deactivated and did not include speciated aerosol iodine or iodide dehalogenation, making it unclear how the updated surface emissions would affect Iy under the new iodine chemistry. Sea surface iodide concentrations from Sherwen et al. (2019), which were used in GEOS-Chem in Pound et al. (2024), have not been implemented in the base iodine emission scheme of GEOS-Chem and are therefore not considered in this work.
Figure 4 shows that adding speciated aerosol iodine and iodide dehalogenation to GEOS-Chem significantly improved agreement between modeled and measured bulk soluble aerosol iodine. While the 14.4 base model overestimated aerosol iodine, the new iodine parameterization brings the model much closer to the 1:1 ratio line (Fig. 4a and b). The model also shows a stronger correlation with observed total aerosol iodine for latitudes less than 30° N, with an r2 values of 0.76 compared to 0.34 for the new model and base model, respectively. The normalized mean bias of total aerosol iodine for latitudes below 30° N is +7.3 %, showing a substantial improvement over the base model, which had a normalized mean bias of +285.9 %. For latitudes >30° N, the model underestimates SOI, iodide, and iodate, suggesting missing iodine sources at mid- to high-latitudes in the Northern Hemisphere (Figs. 4 and C1–C3). Zonal comparisons of modeled and measured SOI, iodide, and iodate (including size-resolved observations) may be found in Figs. C1 and C2.

Figure 4(a–e) Comparisons between annual modeled and measured bulk aerosol iodine (ppt) for the GEOS-Chem base model (v14.4) (a) and new iodine chemistry simulation (b). (c)–(e) show the comparison between modeled and measured SOI (c), iodide (d), and iodate (e) for the new iodine chemistry simulation. The dashed black line represents the 1:1 ratio between the model and observations while the solid black regression line is plotted for latitudes less than 30° N. The colors represent latitude, which are grouped into 4° zonal-mean bins to correspond with the model grid resolution.
In general, the model underestimates mean bulk SOI and iodide by −0.25 ± 1.0 and −0.16 ± 0.2 ppt, respectively (Fig. 4c and d). On the other hand, iodate is overestimated for latitudes <30° N, which compensates for the underestimate in SOI and iodide (Fig. 4e). The normalized mean biases for SOI, iodide, iodate, and total aerosol iodine for all latitudes are −83.3 %, −72.3 %, +21.0 %, and −53.4 %, respectively. The normalized mean bias for total aerosol iodine in the base model for all latitudes is +22.1 %. The lower bias overall is due to the overestimate for aerosol iodine at latitudes less than +30° N, which compensates for the underestimate at latitudes greater than +30° N.
Aerosol iodine interconversion rates could theoretically alter the individual concentrations of the aerosol iodine species to increase the abundances of SOI and iodide relative to iodate. However, by increasing the conversion rate of iodate to iodide, the reactions between HOX and XNO3 with iodide are too efficient to preserve aerosol iodide before it is recycled to the gas-phase in the model. Tuning the aerosol iodine interconversion rates in the model is somewhat arbitrary, since changes in aerosol surface area and acidity in the model can alter the acid-catalysed reaction rates for HOX + iodide. Additionally, it's likely that we are missing sources of SOI that are not derived from primary marine organic aerosol, such as secondary marine organic aerosol and continental organic sources. Global observations from Gómez Martín et al. (2022b) suggest that 20 %–35 % of total aerosol iodine is non-soluble and likely derived from combustion and biomass burning, which is not currently considered in this model. Adding additional SOI sources would also help address the low bias in this study. Speciated aerosol iodine observations are strongly correlated with the amount of observed total soluble iodine at all latitudes, with r2 of 0.76, 0.92, and 0.66 for SOI, iodide, and iodate, respectively (Fig. C3). As this study is the first attempt of modeling speciated aerosol iodine at the global scale, the main goal was to reproduce total soluble iodine observations. The low bias for SOI and iodide and high bias for iodate all suggest that the rates used for aerosol iodine interconversion in this study still need to be refined once quantitative experimental results are available.
The poor model agreement with observed total aerosol iodine shown in Fig. 4 at latitudes higher than 30° N arises from severe underestimates of aerosol iodine in the North Atlantic (mainly at Mace Head, Ireland) and off the coast of Northern Alaska, which is also evident in Fig. 2a–d. The inability of GEOS-Chem to reproduce iodine observations at Mace Head is not surprising. The highly productive algae beds make this site a volcano of reactive iodine emissions to the atmosphere, with a median observed total aerosol iodine of 5.2 ppt and observations up to 37 ppt during the Marine Aerosol Production (MAP) (2006) campaign (Gilfedder et al., 2008). Nearly all of the aerosol iodine at Mace Head is SOI (96 % ± 4 %), with 58 % and 42 % of SOI in the coarse and fine mode, respectively, suggesting that a large portion is primary SOI (Fig. C1) (Gilfedder et al., 2008). Other regions rich in kelp, including The Russian Far East, the West Coast of South America, and the West Coast of Australia (Eger et al., 2023), do not exhibit the same underestimate in aerosol iodine, suggesting that this is not a systematic issue in the model for the lower latitudes (Fig. 2b) The underestimate of SOI at latitudes >30° N, however, does point to the need for more SOI sources in the model (Figs. 4c and C1, C2).
Another known missing source in GEOS-Chem is iodine emissions from snow and ice, which have also previously been demonstrated to be an important bromine source contributing to Arctic O3 depletion events (Raso et al., 2017; Thompson et al., 2015). The snowpack iodine in Brown et al. (2025) appeared to originate from the deposition of aerosol iodine onto the snowpack and subsequent re-emissions through aerosol recycling back to Iy. Since observations suggest that both iodine and bromine are enriched in snowpack relative to seawater, this iodine source should be considered in future work (Brown et al., 2025; Celli et al., 2023; Raso et al., 2017).
3.2 Vertical profiles of speciated aerosol iodine and Iy
Figure 5a–i shows modeled vertical profiles of annual-mean aerosol iodine concentrations and their speciated fractional contributions to total aerosol iodine for the tropics, mid-latitudes, and polar regions. Non-sea-salt aerosol iodine (nssI) observations from the ATom campaign (plotted in silver in Fig. 5a–c) provide a lower limit for fine-mode aerosol iodine in the atmosphere (Schill et al., 2025). During ATom, aerosol iodine was detected in several types of non-sea-salt organic particles including sulfate-rich organic aerosol, biomass burning particles, and metal-rich particles associated with ship emissions (Schill et al., 2025). While the concentration of iodine in non-sea salt particles is reported, the partitioning between SOI, iodide, and iodate in the observations is unknown. Schill et al. (2025) showed that nssI is ubiquitous in the upper atmosphere, despite the distance from the ocean surface, which is the main source of atmospheric iodine. The vertical profile of nssI closely resembles the profile of non-sea salt organic particles (Schill et al., 2025) (Fig. 5a–c). While the speciation of the ATom nssI observations is unknown, one explanation for the correlation between organics and nssI could be the formation or transport of SOI to the upper troposphere.
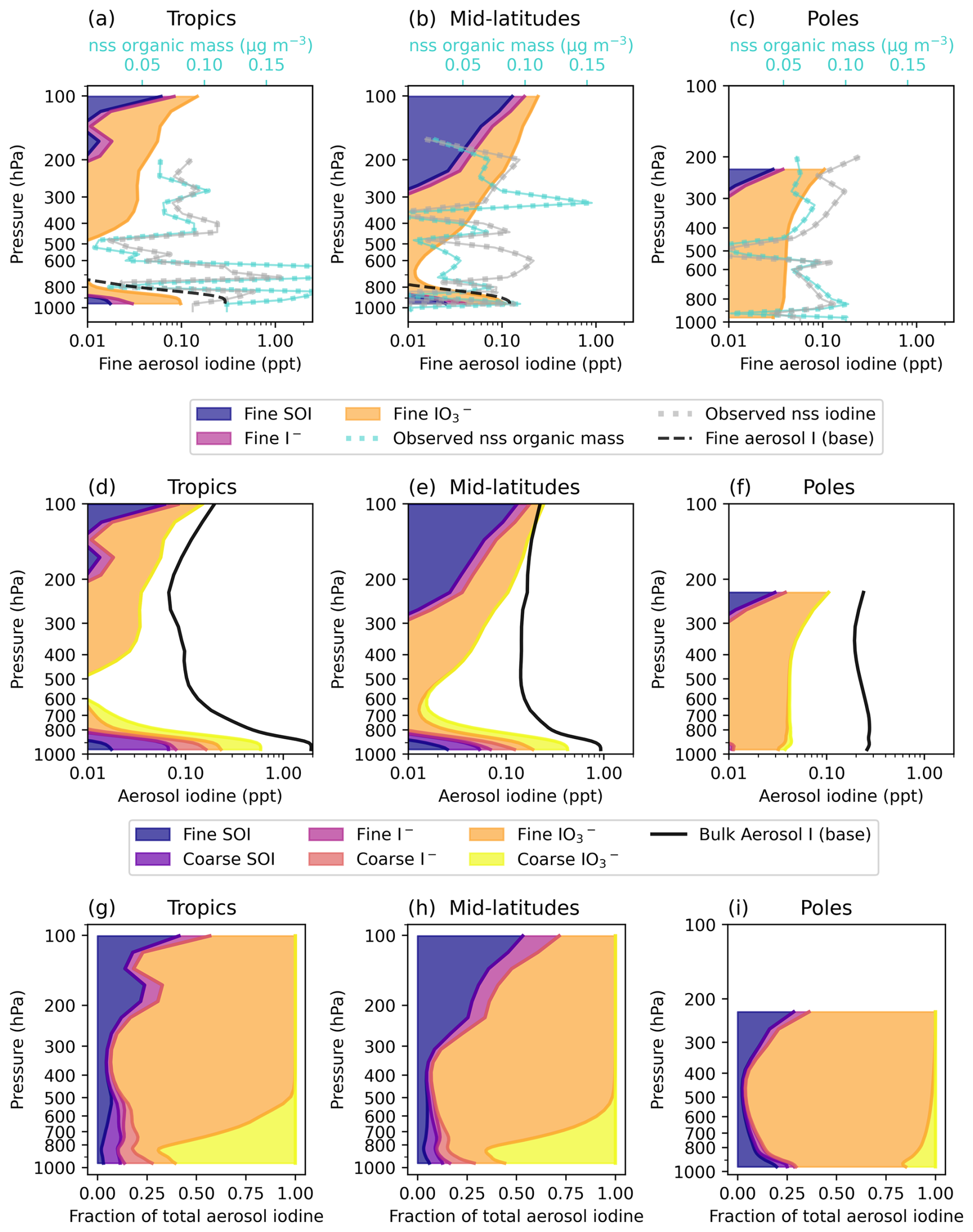
Figure 5Vertical profiles of speciated aerosol iodine in the tropics, mid-latitudes, and poles. (a)–(c) show modeled fine mode SOI (blue), iodide (pink), and iodate (orange) in ppt. The shaded profiles are stacked to represent the sum of fine SOI, iodide, and iodate. The black dashed lines show fine aerosol iodine concentrations in the base model. Average AToM nss iodine (ppt) and nss organic mass observations (µg m−3) are indicated by the silver and teal dashed lines, respectively. Panels (d)–(f) show modeled vertical profiles of both fine and coarse mode aerosol iodine, which are stacked to represent bulk aerosol iodine. The different colors represent the sizes and species of aerosol iodine including fine SOI (dark blue), coarse SOI (dark purple), fine iodide (pink), coarse iodide (coral), fine iodate (orange), and coarse iodate (yellow). The solid black line represents bulk aerosol iodine concentrations in the base model including both the fine and coarse modes. Panels (g)–(i) show the fractional contributions of fine and coarse mode iodine species to bulk total aerosol iodine, where the colors are the same as (d)–(f).
Figure 5a–c shows that fine-mode iodine aerosol is likely underestimated in the model compared to the nssI ATom observations. Fine aerosol iodine in the base model (plotted as a dashed black line) is <0.01 ppt in polar regions, underestimating fine aerosol iodine by more than an order of magnitude (Fig. 5a). Fine aerosol iodine in the base model shows an even steeper decrease in mass with height, resulting in worse agreement with observations in the upper troposphere. Part of the increase in modeled upper-troposphere aerosol iodine compared to the base model is from HIO3 new particle formation in the upper atmosphere. Because SOI and are not directly recycled to Iy in the model, their presence helps preserve the aerosol iodine to allow it to be transported away from the surface. Despite these additions, the amount of fine iodine in the upper troposphere is still underestimated compared to observations. The model's underestimation in the upper troposphere could be due to the underestimated surface emissions with the MacDonald scheme in addition to issues with aerosol iodine transport, deposition, or a combination of these factors. Due to the high abundance of HOX and XNO3 in the lower troposphere, the mean lifetime of fine mode iodide against dehalogenation is only 12 min, resulting in low iodide abundance throughout the troposphere (Figs. 4a–c and C1). Because HOX dehalogenation reactions are acid-catalysed, fine-mode aerosol in the model may be too acidic, making the recycling rate to form Iy too fast. Alternatively, surfactants on the aerosol surface may slow down dehalogenation, which is not currently considered in the model.
Figure 5d–f includes coarse mode aerosol concentrations in addition to the fine mode concentrations to represent total aerosol iodine in the model. Total aerosol iodine peaks at the surface and decreases with height through the mid-troposphere. In the upper troposphere, this trend reverses and aerosol iodine begins to increase with height. Schill et al. (2025) also observed an increase of nssI with height in the upper troposphere during ATom. Figure 5g–i shows that the modeled fraction of SOI increases with height in the tropics and mid-latitudes. This is also consistent with the hypothesis that organics have a stabilizing effect on aerosol iodine in the troposphere by binding with iodide to form adducts, effectively slowing down reactive iodine chemistry by not allowing iodide to be recycled back to the gas phase. While we lack laboratory studies that explicitly investigate reaction rates between iodide and organic aerosol to form adducts, observations from Yu et al. (2019) found that iodide-organic adducts constituted 64 % ± 8 % of aerosol iodine at their inland site 200 km from the coast, which was higher than the coastal site contribution at 31 % ± 16 % of total aerosol iodine. In the aged particles from their study, SOI contributed 76 % ± 7 % of total aerosol iodine, which further supports the hypothesis of organics having a stabilizing effect on aerosol iodine.
Modeled fine mode iodate is the dominant form of aerosol iodine above the boundary layer in the troposphere (Fig. 5g–i). Koenig et al. (2020) found an increase in the fraction of iodate moving upward from the upper-troposphere to the lower stratosphere based on ratios in aerosol mass spectrometry (AMS) observations. They report a mean iodate fraction of 56 % compared to total aerosol iodine in the upper troposphere, however, only one organic iodine aerosol species (5-iodo-2-furfural) was considered, so the contribution of iodate to total aerosol iodine is not easily comparable to this study (Koenig et al., 2020). Obtaining more speciated aerosol iodine observations in the upper atmosphere would shed light on the relative stabilities of SOI, iodate, and iodide as they age in the atmosphere and provide further guidance for model improvement. Additionally, the relative distribution of aerosol iodine in non-sea salt vs. sea salt particles is unknown in the upper atmosphere, though sea salt abundances decrease by around a factor of 10 for every 2 km in altitude, with very low sea salt abundance in the upper troposphere (Murphy et al., 2019). This likely means that nssI aerosol becomes a relatively larger contributor to total aerosol iodine in the upper troposphere, as sea salt particles are more efficiently removed by deposition due to their high solubility and larger size. Quantifying the abundance and speciation of iodine in different particle types as a function of altitude would further aid in model improvement.
Figure 6 shows vertical-mean profiles and zonal-mean surface Iy composition for the base model (a and b), the new iodine chemistry simulation (c and d), and the new iodine chemistry (no HIO3 NPF) simulation (see Table 1 for model configuration information). HIO3 is a large contributor to Iy in the troposphere in the model. The HIO3 concentration increases with height and peaks in the mid-troposphere (around 600 hPa). In the sensitivity study where HIO3 NPF is not included (“no HIO3 NPF”), a large bubble of HIO3 forms in the upper troposphere, with mean concentrations around 1 ppt (Fig. 6e). HIO3 accumulates as a function of altitude without NPF due to the lack of existing aerosol surface area for uptake of HIO3 to form iodate. This makes HIO3 the globally dominant iodine species in the upper atmosphere, contributing almost all of total Iy upward from 800 hPa (Fig. 6e). The difference in the tropospheric burden of HIO3 in the “no HIO3 NPF” and “new iodine chemistry” simulations in Fig. 6 is large, contributing 6.6 and 2.4 Gg I of HIO3, respectively. In the “new iodine chemistry” simulation, HIO3 NPF contributes 24 % of the HIO3 loss rate, with fine aerosol uptake and coarse aerosol uptake contributing 46 % and 25 %, respectively, and deposition contributing the remainder of HIO3 loss rate at 5 %. These results suggest that HIO3 is a critical terminal product of iodine in the gas phase and that the NPF mechanism is an important sink for HIO3 in the upper troposphere in the model, allowing for iodate formation and recycling back to other forms of Iy. This further indicates that global-scale simulations should include HIO3 when assessing iodine's impact on atmospheric oxidizing capacity and particle formation.
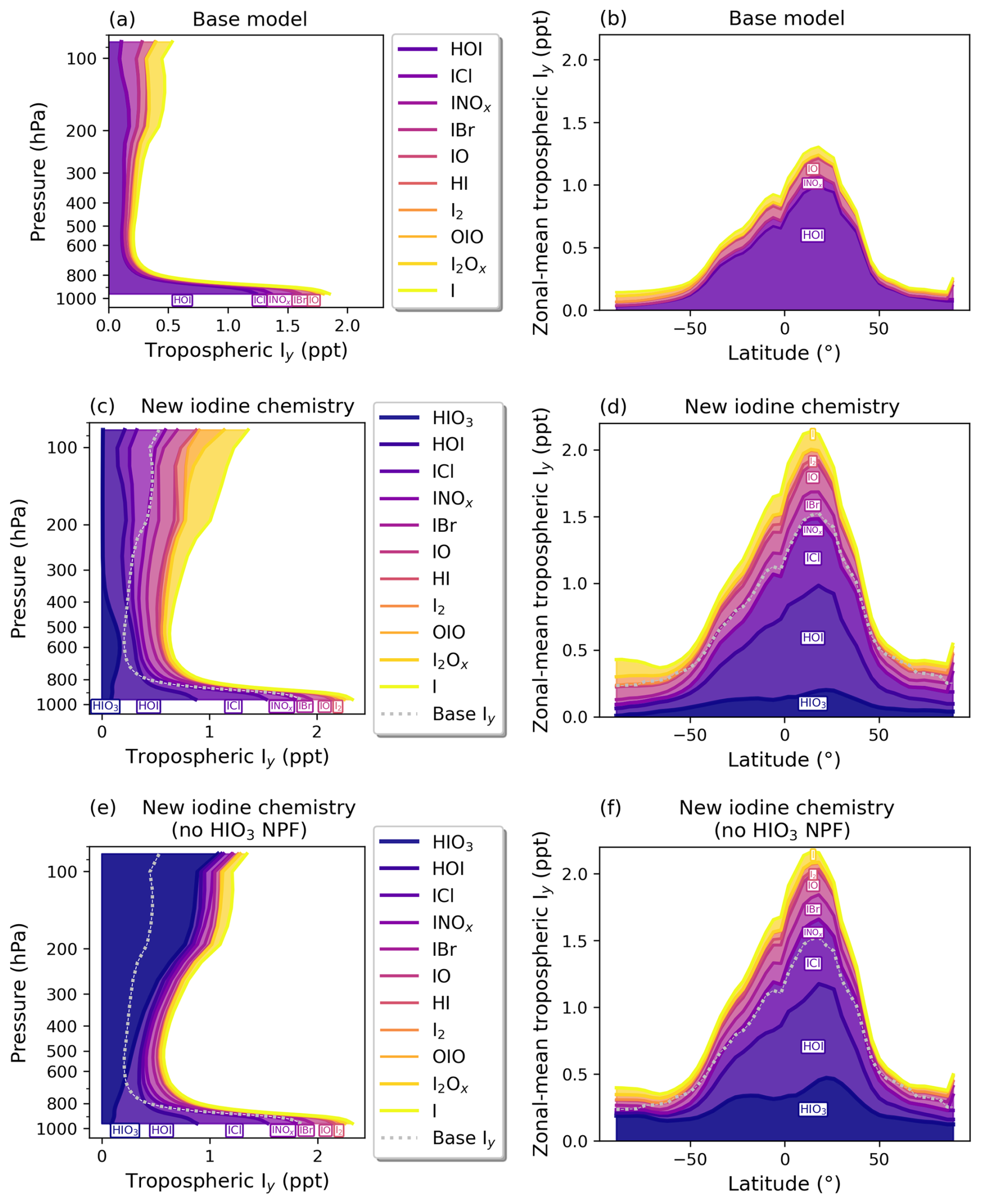
Figure 6Modeled annual-mean vertical and zonal profiles of Iy mixing ratios in ppt for the base model (a, b), new iodine chemistry (c, d), and new iodine chemistry (no NPF) (e, f). The colors represent different reactive iodine species. The silver dashed line in (c)–(f) represents total Iy in the base model for easy comparison with the updated iodine chemistry simulations.
With the added speciated aerosol iodine and HIO3 chemistry, the abundance and composition of Iy changed compared to the base model. In the “new iodine chemistry” simulation, non-HIO3 Iy increased by 75 % while total Iy (including HIO3) increased from 11.5 to 23.1 Gg in the troposphere, more than doubling the total Iy abundance (Tables 2 and D1). The largest increases in Iy were due to iodide dehalogenation to form ICl, IBr, and I2. The total dihalogen iodine concentration increased by a factor of 11, with ICl, IBr, and I2 concentrations increasing by factors of 7, 22, and 47, respectively (Table D1). This corresponded to a 74 % decrease in total aerosol iodine (from 6.9 Gg total aerosol I in the base model to 1.8 Gg in the new iodine chemistry simulation) (Tables 2 and D1).
The updated iodine chemistry in GEOS-Chem increases IO concentrations in the upper-troposphere in the model, which we compare with observations from two aircraft campaigns, TORERO and CONTRAST (Fig. 7a–d) (Koenig et al., 2020; Pan et al., 2017; Volkamer et al., 2015, 2020; Volkamer and Dix, 2017). IO was measured during both campaigns by the CU AMAX-DOAS instrument. The Tropical Ocean Troposphere Exchange of Reactive Halogen Species and Oxygenated VOC (TORERO) campaign in the eastern Pacific measured IO profiles between 1000–200 hPa, observing mean concentrations of 0.19 ± 0.17 ppt (Fig. 7a) (Koenig et al., 2020; Volkamer et al., 2015; Volkamer and Dix, 2017). Selecting gridboxes along the flight path between 12:00–15:00 LT to represent daytime concentrations in the model, the new iodine simulation performs better at reproducing mean IO concentrations during TORERO (0.12 ± 0.12 ppt) compared to the base model (0.06 ± 0.11 ppt) (Fig. 7a). This reduces the magnitude of normalized mean bias of IO in the new iodine chemistry simulation (−39.3 %) compared to the base model (−65.1 %).
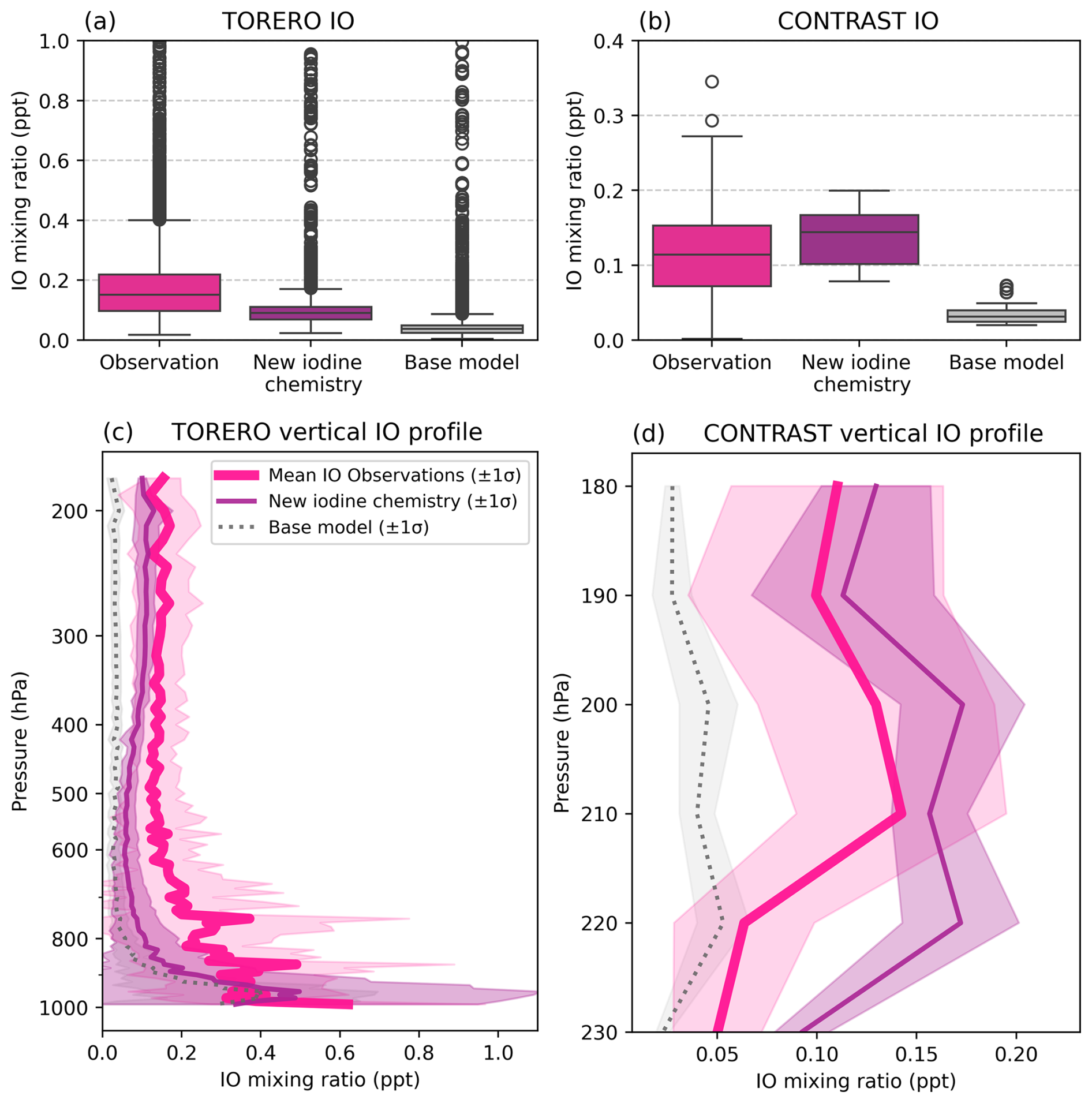
Figure 7Comparison between TORERO (a, c) and CONTRAST (b, d) observations and January GEOS-Chem output between 12:00 and 15:00 LT to reflect daytime concentrations in the model. IO observations are in pink, the new iodine chemistry IO is in purple, and base model IO is in silver.
Similarly, the Convective Transport of Active Species in the Tropics (CONTRAST) campaign provided aircraft-based IO measurements in the Western Pacific upper troposphere (260–180 hPa), reporting mean IO concentrations of 0.12 ± 0.06 ppt. Along the flight paths between 12:00–15:00 LT, January-mean GEOS-Chem IO concentrations were 0.14 ± 0.04 ppt with the new iodine scheme, an improvement over the base model's 0.03 ± 0.02 ppt (Koenig et al., 2020; Pan et al., 2017; Volkamer et al., 2020) (Fig. 7b and d). The normalized mean bias in the new iodine chemistry simulation compared to CONTRAST observations is +29.2 %, which is a slight improvement over the base model −67.4 %, though it also represents a substantial increase in IO in the upper troposphere and leads the model to slightly overestimate compared to observations. The increase in gas-phase iodine in the upper troposphere with the new iodine chemistry is also consistent with Dix et al. (2013), who found that a substantial portion of the IO column resided above the marine boundary layer and hypothesized that its source originated from heterogeneous recycling of iodine from aerosols (Dix et al., 2013).
Above 300 hPa, the new iodine chemistry simulation yields IO levels that are closer to the TORERO and CONTRAST observations than the base model, indicating improved model performance in the upper troposphere (Fig. 7c and d). During TORERO the mean difference in IO concentration above 300 hPa between the model and observations was −0.04 ppt (compared to −0.12 ppt in the base model) and during CONTRAST the mean difference above 300 hPa was +0.02 ppt (compared to −0.08 ppt in the base model). GEOS-Chem IO in version 12.9 compared well with TORERO observations, however, the emission of iodine in the model is extremely sensitive to O3 concentrations in the marine boundary layer (Wang et al., 2021). This means that iodine abundances can vary substantially across GEOS-Chem versions along with changes in VOC, NOx, and oxidant chemistry. Even so, the improvement in modeled IO in the upper atmosphere in this work compared to observations suggests better representation of iodine in the upper troposphere with the new scheme.
3.3 Impact on effective iodine lifetime and global halogen budgets
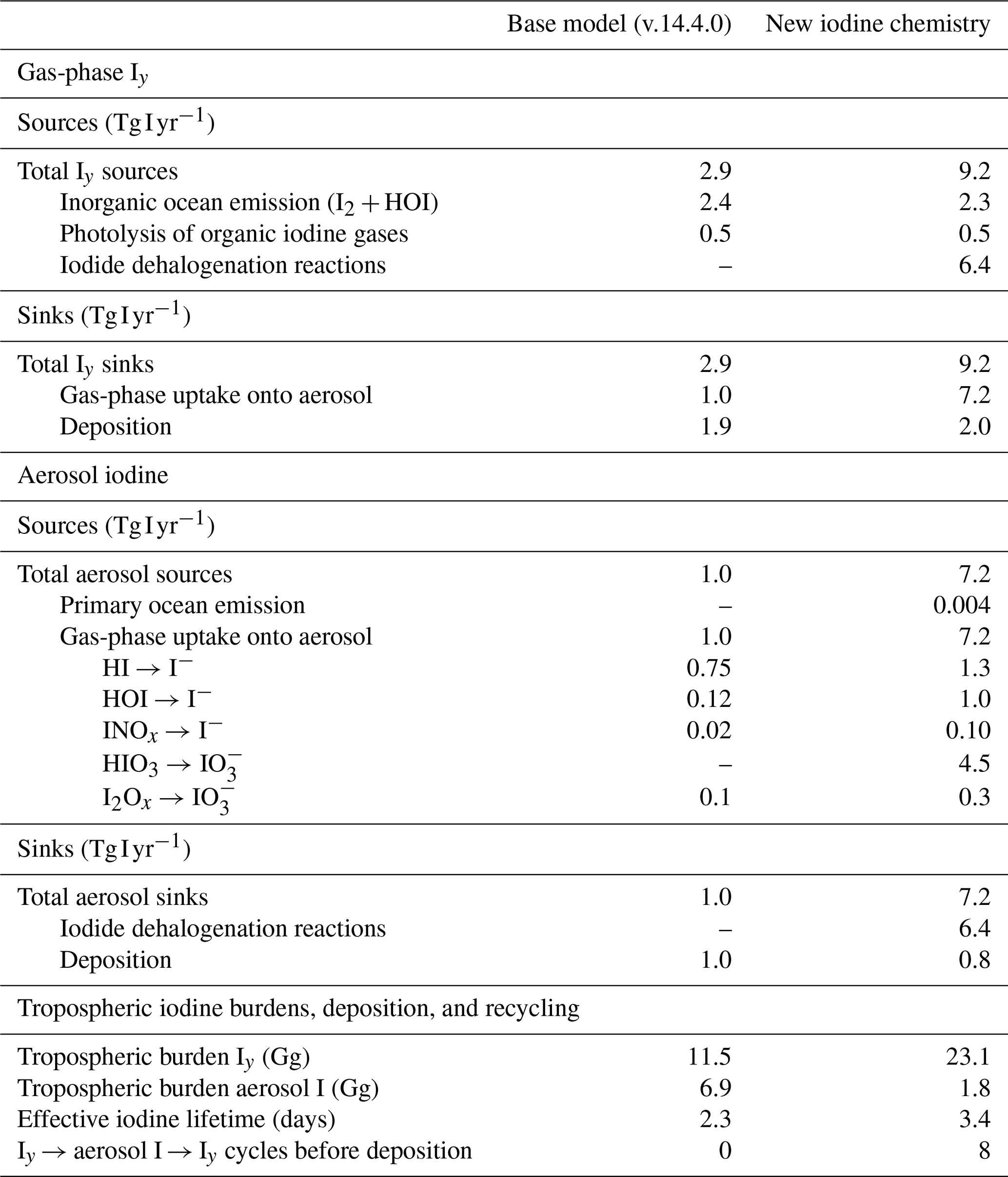
Table 2 and Fig. 8 show the changes in the global iodine sources and sinks after implementing the new chemistry. The total emission of inorganic iodine in the updated model decreases by −0.12 Tg I yr−1 (−5.4 %) due to a decrease in the tropospheric burden of O3 resulting in decreased sea surface reaction between iodide and O3.
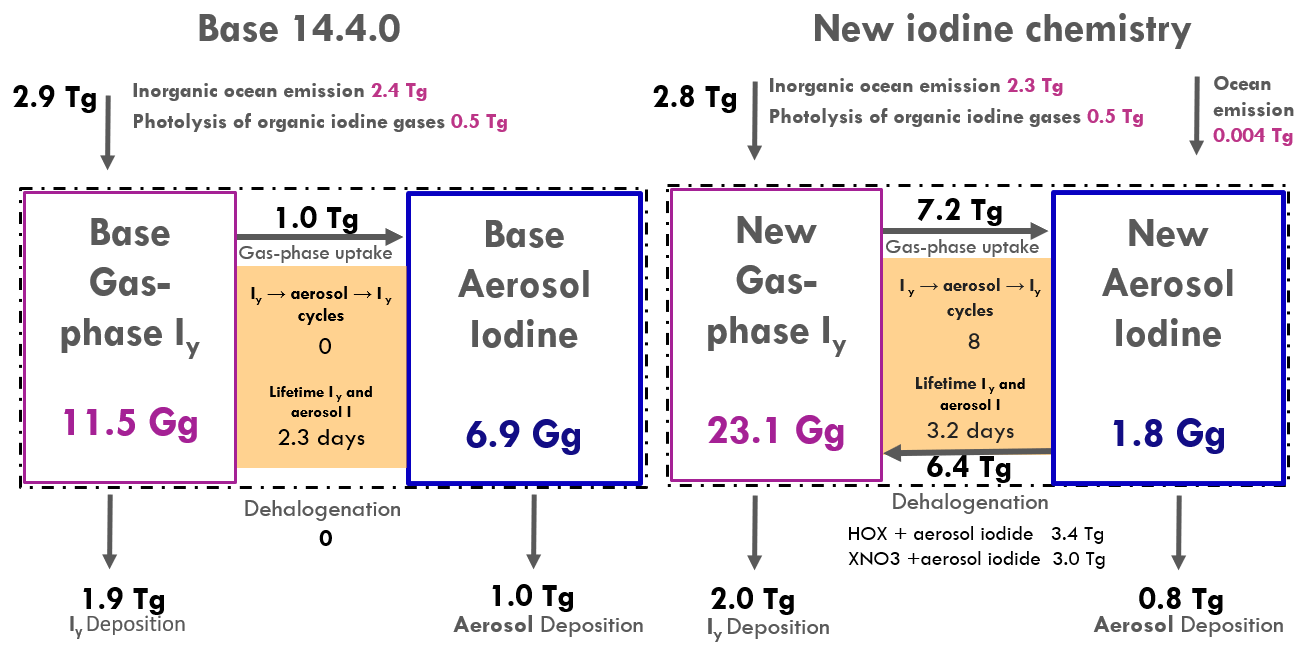
Figure 8Schematic of iodine sources and sinks in the base model (left) and updated model (right). The pink boxes and blue boxes show the tropospheric burdens in Gigagrams of Iy total aerosol iodide, respectively. The dashed black line represents total iodine (including gas and aerosol phases). Arrows moving toward the dashed black line indicate iodine sources while arrows pointing away represent sinks, with rates in Tg yr−1. The black arrows in between the pink and blue boxes show the rates of gas-phase uptake to form aerosol iodide and aerosol iodine dehalogenation to yield Iy, expressed as rates in Tg yr−1. The orange shaded boxes indicate the number of cycles before deposition and the effective lifetime of gas and aerosol iodine in days, which is calculated using the burden of both reservoirs against the total depositional loss rate (Table 2).
The tropospheric ozone burden decreases by −2 % in the new iodine chemistry simulation compared to the base model (see Table E1). On average, surface O3 concentrations decrease by −3 %; however, the spatial pattern of O3 changes are not uniform across the two hemispheres (Fig. E1). Surface O3 increases for latitudes less than −30° S by up to +11 % over the Southern Ocean and Antarctica. The increase in surface O3 over the Southern Ocean coincides with decreases in BrO by −0.6 ppt on average. Lower BrO concentrations reduce the rate of the reaction, which is a major contributor to the ozone loss rate (Bates and Jacob, 2020). At latitudes greater than −30° S, surface O3 decreases by up to −18 %, with the largest decreases over the tropical Pacific and Indian Oceans and in the Arctic (Fig. E1). We discuss the impacts of the new iodine chemistry on oxidants further in a follow-up publication.
The net decrease in surface O3 is responsible for the −0.12 Tg decrease in inorganic iodine emissions in the new iodine chemistry simulation. Figure E2 compares annual-mean surface ozone concentrations in the base model and new iodine chemistry simulations with Tropospheric Ozone Assessment Report II (TOAR II) ship and buoy observations (Kanaya et al., 2025a, b). Overall, both the base model and new iodine chemistry simulations reasonably simulate surface ozone concentrations, with r2 values of 0.84 and 0.90 for the base model and new iodine chemistry simulations, respectively. The decrease in surface ozone for latitudes ° S leads to a slightly more negative normalized mean bias in the new iodine chemistry simulation (−16.0 %) compared to the base model (−10.7 %).
Iodide dehalogenation reactions produce 6.4 Tg I yr−1, which is more than double the total source of Iy in the model from ocean emissions and the photolysis of organic iodine gases, which produce 2.8 Tg I yr−1 (Table 2 and Fig. 8). This suggests that even though the aerosol is not the initial Iy source, the overall distribution and abundance of reactive iodine is strongly mediated by heterogeneous chemistry of aerosol iodine.
As a result of the increased Iy recycling rate, the aerosol uptake rate of Iy substantially increases from 1.0 to 7.2 Tg I yr−1. The majority (62 %) of Iy to aerosol iodine conversion is from HIO3 uptake onto aerosol and new particle formation to form iodate (Table 2). The lifetime of fine mode aerosol iodate in the model is 1 h before it is converted to iodide, which can then be liberated to the gas phase via heterogeneous reactions (Reactions R3 and R4). The short modeled lifetime of fine iodate is consistent with rapid reduction of iodate to yield Iy in Reza et al. (2024) and Li et al. (2024), who observed iodate recycling even in the absence of light (Li et al., 2024; Reza et al., 2024). The overestimate of iodate compared to surface observations suggests that the conversion of iodate to either iodide or Iy is likely underestimated. The uncertainty of HIO3 uptake and subsequent recycling is compounded by the fact that the reactive uptake coefficients () for HIO3 in the updated iodine simulation of 0.03 and 0.10 for the fine and coarse modes, which is on the lower end of the estimates from other studies. Previous modeling studies have set from 0.2 to unity (Pechtl et al., 2007; Zhao et al., 2024). In a follow up paper, we evaluate the sensitivity of the model to prescribing to unity and iodate to iodide conversion rates on the order minutes instead of hours.
The effective lifetime of iodine in the atmosphere can be calculated by dividing the tropospheric burdens of gas and aerosol iodine by their total loss rate from wet and dry deposition to the surface (Eq. 6).
The effective lifetime of iodine in the atmosphere increases by 42 % in the updated model from 2.4 to 3.4 d (Table 2 and Fig. 8). This coincides with the tropospheric burden of Iy increasing from 11.5 to 23.1 Gg (Table 2 and Fig. 8). The increase in the effective iodine lifetime from the speciated iodine chemistry and iodide dehalogenation is responsible for the increase in the Iy burden, where less time is spent in the aerosol phase compared to the gas phase, increasing the amount of time the iodine can persist in the atmosphere before deposition. With the addition of aerosol recycling, the contributions of Iy deposition increases compared to aerosol I deposition; with a larger contribution from Iy in the new iodine chemistry simulation (71 % of total I deposition) compared to the base model (66 % of total I deposition).
The average number of cycles that Iy is converted to aerosol I and back to Iy before deposition can be calculated by using the ratio between the iodide dehalogenation rate and the aerosol iodine deposition rate. On average, this cycle occurs 8 times in the troposphere with the current model configuration. We note that the interconversion rates between SOI → iodide, iodide → SOI, and iodate → iodide effectively dictate the overall iodide dehalogenation rates, where higher SOI and iodate concentrations relative to iodide slows down reactive iodine chemistry and increases the depositional loss from aerosol I relative to Iy. We explore the sensitivity of atmospheric oxidants to iodine parameterizations in GEOS-Chem including the rates of conversion between SOI, iodide, and iodate and model sensitivity to HIO3 NPF and reactive uptake coefficients in a follow-up paper.
3.4 Remaining uncertainties in the measurements and modeling of atmospheric iodine
We have implemented speciated aerosol iodine and heterogeneous chemistry of iodide aerosol in GEOS-Chem, largely relying on the existing network of surface observations presented in Gómez Martín et al. (2022b). We would like to emphasize that observations of the relative abundances of aerosol iodine species are uncertain. This is because speciated aerosol iodine is measured through offline filter extraction into solution, separation via ion chromatography (IC), and then measurement of iodine as a function of retention time (IC-ICPMS) (Gómez Martín et al., 2022b). Given the sensitivity of iodine speciation to pH, the speciation in the sample may be altered the moment the filter is extracted by simply diluting the solution, inducing interconversion between SOI, iodide, and potentially iodate. Several studies have also shown that sonication during sample extraction causes interconversion between SOI and iodide. Sonication can either increase the amount of iodide by causing SOI to dissociate (observed in Yodel and Baker, 2019 ), or increase the amount of SOI as iodide reacts with organics in bulk solution (observed in Baker et al., 2000). These uncertainties in measuring the speciation of aerosol iodine propagate into global modeling uncertainties. Therefore, we call for future endeavours to carry out in situ online measurement of these components to constrain the uncertainties.
We do not represent non-soluble iodine in the model, though it has been observed in the atmosphere and contributes 20 %–35 % of total aerosol iodine (Gómez Martín et al., 2022b). It is hypothesized that non-soluble iodine (NSI) aerosol is formed in hydrophobic aerosol like soot and has a continental origin (Gómez Martín et al., 2022b). Schill et al. (2025) also identified biomass burning as a source of primary aerosol iodine, though neither of these formation processes has been included in the model. NSI may be an important sink for Iy in biomass-burning plumes since they provide a surface for aerosol uptake. Shi et al. (2021) observed large increases in both primary and secondary organic iodine aerosol during the heating season in Beijing, which seemed to come from coal combustion, suggesting that there are likely also anthropogenic sources of organic iodine aerosol (Shi et al., 2021).
There are likely other missing iodine sources in the model. GEOS-Chem performs poorly in reproducing total aerosol iodine observations north of 50° N. This may be improved by transitioning from the MacDonald Iy emission scheme to the Sherwen et al. (2019) scheme, which has substantially higher Arctic emissions. Adding a snow source for iodine may also improve model performance in the Arctic.
The model is also missing continental sources of iodine. Biomass burning and anthropogenic emissions are potentially important sources of iodine (Schill et al., 2025; Shi et al., 2021). Additionally, aircraft observations of IO have indicated that dust is a potential driver of gas-phase iodine, where IO concentrations were enhanced by up to a factor of 10 within lofted dust layers off the coast of Chile, coinciding with lower ozone abundances (Koenig et al., 2021). This is in line with observations in the Canary Islands, where dust events were associated with higher gas-phase iodine abundances, with CH3I concentrations increasing by factors of 2 to 14 compared to background conditions (Puentedura et al., 2012; Williams et al., 2007). At a mountain site in Colorado, Lee et al. (2024) found that GEOS-Chem underestimated IO by about a factor of 3. This bias could be due to overestimated IO sinks, likely in part because of the previous lack of aerosol recycling and shorter effective iodine lifetime in GEOS-Chem (Lee et al., 2024). However, their findings could also indicate missing continental iodine sources. More observations of iodine in inland sites will further aid in identifying the relative importance of non-marine sources in the global iodine budget.
Because almost all speciated iodine observations are from the marine boundary layer, relatively little is known about the processes governing the relative abundance of these species as they age and are transported upwards in the atmosphere. Interconversion rates between aerosol iodine species are uncertain and are currently parameterized by first-order rate constants tuned to global surface observations. Iodate is the dominant aerosol iodine species throughout most of the troposphere in the model, though as discussed before, it is overestimated compared to surface observations (Figs. 4 and 5). It is possible, however, that iodate is the dominant species in the upper troposphere due to the efficiency of HIO3 NPF at low temperatures and its relative stability against dehalogenation compared to iodide (He, 2023; He et al., 2021a).
We do not account for ionic strength and pH in modeling the uptake of Iy onto aerosol because we use a kinetic approach, where the aerosol formation rates from HI and HIO3 are estimated based on the estimated reaction probability and aerosol surface area. In reality, this is a thermodynamic process influenced by temperature, the pH of aerosol liquid water, and the ionic strength of the solution. To better represent and understand gas-to-aerosol iodine partitioning, inclusion of HI-I− and partitioning using a thermodynamic approach may be better.
Given the sensitivity of dehalogenation chemistry to pH, further evaluation of this model parameter is warranted. Cloud water pH in GEOS-Chem was evaluated in Shah et al. (2020), where they found good agreement between global observed and modeled cloud water pH, with mean values of 5.2 ± 0.9 and 5.0 ± 0.8, respectively (Shah et al., 2020). On the other hand, fresh sea salt aerosol has been found to rapidly acidify in the remote marine atmosphere, with aerosol pH ranging between 1.5 and 2.6 within minutes of emission, compared to the seawater pH around 8 (Angle et al., 2021). Observations of aerosol pH in the marine boundary layer are spatially and temporally sparse, making evaluating model performance in simulating aerosol pH difficult. In general, aerosol pH used to calculate dehalogenation rates ranges between −0.3–5.3 and 5–8 for the fine mode and coarse mode, respectively, which is within the ranges estimated in the literature at −1.1 to 5.3 for fine mode and 1.2–8.0 for coarse mode (Angle et al., 2021; Pye et al., 2020). More work is needed in the atmospheric chemistry community to evaluate model performance in simulating the pH of fine and coarse mode aerosol in the remote marine atmosphere.
We have added aerosol iodine dehalogenation to the model using the same rate constants for debromination. Additional BrCl, ICl, and IBr observations will help constrain aerosol dehalogenation rates, since these dihalogen species have only been measured in a handful of studies (Finley and Saltzman, 2008; Tham et al., 2021). Finally, more synchronous gas and aerosol phase observations would help us better understand the role of heterogeneous chemistry in regulating the abundance of Iy and oxidants in the atmosphere.
While there are still many uncertainties in understanding the formation and interconversion between aerosol iodine species, this study serves as a framework for future work. Adding the new SOI and iodate tracers to GEOS-Chem allows for explicit representation of the different types of aerosol iodine and examination of their potential importance in the atmosphere. Additionally, the first-order rate constants used to represent the interconversion rates between aerosol are easily tuneable until a more detailed parameterization informed by new laboratory studies is developed. Previous versions of GEOS-Chem have showed better performance in reproducing IO observations than the 14.4 base model presented in this study. In the absence of aerosol iodide recycling and speciated aerosol iodine chemistry in the base model, the abundance of Iy is largely tied to the surface abundances of ozone, which dictate the release of I2 and HOI from the sea surface. Because of this, iodine chemistry in GEOS-Chem is sensitive to the model version as NOx, VOC, and oxidant abundances are altered. It is possible that performance will vary with future model developments, as we've already observed in comparing the results of previous iodine modeling work in GEOS-Chem, including v10 in Sherwen et al. (2016b), v12.9 in Wang et al. (2021) and Schill et al. (2025), v13.2 in Lee et al. (2024), and v14.4, which served as the base model in this study.
Despite the modeled sensitivity of iodine chemistry to surface ozone concentrations, the large increase in the Iy burden and the substantial contribution of iodide dehalogenation to Iy production demonstrated in this work are expected to persist across model versions. Additionally, the treatment of aerosol iodine here differs fundamentally from previous GEOS-Chem implementations: rather than serving primarily as a depositional sink for Iy, aerosols are shown to be a significant source. This new framework for modeling aerosol iodine will ultimately make for a more robust representation in GEOS-Chem in future model releases because we present additional model parameters that can mediate the abundances and recycling of atmospheric iodine.
Incorporating speciated aerosol iodine and iodide dehalogenation in GEOS-Chem has revealed new insights into the role of heterogeneous chemistry in regulating Iy abundance. Formation of aerosol iodine was previously treated as a depositional sink for Iy in GEOS-Chem. To our knowledge, we have implemented recycling of aerosol iodide back to the gas phase for the first time in a global model. In doing so, we found that aerosol iodide is a large source of gas-phase Iy. The Iy production rate from aerosol is more than double the rate of inorganic gas-phase emissions and organic photolysis combined, suggesting that aerosol iodine is a major control in mediating Iy abundances (Table 2 and Fig. 8). The new iodine chemistry has increased the effective lifetime of total gas and aerosol iodine by 42 % compared to the base model (Table 2 and Fig. 8). This increase in the effective iodine lifetime has profound implications for global halogens since it allows for further transport of iodine away from the marine boundary layer into continental regions and up to the upper troposphere (as shown in Figs. 6 and 7). SOI and iodate act to stabilize the iodine aerosol against dehalogenation, slowing down the interconversion rates of aerosol and gas-phase iodine species and impacting Iy abundance. These findings have implications for global oxidant budgets and new particle formation. We will explore the impact of the changes in iodine abundance and distribution on global oxidant budgets in a follow-up publication.
Model bias was improved compared to the 14.4 base model for IO and aerosol iodine compared to surface and aircraft-based observations, though it's likely that surface Iy concentrations are still underestimated in the model. We also added HIO3 chemistry to this model version, reproducing surface HIO3 observations within a factor of two at most sites. As stated before, transitioning to a higher Iy emission scheme may improve model agreement with surface observations, though a cautious approach is warranted given the efficiency of aerosol recycling and the increased effective lifetime of iodine in the model. The improvement in modeled IO and aerosol iodine in the new iodine chemistry simulation compared to the base model is promising, especially in trying to understand the importance of iodine for global oxidant budgets.
Ice core records from Greenland and the French Alps have previously suggested that iodine concentrations have increased by a factor of three since 1950 (Corella et al., 2022; Cuevas et al., 2018; Legrand et al., 2018; Zhao et al., 2019). This rise has been attributed to a combination of factors, including elevated sea surface temperatures enhancing marine primary productivity, reduced sea ice extent, and increased ambient ozone concentrations boosting gas-phase Iy release from the sea surface (Corella et al., 2022; Cuevas et al., 2018; Legrand et al., 2018; Zhao et al., 2019). Zhai et al. (2023) found that changes in atmospheric acidity can explain observed trends in ice core bromine via its effects on acid-catalyzed aerosol debromination (Zhai et al., 2023, 2024). Due to the importance of acid-catalyzed heterogeneous chemistry for Iy abundances shown here, changes in atmospheric acidity should also be considered as a potential influence on trends in tropospheric reactive iodine.
Appendix A details the parameterization for the sources of speciated aerosol iodine in GEOS-Chem. Primary emissions of aerosol are described in Table A1, secondary inorganic aerosol formation is in Table A2, secondary organic aerosol formation is in Table A3, and interconversion between aerosol iodine species is in Table A4.
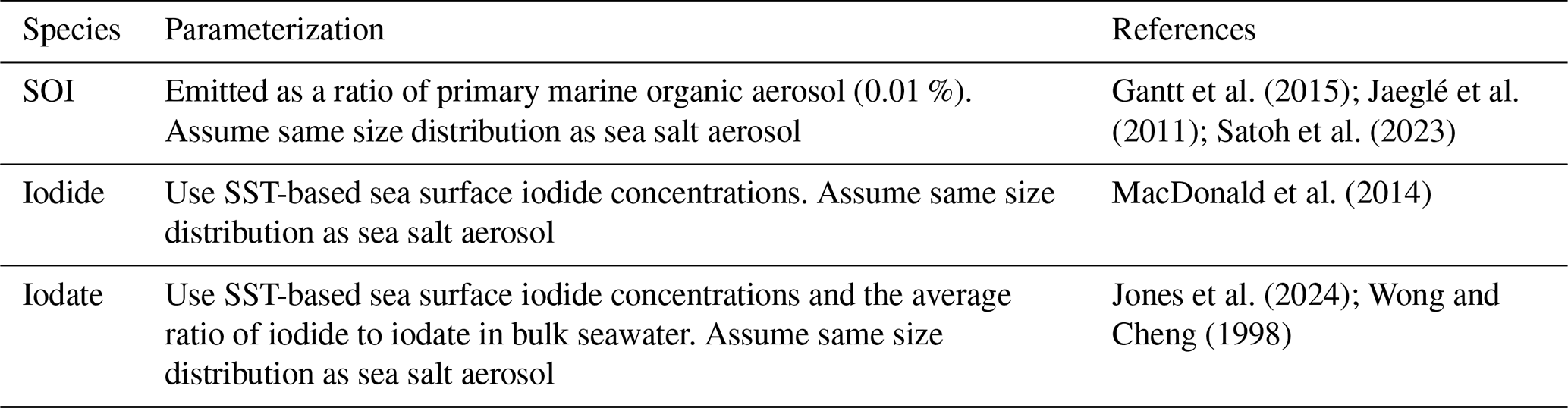
Table A2Secondary formation of inorganic iodine aerosol in GEOS-Chem.
a Reactive uptake only occurs on alkaline sea salt aerosol. b We increase the reactive uptake coefficients of the I2Ox species compared to Sherwen et al. (2016), who estimated a γ of 0.02 for all I2Ox species. The reactive uptake coefficients for I2Ox species have not been measured; however, they are suspected to be significant contributors to new particle formation (Gómez Martín et al., 2022a). Thus, we increase the reactive uptake of I2Ox to be consistent with HI, INOx, and coarse HIO3.
Table A3Secondary formation of organic iodine aerosol in GEOS-Chem.

a Mass accommodation coefficient (unitless) based on HOBr reaction with chloride and bromide from Sherwen et al. (2016). b Gas-phase molecular diffusion coefficient (cm2 s−1) based on HOBr reaction with chloride and bromide from Ammann et al. (2013). c Henry's Law inputs used to calculate the gas phase diffusion of HOI as a function of temperature. kchem is in M s−1 and CR is in K.
Table A4Interconversion between aerosol iodine species in GEOS-Chem.
A1 Secondary inorganic aerosol iodine formation
The conversion rate of iodate to iodide has not been directly measured. We used global iodate observations from Gómez Martín et al. (2022b) to tune the modeled HIO3 uptake and iodate reduction rates. It is possible that the reactive uptake coefficient for HIO3 approaches unity; however, selecting a reactive uptake coefficient less than unity provides a conservative estimate for reactive iodine chemistry. This is because reactive uptake coefficient for HIO3 requires rapid iodate reduction in order to reproduce surface aerosol observations. The rapid conversion from iodate to iodide accelerates aerosol iodide dehalogenation and iodine-induced oxidant loss. We show the sensitivity of modeled Iy and oxidant abundances to the reactive uptake coefficient of HIO3 in a follow up paper.
A2 Secondary organic aerosol iodine formation
Several mechanisms for SOI formation via HOI chemistry in aerosols have been proposed. Yu et al. (2019) investigated SOI composition at coastal sites along the eastern coast of China, finding that SOI accounted for 46 % of total PM2.5 iodine. The study identified iodide-organic adducts, iodoacetic acid, and iodopropenoic acid (or 2-iodomalondialdehyde, as proposed by Spólnik et al., 2020) as significant contributors, making up 31 %, 7 %, and 5 % of total aerosol iodine, respectively (Spólnik et al., 2020; Yu et al., 2019). Iodide-organic adducts are formed from dissolved iodine (likely free iodide or HOI) in aerosols, which can bind with hydroxyl, acid, or keto groups (Lee et al., 2014; Yu et al., 2019). The C-I bond in these compounds is likely relatively weak, leaving the possibility of re-dissociation into free iodide in the atmosphere or during sample extraction.
Iodoacetic acid (C2H3O2I) was identified in 9 out of 10 study samples in Yu et al., including both coastal and inland sites (Yu et al., 2019). Iodoacetic acid had a bimodal size distribution with peaks in both the fine (between 0.1–1 µm) and coarse modes (between 5–10 µm), which the authors attributed to sea salt emission (or chemistry) as its major source (Yu et al., 2019). A possible reaction for the formation of iodoacetic acid in the atmosphere via either HOI is
Iodopropenoic acid (C3H3O2I), another prominent SOI species identified by Yu et al. (2019), exhibited a bimodal size distribution with modes at 0.5 and around 1 µm diameters. 2-Iodomalondialdehyde (C3H3IO2), an isomer to iodopropenoic acid, has also been reported to be an abundant SOI species in precipitation (Spólnik et al., 2020). Spólnik et al. (2020) proposed a mechanism involving the nucleophilic activation of acrolein (C3H4O) by water to form an intermediate diol (C3H6O2). This intermediate reacts with HOI to produce hydroxy-2-iodopropanal (C3H5IO2) and water. Hydroxy-2-iodopropanal can either form iodomalondialdehyde (C3H3IO2, IMDA) if it is oxidized or 2-iodoacrolein (C2H3IO) if it is dehydrated. The latter reaction is reversible so it could still be converted to IMDA later. Spólnik et al. (2020) suggest that HOI is a likely oxidant for IMDA formation (Spólnik et al., 2020).
We incorporate HOI reaction with primary marine organic aerosol to form SOI into GEOS-Chem. Given the uncertain composition of SOI in atmospheric aerosols, the SOI formed from the reaction between HOI and dissolved organic matter (DOM) is not speciated. We hypothesize that these reactions are limited by the gas-phase diffusion of soluble reactive iodine (likely HOI) and competition with other heterogeneous reactions involving HOI, due to the abundance and ubiquity of organics in the atmosphere. The gas-phase diffusion of HOI for the HOI + DOM reaction is calculated the same way the HOI + halide reactions are calculated. We discuss this in detail in Appendix B. Inputs into the reactive probability (γ), which is used for all of the heterogeneous HOI reactions, are included in the table below.
Following Pechtl et al. (2007), the rate constant kchem for HOI + DOM is estimated to compete with the dehalogenation reactions. We use a larger rate constant than Pechtl, which estimated a range between 105 to 107 M s−1. Additionally, Pechtl assumed a fixed ratio of DOM compared to surface aerosol in their box model, while we use an online calculation of primary marine organic aerosol based on satellite-derived chl a in a global chemical transport model. The rate constant for HOI + DOM may be substantially smaller or larger than the constant used in Table A3. This is not a large source of uncertainty since the overall reaction rate (k) is limited by the gas-phase diffusion of HOI onto aerosol. Once HOI enters the aerosol phase, it reacts readily with any of the aerosol reagents (i.e. DOM, halides, and S(IV)). Thus, the overall formation rate of secondary SOI is more sensitive to parameters like aerosol surface area and composition, which is elaborated upon further in Appendix B.
A3 Interconversion between aerosol iodine species
The first-order reaction rates for the interconversion between iodide and SOI are tuned to observations from Gómez Martín et al. (2022b). To better estimate rate constants for these reactions, we may need to know more about the composition and stability of SOI in various aerosol conditions. Iodate reduction in aerosol to form iodide is likely facilitated by aerosol acidity and composition (i.e. the presence of organics), so a more detailed mechanism in GEOS-Chem that incorporates these parameters is warranted once they become available (Baker and Yodle, 2021; Pechtl et al., 2007; Reza et al., 2024; Saunders et al., 2012).
B1 Reaction rates and assumptions for calculating gas-phase diffusion onto aerosol
The rate for heterogeneous loss of the hypohalous acids (HOCl, HOBr, HOI) and halogen nitrates (ClNO3, BrNO3, INO3) onto aerosol, khet, (s−1), is calculated with Eq. (B1), following Jacob (2000).
where a is the particle radius (cm), Dg is the molecular diffusion coefficient (cm2 s−1), v is the molecular speed (cm s−1), A is aerosol surface area per unit volume (cm−1) and γ is the reaction probability calculated with Eq. (B2) (unitless) (Jacob, 2000). The reaction probability, γ, is calculated with Eq. (B2)
where α is a mass accommodation coefficient (unitless), v is the molecular speed (cm s−1), K∗ is the calculated Henry's Law solubility as a function of temperature (M atm−1), R is the universal gas constant (), T is temperature (K), Dl is the molecular diffusion coefficient (cm2 s−1), and kchem is the rate constant for first-order chemical loss of the gas-phase species (e.g., HOX and XNO3 in Tables B1 and B2, respectively) in the aqueous phase (s−1). kchem for the reactions involving the hypohalous acids and halogen nitrates are calculated with Eqs. (B3) and (B4), respectively, where kIII and kII are third-order and second-order reactions, in and , respectively, and X− corresponds to chloride, bromide, and iodide.
Tables B1 and B2 detail the rates used for dehalogenation reactions via the hypohalous acids (HOCl, HOBr, HOI) and halogen nitrates (ClNO3, BrNO3, INO3). Reactions with ∗ indicate new reactions added in this study.
Table B1Heterogeneous reactions with hypohalous acids in GEOS-Chem.
∗ New reaction added to GEOS-Chem in this work. a Occurs in liquid clouds only. b Occurs in liquid clouds and aerosol. c Occurs in ice clouds, liquid clouds, and aerosol. d Used same rate constant as HOBr + bromide. e Used same rate constant as HOCl + bromide. f Reactive uptake of HOI is calculated using Eq. (B2), with the same inputs from Table A3 (α=0.6 and in cm2 s−1).
Table B2Heterogeneous reactions with halogen nitrates in GEOS-Chem.
∗ New reaction added to GEOS-Chem. a Occurs in liquid clouds and aerosol. b Occurs in ice clouds, liquid clouds, and aerosol. c Following Eastham (2014) with mass accommodation coefficient from Deiber et al. (2004). The following parameters were used for calculating. . α=0.063 (unitless), (cm2 s−1). Henry's , Henry's CR=0.0. d BrNO3 + bromide and BrNO3 + chloride were added for liquid clouds and aerosol following the ClNO3 + bromide reaction from Sherwen et al. (2016). e Used same rate constant as BrNO3 + bromide and followed the other BrNO3 + halide reactions. f Based on Eastham (2014) with the same mass accommodation coefficient as bromine from Deiber et al. (2004). The following parameters were used for calculating . α=0.063 and (cm2 s−1).
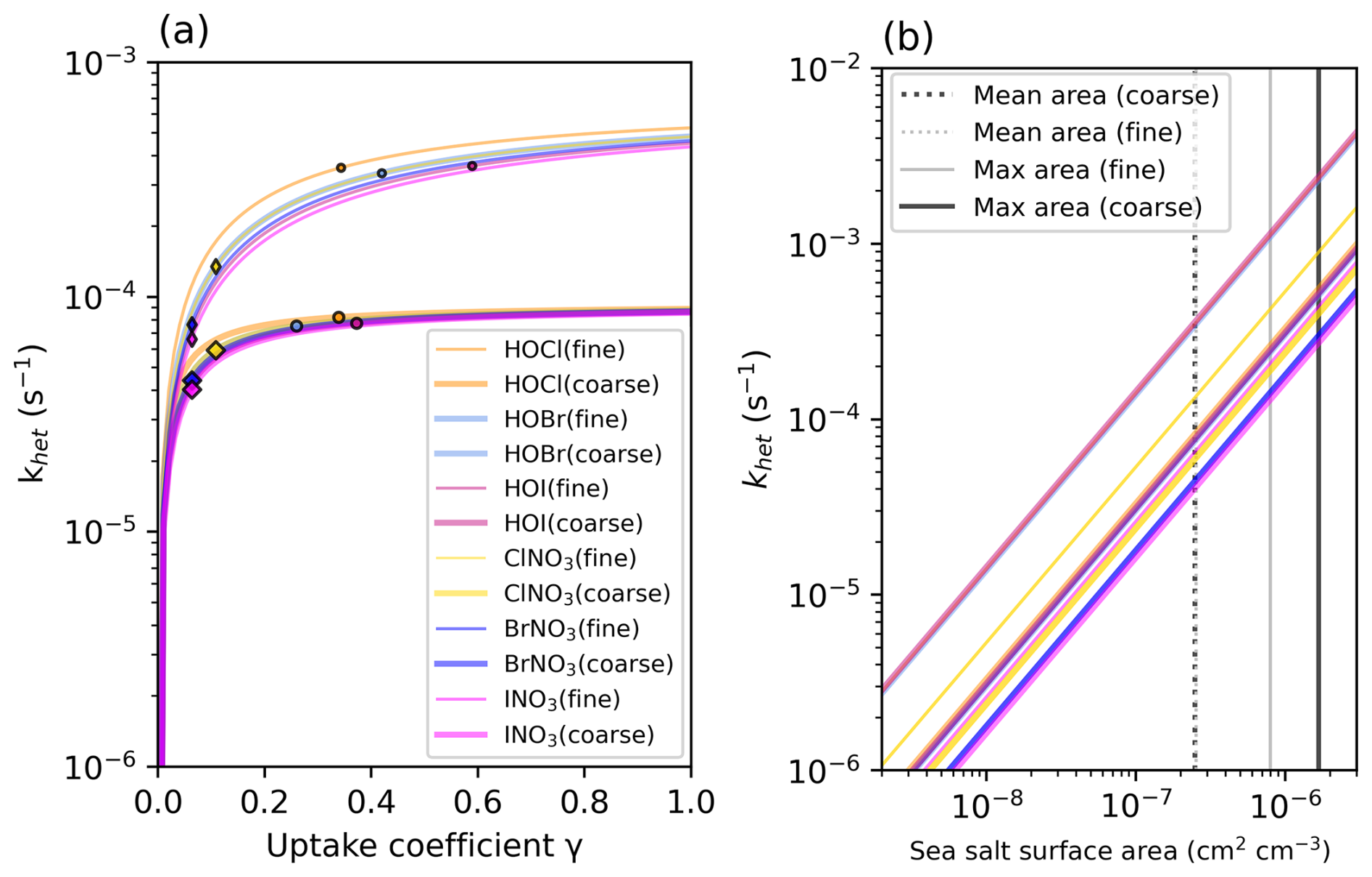
Figure B1Offline calculations showing the relationship between the heterogeneous loss rate constant of the hypohalous acids (HOCl, HOBr, HOI) and halogen nitrates (ClNO3, BrNO3, INO3) onto aerosol (khet) and reaction probability (a) and sea salt aerosol surface area density (b). The points on (a) show mean reaction probability (γ) values with the hypohalous acids indicated by circles and halogen nitrates by diamonds.
Table B3The difference in khet as a function of γ.

a Assume pH=2, temperature=278 K, pressure=101 325 Pa. Air aqueous volume, surface area, and radius are mean surface values from the aerosol diagnostic in GEOS-Chem. b Assume pH=5, temperature=278 K, pressure=101 325 Pa. Air aqueous volume, surface area, and radius are mean surface values from the aerosol diagnostic in GEOS-Chem.
B2 Rate-determining steps in heterogeneous reactions involving hypohalous acids and halogen nitrates
Reactions between HOX and XNO3 with bromide and iodide use the same kchem rate constants. In this section, we show that the overall rate of dehalogenation is limited by the gas-phase diffusion of reactive halogen species onto aerosol. Figure B1a shows the full range in khet for each gas phase species as a function of reaction probability (γ) (Eqs. B1 and B2). For this calculation, we use the mean temperature, particle radius, and particle surface areas in the model, though these parameters change for every gridbox and timestep. By plotting the full range of reaction probabilities, we show the model sensitivity of khet to a full range of kchem rate constants, which have not been measured for many of the reactions in Tables B1 and B2.
In Fig. B1a, there is a threshold for each reaction where the heterogeneous loss rate constant of the heterogeneous reaction (khet) increases as the reaction probability (γ) increases. We show this with an offline calculation, however, these parameters are calculated online in the model in every grid box and timestep. After a reaction probability of 0.2, the overall rate of khet gas loss is insensitive to increases in kchem and γ (Fig. B1a). Table B3 calculates the increase in khet for fine- and coarse-mode dehalogenation using mean offline reaction probability estimates (γ) and the rate for a reaction probability (γ) of 1. We use this calculation to understand the uncertainties in the rates of iodide dehalogenation reactions. The percent increase for the hypohalous acids is relatively minor, ranging between 8 %–48 %. The halogen nitrates have smaller reaction probability (γ) values in the model, thus their rates increase by 47 %–560 % as reaction probabilities increase to unity, with larger increases in khet in the fine mode compared to the coarse mode. The reason why dehalogenation rates become relatively insensitive to the reaction probability is related to the two terms in Eq. (B1). The left term dictates the gas-phase diffusion of the reactant (as a function of particle radius and the gas-phase diffusion coefficient) while the right term incorporates the molecular speed and the reaction probability (γ) calculated in Eq. (B2). As the reaction probability increases, the left term in the reaction and the aerosol surface area become increasingly important, driving the reaction rate to be limited by the gas-phase diffusion onto aerosol.
This suggests that the overall rates of the HOX-induced dehalogenation reactions are limited by gas-phase diffusion instead of the rate constant (kchem) or reaction probability (γ), while fine mode XNO3-induced dehalogenation rates are still sensitive to γ. Aerosol radius and surface area are important terms in Eq. (B1). Figure B1b shows a strong relationship between aerosol surface area density and khet. The mean surface area density values used in Fig. 1a are indicated by the dotted grey and black vertical lines and the maximum surface area density for each bin in the model are indicated by the solid grey and black vertical lines. The difference in the range of khet for (a) and (b) is noteworthy, since it shows how the rate khet becomes faster by orders of magnitude as surface area density increases. In short, increasing kchem for HOX and XNO3 reactions with iodide, as expected since iodide is more reactive than bromide, would not lead to substantial increases in dehalogenation rates, since an increase in reaction probability does not have a large impact of khet. This means that the missing rate constants are a relatively smaller source of uncertainty in this study compared to accurately modeling aerosol surface area, mass, and composition.

Figure C1(a–d) Zonal distribution of size-resolved, speciated aerosol iodine concentrations (a, b) and fractional composition (c, d). Observations are shown in (a) and (c) and are from Gómez Martín et al. (2022b). GEOS-Chem mean model output from the new iodine chemistry simulation is shown in (b) and (d). Fine SOI is in navy, coarse SOI is in purple, fine iodide is in pink, coarse iodide is in salmon, fine iodate is in orange, and coarse iodate is in gold.
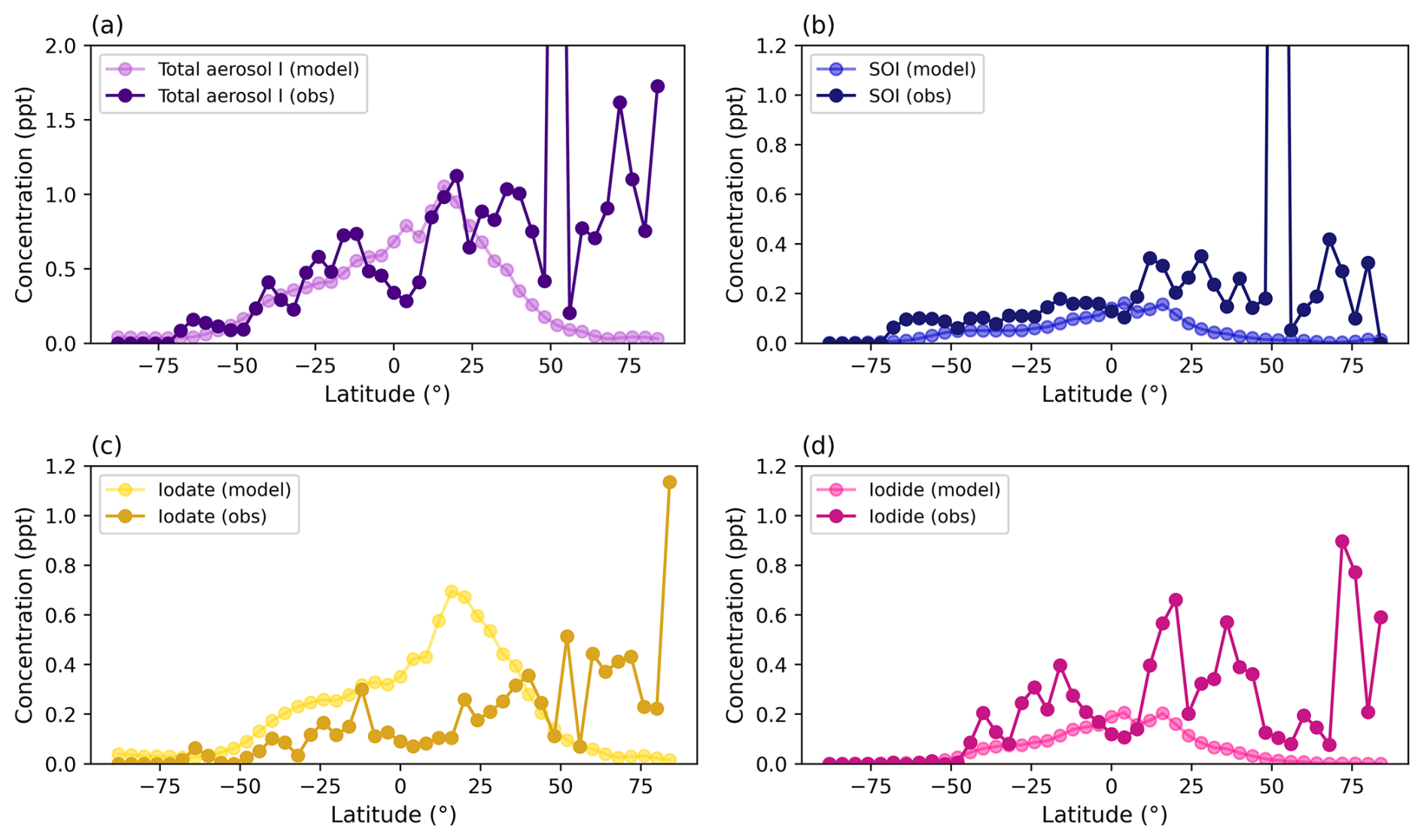
Figure C2Zonal distribution of bulk speciated aerosol iodine concentrations for total soluble iodine (a), SOI (b), iodate (c), and iodide (d). GEOS-Chem model output is plotted in the lighter colors while the darker colors represent mean bulk speciated iodine observations from Gómez Martín et al. (2022b).

Figure C3(a–c) The relationship between bulk total soluble iodine (TSI) and bulk SOI (a), bulk iodide (b), and bulk iodate (c). The colors correspond to latitude. Observations are from Gómez Martín et al. (2022b).
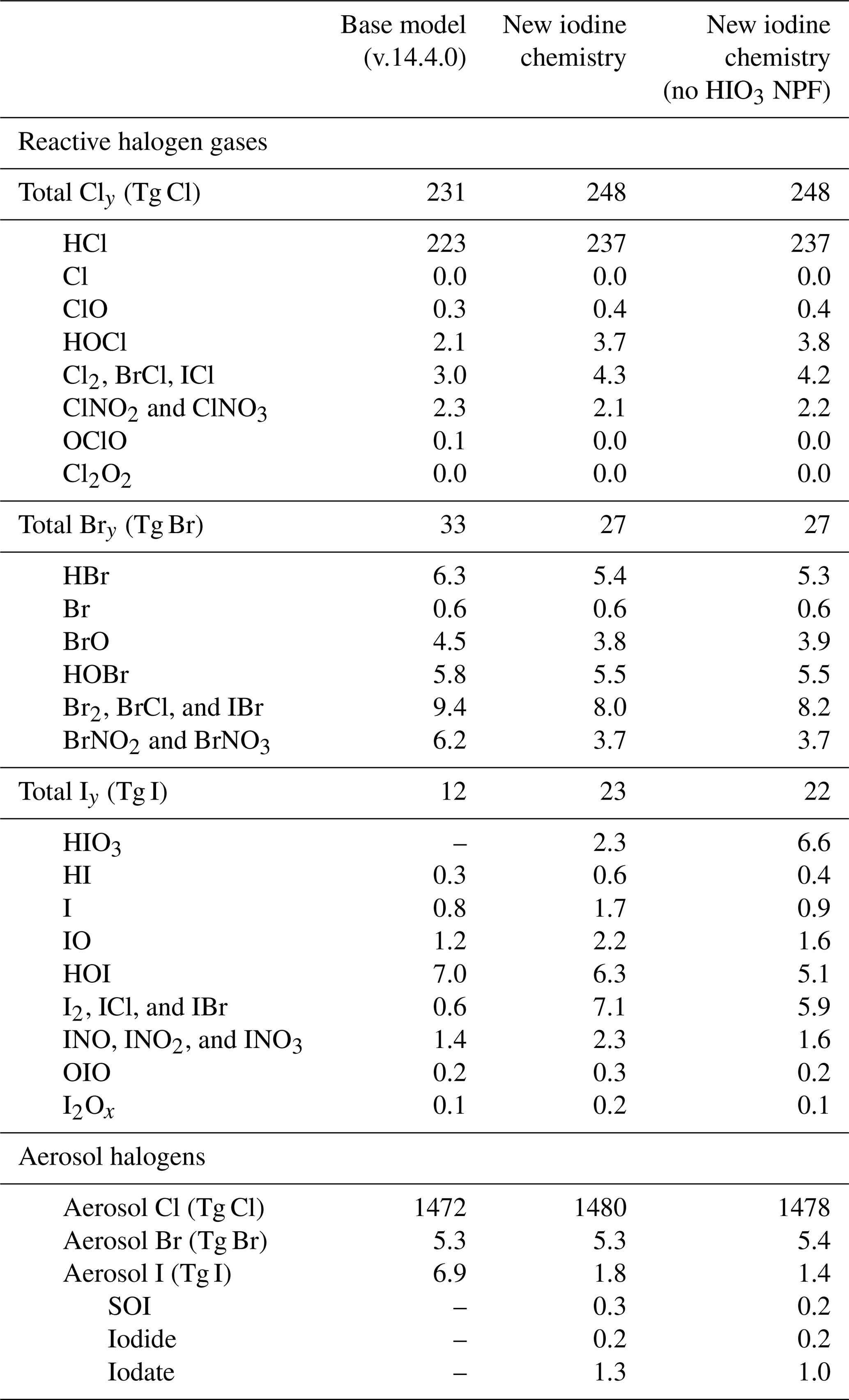
Table E1 shows the tropospheric O3 burden in Tg in the base model and new iodine chemistry simulations. Previous literature values include the mean and interquartile range in tropospheric O3 burden from 49 model studies (Young et al., 2018).
Figure E1 shows the change in surface O3 in the base model and new iodine chemistry simulations. Figure E1a and b shows surface concentrations in the base model (a) and new iodine chemistry simulation (b). Figure E1c and d shows the ppb difference in surface ozone (c) and percent change (d) in surface ozone between the new iodine chemistry simulation and the base model.
Figure E2 shows surface ozone concentrations from the Tropospheric Ozone Assessment Report II (TOAR2) ship and buoy dataset (Kanaya et al., 2025b, a), Annual-mean surface ozone observations are calculated for the years 2012 to 2022, with observations used shown in Fig. E2a along with 2022-mean model output from the new iodine chemistry simulation. Figure E2b and c compares surface ozone between the base model (b) and the new iodine chemistry simulation (c). Overall, there is good agreement between both the base model and the new iodine chemistry simulation with TOAR2 ship and buoy data. The correlation coefficient in the new iodine chemistry simulation is slightly higher (r2=0.90) compared to the base model (r2=0.84). The slope and normalized mean bias in the new iodine chemistry simulation (slope=0.93 and %) are slightly worse than the base model (slope=1.01 and ).
Table E1Tropospheric O3 burden in the new iodine chemistry simulation and base model.

∗ Previous literature values for O3 burden in Tg from 49 model studies, presented as the mean value with the interquartile range in parenthesis (Young et al., 2018).

Figure E1(a, b) show surface O3 concentrations for the base model (a) and new iodine chemistry (b) simulations. (c, d) show changes in ozone in pbb (c) and percent (d).

Figure E2(a–c) Surface ozone concentrations from the second Tropospheric Ozone Assessment Report (TOAR2) ship and buoy dataset. (a) shows surface ozone concentrations from the new iodine chemistry simulation, where the circles on the map represent annual-mean TOAR2 observations. (b) and (c) show the relationship between measured and modeled surface ozone concentrations for the base model and new iodine chemistry simulations, respectively. The points in (b) and (c) represent annual-mean mixing ratio grouped into 4° latitude bins to correspond with the model resolution.
The model code used here will be made available to the community through the GEOS-Chem repository once it has been merged with the most-recent model version. GEOS-Chem simulations from this work are available upon request. Requests for modeling materials should be addressed to Becky Alexander (beckya@uw.edu). Global speciated iodine aerosol observations are available in Gómez Martín (2022b). Surface HIO3 observations from He et al. (2021b) are available at https://doi.org/10.5281/zenodo.4299441. TORERO and CONTRAST IO data are available at https://doi.org/10.26023/DJX0-85VQ-Q80X (Volkamer and Dix, 2014) and https://doi.org/10.5065/D6F769MF (Volkamer et al., 2020), respectively. AToM aerosol data can be found at https://doi.org/10.3334/ORNLDAAC/1925 (Wofsy et al., 2021). Surface IO observations were compiled from the following references: Allan et al. (2000); Butz et al. (2009); Carpenter et al. (2001); Gómez Martín et al. (2013); Grilli et al. (2012, 2013); Großmann et al. (2013); Huang et al. (2010); Inamdar et al. (2020); Mahajan et al. (2010b, a, 2012, 2021); Oetjen (2009); Peters et al. (2005); Prados-Roman et al. (2015a); Read et al. (2008); Saiz-Lopez et al. (2008); Saiz-Lopez and Plane (2004); Stutz et al. (2007). Surface ozone observations are from Tropospheric Ozone Assessment Report II, described in Kanaya et al. (2025b) and publicly available at https://doi.org/10.17596/0004044 (Kanaya et al., 2025a).
ARM and BA implemented the updated iodine chemistry and ran the simulations in GEOS-Chem, with input guiding model development from co-authors (LL, XW, YCC, AF, RP, AL, LM, ME, LJC, JS, JAT, GN, AR, GPS, XCH, HF, MR, RV, KHB). ARM and BA prepared the manuscript with input from all co-authors. LL and XW implemented gas-phase HIO3 chemistry and the new particle formation mechanism for HIO3. ASL and ASM contributed surface IO observations from cruises. X-CH contributed compiled surface HIO3 observations. RV contributed aircraft-based IO observations. GPS contributed aircraft-based non-sea-salt iodine observations.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
A.R.M., B.A., and J.A.T. were supported by NSF AGS 2109323. L.L. and X.W. are supported by the National Natural Science Foundation of China (42375087) and Research Grants Council of the Hong Kong SAR, China (C6001-24Y). J.S. and A.F. were supported by NSF Award #2109331. A.S.L. was supported by the Panorama Natural Environment Research Council (NERC) Doctoral Training Partnership (DTP), under grant NE/S007458/1 and Natural Environment Research Council (NERC) BLEACH grant: NSFGEO-NERC:BLEACH (NE/W00724X/1). L.J.C. acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 program (Grant agreement no. 833290). X.-C.H. acknowledges support from Research Council of Finland grant nos. 359331, 349659, 371185. R.V. acknowledges support from National Science Foundation award AGS-2027252.
This research has been supported by the National Science Foundation (grant nos. AGS 2109323, AGS 2109331, and AGS 2027252), the National Natural Science Foundation of China (grant no. 42375087), the Natural Environment Research Council (grant nos. NE/S007458/1 and NE/W00724X/1), the European Research Council, HORIZON EUROPE European Research Council (grant no. 833290), and the Research Council of Finland (grant nos. 359331, 349659, and 371185). R.V. acknowledges support from National Science Foundation award AGS-2027252.
This paper was edited by Qi Chen and reviewed by Men Xia and one anonymous referee.
Alicke, B., Hebestreit, K., Stutz, J., and Platt, U.: Iodine oxide in the marine boundary layer, Nature, 397, 572–573, https://doi.org/10.1038/17508, 1999.
Allan, B. J., McFiggans, G., Plane, J. M. C., and Coe, H.: Observations of iodine monoxide in the remote marine boundary layer, J. Geophys. Res., 105, 14363–14369, https://doi.org/10.1029/1999JD901188, 2000.
Ammann, M., Cox, R. A., Crowley, J. N., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates, Atmos. Chem. Phys., 13, 8045–8228, https://doi.org/10.5194/acp-13-8045-2013, 2013.
Andreae, M. O.: Emission of trace gases and aerosols from biomass burning – an updated assessment, Atmos. Chem. Phys., 19, 8523–8546, https://doi.org/10.5194/acp-19-8523-2019, 2019.
Angle, K. J., Crocker, D. R., Simpson, R. M. C., Mayer, K. J., Garofalo, L. A., Moore, A. N., Mora Garcia, S. L., Or, V. W., Srinivasan, S., Farhan, M., Sauer, J. S., Lee, C., Pothier, M. A., Farmer, D. K., Martz, T. R., Bertram, T. H., Cappa, C. D., Prather, K. A., and Grassian, V. H.: Acidity across the interface from the ocean surface to sea spray aerosol, Proc. Natl. Acad. Sci. USA, 118, e2018397118, https://doi.org/10.1073/pnas.2018397118, 2021.
Baccarini, A., Karlsson, L., Dommen, J., Duplessis, P., Vüllers, J., Brooks, I. M., Saiz-Lopez, A., Salter, M., Tjernström, M., Baltensperger, U., Zieger, P., and Schmale, J.: Frequent new particle formation over the high Arctic pack ice by enhanced iodine emissions, Nat. Commun., 11, 4924, https://doi.org/10.1038/s41467-020-18551-0, 2020.
Badia, A., Reeves, C. E., Baker, A. R., Saiz-Lopez, A., Volkamer, R., Koenig, T. K., Apel, E. C., Hornbrook, R. S., Carpenter, L. J., Andrews, S. J., Sherwen, T., and von Glasow, R.: Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem, Atmos. Chem. Phys., 19, 3161–3189, https://doi.org/10.5194/acp-19-3161-2019, 2019.
Badia, A., Iglesias-Suarez, F., Fernandez, R. P., Cuevas, C. A., Kinnison, D. E., Lamarque, J., Griffiths, P. T., Tarasick, D. W., Liu, J., and Saiz-Lopez, A.: The Role of Natural Halogens in Global Tropospheric Ozone Chemistry and Budget Under Different 21st Century Climate Scenarios, J. GEOPHYS. RES. Atmospheres, 126, e2021JD034859, https://doi.org/10.1029/2021JD034859, 2021.
Baker, A. R.: Marine Aerosol Iodine Chemistry: The Importance of Soluble Organic Iodine, Environ. Chem., 2, 295, https://doi.org/10.1071/EN05070, 2005.
Baker, A. R. and Yodle, C.: Measurement report: Indirect evidence for the controlling influence of acidity on the speciation of iodine in Atlantic aerosols, Atmos. Chem. Phys., 21, 13067–13076, https://doi.org/10.5194/acp-21-13067-2021, 2021.
Baker, A. R., Thompson, D., Campos, M. L. A. M., Parry, S. J., and Jickells, T. D.: Iodine concentration and availability in atmospheric aerosol, Atmospheric Environment, 34, 4331–4336, https://doi.org/10.1016/S1352-2310(00)00208-9, 2000.
Baker, A. R., Tunnicliffe, C., and Jickells, T. D.: Iodine speciation and deposition fluxes from the marine atmosphere, J. Geophys. Res., 106, 28743–28749, https://doi.org/10.1029/2000JD000004, 2001.
Bates, K. H. and Jacob, D. J.: An Expanded Definition of the Odd Oxygen Family for Tropospheric Ozone Budgets: Implications for Ozone Lifetime and Stratospheric Influence, Geophysical Research Letters, 47, e2019GL084486, https://doi.org/10.1029/2019GL084486, 2020.
Beck, L. J., Sarnela, N., Junninen, H., Hoppe, C. J. M., Garmash, O., Bianchi, F., Riva, M., Rose, C., Peräkylä, O., Wimmer, D., Kausiala, O., Jokinen, T., Ahonen, L., Mikkilä, J., Hakala, J., He, X., Kontkanen, J., Wolf, K. K. E., Cappelletti, D., Mazzola, M., Traversi, R., Petroselli, C., Viola, A. P., Vitale, V., Lange, R., Massling, A., Nøjgaard, J. K., Krejci, R., Karlsson, L., Zieger, P., Jang, S., Lee, K., Vakkari, V., Lampilahti, J., Thakur, R. C., Leino, K., Kangasluoma, J., Duplissy, E., Siivola, E., Marbouti, M., Tham, Y. J., Saiz-Lopez, A., Petäjä, T., Ehn, M., Worsnop, D. R., Skov, H., Kulmala, M., Kerminen, V., and Sipilä, M.: Differing Mechanisms of New Particle Formation at Two Arctic Sites, Geophysical Research Letters, 48, e2020GL091334, https://doi.org/10.1029/2020GL091334, 2021.
Bell, N., Hsu, L., Jacob, D. J., Schultz, M. G., Blake, D. R., Butler, J. H., King, D. B., Lobert, J. M., and Maier-Reimer, E.: Methyl iodide: Atmospheric budget and use as a tracer of marine convection in global models, J. Geophys. Res., 107, https://doi.org/10.1029/2001JD001151, 2002.
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q., Liu, H. Y., Mickley, L. J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res., 106, 23073–23095, https://doi.org/10.1029/2001JD000807, 2001.
Breider, T. J., Mickley, L. J., Jacob, D. J., Ge, C., Wang, J., Payer Sulprizio, M., Croft, B., Ridley, D. A., McConnell, J. R., Sharma, S., Husain, L., Dutkiewicz, V. A., Eleftheriadis, K., Skov, H., and Hopke, P. K.: Multidecadal trends in aerosol radiative forcing over the Arctic: Contribution of changes in anthropogenic aerosol to Arctic warming since 1980, J. GEOPHYS. RES. Atmospheres, 122, 3573–3594, https://doi.org/10.1002/2016JD025321, 2017.
Brown, L. V., Pound, R. J., Jones, M. R., Rowlinson, M. J., Chance, R., Jacobi, H.-W., Frey, M. M., Archer, S. D., Arndt, S., Barten, J. G. M., Blomquist, B. W., Dadic, R., Ganzeveld, L. N., Hannula, H.-R., Helmig, D., Jaggi, M., Krampe, D., Macfarlane, A. R., Miller, S., Schneebeli, M., and Carpenter, L. J.: Iodine speciation in snow during the MOSAiC expedition and its implications for Arctic iodine emissions, Faraday Discuss., 258, 441–472, https://doi.org/10.1039/d4fd00178h, 2025.
Butz, A., Bösch, H., Camy-Peyret, C., Chipperfield, M. P., Dorf, M., Kreycy, S., Kritten, L., Prados-Román, C., Schwärzle, J., and Pfeilsticker, K.: Constraints on inorganic gaseous iodine in the tropical upper troposphere and stratosphere inferred from balloon-borne solar occultation observations, Atmos. Chem. Phys., 9, 7229–7242, https://doi.org/10.5194/acp-9-7229-2009, 2009.
Carpenter, L. J., Hebestreit, K., Platt, U., and Liss, P. S.: Coastal zone production of IO precursors: a 2-dimensional study, Atmos. Chem. Phys., 1, 9–18, https://doi.org/10.5194/acp-1-9-2001, 2001.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nature Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Carpenter, L. J., Chance, R. J., Sherwen, T., Adams, T. J., Ball, S. M., Evans, M. J., Hepach, H., Hollis, L. D. J., Hughes, C., Jickells, T. D., Mahajan, A., Stevens, D. P., Tinel, L., and Wadley, M. R.: Marine iodine emissions in a changing world, Proc. R. Soc. A, 477, 20200824, https://doi.org/10.1098/rspa.2020.0824, 2021.
Celli, G., Cairns, W. R. L., Scarchilli, C., Cuevas, C. A., Saiz-Lopez, A., Savarino, J., Stenni, B., Frezzotti, M., Becagli, S., Delmonte, B., Angot, H., Fernandez, R. P., and Spolaor, A.: Bromine, iodine and sodium along the EAIIST traverse: Bulk and surface snow latitudinal variability, Environmental Research, 239, 117344, https://doi.org/10.1016/j.envres.2023.117344, 2023.
Chance, R., Baker, A. R., Carpenter, L., and Jickells, T. D.: The distribution of iodide at the sea surface, Environ. Sci.: Processes Impacts, 16, 1841–1859, https://doi.org/10.1039/c4em00139g, 2014.
Corella, J. P., Maffezzoli, N., Spolaor, A., Vallelonga, P., Cuevas, C. A., Scoto, F., Müller, J., Vinther, B., Kjær, H. A., Cozzi, G., Edwards, R., Barbante, C., and Saiz-Lopez, A.: Climate changes modulated the history of Arctic iodine during the Last Glacial Cycle, Nat. Commun., 13, 88, https://doi.org/10.1038/s41467-021-27642-5, 2022.
Cuevas, C. A., Maffezzoli, N., Corella, J. P., Spolaor, A., Vallelonga, P., Kjær, H. A., Simonsen, M., Winstrup, M., Vinther, B., Horvat, C., Fernandez, R. P., Kinnison, D., Lamarque, J.-F., Barbante, C., and Saiz-Lopez, A.: Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century, Nat. Commun., 9, 1452, https://doi.org/10.1038/s41467-018-03756-1, 2018.
Deiber, G., George, Ch., Le Calvé, S., Schweitzer, F., and Mirabel, Ph.: Uptake study of ClONO2 and BrONO2 by Halide containing droplets, Atmos. Chem. Phys., 4, 1291–1299, https://doi.org/10.5194/acp-4-1291-2004, 2004.
Dix, B., Baidar, S., Bresch, J. F., Hall, S. R., Schmidt, K. S., Wang, S., and Volkamer, R.: Detection of iodine monoxide in the tropical free troposphere, Proc. Natl. Acad. Sci. USA, 110, 2035–2040, https://doi.org/10.1073/pnas.1212386110, 2013.
Droste, E. S., Baker, A. R., Yodle, C., Smith, A., and Ganzeveld, L.: Soluble Iodine Speciation in Marine Aerosols Across the Indian and Pacific Ocean Basins, Front. Mar. Sci., 8, 788105, https://doi.org/10.3389/fmars.2021.788105, 2021.
Eastham, S. D., Weisenstein, D. K., and Barrett, S. R. H.: Development and evaluation of the unified tropospheric–stratospheric chemistry extension (UCX) for the global chemistry-transport model GEOS-Chem, Atmospheric Environment, 89, 52–63, https://doi.org/10.1016/j.atmosenv.2014.02.001, 2014.
Eger, A. M., Marzinelli, E. M., Beas-Luna, R., Blain, C. O., Blamey, L. K., Byrnes, J. E. K., Carnell, P. E., Choi, C. G., Hessing-Lewis, M., Kim, K. Y., Kumagai, N. H., Lorda, J., Moore, P., Nakamura, Y., Pérez-Matus, A., Pontier, O., Smale, D., Steinberg, P. D., and Vergés, A.: The value of ecosystem services in global marine kelp forests, Nat. Commun., 14, 1894, https://doi.org/10.1038/s41467-023-37385-0, 2023.
Emerson, E. W., Hodshire, A. L., DeBolt, H. M., Bilsback, K. R., Pierce, J. R., McMeeking, G. R., and Farmer, D. K.: Revisiting particle dry deposition and its role in radiative effect estimates, Proc. Natl. Acad. Sci. USA, 117, 26076–26082, https://doi.org/10.1073/pnas.2014761117, 2020.
Finkenzeller, H., Iyer, S., He, X.-C., Simon, M., Koenig, T. K., Lee, C. F., Valiev, R., Hofbauer, V., Amorim, A., Baalbaki, R., Baccarini, A., Beck, L., Bell, D. M., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dada, L., Duplissy, J., Heinritzi, M., Kemppainen, D., Kim, C., Krechmer, J., Kürten, A., Kvashnin, A., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mauldin, R. L., Mentler, B., Müller, T., Petäjä, T., Philippov, M., Ranjithkumar, A., Rörup, B., Shen, J., Stolzenburg, D., Tauber, C., Tham, Y. J., Tomé, A., Vazquez-Pufleau, M., Wagner, A. C., Wang, D. S., Wang, M., Wang, Y., Weber, S. K., Nie, W., Wu, Y., Xiao, M., Ye, Q., Zauner-Wieczorek, M., Hansel, A., Baltensperger, U., Brioude, J., Curtius, J., Donahue, N. M., Haddad, I. E., Flagan, R. C., Kulmala, M., Kirkby, J., Sipilä, M., Worsnop, D. R., Kurten, T., Rissanen, M., and Volkamer, R.: The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source, Nat. Chem., 15, 129–135, https://doi.org/10.1038/s41557-022-01067-z, 2023.
Finley, B. D. and Saltzman, E. S.: Observations of Cl2, Br2, and I2 in coastal marine air, J. Geophys. Res., 113, 2008JD010269, https://doi.org/10.1029/2008JD010269, 2008.
Furneaux, K. L., Whalley, L. K., Heard, D. E., Atkinson, H. M., Bloss, W. J., Flynn, M. J., Gallagher, M. W., Ingham, T., Kramer, L., Lee, J. D., Leigh, R., McFiggans, G. B., Mahajan, A. S., Monks, P. S., Oetjen, H., Plane, J. M. C., and Whitehead, J. D.: Measurements of iodine monoxide at a semi polluted coastal location, Atmos. Chem. Phys., 10, 3645–3663, https://doi.org/10.5194/acp-10-3645-2010, 2010.
Gantt, B., Johnson, M. S., Meskhidze, N., Sciare, J., Ovadnevaite, J., Ceburnis, D., and O'Dowd, C. D.: Model evaluation of marine primary organic aerosol emission schemes, Atmos. Chem. Phys., 12, 8553–8566, https://doi.org/10.5194/acp-12-8553-2012, 2012.
Gantt, B., Johnson, M. S., Crippa, M., Prévôt, A. S. H., and Meskhidze, N.: Implementing marine organic aerosols into the GEOS-Chem model, Geosci. Model Dev., 8, 619–629, https://doi.org/10.5194/gmd-8-619-2015, 2015.
García-Flor, N., Guitart, C., Ábalos, M., Dachs, J., Bayona, J. M., and Albaigés, J.: Enrichment of organochlorine contaminants in the sea surface microlayer: An organic carbon-driven process, Marine Chemistry, 96, 331–345, https://doi.org/10.1016/j.marchem.2005.01.005, 2005.
Gilfedder, B. S., Lai, S. C., Petri, M., Biester, H., and Hoffmann, T.: Iodine speciation in rain, snow and aerosols, Atmos. Chem. Phys., 8, 6069–6084, https://doi.org/10.5194/acp-8-6069-2008, 2008.
Gómez Martín, J. C., Mahajan, A. S., Hay, T. D., Prados-Román, C., Ordóñez, C., MacDonald, S. M., Plane, J. M. C., Sorribas, M., Gil, M., Paredes Mora, J. F., Agama Reyes, M. V., Oram, D. E., Leedham, E., and Saiz-Lopez, A.: Iodine chemistry in the eastern Pacific marine boundary layer, J. GEOPHYS. RES. Atmospheres, 118, 887–904, https://doi.org/10.1002/J. Geophys. Res.d.50132, 2013.
Gómez Martín, J. C., Lewis, T. R., James, A. D., Saiz-Lopez, A., and Plane, J. M. C.: Insights into the Chemistry of Iodine New Particle Formation: The Role of Iodine Oxides and the Source of Iodic Acid, J. Am. Chem. Soc., 144, 9240–9253, https://doi.org/10.1021/jacs.1c12957, 2022a.
Gómez Martín, J. C., Saiz-Lopez, A., Cuevas, C. A., Baker, A. R., and Fernández, R. P.: On the Speciation of Iodine in Marine Aerosol, J. Geophys. Res. Atmospheres, 127, https://doi.org/10.1029/2021JD036081, 2022b.
Gong, T. and Zhang, X.: Determination of iodide, iodate and organo-iodine in waters with a new total organic iodine measurement approach, Water Research, 47, 6660–6669, https://doi.org/10.1016/j.watres.2013.08.039, 2013.
Grilli, R., Méjean, G., Kassi, S., Ventrillard, I., Abd-Alrahman, C., and Romanini, D.: Frequency Comb Based Spectrometer for in Situ and Real Time Measurements of IO, BrO, NO2, and H2CO at pptv and ppqv Levels, Environ. Sci. Technol., 46, 10704–10710, https://doi.org/10.1021/es301785h, 2012.
Grilli, R., Legrand, M., Kukui, A., Méjean, G., Preunkert, S., and Romanini, D.: First investigations of IO, BrO, and NO2 summer atmospheric levels at a coastal East Antarctic site using mode-locked cavity enhanced absorption spectroscopy, Geophysical Research Letters, 40, 791–796, https://doi.org/10.1002/grl.50154, 2013.
Großmann, K., Frieß, U., Peters, E., Wittrock, F., Lampel, J., Yilmaz, S., Tschritter, J., Sommariva, R., von Glasow, R., Quack, B., üger, K., Pfeilsticker, K., and Platt, U.: Iodine monoxide in the Western Pacific marine boundary layer, Atmos. Chem. Phys., 13, 3363–3378, https://doi.org/10.5194/acp-13-3363-2013, 2013.
He, X.-C., Simon, M., Iyer, S., Xie, H.-B., Rörup, B., Shen, J., Finkenzeller, H., Stolzenburg, D., Zhang, R., Baccarini, A., Tham, Y. J., Wang, M., Amanatidis, S., Piedehierro, A. A., Amorim, A., Baalbaki, R., Brasseur, Z., Caudillo, L., Chu, B., Dada, L., Duplissy, J., El Haddad, I., Flagan, R. C., Granzin, M., Hansel, A., Heinritzi, M., Hofbauer, V., Jokinen, T., Kemppainen, D., Kong, W., Krechmer, J., Kürten, A., Lamkaddam, H., Lopez, B., Ma, F., Mahfouz, N. G. A., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Massabò, D., Mauldin, R. L., Mentler, B., Onnela, A., Petäjä, T., Pfeifer, J., Philippov, M., Ranjithkumar, A., Rissanen, M. P., Schobesberger, S., Scholz, W., Schulze, B., Surdu, M., Thakur, R. C., Tomé, A., Wagner, A. C., Wang, D., Wang, Y., Weber, S. K., Welti, A., Winkler, P. M., Zauner-Wieczorek, M., Baltensperger, U., Curtius, J., Kurtén, T., Worsnop, D. R., Volkamer, R., Lehtipalo, K., Kirkby, J., Donahue, N. M., Sipilä, M., and Kulmala, M.: Iodine oxoacids enhance nucleation of sulfuric acid particles in the atmosphere, Science, 382, 1308–1314, https://doi.org/10.1126/science.adh2526, 2023.
He, X.-C., Tham, Y. J., Dada, L., Wang, M., Finkenzeller, H., Stolzenburg, D., Iyer, S., Simon, M., Kürten, A., Shen, J., Rörup, B., Rissanen, M., Schobesberger, S., Baalbaki, R., Wang, D. S., Koenig, T. K., Jokinen, T., Sarnela, N., Beck, L. J., Almeida, J., Amanatidis, S., Amorim, A., Ataei, F., Baccarini, A., Bertozzi, B., Bianchi, F., Brilke, S., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dias, A., Ding, A., Dommen, J., Duplissy, J., El Haddad, I., Gonzalez Carracedo, L., Granzin, M., Hansel, A., Heinritzi, M., Hofbauer, V., Junninen, H., Kangasluoma, J., Kemppainen, D., Kim, C., Kong, W., Krechmer, J. E., Kvashin, A., Laitinen, T., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Möhler, O., Müller, T., Nie, W., Onnela, A., Petäjä, T., Pfeifer, J., Philippov, M., Ranjithkumar, A., Saiz-Lopez, A., Salma, I., Scholz, W., Schuchmann, S., Schulze, B., Steiner, G., Stozhkov, Y., Tauber, C., Tomé, A., Thakur, R. C., Väisänen, O., Vazquez-Pufleau, M., Wagner, A. C., Wang, Y., Weber, S. K., Winkler, P. M., Wu, Y., Xiao, M., Yan, C., Ye, Q., Ylisirniö, A., Zauner-Wieczorek, M., Zha, Q., Zhou, P., Flagan, R. C., Curtius, J., Baltensperger, U., Kulmala, M., Kerminen, V.-M., Kurtén, T., et al.: Role of iodine oxoacids in atmospheric aerosol nucleation, Science, 371, 589–595, https://doi.org/10.1126/science.abe0298, 2021a.
He, X.-C., Tham, Y. J., Dada, L., Wang, M., Finkenzeller, H., Stolzenburg, D., Iyer, S., Simon, M., Kürten, A., Shen, J., Rörup, B., Rissanen, M., Schobesberger, S., Baalbaki, R., Wang, D. S., Koenig, T. K., Jokinen, T., Sarnela, N., Beck, L., Almeida, J., Amanatidis, S., Amorim, A., Ataei, F., Baccarini, A., Bertozzi, B., Bianchi, F., Brilke, S., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dias, A., Ding, A., Dommen, J., Duplissy, J., El Haddad, I., Gonzalez Carracedo, L., Granzin, M., Hansel, A., Heinritzi, M., Hofbauer, V., Junninen, H., Kangasluoma, J., Kemppainen, D., Kim, C., Kong, W., Krechmer, J. E., Kvashnin, A., Laitinen, T., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Möhler, O., Müller, T., Nie, W., Onnela, A., Petäjä, T., Pfeifer, J., Philippov, M., Ranjithkumar, A., Saiz-Lopez, A., Salma, I., Scholz, W., Schuchmann, S., Schulze, B., Steiner, G., Stozhkov, Y., Tauber, C., Tomé, A., Thakur, R. C., Väisänen, O., Vazquez-Pufleau, M., Wagner, A. C., Wang, Y., Weber, S. K., Winkler, P. M., Wu, Y., Xiao, M., Yan, C., Ye, Q., Ylisirniö, A., Zauner-Wieczorek, M., Zha, Q., Zhou, P., Flagan, R. C., Curtius, J., Baltensperger, U., Kulmala, M., Kerminen, V.-M., Kurtén, T., Donahue, N., Volkamer, R., Kirkby, J., Worsnop, D. R., and Sipilä, M.: Role of iodine oxoacids in atmospheric aerosol nucleation: data resources (v1), Zenodo [data set], https://doi.org/10.5281/zenodo.4299441, 2021b.
Hoffmann, T., O'Dowd, C. D., and Seinfeld, J. H.: Iodine oxide homogeneous nucleation: An explanation for coastal new particle production, Geophysical Research Letters, 28, 1949–1952, https://doi.org/10.1029/2000GL012399, 2001.
Holmes, C. D., Prather, M. J., and Vinken, G. C. M.: The climate impact of ship NOx emissions: an improved estimate accounting for plume chemistry, Atmos. Chem. Phys., 14, 6801–6812, https://doi.org/10.5194/acp-14-6801-2014, 2014.
Huang, J. and Jaeglé, L.: Wintertime enhancements of sea salt aerosol in polar regions consistent with a sea ice source from blowing snow, Atmos. Chem. Phys., 17, 3699–3712, https://doi.org/10.5194/acp-17-3699-2017, 2017.
Huang, R.-J., Seitz, K., Buxmann, J., Pöhler, D., Hornsby, K. E., Carpenter, L. J., Platt, U., and Hoffmann, T.: In situ measurements of molecular iodine in the marine boundary layer: the link to macroalgae and the implications for O3, IO, OIO and NOx, Atmos. Chem. Phys., 10, 4823–4833, https://doi.org/10.5194/acp-10-4823-2010, 2010.
Inamdar, S., Tinel, L., Chance, R., Carpenter, L. J., Sabu, P., Chacko, R., Tripathy, S. C., Kerkar, A. U., Sinha, A. K., Bhaskar, P. V., Sarkar, A., Roy, R., Sherwen, T., Cuevas, C., Saiz-Lopez, A., Ram, K., and Mahajan, A. S.: Estimation of reactive inorganic iodine fluxes in the Indian and Southern Ocean marine boundary layer, Atmos. Chem. Phys., 20, 12093–12114, https://doi.org/10.5194/acp-20-12093-2020, 2020.
Jacob, D.: Heterogeneous chemistry and tropospheric ozone, Atmospheric Environment, 34, 2131–2159, https://doi.org/10.1016/S1352-2310(99)00462-8, 2000.
Jaeglé, L., Quinn, P. K., Bates, T. S., Alexander, B., and Lin, J.-T.: Global distribution of sea salt aerosols: new constraints from in situ and remote sensing observations, Atmos. Chem. Phys., 11, 3137–3157, https://doi.org/10.5194/acp-11-3137-2011, 2011.
Jokinen, T., Sipilä, M., Kontkanen, J., Vakkari, V., Tisler, P., Duplissy, E.-M., Junninen, H., Kangasluoma, J., Manninen, H. E., Petäjä, T., Kulmala, M., Worsnop, D. R., Kirkby, J., Virkkula, A., and Kerminen, V.-M.: Ion-induced sulfuric acid–ammonia nucleation drives particle formation in coastal Antarctica, Sci. Adv., 4, eaat9744, https://doi.org/10.1126/sciadv.aat9744, 2018.
Jones, M. R., Chance, R., Bell, T., Jones, O., Loades, D. C., May, R., Tinel, L., Weddell, K., Widdicombe, C., and Carpenter, L. J.: Iodide, iodate & dissolved organic iodine in the temperate coastal ocean, Front. Mar. Sci., 11, 1277595, https://doi.org/10.3389/fmars.2024.1277595, 2024.
Kanaya, Y., Sommariva, R., Saiz-Lopez, A., Mazzeo, A., Koenig, T. K., Kawana, K., Johnson, J. E., Colomb, A., Tulet, P., Molloy, S., Galbally, I. E., Volkamer, R., Mahajan, A., Halfacre, J. W., Shepson, P. B., Schmale, J., Angot, H., Blomquist, B., Shupe, M. D., Helmig, D., Gil, J., Lee, M., Coburn, S. C., Ortega, I., Chen, G., Lee, J., Aikin, K. C., Parrish, D. D., Holloway, J. S., Ryerson, T. B., Pollack, I. B., Williams, E. J., Lerner, B. M., Weinheimer, A. J., Campos, T., Flocke, F. M., Spackman, J. R., Bourgeois, I., Peischl, J., Thompson, C. R., Staebler, R. M., Aliabadi, A. A., Gong, W., Van Malderen, R., Thompson, A. M., Stauffer, R. M., Kollonige, D. E., Gómez Martin, J. C., Fujiwara, M., Read, K., Rowlinson, M., Sato, K., Kurokawa, J., Iwamoto, Y., Taketani, F., Takashima, H., Navarro-Comas, M., Panagi, M., and Schultz, M. G.: Observational ozone data over the global oceans and polar regions: The TOAR-II Oceans data set version 2024, https://doi.org/10.17596/0004044, 2025a.
Kanaya, Y., Sommariva, R., Saiz-Lopez, A., Mazzeo, A., Koenig, T. K., Kawana, K., Johnson, J. E., Colomb, A., Tulet, P., Molloy, S., Galbally, I. E., Volkamer, R., Mahajan, A., Halfacre, J. W., Shepson, P. B., Schmale, J., Angot, H., Blomquist, B., Shupe, M. D., Helmig, D., Gil, J., Lee, M., Coburn, S. C., Ortega, I., Chen, G., Lee, J., Aikin, K. C., Parrish, D. D., Holloway, J. S., Ryerson, T. B., Pollack, I. B., Williams, E. J., Lerner, B. M., Weinheimer, A. J., Campos, T., Flocke, F. M., Spackman, J. R., Bourgeois, I., Peischl, J., Thompson, C. R., Staebler, R. M., Aliabadi, A. A., Gong, W., Van Malderen, R., Thompson, A. M., Stauffer, R. M., Kollonige, D. E., Gómez Martin, J. C., Fujiwara, M., Read, K., Rowlinson, M., Sato, K., Kurokawa, J., Iwamoto, Y., Taketani, F., Takashima, H., Navarro-Comas, M., Panagi, M., and Schultz, M. G.: Observational ozone datasets over the global oceans and polar regions (version 2024), Earth Syst. Sci. Data, 17, 4901–4932, https://doi.org/10.5194/essd-17-4901-2025, 2025b.
Koenig, T. K., Baidar, S., Campuzano-Jost, P., Cuevas, C. A., Dix, B., Fernandez, R. P., Guo, H., Hall, S. R., Kinnison, D., Nault, B. A., Ullmann, K., Jimenez, J. L., Saiz-Lopez, A., and Volkamer, R.: Quantitative detection of iodine in the stratosphere, Proc. Natl. Acad. Sci. USA, 117, 1860–1866, https://doi.org/10.1073/pnas.1916828117, 2020.
Koenig, T. K., Volkamer, R., Apel, E. C., Bresch, J. F., Cuevas, C. A., Dix, B., Eloranta, E. W., Fernandez, R. P., Hall, S. R., Hornbrook, R. S., Pierce, R. B., Reeves, J. M., Saiz-Lopez, A., and Ullmann, K.: Ozone depletion due to dust release of iodine in the free troposphere, Sci. Adv., 7, eabj6544, https://doi.org/10.1126/sciadv.abj6544, 2021.
Lai, S. C., Hoffmann, T., and Xie, Z. Q.: Iodine speciation in marine aerosols along a 30 000 km round-trip cruise path from Shanghai, China to Prydz Bay, Antarctica, Geophys. Res. Lett., 35, L21803, https://doi.org/10.1029/2008GL035492, 2008.
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cycles, 25, https://doi.org/10.1029/2010GB003850, 2011.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, C. F., Elgiar, T., David, L. M., Wilmot, T. Y., Reza, M., Hirshorn, N., McCubbin, I. B., Shah, V., Lin, J. C., Lyman, S. N., Hallar, A. G., Gratz, L. E., and Volkamer, R.: Elevated Tropospheric Iodine Over the Central Continental United States: Is Iodine a Major Oxidant of Atmospheric Mercury?, Geophysical Research Letters, 51, e2024GL109247, https://doi.org/10.1029/2024GL109247, 2024.
Legrand, M., McConnell, J. R., Preunkert, S., Arienzo, M., Chellman, N., Gleason, K., Sherwen, T., Evans, M. J., and Carpenter, L. J.: Alpine ice evidence of a three-fold increase in atmospheric iodine deposition since 1950 in Europe due to increasing oceanic emissions, Proc. Natl. Acad. Sci. USA, 115, 12136–12141, https://doi.org/10.1073/pnas.1809867115, 2018.
Li, D., Nie, W., Liu, Y., Yan, C., Ge, D., Zha, Q., Liu, C., Wang, J., Wang, J., Wang, L., Liu, T., Chi, X., and Ding, A.: Field Evidence of Nocturnal Multiphase Production of Iodic Acid, Environ. Sci. Technol. Lett., 11, 709–715, https://doi.org/10.1021/acs.estlett.4c00244, 2024.
Li, Q., Fernandez, R. P., Hossaini, R., Iglesias-Suarez, F., Cuevas, C. A., Apel, E. C., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Reactive halogens increase the global methane lifetime and radiative forcing in the 21st century, Nat. Commun., 13, 2768, https://doi.org/10.1038/s41467-022-30456-8, 2022a.
Li, Q., Tham, Y. J., Fernandez, R. P., He, X., Cuevas, C. A., and Saiz-Lopez, A.: Role of Iodine Recycling on Sea-Salt Aerosols in the Global Marine Boundary Layer, Geophysical Research Letters, 49, e2021GL097567, https://doi.org/10.1029/2021GL097567, 2022b.
Li, Y., Martin, R. V., Li, C., Boys, B. L., van Donkelaar, A., Meng, J., and Pierce, J. R.: Development and evaluation of processes affecting simulation of diel fine particulate matter variation in the GEOS-Chem model, Atmos. Chem. Phys., 23, 12525–12543, https://doi.org/10.5194/acp-23-12525-2023, 2023.
Liang, Q., Stolarski, R. S., Kawa, S. R., Nielsen, J. E., Douglass, A. R., Rodriguez, J. M., Blake, D. R., Atlas, E. L., and Ott, L. E.: Finding the missing stratospheric Bry: a global modeling study of CHBr3 and CH2Br2, Atmos. Chem. Phys., 10, 2269–2286, https://doi.org/10.5194/acp-10-2269-2010, 2010.
Liu, H., Jacob, D. J., Bey, I., and Yantosca, R. M.: Constraints from 210Pb and 7Be on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields, J. Geophys. Res., 106, 12109–12128, https://doi.org/10.1029/2000JD900839, 2001.
Liu, L.: Modeling the Novel Heterogeneous Pathways of Aerosol Formation from Nitrate Photolysis and Iodine Chemistry in Marine Atmosphere, Doctoral Dissertation, City University of Hong Kong, City University of Hong Kong, 236 pp., 2024.
Liu, T. and Abbatt, J. P. D.: An Experimental Assessment of the Importance of S(IV) Oxidation by Hypohalous Acids in the Marine Atmosphere, Geophysical Research Letters, 47, e2019GL086465, https://doi.org/10.1029/2019GL086465, 2020.
Liu, Q. and Margerum, D. W.: Equilibrium and Kinetics of Bromine Chloride Hydrolysis, Environ. Sci. Technol., 35, 1127–1133, https://doi.org/10.1021/es001380r, 2001.
MacDonald, S. M., Gómez Martín, J. C., Chance, R., Warriner, S., Saiz-Lopez, A., Carpenter, L. J., and Plane, J. M. C.: A laboratory characterisation of inorganic iodine emissions from the sea surface: dependence on oceanic variables and parameterisation for global modelling, Atmos. Chem. Phys., 14, 5841–5852, https://doi.org/10.5194/acp-14-5841-2014, 2014.
Mahajan, A. S., Oetjen, H., Saiz‐Lopez, A., Lee, J. D., McFiggans, G. B., and Plane, J. M. C.: Reactive iodine species in a semi‐polluted environment, Geophysical Research Letters, 36, 2009GL038018, https://doi.org/10.1029/2009GL038018, 2009.
Mahajan, A. S., Shaw, M., Oetjen, H., Hornsby, K. E., Carpenter, L. J., Kaleschke, L., Tian-Kunze, X., Lee, J. D., Moller, S. J., Edwards, P., Commane, R., Ingham, T., Heard, D. E., and Plane, J. M. C.: Evidence of reactive iodine chemistry in the Arctic boundary layer, J. Geophys. Res., 115, 2009JD013665, https://doi.org/10.1029/2009JD013665, 2010a.
Mahajan, A. S., Plane, J. M. C., Oetjen, H., Mendes, L., Saunders, R. W., Saiz-Lopez, A., Jones, C. E., Carpenter, L. J., and McFiggans, G. B.: Measurement and modelling of tropospheric reactive halogen species over the tropical Atlantic Ocean, Atmos. Chem. Phys., 10, 4611–4624, https://doi.org/10.5194/acp-10-4611-2010, 2010b.
Mahajan, A. S., Gómez Martín, J. C., Hay, T. D., Royer, S.-J., Yvon-Lewis, S., Liu, Y., Hu, L., Prados-Roman, C., Ordóñez, C., Plane, J. M. C., and Saiz-Lopez, A.: Latitudinal distribution of reactive iodine in the Eastern Pacific and its link to open ocean sources, Atmos. Chem. Phys., 12, 11609–11617, https://doi.org/10.5194/acp-12-11609-2012, 2012.
Mahajan, A. S., Biswas, M. S., Beirle, S., Wagner, T., Schönhardt, A., Benavent, N., and Saiz-Lopez, A.: Observations of iodine monoxide over three summers at the Indian Antarctic bases of Bharati and Maitri, Atmos. Chem. Phys., 21, 11829–11842, https://doi.org/10.5194/acp-21-11829-2021, 2021.
Meinshausen, M., Vogel, E., Nauels, A., Lorbacher, K., Meinshausen, N., Etheridge, D. M., Fraser, P. J., Montzka, S. A., Rayner, P. J., Trudinger, C. M., Krummel, P. B., Beyerle, U., Canadell, J. G., Daniel, J. S., Enting, I. G., Law, R. M., Lunder, C. R., O'Doherty, S., Prinn, R. G., Reimann, S., Rubino, M., Velders, G. J. M., Vollmer, M. K., Wang, R. H. J., and Weiss, R.: Historical greenhouse gas concentrations for climate modelling (CMIP6), Geosci. Model Dev., 10, 2057–2116, https://doi.org/10.5194/gmd-10-2057-2017, 2017.
Miller, S. J., Makar, P. A., and Lee, C. J.: HETerogeneous vectorized or Parallel (HETPv1.0): an updated inorganic heterogeneous chemistry solver for the metastable-state –Na+–Ca2+–K+–Mg2+–––Cl−–H2O system based on ISORROPIA II, Geosci. Model Dev., 17, 2197–2219, https://doi.org/10.5194/gmd-17-2197-2024, 2024.
Murphy, D. M., Froyd, K. D., Bian, H., Brock, C. A., Dibb, J. E., DiGangi, J. P., Diskin, G., Dollner, M., Kupc, A., Scheuer, E. M., Schill, G. P., Weinzierl, B., Williamson, C. J., and Yu, P.: The distribution of sea-salt aerosol in the global troposphere, Atmos. Chem. Phys., 19, 4093–4104, https://doi.org/10.5194/acp-19-4093-2019, 2019.
Mustaffa, N. I. H., Badewien, T. H., Ribas-Ribas, M., and Wurl, O.: High-resolution observations on enrichment processes in the sea-surface microlayer, Sci. Rep., 8, 13122, https://doi.org/10.1038/s41598-018-31465-8, 2018.
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hämeri, K., Pirjola, L., Kulmala, M., Jennings, S. G., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions, Nature, 417, 632–636, https://doi.org/10.1038/nature00775, 2002.
Oetjen, H.: Measurements of halogen oxides by scattered sunlight differential optical absorption spectroscopy, Universität Bremen, 2009.
Ordóñez, C., Lamarque, J.-F., Tilmes, S., Kinnison, D. E., Atlas, E. L., Blake, D. R., Sousa Santos, G., Brasseur, G., and Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 12, 1423–1447, https://doi.org/10.5194/acp-12-1423-2012, 2012.
Pan, L. L., Atlas, E. L., Salawitch, R. J., Honomichl, S. B., Bresch, J. F., Randel, W. J., Apel, E. C., Hornbrook, R. S., Weinheimer, A. J., Anderson, D. C., Andrews, S. J., Baidar, S., Beaton, S. P., Campos, T. L., Carpenter, L. J., Chen, D., Dix, B., Donets, V., Hall, S. R., Hanisco, T. F., Homeyer, C. R., Huey, L. G., Jensen, J. B., Kaser, L., Kinnison, D. E., Koenig, T. K., Lamarque, J.-F., Liu, C., Luo, J., Luo, Z. J., Montzka, D. D., Nicely, J. M., Pierce, R. B., Riemer, D. D., Robinson, T., Romashkin, P., Saiz-Lopez, A., Schauffler, S., Shieh, O., Stell, M. H., Ullmann, K., Vaughan, G., Volkamer, R., and Wolfe, G.: The Convective Transport of Active Species in the Tropics (CONTRAST) Experiment, Bulletin of the American Meteorological Society, 98, 106–128, https://doi.org/10.1175/BAMS-D-14-00272.1, 2017.
Pechtl, S., Schmitz, G., and von Glasow, R.: Modelling iodide – iodate speciation in atmospheric aerosol: Contributions of inorganic and organic iodine chemistry, Atmos. Chem. Phys., 7, 1381–1393, https://doi.org/10.5194/acp-7-1381-2007, 2007.
Peters, C., Pechtl, S., Stutz, J., Hebestreit, K., Hönninger, G., Heumann, K. G., Schwarz, A., Winterlik, J., and Platt, U.: Reactive and organic halogen species in three different European coastal environments, Atmos. Chem. Phys., 5, 3357–3375, https://doi.org/10.5194/acp-5-3357-2005, 2005.
Pound, R. J., Brown, L. V., Evans, M. J., and Carpenter, L. J.: An improved estimate of inorganic iodine emissions from the ocean using a coupled surface microlayer box model, Atmos. Chem. Phys., 24, 9899–9921, https://doi.org/10.5194/acp-24-9899-2024, 2024.
Prados-Roman, C., Cuevas, C. A., Hay, T., Fernandez, R. P., Mahajan, A. S., Royer, S.-J., Galí, M., Simó, R., Dachs, J., Großmann, K., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Iodine oxide in the global marine boundary layer, Atmos. Chem. Phys., 15, 583–593, https://doi.org/10.5194/acp-15-583-2015, 2015a.
Prados-Roman, C., Cuevas, C. A., Hay, T., Fernandez, R. P., Mahajan, A. S., Royer, S.-J., Galí, M., Simó, R., Dachs, J., Großmann, K., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Iodine oxide in the global marine boundary layer, Atmos. Chem. Phys., 15, 583–593, https://doi.org/10.5194/acp-15-583-2015, 2015b.
Prather, M. J.: Photolysis rates in correlated overlapping cloud fields: Cloud-J 7.3c, Geosci. Model Dev., 8, 2587–2595, https://doi.org/10.5194/gmd-8-2587-2015, 2015.
Puentedura, O., Gil, M., Saiz-Lopez, A., Hay, T., Navarro-Comas, M., Gómez-Pelaez, A., Cuevas, E., Iglesias, J., and Gomez, L.: Iodine monoxide in the north subtropical free troposphere, Atmos. Chem. Phys., 12, 4909–4921, https://doi.org/10.5194/acp-12-4909-2012, 2012.
Pye, H. O. T., Nenes, A., Alexander, B., Ault, A. P., Barth, M. C., Clegg, S. L., Collett Jr., J. L., Fahey, K. M., Hennigan, C. J., Herrmann, H., Kanakidou, M., Kelly, J. T., Ku, I.-T., McNeill, V. F., Riemer, N., Schaefer, T., Shi, G., Tilgner, A., Walker, J. T., Wang, T., Weber, R., Xing, J., Zaveri, R. A., and Zuend, A.: The acidity of atmospheric particles and clouds, Atmos. Chem. Phys., 20, 4809–4888, https://doi.org/10.5194/acp-20-4809-2020, 2020.
Raso, A. R. W., Custard, K. D., May, N. W., Tanner, D., Newburn, M. K., Walker, L., Moore, R. J., Huey, L. G., Alexander, L., Shepson, P. B., and Pratt, K. A.: Active molecular iodine photochemistry in the Arctic, Proc. Natl. Acad. Sci. USA, 114, 10053–10058, https://doi.org/10.1073/pnas.1702803114, 2017.
Read, K. A., Mahajan, A. S., Carpenter, L. J., Evans, M. J., Faria, B. V. E., Heard, D. E., Hopkins, J. R., Lee, J. D., Moller, S. J., Lewis, A. C., Mendes, L., McQuaid, J. B., Oetjen, H., Saiz-Lopez, A., Pilling, M. J., and Plane, J. M. C.: Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 453, 1232–1235, https://doi.org/10.1038/nature07035, 2008.
Reza, M., Iezzi, L., Finkenzeller, H., Roose, A., Ammann, M., and Volkamer, R.: Iodine Activation from Iodate Reduction in Aqueous Films via Photocatalyzed and Dark Reactions, ACS Earth Space Chem., 8, 2495–2508, https://doi.org/10.1021/acsearthspacechem.4c00224, 2024.
Roberts, T. J., Jourdain, L., Griffiths, P. T., and Pirre, M.: Re-evaluating the reactive uptake of HOBr in the troposphere with implications for the marine boundary layer and volcanic plumes, Atmos. Chem. Phys., 14, 11185–11199, https://doi.org/10.5194/acp-14-11185-2014, 2014.
Saiz-Lopez, A. and Plane, J. M. C.: Novel iodine chemistry in the marine boundary layer, Geophysical Research Letters, 31, 2003GL019215, https://doi.org/10.1029/2003GL019215, 2004.
Saiz-Lopez, A., Mahajan, A. S., Salmon, R. A., Bauguitte, S. J.-B., Jones, A. E., Roscoe, H. K., and Plane, J. M. C.: Boundary Layer Halogens in Coastal Antarctica, Science, 317, 348–351, https://doi.org/10.1126/science.1141408, 2007.
Saiz-Lopez, A., Plane, J. M. C., Mahajan, A. S., Anderson, P. S., Bauguitte, S. J.-B., Jones, A. E., Roscoe, H. K., Salmon, R. A., Bloss, W. J., Lee, J. D., and Heard, D. E.: On the vertical distribution of boundary layer halogens over coastal Antarctica: implications for O3, HOx, NOx and the Hg lifetime, Atmos. Chem. Phys., 8, 887–900, https://doi.org/10.5194/acp-8-887-2008, 2008.
Saiz-Lopez, A., Plane, J. M. C., Baker, A. R., Carpenter, L. J., Von Glasow, R., Gómez Martín, J. C., McFiggans, G., and Saunders, R. W.: Atmospheric Chemistry of Iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012.
Saiz-Lopez, A., Fernandez, R. P., Ordóñez, C., Kinnison, D. E., Gómez Martín, J. C., Lamarque, J.-F., and Tilmes, S.: Iodine chemistry in the troposphere and its effect on ozone, Atmos. Chem. Phys., 14, 13119–13143, https://doi.org/10.5194/acp-14-13119-2014, 2014.
Satoh, Y., Otosaka, S., Suzuki, T., and Nakanishi, T.: Factors regulating the concentration of particulate iodine in coastal seawater, Limnology & Oceanography, 68, 1580–1594, https://doi.org/10.1002/lno.12369, 2023.
Saunders, R. W., Kumar, R., MacDonald, S. M., and Plane, J. M. C.: Insights into the Photochemical Transformation of Iodine in Aqueous Systems: Humic Acid Photosensitized Reduction of Iodate, Environ. Sci. Technol., 46, 11854–11861, https://doi.org/10.1021/es3030935, 2012.
Schill, G. P., Froyd, K. D., Murphy, D. M., Williamson, C. J., Brock, C. A., Sherwen, T., Evans, M. J., Ray, E. A., Apel, E. C., Hornbrook, R. S., Hills, A. J., Peischl, J., Ryerson, T. B., Thompson, C. R., Bourgeois, I., Blake, D. R., DiGangi, J. P., and Diskin, G. S.: Widespread trace bromine and iodine in remote tropospheric non-sea-salt aerosols, Atmos. Chem. Phys., 25, 45–71, https://doi.org/10.5194/acp-25-45-2025, 2025.
Schwehr, K. A. and Santschi, P. H.: Sensitive determination of iodine species, including organo-iodine, for freshwater and seawater samples using high performance liquid chromatography and spectrophotometric detection, Analytica Chimica Acta, 482, 59–71, https://doi.org/10.1016/S0003-2670(03)00197-1, 2003.
Shah, V., Jacob, D. J., Moch, J. M., Wang, X., and Zhai, S.: Global modeling of cloud water acidity, precipitation acidity, and acid inputs to ecosystems, Atmos. Chem. Phys., 20, 12223–12245, https://doi.org/10.5194/acp-20-12223-2020, 2020.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016a.
Sherwen, T., Chance, R. J., Tinel, L., Ellis, D., Evans, M. J., and Carpenter, L. J.: A machine-learning-based global sea-surface iodide distribution, Earth Syst. Sci. Data, 11, 1239–1262, https://doi.org/10.5194/essd-11-1239-2019, 2019.
Sherwen, T. M., Evans, M. J., Spracklen, D. V., Carpenter, L. J., Chance, R., Baker, A. R., Schmidt, J. A., and Breider, T. J.: Global modeling of tropospheric iodine aerosol, Geophysical Research Letters, 43, 10012–10019, https://doi.org/10.1002/2016GL070062, 2016b.
Shi, X., Qiu, X., Chen, Q., Chen, S., Hu, M., Rudich, Y., and Zhu, T.: Organic Iodine Compounds in Fine Particulate Matter from a Continental Urban Region: Insights into Secondary Formation in the Atmosphere, Environ. Sci. Technol., 55, 1508–1514, https://doi.org/10.1021/acs.est.0c06703, 2021.
Simone, N. W., Stettler, M. E. J., and Barrett, S. R. H.: Rapid estimation of global civil aviation emissions with uncertainty quantification, Transportation Research Part D: Transport and Environment, 25, 33–41, https://doi.org/10.1016/j.trd.2013.07.001, 2013.
Sipilä, M., Sarnela, N., Jokinen, T., Henschel, H., Junninen, H., Kontkanen, J., Richters, S., Kangasluoma, J., Franchin, A., Peräkylä, O., Rissanen, M. P., Ehn, M., Vehkamäki, H., Kurten, T., Berndt, T., Petäjä, T., Worsnop, D., Ceburnis, D., Kerminen, V.-M., Kulmala, M., and O'Dowd, C.: Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3, Nature, 537, 532–534, https://doi.org/10.1038/nature19314, 2016.
Spólnik, G., Wach, P., Wróbel, Z., and Danikiewicz, W.: 2-Iodomalondialdehyde is an abundant component of soluble organic iodine in atmospheric wet precipitation, Science of The Total Environment, 730, 139175, https://doi.org/10.1016/j.scitotenv.2020.139175, 2020.
Stemmler, I., Hense, I., Quack, B., and Maier-Reimer, E.: Methyl iodide production in the open ocean, Biogeosciences, 11, 4459–4476, https://doi.org/10.5194/bg-11-4459-2014, 2014.
Stutz, J., Pikelnaya, O., Hurlock, S. C., Trick, S., Pechtl, S., and Von Glasow, R.: Daytime OIO in the Gulf of Maine, Geophysical Research Letters, 34, 2007GL031332, https://doi.org/10.1029/2007GL031332, 2007.
Thakur, R. C., Dada, L., Beck, L. J., Quéléver, L. L. J., Chan, T., Marbouti, M., He, X.-C., Xavier, C., Sulo, J., Lampilahti, J., Lampimäki, M., Tham, Y. J., Sarnela, N., Lehtipalo, K., Norkko, A., Kulmala, M., Sipilä, M., and Jokinen, T.: An evaluation of new particle formation events in Helsinki during a Baltic Sea cyanobacterial summer bloom, Atmos. Chem. Phys., 22, 6365–6391, https://doi.org/10.5194/acp-22-6365-2022, 2022.
Tham, Y. J., He, X.-C., Li, Q., Cuevas, C. A., Shen, J., Kalliokoski, J., Yan, C., Iyer, S., Lehmusjärvi, T., Jang, S., Thakur, R. C., Beck, L., Kemppainen, D., Olin, M., Sarnela, N., Mikkilä, J., Hakala, J., Marbouti, M., Yao, L., Li, H., Huang, W., Wang, Y., Wimmer, D., Zha, Q., Virkanen, J., Spain, T. G., O'Doherty, S., Jokinen, T., Bianchi, F., Petäjä, T., Worsnop, D. R., Mauldin, R. L., Ovadnevaite, J., Ceburnis, D., Maier, N. M., Kulmala, M., O'Dowd, C., Dal Maso, M., Saiz-Lopez, A., and Sipilä, M.: Direct field evidence of autocatalytic iodine release from atmospheric aerosol, Proc. Natl. Acad. Sci. USA, 118, e2009951118, https://doi.org/10.1073/pnas.2009951118, 2021.
Thompson, C. R., Shepson, P. B., Liao, J., Huey, L. G., Apel, E. C., Cantrell, C. A., Flocke, F., Orlando, J., Fried, A., Hall, S. R., Hornbrook, R. S., Knapp, D. J., Mauldin III, R. L., Montzka, D. D., Sive, B. C., Ullmann, K., Weibring, P., and Weinheimer, A.: Interactions of bromine, chlorine, and iodine photochemistry during ozone depletions in Barrow, Alaska, Atmos. Chem. Phys., 15, 9651–9679, https://doi.org/10.5194/acp-15-9651-2015, 2015.
Thurlow, M. E., Co, D. T., O’Brien, A. S., Hannun, R. A., Lapson, L. B., Hanisco, T. F., and Anderson, J. G.: The development and deployment of a ground-based, laser-induced fluorescence instrument for the in situ detection of iodine monoxide radicals, Review of Scientific Instruments, 85, 044101, https://doi.org/10.1063/1.4869857, 2014.
Tinel, L., Adams, T. J., Hollis, L. D. J., Bridger, A. J. M., Chance, R. J., Ward, M. W., Ball, S. M., and Carpenter, L. J.: Influence of the Sea Surface Microlayer on Oceanic Iodine Emissions, Environ. Sci. Technol., 54, 13228–13237, https://doi.org/10.1021/acs.est.0c02736, 2020.
Troy, R. C. and Margerum, D. W.: Non-metal redox kinetics: hypobromite and hypobromous acid reactions with iodide and with sulfite and the hydrolysis of bromosulfate, Inorg. Chem., 30, 3538–3543, https://doi.org/10.1021/ic00018a028, 1991.
Vinken, G. C. M., Boersma, K. F., Jacob, D. J., and Meijer, E. W.: Accounting for non-linear chemistry of ship plumes in the GEOS-Chem global chemistry transport model, Atmos. Chem. Phys., 11, 11707–11722, https://doi.org/10.5194/acp-11-11707-2011, 2011.
Vogt, R., Sander, R., Von Glasow, R., and Crutzen, P. J.: Iodine Chemistry and its Role in Halogen Activation and Ozone Loss in the Marine Boundary Layer: A Model Study, Journal of Atmospheric Chemistry, 32, 375–395, https://doi.org/10.1023/A:1006179901037, 1999.
Volkamer, R. and Dix, B.: GV AMAX-DOAS Data, Version 5.0 (5.0), NCAR [data set], https://doi.org/10.26023/DJX0-85VQ-Q80X, 2017.
Volkamer, R., Baidar, S., Campos, T. L., Coburn, S., DiGangi, J. P., Dix, B., Eloranta, E. W., Koenig, T. K., Morley, B., Ortega, I., Pierce, B. R., Reeves, M., Sinreich, R., Wang, S., Zondlo, M. A., and Romashkin, P. A.: Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2−O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements, Atmos. Meas. Tech., 8, 2121–2148, https://doi.org/10.5194/amt-8-2121-2015, 2015.
Volkamer, R., Koenig, T., Baidar, S., and Dix, B.: Airborne Multi-AXis Differential Optical Absorption Spectroscopy (AMAX-DOAS) Data, Version 2.0 (2.0), NCAR [data set], https://doi.org/10.5065/D6F769MF, 2020.
Wada, R., Beames, J. M., and Orr-Ewing, A. J.: Measurement of IO radical concentrations in the marine boundary layer using a cavity ring-down spectrometer, J. Atmos. Chem., 58, 69–87, https://doi.org/10.1007/s10874-007-9080-z, 2007.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Wang, X., Jacob, D. J., Downs, W., Zhai, S., Zhu, L., Shah, V., Holmes, C. D., Sherwen, T., Alexander, B., Evans, M. J., Eastham, S. D., Neuman, J. A., Veres, P. R., Koenig, T. K., Volkamer, R., Huey, L. G., Bannan, T. J., Percival, C. J., Lee, B. H., and Thornton, J. A.: Global tropospheric halogen (Cl, Br, I) chemistry and its impact on oxidants, Atmos. Chem. Phys., 21, 13973–13996, https://doi.org/10.5194/acp-21-13973-2021, 2021.
Whalley, L. K., Furneaux, K. L., Gravestock, T., Atkinson, H. M., Bale, C. S. E., Ingham, T., Bloss, W. J., and Heard, D. E.: Detection of iodine monoxide radicals in the marine boundary layer using laser induced fluorescence spectroscopy, J. Atmos. Chem., 58, 19–39, https://doi.org/10.1007/s10874-007-9075-9, 2007.
Williams, J., Gros, V., Atlas, E., Maciejczyk, K., Batsaikhan, A., Schöler, H. F., Forster, C., Quack, B., Yassaa, N., Sander, R., and Van Dingenen, R.: Possible evidence for a connection between methyl iodide emissions and Saharan dust, J. Geophys. Res., 112, 2005JD006702, https://doi.org/10.1029/2005JD006702, 2007.
Wofsy, S. C., Afshar, S., Allen, H. M., Apel, E. C., Asher, E. C., Barletta, B., Bent, J., Bian, H., Biggs, B. C., Blake, D. R., Blake, N., Bourgeois, I., Brock, C. A., Brune, W. H., Budney, J. W., Bui, T. P., Butler, A., Campuzano-Jost, P., Chang, C. S., and Vieznor, N.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, Version 2 (Version 2.0), ORNL Distributed Active Archive Center [data set], https://doi.org/10.3334/ORNLDAAC/1925, 2021.
Wong, G. T. F. and Cheng, X.-H.: Dissolved organic iodine in marine waters: Determination, occurrence and analytical implications, Marine Chemistry, 59, 271–281, https://doi.org/10.1016/S0304-4203(97)00078-9, 1998.
Wurl, O. and Obbard, J. P.: A review of pollutants in the sea-surface microlayer (SML): a unique habitat for marine organisms, Marine Pollution Bulletin, 48, 1016–1030, https://doi.org/10.1016/j.marpolbul.2004.03.016, 2004.
Xavier, C., De Jonge, R. W., Jokinen, T., Beck, L., Sipilä, M., Olenius, T., and Roldin, P.: Role of Iodine-Assisted Aerosol Particle Formation in Antarctica, Environ. Sci. Technol., 58, 7314–7324, https://doi.org/10.1021/acs.est.3c09103, 2024.
Yodle, C. and Baker, A. R.: Influence of collection substrate and extraction method on the speciation of soluble iodine in atmospheric aerosols, Atmospheric Environment: X, 1, 100009, https://doi.org/10.1016/j.aeaoa.2019.100009, 2019.
Young, P. J., Naik, V., Fiore, A. M., Gaudel, A., Guo, J., Lin, M. Y., Neu, J. L., Parrish, D. D., Rieder, H. E., Schnell, J. L., Tilmes, S., Wild, O., Zhang, L., Ziemke, J., Brandt, J., Delcloo, A., Doherty, R. M., Geels, C., Hegglin, M. I., Hu, L., Im, U., Kumar, R., Luhar, A., Murray, L., Plummer, D., Rodriguez, J., Saiz-Lopez, A., Schultz, M. G., Woodhouse, M. T., and Zeng, G.: Tropospheric Ozone Assessment Report: Assessment of global-scale model performance for global and regional ozone distributions, variability, and trends, Elementa: Science of the Anthropocene, 6, 10, https://doi.org/10.1525/elementa.265, 2018.
Yu, H., Ren, L., Huang, X., Xie, M., He, J., and Xiao, H.: Iodine speciation and size distribution in ambient aerosols at a coastal new particle formation hotspot in China, Atmos. Chem. Phys., 19, 4025–4039, https://doi.org/10.5194/acp-19-4025-2019, 2019.
Zhai, S., Swanson, W., McConnell, J. R., Chellman, N., Opel, T., Sigl, M., Meyer, H., Wang, X., Jaeglé, L., Stutz, J., Dibb, J. E., Fujita, K., and Alexander, B.: Implications of Snowpack Reactive Bromine Production for Arctic Ice Core Bromine Preservation, J. GEOPHYS. RES. Atmospheres, 128, e2023JD039257, https://doi.org/10.1029/2023JD039257, 2023.
Zhai, S., McConnell, J. R., Chellman, N., Legrand, M., Opel, T., Meyer, H., Jaeglé, L., Confer, K., Fujita, K., Wang, X., and Alexander, B.: Anthropogenic Influence on Tropospheric Reactive Bromine Since the Pre-industrial: Implications for Arctic Ice-Core Bromine Trends, Geophysical Research Letters, 51, e2023GL107733, https://doi.org/10.1029/2023GL107733, 2024.
Zhang, B., Shen, H., Yun, X., Zhong, Q., Henderson, B. H., Wang, X., Shi, L., Gunthe, S. S., Huey, L. G., Tao, S., Russell, A. G., and Liu, P.: Global Emissions of Hydrogen Chloride and Particulate Chloride from Continental Sources, Environ. Sci. Technol., 56, 3894–3904, https://doi.org/10.1021/acs.est.1c05634, 2022.
Zhang, Y., Li, D., He, X.-C., Nie, W., Deng, C., Cai, R., Liu, Y., Guo, Y., Liu, C., Li, Y., Chen, L., Li, Y., Hua, C., Liu, T., Wang, Z., Xie, J., Wang, L., Petäjä, T., Bianchi, F., Qi, X., Chi, X., Paasonen, P., Liu, Y., Yan, C., Jiang, J., Ding, A., and Kulmala, M.: Iodine oxoacids and their roles in sub-3 nm particle growth in polluted urban environments, Atmos. Chem. Phys., 24, 1873–1893, https://doi.org/10.5194/acp-24-1873-2024, 2024.
Zhao, B., Donahue, N. M., Zhang, K., Mao, L., Shrivastava, M., Ma, P.-L., Shen, J., Wang, S., Sun, J., Gordon, H., Tang, S., Fast, J., Wang, M., Gao, Y., Yan, C., Singh, B., Li, Z., Huang, L., Lou, S., Lin, G., Wang, H., Jiang, J., Ding, A., Nie, W., Qi, X., Chi, X., and Wang, L.: Global variability in atmospheric new particle formation mechanisms, Nature, 631, 98–105, https://doi.org/10.1038/s41586-024-07547-1, 2024.
Zhao, X., Hou, X., and Zhou, W.: Atmospheric Iodine (127I and 129I) Record in Spruce Tree Rings in the Northeast Qinghai–Tibet Plateau, Environ. Sci. Technol., 53, 8706–8714, https://doi.org/10.1021/acs.est.9b01160, 2019.
- Abstract
- Introduction
- Methods
- Results
- Atmospheric implications and conclusions
- Appendix A: Model configuration and assumptions for speciated aerosol iodine
- Appendix B: Model configuration and assumptions for dehalogenation reactions
- Appendix C: Comparisons between modeled and measured iodine aerosol
- Appendix D: Comparisons between modeled and measured gas-phase iodine
- Appendix E: Ozone comparisons between new iodine chemistry simulation and base model
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Abstract
- Introduction
- Methods
- Results
- Atmospheric implications and conclusions
- Appendix A: Model configuration and assumptions for speciated aerosol iodine
- Appendix B: Model configuration and assumptions for dehalogenation reactions
- Appendix C: Comparisons between modeled and measured iodine aerosol
- Appendix D: Comparisons between modeled and measured gas-phase iodine
- Appendix E: Ozone comparisons between new iodine chemistry simulation and base model
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References