the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Jul 2026
| 16 Jul 2026
High yields of formic acid and acetic acid during multi-generational oxidation of toluene
Hengqing Shen
Min Zhao
Yu Yang
Huan Li
Huihui Wu
Zhongming Chen
Formic acid and acetic acid are the most abundant gas-phase organic acids in the atmosphere, yet their concentrations are substantially underestimated by both global and regional atmospheric models across diverse environments. In this study, we report unexpectedly high yields of formic acid and acetic acid during the multi-generational photooxidation of toluene, a canonical anthropogenic volatile organic compound. Their yields exhibit a strong dependence on hydroxyl radical (•OH) exposure ([•OH] × residence time), increasing from 25 % and 24 % under low exposure (< 0.2 equivalent days) to 74 % and 40 % under elevated exposure (1–3 equivalent days) for formic and acetic acid, respectively. The formation of these organic acids is not significantly affected by NOx concentrations. A modified box model based on MCM v3.3.1 underestimates the peak concentrations of both acids by approximately a factor of five, indicating substantial gaps in current mechanistic understanding. Although both secondary aerosol formation and organic acid production increase with aging to a certain degree of oxidation, their distinct temporal evolutions suggest that particle photodegradation is not the dominant pathway. The contrasting •OH exposure dependence between organic acids and primary carbonyl compounds further implies that these acids are predominantly multi-generational oxidation products. These findings demonstrate that multi-generational oxidation of aromatic compounds is an important and previously underappreciated source of atmospheric organic acids. The omission of organic acid formation from aromatic oxidation in current chemical mechanisms likely contributes to their widespread underestimation in models, highlighting the need for detailed laboratory studies and updated chemical mechanisms.
- Article
(1300 KB) - Full-text XML
-
Supplement
(3969 KB) - BibTeX
- EndNote




Organic acids are ubiquitous in the troposphere and are important contributors to precipitation acidity (Stavrakou et al., 2012). Among them, formic acid (HCOOH) and acetic acid (CH3COOH) are the most abundant gas-phase organic acids in various atmospheric environments, with mixing ratios spanning from several pptv to tens of ppbv (Chebbi and Carlier, 1996; Ding et al., 2025; Mungall et al., 2018; Veres et al., 2011). Formic acid alone can contribute to more than half of rainwater acidity in many continental regions (Stavrakou et al., 2012). In addition, these organic acids can also affect secondary aerosol formation by altering aerosol pH and gas-particle partitioning processes (Shen et al., 2018; Tao and Murphy, 2019).
On a global scale, the dominant source of formic acid and acetic acid is considered to be secondary production from the photooxidation of biogenic volatile organic compounds (VOCs), particularly isoprene and its oxidation products (Khan et al., 2018; Link et al., 2020; Paulot et al., 2011). However, rapid secondary production and high concentrations of formic acid and acetic acid have also been observed in anthropogenic air masses and urban environments (Liggio et al., 2017; Millet et al., 2015; Veres et al., 2011; Yuan et al., 2015), suggesting that anthropogenic VOCs may play a significant role in the formation of formic acid and acetic acid in polluted areas. Despite this, the sources and detailed formation pathways of atmospheric organic acids remain poorly understood. This is evidenced by the fact that current atmospheric chemical models substantially underestimate (up to > 10 times) their field-observed concentrations on both global (Chaliyakunnel et al., 2016; Khan et al., 2018; Paulot et al., 2011; Stavrakou et al., 2012) and regional scales (Le Breton et al., 2012; Liggio et al., 2017; Millet et al., 2015; Yuan et al., 2015) in the free troposphere (Millet et al., 2015; Paulot et al., 2011), the forest (Schobesberger et al., 2016; Stavrakou et al., 2012), the urban (Bannan et al., 2017; Khan et al., 2018; Yuan et al., 2015), the polar region (Mungall et al., 2018; Paulot et al., 2011), and the oil sands region (Liggio et al., 2017; Yuan et al., 2015). Such widespread underestimation implies the existence of large and pervasive missing or underestimated sources in current models (Liggio et al., 2017; Yuan et al., 2015). Numerous hypotheses have been proposed to reconcile this model–observation gap, including previously unrecognized contributions from biomass burning (Chaliyakunnel et al., 2016; Paulot et al., 2011), unmeasured anthropogenic intermediate VOCs (Le Breton et al., 2012; Liggio et al., 2017), acetaldehyde tautomerization (Shaw et al., 2018), cloud-mediated methanediol oxidation (Franco et al., 2021), and chemical aging of organic aerosols (Bates et al., 2023; Jiang et al., 2023; Malecha and Nizkorodov, 2016, 2017; Paulot et al., 2011). Nevertheless, none of these processes alone can fully account for the high ambient concentrations of formic acid and acetic acid observed in the atmosphere (Franco et al., 2021; Liggio et al., 2017; Yuan et al., 2015).
Recent studies have suggested that resolving this discrepancy requires much higher effective molar yields of formic acid from the oxidation of monoterpenes or anthropogenic VOCs than currently assumed (Liggio et al., 2017; Stavrakou et al., 2012). For example, Liggio et al. (2017) estimated that ∼ 50 % of formic acid yield would be necessary to reconcile observations and modeling results in the oil sands region (Liggio et al., 2017). In contrast, laboratory-reported yields of formic acid and acetic acid from VOC photooxidation are typically below 15 % (Baltensperger et al., 2005; Berndt and Böge, 2001; Paulot et al., 2011; Yuan et al., 2015). Notably, most of these yields were measured under relatively low •OH exposures ([•OH] × residence time), corresponding to less than one equivalent atmospheric photochemical day (Praplan et al., 2014). However, recent studies have revealed that molecular fragmentation reactions become increasingly important with oxidative aging (Isaacman-VanWertz et al., 2018; Lambe et al., 2012; Ortega et al., 2013), potentially enhancing the formation of small oxygenated species. Consistent with this hypothesis, substantial increases in gas-phase organic acids have been observed during the aging of biomass-burning emissions (Bruns et al., 2017; Ortega et al., 2013; Permar et al., 2023) and diesel exhaust (Friedman et al., 2017). For instance, Bruns et al. (2017) reported that formic acid concentrations increased by factors of ∼ 5–50 with aging. Friedman and Farmer (2018) further demonstrated that the molar yield of organic acids strongly depends on •OH concentrations during the photooxidation of monoterpenes. These findings suggest that the persistent model–observation discrepancy may be owing to an incomplete mechanistic and quantitative understanding of organic acid formation during multi-generational oxidation under atmospherically relevant conditions.
An oxidation flow reactor (OFR) provides a powerful complement to traditional environmental chambers by enabling the simulation of oxidation equivalent to multiple days of atmospheric aging within residence times of seconds to minutes through the use of elevated oxidant concentrations (Lambe et al., 2015). A key advantage of the OFR is its ability to achieve high photochemical exposure while minimizing wall losses of gases and particles, which can be substantial in Teflon chambers due to the typically long residence times employed (Brune, 2019; Zhang et al., 2014). The validity of OFRs has been demonstrated in numerous studies (Lambe et al., 2015; Peng et al., 2015), and they are increasingly used to investigate the effects of photochemical aging on gas-phase chemistry (Friedman and Farmer, 2018; Lambe et al., 2015), secondary organic aerosol (SOA) formation. (Bruns et al., 2015), and aerosol optical properties (Lambe et al., 2013). In this study, we employ an OFR to investigate the formation of formic acid and acetic acid during the photooxidation of toluene, a canonical and highly abundant anthropogenic VOC, over a wide range of •OH exposures, and examine how these processes are affected by relative humidity (RH), initial toluene concentration, and NOx concentrations. By combining laboratory experiments with model simulations, we further explore the underlying mechanisms for the formation of formic acid and acetic acid.
2.1 Oxidation flow reactor
Toluene photooxidation experiments were conducted in a custom-built quartz oxidation flow reactor (OFR) with a total volume of 2 L (1 m in length and 5 cm inner diameter; Fig. 1). The reactor temperature was maintained at 298 ± 1 K by a continuously circulating water jacket. No systematic temperature increase was observed with increasing lamp number or •OH exposure, even when all eight lamps were illuminated. Low-pressure mercury lamps (primary emission at 254 nm) were evenly distributed around the reactor, with up to eight lamps operating simultaneously. The •OH radicals in the OFR were generated via the reaction O(1D) + H2O → 2•OH, where O(1D) was produced by the photolysis of externally introduced O3. O3 was generated upstream of the OFR using a custom-built O3 generator in which O2 was irradiated at 185 nm. Photons at 185 nm were effectively filtered by the quartz lamp sleeves, the quartz reactor walls, and the water jacket, thereby minimizing in situ O3 formation inside the OFR (< 10 ppbv). A movable sampling port (6 mm outer diameter) enabled gas and particle sampling at different axial positions along the OFR. The •OH exposure within the reactor was determined using the box model described in Sect. 2.3. At 20 % RH, the modeled •OH exposures ranged from 2.03 × 1010 to 1.97 × 1012 molecules cm−3 s, corresponding to 0.07–6.34 d of equivalent atmospheric aging assuming a 24 h average •OH concentration of 3.6×106 molecules cm−3 in urban Beijing (Tan et al., 2018). At 70 % RH, the equivalent atmospheric aging times in the experiments ranged from 0.20 to 15.64 d.
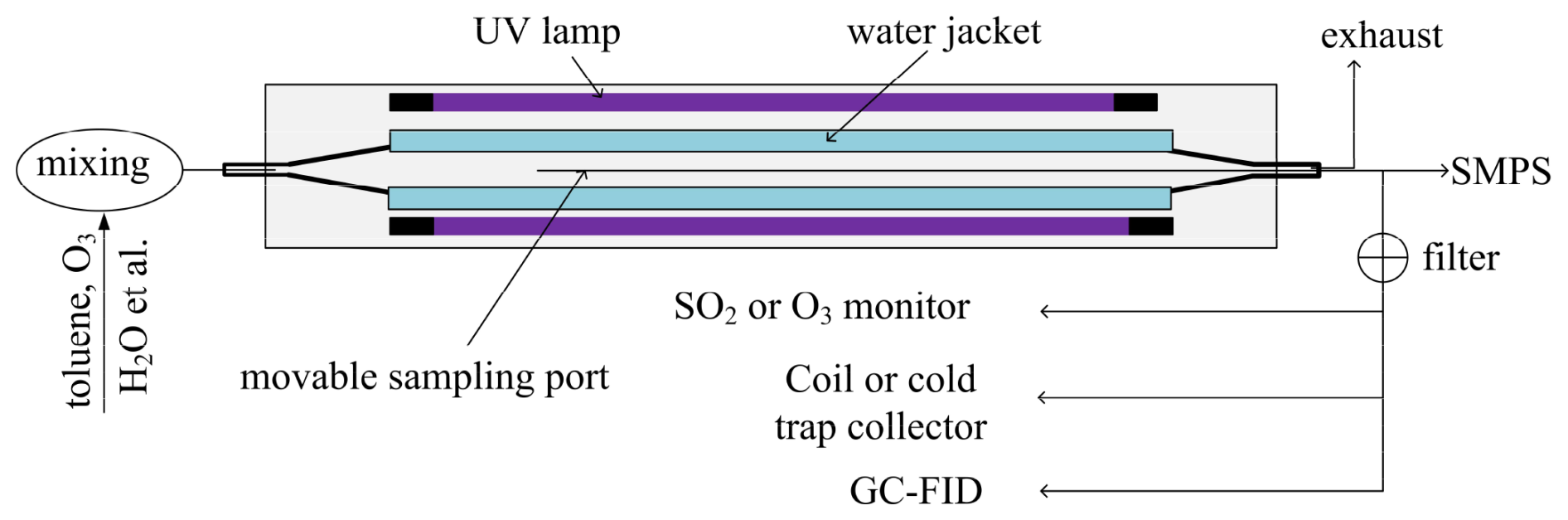
2.2 Experimental procedures
Toluene gas with an initial concentration of ∼94 ppbv was generated by passing a flow of N2 (99.999 %, Beijing Haipubeifen Gas) over liquid toluene (Merck, ≥ 99.9 %) in a temperature-controlled diffusion tube. RH in the OFR was adjusted by introducing water vapor generated using a bubbler. The gas mixture of toluene, O3, and water vapor was continuously introduced into the OFR at a total flow rate of 2 L min−1, and the initial concentration of O3 was about 1580 ppbv. Samples were collected at axial positions corresponding to 5 %, 25 %, 45 %, 65 %, and 85 % of the reactor length using the movable sampling port. Under laminar flow conditions, these positions corresponded to residence times of 3.9, 15.8, 27.5, 39.3, and 51.0 s, respectively. Additional experiments were conducted with 2, 4, 6, or 8 lamps to achieve higher •OH exposures. The influence of NOx on organic acid formation was investigated by adding N2O to the OFR. NO and NO2 were produced via the reaction O(1D) + N2O → 2NO, followed by NO + O3→ NO2+ O2 (Lambe et al., 2017). The NO concentration was systematically varied by adjusting the N2O flow rate to 0, 20, 40, 80, 120, 160, and 200 mL min−1, corresponding to N2O mixing ratios of 0 %, 1 %, 2 %, 4 %, 6 %, 8 %, and 10 %, respectively. All NOx experiments were conducted using a single lamp, with sampling at the reactor outlet corresponding to a residence time of 60 s.
Toluene concentrations were quantified using gas chromatography with a flame ionization detector (GC-FID, Agilent 7890A) (Wu et al., 2017). O3 and SO2 were measured using an O3 analyzer (model 202, 2B Technologies, USA) and an SO2 analyzer (model 43i-TLE, Thermo), respectively. Particle number size distributions were measured using a scanning mobility particle sizer (SMPS, DMA 3081 connected to CPC 3776, TSI Inc.). Carbonyl compounds were collected using the cold trap method (Huang et al., 2013) and analyzed by high-performance liquid chromatography (HPLC) with ultraviolet detection (Shen et al., 2018, 2024). All experiments were conducted in the absence of seed particles. Gas-phase organic acids were collected using a temperature-controlled scrubbing glass coil maintained at 277 K and extracted with ultrapure water as the stripping solution (Wu et al., 2017; Xing et al., 2018). Gas and stripping solution flow rates were 0.6 L min−1 and 0.2 mL min−1, respectively. The sample collection efficiencies were measured as approximately 85 % for acetic acid, 95 % for formic acid, and 99 % for pyruvic acid. The eluent was collected for 15 min, and then the sample was analyzed immediately using ion chromatography (DIONEX ICS–2000) (Shen et al., 2018). The concentrations of organic acids were quantified using a mixed liquid standard solution containing formic acid, acetic acid, and pyruvic acid. To evaluate potential artifacts associated with aqueous-phase formation in the stripping solution, five additional gas-phase samples were collected using Horibe tubes with an ethanol cold trap maintained at 183 K. Concentrations measured using the scrubbing coil and Horibe tubes agreed within 10 %, indicating that aqueous-phase artifacts had a negligible influence on measurements. The molar yields of formic acid and acetic acid were calculated relative to the amount of toluene consumed at each sampling position, as shown in Eq. (1):
where Δ[organic acid] is the concentration of formic acid or acetic acid (ppbv) produced at a given sampling position, and Δ[toluene] is the corresponding amount of toluene consumed (ppbv) at the same position. It should be noted that the reported yields are apparent yields and were not corrected for the subsequent reactions of organic acids with •OH.
A series of control experiments was conducted to ensure data quality and reproducibility. (1) Before each experiment, high concentrations of O3 were introduced into the OFR with two lamps illuminated until particle concentrations measured by the SMPS were negligible (< 50 cm−3 or 0.1 µg m−3) and no impurities were detected by GC-FID. (2) O3 photolysis experiments were performed before each toluene photooxidation experiment to verify consistent irradiation conditions based on comparable O3 decay rates. (3) Blank experiments were conducted without toluene, with toluene but without O3, and with both toluene and O3 but without water vapor. Formic and acetic acid concentrations observed in these blank experiments were low relative to those measured during toluene photooxidation (< 15 %). All reported data were corrected for blank contributions. In addition, the wall loss rates of formic acid and acetic acid were measured to be less than 5 % of their respective •OH reaction rates under our experimental conditions and were therefore not corrected for in the yield calculations.
2.3 Model description and determination of photon flux
A zero-dimensional box model based on the Master Chemical Mechanism (MCM) v3.3.1 (https://mcm.york.ac.uk/MCM, last access: 15 July 2026) and modified according to previous work (Li et al., 2015) was employed to simulate toluene photooxidation and organic acid formation in the OFR (Jenkin et al., 2015). Rate coefficients for additional reactions were adopted from the latest JPL kinetic evaluation (Li et al., 2015). Photolysis cross sections and quantum yields were taken from Keller-Rudek et al. (2013). The detailed chemical reactions added to the model and their corresponding rate constants are provided in Table S1 in the Supplement. The initial concentrations of toluene, O3, water vapor, and N2O, together with temperature, residence time, and J(O3→O(1D)), were set according to the experimental conditions summarized in Table S2.
Accurate quantification of the photon flux in the OFR is essential for determining photolysis rates in the model. Direct measurement of photon flux is challenging due to attenuation by the water jacket and quartz reactor walls. Therefore, photon flux in the OFR was determined by combining model simulations with direct measurements of O3 decay from experiments conducted in the absence of toluene. The photon flux was iteratively adjusted in the model until the simulated O3 decay rate matched the observed decay. Using this approach, the photon flux with one lamp illuminated was estimated to be ∼ 2.9×1015 photons cm−2 s−1, which is consistent with photon fluxes commonly used in OFR studies (Peng et al., 2016). Photon fluxes corresponding to different numbers of illuminated lamps (up to eight) were determined using the same method, and the results are shown in Fig. S1. The validity of this approach for photon flux determination, as well as the applicability of the box model to the OFR system, was further evaluated by comparing simulated and measured SO2 decay rates (Fig. S2). The modeled •OH exposures under different experimental conditions are summarized in Table S3, and their comparison with •OH exposures derived from SO2 decay and toluene decay is shown in Fig. S3. It should be noted that although both the O3 concentration and the 254 nm photon flux in the OFR substantially exceed typical ambient levels, •OH concentration is enhanced to an even greater extent, ensuring that •OH-initiated reactions remain the dominant oxidation pathway. The relative contributions of O3 reactions and direct 254 nm photolysis were evaluated by comparing their pseudo-first-order loss rates with the corresponding •OH reaction rates, as displayed in Figs. S4 and S5.
3.1 Production of organic acids in toluene photooxidation
3.1.1 •OH Exposure dependence
The major gas-phase organic acids observed during the photooxidation of toluene were formic acid, acetic acid, and pyruvic acid, with minor contributions from lactic acid and oxalic acid at both 20 % and 70 % RH. Given that formic acid and acetic acid together accounted for approximately 90 % of the total measured gas-phase organic acids, the following analysis focuses primarily on these two species. Figure 2 illustrates the dependence of the concentrations and molar yields of formic acid and acetic acid on •OH exposure. As •OH exposure increased, their concentrations initially increased and subsequently decreased, reaching maxima at approximately 1–3 equivalent days under both RH conditions. The similar trends observed for formic acid and acetic acid suggest that their formation is governed by closely related chemical pathways during toluene photooxidation. The decrease in organic acid concentrations at higher •OH exposure is likely due to kinetic competition between organic acid formation and subsequent acid degradation (including direct reaction with •OH and depletion of their precursors) as •OH exposure increases. At the highest •OH exposure investigated (15.64 equivalent days), the concentrations of both acids decreased to about 25 % of their respective peak values. As shown in Fig. 2a and c, the evolution of both formic acid and acetic acid concentrations with increasing •OH exposure at 20 % RH is similar to that at 70 % RH. However, it is found that organic acid concentrations were consistently higher at low RH (20 %) than at high RH (70 %) under identical •OH exposures, suggesting that reactions involving water vapor may play a limited role in organic acid formation in this study, in contrast to the substantial contribution of stabilized Criegee intermediate–water reactions commonly observed during alkene ozonolysis (Kang et al., 2025; Neeb et al., 1997; Orzechowska and Paulson, 2005).
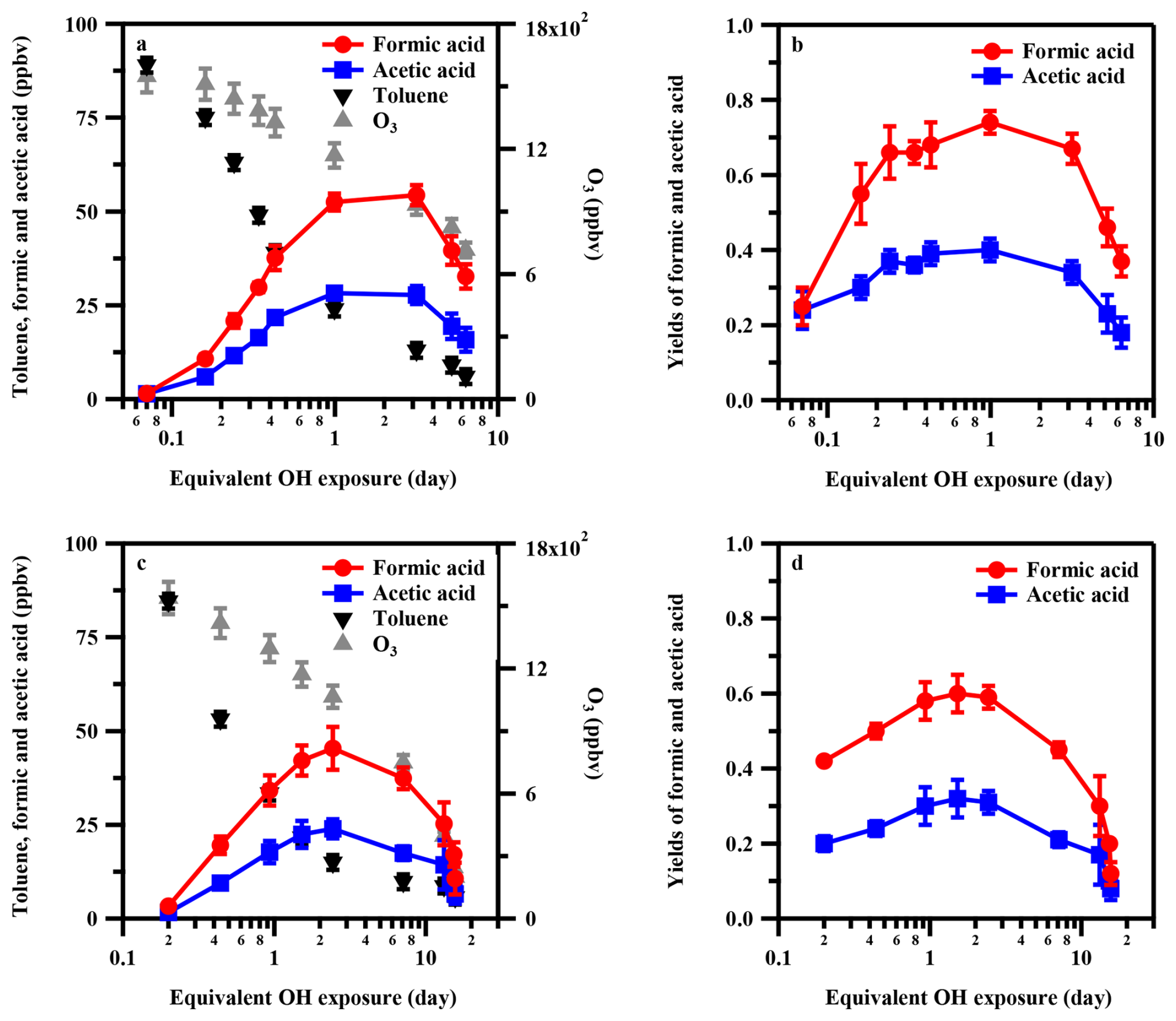
Figure 2Production of formic acid and acetic acid during the photooxidation of toluene as a function of •OH exposure. Panels (a) and (b) correspond to 20 % RH, and panels (c) and (d) correspond to 70 % RH. The initial toluene concentration was ∼ 94 ppbv. Error bars represent standard deviations of repeated experiments under identical conditions. Note that the first data point in each panel corresponds to a condition where measurable toluene consumption has already occurred, with •OH exposures of 0.07 equivalent days at 20 % RH and 0.2 equivalent days at 70 % RH, respectively.
Figure 2b and d show that the molar yields of formic acid and acetic acid strongly depended on •OH exposure at both 20 % and 70 % RH. The yields increase from 25 % (formic acid) and 24 % (acetic acid) at low •OH exposures (< 0.2 equivalent days) to maximum values of 74 % and 40 %, respectively, at intermediate •OH exposures (1–3 equivalent days). Previous studies have shown that molecular fragmentation reactions become increasingly important under conditions of elevated •OH exposure (Lambe et al., 2012; Ortega et al., 2013), conditions that are difficult to achieve in traditional environmental chamber experiments that typically span less than one equivalent day of atmospheric aging. The pronounced increase in organic acid yields with •OH exposure suggests that molecular fragmentation processes likely play an important role in formic acid and acetic acid formation.
We further compared the yields of formic acid and acetic acid measured from the photooxidation of toluene with literature results. Seuwen and Warneck (1996) quantified the yield of formic acid during toluene oxidation at approximately 13 %, which is ∼ 5 times lower than the peak value observed in our study. In addition to toluene, the yields of formic and acetic acid produced from other aromatic compounds reported in previous studies were also lower than our results (Bandow et al., 1985; Bandow and Washida, 1985; Berndt et al., 1999; Gery et al., 1985; Müller et al., 2012; Wyche et al., 2009). For example, Berndt et al. (1999) reported a formic acid yield of ∼ 13 % from benzene oxidation, and Wyche et al. (2009) observed a formic acid yield of 14 % from 1,3,5-trimethylbenzene under high-NOx conditions (Berndt et al., 1999; Wyche et al., 2009). This discrepancy may be ascribed to the differences in •OH exposure levels. Previous chamber studies were typically conducted under •OH exposures of < 0.1 equivalent days (Seuwen and Warneck, 1996; Wyche et al., 2009), which are substantially lower than those (1–3 equivalent days) at which peak organic acid yields were observed in our study. Under such limited •OH exposures, primary oxidation products dominate, whereas formic and acetic acids likely form as secondary or later-generation products requiring higher •OH exposures (Drozd et al., 2019; Isaacman-VanWertz et al., 2018; Lambe et al., 2012). Evidence supporting the role of higher •OH exposures in organic acid formation comes from two previous studies. Wyche et al. (2009) observed formic acid and acetic acid yields of 49 % and 33 %, respectively, during the photooxidation of 1,3,5-trimethylbenzene under low-NOx conditions, where the aromatic precursor was nearly fully consumed. Similarly, Murschell and Farmer (2018) reported formic acid yields as high as 43 % during the oxidation of the chlorinated aromatic herbicide 2-methyl-4-chlorophenoxyacetic acid under very high •OH exposures. Consistent with these findings, combustion studies, which effectively accelerate atmospheric oxidation, also reported enhanced formation of formic acid and acetic acid from toluene relative to non-aromatic compounds such as propane and isooctane (Battin-Leclerc et al., 2007; Zervas, 2005). Additionally, previous experiments often used very high concentrations of aromatic compounds (hundreds of ppbv to ppmv levels), which may suppress •OH concentrations and limit effective oxidative aging. Our experiments similarly show that increasing toluene concentrations suppressed the formation of organic acids (Fig. S6). At 20 % RH, the yield of formic acid decreased from 74 % to 48 % as the initial toluene concentration increased from 94 to 381 ppbv. Together, these results highlight the critical role of •OH exposure in controlling organic acid formation during aromatic oxidation. Further discussion of these yield discrepancies is provided in Sect. 3.2 and 3.3.
3.1.2 NOx Dependence
Small organic acids, such as formic acid and acetic acid, are commonly thought to form via reactions of peroxyacyl radicals with HO2 RO2 radicals (HO2• RO2•). The presence of NOx is expected to suppress this pathway by preferentially reacting with peroxy radicals, diverting them toward NO reaction channels that favor nitrate and carbonyl formation over organic acid production. To investigate the influence of NOx on organic acid formation, N2O was introduced into the OFR to generate NOx, resulting in NO concentrations of 0–29 ppbv. It should be noted that NO and NO2 were produced simultaneously in our experiments (via O(1D) + N2O → 2NO, followed by NO + O3→ NO2+ O2), and model simulations indicate that their concentrations were strongly correlated (R2= 0.98). NO primarily reacts with RO2• to form alkoxy radicals (RO•) (Jenkin et al., 2019; Peng et al., 2019), which can divert the traditional organic acid formation pathway toward carbonyl products. NO2 can react with peroxyacyl radicals to form peroxyacyl nitrates (PANs), potentially sequestering acyl peroxy intermediates, and can also react with •OH, thereby affecting •OH concentration. As shown in Fig. 3, molar yields of formic acid and acetic acid were not significantly affected by NO concentration at either 20 % or 70 % RH. Even at the highest NO level (∼ 29 ppbv at 20 % RH), where the modeled NO : HO2• ratio approached 1000 (Table S4), substantial production of organic acids (∼ 40 % for formic acid and ∼ 20 % for acetic acid at 20 % RH) was still observed. If organic acid formation were dominated by the peroxy radical pathway, such high NO : HO2• ratios would be expected to substantially suppress their formation. However, at 70 % RH, organic acid yields even increased slightly with increasing NO concentrations. These results suggest that neither the NO-mediated RO2• diversion pathway nor the NO2-related pathways significantly control organic acid production.
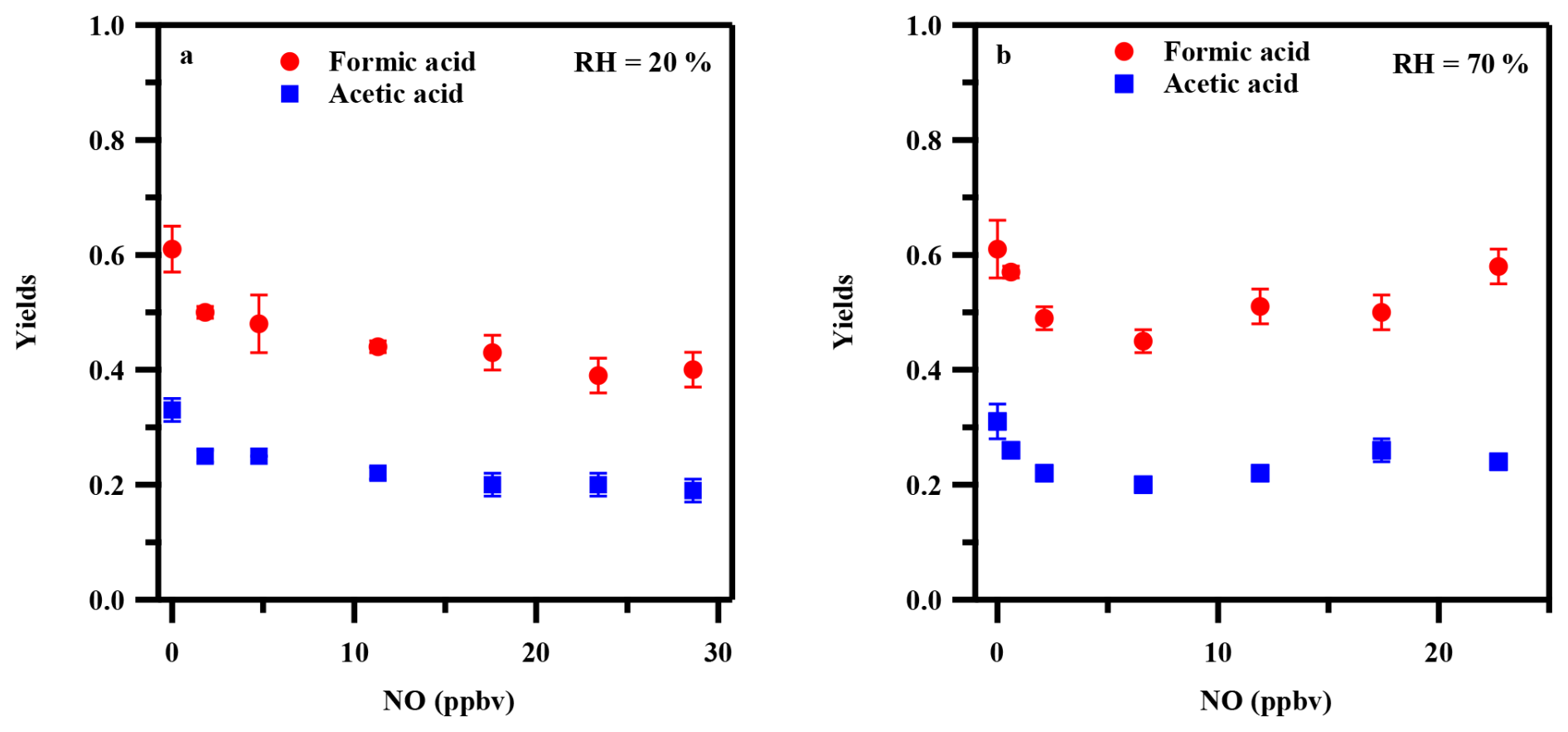
Figure 3NOx dependence of formic acid and acetic acid yields under 20 % RH (a) and 70 % RH (b) conditions. Error bars represent standard deviations from repeated experiments under identical conditions.
The limited suppression by NOx therefore supports the presence of a NOx-insensitive pathway for organic acid production during toluene photooxidation. Moreover, the persistence of high organic acid yields despite the substantial decrease in simulated •OH concentration from 2.92×109 to 1.80×108 molecules cm−3 and the corresponding decrease in toluene consumption from ∼ 60 to ∼ 25 ppbv with increasing N2O addition (Table S4 and Fig. S7) further supports this conclusion and suggests that NO may even facilitate organic acid production through enhanced alkoxy radical formation and subsequent fragmentation. This finding is consistent with field observations of rapid organic acid formation in polluted environments, where NOx levels are typically elevated (Liggio et al., 2017; Millet et al., 2015; Veres et al., 2011; Yuan et al., 2015), and offers a plausible explanation for the persistent underestimation of organic acid concentrations in atmospheric models under both pristine and polluted conditions (Bannan et al., 2017; Khan et al., 2018; Stavrakou et al., 2012; Yuan et al., 2015).
3.2 Organic acid yields simulated by chemical models
To further assess the ability of current chemical mechanisms to reproduce observed organic acid formation, we compared our experimental results with simulations using the MCM v3.3.1 model. For acetic acid, two formation pathways are included in MCM v3.3.1 (Reactions R1 and R2):
Model simulations indicate that Reaction (R2) (CH3CO3• + RO2•) dominates acetic acid production (> 90 %). While the model can reproduce the general trend of acetic acid concentrations, which initially increase and then decrease with •OH exposure, it substantially underestimates the observed values. The maximum modeled yield (∼ 7 % at ∼ 3 equivalent days) is roughly five times lower than the measured yields under both RH conditions (Fig. 4a–b). This discrepancy suggests that additional formation pathways, currently absent from the model, may significantly contribute to acetic acid production during toluene photooxidation.
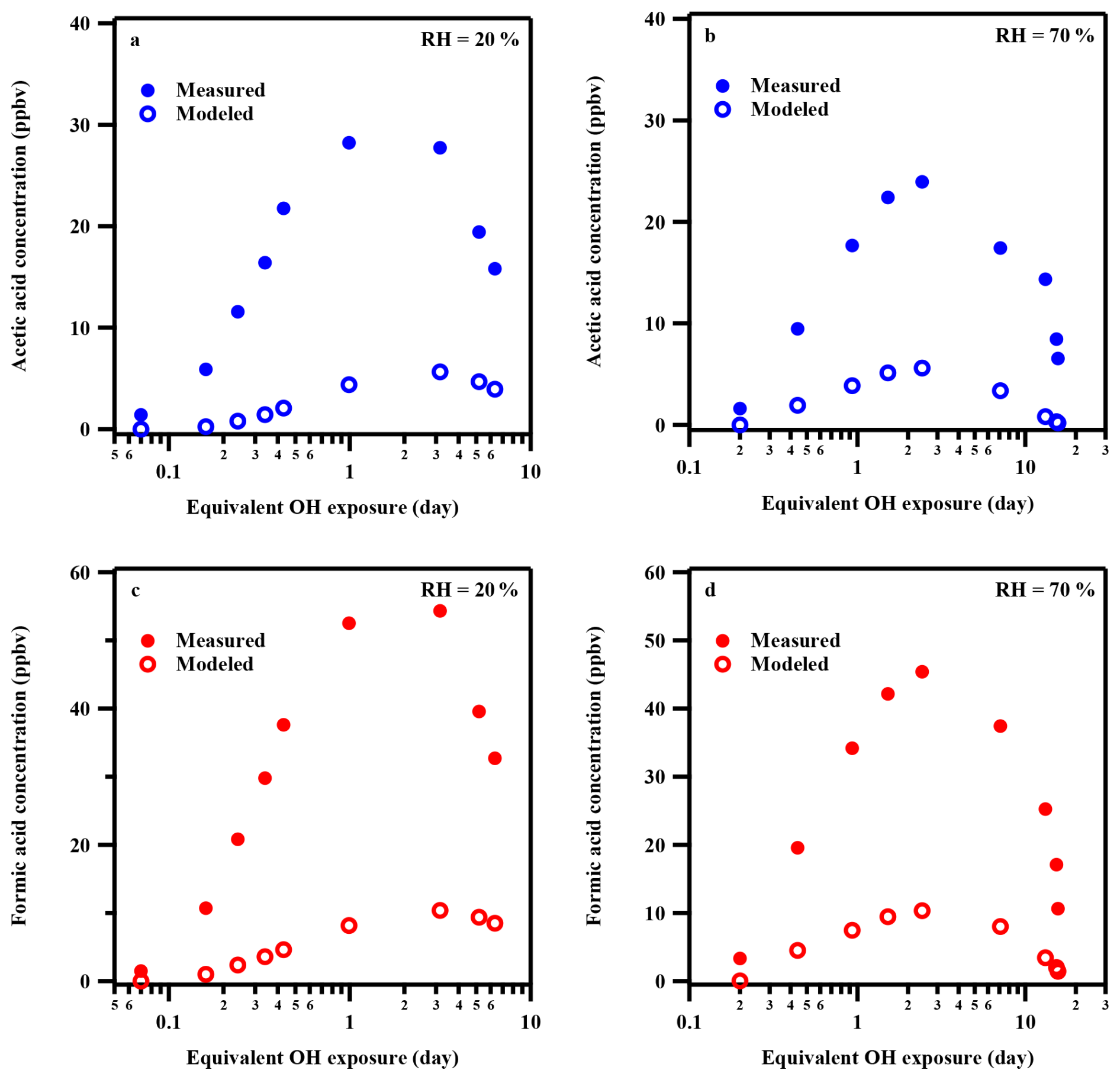
Figure 4Comparison of measured and modeled concentrations of formic acid and acetic acid at different •OH exposures. (a) Acetic acid at 20 % RH; (b) Acetic acid at 70 % RH; (c) Formic acid at 20 % RH; (d) Formic acid at 70 % RH.
It should be noted that formic acid is not considered a product of •OH oxidation of aromatic compounds, including toluene, in the current MCM v3.3.1 model. Therefore, before model simulation, we implemented four candidate pathways based on previous studies (Archibald et al., 2007; Fittschen et al., 2014; Shaw et al., 2018; Yetter et al., 1989; Yuan et al., 2015). (1) HOCH2OO• chemistry: Analogous to the acetic acid mechanism, formic acid can also be formed from either the reaction of HOCH2OO• with HO2• or the bimolecular reaction of HOCH2OO• (Jenkin et al., 2008; Yuan et al., 2015). (2) Vinyl alcohol (CH2=CHOH) oxidation: Formic acid formation via •OH-initiated oxidation of vinyl alcohol, which is generated through acetaldehyde tautomerization. Both photo-induced and organic-acid-catalyzed tautomerization processes were included (Archibald et al., 2007; Shaw et al., 2018; Yuan et al., 2015). (3) HCHO + •OH reaction: Direct formation through HCHO + •OH → HCOOH + H, with a rate coefficient of 2.0× 10−13 cm3 molecule−1 s−1 (Yetter et al., 1989). (4) CH3O2• + •OH reaction: Formic acid formation via CH3O2• + •OH → HCOOH + H2O, with a very high rate coefficient of cm3 molecule−1 s−1 (Fittschen et al., 2014). It should be noted that the branching ratio for formic acid formation from this reaction remains uncertain; thus, a yield of 100 % was assumed in our model, representing an upper-limit estimation. Model evaluation results suggest that pathways 1–3 contribute minimally (< 1 %). Pathway 1 (HOCH2OO• chemistry) exhibits a much lower yield than the analogous acetic acid mechanism due to the low stability and consequently low concentration of HOCH2OO• relative to CH3CO3 radicals. Pathways 2 and 3 involve aldehydes as precursors, which also limit formic acid production. Only pathway 4 (CH3O2• + •OH) can produce a substantial amount of formic acid, with a maximum modeled yield of ∼ 13 % at ∼ 3 equivalent days. Even under this upper-limit assumption, the modeled concentrations of formic acid are still approximately five times lower than the experimental measurements (Fig. 4c–d). It is noted that pathway 4 is highly sensitive to NOx. Given that our experimental results show that organic acid formation is insensitive to NOx levels, this pathway is unlikely to be the dominant mechanism for organic acid production. Overall, these results indicate that known pathways account for only a small fraction of the observed formic and acetic acid production, implying the existence of additional, yet unidentified, formation mechanisms.
3.3 Organic acids formed from multi-generational oxidation
Previous modeling studies have suggested that the aging of SOA could significantly contribute to the atmospheric budgets of organic acids (Ervens et al., 2011; Paulot et al., 2011), and laboratory experiments have confirmed the production of organic acids during SOA photodegradation (Jiang et al., 2023; Malecha and Nizkorodov, 2016, 2017). In this study, we analyzed the relationship between SOA particle mass and organic acid production to evaluate this potential pathway. However, the distinctly different temporal profiles of gas-phase organic acid production and SOA formation (Fig. 5) imply that SOA photodegradation is not the primary source of the observed high organic acid yields. First, SOA formation exhibited a delay relative to the rapid production of organic acids, consistent with the “incubation period” for aromatic SOA formation (Ng et al., 2007; Wyche et al., 2009). Substantial organic acid yields were observed even when the SOA mass concentration was negligible (Figure 5a), effectively ruling out SOA photodegradation as the primary source. Second, as gas-phase organic acid concentrations began to decline, SOA mass continued to increase (Fig. 5), indicating decoupled formation and removal pathways for SOA and organic acids. At very high •OH exposures (Fig. 5b), organic acid concentrations decreased while the SOA mass remained nearly unchanged. This suggests that gas-phase organic acids are more reactive toward gas-phase oxidants than the more persistent SOA, consistent with the generally longer lifetimes of aerosol-phase organics against oxidation (Isaacman-VanWertz et al., 2018). Although SOA photodegradation can produce small oxygenated VOCs (including formic acid and acetic acid), previous studies showed that aromatic-derived SOA generates much lower yields of such products compared to biogenic SOA (e.g., derived from isoprene or α-pinene) (Malecha and Nizkorodov, 2016). It should be noted that this conclusion is specific to toluene-derived SOA under our experimental conditions. In the real atmosphere, SOA formed from other precursors (e.g., biogenic VOCs or biomass burning emissions) may exhibit substantially higher organic acid yields upon photodegradation, and the contribution of aerosol-phase chemistry to the overall organic acid budget cannot be excluded based on our results alone. Additionally, it is worth noting that a greater aerosol mass was formed at 70 % RH than at 20 % RH under comparable •OH exposures (Fig. 5). This enhanced SOA formation at higher humidity may partially account for the lower organic acid yields observed under high-RH conditions. Specifically, greater partitioning of water-soluble intermediates into the particle phase could reduce their availability for gas-phase fragmentation into organic acids, and enhanced peroxide formation may divert intermediates toward non-acid-producing pathways.
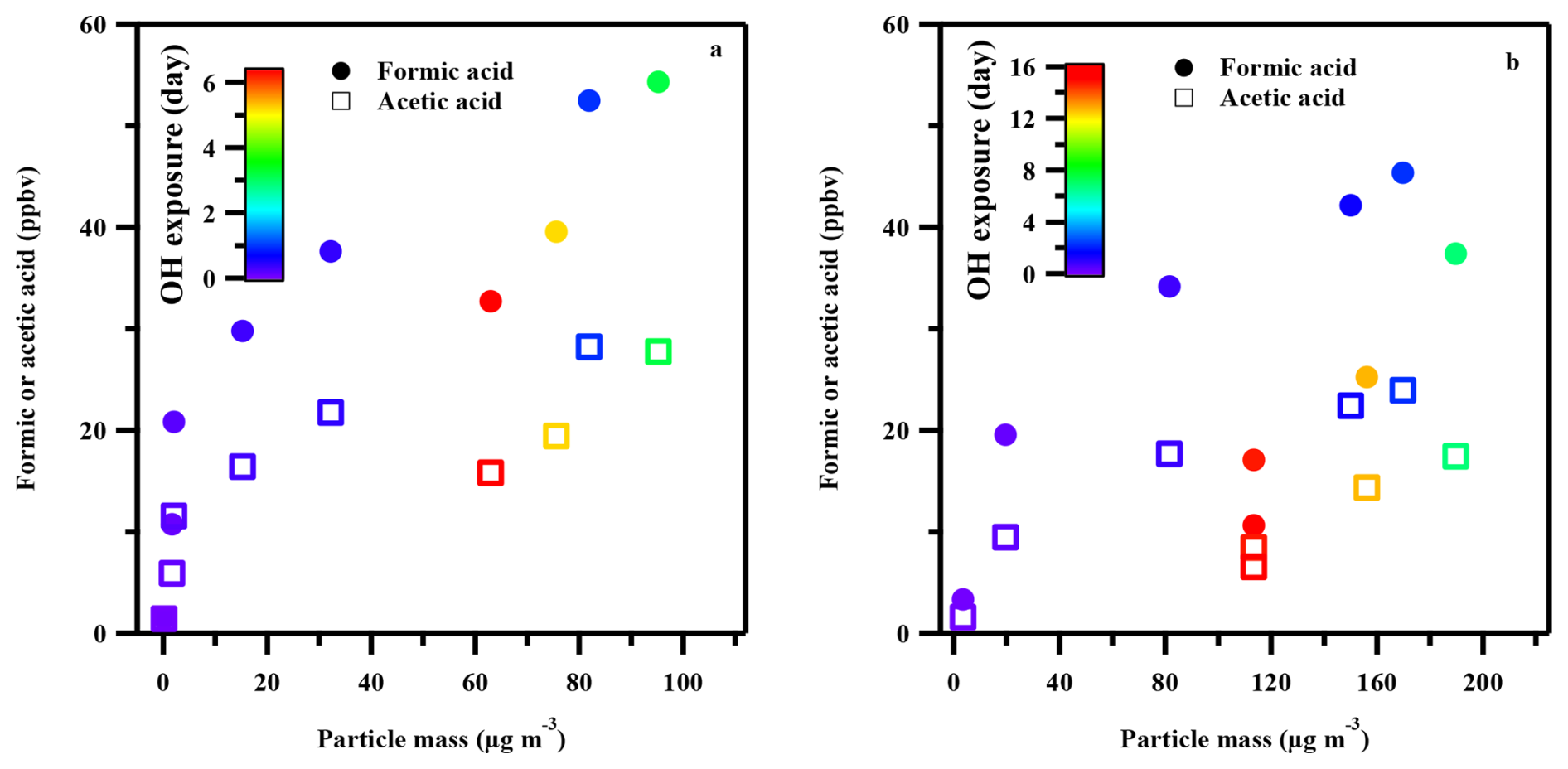
Figure 5Correlations of particle mass concentration with gas-phase formic acid and acetic acid during the photooxidation of toluene at different •OH exposure. (a) 20 % RH; (b) 70 % RH. Marker color represents different •OH exposure levels.
To further investigate the origin of the high organic acid yields observed during toluene photooxidation, we first examined key first-generation ring-opened products, namely glyoxal and methylglyoxal, which are well-known primary products in aromatic photooxidation (Volkamer et al., 2001). In our experiments, the molar yields of these carbonyl compounds at low •OH exposure were 11 % and 10 %, respectively, consistent with results reported in previous studies (Bandow et al., 1985; Gery et al., 1985). Figure 6 shows that the yields of glyoxal and methylglyoxal decreased rapidly with increasing •OH exposure, dropping to approximately 1 % at higher exposures. In contrast, the yields of formic acid and acetic acid showed distinct maxima at intermediate •OH exposures (1–3 equivalent days). Previous studies have proposed that oxidation of glyoxal and methylglyoxal can contribute to formic acid and acetic acid formation (Paulot et al., 2011). However, these pathways are unlikely to dominate given the substantially lower yields of glyoxal and methylglyoxal observed. Moreover, the poor linear correlation between the yields of formic acid and acetic acid and the first-generation ring-opened products suggests that these acids are secondary or later-generation products formed through multistep oxidation processes instead of being the primary oxidation products. A previous study suggested that the fragmentation and further oxidation of five-membered oxygen-containing heterocyclic compounds, which are important intermediates in the oxidation of aromatic compounds, can yield acetic acid (Bahreini et al., 2005; Forstner et al., 1997; Walavalkar et al., 2017). However, this pathway is still controversial due to the lack of robust evidence. The major precursors and detailed mechanisms underlying the multigenerational oxidation that results in the high yields of formic acid and acetic acid during the photooxidation of toluene warrant further investigation.
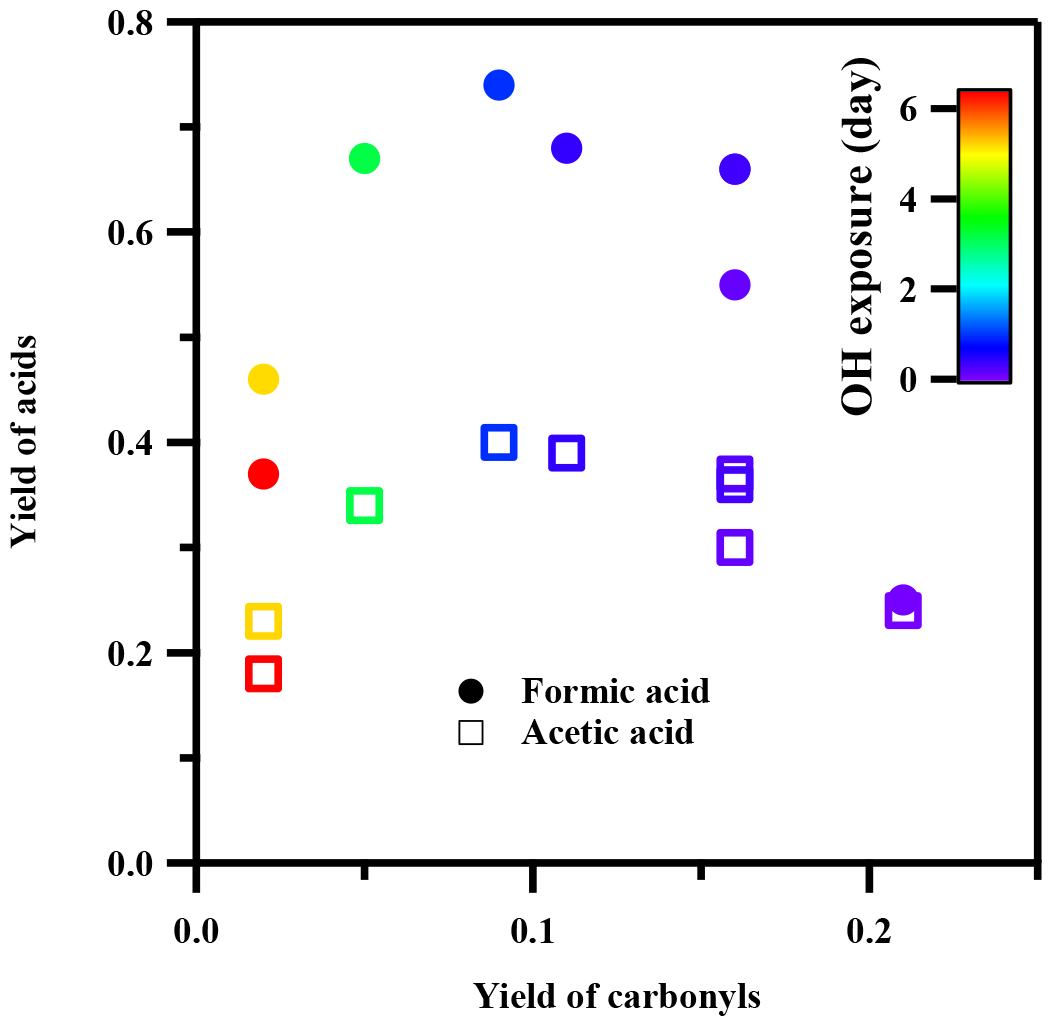
Figure 6Correlations of yields of carbonyl compounds (sum of glyoxal and methylglyoxal) with yields of gas-phase formic acid and acetic acid during the photooxidation of toluene at different •OH exposures (20 % RH). Markers are color-coded to represent different •OH exposure levels.
This study reveals unexpectedly high yields of formic acid (up to 74 %) and acetic acid (up to 40 %) during the multi-generational photooxidation of toluene, achieved over equivalent atmospheric aging times of 1–3 d in the OFR experiments. The formation of these organic acids exhibited minimal sensitivity to NOx concentrations and RH. The up-to-date MCM v3.3.1 model substantially underestimated the observed organic acid concentrations in the OFR. Although both small gas-phase organic acids and low-volatility particulate matter increased with atmospheric aging (within a certain degree of oxidation), their differing production timescales suggest that photodegradation of SOA may not be the dominant formation pathway for organic acids. Combined with measurements of carbonyl compounds, our results suggest that the observed organic acids predominantly arise from the multi-generational oxidation of toluene. A mechanistic understanding of this process, particularly the photooxidation of intermediate products such as maleic anhydride, requires further investigation (Lu et al., 2024; Wang et al., 2024).
Our findings also provide new insight into understanding organic acid budgets in urban atmospheres. Previous studies have observed rapid secondary production of organic acids in urban air masses (Friedman and Farmer, 2018; Veres et al., 2011), but model simulations have significantly underestimated their concentrations (Le Breton et al., 2012; Yuan et al., 2015). If the high yields of organic acids observed during the multi-generational oxidation of toluene are broadly applicable to other aromatics, the elevated yields of formic and acetic acids at high •OH exposures reported in this study may help explain these discrepancies. Liggio et al. (2017) found that the underestimation of organic acids increased with reaction time, while Yuan et al. (2015) reported that the peak concentrations of formic acid occurred 4–6 d after the peak benzene levels (Liggio et al., 2017; Yuan et al., 2015). These studies evaluated the contributions of aromatics to formic acid, assuming a yield of 15 %, concluding that aromatics can contribute about 11 %–12 % of the total formic acid production. If a yield of 74 % is adopted, the contributions of aromatics would rise to as high as 55 %–60 %. This outcome may explain the remaining 50 % of missing sources in the model after known sources are considered (Liggio et al., 2017; Yuan et al., 2015). The large amounts of small gas-phase organic acids produced from the photooxidation of aromatics in urban areas may further affect aerosol pH and aerosol formation. It is noted that biomass burning emissions also contain substantial amounts of aromatic compounds, and multi-generational oxidation within biomass burning plumes may similarly produce significant quantities of organic acids, as supported by both laboratory experiments and field observations (Bruns et al., 2017; Permar et al., 2023). Additionally, while small organic acids such as formic and acetic acid have long been recognized as useful tracers for atmospheric VOC oxidation and SOA aging (Hansen et al., 2014; Munger et al., 1986), our results suggest that their utility as reliable tracers for SOA production is limited to a relatively narrow •OH exposure window (approximately 0.2–3 equivalent days). Beyond this range, their concentrations decouple from SOA mass due to differences in reactivity and removal pathways (Isaacman-VanWertz et al., 2018).
The data used in this study are available upon request from the corresponding authors.
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-10029-2026-supplement.
ZC, LH, and HS designed the research. HS performed the experiments, analyzed the data, and drafted the initial manuscript. LH and ZC revised the manuscript. MZ and YY assisted with the model simulation. YZ, HL, and HW provided valuable comments and suggestions for the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We sincerely thank the three anonymous reviewers for their constructive comments and suggestions during the review process, which substantially improved the quality of this manuscript. We are also grateful to Prof. Zhe Peng from Jinan University for providing the program used to evaluate the relative importance of ozone, photolysis, and OH radical pathways as presented in Figs. S4 and S5.
This research has been supported by the National Key Research and Development Program of China (grant no. 2022YFC3701202).
This paper was edited by Kelvin Bates and reviewed by three anonymous referees.
Archibald, A. T., McGillen, M. R., Taatjes, C. A., Percival, C. J., and Shallcross, D. E.: Atmospheric transformation of enols: A potential secondary source of carboxylic acids in the urban troposphere, Geophys. Res. Lett., 34, 2007GL031032, https://doi.org/10.1029/2007GL031032, 2007.
Bahreini, R., Keywood, M. D., Ng, N. L., Varutbangkul, V., Gao, S., Flagan, R. C., Seinfeld, J. H., Worsnop, D. R., and Jimenez, J. L.: Measurements of Secondary Organic Aerosol from Oxidation of Cycloalkenes, Terpenes, and m-Xylene Using an Aerodyne Aerosol Mass Spectrometer, Environ. Sci. Technol., 39, 5674–5688, https://doi.org/10.1021/es048061a, 2005.
Baltensperger, U., Kalberer, M., Dommen, J., Paulsen, D., Alfarra, M. R., Coe, H., Fisseha, R., Gascho, A., Gysel, M., Nyeki, S., Sax, M., Steinbacher, M., Prevot, A. S. H., Sjögren, S., Weingartner, E., and Zenobi, R.: Secondary organic aerosols from anthropogenic and biogenic precursors, Faraday Discuss., 130, 265–278, https://doi.org/10.1039/B417367H, 2005.
Bandow, H. and Washida, N.: Ring-cleavage Reactions of Aromatic Hydrocarbons Studied by FT–IR Spectroscopy. III. Photooxidation of 1,2,3-, 1,2,4-, and 1,3,5-Trimethylbenzenes in the NOx–Air System, B. Chem. Soc. Jpn., 58, 2549–2555, https://doi.org/10.1246/bcsj.58.2549, 1985.
Bandow, H., Washida, N., and Akimoto, H.: Ring-cleavage Reactions of Aromatic Hydrocarbons Studied by FT–IR Spectroscopy. I. Photooxidation of Toluene and Benzene in the NOx–Air System, B. Chem. Soc. Jpn., 58, 2531–2540, https://doi.org/10.1246/bcsj.58.2531, 1985.
Bannan, T. J., Murray Booth, A., Le Breton, M., Bacak, A., Muller, J. B. A., Leather, K. E., Khan, M. A. H., Lee, J. D., Dunmore, R. E., Hopkins, J. R., Fleming, Z. L., Sheps, L., Taatjes, C. A., Shallcross, D. E., and Percival, C. J.: Seasonality of formic acid (HCOOH) in London during the ClearfLo campaign, J. Geophys. Res.-Atmos., 122, 12488–12498, https://doi.org/10.1002/2017JD027064, 2017.
Bates, K. H., Jacob, D. J., Cope, J. D., Chen, X., Millet, D. B., and Nguyen, T. B.: Emerging investigator series: aqueous oxidation of isoprene-derived organic aerosol species as a source of atmospheric formic and acetic acids, Environ. Sci.-Atmos., 3, 1651–1664, https://doi.org/10.1039/D3EA00076A, 2023.
Battin-Leclerc, F., Konnov, A. A., Jaffrezo, J. L., and Legrand, M.: To Better Understand the Formation of Short-Chain Acids in Combustion Systems, Combust. Sci. Technol., 180, 343–370, https://doi.org/10.1080/00102200701740782, 2007.
Berndt, T. and Böge, O.: Gas-phase reaction of OH radicals with benzene: products and mechanism, Phys. Chem. Chem. Phys., 3, 4946–4956, https://doi.org/10.1039/B106667F, 2001.
Berndt, T., Böge, O., and Herrmann, H.: On the formation of benzene oxide/oxepin in the gas-phase reaction of OH radicals with benzene, Chem. Phys. Lett., 314, 435–442, https://doi.org/10.1016/S0009-2614(99)01041-6, 1999.
Brune, W. H.: The Chamber Wall Index for Gas–Wall Interactions in Atmospheric Environmental Enclosures, Environ. Sci. Technol., 53, 3645–3652, https://doi.org/10.1021/acs.est.8b06260, 2019.
Bruns, E. A., El Haddad, I., Keller, A., Klein, F., Kumar, N. K., Pieber, S. M., Corbin, J. C., Slowik, J. G., Brune, W. H., Baltensperger, U., and Prévôt, A. S. H.: Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition, Atmos. Meas. Tech., 8, 2315–2332, https://doi.org/10.5194/amt-8-2315-2015, 2015.
Bruns, E. A., Slowik, J. G., El Haddad, I., Kilic, D., Klein, F., Dommen, J., Temime-Roussel, B., Marchand, N., Baltensperger, U., and Prévôt, A. S. H.: Characterization of gas-phase organics using proton transfer reaction time-of-flight mass spectrometry: fresh and aged residential wood combustion emissions, Atmos. Chem. Phys., 17, 705–720, https://doi.org/10.5194/acp-17-705-2017, 2017.
Chaliyakunnel, S., Millet, D. B., Wells, K. C., Cady-Pereira, K. E., and Shephard, M. W.: A Large Underestimate of Formic Acid from Tropical Fires: Constraints from Space-Borne Measurements, Environ. Sci. Technol., 50, 5631–5640, https://doi.org/10.1021/acs.est.5b06385, 2016.
Chebbi, A. and Carlier, P.: Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review, Atmos. Environ., 30, 4233–4249, https://doi.org/10.1016/1352-2310(96)00102-1, 1996.
Ding, Z., Zhu, Y., Wang, G., Li, H., Xu, J., Tian, M., Liu, Z., Chen, J., Yun, L., Zheng, H., Gui, H., Liu, J., Li, R., Deng, C., and Huang, K.: Coastal eutrophic ecosystem as an overlooked pool of atmospheric formic acid: disentangling biogenic and abiotic contributions, Environ. Sci. Tech. Lett., 12, 1402–1410, https://doi.org/10.1021/acs.estlett.5c00894, 2025.
Drozd, G. T., Zhao, Y., Saliba, G., Frodin, B., Maddox, C., Oliver Chang, M.-C., Maldonado, H., Sardar, S., Weber, R. J., Robinson, A. L., and Goldstein, A. H.: Detailed Speciation of Intermediate Volatility and Semivolatile Organic Compound Emissions from Gasoline Vehicles: Effects of Cold-Starts and Implications for Secondary Organic Aerosol Formation, Environ. Sci. Technol., 53, 1706–1714, https://doi.org/10.1021/acs.est.8b05600, 2019.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Fittschen, C., Whalley, L. K., and Heard, D. E.: The reaction of CH3O2 radicals with OH radicals: a neglected sink for CH3O2 in the remote atmosphere, Environ. Sci. Technol., 48, 7700–7701, https://doi.org/10.1021/es502481q, 2014.
Forstner, H. J. L., Flagan, R. C., and Seinfeld, J. H.: Secondary Organic Aerosol from the Photooxidation of Aromatic Hydrocarbons: Molecular Composition, Environ. Sci. Technol., 31, 1345–1358, https://doi.org/10.1021/es9605376, 1997.
Franco, B., Blumenstock, T., Cho, C., Clarisse, L., Clerbaux, C., Coheur, P.-F., De Mazière, M., De Smedt, I., Dorn, H.-P., Emmerichs, T., Fuchs, H., Gkatzelis, G., Griffith, D. W. T., Gromov, S., Hannigan, J. W., Hase, F., Hohaus, T., Jones, N., Kerkweg, A., Kiendler-Scharr, A., Lutsch, E., Mahieu, E., Novelli, A., Ortega, I., Paton-Walsh, C., Pommier, M., Pozzer, A., Reimer, D., Rosanka, S., Sander, R., Schneider, M., Strong, K., Tillmann, R., Van Roozendael, M., Vereecken, L., Vigouroux, C., Wahner, A., and Taraborrelli, D.: Ubiquitous atmospheric production of organic acids mediated by cloud droplets, Nature, 593, 233–237, https://doi.org/10.1038/s41586-021-03462-x, 2021.
Friedman, B. and Farmer, D. K.: SOA and gas phase organic acid yields from the sequential photooxidation of seven monoterpenes, Atmos. Environ., 187, 335–345, https://doi.org/10.1016/j.atmosenv.2018.06.003, 2018.
Friedman, B., Link, M. F., Fulgham, S. R., Brophy, P., Galang, A., Brune, W. H., Jathar, S. H., and Farmer, D. K.: Primary and Secondary Sources of Gas-Phase Organic Acids from Diesel Exhaust, Environ. Sci. Technol., 51, 10872–10880, https://doi.org/10.1021/acs.est.7b01169, 2017.
Gery, M. W., Fox, D. L., Jeffries, H. E., Stockburger, L., and Weathers, W. S.: A continuous stirred tank reactor investigation of the gas-phase reaction of hydroxyl radicals and toluene, Int. J. Chem. Kinet., 17, 931–955, https://doi.org/10.1002/kin.550170903, 1985.
Hansen, A. M. K., Kristensen, K., Nguyen, Q. T., Zare, A., Cozzi, F., Nøjgaard, J. K., Skov, H., Brandt, J., Christensen, J. H., Ström, J., Tunved, P., Krejci, R., and Glasius, M.: Organosulfates and organic acids in Arctic aerosols: speciation, annual variation and concentration levels, Atmos. Chem. Phys., 14, 7807–7823, https://doi.org/10.5194/acp-14-7807-2014, 2014.
Huang, D., Chen, Z. M., Zhao, Y., and Liang, H.: Newly observed peroxides and the water effect on the formation and removal of hydroxyalkyl hydroperoxides in the ozonolysis of isoprene, Atmos. Chem. Phys., 13, 5671–5683, https://doi.org/10.5194/acp-13-5671-2013, 2013.
Isaacman-VanWertz, G., Massoli, P., O'Brien, R., Lim, C., Franklin, J. P., Moss, J. A., Hunter, J. F., Nowak, J. B., Canagaratna, M. R., Misztal, P. K., Arata, C., Roscioli, J. R., Herndon, S. T., Onasch, T. B., Lambe, A. T., Jayne, J. T., Su, L., Knopf, D. A., Goldstein, A. H., Worsnop, D. R., and Kroll, J. H.: Chemical evolution of atmospheric organic carbon over multiple generations of oxidation, Nat. Chem., 10, 462–468, https://doi.org/10.1038/s41557-018-0002-2, 2018.
Jenkin, M. E., Hurley, M. D., and Wallington, T. J.: Investigation of the radical product channel of the CH3C(O)CH2O2+ HO2 reaction in the gas phase, Phys. Chem. Chem. Phys., 10, 4274–4280, https://doi.org/10.1039/B802898B, 2008.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jiang, Y., Xia, M., Wang, Z., Zheng, P., Chen, Y., and Wang, T.: Photochemical ageing of aerosols contributes significantly to the production of atmospheric formic acid, Atmos. Chem. Phys., 23, 14813–14828, https://doi.org/10.5194/acp-23-14813-2023, 2023.
Kang, M., Zhang, H., and Ying, Q.: Sulfate or organic acids: the atmospheric fate of stabilized criegee intermediates, J. Geophys. Res.-Atmos., 130, e2025JD044867, https://doi.org/10.1029/2025JD044867, 2025.
Keller-Rudek, H., Moortgat, G. K., Sander, R., and Sörensen, R.: The MPI-Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest, Earth Syst. Sci. Data, 5, 365–373, https://doi.org/10.5194/essd-5-365-2013, 2013.
Khan, M. A. H., Lyons, K., Chhantyal-Pun, R., McGillen, M. R., Caravan, R. L., Taatjes, C. A., Orr-Ewing, A. J., Percival, C. J., and Shallcross, D. E.: Investigating the tropospheric chemistry of acetic acid using the global 3-D chemistry transport model, STOCHEM-CRI, J. Geophys. Res.-Atmos., 123, 6267–6281, https://doi.org/10.1029/2018JD028529, 2018.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D)+ N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Lambe, A. T., Onasch, T. B., Croasdale, D. R., Wright, J. P., Martin, A. T., Franklin, J. P., Massoli, P., Kroll, J. H., Canagaratna, M. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Transitions from Functionalization to Fragmentation Reactions of Laboratory Secondary Organic Aerosol (SOA) Generated from the OH Oxidation of Alkane Precursors, Environ. Sci. Technol., 46, 5430–5437, https://doi.org/10.1021/es300274t, 2012.
Lambe, A. T., Cappa, C. D., Massoli, P., Onasch, T. B., Forestieri, S. D., Martin, A. T., Cummings, M. J., Croasdale, D. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Relationship between Oxidation Level and Optical Properties of Secondary Organic Aerosol, Environ. Sci. Technol., 47, 6349–6357, https://doi.org/10.1021/es401043j, 2013.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Le Breton, M., McGillen, M. R., Muller, J. B. A., Bacak, A., Shallcross, D. E., Xiao, P., Huey, L. G., Tanner, D., Coe, H., and Percival, C. J.: Airborne observations of formic acid using a chemical ionization mass spectrometer, Atmos. Meas. Tech., 5, 3029–3039, https://doi.org/10.5194/amt-5-3029-2012, 2012.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., De Gouw, J. A., and Jimenez, J. L.: Modeling the Radical Chemistry in an Oxidation Flow Reactor: Radical Formation and Recycling, Sensitivities, and the OH Exposure Estimation Equation, J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Liggio, J., Moussa, S. G., Wentzell, J., Darlington, A., Liu, P., Leithead, A., Hayden, K., O'Brien, J., Mittermeier, R. L., Staebler, R., Wolde, M., and Li, S.-M.: Understanding the primary emissions and secondary formation of gaseous organic acids in the oil sands region of Alberta, Canada, Atmos. Chem. Phys., 17, 8411–8427, https://doi.org/10.5194/acp-17-8411-2017, 2017.
Link, M. F., Nguyen, T. B., Bates, K., Müller, J.-F., and Farmer, D. K.: Can Isoprene Oxidation Explain High Concentrations of Atmospheric Formic and Acetic Acid over Forests?, ACS Earth Space Chem., 4, 730–740, https://doi.org/10.1021/acsearthspacechem.0c00010, 2020.
Lu, R., Zhou, P., Ma, F., Zhao, Q., Peng, X., Chen, J., and Xie, H.-B.: Multi-generation oxidation mechanism of M-xylene: unexpected implications for secondary organic aerosol formation, Atmos. Environ., 327, 120511, https://doi.org/10.1016/j.atmosenv.2024.120511, 2024.
Malecha, K. T. and Nizkorodov, S. A.: Photodegradation of Secondary Organic Aerosol Particles as a Source of Small, Oxygenated Volatile Organic Compounds, Environ. Sci. Technol., 50, 9990–9997, https://doi.org/10.1021/acs.est.6b02313, 2016.
Malecha, K. T. and Nizkorodov, S. A.: Feasibility of photosensitized reactions with secondary organic aerosol particles in the presence of volatile organic compounds, J. Phys. Chem. A, 121, 4961–4967, https://doi.org/10.1021/acs.jpca.7b04066, 2017.
Millet, D. B., Baasandorj, M., Farmer, D. K., Thornton, J. A., Baumann, K., Brophy, P., Chaliyakunnel, S., de Gouw, J. A., Graus, M., Hu, L., Koss, A., Lee, B. H., Lopez-Hilfiker, F. D., Neuman, J. A., Paulot, F., Peischl, J., Pollack, I. B., Ryerson, T. B., Warneke, C., Williams, B. J., and Xu, J.: A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 15, 6283–6304, https://doi.org/10.5194/acp-15-6283-2015, 2015.
Müller, M., Graus, M., Wisthaler, A., Hansel, A., Metzger, A., Dommen, J., and Baltensperger, U.: Analysis of high mass resolution PTR-TOF mass spectra from 1,3,5-trimethylbenzene (TMB) environmental chamber experiments, Atmos. Chem. Phys., 12, 829–843, https://doi.org/10.5194/acp-12-829-2012, 2012.
Mungall, E. L., Abbatt, J. P. D., Wentzell, J. J. B., Wentworth, G. R., Murphy, J. G., Kunkel, D., Gute, E., Tarasick, D. W., Sharma, S., Cox, C. J., Uttal, T., and Liggio, J.: High gas-phase mixing ratios of formic and acetic acid in the High Arctic, Atmos. Chem. Phys., 18, 10237–10254, https://doi.org/10.5194/acp-18-10237-2018, 2018.
Munger, J. W., Tiller, C., and Hoffmann, M. R.: Identification of hydroxymethanesulfonate in fog water, Science, 231, 247–249, https://doi.org/10.1126/science.231.4735.247, 1986.
Murschell, T. and Farmer, D. K.: Atmospheric OH oxidation of three chlorinated aromatic herbicides, Environ. Sci. Technol., 52, 4583–4591, https://doi.org/10.1021/acs.est.7b06025, 2018.
Neeb, P., Sauer, F., Horie, O., and Moortgat, G. K.: Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour, Atmos. Environ., 31, 1417–1423, https://doi.org/10.1016/S1352-2310(96)00322-6, 1997.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Orzechowska, G. E. and Paulson, S. E.: Photochemical sources of organic acids. 1. Reaction of ozone with isoprene, propene, and 2-butenes under dry and humid conditions using SPME, J. Phys. Chem. A, 109, 5358–5365, https://doi.org/10.1021/jp050166s, 2005.
Paulot, F., Wunch, D., Crounse, J. D., Toon, G. C., Millet, D. B., DeCarlo, P. F., Vigouroux, C., Deutscher, N. M., González Abad, G., Notholt, J., Warneke, T., Hannigan, J. W., Warneke, C., de Gouw, J. A., Dunlea, E. J., De Mazière, M., Griffith, D. W. T., Bernath, P., Jimenez, J. L., and Wennberg, P. O.: Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 11, 1989–2013, https://doi.org/10.5194/acp-11-1989-2011, 2011.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Peng, Z., Lee-Taylor, J., Orlando, J. J., Tyndall, G. S., and Jimenez, J. L.: Organic peroxy radical chemistry in oxidation flow reactors and environmental chambers and their atmospheric relevance, Atmos. Chem. Phys., 19, 813–834, https://doi.org/10.5194/acp-19-813-2019, 2019.
Permar, W., Jin, L., Peng, Q., O'Dell, K., Lill, E., Selimovic, V., J. Yokelson, R., S. Hornbrook, R., J. Hills, A., C. Apel, E., Ku, I.-T., Zhou, Y., C. Sive, B., P. Sullivan, A., L. Collett, J., B. Palm, B., A. Thornton, J., Flocke, F., V. Fischer, E., and Hu, L.: Atmospheric OH reactivity in the western United States determined from comprehensive gas-phase measurements during WE-CAN, Environ. Sci.-Atmos., 3, 97–114, https://doi.org/10.1039/D2EA00063F, 2023.
Praplan, A. P., Hegyi-Gaeggeler, K., Barmet, P., Pfaffenberger, L., Dommen, J., and Baltensperger, U.: Online measurements of water-soluble organic acids in the gas and aerosol phase from the photooxidation of 1,3,5-trimethylbenzene, Atmos. Chem. Phys., 14, 8665–8677, https://doi.org/10.5194/acp-14-8665-2014, 2014.
Schobesberger, S., Lopez-Hilfiker, F. D., Taipale, D., Millet, D. B., D'Ambro, E. L., Rantala, P., Mammarella, I., Zhou, P., Wolfe, G. M., Lee, B. H., Boy, M., and Thornton, J. A.: High upward fluxes of formic acid from a boreal forest canopy, Geophys. Res. Lett., 43, 9342–9351, https://doi.org/10.1002/2016GL069599, 2016.
Seuwen, R. and Warneck, P.: Oxidation of toluene in NOx free air: product distribution and mechanism, Int. J. Chem. Kinet., 28, 315–332, https://doi.org/10.1002/(SICI)1097-4601(1996)28:5<315::AID-KIN1>3.0.CO;2-Y, 1996.
Shaw, M. F., Sztáray, B., Whalley, L. K., Heard, D. E., Millet, D. B., Jordan, M. J. T., Osborn, D. L., and Kable, S. H.: Photo-tautomerization of acetaldehyde as a photochemical source of formic acid in the troposphere, Nat. Commun., 9, 2584, https://doi.org/10.1038/s41467-018-04824-2, 2018.
Shen, H., Chen, Z., Li, H., Qian, X., Qin, X., and Shi, W.: Gas-particle partitioning of carbonyl compounds in the ambient atmosphere, Environ. Sci. Technol., 52, 10997–11006, https://doi.org/10.1021/acs.est.8b01882, 2018.
Shen, H., Huang, L., Qian, X., Qin, X., and Chen, Z.: Positive Feedback between Partitioning of Carbonyl Compounds and Particulate Sulfur Formation during Haze Episodes, Environ. Sci. Technol., 58, 21286–21294, https://doi.org/10.1021/acs.est.4c07278, 2024.
Stavrakou, T., Müller, J.-F., Peeters, J., Razavi, A., Clarisse, L., Clerbaux, C., Coheur, P.-F., Hurtmans, D., De Mazière, M., Vigouroux, C., Deutscher, N. M., Griffith, D. W. T., Jones, N., and Paton-Walsh, C.: Satellite evidence for a large source of formic acid from boreal and tropical forests, Nat. Geosci., 5, 26–30, https://doi.org/10.1038/ngeo1354, 2012.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tao, Y. and Murphy, J. G.: Evidence for the Importance of Semivolatile Organic Ammonium Salts in Ambient Particulate Matter, Environ. Sci. Technol., 53, 108–116, https://doi.org/10.1021/acs.est.8b03800, 2019.
Veres, P. R., Roberts, J. M., Cochran, A. K., Gilman, J. B., Kuster, W. C., Holloway, J. S., Graus, M., Flynn, J., Lefer, B., Warneke, C., and de Gouw, J.: Evidence of rapid production of organic acids in an urban air mass, Geophys. Res. Lett., 38, https://doi.org/10.1029/2011GL048420, 2011.
Volkamer, R., Platt, U., and Wirtz, K.: Primary and Secondary Glyoxal Formation from Aromatics: Experimental Evidence for the Bicycloalkyl-Radical Pathway from Benzene, Toluene, and p-Xylene, J. Phys. Chem. A, 105, 7865–7874, https://doi.org/10.1021/jp010152w, 2001.
Walavalkar, M. P., Sharma, A., Dhanya, S., and Naik, P. D.: Reactions of lactones with tropospheric oxidants: A kinetics and products study, Atmos. Environ., 161, 18–26, https://doi.org/10.1016/j.atmosenv.2017.04.028, 2017.
Wang, Y., Li, C., Zhang, Y., Li, Y., Yang, G., Yang, X., Wu, Y., Yao, L., Zhang, H., and Wang, L.: Secondary reactions of aromatics-derived oxygenated organic molecules lead to plentiful highly oxygenated organic molecules within an intraday OH exposure, Atmos. Chem. Phys., 24, 7961–7981, https://doi.org/10.5194/acp-24-7961-2024, 2024.
Wu, H., Wang, Y., Li, H., Huang, L., Huang, D., Shen, H., Xing, Y., and Chen, Z.: The OH-initiated oxidation of atmospheric peroxyacetic acid: Experimental and model studies, Atmos. Environ., 164, 61–70, https://doi.org/10.1016/j.atmosenv.2017.05.038, 2017.
Wyche, K. P., Monks, P. S., Ellis, A. M., Cordell, R. L., Parker, A. E., Whyte, C., Metzger, A., Dommen, J., Duplissy, J., Prevot, A. S. H., Baltensperger, U., Rickard, A. R., and Wulfert, F.: Gas phase precursors to anthropogenic secondary organic aerosol: detailed observations of 1,3,5-trimethylbenzene photooxidation, Atmos. Chem. Phys., 9, 635–665, https://doi.org/10.5194/acp-9-635-2009, 2009.
Xing, Y., Li, H., Huang, L., Wu, H., Shen, H., and Chen, Z.: The production of formaldehyde and hydroxyacetone in methacrolein photooxidation: New insights into mechanism and effects of water vapor, J. Environ. Sci., 66, 1–11, https://doi.org/10.1016/j.jes.2017.05.037, 2018.
Yetter, R. A., Rabitz, H., Dryer, F. L., Maki, R. G., and Klemm, R. B.: Evaluation of the rate constant for the reaction OH+H2CO: Application of modeling and sensitivity analysis techniques for determination of the product branching ratio, J. Chem. Phys., 91, 4088–4097, https://doi.org/10.1063/1.456838, 1989.
Yuan, B., Veres, P. R., Warneke, C., Roberts, J. M., Gilman, J. B., Koss, A., Edwards, P. M., Graus, M., Kuster, W. C., Li, S.-M., Wild, R. J., Brown, S. S., Dubé, W. P., Lerner, B. M., Williams, E. J., Johnson, J. E., Quinn, P. K., Bates, T. S., Lefer, B., Hayes, P. L., Jimenez, J. L., Weber, R. J., Zamora, R., Ervens, B., Millet, D. B., Rappenglück, B., and de Gouw, J. A.: Investigation of secondary formation of formic acid: urban environment vs. oil and gas producing region, Atmos. Chem. Phys., 15, 1975–1993, https://doi.org/10.5194/acp-15-1975-2015, 2015.
Zervas, E.: Formation of organic acids from propane, isooctane and toluene/isooctane flames, Fuel, 84, 691–700, https://doi.org/10.1016/j.fuel.2004.11.012, 2005.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.