the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Apr 2025
| 16 Apr 2025
Monoterpene oxidation pathways initiated by acyl peroxy radical addition
Dominika Pasik
Thomas Golin Almeida
Emelda Ahongshangbam
Siddharth Iyer
Monoterpenes are released into the atmosphere in significant quantities, where they undergo various oxidation reactions. Despite extensive studies in this area, there are still gaps that need to be addressed to fully understand the oxidation processes occurring in the atmosphere. Recent findings suggest that reactions between alkenes and acyl peroxy radicals (APRs) can be competitive under atmospheric conditions. In this study, we investigate the oxidation reactions of seven monoterpenes with the CH3C(O)OO⋅ radical and subsequent diverse reactions, including accretion, alkyl radical rearrangements, H-shift, and β-scission reactions. The accretion reaction leads to the release of excess energy, which is sufficient to open small secondary rings in the alkyl radical structures. This reaction is most significant for sabinene (31 %) and α-thujene (18 %). A competing reaction is O2 addition, which the majority of alkyl radicals undergo, subsequently leading to the formation of peroxy radicals. These then react further, forming alkoxy radicals that can subsequently undergo β-scission reactions. Our calculations show that for β-pinene, camphene, and sabinene, β-scission rearrangements result in radicals capable of undergoing further propagation of the oxidative chain, while for limonene, α-pinene, and α-thujene, scissions leading to closed-shell products that terminate the oxidative chain are preferred. The results indicate that if reactions of monoterpenes with APRs are indeed competitive under atmospheric conditions, their oxidation would lead to more oxygenated compounds with a higher molar mass, potentially contributing to secondary organic aerosol yields. Moreover, this study highlights the significance of stereochemistry in controlling the oxidation of monoterpenes initiated by APRs.
- Article
(3765 KB) - Full-text XML
-
Supplement
(798 KB) - BibTeX
- EndNote




Monoterpenes (C10H16) represent a diverse group of compounds emitted by plants and trees, comprising roughly 15 % of biogenic volatile organic compound emissions into the atmosphere (Friedman and Farmer, 2018; Hantschke et al., 2021; Guenther et al., 2012; Lun et al., 2020). Due to their high reactivity, they undergo numerous oxidation reactions in the atmosphere, which can result in compounds with higher molar mass and lower volatility (Iyer et al., 2021, 2018; Kanakidou et al., 2005; Boyd et al., 2015; Bianchi et al., 2019; Zhao et al., 2015; Vereecken and Nozière, 2020). These, in turn, contribute to the production of secondary organic aerosols (SOAs), which have significant impacts on climate, air quality, and human health (Friedman and Farmer, 2018; Ehn et al., 2014; Xu et al., 2015, 2018). While it is well established that aerosol particles play a crucial role in the atmosphere by influencing Earth's radiative balance and serving as cloud condensation nuclei (CCN), and extensive studies have been conducted to understand their effects, there is still high uncertainty in global climate models, largely due to the limited understanding of SOA formation from biogenic volatile organic compounds (VOCs) (Glasius and Goldstein, 2016; Jokinen et al., 2012; Shrivastava et al., 2017; Dada et al., 2023). Numerous studies estimate that monoterpenes contribute to approximately half of the global biogenic SOA source, primarily through reactions with OH radicals, ozone, and NO3 radicals (Spracklen et al., 2011; Day et al., 2022; Zhang et al., 2018; Presto et al., 2005). The oxidized organic compounds formed in these reactions have a lower volatility compared to their parent monoterpene and thus can participate in aerosol particle formation or growth by either creating new particles through homogeneous nucleation or increasing the size of existing newly formed particles through condensation.
Despite their structural similarities, monoterpenes exhibit different oxidation reactivity and thus different SOA yields. For instance, Lee et al. (2006) showed that the ozonolysis of monoterpenes resulted in SOA yields ranging from 17 % for β-pinene to 41 % for α-pinene. Moreover, the trends in SOA yields vary depending on the oxidant (Draper et al., 2015; Liu et al., 2022). Recently, Draper et al. (2024) investigated the reactions of monoterpenes with NO3 followed by ring-opening reactions and bond scissions. Their research not only provided evidence that each monoterpene possesses a specific reactivity towards the same oxidation pathways but also presented rearrangement reactions that open up possibilities for further oxidation, thereby contributing to SOA yields. Their calculations indicate that the alkyl radical rearrangement outcompetes O2 addition in the case of sabinene, while it is minor but competitive in the case of α-thujene and β-pinene and negligible in the case of camphene. These ring-opening rearrangements can promote further propagation of the oxidative chain.
Peroxy radical (RO2) reactions with VOCs were initially thought to be negligible at room temperature and were thus overlooked in atmospheric chemistry. However, recent studies by Nozière and Fache (2021) revealed that unsaturated hydrocarbons can indeed react with RO2, and this reaction was initially thought to lead to epoxides like in combustion conditions. Later Nozière et al. (2023) continued kinetic experiments of RO2 with unsaturated hydrocarbons and suggested an accretion pathway for this reaction. Additionally, Pasik et al. (2024a) demonstrated that reactions between isoprene and monoterpenes with acyl peroxy radicals (APRs) occur at rates significant enough to impact atmospheric chemistry. For example, reaction of limonene with CH3C(O)OO⋅ is fast enough to compete in the atmosphere, potentially accounting for up to 0.1 % of the limonene sink. Moreover, monoterpene + APR reactions result in the formation of new and more functionalized products as in addition of oxidation reactions, which is a sequential and multi-step process of intra-molecular reaction followed by O2 addition. Furthermore, oxidation by APR also increases the total number of carbon atoms, and this, along with autoxidation, leads to lower-volatility compounds. APRs as novel oxidants open up numerous new reaction pathways, potentially elucidating missing data regarding SOA formation.
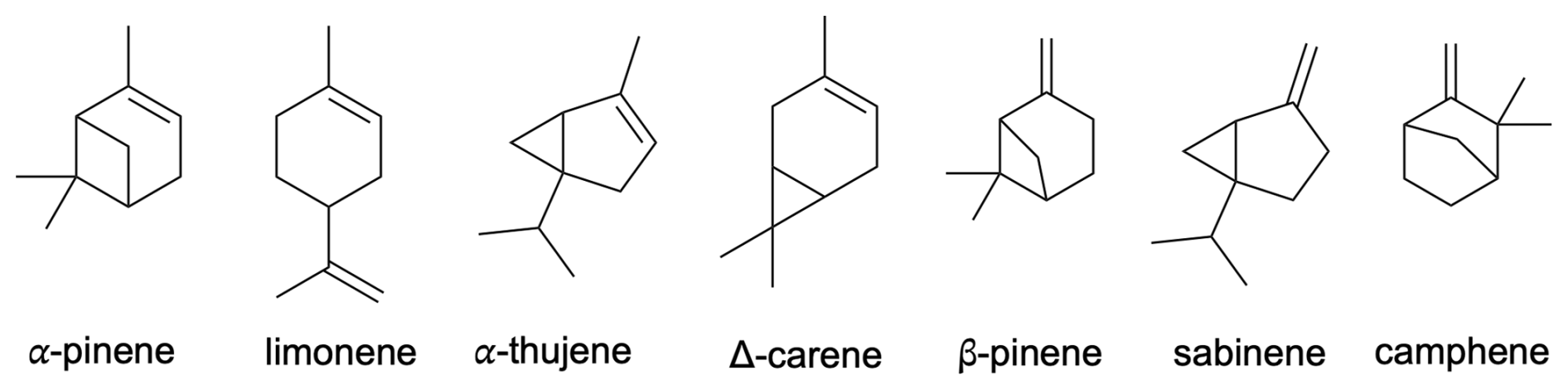
Building upon the research conducted by Draper et al. (2024) and Pasik et al. (2024a), in this study, we investigate the oxidation pathways of monoterpenes + APR through alkyl ring-opening reactions and alkoxy radical bond scissions. Utilizing computational methods, we explore these reactions in the CH3C(O)OO⋅-initiated oxidation of seven monoterpenes: limonene, α-pinene, β-pinene, camphene, sabinene, α-thujene, and Δ3-carene (see Fig. 1). The acetyl peroxy radical (CH3C(O)OO⋅) is a small system to model; moreover it was recently shown to be a good counterpart for bimolecular reactions due to its slow unimolecular reactions (Seal et al., 2023; Karppinen et al., 2025). This research aims to solve the processes leading to oxygenated multifunctional organic compounds possibly contributing to the formation of secondary organic aerosols. Understanding these processes is crucial for addressing the enduring knowledge gap in the formation of new particles.
2.1 Computational details
To investigate the accretion reactions between APRs and monoterpenes as well as subsequent bond-scission reactions, the first step involved locating the reactants, products, and corresponding transition states using density functional theory (DFT) at the ωB97X-D/6-31+G* level of theory (Pasik et al., 2024b; Chai and Head-Gordon, 2008; Myllys et al., 2016b; Hariharan and Pople, 1973). These served as input for conformational sampling, which was carried out using Conformer-Rotamer Ensemble Sampling Tool (CREST) software at the GFN2-xTB level (Pracht et al., 2020; Bannwarth et al., 2019). For sabinene, camphene, Δ-carene, and α-thujene, only the dominant APR addition pathway leading to the tertiary alkyl radical was taken into account. For β-pinene and limonene, we utilized the results published in our previous work (Pasik et al., 2024a). For α-pinene, a comprehensive analysis was conducted (see Supplement for details). Since the electronic energies obtained at the xTB level correlate well with DFT energies, we further optimized conformers within 2.5 kcal mol−1 relative to the lowest-energy conformer. For reactants and products, a reduced number of structures were optimized at the ωB97X-D/6-31+G* level of theory. Based on electronic energy, dipole moment, and squared rotational constant values, duplicate structures were removed, and only unique conformers were considered. The generated conformers for transition state (TS) structures were optimized at the DFT level, while keeping the broken/created bond distance frozen. Following this, the full TS optimization and frequency calculation was conducted. Transition states were verified by the presence of exactly one imaginary frequency. To calculate the reaction energy barrier more accurately, on top of the DFT structures linear-scaling coupled-cluster with single and double excitations as well as perturbative-inclusion-of-triples single-point calculations were performed using the DLPNO-CCSD(T)/aug-cc-pVTZ method (Riplinger et al., 2013, 2016; Myllys et al., 2016a; Draper et al., 2019; Dunning Jr., 1989). For all conformers, the zero-point-corrected energies were calculated. The barrier heights were computed as the difference between the energies of the lowest-energy conformers of TS and the reactants.
The reaction rate coefficients were calculated using the multiconformer transition state theory (MC-TST) approach for bimolecular reactions with the following expression (Vereecken and Peeters, 2003; Viegas, 2023):
where κt is the quantum-mechanical tunnelling coefficient (κt=1 is used for the reactions involving atoms other than hydrogen), T is the temperature (=298.15 K), h is Planck's constant, Pref is the reference pressure (=1 atm, here we used 2.45×1019 cm−3 value), and kB is Boltzmann’s constant. QR,j and QTS,i are the partition functions of the reactant (acetyl peroxyl radical) and transition state conformers, respectively. QMT is the partition function of the monoterpene. Most monoterpenes only have one conformer. For sabinene and α-thujene, we found three conformers each, and all three were included in the equation. ΔEj and ΔEi correspond to the zero-point-corrected electronic energies of the reactant and transition state conformers relative to the lowest-energy conformers, respectively. ER and ETS are the zero-point-corrected electronic energies of the lowest-energy reactant and transition state conformers. The partition functions were calculated at the ωB97X-D/6-31+G* level of theory, while the reaction barrier (ETS−ER) includes the DLPNO-CCSD(T)/aug-cc-pVTZ energy correction.
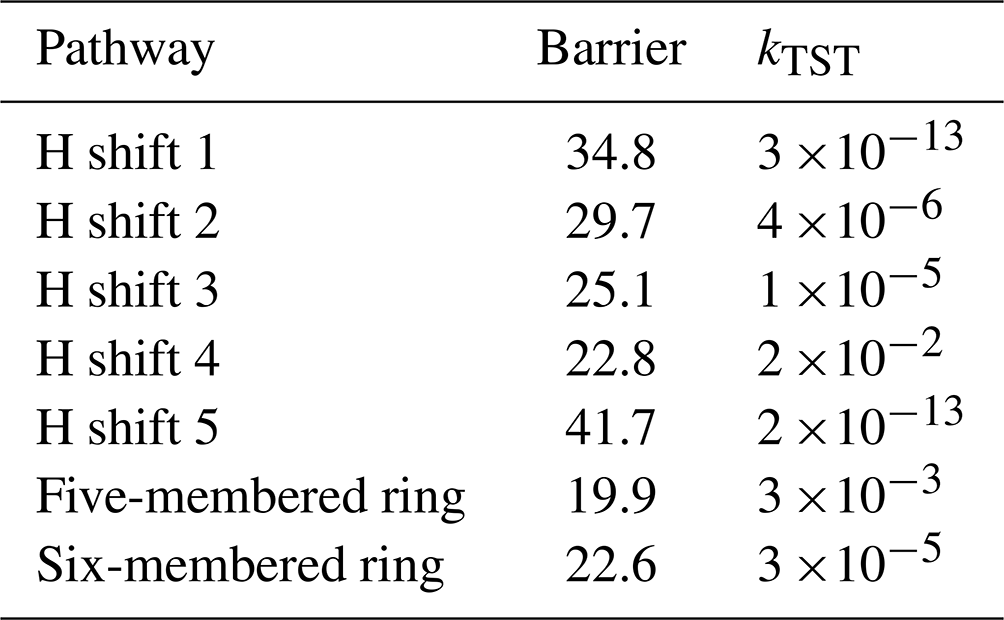
For the hydrogen shift reactions and monoterpene + APR-derived alkoxy radical bond scissions, the unimolecular reaction rate coefficients were calculated following this equation (Møller et al., 2016; Zhao et al., 2022):
Additionally, intrinsic reaction coordinate (IRC) calculations were performed to ensure that the transition states are connected to the correct reactant and product wells. To calculate the tunnelling coefficient κt, we utilized the Eckart tunnelling method. This approach involves a one-dimensional calculation in which the tunnelling coefficient is obtained by solving a three-parameter equation for an asymmetric one-dimensional potential, referred to as the Eckart potential. The calculation requires the transition state energy, along with the energies of the reactant and product connected to the lowest transition state structure, as well as the imaginary frequency. The Eckart tunnelling method has been successfully applied in previous studies to calculate hydrogen shift reaction rates in similar systems (Hasan et al., 2020; Zhang and Dibble, 2011; Skodje et al., 1981).
To calculate alkyl radical ring-opening rearrangement we adopt the methodology outlined in the work by Draper et al. (2024). For the bicyclic monoterpenes where the three-, four-, or five-membered ring-opening rearrangement is possible (α-pinene, α-thujene, sabinene, β-pinene, and camphene), the reaction kinetics were simulated using the Master Equation Solver for Multi Energy-well Reactions (MESMER) software (Glowacki et al., 2012). Vibrational frequencies and rotational constants were calculated at the DFT level, while the zero-point-corrected energies were calculated at the DLPNO-CCSD(T)/aug-cc-pVTZ//ωB97X-D/6-31+G* level. The reactions in question were modelled using the following methods: the monoterpene + APR − addition reaction was simulated using the MESMER inverse Laplace transform (MesmerILT) method, assuming a barrierless association and using realistic bimolecular rate coefficients (kTST-1 was used as a pre-exponential factor in ILT reactions). O2-addition reactions were attributed to the initial APR-alkyl radical and its ring-opening product, treating them as bimolecular sinks. The ring-break reactions, whose TS energetics were calculated, were modelled using simple Rice–Ramsperger–Kassel–Marcus (RRKM) theory with Eckart tunnelling correction (kMESMER) and compared with rates calculated with MC-TST (kTST-2). Bimolecular rate coefficients used were cm3 molecule−1 s−1 for O2 addition with O2 concentration of 5.16×1018 molecules cm−3 (0.21 atm). These values have previously been used for O2-addition reactions to carbon-centred radicals (Draper et al., 2024; Berndt et al., 2015; Kurtén et al., 2017). The respective accretion reaction rate coefficients (used as pre-exponential factor in ILT model) for each monoterpene are listed in Table 1 (kTST-1). Modelling of the ring-opening reaction was conducted according to the procedure described by Draper et al. (2024). We used an exponential-down parameter ΔEdown=225 cm−1, and for each structure we estimated Lennard-Jones parameters based on Joback and Reid (1987) work. N2 was assigned as the bath gas with Lennard-Jones parameters K and σ=3.919 Å. We used a grain size of 30 cm−1, and the span of grains above the highest-energy transition state value was 50 kBT (see Supplement for full analysis). The concentration of CH3C(O)OO⋅ used was 1015 cm−3 to ensure rapid formation of the modelled monoterpene–APR intermediate (R-APR). The Lennard-Jones parameter values were estimated using the procedure described by Gao et al. (2016) and Tee et al. (1966) based on the group-additivity method for deriving pure-compound critical properties by Joback and Reid (1987) and are available in the Supplement.
3.1 Monoterpene-derived alkyl ring-opening reactions
In the first step, we considered the reaction between CH3C(O)OO⋅ and selected monoterpenes (see Fig. 2). In our previous study, we calculated reaction barriers for CH3C(O)OO⋅ + limonene, α-pinene, and β-pinene (Pasik et al., 2024a) according to which, of the two possible addition sites, APR addition leading to a secondary alkyl radical was preferred for α-pinene. Since we expect that the formation of tertiary radicals should be faster, the analysis for α-pinene was repeated, taking into account the stereochemistry of the radical addition to the double bond. Our results show that this has a significant impact on the kinetics of the reaction, and the addition of APR from the opposite side of the secondary ring and methyl groups (R isomer) proceeds with a significantly lower barrier of 1 kcal mol−1 and a rate of cm3 s−1 than if the attack occurs from the same side as the methyl groups (S isomer, barrier showed by Pasik et al., 2024a). Detailed analysis is described in the Supplement. These new findings underscore the impact of stereochemistry on the reactions between monoterpenes and peroxy radicals.
Reaction barriers and calculated rate coefficients for CH3C(O)OO⋅ + limonene and β-pinene (Pasik et al., 2024a) were adopted to this study. All barrier heights and calculated reaction rate coefficients (kTST-1) are summarized in Table 1. As can be expected based on our previous study (Pasik et al., 2024a), all reaction barriers were low (0.2–3.1 kcal mol−1), and the reaction rate coefficients were on the order of 10−15–10−17 cm3 s−1. The reaction rate coefficients for these reactions are sufficiently high to be considered feasible under atmospheric conditions with realistic APRs concentrations. Similarly to α-pinene, for the stereoisomers of α-thujene, the results vary significantly, indicating a structural dependency on reactivity. According to Table 1, the addition of APR to the α-thujene resulting in R-acetyl product isomer proceeds about 1 order of magnitude faster than for the S-acetyl product isomer.
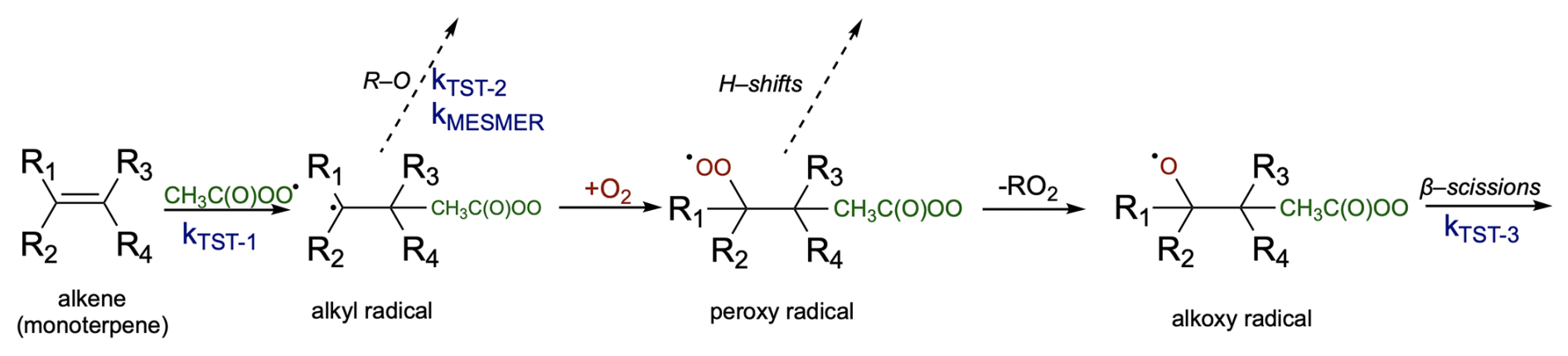
Table 1Zero-point-corrected energies [kcal/mol] for the monoterpene + CH3C(O)OO⋅ accretion reaction barrier (ΔE1) and its excess energy (ΔEex) followed by the ring-opening reaction (ΔE2) calculated at DLPNO-CCSD(T)/aug-cc-pVTZ//ωB97X-D/6-31+G*. Corresponding reaction rate coefficients for the monoterpene + CH3C(O)OO⋅ accretion reaction (kTST-1) [cm3 s−1] as well as the ring-opening reaction calculated at the MC-TST (kTST-2 [s−1]) and RRKM-ME (kMESMER) [s−1]. Yield [ %] of ring-opening products calculated with MC-TST ( %TST-2) and RRKM-ME (%MESMER). RRKM-ME yields were taken from time profiles and MC-TST yields were calculated assuming a rate coefficient of cm3 molecule−1 s−1 with O2 concentration of 5.16×1018 molecules cm−3 for the competing R-APR + O2 reaction. Excess energy (ΔEex) [kcal mol−1] calculated as the difference between reactant and APR–monoterpene adduct energies.
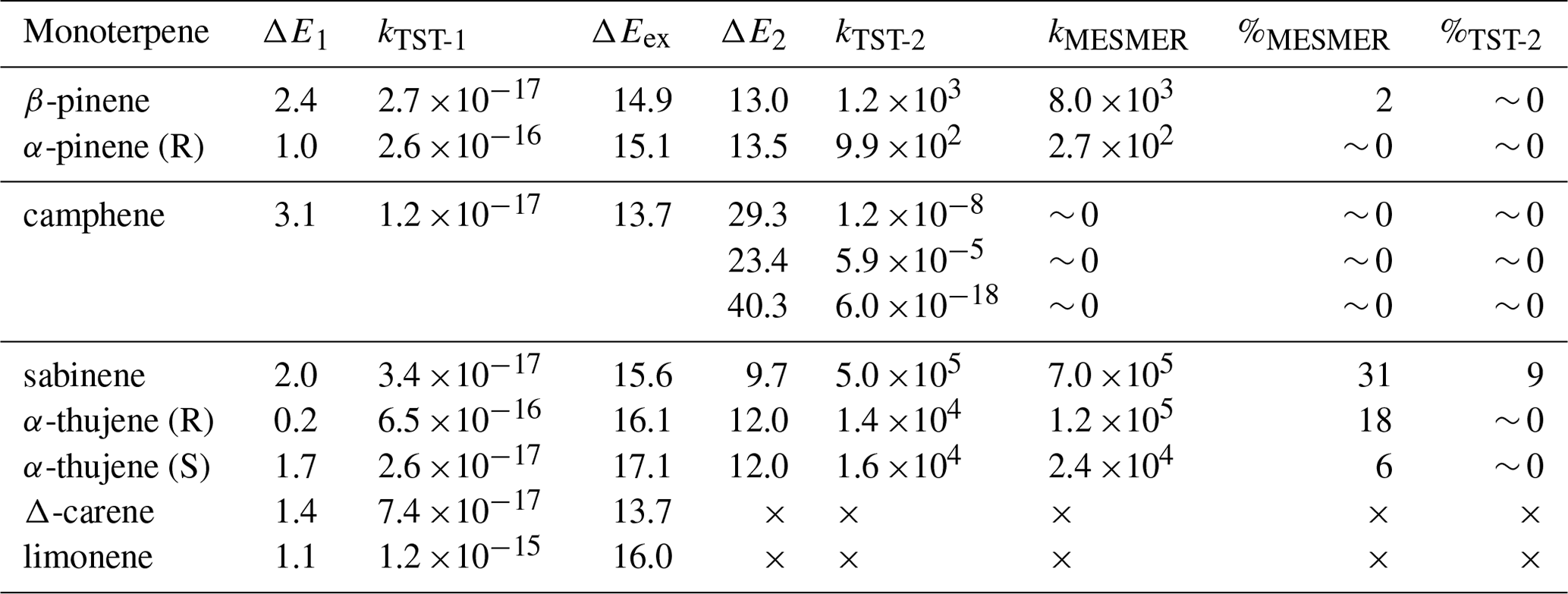
The resulting alkyl radical can react with molecular oxygen, which, due to its high concentration in the atmosphere, is considered the primary pathway for VOC oxidation. However, the addition of the acetyl peroxy radical to the double bond in monoterpene, as indicated by Table 1, is an exothermic process. The energy released in this reaction can contribute to overcoming the barrier for a subsequent unimolecular ring-opening reaction, a reaction pathway that can lead to highly functionalized oxidized products. Table 1 highlights the importance of considering energy released in reaction (excess energy, ΔEex). There is a noticeable difference between the reaction rate coefficients calculated and, thus, ring-opening yields, using TST (which does not account for excess energy) and simulations conducted with standard RRKM theory with Eckart tunnelling correction (kRRKM). This is particularly evident for sabinene, where accounting for excess energy increases the yield of ring opening from 9 % to 31 %. Additionally, the calculations show that the ring-opening reaction is significant only for sabinene (31 %) and α-thujene (up to 18 %). For β-pinene (2 %) it is minor, and for α-pinene and camphene, it is negligible. For Δ-carene, we do not predict ring-opening reactions as alkyl radical centre is too far from the opening ring structure. It is worth noting that the ring-opening reactions are pressure dependent, with the ring-opened product yield increasing with decreasing pressure for the sabinene system, indicating that the acetyl peroxy–terpene adducts are chemically activated (see Supplement, Table S5).
It is worth noting that MESMER-derived rate coefficients tend to be higher than MC-TST rate coefficients as the former do not implicitly account for multiple reactant and TS conformers. A comparison with single-conformer TST-equation-derived rate coefficients, however, showed that excess energy is still driving the formation of ring opened products. Details are provided in the Supplement. The ring-opening reaction is particularly interesting from an oxidation chemistry point of view as it leads to an alkyl radical with less steric constraints than the original “unbroken” counterpart. This promotes more efficient autoxidation and consequently higher molecular functionalization (see Fig. 3).
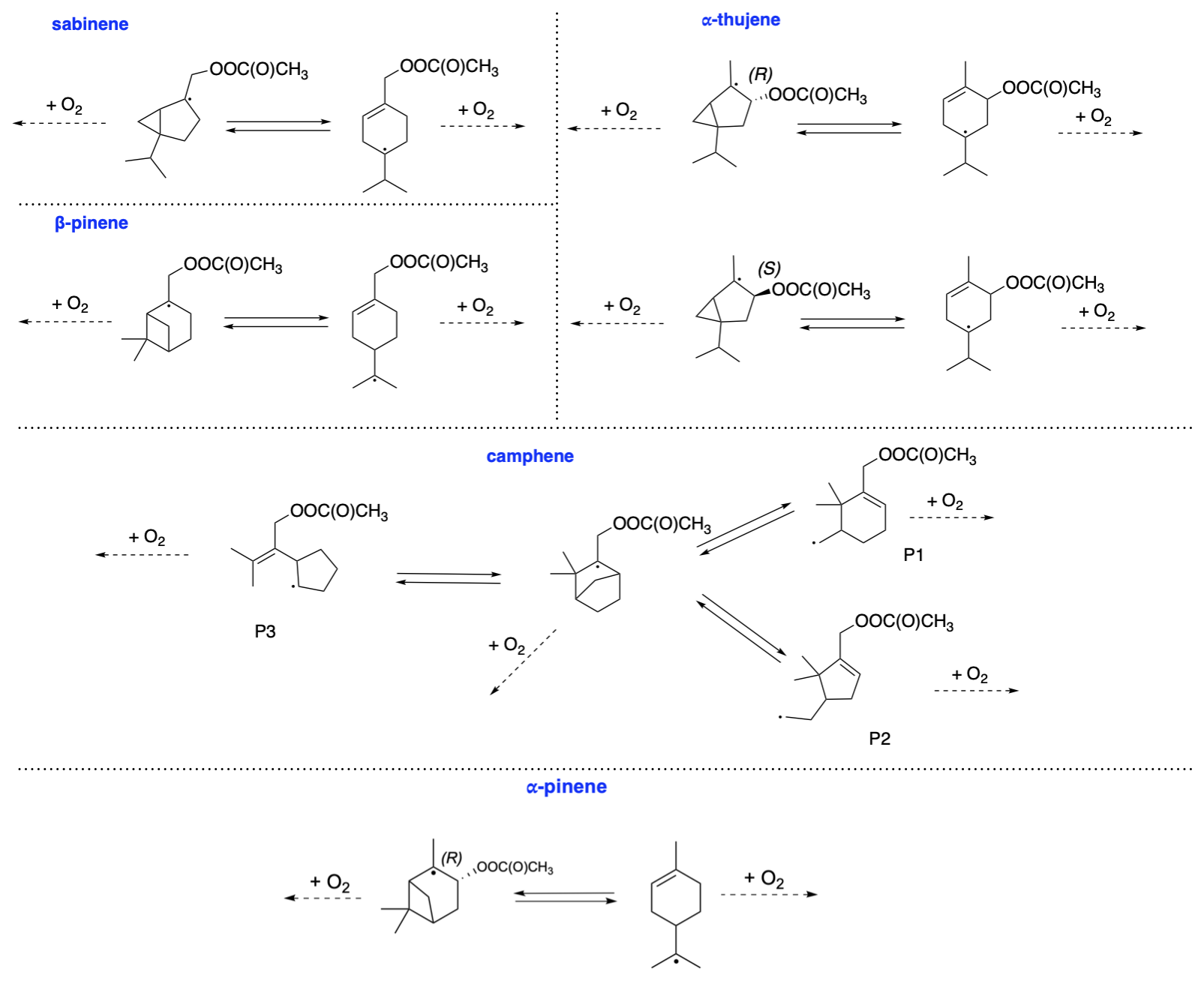
Figure 3Ring-opening radical rearrangement reactions for acetyl–alkyl radicals from bicyclic monoterpenes.
Simulated time profiles of species distributions for bicyclic alkyl radicals reacting via ring opening versus O2 addition are presented in Fig. 4. The results are consistent with the reactions of monoterpenes with NO3 radicals obtained by Draper et al. (2024). However, ring opening for APR constitutes a smaller sink for the respective monoterpenes compared to the NO3 case. For example, the ring-opening reaction accounts for 86 % in the case of sabinene with the nitrooxy–alkyl radical and 31 % for sabinene with the acetyl radical. The ring-opening reaction becomes more competitive as the ring size decreases. For sabinene (31 %) and α-thujene (18 % for R isomer and 6 % for S isomer), which contain a three-membered ring, this competition is the greatest, while it is minor for β-pinene (2 %) containing the four-membered ring and negligible for the five-membered camphene. There is also a noticeable difference between sabinene and α-thujene, which differ only in the position of the double bond and, consequently, the site of APR addition. This suggests that stereo-electronic effects must influence the reactivity towards ring opening (Draper et al., 2024).
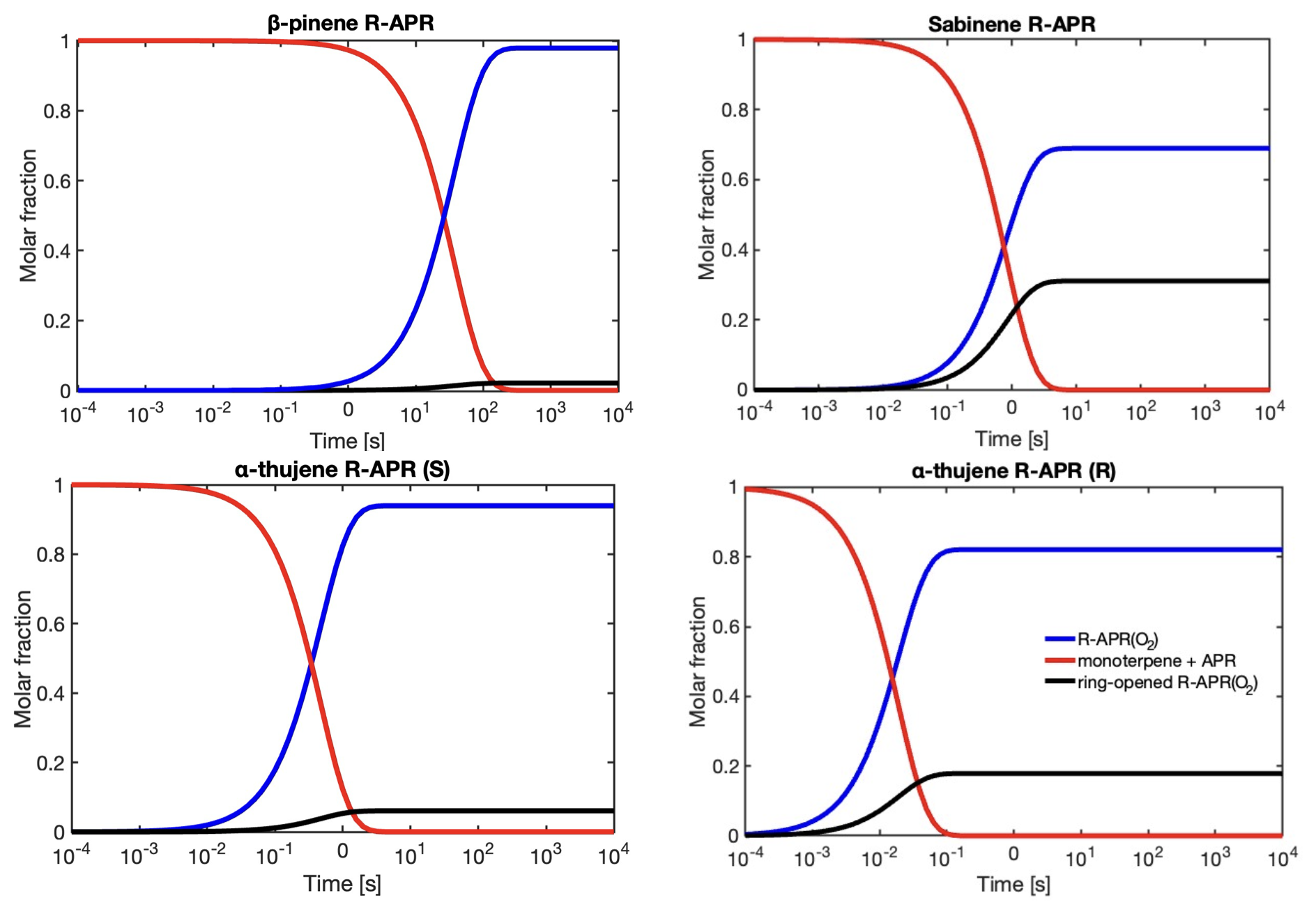
Figure 4RRKM-ME-simulated time profiles of species distributions for bicyclic alkyl radicals reacting via ring opening versus O2 addition. The red line represents the reactant (monoterpene + CH3C(O)OO⋅) for the ring-opening reaction, the black line corresponds to products of O2 addition to the intact alkyl radical (R-APR(O2)), and the blue line represents the remaining non-ring-opened product that undergoes further oxidation with O2.
3.2 APR-initiated autoxidation
The reaction of monoterpenes with CH3C(O)OO⋅ results in a compound with a carbon-centred radical, to which O2 can add and subsequently perform a H-shift reaction. Additionally, for monoterpenes that undergo ring opening, carbon-centred radicals are also formed, allowing for further oxidation. To complete the oxidation pathways of the studied monoterpenes, H-shift reactions for selected intermediate systems were calculated.
Limonene, as it is not a bicyclic monoterpene, cannot undergo alkyl radical ring-opening reactions. Formed in accretion reaction with APR, the alkyl radical adds oxygen, and subsequently it can react via bimolecular reactions, performing HO2-loss or H-shift reactions. In the case of H-shift reactions, these would result in the formation of another carbon-centred radical. The considered reactions are illustrated by labels in Fig. 5, and corresponding barrier heights and reaction rate coefficients are presented in Table 2.
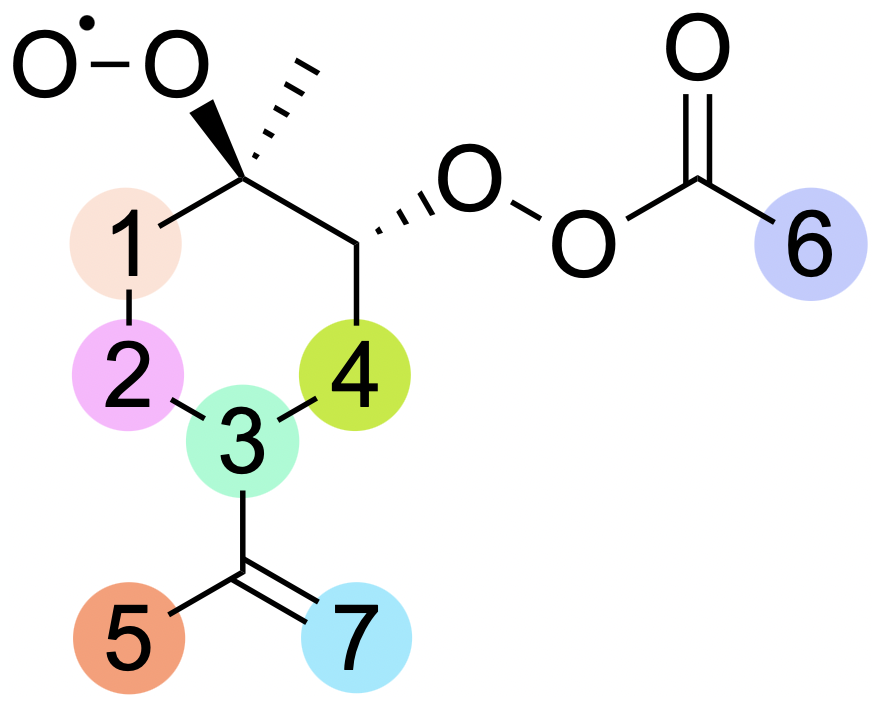
Figure 5Labelling for H-shift reactions of the limonene peroxyl radical. The numbers on the scheme correspond to the numbering in Table 2.
Table 2Energy barrier heights [kcal mol−1] and calculated TST reaction rate coefficients [kTST s−1] for studied H-shift reactions of the limonene-derived peroxyl radical.
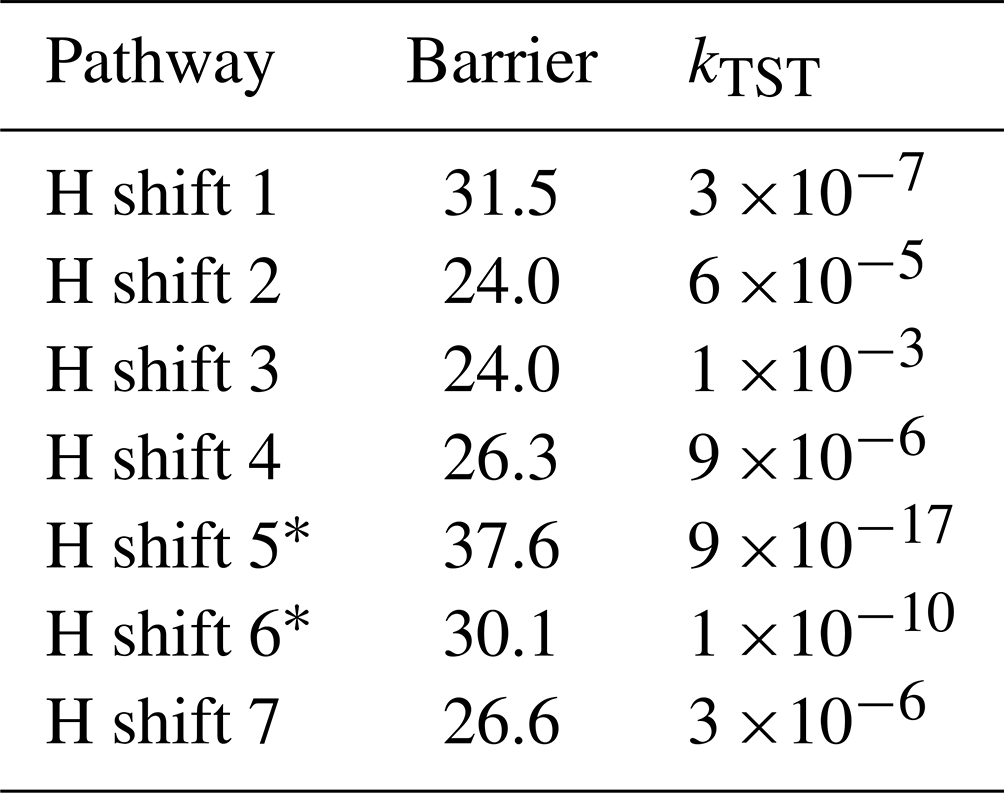
* This rates were calculated using the LC-TST equation (see Supplement).
The lowest barriers are exhibited by H shift 2 (24 kcal mol−1) and H shift 3 (24 kcal mol−1). Although the barriers are identical, the reaction rate coefficient calculated using MC-TST is the highest for H shift 3, on the order of 10−3 s−1. This difference can be explained by a significantly higher tunnelling factor for this reaction (κt=1192) compared to H shift 2 (κt=54). This value is too low for the H-shift reaction to compete with bimolecular reactions that take place in the atmosphere. The calculations presented here draw a direct analogy to the research carried out by Pasik et al. (2024a) concerning the reaction of isoprene with APR radicals. The findings from our current study extend these concepts to limonene. As in the case of isoprene, our results indicate that the H-shift reactions calculated for limonene occur at a rate that is insufficient to drive an autoxidation mechanism. The slow rate of H-shift reactions implies that alternative pathways (for example with RO2) might be more dominant in the oxidation process of APR + limonene-derived RO2.
As mentioned in Sect. 3.1, the addition of CH3C(O)OO⋅ to the double bond in sabinene results in the release of energy high enough to lead to secondary ring opening. RRKM-ME calculations show a 31 % yield of the alkyl ring-opening reaction in the case of sabinene. As a result, a carbon-centred radical is formed, to which molecular oxygen adds, forming an RO2 radical that could undergo H-shift reactions as indicated in Fig. 6.
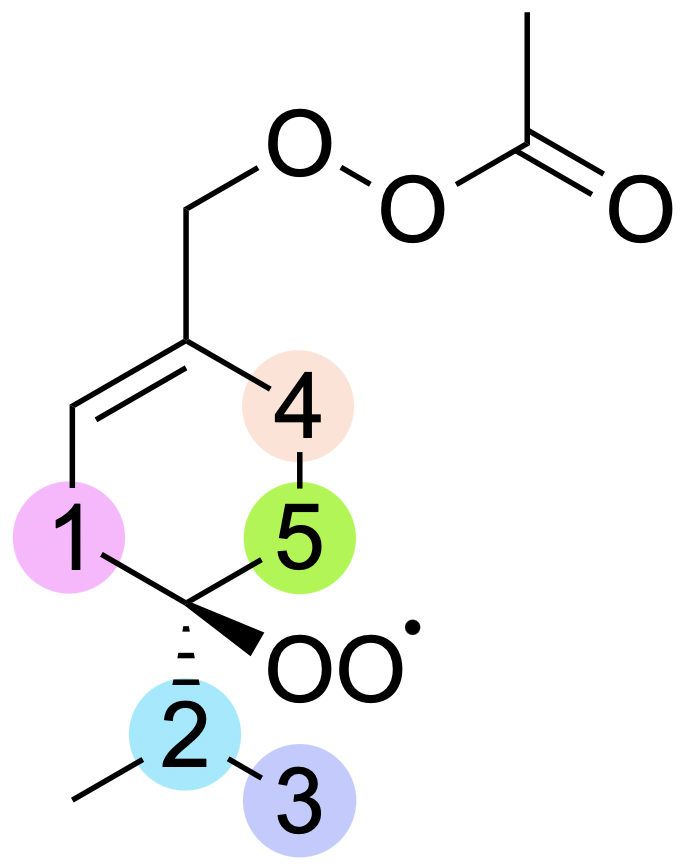
Figure 6Labelling for H-shift reactions of ring-opened sabinene acetyl–peroxyl radical. The numbers on the scheme correspond to the numbering in Table 3.
For this system, we tested some possible H-shift reactions, as indicated in Fig. 6. Also the five- and six-membered ring formation reactions were investigated. The calculated energy barriers and unimolecular reaction rate coefficients are summarized in Table 3. The data indicate that the fastest reaction occurs for H shift 4, with a rate constant of 2 s−1. This rate is sufficiently high to compete with bimolecular reactions in a relatively clean atmosphere with low NOx, suggesting that this mechanism could represent APR-initiated autoxidation. The H-shift reaction leads to a carbon-centred radical capable of adding O2. At this stage, this reaction, along with a competitive ring-opening pathway and subsequent oxidation, produces a compound with 12 carbon atoms and 7 oxygen atoms. Further oxidation can eventually result in a compound with a carbon-to-oxygen ratio near to 1:1.
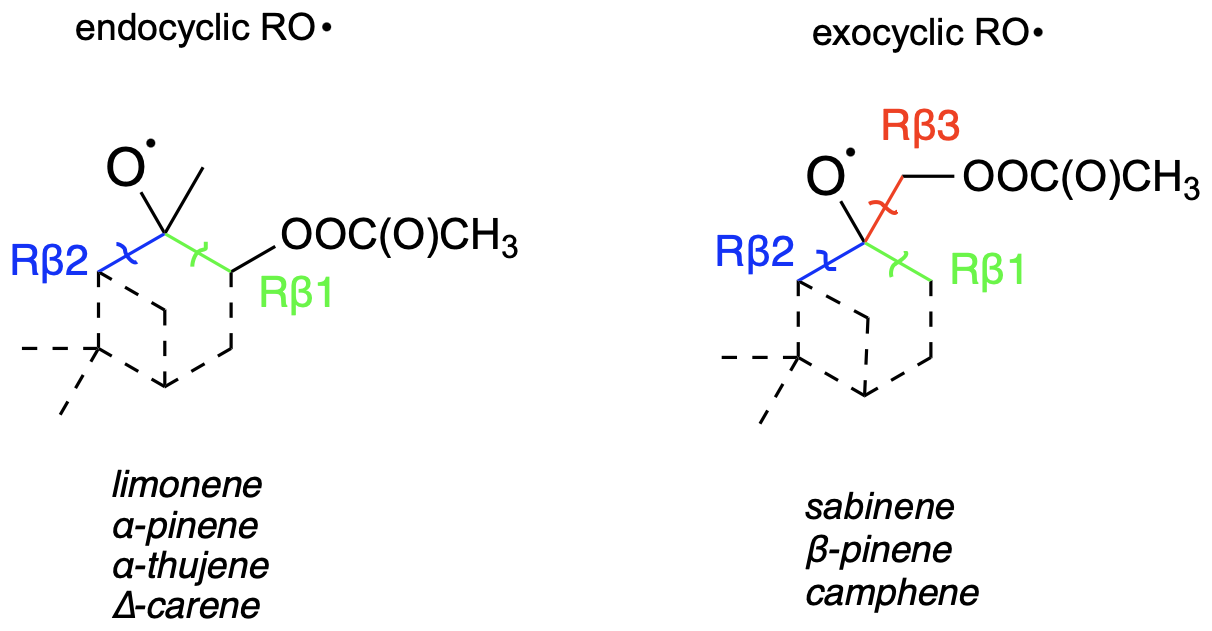
For the ring closure reactions, the obtained rate coefficients are significantly slower compared to the results from similar studies (Nozière and Vereecken, 2024; Vereecken et al., 2021). In these studies, the six-membered ring closure proceeds with a rate on the order of 10−2 s−1. However, these results refer to unsaturated acyclic RO2 radicals, whereas in the case of sabinene-derived RO2, a constrained ring is present. This may account for the discrepancies observed between the values obtained in these studies. In studies by Møller et al. (2020), the authors investigated H-shift and endoperoxide cyclization reactions for OH-derived oxidation products of different monoterpenes. Among these, terpinolene + OH + O2 peroxy radical five- and six-membered ring closures were examined, which have a similar structure to the system we studied. The authors reported rates coefficients for five-membered endoperoxide cyclization as 10−2 s−1. The six-membered ring formation was calculated on a lower level of theory, yielding a rate of 10−3 s−1. Comparing our results with those of Møller et al. (2020), the formation of the five-membered ring is generally faster than that of the six-membered ring, which can be attributed to the formation of a stabilized tertiary carbon-centred radical in the five-membered case. Differences in the calculated rates may be due to different reactant structures.
Table 3Energy barrier heights [kcal mol−1] and calculated TST reaction rate coefficients [kTST s−1] for studies H shift and ring formation reactions of sabinene-derived peroxyl radical.
3.3 Alkoxy radical β-scission reactions
Competitively to H shifts, formed peroxy radical can undergo bimolecular reactions with radicals present in the atmosphere like RO2 or HO2 to form alkoxy radical and subsequently undergo β-scission reactions (see Fig. 2 for overview). For endocyclic monoterpenes, both “right” (Rβ1) and “left” (Rβ2) β-scissions were considered, whereas for exocyclic monoterpenes, “right”, “left”, and “top” (Rβ3) C−C bond scissions were analysed (see Fig. 7). Reaction with HO2 could also lead to the formation of hydroperoxides and molecular oxygen following . However, this process was not investigated in this study.
The stereochemistry was included according to Fig. 8. Table 4 presents the calculated barriers for the corresponding C−C bond scissions, MC-TST reaction rate coefficients, and product yields for the reactions for seven monoterpenes.
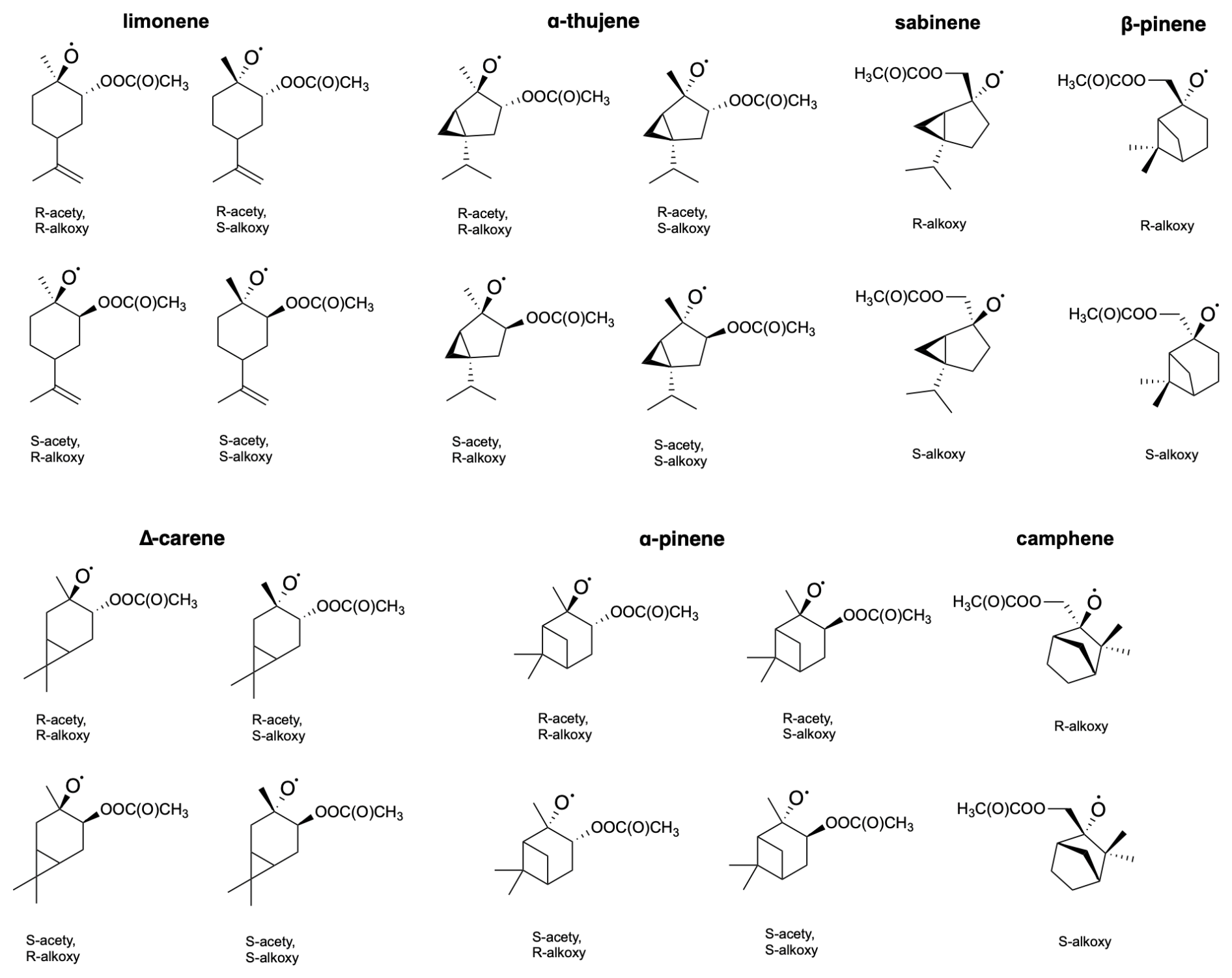
Figure 8Molecular structures of examined acetyl–alkoxyl stereoisomers of limonene, α-thujene, sabinene, β-pinene, Δ-carene, α-pinene and camphene.
Table 4Zero-point-corrected barrier heights (ΔE) for considered acetyl–alkoxy β-scission reactions, corresponding MC-TST rate coefficients and product yields.
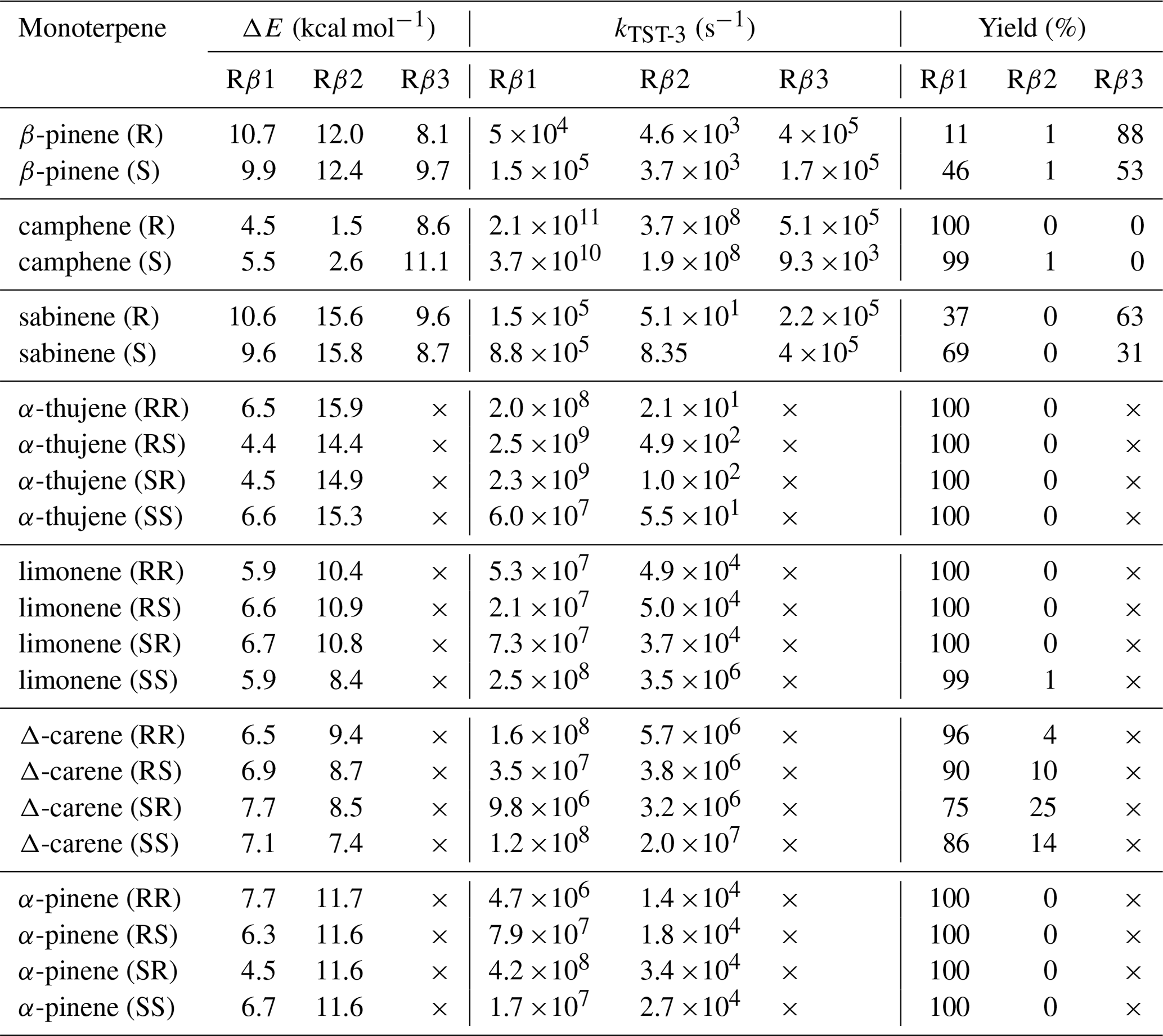
From the data we can draw general conclusions: for all monoterpenes, C−C bond-scission reactions that result in a radical centre on a three-, four-, or five-membered ring structure exhibit the highest energy barriers. These findings align with the results reported by Draper et al. (2024) throughout for nitrooxy alkoxyl β-scission reactions. Interestingly, for the exocyclic monoterpenes sabinene and β-pinene, the preference for the β-scission pathway significantly depends on stereochemistry. This is particularly evident for sabinene, where the R isomer predominantly follows the Rβ3 pathway, whereas the S isomer prefers the Rβ1 pathway. For β-pinene, both isomers predominantly undergo a reaction leading to a closed-shell ketone (Rβ3), but this is more preferred in the R-alkoxyl isomer (88 %) compared to the S-alkoxyl isomer (53 %). For camphene, which is also an exocyclic monoterpene, a clear preference for right-side C−C bond cleavage (Rβ1) is observed. For α-pinene, α-thujene, and limonene, regardless of stereoisomerism, a pronounced major product of Rβ1 C−C bond scission is consistently observed. This pathway leads to termination of the oxidative chain reaction. In contrast to the other investigated endocyclic monoterpenes, Δ-carene exhibits competitive pathways, with each stereoisomer showing its own preference.
Figures 9–15 present the proposed fate for each studied monoterpene. Despite their structural similarities and belonging to the same group of compounds, each studied compound exhibits its own unique reactivity. Consequently, the potential contribution to the formation and growth of organic aerosols will differ for each monoterpene. This underscores the importance of these studies, as differences that arise early in the oxidation pathways can significantly impact the overall outcomes.
3.3.1 Limonene
Figure 9 presents the proposed fate in the CH3C(O)OO⋅-initiated oxidation pathway of limonene. Since limonene does not have a bicyclic structure, it is incapable of undergoing alkyl radical ring-opening reactions. Therefore, after the accretion reaction with APR, where an alkyl radical is formed, it most likely undergoes O2 addition. As shown in Sect. 3.2, H-shift reactions for this intermediate are too slow, making it most likely to react bimolecularly to form an alkoxy radical. Further calculations demonstrate a clear dominance of C−C bond scissions following the Rβ1 pathway, leading to the α-QOOR alkyl radical. Multiple studies indicate that α-QOOR radicals are unstable and undergo decomposition (Vereecken et al., 2004; Anglada et al., 2016; Rissanen et al., 2014). In the case of limonene, this leads to the detachment of the CH3C(O)OO⋅ radical and the formation of closed-shell endolim. This signifies the termination of oxidation chain, although the formed endolim has a C=C double bond capable of reacting in a second-generation oxidation pathway. A relatively simple and unbranched first-generation oxidation pathway would most likely lead to relatively small SOA yields in APR-initiated oxidation. This is only a hypothesis, and further research is needed to confirm the exact mechanisms and factors contributing to the SOA yields.
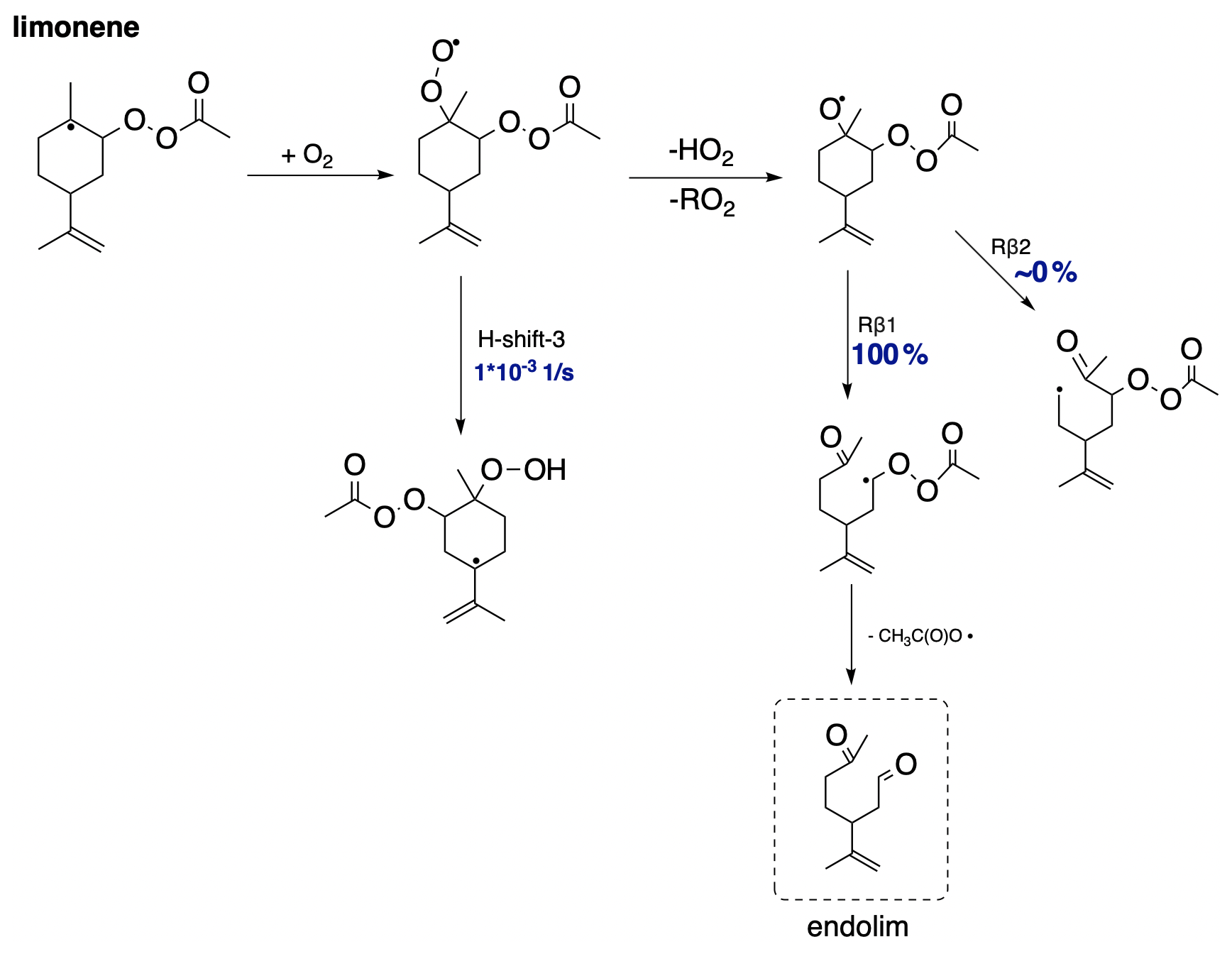
Figure 9Proposed fate of limonene + CH3C(O)OO⋅. The dashed box presents the closed-shell product that terminates the oxidation pathway.
3.3.2 α-Pinene
Next, we propose the oxidation mechanism for α-pinene (see Fig. 10). As mentioned earlier, the in-depth analysis of the addition of CH3C(O)OO⋅ to the double bond in α-pinene revealed a new, lower barrier compared to Pasik et al. (2024a) that proceeds through the tertiary radical formation pathway. This finding arises from considering the side from which the radical attacks the double bond. The analysis shows that the lowest barrier occurs through the formation of a tertiary radical in the R stereoisomer of the α-pinene + APR product.
The opening of the secondary ring rearrangement was considered. Calculations show that about 100 % of the accretion product undergoes O2 addition, and ring opening does not occur. Assuming the formation of an alkoxy radical in the subsequent step, both right and left C−C bond scissions were examined. Similar studies of α-pinene + NO3 were conducted by Kurtén et al. (2017) and subsequently reviewed by Draper et al. (2024). According to their calculations, the reaction between α-pinene and NO3 proceeds with the formation of a tertiary radical, and nitrooxy alkoxy β-scissions lead to the main product, α-pinoaldehyde, through left-side C−C bond scission. TST reaction rate coefficient calculations show that for all stereoisomers of α-pinene-derived RO radicals, the main product is α-pinoaldehyde formed via Rβ1. Similarly to the findings of Kurtén et al. (2017), the oxidation pathway of α-pinene leads to a closed-shell compound, resulting in the termination of the oxidation chain. This would lead to the small secondary organic aerosol (SOA) yields of α-pinene.
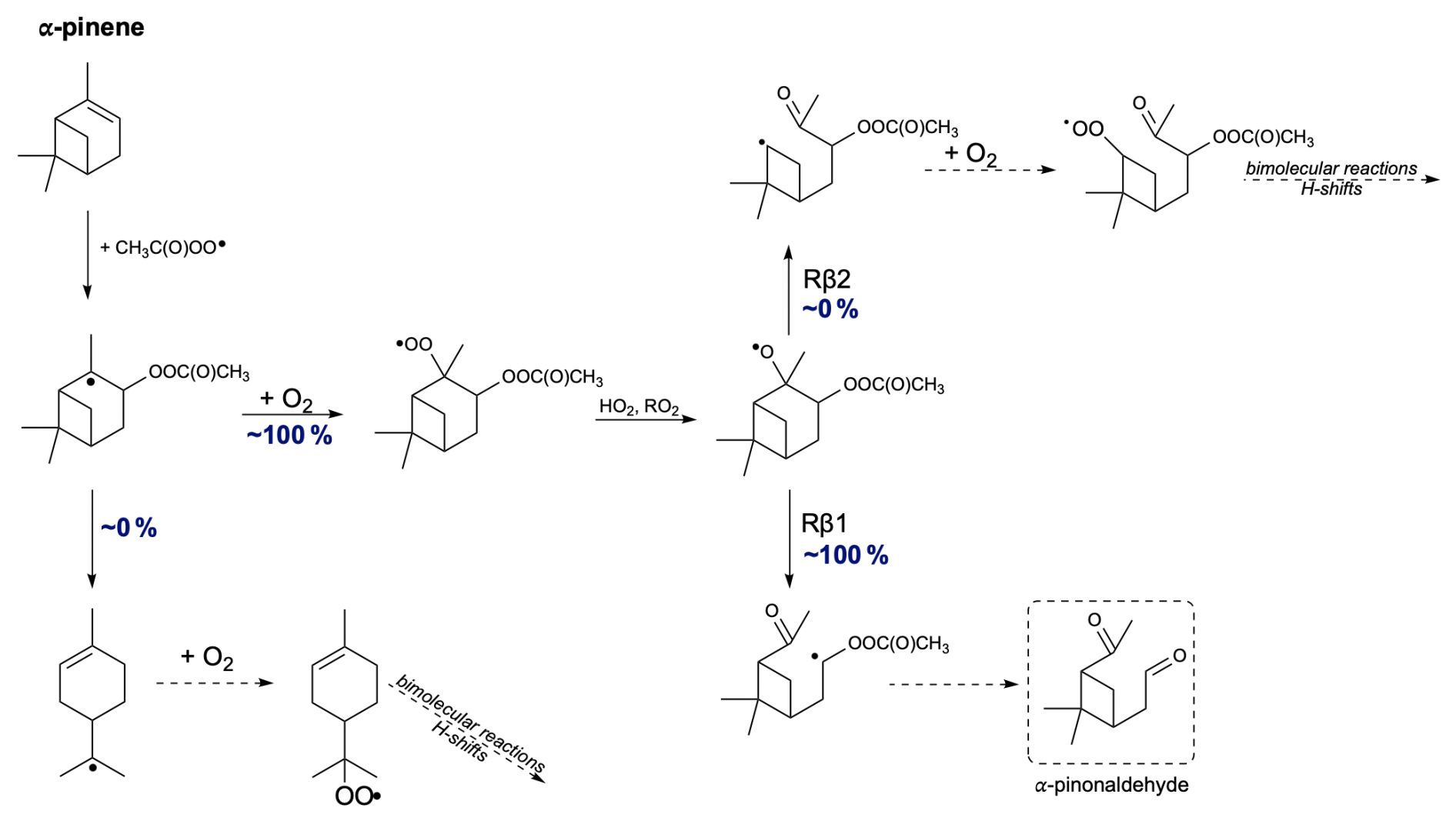
Figure 10Proposed fate of α-pinene + CH3C(O)OO⋅. The dashed box presents the closed-shell product that terminates the oxidation pathway.
3.3.3 β-Pinene
We now discuss the oxidation pathway initiated by APR for β-pinene, as shown in Fig. 11. Calculations indicate that the alkyl radical ring-opening rearrangement accounts for a minor yield of 2 %. This means that nearly all alkyl radicals proceed without ring opening and undergo O2 addition, leading to further reactions that result in the formation of the alkoxy radical. β-pinene is unique among exocyclic monoterpenes in that it provides significant branching ratios for all three possible C−C bond scissions. The most favoured pathway is Rβ3 for both stereoisomers but with different yields – 88 % for the R-alkoxy isomer and significantly less, 53 %, for the S-alkoxy isomer. This reaction leads to the formation of closed-shell nopinone. Similarly, for both stereoisomers, the least favoured pathway is the one leading to the left-side scission (Rβ2). Despite its similarity to α-pinene, β-pinene has the potential for various C−C bond scissions, whereas α-pinene favours the pathway leading to the closed-shell α-pinonaldehyde. Although for β-pinene, the analogous pathway leading to nopinone is the most favoured, other pathways are also competitive. This would result in the higher SOA yields observed for β-pinene compared to α-pinene.
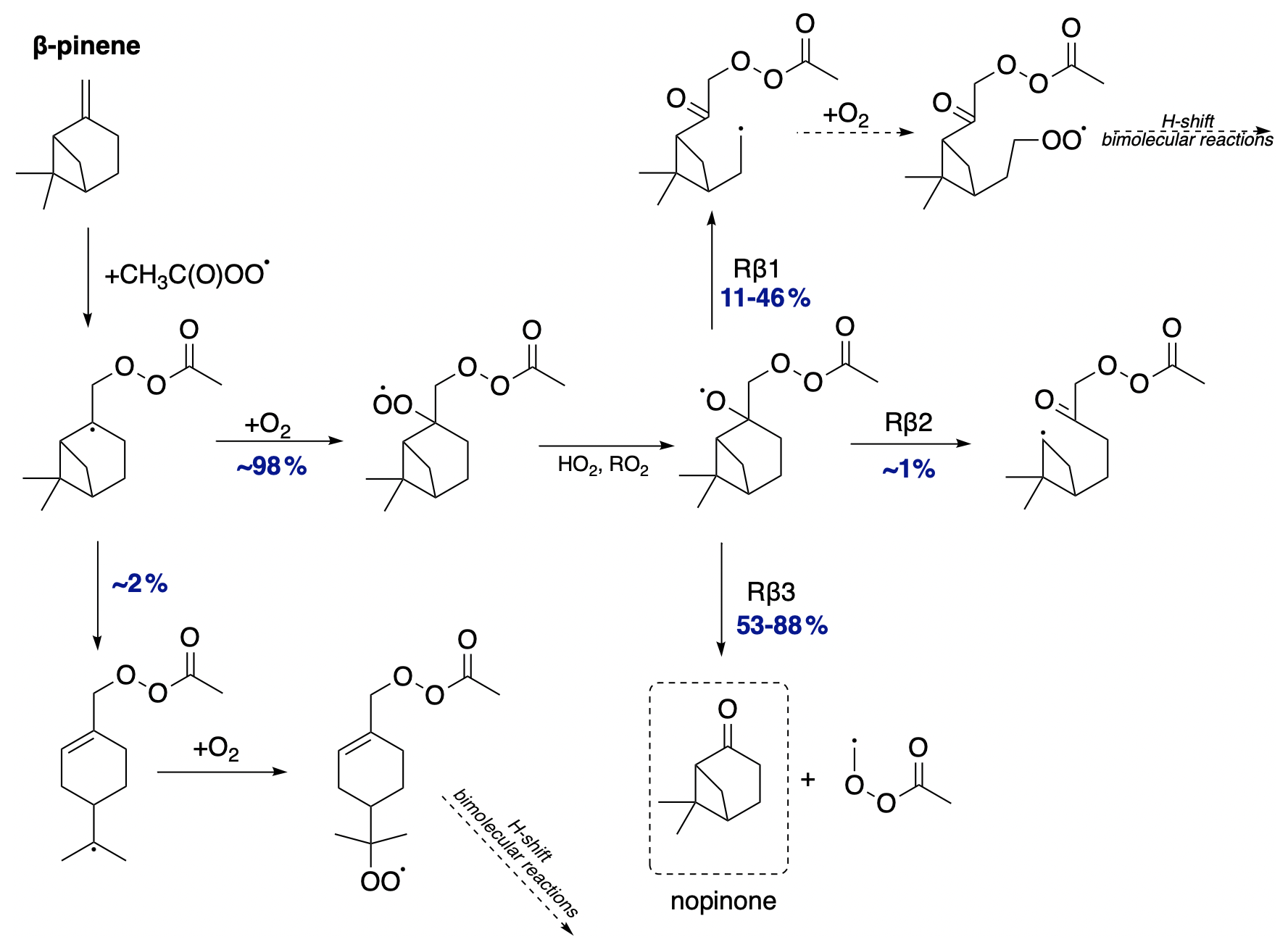
3.3.4 Camphene
The mechanism for camphene CH3C(O)OO⋅-initiated oxidation is shown in Fig. 12. Due to the presence of a five-membered ring in its structure, the ring-opening reaction contributes to smaller strain relief than in smaller-sized rings, directly resulting in a high energy barrier for this reaction. Consequently, ring-opening reactions are not competitive with O2 addition. For alkoxy β-scissions, the calculated reaction rate coefficients indicate that the vast majority (99 %–100 %) of camphene intermediates will favour right-sided scission (Rβ1), leading to a carbon-centred radical capable of further oxidation. The minor pathway, Rβ2, also leads to an intermediate whose oxidation pathway does not terminate early. It is noteworthy that the calculated reaction rate coefficients for both β-scissions of camphene radicals are faster than those for any other monoterpenes studied here. This is consistent with the results obtained by Draper et al. (2024). Moreover, the reaction leading to the formation of closed-shell camphenilone (Rβ3) is negligible, indicating no early oxidative chain termination and a potential for high SOA yields from camphene.
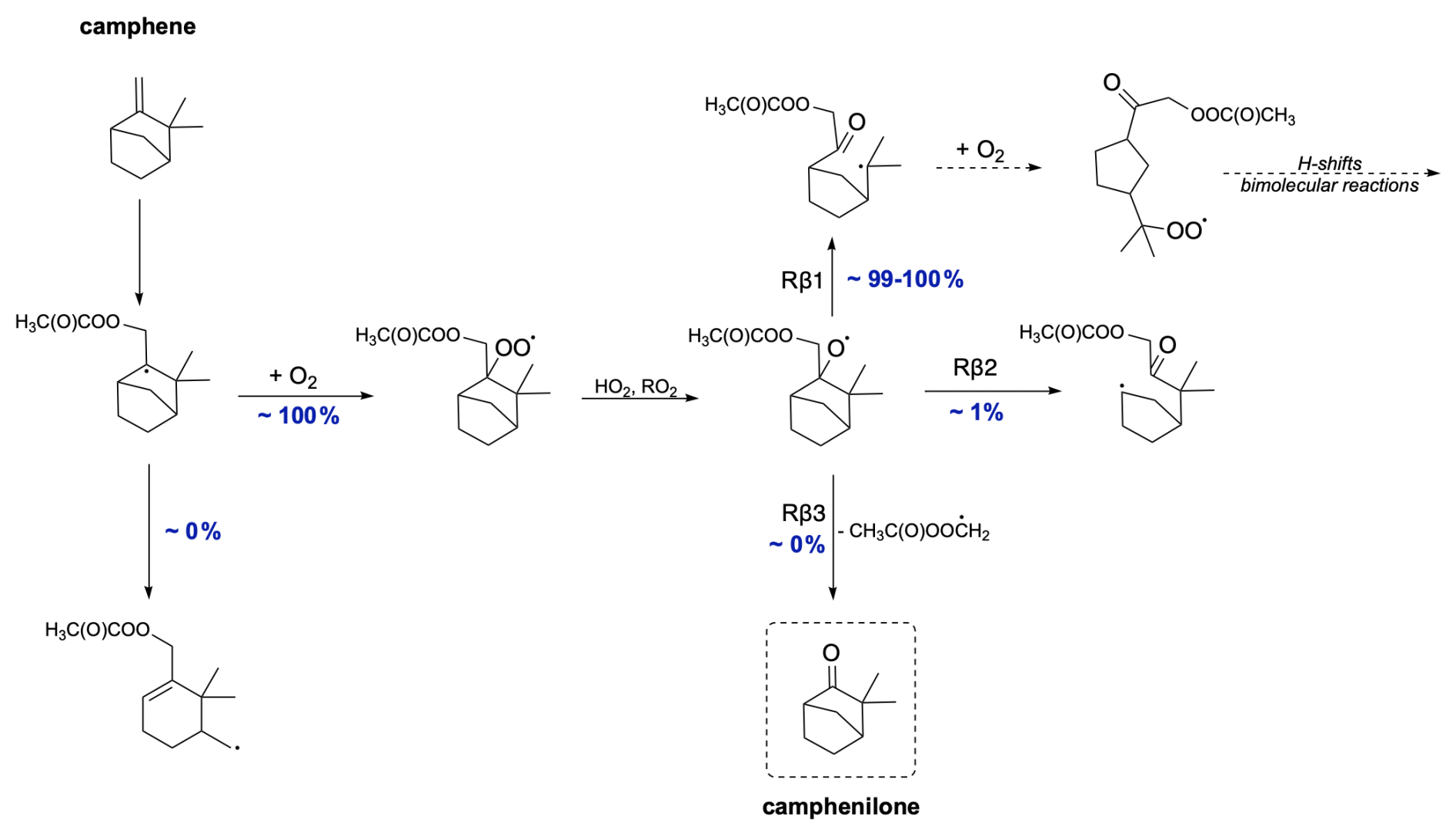
Figure 12Proposed fate of camphene + CH3C(O)OO⋅. The dashed box presents the closed-shell product that terminates the oxidation pathway.
3.3.5 α-Thujene
The mechanism for α-thujene oxidation is shown in Fig. 13. Due to the presence of a three-membered ring, the additional ring strain is significant enough that the earlier alkyl ring-opening radical rearrangement reaction becomes competitive with O2 addition, with a small but not negligible yield of 6 %–18 % (depending on the stereoisomer). The ring-opening reaction allows for an additional branch in the oxidation pathway of α-thujene. The resulting rearranged radical most likely undergoes O2 addition, forming a peroxy radical with the potential for further propagation reactions via H shifts, cyclization, bimolecular reactions, or addition to the newly formed double bond. Besides the unimolecular ring-opening rearrangement, the dominant pathway proceeds through O2 addition and the formation of an monoterpene + APR-derived alkoxy radical. For the two possible alkoxy scissions, we observe the exclusive formation of the Rβ1 product regardless of the stereoisomer. This results in the formation of an α-QOOR alkyl radical, which decomposes to α-thujonaldehyde, providing an additional source of this compound in the atmosphere.
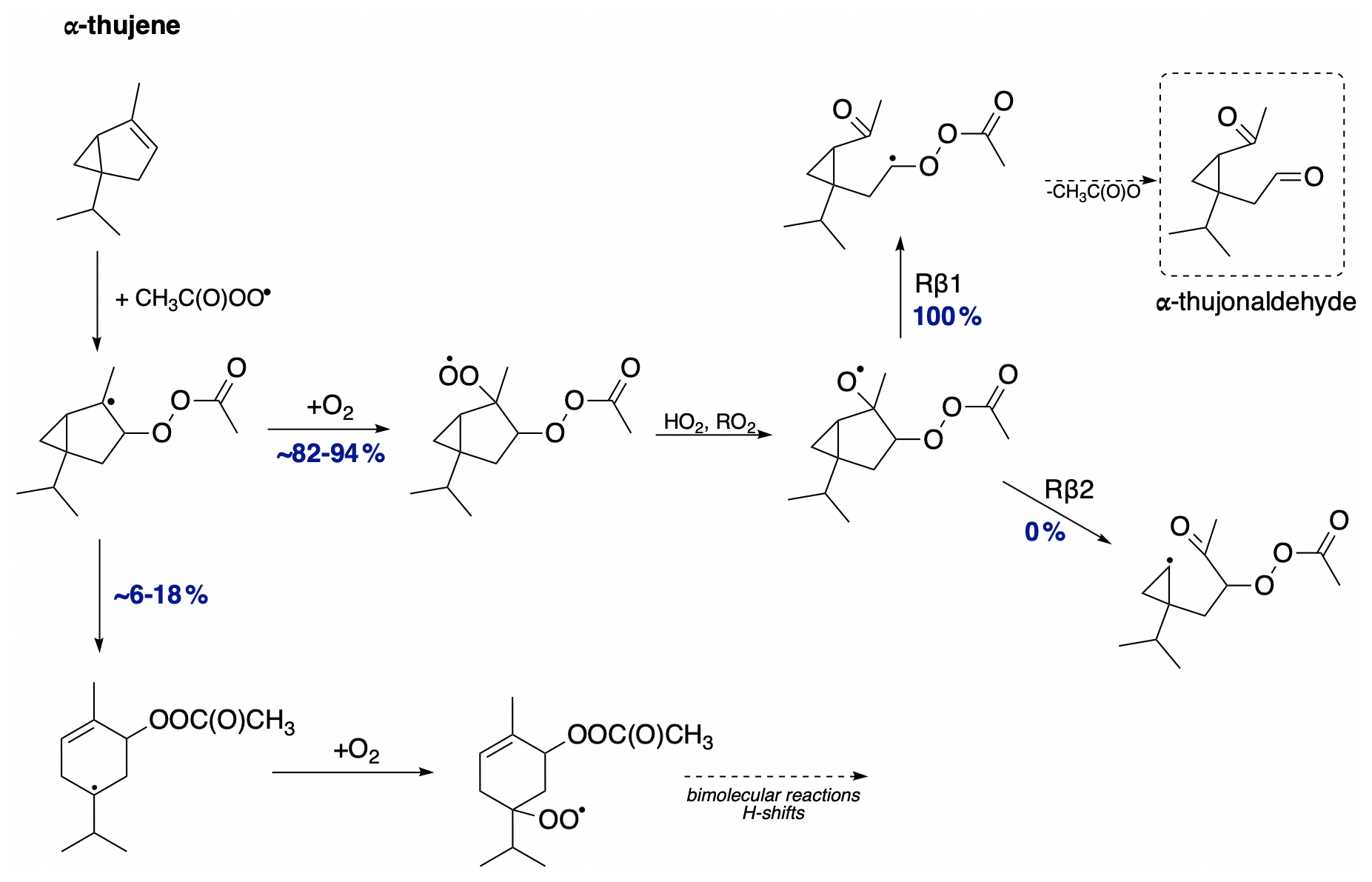
Figure 13Proposed fate of α-thujene + CH3C(O)OO⋅. The dashed box presents the closed-shell product that terminates the oxidation pathway.
3.3.6 Sabinene
The most interesting case is represented by sabinene. Among all bicyclic monoterpenes investigated, it was found that this monoterpene undergoes a competitive reaction with alkyl radical ring opening, with a yield of 31 % compared to direct O2 addition. This is significant enough to impact the chemistry of this compound and its oxidation pathways. Calculations show that the radical formed in this reaction can add O2 and perform a further H-shift reaction with the rate constant 2 s−1, leading to a more oxidized compound able to undergo additional propagation steps (via RO2 bimolecular reactions or unimolecular reactions) and leading to a highly oxidized compound with the potential to participate in SOA formation.
Regarding the further oxidation of the 69 % that proceeds without ring opening, the most favourable pathway for the alkoxy radical channels differs for the two R-APR stereoisomers. The R-alkoxy isomer favours the Rβ3 pathway, leading to closed-shell sabinaketone (63 %) and termination of the oxidative chain. On the other hand, the majority of the S-alkoxy isomer favours the Rβ1 scission (69 %), leading to a radical capable of further propagation. Additionally, the left-side β-scissions showed 0 % yield for both stereoisomers.
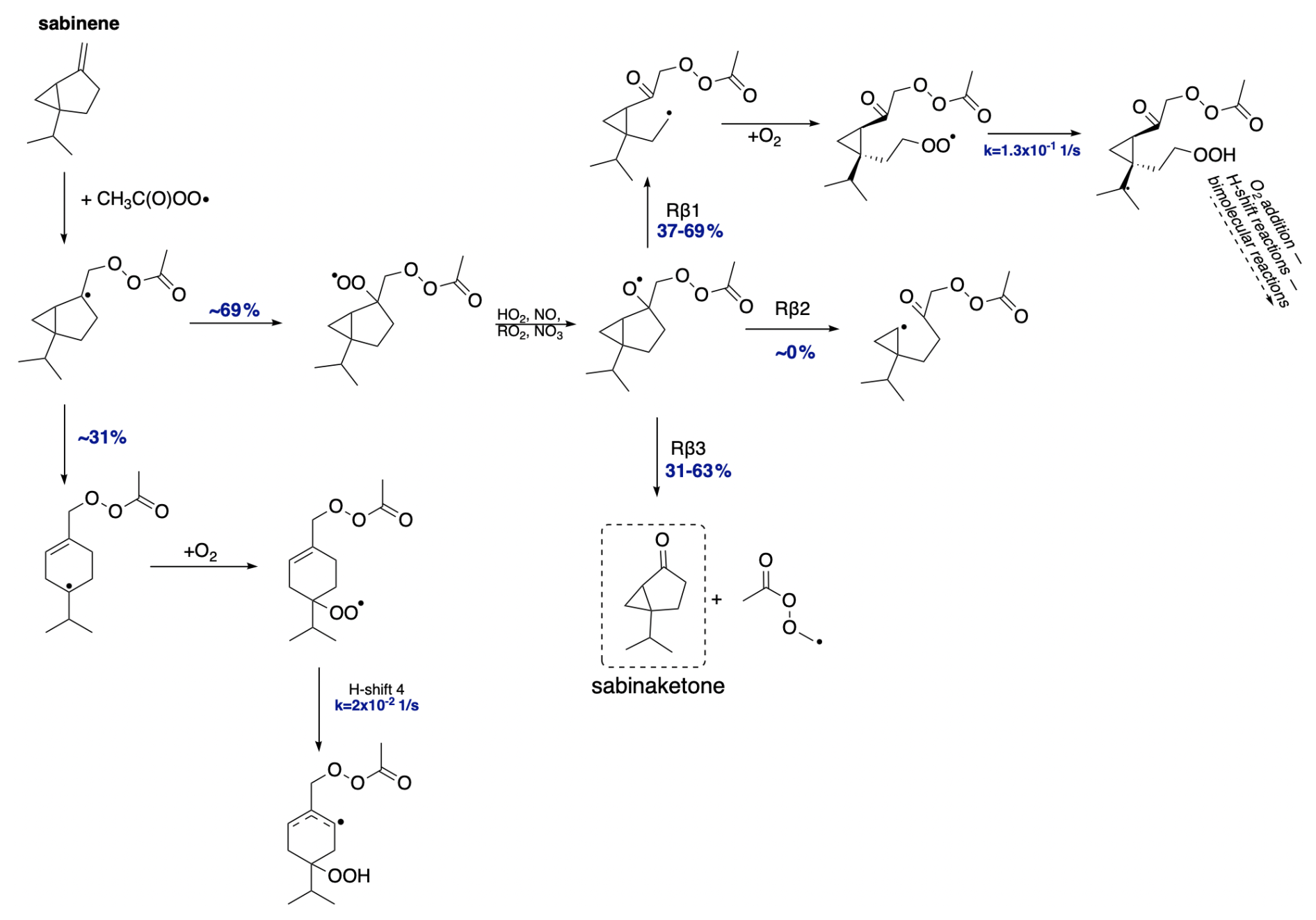
Figure 14Proposed fate of sabinene + CH3C(O)OO⋅. The dashed box presents the closed-shell product that terminates the oxidation pathway. The indicated yields for β-scission reactions depend on the stereoisomer (see Table 4).
For the right-side reaction, which facilitates further propagation, H-shift reactions for the RO2 radical were investigated (see Supplement for overview). The fastest of these reactions proceeds with a rate constant of 1.3 s−1, sufficient to compete with bimolecular reactions or the alkyl radical three-membered ring opening. Moreover, this reaction produces another radical that can undergo further oxidation. Along with alkyl ring-opening rearrangement, this suggests high SOA yields from sabinene + APR.
3.3.7 Δ-Carene
The proposed oxidation pathway for Δ-carene is shown in Fig. 15. Δ-carene exhibits a unique oxidation pathway. For the two possible C−C bond scissions, Δ-carene most prefers the right-side alkoxy β-scission but with varying branching ratios for different stereoisomers. This contrasts with the NO3-initiated oxidation chain published by Kurtén et al. (2017) and later by Draper et al. (2024), where their calculations showed that Δ-carene prefers left-side alkoxy scissions. This confirms the uniqueness of Δ-carene among monoterpenes and highlights the necessity for further investigation into its distinct oxidation behaviour. In the case of Rβ1 bond scission, an α-QOOR alkyl radical is formed, which then decomposes to caronaldehyde. The less preferred but still possible left-side C−C bond scission leads to the formation of a carbon-centred radical, which can undergo further secondary ring-opening reactions or O2 addition. This opens an oxidation pathway for Δ-carene and would lead to the relatively high SOA yields.
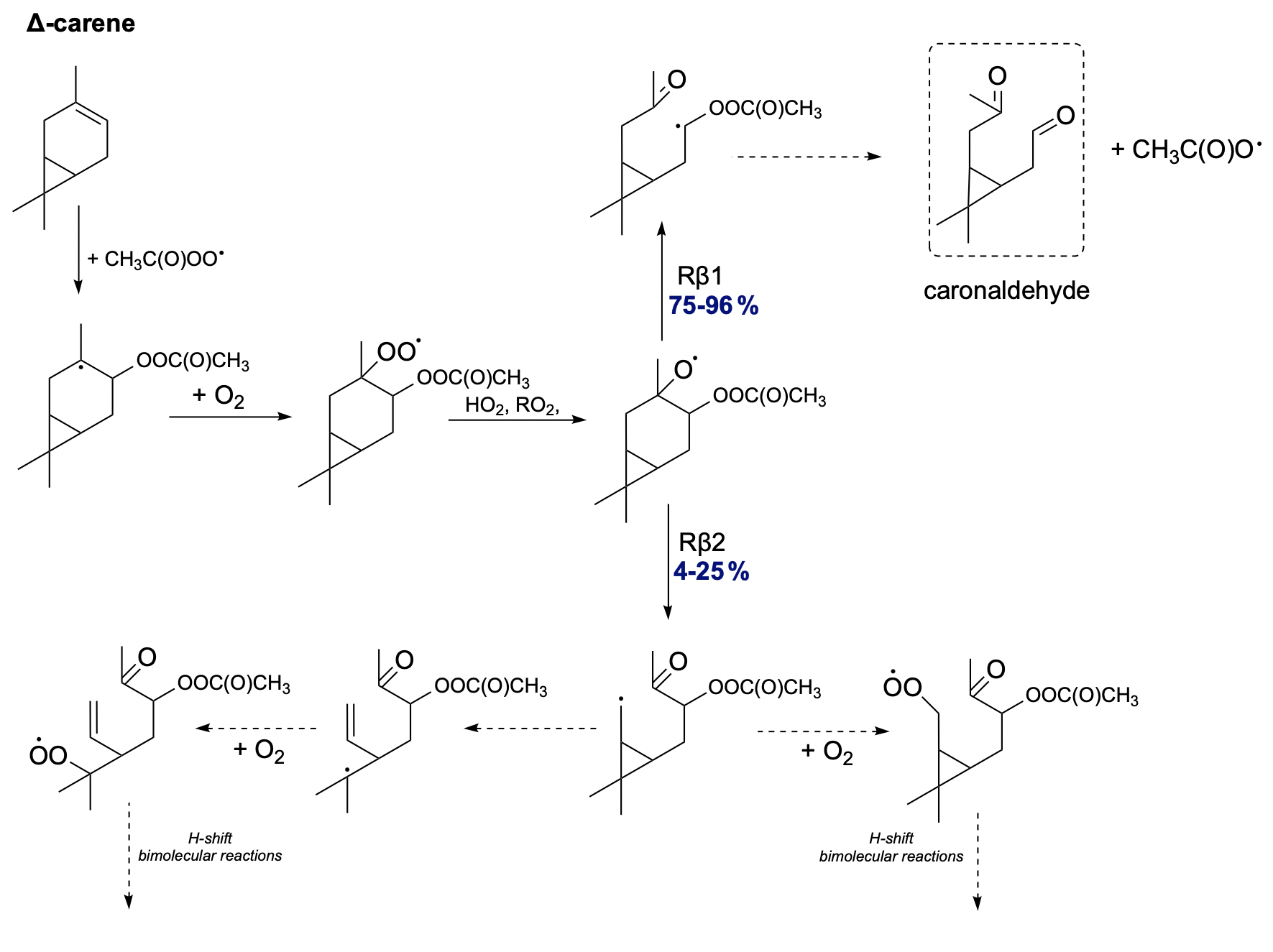
Figure 15Proposed fate of Δ-carene + CH3C(O)OO⋅. The dashed box presents the closed-shell product that terminates the oxidation pathway.
In this study, we investigated the possible oxidation pathways for CH3C(O)OO⋅-initiated oxidation of seven atmospherically relevant monoterpenes. Building upon research by Pasik et al. (2024a), Draper et al. (2024), and Kurtén et al. (2017), we identified oxidation pathways that could lead to the high SOA yields for certain monoterpenes and low SOA yields for others.
Our calculations indicate that alkyl radical rearrangement is significant primarily for sabinene and α-thujene, where this pathway competes with O2 addition. For the other monoterpenes (β-pinene, α-pinene, camphene), this channel is minor (0 %–1 %). This is consistent with observations by Draper et al. (2024), who noted that the preference for ring opening is partly dependent on ring size and the resulting ring strain released – this effect being more pronounced for smaller rings. The variance in ring-opening yields seen in sabinene and α-thujene, both featuring a three-membered ring, implies that stereoelectronic effects and the APR's position may significantly influence reactivity. For sabinene, further O2 addition and H-shift reactions were investigated. The fastest H shift, which leads to a resonance-stabilized allylic radical, has a reaction rate coefficient on the order of 0.01 s−1, making it competitive under atmospheric conditions. This oxidation pathway may represent an APR-initiated autoxidation mechanism proposed by Pasik et al. (2024a).
Our calculations show that each monoterpene has its unique reactivity towards oxidation pathways. The probable oxidation pathway for limonene leads to the formation of closed-shell endolim, which terminates the oxidation chain. This would lead to low SOA yield of APR-initiated oxidation of limonene, as the termination prevents further radical propagation and aerosol formation. Despite having similar structures, the β-scission rearrangements for α- and β-pinene differ significantly due to the position of the double bond. For α-pinene, the most favourable pathway leads to the formation of an α-QOOR radical that decomposes to α-pinonaldehyde. In contrast, for β-pinene, this pathway competes with C−C bond scission that allows for further radical propagation steps. For camphene with a five-membered secondary ring, alkoxy rearrangements predominantly lead to right-sided scission (Rβ1), resulting in a carbon-centred radical capable of further oxidation. In the case of α-thujene and sabinene, which differ only in the position of the double bond, significant differences in preferred oxidation pathways are observed. While α-thujene consistently undergoes complete right-side β-scission, leading ultimately to α-thujonaldehyde, sabinene exhibits two competitive pathways with varying stereochemical preferences. These intriguing observations indicate that steric effects and stereochemistry clearly play a significant role in controlling oxidative chains. For Δ-carene, a major preference for right-side β-scission is observed, varying between 75 % and 96 % depending on the stereoisomer. Results suggest a general conclusion that all endocyclic monoterpenes prefer right-side C−C bond scission, leading to closed-shell products. However, exocyclic monoterpenes do not exhibit a consistent trend, but their oxidation pathways proceed via intermediate products, allowing for further propagation steps and higher SOA contribution.
This work highlights the importance of APRs in atmospheric oxidation chemistry, particularly in the formation of oxygenated multifunctional compounds. However, it should be noted that although the rearrangements presented above exhibit feasible rate constants under atmospheric conditions, the overall significance of these reactions is highly dependent on the initial step, which is the addition of APR to the double bond in a monoterpene. While calculations may indicate a small percentage compared to OH oxidation, the actual results are heavily dependent on the real concentrations of APRs in the atmosphere and the actual reaction rate constants. Furthermore, as shown by multiple studies, the main sink for APR is its reaction with NOx to form PANs. Consequently, the reactions of APRs with monoterpenes (or unsaturated hydrocarbons in general) will be most significant in areas with low concentrations of NOx (Orlando and Tyndall, 2012; Zheng et al., 2011; Cleary et al., 2007; Liu et al., 2021; Rissanen, 2018). With this work, authors aim to emphasize the importance of further research in this direction, particularly experimental studies, to determine whether APR-initiated oxidation can indeed impact the atmospheric chemistry of unsaturated hydrocarbons.
The optimized structures and calculation output files of all relevant compounds that support the findings of the paper are available in the Zenodo repository at https://doi.org/10.5281/zenodo.14892593 (Pasik, 2025).
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-4313-2025-supplement.
DP performed the calculations and wrote the manuscript; TGA assisted with the MESMER calculations; EA carried out the calculations for Sect. 3.2 for sabinene; and TGA, SI, and NM contributed to the analysis. The study was designed and supervised by NM. All authors proofread the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We acknowledge the CSC-IT Center for Science in Espoo, Finland, for computational resources.
Dominika Pasik thanks the Doctoral Programme in Atmospheric Sciences (ATM-DP) at the University of Helsinki for the provided funding.
This research has been supported by the Research Council of Finland (grant nos. 347775 and 355966).
Open-access funding was provided by the Helsinki University Library.
This paper was edited by Harald Saathoff and reviewed by two anonymous referees.
Anglada, J. M., Crehuet, R., and Francisco, J. S.: The stability of α-hydroperoxyalkyl radicals, Chemistr, 22, 18092–18100, 2016. a
Bannwarth, C., Ehlert, S., and Grimme, S.: GFN2-xTB-An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comput., 15, 1652–1671, 2019. a
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-phase ozonolysis of cycloalkenes: formation of highly oxidized RO2 radicals and their reactions with NO, NO2, SO2, and other RO2 radicals, J. Phys. Chem. A, 119, 10336–10348, 2015. a
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M., Roldin, P., Berndt, T., Crounse, J., Wennberg, P., Mentel, T., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D., Thornton, J., Donahue, N., Kjaergaard, H., and Ehn, M.: Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: A key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, 2019. a
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015. a
Chai, J.-D. and Head-Gordon, M.: Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 10, 6615–6620, 2008. a
Cleary, P. A., Wooldridge, P. J., Millet, D. B., McKay, M., Goldstein, A. H., and Cohen, R. C.: Observations of total peroxy nitrates and aldehydes: measurement interpretation and inference of OH radical concentrations, Atmos. Chem. Phys., 7, 1947–1960, https://doi.org/10.5194/acp-7-1947-2007, 2007. a
Dada, L., Stolzenburg, D., Simon, M., Fischer, L., Heinritzi, M., Wang, M., Xiao, M., Vogel, A., Ahonen, L., Amorim, A., Baalbaki, R., Baccarini, A., Baltensperger, U., Bianchi, F., Dällenbach, K., DeVivo, J., Dias, A., Dommen, J., Duplissy, J., Finkenzeller, H., Hansel, A., He, X.-C., Hofbauer, V., Hoyle, C., Kangasluoma, J., Kim, C., Kürten, A., Kvashnin, A., Mauldin, R., Makhmutov, V., Marten, R., Mentler, B., Nie, W., Petäjä, T., Quéléver, L., Saathoff, H., Tauber, C., Tome, A., Molteni, U., Volkamer, R., Wagner, R., Wagner, A., Wimmer, D., Winkler, P., Yan, C., Zha, Q., Rissanen, M., Gordon, H., Curtius, J., Worsnop, D., Lehtipalo, K., Donahue, N., Kirkby, J., Haddad, I., and Kulmala, M.: Role of sesquiterpenes in biogenic new particle formation, Sci. Adv., 9, eadi5297, https://doi.org/10.1126/sciadv.adi5297, 2023. a
Day, D. A., Fry, J. L., Kang, H. G., Krechmer, J. E., Ayres, B. R., Keehan, N. I., Thompson, S. L., Hu, W., Campuzano-Jost, P., Schroder, J. C., Stark, H., DeVault, M. P., Ziemann, P. J., Zarzana, K. J., Wild, R. J., Dubè, W. P., Brown, S. S., and Jimenez, J. L.: Secondary organic aerosol mass yields from NO3 oxidation of α-pinene and Δ-carene: Effect of RO2 radical fate, J. Phys. Chem. A, 126, 7309–7330, 2022. a
Draper, D. C., Farmer, D. K., Desyaterik, Y., and Fry, J. L.: A qualitative comparison of secondary organic aerosol yields and composition from ozonolysis of monoterpenes at varying concentrations of NO2, Atmos. Chem. Phys., 15, 12267–12281, https://doi.org/10.5194/acp-15-12267-2015, 2015. a
Draper, D., Almeida, T. G., Iyer, S., Smith, J. N., Kurtén, T., and Myllys, N.: Unpacking the diversity of monoterpene oxidation pathways via nitrooxy–alkyl radical ring-opening reactions and nitrooxy–alkoxyl radical bond scissions, J. Aerosol Sci., 179, 106379, https://doi.org/10.1016/j.jaerosci.2024.106379, 2024. a, b, c, d, e, f, g, h, i, j, k, l, m
Draper, D. C., Myllys, N., Hyttinen, N., Møller, K. H., Kjaergaard, H. G., Fry, J. L., Smith, J. N., and Kurtén, T.: Formation of Highly Oxidized Molecules from NO3 Radical Initiated Oxidation of Δ-3-Carene: A Mechanistic Study, ACS Earth and Space Chem., 3, 1460–1470, 2019. a
Dunning Jr., T. H.: Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 90, 1007–1023, 1989. a
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, 2014. a
Friedman, B. and Farmer, D. K.: SOA and gas phase organic acid yields from the sequential photooxidation of seven monoterpenes, Atmos. Environ., 187, 335–345, 2018. a, b
Gao, C. W., Allen, J. W., Green, W. H., and West, R. H.: Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms, Comput. Phys. Commun., 203, 212–225, 2016. a
Glasius, M. and Goldstein, A. H.: Recent discoveries and future challenges in atmospheric organic chemistry, Environ. Sci. Technol., 50, 2754–2764, 2016. a
Glowacki, D. R., Liang, C.-H., Morley, C., Pilling, M. J., and Robertson, S. H.: MESMER: an open-source master equation solver for multi-energy well reactions, J. Phys. Chem. A, 116, 9545–9560, 2012. a
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a
Hantschke, L., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., Glowania, M., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Atmospheric photooxidation and ozonolysis of Δ3-carene and 3-caronaldehyde: rate constants and product yields, Atmos. Chem. Phys., 21, 12665–12685, https://doi.org/10.5194/acp-21-12665-2021, 2021. a
Hariharan, P. C. and Pople, J. A.: The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 28, 213–222, 1973. a
Hasan, G., Salo, V.-T., Valiev, R. R., Kubecka, J., and Kurten, T.: Comparing reaction routes for 3 (ROOR') intermediates formed in peroxy radical self-and cross-reactions, The J. Phys. Chem. A, 124, 8305–8320, 2020. a
Iyer, S., Reiman, H., Møller, K. H., Rissanen, M. P., Kjaergaard, H. G., and Kurten, T.: Computational investigation of RO2 + HO2 and RO2 + RO2 reactions of monoterpene derived first-generation peroxy radicals leading to radical recycling, J. Phys. Chem. A, 122, 9542–9552, 2018. a
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 12, 878, https://doi.org/10.1038/s41467-021-21172-w, 2021. a
Joback, K. G. and Reid, R. C.: Estimation of pure-component properties from group-contributions, Chem. Eng. Commun., 57, 233–243, 1987. a, b
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012. a
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005. a
Karppinen, I., Pasik, D., Ahongshangbam, E., and Myllys, N.: The impact of unimolecular reactions on the possibility of acyl peroxy radical initiated isoprene oxidation, Aerosol Res., 3, 175–183, https://doi.org/10.5194/ar-3-175-2025, 2025. a
Kurtén, T., Møller, K. H., Nguyen, T. B., Schwantes, R. H., Misztal, P. K., Su, L., Wennberg, P. O., Fry, J. L., and Kjaergaard, H. G.: Alkoxy radical bond scissions explain the anomalously low secondary organic aerosol and organonitrate yields from α-pinene + NO3, J. Phys. Chem. Lett., 8, 2826–2834, 2017. a, b, c, d, e
Lee, A., Goldstein, A. H., Kroll, J. H., Ng, N. L., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes, J. Geophys. Res.-Atmos., 111, https://doi.org/10.1029/2005JD006437, 2006. a
Liu, J., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Bell, D. M., Zaveri, R. A., Zelenyuk, A., Thornton, J. A., and Shilling, J. E.: Monoterpene Photooxidation in a Continuous-Flow Chamber: SOA Yields and Impacts of Oxidants, NOx, and VOC Precursors, Environ. Sci. Technol., 56, 12066–12076, 2022. a
Liu, Y., Shen, H., Mu, J., Li, H., Chen, T., Yang, J., Jiang, Y., Zhu, Y., Meng, H., Dong, C., Wang, W., and Xue, L.: Formation of peroxyacetyl nitrate (PAN) and its impact on ozone production in the coastal atmosphere of Qingdao, North China, Sci. Total Environ., 778, 146265, https://doi.org/10.1016/j.scitotenv.2021.146265, 2021. a
Lun, X., Lin, Y., Chai, F., Fan, C., Li, H., and Liu, J.: Reviews of emission of biogenic volatile organic compounds (BVOCs) in Asia, J. Environ. Sci., 95, 266–277, 2020. a
Møller, K. H., Otkjær, R. V., Hyttinen, N., Kurtén, T., and Kjaergaard, H. G.: Cost-effective implementation of multiconformer transition state theory for peroxy radical hydrogen shift reactions, J. Phys. Chem. A, 120, 10072–10087, 2016. a
Møller, K. H., Otkjær, R. V., Chen, J., and Kjaergaard, H. G.: Double bonds are key to fast unimolecular reactivity in first-generation monoterpene hydroxy peroxy radicals, J. Phys. Chem. A, 124, 2885–2896, 2020. a, b
Myllys, N., Elm, J., Halonen, R., Kurtén, T., and Vehkamäki, H.: Coupled cluster evaluation of the stability of atmospheric acid–base clusters with up to 10 molecules, J. Phys. Chem. A, 120, 621–630, 2016a. a
Myllys, N., Elm, J., and Kurten, T.: Density functional theory basis set convergence of sulfuric acid-containing molecular clusters, Computat. Theor. Chem., 1098, 1–12, 2016b. a
Nozière, B. and Fache, F.: Reactions of organic peroxy radicals, RO2, with substituted and biogenic alkenes at room temperature: unsuspected sinks for some RO2 in the atmosphere?, Chem. Sci., 12, 11676–11683, 2021. a
Nozière, B. and Vereecken, L.: H-shift and cyclization reactions in unsaturated alkylperoxy radicals near room temperature: propagating or terminating autoxidation?, Phys. Chem. Chem. Phys., 26, 25373–25384, 2024. a
Nozière, B., Durif, O., Dubus, E., Kylington, S., Emmer, Å., Fache, F., Piel, F., and Wisthaler, A.: The reaction of organic peroxy radicals with unsaturated compounds controlled by a non-epoxide pathway under atmospheric conditions, Phys. Chem. Chem. Phys., 25, 7772–7782, 2023. a
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, 2012. a
Pasik, D.: Monoterpene oxidation pathways initiated by acyl peroxy radical addition, Zenodo [data set], https://doi.org/10.5281/zenodo.14892593, 2025. a
Pasik, D., Frandsen, B. N., Meder, M., Iyer, S., Kurtén, T., and Myllys, N.: Gas-Phase Oxidation of Atmospherically Relevant Unsaturated Hydrocarbons by Acyl Peroxy Radicals, J. Am. Chem. Soc., 146, 13427–13437, 2024a. a, b, c, d, e, f, g, h, i, j, k
Pasik, D., Iyer, S., and Myllys, N.: Cost-effective approach for atmospheric accretion reactions: a case of peroxy radical addition to isoprene, Phys. Chem. Chem. Phys., 26, 2560–2567, 2024b. a
Pracht, P., Bohle, F., and Grimme, S.: Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 22, 7169–7192, 2020. a
Presto, A. A., Huff Hartz, K. E., and Donahue, N. M.: Secondary organic aerosol production from terpene ozonolysis. 1. Effect of UV radiation, Environ. Sci. Technol., 39, 7036–7045, 2005. a
Riplinger, C., Sandhoefer, B., Hansen, A., and Neese, F.: Natural Triple Excitations in Local Coupled Cluster Calculations with Pair Natural Orbitals, J. Chem. Phys., 139, 134101, https://doi.org/10.1063/1.4821834, 2013. a
Riplinger, C., Pinski, P., Becker, U., Valeev, E. F., and Neese, F.: Sparse Maps–A Systematic Infrastructure for Reduced-Scaling Electronic Structure Methods. II. Linear Scaling Domain Based Pair Natural Orbital Coupled Cluster Theory, J. Chem. Phys., 144, 024109, https://doi.org/10.1063/1.4939030, 2016. a
Rissanen, M. P.: NO2 suppression of autoxidation–inhibition of gas-phase highly oxidized dimer product formation, ACS Earth and Space Chemistry, 2, 1211–1219, 2018. a
Rissanen, M., Kurten, T., Sipilä, M., Thornton, J., Kangasluoma, J., Sarnela, N., Junninen, H., Jorgensen, S., Schallhart, S., Kajos, M., Taipale, R., Springer, M., Mentel, T., Ruuskanen, T., Petäjä, T., Worsnop, D., Kjaergaard, H., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, 2014. a
Seal, P., Barua, S., Iyer, S., Kumar, A., and Rissanen, M.: A systematic study on the kinetics of H-shift reactions in pristine acyl peroxy radicals, Phys. Chem. Chem. Phys., 25, 28205–28212, 2023. a
Shrivastava, M., Cappa, C., Fan, J., Goldstein, A., Guenther, A., Jimenez, J., Kuang, C., Laskin, A., Martin, S., Ng, N., Petäjä, T., Pierce, J., Rasch, P., Roldin, P., Seinfeld, J., Shilling, J., Smith, J., Thornton, J., Volkamer, R., Wang, J., Worsnop, D., Zaveri, R., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, 2017. a
Skodje, R. T., Truhlar, D. G., and Garrett, B. C.: A general small-curvature approximation for transition-state-theory transmission coefficients, J. Phys. Chem., 85, 3019–3023, 1981. a
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011. a
Tee, L. S., Gotoh, S., and Stewart, W. E.: Molecular parameters for normal fluids. Lennard-Jones 12-6 Potential, Ind. Eng. Chem. Fund., 5, 356–363, 1966. a
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020. a
Vereecken, L. and Peeters, J.: The 1, 5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system, J. Chem. Phys., 119, 5159–5170, 2003. a
Vereecken, L., Nguyen, T., Hermans, I., and Peeters, J.: Computational study of the stability of α-hydroperoxyl-or α-alkylperoxyl substituted alkyl radicals, Chem. Phys. Lett., 393, 432–436, 2004. a
Vereecken, L., Vu, G., Wahner, A., Kiendler-Scharr, A., and Nguyen, H.: A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 23, 16564–16576, 2021. a
Viegas, L.: A Multiconformational Transition State Theory Approach to OH Tropospheric Degradation of Fluorotelomer Aldehydes, Chem. Phys. Chem., 24, https://doi.org/10.1002/cphc.202300259, 2023. a
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, 2015. a
Xu, L., Pye, H. O. T., He, J., Chen, Y., Murphy, B. N., and Ng, N. L.: Experimental and model estimates of the contributions from biogenic monoterpenes and sesquiterpenes to secondary organic aerosol in the southeastern United States, Atmos. Chem. Phys., 18, 12613–12637, https://doi.org/10.5194/acp-18-12613-2018, 2018. a
Zhang, F. and Dibble, T. S.: Impact of tunneling on hydrogen-migration of the n-propylperoxy radical, Phys. Chem. Chem. Phys., 13, 17969–17977, 2011. a
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, 2018. a
Zhao, D. F., Kaminski, M., Schlag, P., Fuchs, H., Acir, I.-H., Bohn, B., Häseler, R., Kiendler-Scharr, A., Rohrer, F., Tillmann, R., Wang, M. J., Wegener, R., Wildt, J., Wahner, A., and Mentel, Th. F.: Secondary organic aerosol formation from hydroxyl radical oxidation and ozonolysis of monoterpenes, Atmos. Chem. Phys., 15, 991–1012, https://doi.org/10.5194/acp-15-991-2015, 2015. a
Zhao, Q., Møller, K. H., Chen, J., and Kjaergaard, H. G.: Cost-effective implementation of multiconformer transition state theory for alkoxy radical unimolecular reactions, J. Phys. Chem. A, 126, 6483–6494, 2022. a
Zheng, W., Flocke, F. M., Tyndall, G. S., Swanson, A., Orlando, J. J., Roberts, J. M., Huey, L. G., and Tanner, D. J.: Characterization of a thermal decomposition chemical ionization mass spectrometer for the measurement of peroxy acyl nitrates (PANs) in the atmosphere, Atmos. Chem. Phys., 11, 6529–6547, https://doi.org/10.5194/acp-11-6529-2011, 2011. a