the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Nov 2025
| 17 Nov 2025
Formation of highly oxygenated organic molecules from α-pinene photooxidation: evidence for the importance of highly oxygenated alkoxy radicals
Sungah Kang
Jürgen Wildt
Iida Pullinen
Luc Vereecken
Andreas Wahner
Thomas F. Mentel
Highly oxygenated organic compounds (HOMs) from α-pinene oxidation are of great interest because of their importance in secondary organic aerosol (SOA) formation. Despite intensive investigations, the mechanisms of HOM formation from first-generation peroxy radicals to HOM-peroxy radicals (HOM-RO2•) and to HOM-closed shell products are not well understood. One reason is that HOM-alkoxy radicals (HOM-RO•) are likely to contribute to the propagation of oxidative radical chains (alkoxy-peroxy pathway) because isomerization of functionalized alkoxy radicals can compete with their fragmentation (and reaction with O2), as shown by theoretical kinetics. However, HOM-RO• reaction steps are difficult to verify in mechanisms. In this work, we have investigated HOM formation by varying the significance of the alkoxy-peroxy pathway as a function of NOx, OH•, and CO. HOM-RO• are likely formed with high branching ratios in reactions of HOM-RO2• with peroxy radicals and NO in analogy to simpler alkoxy radicals. We provide experimental evidence that for HOM-RO• the branching into isomerization is about 50 % (±14 %). Thus, HOM-RO• can play a central role in HOM formation, since alkoxy-peroxy pathways can compete with direct autoxidation. We observed significant concentrations of HOM-RO2•, despite fast termination by NO, and shifts to higher for HOM-RO2• and termination products with increasing NO. At NO concentrations > 1.5 ppb, the alkoxy-peroxy pathway may even prevail in propagating the oxidative radical chain leading to HOM formation. The increasing sink of HOM-RO2• with increasing concentration of peroxy radicals and NO is compensated by an increasing source via the alkoxy-peroxy pathway.
This manuscript is dedicated to Astrid Kiendler-Scharr (deceased, 6 February 2023).
- Article
(3082 KB) - Full-text XML
-
Supplement
(1589 KB) - BibTeX
- EndNote




Volatile organic compounds (VOCs) play a key role and fuel the oxidation cycle in the atmosphere. Biogenic and anthropogenic sources contribute to atmospheric VOCs; on a global scale, biogenic VOCs are considered more important than the anthropogenic VOCs, accounting for about 90 % of total VOC emissions (Atkinson and Arey, 2003a; Guenther et al., 2012; Lamarque et al., 2010). Among biogenic VOCs isoprene is the most dominant nonmethane emission, followed by α-pinene, which is the most emitted monoterpene with about 32 Tg of C yr−1, contributing 34 % to the total terpene emission (Guenther et al., 2012; Sindelarova et al., 2014). In the atmosphere α-pinene is oxidized by ozone (O3), hydroxyl radicals (OH•), and nitrate radicals (NO3•), producing less volatile oxidized organic compounds with varying oxygen content (Atkinson and Arey, 2003a). Assuming a daytime [OH•] of about 2 × 106 molec. cm−3 and an [O3] of about 30 ppbv, the OH• radical is the main oxidant of α-pinene during the day. (Note: in the following we denote the concentration or mixing ratio of a compound X by [X].) Although oxidation of α-pinene with the OH• radical has been well studied, the complete oxidation mechanism has not yet been established (Aschmann et al., 2002; Berndt et al., 2016; Berndt, 2021; Capouet et al., 2008; Eddingsaas et al., 2012a; Ehn et al., 2017; Nozière et al., 1999; Peeters et al., 2001; Xu et al., 2019). As shown by Ehn et al. (2014) and Berndt et al. (2016), α-pinene oxidation can lead to the formation of highly oxygenated organic molecules (HOM). Here, we define HOM as molecules that have more than six oxygen atoms and are formed by a process called autoxidation (Bianchi et al., 2019).
It is important to disentangle HOM formation from α-pinene and other monoterpenes since the oxidation products of monoterpenes, including HOMs, contribute significantly to secondary organic aerosol (SOA) formation (Hallquist et al., 2009; Kanakidou et al., 2005; Pye et al., 2010). Among all oxidation products, HOMs are potentially crucial precursors for SOA formation due to their low to extremely low volatility. HOMs participate in the formation of new particles and contribute to particle growth (Ehn et al., 2014; Jokinen et al., 2015; McFiggans et al., 2019; Mutzel et al., 2015; Stolzenburg et al., 2018; Tröstl et al., 2016). Highly oxidized accretion products formed by HOM-peroxy radicals (HOM-RO2•) may have been an important source of new particles in the absence of sulfuric acid in the pre-industrial era (Kirkby et al., 2016; Bianchi et al., 2016; Jokinen et al., 2017). Therefore, understanding HOM formation is crucial for describing the impact of secondary organic aerosols on climate and human health (Davidson et al., 2005; Hallquist et al., 2009; Von Schneidemesser et al., 2015).
A key in HOM formation is the autoxidation of peroxy radicals. Autoxidation is known to be an important chemical process in low-temperature combustion (Cox and Cole, 1985). However, only recently researchers have discovered the importance of autoxidation for atmospheric oxidation processes (Bianchi et al., 2019; Crounse et al., 2012; Crounse et al., 2013; Ehn et al., 2014). Autoxidation of peroxy radicals (Reaction R1) starts with an intramolecular hydrogen shift from a C-H bond to the peroxy radical group, forming a carbon-centered radical and a hydroperoxide group followed by the addition of an oxygen molecule to the carbon radical center. Autoxidation rapidly and efficiently generates series of peroxy radicals (HOM-RO2•) with oxygen to carbon ratios of up to one or even larger. HOM-RO2• react with other reactants such as hydroperoxyl radicals (HO2•), peroxy radicals (RO2•), and NO to form stable closed-shell multifunctional compounds. Herein the termination groups can be hydroperoxides, carbonyls, alcohols, or organic nitrates (Reactions R2, R3a, b, R4, R4a). In addition, carbonyl compounds can be formed by a unimolecular termination reaction of HOM-RO2• (Reaction R5, Rissanen et al., 2014).
Recent studies indicate that RO2• could also react with OH• radicals (summarized in Fittschen, 2019). Theoretical calculations suggest that larger peroxy radicals could form stable trioxides under atmospheric conditions, while formation of alkoxy radicals is negligible (Assaf et al., 2018).
In addition, many studies have observed the formation of accretion products presumably by the recombination of two peroxy radicals via a tetroxide HOM-R-OOOO-R′-HOM (Reaction R6, Berndt et al., 2018a, b; Ehn et al., 2014; Hasan et al., 2020; McFiggans et al., 2019; Pullinen et al., 2020; Valiev et al., 2019).
As side products of Reaction (R6) HOM-C19-Esters can be formed from the tetroxide by elimination of formaldehyde (Peräkylä et al., 2023).
The bimolecular reactions of HOM-RO2 with NO, RO2, and possibly HO2• also lead to HOM alkoxy radicals (HOM-RO•) analogous to simple peroxy radicals (Reactions R7, R8, R9). The branching ratio of alkoxy radical formation from peroxy radicals varies strongly depending on their structure; for example, for primary and secondary RO2• 60 % are given for Reaction (R7) by Jenkin et al. (2019). For Reaction (R8) branching ratios supposedly vary between 70 % and 90 % (MCMv3.1.1). For specific peroxy radicals even reaction with HO2 can lead to significant branching into alkoxy formation (Jenkin et al., 2019). Alkoxy intermediates play an important role in the atmospheric degradation of VOCs (Färber et al., 2024; Jaoui et al., 2021; Yang et al., 2025). Due to the fast reaction of peroxy radicals with NO, formation of alkoxy radicals is particularly efficient in the presence of NO.
All of these Reactions (R1)–(R9) above, including accretion product formation and alkoxy formation, compete with each other and the relative importance of each channel varies, depending on the concentration of reaction partners and the molecular structure of the precursors (Berndt et al., 2015, 2016; Berndt, 2021; Iyer et al., 2018, 2019, 2021; Jenkin et al., 2019; Peräkylä et al., 2023; Rissanen, 2018).
NOx is often so abundant in the atmosphere that peroxy radicals mostly react with NO to form organic nitrates and alkoxy radicals. Understanding the role of NOx in VOC oxidation is a key to quantitatively describe atmospheric SOA mass and SOA yield (Eddingsaas et al., 2012b; Kiendler-Scharr et al., 2016; Lee et al., 2016; Ng et al., 2007; Rollins et al., 2010; Sarrafzadeh et al., 2016; Zhao et al., 2018). Some studies have investigated the influence of NO on the gas phase chemistry of SOA precursors, but with emphasis on the less oxidized molecules (Aschmann et al., 2002; Eddingsaas et al., 2012a; Ng et al., 2007).
Pullinen et al. (2020) investigated the impact of increasing NOx on the HOM-RO2• chemistry for α-pinene and β-pinene, which resulted in a suppression of HOM accretion products (HOM-ACC) and thus a reduced SOA formation potential (Pullinen et al., 2020). Along with an increasing fraction of HOM organic nitrates (HOM-ON) they observe an increasing fraction of HOM products with carbon numbers less than ten. These HOMs must have undergone a fragmentation step during their formation. The latter points to an increasing importance of HOM-RO• with increasing NOx. A recent study demonstrated that increasing HOM-RO• chemistry plays an important role in SOA formation and increases SOA yields (Nie et al., 2023). A potential role for RO• in HOM formation has also been indicated for alkane oxidation by Wang et al. (2021).
Nevertheless, detailed studies about the role of (HOM-) alkoxy radicals in the HOM formation mechanism are rare. One reason is extremely low concentrations of the highly reactive alkoxy intermediates which cannot be measured directly. Their impact must therefore be deduced from variations in the measurable product distributions in response to varying boundary conditions that favor or reduce alkoxy radicals.
In general, alkoxy radicals are energy rich entities and tend to fragment, which on one hand propagates radical chain reactions, and on the other hand leads to intermediates and products with fewer carbon atoms than the precursor (Atkinson and Arey, 2003b; Vereecken and Peeters, 2009). However, Vereecken and Peeters (2010) show that especially functionalized alkoxy radicals can also isomerize by intramolecular H-shifts, forming peroxy radicals and subsequent products while preserving the carbon backbone. Since the alkoxy channels for HOM-RO2• lead to highly functionalized alkoxy radicals (HOM-RO•) and the likelihood of isomerization for alkoxy radicals can increase with the degree of substitution, isomerization can become particularly important in the case of HOM-RO•.
Here we performed steady-state experiments of α-pinene oxidation by OH• radicals in the presence of varying amounts of NOx. Increased formation of alkoxy radicals and its effect on the oxidation mechanism for non-fragmented C10 molecules will be investigated in this work. We will take advantage of the fact that high-resolution mass spectrometry allows for the direct observation of the product distribution at the level of chemical formula composition in the gas phase, including HOM-RO2• , the potential precursors of HOM-RO•. We will classify C10-HOM peroxy radicals, C10-, and C20- closed-shell HOMs according to their number of hydrogen atoms and analyze their distribution as a function of steady-state [NOx]ss, where ss indicates that the concentrations (mixing ratios) were observed at steady state. In addition, measurements with increasing [OH•]ss and CO addition were carried out to perturb the chemical system and to observe the response in the mass spectrometric pattern of C10 and C20 HOM compounds. The amount of fragmented HOM products as well as the parity of the number of oxygen atoms in HOM-RO2• are utilized as parameters to explore the role of HOM-RO• in the context of autoxidation.
2.1 Chamber experiments
The experiments were carried out in the Jülich Plant Atmosphere Chamber (JPAC, Mentel et al., 2009). Recent modifications of JPAC have been described in detail previously (Mentel et al., 2015; McFiggans et al., 2019) and only a summary will be given here. The reactor in JPAC is made of borosilicate glass, has a volume of 1.45 m3 and is housed in a thermostat. During the experiments a total flow of about 30 L min−1 of purified and humidified air passed through the reactor, resulting in a residence time of about 50 min. The chamber was operated as a continuously stirred tank reactor with a typical mixing time of 2 min. The total flow was divided approximately equally into two separate inlet lines, the first providing ozone and the second α-pinene and NO, to prevent ozone reactions in the lines before the gases reached the reactor. The temperature and relative humidity were maintained at 289 ± 1 K (16 ± 1 °C) and to 63 ± 2 %, respectively, throughout the experiments. The reactor is equipped with two different UV light sources. The first source is a UV-C lamp (Philips, TUV 40 W, λmax = 254 nm). At the wavelength of 254 nm ozone is photolyzed to generate O1D atoms, which then react with water vapor to produce OH• radicals. The UV-C lamp is housed in a quartz tube across the inside of the reactor and covered by two movable glass tubes that shield the UV-C radiation. Changing the gap between them changes the fraction of lamp exposure and thus the photolysis frequency for ozone (j(O1D)) and the OH• radical source. The photolysis frequency of the UV-C lamp was determined by an actinometric experiment to be 2.9 × 10−3 s−1 for a reference gap of 23 cm. The second light source consists of twelve discharge lamps emitting UV-A light (Philips, TL 60 W/10-R, 60 W, λmax = 365 nm) to produce NO by NO2 photolysis in the NOx experiments. The photolysis frequency achieved for NO2 was 1.3 × 10−3 s−1 in the experiments described here. In addition, two discharge lamps (HQI400 W/D, Osram) are used as visible light sources.
The α-pinene (Sigma Aldrich, 95 %) was added to one of the reactor inflows using a temperaturecontrolled diffusion source. NOx was provided by adding a controlled flow of NO in N2 to the same inflow line (Linde, 90 ppm NO in N2). In the reactor, most of the added NO was converted to NO2 by reaction with ozone. Due to the use of ambient air treated by catalytic oxidation and adsorption drying, memory effects from Teflon parts, and low volatile organic nitrates residing on the wall, approximately 300 ppt NOx and 15 ppb CO were always present as a background in the chamber (see Pullinen et al., 2020). To reduce NOx memory effects in the chamber, we allowed at least one day between NO addition experiments. Supplementary control and reference experiments without NOx were conducted between two NOx addition experiments. The background level of NOx was more than an order of magnitude smaller than the NOx added in the experiments. Since sufficient O3 was always present, background NO was mainly converted to NO2, and thus did not significantly contribute to the NOx chemistry within the system.
Direct measurements of [OH•] were not performed during the experiments. Therefore, [OH•]ss was calculated from the consumption of α-pinene in the chamber as previously described by Pullinen et al. (2020).
Chamber experiments were performed as follows. The two flows containing α-pinene and ozone were mixed in the clean chamber. Once steady state was reached, reference data for α-pinene ozonolysis were recorded. Subsequently, photochemical oxidation of α-pinene was initiated by switching on the UV-C lamp. As soon as OH• radical production started, a rapid increase in HOM production was observed.
In the photochemical experiments without NOx and CO addition, [OH•]ss was varied by altering the gap of the UV-C lamp and thus the photolysis frequency of ozone. Following each alteration, steady state was attained within a few hours. The time traces for the reference experiment can be found in Fig. S1 in the Supplement.
In case of experiments with CO and NOx, CO and NO were added after the α-pinene ozonolysis steady state and prior to the activation of the UV-C lamps. For the NOx addition experiments, the UV-A lights were always on, irrespective of the addition of NOx to the chamber. Steady state data were collected when all parameters remained (nearly) constant, typically after three residence times. A typical example of a NOx experiment is given in Fig. S2.
For direct comparison of the HOM product distribution for varying NOx, we considered that NOx can affect [HOM] indirectly by affecting [OH•]ss (Sarrafzadeh et al., 2016) and by suppressing new particle formation (Wildt et al., 2014). Levels of [OH•]ss were constrained to a range of less than a factor of two (4–7 × 107 cm−3) by accordingly adjusting the concentration of O3 to the varied NO levels. We also conducted the experiments at as low as possible α-pinene concentrations to largely suppress new particle formation. However, at lower [NOx]ss (and the highest [OH•]ss in the reference experiments) particle formation could not be fully suppressed. The experimental conditions at steady state are given in Table S1 in the Supplement.
2.2 Instrumentation
JPAC was equipped with various instruments for gas-phase measurements. The concentration of α-pinene, [α-pinene], was monitored by gas chromatography mass spectrometry (GC-MS, Agilent GC/MSD system with HP6890 GC and 5973 MSD) and by proton-transfer-reaction mass spectrometry (HR-PTR-MS, Ionicon, Innsbruck, Austria). Prior to each NOx experiment, the instruments were calibrated by switching them to a diffusion source with known α-pinene output. [O3] was measured by an ozone monitor (UV absorption, Thermo, Environmental 49). [NO] and [NO2] were monitored by NO chemiluminescence (Eco Physics, CLD 770 AL ppt, photolytic converter Eco Physics, PLC 760). Calibration of the NOx chemiluminescence detector was made by switching the device to air with known NO and NO2 concentration. Water vapor was measured with a dew point mirror (TP-2, Walz).
HOMs were measured by a chemical ionization atmospheric pressure interface time-of-flight mass spectrometer using nitrate (NO) as reagent ion (CI-APi-TOF, Aerodyne Research and TOFWERK AG, Ehn et al., 2012; Jokinen et al., 2012; Junninen et al., 2010). The chemical ionization device is similar to those described by Eisele and Tanner (1993) and Kurtén et al. (2011). The Eisele inlet was connected to the chamber via a 20 cm-long tube with an internal diameter of 3/4′′. The inlet was operated with a sheath air flow of 20 L min−1, and a total flow of 30 L min−1 was extracted from the instrument resulting in a 10 L min−1 sample flow from the chamber through the inlet. Nitrate ions were produced by exposing HNO3 contained in the sheath air flow to alpha radiation from a 10 MBq 241Am source. Nitrate ions are commonly used to study HOMs due to strong cluster formation ability with, and high selectivity towards, HOMs. The relative transmission curve of the instrument was nearly mass independent in the mass range from 230 to 600 Da (Pullinen et al., 2020).
In this work, the observed MS signals, normalized to the total signal, were used for the interpretation of the data since the relative changes of the HOM product distribution for the different reaction conditions were more relevant than the absolute concentrations. We refer to it as “signal” with the dimensionless unit “ncps” (normalized counts). The 3σ detection limits were 2–5 × 10−6 ncps in the reference experiments (Table S1, J1–J12) and 5 × 10−7–1 × 10−5 ncps in the NOx experiments (Table S2, N1–N11), determined by the noise of the background for several .
As shown by previous quantum mechanical calculations, HOMs with more than six oxygen atoms are detected with comparable sensitivity by NO-CI-API-TOF (Hyttinen et al., 2017). As consequence compounds with less oxygen atoms cannot be detected with the same sensitivity. For example, the first generation of conventional peroxy radicals cannot be detected at all. Currently, absolute calibration for HOMs does not exist. Assuming clustering of HOMs with NO takes place at the kinetic limit it is common to apply the sensitivity towards sulfuric acid. This sensitivity was determined to be 3.7(±1.2) × 1010 molec. cm−3 ncps−1 and applied to convert the MS signal to a concentration (see Pullinen et al., 2020).
Data processing and peak identification was achieved using the program Tofware v2.5.11 (Tofwerk AG/Aerodyne Inc.) in Igor Pro (WaveMetrics Inc.). We identified about 700 compounds in the NOx experiments and about 350 compounds in the no-NOx experiments.
In the analysis here we focused on the subset of C10 and C20 compounds (monomers and accretion products), and on the sum of compounds with C5–C9 and C11–C19. Since these latter two groups must have undergone a fragmentation step in their formation, we denote C5–C9 compounds as fragmented monomers or fragmented HOM-RO2•, where it applies, and C11–C19 compounds as fragmented accretion products. The C10 and C20 compounds we denote accordingly as non-fragmented monomers, HOM-RO2•, and accretion products.
For direct intercomparison of the experiments with respect to the impact of NOx on HOM peroxy radical chemistry specifically, remaining dependencies on OH• and on condensation to new formed particles were eliminated by normalizing the HOM signal to a particle-free condition and to a reference turnover at a reference [OH•]ss in the absence of NOx. The correction procedure is described in Sect. S2 of the Supplement. By using the experiments wherein we varied [OH•]ss, also presented here, we re-evaluated the dependence of the HOM production on the α-pinene turnover and achieved a nearly quadratic power law dependency (Fig. S3). As reference turnover we chose 6.7 × 107 cm−3 s−1, representing the highest turnover in the experiments wherein the OH• source strength was varied (see supporting information).
The particle correction factors varied between 1 and 1.1 in the OH• reference experiments (Table S1, J1–J12) and between 1 and 1.25 in the NOx experiments (Table S2, N1–N11). The OH• correction factor of the latter scattered around 0.2, with the exception of the experiment at the highest NOx concentration (N11), where it reached 0.31. The combined correction factors in N1–N11 varied between 0.19 and 0.31.
2.3 Model calculations
To address certain mechanistic considerations, it is necessary to estimate the radical concentrations of HO2• and RO2• in our experiments. For that we performed box model calculations adapting the gas-phase mechanism for α-pinene from MCMv3.3.1 (Saunders et al., 2003, https://mcm.york.ac.uk/MCM/, last access: 11 November 2021) to the boundary and initial conditions of JPAC (Sect. 3). In order to assess the model performance, we compared the model output to the measured [α-pinene]ss, [O3]ss, and [OH•]ss, as well as to [NO]ss and [NO2]ss where applicable (Fig. S4).
2.4 Generic mechanistic framework for HOM formation
In our perception HOM species behave chemically similar to less oxidized organic compounds (Bianchi et al., 2019), and we will further assume analogy to generic atmospheric organic chemistry, as represented by the Master Chemical Mechanism (Saunders et al., 2003) under consideration of recent developments (Berndt et al., 2018a, b; Jenkin et al., 2018, 2019). Reaction of HOM-RO2• with OH• (Assaf et al., 2018; Fittschen, 2019) could contribute somewhat to the HOM-termination products at our reaction conditions but it will not be considered, since it cannot compete with the particular major termination pathways in the experiments.
For about 90 non-fragmented C10- and C20-compounds we used the number of hydrogen and oxygen atoms to rationalize the mechanism of their formation in α-pinene photooxidation. In addition, we considered two sums of HOMs with C5–C9 and C11–C19 as indicator for fragmentation steps via alkoxy radicals.
To classify the mass spectrometric derived formulas of the C10 and C20 compounds we use a framework, wherein HOMs belong to the same family when they have the same number of carbon and hydrogen atoms in the molecule, differing only in the number of oxygen atoms. For example, all the molecules with 10 carbon and 14 hydrogen atoms will be referred to as C10H14OX family, independently of the number of oxygen atoms. This is an obvious choice if one thinks of autoxidation chains, which start from a given peroxy radical and undergo multiple additions of O2 to the molecule. The hydrogen number (for a given C number) allows tracing the peroxy radical that is the precursor of the product, formed by certain formation and termination reactions.
Photooxidation of α-pinene is expected to produce C10H17OX and C10H15OX as major HOM-RO2• families. The C10H17OX family is initiated by the dominant addition of OH• to the α-pinene double bond, and by addition of OH• to α-pinene under opening of the 4-membered ring (Berndt et al., 2016; Lee et al., 2023; Piletic and Kleindienst et al., 2022; Vereecken and Peeters, 2004; Xu et al., 2019). The C10H15OX family, on the other hand, arises from H-abstraction, either from α-pinene itself (Shen et al., 2022) or from first generation products like pinonaldehyde. C10H15OX is also formed via the vinylhydroperoxide pathway in α-pinene ozonolysis (Johnson and Marston, 2008; Rissanen et al., 2014; Mentel et al., 2015). The C10H15OX family can be terminated to closed shell products C10H14OX and C10H16OX as carbonyl and alcohol products from the disproportionation Reaction (R3). In addition, C10H14OX can be formed from C10H15OX by ring closure reaction (Vereecken et al., 2007) or unimolecular termination reaction (Reaction R5, Rissanen et al., 2014).
While C10H14OX products are uniquely related to the C10H15OX peroxy radical family, three C10H15OX pathways can contribute to C10H16OX products. The reaction of C10H15OX+1 peroxy radicals with other RO2• can produce alcohols (C10H16OX; Reaction R3b), and C10H15OX can react with hydroperoxyl radicals, Reaction (R2) resulting in the hydroperoxide C10H16OX. Moreover, C10H17OX+1 peroxy radicals can also undergo disproportionation Reactions (R3a) and (R3b) and produce C10H16OX carbonyl compounds (Reaction R3a). Therefore, the C10H16OX family has contributions from both radical families, which complicates the analysis.
For the C10H18OX family, it is again simpler to relate the products to precursor peroxy radicals since C10H18OX can only be produced as alcohol (Reaction R3b) or hydroperoxide (Reaction R2) from C10H17OX+1 or C10H17OX peroxy radicals, respectively. As described above, with the exception of C10H14OX, which must be carbonyls, one cannot conclude from the hydrogen number alone which functional group was formed by the termination reaction.
If NOx is part of the reaction system, C10H15OX and C10H17OX can form organic nitrates (HOM-NO3) via Reaction (R4). In case of HOM compounds containing a single N-atom, their hydrogen number is odd, C10H15NOx or C10H17NOx, and directly relates to the precursor peroxy radical family.
In general, one can expect HOMs to have multi-functionality besides the termination group due to their formation process by autoxidation. Still, classifying C10 and C20 molecules by family may help to understand which HOM-RO2• were involved in their formation, especially in case of C10H14OY and C10H15-HOM-NO3, which are exclusively produced by C10H15OX, and for C10H18OY and C10H17-HOM-NO3, which are exclusively formed from C10H17OX.
In case of high [RO2•], or in presence of [NO] and potentially also with HO2•, HOM-RO2• can form alkoxy radicals, HOM-RO• (Reactions R7, R8, R9). This is of interest for HOM formation, as also HOM-RO• may undergo isomerization under retaining the carbon backbone, in addition to fragmentation by bond scission. (The H-abstraction by O2 is typically unimportant for larger organic molecules and will be neglected here; Atkinson, 2007.) HOM-RO2• (A) (Fig. 1) can undergo H-shift and proceed by autoxidation Reaction (R1), or, as schematically sketched in Fig. 1, can undergo bimolecular reaction with NO or RO2• and produce HOM-RO• (B). For highly functionalized alkoxy radicals, such as HOM-RO• , isomerization can compete with fragmentation (Vereecken and Peeters, 2009, 2010). Isomerization of alkoxy radicals generates a hydroxyl group and a carbon centered radical site (C), and fast addition of O2 produces a new HOM-RO2• (D). In this case, HOM-RO2• (D) has one more oxygen than the originating peroxy radical (A) of the alkoxy radical (B). If the migrating hydrogen is abstracted from an HO2-group, the new HOM-RO2• (E) is produced directly and contains one less oxygen than the precursory HOM-RO2• (A). The parity of oxygen number changes in both cases. Therefore, the parity change in oxygen number indicates an alkoxy step in the radical chain. We denote the combination of HOM-RO• formation, subsequent isomerization, and O2 addition as the “alkoxy-peroxy pathway” (Dames and Green, 2016; Mentel et al., 2015; Vereecken and Peeters, 2010).
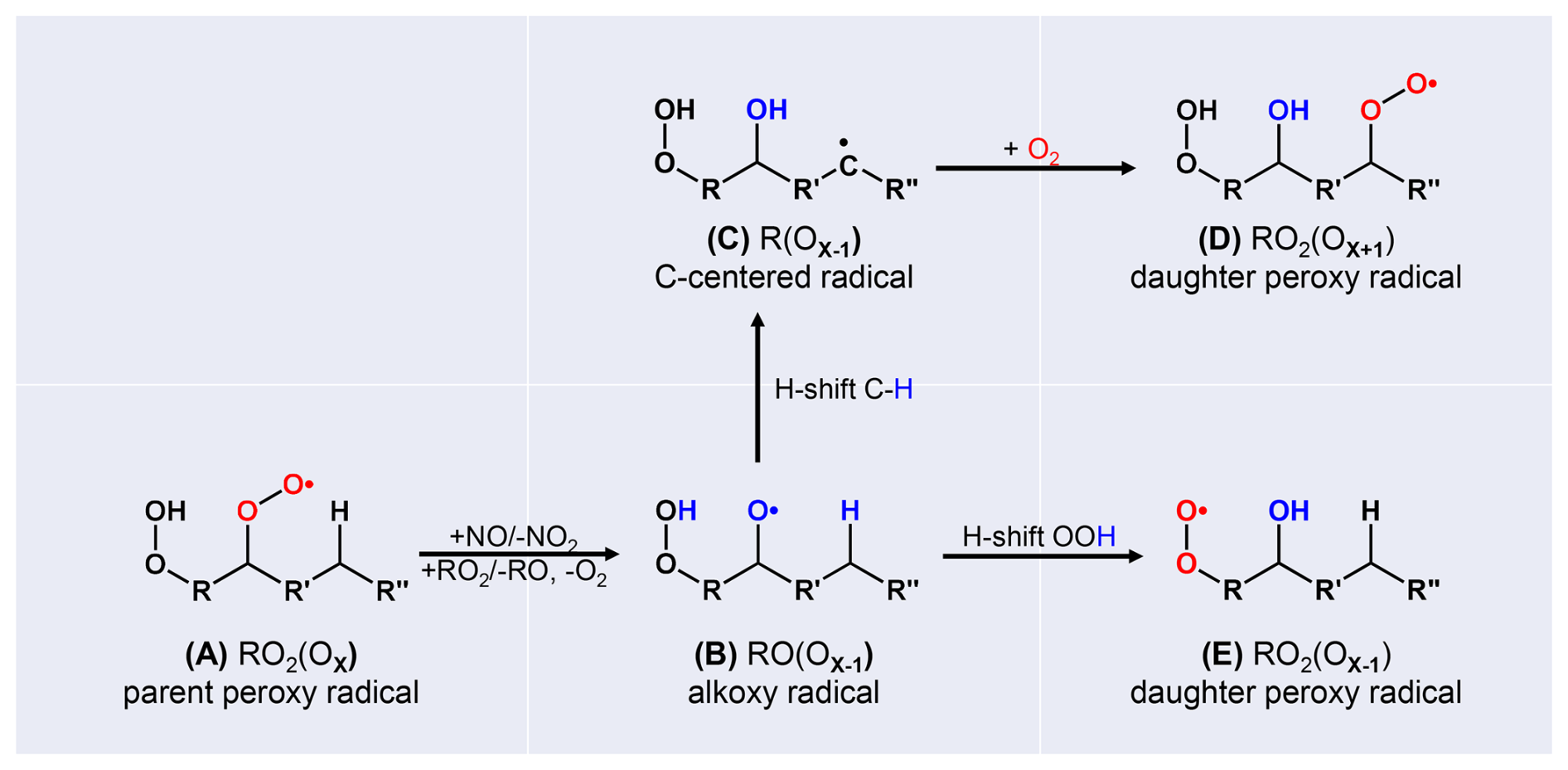
Figure 1Schematic sketch of the alkoxy-peroxy pathways (A, B, C, D) and (A, B, E), considering fast isomerization of the alkoxy radical (B) by H-shift from a C-H bond, or by H-shift from a HO2• group. The resulting peroxy radicals (D), (E) have one more or one less oxygen atom compared to the parent peroxyradical (A); thus, the parity of the oxygen number is changed.
The fate of HOM-RO• depends of course on the detailed molecular structure R, such as the functional groups adjacent to the alkoxy radical, or the span and substitutions for available hydrogen migration (Vereecken and Peeters, 2010). However, multifunctional HOM-RO• should have quite a potential for hydrogen rearrangement since they are highly functionalized, and functionalization facilitates isomerization (Vereecken and Peeters, 2010). Note that we are using HR-MS data, which allow for determining the compounds formulas but not the speciation. As a consequence, more than one structure isomer can contribute to the signal for one formula. We have thus no direct handle on the structure of R. The advantage of HR-MS is that we can observe the steady state abundance of 100's of compounds simultaneously, so that we capture quite a large wealth of R's. Nevertheless, our approach is limited to relations between formulas, which e.g. represent HOM-RO2• and their termination products. We assume that at least one of the structure isomers represented by the reactant formula has the right structure of R for a fast isomerization step to react to an isomer represented by the product formula. Our suggestions/conclusions are thus not to interpret in a statistical sense but in terms of “there must be one or more isomers” with the given formula that are able to provide the observed conversion to the product formula. Using this approach, we will show that isomerization can become dominant in cases of large, highly functionalized molecules, such as α-pinene derived HOM-RO• radicals.
For the H-abstraction channel, Shen et al. (2022) showed that the first generation peroxy radical, which can undergo autoxidation, has the molecular formula C10H15O4. In secondary oxidation channels, the OH• radical abstracts a hydrogen atom from a first-generation product with formula C10H16O2, such as pinonaldehyde, and fast O2 addition to the alkyl radical site results also in compounds with the formula C10H15O4. By autoxidation of these peroxy radicals we expect families with the general formula C10H15O2n, i.e., HOM-RO2• with even oxygen parity. However, if a HOM-RO• step is involved and the HOM-RO• undergoes isomerization, the oxygen parity in the radical chain changes from even to odd with the general family formula C10H15O2n+1.
In case of OH addition to the endocyclic double bond of α-pinene or by addition under four-membered ring opening (Vereecken and Peeters, 2004, 2007; Xu et al., 2019), the first-generation peroxy radical is C10H17O3, and a HOM-RO2• family is produced by autoxidation with odd oxygen parity and the general formula C10H17O2n+1. An alkoxy step will change parity of the oxygen number of this HOM-RO2• family from odd to even, with a general formula of C10H17O2n.
Since two steps of alkoxy radical formation will reverse the oxygen number parity again, we are not able to distinguish between HOM-RO2• which did not undergo alkoxy radical formation, versus those which underwent two alkoxy steps. Nevertheless, oxygen number parity is useful information when the likelihood of multiple alkoxy steps is small. We will provide evidence that the alkoxy-peroxy pathway can compete with the fragmentation of the HOM-RO•, and that it enables for propagation of the autoxidation chain in cases where the autoxidation of the parent peroxy radical itself is non-efficient or cannot compete with bimolecular reactions with NO or RO2• . Therefore, we will put some focus on the observation of the oxygen parity in the peroxy radical families. Parity analysis and fraction of fragmented HOM-monomers and accretion products will serve in the following as indicators for alkoxy steps.
3.1 Base case: α-pinene photooxidation as a function of OH• source strength
Figure 2 shows the HOM-RO2• families (Fig. 2a) as a function of the α-pinene turnover, as well as the families of closed shell products, non-fragmented C10-monomers (Fig. 2a), non-fragmented C20 accretion products (Fig. 2c), and the sums of fragmented monomers and accretion products (Fig. 2d). The turnover represents the primary production of peroxy radicals and is calculated as kOH• [OH•]ss•[α−pinene]ss, where “ss” stands for steady state. Individual original data of all C10 and C20 HOMs are documented in Fig. S5. The first two points at low turnover result from the dark ozonolysis phase without photolyzing ozone for OH• radical production. Nevertheless, OH• radicals are formed in the ozonolysis phase (dark OH•) as a product of fragmentation of Criegee intermediates, i.e. in the vinylhydroperoxide pathway (Johnson and Marston, 2008; Paulson et al., 1998). Since we did not scavenge the dark OH•, the oxidation of α-pinene in the dark is characterized by about 60 % ozonolysis and about 40 % of reaction with OH• radicals.
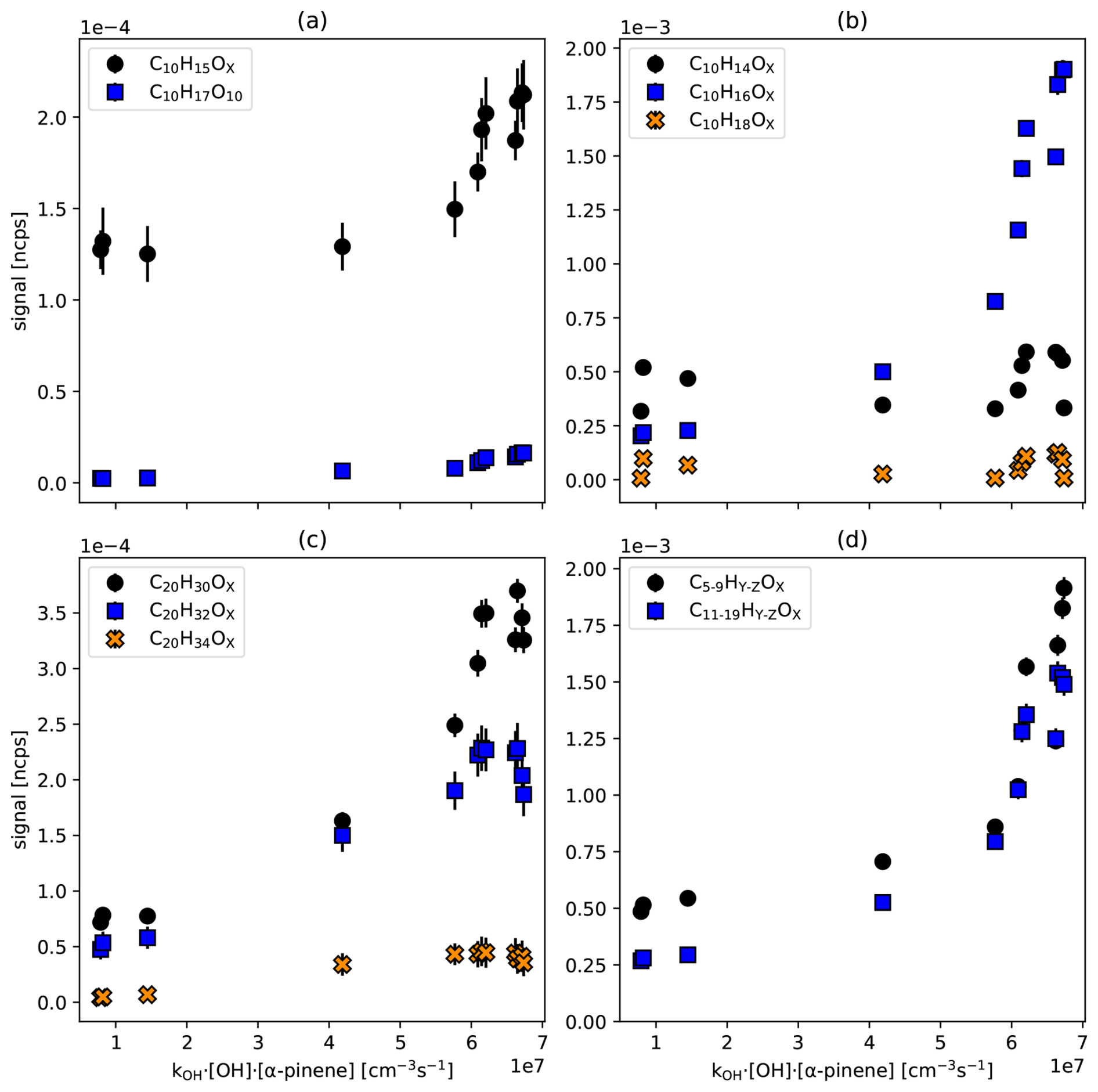
Figure 2Signal of HOM peroxy radicals, non-fragmented monomer and accretion product families, as well as the sums of fragmented families versus turnover. All signals have been particle corrected. (a) C10-HOM-RO2• (C10H15OX: black circles, C10H17OX: blue squares) (b) C10 closed-shell HOM-families. (C10H14OX: black circles, C10H16OX: blue squares, C10H18OX: orange crosses) (c) C20 Accretion products (C20H30OX: black circles, C20H32OX: blue squares, C20H34OX: orange crosses) (d) sums of fragmented monomers C5–C9 (black circles) and fragmented accretion products C11 to C19 (blue squares). Standard deviations (1σ) for averaging over the steady state interval are shown, whenever larger than symbol size.
Figure 2a shows the particle corrected signal in ncps of the C10H15OX family (black circles) and C10H17O10 (blue squares) plotted against turnover. In the ozonolysis phase, C10H15OX turned out as the major peroxy radical family, as expected. Although 40 % of the α-pinene oxidation is contributed by OH• radicals, we could detect only one member of the C10H17OX peroxy radical family (C10H17O10) with low signal. All other C10H17OX radicals were below the detection limit (3σ) of about 2–5 × 10−6 ncps. More surprisingly, C10H15OX remains the major peroxy radical family even when oxidation by OH• contributes to more than 90 %, i.e., for kOH• [OH•]ss•[α-pinene]ss> 4 × 107 cm−3 s−1. C10H17O10 remains the only detectable member of the C10H17OX HOM-RO2• family. This result is unexpected since the OH• addition reaction at the double bond or under opening of the four-membered ring leading to C10H17OX peroxy radicals accounts for about 90 % of the α-pinene oxidation by OH• radicals.
Figure 2b shows that the C10H14OX family (black circles) has about a factor of four higher signal than the C10H18OX family (orange crosses). This means that products formed exclusively by C10H15OX HOM-RO2• show distinctively higher signals than products that solely originate from C10H17OX HOM-RO2•. Compounds with the formula C10H16OX can arise from both radical families, therefore a contribution of C10H17OX HOM-RO2• to C10H16OX via Reaction (R3a) cannot be excluded. However, this contribution must be small, as can be deduced from the following considerations.
The large increase in C10H16OX is dominated by C10H16O7, which accounts for 57 % of the family at the largest turnover point. C10H16O7 can be formed from C10H17O8 by Reaction (R3a) or Reaction (R5). Assuming a [C10H17O8]ss at the detection limit (≈ 1 × 105 cm−3), taking an [RO2•]ss of about 7 × 109 cm−3 from the model calculation, and applying a rate coefficient = 5 × 10−12 cm3 s−1 (estimated by us based on Table 10 in Jenkin et al., 2019), we calculate a production rate of C10H16O7 by Reaction (R3a) of about 3500 cm−3 s−1. Assuming that the lifetime of 120 s due to wall loss, as determined for C10H16O7, is also valid for the precursor peroxy radical, and that the wall loss is the major sink of C10H16O7, we can calculate an upper limit of the expected steady-state concentration of 4.2 × 105 cm−3 for C10H16O7 contributed by the C10H17O8 carbonyl channel (Reaction R3a). In addition, internal termination (Reaction R5) with an intramolecular rate coefficient of about 0.01 s−1 can contribute another 6 × 105 cm−3 of C10H16O7. Consistent with this estimation we observe about the same concentration of 1.1 × 106 cm−3 of C10H18O7, which can be the corresponding alcohol (Reaction R3b) or a hydroperoxide (Reaction R2). The concentration of the latter is estimated to be only minor with 2 × 104 cm−3 because of low [HO2] ([HO2•]ss = 8 × 107 cm−3, = 2 × 10−11 cm3 s−1, wall loss coefficient 1/120 s−1 = 0.0083 s−1). This means that formation of C10H16OX from C10H17OX precursors is low and contributes only a few percent to the observed signal of the C10H16OX family. C10H16O7 is thus likely formed from C10H15O8 or C10H15O7 which show high abundances (Fig. S5). The large contribution of C10H16O7 to the C10H16OX family indicates specifically efficient termination reactions of C10H15O8 + RO2 or C10H15O7 + HO2. From this low contribution and from the overall much higher signals of C10H14OX-HOMs compared to C10H18OX HOMs, we conclude that α-pinene HOM chemistry must be dominated by C10H15OX radical chemistry, with minor contributions from C10H17OX.
Observations of the accretion products support that the C10H15OX HOM peroxy radical family is the major player in photochemical HOM formation initiated by OH•. C20H30OX (formed from the recombination of two C10H15OX peroxy radicals) and C20H32OX (formed from one C10H15OX and one C10H17OX peroxy radical) show the highest signals among C20 accretion products, and both contain C10H15OX contributions (Fig. 2c). The C20H34OX accretion product family (formed by the recombination of two C10H17OX peroxy radicals) has the lowest abundance, which supports that C10H17OX cannot be the major HOM peroxy radical family. On the other hand, the C20H32OX family shows a relatively high contribution, which indicates the importance of C10H17OX peroxy radicals in accretion product formation, despite the low importance of the HOM-C10H17OX and related HOM monomers. This supports that HOM-ACC can also be formed by HOM-RO2• + RO2•, for example also with C10H17O3• (Berndt et al., 2018a; McFiggans et al., 2019; Pullinen et al., 2020). In conventional α-pinene chemistry C10H17O3 radicals dominate RO2• by far and therefore the high relative abundance of C10H32OX is understandable.
Our observation of the dominance of the C10H15OX peroxy radical family in HOM formation by OH• is in contrast to the frequently reported C10H17OX as the major peroxy radical family (Berndt et al., 2016; Berndt, 2021; Kirkby et al., 2016; Lee et al., 2023; Roldin et al., 2019; Xu et al., 2019). The dominance of C10H15OX is unexpected as OH• addition represents the major pathways in α-pinene photooxidation, either as direct addition to the endocyclic double bond (≈ 90 %) or under opening the four-membered ring (≈ 10 %) (Saunders et al., 2003; Vereecken et al., 2007). Both OH• addition pathways comprise the C10H17OX peroxy radical family, and Xu et al. (2019) showed that efficient autoxidation and HOM formation start with opening the four-membered ring.
For an explanation of our observations, we refer firstly to a recent study by Shen et al. (2022) and secondly to the style of our experiments. Shen et al. (2022) demonstrate that hydrogen abstraction from α-pinene by OH• can be an important source of HOMs in α-pinene photooxidation. This minor initiation pathway, with an estimated branching ratio of approximately 10 %, effectively produces the C10H15OX peroxy radical family via two alkoxy steps. These steps involve breaking the six-membered ring and the four-membered ring of α-pinene. After ring breaking, autoxidation can become very fast. Note that HOMs are minor products in OH• oxidation (and ozonolysis) with molecular yields of a few percent. Therefore, minor yet efficient pathways can easily dominate HOM formation. As proposed by Shen et al. (2022), H-abstraction from α-pinene seems to be such an efficient minor pathway. Meanwhile, this has also been confirmed for HOMs formed from limonene (Luo et al., 2023).
Secondly, we conducted our experiment under steady-state conditions, with a residence time of about one hour and OH• concentrations of several times 107 cm−3. Under these conditions, secondary oxidation can become an important pathway for the formation of C10H15OX radicals, too, as hydrogen abstraction from first-generation oxidation products with 16 hydrogen atoms, e.g. pinonaldehyde, will also produce C10H15OX peroxy radicals. Note that the formation of second-generation compounds via OH•, such as pinonaldehyde, involves also at least one alkoxy step.
Given that our experiments presented here always started with ozonolysis, thereby introducing pinonaldehyde into the system, we currently cannot distinguish whether H-abstraction or secondary oxidation is the major channel for the C10H15OX chemistry in our system. Estimates from steady-state concentrations of α-pinene, pinonaldehyde (model result), and OH• indicate that both H-abstraction and secondary oxidation could be important and could contribute with similar fractions to C10H15OX related chemistry (Fig. S6). It seems that under conditions of our experiments C10H15OX related chemistry is more efficient in HOM formation than C10H17OX related chemistry. It seems even to outcompete the pathway induced by OH addition under opening of the four-membered ring (Berndt et al., 2016; Xu et al., 2019), which contributes also about 10 % to the α-pinene turnover (Fig. S6, grey spheres). Despite the line of arguments above, we are not able to finally clarify why we observe only a minor contribution of C10H17OX chemistry to HOM formation in our experiments. It had at least some significance as indicated by the observed termination products C10H18OY and C10H32,34OY.
The formation of fragmented monomers and accretion products are also indicators for alkoxy radical formation, since fragmentation is an important pathway of alkoxy radicals. Figure 2d indicates, that the abundance of fragmented compounds indeed increases over-proportional with increasing [OH•]ss. This is in accordance with an increasing importance of alkoxy steps with increasing [OH•]ss.
3.2 Experimental evidence for the importance of HOM-RO• formation
3.2.1 Effect of increasing oxidation by OH
In Fig. 3 we show the particle corrected signal of all C10H15OX (black circles) as a function of the turnover, along with the contributions of C10H15O2n with even oxygen numbers (red crosses) and C10H15O2n+1 with odd oxygen numbers (blue squares) (Fig. 3a), as well as the fractions C10H15O2n C10H15OX (red) and C10H15O2n+1 C10H15OX (blue) (Fig. 3b). The individual contributors to C10H15O2n and C10H15O2n+1 are shown in Fig. 3c and d. The data in Fig. 3c and d have been normalized by their Frobenius norm to highlight the similarity and dissimilarity of their shapes as a function of turnover, despite their differences in signal strength.
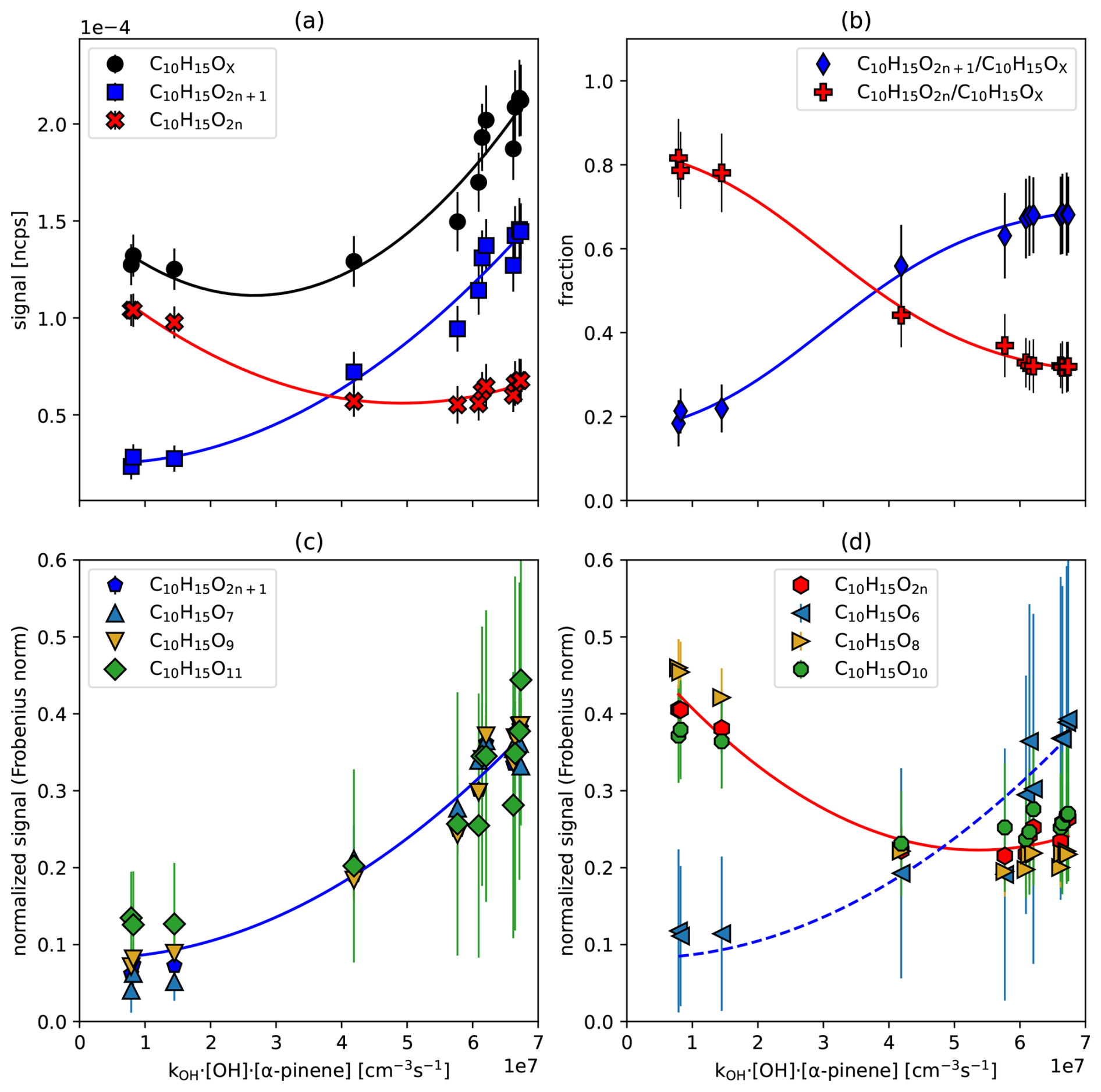
Figure 3C10H15OX peroxy radicals as a function for α-pinene turnover by OH•. (a) Signal of the sum of C10H15OX (black circles), C10H15O2n (blue squares), C10H15O2n+1 (red crosses) as a function of turnover. (b) The fraction of C10H15O2n (red) and C10H15O2n+1 (blue) as a function of turnover. (c, d) Sum of C10H15O2n+1, or C10H15O2n and respective individual peroxy radical signals, normalized by their Frobenius norm and plotted against turnover. Error bars are derived by error propagation based on the standard deviations (1σ) for averaging over the steady state periods. Lines are shown for the guidance of the eyes only.
The group of C10H15O2n radicals exhibits a high signal during the ozonolysis phase, which dropped with increasing oxidation by OH•. However, when oxidation by OH• dominates at kOH• [OH•]ss•[α−pinene]ss > 4 × 107 cm−3 s−1, C10H15O2n remains at a consistent level (Fig. 3a). In contrast, C10H15O2n+1 shows an increasing signal with turnover (Fig. 3a), resulting in a continuous increase in the C10H15O2n+1 fraction (Fig. 3b). Consequently, the C10H15O2n+1 peroxy radical family shows a strong increase when OH• oxidation becomes the major oxidation reaction.
This conclusion is further confirmed by the behavior of individual family members. As turnover with OH• increases, C10H15O7 and C10H15O9 become the dominant HOM-RO2• (Fig. S5), shaping the overall trend of C10H15O2n+1 in Fig. 3c. On the other hand, C10H15O8 and C10H15O10 peroxy radicals are dominant in the ozonolysis phase in absolute terms (Fig. S5) and determine the overall trend of C10H15O2n (Fig. 3d). Since C10H15O8 and C10H15O10 strongly decrease and become nearly constant with increasing turnover, while C10H15O7 and C10H15O9 strongly increase, one can observe the large fraction of C10H15O2n+1 at higher turnovers (Fig. 3b).
We performed shape analysis for C10H17OX• and the observed members of the C10H18OX family, which can be solely formed via OH oxidation (Fig. S7). Comparison to the behavior of the C10H15O2n+1 family (Fig. 3c) confirms that the C10H15O2n+1 family mainly results from OH oxidation.
It is noteworthy that C10H15O6, for which we observed the lowest absolute signals, shows a deviating shape (blue triangles in Fig. 3d) that resembles the behavior of the C10H15O2n+1 peroxy radical group (Fig. 3d) and the OH initialized C10H18OX family in Fig. S7. Different from C10H15O8 and C10H15O10, formation of C10H15O6 is efficiently initialized by OH• oxidation. This observation is consistent with the HOM pathways in α-pinene ozonolysis proposed by Iyer et al. (2021) wherein an efficient 1,4 shift to C10H15O8 should lead to a small concentration of C10H15O6 in bare ozonolysis. The fraction of C10H15O6, which in Meder's et al. (2023, 2025) experiments could not be explained by the Iyer mechanism, could arise in parts from dark OH as in our experiments.
In terms of our generic framework detailed in the Methods Sect. 2.4, the increased fraction of C10H15O2n+1 at elevated levels of OH• oxidation should be the result of one alkoxy radical step within the autoxidation chain. The increase of alkoxy radical formation at high turnovers is the result of the increased importance of HOM-RO2• + RO2• reactions (Reaction R7), which should produce alkoxy radicals with a branching ratio of approximately 0.6 (Jenkin et al., 2019). A parity shift in oxygen number would occur for every odd number of alkoxy steps. However, three bimolecular reaction steps of HOM-RO2• + RO2• are less likely to occur than a single step, since [RO2•]ss increases cum grano salis linearly with the OH• turnover. The chance for a HOM-RO2• produced via Reaction (R7) to have encountered one alkoxy step is given by the branching ratio in HOM-RO• times the branching ratio for isomerization of HOM-RO• by H-shift. If we assume 0.6 for the branching into HOM-RO• and 0.5 for HOM-RO• to isomerize the chance to encounter one alkoxy step is 0.3 while the chances to encounter two or three alkoxy steps are 0.09 and 0.0027, respectively. This also would mean that a certain fraction of the alkoxy steps – about 10 % – are hidden, as two steps do not result in an oxygen parity change. Note, that the applied branching ratios of 0.6 (Jenkin et al., 2019 for primary and secondary RO2•) and 0.5 for HOM-RO isomerization (see below and Sect. S6) comprise a certain degree of lumping and serve here to demonstrate the possibility of alkoxy-peroxy steps. Individual branching ratios and thus alkoxy-peroxy steps for specific HOM-RO2• and HOM-RO• are strongly dependent on the structure of R and can deviate quite far from the chosen values. With respect to the C10H17OX family, only one member, namely C10H17O10, has been detected by our measurements. This falls into the C10H17O2n group, where one step of alkoxy radical formation would be needed if the autoxidation starts from C10H17O5, as proposed by Xu et al. (2019).
On the other hand, the group of C10H15O2n peroxy radicals starts at a high level in the ozonolysis phase and decreases as the fraction of oxidation by OH• increases. However, it always remains important at a nearly constant level despite of decreasing [O3]ss due to increased j(O1D) for increasing OH production (Table S1). From the constancy of [C10H15O2n•] despite of decreasing [O3] at turnovers above ∼ 4 × 107 cm−3 s−1, we conclude that autoxidation initialized by OH• oxidation without alkoxy steps (or with two alkoxy steps) must be a pathway to C10H15O8,10 peroxy radicals besides ozonolysis.
This observation, in conjunction with the deviating behavior of C10H15O6, could indicate that alkoxy radical isomerization and, consequently, the parity changes to C10H15O2n occur preferably for peroxy radicals that have more than six oxygen atoms, in line with Vereecken and Peeters (2009, 2010) finding that functionalization supports alkoxy isomerization. In other words, C10H15O4 undergoes one or two oxygen addition steps, producing C10H15O6,8 peroxy radicals. These peroxy radicals can subsequently undergo autoxidation, including an alkoxy-peroxy step to produce C10H15O7,9, which are the major peroxy radicals in the OH• oxidation phase. However, further studies are necessary to elucidate the explicit formation mechanism of C10H15O6 and other peroxy radicals.
In summary, the parity of the oxygen number in the peroxy radicals as a function of the turnover of α-pinene with OH• indicates the importance of alkoxy steps. Furthermore, shape analysis of the data, normalized by their vector norm, shows that the formation of all C10H15O2n+1 members and the C10H15O6 peroxy radical was initialized by OH• oxidation. C10H15O2n family members, except the C10H15O6 peroxy radical, are the major peroxy radicals from ozonolysis reactions. Parity shift and importance of alkoxy steps in ozonolysis are small, since [RO2•]ss is about a factor of 4–5 smaller than at the largest OH• turnover as shown by the model calculations. Nevertheless, the formation of certain autoxidation products in ozonolysis can be only explained by alkoxy-peroxy steps (Mentel et al., 2015).
3.2.2 Effect of CO on formation of HOM-RO•
Carbon monoxide (CO) was added to the photooxidation system of α-pinene to investigate the alkoxy radical formation in a chemical regime where HOM-RO2• + HO2• reactions are more important than HOM-RO2• + RO2• reactions. In HOM-RO2• + HO2• reactions, HOM-ROOH is essentially formed by H transfer (Reaction R2). The branching into alkoxy radicals (Reaction R9) is small (see Jenkin et al., 2019), therefore effects of alkoxy radicals should be suppressed in presence of CO.
Here, we are comparing two steady states with and without CO addition at a comparable kOH• [OH•]ss• [α−pinene]ss of 6–7 × 107 cm−3 s−1. Turnover by O3 contributes 14 % in case of CO and 3 % in case of the OH reference experiment. The added CO reacted with OH• radicals and produced HO2• radicals. From our model calculations for the system with [CO]ss ≈ 5 ppm we obtained [HO2•]ss ≈ 2.8 × 109 and [RO2•]ss ≈ 1.4 × 109 cm−3 (C1 in Table`S3). This compares to [HO2•]ss ≈ 5 × 107 and [RO2•]ss ≈ 5 × 109 cm−3 in the reference experiment without CO addition (J5 in Table S3). Taking into account that bimolecular termination reactions of peroxy radicals with HO2• are about an order of magnitude faster than the permutation reactions between peroxy radicals, the system including CO is indeed characterized by HOM-RO2• + HO2• reactions at the expense of RO2 + RO2 reactions.
As for conventional RO2• the increased [HO2•]ss led to a reduction of [C10H15OX•]ss by about a factor of two. Our focus here is the effect of [HO2•]ss on the alkoxy radical formation. As indicators we use again the parity of C10H15OX peroxy radicals and the fraction of decomposition products. As shown in Fig. 4a, C10H15O2n+1 peroxy radicals with odd oxygen numbers account for 70 % of the whole C10H15OX family in the reference experiment without CO addition, where HOM-RO2• + RO2• reactions dominate (compare Fig. 3b). In the presence of CO, the fraction of C10H15O2n+1 decreases to 20 % whereas C10H15O2n accounts for 80 % of the total C10H15OX family. As described before, C10H15O2n peroxy radicals result from straight autoxidation of the initial peroxy radicals, C10H15O4, while C10H15O2n+1 peroxy radicals mainly experienced one alkoxy step during their formation. Therefore, suppression of C10H15O2n+1 in the presence of high CO must be the result of the suppression in alkoxy radical formation in the chemical regime, where HOM-RO2• + HO2• dominates. Regarding the reaction HOM-RO2• + HO2• (Reaction R9) a production of HOM-RO• cannot be excluded; however, from our it seems to be less efficient than HOM-ROOH formation (Reaction R2), though.
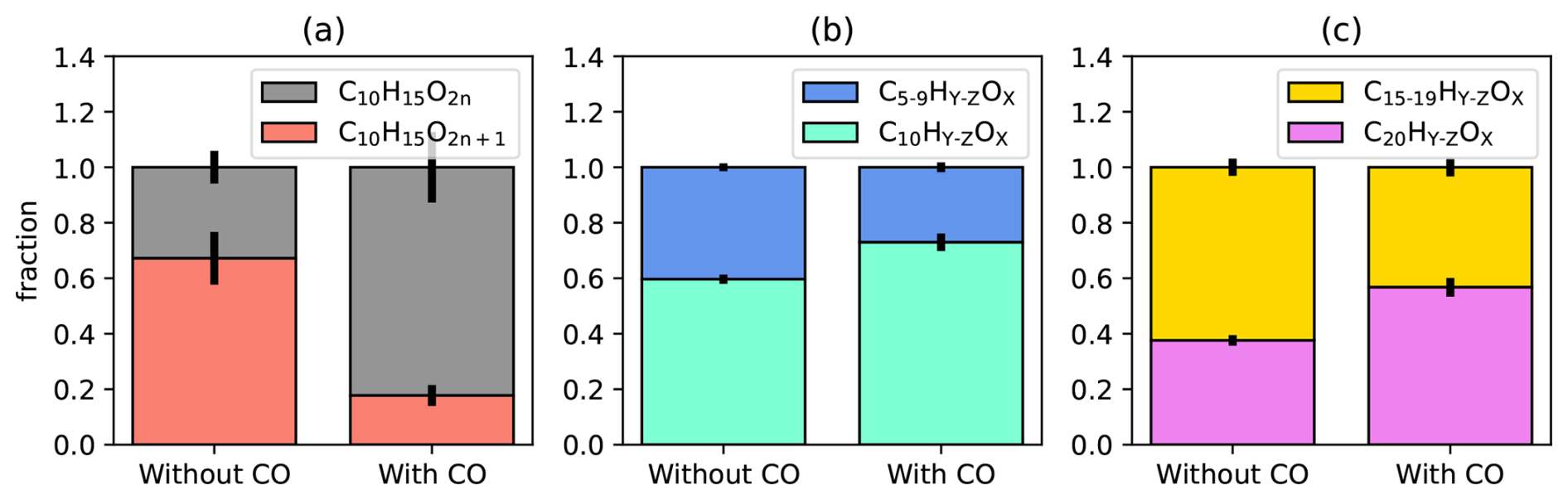
Figure 4Impact of enhanced [HO2•]ss on oxygen parity of C10H15OX• radicals and on fragmented products. (a) The fraction of C10H15O2n (black) and C10H15O2n+1 (red) for the cases without and with CO addition. (b) Fraction of fragmented monomers (C5−9, blue) and C10 monomers (mint). (c) Fraction of fragmented accretion products (yellow) and C20 accretion products (magenta). Error bars are calculated by error propagation based on the standard deviations for averaging over the steady state intervals.
The suppression of alkoxy steps can also be detected in the reduction of signals for compounds that underwent a fragmentation step during their formation (“fragmented compounds”). We summed up the signals C5−9-HOM monomers and C11−19-HOM accretion products with as an indicator for fragmentation and thus for the potential influence of alkoxy radical formation, shown in Fig. 4b and c, where it can be seen that the importance of fragmentation is decreased for both monomer and accretion products in the presence of CO.
In summary, the enhanced importance of HOM-RO2• + HO2• reactions compared to HOM-RO2• + RO2• reactions in the CO experiment led to a general suppression of the abundance of HOM-RO2• . Hereby concentrations of HOM-C10H15O2n+1 and fragmented compounds which are related to alkoxy steps were disproportionally stronger suppressed. This suggests that alkoxy-peroxy steps must play an important role in chemical regimes where HOM-RO2• + RO2• reactions are dominant. This includes those observed in our chamber experiments, where the concentration of precursors and OH• were chosen as low as possible but are still high compared to atmospheric conditions. High [HO2•] regimes are atmospherically relevant in remote and pristine areas, where NO concentrations are very low. In such cases, permutation reactions of peroxy radicals and thus alkoxy-peroxy steps should be less relevant. However, in the presence of sufficient NO, i.e., in chemical regimes where HOM-RO2• + NO reactions dominate, alkoxy-peroxy steps should be much more important, which we will investigate in the next sections.
3.2.3 Effect of NOx on formation of HOM-RO•
HOM peroxy radicals react with NO or NO2 forming organic nitrates (Reaction R4) and peroxy acyl nitrates (Reaction R4a) as stable closed-shell products. HOM that are organic nitrates (HOM-NO3) are mechanistically of diagnostic value, since the number of hydrogens in C10H15NOx does not change compared to the precursor peroxy radical C10H15OX−1. However, for this study the most interesting products of the HOM-RO2• + NO reaction are highly oxidized alkoxy radicals (HOM-RO•). A faster production of HOM-RO• by HOM-RO2• + NO reactions compared to HOM-RO2• + RO2• reactions can be expected by analogy to conventional radical chemistry (e.g., MCMv3.3.1, Saunders et al., 2003; Jenkin et al., 2019). Therefore, more alkoxy-peroxy steps should lead more often to changes of the parity of the oxygen numbers for C10H15OX peroxy radicals in the presence of NOx.
In Fig. 5, we show the effect of increasing [NOx]ss on the HOM peroxy radical distribution with the purpose of elucidating the importance of alkoxy-peroxy steps in the oxidation chain, indicated by the parity change of the oxygen number in the HOM-RO2•. Despite increasing [O3]ss with increasing [NOx]ss (Table S1), the turnover of α-pinene by OH contributed more than 90 % with exception of the highest [NOx] where it still contributed 86 %. The data in Fig. 5a are corrected to a reference state without particles and a turnover of 6.7 × 107 cm−3 s−1, which was the highest turnover rate observed in the OH• variation experiments (for details, see Sect. S2 and Pullinen et al., 2020). For the case without NOx addition, we observe similar fractions of C10H15O2n+1 and C10H15O2n (Fig. 5b) as for the highest turnover in the OH• variation experiment (Fig. 3b). Due to the OH• turnover of ≈ 1.5 × 108 cm−3 s−1 chosen in the experimental series, we start already with a substantial fraction of C10H15O2n+1 (≈ 0.8) without NOx addition. The large fraction of C10H15O2n+1 reflects the importance of alkoxy-peroxy steps, which are already caused by high RO2• concentrations, as discussed before. With increasing NOx, the fraction of C10H15O2n+1 decreased while the fraction with an even number of oxygen atoms, C10H15O2n, increased. We explain this observation by the fact that the high concentrations of RO2• and HOM-RO2• already favors the first alkoxy-peroxy step so strongly that nearly all accessible HOM-RO2• have undergone an alkoxy-peroxy step in their formation, leading to a dominance of odd oxygen numbers. The changes with NOx in Fig. 5b would then reflect the probability of encountering a second alkoxy-peroxy step, producing even oxygen numbers. In other words, within the residence time in our chamber C10H15O2n peroxy radicals undergo one alkoxy-peroxy step already in the absence of NOx and tend to undergo a second alkoxy-peroxy steps when [NOx] increases. Two alkoxy-peroxy steps result eventually in an increasing fraction of C10H15O2n peroxy radicals.
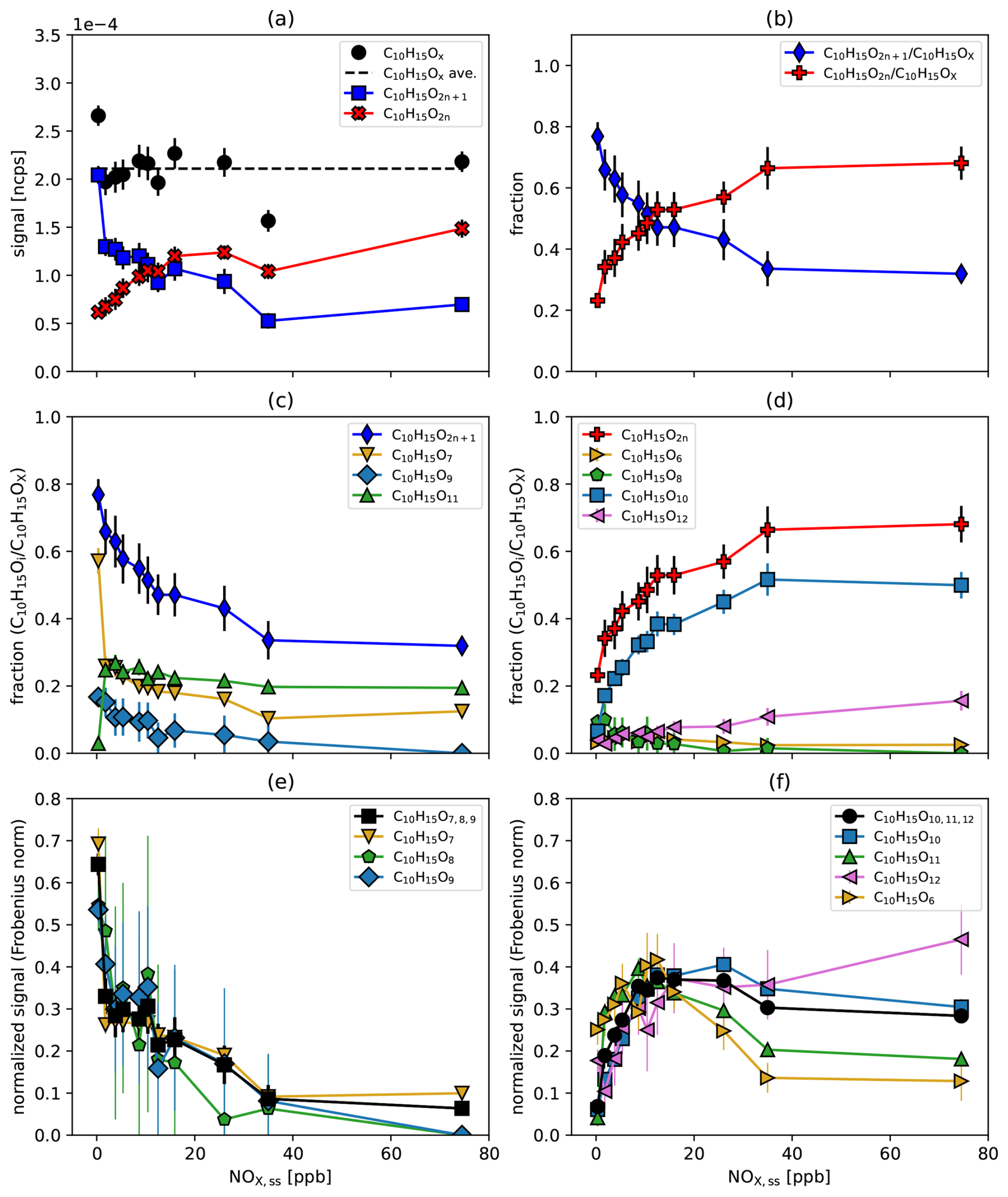
Figure 5C10H15OX peroxy radicals as a function of the steady state NOx concentration. (a) Particle and turnover corrected signal of the sum of the C10H15OX peroxy radicals and their average over all [NOx]ss (black) and the summed signals of C10H15O2n (red) and C10H15O2n+1 (blue). (b) The fractions of C10H15O2n (red) and C10H15O2n+1 (blue). (c, d) Contribution of the individual members to the fractions of C10H15O2n+1 and of C10H15O2n. (e, f) Signal of the individual family members, normalized by the Frobenius norm to highlight typical shapes as a function of increasing [NOx]. Also shown is the sum of the family members (black). The abundance of less oxidized C10H15O7−9 decreases with increasing [NOx] (e), while that of more oxidized C10H15O10−12 first increases and then remains at a high level with increasing [NOx] (f). C10H16OX (right pointing golden triangles) shows more similarity to the higher oxidized peroxy radicals. Errors are calculated by error propagation based on the standard deviations for averaging over the steady state intervals. The data in the range of 5–10 ppb NOx vary since similar [NO] in the system can be achieved through various combinations of NO source strength and O3 input.
The increasing HOM-RO• production with increasing [NOx]ss was confirmed by the increased fraction of fragmented HOM monomers (C5−9), which involve alkoxy decomposition in their formation process (Fig. S8a). Fragmented HOM accretion products (C11−19) decreased with [NOx]ss like C20-HOM-ACC as shown by Pullinen et al. (2020), but their relative importance increased compared to C20-HOM-ACC (Fig. S8b).
There is no common pattern for the fraction of each member as a function of [NOx]ss for both oxygen parities (Fig. 5c and d). C10H15O2n is essentially determined by C10H15O10 with minor contributions from the other three family members with different patterns (Fig. 5d), while C10H15O2n+1 has three significant contributions with varying patterns (Fig. 5c). Figure 5e and f highlight the typical shapes for the function signal versus [NOx]ss by dividing the signals by their Frobenius norm and grouping the C10H15OX peroxy radicals by oxygen number. The less oxidized peroxy radicals C10H15O strongly decrease with [NOx]ss (Fig. 5e), while the more oxidized C10H15O peroxy radicals strongly increase with [NOx]ss at low [NOx]ss and level off remaining high at high [NOx]ss (Fig. 5f). Apparently, the degree of oxidation of HOM-RO2• increases as the importance of the alkoxy-peroxy pathway increases. We conclude that the reaction with NO may compete with moderate fast autoxidation (see forthcoming Fig. 9b), but the alkoxy-peroxy steps, i.e., the isomerization of HOM-RO•, can continue the oxidation chain and lead to even higher oxidized products.
We interpret this as an indication that alkoxy steps must open additional channels to continue “the autoxidation chain”. Autoxidation in its pure form is characterized by a series of unimolecular H-shifts in a bath of a huge number of O2 molecules, which can be arbitrarily fast, but is strictly limited to molecular configurations that are structurally and energetically suitable. The formation of HOM-RO• is a bimolecular process, which is limited by the maximum number of reactive collisions. However, because alkoxy radicals are highly reactive, once formed they may have more options for unimolecular isomerization reactions than peroxy radicals with similar structures. Thus, it is possible that HOM-RO• extent the oxidation chain whenever direct autoxidation cannot compete with bimolecular alkoxy formation and bimolecular termination reactions. If a peroxy radical is then re-generated by rearrangement of HOM-RO• (and O2 addition), the autoxidation process can continue from there until an alkoxy step can compete again, and so on and so forth.
The effective production of HOM-RO2• by the alkoxy-peroxy pathway is confirmed by comparing the sum of [HOM−RO2•]ss to the sum of conventional C10-RO2• as a function of [NOx]ss. As shown before the C10H15OX signal remained high and was almost independent of NOx after correction for particle formation and OH• turnover (Fig. 5a, black circles). Directly observed data for the sum of all C10-HOM-RO2• without turnover corrections are shown in Fig. 6. The C10-HOM-RO2• show an increase with [NOx]ss up to 15 ppb and then a decrease with increasing [NOx]ss (Fig. 6, spheres). The maximum is related to the maximum in [OH•]ss as indicated by the color code and is the result of NOx controlling the HOX cycle. At lower [NOx]ss, [OH•]ss increases due to the reaction of NO + HO2• and decreases due to the reaction of NO2 + OH• producing HNO3 at higher [NOx] (Ehhalt, 1999). (This effect was removed in Fig. 5 by correcting for α-pinene turnover; Sarrafzadeh et al., 2016; Pullinen et al., 2020). However, at the highest [NOx]ss, the HOM-RO2• signal does not decrease completely, but still remains as high as in the no-NOx reference case. As can be seen from the color code, this is the case even though [OH•]ss is much lower than in the no-NOx reference case.
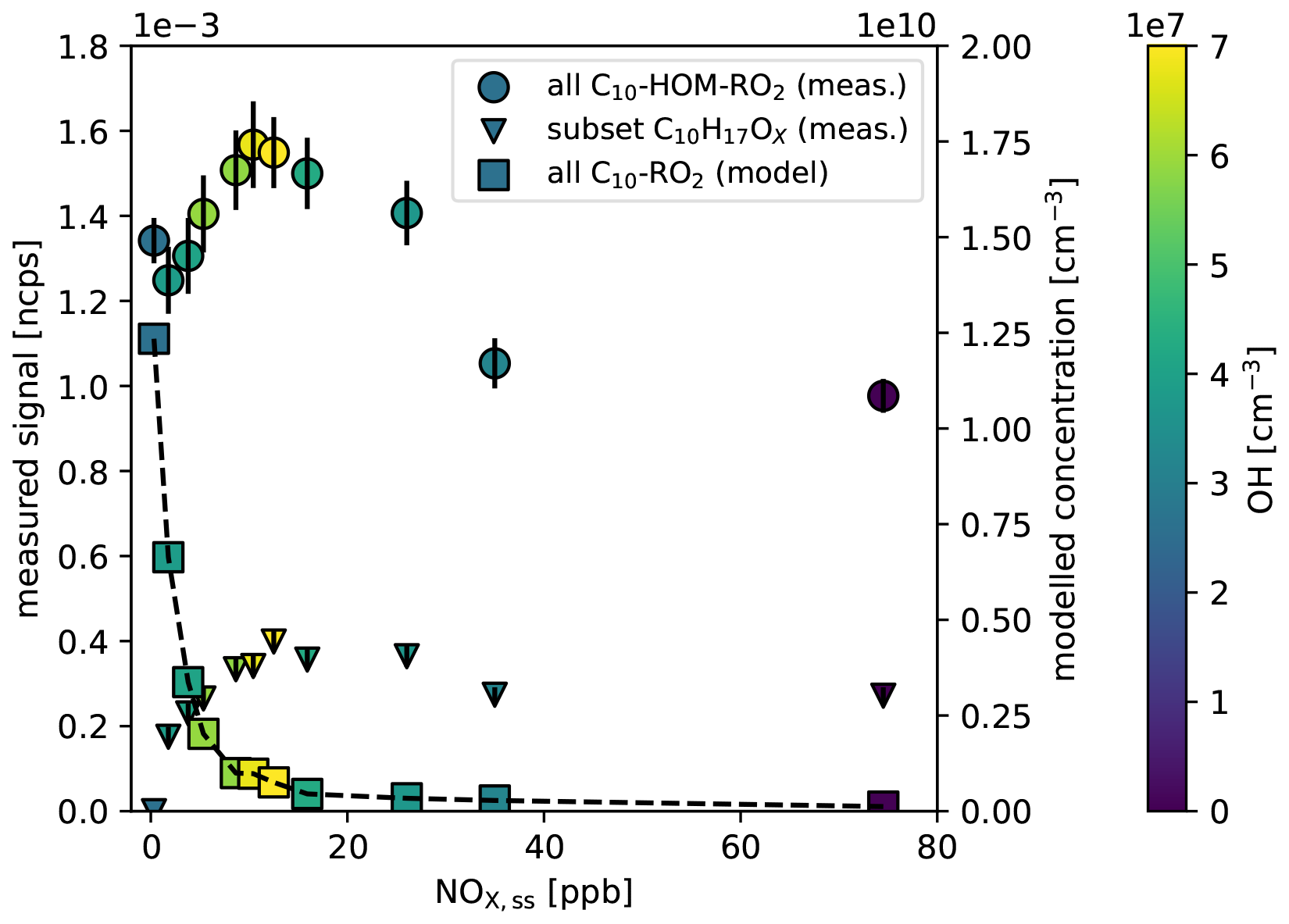
Figure 6Comparison of the dependence on [NOx]ss for observed C10-HOM-RO2• signal (spheres, left axis) and for modelled concentration of conventional C10-RO2• (squares, right axis), which is a fraction of the peroxy radical sum termed “RO2” in MCMv3.1.2. The color code indicates the actual [OH•]ss with a maximum of 6.9 × 107 cm−3 at about 15 ppb [NOx]ss. While the concentration of conventional, less oxygenated C10-RO2• drops quickly with [NOx]ss because of the strong sink provided by reaction with NO, the remains at a significant level even at the highest [NOx]ss. The triangles show the signal of the subset of C10H17OX HOM-peroxy radicals (left axis) which remain also high at high [NOx]ss. Errors for the experimental data are calculated by error propagation based on the standard deviations for averaging over the steady state intervals.
The fact that the C10-HOM-RO2• signal remains at a substantial level at high [NOx]ss is in contrast to the results to our model calculations for the sum of conventional C10-RO2•. This sum decreases rapidly with increasing [NOx]ss despite the intermediate increase in [OH]ss (Fig. 6, squares). The master chemical mechanism applied in the model calculation does neither allow for autoxidation of peroxy radicals nor for isomerization of alkoxy radicals, only for their fragmentation. (Note, the concentration of HOM-RO2• is less affected by increasing [NOx] compared to conventional RO2• since the autoxidation of specific peroxy radicals is still able to compete with bimolecular termination by NO.) As a consequence, the increasing formation of RO• and its fragmentation with increasing [NOx]ss provides a strong sink for RO2. Since the reaction with NO also becomes the fastest for HOM-RO2•, the NO sink must also be in operation for HOM-RO2•. As a consequence, there must be an efficient source for HOM-RO2•, which is not implemented in MCMv3.3.1, and compensates for the loss. Our findings suggest that isomerization of HOM-RO• radicals produced from HOM-RO2• + NO plays a key role in maintaining HOM-RO2• at high [NOx]ss. Indeed, HOM-RO• appear to rearrange by H-shift and add O2, as proposed by Vereecken and Peeters (2010) and sketched in Fig. 1, thus acting as a source of HOM-RO2•.
Additional production of HOM-peroxy radicals by the HOM-RO2• + NO reaction was also observed by Berndt et al. (2015). They show that even at 300 times higher [NO], HOM yield and peroxy radical (C10H17O7) yield were only suppressed by a factor of two (compare to our C10H17OX in Fig. 6, triangles). This demonstrates that HOM-RO• is also recycled to HOM-RO2• under conditions other than in our experiments. The alkoxy-peroxy pathway may be an important process in urban atmospheres, where the VOC to NOx ratio is often low due to the presence of high NOx concentrations.
3.3 Importance of alkoxy-peroxy steps for the ratio
We showed that higher oxygenated HOM-RO2• substantially gained importance with increasing [NOx]ss and remained at high levels at high NOx mixing ratios (Fig. 5f, black circles). In the following we investigate how the effect of alkoxy-peroxy steps is reflected in the ratio of the HOMs. We calculate concentration weighted for each family wherein the abundance of each family member i was considered as given in Eq. (1).
Here nO,i is the oxygen number and nC,i is the carbon number, while si is the signal of the family member and Σjsj is the summed signal of the whole family. For organic nitrates, one oxygen atom was excluded from the calculation to compensate for the extra addition of an O atom by the addition of NO to the molecule. With Eq. (1) also an overall ratio can be calculated by summing over all individual HOMs.
In the reference experiments without NOx addition (Table S1, J5–J11) the overall ratio is relatively invariant despite increasing turnover and [OH•] (background NOx 0.3 ppb, yellow circles, Fig. 7b). Only data for steady states are shown where OH• oxidation dominated and the contribution by ozonolysis was < 10 %. If at all, the tends to decrease from 0.77 to 0.76 with increasing turnover. We would argue that in absence of NOx the constant with increasing turnover despite increasing RO2• must be in parts due to an increasing importance of alkoxy steps compensating increasing RO2• + RO2• termination.
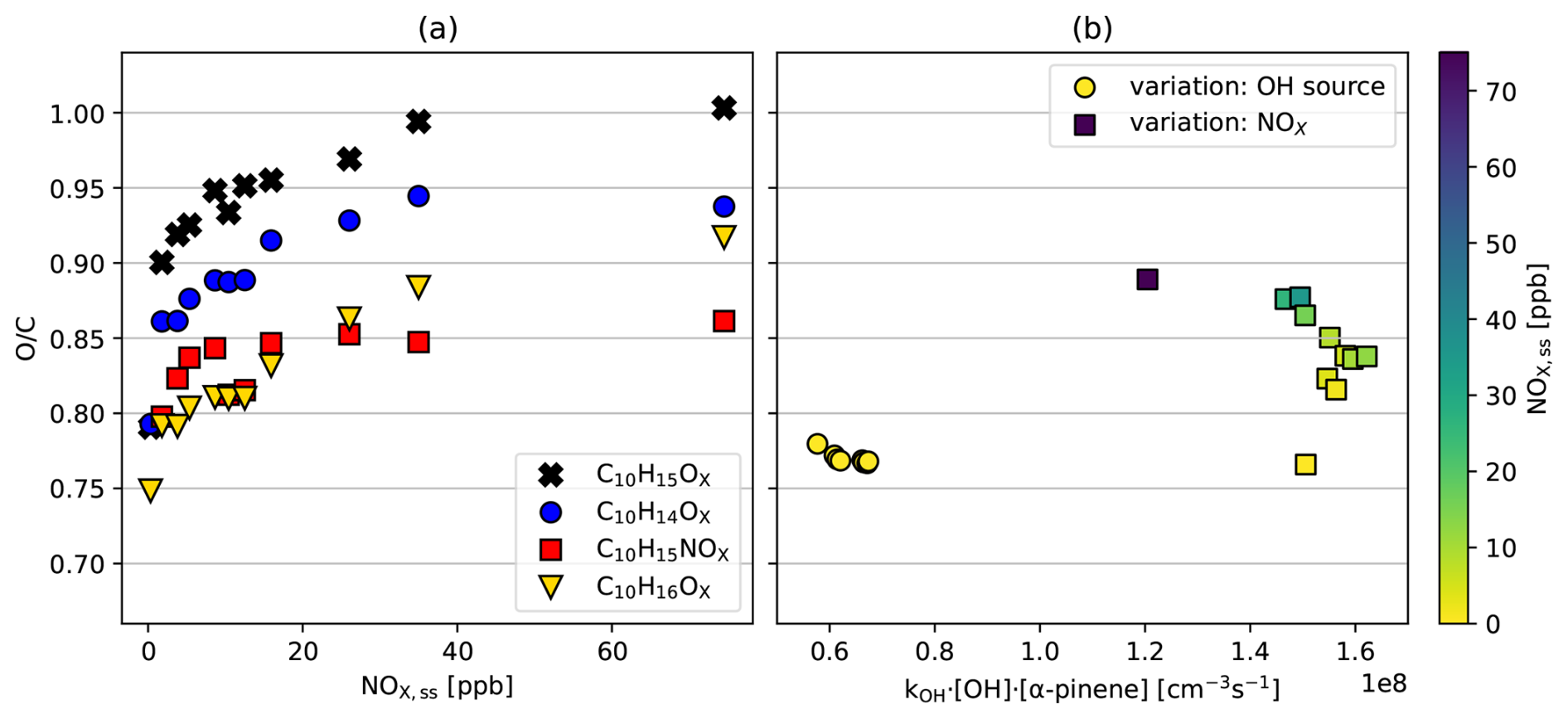
Figure 7(a) Averaged ratio of the HOM families as a function of [NOx]ss in the NOx experiments. (b) Overall as a function of turnover for C10 monomer compounds in the reference experiments (circles) and the NOx experiments series (squares). The color scale represents the level of steady-state concentration [NOx]ss in the chamber.
When NOx is added to system ratios increase with [NOx]ss as demonstrated in Fig. 7a for the ratio of the C10H15OX peroxy radical family and related termination products. The oxygen content of all families increased with [NOx]ss, partly because highly oxidized family members gain relative importance, as shown for C10H15OX in Fig. 5e and f, and partly because higher oxidation states were achieved within a given family. For example, the most oxidized molecule detected in the C10H16OX family was C10H16O12 in the no-NOx reference case, while it was C10H16O15 in the NOx experiments.
The increase of over the whole range of NOx concentrations is strongest with 0.2 for the HOM-RO2• themselves (≈ 0.8 increasing to ≈ 1.0). With ≈ 0.15 it is also strong for the C10H14OX- (≈ 0.8 increasing to 0.95) and C10H16OX families (0.75 to 0.92). The increase of 0.06 for HOM-NO3 (0.8 to 0.86) is weaker. This lower increase makes sense, since a part of the HOM-RO• produced in the reaction with NO terminates to HOM-NO3 and only the other part can continue the oxidation chain to higher oxygenated HOM products. Figure 7b shows the overall oxidation state for the families in Fig. 7c as a function of the turnover of α-pinene with the OH• radicals (squares). Notably, the reference data point without NOx addition (N1, yellow square) for the NOx experiments shows about the same as the reference experiments (spheres), although it was taken at 2.5 times higher turnover (highest [OH•]ss and higher [α-pinene]ss). On the other hand, as [NOx]ss increased, the ratio also increased from 0.76 (background 0.3 ppb of NOx) to 0.89 (72 ppb of NOx). This corresponds to more than one additional oxygen atom in the products on average.
In ozonolysis experiments including dark OH•, Molteni et al. (2019) found that the increase in the concentration of peroxy radicals inhibits autoxidation and lowers the average HOM ratio. They attributed this to the competition between the RO2• + RO2• reaction and autoxidation. In our photochemical system, we do not observe such effects for a variation of RO2• (from the model) and HOM-RO2• by more than a factor of 3. We observed a slight decrease in with increasing turnover, but it was very minor. For our photooxidation conditions the combination of autoxidation and alkoxy-peroxy chain is efficient enough to compete with termination and is always able to proceed to the highest possible degree of oxidation in HOM-RO2• (though with some shift in the distribution of termination products). In our opinion, this due to increasing HOM-RO• formation and subsequent isomerization which are a source of HOM-RO2• and compensate for increased loss of HOM-RO2• by increased [RO2•].
If HOM-RO2• + RO2• would suppress autoxidation by termination at the [RO2•]ss levels of about 109–1010 cm−3 observed in our reference experiments, one would expect a much stronger effect by NO addition because of the faster reaction of peroxy radicals with NO. A higher degree of autoxidation suppression and lower ratio should be observed. However, we observed that the presence of NOx boosted the degree of oxidation in the HOM-RO2• and the related HOM closed shell products.
There are two possible explanations for the increase in the ratio in the case of NOx. In general, the formation and isomerization of HOM-RO• continue the radical chain. Especially, when the alkoxy-peroxy pathway occurs via H-migration from a C-atom, a new HOM-RO2• can be formed that has one oxygen atom more than parent peroxy radical. In addition, the isomerization of HOM-RO• with either H-migration from C-H or -OOH opens more oxidation pathways and thus enables continuation of the autoxidation itself. The reason could be that in HOM-RO• also H-shifts with higher energy barriers can be realized which would not be accessible in HOM-RO2•. This is supported by quantum chemical calculations showing that H-shifts in alkoxy radicals can be orders of magnitude faster than in peroxy radicals, and can migration H-atoms that are not accessible to RO2 radical migrations (Jaoui et al., 2021; Vereecken and Nozière, 2020; Vereecken and Peeters, 2010).
The stronger increase in for the C10H15OX, C10H14OX, and C10H16OX families compared to HOM-NO3 (C10H15NOX) (Fig. 7a) suggests that the presence of NO leads to termination on one hand but also generates HOM-RO• that are able to isomerize to a large extend and continue the oxidation chain. However, quantum chemical calculations considering H-shift for molecules with high HO2-functionalization as HOMs have not yet been performed. The strong increase in caused by NOx addition is explainable by impacts of HOM-RO• produced by reactions of NO with the precursor HOM RO2•.
3.4 Competition between alkoxy-peroxy steps and direct autoxidation
Our findings so far are in support of isomerizations of HOM-RO• preserving the number of carbon atoms. We suggest a mechanistic scheme based on the quantum chemical considerations for RO• rearrangements by Vereecken and Peeters (2010) (Fig. 8), where the basic principle of the hydrogen migration in HOM-RO• was already shown in Fig. 1. There are two alkoxy-peroxy pathways besides isomerization for a parent HOM-RO2(OX)• (A) containing X oxygen atoms: if isomerization is competitive with NO and RO2• reactions, (A) will take the direct autoxidation route 1. An H-shift with subsequent O2 addition will lead to the next autoxidation product (B). However, the alkoxy route 2 may compete with direct autoxidation under formation of the intermediate HOM-RO• (C). By isomerization of (C) via route 3 HOM-RO2(OX+1)• (D) is formed with one oxygen more and a different oxygen parity than the parent HOM-RO2(OX)• (A). The HOM alkoxy radical (C) may also fragment (not shown), and the efficiency of isomerization compared to fragmentation will determine the importance of the alkoxy-peroxy step for further HOM formation. (Carbonyl formation by (C) with O2 is negligible.) The new peroxy radical HOM-RO2(OX+1)• (D) can either repeat the alkoxy-peroxy pathway via routes 4 and 5 or continue by autoxidation (route 6), contributing to the family of peroxy radicals with odd oxygen numbers. Route 4, 5 will lead via HOM-RO(OX)• (E) to HOM-RO(OX+2)• (B) with the same chemical formula and parity as the autoxidation product (A) via route 1. The structure of both species (B) is most likely different and mass spectrometry alone would not be able to distinguish between them. Peroxy radicals (B) can continue the autoxidation chain 7.
If HOM-RO• (C) is isomerized by shifting an H-atom from an HO2-group (route 8), a peroxy radical (F) is immediately formed with one oxygen atom less than the parent peroxy radical. This can also contribute by autoxidation to the HOM-RO2• family with odd oxygen numbers (route 9). Similarly, HOM-RO• (E) can form HOM-RO2• via step 10 with the same formula as (A). Of course, each HOM-RO2• in the scheme could be transformed into HOM-RO• and the steps described above could be repeated as indicated by route 11.
From Fig. 8 we can recognize that alkoxy-peroxy steps can directly compete with pure autoxidation by providing alternative routes to peroxy radicals of different (single alkoxy-peroxy step) or the same oxygen parity (two alkoxy-peroxy steps). It is also understandable that an alkoxy step can enable the continuation of the autoxidation process in competition with termination when isomerization is not possible or slow for a specific HOM-RO2• . This excursion via HOM-RO• will eventually form new HOM-RO2• capable of continuing the fast autoxidation processes (via hydrogen shifts in the new HOM-RO2•). However, alkoxy formation always involves a bimolecular step, which is kinetically limited by the reactive collision rate, while intramolecular rearrangements can exceed even the collision limit.
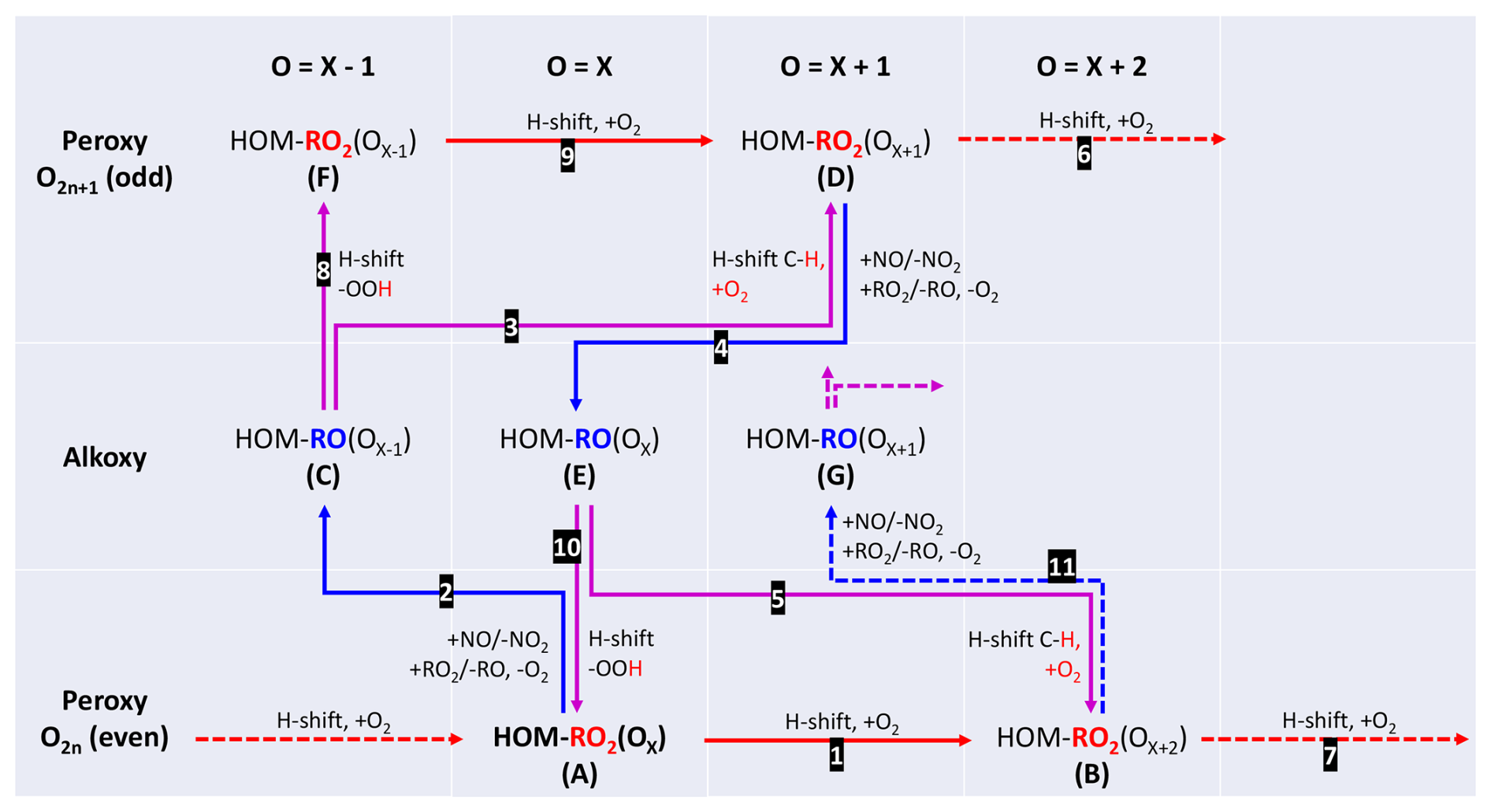
Figure 8Scheme of alkoxy-peroxy pathways. Red arrows indicate autoxidation of peroxy radicals, blue arrows indicate formation of alkoxy radicals in reactions of the peroxy radicals with other RO2 or with NO, and lilac arrows indicate isomerization of alkoxy radicals with subsequent O2 addition, where it applies.
The scheme in Fig. 8 demonstrates that whenever an autoxidation step is hindered, alkoxy isomerization can step in and provide a new higher (differently) oxygenated HOM-RO2• and thus opens more potential reaction channels. So, a higher degree of oxidation can be expected for HOM-RO2• and their termination products, as was indeed observed (Fig. 7).
Besides the bimolecular reaction rates for the reactions HOM-RO2• + RO2• and HOM-RO2• + NO, the branching ratio into isomerization by H-shift for HOM-RO• (fiso) determines the efficiency of alkoxy-peroxy pathways in competition to fragmentation. It is not unlikely that larger and more functionalized HOM-RO•, such as those produced by α-pinene photooxidation, tend to undergo isomerization rather than fragmentation (Jaoui et al., 2021; Vereecken and Peeters, 2010). By considering the fraction of C10H15O2n+1 as a measure for alkoxy steps that have taken place and the distribution of non-fragmented and fragmented HOM monomers as well as non-fragmented and fragmented entities in HOM-ACC, we estimated the average fiso for α-pinene to be approximately 0.5 with an estimated uncertainty range of 0.36–0.64 (details in Sect. S6).
To ensure that reactions of HOM-RO2• with NO occur with comparable reaction rates to conventional chemistry, we calculated the rate coefficient for reaction R4 from the seven pairs of HOM-RO2• HOM-NO3 that we could observe (Sect. S6, Fig. S10). Note is not referring to an elementary reaction, since each stoichiometric compound – detectable by mass spectrometry – can comprise several structure isomers. The calculated for the individual couples varied with NO, with a tendency towards higher values at lower NO. The average of the is 1.15(±1.0) × 10−12 cm3 s−1, the relatively large variation results from lumping within and across all formula compounds (Fig. S10). Applying the branching into organic nitrates of 0.36 (Pullinen et al., 2020) we yield an average rate coefficient of 3.2 × 10−11 cm3 s−1 for the reaction of HOM-RO2• + NO.
For the following comparison of the relative importance of autoxidation and bimolecular pathways we used the values determined by us. The rate coefficient into the alkoxy path will then amount to 0.64 × 3.2 × 10−11 cm3 s−1. For the branching of HOM-RO• into isomerization, we will apply fiso = 0.5 as determined by us. Furthermore, we apply a rate coefficient for HOM-RO2• + RO2• of 5 × 10−12 cm3 s−1, and a branching ratio into HOM-RO• + RO• + O2 (Reaction R7) of 0.6 (Jenkin et al., 2019). Note, we are dealing here with mass spectrometric data, and several isomers are possibly lumped under the same formula. The values for rate coefficients and branching ratios we derived and chose for the following calculations are suited for the intended plausibility considerations and should be handled with care when transferred to other systems or used for the purpose of strict quantification.
Figure 9 compares the relative importance of autoxidation (green dashed line), alkoxy-peroxy path (red diamonds and pink triangles), and termination (black spheres) as a function of [OH•]ss for the reference case (Table S1, J1–J12, Fig. 9a) and as a function of [NOx]ss for the NOx addition case (Table S2, N1–N11, Fig. 9a). For autoxidation an average unimolecular autoxidation rate coefficient of 0.1 is shown, which can vary a lot, indicated by a factor of 10 range shaded in green (compare Praske et al., 2018; Nie et al., 2023). In addition, wall loss (τwall = 120 s) is depicted (black dashed line).
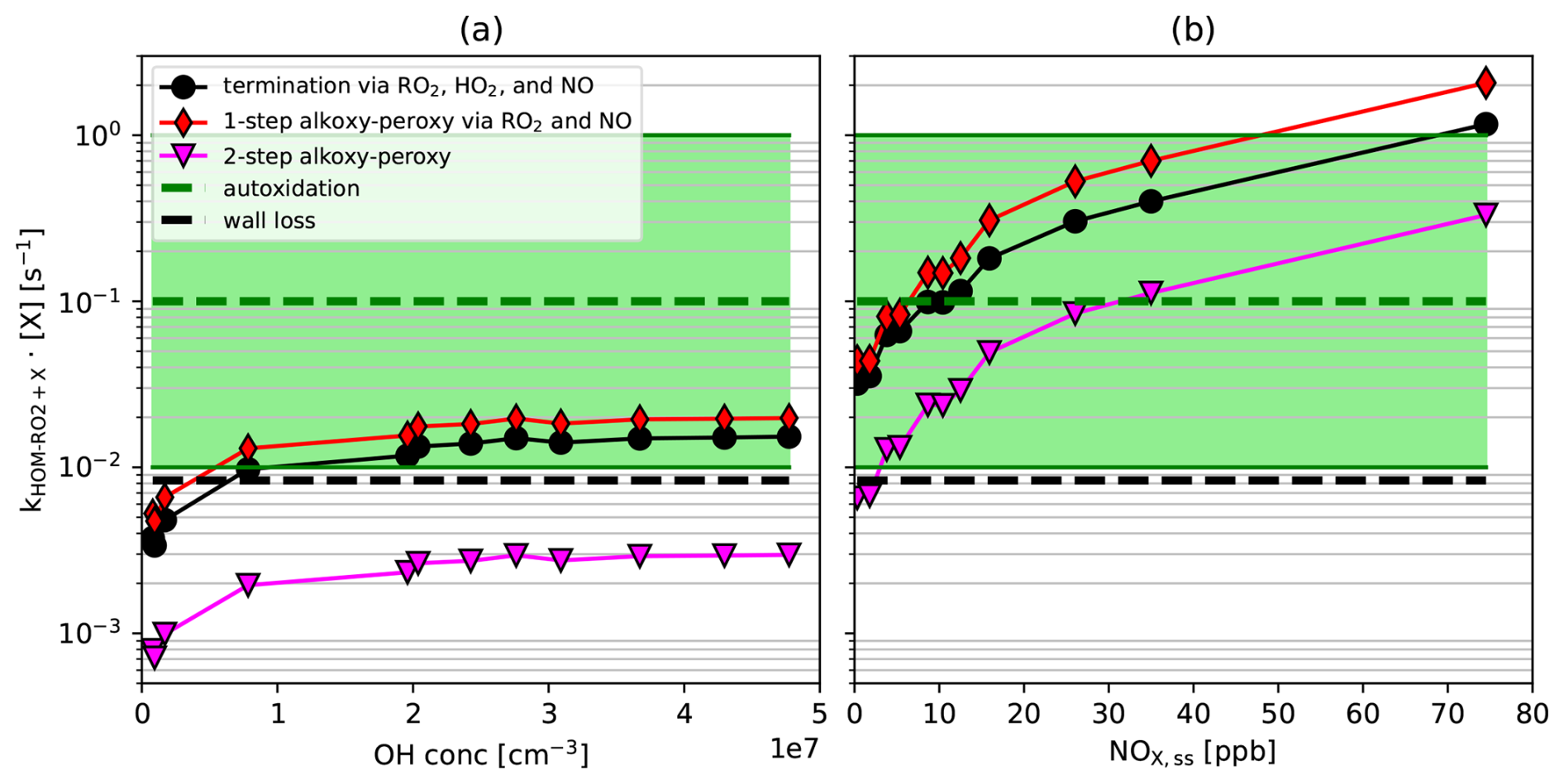
Figure 9First order rate coefficients (rate coefficient multiplied by the concentration of the reaction partner for bimolecular reaction or rate coefficient for unimolecular reaction) versus steady-state [OH•]ss (a) and [NOx]ss (b). Black spheres represent the sum of termination reactions by RO2•, HO2•, and NO, where it applies. For the termination rate coefficients of the reactions with RO2• and NO branching ratios 0.4 and 0.36 were taken into account. Red diamonds represent the alkoxy-peroxy path with branching ratios 0.6 and 0.64 for HOM-RO• formation taken into account and fiso = 0.5 for isomerization of HOM-RO• under retaining the C10-backbone. Loss to the wall (black dotted line) and typical autoxidation (green dotted line) with factor 10 upper and lower bands (green) are also shown in both plots. The importance of the pathway with two alkoxy-peroxy steps in series is shown by pink triangles.
Termination Reactions (R2), (R3), and (R4) were considered and rate coefficients were multiplied by the according branching ratios into alcohols carbonlys (0.4) and organic nitrates (0.36) and by the respective steady state concentrations of [RO2•]ss (model), [HO2•]ss (model), and [NO]ss (measured). For Reaction (R2) we applied the generic rate coefficient KRO2HO2 from MCMv3.1.1, as used in the model calculations. The contributions of Reactions (R2), (R3), and (R4) were summed (Fig. 9, black spheres).
The alkoxy-peroxy path cannot be faster than the formation of HOM-RO•. Under the assumption that this entrance channel is the rate limiting step, multiplication by fiso determines the fraction that is recovered as HOM-RO2• with intact C10 backbone. The rate coefficients for Reactions (R7) and (R8) were thus multiplied with the according branching ratio into HOM-RO• of 0.6 and 0.64, the respective steady state concentrations of [RO2•]ss (model) and [NO]ss (measured), and fiso for isomerization of HOM-RO•. Both contributions were summed up (red diamonds). The efficiency of the pathway with two alkoxy-peroxy steps which produces the same formula compounds as a single autoxidation step is calculated by taking advantage of the steady-state conditions under assumption that autoxidation of the intermediate HOM-RO2• is negligible (details are described in Sect. S6). The triangles show the result applying our fiso = 0.5, i.e. 50 % of the alkoxy radicals undergo the alkoxy-peroxy pathway and 50 % undergo fragmentation.
For the reference case without NOx addition, autoxidation is on average by far the most efficient process (Fig. 9a, green dashed line) for all [OH•]ss. The one-step alkoxy-peroxy pathway (Fig. 9a red diamonds) is by about a factor of five less important. Alkoxy-peroxy steps can become competitive for specific HOM-RO2• with autoxidation rate coefficients < 0.03 s−1. The alkoxy-peroxy path is in any case competitive compared to closed-shell formation from Reactions (R3) and (R2) (Fig. 9a, black spheres). The formation of HOMs by two subsequent alkoxy-peroxy steps is unimportant (Fig. 9a, pink triangles). As a result, in the reference case HOM-RO2• were likely formed by the autoxidation supported by single alkoxy peroxy steps. The latter was shown by the increasing fraction of C10H15O2n+1 peroxy radicals, with changed oxygen parity (Fig. 3b). The dominance of autoxidation over termination explains also why the is invariant with [OH•]ss.
In the presence of NOx, the formation of closed shell products is increasing strongly with [NOx]ss (Fig. 9b, black spheres) and is due to increasing HOM-NO3 formation. Termination by NO is able to compete with average autoxidation. At the same time the importance of the alkoxy-peroxy path (Fig. 9b, red diamonds) is also increasing with [NOx]ss and can compete with average autoxidation as well with termination. Even the two-step alkoxy-peroxy path can compete with autoxidation at the highest [NOx]ss. In other words, as [NOx]ss increases, the importance of the alkoxy-peroxy pathway increases and may even surpass autoxidation (via isomerization of HOM-RO2•) as carrier of the radical chain. As proposed by Shen et al. (2022), in earlier work by Vereecken and Peeters (2004), and Xu et al. (2019), the alkoxy step may also occur very early before the autoxidation chain, e.g. for ring opening.
It is further interesting that for the chemical systems and rate coefficients determined and used here, with and without NOx addition, termination and one-step alkoxy-peroxy path are of about the same magnitude. Introduction of the alkoxy-peroxy pathway thus enabled the compensation of the increasing HOM-RO2• sink, because of increasing bimolecular termination, by increasing importance of alkoxy-peroxy steps as a source of HOM-RO2•. This is consistent with our observations (see Fig. 6). In case of NOx addition where the turnover was about constant the [HOM-RO2•]ss became relatively invariant of [NOx]ss. In the reference case the sink and alkoxy source of HOM-RO2• are also about balanced (Fig. 9a) but [HOM-RO2•]ss still increased (Fig. 2a) because of increasing production of peroxy radicals by increasing [OH]ss.
A series of α-pinene photooxidation experiments were carried out under different chemical regimes to investigate the importance of highly oxygenated alkoxy radicals for HOM formation. At our conditions, i.e. steady state with one hour residence time, we observed that the major contribution to the HOM chemistry is related to C10H15OX peroxy radicals formed from H-abstraction, although OH addition to α-pinene is its major degradation pathway and HOM chemistry related to C10H17OX would be expected to dominate. The direct observation of a higher fraction of C10H15OX compared to C10H17OX as well as the product distribution of HOM monomers and HOM accretion products provided evidence that C10H15OX peroxy radicals act as major peroxy radicals for HOM formation. This result is valid regardless of the chemical regime, independently whether HOM peroxy radicals react mainly with RO2•, HO2• or NO. There are probably two reasons for this: (a) in the evolved chemical system at steady state, H-abstraction by OH• from primary C10H16O2 products may contribute significantly to HOM chemistry, and (b) H-abstraction from α-pinene by OH• (instead of OH• addition), leading to C10H15OX• peroxy radicals, may be a major path to HOM, as proposed by Shen et al. (2022). Both pathways could be of similar importance (Fig. S6), and this would be also valid for the chemically evolved ambient atmosphere.
The change in parity of the oxygen number within the C10H15OX family proved to be a good indicator of the involvement of formation steps via isomerization of HOM alkoxy radicals. By parity analysis, we detected increasing HOM-RO• formation with increasing importance of RO2• + RO2• or RO2• + NO reactions. Increasing RO2• + HO2• reaction showed the suppression of HOM-RO• formation. Moreover, the corresponding change of HOMs with C<10 and HOM-ACC with C11−19, which went through fragmentation steps in their formation process, agrees with the result of the parity analysis.
Based on our findings we provided a generic scheme that illustrates alkoxy-peroxy pathways (Fig. 8). Alkoxy-peroxy pathways can compete with or even outperform the autoxidation at high NOx concentrations. The formation of HOM-RO• in bimolecular reactions and its isomerization also allows the oxidation chain to continue when the autoxidation of the preceding HOM-RO2• cannot compete with bimolecular reactions. The formation of HOM-RO• opens additional channels to achieve high in atmospheric oxidation of VOC. Even in absence of NOx, where RO2• + RO2• reactions dominate single alkoxy steps may take place, in our case if the actual autoxidation rate is about a factor of five lower than an average autoxidation rate of 0.1 s−1. We conclude that in RO2• + RO2• and particularly in RO2• + NO dominated regimes, HOM-RO• formation, HOM-RO• isomerization, and subsequent peroxy radical formation are important steps during HOM formation. Especially in NOx regimes with enhanced RO2• + NO reaction, HOM-RO• formation is very efficient, and it can contribute significantly to HOM-RO2• recycling after undergoing the alkoxy-peroxy pathway (Figs. 5a, 5f, 6).
In pristine regions, where the main bimolecular reaction of HOM-RO2• is the reaction with HO2• , the formation of HOM-RO• and HOM-RO2• by the alkoxy-peroxy pathway may be less important. However, in the ambient atmosphere the fate of peroxy radicals is also often controlled by NOx. In highly polluted urban areas where the NOx concentration can be high, the formation of HOM-RO• and the higher degree of oxidation by the alkoxy-peroxy pathway can be an important source of HOMs. In the NOx regime, HOM-RO• formation via the rate limiting bimolecular HOM-RO2• + NO reaction is competitive with autoxidation and can become easily an order of magnitude faster than autoxidation steps of ∼ 0.01 s−1. Under high NOx conditions even a two-step alkoxy-peroxy chain can become competitive with a single autoxidation step (Fig. 9b). Under NOx regimes enhancement of and the autoxidation chain via HOM-RO• can be important for particle formation and growth. However, this is somewhat offset by the suppression of accretion product formation, which is the reason for the reduced new particle formation for SOA mass suppression in the presence of NOx as often observed in laboratory experiments.
The available studies on the role of alkoxy-peroxy steps in autoxidation are limited to bicyclic compounds (e.g. this work, Shen et al. 2022, Nie et al. 2023), alkanes (e.g. Wang et al. 2021), and some other molecules with specific functionalities. The analysis of the contribution of alkoxy-peroxy steps in this work, however, does not rely on specific chemistry of α-pinene but also holds for a variety of atmospheric VOC. Prerequisite is a backbone of a certain minimum length since H-shift are largely determined by formation of cyclic structures in the transition states. Consideration of alkoxy-peroxy steps enables to derive a consistent interpretation across a wide variety of reaction conditions based solely on the reactivity trends captured in generalized Structure-Activity Relationships applicable to all peroxy- and alkoxy radicals (e.g. Jenkin et al. 2019, Vereecken and Peeters, 2009, 2010). This forces upon us the realization that the alkoxy-peroxy step is likely to be a general feature of many VOC autoxidation chains, and that the prevalent view that autoxidation and HOM formation is chemistry based on peroxy radicals is highly incomplete. Indeed, the ability of H-migration in alkoxy radicals to restart the RO2• autoxidation chain makes the autoxidation process much more resilient against bimolecular termination reactions, or against simply reaching an RO2• intermediate not amenable to autoxidation. Without alkoxy-peroxy steps, autoxidation chains may prove to be rather short. This might even imply that the pristine environment, with longer RO2• lifetimes but dominated by HO2• termination, could be the less favorable regime for HOM formation compared to more polluted reaction mixtures favoring alkoxy formation in RO2• + NO, RO2• , or NO3• bimolecular “termination” reactions. This in turn shifts our perception in which atmospheric regimes HOM and aerosol formation through autoxidation will play its most dominant role. Given the molecular structure of the most commonly emitted VOCs, i.e. (bi)cyclic and unsaturated terpenoids, even bond breaking in the alkoxy radicals may prove to be a key feature in the atmosphere, as it enhances autoxidation by breaking of cyclic structures hindering H-migration (see e.g. Shen et al. 2022), for both, rings present in the VOC as well as rings formed in e.g. the fast ring closure reactions in unsaturated RO2• (Vereecken et al., 2021). The qualitative and quantitative interpretation of experimental observations on HOM formation and aerosols, as well as the formulation of chemical mechanisms describing these processes, seems impossible without the systematic inclusion of HOM-alkoxy radical chemistry for all relevant VOCs.
Information about all steady-state conditions utilized and the data necessary to reproduce the figures in this study are available on Jülich DATA (https://doi.org/10.26165/JUELICH-DATA/0MTPDO, Kang et al., 2025).
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-15715-2025-supplement.
Author contributions. SK, JW, and TFM prepared the manuscript with contributions by LV and SRZ. IP, CW and JW performed the measurements. SK, IP and CW analyzed the data. SK provided the high resolution CIMS data. TFM performed the model calculations. The compiled data set was interpreted by SK, TFM, and JW. All co-authors discussed the results and commented on the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We acknowledge the helpful discussions with Lukas Pichelstorfer and Gordon McFiggans regarding the formulation of mechanistic aspects in HOM formation. We thank Franz Rohrer for providing the NOx measurements and Stefanie Andres for measuring particle data. A special thanks to Einhard Kleist for helping with the JPAC operation and providing JPAC parameters. We thank both reviewers for their helpful comments.
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Mingyi Wang and reviewed by two anonymous referees.
Aschmann, S. M., Atkinson, R., and Arey, J.: Products of reaction of OH radicals with α-pinene, J. Geophys. Res.-Atmos., 107, 4191, https://doi.org/10.1029/2001jd001098, 2002.
Assaf, E., Schoemaecker, C., Vereecken, L., and Fittschen, C.: Experimental and theoretical investigation of the reaction of RO2 radicals with OH radicals: Dependence of the HO2 yield on the size of the alkyl group, Int. J. Chem. Kin., 50, 670–680, https://doi.org/10.1002/kin.21191, 2018.
Atkinson, R.: Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 41, 8468–8485, https://doi.org/10.1016/j.atmosenv.2007.07.002, 2007.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, S197–S219, 2003a.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003b.
Berndt, T.: Peroxy Radical Processes and Product Formation in the OH Radical-Initiated Oxidation of α-Pinene for Near-Atmospheric Conditions, J. Phys. Chem. A, 125, 9151–9160, https://doi.org/10.1021/acs.jpca.1c05576, 2021.
Berndt, T., Richters, S., Kaethner, R., Voigtlaender, J., Stratmann, F., Sipilae, M., Kulmala, M., and Herrmann, H.: Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurten, T., Otkjaer, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipila, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Mender, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bianchi, F., Trostl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kurten, A., Manninen, H. E., Munch, S., Perakyla, O., Petaja, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, https://doi.org/10.1126/science.aad5456, 2016.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Capouet, M., Müller, J.-F., Ceulemans, K., Compernolle, S., Vereecken, L., and Peeters, J.: Modeling aerosol formation in α-pinene photo-oxidation experiments, J. Geophys. Res., 113, D02308, https://doi.org/10.1029/2007JD008995, 2008.
Cox, R. A. and Cole, J. A.: Chemical aspects of the autoignition of hydrocarbon-air mixtures, Combustion and Flame, 60, 109–123, https://doi.org/10.1016/0010-2180(85)90001-x, 1985.
Crounse, J. D., Knap, H. C., Ornso, K. B., Jorgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O-2, Journal of Physical Chemistry A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012.
Crounse, J. D., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, Journal of Physical Chemistry Letters, 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Dames, E. E. and Green, W. H.: The Effect of Alcohol and Carbonyl Functional Groups on the Competition between Unimolecular Decomposition and Isomerization in C4 and C5 Alkoxy Radicals, Int. J. Chem. Kinet., 48, 544–555, https://doi.org/10.1002/kin.21015, 2016.
Davidson, C. I., Phalen, R. F., and Solomon, P. A.: Airborne particulate matter and human health: A review, Aerosol Sci. Tech., 39, 737–749, https://doi.org/10.1080/02786820500191348, 2005.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 1: Gas-phase composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 6489–6504, https://doi.org/10.5194/acp-12-6489-2012, 2012a.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012b.
Ehhalt, D. H.: Chemical reactions in the troposphere, in: Global Aspects of Atmospheric Chemistry, edited by: Zellner, R. G. E., Topics in Physical Chemistry, Steinkopff, Darmstadt, 60–94, ISBN 978-3798511279, 1999.
Ehn, M., Kleist, E., Junninen, H., Petäjä, T., Lönn, G., Schobesberger, S., Dal Maso, M., Trimborn, A., Kulmala, M., Worsnop, D. R., Wahner, A., Wildt, J., and Mentel, Th. F.: Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 12, 5113–5127, https://doi.org/10.5194/acp-12-5113-2012, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ehn, M., Berndt, T., Wildt, J., and Mentel, T.: Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 49, 821–831, https://doi.org/10.1002/kin.21130, 2017.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas-phase concentration of H2SO4 and Methane Sulfonic Acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.-Atmos., 98, 9001–9010, https://doi.org/10.1029/93jd00031, 1993.
Färber, M., Fuchs, H., Bohn, B., Carlsson, P. T., Gkatzelis, G. I., Marcillo Lara, A. C., Rohrer, F., Vereecken, L., Wedel, S., and Wahner, A.: Effect of the Alkoxy Radical Chemistry on the Ozone Formation from Anthropogenic Organic Compounds Investigated in Chamber Experiments, ACS ES&T Air, 1, 1096–1111, 2024.
Fittschen, C.: The reaction of peroxy radicals with OH radicals, Chem. Phys. Lett., 725, 102–108, https://doi.org/10.1016/j.cplett.2019.04.002, 2019.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hasan, G., Salo, V. T., Valiev, R. R., Kubecka, J., and Kurtén, T.: Comparing Reaction Routes for 3(RO•••OR′) Intermediates Formed in Peroxy Radical Self- and Cross-Reactions, J. Phys. Chem. A, 124, 8305–8320, https://doi.org/10.1021/acs.jpca.0c05960, 2020.
Hyttinen, N., Rissanen, M. P., and Kurtén, T.: Computational Comparison of Acetate and Nitrate Chemical Ionization of Highly Oxidized Cyclohexene Ozonolysis Intermediates and Products, J. Phys. Chem. A, 121, 2172–2179, https://doi.org/10.1021/acs.jpca.6b12654, 2017.
Iyer, S., Reiman, H., Møller, K. H., Rissanen, M. P., Kjaergaard, H. G., and Kurtén, T.: Computational Investigation of RO2 + HO2 and RO2 + RO2 Reactions of Monoterpene Derived First-Generation Peroxy Radicals Leading to Radical Recycling, J. Phys. Chem. A, 122, 9542–9552, https://doi.org/10.1021/acs.jpca.8b09241, 2018.
Iyer, S., Rissanen, M. P., and Kurtén, T.: Reaction between Peroxy and Alkoxy Radicals Can Form Stable Adducts, J. Phys. Chem. Lett., 10, 2051–2057, https://doi.org/10.1021/acs.jpclett.9b00405, 2019.
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 12, 878, https://doi.org/10.1038/s41467-021-21172-w, 2021.
Jaoui, M., Piletic, I. R., Szmigielski, R., Rudzinski, K. J., Lewandowski, M., Riedel, T. P., and Kleindienst, T. E.: Rapid production of highly oxidized molecules in isoprene aerosol via peroxy and alkoxy radical isomerization pathways in low and high NOx environments: Combined laboratory, computational and field studies, Sci. Total Environ., 775, 145592, https://doi.org/10.1016/j.scitotenv.2021.145592, 2021.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/b704260b, 2008.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V. M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Jokinen, T., Kontkanen, J., Lehtipalo, K., Manninen, H. E., Aalto, J., Porcar-Castell, A., Garmash, O., Nieminen, T., Ehn, M., Kangasluoma, J., Junninen, H., Levula, J., Duplissy, J., Ahonen, L. R., Rantala, P., Heikkinen, L., Yan, C., Sipilä, M., Worsnop, D. R., Bäck, J., Petäjä, T., Kerminen, V. M., and Kulmala, M.: Solar eclipse demonstrating the importance of photochemistry in new particle formation, Scientific Reports, 7, https://doi.org/10.1038/srep45707, 2017.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kang, S., Wildt, J., Pullinen, I., Vereecken, L., Wu, C., Wahner, A., Zorn, S. R., and Mentel, T. F.: Supplementary data for Formation of highly oxygenated organic molecules from α-pinene photooxidation: evidence for the importance of highly oxygenated alkoxy radicals, Jülich DATA [data set], https://doi.org/10.26165/JUELICH-DATA/0MTPDO, 2025.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016gl069239, 2016.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Trostl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X. M., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kurten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Perakyla, O., Piel, F., Petaja, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipila, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tome, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P. L., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Kurtén, T., Petäjä, T., Smith, J., Ortega, I. K., Sipilä, M., Junninen, H., Ehn, M., Vehkamäki, H., Mauldin, L., Worsnop, D. R., and Kulmala, M.: The effect of H2SO4 – amine clustering on chemical ionization mass spectrometry (CIMS) measurements of gas-phase sulfuric acid, Atmos. Chem. Phys., 11, 3007–3019, https://doi.org/10.5194/acp-11-3007-2011, 2011.
Lamarque, J.-F., Bond, T. C., Eyring, V., Granier, C., Heil, A., Klimont, Z., Lee, D., Liousse, C., Mieville, A., Owen, B., Schultz, M. G., Shindell, D., Smith, S. J., Stehfest, E., Van Aardenne, J., Cooper, O. R., Kainuma, M., Mahowald, N., McConnell, J. R., Naik, V., Riahi, K., and van Vuuren, D. P.: Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: methodology and application, Atmos. Chem. Phys., 10, 7017–7039, https://doi.org/10.5194/acp-10-7017-2010, 2010.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, 2016.
Lee, B. H., Iyer, S., Kurtén, T., Varelas, J. G., Luo, J. Y., Thomson, R. J., and Thornton, J. A.: Ring-opening yields and auto-oxidation rates of the resulting peroxy radicals from OH-oxidation of α-pinene and β-pinene (vol 3, pg 399, 2023), Environmental Science-Atmospheres, 3, 1847–1847, https://doi.org/10.1039/d3ea90045b, 2023.
Luo, H., Vereecken, L., Shen, H., Kang, S., Pullinen, I., Hallquist, M., Fuchs, H., Wahner, A., Kiendler-Scharr, A., Mentel, T. F., and Zhao, D.: Formation of highly oxygenated organic molecules from the oxidation of limonene by OH radical: significant contribution of H-abstraction pathway, Atmos. Chem. Phys., 23, 7297–7319, https://doi.org/10.5194/acp-23-7297-2023, 2023.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Meder, M., Peräkylä, O., Varelas, J. G., Luo, J., Cai, R., Zhang, Y., Kurtén, T., Riva, M., Rissanen, M., Geiger, F. M., Thomson, R. J., and Ehn, M.: Selective deuteration as a tool for resolving autoxidation mechanisms in α-pinene ozonolysis, Atmos. Chem. Phys., 23, 4373–4390, https://doi.org/10.5194/acp-23-4373-2023, 2023.
Meder, M., Graeffe, F., Luo, Y. Y., Luo, J. Y., Iyer, S., Valiev, R., Cai, R. L., Rissanen, M., Kurtén, T., Varelas, J. G., Geiger, F. M., Thomson, R. J., and Ehn, M.: Selective Deuteration Reveals the Importance of Multiple Branching Pathways in α-Pinene Autoxidation, J. Am. Chem. Soc., 147, 14131–14138, https://doi.org/10.1021/jacs.4c14462, 2025.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Mentel, Th. F., Wildt, J., Kiendler-Scharr, A., Kleist, E., Tillmann, R., Dal Maso, M., Fisseha, R., Hohaus, Th., Spahn, H., Uerlings, R., Wegener, R., Griffiths, P. T., Dinar, E., Rudich, Y., and Wahner, A.: Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 9, 4387–4406, https://doi.org/10.5194/acp-9-4387-2009, 2009.
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kurten, A., Lehtipalo, K., Makhmutov, V., Petaja, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tome, A., Trostl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of Highly Oxygenated Organic Molecules from α-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, ACS Earth and Space Chemistry, 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Mutzel, A., Poulain, L., Berndt, T., Iinuma, Y., Rodigast, M., Boege, O., Richters, S., Spindler, G., Sipila, M., Jokinen, T., Kulmala, M., and Herrmann, H.: Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study, Environ. Sci. Technol., 49, 7754–7761, https://doi.org/10.1021/acs.est.5b00885, 2015.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Nie, W., Yan, C., Yang, L. W., Roldin, P., Liu, Y. L., Vogel, A. L., Molteni, U., Stolzenburg, D., Finkenzeller, H., Amorim, A., Bianchi, F., Curtius, J., Dada, L., Draper, D. C., Duplissy, J., Hansel, A., He, X. C., Hofbauer, V., Jokinen, T., Kim, C., Lehtipalo, K., Nichman, L., Mauldin, R. L., Makhmutov, V., Mentler, B., Mizelli-Ojdanic, A., Petäjä, T., Quéléver, L. L. J., Schallhart, S., Simon, M., Tauber, C., Tomé, A., Volkamer, R., Wagner, A. C., Wagner, R., Wang, M. Y., Ye, P. L., Li, H. Y., Huang, W., Qi, X. M., Lou, S. J., Liu, T. Y., Chi, X. G., Dommen, J., Baltensperger, U., El Haddad, I., Kirkby, J., Worsnop, D., Kulmala, M., Donahue, N. M., Ehn, M., and Ding, A. J.: NO at low concentration can enhance the formation of highly oxygenated biogenic molecules in the atmosphere, Nat. Commun., 14, https://doi.org/10.1038/s41467-023-39066-4, 2023.
Nozière, B., Barnes, I., and Becker, K. H.: Product study and mechanisms of the reactions of α-pinene and of pinonaldehyde with OH radicals, J. Geophys. Res.-Atmos., 104, 23645–23656, https://doi.org/10.1029/1999jd900778, 1999.
Paulson, S. E., Chung, M., Sen, A. D., and Orzechowska, G.: Measurement of OH radical formation from the reaction of ozone with several biogenic alkenes, J. Geophys. Res.-Atmos., 103, 25533–25539, 1998.
Peeters, J., Vereecken, L., and Fantechi, G.: The detailed mechanism of the OH-initiated atmospheric oxidation of α-pinene: a theoretical study, Phys. Chem. Chem. Phys., 3, 5489–5504, https://doi.org/10.1039/b106555f, 2001.
Peräkylä, O., Berndt, T., Franzon, L., Hasan, G., Meder, M., Valiev, R. R., Daub, C. D., Varelas, J. G., Geiger, F. M., Thomson, R. J., Rissanen, M., Kurtén, T., and Ehn, M.: Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 145, 7780–7790, https://doi.org/10.1021/jacs.2c10398, 2023.
Piletic, I. R. and Kleindienst, T. E.: Rates and Yields of Unimolecular Reactions Producing Highly Oxidized Peroxy Radicals in the OH-Induced Autoxidation of α-Pinene, β-Pinene, and Limonene, J. Phys. Chem. A, 126, 88–100, https://doi.org/10.1021/acs.jpca.1c07961, 2022.
Praske, E., Otkjaer, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Rissanen, M. P.: NO2 Suppression of Autoxidation-Inhibition of Gas-Phase Highly Oxidized Dimer Product Formation, ACS Earth and Space Chemistry, 2, 1211–1219, https://doi.org/10.1021/acsearthspacechem.8b00123, 2018.
Rissanen, M. P., Kurten, T., Sipila, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jorgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petaja, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Roldin, P., Ehn, M., Kurten, T., Olenius, T., Rissanen, M. P., Sarnela, N., Elm, J., Rantala, P., Hao, L. Q., Hyttinen, N., Heikkinen, L., Worsnop, D. R., Pichelstorfer, L., Xavier, C., Clusius, P., Ostrom, E., Petaja, T., Kulmala, M., Vehkamaki, H., Virtanen, A., Riipinen, I., and Boy, M.: The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system, Nat. Commun., 10, https://doi.org/10.1038/s41467-019-12338-8, 2019.
Rollins, A. W., Smith, J. D., Wilson, K. R., and Cohen, R. C.: Real Time In Situ Detection of Organic Nitrates in Atmospheric Aerosols, Environ. Sci. Technol., 44, 5540–5545, https://doi.org/10.1021/es100926x, 2010.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Shen, H., Vereecken, L., Kang, S., Pullinen, I., Fuchs, H., Zhao, D., and Mentel, T. F.: Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Science Advances, 8, eabp8702, https://doi.org/10.1126/sciadv.abp8702, 2022.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simon, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Mazon, S. B., Chen, D. X., Dias, A., Draper, D. C., Duplissy, J., Haddad, I., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kirkby, J., Kontkanen, J., Kuerten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petaja, T., Quelever, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, C., Tome, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, P. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kurten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Mohler, O., Nieminen, T., Onnela, A., Petaja, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipila, M., Smith, J. N., Steiner, G., Tome, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P. L., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Valiev, R. R., Hasan, G., Salo, V. T., Kubecka, J., and Kurten, T.: Intersystem Crossings Drive Atmospheric Gas-Phase Dimer Formation, J. Phys. Chem. A, 123, 6596–6604, https://doi.org/10.1021/acs.jpca.9b02559, 2019.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Vereecken, L. and Peeters, J.: Nontraditional (per)oxy ring-closure paths in the atmospheric oxidation of isoprene and monoterpenes, J. Phys. Chem. A, 108, 5197–5204, https://doi.org/10.1021/jp049219g, 2004.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – part I: a generalized structure-activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/b909712k, 2009.
Vereecken, L. and Peeters, J.: A structure-activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 12, 12608–12620, https://doi.org/10.1039/c0cp00387e, 2010.
Vereecken, L., Mueller, J. F., and Peeters, J.: Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of α-pinene: impact of non-traditional peroxyl radical chemistry, Phys. Chem. Chem. Phys., 9, 5241–5248, https://doi.org/10.1039/b708023a, 2007.
Vereecken, L., Vu, G., Wahner, A., Kiendler-Scharr, A., and Nguyen, H. M. T.: A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 23, 16564–16576, https://doi.org/10.1039/d1cp02758a, 2021.
von Schneidemesser, E., Monks, P. S., Allan, J. D., Bruhwiler, L., Forster, P., Fowler, D., Lauer, A., Morgan, W. T., Paasonen, P., Righi, M., Sindelarova, K., and Sutton, M. A.: Chemistry and the Linkages between Air Quality and Climate Change, Chem. Rev., 115, 3856–3897, https://doi.org/10.1021/acs.chemrev.5b00089, 2015.
Wang, Z. D., Ehn, M., Rissanen, M. P., Garmash, O., Quéléver, L., Xing, L. L., Monge-Palacios, M., Rantala, P., Donahue, N. M., Berndt, T., and Sarathy, S. M.: Efficient alkane oxidation under combustion engine and atmospheric conditions, Communications Chemistry, 4, https://doi.org/10.1038/s42004-020-00445-3, 2021.
Wildt, J., Mentel, T. F., Kiendler-Scharr, A., Hoffmann, T., Andres, S., Ehn, M., Kleist, E., Müsgen, P., Rohrer, F., Rudich, Y., Springer, M., Tillmann, R., and Wahner, A.: Suppression of new particle formation from monoterpene oxidation by NOx, Atmos. Chem. Phys., 14, 2789–2804, https://doi.org/10.5194/acp-14-2789-2014, 2014.
Xu, L., Moller, K. H., Crounse, J. D., Otkjwr, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular Reactions of Peroxy Radicals Formed in the Oxidation of α-Pinene and β-Pinene by Hydroxyl Radicals, J. Phys. Chem. A, 123, 1661–1674, https://doi.org/10.1021/acs.jpca.8b11726, 2019.
Yang, H., Raucci, U., Iyer, S., Hasan, G., Almeida, T. G., Barua, S., Savolainen, A., Kangasluoma, J., Rissanen, M., Vehkamäki, H., and Kurtén, T.: Molecular dynamics-guided reaction discovery reveals endoperoxide-to-alkoxy radical isomerization as key branching point in α-pinene ozonolysis, Nat. Commun., 16, https://doi.org/10.1038/s41467-025-55985-w, 2025.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.