the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Jan 2025
| 31 Jan 2025
Atmospheric oxidation of 1,3-butadiene: influence of seed aerosol acidity and relative humidity on SOA composition and the production of air toxic compounds
Klara Nestorowicz
Krzysztof J. Rudzinski
Michael Lewandowski
Tadeusz E. Kleindienst
Julio Torres
Ewa Bulska
Witold Danikiewicz
This study investigated the effect of relative humidity (RH) on the chemical composition of gas and particle phases formed from the photooxidation of 1,3-butadiene (13BD) in the presence of NOx under acidified and non-acidified seed aerosol. The experiments were conducted in a 14.5 m3 smog chamber operated in a steady-state mode. Products were identified by high-performance liquid chromatography, gas chromatography–mass spectrometry, and ultrahigh-performance liquid chromatography coupled with high-resolution mass spectrometry. More than 50 oxygenated products were identified, including 33 oxygenated organics, 10 organosulfates (OSs), PAN, APAN, glyoxal, formaldehyde, and acrolein. Secondary organic aerosol (SOA) mass and reaction products formed depended on RH and on the acidity of the seed aerosol. Based on the Extended Aerosol Inorganics Model (E-AIM), the seed aerosol originated from the acidified and non-acidified solutions was found to exist under aqueous and solid phases, respectively. Although the terms “acidified” and “non-acidified” are true for the solutions from which the seeds were atomized, there are far more fundamental differences between the phase states in which species partition to or from (aqueous/solid), which considerably affects their partitioning and formation mechanisms. SOA mass and most SOA products (i) were higher under acidified seed conditions, where the aerosol particles were deliquescent, than under non-acidified seed conditions, where the aerosol particles did not contain any aqueous phase; (ii) increased with the acidity of the aerosol aqueous phase in the experiments under acidified seed conditions; and (iii) decreased with increasing RH. Glyceric acid, threitols, threonic acids, four dimers, three unknowns, and four organosulfates were among the main species measured under either acidified or non-acidified conditions across all RH levels. Total secondary organic carbon and carbon yield decreased with increasing RH under both acidified and non-acidified seed conditions. The photochemical reactivity of 13BD in our systems decreased with increasing RH and was faster under non-acidified than acidified seed conditions. To determine the contribution of 13BD products to ambient aerosol, we analyzed PM2.5 samples collected at three European monitoring stations located in Poland. The occurrence of several 13BD SOA products (e.g., glyceric acid, tartronic acid, threonic acid, tartaric acid, and OSs) in the field samples suggests that 13BD could contribute to ambient aerosol formation.
- Article
(8353 KB) - Full-text XML
-
Supplement
(2934 KB) - BibTeX
- EndNote





Atmospheric particulate matter, especially particles with diameters less than 2.5 µm (PM2.5), plays an important role in Earth's climate through direct scattering and absorption of incoming solar radiation, trapping of outgoing longwave radiation (Charlson et al., 1992), and altering cloud formation and lifetime (Shrivastava et al., 2017; Fletcher et al., 2018). PM2.5 is considered harmful to humans and the environment (Majewska et al., 2021; Manisalidis et al., 2020), but its impact on climate, air quality, health, and the chemistry of the atmosphere is still not adequately understood (Myhre et al., 2013; Noziere et al., 2015). The recent literature suggests that secondary organic aerosol (SOA) contributes significantly to ambient PM2.5, and it remains a relevant subject of research (Jimenez et al., 2009; Shrivastava et al., 2017; Srivastava et al., 2022). A substantial number of studies have been conducted to investigate SOA chemical composition, formation mechanisms and contribution to ambient PM2.5. SOA is formed through the oxidation reactions of volatile organic compounds (VOCs) that are emitted directly into the atmosphere from anthropogenic and biogenic sources, producing highly oxidized species of low volatility, which can form new particles or condense onto existing particle surfaces. Precursor VOCs emitted in urban and industrial areas have a significant influence on aerosol burden either through direct SOA formation or through the enhancement of SOA originated from naturally emitted precursors (Jaoui et al., 2008; Hoyle et al., 2011).
The mechanism of SOA formation is complex and depends on several environmental parameters, including temperature, relative humidity (RH), acidity of the aqueous particles, solar irradiance, type of oxidant, and the concentration of precursor VOCs that controls peroxy radicals (RO2) and NOx concentrations. All these parameters can affect SOA chemical composition, an important characteristic of atmospheric particles that may provide key insights on properties governing the effect of particles on climate, air quality, and human health. While considerable effort has been expended studying acidity and RH effects on biogenic precursors, far less effort has been made to examine such effects on anthropogenic hydrocarbons, including reactive hazardous air pollutant (HAP) species such as 1,3-butadiene (13BD) and benzene. RH affects the quantity of water available in the system by influencing the processes in which water acts as a reactant, catalyst, product, or solvent. Atmospheric water also has a strong effect on the acidity of aqueous particles (Tilgner et al., 2021; Lei et al., 2022; Cooke et al., 2024). While the effect of RH on biogenic and anthropogenic SOA bulk properties (e.g., SOA yield) has been the subject of a number of studies (Edney et al., 2000; Cocker et al., 2001; Zhou et al., 2011; Nguyen et al., 2011; Zhang et al., 2011; Kamens et al., 2011; Lewandowski et al., 2015; Riva et al., 2016; Faust et al., 2017; Hinks et al., 2018; S. Liu et al., 2019; Healy et al., 2009; Jia and Xu, 2014, 2018; Chen et al., 2021), the effect of RH on SOA chemical composition is complex and has been reported only in a few studies (Nestorowicz et al., 2018; Wang et al., 2020; Zhang et al., 2021; Klodt et al., 2023; Luo et al., 2024; Thomsen et al., 2024).
Although significant progress has been made over the past 3 decades in improving our understanding of the atmospheric chemistry controlling the formation and transformation of air pollutants, significant uncertainties remain, as the suite of chemical compounds involved is ever expanding. HAPs, also known as air toxics, are an increasingly studied group of chemicals because they are known to be potentially carcinogenic and dangerous for human health and the environment (Scheffe et al., 2016; Kao, 1994). HAPs are either emitted directly (primary HAPs, or PHAPs) or formed through secondary reactions in the atmosphere (SHAPs). For example, acrolein, formaldehyde, and acetaldehyde are important HAPs from a health and atmospheric chemistry standpoint, yet their sources and reactions remain uncertain. Regulatory policies for HAPs target the sources of direct emissions; however, several aromatic, nitrogenated, and oxygenated HAPs can be formed in the atmosphere through chemical reactions. Numerous PHAPs undergo atmospheric transformation following their emissions by reacting with atmospheric oxidants, leading to the formation of SOA and SHAPs. For example, formaldehyde is produced from almost every atmospheric photooxidation reaction (Kao, 1994).
1,3-Butadiene, a structural analog of isoprene, is an important anthropogenic PHAP released into the atmosphere through industrial petroleum processing for synthetic rubber, resins and plastics production, combustion of vehicle fuels, tobacco smoke, and biomass combustion (Berndt and Böge, 2007; Anttinen-Klemetti et al., 2006; Dollard et al., 2001; Eatough et al., 1990; Pankow et al., 2004; Penn and Snyder, 1996; Ye et al., 1998; Thornton-Manning et al., 1997; Sorsa et al., 1996; Hurst, 2007). 13BD is classified as a hazardous (U.S. EPA, 1996), carcinogenic, toxic, mutagenic, and genotoxic pollutant in humans and other mammals (Acquavella, 1996; IARC, 2015; IPCS, 2001; U.S. EPA, 2002). The annual emission of 13BD into the atmosphere is estimated at 6×106 t worldwide (Berndt and Böge, 2007). A recent paper by Chen and Zhang (2022) provides an update on the properties of 13BD from newly available data. 13BD reacts in the atmosphere with OH, O3, NO3, and Cl and can produce many potential toxic products, including acrolein and formaldehyde (U.S. EPA, 1996; Angove et al., 2006; Doyle et al., 2004; Notario et al., 1997; Kramp and Paulson, 2000; Liu et al., 1999). Previous research efforts have made significant advances in 13BD oxidation product characterization and its role in SOA formation (Angove et al., 2006; Berndt and Böge, 2007; Kramp and Paulson, 2000; Liu et al., 1999; Sato, 2008; Sato et al., 2011; Jaoui et al., 2014); however, 13BD SOA composition has not been adequately characterized and remains largely unknown. To date, few smog chamber studies have focused on 13BD SOA formation (Angove at al., 2006; Sato, 2008; Sato et al., 2011; Jaoui et al., 2014), and only a limited number of studies have focused on the effects of seed aerosol acidity and relative humidity on SOA formation (Lewandowski et al., 2015). Our previous work (Jaoui et al. 2014) reported a detailed chemical characterization of gas and particle phases formed from the oxidation of 13BD in the presence (RH = 30 %) and absence (RH < 3 %: dry conditions) of NOx. We identified several oxygenated organic compounds using GC-MS analysis, including threitol, erythritol, glyceric acid, malic acid, tartaric acid, and threonic acid. Many of these compounds were detected in ambient aerosol. Our previous work (Jaoui et al., 2014) focused on non-sulfate species analysis due to the capabilities of the analytical method used; therefore additional characterization of SOA from 13BD oxidation is still needed to characterize organosulfates (OSs) and nitro- or nitroso-organosulfates (NOSs).
The objective of the present study is to assess the impact of aerosol water content on acidity-influenced SOA formation during NOx-mediated irradiation of 13BD. It extends our work previously reported showing the critical role RH plays in SOA composition from the irradiated isoprene/NOx system (Lewandowski et al., 2015; Nestorowicz et al., 2018). Our previous work focused mainly on the role of RH (< 5 % and 30 %) on aerosol bulk parameters, that is, secondary organic carbon (SOC) yields and (Lewandowski et al., 2015; Jaoui et al., 2014). Here we explored the effect of RH on a wide range of 13BD oxygenated products in the presence of acidified and non-acidified sulfate seed aerosol and incorporated a new method to analyze organosulfates (OSs) using a liquid chromatography–mass spectrometry (LC-MS) technique (Szmigielski, 2016; Rudziński et al., 2009; Darer et al., 2011). Non-sulfate SOA oxygenates were also examined using silylation derivatization followed by GC-MS analysis (Jaoui et al., 2004). We also analyzed the influence of RH and acidity of seed aerosol on HAP formation, as well as glyoxal, peroxyacetyl nitrate (PAN), and peroxyacryloyl nitrate (APAN). The results were compared to the chemical analysis of PM2.5 field samples to substantiate the importance of the laboratory findings and their role in ambient SOA formation. Numerical calculations using the Extended Aerosol Inorganics Model (E-AIM) were used to examine the response of RH in aerosols upon the addition of sulfuric acid. The results of the present study contribute to further development of acidity-influenced SOA chemistry in air quality models.
All chemicals used in the smog chamber experiments and GC-MS analysis, including 13BD, external standards used for calibration curves, and the derivatization agent N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) with 1 % of trimethylchlorosilane (TMCS) as a catalyst, were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA) at the highest purity available and were used without further purification. Solvents with GC2 quality were purchased from Burdick & Jackson (Muskegon, MI, USA). Standard compounds for LC-MS analysis – DL-tartaric acid (99 %), D-threonolactone (≥ 95 %), tartronic acid (> 97 %), DL-malic acid (≥ 99 %), and sodium 1-pentyl sulfate (99 %) – were purchased from Sigma-Aldrich, Poland, at the highest available purity and used without further purification. Aerosol extracts and LC-MS mobile phases were prepared using high-purity water (resistivity 18.2 MΩ cm−1) from a Milli-Q Advantage water purification system (Merck, Poland), and high-purity methanol (LC-MS Chromasolv grade) was purchased from Sigma-Aldrich, Poland. D-threonic acid was prepared in our laboratory according to the procedure described in Jaoui et al. (2019).
2.1 Smog chamber experiments
The experiments were conducted in a 14.5 m3 fixed-volume chamber with a stainless-steel frame and interior walls fused with a 40 µm PTFE Teflon coating. The chamber operation, sample collection, derivatization procedure, and GC-MS and LC-MS analysis methods used in this study are described in detail elsewhere (Jaoui et al., 2004, 2014; Kleindienst et al., 2006; Nestorowicz et al., 2018). The chamber was operated in flow mode as a continuous stirred tank reactor with a residence time of 4 h to produce a steady-state, constant aerosol distribution which could be repeatedly sampled at various seed aerosol acidities and relative humidities. A set of UV fluorescent bulbs was used in the chamber to produce radiation in the actinic region of the spectrum at 300–400 nm, photolytically comparable to that of solar radiation (Black et al., 1998).
13BD and NO were added continuously from high-pressure cylinders, each diluted with N2, through flow controllers into the inlet manifold, where they were diluted and mixed prior to the introduction to the chamber. Inorganic seed aerosol was added continuously to the chamber by nebulizing (TSI model 9302, Shoreville, MN) a dilute aqueous solution of ammonium sulfate (non-acidified seed aerosol) or an aqueous solution of ammonium sulfate and sulfuric acid (acidified seed aerosol), with the total sulfate concentration held constant to maintain stable inorganic concentrations in the chamber (Lewandowski et al., 2015). The seed aerosol stream was equilibrated to the computer-controlled relative humidity designated for a particular experiment. The reaction was run for about 24 h to reach steady-state conditions. The concentrations of 13BD in the inlet manifold and the chamber were measured using a gas chromatograph with flame ionization detection (Hewlett-Packard, model 5890 GC). NO and NOx were measured with a chemiluminescent analyzer (TECO model 42C, Franklin, MA), and O3 was measured with a chemiluminescent ozone monitor (Bendix model 8002, Lewisburg, WV). Temperature and relative humidity were measured with a thermohydrometer (model RH411, Omega Engineering, Inc., Stamford, CT). The effect of relative humidity on 13BD SOA composition was examined with two sets of experiments, each with multiple stages. In the first set of experiments, the 13BD/NO system was examined at multiple humidity levels using a low concentration (1 µg m−3) of ammonium sulfate seed aerosol (ER666; Table 1). The relative humidity was varied in stages from roughly 11 % to 60 % (Table 1). The overall average temperature was ∼ 25 °C. This set provided a base case for exploring the changes in aerosol composition in the absence of significant seed aerosol acidity. In the second set of experiments, the same system (13BD/NO) was examined in the presence of an acidified inorganic sulfate seed aerosol, generated using a solution incorporating sulfuric acid solution and ammonium sulfate solution (Table 1), in which sulfate concentration was held constant across the full range of humidity examined. Additional bulk aerosol analysis associated with these experiments was reported by Lewandowski et al. (2015). Secondary organic aerosol produced in the chamber at each stage was collected for 24 h at 16.7 L min−1 on 47 mm glass-fiber (GF) filters (Pall Corporation, Ann Arbor, MI).
Table 113BD photooxidation smog chamber experiments in the presence of acidified and non-acidified seed aerosol: initial and steady-state conditions, reaction and 13BD conversion, and yields data. The initial NOx was entirely nitric oxide. The non-acidified experiments were conducted with ammonium sulfate seed at ∼ 1 µg m−3. The acidified experiments were conducted with inorganic seed generated from a nebulized solution for which two-thirds of the sulfate mass was derived from sulfuric acid and the other one-third was derived from ammonium sulfate, giving a constant aerosol sulfate concentration at ∼ 35 µg m−3 (Lewandowski et al., 2015). For a wide range of particles, the wall loss rate is 0.063 h−1. However, with the chamber running in a flow mode, the wall loss rate is subsumed in the observed decrease from the input reactants and the steady-state concentrations. The steady-state SOCs for both experiments are wall-loss-corrected.
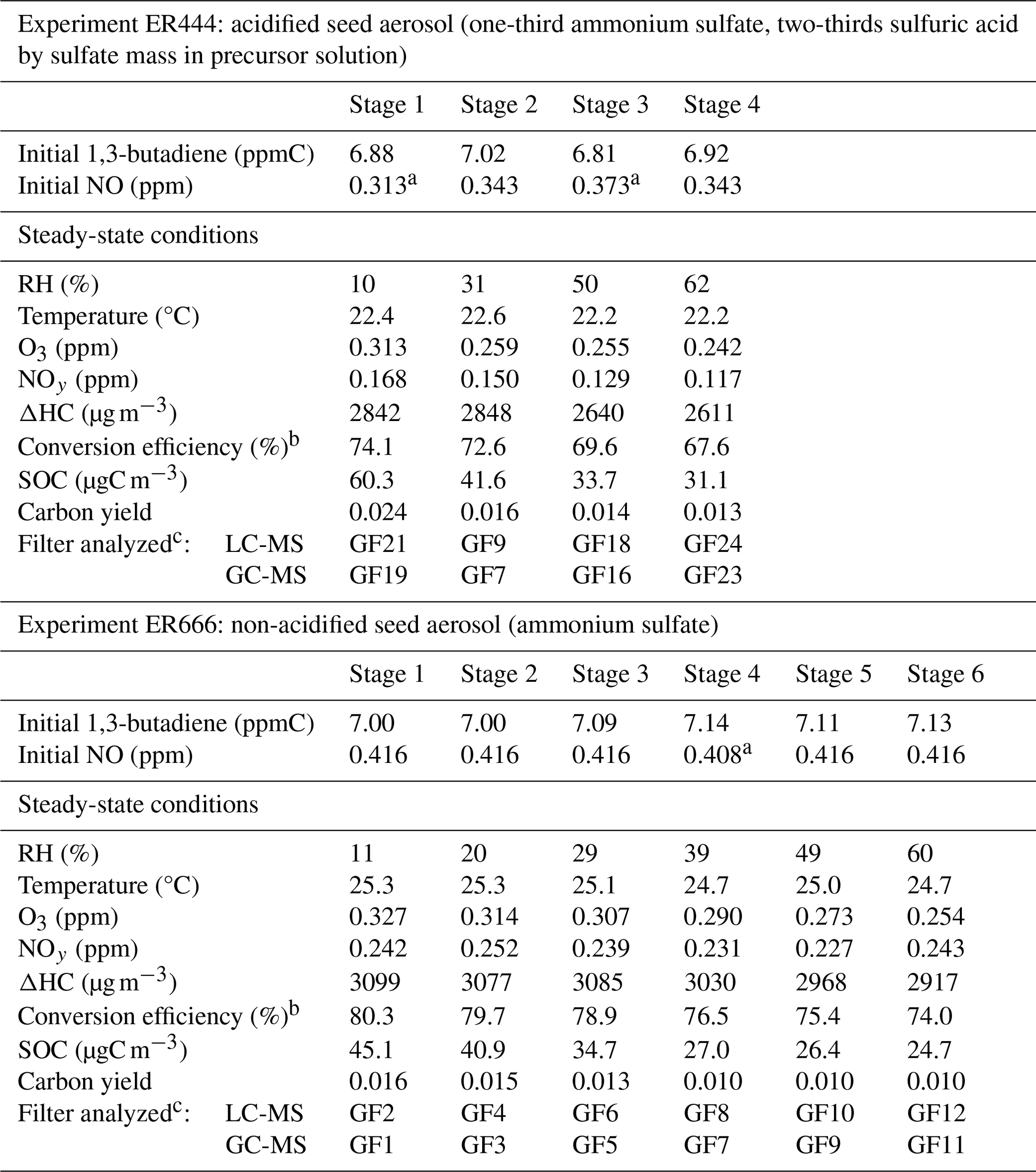
a NO-explicit measurement specific to that stage is reported; for the remaining stages, a full-experiment (all stages) global NO average is reported. b The conversion efficiency is defined as in Y. Liu et al. (2019). c For filter numbering, see Sect. 2.3. [H+] air was reported for ER444 (Lewandowski et al., 2015).
2.2 Ambient aerosol samples
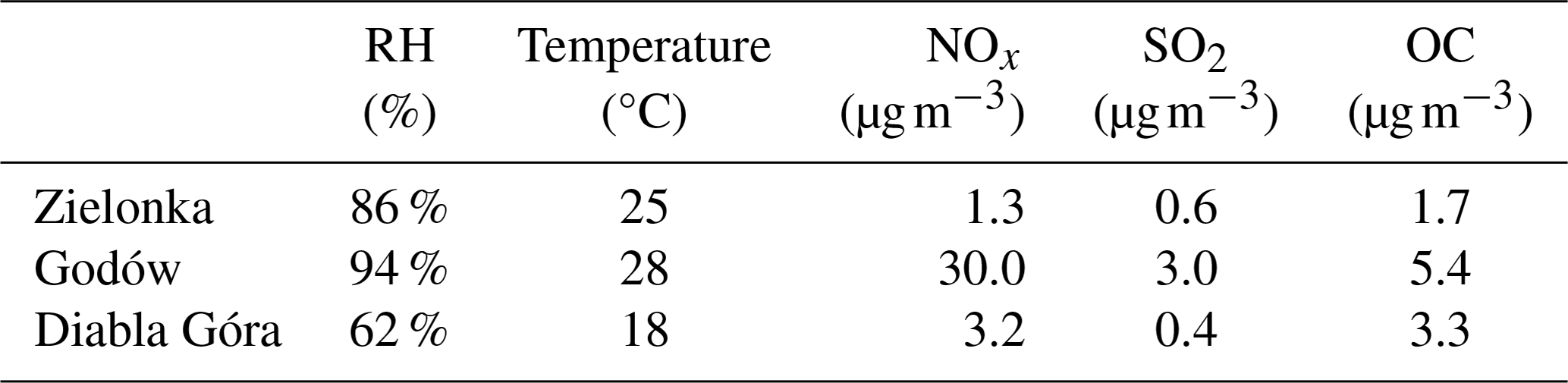
Ambient PM2.5 samples were collected on pre-baked quartz fiber filters with a high-volume aerosol sampler (DH-80, Digitel) at the regional background monitoring stations in (1) Zielonka (Kuyavian–Pomeranian Province, Poland; 53°39′ N, 17°55′ E), (2) Diabla Góra (Warmian–Masurian Province in northern Poland; 54°07′ N, 22°02′ E) during the summer 2016 campaigns, and (3) the regional monitoring station in Godów (Silesian Province, Poland; 49°55′ N 18°28′ E) in summer 2014. All sites are located in forested areas; however, the Godów site is close to major industrial cities of the Silesian region in Poland and a coal-fired power station in Dětmarovice, Czechia, so samples collected at this site were highly influenced by anthropogenic sources. Additional characteristics of Zielonka and Godów sites were reported in Nestorowicz et al. (2018). Diabla Góra is surrounded by boreal forests and highly influenced by biogenic precursors, e.g., isoprene and α-pinene. The anthropogenic emissions are seasonal and limited to biomass burning and vehicle exhausts. Meteorological and pollution characteristics of the sampling sites are presented in Table 2.
Table 2Characteristics of sampling sites in Poland: meteorological data (average temperature and RH), ambient air parameters (NOx, SO2), and organic carbon (OC) levels in PM2.5.
2.3 Secondary organic aerosol analysis
2.3.1 EC-OC analysis
Organic carbon (OC) formed in each stage of the experimental studies was measured using a semi-continuous elemental carbon–organic carbon (EC-OC) instrument (Sunset Laboratories, Tigard, OR). The instrument operates with a quartz filter positioned within the oven housing used for the analysis. The pumping system drew chamber effluent through the filter at a rate of 8 L min−1. A carbon strip denuder was placed in-line before the quartz filter to remove gas-phase organic compounds in the effluent that might interfere with the OC measurements. With a sample collection time of 0.5 h and an analysis time of 0.25 h, the duty cycle for the measurement of OC was 0.75 h (Lewandowski et al., 2015).
2.3.2 GC-MS analysis
SOA formed in each stage of each experiment (Table 1) was analyzed for individual organic compounds by extracting one GF filter from each stage using sonication in a 1:1 dichloromethane methanol mixture for 1 h. Prior to extraction, cis-ketopinic acid (KPA; 21.4 µg) and d50-tetracosane (20.8 µg) were added to each filter as internal standards. Filter extracts were concentrated to dryness and then derivatized with bis(trimethylsilyl) trifluoroacetamide (BSTFA) containing 1 % trimethylchlorosilane as a catalyst in the presence of 100 µL pyridine. Samples were heated to 70 °C to complete the reaction (Jaoui et al., 2018). The derivatized extracts were analyzed using a ThermoQuest GC (Austin, TX, USA) coupled to an ion trap mass spectrometer (ITMS). The injector, heated to 270 °C, was operated in splitless mode. Compounds were separated on a 60 m long, 0.25 mm inner diameter RTx-5MS column (Restek, Inc., Bellefonte, PA, USA) with a 0.25 µm film thickness. The GC oven temperature program for the analysis started isothermally at 84 °C for 1 min, followed by a temperature ramp of 8 °C min−1 to 200 °C and a 2 min hold, and was then ramped at 10 °C min−1 to 300 °C. The ion source, ion trap, and interface temperatures were 200, 200, and 300 °C, respectively. Mass spectra were collected in both the methane chemical ionization (CI) and electron ionization (EI) modes (Jaoui et al., 2014). Jaoui et al. (2004) determined the efficiency of BSTFA derivatization to be greater than 95 %. In addition to important pieces of information used for structural elucidation of unknown oxygenated organic species, the silylation method can be used for quantitative analysis using recovery and internal standards. Previous quantitative analysis was performed on nitroaromatic and other oxygenated organic compounds containing hydroxyl and/or carboxylic groups (Jaoui et al., 2004, 2018). In this study, calibration curves were prepared by diluting standard mixtures (Sect. S1 in the Supplement). The compounds listed in Tables S1 and S2 in the Supplement were chosen due to their structural diversity and similarity to products detected in chamber and ambient samples. The method was evaluated previously by our laboratory (Jaoui et al., 2004, 2018). Calibration curves were developed for silylated compounds as reported in Tables S1 and S2 by plotting the relative integrated peak areas versus the amount relative to the internal standard (cis-ketopinic acid) as shown in Fig. S1 in the Supplement. Figures S2 and S3 show examples of TIC chromatogram and mass spectra recorded for selected standards, respectively.
2.3.3 LC-MS analysis (GF and ambient filters)
For LC-MS analysis, one GF filter from each stage was used as shown in Table 1. The procedure of filter extraction was similar to the one described in Nestorowicz et al. (2018). From each filter, two 1.5 cm2 punches were taken, extracted with 15 mL aliquots of methanol for 60 min using a Multi-functional Orbital Shaker (PSU-20i, Biosan). The extracts were concentrated on a rotary evaporator (Rotavapor® R-215, Buchi; temperature 30 °C, vapor pressure 150 mbar), then filtered with disposable 0.2 µm PTFE syringe filters and dried to dryness under a gentle stream of nitrogen at ambient temperature. The residues were reconstituted with 200 µL of 1:1 methanol water mixture and shaken for 1 min. The qualitative UHPLC-ESI-MS/MS analyses used are described elsewhere (Nestorowicz et al., 2018; Spolnik et al., 2018). A Waters ACQUITY UPLC I-Class chromatograph coupled with a Waters SYNAPT G2-S high-resolution mass spectrometer was used. The chromatographic separations were performed using an ACQUITY HSS T3 column (2.1 × 100 mm, 1.8 µm particle size) at room temperature. The mobile phases consisted of 10 mM ammonium acetate (eluent A) and methanol (eluent B). An injection volume of 1 µL was used. The SYNAPT G2-S spectrometer equipped with an electrospray ionization source (ESI) was operated in negative-ion mode. Optimal ESI source conditions were 3 kV capillary voltage with a 20 V sampling cone and a full-width-at-half-maximum mass resolving power of 20 000. High-resolution mass spectra were recorded from 50 to 600 in MS or MS/MS mode. All data were recorded and analyzed with the Waters MassLynx V4.1 software package.
Quantitative UHPLC-ESI-MS/MS analysis was performed using an Agilent 1290 coupled with a 6540 UHD Accurate-Mass Q-TOF spectrometer (Agilent Technologies) equipped with an ESI source. Compounds were separated with the same ACQUITY UPLC HSS T3 column at 40 °C. The mobile phases were used as follows: water with 10 mM ammonium formate and methanol with 10 mM ammonium formate as solvents A and B, respectively. Samples were loaded directly onto the analytical column at a flow rate of 0.3 mL min−1 with 100 % solvent A. The injection volume was 2 and 8 µL for survey and MS/MS scans, respectively. Compounds were eluted from the column at a flow rate of 0.3 mL min−1 with the following linear gradient: 0 min with 100 % A, 3 min with 100 % A, 6 min with 100 % B, 9 min with 100 % B, and 12 min with 100 % A. The eluted compounds were ionized in negative-ion mode with a capillary voltage of 2.5 kV in the ESI source. Nitrogen was used as a sheath and auxiliary gas (5 L min−1, 150 °C, 20 psi). Survey scans were recorded in the Q-TOF mass analyzer at a resolving power of 20 000 in the range of 50–500. MS/MS spectra were recorded in a cycle time of 2 s. Collision energy values were optimized to obtain appropriate fragmentations of the studied compounds. Survey scans and MS/MS spectra were evaluated manually using MassHunter software (Agilent Technologies, Santa Clara, USA). Quantitative analysis performed for OSs and NOSs of 13BD SOA was based on a surrogate standard compound: sodium 1-pentyl sulfate (SP-OS). A stock solution was prepared with a concentration of 0.1 mg mL−1. Tables S3 and S4 present the mean peak area, recovery, and LOD and LOQ levels obtained for pentyl sulfate. All calculations were performed for deprotonated ions (Fig. S4).
2.4 Chamber secondary HAP, glyoxal, PAN, and APAN measurements
Selected low-molecular-weight carbonyls and dicarbonyls (e.g., HAPs) were quantified by derivatization using 2,4-dinitrophenylhydrazine (DNPH; Smith et al., 1989). Samples were collected at 0.5 L min−1 for 20 min and derivatized in a 4 mL solution of acidified DNPH and then heated for 40 min at 70 °C. Air samples were drawn through an impinger containing the DNPH solution in acetonitrile. The resulting solutions were analyzed by high-performance liquid chromatography with an ultraviolet detector (HPLC/UV) (Smith et al., 1989). A 15-component hydrazone standard (comprising formaldehyde, acetaldehyde, acrolein, acetone, propionaldehyde, crotonaldehyde, methacrolein, butyraldehyde, 2-butanone, benzaldehyde, glyoxal, valeraldehyde, m-tolualdehyde, methylglyoxal, and hexaldehyde; AccuStandard, Inc.) at a free-carbonyl concentration of 30 µg cm−3 for each component was used for calibration. Separate dihydrazone standards of glyoxal-DNPH and methylglyoxal-DNPH were also formulated. Carbonyls were separated using a Hewlett-Packard (HP) 1100 HPLC system with an Agilent Zorbax ODS 4.6 × 250 mm, 5 µm column maintained at 30 °C eluted with binary acetonitrile–water gradient. A 10 µL injection volume was used for all standards and samples. Carbonyls were quantified by UV absorption with a diode array detector set to 360 nm. Control and sample processing were managed with HP ChemStation software. Peroxyacetyl nitrate (PAN) and peroxyacryloyl nitrate (APAN) concentrations were determined using a gas chromatograph–pulse discharge electron capture detector (GC-PDECD; model D-2, Valco Instruments Co., Houston, TX). The column made of fused silica had a 30 m × 0.53 mm inner diameter with a 1.0 µm Rtx-200MS liquid phase (Restek, Bellefonte, PA). It was operated at a carrier flow of 11.5 cm3 min−1. Samples were injected onto the head of the GC through a 1 cm3 loop. Gas flows were adjusted for proper operation of the PDECD, that is, He at 30 cm3 min−1 with a 3 cm3 min−1 flow of a 5 % , which creates the standing current. The GC-PDECD system was operated at 60 °C, and the total flow through the ECD was 44.5 cm3 min−1.
2.5 Aerosol acidity and liquid water content
Liquid water content (LWC) and the acidity of the aerosol aqueous phase generated during the smog chamber experiments were estimated using the Extended AIM Thermodynamic Model (Wexler and Clegg, 2002; Carslaw et al., 1995; Clegg et al., 1992, 1998; Clegg and Pitzer, 1994; Clegg and Brimblecombe, 1995). The model was evaluated and used extensively for atmospheric applications (Pye et al., 2020). E-AIM does quantitative thermodynamic modeling of liquid/solid, liquid/gas, and solid/gas equilibrium partitioning of aerosol components in an atmospheric system. We used the Model II Comprehensive variant of E-AIM, designed for systems containing . Nitrite ions were set to zero and no organic components were considered because of the uncertain stoichiometry of dissociable hydrogen ion production during their formation and the lack of reliable dissociation constants. Our input data included temperature, relative humidity, and formal molar composition of seed aerosol (NH, SO, H+).
3.1 Chemical characterization
3.1.1 Gas-phase (GP) products
The chamber conditions, including temperature and RH, are shown in Table 1. Table 1 also shows initial 13BD and NO concentrations, reacted 13BD (ΔHC), steady-state concentrations of O3, and NOy (sum of NO, NO2, and all oxidized odd-nitrogen species). Once the 13BD/NO mixture reached steady state under acidified or non-acidified conditions, NO had completely reacted, while only a fraction of the initial 13BD was consumed. The reacted 13BD depended on RH and seed aerosol acidity (Table 1). Under these conditions, the steady-state concentrations of NOy and O3 given in Table 1 also depend on RH and seed aerosol acidity. O3 and NOy produced during the reactions show a steady decrease with increasing RH (Fig. 1a), except for NOy under non-acidified conditions, which did not change much across the RH used in this study. The results show that the increase in RH can reduce the maximum O3 under both acidified and non-acidified seed aerosol (Fig. 1a). Ozone was higher under non-acidified than acidified conditions across all levels of RH used in this study. Figure 1b shows 13BD conversion efficiency (CE) (Y. Liu et al., 2019) as a function of RH for both experiments, ER444 (acidified) and ER666 (non-acidified). Once the 13BD/NO mixture reached steady state, 13BD CE was found to be lower under acidified than non-acidified conditions, at ∼ 8 % lower across all levels of RH used in this study. A maximum of 80 % and 74 % CE was reached at lower RH (11 %–30 %), and a maximum of 74 % and 68 % was reached at higher RH (49 %–60 %) under non-acidified and acidified conditions, respectively (Table 1, Fig. 1b). Wang et al. (2020) reported that the RH and chemical make-up of the gas phase where the photooxidation occurs can influence the CE of 13BD. They found that 13BD photochemical reactivity first increased and then decreased with increased RH. Our study shows that the photochemical conversion of 13BD, either under acidified or non-acidified conditions, decreased with increasing RH across all levels of RH used in our study. The slight discrepancy at low RH between the present study and the findings of Wang et al. (2020) is probably due to the experimental conditions used in both studies, including chamber mode (static vs. dynamic), chemical make-up of the GP where oxidation occurs, RH (constant in our study vs. only initial RH provided in Wang et al., 2020 experiments), and CE values taken after 24 h reaction time vs. ∼ 4 h in our study. It has been reported that RH changes promote changes in OH-radical concentrations (Hu et al., 2011). These changes may be some of the reasons for the RH affecting the photochemical conversion of 13BD in our study.
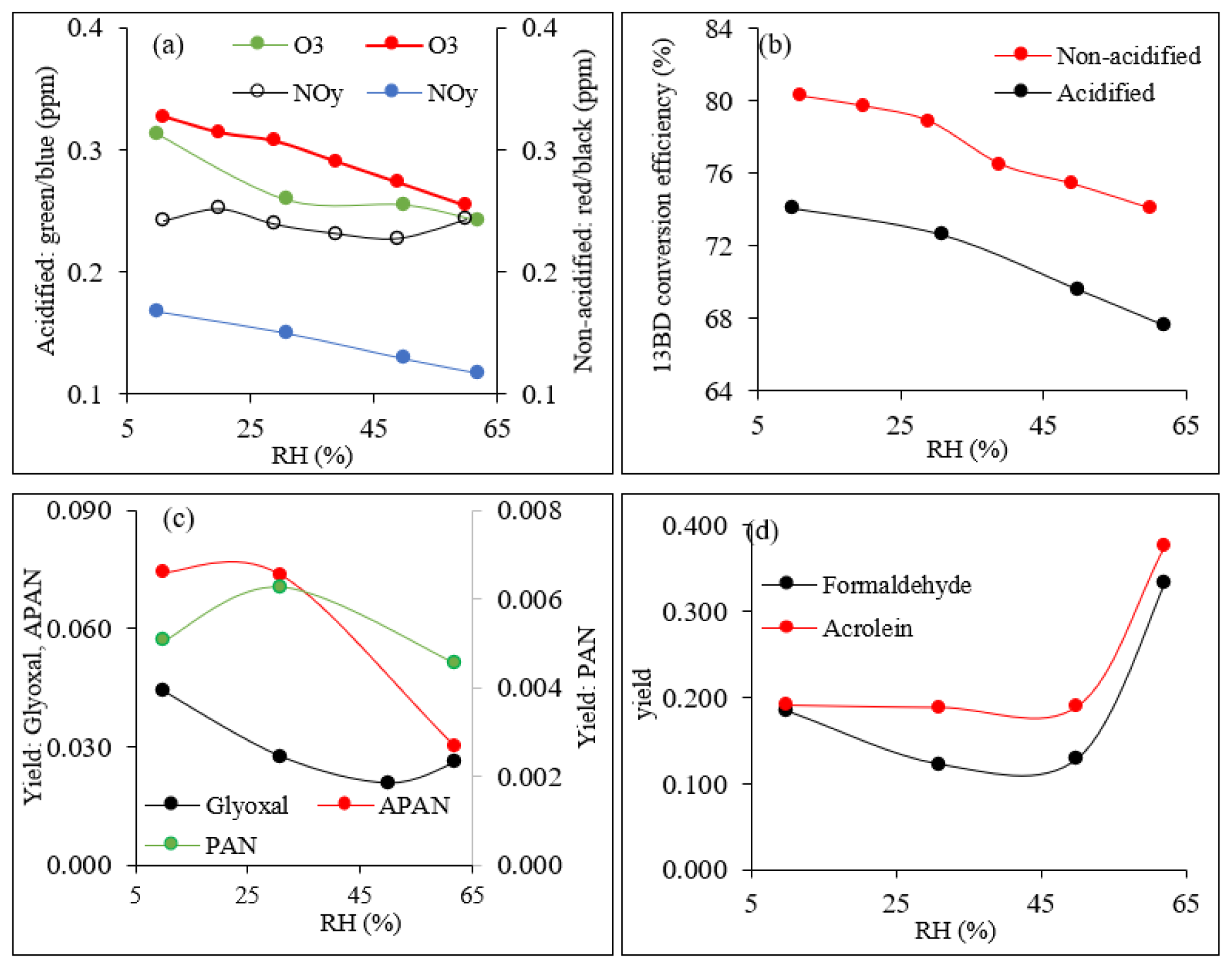
Figure 1Influence of relative humidity on (a) O3 and NOy concentrations (ppm) under acidified and non-acidified conditions; (b) 1,3BD conversion efficiency (%) under acidified and non-acidified conditions; (c) glyoxal, PAN, and APAN yield under acidified conditions; and (d) formaldehyde and acrolein yields under acidified conditions. The smoothed lines in these plots are used solely as a visual guide.
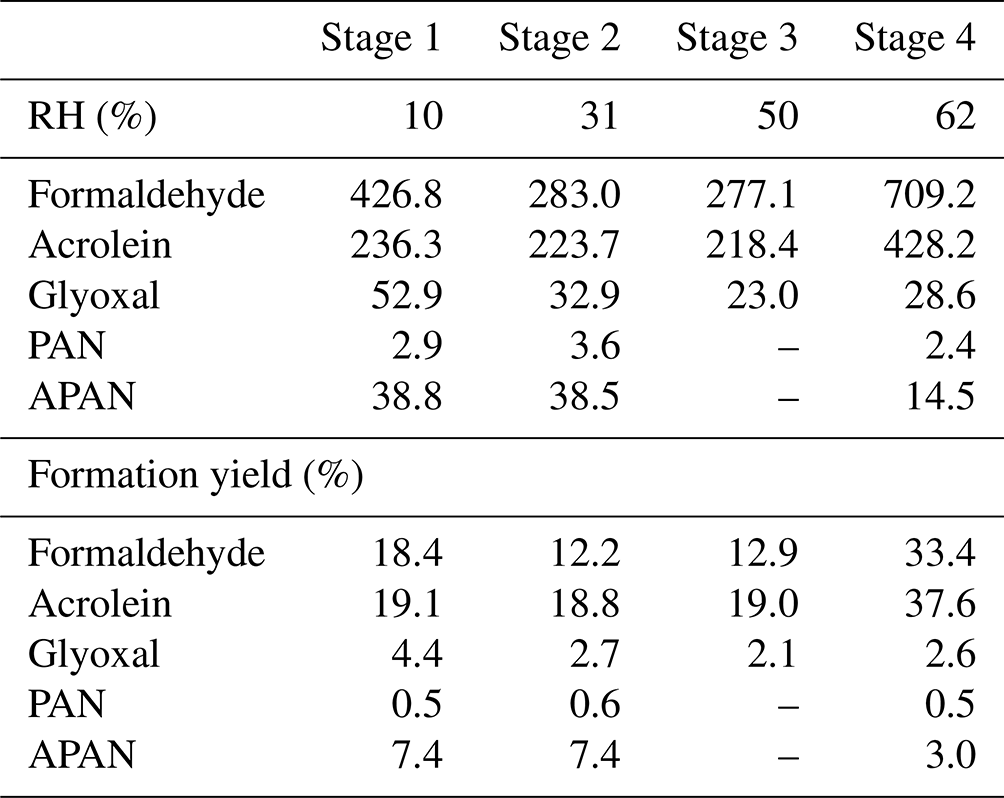
Steady-state concentrations and formation yields of formaldehyde, acrolein, glyoxal, PAN, and APAN were measured only in experiment ER444 (Table 3), as the instruments were not working during experiment ER666. Under the conditions shown in Table 1 (ER444), high concentrations were observed for formaldehyde and acrolein and, to a lesser extent, glyoxal, PAN, and APAN (Table 3). This is the first time that experimental data have been presented for secondary HAPs from the oxidation of 13BD under a range of RH levels. Figure 1c–d show changes in the mass yield of these five species as a function of RH. The product mass yield was calculated as the product mass (µg m−3) formed divided by the mass of reacted 13BD (µg m−3). The reacted 13BD was calculated from the difference between the initial and steady-state concentrations. The concentrations of gas-phase species were corrected for background and blank. Glyoxal and formaldehyde yields first exhibited a decrease, followed by a sharp increase as the RH increased. However, the yields of APAN and PAN first exhibited a slight increase, followed by a decrease as the RH increased. Meanwhile, the yield of acrolein remained relatively steady as the RH increased, then it increased sharply at 60 % RH. This is consistent with previous findings for glyoxal formed from benzene and ethylbenzene (Jia and Xu, 2014). Glyoxal was found to partition into the particle phase and become hydrated at high RH and enhance the formation of SOA (Jia and Xu, 2014). The wall loss of particles is ∼ 6 % h−1. We expect gas-phase products and the 13BD wall to be negligible as the experiments were conducted at steady-state conditions and in flow mode. The yield of formaldehyde and acrolein from the photooxidation of 13BD has been examined in a limited number of laboratory studies in the presence and absence of oxides of nitrogen. Berndt and Böge (2007) reported yields of formaldehyde (0.64 ± 0.08) and acrolein (0.98 ± 0.12) from OH radical reactions with 13BD conducted in a flow reactor. Kramp and Paulson (2000) reported the formation of acrolein from the ozonolysis of 13BD with a yield of 52 ± 7 %. In addition to primary emission, formaldehyde has been reported with high yields from the oxidation of most hydrocarbon photooxidation reactions, including 13BD (Bauwens et al., 2016; Wolfe et al., 2016). The formaldehyde yield reported in our study (Table 3) represents the overall yield (not only first-generation-derived yield) considering all branches that can produce formaldehyde (second-generation pathways and further) and consume formaldehyde (photolysis and reaction with OH radicals and other species, partitioning in the particle phase, etc.). Therefore, our yield is lower than those reported in the literature. Formaldehyde is used to estimate the emission rate of atmospheric VOCs (Bauwens et al., 2016; Cho et al., 2021, and references therein). High emissions of 13BD in urban areas could be a significant source of secondary acrolein and formaldehyde and could influence urban atmospheric chemistry in these areas. PAN and APAN were also formed in this study with a yield of ∼ 0.5 for PAN and up to 7 % for APAN at lower RH. A mass yield of 4 % was measured for glyoxal at 10 % RH and decreased as RH increased.
Table 3Steady-state gas-phase concentrations (ppb) and formation mass yields (%) during 13BD photooxidation under acidified seed aerosol (ER444: one-third ammonium sulfate, two-thirds sulfuric acid by sulfate mass in precursor solution).
Insights from the present study can help elucidate the chemistry of other atmospherically relevant compounds, especially isoprene, the major non-methane hydrocarbon emitted globally (Guenther et al., 1995). While past work has identified reaction products from 13BD oxidation, we still do not understand explicit secondary HAP formation and other oxygenated products, particularly over a range of seed aerosol acidity and RH. In addition, the uptake of some of these compounds (e.g., glyoxal) in the aerosol phase followed by heterogeneous sulfur chemistry can lead to SOA formation (Carlton et al., 2007; Liggio et al., 2005; Chan et al., 2010). The role of APAN in ambient SOA formation could be important and might follow chemistry similar to MPAN, which only recently was found to play a role in SOA formation from isoprene (Tanimoto and Akimoto, 2001; Surratt et al., 2010).
3.1.2 Particle-phase products
In this study, up to six glass-fiber (GF) filters were collected at each stage associated with ER444 and ER666 (Table 1). One filter from each stage was analyzed by UHPLC without derivatization. A second filter from each stage was analyzed by GC-MS following the derivatization procedure described above. The analysis at the molecular level of 13BD SOA generated from smog chamber experiments was based on the interpretation of GC-MS (in EI and CI modes) and UHPLC/ESI(-) HRMS mass spectra of derivatized and underivatized SOA products, respectively.
GC-MS analysis
The identification of reaction products from GC-MS analysis was based on comparisons between the mass spectra of the derivatized samples and the authentic spectra in CI and/or EI mode and on retention times, with the exception of compounds with no authentic standards. In this case, the identification was made based on the interpretation of fragment/adduct peaks in CI mode that permit determination of the number and identity of functional groups and the molecular weight of the derivative. A peak was associated with a reaction product only if its corresponding mass spectrum was consistent with the fragmentation pattern of the BSTFA derivatization reagent and was not detected in blank and background samples. In addition, an extensive evaluation was made by comparing products identified in this study with those identified in our previous work or other laboratories to determine the degree of consistency and to provide information on which chemical systems might be subject to the greatest uncertainties. Except for products bearing nitrate and sulfate groups, the BSTFA single-derivatization technique provides good qualitative and quantitative analysis due to both its simplicity and its efficiency (Jaoui et al., 2004). We previously reported the characteristic ions associated with BSTFA derivatization (see Jaoui et al., 2014, for more information). In this GC-MS analysis, our main purpose was to compare and highlight the effect of relative humidity on the chemical characteristics of SOA products generated under acidified and non-acidified conditions. Additional analysis using LC-MS was carried out focusing mainly on products bearing sulfate and nitrate groups and reported in the LC-MS section below.
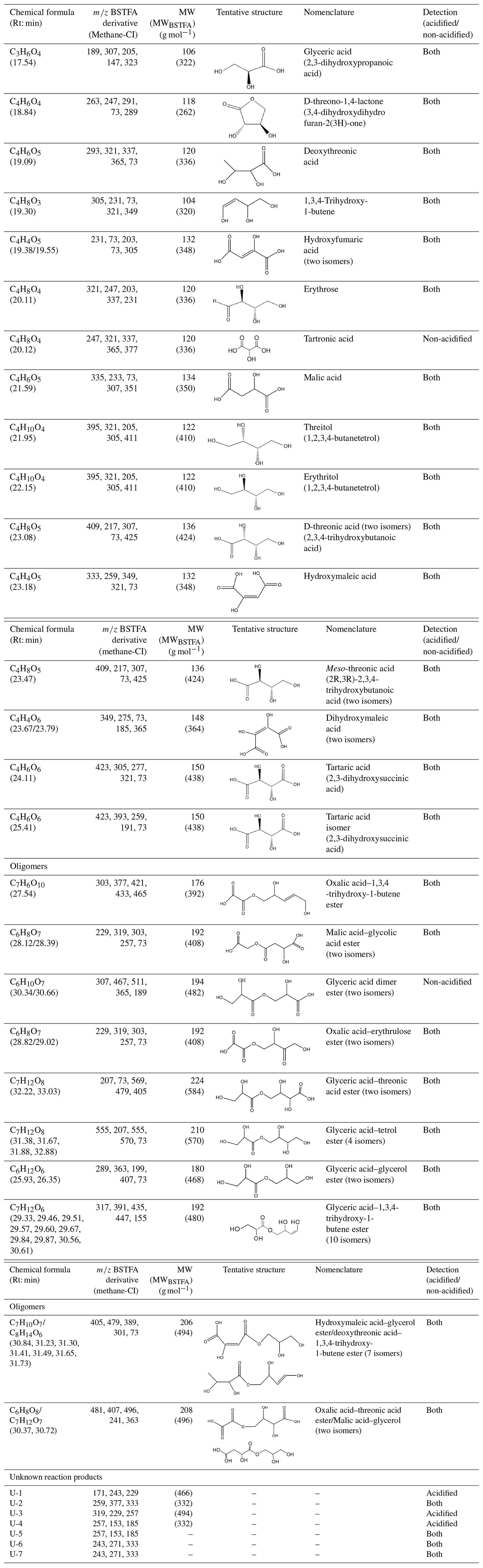
Figures 2, 3, and 4 present GC-MS extracted-ion chromatograms (EICs) associated with 13BD/NOx photooxidation experiments ER444 (Figs. 2, 3) and ER666 (Fig. 4), respectively. Each figure contains EICs associated with each RH used in this study. According to obtained chromatograms, several compounds can be distinguished, i.e., five isomers of threonic acid, tartaric acid, threitol, meso-threitol, glyceric acid, and erythrose. Tartronic acid and erythrose (MW 120) both have three OH active groups co-eluted under the GC-MS conditions used in this study, and their mass spectra were similar. The chromatograms in Figs. 2, 3, and 4 can be directly compared because the chamber air sampled and the extraction and analysis were the same for each filter, although the relative abundance in the y axis is different. Therefore, a relative contribution of products to SOA masses at various RH levels could be visually gained through these EICs. Two EICs were provided for the acidified experiment. They highlight oxygenated organic species (OOSs) eluted early in the chromatograms (Fig. 2) and several unknown (U1 through U7) products eluted later in the chromatograms (Fig. 3). EICs of non-acidified smog chamber experiments highlight OOSs eluted before 27 min and dimers eluted after 27 min (Fig. 4). Compounds identified using GC-MS analysis are summarized in Table 4, which contains proposed structures for products identified (when possible), chemical formula, retention time, nomenclature, and molecular weights of the derivatized (MWBSTFA) and underivatized compounds (MW), as well as if the product is detected under acidified and/or non-acidified conditions. All of these products contain one or more of the flowing functional groups: alcoholic (-OH) groups, acidified (-COOH) groups, or both. We previously reported the formation of 13BD SOA products, such as threitol, erythritol, glyceric acid, threonic acid, and tartaric acid, in field samples and chamber studies under low- and high-NOx conditions (Jaoui et al., 2014). Therefore, their identification is discussed only briefly in this study. Figure S5 presents mass spectra in CI mode of selected reaction monomer products observed in this study.
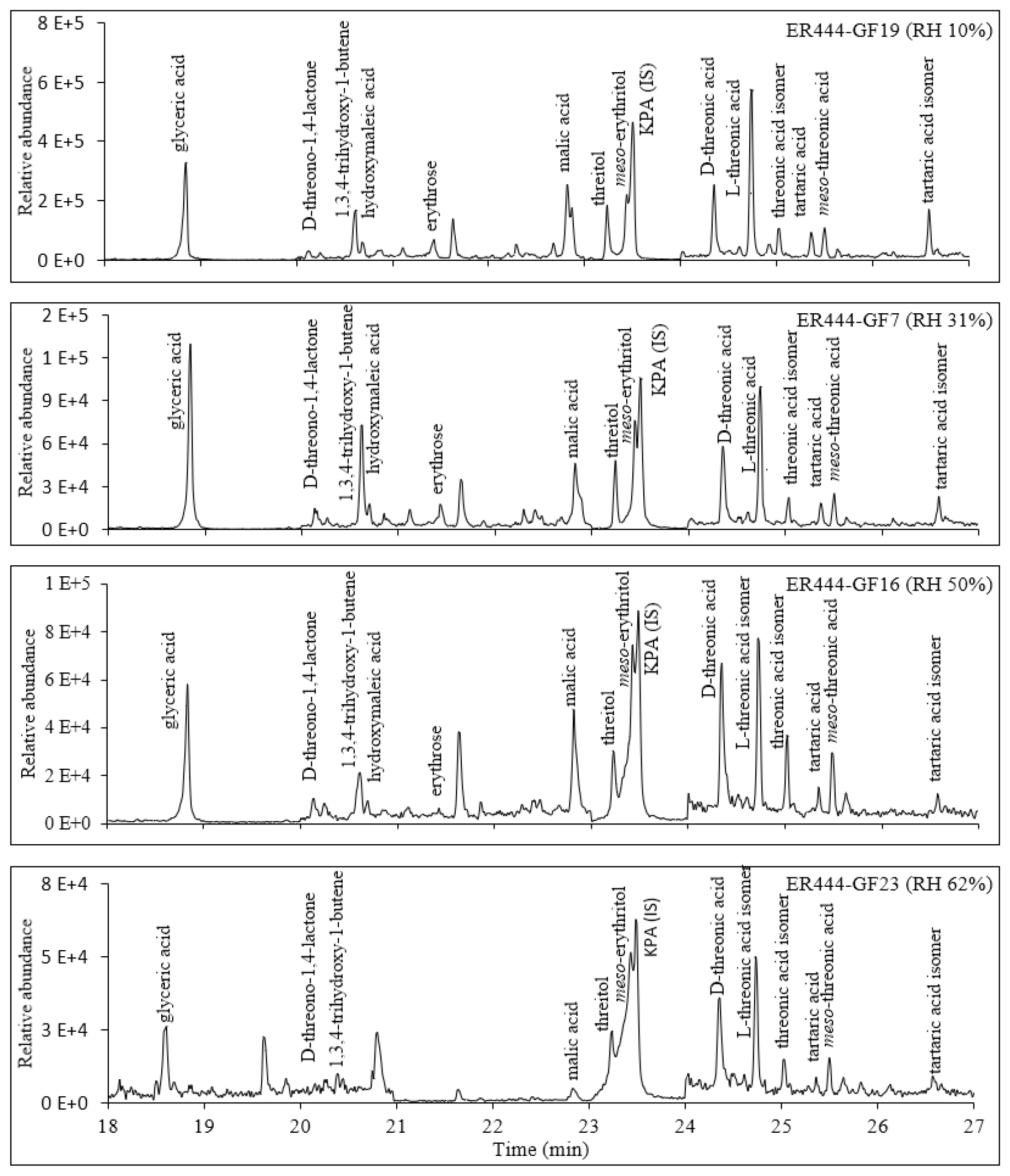
Figure 2Extracted-ion chromatograms – 165 (ketopinic acid (KPA)), 307 (glyceric acid), 409 (threonic acid, five isomers at 24.36, 24.53, 24.75, 25.04, and 25.51 min), 321 (erythrose), 395 (threitol, two isomers), 247 (D-threono-1,4-lactone), and 423 (tartaric acid) – for 1,3-butadiene/NOx photooxidation experiment ER444 (acidified seed) as a function of RH. Compounds detected as silylated derivatives. The intensity was multiplied by 5 between 20–23 and 24–27 min.
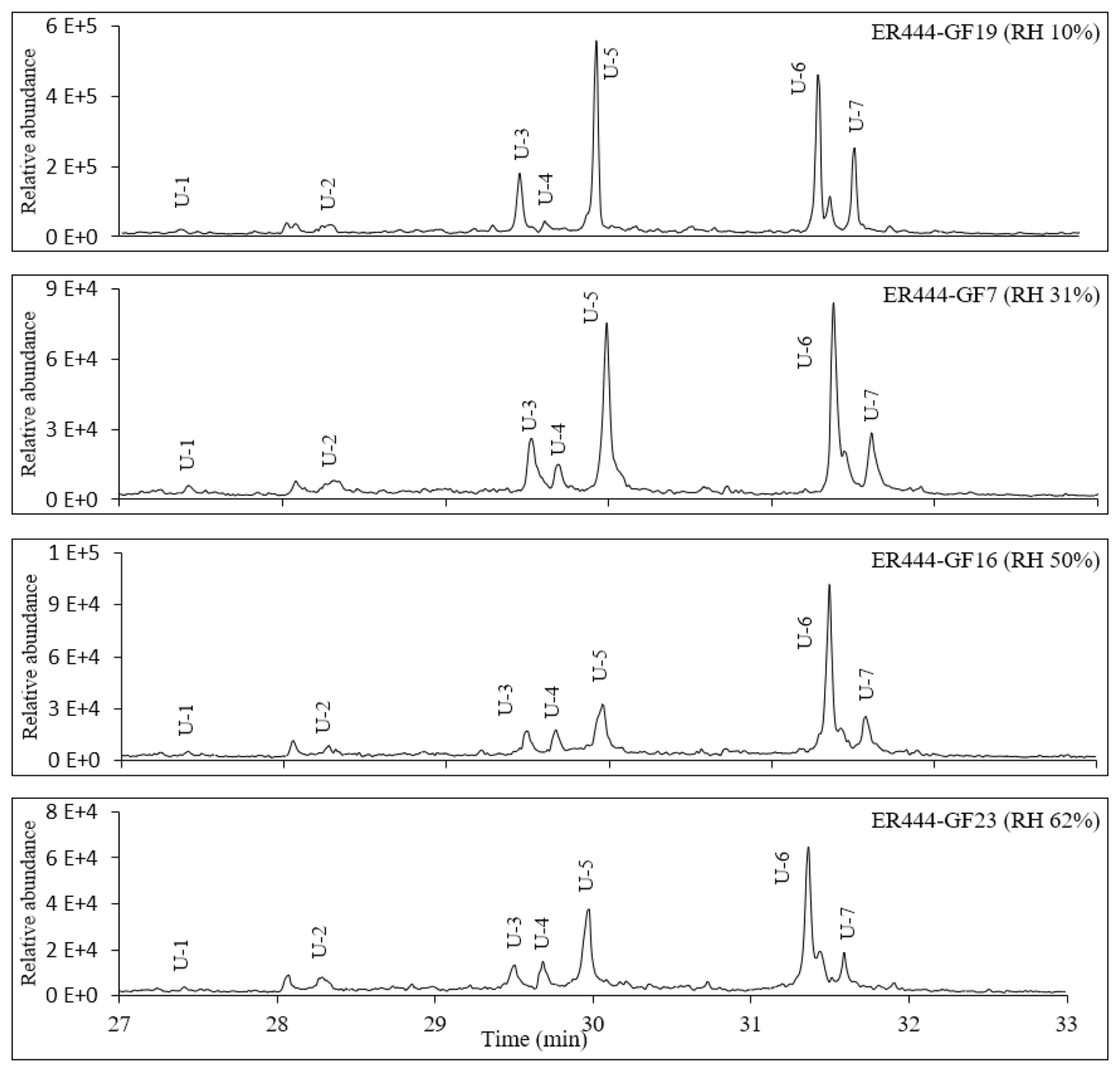
Figure 3Portion of extracted-ion chromatograms at 153, 229, 243, 259, 317, 405, and 481 for 1,3-butadiene/NOx photooxidation experiment ER444 under acidified seed aerosol as a function of RH (retention time between 27 and 32 min). Compounds detected as silylated derivatives. Only limited species are provided in this figure for clarity.
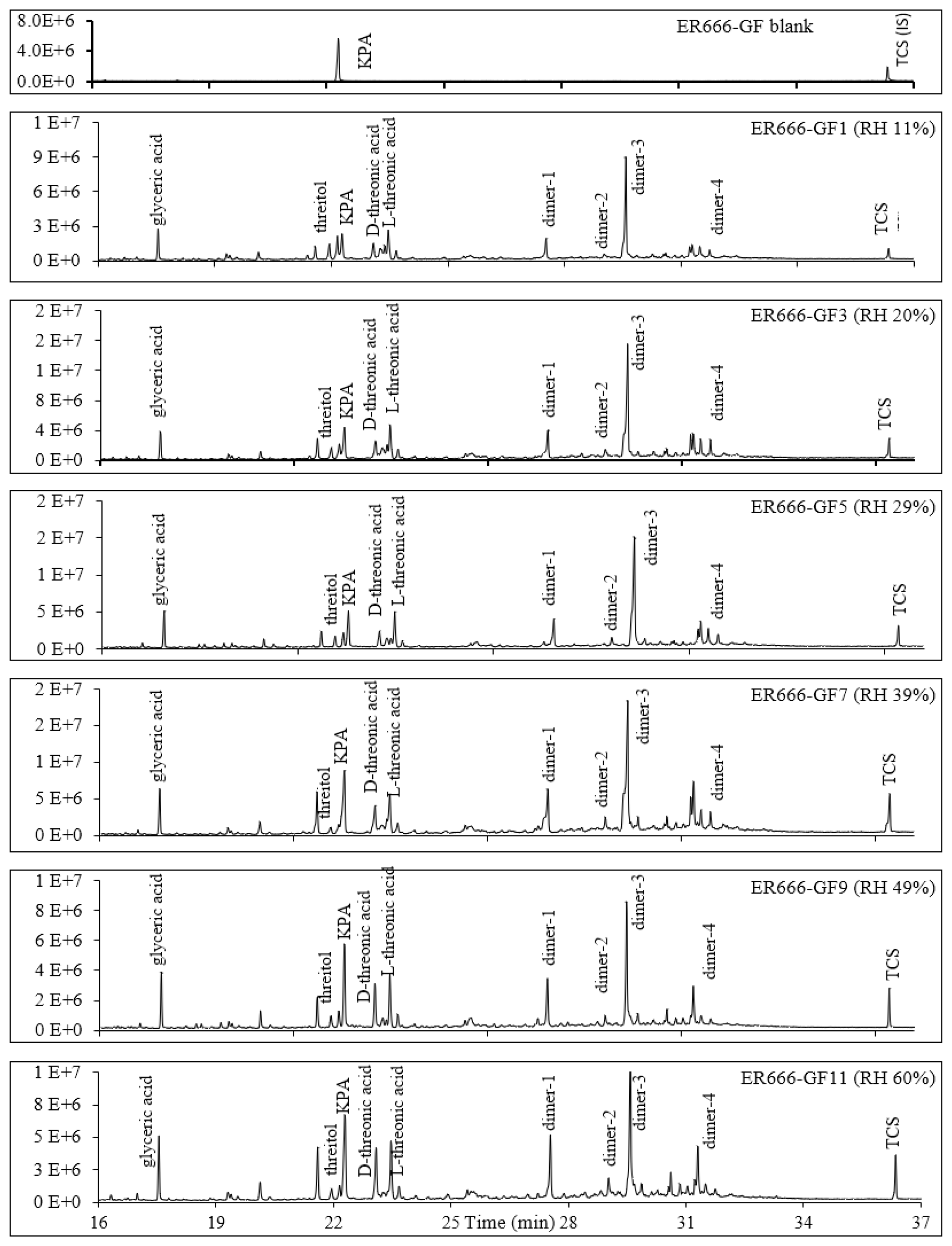
Figure 4Extracted-ion chromatograms showing monomeric compounds eluting before 25 min and dimers eluting between 26 and 33 min for 1,3-butadiene/NOx photooxidation experiment ER666 as a function of RH. Dimer 1: oxalic acid-1,3,4-trihydroxy-1-butene ester; dimer 2: oxalic acid–erythrulose ester; dimer 3: glyceric acid–1,3,4-trihydroxy-1-butene ester; dimer 4: hydroxymaleic acid–glycerol. Compounds detected as silylated derivatives. For clarity, not all identified compounds are included in Fig. 3.
Two groups of unknown products were detected in both systems and were absent in the background chromatograms. Although no structural information could be obtained, the molecular weights of selected unknowns were tentatively obtained (Table 4). Their mass spectra were consistent with compounds bearing OH and COOH groups. Groups 1 and 2 consist of seven monomers (Fig. 3, Table 4) and several dimers with mass spectra similar to those dimers (dimers 1–4) reported in Fig. 3 (not shown in Table 4), respectively. Figure S6 shows mass spectra associated with these unknown dimers in CI mode. The formation mechanism for selected compounds under low-NOx conditions is possible given the reactive uptake of 2,3-epoxy-1,4-butanediol (BEPOX) onto acidified aerosol seeds and under high-NOx conditions by further oxidation of acryloyl peroxynitrate (APAN) to acrylic acid epoxide (AAE) (Jaoui et al., 2014). Additional organic compounds were present in the SOA collected that were not detected by the GC-MS derivative method, as can be seen from the presence of organonitrates and OSs using LC-MS. A detailed analysis is reported in the section below (LC-MS analysis).
Table 4Selected SOA reaction products identified from 13BD/NOx photooxidation under acidified and non-acidified conditions using GC-MS. Additional products are reported in Jaoui et al. (2014). Although dimers were detected under non-acidified conditions, they were more pronounced in the presence of acidified seed aerosol. Retention times (min) are those associated with ER666-GF7. MW: molecular weight of the underivatized compound; MWBSTFA: molecular weight of the silylated derivatized compound. Unknown products are those where a formula has been obtained but the structure was not provided. Some organic acids were also identified using LC-MS.
LC-MS analysis
In this section, we further explore the SOA analysis of smog chamber samples in order to confirm (1) the structures obtained for selected organic acids by GC-MS analysis (see Organic acids section below) and (2) identification of OSs. The LC-MS analyses focused mainly on the identification of a variety of OSs and NOSs, as described below.
Organic acids
A set of the main organic acids, including tartaric acid (TA), D-threonic acid (TrA), tartronic acid (TrtA), and malic acid (MA), were detected in the particle phase at all RH levels under acidified conditions, non-acidified conditions, or both using the LC-MS analysis. This is consistent with the results reported above using the derivatization technique followed by GC-MS analysis (Table 4, last column). The identification was based on comparing the results of smog chamber experiments ER444 and ER666 with standard compounds for these organic acids. Figure 5 shows EICs of TA, TrA, TrtA, and MA recorded for SOA samples (ER666) and authentic standards. The retention times and fragmentation patterns obtained for these SOA components are in good agreement with reference compounds. Recent studies suggest that TA and other carboxylic acids undergo a heterogeneous OH reaction in aqueous solution (Cheng et al., 2016). Figures 6–9 show representative MS/MS spectra of TA, TrA, TrtA, and MA, along with proposed fragmentation patterns. Molecular formulas of the fragment ions were confirmed by mass measurements, but their structures were only tentative. Figure 6a and b show two high-resolution product ion mass spectra of tartaric acid with characteristic fragment ions at 149 [MC – 1], 131 [MC – 18], 103 [MC – 46], 73 [MC – 76], and 59 [MC – 90] (MC is the molecular weight of the compound) recorded for both the smog chamber sample and the standard compound. The fragmentation pathway in Fig. 6 shows that fragment ions with 131 and 87 are consistent with the loss of H2O and of H2O and CO2 from the deprotonated ion of TA ( 149), respectively. Fragment ions with 105 and 73 are consistent with loss of CO2 and C2H4O3, respectively. Finally, fragment ions with 103 and 59 are consistent with the loss of (H2O+CO) and , respectively. High-resolution product ion mass spectra associated with TrA, TrtA, and MA are shown in Figs. 7, 8, and 9, respectively.
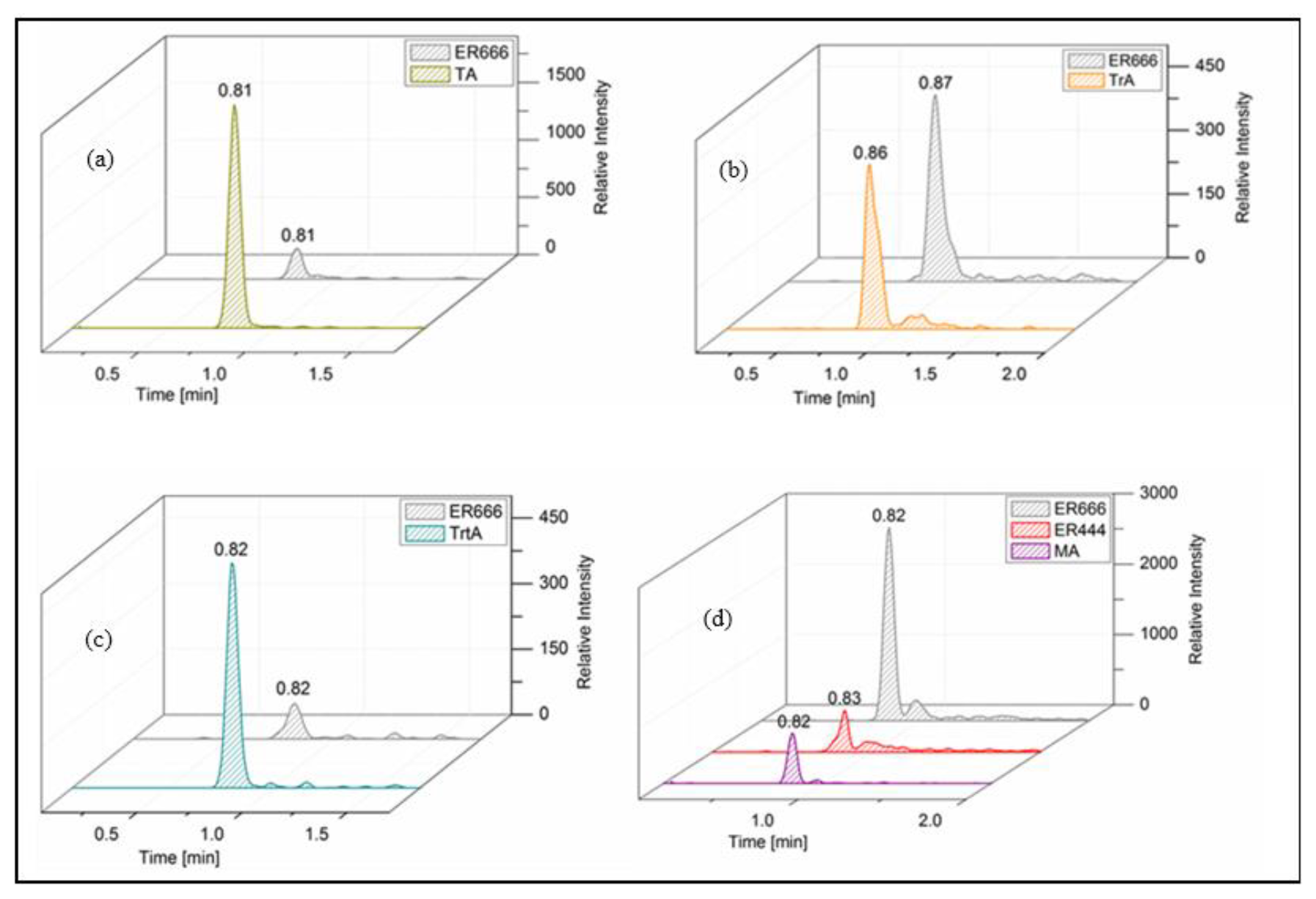
Figure 5Extracted-ion chromatograms of (a) tartaric acid (TA: MW 150), (b) threonic acid (TrA: MW 136), (c) tartronic acid (TrtA: MW 120), and (d) malic acid (MA: MW 134) from smog chamber experiments and reference compounds.
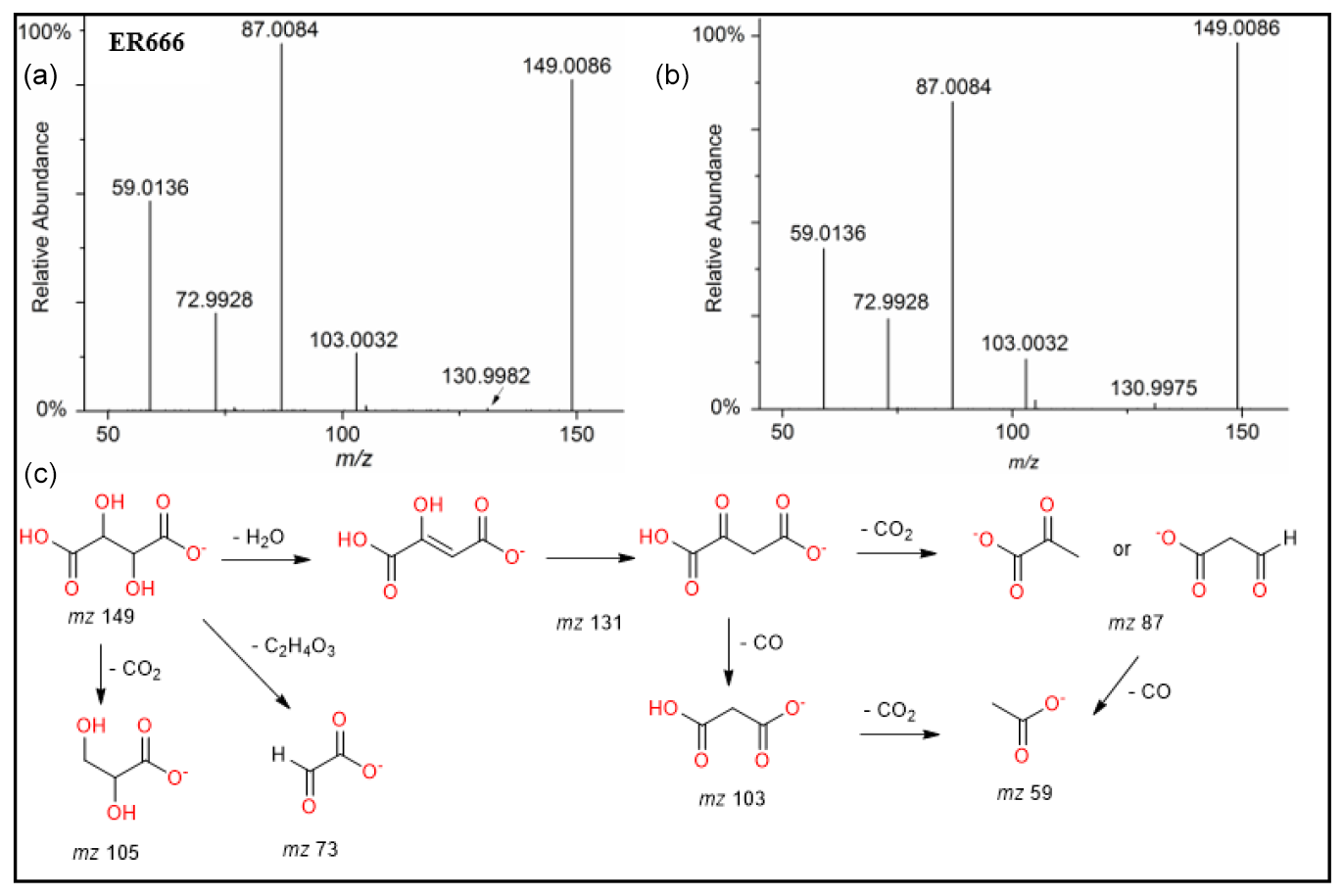
Figure 6ESI(-) product ion mass spectra of tartaric acid (MW 150) eluting at RT = 0.81 min, registered for the ER666 non-acidified seed aerosol sample (a) and the standard compound (b) along with a proposed fragmentation pathway (c).
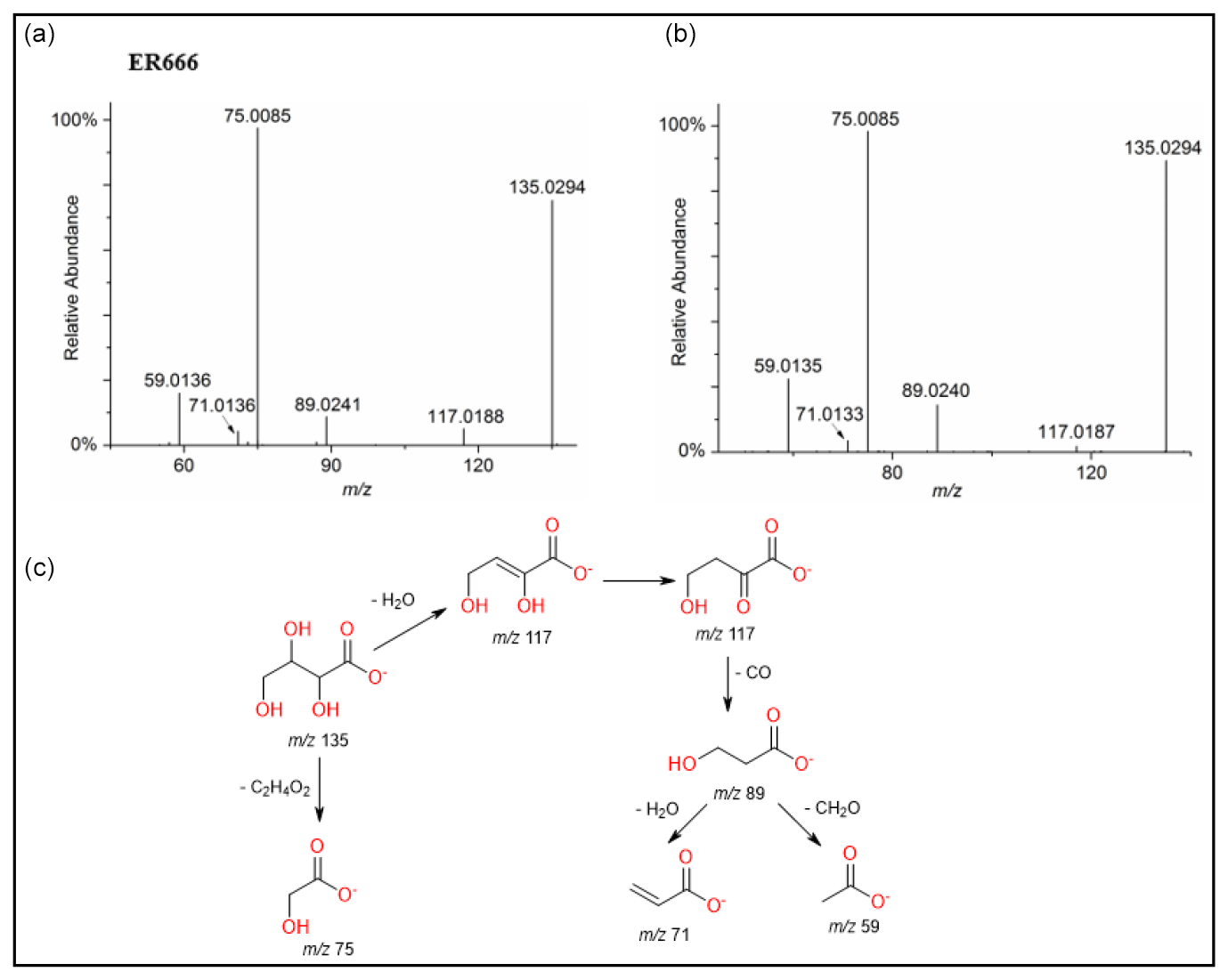
Figure 7ESI(-) product ion mass spectra of threonic acid (MW 136) eluting at RT = 0.87 min, registered for a sample from the non-acidified ER666 experiments (a) and for the reference compound obtained by alkaline hydrolysis of D-threonolactone (b) along with a proposed fragmentation pathway (c).
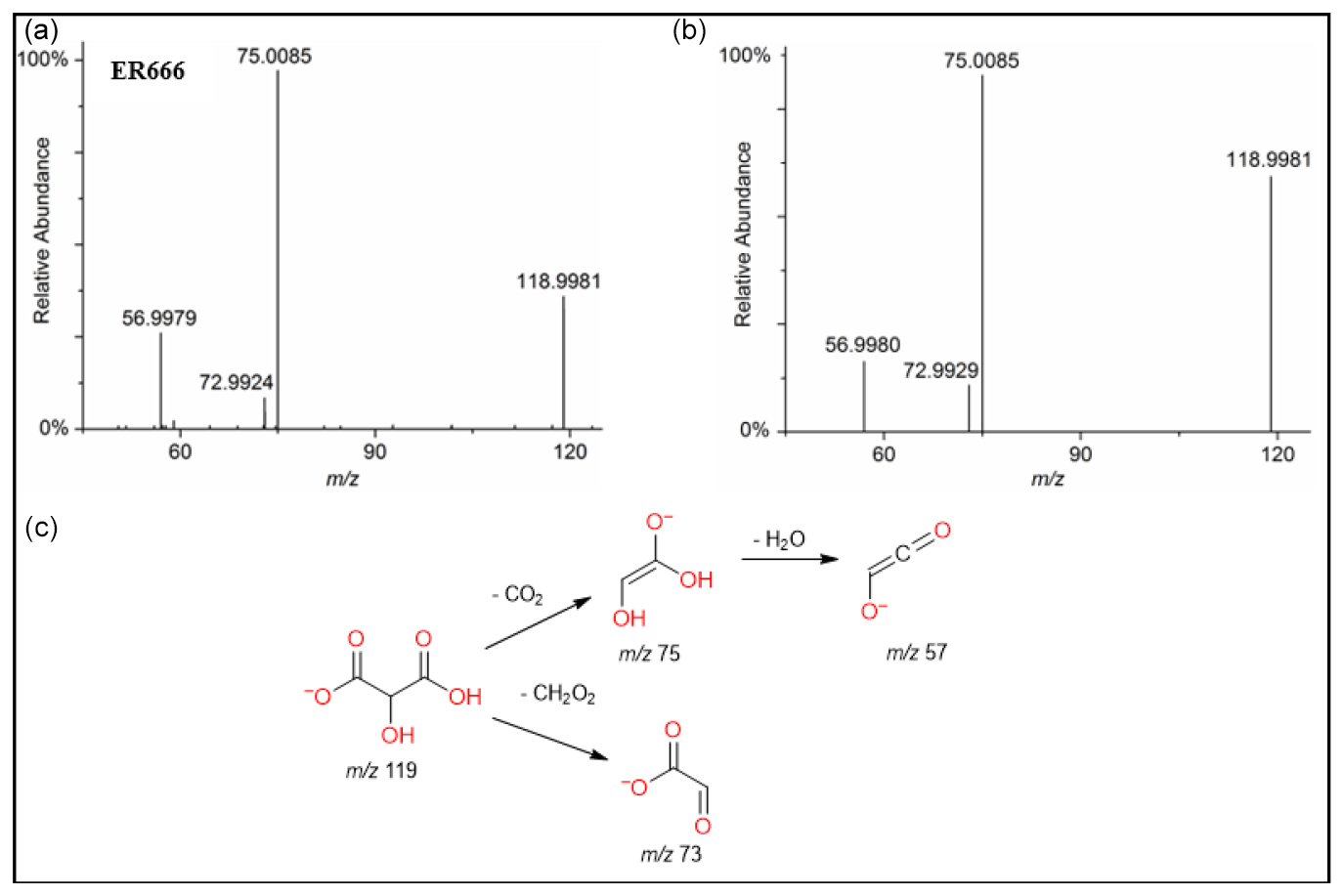
Figure 8ESI(-) product ion mass spectra of tartronic acid (MW 120) eluting at RT = 0.82 min, registered for a sample from the non-acidified ER666 experiments (a) and the reference standard compound (b) along with a proposed fragmentation pathway (c).
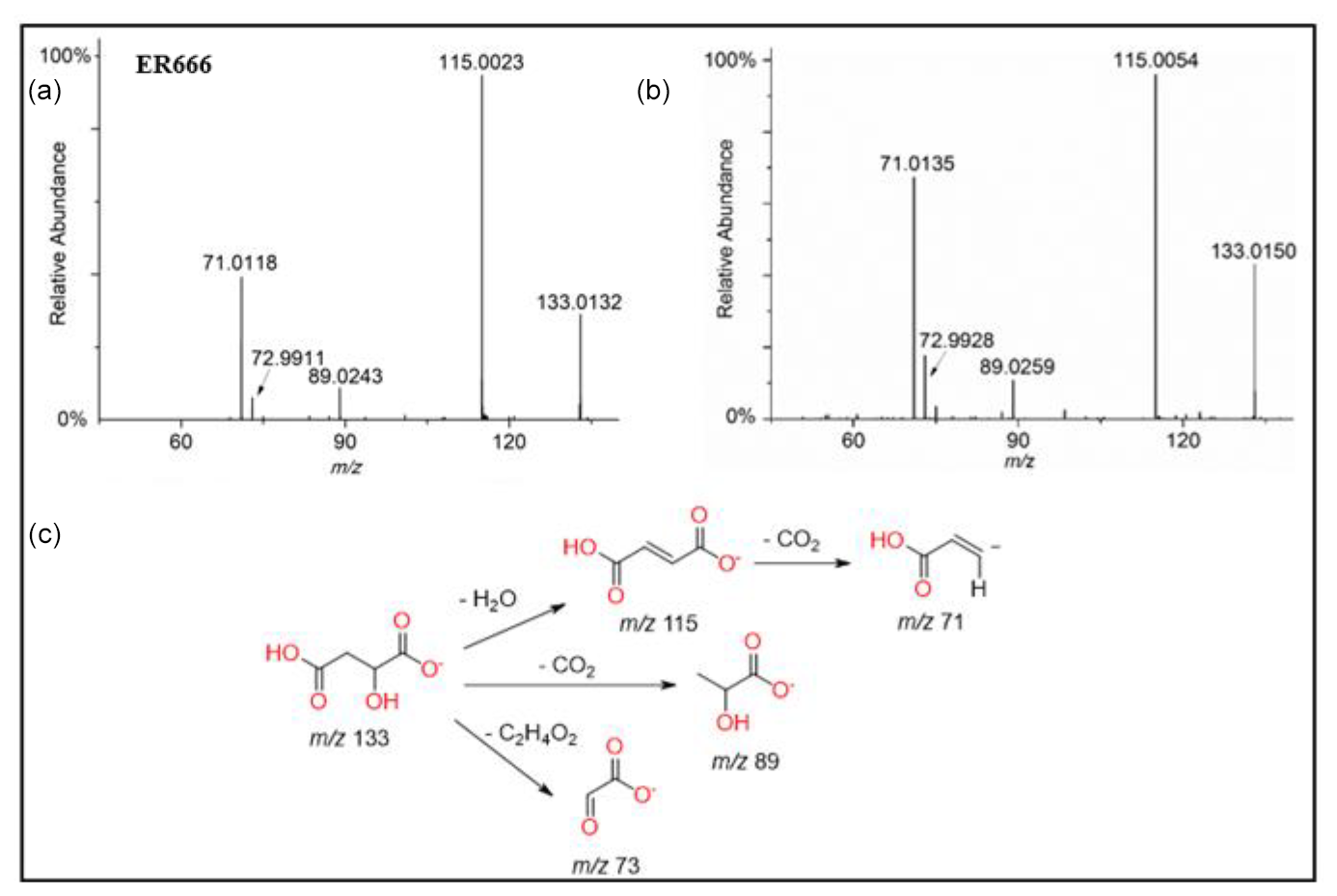
Figure 9ESI(-) product ion mass spectra of malic acid (MW 134) eluting at RT = 0.82 min, registered for the ER666 non-acidified seed aerosol sample (a) and the standard compound (b) along with a proposed fragmentation pathway (c).
Organosulfates
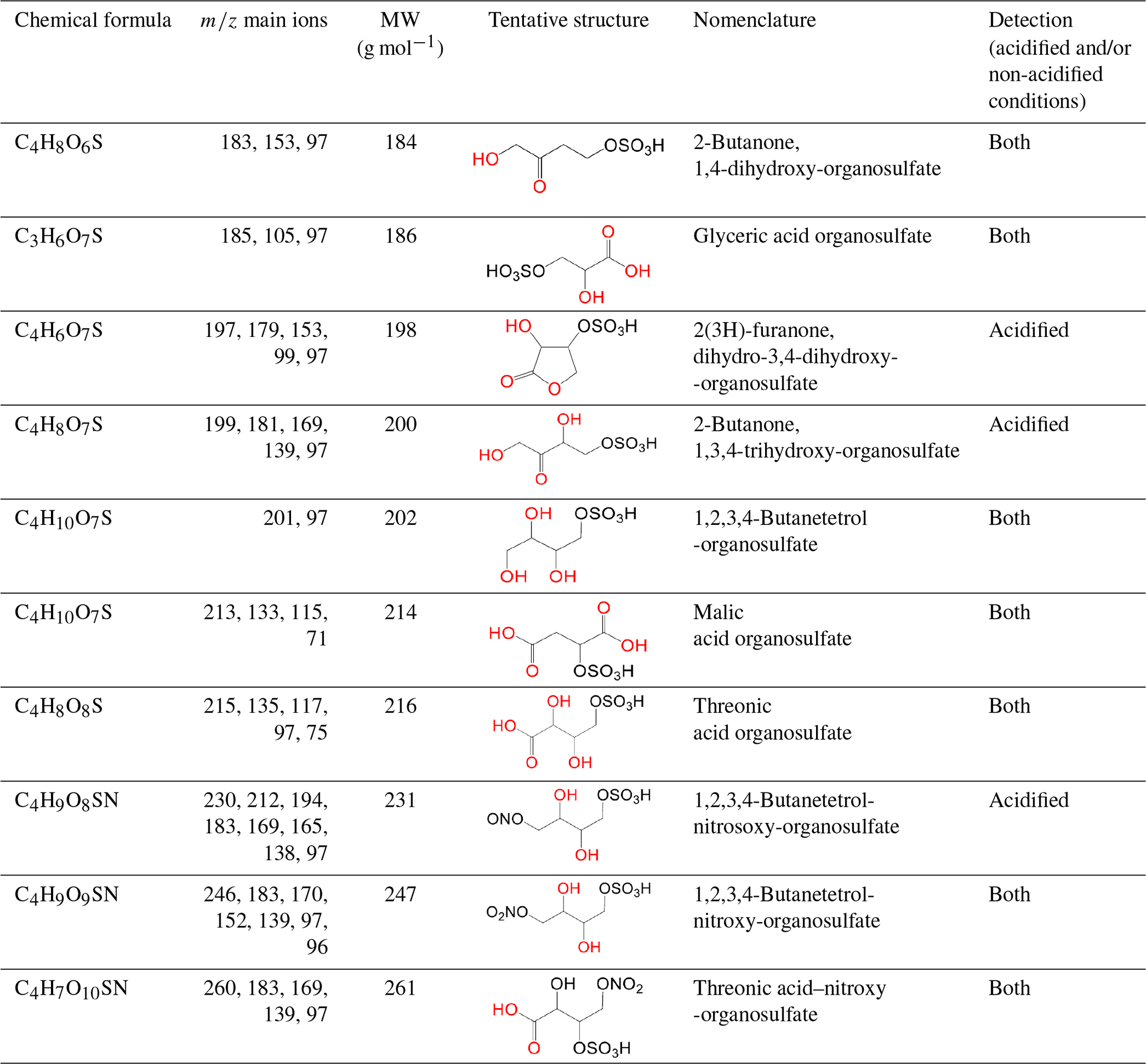
In this section, a tentative identification of OSs and NOSs is proposed based on accurate high-resolution mass data and MS/MS spectra, following the assumptions described in Nestorowicz et al. (2018). Briefly, this analysis was based on the deprotonated ions [M-H]− and the corresponding fragmentation pathways. OSs were recognized by the loss of characteristic ions at 80 (SO) and 96 (SO) and by the presence of the 97 ion (HSO) (Darer et al., 2011; Szmigielski 2016). The nitroxy-organosulfates and nitrosoxy-organosulfates were identified based on the neutral loss of 63 (HNO3) and 47 (HNO2), respectively. All OSs identified have the same carbon backbone of butane, except glyceric acid organosulfate (GA-OS). The presence of the 97 ion in a spectrum, corresponding to HSO, indicates that the hydrogen atom or a hydroxyl group is present at the carbon atom next to that bearing HO–SO2–O– moiety (Attygalle et al., 2001; Wach et al., 2020). For nitrosoxy- and nitroxy-OSs, the elimination of the HNO2 and HNO3 from the precursor ion was used for their identification. Figure 10 (left-hand panels) shows EICs associated with SOA samples collected from ER444 and/or ER666 experiments and highlights the selected newly identified OSs also present in ambient aerosol. Figure 10 (right-hand panels) shows high-resolution product ion mass spectra associated with peaks of OSs shown in the corresponding left-hand panels. Figure 11 shows proposed fragmentation patterns associated with the mass spectra shown in Fig. 10. As an example, and for illustration purposes, the top row of Fig. 10 shows the high-resolution product ion mass spectrum of GA-OS with characteristic fragment ions at 185 [MC – 1], 105 [MC – 80], 97 [MC – 88], and 75 [MC – 111]. The fragment ions detected at 97 and 105 are consistent with the diagnostic ion HSO and the loss of SO3 from the deprotonated GA-OS ion, respectively (Fig. 11, top). For the remaining OSs shown in Fig. 10, high-resolution product ion mass spectra associated with 2Fur-OS (two peaks), MA-OS, and TrA-OS are presented in Fig. 11. Additional newly identified OSs, including nitroxy- and nitrosoxy-OSs, detected solely in smog chamber experiments, are presented in the Supplement, along with proposed fragmentation schemes (Figs. S7–S12). Table 5 shows SOA organic products bearing sulfate and nitrate groups detected from 13BD/NOx photooxidation under acidified and non-acidified conditions using UHPLC-ESI-MS/MS. It contains a proposed structure for products identified, chemical formula, nomenclature, molecular weights of the compound, and the main ions , as well as if the product is detected under acidified and/or non-acidified conditions. Organosulfates were detected mainly under acidified conditions; however, some of them were also detected under non-acidified conditions at low intensity, which may have originated from aerosol heterogenous reactions involving ammonium sulfate present in the seed aerosol (Duporté et al., 2020).
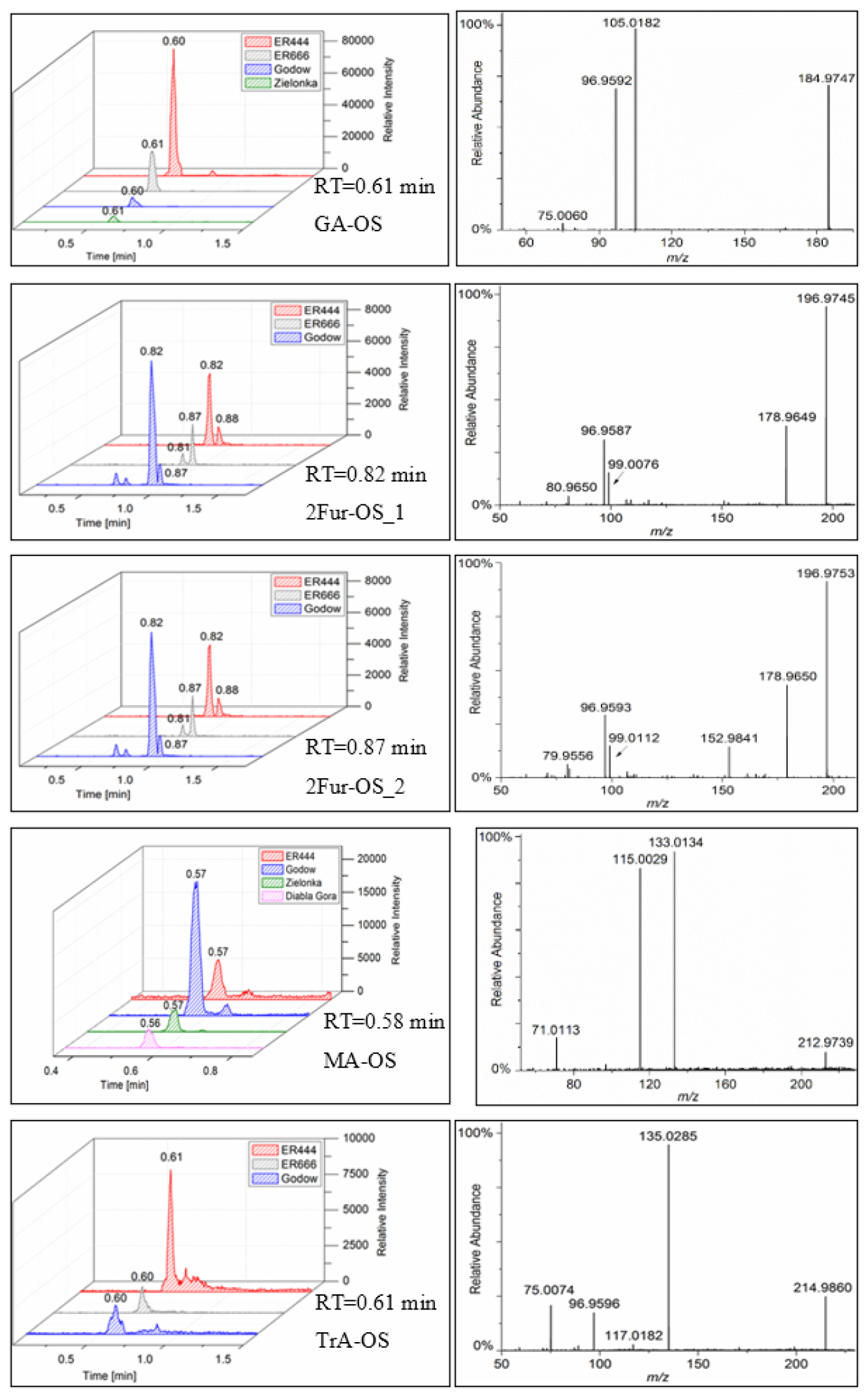
Figure 10Extracted-ion chromatograms of selected organosulfates and their corresponding high-resolution product ion mass spectra, MW 186 (glyceric acid organosulfate: GA-OS), MW 198 (2(3H)-furanone, dihydro-3,4-dihydroxy-organosulfate: 2Fur-OS_1 and 2Fur-OS_2; two isomers), MW 214 (malic acid organosulfate: MA-OS), and MW 216 (threonic acid organosulfate: TrA-OS), obtained from smog chamber experiment ER444 and ambient samples (see Sect. 3.3 for field sample analysis).
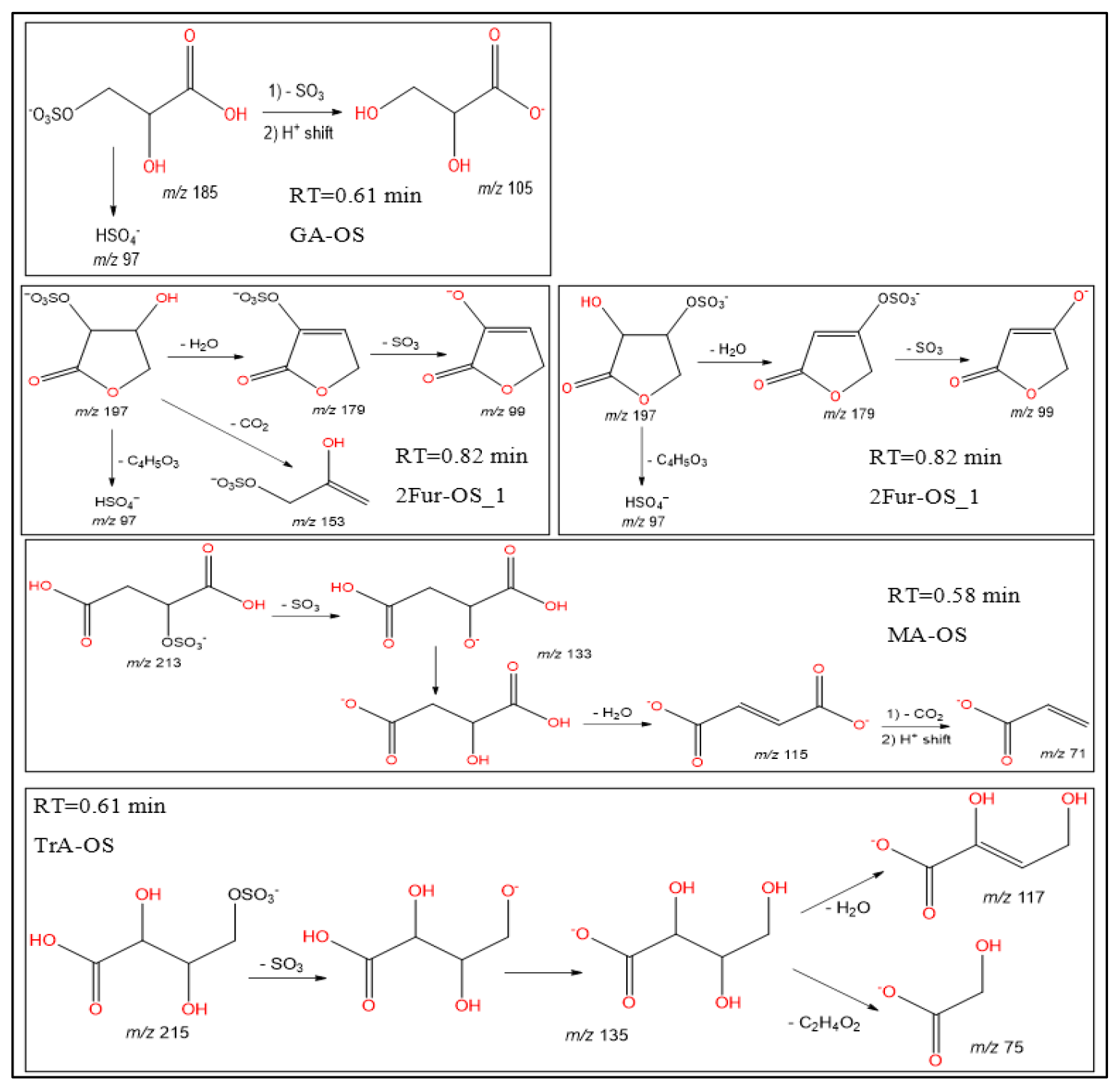
Figure 11Proposed fragmentation pathways of organosulfates identified in Fig. 10: MW 186 (glyceric acid organosulfate: GA-OS), MW 198 (2(3H)-furanone, dihydro-3,4-dihydroxy-organosulfate: 2Fur-OS_1 and 2Fur-OS_2; two isomers), MW 214 (malic acid organosulfate: MA-OS), and MW 216 (threonic acid organosulfate: TrA-OS).
Table 5Selected SOA reaction products bearing sulfate and nitrate groups detected from 13BD/NOx photooxidation under acidified and non-acidified conditions using UHPLC-ESI-MS/MS.
3.2 Quantitative/semi-quantitative analysis
3.2.1 Effect of relative humidity on SOA products
GCM-S analysis
The concentrations of selected reaction products reported in Table 4 were measured. The quantitative analysis was conducted using D-threitol as a surrogate standard and cis-ketopinic acid (KPA) as an internal standard, except for malic acid and tartaric acid because authentic standards were not commercially available. Although calibration curves were obtained for several surrogates (Tables S1, S2; Fig. S1), D-threitol was used because its mass spectrum shares several fragment ions with the majority of reaction products identified with GC-MS (Table 4). Although quantitative information is very important for source apportionment and health effect purposes, due to the non-availability of commercial authentic standards in this study, our main objective was to gain insight into the identification and distribution of 13BD SOA products and their relative contribution as a function of RH and seed aerosol acidity. Therefore, the quantitative/semi-quantitative analysis either with GC-MS or LC-MS should be regarded as such. Figures 12 and 13 display a semi-quantitative analysis of selected reaction products identified under acidified (ER444) and non-acidified (ER666) conditions across the RH used in this study (Table 4). Figure 12 shows estimated concentrations of 14 monomers (top), 4 dimers (middle), and 7 unknowns (bottom) identified in ER444 as a function of RH. The total SOC mass concentration during the ER444 experiment ranged from 60.3 at RH = 10 % to 31.1 µg m−3 at RH = 62 %. The concentrations of individual products were as follows: (1) monomers ranged between below detection limit to about 1064 ng m−3, (2) dimers ranged between 16 and 497 ng m−3, and (3) unknowns ranged between 18 and 555 ng m−3. These values are typical of the range of values often seen for individual compounds detected in SOA chamber samples from the oxidation of other hydrocarbons. The concentration of the majority of products decreased with increasing RH with a few exceptions. The major contributors to 13BD SOA across all RH levels were glyceric acid reaching up to 1064 ng m−3 at 31 % RH, followed by glyceric acid–1,3,4-trihydroxy-1-butene ester dimer (second major component detected) reaching an estimated value of 497 ng m−3 at 31 % RH. The three major monomers observed after glyceric acid were threitol (all isomers), threonic acid (all isomers), and 1,3,4-trihydroxy-1-butene (Fig. 12, top). All dimers and unknowns (U1–U7) were detected at a relatively high concentration (Fig. 12, middle and bottom). Similarly to aerosol production and monomers, they were found to be highly sensitive to the initial conditions, particularly RH.
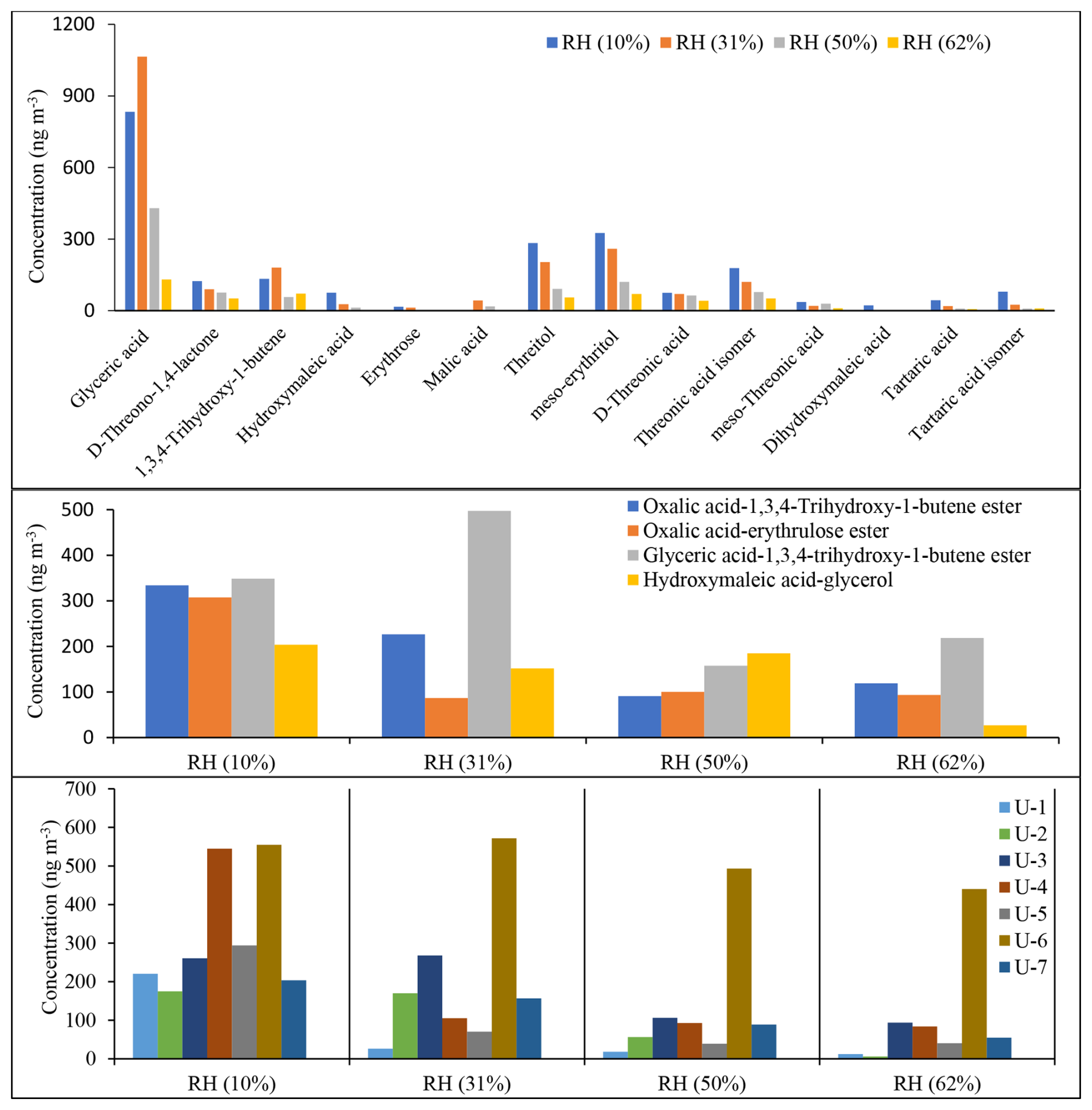
Figure 12Estimated concentrations of selected SOA products as a function of RH obtained from the ER444 experiment using BSTFA derivatization. D-threitol was used as an external surrogate standard for the semi-quantitative analysis for all products, except D-threitol, tartaric acid, and malic acid, for which authentic standards were used. Cis-ketopinic acid was used as an internal standard for all reaction products. See Table 3 and its description for additional information about the identity of these products.
Figure 13 shows the estimated concentrations of 14 monomer (top), 4 dimer (middle), and 4 unknown (bottom) products identified in ER666 as a function of RH using BSTFA derivatization. The total SOC mass concentration during the ER666 experiment ranged from 45.1 µg m−3 at RH = 11 % to 24.7 µg m−3 at RH = 60 %, values lower than those measured under acidified seed aerosol. The concentrations of individual products were as follows: (1) monomers ranged between below detection limit to about 207 ng m−3; (2) dimers ranged between 10 and 178 ng m−3, except for glyceric acid–1,3,4-trihydroxy-1-butene ester, which was observed at very high concentrations between 142 (RH = 60 %) and 798 (RH = 11 %) ng m−3; and (3) unknowns ranged between 18 and 555 ng m−3. Note that only U3, U6, and U7 were observed under non-acidified conditions (Fig. 13, bottom) and at relatively low concentrations compared to those under acidified conditions. Again, these values are typical of the range of values often seen for individual compounds detected in SOA chamber samples from the oxidation of other hydrocarbons. The concentration of the majority of products decreased with the increase in RH. Threonic acid isomers and threitol isomers, followed by glyceric acid, were among the most abundant species observed under non-acidified conditions, respectively. For example, the concentrations of meso-threonic acid ranged between 82 ng m−3 at 60 % RH and 207 ng m−3 at 11 % RH, and they increased with decreasing RH. The formation of individual compounds as under acidified conditions was found to be highly sensitive to the initial conditions, particularly RH.
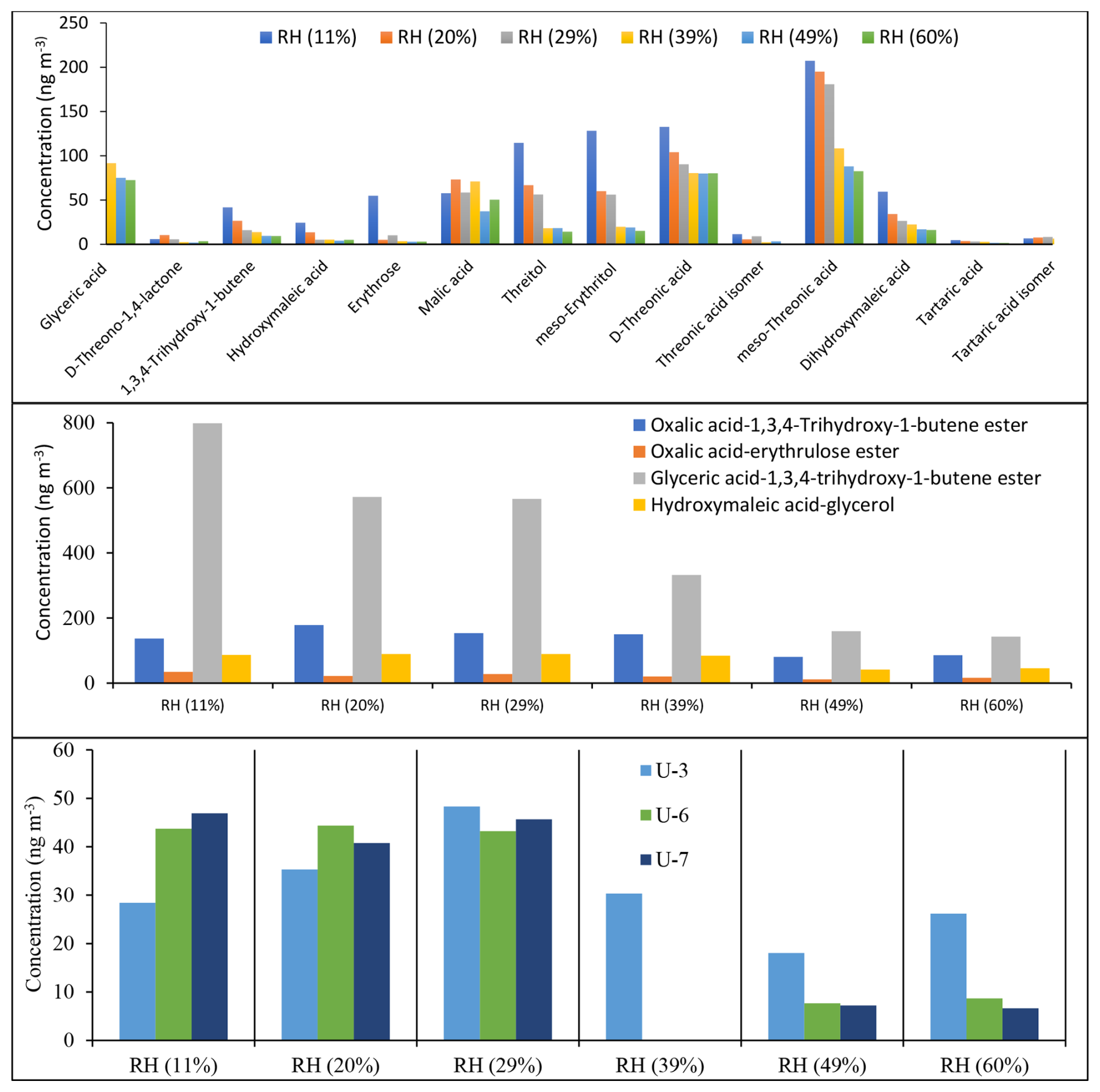
Figure 13Estimated concentrations of selected SOA products as a function of RH obtained from the ER666 experiment using BSTFA derivatization. D-threitol was used as an external surrogate standard for the semi-quantitative analysis for all products, except D-threitol, tartaric acid, and malic acid, for which authentic standards were used. Cis-ketopinic acid was used as an internal standard for all reaction products. See Table 3 and its description for additional information about the identity of these products.
LC-MS analysis
The concentrations of organosulfates shown in Table 5 were measured using LC-MS analysis, except for C4H7O10SN ( 261). The concentrations were obtained using sodium 1-pentyl sulfate (SP-OS) as a surrogate because authentic standards of 13BD OS are not commercially available. The recovery, limit of detection (LOD), and limit of quantification (LOQ) are reported for SP-OS in Table S4. Figure S4 displays calibration curves used for quantitative analysis associated with SP-OS. Figure 14 displays the concentrations measured using SP-OS as a surrogate for these OSs under acidified (top) and non-acidified (bottom) conditions as a function of RH. High concentrations across all RH levels were measured for 1,2,3,4-butaneterol-NOS ( 246) and at relatively moderate concentrations for 1,2,3,4-butanetetrol-OS ( 201), glyceric acid OS ( 185), and 2-butanone,1,4-dihydroxy OS ( 183). The highest values were observed under the acidified seed experiment (ER444) at low RH and decreased with increasing RH, exceeding 3000 ng m−3 at 30 % RH (Fig. 14). Similar mechanistic pathways can lead to the formation of these OSs through acid-catalyzed multiphase chemistry of BEPOX (e.g., for 1,2,3,4-butaneterol-OS, 201; 1,2,3,4-butanetetrol-NOS, 246) and AAE (e.g., for glyceric acid OS, 185), the same as those proposed for isoprene (Nestorowicz et al., 2018). In general, the increase in RH in the chamber resulted in decreased concentrations of OSs, although some OSs observed were not RH-dependent (e.g., malic acid OS: 213; glyceric acid OS: 185). These results are consistent with previously reported data for isoprene SOA components (Nestorowicz et al., 2018). 2-Butanone, 1,4-dihydroxy-OS ( 183); (3H)-furanone, dihydro-3,4-dihydroxy-OS ( 197); 2-butanone, 1,3,4-trihydroxy-OS ( 199); and 1,2,3,4-butanetetrol nitrosoxy-organosulfate ( 230) and are found to be considerably abundant under acidified conditions. Glyceric acid OS was found to be RH-dependent under acidified conditions only, results consistent with methyl-glyceric acid OS reported in isoprene SOA (Nestorowicz et al., 2018). 1,2,3,4-Butanetetrol-OS ( 201) was detected under both conditions and RH-dependent, though with high abundance under acidified conditions. Malic acid OS ( 213) and threonic acid OS ( 215) were detected in trace amounts under both conditions, acidified and non-acidified. Threonic acid NOS ( 260) was also detected under both conditions in trace amounts but at a level below LOD (not shown in Fig. 14). The most abundant compound produced under acidified and non-acidified conditions was 1,2,3,4-butanetetrol-NOS ( 246). The estimated concentrations were 10-fold higher than those obtained for other 13BD SOA components. The amounts at the lowest RH reached ∼ 3200 and ∼ 450 ng m−3 under acidified and non-acidified seed conditions, respectively. The estimated concentrations of detected 13BD SOA components established by LC-MS and GC-MS techniques are presented in the Supplement (Tables S5–S8).
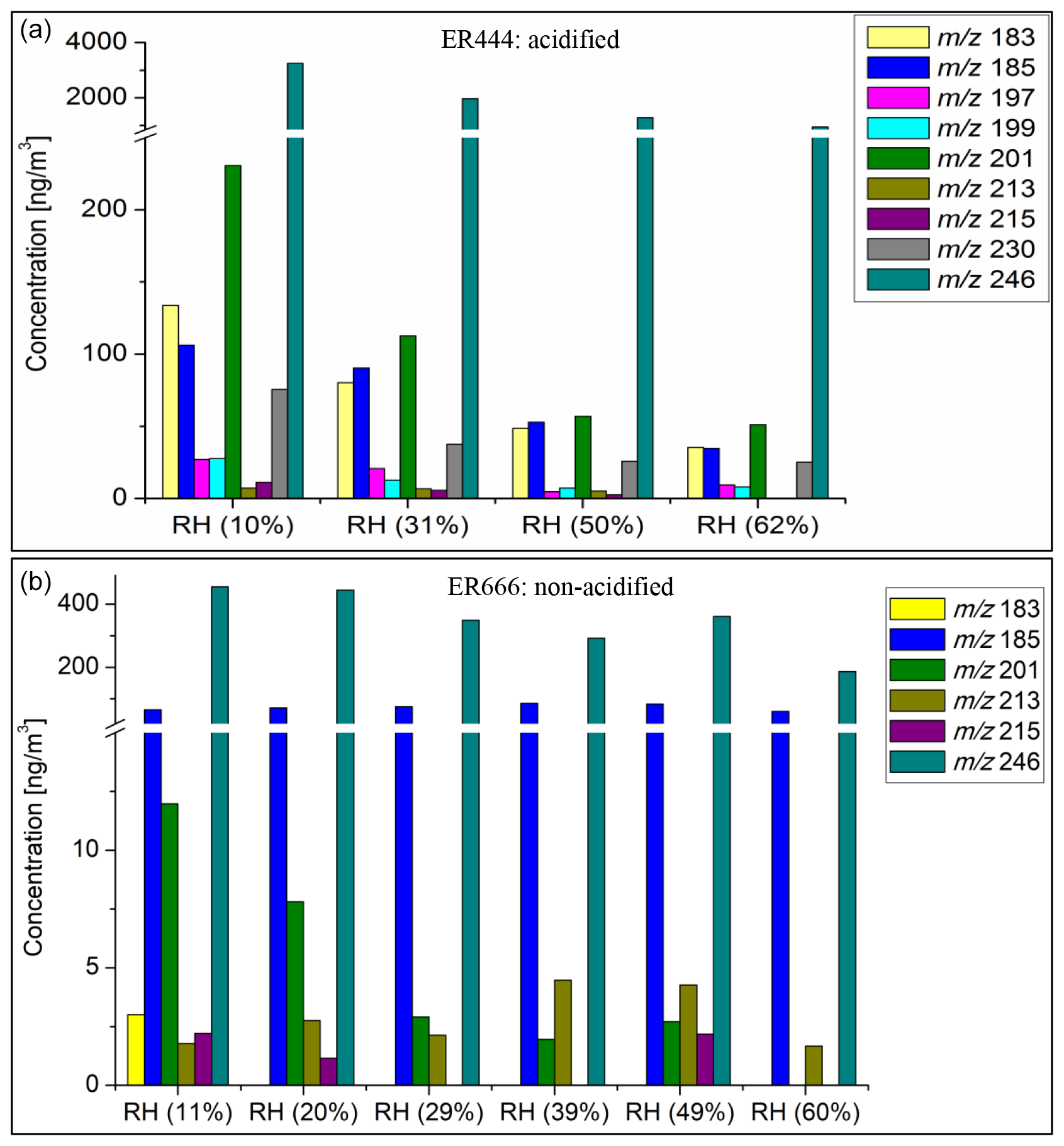
Figure 14Estimated LC-MS concentrations (ng m−3) of OS and NOS reaction products detected in SOA (Table 5) under acidified (ER444: a) and non-acidified (ER666: b) conditions as a function of RH. The chemical names associated with are provided in Table 5.
3.2.2 Effect of seed aerosol acidity on 13BD SOA products
The TIC chromatograms shown in Fig. 15 can be compared because (1) the sampling time, flow rate, and analytical processes were the same for each experiment; (2) they were originated from two stages of experiments ER444 (blue: acidified seed) and ER666 (red: non-acidified seed) conducted under similar RH (50 %), though at slightly different temperatures (Table 1); and (3) delta-13BD was the same for each stage. Figure 15 displays parts of the TICs associated with SOA originated from the photooxidation of 13BD at similar relative humidity (∼ 50 %) under acidified (blue) and non-acidified (red) seed aerosol for the portions between 17–27 min (top) and 27–33 min (bottom). A detailed examination of mass spectra associated with each peak detected in these chromatograms reveals significant changes in the composition of particles depending on the acidity of the original seed aerosol. As shown in Table 4 (last column), the majority of compounds were detected in both chromatograms, except in the blank, although at variable abundance. The presence of sulfuric acid in the seed aerosol (ER444) resulted in a relatively substantial (1) increase in the intensity of the majority of peaks detected after 27 min and (2) decrease in the intensity of the majority of peaks detected below 27 min. Compounds eluted before 27 min were more abundant under acidified conditions (right y axis: red) than under non-acidified conditions (left y axis: blue). This is consistent with the occurrence of chemical reactions probably in the particle phase in the presence of acidified seed aerosol. The changes in seed aerosol acidity content between ER444 and ER666 resulted in significant decreases in product concentrations associated with monomers, as shown in Figs. 12 and 13 (top panels: e.g., threitols, threonic acid), and an increase in the intensity and formation of dimers (Figs. 12, 13: middle panels) and organosulfates (Fig. 14). It is likely that the production of dimers and organosulfates is partially suppressed in the absence of acidified seed aerosol in the system. The presence of acidified seed aerosol also resulted in a pronounced increase in the concentration of unknown compounds (Figs. 12 and 13, bottom panels).
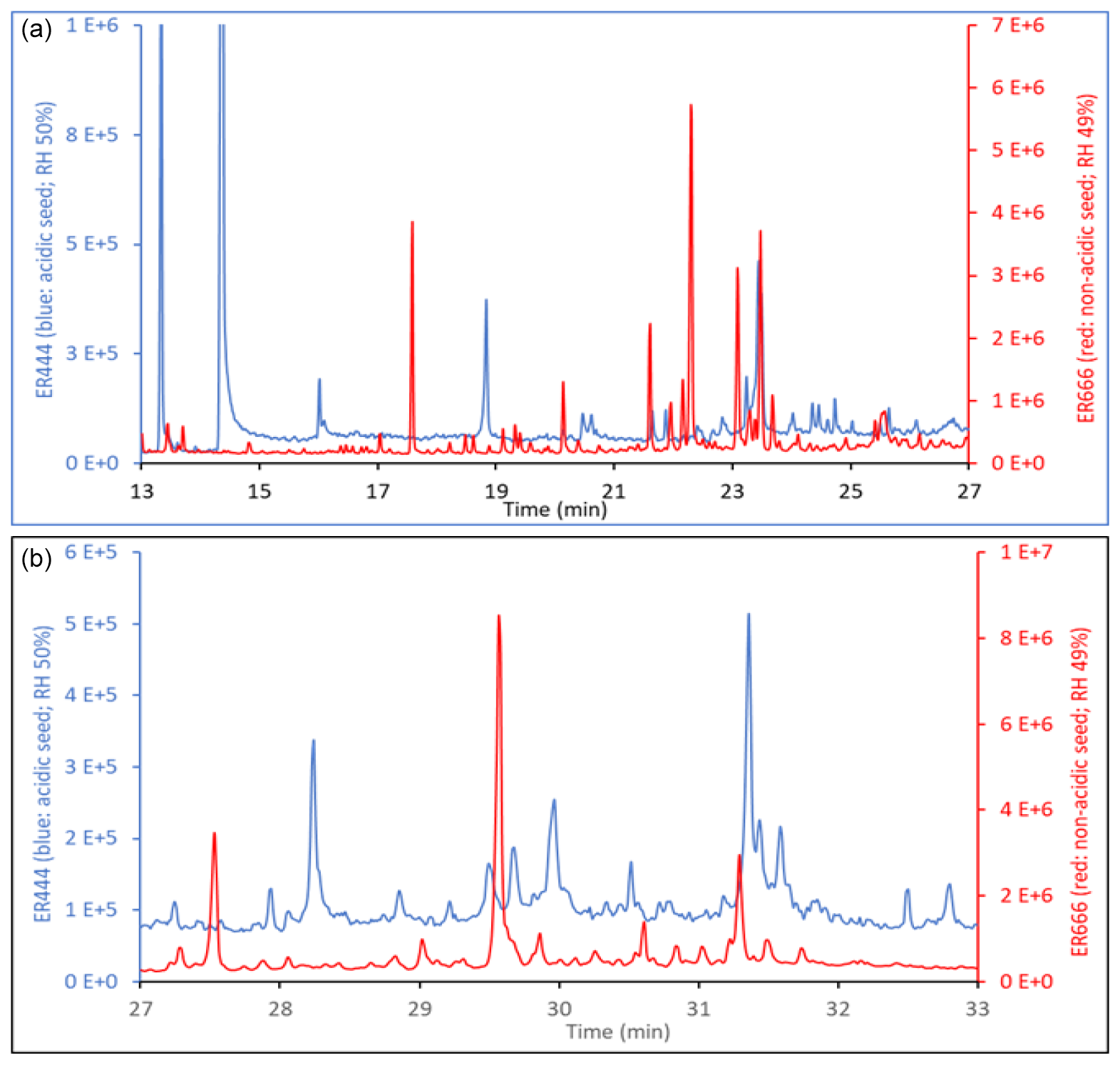
Figure 15Parts of the total ion chromatograms associated with SOA originated from the photooxidation of 13BD at similar relative humidity (∼ 50 %) under acidified (blue) and non-acidified (red) seed aerosol for portions between 17–27 min (a) and 27–33 min (b).
3.2.3 Relation of humidity and acidity of the particle aqueous phase
The acidity of the particle aqueous phase was estimated using E-AIM with input data given in the Supplement (Sect. S2). The aqueous phase in the ER666 experiment (non-acidified seed) was absent across the whole RH range; therefore only the organic phase was present. In the ER444 experiment (acidified seed), the particle aqueous phase was always present. Table 6 shows the H+ activity coefficient, the activity (aH+), and the LWC associated with the particle aqueous phase across the whole RH range in ER444. The acidity of the aqueous phase decreased and LWC increased with increasing RH. The pH (aH+) of the aqueous phase increased from −1.79 at 10 % RH to ∼ 0.40 at 62 % RH. The contribution of organic acids to the formation of the particle aqueous phase and its acidity was not considered. It could be only marginal in acidified seed experiments, where the highest possible amount of dissociable hydrogen ions from organic acids was less than 3 % of the amount contributed by the sulfuric acid used to prepare seed aerosol. In the non-acidified seed experiments, organic acids could have a more profound influence, so the results of our modeling should be taken as a first approximation and verified as soon as the precise stoichiometry of the acid formation and reliable dissociation constants of those acids are available.
Table 6Water contents, pH, and acidity of the particle aqueous phase in the ER444 smog chamber experiment.
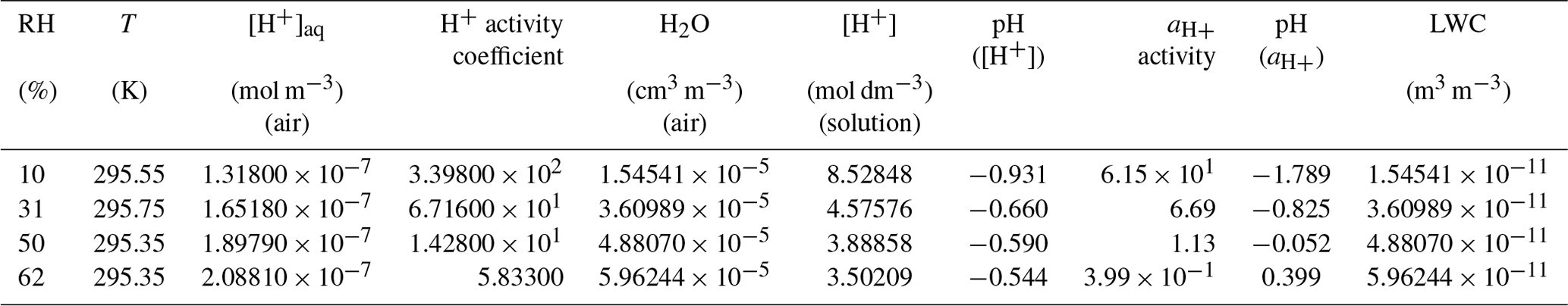
The SOC concentration and carbon yield (Table 1) increased with decreasing RH under acidified and non-acidified conditions. The carbon yield was higher under acidified conditions than non-acidified conditions and increased with increasing acidity of the aerosol aqueous phase. On the contrary, the 13BD conversion efficiency was higher at non-acidified conditions than acidified (Fig. 1b), indicating increased non-reactive uptake of 13BD by non-acidified seeds which contained no aqueous phase. Figure 16 shows the effect of seed aerosol acidity and RH on the SOC concentrations associated with the ER444 and ER666 experiments.
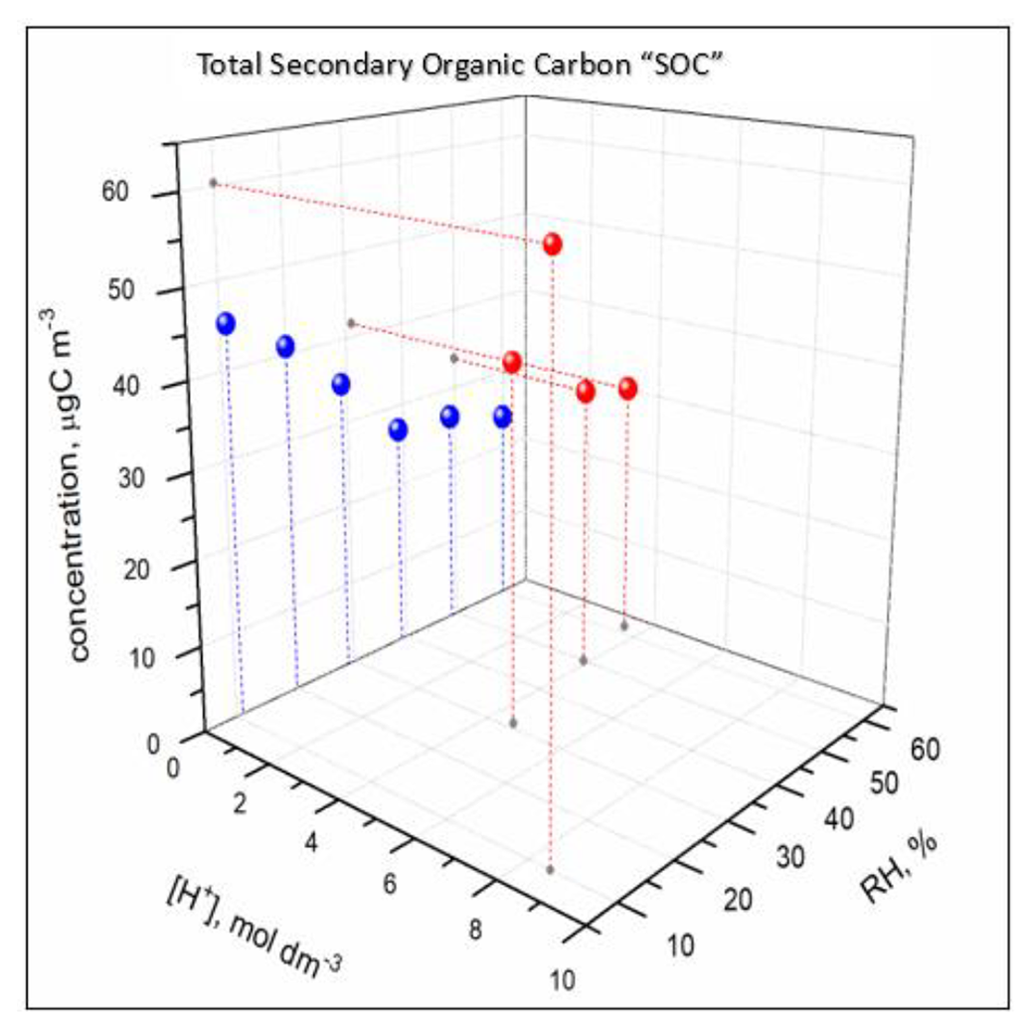
Figure 16Effect of RH and seed aerosol acidity on SOC concentrations under acidified (ER444: red) and non-acidified (ER666: blue) conditions.
The concentration of the majority of reaction products (1) was higher under acidified seed aerosol conditions, where the aerosol particles contained aqueous phase, than under non-acidified seed conditions, where the aerosol particles did not contain any aqueous phase, across the RH range used in this study; (2) increased with the acidity of the aerosol aqueous phase in the experiments under acidified seed conditions, with aqueous phase always present; and (3) decreased with increasing RH under either acidified or non-acidified seed conditions (Figs. 12, 13, 14, 17, and S13–S43). However, there were a few significant deviations. (1) In non-acidified conditions, the concentrations of meso-threonic, D-threonic, and dihydroxymaleic acids were higher at all RH values (Figs. 17, S23, S25, S36); the concentrations of glyceric acid OS ( 185 [M-H]−), malic acid OS ( 213 [M-H]−), and erythrose were higher at RH = 40 %–61 % (Figs. 17, S14, S18, S26); and the concentration of glyceric acid–1,3,4-trihydroxy-1-butene ester was higher at RH = 10 %–30 % (Figs. 17, S32). (2) In non-acidified experiments, the concentration of 1,3,4-trihydroxy-1-butene was minimal at RH = 50 % (Figs. 17, S34), and that of malic acid OS ( 213 [M-H]−) was maximal at RH = 40 % (Fig. S18). In acidified experiments, the concentrations of glyceric acid–1,3,4-trihydroxy-1-butene ester and 1,3,4-trihydroxy-1-butene were maximal at RH = 30 % (Figs. S32, S34), and the concentration of oxalic acid–erythrulose ester was minimal at RH = 30 % (Fig. S31). The yield trends observed in acidified seed experiments (ER444), in which the aqueous particle phase was always present, indicate that acidity enhances the formation of most products of 13BD oxidation in the aqueous phase. The concentration trends observed in non-acidified seed experiments (ER666), in which the particles contained no aqueous phase, may indicate that the phase state and/or gaseous H2O limited the access of reactants to particle surfaces and hindered the formation of most products. A few exceptions to the major yield trend may indicate more complex formation and partitioning mechanisms involved, which cannot be explained based on current work. In addition, the “acidified” and “non-acidified” terms are associated with seed aerosol solutions, and fundamental differences in chemical mechanism may be responsible for the reaction products formed in this study due to the phase states in which the particles themselves exist, liquid (ER444) or solid (ER666). The observed differences between the acidified (ER444) and non-acidified (ER666) seed aerosol experiments may be due to the acidity of the seed aerosol, the liquid water content, and/or the phase state of the aerosol particles. Furthermore, more work is needed to gain insight into mechanisms of reactant formation and partitioning to deliquescent particles and to particles lacking the aqueous phase.
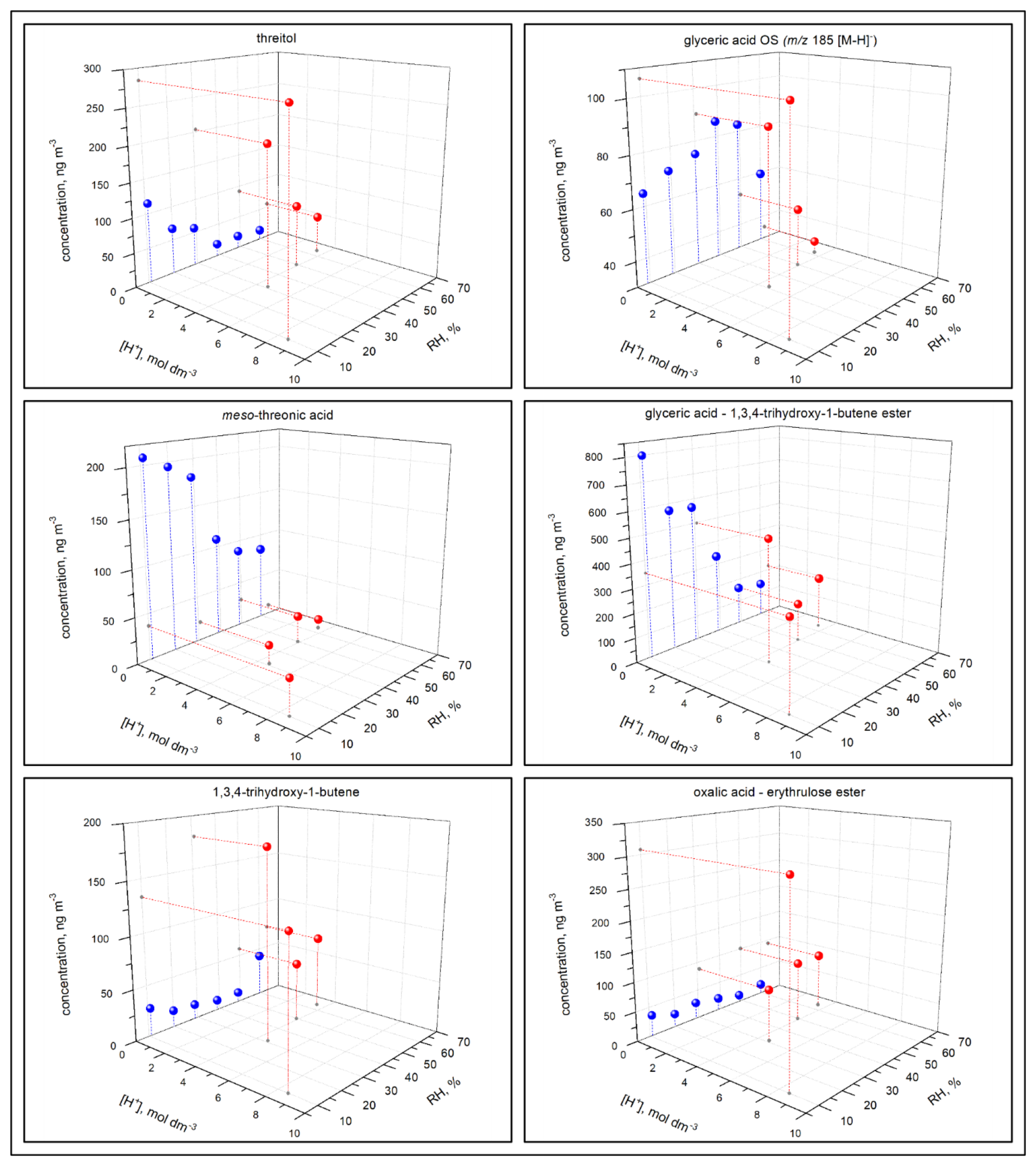
Figure 17Effect of RH and seed aerosol acidity on the concentration of threitol, glyceric acid OS ( 185 [M-H]−), meso-threonic acid, glyceric acid–1,3,4-trihydroxy-1-butene ester, 1,3,4-trihydroxy-1-butene ester, and oxalic acid–erythrulose ester produced in non-acidified (blue) and acidified (red) experiments.
The preparation of non-acidified and acidified seed aerosol by nebulizing aqueous solutions of NH4SO4 and NH4SO4+H2SO4, respectively, has been used in chamber experiments for many years (Czoschke et al., 2003; Deng et al., 2021; Zhang et al., 2023). As stated above, the LWC and acidity of the aerosol were estimated using E-AIM, which assumes the vapor–particle partitioning equilibrium in the system. The non-acidified seed aerosols were dry based on E-AIM. This may be a major difference in the acidified and non-acidified conditions and probably one of the main drivers of differences in the chemistry between the two systems. Note that E-AIM is an equilibrium model, which has its own limitations. Therefore, it is important to consider that, under our experimental conditions, the system should be at chemical equilibrium, which can be difficult to characterize. In our experimental system, we are going from liquid solution in the nebulizer reservoir to ammonium sulfate particles in air of nearly 100 % RH exiting the nebulizer to chamber dry air in less than 1 min. Then the particles have a chance to equilibrate in the chamber for a matter of hours. The equilibrium time needed for acidified wet aerosols to adjust their LWC to the chamber RH conditions is probably on the order of minutes to tens of minutes (Saleh et al., 2013), so they are probably close to their E-AIM-predicted LWCs in the chamber. However, the time needed for the wet neutral aerosols to completely dry out at room temperature may be on the order of hours. It is probable that, under our experimental conditions, the wet neutral aerosols do not reach the E-AIM-predicted dryness on the timescale of the chamber residence time. This may explain why the two systems are not as chemically drastically different as expected.
3.3 Field measurements
We compared the results of chamber experiments conducted under acidified (ER444) and non-acidified (ER666) conditions with ambient PM2.5 samples collected at the Godów, Zielonka, and Diabla Góra monitoring stations in Poland (Sect. 2.2). The purpose of presenting the field results was to show that some compounds observed in smog chamber experiments also occur in the real ambient aerosol. Four products, which did not contain sulfate groups, were detected both in the chamber SOA and in ambient samples: tartronic acid (MW 120), malic acid (MW 134), threonic acid (MW 136), and tartaric acid (MW 150). The same analytical LC-MS method was used for the chamber and field samples. Figure 18 shows the extracted-ion chromatograms associated with the ER444 and ER666 chamber experiments and with the PM2.5 samples collected during 2014 and 2016 field studies (Sect. 2.2). Several species (e.g., glyceric acid, malic acid, threitol, erythritol, and tartaric acid) were previously observed in ambient samples collected in several places around the USA and were also associated with 13BD oxidation (Jaoui et al., 2014). In this work, it now appears that these compounds could be generated in the atmosphere from the oxidation of 13BD in another part of the world (Poland).
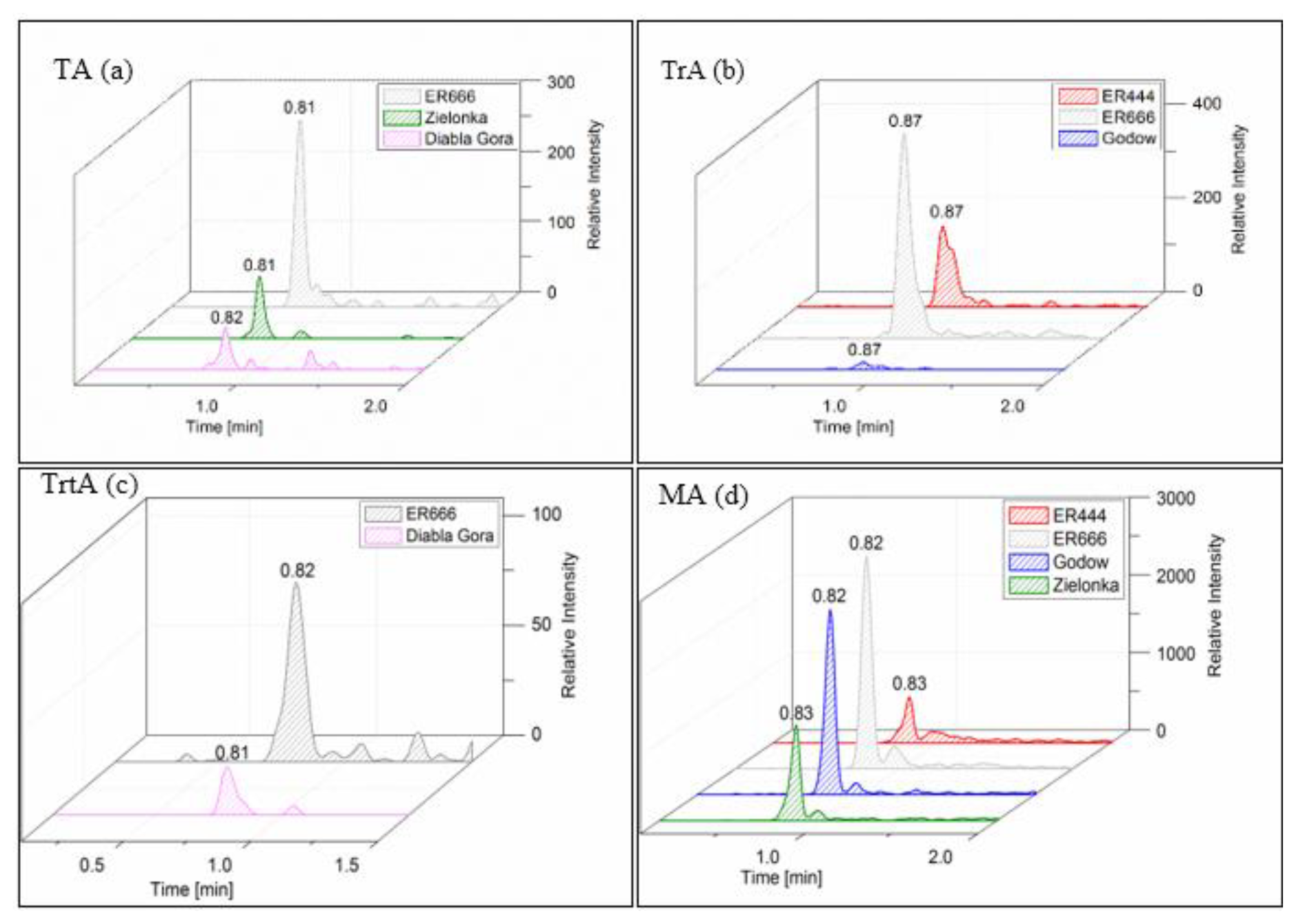
Figure 18Extracted-ion chromatograms recorded for samples from field studies (Zielonka, Godów, and Diabla Góra) and smog chamber experiments (ER444 and ER666) of (a) tartaric acid (TA; MW 150), (b) threonic acid (TrA; MW 136), (c) tartronic acid (TrtA; MW 120), and (d) malic acid (MA; MW 134).
Note that samples from chamber experiments conducted in our laboratory involving the oxidation of biogenic (e.g., isoprene, monoterpenes, sesquiterpenes, 2-methyl-3-buten-2-ol), aromatic (e.g., toluene, 1,3,5-trimethylbenzene, benzene), and polycyclic aromatic hydrocarbons (PAHs) (e.g., naphthalene) did not contain the compounds identified in this section, except malic acid and tartaric acid, using the same experimental and analytical procedures. A second comparison of chamber SOA samples from the ER444 and ER666 experiments with ambient PM2.5 samples was conducted using the LC-MS analysis focused on OSs. Extracted-ion chromatograms in Fig. 10 show that several OSs occurred in chamber SOA and ambient PM2.5, including glyceric acid organosulfate; 2(3H)-furanone, dihydro-3,4-dihydroxy organosulfate (two isomers); malic acid organosulfate; and threonic acid organosulfate.
Interestingly, some organic compounds observed in the gas phase in chamber experiments (Fig. 1, Table 3) were reported to be ubiquitous in ambient air. High concentrations of formaldehyde and, to a lesser extent, glyoxal, acrolein, PAN, and APAN ,were observed from 13BD oxidation under both acidified and non-acidified seed aerosol across the whole RH range considered in this study. These compounds are ubiquitous in urban ambient samples, so 13BD may be their precursor in areas with considerable 13BD emission. The presence of secondary formaldehyde and acrolein (S-HAPs) and the effect of seed aerosol acidity and RH on their formation highlight the importance of this study mainly for health effect studies.
While it is impossible to cover and analyze all reaction products associated with 13BD oxidation, this study addresses many of the main products that form under various acidified and non-acidified seed aerosol and relative humidity conditions. We identified more than 50 oxygenated organic compounds in the gas and particle phases obtained from chamber oxidation of 13BD. Some reaction products have been reported in the literature, but several others were identified for the first time: glyceric acid-organosulfate, 2(3H)-furanone, dihydro-3,4-dihydroxy-organosulfate (two isomers), threonic acid-organosulfate, 1,2,3,4-butanetetrol nitrosoxy-organosulfate, 1,2,3,4-butanetetrol nitroxy-organosulfate, malic acid-organosulfate, and threonic acid nitroxy-organosulfate. The yield trends are observed in acidified acid organosulfate, 2(3H)-furanone, dihydro-3,4-dihydroxy-organosulfate (two isomers), threonic acid organosulfate, 1,2,3,4-butanetetrol nitrosoxy-organosulfate, 1,2,3,4-butanetetrol nitroxy-organosulfate, malic acid organosulfate, and threonic acid nitroxy-organosulfate. The quantitative analysis showed that glyceric acid; threitol; erythritol; threonic acid; and several dimers, unknowns, and organosulfates were the most abundant components of 13BD SOA under acidified and non-acidified seed conditions. Other components contributing to SOA mass were diols, mono- and dicarboxylic acids, organosulfates, dimers, and nitroxy- and nitrosoxy-organosulfates. Several carboxylic acids, organosulfates, and nitroxy-organosulfates identified in chamber samples were also detected in ambient aerosol samples collected at various sites in Poland. Such consistency reinforces the relevance of the chamber findings even though some components were found only in chamber experiments.
Comparison between experiments conducted with acidified and non-acidified seed aerosol at various RH levels revealed that the acidity of seed aerosol (ER444), the aerosol phase state (ER444 vs. ER666), and the RH (ER444 and ER666) influenced the production of SOA and the number and molecular structure of products formed in the gas and particle phases. Total SOC decreased with increasing RH, but the levels of individual components varied, though the majority followed similar trends to the SOC. Ozone production and conversion efficiency of 13BD depended on the RH, the aerosol phase state, and the acidity of the seed aerosol similarly to SOC. The concentrations of most compounds decreased as RH increased, but few products were most abundant at intermediate RH around 30 % (e.g., malic acid, glyceric acid OS). This is also true for gas-phase formaldehyde and acrolein, whose yields increased significantly at higher RH. However, at high humidity, the difference was relatively small. For the range of RH considered, the acidified seed experiments, in which the aerosol particles were deliquescent, enhanced SOC production compared to non-acidified seed experiments, in which the aerosol particles did not contain aqueous phase. SOC production increased with the acidity of the aqueous phase in the acidified seed experiment ER444. Yields of the individual SOA components followed the same pattern as SOC, while a few were more abundant in non-acidified seed experiments (e.g., unknowns) or behaved in an inconsistent manner. Although the terms “acidified” and “non-acidified” are true for the solutions from which the seeds were atomized, there are far more fundamental differences between the phase states in which species partition to or from (aqueous/solid), which considerably affects their partitioning and formation mechanisms. Therefore, further research is needed to elucidate the mechanisms of their formation and partitioning in the atmosphere, both for dry and deliquescent particles. Overall, the 13BD results are consistent with those reported by Nestorowicz et al. (2018) for isoprene.
The present study employed analytical methods suitable for a wide range of 13BD reaction products in the particle and gas phases at various RH levels and seed aerosol acidity. It provided a solid identification of a wide variety of reaction products, including HAPs, dimers ,and organosulfates, and quantitative changes in their formation as a function of RH and seed aerosol acidity. Certainly, this study includes some uncertainties that need to be addressed in future work. Perhaps the most significant improvement would be the use of authentic standards to better quantify the contribution of individual products to SOA at various RH levels and seed aerosol acidity. This study shows that SOA and SOC consistently increased with decreasing RH and were higher under acidified than non-acidified seed conditions. Similar trends were observed for the majority of reaction products reported in this study. To assess that observation, the LWC and other thermodynamic properties of the aerosol phase were estimated as a function of RH using the thermodynamic model E-AIM. Organic acids could have more profound influence, especially for the non-acidified seed experiment, so the results of our modeling should be taken as a first approximation and verified as soon as the precise stoichiometry of the acid formation and reliable dissociation constants of those acids are available.
13BD does not contribute more to atmospheric aerosol than isoprene, which is emitted to the atmosphere in much larger amounts and plays a significant role in atmospheric SOA modeling. However, 13BD emissions are less understood than isoprene, and current increases in wildfires and acreages burned due to climate issues can increase 13BD emissions and therefore the production of 13BD SOA, which may have implications for air quality, health, and well-being.
The data used in this study can be found at https://catalog.data.gov/dataset?q=jaoui (Jaoui, 2024). All data presented in this paper can be requested by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-25-1401-2025-supplement.
MJ: study conceptualization, standards and chamber sample analysis, data collection, results interpretation, writing (original and revised drafts), and communication. ML: conduction of chamber experiments and data collection and analysis. TEK: supervision, study conceptualization, results interpretation, and editing drafts. KN, KJR, JT, EB, WD, and RS: LC-MS analysis, results interpretation, and editing drafts.
The contact author has declared that none of the authors has any competing interests.
This work has been subjected to the U.S. Environmental Protection Agency's administrative review and approved for publication. The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Mention of trade names does not constitute endorsement or recommendation of a commercial product by the U.S. EPA.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The work of the Polish researchers, i.e., Rafal Szmigielski and Krzysztof J. Rudzinski, was partially supported by funds from the National Science Centre, Poland, through an OPUS21 grant scheme (no. 2021/41/B/ST10/02748).
This paper was edited by Kelvin Bates and reviewed by two anonymous referees.
Acquavella, J. F.: Butadiene epidemiology: a summary of results and outstanding issues, Toxicology, 113, 148–156, https://doi.org/10.1016/0300-483X(96)03440-3, 1996.
Angove, D. E., Fookes, C. J. R., Hynes, R. G., Walters, C. K., and Azzi, M.: The characterisation of secondary organic aerosol formed during the photodecomposition of 1,3-butadiene in air containing nitric oxide, Atmos. Environ., 40, 4597–4607, https://doi.org/10.1016/j.atmosenv.2006.03.046, 2006.
Anttinen-Klemetti, T., Vaaranrinta, R., Mutanen, P., and Peltonen, K.: Inhalation exposure to 1,3-butadiene and styrene in styrene–butadiene copolymer production, Int. J. Hyg. Envir. Heal., 209, 151–158, https://doi.org/10.1016/j.ijheh.2005.09.006, 2006.
Attygalle, A. B., García-Rubio, S., Ta, J., and Meinwald, J.: Collisionally-induced dissociation mass spectra of organic sulfate anions, J. Chem. Soc. Perk. T. 2, 498–506, https://doi.org/10.1039/B009019K, 2001.
Bauwens, M., Stavrakou, T., Müller, J.-F., De Smedt, I., Van Roozendael, M., van der Werf, G. R., Wiedinmyer, C., Kaiser, J. W., Sindelarova, K., and Guenther, A.: Nine years of global hydrocarbon emissions based on source inversion of OMI formaldehyde observations, Atmos. Chem. Phys., 16, 10133–10158, https://doi.org/10.5194/acp-16-10133-2016, 2016.
Berndt, T. and Böge, O.: Atmospheric Reaction of OH Radicals with 1,3-Butadiene and 4-Hydroxy-2-butenal, J. Phys. Chem. A, 111, 12099–12105, https://doi.org/10.1021/jp075349o, 2007.
Black, F., Tejada, S., and Kleindienst, T.: Preparation of automobile organic emission surrogates for photochemical model validation, Atmos. Environ., 32, 2443–2451, https://doi.org/10.1016/S1352-2310(98)00045-4, 1998.
Carlton, A. G., Turpin, B. J., Altieri, K. E., Seitzinger, S., Reff, A., Lim, H.-J., and Ervens, B.: Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments, Atmos. Environ., 41, 7588–7602, https://doi.org/10.1016/j.atmosenv.2007.05.035, 2007.
Carslaw, K. S., Clegg, S. L., and Brimblecombe, P.: A Thermodynamic Model of the System HCl-HNO3-H2SO4-H2O, Including Solubilities of HBr, from < 200 to 328 K, J. Phys. Chem., 99, 11557–11574, https://doi.org/10.1021/j100029a039, 1995.
Chan, A. W. H., Chan, M. N., Surratt, J. D., Chhabra, P. S., Loza, C. L., Crounse, J. D., Yee, L. D., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation, Atmos. Chem. Phys., 10, 7169–7188, https://doi.org/10.5194/acp-10-7169-2010, 2010.
Charlson, R. J., Schwartz, S. E., Hales, J. M., Cess, R. D., Coakley, J. A., Hansen, J. E., and Hofmann, D. J.: Climate Forcing by Anthropogenic Aerosols, Science, 255, 423–430, https://doi.org/10.1126/science.255.5043.423, 1992.
Chen, T., Chu, B., Ma, Q., Zhang, P., Liu, J., and He, H.: Effect of relative humidity on SOA formation from aromatic hydrocarbons: Implications from the evolution of gas- and particle-phase species, Sci. Total Environ., 773, 145015, https://doi.org/10.1016/j.scitotenv.2021.145015, 2021.
Chen, W.-Q. and Zhang, X.-Y.: 1,3-Butadiene: a ubiquitous environmental mutagen and its associations with diseases, Genes and Environment, 44, 3, https://doi.org/10.1186/s41021-021-00233-y, 2022.
Cheng, C. T., Chan, M. N., and Wilson, K. R.: Importance of Unimolecular HO2 Elimination in the Heterogeneous OH Reaction of Highly Oxygenated Tartaric Acid Aerosol, J. Phys. Chem. A, 120, 5887–5896, https://doi.org/10.1021/acs.jpca.6b05289, 2016.
Cho, C., Clair, J. M. S., Liao, J., Wolfe, G. M., Jeong, S., Kang, D. i., Choi, J., Shin, M.-H., Park, J., Park, J.-H., Fried, A., Weinheimer, A., Blake, D. R., Diskin, G. S., Ullmann, K., Hall, S. R., Brune, W. H., Hanisco, T. F., and Min, K.-E.: Evolution of formaldehyde (HCHO) in a plume originating from a petrochemical industry and its volatile organic compounds (VOCs) emission rate estimation, Elementa: Science of the Anthropocene, 9, 00015, https://doi.org/10.1525/elementa.2021.00015, 2021.
Clegg, S. L. and Brimblecombe, P.: Application of a Multicomponent Thermodynamic Model to Activities and Thermal Properties of 0–40 mol kg−1 Aqueous Sulfuric Acid from < 200 to 328 K, J. Chem. Eng. Data, 40, 43–64, https://doi.org/10.1021/je00017a012, 1995.
Clegg, S. L. and Pitzer, K. S.: Thermodynamics of multicomponent, miscible, ionic solutions: generalized equations for symmetrical electrolytes. [Erratum to document cited in CA116(20):202049x], J. Phys. Chem., 98, 1368–1368, https://doi.org/10.1021/j100055a052, 1994.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: Thermodynamic Model of the System H+–NH–Na+-SO–NO–Cl−–H2O at 298.15 K, J. Phys. Chem. A, 102, 2155–2171, https://doi.org/10.1021/jp973043j, 1998.
Clegg, S. L., Pitzer, K. S., and Brimblecombe, P.: Thermodynamics of multicomponent, miscible, ionic solutions. Mixtures including unsymmetrical electrolytes, J. Phys. Chem., 96, 9470–9479, https://doi.org/10.1021/j100202a074, 1992.
Cocker III, D. R., Clegg, S. L., Flagan, R. C., and Seinfeld, J. H.: The effect of water on gas–particle partitioning of secondary organic aerosol. Part I: á-pinene/ozone system, Atmos. Environ., 35, 6049–6072, https://doi.org/10.1016/S1352-2310(01)00404-6, 2001.
Cooke, M. E., Armstrong, N. C., Fankhauser, A. M., Chen, Y., Lei, Z., Zhang, Y., Ledsky, I. R., Turpin, B. J., Zhang, Z., Gold, A., McNeill, V. F., Surratt, J. D., and Ault, A. P.: Decreases in Epoxide-Driven Secondary Organic Aerosol Production under Highly Acidic Conditions: The Importance of Acid–Base Equilibria, Environ. Sci. Technol., 58, 10675–10684, https://doi.org/10.1021/acs.est.3c10851, 2024.
Czoschke, N. M., Jang, M., and Kamens, R. M.: Effect of acidic seed on biogenic secondary organic aerosol growth, Atmos. Environ., 37, 4287–4299, https://doi.org/10.1016/S1352-2310(03)00511-9, 2003.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
Deng, Y., Inomata, S., Sato, K., Ramasamy, S., Morino, Y., Enami, S., and Tanimoto, H.: Temperature and acidity dependence of secondary organic aerosol formation from α-pinene ozonolysis with a compact chamber system, Atmos. Chem. Phys., 21, 5983–6003, https://doi.org/10.5194/acp-21-5983-2021, 2021.
Dollard, G. J., Dore, C. J., and Jenkin, M. E.: Ambient concentrations of 1,3-butadiene in the UK, Chem.-Biol. Interact., 135–136, 177–206, https://doi.org/10.1016/S0009-2797(01)00190-9, 2001.
Doyle, M., Sexton, K. G., Jeffries, H., Bridge, K., and Jaspers, I.: Effects of 1,3-Butadiene, Isoprene, and Their Photochemical Degradation Products on Human Lung Cells, Environ. Health Persp., 112, 1488–1495, https://doi.org/10.1289/ehp.7022, 2004.
Duporté, G., Flaud, P. M., Kammer, J., Geneste, E., Augagneur, S., Pangui, E., Lamkaddam, H., Gratien, A., Doussin, J. F., Budzinski, H., Villenave, E., and Perraudin, E.: Experimental Study of the Formation of Organosulfates from á-Pinene Oxidation. 2. Time Evolution and Effect of Particle Acidity, J. Phys. Chem.-A, 124, 409–421, https://doi.org/10.1021/acs.jpca.9b07156, 2020.
Eatough, D. J., Hansen, L. D., and Lewis, E. A.: The chemical characterization of environmental tobacco smoke, Environ. Technol., 11, 1071–1085, https://doi.org/10.1080/09593339009384961, 1990.
Edney, E. O., Driscoll, D. J., Speer, R. E., Weathers, W. S., Kleindienst, T. E., Li, W., and Smith, D. F.: Impact of aerosol liquid water on secondary organic aerosol yields of irradiated toluene/propylene/NOx/(NH4)2SO4/air mixtures, Atmos. Environ., 34, 3907–3919, https://doi.org/10.1016/S1352-2310(00)00174-6, 2000.
Faust, J. A., Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Role of Aerosol Liquid Water in Secondary Organic Aerosol Formation from Volatile Organic Compounds, Environ. Sci. Technol., 51, 1405–1413, https://doi.org/10.1021/acs.est.6b04700, 2017.
Fletcher, C. G., Kravitz, B., and Badawy, B.: Quantifying uncertainty from aerosol and atmospheric parameters and their impact on climate sensitivity, Atmos. Chem. Phys., 18, 17529–17543, https://doi.org/10.5194/acp-18-17529-2018, 2018.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res.-Atmos., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995.
Healy, R. M., Temime, B., Kuprovskyte, K., and Wenger, J. C.: Effect of Relative Humidity on Gas/Particle Partitioning and Aerosol Mass Yield in the Photooxidation of p-Xylene, Environ. Sci. Technol., 43, 1884–1889, https://doi.org/10.1021/es802404z, 2009.
Hinks, M. L., Montoya-Aguilera, J., Ellison, L., Lin, P., Laskin, A., Laskin, J., Shiraiwa, M., Dabdub, D., and Nizkorodov, S. A.: Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene, Atmos. Chem. Phys., 18, 1643–1652, https://doi.org/10.5194/acp-18-1643-2018, 2018.
Hoyle, C. R., Boy, M., Donahue, N. M., Fry, J. L., Glasius, M., Guenther, A., Hallar, A. G., Huff Hartz, K., Petters, M. D., Petäjä, T., Rosenoern, T., and Sullivan, A. P.: A review of the anthropogenic influence on biogenic secondary organic aerosol, Atmos. Chem. Phys., 11, 321–343, https://doi.org/10.5194/acp-11-321-2011, 2011.
Hu, G., Xu, Y., and Jia, L.: Effects of relative humidity on the characterization of a photochemical smog chamber, J. Environ. Sci., 23, 2013–2018, 2011.
Hurst, H. E.: Toxicology of 1,3-Butadiene, Chloroprene, and Isoprene, in: Reviews of Environmental Contamination and Toxicology: Continuation of Residue Reviews, edited by: Whitacre, D. M., Ware, G. W., Nigg, H. N., Doerge, D. R., Albert, L. A., de Voogt, P., Gerba, C. P., Hutzinger, O., Knaak, J. B., Mayer, F. L., Morgan, D. P., Park, D. L., Tjeerdema, R. S., Yang, R. S. H., and Gunther, F. A., Springer New York, New York, NY, 131–179, https://doi.org/10.1007/978-0-387-35368-5_6, 2007.
IARC: Preamble, in: IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, WHO, Lyon, France, 2015.
IPCS: 1,3-Butadiene: Human health aspects, WHO, Geneva, ISBN 92 4 153030 8, ISSN 1020-6167, https://www.inchem.org/documents/cicads/cicads/cicad30.htm (last access: 14 November 2024), 2001.
Jaoui, M.: Datasets, Data.gov, https://catalog.data.gov/dataset?q=jaoui, last access: 14 November 2024.
Jaoui, M., Kleindienst, T. E., Lewandowski, M., and Edney, E. O.: Identification and Quantification of Aerosol Polar Oxygenated Compounds Bearing Carboxylic or Hydroxyl Groups. 1. Method Development, Anal. Chem., 76, 4765–4778, https://doi.org/10.1021/ac049919h, 2004.
Jaoui, M., Edney, E. O., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Surratt, J. D., and Seinfeld, J. H.: Formation of secondary organic aerosol from irradiated α-pinene/toluene/NOx mixtures and the effect of isoprene and sulfur dioxide, J. Geophys. Res.-Atmos., 113, D09303, https://doi.org/10.1029/2007JD009426, 2008.
Jaoui, M., Lewandowski, M., Docherty, K., Offenberg, J. H., and Kleindienst, T. E.: Atmospheric oxidation of 1,3-butadiene: characterization of gas and aerosol reaction products and implications for PM2.5, Atmos. Chem. Phys., 14, 13681–13704, https://doi.org/10.5194/acp-14-13681-2014, 2014.
Jaoui, M., Lewandowski, M., Offenberg, J. H., Colon, M., Docherty, K. S., and Kleindienst, T. E.: Characterization of aerosol nitroaromatic compounds: Validation of an experimental method, J. Mass Spectrom., 53, 680–692, https://doi.org/10.1002/jms.4199, 2018.
Jaoui, M., Szmigielski, R., Nestorowicz, K., Kolodziejczyk, A., Sarang, K., Rudzinski, K. J., Konopka, A., Bulska, E, Lewandowski, M., and Kleindienst, T. E.: Organic hydroxy acids as highly oxygenated molecular (HOM) tracers for aged isoprene aerosol, Environ. Sci. Technol., 53, 14516–14527, https://doi.org/10.1021/acs.est.9b05075, 2019.
Jia, L. and Xu, Y.: Effects of Relative Humidity on Ozone and Secondary Organic Aerosol Formation from the Photooxidation of Benzene and Ethylbenzene, Aerosol Sci. Tech., 48, 1-12, https://doi.org/10.1080/02786826.2013.847269, 2014.
Jia, L. and Xu, Y.: Different roles of water in secondary organic aerosol formation from toluene and isoprene, Atmos. Chem. Phys., 18, 8137–8154, https://doi.org/10.5194/acp-18-8137-2018, 2018.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kamens, R. M., Zhang, H., Chen, E. H., Zhou, Y., Parikh, H. M., Wilson, R. L., Galloway, K. E., and Rosen, E. P.: Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: Water and particle seed effects, Atmos. Environ., 45, 2324–2334, https://doi.org/10.1016/j.atmosenv.2010.11.007, 2011.
Kao, A. S.: Formation and Removal Reactions of Hazardous Air Pollutants, Air & Waste, 44, 683–696, https://doi.org/10.1080/1073161X.1994.10467272, 1994.
Kleindienst, T. E., Edney, E. O., Lewandowski, M., Offenberg, J. H., and Jaoui, M.: Secondary Organic Carbon and Aerosol Yields from the Irradiations of Isoprene and α-Pinene in the Presence of NOx and SO2, Environ. Sci. Technol., 40, 3807–3812, https://doi.org/10.1021/es052446r, 2006.
Klodt, A. L., Aiona, P. K., MacMillan, A. C., Lee, H. J., Zhang, X., Helgestad, T., Novak, G. A., Lin, P., Laskin, J., Laskin, A., Bertram, T. H., Cappa, C. D., and Nizkorodov, S. A.: Effect of relative humidity, NOx, and ammonia on the physical properties of naphthalene secondary organic aerosols, Environmental Science: Atmospheres, 3, 991–1007, https://doi.org/10.1039/D3EA00033H, 2023.
Kramp, F. and Paulson, S. E.: The gas phase reaction of ozone with 1,3-butadiene: formation yields of some toxic products, Atmos. Environ., 34, 35–43, https://doi.org/10.1016/S1352-2310(99)00327-1, 2000.
Lei, Z., Chen, Y., Zhang, Y., Cooke, M. E., Ledsky, I. R., Armstrong, N. C., Olson, N. E., Zhang, Z., Gold, A., Surratt, J. D., and Ault, A. P.: Initial pH Governs Secondary Organic Aerosol Phase State and Morphology after Uptake of Isoprene Epoxydiols (IEPOX), Environ. Sci. Technol., 56, 10596–10607, https://doi.org/10.1021/acs.est.2c01579, 2022.
Lewandowski, M., Jaoui, M., Offenberg, J. H., Krug, J. D., and Kleindienst, T. E.: Atmospheric oxidation of isoprene and 1,3-butadiene: influence of aerosol acidity and relative humidity on secondary organic aerosol, Atmos. Chem. Phys., 15, 3773–3783, https://doi.org/10.5194/acp-15-3773-2015, 2015.
Liggio, J., Li, S.-M., and McLaren, R.: Heterogeneous Reactions of Glyoxal on Particulate Matter: Identification of Acetals and Sulfate Esters, Environ. Sci. Technol., 39, 1532–1541, https://doi.org/10.1021/es048375y, 2005.
Liu, S., Jiang, X., Tsona, N. T., Lv, C., and Du, L.: Effects of NOx, SO2 and RH on the SOA formation from cyclohexene photooxidation, Chemosphere, 216, 794–804, https://doi.org/10.1016/j.chemosphere.2018.10.180, 2019.
Liu, X., Jeffries, H. E., and Sexton, K. G.: Hydroxyl radical and ozone initiated photochemical reactions of 1,3-butadiene, Atmos. Environ., 33, 3005–3022, https://doi.org/10.1016/S1352-2310(99)00078-3, 1999.
Liu, Y., Li, Q., Su, G., Wei, D., Zheng, M., Gao, L., Liu, W., and Liu, G.: Photochemical conversion of toluene in simulated atmospheric matrix and characterization of large molecular weight products by +APPI FT-ICR MS, Sci. Total Environ., 649, 111–119, https://doi.org/10.1016/j.scitotenv.2018.08.293, 2019.
Luo, H., Guo, Y., Shen, H., Huang, D. D., Zhangabcef, Y., and Zhao, D.: Effect of relative humidity on the molecular composition of secondary organic aerosols from a-pinene ozonolysis, Environmental Science: Atmospheres, 4, 519–530, https://doi.org/10.1039/D3EA00149K, 2024.
Majewska, M., Khan, F., Pieta, I. S., Wróblewska, A., Szmigielski, R., and Pieta, P.: Toxicity of selected airborne nitrophenols on eukaryotic cell membrane models, Chemosphere, 266, 128996, https://doi.org/10.1016/j.chemosphere.2020.128996, 2021.
Manisalidis, I., Stavropoulou, E., Stavropoulos, A., and Bezirtzoglou, E.: Environmental and Health Impacts of Air Pollution: A Review, Frontiers in Public Health, 8, 14, https://doi.org/10.3389/fpubh.2020.00014, 2020.
Myhre, G., Lund Myhre, C. E., Samset, B. H., and Storelvmo, T.: Aerosols and their Relation to Global Climate and Climate Sensitivity, Nature Education Knowledge, 4, 7, 2013.
Nestorowicz, K., Jaoui, M., Rudzinski, K. J., Lewandowski, M., Kleindienst, T. E., Spólnik, G., Danikiewicz, W., and Szmigielski, R.: Chemical composition of isoprene SOA under acidic and non-acidic conditions: effect of relative humidity, Atmos. Chem. Phys., 18, 18101–18121, https://doi.org/10.5194/acp-18-18101-2018, 2018.
Nguyen, T. B., Roach, P. J., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 11, 6931–6944, https://doi.org/10.5194/acp-11-6931-2011, 2011.
Notario, A., Le Bras, G., and Mellouki, A.: Kinetics of Cl atom reactions with butadienes including isoprene, Chem. Phys. Lett., 281, 421–425, https://doi.org/10.1016/S0009-2614(97)01303-1, 1997.
Noziere, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decesari, S., Finessi, E., Glasius, M., Grgic, I., Hamilton, J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The molecular identification of organic compounds in the atmosphere: state of the art and challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015.
Pankow, J. F., Luo, W., Tavakoli, A. D., Chen, C., and Isabelle, L. M.: Delivery Levels and Behavior of 1,3-Butadiene, Acrylonitrile, Benzene, and Other Toxic Volatile Organic Compounds in Mainstream Tobacco Smoke from Two Brands of Commercial Cigarettes, Chem. Res. Toxicol., 17, 805–813, https://doi.org/10.1021/tx0342316, 2004.
Penn, A. and Snyder, C. A.: 1,3 Butadiene, a Vapor Phase Component of Environmental Tobacco Smoke, Accelerates Arteriosclerotic Plaque Development, Circulation, 93, 552–557, https://doi.org/10.1161/01.CIR.93.3.552, 1996.
Pye, H. O. T., Nenes, A., Alexander, B., Ault, A. P., Barth, M. C., Clegg, S. L., Collett Jr., J. L., Fahey, K. M., Hennigan, C. J., Herrmann, H., Kanakidou, M., Kelly, J. T., Ku, I.-T., McNeill, V. F., Riemer, N., Schaefer, T., Shi, G., Tilgner, A., Walker, J. T., Wang, T., Weber, R., Xing, J., Zaveri, R. A., and Zuend, A.: The acidity of atmospheric particles and clouds, Atmos. Chem. Phys., 20, 4809–4888, https://doi.org/10.5194/acp-20-4809-2020, 2020.
Riva, M., Bell, D. M., Hansen, A.-M. K., Drozd, G. T., Zhang, Z., Gold, A., Imre, D., Surratt, J. D., Glasius, M., and Zelenyuk, A.: Effect of Organic Coatings, Humidity and Aerosol Acidity on Multiphase Chemistry of Isoprene Epoxydiols, Environ. Sci. Technol., 50, 5580–5588, https://doi.org/10.1021/acs.est.5b06050, 2016.
Rudziński, K. J., Gmachowski, L., and Kuznietsova, I.: Reactions of isoprene and sulphoxy radical-anions – a possible source of atmospheric organosulphites and organosulphates, Atmos. Chem. Phys., 9, 2129–2140, https://doi.org/10.5194/acp-9-2129-2009, 2009.
Saleh, R., Donahue, N. M., and Robinson, A. L.: Time Scales for Gas-Particle Partitioning Equilibration of Secondary Organic Aerosol Formed from Alpha-Pinene Ozonolysis, Environ. Sci. Technol., 47, 5588–5594, https://doi.org/10.1021/es400078d, 2013.
Sato, K.: Detection of nitrooxypolyols in secondary organic aerosol formed from the photooxidation of conjugated dienes under high-NOx conditions, Atmos. Environ., 42, 6851–6861, https://doi.org/10.1016/j.atmosenv.2008.05.010, 2008.
Sato, K., Nakao, S., Clark, C. H., Qi, L., and Cocker III, D. R.: Secondary organic aerosol formation from the photooxidation of isoprene, 1,3-butadiene, and 2,3-dimethyl-1,3-butadiene under high NOx conditions, Atmos. Chem. Phys., 11, 7301–7317, https://doi.org/10.5194/acp-11-7301-2011, 2011.
Scheffe, R. D., Strum, M., Phillips, S. B., Thurman, J., Eyth, A., Fudge, S., Morris, M., Palma, T., and Cook, R.: Hybrid Modeling Approach to Estimate Exposures of Hazardous Air Pollutants (HAPs) for the National Air Toxics Assessment (NATA), Environ. Sci. Technol., 50, 12356–12364, https://doi.org/10.1021/acs.est.6b04752, 2016.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Smith, D. F., Kleindienst, T. E., and Hudgens, E. E.: Improved high-performance liquid chromatographic method for artifact-free measurements of aldehydes in the presence of ozone using 2,4-dinitrophenylhydrazine, J. Chromatogr. A, 483, 431–436, https://doi.org/10.1016/S0021-9673(01)93146-2, 1989.
Sorsa, M., Peltonen, K., Anderson, D., Demopoulos, N. A., Neumann, H., and Osterman Golkar, S.: Assessment of environmental and occupational exposures to butadiene as a model for risk estimation of petrochemical emission, Mutagenesis, 11, 9–17, https://doi.org/10.1093/mutage/11.1.9, 1996.
Spolnik, G., Wach, P., Rudzinski, K. J., Skotak, K., Danikiewicz, W., and Szmigielski, R.: Improved UHPLC-MS/MS Methods for Analysis of Isoprene-Derived Organosulfates, Anal. Chem., 90, 3416–3423, https://doi.org/10.1021/acs.analchem.7b05060, 2018.
Srivastava, D., Vu, T. V., Tong, S., Shi, Z., and Harrison, R. M.: Formation of secondary organic aerosols from anthropogenic precursors in laboratory studies, npj Climate and Atmospheric Science, 5, 22, https://doi.org/10.1038/s41612-022-00238-6, 2022.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Szmigielski, R.: Evidence for C5 organosulfur secondary organic aerosol components from in-cloud processing of isoprene: Role of reactive SO4 and SO3 radicals, Atmos. Environ., 130, 14–22, https://doi.org/10.1016/j.atmosenv.2015.10.072, 2016.
Tanimoto, H. and Akimoto, H.: A new peroxycarboxylic nitric anhydride identified in the atmosphere: CH2=CHC(O)OONO2 (APAN), Geophys. Res. Lett., 28, 2831–2834, https://doi.org/10.1029/2001GL012998, 2001.
Thomsen, D., Iversen, E. M., Skønager, J. T., Luo, Y., Li, L., Roldin, P., Priestley, M., Pedersen, H. B., Hallquist, M., Ehn, M., Bilde, M., and Glasius, M.: The effect of temperature and relative humidity on secondary organic aerosol formation from ozonolysis of Δ3-carene, Environmental Science: Atmospheres, 4, 88, https://doi.org/10.1039/D3EA00128H, 2024.
Thornton-Manning, J. R., Dahl, A. R., Bechtold, W. E., Griffith Jr., W. C., and Henderson, R. F.: Comparison of the disposition of butadiene epoxides in Sprague–Dawley rats and B6C3F1 mice following a single and repeated exposures to 1,3-butadiene via inhalation, Toxicology, 123, 125–134, https://doi.org/10.1016/S0300-483X(97)00112-1, 1997.
Tilgner, A., Schaefer, T., Alexander, B., Barth, M., Collett Jr., J. L., Fahey, K. M., Nenes, A., Pye, H. O. T., Herrmann, H., and McNeill, V. F.: Acidity and the multiphase chemistry of atmospheric aqueous particles and clouds, Atmos. Chem. Phys., 21, 13483–13536, https://doi.org/10.5194/acp-21-13483-2021, 2021.
U.S. EPA: Locating and estimating air emissions from sources of 1,3-butadiene, United States Environmental Protection Agency, Office of Air Quality Planning and Standards, Research Triangle Park, NC, EPA-454/R-96-008, 1996.
U.S. EPA: Health Assessment Of 1,3-Butadiene, U.S. Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment, Washington Office, Washington, DC, EPA/600/P-98/001F, 2002.
Wach, P., Spolnik, G., Surratt, J. D., Blaziak, K., Rudzinski, K. J., Lin, Y. H., Maenhaut, W., Danikiewicz, W., Claeys, M., and Szmigielski, R.: Structural Characterization of Lactone-Containing MW 212 Organosulfates Originating from Isoprene Oxidation in Ambient Fine Aerosol, Environ. Sci. Technol., 54, 1415–1424, https://doi.org/10.1021/acs.est.9b06190, 2020.
Wang, Q., Li, Q., Wei, D., Su, G., Wu, M., Li, C., Sun, B., and Dai, L.: Photochemical reactions of 1,3-butadiene with nitrogen oxide in different matrices: Kinetic behavior, humidity effect, product and mechanisms, Sci. Total Environ., 721, 137747, https://doi.org/10.1016/j.scitotenv.2020.137747, 2020.
Wexler, A. S. and Clegg, S. L.: Atmospheric aerosol models for systems including the ions H+, NH, Na+, SO, NO, Cl−, Br−, and H2O, J. Geophys. Res.-Atmos., 107, ACH 14-11–ACH 14-14, https://doi.org/10.1029/2001JD000451, 2002.
Wolfe, G. M., Kaiser, J., Hanisco, T. F., Keutsch, F. N., de Gouw, J. A., Gilman, J. B., Graus, M., Hatch, C. D., Holloway, J., Horowitz, L. W., Lee, B. H., Lerner, B. M., Lopez-Hilifiker, F., Mao, J., Marvin, M. R., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., Thornton, J. A., Veres, P. R., and Warneke, C.: Formaldehyde production from isoprene oxidation across NOx regimes, Atmos. Chem. Phys., 16, 2597–2610, https://doi.org/10.5194/acp-16-2597-2016, 2016.
Ye, Y., Galbally, I. E., Weeks, I. A., Duffy, B. L., and Nelson, P. F.: Evaporative emissions of 1,3-butadiene from petrol-fuelled motor vehicles, Atmos. Environ., 32, 2685–2692, https://doi.org/10.1016/S1352-2310(97)00379-8, 1998.
Zhang, H., Surratt, J. D., Lin, Y. H., Bapat, J., and Kamens, R. M.: Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions, Atmos. Chem. Phys., 11, 6411–6424, https://doi.org/10.5194/acp-11-6411-2011, 2011.
Zhang, J., Shrivastava, M., Zelenyuk, A., Zaveri, R. A., Surratt, J. D., Riva, M., Bell, D., and Glasius, M.: Observationally Constrained Modeling of the Reactive Uptake of Isoprene-Derived Epoxydiols under Elevated Relative Humidity and Varying Acidity of Seed Aerosol Conditions, ACS Earth Space Chem., 7, 788–799, https://doi.org/10.1021/acsearthspacechem.2c00358, 2023.
Zhang, T., Shen, Z. X., Su, H., Liu, S. X., Zhou, J. M., Zhao, Z. Z., Wang, Q. Y., Prévôt, A. S. H., and Cao, J. J.: Effects of Aerosol Water Content on the formation of secondary inorganic aerosol during a Winter Heavy PM2.5 Pollution Episode in Xi'an, China, Atmos. Environ., 252, 118304, https://doi.org/10.1016/j.atmosenv.2021.118304, 2021.
Zhou, Y., Zhang, H., Parikh, H. M., Chen, E. H., Rattanavaraha, W., Rosen, E. P., Wang, W., and Kamens, R. M.: Secondary organic aerosol formation from xylenes and mixtures of toluene and xylenes in an atmospheric urban hydrocarbon mixture: Water and particle seed effects (II), Atmos. Environ., 45, 3882–3890, https://doi.org/10.1016/j.atmosenv.2010.12.048, 2011.