the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Apr 2024
| 22 Apr 2024
Impact of HO2∕RO2 ratio on highly oxygenated α-pinene photooxidation products and secondary organic aerosol formation potential
Yarê Baker
Sungah Kang
Rongrong Wu
Jian Xu
Annika Zanders
Quanfu He
Thorsten Hohaus
Till Ziehm
Veronica Geretti
Thomas J. Bannan
Simon P. O'Meara
Aristeidis Voliotis
Mattias Hallquist
Gordon McFiggans
Sören R. Zorn
Andreas Wahner
Highly oxygenated molecules (HOMs) from the atmospheric oxidation of biogenic volatile organic compounds are important contributors to secondary organic aerosol (SOA). Organic peroxy radicals (RO2) and hydroperoxy radicals (HO2) are key species influencing the HOM product distribution. In laboratory studies, experimental requirements often result in overemphasis on RO2 cross-reactions compared to reactions of RO2 with HO2. We analyzed the photochemical formation of HOMs from α-pinene and their potential to contribute to SOA formation under high (≈1/1) and low (≈1/100) conditions. As > 1 is prevalent in the daytime atmosphere, sufficiently high is crucial to mimic atmospheric conditions and to prevent biases by low on the HOM product distribution and thus SOA yield. Experiments were performed under steady-state conditions in the new, continuously stirred tank reactor SAPHIR-STAR at Forschungszentrum Jülich. The ratio was increased by adding CO while keeping the OH concentration constant. We determined the HOM's SOA formation potential, considering its fraction remaining in the gas phase after seeding with (NH4)2SO4 aerosol. An increase in led to a reduction in SOA formation potential, with the main driver being a ∼ 60 % reduction in HOM-accretion products. We also observed a shift in HOM-monomer functionalization from carbonyl to hydroperoxide groups. We determined a reduction of the HOM's SOA formation potential by ∼ 30 % at ≈1/1 compared to ≈ 1/100. Particle-phase observations measured a similar decrease in SOA mass and yield. Our study shows that too low ratios compared to the atmosphere can lead to an overestimation of SOA yields.
- Article
(3642 KB) - Full-text XML
-
Supplement
(800 KB) - BibTeX
- EndNote




Dedicated to Astrid Kiendler-Scharr, deceased 6 February 2023.
In the atmosphere, highly oxidized products from the oxidation of biogenic or anthropogenic volatile organic compounds (VOCs) are an important source of secondary organic aerosol (SOA) (Roldin et al., 2019; Mohr et al., 2019). SOA is an important contributor to the overall ambient aerosol and of interest because of its impact on climate, visibility and human health (Hallquist et al., 2009).
Recently, many studies (Pullinen et al., 2020; Berndt et al., 2016; Bianchi et al., 2017) have focused on understanding the oxidation pathways of VOCs that yield highly oxygenated molecules (HOMs), as these are expected to be of low enough volatility to condense into the particle phase. One important tool for the investigation of VOC degradation and SOA formation is the utilization of experiments in atmospheric simulation chambers (Hidy, 2019). Such experiments have also helped to elucidate key processes in the HOM formation, i.e., the process of autoxidation.
After an initial oxidant attack and the formation of a peroxy radical (RO2), autoxidation adds oxygen to the molecule via an internal H shift to the peroxy group, forming a hydroxy peroxide group and an alkyl radical, to which O2 immediately adds, re-establishing the peroxy functionality. This process can be repeated multiple times, yielding almost instantaneously highly oxygenated peroxy radicals (HOM-RO2) which are terminated to a series of HOM closed-shell products (Bianchi et al., 2019; Ehn et al., 2014; Crounse et al., 2013).
Chamber studies often work with a singular compound and operate at higher precursor concentrations than those observed in the atmosphere for experimental reasons. These experiments cannot represent the complex mixture of VOCs and oxidized VOCs present in the atmosphere (McFiggans et al., 2019). Higher precursor concentrations can lead, per se, to higher SOA yields than observed in the atmosphere (a well-characterized phenomenon; see Henry et al., 2012, and Shilling et al., 2009) and to a general preference for higher-order processes which may not be important in the atmosphere. One example is that chamber studies tend to overestimate the role of cross-reactions between organic peroxy radicals (RO2), owing to high precursor concentrations of a single VOC. In chambers, reactions of HOM-RO2 with other organic peroxy radicals terminate the autoxidation chain, leading typically to multifunctional carbonyl and alcohol compounds. In comparison, in the atmosphere termination by HO2 is more likely, leading to multifunctional hydroperoxides. In the presence of sufficient NO, termination to multifunctional organic nitrates may be more important (Schervish and Donahue, 2021).
Another possible termination reaction of HOM-RO2 with HOM-RO2 and less oxidized RO2 leads to the formation of accretion products, which are expected to be extremely low volatility organic compounds (ELVOCs) and are therefore expected to contribute to new particle formation and SOA formation (Ehn et al., 2014; Berndt et al., 2018). Schervish and Donahue (2021) raised awareness that chamber studies could overestimate the SOA formation potential from the oxidation of terpenes such as α-pinene compared to the atmosphere because of missing HO2 and low-molecular-weight RO2 (e.g., CH3O2), which favors accretion product formation. Previous studies of VOC ozonolysis with different OH scavengers by Docherty and Ziemann (2003) and Keywood et al. (2004) indicated a significant impact of the ratio on SOA yields.
In chamber studies, the use of higher VOC concentrations is often an unavoidable necessity either to match the sensitivity of the analytical instrumentation or to overcome chamber-related effects. The question remains, how can conditions dictated by the chamber be steered towards more realistic chemical pathways and higher atmospheric relevance?
In this study, we address this overestimated importance of peroxy radical cross-reactions. We studied the photooxidation of α-pinene in a series of steady-state experiments in the newly built, continuously stirred tank reactor SAPHIR-STAR (a modernized version of JPAC; see Mentel et al., 2009).
We compared two experimental conditions, a pure α-pinene photooxidation case, leading to low ratios and high importance of RO2 cross-reactions, and a high case, representing more atmospherically relevant conditions with high importance of RO2+ HO2 reactions. One important concept of the conducted experiments is the constant OH availability to α-pinene in order to prevent effects of different oxidant levels and allow for a direct comparison between the two chemical regimes. To this end, the OH concentration in the experiments was adjusted to keep the α-pinene OH turnover constant and to avoid changes due to oxidant scavenging.
Furthermore, the addition of seed particles ((NH4)2SO4) allowed us to observe the condensation behavior of the HOM products and to compare our gas-phase observations directly with particulate-phase measurements of the condensed organic mass.
In this study, we will address two central questions. How does the shift in impact the oxidation mechanism of α-pinene, especially the HOM formation pathway? And what is the subsequent impact on the SOA formation potential of the α-pinene photooxidation system? As the central analysis tool, we will use high-resolution time-of-flight mass spectrometry with chemical ionization (HR-TOF-CIMS) with nitrate (NO reagent ions as this ionization scheme is selective towards HOM compounds (Hyttinen et al., 2018).
2.1 Generic α-pinene HOM peroxy radical chemistry
The chemical mechanistic information for the basic oxidation scheme of α-pinene was taken from the Master Chemical Mechanism (MCM) v3.3.1 (Jenkin et al., 1997; Saunders et al., 2003) (http://mcm.york.ac.uk, last access: 20 October 2023). The main peroxy radicals expected from α-pinene photooxidation are C10H17Ox and C10H15Ox. C10H17Ox is formed by the addition of OH to α-pinene, followed by O2 (starting RO2: C10H17O3) (MCM v3.3.1 (Jenkin et al., 1997; Saunders et al., 2003)). Studies showed that the autoxidation can start from C10H17O3 with the four-member ring in α-pinene opened (Berndt, 2021; Xu et al., 2019).
For C10H15Ox, the autoxidation chain is assumed to start with C10H15O4, which can be formed directly from ozonolysis via the vinyl hydroperoxide path (Johnson and Marston, 2008; Iyer et al., 2021) or via H abstraction from first-generation oxidation products such as pinonaldehyde (C10H16O2) (MCM v3.3.1; Jenkin et al., 1997; Saunders et al., 2003; Fantechi et al., 2002). A recent study suggests direct H abstraction by OH from α-pinene (Shen et al., 2022) as a starting point for the autoxidation chain.
The autoxidation process is rapid with H-shift rates of about 0.01–0.1 s−1 and faster (Piletic and Kleindienst, 2022; Berndt, 2021; Xu et al., 2019; Vereecken et al., 2007). The autoxidation chain will run quickly, adding more oxygen to the molecule, until bimolecular termination reactions are able to compete with all available H-shift rates. The rate of an H shift is determined by the hydrogen's position in relation to the peroxy radical and the functional groups near the hydrogen and peroxy radical (Otkjaer et al., 2018; Vereecken and Nozière, 2020). In the absence of NOx, the peroxy radicals have two major bimolecular termination channels: the reaction with another RO2 or with HO2. A third pathway is the intramolecular termination (Rissanen et al., 2014).
Based on the considerations above, we apply a simplified generic reaction scheme to analyze our observations. Figure 1 shows an overview of the reaction pathways for the main peroxy radical families in the α-pinene photooxidation and the resulting product groups and families. The compounds can be separated into four classes; peroxy radicals (HOM-RO2), monomers (HOM-Mon), accretion products (HOM-Acc) and fragments (HOM-Frag). The HOM-RO2 class consists of all detected HOM-RO2, with special focus on the analysis of the C10 HOM-RO2 families. The HOM-Mon class contains the closed-shell HOM-C10 products. The compounds in the fragment class contain less than 10 carbon atoms, while all HOM-Acc compounds contain more than 10 carbon atoms. The compound classes are further divided into groups and families. Here, the term group is used for compounds with the same carbon number, while a family contains all compounds with the same carbon and hydrogen number but a varying oxygen number.
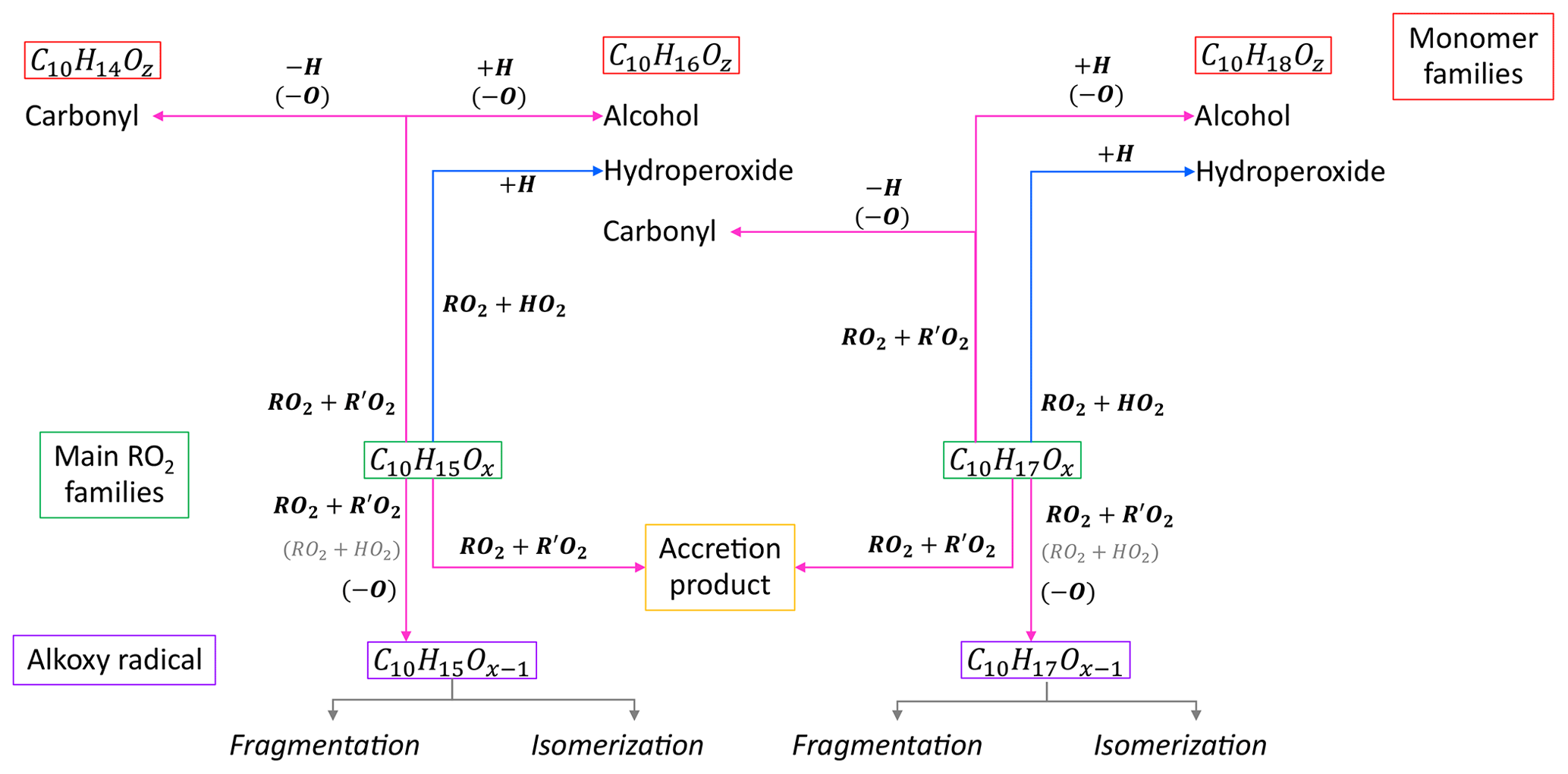
The termination of RO2 with HO2 will lead to hydroperoxide formation:
In the case of C10H15Ox, Reaction (R1) will lead to multifunctional C10H16Oz hydroperoxides (wherein the notation “hydroperoxides”, “carbonyls”, or “alcohols”, etc., here and in the following relates to the functionality of the group formed by the termination reaction). For C10H17Ox, it will lead to the formation of C10H18Oz hydroperoxides. The termination via RO2+RO2 can either result in the formation of accretion products or in the formation of carbonyls and alcohols. For the accretion product formation, it is assumed that the two RO2 chemically bond, eliminating O2 from the molecule:
Recombination reactions of the main peroxy radical families C10H15Ox and C10H17Ox lead to the product families C20H30Oz (combination of two C10H15Ox), C20H32Oz (combination of C10H15Ox and C10H17Ox), and C20H34Oz (combination of two C10H17Ox).
However, due to reactions with smaller peroxy radicals, HOM-Acc families with smaller carbon and hydrogen numbers are also observed. Indeed, one reason why the RO2+R′O2 termination is expected to affect the SOA formation potential is the formation of accretion products by scavenging of less oxidized and low-molecular-weight RO2 by HOM-RO2. Thus, the smaller RO2 will also contribute to the SOA mass which would otherwise not be the case. For the HOM-RO2 itself, it is expected that they contribute to SOA formation independently of the termination pathway, due to the low volatility of its expected termination products (Pullinen et al., 2020; McFiggans et al., 2019).
The second RO2+R′O2 termination pathway is the formation of a carbonyl and alcohol compound:
In this reaction, both radicals lose an oxygen atom, and a hydrogen atom is transferred to the RO2, forming the alcohol termination group. Preferences of RO2 to form an alcohol or carbonyl compound are possible for individual reactions, but statistically carbonyl and alcohols should be formed with the same fractions. Since mass spectrometry can only determine formula composition, we cannot distinguish alcohols and hydroperoxides, which arise from RO2 differing by one O atom. Therefore, details of balance of alcohol and carbonyl formation cannot be detected.
However, the formula composition can help to differentiate certain formation pathways. The C10H14Oz family contains only carbonyl formed from a C10H15Ox RO2, while the alcohol will be part of the C10H16Oz family. The C10H16Oz family also contains the carbonyl produced from the RO2+R′O2 monomer termination of C10H17Ox, while the alcohol from this RO2 family will be found in the C10H18Oz family. So, from a diagnostic point of view, C10H14Oz as well as C10H18Oz are uniquely related to a precursor radical family.
The classification of the formation pathways of the monomers is helpful to analyze the effect of the ratio shift in the experiments. Considering the termination pathways, a decrease in the C10H14Oz family and an increase in the C10H18Oz family are expected with increasing because of increasing termination by HO2 and decreasing termination by RO2. In the case of C10H18Oz, the increase in hydroperoxides is partially compensated by a decrease in the alcohol channel. For C10H16Oz, the situation is more complicated as it contains contributions from all termination pathways.
Besides closed-shell products, HOM-RO2 can also form alkoxy radicals (HOM-RO). In general, alkoxy radicals (RO) are important intermediates in the oxidation scheme of organics and are formed via (Reaction R4) and probably also via (Reaction R5) for specific RO2 (Jenkin et al., 2019):
In Reaction (R5), OH will be formed. The importance of Reaction (R5) compared to Reaction (R1) is still unclear in the literature, but functionalization of the RO2 close to the peroxy functionality possibly enables this reaction (Iyer et al., 2018; Eddingsaas et al., 2012; Hasson et al., 2005; Jenkin et al., 2019). If Reaction (R5) is of negligible importance, the reaction scheme will simplify, and the effect of increased is easier to diagnose.
We are interested in the importance of alkoxy radical formation as (HOM)-RO tends to fragment, leading to the formation of smaller products (Vereecken et al., 2007). In the context of SOA formation, these fragments are less likely to contribute to SOA mass because of their higher volatility. Since alkoxy radicals are too unstable to be detected directly, we use two diagnosis tools to judge the importance of HOM-RO. Firstly, HOM-RO fragmentation can lead to HOM-RO2 with less than 10 carbon atoms, which may also continue the autoxidation chain. Therefore, the abundance of HOM with less than 10 carbon atoms (HOM-Frag) indicates the importance of alkoxy steps. Secondly, with increasing functionalization, H shifts retaining the carbon backbone become more likely (Vereecken et al., 2007), which will lead to the generation of new C10-HOM-RO2. Such alkoxy peroxy steps can continue the autoxidation chain (Mentel et al., 2015). Interestingly, by coupling of an alkoxy and a peroxy step, the parity of the number of oxygen atoms in the HOM-RO2 changes, while in pure autoxidation steps the oxygen parity remains the same. Therefore, a parity change of the oxygen number can be used as an indication of alkoxy step abundance (Kang, 2021).
In summary, we will use the changes in contribution and relative signal of the different families and classes to judge the impact of shifting from low to high on the α-pinene photooxidation pathway.
2.2 Control of α-pinene OH turnover
After the initial α-pinene photooxidation phase as a reference, CO was added to the oxidation system. The idea is to represent small, oxidized VOCs in the atmosphere that can produce HO2 by reaction with OH (compare Schervish and Donahue, 2021). The presence of CO shifts the ratio, increasing the importance of the termination of RO2 by HO2. McFiggans et al. (2019) showed that one limiting factor in mixture experiments is oxidant scavenging: the products and their yields in mixed systems change, because there is less OH available to the individual VOCs. Thus, after the CO addition the OH production in the chamber was increased to compensate for the OH consumed by CO. The OH levels in the system before and after the CO addition were approximately the same, keeping the α-pinene OH turnover approximately constant. This OH adjustment ensures that the primary α-pinene chemistry was kept the same, avoiding effects by different oxidant levels and enabling a direct comparison.
However, since experiments could only be performed at about the same OH levels, a normalization by the actual α-pinene OH turnover is applied to the data. This compensates for the slight experimental imperfections and enables better comparison of experiment series with different boundary conditions. The turnover in steady state is given in Eq. (1). Here the subscript “SS” denotes steady-state condition for the concentrations of α-pinene and OH, and kOH is the α-pinene OH reaction rate constant.
This normalization also directly shows the yield of certain oxidation products or product groups per α-pinene consumed by OH.
2.3 Derivation of effect on condensable mass from gas-phase measurement
A simple proxy for the condensable mass from HOM products can be calculated from the steady-state HOM signals measured by the NO3-CIMS, assuming condensation for all low-volatility HOM compounds and no back-evaporation into the gas phase. To only take low-volatility products into account, we used all detected formula compositions with M>230 g mol−1 and weighted them with their molar mass. The reasoning behind this threshold can be found in Sect. 4.4. All contributions were summed up and normalized with the α-pinene OH turnover for the comparison between the low and high cases (Eq. 2).
We also estimated the expected SOA mass formed using the calibration factor obtained for sulfuric acid for our NO3-CIMS instrument in a calibration setup (see Supplement Sect. S1). From this, we calculated an upper boundary concentration of detected HOM compounds in the gas phase under the assumption that sulfuric acid clusters with nitrate at the collision limit, yielding maximum sensitivity (a common approach; see, for example, Ehn et al., 2014, and Pullinen et al., 2020).
The calculated gas-phase concentration was then used in the steady-state equation describing the relationship between gas- and particle-phase concentrations of a single compound i shown in Eq. (3).
Equation (3) shows that the steady-state particle-phase (mass) concentration mi,seed(p) of compound i in the presence of seed particles in the chamber is only dependent on the steady-state gas-phase concentration mi,seed (g), the condensation rate and evaporation rate constants kcond,i and kevap,i of i (to and from the particles) and the particle-loss rate constant kparticleLoss in the chamber. The condensation rate can be calculated (see Supplement Sect. S8), and the particle-loss rate constant was measured by observation of the particle loss in the chamber after ending the seed addition (details in the Supplement Sect. S2). The evaporation rate was assumed to be negligible for the investigated HOM compounds.
For the SOA yield calculation, we calculated a corrected organic mass mSOA from the organic mass mAMS measured by aerosol mass spectrometry (AMS) and the fraction expected to be lost on the seed particles compared to the overall loss on particles and chamber wall as shown in Eq. (4) (McFiggans et al., 2019).
In Eq. (4), we use the condensation rate constant kcond calculated for one major HOM product (C10H16O7) and the average HOM-Mon wall loss rate kwall, which was determined by switching off the UV-C light and observing the decay of photooxidation products in the NO3-CIMS. The wall loss determination, as well as SOA mass correction, was described before in Sarrafzadeh et al. (2016) and McFiggans et al. (2019).
3.1 Chamber setup
Experiments were conducted in the Jülich SAPHIR-STAR chamber, which is the modern successor of the JPAC setup (Mentel et al., 2009). The basic concepts are the same as in JPAC, but each parameter is set, controlled and monitored in a program. The chamber was operated as a continuously stirred tank reactor. It is a borosilicate glass cylinder (l=2.5 m, d=1 m) with a volume of close to 2000 L, and all equipment inside the chamber is either glass or glass-coated steel (SilcoTek GmbH).
With an inflow of 32 L min−1, the residence time in the chamber was approximately 61 min with a fan ensuring mixing within minutes. In contrast to the JPAC chamber, the stirring is conducted perpendicular to the cylinder axis, as opposed to coaxial. Chamber inflow is split into two humidified clean air flows (mixed from N2 and O2) of about equal volume: one with added oxidant (here O3) and the other with added VOC and other trace gases (here α-pinene and CO). All experiments were performed at a relative humidity of 50 % and temperature of 20 °C. Temperature stability is ensured by the climate-controlled surroundings of the chamber.
α-Pinene (≥ 99 % purity, Sigma-Aldrich, Merck KGaA) was introduced via liquid injection with a syringe pump (Fusion 4000, CHEMYX Inc.) into a heated glass bulb and flushed by a 1 L min−1 N2 stream into the chamber. CO was added from a gas bottle (10 % CO in N2, Messer SE & Co. KGaA). Ozone was directly produced photolytically before injection with a self-built ozone generator.
OH is produced in the chamber by ozone photolysis using two UV-C lamps with a wavelength of 254 nm and subsequent reaction of O(1D) with water vapor. The lamps are mounted in closed quartz cylinders in the middle of the chamber, vertically to the cylinder axis, and light intensity can be varied with a movable shielding installed around the lamps. The shielding allows for an exact percentage of the lamp to be covered, thus controlling the amount of OH produced in the chamber.
The OH radical concentration after CO addition was adjusted by setting the shielding of the UV-C lamps and a slight adjustment of O3 inflow. The applied J(O1D) values in different phases were calculated to be in the range of to s−1.
In some of the experiments, ammonium sulfate (≥ 99 % purity, Merck KGaA) seed particles were added to the system to provide a surface for the condensation of organic material. The aerosol was produced with a modified TSI atomizer (model 3076, TSI GmbH) and dried to 50 % relative humidity.
VOC concentrations in the chamber were measured using proton-transfer-reaction mass spectrometry (PTR-TOF-MS; Ionicon GmbH). CO2, CO and H2O (G2401 cavity ring-down spectrometer, Picarro Inc.); NO and NOx (NCLD899, Eco Physics GmbH with a home-built photolytic converter); and O3 (O342e, Envea GmbH) were additionally monitored. Particle distribution and concentration were measured with a condensation particle counter (CPC, model 3788, TSI GmbH) and a scanning mobility particle sizer (SMPS; model 3080, TSI GmbH) with a CPC (model 3788, TSI GmbH). The aerosol composition was measured with a high-resolution aerosol mass spectrometer (HR-TOF AMS; Aerodyne Inc.).
In all experiments, VOC, O3 and SMPS + CPC sampling switched between inlet and outlet of the chamber to measure the input concentrations as well as the concentrations in the reactor. The flow control system of the chamber adapts to these switches so that the inflow into the chamber stays constant.
All results discussed here were observed under steady-state conditions when all parameters were constant. For each steady state, the OH concentration was calculated from the decay of α-pinene as described by Kiendler-Scharr et al. (2009). Equation (5) is derived from the mass balance of α-pinene at steady state. The steady-state OH concentration [OH]SS depends on the amount of α-pinene consumed by reaction with OH and the reaction with O3, as well as the flush out.
Here, F is the total flow and V the volume of the chamber. The subscript “SS” indicates steady-state concentrations, while [VOC]in represents the α-pinene concentration entering the chamber. kO3 and kOH represent the reaction rate constants of α-pinene with the corresponding oxidant. We applied rate coefficients of cm3 s−1 (Atkinson and Arey, 2003) and cm3 s−1 (Cox et al., 2020) at 20 °C. The uncertainty of the OH calculation was estimated as 20 % by Wildt et al. (2014).
3.2 Experiment conditions
An overview of the experiments and their boundary conditions can be found in Table 1. Four experiments were performed in total, leading to one repetition of each studied condition. In two of the experiments, ammonium sulfate seeds were added, leading to a total particle surface in the chamber on the order of m2 m−3 and organic loadings of about 3 µg m−3 in the photooxidation stage. In the unseeded experiments, no significant nucleation was observed, leading to pure gas-phase conditions. The Exp2 experiment is a consecutive combination of a seeded followed by a non-seeded experiment to provide direct insight into the effect of the seed presence on the system.
As the OH radical is produced by photolysis of ozone and as α-pinene reacts with ozone, it is important to know the relative contribution of the α-pinene consumption by OH and by O3. This is achieved by comparing the turnover of α-pinene with OH and O3, respectively. The results can be found in Table 1. The listed results are for the low conditions, but nearly identical values were reached after the shift.

3.3 Experimental procedure
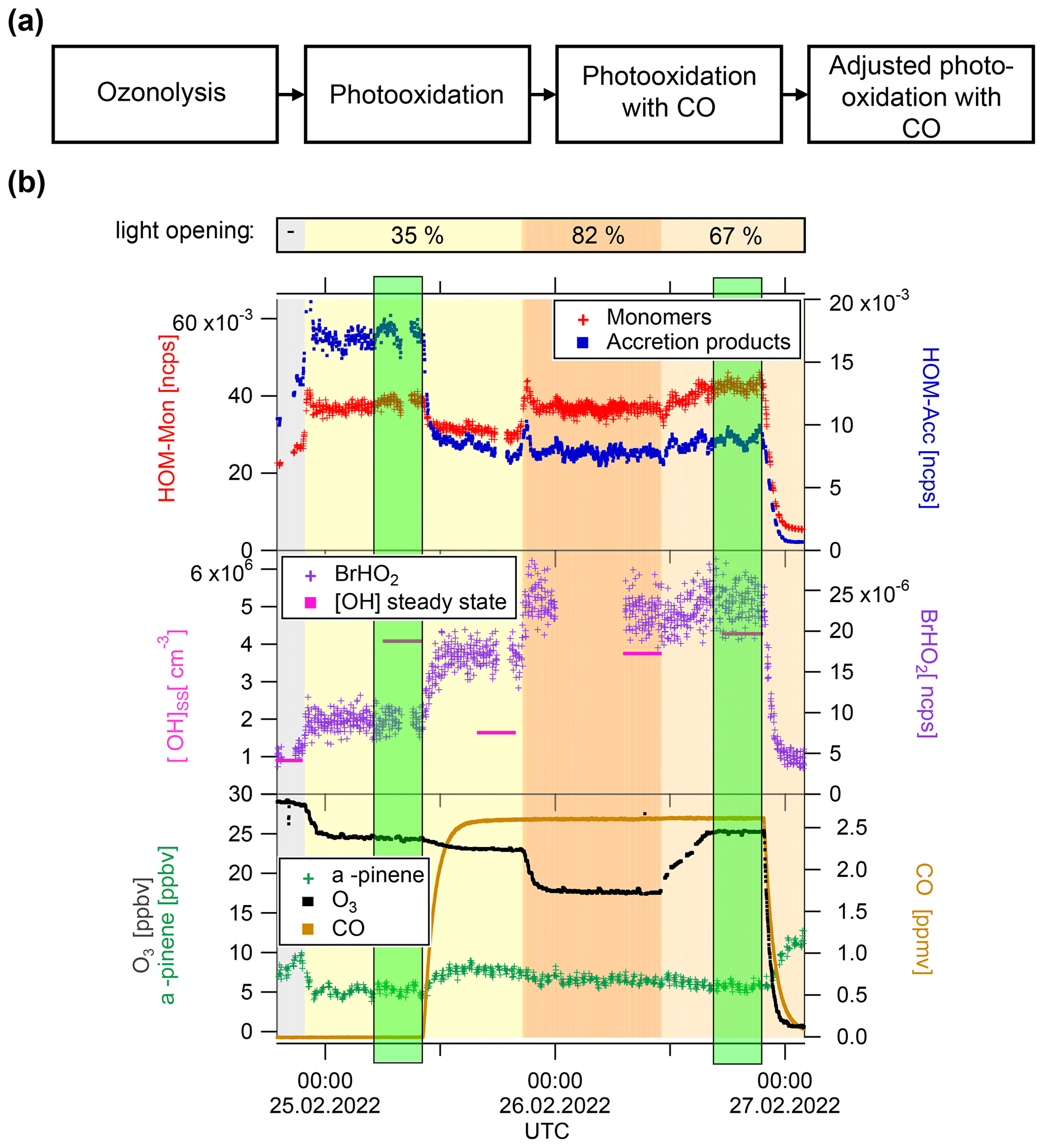
Figure 2(a) Experiment flow scheme. (b) Exemplary time series of Exp1, showing HOM-Mon and HOM-Acc product sum (top panel); calculated OH concentration and BrHO2 signal (middle panel), and ozone, α-pinene, and CO concentrations (bottom panel). Background color represents light intensity. Highlighted in green are the low steady state and the steady state at high (addition of CO and adjusted oxidant level).
All experiments started with α-pinene ozonolysis followed by illumination with the UV-C lights to induce the reaction with OH. A general flow scheme of the experiment can be found in Fig. 2, together with one exemplary time series of the unseeded experiment (Exp1). After the photooxidation steady state, CO was added to the system. In the displayed Exp1, the OH level was adjusted in three steps to approach the same concentration as before the CO addition. First the UV-C light opening was adjusted and then O3 was added, and the UV-C light opening was adjusted again. In some experiments initially the effect of CO on the unchanged system was observed before the adjustment of OH. In other experiments (Exp2.2, Exp3), the adjustment of the α-pinene OH turnover via ozone concentration and UV-C light opening were made simultaneously with the CO addition. Highlighted in green are the steady states with the “same” OH concentration characterized by low and high , which were used for analysis and interpretation.
3.4 Model calculation for ratio estimation
Box-model calculations were performed by applying the MCM v3.3.1 chemistry (Jenkin et al., 1997; Saunders et al., 2003) under the boundary conditions of the SAPHIR-STAR chamber. All calculations were performed with the institute software package EASY, which uses FACSIMILE to solve the differential equations (EASY Version 5.69b). More details about the model parameters can be found in the Supplement Sect. S3. The model calculations reproduced the primary observables α-pinene, O3, CO and OH within the experimental uncertainties. The box-model results were used to characterize the ratio of the chemical systems, as no direct measurement of these parameters was available. The observed cluster signal BrHO follows the modeled HO2 concentration (Fig. 3).
The model predicts a shift of the ratio from about 0.01 to about 1 by CO addition and oxidant adjustment, an increase by 2 orders of magnitude. Owing to a lack of observations to verify model results, we will consider only the magnitude of here. The model results show that indeed a major shift from RO2 + RO2 to RO2 + HO2 reactions can be expected.
We further used the modeled RO2 and HO2 concentrations to estimate the relative importance of pathways for individual (observed) HOM-RO2. For that, we applied two generic rate coefficients kRO2HO2 and kRO2RO2. As the rate coefficient for the RO2 + HO2 termination to a hydroperoxide (kRO2HO2), we used the value specified in the MCM ( cm3 s−1 at 20 °C (Jenkin et al., 1997; Saunders et al., 2003)). We chose a kRO2RO2 of cm3 s−1 as the approximated reaction rate of the RO2+ RO2 reactions. This value applies to all possible reactions (accretion product, monomer, and alkoxy formation) and is in the range of kRO2RO2 utilized by Roldin et al. (2019) in the PRAM model.
3.5 Determination of oxidized VOCs, HOMs and HO2
Chemical ionization mass spectrometry (HR-TOF-CIMS) techniques were used to detect a range of gaseous compounds. For this, two atmospheric-pressure-interface time-of-flight mass spectrometers (APi-TOF-MS; Tofwerk AG) with different inlet systems were used simultaneously. General information about the APi-TOF-MS instrument can be found in Junninen et al. (2010).
A long TOF (LTOF, Tofwerk AG) (resolution of ∼ 8500 for peaks at 200) was coupled with the multi-scheme ionization inlet (MION; Karsa Oy). The setup of the inlet is described in detail by Rissanen et al. (2019). The distinctive feature of the MION inlet is the switching between two reagent ions. Here, nitrate was used to detect closed-shell HOMs, as well as HOM-RO2. Bianchi et al. (2019) suggested to define HOMs as products stemming from autoxidation containing more than six oxygen atoms. In our overall analysis, we decided to also include fragments and monomers containing five or in a few cases four oxygens (see the peak list in Supplement Sect. S5) as we are interested to see if the importance of these less oxidized (but still with NO3-CIMS detectable) products increases at higher . However, in all considerations regarding SOA formation, we furthermore set a molar weight threshold which automatically excluded any products with less than six oxygens.
As the second reagent ion, bromide was used to detect less oxidized products and the HO2 radical (Albrecht et al., 2019; Sanchez et al., 2016). The nitrate ion source had a reaction time of 600 ms, while the bromide ion source had a shorter reaction time of 60 ms. For all experiments, an inlet flow of 10 L min−1 was used, and the ionization scheme was switched every 10 min.
In the data evaluation, the first step was the separation of the time series of the two reagent ions. The data were subsequently processed with Tofware (Version 3.2.3, Tofwerk AG), using the high-resolution time series workflow. No transmission correction was performed as previous measurements showed an approximately flat relative transmission curve in the mass region of interest. The analyte signals were normalized with the reagent ion signal (NO and HNO3NO for nitrate and Br− and BrH2O− for bromide).
Since no direct HO2 calibration was available, the HO2 signal in the Br-MION-CIMS was used to compare the levels of HO2 relative to each other in the different phases of the experiment. The comparison of the measured HO2 signal to the modeled HO2 concentration shows a good linear relation between the model predictions and observations.
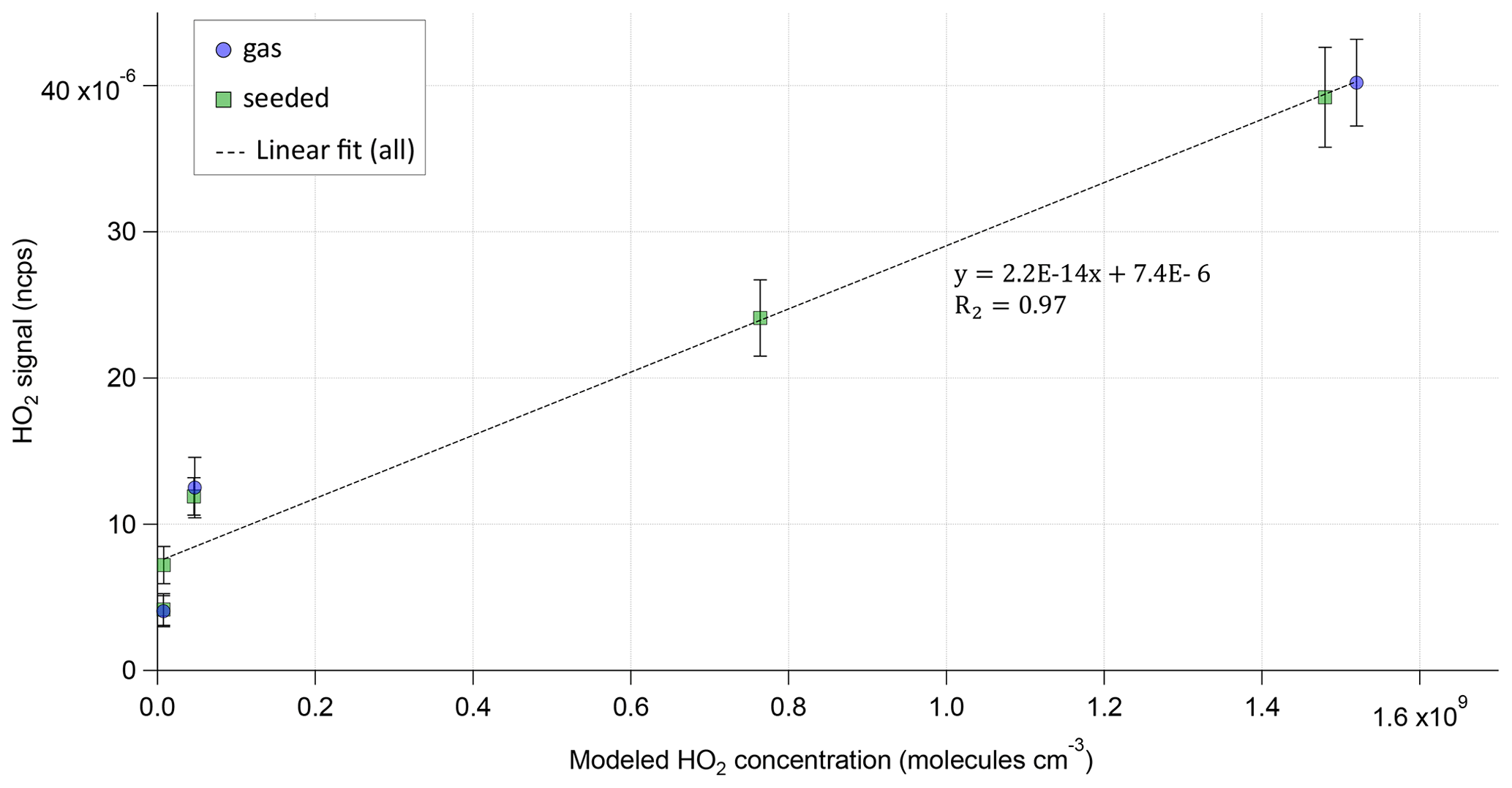
Figure 3Modeled HO2 concentration vs. normalized HO2 signal for each steady state of Exp2. HO2 is measured as the BrHO2 cluster and is normalized with the sum of the reagent ion Br− and its water cluster. The dotted line shows the linear fit to all (gas phase and seeded) measurement points.
Figure 3 illustrates this for the example of the Exp2 experiment. A background signal of around ∼ is observed as soon as VOC and ozone are present in the reactor. The background HO2 signal was not observed when only O3 or only VOC were in the system. As shown by the MCM modeling results, HO2 production of this strength is not expected in the α-pinene ozonolysis phase, but this background phenomenon was observed before (Albrecht et al., 2019) and is not fully understood.
For the HOM molecules measured by the NO3-MION-CIMS, the relative changes between different experiment phases are compared. For all detected HOM products, the same detection sensitivity is assumed. Hyttinen et al. (2018) showed in quantum chemical calculations that HOMs containing six or more oxygen atoms have comparable sensitivity with the nitrate reagent ion. At this degree of oxidation, it can be expected that the HOMs already contain multiple hydroperoxyl and/or hydroxy functional groups (Bianchi et al., 2019) prior to the termination step, making it unlikely that the sensitivity is strongly influenced by the termination group. Thus, the signal strength reflects the correct ranking of the observations, and relative comparisons do not require calibration. Pullinen et al. (2020) studied the mass balance between condensable HOMs and formed particle mass and were able to find closure within a factor of 2.
A second CI-APi-TOF was used to measure less oxidized species. It was configured with a CI inlet based on the design of Eisele and Tanner (1993) coupled to a TOF-MS (HTOF Tofwerk AG, resolution ∼ 2700 for peaks at 200) and was operated in positive mode with propylamine (C3H7NH2, Sigma-Aldrich, purity ≥ 99 %) to detect the early-generation RO2 and oxidation products (Berndt et al., 2018). The propylamine was purified and added as an amine–N2 mixture (flow: 0.12 mL min−1) to the 30 L min−1 sheath flow. Furthermore, the sheath flow air is humidified to optimize ionization. The instrument sampled 0.1 L min−1 from the chamber, which was diluted with 9.9 L min−1 for a sample flow of 10 L min−1. The dilution was necessary to reduce depletion of the primary ion (Hantschke, 2022).
In order to understand the effect of on the gas-phase product composition, we will present and compare two cases: the steady state without CO (low ) and the steady state with CO addition and OH adjustment by J(O1D) and O3 (high ). The modeling results predicted of about 1/100 and of about 1/1 for these two cases, respectively. The modeled concentrations can be found in Supplement Sect. S4. The modeling results show that the ratio changes by 2 orders of magnitude, because [RO2] was reduced by about a factor of 3, while [HO2] was increased by a factor of 30. Consequently, HO2 reactions were almost negligible at low while RO2+ RO2 reactions can still contribute at high .
ratios of around 1 are highly relevant for atmospheric conditions with significant OH oxidation, though it should be kept in mind that in atmospheric conditions the methyl peroxy radical and other small RO2 contribute a significant portion to the total of peroxy radicals (Khan et al., 2015). Field studies reporting HO2 and RO2 measurements for different environments can be found in the Supplement Table S5. These exemplary studies show that ratios around 1 are relevant in remote to urban environments with different VOC sources and NOx levels.
Assuming correctly modeled [HO2] and [RO2], we calculated the competition between HO2 and RO2 reactions for each (observed) RO2 expressed in the form of pseudo-first-order rate coefficients in kRO2HO2 ⋅ [HO2] or kRO2RO2 ⋅ [RO2]. Herein [RO2] is the sum of all RO2 species as defined in the MCM v3.3.1. For all experiments, the results of our calculations indicate that the sink for HOM-RO2 is dominated by RO2+ RO2 reactions at low (∼ 97 % contribution), while at high RO2+ HO2 contributed ∼ 80 %. As the rate coefficients are not well known and as we cannot verify the modeling results for HO2 and RO2, our calculations serve solely as an indication of expected trends in the chemical system.
4.1 Impact on overall HOM formation
The top panel of Fig. 2 shows the time series of HOM-Mon and HOM-Acc products. The HOM-Mon signal recovers after the oxidant adjustment, while the HOM-Acc signal is significantly suppressed at high . This indicates that the shift from low to high substantially impacts the termination reactions, shifting formation from the HOM-Acc product channel (RO2+RO2) to the HOM-Mon channel.
An overview of the results for the product classes defined in the method section is shown in Fig. 4. Plotted are the average ratios of signal in the NO3-CIMS in the high steady state compared to the low steady state. For better comparison, all experiment phases were normalized to the actual α-pinene OH turnover. The overall HOM-signal was lower at high , showing a reduction of about 20 %. Most distinctive, the HOM-Acc compounds were strongly reduced by about 60 %. A reduction of HOM-Acc by addition of CO was observed before by McFiggans et al. (2019); however, there the OH concentration was not kept constant. The HOM-Frag signal (5 ≥ C < 10) also shows a reduction of about 20 %. At high , the C10-HOM-RO2 signal was also reduced significantly by about 40 %.
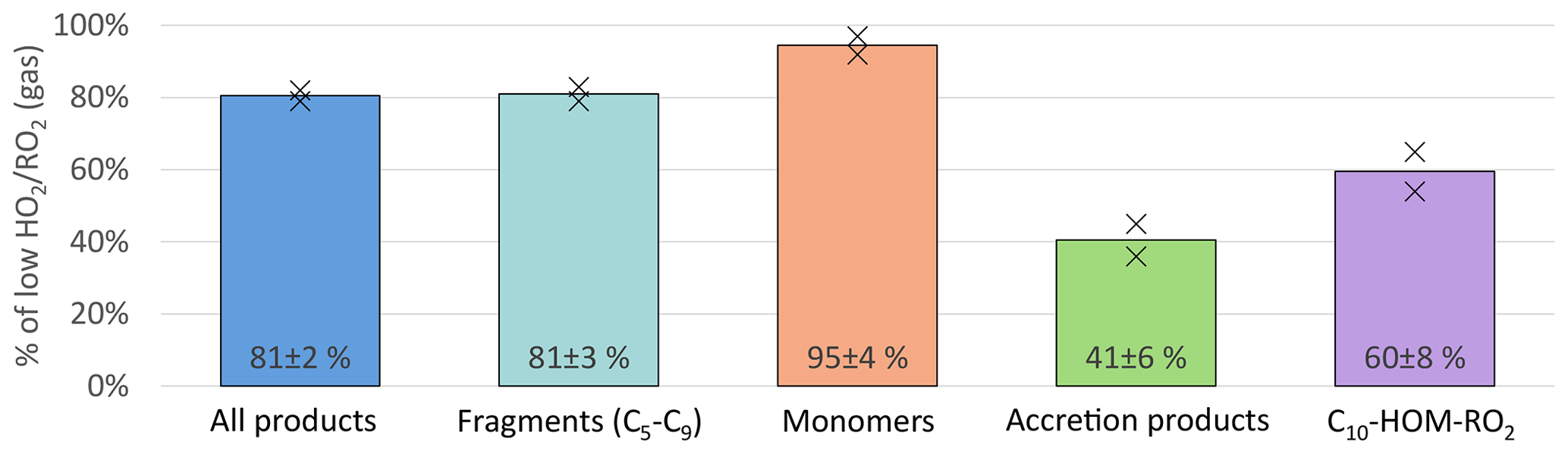
Figure 4Overview of average, relative change in product classes detected in NO3-CIMS between low and high cases (both normalized to α-pinene OH turnover) for unseeded experiments. Bars represent the average of the two experiments; markers represent individual experiments.
The HOM-Mon signal level remained about the same at low and high . Without changes in the rates and contributions of the different termination reactions, the observed reduction in the HOM-RO2 precursors should lead to nearly the same reduction in HOM-Mon. However, the decrease in accretion product formation and fragmentation should lead to an increase in HOM-Mon. The presence of HO2 could reduce the alkoxy formation and thus fragmentation of HOM-RO2. This missing sink could lead to an additional HOM-Mon source compared to the low case. However, the distribution of the product classes at low and high (Fig. 5) shows that contributions are shifted from HOM-Acc to HOM-Mon, while the contribution of HOM-Frag remains constant. Each HOM-Acc is formed from one HOM-RO2 (HOM-RO2+ RO2) or potentially even two HOM-RO2 compounds (HOM-RO2+ HOM-RO2) and therefore each HOM-Acc not formed will lead to at least one HOM-Mon.
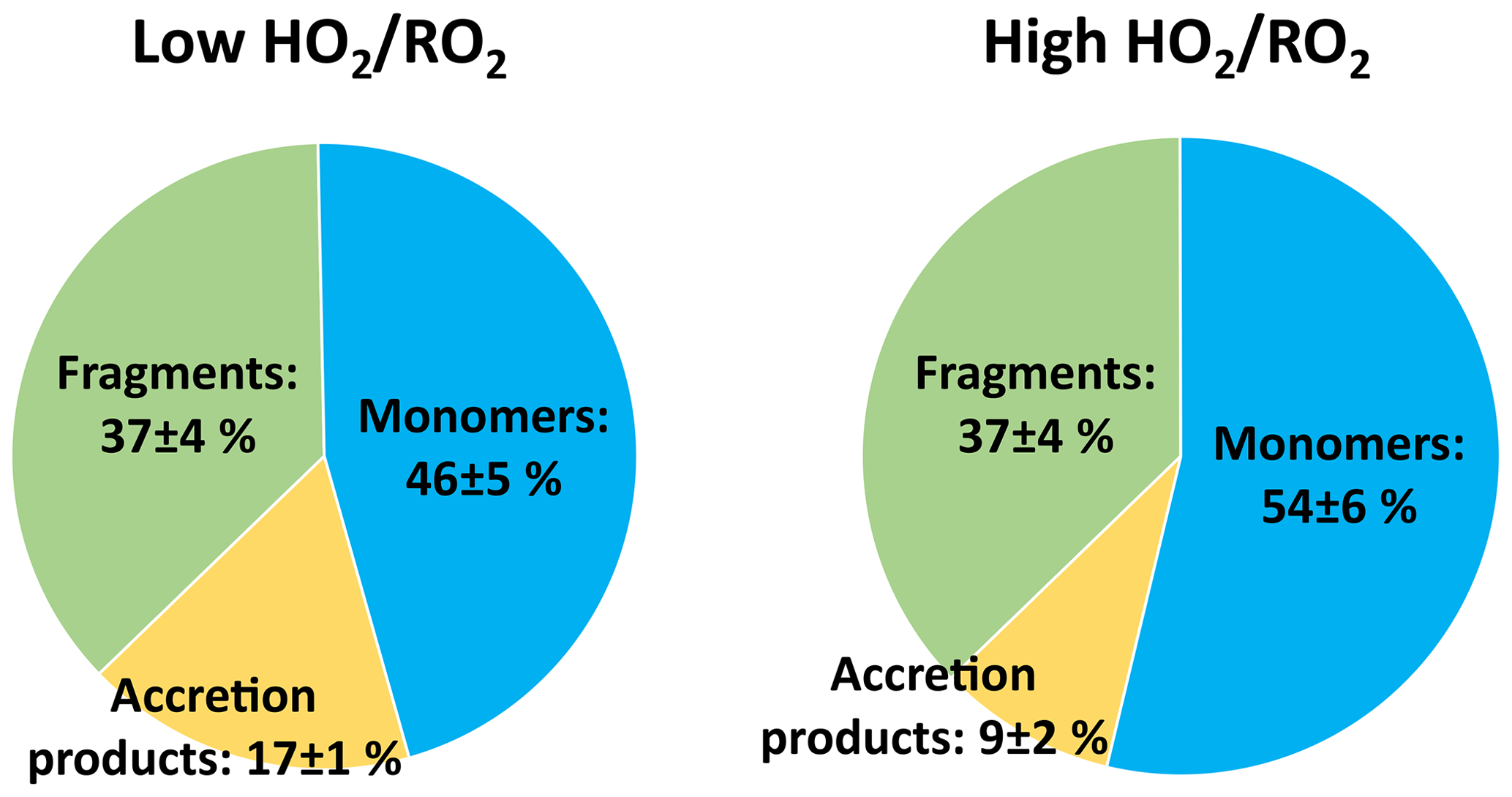
Figure 5Average contribution of the closed-shell product classes to overall HOM-product signal in the low and high cases (unseeded experiments).
Further changes in the product distribution become evident when considering the individual HOM-Mon families as shown in Fig. 6. The C10H15Ox peroxy radical family and the related C10H14Oz family (carbonyl compounds) show the strongest suppression with a decrease of about 40 % at high . For the C10H17Ox peroxy radical family, the suppression was less pronounced with a 18 % reduction. In contrast, the C10H16Oz family remained about the same, while the C10H18Oz family showed a strong increase at high .
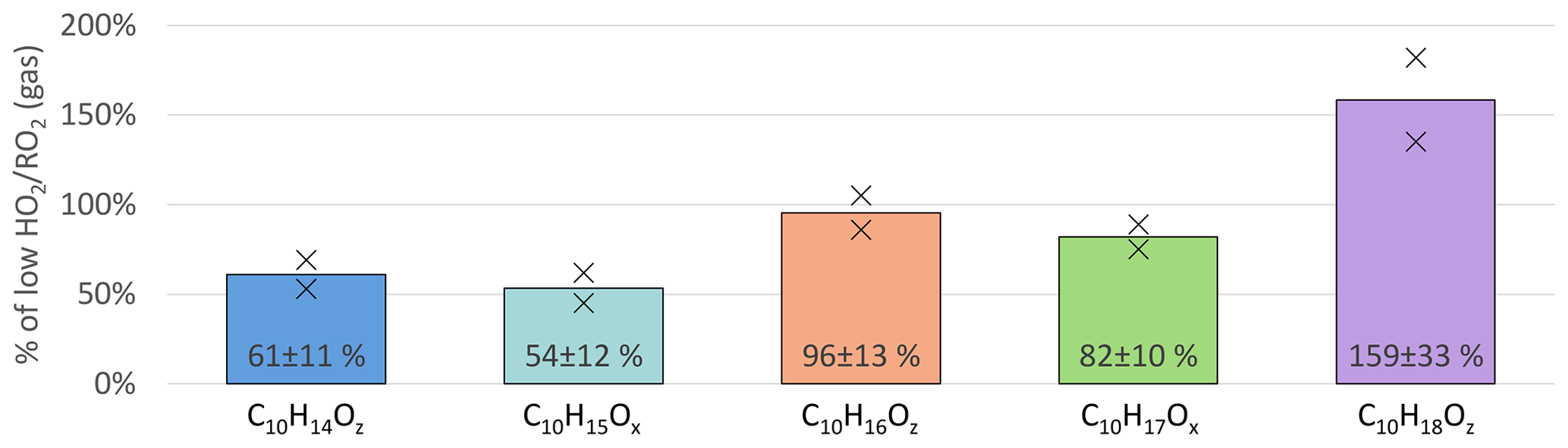
Figure 6Overview of average, relative change in monomer families detected in NO3-CIMS between low and high cases (both normalized to α-pinene OH turnover) for unseeded experiments. Bars represent the average of the two experiments; markers represent individual experiments.
The suppression of C10-HOM-RO2 of only about 40 % compared to the reduction of overall [RO2] by ∼70 % in the model calculations (for the modeled concentrations, see the Supplement Sect. S4) shows that in many instances the autoxidation is too efficient to be out-competed by the RO2+ HO2 termination reaction, which is several times faster than RO2+ RO2 reactions.
Furthermore, the signal-weighted ratio of the monomer class does not change between low and high (0.70±0.01). If the HO2 termination would interrupt the autoxidation chain, a lower oxidation level would be expected at high . The unchanged oxidation level and the suppression of HOM-Acc indicate that the average autoxidation rate must be faster than kRO2HO2⋅ [HO2], while the average accretion rate for [RO2] must be slower. In conclusion, the change in should essentially impact the distribution of the HOM-RO2 termination products.
4.2 Impact on HOM-RO2
C10-HOM-RO2 compounds are key to understand the changes in the HOM product distribution. Therefore, we will first discuss the changes in the HOM-RO2 products and then the changes in the closed-shell products.
The C10 peroxy radical class consists of the C10H15Ox and C10H17Ox families, which were reduced to 54 % and 82 %, respectively, when comparing the high and low cases (Fig. 6, light blue and green bars). The observed reduction in C10-HOM-RO2 is significantly smaller than the overall RO2 concentration reduction predicted by the MCM model results (reduction to ∼ 30 %). In the following paragraphs, we present a plausibility consideration to assess if these observed changes are consistent with our expectations from modeling results and reaction rates.
The change in the steady-state concentration of a compound is always defined by the changes in its sources and sinks. The source of a HOM-RO2 is the intramolecular reaction of a precursor RO2; thus, the HOM-RO2's source is reduced if the steady-state concentration of the precursor RO2 is reduced. However, assuming the source term of the precursor RO2 is the same in low and high (due to the constant α-pinene OH turnover) and the precursor RO2's sink term is dominated by the fast autoxidation in both cases, then the RO2's steady-state concentration would not be significantly changed. This consideration is only applicable for RO2 where autoxidation dominates the sink term at low and high . However, the unchanged oxidation level of the HOM-Mon indicates that once the autoxidation is initiated it out-competes the possible termination reactions.
In this case, the change in steady-state concentration of the HOM-RO2 will be defined by the changes in the sink terms. Owing to the faster reaction of RO2+ HO2 compared to RO2+ RO2 the chemical sink for all RO2 including HOM-RO2 with slower autoxidation rates increased, which leads to a reduction in the steady-state concentration of RO2 in general, despite holding the primary RO2 source term constant.
For steady-state conditions, we can estimate the expected effect on the RO2 ratio between high and low conditions for those HOM-RO2 with production directly linked to the primary production (kOH⋅ [OH] ⋅ [α-pinene]) with negligible further autoxidation. The necessary equations and assumptions can be found in Supplement Sect. S7. We assume the same primary production at low and high and that the reaction with HO2, the reaction with RO2 and the wall loss are the only significant loss pathways. At high , a reduction to 80 % is expected if the chosen bulk rate constants are used ( cm3 s−1 at 20 °C (Jenkin et al., 1997; Saunders et al., 2003); cm3 s−1). A reduction to 60 % is expected if kRO2HO2 is around 7 times faster than kRO2RO2 ( cm3 s−1). These reductions are in the range of what is observed for the C10-HOM-RO2. Of course, the approach of using generalized bulk rate constants is limited, but the resulting values for kRO2RO2 were clearly within the range of rate coefficients expected for HOM-RO2+ RO2 reactions (Roldin et al., 2019), showing that the increased chemical sink is a plausible explanation for our observations.
The C10H15Ox family is on average reduced by around 30 % more than the C10H17Ox family (see Fig. 6). C10H15Ox peroxy radicals are either formed by sequential oxidation of α-pinene, e.g., from oxidation products like pinonaldehyde, or directly from α-pinene via the H-abstraction pathway (Shen et al., 2022). Formation of pinonaldehyde and even more so HOM formation via the H-abstraction channel involve alkoxy steps. However, alkoxy radicals should be reduced at high since they are mainly formed by RO2+ RO2 reactions in the absence of NOx. Thus, missing source terms add to the increased chemical sink by HO2 for C10H15Ox peroxy radicals.
Amine CIMS measurements enabled detection of the formula composition C10H16O2 (e.g., pinonaldehyde). C10H16O2 was reduced on average to 70 % ± 1 % at high compared to low . This supports that a fraction of the C10H15Ox radical decrease at high arose from suppression of C10H16O2 first generation products. In addition, a further suppression of HOM formation via the H-abstraction channel is likely. It should be noted that the reduction of C10H16O2 is smaller than that expected by the MCM model results. Modeling results can be found in Supplement Sect. S4. This might indicate that HO2 can also enable alkoxy radical steps to a certain degree as summarized by Jenkin et al. (2019) and postulated by e.g., Eddingsaas et al. (2012) as a source of pinonaldehyde in HO2 dominated systems.
According to the model calculations, the pseudo-first-order rate coefficient kRO2HO2⋅ [HO2] is expected to be about 0.04 s−1 for the RO2+ HO2 reaction at high . Consequently, only such HOM-RO2 with autoxidation rates of ≤ 0.04 s−1 will be significantly lost by reaction with HO2 at the higher HO2 concentrations. However, typical isomerization rates of peroxy radicals in autoxidation are on the order of 0.1 s−1, and many are faster (Piletic and Kleindienst, 2022; Berndt, 2021). Therefore, reduction in a HOM-RO2 is only expected when the faster termination rate of kRO2HO2⋅ [HO2] can compete with the autoxidation rate, i.e. when the autoxidation slows as the degree of oxidation increases on the specific HOM-RO2. This consideration shows that the smaller reduction in HOM-RO2 compared to the less oxidized RO2 in the model is compatible with fast autoxidation reactions that are missing in the MCM.
The increase in chemical sink strength by going from RO2 termination to HO2 termination is the main expected reason for the decrease in C10H17Ox. As discussed, the C10H15Ox family is subject to an additional decrease in the precursors due to the alkoxy steps necessary in the formation pathway. Since C10H15Ox compounds were the main contributors to the C10-HOM-RO2 class, their stronger reduction is reflected in the overall reduction of C10-HOM-RO2.
4.2.1 Contribution of C10H15Ox and C10H17Ox families to HOM-RO2
In the unseeded, pure gas-phase experiments, the contribution of the C10H17Ox family to the C10-HOM-RO2 class is 23 % ± 2 % on average in the low case. In the high case, the contribution increases to 31 % ± 4 % on average. As discussed above, the suggested pathways to C10H15Ox HOM-RO2 may be additionally suppressed due to a decrease in alkoxy steps at high , reducing the entry channel into C10H15Ox HOM-RO2.
Nevertheless, the contribution of C10H15Ox is substantial in both experiment stages. Kang (2021) and Shen et al. (2022) reported that, in the photooxidation of α-pinene, the HOM-RO2 compounds detected by NO3-CIMS are dominated by the C10H15Ox family, while C10H17Ox formation is the main expected OH reaction pathway described in the literature (Berndt, 2021; Berndt et al., 2016; Xu et al., 2019).
This hints towards an effective pathway to HOM via C10H15Ox. A reason may be the fast opening of both carbon rings in the bicyclic α-pinene (Shen et al., 2022) or a four-ring opening in pinonaldehyde or similar compounds for easy autoxidation. From our observations, increasing the ratio does increase the relative importance of the C10H17Ox family, but the change is less than 10 % in contribution.
Contribution of the two peroxy radical families to the HOM formation is also reflected in the composition of C20 HOM-Acc. Figure 7 shows the average contributions of the C20H30Oz, C20H32Oz and C20H34Oz families in the low and high cases. Although the absolute amount of HOM-Acc was suppressed by 60 % the family distribution was similar, C20H32Oz dominated, while C20H30Oz was lowest. C20H30Oz is formed from two members of the C10H15Ox family, while C20H34Oz is formed by two members of the C10H17Ox family. C20H32Oz is then a combination of a C10H15Ox-RO2 and C10H17Ox-RO2.
Families that require one or two C10H17Ox peroxy radicals for their formation have a higher contribution than the C10H17Ox family's contribution to C10-HOM-RO2. Here, it is important to note that not only HOM-RO2 can participate in HOM-Acc formation but also traditional, less oxidized RO2 radicals (Berndt et al., 2018; Pullinen et al., 2020; McFiggans et al., 2019), which are not detectable by NO3-CIMS. However, more oxidized peroxy radicals exhibit faster accretion rates (Berndt et al., 2018).
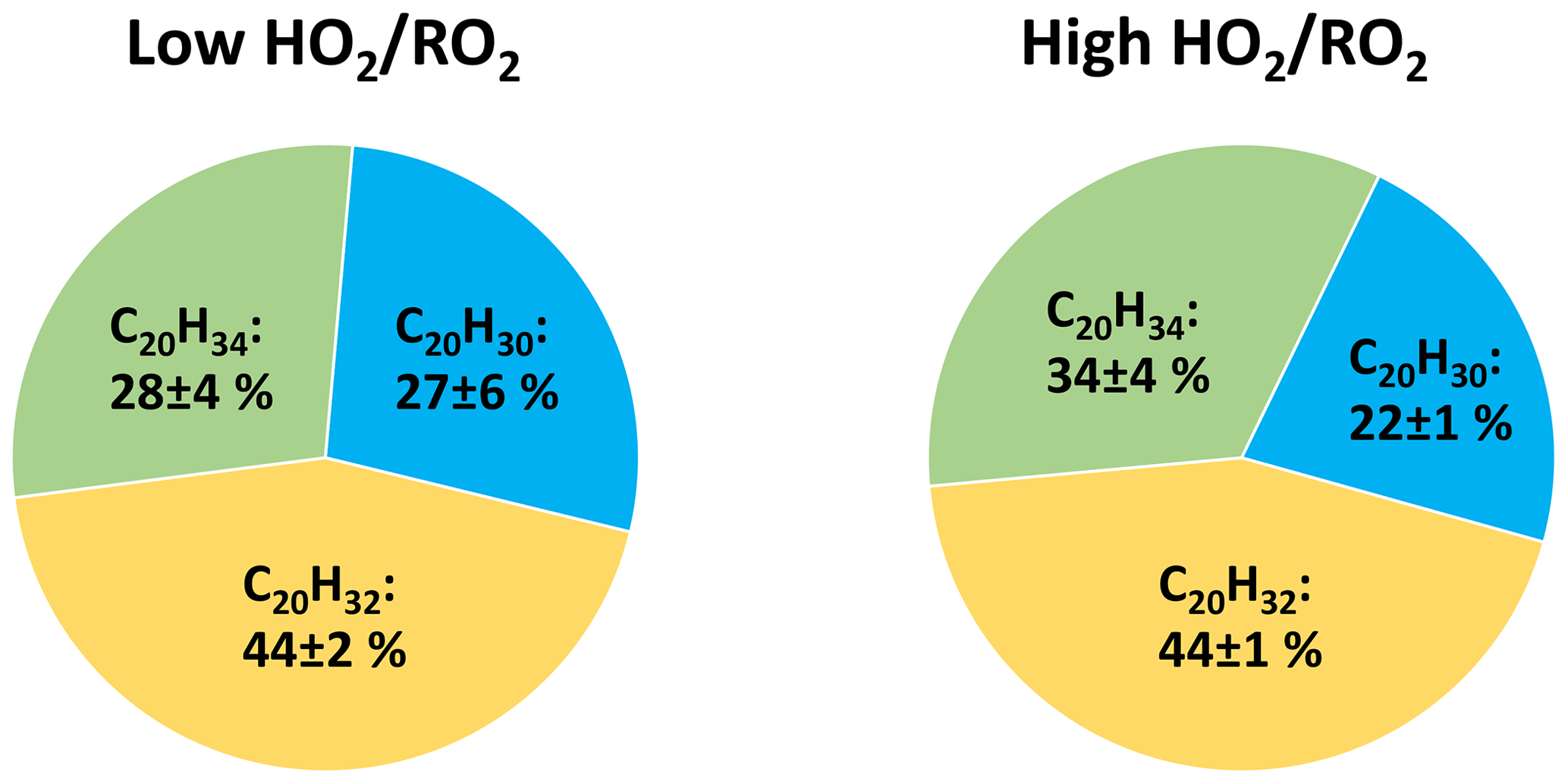
Figure 7Average contribution of the C20H30Oz, C20H32Oz and C20H34Oz families to the C20 HOM-Acc group signal in the low and high cases (unseeded experiments). Not pictured is C20H28Oz due to its negligible signal (contribution ∼ 1 %).
The large contributions of C20H32Oz and C20H34Oz thus clearly show the general importance of the C10H17Ox peroxy radicals. The largest fraction, the C20H32Oz family, indicates the importance of HOM-C10H15Ox and a high abundance of less oxidized C10H17Ox peroxy radicals. Less oxidized C10H17Ox-RO2 compounds were recently measured by Berndt (2021). The fraction of C20H34Oz is smaller because their formation requires HOM-C10H17Ox radicals which are less abundant compared to HOM-C10H15Ox, while the small fraction of C20H30Oz indicates that, despite the importance of HOM-C10H15Ox, less oxidized C10H15Ox are less important.
These results indicate the importance of mixed HOM-Acc formation by cross-reactions of HOM-RO2 and a less oxidized RO2. The importance of mixed HOM-Acc is supported by the relatively small fractions of HOM-Acc products with very high oxygen numbers, which more likely stem from HOM-RO2+HOM-RO2. For example, C20-HOM-Acc compounds with 12 or more oxygen atoms contribute only around 30 % (low : 26 % ± 4 %; high : 31 % ± 2 %) of the signal in the product group.
Although the effect of the changed ratio is small, a tendency to higher C20H34Oz contribution was observed. This is consistent with the observation of a slightly higher C10H17Ox contribution to the C10-HOM RO2. The stronger suppression of the C10H15Ox family at high is the first indication for, and can be explained by a reduction in the alkoxy radical formation.
4.2.2 Impact on HOM-alkoxy radical formation
Alkoxy radicals (RO) are the second important radical type in the oxidation chain of α-pinene. RO compounds cannot be detected directly as they are highly unstable and thus have very low concentrations. However, as explained in Sect. 2.2 the parity change in the HOM-RO2 families can be used as a diagnosis tool for the abundance of alkoxy steps (Kang, 2021). A second indicator for alkoxy steps is the abundance of HOM products with less than 10 carbon atoms.
Figure 8 shows the average contribution of C10H15Ox and C10H17Ox with an even and odd number of oxygens at low and high . C10H15Ox radicals with an even number of oxygens contribute on average 32 % at low . For C10H15Ox, the autoxidation chain is expected to start from an even number of oxygens either from C10H15O4 (pinonaldehyde-like) (MCM v3.3.1 (Jenkin et al., 1997; Saunders et al., 2003) or from C10H15O2 (C10H16 H abstraction) (Berndt, 2021; Shen et al., 2022). Therefore, without the involvement of an alkoxy step, the parity of the oxygen number in the observed C10H15Ox HOM-RO2 is expected to be even. Due to the average contribution of C10H15Oodd of 69 %, we conclude that at least one alkoxy step (or any odd number of alkoxy steps) must have taken place in most of the cases at low .
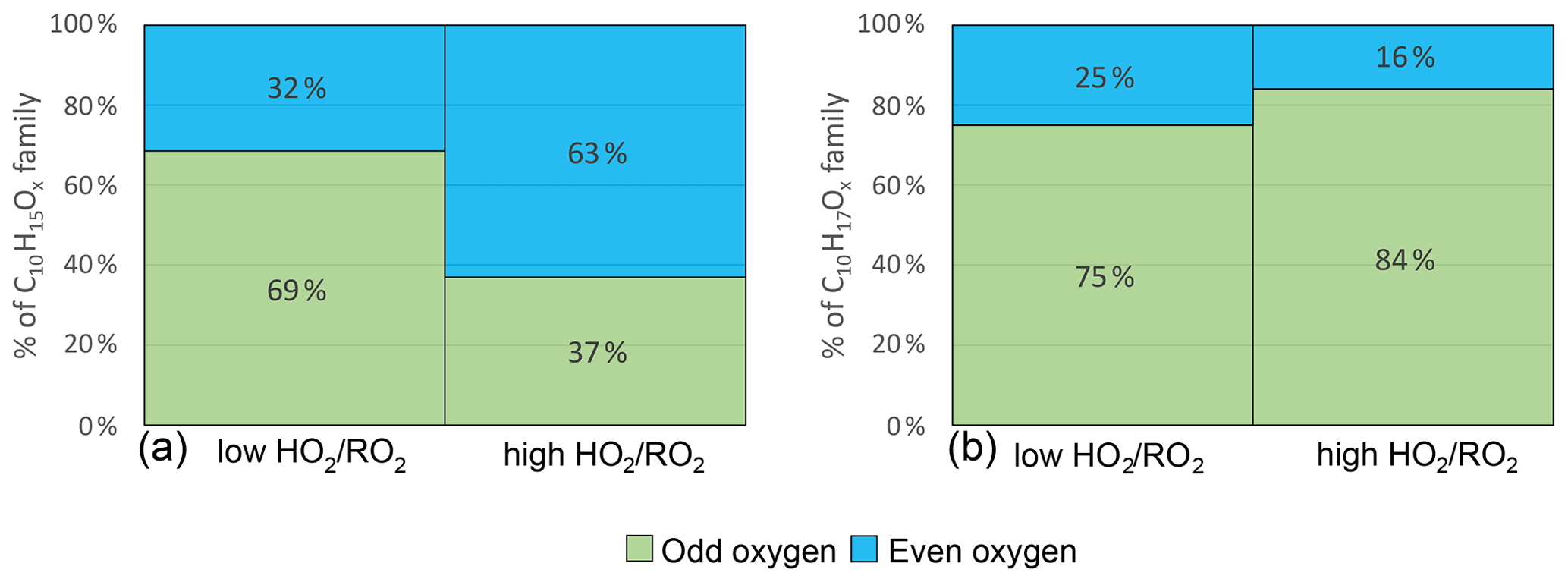
Figure 8Average contribution of Oodd and Oeven to the HOM-RO2 families C10H15Ox (a) and C10H17Ox (b) signal in the low and high cases (unseeded experiments).
At high , C10H15Oeven contributed 63 %, and the C10H15Oodd contribution was reduced to 37 %. This demonstrates a change in the number of alkoxy steps along the formation pathway of the observed HOM-RO2 radicals. The increased contribution of C10H15Oeven at high lets us infer an even number of alkoxy steps as more common (0, 2, 4, ...). In the simplest case, one alkoxy step takes place at low due to HOM-RO formation from HOM-RO2+RO2 reactions, while no alkoxy steps take place at high , because HOM-RO2+HO2 produces none or less HOM-RO than HOM-RO2+RO2.
For C10H17Ox, the entry channel into autoxidation is C10H17O3 with an odd number of oxygen atoms. Therefore, in autoxidation without alkoxy steps the oxygen parity is expected to be odd. At low C10H17Oodd species contribute 75 % to the total C10H17Ox signal indicating that either none or an even number (2, 4, ...) of alkoxy steps occurred. At high the odd contribution increases to 84 % (see Fig. 8). This result could indicate a low occurrence of alkoxy steps even at low , with a further decrease in alkoxy formation at high . However, the observed shift is minor.
In any case, the different responses of the C10H15Ox and C10H17Ox families to the reduction of HOM-RO2 formation from HOM-RO2+RO2 at high indicate that there could be fundamental differences in the autoxidation chains of C10H15Ox and C10H17Ox (or the limit of the parity analysis). The parity analysis indicates a decrease in alkoxy steps at high , but it cannot be directly inferred with certainty. However, the decrease in alkoxy steps at high is supported by the observation of changes in HOM-Frag products.
On average the sum of all HOM-Frag products (detected compounds with 5≥ C < 10 by NO3-CIMS) showed a reduction of around 20 % (unseeded experiments; see Fig. 4). Further trends become recognizable when separating the species according to their carbon number. Figure 9 shows the C5, C7, C8 and C9 HOM-Frag compounds at high compared to the low case, normalized to the α-pinene OH turnover. The fragment group with C6 compounds is not included, as it contributed less than 5 % of the fragment signal and contained few detected compounds.
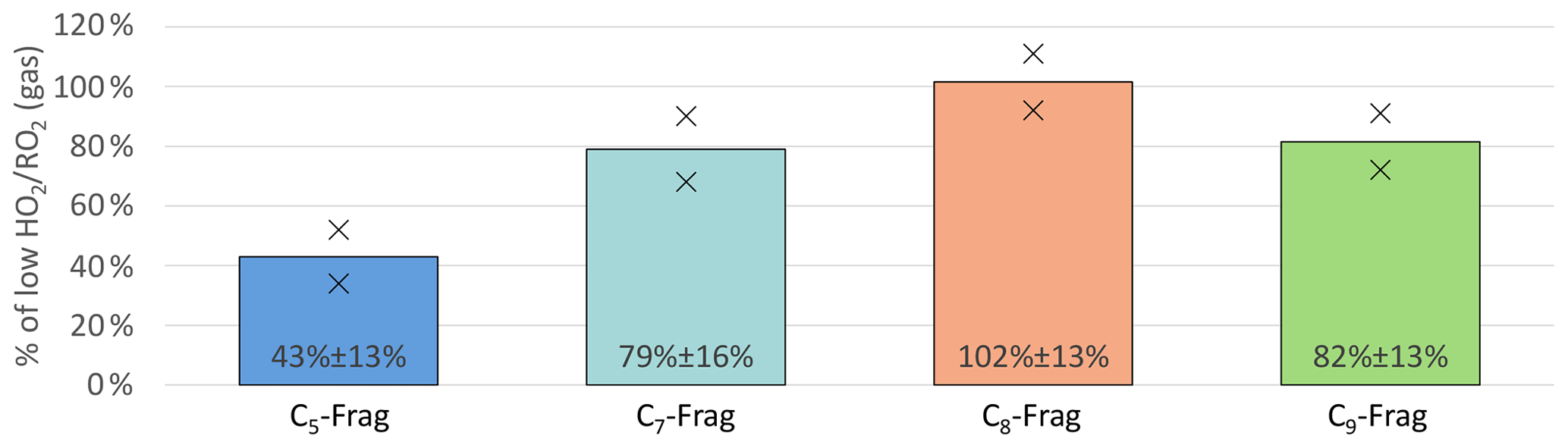
Figure 9Overview of average, relative change in C5, C7, C8 and C9 fragment groups detected in NO3-CIMS between high and low cases (both normalized to α-pinene OH turnover) for unseeded experiments. Bars represent the average of the two experiments; markers represent individual experiments.
Figure 9 shows a significant reduction in HOM-Frag with shorter carbon chain length: C5 HOM-Frag compounds are reduced by around 60 % compared to the low case. When we assume that the fragmentation of C10 compounds happens in consecutive steps via scission of HOM-RO radicals (analogously to the MCM), this observation is in accordance with decreasing importance of alkoxy radical formation at high .
Overall, all observations indicate strong involvement of RO in HOM formation as well as a reduced, but still significant, involvement of RO at high , when HO2 chemistry dominates. This is supported by the change of the oxygen parity in C10-HOM-RO2 and the decrease of fragmentation products, especially with lower carbon number, as well as the only moderate reduction in the observed C10H16O2 product (pinonaldehyde) and the still substantial importance of the C10H15Ox HOM-RO2 family at high .
4.3 Impact on carbonyl and hydroperoxide formation
Increased should shift the product distribution by reduction of alcohol and carbonyl compounds from the so-called molecular channel in the RO2+RO2 reaction (see Reaction R3) in favor of hydroperoxide formation from RO2+HO2 termination (Reaction R1). This effect can be best observed in the C10H18Oz family, which contains the hydroperoxide and alcohol termination products arising from C10H17Ox. C10H18Oz significantly increased to, on average, 159 % (see Fig. 6). This supports an increased hydroperoxide formation but with some uncertainty due to the alcohol termination products from C10H17Ox (by reaction with RO2). To elucidate this further, the contribution of individual species to the C10H18Oz family was examined.
Formation of an alcohol via the molecular path (Reaction R3) leads to the loss of one oxygen atom compared to the precursor C10H17Ox radical, while in the hydroperoxide formation (Reaction R1) the oxygen number remains the same. The most abundant member of the C10H17Ox family was C10H17O7 with a contribution of 72 % ± 6 % at low and a contribution of 82 % ± 1 % at high . C10H17O7 terminates to C10H18Oz products either as an alcohol with sum formula C10H18O6 or as a hydroperoxide with sum formula C10H18O7. These products have additional sources from C10H17O6 and C10H17O8, but due to the dominant contribution of C10H17O7 to the C10H17Ox family we expect any other production channels to be of minor importance.
Figure 10 shows the HOM product distribution within the C10H18Oz family at low and high . The sum of the O6 and O7 product did not change significantly in the two regimes (about 88 %), showing that these are the major products and agreeing well with the observation of C10H17O7 being the major C10H17Ox HOM-RO2. At low , the O6 product has a larger contribution of 64 % ± 8 %, while at high ∼ 30 % of signal is shifted to the O7 product. This shows that the increase in C10H18Oz is matched with an increase in hydroperoxide formation.
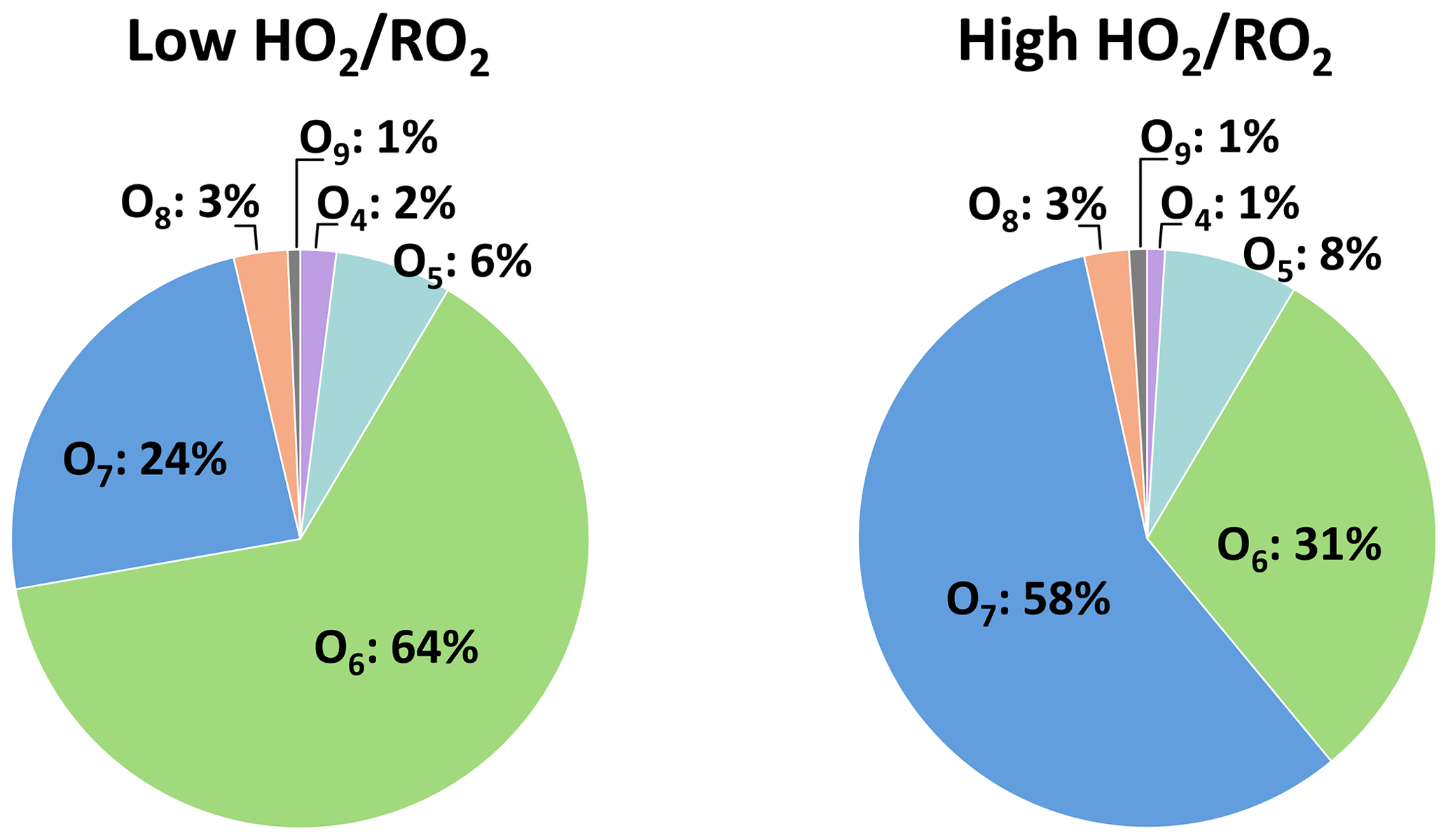
Figure 10Average contribution of the individual compounds to the C10H18Oz family signal at low and high (unseeded experiments).
An indicator for carbonyl formation is the C10H14Oz family as it only contains the carbonyl products arising from C10H15OxRO2. The C10H14Oz family was reduced on average to 61 % at high ; however, this decrease matches the decrease in the C10H15Ox precursor family. If the reaction of a C10H15Ox-HOM-RO2 with a second RO2 were the main formation pathway for C10H14Oz, a stronger reduction would be expected as both precursor species were decreased significantly. Instead, it appears that C10H14Oz is mainly impacted by the decrease in C10H15Ox as their reductions are similar. A possible explanation could be that intramolecular termination is a major reaction pathway for C10H15Ox-RO2, forming C10H14Ox carbonyls. Intramolecular termination of the autoxidation chain has been discussed in the literature for different VOCs (Shen et al., 2021; Guo et al., 2022). Rissanen et al. (2014) discussed the possible importance of the unimolecular termination via an H shift, followed by formation of a carbonyl functional group and OH loss in the autoxidation chain of cyclohexene. Piletic and Kleindienst (2022) calculated fast reaction rate constants in the range of 1–30 s−1 for such intramolecular termination reactions to carbonyls for some C10H17O5 in the α-pinene photooxidation, indicating that this pathway could also be significant for C10H15Ox. However, more investigation is necessary.
The overall contributions of the C10H14Oz, C10H16Oz and C10H18Oz families to the HOM-Mon class at high are shifted as shown in Fig. 11.
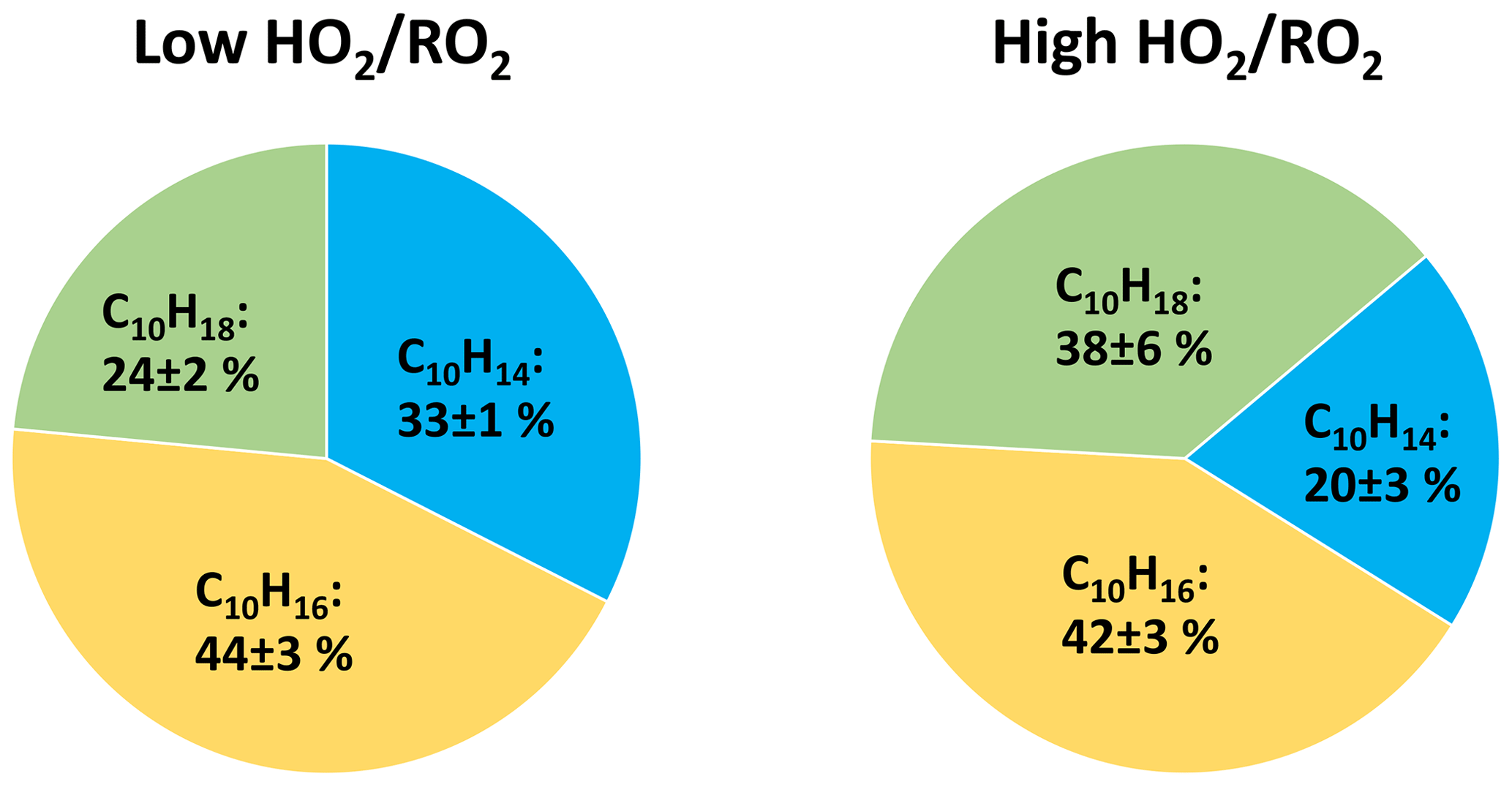
Figure 11Average contribution of the C10H14Oz, C10H16Oz, and C10H18Oz family to the monomer class signal at low and high (unseeded experiments).
The contribution of C10H16Oz is largest and remains similar in both cases, matching the already shown unchanged signal level in Fig. 6. This is the case because the C10H16Oz family contains the alcohols from C10H15Ox+RO2, carbonyls from C10H17Ox+RO2 and hydroperoxides from C10H15Ox+HO2 (see Fig. 1). A separation of the effects of enhanced HO2 on this monomer family is difficult, as for the case where RO2 termination dominates versus the case where HO2 termination dominates, the loss of carbonyls and alcohols is partially compensated by the gain of hydroperoxides. A strong gain in hydroperoxides is clearly reflected in the strong increase in C10H18Oz at high .
Inspection of the C10H14Oz and C10H18Oz families shows that ∼ 13 % of the contribution by C10H14Oz is lost (carbonyls, 33 % at low ) and is present instead as C10H18Oz (hydroperoxides), giving C10H18Oz a contribution of 38 % at high .
4.4 Impact on condensable organic mass
In the previous sections, we demonstrated a shift of the product distribution by the shift from low to high conditions. We also showed that the changes could be rationalized by generic mechanistic considerations. We added (NH4)2SO4 seed aerosol in two experiments to determine how the shift in the product distribution affects the condensable organic mass by determining the fraction which remained in the gas phase after seeding.
Figure 12 shows the fraction remaining for the sum of all products as well as for the individual product classes for the high and the low case. In both cases a significant reduction of products in the gas phase was observed with seed present. Overall, the sum of all products was reduced by around 60 %, with a slightly higher reduction in the low case. This can be attributed to the larger importance of HOM-Acc in the low case, as well as to a 10 % lower reduction of the HOM-Frag in the high case. In both cases a reduction of the HOM-RO2 is observed, which indicates that the provided particle sink could have affected HOM formation chemistry but only moderately.
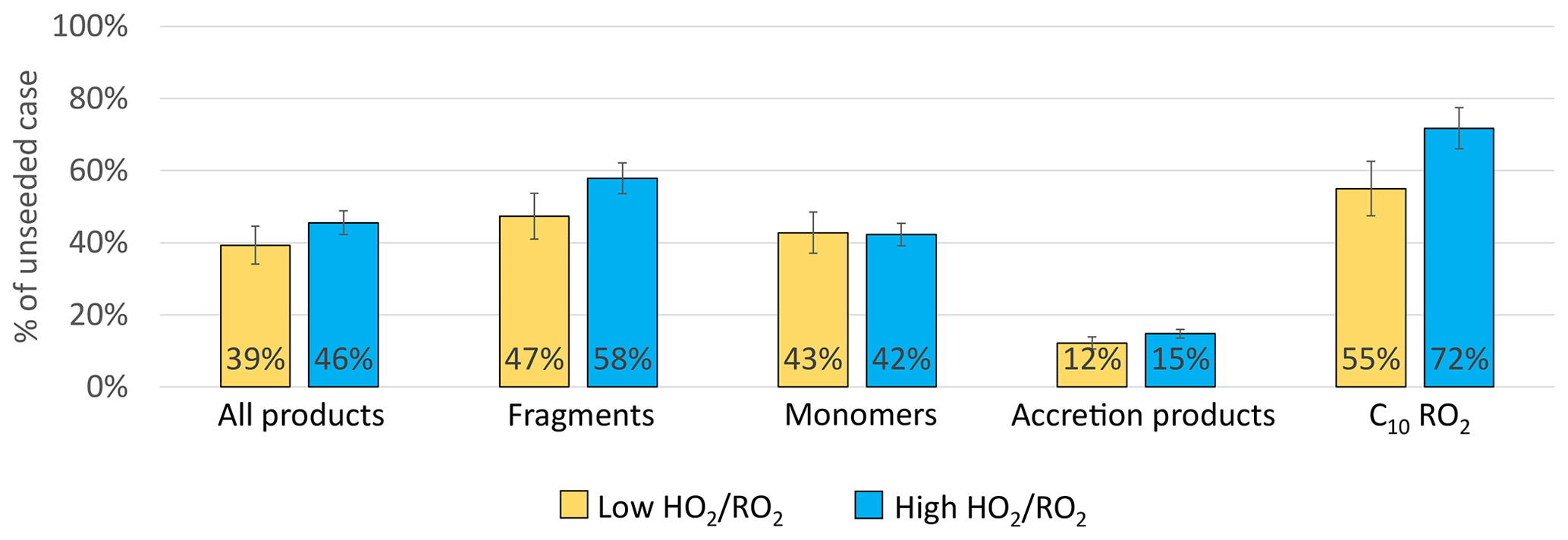
Figure 12Overview of average, relative change in product classes signal between gas phase only and seeded system. Blue shows the high case, yellow the low case. (All are normalized to α-pinene OH turnover, Exp2 experiment).
The total organic particulate mass was determined by AMS measurements and was 2.0 and 3.4 µg m−3 at high and low in the experiment (Exp2) displayed in Fig. 12. A reduction of condensed organic mass to 73 % ± 3 % at high (orange bar in Fig. 14) was observed on average. Since non-seeded and seeded experiments were conducted at otherwise the same conditions and we did not observe significant new particle formation, the gas-phase compositions can be directly compared. Therefore, we conclude that the shift in the product distribution led to a reduction of condensable material at the same α-pinene turnover with OH (and O3).
We calculated the wall-loss-corrected SOA yields with the corrected SOA mass as shown in Eq. (4) and as described by Sarrafzadeh et al. (2016). To this end, we used C10H16O7 as the lead HOM compound. In the two experiments with seed present (Exp2.1 and Exp3) we had SOA yields of 7.3 % and 10.0 % at high and 10.0 % and 12.8 % at low . The difference in the SOA yields between experiments can be explained by the slightly different OH concentrations and subsequent difference in contribution by photooxidation (see Table 1). Overall, our yields are in the lower range in comparison with the SOA yields reported by McFiggans et al. (2019) for the α-pinene photooxidation. However, our experiments were also performed at 5 °C higher temperature (20 °C) compared to 15 °C in McFiggans et al. (2019). The SOA yields show an absolute reduction of ∼ 3 % at high compared to low (relative a reduction of about 30 %). A reduction of the SOA yield of α-pinene by addition of CO was described before by McFiggans et al. (2019); however, there the α-pinene OH turnover was not held constant.
The change from the low to high regime favored termination reactions to protic termination groups, as we observed less carbonyl compounds and more hydroperoxides. This could, overall, shift the product distribution to products with lower vapor pressures and favor SOA formation, since protic groups can act as hydrogen bond donors as well as hydrogen bond acceptors (as exemplified by the comparison of ethanol (boiling point (b.p.) 78 °C) and ethane hydroperoxide (b.p. 93–97 °C) with acetaldehyde (b.p. 20 °C) (Richter et al., 1955)). However, the effect of the termination group should be small for HOMs as they likely contain multiple hydroperoxide groups (compare Pullinen et al., 2020). The reduction in HOM-Acc is expected to decrease the condensable mass, since the HOM-Acc scavenge non-HOM-RO2 that would otherwise not partition into the particle phase.
Which of the measured compounds contribute significantly to the organic particle mass can be inferred by comparing their signal from the pure gas-phase, unseeded cases to their signal with seed in the system. Under the assumptions that for most HOM compounds re-evaporation to the gas phase is negligible and that the precursor chemistry is not substantially disturbed by seed addition, the fraction of signal remaining with seed in the system reflects to which degree the compound is condensing. Figure 13 shows the fraction remaining with seed in the system plotted against the molar mass of each individual compound. The plot includes all closed-shell products that were measured with a relative standard deviation of less than 30 % for all measurement phases and depicts the results for both the high and low cases.
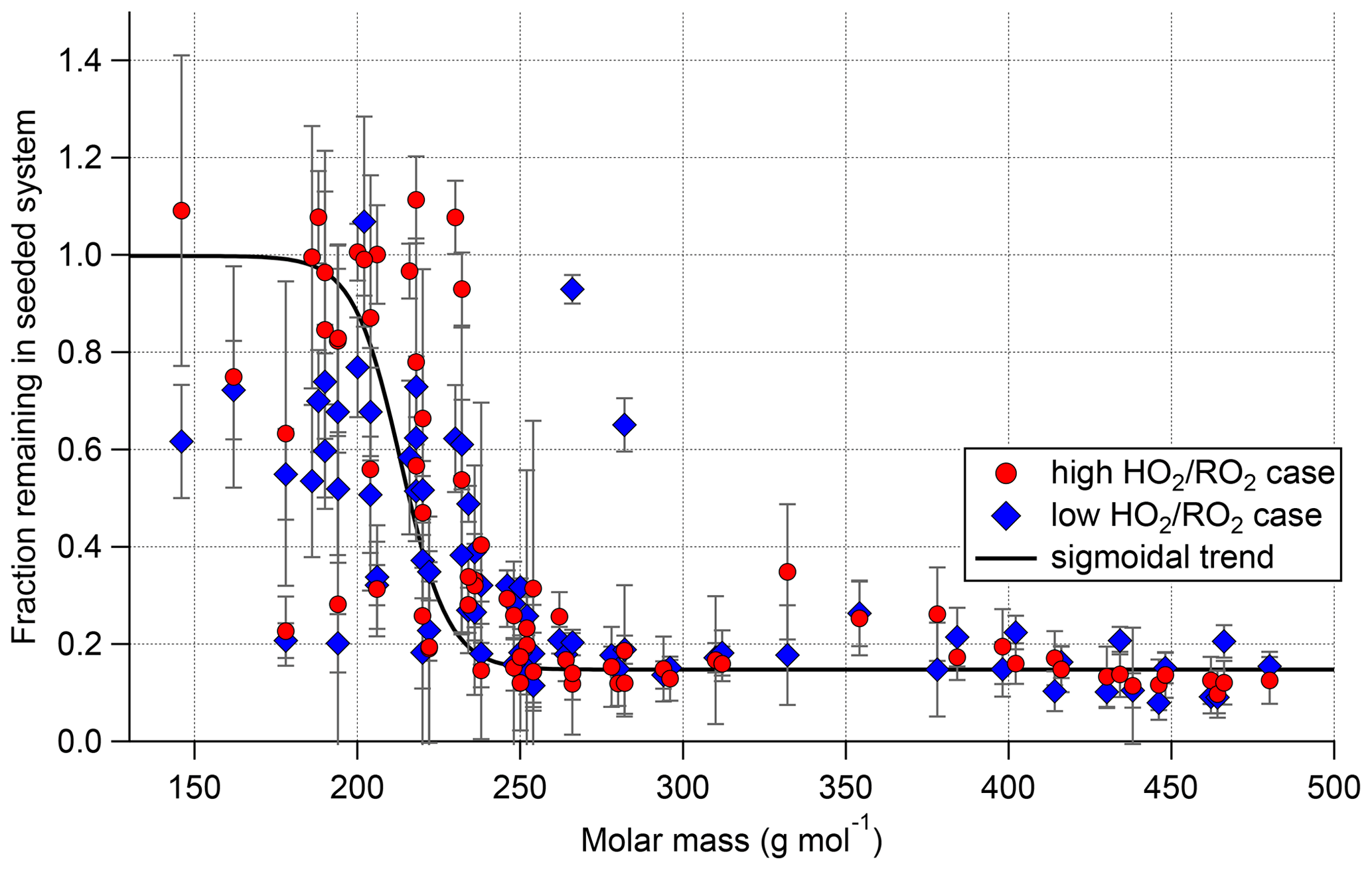
Figure 13Gas-phase fraction remaining in the presence of seed (normalization of all data with α-pinene OH turnover) for the low (blue) and high (red) cases. Displayed points represent all closed-shell compounds that were detected with relative standard deviation <30 % in all four experiment phases. Error bars represent result of error propagation (see Supplement Sect. S9).
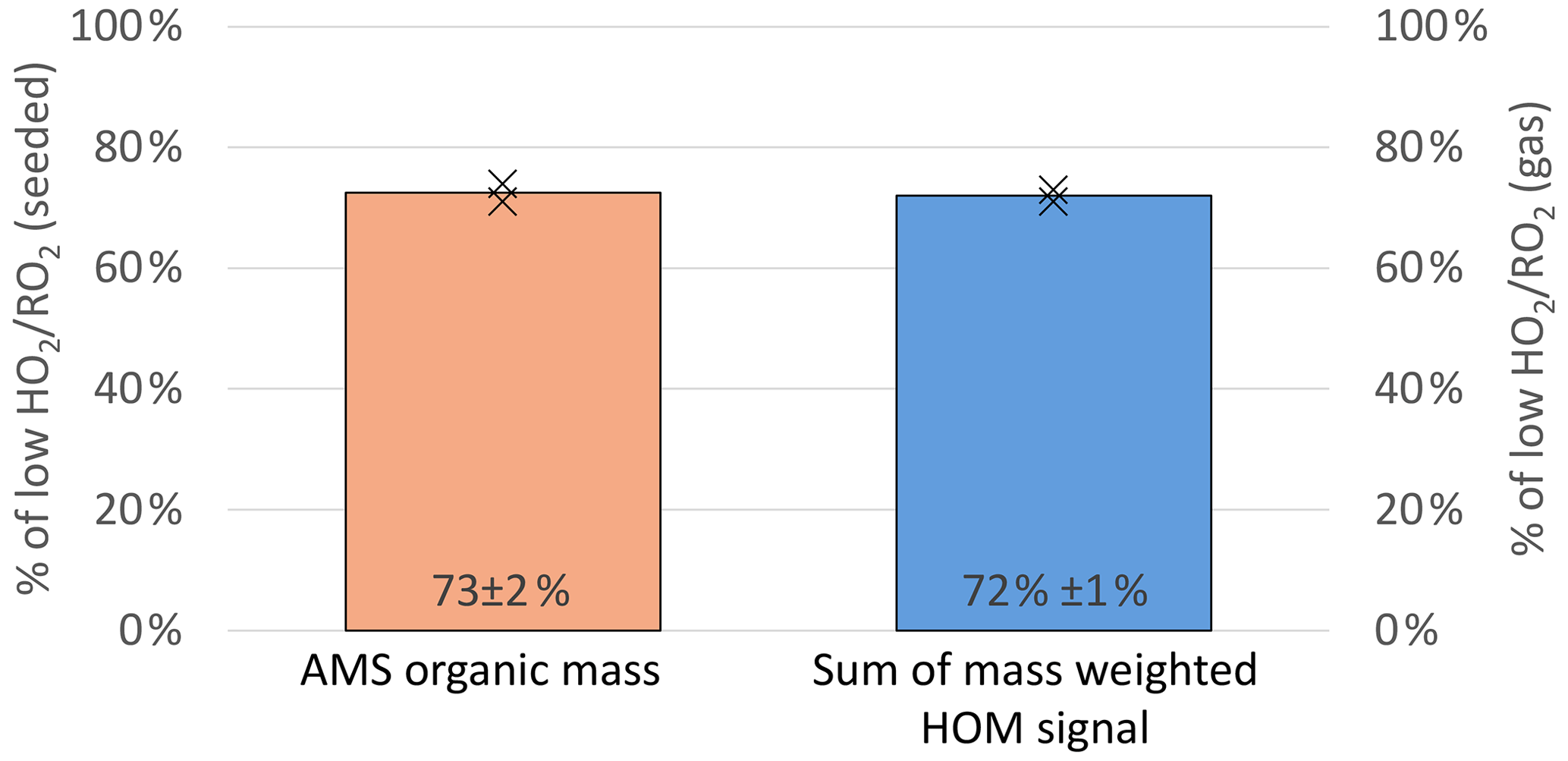
Figure 14Overview of the average, relative change in organic mass observed in the AMS (left y axis, seeded experiments) and the mass-weighted HOM signal observed in the NO3-CIMS (right y axis, unseeded experiments) between the low and high cases (both normalized to α-pinene OH turnover).
Overall, in both cases we observed the same trend. Lighter compounds are not affected by the presence of seed particles, but with increasing molar mass the fraction remaining in the gas phase is reduced. A difference between the low and high cases can be observed in the low molar mass range: in the high case many fragmentation products show a higher gas-phase fraction remaining up to 1. (In some cases, values larger than 1 were observed but within the error limits. For the error estimation, see Supplement Sect. S9.) Fractions remaining larger than 1 beyond error could be an indication that such products have a particle-phase production source. Figure 13 also shows a critical semi-volatile to low-volatility organic compound molar mass region for molar masses between 175 and 250 g mol−1 where neither a fraction remaining of 1 nor complete condensation was observed. The position of this region on the molar mass scale depends on the provided organic mass concentration. The large variation of the fraction remaining in this small range of molar masses shows that the partitioning coefficients are dependent on the detailed structure of the compounds and not simply on their molar mass. The semi-volatile and low-volatility products represent mainly higher oxidized fragments and HOM-Mon with less than eight oxygens.
For compounds with a molar mass larger than 250 g mol−1, a constant fraction remaining is reached in steady state, which is due to an ongoing production of the compounds. From the condensation behavior shown in Fig. 13, we conclude that the compounds heavier than 230 g mol−1 are expected to be of sufficiently low volatility to be mainly found in the particle phase for the organic mass present in the system and therefore contribute significantly to the SOA mass formation. Our finding agrees with the threshold used for low-volatility HOM products in Pullinen et al. (2020).
Therefore, the signal of all compounds with a molar mass heavier than 230 g mol−1 was weighted with their molar mass and summed (see Eq. 2). The ratio of this weighted signal sum at low and high is then a measure of expected SOA mass loss. The calculation leads to an expected reduction to 72 % (blue bar, Fig. 14). This simplified approach leads to a good agreement with the AMS measurements and can thus explain the reduced particulate organic mass within the errors.
To test for closure between HOM lost and particulate organic mass measured, we approximated the upper limit of HOM concentration in the condensed phase. For this calculation, we used the calibration factor determined for sulfuric acid for our NO3-MION-CIMS (7.0×109 molecules cm−3 ncps−1, where ncps represents normalized counts per second) and the relationship between gas and particulate concentration of a compound in the SAPHIR-STAR chamber described in Eq. (3). Again, we considered all compounds with M>230 g mol−1 in our calculation. The summed mass concentration lost from the gas phase was then compared to the SOA mass measured in the AMS. This comparison yields a good agreement within the uncertainties. The detailed calculation results can be found in the Supplement (Fig. S3). Overall, an agreement within 40 % is achieved for all measurement stages.
The comparisons presented above show that we understand the processes governing the SOA formation in our chamber and that the NO3-CIMS measurements are well suited to observe the critical changes to understand the reduction in condensable organic material when shifting from low to high .
In the presented series of experiments, we achieved a shift from a RO2+RO2-dominated chemistry to a more atmospherically relevant ratio under constant α-pinene OH turnover. It was shown that moving towards atmospheric ratio affected the SOA formation potential, with the observed organic mass being reduced at high . This is in support of the potential bias towards high SOA yields in chamber studies at low as discussed by Schervish and Donahue (2021). Our results confirm that too low is one important parameter that can lead to an overestimated SOA yield in laboratory studies. In a broader picture, the results show how important it is to consider the different contributions to the HOM-RO2 sink (e.g., HO2, RO2, NO) when designing experiments and transferring laboratory results to the real atmosphere.
The gas-phase observations showed that the SOA reduction at high was mainly due to a reduced HOM-Acc formation, which were formed by RO2+RO2 cross-reactions in the low case. This prevented contribution to SOA by less oxidized RO2 compounds which were scavenged in the HOM-Acc at low . Under atmospheric conditions, such cross-reactions are less important, and such (mixed) accretion products would contribute less to SOA.
The overall observed HOM products were reduced slightly, showing that under certain circumstances RO2+HO2 termination can impede the HOM formation, mainly by reducing the precursor RO2 levels and less by impeding the autoxidation itself. The autoxidation chain (once initiated) runs to a similar oxidation level at both high and low . The observed HOM-Mon products shift significantly between monomer families due to the different termination reactions. A decrease in carbonyl and alcohol formation from RO2+RO2 and an increase in hydroperoxide formation from RO2+HO2 were observed at high .
Furthermore, a reduction in HOM-Frag products, especially with lower carbon numbers, and a reduction in the parity of the C10H15Ox HOM-RO2 show a reduction in alkoxy radical formation at high . The moderate reduction in larger HOM-Frag products and pinonaldehyde, however, suggests that some alkoxy radical steps are still important. This raises the question of whether alkoxy radical formation can be facilitated by HO2. In the atmosphere such effects are most often overcome whenever RO2+NO is the major alkoxy radical source.
Overall, the observed changes in the gas phase could be well explained with the presented generic mechanistic understanding of HOM formation in the α-pinene system. The addition of seed particles demonstrated that the shift towards high reduced the condensable organic mass, stressing the importance of controlling higher-order reactions of peroxy radicals which lead to overemphasis on HOM-Acc product formation at low ratios.
Furthermore, the seed addition allowed us to determine which products were contributing to the SOA formation and show that their volatility is a function of molar mass and detailed molecular structure. This revealed a critical molar mass region in which compounds have significant fractions in gas and particulate phases. Based on absorptive partitioning theory, the volatilities at which this critical region is found should depend on the organic mass present in the system.
Valuable insight about the condensed phase can be gained from HOM gas-phase measurements. We inferred conclusions about the particulate phase from the gas-phase measurements and compared them to the direct particle-phase observations, finding good agreements between our expectations and the measurements.
Information about all steady-state conditions utilized and the data necessary to reproduce the figures in this study are available at https://doi.org/10.26165/JUELICH-DATA/R8ITFF (Baker et al., 2024).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-4789-2024-supplement.
TFM, MH and GM conceptualized the study, and TFM, YB, SK and SRZ designed the experiments and developed the analysis methodology. The experiments were performed by YB, SK, VG and SRZ. Instrument deployment and/or data analysis were performed by YB, SK, HW, RW, JX, AZ, QH, TZ and VG. YB did model calculations of the experiments. AV, SPO, TJB, MG and MH provided counsel on experiment design and data interpretation. The compiled data set was interpreted by YB and TFM, and the results were discussed by all co-authors. YB visualized the data, and YB and TFM prepared the manuscript. All co-authors reviewed the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This research has received funding from the European Union's Horizon 2020 research and innovation programme under the FORCeS project under grant agreement no. 821205, the German Federal Ministry of Education and Research (BMBF) under the FONA Strategy “Research for Sustainability” as part of the implementation of ACTRIS-D under the funding code 01LK200010, Vetenskapsrådet (VR, grant agreement no. 2018-04430), Svenska Forskningsrådet Formas (grant agreement no. 2019-586) and the UK Natural Environment Research Council (NERC) under grant agreement no. NE/V012665/1.
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Sergey A. Nizkorodov and reviewed by two anonymous referees.
Albrecht, S. R., Novelli, A., Hofzumahaus, A., Kang, S., Baker, Y., Mentel, T., Wahner, A., and Fuchs, H.: Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 12, 891–902, https://doi.org/10.5194/amt-12-891-2019, 2019.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Baker, Y., Kang, S., Wang, H., Wu, R., Xu, J., Zanders, A., He, Q., Hohaus, T., Ziehm, T., Geretti, V., Bannan, T. J., O'Meara, S. P., Voliotis, A., Hallquist, M., McFiggans, G., Zorn, S. R., Wahner, A., Mentel, T.: ”Supplementary data for “Impact of HO2/RO2 ratio on highly oxygenated α-pinene photooxidation products and secondary organic aerosol formation potential”, Jülich DATA [data set], V1, https://doi.org/10.26165/JUELICH-DATA/R8ITFF, 2024.
Berndt, T.: Peroxy Radical Processes and Product Formation in the OH Radical-Initiated Oxidation of α-Pinene for Near-Atmospheric Conditions, J. Phys. Chem. A, 125, 9151–9160, https://doi.org/10.1021/acs.jpca.1c05576, 2021.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjaer, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipila, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion product formation from ozonolysis and OH radical reaction of α-pinene: mechanistic insight and the influence of isoprene and ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018.
Bianchi, F., Garmash, O., He, X., Yan, C., Iyer, S., Rosendahl, I., Xu, Z., Rissanen, M. P., Riva, M., Taipale, R., Sarnela, N., Petäjä, T., Worsnop, D. R., Kulmala, M., Ehn, M., and Junninen, H.: The role of highly oxygenated molecules (HOMs) in determining the composition of ambient ions in the boreal forest, Atmos. Chem. Phys., 17, 13819–13831, https://doi.org/10.5194/acp-17-13819-2017, 2017.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Cox, R. A., Ammann, M., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 20, 13497–13519, https://doi.org/10.5194/acp-20-13497-2020, 2020.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Docherty, K. S. and Ziemann, P. J.: Effects of stabilized criegee intermediate and OH radical scavengers on aerosol formation from reactions of β-pinene with O3, Aerosol Sci. Tech., 37, 877–891, https://doi.org/10.1080/02786820300930, 2003.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 1: Gas-phase composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 6489–6504, https://doi.org/10.5194/acp-12-6489-2012, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Eisele, F. and Tanner, D.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.-Atmos., 98, 9001–9010, https://doi.org/10.1029/93JD00031, 1993.
Fantechi, G., Vereecken, L., and Peeters, J.: The OH-initiated atmospheric oxidation of pinonaldehyde: Detailed theoretical study and mechanism construction, Phys. Chem. Chem. Phys., 4, 5795–5805, https://doi.org/10.1039/B205901K 2002.
Guo, Y., Shen, H., Pullinen, I., Luo, H., Kang, S., Vereecken, L., Fuchs, H., Hallquist, M., Acir, I.-H., Tillmann, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., Zhao, D., and Mentel, T. F.: Identification of highly oxygenated organic molecules and their role in aerosol formation in the reaction of limonene with nitrate radical, Atmos. Chem. Phys., 22, 11323–11346, https://doi.org/10.5194/acp-22-11323-2022, 2022.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hantschke, L. L.: Oxidation of monoterpenes studied in atmospheric simulation chambers, Forschungszentrum Jülich GmbH, Zentralbibliothek, Verlag, ISBN 978-3-95806-653-3, 2022.
Hasson, A. S., Kuwata, K. T., Arroyo, M. C., and Petersen, E. B.: Theoretical studies of the reaction of hydroperoxy radicals (HO2) with ethyl peroxy (CH3CH2O2), acetyl peroxy (CH3C(O)O2), and acetonyl peroxy (CH3C(O)CH2O2) radicals, J. Photochem. Photobiol. A, 176, 218–230, https://doi.org/10.1016/j.jphotochem.2005.08.012, 2005.
Henry, K. M., Lohaus, T., and Donahue, N. M.: Organic aerosol yields from α-pinene oxidation: bridging the gap between first-generation yields and aging chemistry, Environ. Sci. Technol., 46, 12347–12354, https://doi.org/10.1021/es302060y, 2012.
Hidy, G.: Atmospheric chemistry in a box or a bag, Atmos., 10, 401, https://doi.org/10.3390/atmos10070401, 2019.
Hyttinen, N., Otkjær, R. V., Iyer, S., Kjaergaard, H. G., Rissanen, M. P., Wennberg, P. O., and Kurtén, T.: Computational comparison of different reagent ions in the chemical ionization of oxidized multifunctional compounds, J. Phys. Chem. A, 122, 269–279, https://doi.org/10.1021/acs.jpca.7b10015, 2018.
Iyer, S., Reiman, H., Møller, K. H., Rissanen, M. P., Kjaergaard, H. G., and Kurtén, T.: Computational investigation of RO2+ HO2 and RO2+ RO2 reactions of monoterpene derived first-generation peroxy radicals leading to radical recycling, J. Phys. Chem. A, 122, 9542–9552, https://doi.org/10.1021/acs.jpca.8b09241, 2018.
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 12, 878, https://doi.org/10.1038/s41467-021-21172-w, 2021.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/B704260B 2008.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kang, S.: Formation of highly oxygenated organic molecules from α-pinene photochemistry, Forschungszentrum Jülich GmbH, 156 pp., https://doi.org/10.25926/rd20-nb07, 2021.
Keywood, M., Kroll, J., Varutbangkul, V., Bahreini, R., Flagan, R., and Seinfeld, J.: Secondary organic aerosol formation from cyclohexene ozonolysis: Effect of OH scavenger and the role of radical chemistry, Environ. Sci. Technol., 38, 3343–3350, https://doi.org/10.1021/es049725j, 2004.
Khan, M., Cooke, M., Utembe, S., Archibald, A., Derwent, R., Jenkin, M. E., Morris, W., South, N., Hansen, J., Francisco, J., Percival, C. J., and Shallcross, D. E.: Global analysis of peroxy radicals and peroxy radical-water complexation using the STOCHEM-CRI global chemistry and transport model, Atmos. Environ., 106, 278–287, https://doi.org/10.1016/j.atmosenv.2015.02.020, 2015.
Kiendler-Scharr, A., Wildt, J., Maso, M. D., Hohaus, T., Kleist, E., Mentel, T. F., Tillmann, R., Uerlings, R., Schurr, U., and Wahner, A.: New particle formation in forests inhibited by isoprene emissions, Nature, 461, 381–384, https://doi.org/10.1038/nature08292, 2009.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, A. M., Simpson, D., Bergstrom, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Mentel, Th. F., Wildt, J., Kiendler-Scharr, A., Kleist, E., Tillmann, R., Dal Maso, M., Fisseha, R., Hohaus, Th., Spahn, H., Uerlings, R., Wegener, R., Griffiths, P. T., Dinar, E., Rudich, Y., and Wahner, A.: Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 9, 4387–4406, https://doi.org/10.5194/acp-9-4387-2009, 2009.
Mohr, C., Thornton, J. A., Heitto, A., Lopez-Hilfiker, F. D., Lutz, A., Riipinen, I., Hong, J., Donahue, N. M., Hallquist, M., Petaja, T., Kulmala, M., and Yli-Juuti, T.: Molecular identification of organic vapors driving atmospheric nanoparticle growth, Nat. Commun., 10, 4442, https://doi.org/10.1038/s41467-019-12473-2, 2019.
Otkjaer, R. V., Jakobsen, H. H., Tram, C. M., and Kjaergaard, H. G.: Calculated Hydrogen Shift Rate Constants in Substituted Alkyl Peroxy Radicals, J. Phys. Chem. A, 122, 8665–8673, https://doi.org/10.1021/acs.jpca.8b06223, 2018.
Piletic, I. R. and Kleindienst, T. E.: Rates and yields of unimolecular reactions producing highly oxidized peroxy radicals in the OH-induced autoxidation of α-pinene, β-pinene, and limonene, J. Phys. Chem. A, 126, 88–100, https://doi.org/10.1021/acs.jpca.1c07961, 2022.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Richter, F., Ostertag, R., Ammerlahn, G., Behrle, E., Baumann, M., and Kobel, M. (Eds.): Beilstein's handbook of organic chemistry. Third supplement, covering the literature from 1930–1949, Beilstein's handbook of organic chemistry, Springer, Berlin, 1955.
Rissanen, M. P., Mikkilä, J., Iyer, S., and Hakala, J.: Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications, Atmos. Meas. Tech., 12, 6635–6646, https://doi.org/10.5194/amt-12-6635-2019, 2019.
Rissanen, M. P., Kurtén, T., Sipila, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jorgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petaja, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Roldin, P., Ehn, M., Kurtén, T., Olenius, T., Rissanen, M. P., Sarnela, N., Elm, J., Rantala, P., Hao, L., Hyttinen, N., Heikkinen, L., Worsnop, D. R., Pichelstorfer, L., Xavier, C., Clusius, P., Öström, E., Petäjä, T., Kulmala, M., Vehkamäki, H., Virtanen, A., Riipinen, I., and Boy, M.: The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system, Nat. Commun., 10, 4370, https://doi.org/10.1038/s41467-019-12338-8, 2019.
Sanchez, J., Tanner, D. J., Chen, D., Huey, L. G., and Ng, N. L.: A new technique for the direct detection of HO2 radicals using bromide chemical ionization mass spectrometry (Br-CIMS): initial characterization, Atmos. Meas. Tech., 9, 3851–3861, https://doi.org/10.5194/amt-9-3851-2016, 2016.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schervish, M. and Donahue, N. M.: Peroxy radical kinetics and new particle formation, Environ. Sci. Atmos., 1, 79–92, https://doi.org/10.1039/d0ea00017e, 2021.
Shen, H., Zhao, D., Pullinen, I., Kang, S., Vereecken, L., Fuchs, H., Acir, I. H., Tillmann, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Highly Oxygenated Organic Nitrates Formed from NO(3) Radical-Initiated Oxidation of β-Pinene, Environ. Sci. Technol., 55, 15658–15671, https://doi.org/10.1021/acs.est.1c03978, 2021.
Shen, H., Vereecken, L., Kang, S., Pullinen, I., Fuchs, H., Zhao, D., and Mentel, T. F.: Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Sci. Adv., 8, eabp8702, https://doi.org/10.1126/sciadv.abp8702, 2022.
Shilling, J. E., Chen, Q., King, S. M., Rosenoern, T., Kroll, J. H., Worsnop, D. R., DeCarlo, P. F., Aiken, A. C., Sueper, D., Jimenez, J. L., and Martin, S. T.: Loading-dependent elemental composition of α-pinene SOA particles, Atmos. Chem. Phys., 9, 771–782, https://doi.org/10.5194/acp-9-771-2009, 2009.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Vereecken, L., Müller, J.-F., and Peeters, J.: Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of α-pinene: impact of non-traditional peroxyl radical chemistry, Phys. Chem. Chem. Phys., 9, 5241–5248, https://doi.org/10.1039/b708023a, 2007.
Wildt, J., Mentel, T. F., Kiendler-Scharr, A., Hoffmann, T., Andres, S., Ehn, M., Kleist, E., Müsgen, P., Rohrer, F., Rudich, Y., Springer, M., Tillmann, R., and Wahner, A.: Suppression of new particle formation from monoterpene oxidation by NOx, Atmos. Chem. Phys., 14, 2789–2804, https://doi.org/10.5194/acp-14-2789-2014, 2014.
Xu, L., Møller, K. H., Crounse, J. D., Otkjær, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular reactions of peroxy radicals formed in the oxidation of α-pinene and β-pinene by hydroxyl radicals, J. Phys. Chem. A, 123, 1661–1674, https://doi.org/10.1021/acs.jpca.8b11726, 2019.