the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Apr 2024
| 17 Apr 2024
Formation and loss of light absorbance by phenolic aqueous SOA by ●OH and an organic triplet excited state
Stephanie Arciva
Lan Ma
Camille Mavis
Chrystal Guzman
Brown carbon (BrC) is an important component of biomass-burning (BB) emissions that impacts Earth's radiation budget. BB directly emits primary BrC as well as gaseous phenolic compounds (ArOH), which react in the gas and aqueous phases with oxidants – such as hydroxyl radical (•OH) and organic triplet excited states (3C∗) – to form light-absorbing secondary organic aerosol (SOA). These reactions in atmospheric aqueous phases, such as cloud/fog drops and aerosol liquid water (ALW), form aqueous SOA (aqSOA), i.e., low-volatility, high-molecular-weight products. While these are important routes of aqSOA formation, the light absorption and lifetimes of the BrC formed are poorly characterized. To study these aspects, we monitored the formation and loss of light absorption by aqSOA produced by reactions of six highly substituted phenols with •OH and 3C∗. While the parent phenols absorb very little tropospheric sunlight, they are oxidized to aqSOA that can absorb significant amounts of sunlight. The extent of light absorption by the aqSOA depends on both the ArOH precursor and oxidant: more light-absorbing aqSOA is formed from more highly substituted phenols and from triplet reactions rather than •OH. Under laboratory conditions, extended reaction times in •OH experiments diminish sunlight absorption by aqSOA on timescales of hours, while extended reaction times in 3C∗ experiments reduce light absorption much more slowly. Estimated lifetimes of light-absorbing phenolic aqSOA range from 3 to 17 h in cloud/fog drops, where •OH is the major sink, and from 0.7 to 8 h in ALW, where triplet excited states are the major sink.
- Article
(1301 KB) - Full-text XML
-
Supplement
(1934 KB) - BibTeX
- EndNote





Organic aerosols (OAs) account for 20 % to 90 % of airborne particulate mass (Jimenez et al., 2009; Kanakidou et al., 2005), with significant effects on climate (Heald et al., 2008; Shrivastava et al., 2017) and human health (Aguilera et al., 2021; Kim et al., 2015; Zhou et al., 2021). Types of OAs include primary OA (POA), which is directly emitted from sources such as biomass burning and fossil fuel combustion (Andreae, 2019; Bond et al., 2004), and secondary OA (SOA), which is formed from atmospheric aging in the gas or condensed phases (Graber and Rudich, 2006; Jimenez et al., 2009; Powelson et al., 2014; Volkamer et al., 2006). SOA usually accounts for a dominant fraction of the OA budget (Hallquist et al., 2009; Robinson et al., 2007). Biomass burning (BB) is a significant source of POA as well as reactive organic gases that form SOA (Akagi et al., 2011; Andreae, 2019; Bond et al., 2004; Hallquist et al., 2009), which together affect air quality, atmospheric composition, and climate (Garofalo et al., 2019; Kleinman et al., 2020; O'Dell et al., 2021).
While most OA scatters sunlight, brown carbon (BrC, i.e., light-absorbing organic carbon) absorbs sunlight and possibly causes warming (Jo et al., 2016; Reid et al., 2005; Zhang et al., 2020). Compared to black carbon, which strongly absorbs sunlight across the UV–Vis spectrum, light absorption by BrC is strongly wavelength dependent, increasing from the visible to the UV (Hecobian et al., 2010; Laskin et al., 2015). BB is an important source of BrC that strongly absorbs light (Di Lorenzo et al., 2017; Kirchstetter and Thatcher, 2012; Palm et al., 2020; Zhong and Jang, 2014). POA and SOA formed from smog chamber experiments of biomass burning emissions show evidence of light absorption by BrC, especially in the near-UV region (Saleh et al., 2013). The absorption of BrC is affected by atmospheric oxidation, exposure to sunlight, and pH, as well as brown carbon source (Grieshop et al., 2009; Hennigan et al., 2011; Liu et al., 2021; Ortega et al., 2013). Also, atmospheric aging can increase both light absorption by forming chromophores and photo-bleach chromophoric molecules (Fleming et al., 2020; Hems et al., 2020, 2021; Schnitzler et al., 2022; Zhao et al., 2015; Zhong and Jang, 2014).
One important class of reactive organic gases potentially important for secondary BrC formation is phenols, which we abbreviate as ArOH (Andreae, 2019; Chang and Thompson, 2010). ArOH are abundant emissions from BB, with high emissions of simple phenols and smaller amounts of highly substituted ArOH (Andreae, 2019; Schauer et al., 2001). ArOH typically have strong absorbance peaks under 300 nm that fall exponentially at longer wavelengths (Kaeswurm et al., 2021), although phenols with carbonyl or nitro substituents can absorb sunlight (Smith et al., 2016; Wang et al., 2022). Phenols rapidly undergo photochemical transformations both in the gas and aqueous phases with various oxidants to produce low-volatility compounds (Berndt and Böge, 2003; Gurol and Nekouinaini, 1984; Ma et al., 2021; Sun et al., 2010; Yee et al., 2013; Yu et al., 2014). ArOH from BB have a wide range of Henry's law constants (KH). Highly substituted ArOH with modest to high KH values (103–108 M atm−1) will have a higher tendency to partition into the aqueous phase (McFall et al., 2020), where they can be oxidized to form large low-volatility products (Smith et al., 2016; Yu et al., 2016).
Atmospheric aqueous oxidants that rapidly oxidize ArOH during the daytime include hydroxyl radical (•OH) and organic triplet excited states (3C∗) (Smith et al., 2015). Both simple and highly substituted phenols react rapidly with •OH and 3C∗, with second-order rate constants of roughly 109 and 108 M−1 s−1 at pH 5, respectively (Arciva et al., 2022; Buxton et al., 1988; Ma et al., 2021; Smith et al., 2015). Additionally, both the •OH and 3C∗ reactions efficiently form aqueous SOA (aqSOA), with average (±1σ) mass yields of 82(±12) % and 83(±14) %, respectively (Arciva et al., 2022; Ma et al., 2021). SOA mass yields for ArOH oxidation in the aqueous phase are often higher than the parallel gas-phase reactions (Arciva et al., 2022; Berndt and Böge, 2003; Coeur-Tourneur et al., 2010; Ma et al., 2021), a result of more efficient oligomerization and functionalization reactions in the aqueous phase (Yu et al., 2016) as opposed to the more common fragmentation in the gas phase. Additionally, aqueous reactions produce oligomers, i.e., low-volatility, conjugated products that can significantly enhance light absorption (Chang and Thompson, 2010; Huang et al., 2018; Li et al., 2014; Yu et al., 2016).
While the processing of ArOH is a significant source of light-absorbing aqSOA, little is known about how phenol-derived brown carbon is photobleached with continued reaction. In part, this likely depends on which oxidant produced the aqSOA, as reaction products can be quite different. Initial oxidation of ArOH by •OH and 3C∗ forms aqSOA that absorbs light above 300 nm (Jiang et al., 2023b; Li et al., 2022). ArOH reactions with •OH initially produce abundant hydroxylated analogues, while 3C∗ oxidation of ArOH dominantly forms oligomers (Misovich et al., 2021; Yu et al., 2016). However, the extended aging processes for phenolic brown carbon may vary since chromophores of different chemical composition react differently to photooxidation (Li et al., 2022). In cloud and fog conditions, extended •OH reactions transform phenolic aqSOA chromophores more rapidly compared to reactions with 3C∗ (Jiang et al., 2023b). However, in more concentrated conditions of particle water, the concentrations of •OH and 3C∗ are different than under the dilute conditions of cloud and fog drops (Ma et al., 2023, 2024), which likely affects the lifetimes of phenolic brown carbon.
In this study, we monitored the formation and loss of light absorption by aqueous SOA formed by reactions of six highly substituted BB phenols with •OH and 3C∗. We determined mass absorption coefficients (MACs) for the parent ArOH and for the evolving aqSOA as a function of reaction time. We also calculated the rate of sunlight absorption by aqSOA throughout the course of each reaction and the lifetimes of this absorbance in our experiments as well as for estimated cloud and particle water conditions.
2.1 Chemicals
Chemicals were used as received. Hydrogen peroxide (H2O2, 30 % solution in water), 3,4-dimethoxybenzaldehyde (DMB, 99 %), 4-hydroxy-3-methoxyphenylacetone (guaiacyl acetone, GA, 96 %), 4-(hydroxymethyl)-2-methoxyphenol (vanillyl alcohol, VAL, ≥ 98 %), 2-(4-hydroxyphenyl)ethanol (tyrosol, TYR, 98 %), 4-hydroxy-3,5-dimethoxybenzoic acid (syringic acid, SyrAcid, ≥ 95 %), 3-(4-hydroxy-3-methoxyphenyl)prop-2-enoic acid (trans-ferulic acid, FA, 99 %), and 2-nitrobenzaldehyde (2NB, 98 %) were purchased from Sigma-Aldrich. Upon illumination, trans-FA photoisomerizes to a 41:58 mixture of the cis- and trans-isomers; our experiments were performed on this mixture. (3,5-dimethoxy-4-hydroxyphenyl)acetone (syringyl acetone, SA, 82 %) was synthesized by Carbosynth LLC. Sulfuric acid (trace metal grade) and acetonitrile (Optima LC-MS grade) were from Fisher Scientific. Chemical solutions were prepared in air-saturated ultrapure Milli-Q water (≥ 18.2 MΩ cm) from a Millipore Advantage A10 system with an upstream Barnstead activated carbon filter.
2.2 Reaction solutions
Fresh air-saturated solutions were prepared daily containing 50 or 100 µM ArOH and either 5 or 10 mM H2O2 (as an •OH precursor) or 5 or 10 µM DMB (as a 3C∗ precursor) (Table S1 in the Supplement). Solutions were adjusted to pH 5 using sulfuric acid, and ∼ 20 mL was transferred to an airtight 5 cm quartz cuvette. Solutions were illuminated at 20 °C with constant stirring in a solar simulator equipped with a 1000 W Xenon lamp with three downstream optical filters: a water filter, an AM1.0 air mass filter (AM1D-3L, Sciencetech), and a 295 nm long-pass filter (20CGA-295, Thorlabs); see Fig. S1 in the Supplement for a typical photon flux.
During illumination we periodically removed aliquots of sample and measured the ArOH concentration using high-performance liquid chromatography (HPLC: Shimadzu LC-20AB pump, Thermo Scientific Accucore XL C18 column (50×3 mm, 4 µm bead), and Shimadzu SPD-M20A photodiode array detector). HPLC conditions (eluent, flow rate, detection wavelengths) are described elsewhere (Arciva et al., 2022). We sampled to approximately three phenol half-lives, i.e., to , when roughly 12 % of the initial phenol remained. We report times for each aliquot removal in Table S1 and show phenol decay kinetics in Fig. S2. We measured dark controls in a separate temperature-controlled dark chamber at 20 °C with constant stirring; there was no phenol loss in the dark. Additionally, three phenols (SA, SyrAcid, and FA) undergo direct photodegradation, which contributed to 6 % to 35 % of measured ArOH loss (Fig. S3).
At each time point we also measured light absorption by removing the sample cell from the illumination system and taking a UV–Vis spectrum in a Shimadzu UV-2501PC spectrophotometer baselined with Milli-Q water adjusted to pH 5 with sulfuric acid. Absorbance measurements were baseline corrected and represent the absorbance of reaction mixtures (i.e., parent ArOH, oxidant precursor, and aqSOA formed). Molar absorption coefficients for the six phenols are in Table S2 and shown graphically in Fig. S4. Absorbance of the mixtures during reaction is shown in Fig. S5 for the •OH reactions and Fig. S6 for the 3C∗ reactions.
2.3 Oxidant concentrations
Using measured ArOH decay kinetics, we estimated the steady-state concentration of oxidant in each experiment. We first determined the pseudo-first-order decay rate constant ( for phenol loss by •OH or 3C∗ as the negative value of the slope of a plot ln versus reaction time, where [ArOH]t and [ArOH]0 are the phenol concentrations at times t and zero (Fig. S2). Next, we normalized to sunlight conditions at midday on the winter solstice at Davis and corrected for any direct photodegradation using
In this equation, is the normalized rate constant for phenol loss, j2NB is the first-order decay rate constant for loss of 2NB (our actinometer) on the day of the phenol experiment, j2NB,win is the value measured at midday on the winter solstice in Davis, 0.0070 s−1 (Anastasio and McGregor, 2001), and jArOH is the rate constant for direct photodegradation of ArOH. Rate constants are summarized in Table S3 and S4. We then estimated the concentration of oxidant in each solution by dividing by the second-order rate constant for phenol reacting with •OH or 3C∗, which are from our previous works (Arciva et al., 2022; Ma et al., 2021). Since triplet solutions can also form •OH and singlet molecular oxygen (1O2*), we estimated the potential contributions of these secondary oxidants to phenol loss. As described in Sect. S1 of the Supplement, these oxidants appear to be negligible sinks for ArOH in our triplet experiments.
2.4 Mass absorption coefficients, rate of sunlight absorption by aqSOA, and lifetime of aqSOA light absorbance
Mass absorption coefficients (MACs; m2 g−1) for the aqSOA at each sampling time were determined at each wavelength λ using
where 2.303 converts the absorbance from base-10 to base-e; AbsaqSOA,λ is the absorbance of the solution at wavelength λ, corrected to remove the absorbances from the oxidant precursor and remaining parent phenol (see Sect. S2); l is the path length of our cuvette (5 cm); YSOA is the aqSOA mass yield during oxidation of ArOH with •OH or 3C∗ (Arciva et al., 2022; Ma et al., 2021), which were determined using high-resolution aerosol mass spectrometry (AMS) and are summarized in Table S5; and Δ[ArOH] is the decrease in phenol mass concentration (in g L−1) between times zero and t. The factor of 103 is converted from L to cm3, while the factor of 10−4 is converted from cm2 to m2. The MAC values for each of the six highly substituted parent phenols were calculated as described in Sect. S2 of the Supplement.
We calculated the overall rate of sunlight absorption by aqSOA (Rabs) at a given time point by multiplying the corresponding MACaqSOA by the modeled actinic flux and summing from 280 to 800 nm:
Here, Iλ is the actinic flux (photons cm−2 s−1 nm−1) at midday in Davis on the winter solstice from the Tropospheric Ultraviolet and Visible (TUV) Radiation Model version 5.3 (Fig. S1), Δλ is the interval between TUV wavelengths (1 nm), NA is Avogadro's number, and the factor of 104 is converted from m2 to cm2. We calculated the first-order rate constant for loss of sunlight absorption by aqSOA during continued reaction as the negative of the slope of a plot of the natural log of Rabs versus reaction time. These rate constants were normalized to Davis wintertime solstice conditions to determine , the pseudo-first-order rate constant for the loss of sunlight absorption by aqSOA. From this we calculated the lifetime for aqSOA sunlight absorption under our experimental conditions using Eq. (4):
This is the timescale for the loss of sunlight absorption during continued illumination, which we refer to as the lifetime of light absorption by brown carbon or, more simply, the lifetime of brown carbon. To extrapolate these calculated lifetimes of aqSOA BrC light absorbance from laboratory to ambient oxidant conditions, we first calculated the value of , which is the ambient-to-lab ratio of •OH concentration, and the analogous ratio for 3C∗. We then multiplied by this ratio to determine τBrC,ambient, the pseudo-first-order rate constant for loss of BrC absorbance due to that oxidant under ambient conditions. Aqueous concentrations of •OH and 3C∗ were taken from Ma et al. (2024) based on measurements of extracts from four different types of particles (winter–spring, summer–fall, fresh wildfire, and aged wildfire) from Davis, California, and including mass transport of gas-phase •OH. We calculated BrC lifetimes for two conditions: cloud/fog drops and aerosol liquid water (ALW), with PM mass / water mass ratios of and 1 µg of PM/µg of water, respectively. Average aqueous oxidant concentrations (±1σ) were [•OH] = 7.6 (±4.3) M for cloud and fog drops and 6.8 (±1.9) × 10−15 M for ALW and [3C∗] = 3.6 (± 2.6) M for cloud/fog drops and 5.8 (±3.9) M for ALW. We also calculated the overall ambient lifetime of brown carbon with respect to both •OH and 3C∗ under these two conditions using Eq. (5):
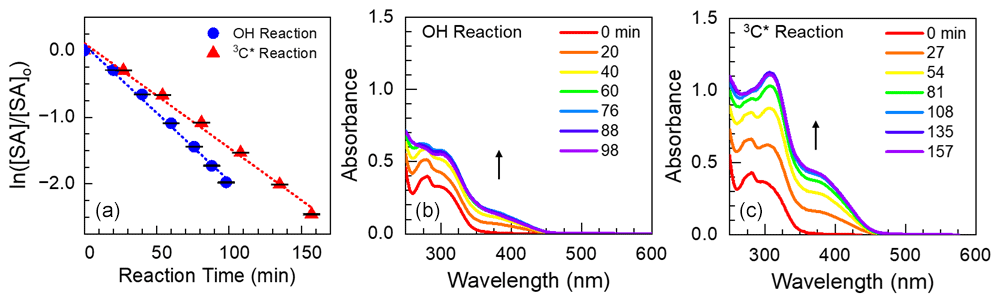
Figure 1(a) Decay kinetics for the loss of syringyl acetone (SA) reacting with •OH (blue circles) or 3C∗ (red triangles). Error bars represent 1 standard deviation, estimated from the average relative standard deviations from the corresponding dark controls. Panels (b) and (c) denote absorbance (in a 5 cm cell) of the reaction mixtures (i.e., oxidant precursor, starting phenol, and products) at various reaction times. Arrows represent the time trends in absorbance.
3.1 Aqueous phenol oxidation by •OH and 3C∗
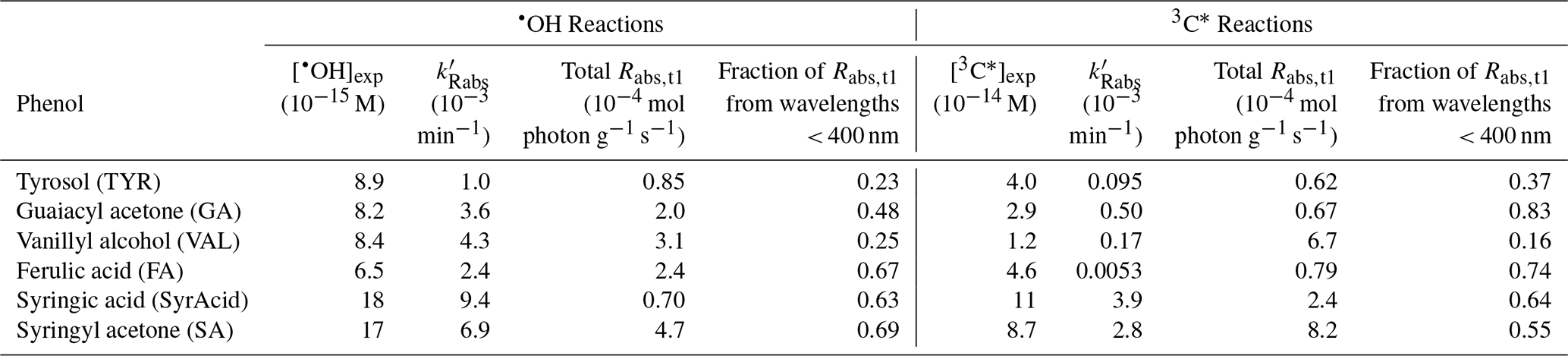
In each experiment, we measured both the aqueous oxidation of ArOH and changes in light absorption by the reaction mixture. As illustrated in Fig. 1, the photooxidation of each phenol follows pseudo-first-order kinetics, with rate constants for phenol loss ( in the range of (1.2–4.1) s−1 for •OH experiments (Table S3) and (0.19–2.4) s−1 for 3C∗ experiments (Table S4). Differences in kinetics were largely driven by starting conditions, such as initial reactant and oxidant concentrations and small changes in lamp intensity (see j2NB values in Tables S3 and S4). We used values to estimate the steady-state concentration of oxidant in each solution (Sect. 2.3). As shown in Table 1, concentrations of •OH were in the range of (6.5 to 18) M, similar to the typical [•OH] range in cloud/fog drops and ALW (Kaur et al., 2019a; Ma et al., 2023). For 3C∗, our experimental concentrations were (1.2–11) M, in the range of cloud/fog drops but lower than the average ALW value of roughly 10−13 M described in Sect. 2.4 (Ma et al., 2023).
Each of the parent phenols has an absorption peak between 260 and 290 nm, but only FA, SyrAcid, and SA have any significant absorbance above 300 nm (Fig. S3). These three phenols undergo direct photodegradation (Ma et al., 2021), which accounted for 6 %–35 % of ArOH loss (Fig. S3) and might be a source of light-absorbing products in our experiments. The amount of light absorption by the reaction mixture after illumination varies significantly between different solutions (Figs. S5 and S6). General trends of absorption by phenolic reaction mixtures throughout the course of illumination vary, but there is always more absorption at longer wavelengths compared to the starting phenol. Oxidant type also impacts light absorbance by the aqSOA: after the initial time point, continued •OH oxidation generally decreases absorbance below wavelengths of 300 nm but increases absorbance above 300 nm (Figs. 1 and S5), while 3C∗ oxidation mostly increases absorbance across all wavelengths (Figs. 1 and S6).
Table 1Oxidant concentrations, pseudo-first-order rate constants for the loss of BrC light absorption (, the total rate of sunlight absorption at the first illumination time point (total Rabs,t1) for each reaction system, and the fraction of light absorption from 280 to 400 nm.
3.2 Mass absorption coefficients (MACs)
To compare light absorption by aqSOA as a function of phenol, oxidant, and illumination time, we determined mass absorption coefficients (MACs) for each experiment. aqSOA MAC values were determined by correcting measured absorbance values for the contributions from both the remaining starting phenol and the oxidant precursor (Sect. S2). Generally, across tropospherically relevant wavelengths (i.e., above 300 nm), the MAC curves of aqSOA are highest around 300 nm (Figs. 2 and S7) and tail at longer wavelengths. The MAC values of the starting phenols (MACArOH) are typically low compared to values of the resulting aqSOA (MACaqSOA), especially for the first oxidation time point (MACaqSOA,t1) for both •OH and 3C∗ (Table S6). The one exception is FA, which has a high parent absorbance that decreases during oxidation. After the first illumination time point, continued reaction generally decreases light absorbance by the aqSOA, but there are some exceptions (e.g., TYR in Fig. 2 and VAL with 3C∗ in Fig. S7).
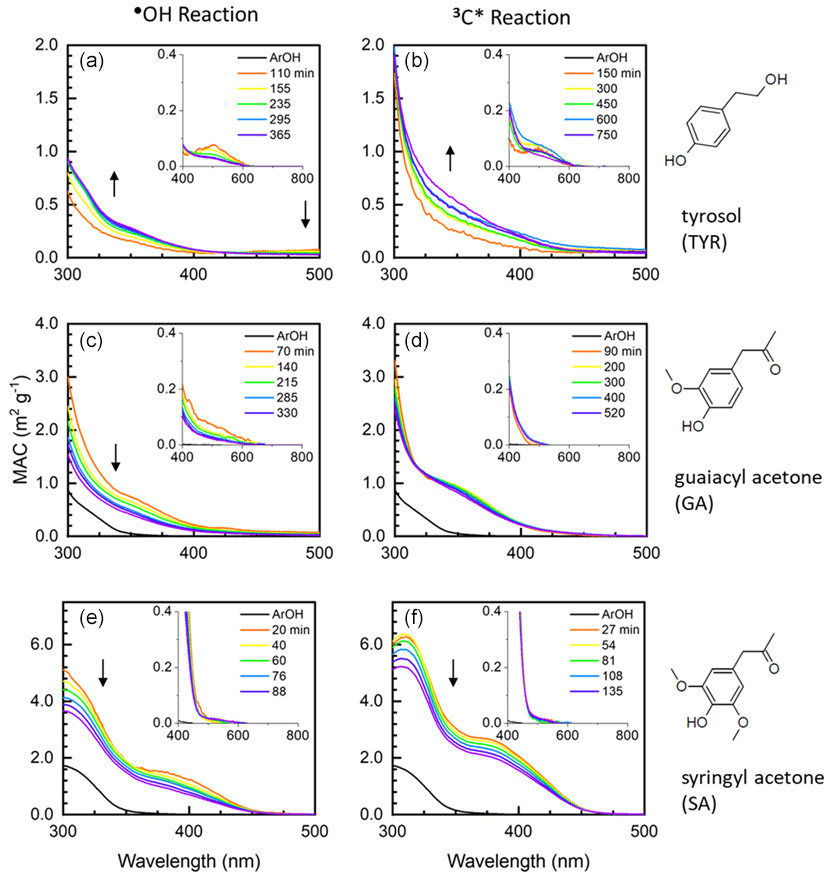
Figure 2Mass absorption coefficients for aqSOA formed via reactions with •OH (a, c, e) and 3C∗ (b, d, f) for tyrosol (top plots), guaiacyl acetone (middle plots), and syringyl acetone (bottom plots). For a given phenol, each colored line represents a different illumination time (see legend for times). Arrows represent the time trends of aqSOA MAC values after the initial illumination time point. The black line in each panel is the MAC for the parent phenol; the absorbance of the remaining parent phenol was removed from the aqSOA MAC values at each illumination time (Supplement Sect. S2). The MAC for TYR is zero in this wavelength range.
Figure 2 shows MAC values for three of the phenols we studied – tyrosol, guaiacyl acetone, and syringyl acetone – and their resulting aqSOA. These phenols are substituted versions of the three most abundant phenols from BB: phenol, guaiacol (2-methoxyphenol), and syringol (2,6-dimethoxyphenol). Results for the other three phenols we studied, which are derivatives of guaiacol and syringol, are shown in Fig. S7. For each solution, we first sampled the illumination solution when approximately 25 % of the parent phenol had decayed (i.e., 0.5 , so the corresponding spectrum likely does not capture the maximum absorbance exhibited by the solution. For most of the •OH reactions with the six phenols, the MAC of aqSOA increases from the parent ArOH (which typically has little to no absorbance in the solar wavelengths) to the initial aqSOA. This is because •OH reactions with ArOH form hydroxylated products, which can be important contributors to light absorption (Sun et al., 2010; Yu et al., 2014). Additionally, the formation of quinones, which can contain extensive conjugated pi electrons, might explain our observation of increased aqSOA light absorption at longer wavelengths (Dulo et al., 2021). The aqSOA of TYR is the least absorbing, and aqSOA MAC tends to increase with increased methoxy substitution on the aromatic ring (Figs. 2 and S7). For example, the MACaqSOA,t1 of SyrAcid is greater than that of TYR by a factor of approximately 10 at 300 nm. FA oxidation is somewhat of an exception since the MAC of FA is higher than that of its aqSOA at wavelengths below 350 nm (Fig. S7). The decrease in absorbance during oxidation for FA is probably because of a loss of unsaturation in the acrylic acid substituent, which decreases conjugation and light absorption.
For 3C∗ reactions, the trends in absorbance are similar to the •OH results above, including a general increase in MAC from the parent phenol to the initial daughter aqSOA, as shown in Fig. 2 for TYR, GA, and SA (and Fig. S7 for VAL, FA, and SyrAcid). More highly substituted ArOH (FA, SA, and SyrAcid) produce aqSOA that absorbs more strongly compared to the less substituted phenols (TYR, GA, and VAL). Also, 3C∗-derived aqSOA absorbs more light compared to aqSOA from the corresponding •OH reactions. The MACaqSOA at 300 nm for SyrAcid at this condition was 9.0 m2 g−1, the highest MAC we observed for both •OH and 3C∗ reactions. One reason for higher MAC values in 3C∗-produced aqSOA may be because triplet reactions produce more oligomers, which can strongly absorb light, compared to •OH reactions (Yu et al., 2014; Jiang et al., 2021).
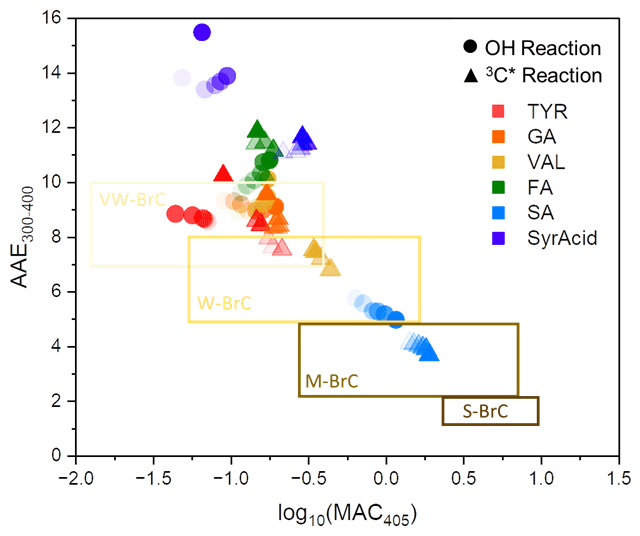
Figure 3aqSOA light absorption based on the brown carbon (BrC) classification scheme (Saleh, 2020). The boxes from top to bottom represent very weakly absorbing BrC (VW-BrC), weakly absorbing BrC (W-BrC), moderately absorbing BrC (M-BrC), and strongly absorbing BrC (S-BrC). Triangles represent aqSOA formed via reactions of each phenol with the triplet state of DMB, while circles are data from reactions of each phenol with hydroxyl radical. These values are summarized in Table S7 in the Supplement. The time series of each reaction follows a gradient: the first time point is represented as the darkest marker followed by a decrease in color with increasing time.
In Fig. 3 we plot our MAC data at 405 nm in the BrC classification scheme from Saleh (2020). Most of the phenolic BrC is very weakly absorbing, but the products from SA (our most strongly absorbing aqSOA at 405 nm) are weakly to moderately absorbing. The magnitudes of absorption Ångström exponent (AAE) from 300 to 400 nm (3.7 to 15) and MAC405 (0.05 to 1.9) (Table S7) depend on parent ArOH and generally increase with more substituted ArOH. BrC from SA is an exception because the MACs at both 300 and 400 nm are high and do not change drastically across these wavelengths compared to BrC from the other phenols. Oxidant identity also plays an important role in the extent of light absorption by the BrC formed. Most of the BrC formed by •OH oxidation was very weakly absorbing, while BrC formed by triplet oxidation had a broader range of absorption, spanning very weakly to moderately absorbing. Also, in most cases, continued oxidation photobleaches BrC towards the very weakly absorbing bin (Fig. 3). Our observation that most phenolic BrC is very weak to weakly absorbing is consistent with results for water-soluble BrC in ambient particles impacted by residential wood combustion and wildfires (Jiang et al., 2023a; Ma et al., 2024).
3.3 Rates of sunlight absorption
We next calculated Rabs, the rate of sunlight absorption, for the parent phenol and resulting aqSOA at each time point, as shown in Fig. 4 for TYR, GA, and SA (and Fig. S8 for VAL, FA, and SyrAcid). Except for FA, the parent phenols generally absorb very weakly at short solar wavelengths (i.e., below 400 nm) or do not absorb any sunlight (i.e., TYR and VAL). As such, the rate of sunlight absorption by aqSOA is generally much higher than that of the parent ArOH, with the exception of FA. For example, going from ArOH to aqSOA, Rabs values for GA and SA increase by factors of 14 to 17 for •OH reactions and 5.5 to 24 for triplet reactions. The increase for the Rabs of SyrAcid aqSOA is much higher, with factors of 74 to 250 for •OH and triplet reactions, respectively, due to the very weak absorption by the parent phenol. In contrast, ferulic acid aqSOA shows very small changes compared to FA, with factors of 1.6 for •OH reactions and 0.52 for triplet reactions. For the entire set of six phenols, continued oxidation after the first illumination time point can either increase or decrease Rabs. For •OH reactions, the rate of sunlight absorption for aqSOA from VAL, GA, FA, and SA decreases with increasing oxidation; i.e., continued aging produces less-absorbing aqSOA, possibly because of fragmentation by •OH (Sun et al., 2010). Continued 3C∗ reactions generally decrease Rabs for aqSOA but much more slowly than •OH reactions. This may be because triplets are less effective at oxidizing aqSOA than is •OH (Jiang et al., 2021).
We also calculated the fraction of sunlight absorption due to UV wavelengths, i.e., those below 400 nm. As summarized in Table 1 for the initial time point, the fraction of Rabs due to UV wavelengths varies significantly across different ArOH but also, to a smaller extent, across the two oxidants. Fractions of Rabs due to UV wavelengths for each illumination time point are shown in Table S7. Generally, more substituted ArOH produce aqSOA where shorter wavelengths dominate sunlight absorption, and this remains throughout the course of reaction. In contrast, for simpler ArOH (i.e., TYR, GA, and VAL), the fraction of Rabs due to wavelengths below 400 nm generally increases with reaction time (Table S7). Also, except for VAL, the fraction of Rabs contributed by short wavelengths is always higher for the triplet reaction compared to the •OH reaction. Interestingly, the VAL–aqSOA formed by both oxidants (Fig. S8, top plot) and TYR–aqSOA by •OH (Fig. 4, top plot) contain characteristic peaks at 500 nm, possibly due to quinones. In the case of VAL, this peak behaves differently for the two oxidants. For VAL reaction with •OH, aqSOA light absorption diminishes across the entire wavelength range due to photobleaching, with the fastest loss between 400 to 800 nm, possibly due to faster direct photodegradation of chromophores. In contrast, during reactions with triplets, the VAL–aqSOA becomes more absorbing between 300 and 425 nm but photobleaches at longer wavelengths. We observe similar behavior for TYR–aqSOA with •OH. This indicates that during the evolution of aqSOA light absorption can simultaneously increase in some wavelength regions and decrease in others.
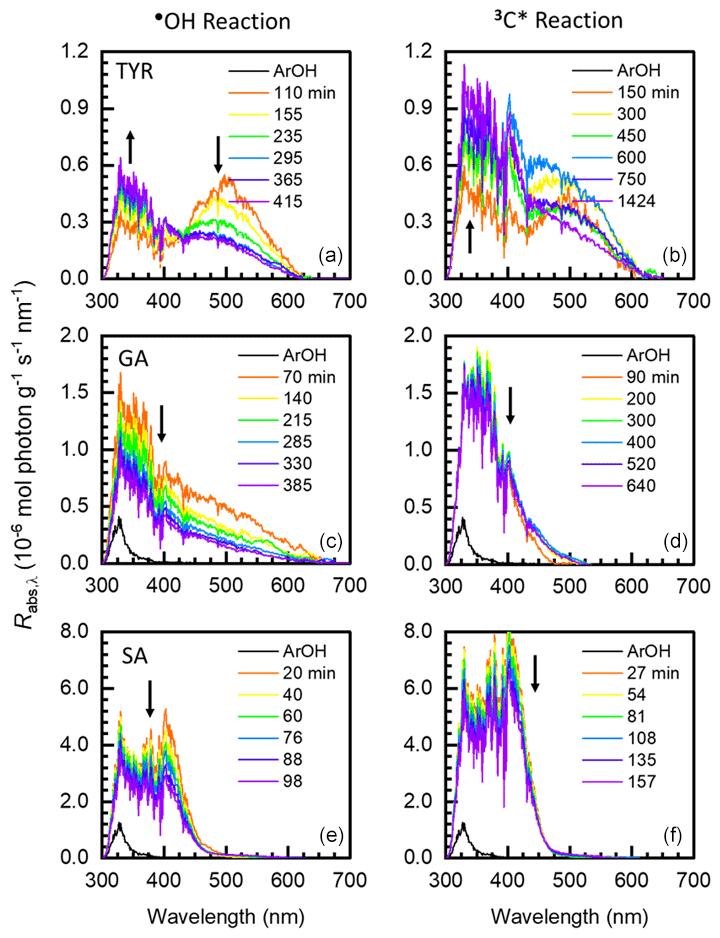
Figure 4Wavelength-specific rates of sunlight absorption by aqSOA formed via reactions with •OH (a, c, e) and 3C∗ (b, d, f) for tyrosol (top plots), guaiacyl acetone (middle plots), and syringyl acetone (bottom plots). For a given phenol, the black line represents sunlight absorption by the parent ArOH and colored lines represent absorption for aqSOA at different illumination times (see legend). Arrows represent the time trends of aqSOA MAC values after the initial illumination time point. Rates of aqSOA sunlight absorption for vanillyl alcohol, ferulic acid, and syringic acid are shown in Fig. S8.
3.4 Lifetimes of light-absorbing aqSOA
We calculated the lifetime of light absorption by aqSOA (i.e., the lifetime of phenolic BrC) by monitoring the decline in Rabs with continued illumination, as shown in Fig. 5 for SA. We show the equivalent figures for the other five phenols in Figs. S9 and S10. Plotting the natural log of Rabs versus reaction time yields the experimental first-order decay rate constant for loss of light absorption (. In some cases, the natural log of Rabs increases at short reaction times (e.g., the triplet reactions of TYR, GA, and FA) before decreasing; we did not include these increasing points in our determination of . We then normalized this rate constant to Davis midday winter solstice sunlight to obtain ( (Table S8); values are summarized in Table 1. for •OH reactions are in the range 1.0 to 9.4 min−1, while rate constants for triplet reactions are lower and range from 0.0053 to 3.9 min−1; i.e., the loss of light-absorbing aqSOA by triplets is much slower than with •OH under our experimental conditions. Jiang et al. (2023b) also measured the first-order decay rate constant for loss of light absorption for BrC formed via reactions of GA with •OH and 3C∗. Compared to their experiments (with no additional oxidant), the rate of decay of 3C∗–aqSOA in Jiang et al. (2023b) is higher than our value because their [3C∗] is higher by a factor of 3.8. In contrast, for •OH–aqSOA, our decay value is higher than that of Jiang et al. (2023) because our [•OH] is higher by a factor of 3.2. However, despite these differences in oxidant concentrations, the observed trends for aqSOA are similar. That is, initial photooxidation forms light-absorbing aqSOA, but during extended aging •OH–aqSOA photobleaches rapidly, while 3C∗–aqSOA photobleaches more slowly. It is possible that molecular differences in the aqSOA formed by 3C∗ and •OH play a role in the different rates of photobleaching. Triplet-mediated reactions efficiently form oligomers, while •OH-mediated reactions tend to form hydroxylated products that eventually fragment (Jiang et al., 2023a; Yee et al., 2013). Also, during continuous oxidation triplets are less reactive towards aqSOA, possibly because the BrC molecules do not have phenolic hydroxyl groups or aliphatic double bonds, which are both reactive with triplets (Kaur et al., 2019b; Ma et al., 2021). In contrast, •OH, which reacts rapidly with most organic molecules, more quickly degrades phenolic aqSOA.
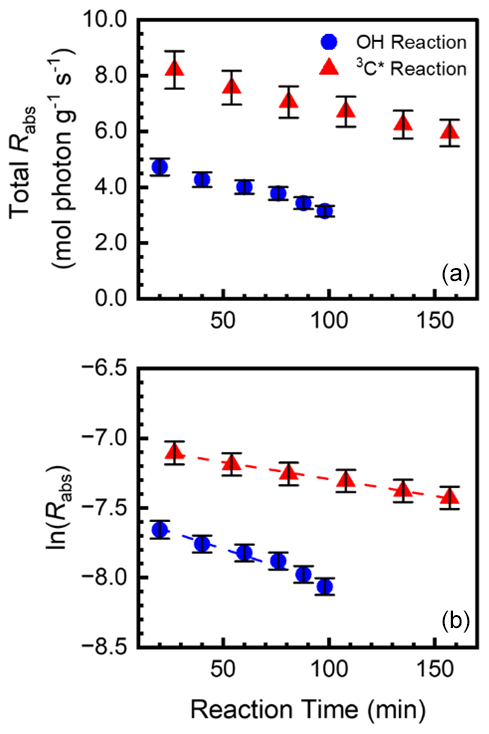
Figure 5(a) Total rate of aqSOA sunlight absorption (Rabs, 10−4 mol photon g−1 s−1) as a function of reaction time during the oxidation of SA by •OH (blue circles) and 3C∗ (red triangles). (b) Natural log of Rabs versus reaction time, which was used to determine rate constants for the loss of light absorbance by aqSOA. Error bars represent 1 standard deviation propagated from the standard deviations of the dark controls and aqSOA mass yields.
The inverse of is the lifetime of sunlight absorption by phenolic aqSOA during continued •OH and 3C∗ reactions under our experimental conditions, i.e., the timescale for loss of absorbance by brown carbon compounds in the aqueous SOA. Under our experimental conditions, these BrC lifetimes range from 1.8 to 16 h for •OH solutions and 4.3 to 3200 h for 3C∗ experiments (Fig. S11). Next, we extrapolate these experimental lifetimes to ambient lifetimes by assuming that the concentration of oxidant (•OH or 3C∗) is driving the loss of brown carbon. We consider two ambient conditions: cloud/fog drops (with a PM mass / liquid water mass ratio of µg of PM/µg of water) and ALW (1 µg of PM/1 µg of water). Concentrations of the two oxidants under our experimental conditions, as well as values for cloud/fog drops and ALW, are described in Tables S9 and S10; the cloud/fog and ALW oxidant concentrations are based on measurements and extrapolations in Ma et al. (2024).
Because the aqueous concentration of •OH is similar in our experimental solutions, in cloud/fog drops, and in ALW, we find similar lifetimes for sunlight absorption by aqSOA under all three conditions for •OH, as shown in Fig. S11. Lifetimes of BrC during reactions in the •OH solutions range from 4.2 to 21 h across both ambient conditions for aqSOA from all six highly substituted phenols (Fig. S11). Differences in ArOH substitution do not significantly influence the lifetime of BrC formed from •OH, with lifetimes mostly within a factor of 2. Even for TYR–aqSOA, which forms longer-lived absorbing aqSOA, the BrC lifetime only differs by a factor of 4 compared to that for the other phenols. As mentioned above for TYR–aqSOA, this is complicated by the simultaneous occurrence of both photobleaching and photoenhancement in different wavelength regions.
Lifetimes for the light-absorbing aqSOA in the triplet solutions differ from the •OH-associated values in two main ways. First, extrapolation to ambient conditions results in a broader range of lifetimes (Fig. S11) due to the 16 times increase in triplet concentrations as we move from cloud/fog drops to particle water. The higher concentration of triplets in ALW makes for shorter lifetimes of light-absorbing aqSOA, ranging from 0.83 to 12 h (except for FA, which is 250 h). However, under cloud/fog conditions, where triplet concentrations are lower, lifetimes of phenolic BrC with respect to triplet reactions are much longer, ranging from 13 to 200 h. The exception again is FA, where the light-absorbing aqSOA had a lifetime of 4000 h. This resistance of FA–aqSOA to 3C∗ oxidation is possibly because a large fraction of FA–aqSOA interactions with triplets result in physical quenching rather than chemical transformation of the aqSOA, likely because the unsaturated side chains from FA undergo isomerization readily (Ma et al., 2021).
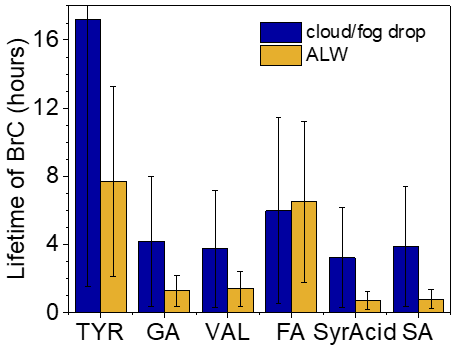
Figure 6Overall lifetimes of brown carbon (BrC) with respect to photooxidation by •OH and 3C∗ under cloud/fog drop and aerosol liquid water (ALW) conditions calculated using Eq. (5). Lifetimes with respect to photooxidation from the two oxidants individually are shown in Fig. S11. Error bars represent 1 standard deviation, primarily representing the variability in photooxidant concentrations averaged across the four different sample types (Ma et al., 2024), but also include the uncertainty in the rate constant for loss of brown-carbon light absorption.
We next calculate the overall lifetime of light absorption by phenolic BrC with respect to both •OH and 3C∗ for the two atmospheric conditions. As shown in Fig. 6, for cloud/fog drops the phenolic BrC lifetimes are typically a few hours but range up to 17 h for TYR aqSOA; these lifetimes are controlled by •OH (Fig. S11). In particle water, phenolic BrC lifetimes range from 0.70 to 7.7 h; these are typically shorter than in cloud/fog drops and are generally controlled by reactions with 3C∗. The exception is FA, where the lifetimes are controlled by •OH, and thus the lifetime is shorter in cloud/fog drops. While many of the light-absorbing aqSOA lifetimes are under 4 h for the two conditions, the longer lifetimes for brown carbon from TYR and FA show that identity of the precursor phenol also plays a role.
Lastly, we compare the lifetimes of light-absorbing aqSOA across a continuum of liquid water content (LWC), from cloud/fog conditions to ALW (Fig. 7). Combining winter •OH and 3C∗ concentrations as a function of liquid water content from Ma et al. (2023, 2024), we calculated the lifetimes of light-absorbing aqSOA with respect to the two oxidants individually. Lifetimes of phenolic aqSOA with respect to oxidation by •OH change relatively little across the enormous range of LWC, a consequence of the predicted small change in hydroxyl radical concentrations (Ma et al., 2023). In contrast, lifetimes of aqSOA produced and oxidized by 3C∗ show a much larger variation. In the dilute conditions of cloud/fog drops, aqSOA is less susceptible to aging by 3C∗, resulting in some of the longer-lived aqSOA across the liquid water content range. But as solutions become more concentrated, the triplet-mediated BrC lifetime drops quickly and then plateaus at a PM mass / water mass ratio of approximately 10−2 and above (Fig. 7). This reflects changes in predicted triplet concentrations, which increase by approximately a factor of 16 from cloud/fog to ALW. The net result is that lifetimes of light absorbance by aqSOA are generally controlled by •OH under the more dilute conditions of cloud/fog drops, while triplets dominate phenolic BrC lifetimes under more concentrated conditions, including in ALW. The clear exception again is FA, where the BrC lifetime is always controlled by •OH, while TYR follows the trend of the other phenols but with a less dramatic shift from •OH to 3C∗ controlling the lifetime as we move from cloud/fog drops to ALW (Fig. 7). Otherwise, the other four phenols behave as a group, with no clear trend between ArOH substitution and aqSOA lifetime. One caveat to our calculations is that we assume the phenolic BrC lifetimes are governed by reactions with •OH and 3C∗ rather than direct photodegradation. But the latter pathway might also be important, especially for BrC species that absorb at longer wavelengths where the photon flux is higher (Fig. S8). Although it is difficult for us to constrain this, any contribution of direct photodegradation to BrC loss under wintertime Davis conditions would be similar to that in our experimental conditions since our laboratory photon fluxes are similar to ambient values (Fig. S1).
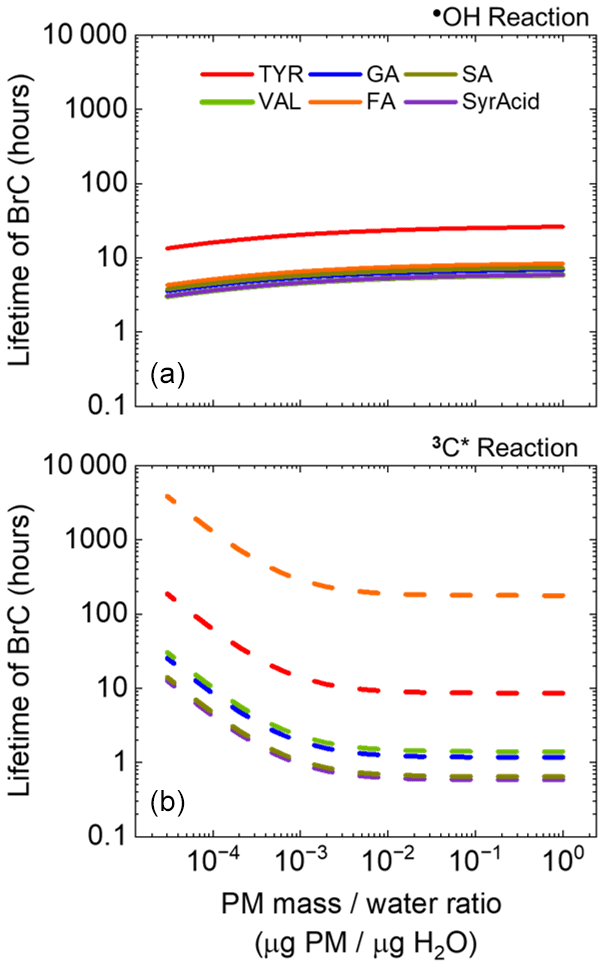
Figure 7Lifetimes of light-absorbing aqSOA (i.e., BrC) as a function of particle-mass-to-water-mass ratio for TYR (red), GA (blue), SA (mustard), VAL (green), FA (orange), and SyrAcid (purple). Solid and dashed lines indicate lifetimes with respect to reaction with •OH (a) and 3C∗ (b), respectively. For a PM concentration of 10 µg m−3, the range of PM mass / water mass ratios corresponds to liquid water contents of 1 to g m−3, i.e., conditions of cloud/fog drops to ALW, respectively.
We examined light absorption by the aqueous secondary organic aerosol formed from reactions of phenols with two aqueous oxidants: hydroxyl radical (•OH) and a triplet excited state (3C∗). These two routes are important for ArOH processing and significant contributors to aqSOA formation, but the light absorption trends with continuous reaction had not been evaluated previously. Initial reactions of highly substituted phenols with both oxidants produced aqSOA that absorbs much more sunlight compared to the parent phenol, a result of functionalization and/or oligomerization. Continued photo-aging alters the light absorption by aqSOA. Overall, continued •OH oxidation more rapidly causes photobleaching, while the aqSOA formed and oxidized by triplet excited states loses its light absorption much more slowly. Our reactions are restricted with respect to each oxidant; i.e., we did not study how susceptible aqSOA formed by •OH is to triplet oxidation or vice versa.
We also extrapolated from our experimental conditions to ambient conditions based on previously measured and estimated ambient oxidant concentrations. Lifetimes of phenolic secondary BrC range from 3.2 to 17 h for cloud/fog drops, where •OH is the major sink, and from 0.70 to 7.7 for particle water, where triplet excited states are the major sink. Overall, our results will help constrain atmospheric lifetimes of phenolic BrC, which should aid predictions of the global impact of BrC from biomass burning.
All data are available upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-4473-2024-supplement.
CA and SA developed the research goals and designed the experiments. SA and CM performed the •OH oxidation experiments, while LM and CG performed the triplet oxidation experiments. SA analyzed the data and prepared the manuscript. CA reviewed and edited the manuscript and provided supervision and oversight during the experiments and writing.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We appreciate the comments and efforts of the four anonymous reviewers.
This research has been supported by the National Science Foundation (grant nos. AGS-1649212 and AGS-2220307) and the University of California, Davis (Jastro-Shields Research Award and Donald G. Crosby Fellowship grant).
This paper was edited by Theodora Nah and reviewed by four anonymous referees.
Aguilera, R., Corringham, T., Gershunov, A., and Benmarhnia, T.: Wildfire smoke impacts respiratory health more than fine particles from other sources: observational evidence from Southern California., Nat. Commun., 12, 1493, https://doi.org/10.1038/s41467-021-21708-0, 2021.
Akagi, S. K., Yokelson, R. J., Wiedinmyer, C., Alvarado, M. J., Reid, J. S., Karl, T., Crounse, J. D., and Wennberg, P. O.: Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 11, 4039–4072, https://doi.org/10.5194/acp-11-4039-2011, 2011.
Anastasio, C. and McGregor, K. G.: Chemistry of fog waters in California's Central Valley: 1. In situ photoformation of hydroxyl radical and singlet molecular oxygen, Atmos. Environ., 35, 1079–1089, https://doi.org/10.1016/S1352-2310(00)00281-8, 2001.
Andreae, M. O.: Emission of trace gases and aerosols from biomass burning – an updated assessment, Atmos. Chem. Phys., 19, 8523–8546, https://doi.org/10.5194/acp-19-8523-2019, 2019.
Arciva, S., Niedek, C., Mavis, C., Yoon, M., Sanchez, M. E., Zhang, Q., and Anastasio, C.: Aqueous • OH oxidation of highly substituted phenols as a source of secondary organic aerosol, Environ. Sci. Technol., 56, 9959–9967, https://doi.org/10.1021/acs.est.2c02225, 2022.
Berndt, T. and Böge, O.: Gas-phase reaction of OH radicals with phenol, Phys. Chem. Chem. Phys., 5, 342–350, https://doi.org/10.1039/B208187C, 2003.
Bond, T. C., Streets, D. G., Yarber, K. F., Nelson, S. M., Woo, J.-H., and Klimont, Z.: A technology-based global inventory of black and organic carbon emissions from combustion, J. Geophys. Res.-Atmos., 109, D14203, https://doi.org/10.1029/2003JD003697, 2004.
Buxton, G. V., Greenstock, C. L., Helman, W. P., and Ross, A. B.: Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O- in Aqueous Solution, J. Phys. Chem. Ref. Data, 17, 513–886, https://doi.org/10.1063/1.555805, 1988.
Chang, J. L. and Thompson, J. E.: Characterization of colored products formed during irradiation of aqueous solutions containing H2O2 and phenolic compounds, Atmos. Environ., 44, 541–551, https://doi.org/10.1016/j.atmosenv.2009.10.042, 2010.
Coeur-Tourneur, C., Cassez, A., and Wenger, J. C.: Rate coefficients for the gas-phase reaction of hydroxyl radicals with 2-methoxyphenol (guaiacol) and related compounds., J. Phys. Chem. A, 114, 11645–11650, https://doi.org/10.1021/jp1071023, 2010.
Di Lorenzo, R. A., Washenfelder, R. A., Attwood, A. R., Guo, H., Xu, L., Ng, N. L., Weber, R. J., Baumann, K., Edgerton, E., and Young, C. J.: Molecular-Size-Separated Brown Carbon Absorption for Biomass-Burning Aerosol at Multiple Field Sites, Environ. Sci. Technol., 51, 3128–3137, https://doi.org/10.1021/acs.est.6b06160, 2017.
Dulo, B., Phan, K., Githaiga, J., Raes, K., and De Meester, S.: Natural quinone dyes: A review on structure, extraction techniques, analysis and application potential, Waste Biomass Valor., 12, 6339–6374, https://doi.org/10.1007/s12649-021-01443-9, 2021.
Fleming, L. T., Lin, P., Roberts, J. M., Selimovic, V., Yokelson, R., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Molecular composition and photochemical lifetimes of brown carbon chromophores in biomass burning organic aerosol, Atmos. Chem. Phys., 20, 1105–1129, https://doi.org/10.5194/acp-20-1105-2020, 2020.
Garofalo, L. A., Pothier, M. A., Levin, E. J. T., Campos, T., Kreidenweis, S. M., and Farmer, D. K.: Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States, ACS Earth Space Chem., 3, 1237–1247, https://doi.org/10.1021/acsearthspacechem.9b00125, 2019.
Graber, E. R. and Rudich, Y.: Atmospheric HULIS: How humic-like are they? A comprehensive and critical review, Atmos. Chem. Phys., 6, 729–753, https://doi.org/10.5194/acp-6-729-2006, 2006.
Grieshop, A. P., Logue, J. M., Donahue, N. M., and Robinson, A. L.: Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 1: measurement and simulation of organic aerosol evolution, Atmos. Chem. Phys., 9, 1263–1277, https://doi.org/10.5194/acp-9-1263-2009, 2009.
Gurol, M. D. and Nekouinaini, S.: Kinetic behavior of ozone in aqueous solutions of substituted phenols, Ind. Eng. Chem. Fund., 23, 54–60, https://doi.org/10.1021/i100013a011, 1984.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Heald, C. L., Henze, D. K., Horowitz, L. W., Feddema, J., Lamarque, J. F., Guenther, A., Hess, P. G., Vitt, F., Seinfeld, J. H., Goldstein, A. H., and Fung, I.: Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change, J. Geophys. Res., 113, D05211, https://doi.org/10.1029/2007JD009092, 2008.
Hecobian, A., Zhang, X., Zheng, M., Frank, N., Edgerton, E. S., and Weber, R. J.: Water-Soluble Organic Aerosol material and the light-absorption characteristics of aqueous extracts measured over the Southeastern United States, Atmos. Chem. Phys., 10, 5965–5977, https://doi.org/10.5194/acp-10-5965-2010, 2010.
Hems, R. F., Schnitzler, E. G., Bastawrous, M., Soong, R., Simpson, A. J., and Abbatt, J. P. D.: Aqueous photoreactions of wood smoke brown carbon, ACS Earth Space Chem., 4, 1149–1160, https://doi.org/10.1021/acsearthspacechem.0c00117, 2020.
Hems, R. F., Schnitzler, E. G., Liu-Kang, C., Cappa, C. D., and Abbatt, J. P. D.: Aging of atmospheric brown carbon aerosol, ACS Earth Space Chem., 5, 722–748, https://doi.org/10.1021/acsearthspacechem.0c00346, 2021.
Hennigan, C. J., Miracolo, M. A., Engelhart, G. J., May, A. A., Presto, A. A., Lee, T., Sullivan, A. P., McMeeking, G. R., Coe, H., Wold, C. E., Hao, W.-M., Gilman, J. B., Kuster, W. C., de Gouw, J., Schichtel, B. A., Collett Jr., J. L., Kreidenweis, S. M., and Robinson, A. L.: Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber, Atmos. Chem. Phys., 11, 7669–7686, https://doi.org/10.5194/acp-11-7669-2011, 2011.
Huang, D. D., Zhang, Q., Cheung, H. H. Y., Yu, L., Zhou, S., Anastasio, C., Smith, J. D., and Chan, C. K.: Formation and Evolution of aqSOA from Aqueous-Phase Reactions of Phenolic Carbonyls: Comparison between Ammonium Sulfate and Ammonium Nitrate Solutions., Environ. Sci. Technol., 52, 9215–9224, https://doi.org/10.1021/acs.est.8b03441, 2018.
Jiang, W., Misovich, M. V., Hettiyadura, A. P. S., Laskin, A., McFall, A. S., Anastasio, C., and Zhang, Q.: Photosensitized Reactions of a Phenolic Carbonyl from Wood Combustion in the Aqueous Phase-Chemical Evolution and Light Absorption Properties of AqSOA, Environ. Sci. Technol., 55, 5199–5211, https://doi.org/10.1021/acs.est.0c07581, 2021.
Jiang, W., Ma, L., Niedek, C., Anastasio, C., and Zhang, Q.: Chemical and Light-Absorption Properties of Water-Soluble Organic Aerosols in Northern California and Photooxidant Production by Brown Carbon Components, ACS Earth Space Chem., 7, 1107–1119, https://doi.org/10.1021/acsearthspacechem.3c00022, 2023a.
Jiang, W., Niedek, C., Anastasio, C., and Zhang, Q.: Photoaging of phenolic secondary organic aerosol in the aqueous phase: evolution of chemical and optical properties and effects of oxidants, Atmos. Chem. Phys., 23, 7103–7120, https://doi.org/10.5194/acp-23-7103-2023, 2023b.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jo, D. S., Park, R. J., Lee, S., Kim, S.-W., and Zhang, X.: A global simulation of brown carbon: implications for photochemistry and direct radiative effect, Atmos. Chem. Phys., 16, 3413–3432, https://doi.org/10.5194/acp-16-3413-2016, 2016.
Kaeswurm, J. A. H., Scharinger, A., Teipel, J., and Buchweitz, M.: Absorption coefficients of phenolic structures in different solvents routinely used for experiments, Molecules, 26, 4656, https://doi.org/10.3390/molecules26154656, 2021.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kaur, R., Labins, J. R., Helbock, S. S., Jiang, W., Bein, K. J., Zhang, Q., and Anastasio, C.: Photooxidants from brown carbon and other chromophores in illuminated particle extracts, Atmos. Chem. Phys., 19, 6579–6594, https://doi.org/10.5194/acp-19-6579-2019, 2019a.
Kaur, R., Hudson, B. M., Draper, J., Tantillo, D. J., and Anastasio, C.: Aqueous reactions of organic triplet excited states with atmospheric alkenes, Atmos. Chem. Phys., 19, 5021–5032, https://doi.org/10.5194/acp-19-5021-2019, 2019b.
Kim, K.-H., Kabir, E., and Kabir, S.: A review on the human health impact of airborne particulate matter, Environ. Int., 74, 136–143, https://doi.org/10.1016/j.envint.2014.10.005, 2015.
Kirchstetter, T. W. and Thatcher, T. L.: Contribution of organic carbon to wood smoke particulate matter absorption of solar radiation, Atmos. Chem. Phys., 12, 6067–6072, https://doi.org/10.5194/acp-12-6067-2012, 2012.
Kleinman, L. I., Sedlacek III, A. J., Adachi, K., Buseck, P. R., Collier, S., Dubey, M. K., Hodshire, A. L., Lewis, E., Onasch, T. B., Pierce, J. R., Shilling, J., Springston, S. R., Wang, J., Zhang, Q., Zhou, S., and Yokelson, R. J.: Rapid evolution of aerosol particles and their optical properties downwind of wildfires in the western US, Atmos. Chem. Phys., 20, 13319–13341, https://doi.org/10.5194/acp-20-13319-2020, 2020.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of atmospheric brown carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Liu, D., Li, S., Hu, D., Kong, S., Cheng, Y., Wu, Y., Ding, S., Hu, K., Zheng, S., Yan, Q., Zheng, H., Zhao, D., Tian, P., Ye, J., Huang, M., and Ding, D.: Evolution of Aerosol Optical Properties from Wood Smoke in Real Atmosphere Influenced by Burning Phase and Solar Radiation, Environ. Sci. Technol., 55, 5677–5688, https://doi.org/10.1021/acs.est.0c07569, 2021.
Li, X., Tao, Y., Zhu, L., Ma, S., Luo, S., Zhao, Z., Sun, N., Ge, X., and Ye, Z.: Optical and chemical properties and oxidative potential of aqueous-phase products from OH and 3C∗-initiated photooxidation of eugenol, Atmos. Chem. Phys., 22, 7793–7814, https://doi.org/10.5194/acp-22-7793-2022, 2022.
Li, Y. J., Huang, D. D., Cheung, H. Y., Lee, A. K. Y., and Chan, C. K.: Aqueous-phase photochemical oxidation and direct photolysis of vanillin – a model compound of methoxy phenols from biomass burning, Atmos. Chem. Phys., 14, 2871–2885, https://doi.org/10.5194/acp-14-2871-2014, 2014.
Ma, L., Guzman, C., Niedek, C., Tran, T., Zhang, Q. and Anastasio, C.: Kinetics and mass yields of aqueous secondary organic aerosol from highly substituted phenols reacting with a triplet excited state, Environ. Sci. Technol., 55, 5772–5781, https://doi.org/10.1021/acs.est.1c00575, 2021.
Ma, L., Worland, R., Jiang, W., Niedek, C., Guzman, C., Bein, K. J., Zhang, Q., and Anastasio, C.: Predicting photooxidant concentrations in aerosol liquid water based on laboratory extracts of ambient particles, Atmos. Chem. Phys., 23, 8805–8821, https://doi.org/10.5194/acp-23-8805-2023, 2023.
Ma, L., Worland, R., Heinlein, L., Guzman, C., Jiang, W., Niedek, C., Bein, K. J., Zhang, Q., and Anastasio, C.: Seasonal variations in photooxidant formation and light absorption in aqueous extracts of ambient particles, Atmos. Chem. Phys., 24, 1–21, https://doi.org/10.5194/acp-24-1-2024, 2024.
McFall, A. S., Johnson, A. W., and Anastasio, C.: Air-Water Partitioning of Biomass-Burning Phenols and the Effects of Temperature and Salinity, Environ. Sci. Technol., 54, 3823–3830, https://doi.org/10.1021/acs.est.9b06443, 2020.
Misovich, M. V., Hettiyadura, A. P. S., Jiang, W., Zhang, Q., and Laskin, A.: Molecular-Level Study of the Photo-Oxidation of Aqueous-Phase Guaiacyl Acetone in the Presence of 3C*: Formation of Brown Carbon Products, ACS Earth Space Chem., https://doi.org/10.1021/acsearthspacechem.1c00103, 2021.
O'Dell, K., Bilsback, K., Ford, B., Martenies, S. E., Magzamen, S., Fischer, E. V., and Pierce, J. R.: Estimated mortality and morbidity attributable to smoke plumes in the united states: not just a western US problem, GeoHealth, 5, e2021GH000457, https://doi.org/10.1029/2021GH000457, 2021.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Palm, B. B., Peng, Q., Fredrickson, C. D., Lee, B. H., Garofalo, L. A., Pothier, M. A., Kreidenweis, S. M., Farmer, D. K., Pokhrel, R. P., Shen, Y., Murphy, S. M., Permar, W., Hu, L., Campos, T. L., Hall, S. R., Ullmann, K., Zhang, X., Flocke, F., Fischer, E. V., and Thornton, J. A.: Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, P. Natl. Acad. Sci. USA, 117, 29469–29477, https://doi.org/10.1073/pnas.2012218117, 2020.
Powelson, M. H., Espelien, B. M., Hawkins, L. N., Galloway, M. M., and De Haan, D. O.: Brown carbon formation by aqueous-phase carbonyl compound reactions with amines and ammonium sulfate, Environ. Sci. Technol., 48, 985–993, https://doi.org/10.1021/es4038325, 2014.
Reid, J. S., Koppmann, R., Eck, T. F., and Eleuterio, D. P.: A review of biomass burning emissions part II: intensive physical properties of biomass burning particles, Atmos. Chem. Phys., 5, 799–825, https://doi.org/10.5194/acp-5-799-2005, 2005.
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking organic aerosols: semivolatile emissions and photochemical aging., Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007.
Saleh, R.: From Measurements to Models: Toward Accurate Representation of Brown Carbon in Climate Calculations, Curr. Pollut. Reports, 6, 90–104, https://doi.org/10.1007/s40726-020-00139-3, 2020.
Saleh, R., Hennigan, C. J., McMeeking, G. R., Chuang, W. K., Robinson, E. S., Coe, H., Donahue, N. M., and Robinson, A. L.: Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions, Atmos. Chem. Phys., 13, 7683–7693, https://doi.org/10.5194/acp-13-7683-2013, 2013.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R.: Measurement of emissions from air pollution sources. 3. C1-C29 organic compounds from fireplace combustion of wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Schnitzler, E. G., Gerrebos, N. G. A., Carter, T. S., Huang, Y., Heald, C. L., Bertram, A. K., and Abbatt, J. P. D.: Rate of atmospheric brown carbon whitening governed by environmental conditions, P. Natl. Acad. Sci. USA, 119, e2205610119, https://doi.org/10.1073/pnas.2205610119, 2022.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Smith, J. D., Kinney, H., and Anastasio, C.: Aqueous benzene-diols react with an organic triplet excited state and hydroxyl radical to form secondary organic aerosol, Phys. Chem. Chem. Phys., 17, 10227–10237, https://doi.org/10.1039/c4cp06095d, 2015.
Smith, J. D., Kinney, H., and Anastasio, C.: Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products, Atmos. Environ., 126, 36–44, https://doi.org/10.1016/j.atmosenv.2015.11.035, 2016.
Sun, Y. L., Zhang, Q., Anastasio, C., and Sun, J.: Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry, Atmos. Chem. Phys., 10, 4809–4822, https://doi.org/10.5194/acp-10-4809-2010, 2010.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006GL026899, 2006.
Wang, Y., Huang, W., Tian, L., Wang, Y., Li, F., Huang, D. D., Zhang, R., Go Mabato, B. R., Huang, R.-J., Chen, Q., Ge, X., Du, L., Ma, Y. G., Gen, M., Hoi, K. I., Mok, K. M., Yu, J. Z., Chan, C. K., Li, X., and Li, Y. J.: Decay Kinetics and Absorption Changes of Methoxyphenols and Nitrophenols during Nitrate-Mediated Aqueous Photochemical Oxidation at 254 and 313 nm, ACS Earth Space Chem., 6, 1115–1125, https://doi.org/10.1021/acsearthspacechem.2c00021, 2022.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Yu, L., Smith, J., Laskin, A., George, K. M., Anastasio, C., Laskin, J., Dillner, A. M., and Zhang, Q.: Molecular transformations of phenolic SOA during photochemical aging in the aqueous phase: competition among oligomerization, functionalization, and fragmentation, Atmos. Chem. Phys., 16, 4511–4527, https://doi.org/10.5194/acp-16-4511-2016, 2016.
Zhang, A., Wang, Y., Zhang, Y., Weber, R. J., Song, Y., Ke, Z., and Zou, Y.: Modeling the global radiative effect of brown carbon: a potentially larger heating source in the tropical free troposphere than black carbon, Atmos. Chem. Phys., 20, 1901–1920, https://doi.org/10.5194/acp-20-1901-2020, 2020.
Zhao, R., Lee, A. K. Y., Huang, L., Li, X., Yang, F., and Abbatt, J. P. D.: Photochemical processing of aqueous atmospheric brown carbon, Atmos. Chem. Phys., 15, 6087–6100, https://doi.org/10.5194/acp-15-6087-2015, 2015.
Zhong, M. and Jang, M.: Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight, Atmos. Chem. Phys., 14, 1517–1525, https://doi.org/10.5194/acp-14-1517-2014, 2014.
Zhou, X., Josey, K., Kamareddine, L., Caine, M. C., Liu, T., Mickley, L. J., Cooper, M., and Dominici, F.: Excess of COVID-19 cases and deaths due to fine particulate matter exposure during the 2020 wildfires in the United States, Sci. Adv., 7, eabi8789, https://doi.org/10.1126/sciadv.abi8789, 2021.