the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Jan 2024
| 31 Jan 2024
Impact of acidity and surface-modulated acid dissociation on cloud response to organic aerosol
Gargi Sengupta
Minjie Zheng
Acid dissociation of the organic aerosol fraction has the potential to impact cloud-activating properties by altering aqueous-phase H+ concentrations and water activity but is currently overlooked in most atmospheric aerosol models. We implemented a simple representation of organic acid dissociation in the aerosol–chemistry–climate box model ECHAM6.3–HAM2.3 and investigated the impact on aerosol-forming aqueous sulfur chemistry, cloud droplet number concentrations, and the shortwave radiative effect. Many atmospheric organic acids are also surface-active and may be strongly adsorbed at the surface of small aqueous droplets. The degree of dissociation has recently been observed for several atmospheric surface-active organics with Brönsted acid character to be significantly shifted in the surface, compared to the bulk aqueous solution. In addition to the well-known bulk acidity, we therefore introduced an empirical account of this surface-modulated dissociation to further explore the potential impact on aerosol climate effects. Malonic acid and decanoic acid were used as proxies for atmospheric organic aerosols of different surface-active and acid strengths. Both acids were found to yield sufficient hydrogen ion concentrations from dissociation in an aqueous droplet population to strongly influence aqueous aerosol sulfur chemistry, leading to enhanced cloud droplet number concentrations and a cooling shortwave radiative effect. Further considering the surface modulation of organic acid dissociation, the impact on cloud microphysics was smaller than according to the well-known bulk solution acidity but still significant. Our results show that organic aerosol acid dissociation can significantly influence predictions of aerosol and cloud droplet formation and aerosol–cloud–climate effects and that, even for a well-known bulk solution phenomenon such as acidity, it may be important to also consider the specific influence of surface effects when surface-active acids comprise a significant fraction of the total organic aerosol mass.
- Article
(2651 KB) - Full-text XML
-
Supplement
(1422 KB) - BibTeX
- EndNote





Atmospheric aerosols are an important contributor to Earth’s climate. They may either absorb or reflect heat and sunlight, directly affecting Earth’s energy budget (IPCC, 2013; Masson-Delmotte et al., 2021). Aerosols also contribute to the global climate through indirect effects where they serve as the necessary seeds for cloud formation (Twomey, 1977; Lohmann and Lesins, 2002). The chemical composition of aerosols is complex and includes numerous organic and inorganic species (O'Dowd et al., 2004; Putaud et al., 2010; Murphy et al., 2006). Organic compounds have been reported to comprise approximately 20 %–50 % of the total aerosol mass at mid-latitude regions (Saxena and Hildemann, 1996; Putaud et al., 2004) and a much higher proportion (approximately 90 %) in tropical forests (Andreae and Crutzen, 1997; Roberts et al., 2001; Kanakidou et al., 2005). Significant amounts of organic aerosols (approximately 70 % of the total aerosol mass) are also reported in the middle troposphere (Huebert et al., 2004). Despite their abundance and importance, the organic fraction is the least understood component of atmospheric aerosols and the uncertainty around organic aerosols, and their interaction with clouds remains one of the largest overall sources of uncertainty in climate projections (IPCC, 2013; Seinfeld et al., 2016; Legg, 2021). The size, chemical composition, and phase state of organic aerosol (OA) are known to directly impact its cloud droplet formation potential (Hallquist et al., 2009; McFiggans et al., 2006) and the radiative effect of clouds (Turnock et al., 2019). However, climate models often have a limited representation of OA processes and properties, such as acidity and surface activity of the organic components (Kanakidou et al., 2005; Prisle et al., 2012a; Freedman et al., 2018; Pye et al., 2020), which contribute to the overall uncertainty in predictions of OAs and their interactions with clouds.
Organic aerosols contain a substantial fraction of species exhibiting Brönsted acid character (Jacob, 1986; Millet et al., 2015; Keene and Galloway, 1984; Chebbi and Carlier, 1996; Chen et al., 2021b; Angelis et al., 2012; Mochizuki et al., 2016; Wu et al., 2020; Kawamura et al., 1985). The concentrations of acidic species in aqueous aerosols directly affect the aerosol pH by modifying the H+ concentrations within the aerosol (Pye et al., 2020; Ault, 2020). This pH affects the dissociation of individual acidic species with significant consequences for aerosol chemistry (Hung et al., 2018; Wang et al., 2018) and phase state (Liu et al., 2019). For example, pH-dependent sulfur oxidation (Liu et al., 2020) and salt formation by acidic or basic OA (Yli-Juuti et al., 2013) can each lead to significant mass formation and alter the overall chemical composition of aerosols. The chemical form (protonated or deprotonated) of acidic OA and contributions to the number of solute species in the aqueous aerosol phase can strongly affect water activity and condensation–evaporation equilibrium (Prisle, 2006; Prisle et al., 2008; Frosch et al., 2011; Michailoudi et al., 2019).
Many atmospheric organic acids also exhibit surface activity in aqueous solutions, such as aqueous aerosols and cloud droplets (Prisle, 2024). Surface-active organics (surfactants) have been reported in atmospheric aerosols from many different regions and environments (Gérard et al., 2016; Petters and Petters, 2016; Nozière et al., 2017; Kroflič et al., 2018; Gérard et al., 2019a). Surfactants adsorb at the aqueous surface, leading to enhanced surface concentrations compared to the interior (bulk) of a solution. In microscopic and submicron-sized aerosols and droplets, the surface adsorption can result in significant redistribution of surface-active OA mass from the bulk to the surface phase, so-called bulk–surface partitioning, as a consequence of the very high ratio of surface area (A) to bulk volume (V) in these size ranges (Prisle et al., 2008, 2010b). For spherical droplets of diameter Dwet=0.1, 1, and 10 µm, is 60, 6, and 0.6 µm−1, respectively (Prisle, 2021). Thermodynamic calculations have shown that for aerosol particles containing surfactant fatty acids and their salts, organosulfates, di- and polycarboxylic acids, and complex fulvic acids, a large fraction of the surface-active OA is partitioned to the surface during major parts of hygroscopic growth and cloud droplet activation (Prisle et al., 2010b, 2011; Hansen et al., 2015; Malila and Prisle, 2018; Lin et al., 2018, 2020; Prisle, 2021; Vepsäläinen et al., 2022, 2023). Consequently, the chemical and physical state of the surface may significantly contribute to determining the overall aerosol properties (Prisle et al., 2012b; Bzdek et al., 2020; Prisle, 2021, 2024).
Highly surface sensitive synchrotron radiation excited X-ray photoelectron spectroscopy (XPS) measurements have been used to investigate the acid–base speciation of surface-active atmospheric Brönsted acids and bases in the surface region of aqueous solutions (Prisle et al., 2012b; Werner et al., 2018; Öhrwall et al., 2015). In these experiments, the protonated form of each conjugate pair was found at an extraordinarily large fraction, compared to that expected from their acidity and the bulk solution pH. Werner et al. (2018) observed a shift in the degree of protonation for simple mono-carboxylic acids at the surface in dilute aqueous solutions (50 mM butyric or pentanoic acid) corresponding to activating cloud droplets. The acid–base equilibrium in the surface was shifted systematically across a very wide range of solution pH, overall corresponding to an apparent shift in pKa on the order of 1–2 pH units, compared to the well-known bulk acidity, for each of the acids. Shifts in surface protonation degree of similar magnitude were also previously observed using XPS for dilute aqueous solutions of 10–25 mM decanoate/decanoic acid (Prisle et al., 2012b), 0.1 M propanoate/propanoic acid, and 0.1 M octanoate/octanoic acid (Öhrwall et al., 2015) at near-neutral pH. XPS measurements on aqueous solutions of succinic acid, a moderately surface-active dicarboxylic acid, over a range of concentrations from 0.05–0.5 M and solution pH from 2.0–12.9 also indicated a shifted acid–base equilibrium in the surface compared to the bulk, where the protonated form showed a considerably higher propensity to reside in the aqueous surface region than its conjugate deprotonated form (Werner et al., 2014a). These observations are further supported by experiments by Wellen et al. (2017), who used surface tension titration and infrared reflection absorption spectroscopy to obtain pH-dependent aqueous surface tension of nonanoic and decanoic acids at their aqueous solution surfaces. They inferred that the so-called surface pKa was greater than the well-known bulk pKa by 1 pH unit for nonanoic acid and 2 pH units for decanoic acid, suggesting that the organic acid dissociation response to a given aqueous bulk pH is different in the surface, compared to the bulk.
The general behavior of acidic compounds at the aqueous interface is still not well constrained (Saykally, 2013). Petersen and Saykally (2005, 2008) observed an enhanced surface concentration of hydronium ions in aqueous solutions of hydroiodic acid, alkali iodides, and alkali hydroxides using second harmonic generation spectroscopy experiments. This was in contrast to previous macroscopic bubble and droplet experiments, which were interpreted to indicate that hydroxide ions were enhanced at the air–water interface (Graciaa et al., 1995; Takahashi, 2005; Karraker and Radke, 2002; Creux et al., 2007). Enami et al. (2010) also made similar observations of enhanced hydronium ions in the surface of trimethylamine solutions using electrospray mass spectrometry. Recently, Gong et al. (2023) used stimulated Raman scattering microscopy to observe enhanced concentrations of sulfate and bisulfate anions, with bisulfate being more surface enriched than sulfate, in the surface of 2.9 µm aerosol droplets generated from an aqueous solution with 300 mM NaHSO4 and 50 mM Na2SO4 at the same pH. They interpret this as an enhancement of acidity, with an approximately 3-fold increase in the hydrogen ion concentration, at the droplet edge, compared to the center of the droplet. Previous observations by Margarella et al. (2013) on the dissociation of sulfuric acid at the water interface, using liquid-jet photoelectron spectroscopy, have also reported that the ratio of bisulfate-to-sulfate anions was higher in the surface region.
In this work, we use an aerosol–chemistry–climate box model to investigate the potential impact on aerosol-forming aqueous-phase sulfur chemistry, cloud droplet activation, and aerosol–cloud–climate parameters of organic acid dissociation in aqueous aerosols and its additional surface modulation for surfactant acidic OA. Very few studies have previously addressed the dissociation of organic components in aerosols in relation to cloud chemistry and microphysics (Tilgner et al., 2021a; Angle et al., 2021). Tilgner et al. (2021a) compiled a kinetic data set to study the implications of varying aerosol acidity for the oxidation of the protonated and deprotonated forms of atmospheric organic acids with aqueous-phase oxidants, such as OH radicals, NO3 radicals, or O3. They showed that acidity strongly affects the chemical processing of dissociating organic compounds but did not provide a direct correlation between organic dissociation and cloud activation. Angle et al. (2021) measured the pH of freshly emitted (nascent) sea spray aerosol using a Micro-Orifice Uniform Deposit Impactor (MOUDI) and impacting the aerosols onto colorimetric pH strips. They found that the pH of nascent sea spray aerosols was approximately 4 pH units lower than that of sea water. The dissociation of organic acids in the aerosols is proposed as a possible factor contributing to the low nascent sea spray aerosol pH. Angle et al. (2021) note that for a nascent sea spray aerosol with a diameter of 200 nm and surface layer of palmitic acid, only 4.4 % acid dissociation would be required to lower the aerosol pH from 8 to 2. However, they do not provide any details on how the organic acid dissociation would affect the aerosol properties. To the best of our knowledge, the organic acid dissociation in aqueous aerosols, let alone accounting for its surface-specific modulation, has never been studied in the context of a cloud activation model.
We first introduce an account of well-known organic acid dissociation in bulk aqueous solution and then augment it with a simple empirical representation to further include surface-driven suppression of dissociation according to observations from XPS measurements. We use ECHAM6.3–HAM2.3 (here referred to as HAMBOX), which is a box model version of the aerosol–chemistry–climate model ECHAM–HAMMOZ (Tegen et al., 2019), to calculate the total aerosol population sulfate mass and cloud droplet number concentrations (CDNCs) for an air parcel and the shortwave radiative effect (RE) from cloud formation, as examples of key processes taking place in aqueous organic aerosols and droplets. The impact of organic aerosol bulk acidity and surface-modulated suppressed dissociation on aerosol sulfur chemistry and cloud microphysics is assessed by comparing to predictions for identical conditions without accounting for organic acid dissociation. The simulation time for all calculations was 1 h, with 1 s time steps.
2.1 Aerosol module in HAMBOX
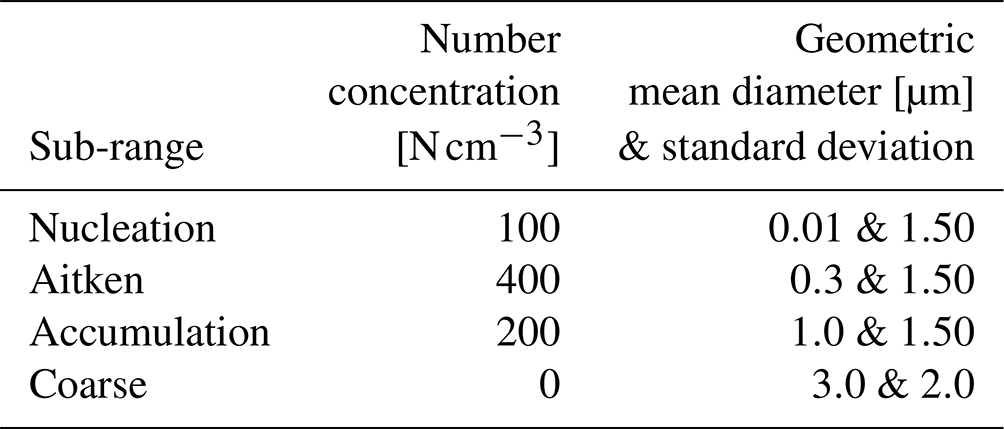
HAMBOX uses the SALSA2.0 aerosol module (Kokkola et al., 2018), where the aerosol size distribution is calculated using the sectional approach (Jacobson, 2005) and represented using 10 size bins i. Here, we group the 10 size bins into four sub-ranges: nucleation (i=1 and 2) with mean particle diameter, nm; Aitken (i=3, 4 and 5) with nm; accumulation (i=6, 7 and 8) with nm, and coarse (i=9 and 10) with µm. The initial number concentration in each sub-range (Table 1) used for all HAMBOX simulations is representative of clean environments, such as European villages (Tunved et al., 2005, 2008). As a property of the sectional approach, when particles grow or shrink out of the boundaries of their size bins, they are redistributed to new size bins, and the new aerosol size distribution is calculated at each simulation time step.
Table 1Initial aerosol number concentration in each size sub-range used in HAMBOX–SALSA2.0, representative of clean environments (Tunved et al., 2005, 2008).
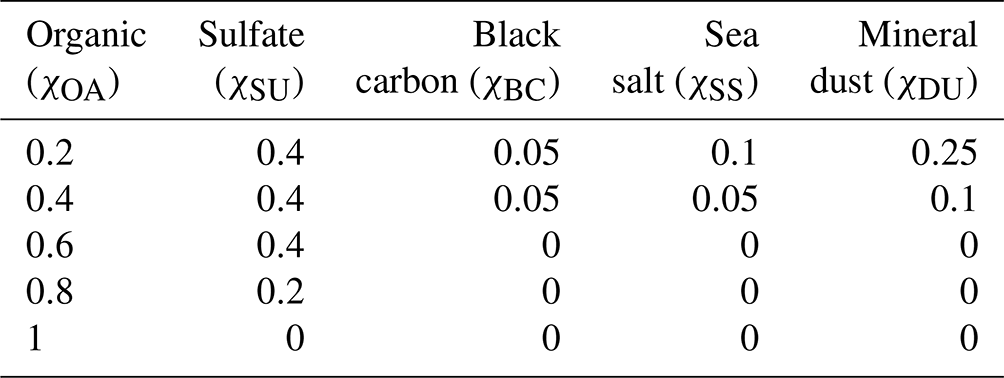
A sulfur chemistry module (Feichter et al., 1996a) is coupled to the aerosol growth module in SALSA2.0. At each time step, the sulfur chemistry module feeds the calculated sulfate mass fraction in the aerosol population into the growth module, which undergoes an aerosol redistribution and feeds back the new aerosol size distribution and chemical composition to the sulfur chemistry module. The aerosol chemical composition in SALSA2.0 is represented by the model compound classes “sulfate” (SU), “organic aerosol” (OA), “sea salt” (SS), “black carbon” (BC), and “mineral dust” (DU). Of these model compounds, sulfate, organic aerosol, and sea salt constitute the soluble species and are considered internally mixed in each size bin of the aerosol population. Black carbon and mineral dust are insoluble species which are externally mixed in each size bin with the soluble species, as described by Kokkola et al. (2018). We consider five different initial conditions with different OA mass fractions to represent different environments where OA has been reported in varying concentrations. For example, in boreal forest environments, the OA mass fraction is reported to be around 0.6 (Äijälä et al., 2019) and in marine environments around 0.2 (O'Dowd et al., 2004). χOA=1 is a hypothetical extreme where we consider OA to comprise the entire aerosol phase. The initial mass fractions of all aerosol model compounds in these five conditions are shown in Table 2.
The sulfur chemistry module is used to calculate the aqueous-phase secondary sulfate mass from oxidation of sulfur dioxide in the aerosol population based on varying hydrogen ion concentration in the aqueous aerosol, considering no organic acid dissociation, organic acid dissociation according to well-known bulk acidity, and surface-modulated organic acid dissociation (Sect. 2.3 below).
Table 2Initial aerosol mass fractions of all model compounds in the five different environmental scenarios considered.
2.2 Cloud microphysics in HAMBOX
From the total aerosol mass and composition, the resulting cloud droplet number concentrations and consequent shortwave radiative effect are calculated with the HAMBOX cloud microphysics module. The HAMBOX cloud microphysics used in this work includes the calculation of critical supersaturation (Si) and activated fraction (ni) for each aerosol size bin i. A detailed description of the parameterizations and equations used to calculate these cloud activation factors is available from Abdul-Razzak (2002) and Abdul-Razzak et al. (1998) and briefly summarized here.
First, the maximum critical supersaturation for the air parcel is calculated as
where η is the surface tension correction factor; ζ is the correction factor for the Kelvin term in the Köhler curve (for details, see Eqs. (5) and (6) in Abdul-Razzak and Ghan, 2002); and Se is the effective critical supersaturation for the air parcel,
Here, I=10 is the total number of aerosol size bins, Ni is the number of particles in each bin i, and Si is the critical supersaturation for each bin, given by
where Dwet and dp are droplet and dry particle diameters, respectively. The terms A and B are calculated as
where Mw=0.018 kg mol−1 is the molecular weight of water; σw=0.073 N m−1 is the surface tension of pure water; ρw=1000 kg m−3 is the density of water; R=8.314 is the ideal gas constant; T=293 K is the temperature; and ns is the number of moles of solute obtained from the mass fractions of soluble species χOA, χSU, and χSS.
With the maximum supersaturation of the air parcel, cloud droplet activation in each bin is determined by comparing Smax with Sil and Siu (the lower and upper critical supersaturation bounds of the bin). The number of activated particles in each size bin i is given by
and
Sil and Siu are obtained using Eq. (3) for the diameters of the smallest (dil) and largest (diu) particles in each size bin. The average activated fraction for all size bins is then calculated by
The total number of activated particles is given by the cloud droplet number concentration, which is calculated using the number of activating particles within each size bin, as
The CDNC calculated using Eq. (9) considering organic bulk acidity and surface-modulated organic acid dissociation is denoted by CDNCHA and the change with respect to the reference condition of no organic acid dissociation (CDNC0) is
We estimate the change in shortwave radiative effect (RE) from including organic bulk acidity and surface-modulated organic acid dissociation, respectively, using the method given by Bzdek et al. (2020). The change in cloud-top albedo at constant cloud liquid water content (LWC=0.03 g m−3, Zheng and Ming, 2022) is calculated from ΔCDNC as
The shortwave radiative effect is then calculated as
where F0=340 W m−2 is the incoming solar flux at the top of the atmosphere; ELWC=0.3 is the fractional coverage of different types of clouds; and TLWC=0.76 is the transmittance of the atmosphere at visible wavelengths, which is assumed to be constant for all simulations.
For all HAMBOX cloud microphysics calculations, we assume a constant cloud temperature of 271 K, cloud pressure of 101 kPa, cloud fraction of 0.3, saturation ratio of gas-phase water of 0.3, and updraft velocity of 0.3 m s−1, consistent with Tegen et al. (2019).
2.3 Sulfur chemistry in HAMBOX
We use the aqueous sulfur chemistry module of Feichter et al. (1996b), with modifications described below, to calculate the aqueous-phase secondary sulfate concentration in the aerosol population formed from the oxidation of SO2 by H2O2 and O3 in aqueous droplets. The reaction rate for the H2O2 oxidation pathway can be written as
where the rate constant k4 is calculated by
where T is the cloud temperature = 271 K. Equation (13) is known to be pH-insensitive (Liu et al., 2020) and is used in this work to determine the aqueous secondary sulfate concentration from the H2O2 oxidation for simulations where organic acid dissociation is not considered.
To calculate from the H2O2 oxidation pathway accounting for pH dependency arising from organic bulk acidity, we follow the procedure given by Liu et al. (2020), which is valid for pH>2. Here, we use the general acid catalysis reaction mechanism, where SO2 in an aqueous environment exists as the anion and reacts with H2O2 in the presence of an organic acid (HA) catalyst, which acts as a proton donor (Maaß et al., 1999; McArdle and Hoffmann, 1983). Briefly, the overall reaction mechanism is represented as
and
The rate expression for (R1)–(R2) is
where Ka1 is the thermodynamic dissociation constant (corresponding to the first acid dissociation constant as defined for organic acids in Reaction R4 below) of H2SO3, and k is a constant derived from the reaction rate coefficient and the thermodynamic equilibrium constants. kHA is the overall rate constant for the general acid catalysis mechanism approximated by (pKa)+6.83 (Liu et al., 2020; Drexler et al., 1991). This approximation for kHA in relation to the pKa of an organic acid was derived by Liu et al. (2020) for an ionic strength of I=0.5 mol kg−1. Therefore, we assume this same ionic strength for aqueous droplets in all our calculations.
The secondary sulfate concentration from O3 oxidation is given by
where rate constants k51 and k52 are calculated from
and
Sulfate concentrations thus calculated in the aqueous-phase sulfur chemistry module are distributed to pre-existing aerosol size bins in SALSA2.0.
When no organic aerosol acid dissociation (“no diss”) is considered, the default H+ concentration in HAMBOX is denoted by [H+]0 and obtained from water and aqueous-phase sulfate concentrations as
where LWC is the cloud liquid water content (in [g m−3]), is the molar weight of the sulfate anion, and the soluble sulfate concentration is obtained from the summation of soluble sulfate (obtained from χSU, Table 2) in all bins. mol L−1 is the hydrogen ion concentration obtained from the cloud pH=5, which is assumed to be uniform for all size bins and consistent with the pH of warm low-lying tropospheric clouds (Pye et al., 2020). For all simulations in the sulfur chemistry module, we assume [SO2], [H2O2], and [O3] in cloud are fixed at 5, 1, and 50 ppb, respectively (Tilgner et al., 2021b).
We introduce organic acid dissociation to the sulfur chemistry module by modifying Eq. (19) to obtain the total hydrogen ion concentration in the aerosol population as
where [H+]HA is the concentration of the hydrogen ions dissociated by the acidic organic aerosol components. The calculated [H+]tot is then used to obtain the aqueous-phase secondary sulfate concentration in the aerosol population from SO2 oxidation by H2O2 (Eq. 15) and O3 (Eq. 16), for varying conditions of organic acid dissociation. The sulfate concentrations thus obtained give a modified sulfate mass fraction (χSU, Sect. 2.1 and Table 2) in the entire aerosol population.
2.4 Organic acid dissociation
We assume the entire OA fraction is comprised of an organic acid and consider two different acids, malonic acid (a diprotic acid) and decanoic acid (a monoprotic acid), as examples of important organic aerosol components in the atmosphere (Yassaa et al., 2001; Narukawa et al., 2002; Mochida et al., 2003; Graham et al., 2003; Cheng et al., 2004; Li and Yu, 2005; Tedetti et al., 2006) with different well-known aqueous bulk acidity and prominent examples of moderately and strongly surface-active organic species, respectively (Vepsäläinen et al., 2022, 2023). The acid dissociation constants for aqueous bulk solutions are here denoted as p to distinguish the well-known bulk dissociation behavior from surface-modulated dissociation introduced in Sect. 2.4.3 below. The molecular weight (MW), density (ρ), and bulk acid constants for decanoic and malonic acids used in our calculations are given in Table 3. For the monoprotic decanoic acid, p is the reported first pKa readily available from the literature, whereas for the diprotic malonic acid, p is taken as the sum of the first and second pKa reported in the literature (same as β in Eq. 29 below). The dissociation behavior of monoprotic and diprotic acids in similar aqueous environments differ greatly, and we use different kinetic equations to describe both treatments of organic bulk and surface-modulated acid dissociation, presented in Sects. 2.4.1, 2.4.2, and 2.4.3.
2.4.1 Monoprotic acids
The dissociation of a monoprotic organic acid (HA) in aqueous solution is represented by the equilibrium
where H+ from the dissociation of the organic acid is considered to be fully hydrated, such that the concentration of H3O+ is equivalent to the concentration of hydrogen ions from dissociation of HA. The equilibrium acid dissociation constant is
where , and aHA are the activities of H3O+ cations, A− anions, and HA molecules, respectively. Equation (21) can be approximated in terms of the molar concentrations and ideal-dilute molar-concentration-based activity coefficients (γi) of each species i as
Since a monoprotic acid HA has only one ionizable hydrogen in Eq. (22),
where [HA]tot is the total concentration of the organic acid.
The acid dissociation degree α is defined as
Combining Eqs. (22) and (24) and approximating with the mean activity coefficient γ±,
For a highly dilute solution (e.g., [HA]tot<0.001 mol L−1), and Eq. (25) can be written as
With [HA]tot known from χOA and other properties of the aerosol population, the hydrogen ion concentration from organic acid dissociation [H+]HA is obtained as [H3O+] in Eq. (24) by
where the acid dissociation degree α is given by Eq. (26).
2.4.2 Diprotic acids
For a diprotic organic acid (H2A), the dissociation of H+ ions in an aqueous solution can be considered to occur in two stages,
and
The dissociation constant for (R4) is the first dissociation constant of the diprotic acid, denoted as Ka1, and the dissociation constant for (R5) is the second dissociation constant of the diprotic acid, denoted as Ka2. The overall dissociation constant of H2A is
and, therefore,
Using similar assumptions as for the monoprotic acid, for a highly dilute solution, the acid dissociation degree α for a diprotic acid can be derived as
where [H2A]tot is the total concentration of the diprotic acid. Analogously to the case of a monoprotic acid, the hydrogen ion concentration from dissociation of a diprotic organic acid [H+]HA is given by
where the acid dissociation degree α is now given by Eq. (30).
2.4.3 Surface-modulated organic acid dissociation
We now introduce a simple empirical representation of the shift in organic acid dissociation previously observed in surface-sensitive XPS experiments. Werner et al. (2018) found that the surface-specific dissociation state of surface-active mono-carboxylic acids was significantly suppressed in dilute aqueous solutions across a very wide range of solution pH=2–12. Similar suppressed dissociation states were also found for other mono- and dicarboxylic acids of both stronger and weaker surface activity, in aqueous solutions closer to neutral pH (Prisle et al., 2012b; Werner et al., 2014b; Öhrwall et al., 2015). The shifted dissociation states are attributed to both increased concentrations of the surface-active organic acids in the surface and increased non-ideality (higher activity coefficients) of the charged deprotonated conjugate species A− and hydronium ions, compared to the neutral molecular acid HA, in the organic-rich air–solution interfacial region (Werner et al., 2018; Prisle, 2024). From Eq. (22), this corresponds to an apparent shift of the acid pKa at the surface,
compared to the well-known bulk acidity p obtained for dilute aqueous solutions, where all activity coefficients are assumed to be ideal, γi=1 (Prisle, 2024).
The dissociation states observed with XPS are broadly consistent with a magnitude of the apparent shift in pKa of –2 pH units across the surface titration curve (Prisle, 2024). Here we introduce the effect of surface-modulated acid dissociation by shifting the well-known bulk pKa of each organic acid according to these shifts of the surface titration curves. We consider two magnitudes of this apparent shift, covering the range of experimental observations from XPS. For a monoprotic acid, we consider p and p, where p is the well-known pKa of the organic acid in aqueous bulk solution. To represent the surface shifted dissociation of both carboxylic groups in a diprotic acid, we increase both the first and second acid constant, by 1 or 2 pH units, to similarly obtain p and p. Here we refer to the shifted pKa values as the surface-modulated apparent pKa. However, we strongly emphasize that the pKa, which is an intrinsic property of each organic acid in bulk aqueous solution, is not itself changed. Only the dissociation responses of the organic acids to a given pH of the solution (here, the cloud pH) are changed in the surface (Prisle, 2024).
For both mono- and diprotic acids, the values used for surface-modulated apparent pKa are given in Table 3. For each pKa, the corresponding acid dissociation degree α is calculated for the monoprotic acid using Eq. (26) and for the diprotic acid using Eq. (30). The value for α decreases with increasing pKa, such that the increased apparent pKa values represent suppressed dissociation of the organic acid in the surface. The surface modulation of organic dissociation is most pronounced in a range of several pH units around the bulk pKa. At very low and very high pH, the surface dissociation states collapse onto the well-known bulk solution dissociation behavior (Werner et al., 2018; Prisle, 2024). For both the organic acids used here, the p is within a few pH units of the cloud pH.
Although we implement the effect of surface-modulated acid dissociation as a consequence of simultaneous surface activity of the organic acid, we do not explicitly consider the bulk–surface partitioning of organic acids in our calculations. Our simple empirical representation by shifting the apparent pKa for the organic acid corresponds to assuming that the overall dissociation state in aqueous aerosols and droplets is described by the surface-modulated properties. This is closely representative of aerosols and droplets where the majority of organic aerosol components are partitioned to the aqueous surface, as a consequence of strong organic surface activity or high in the microscopic and submicron size ranges (Prisle, 2021, 2024). In real atmospheric aerosol and droplet mixtures of both surface-active and more water-soluble OA, organic species will be partially partitioned to the surface, and the overall dissociation state should be described as a combination of both well-known bulk acidity and surface-modulated states. The present simple empirical representation therefore gives an upper bound of the potential effects of surface-modulated acid dissociation according to the previous observations from XPS experiments.
When surface-modulated organic acid dissociation is considered, these properties are assumed to remain consistent throughout the 1 h simulations. Prisle et al. (2008) and Prisle (2021) estimated that surface adsorption of typical atmospheric surfactants equilibrates within a timescale of a second in micron-sized droplets. Lin et al. (2020) investigated the impact of surface adsorption dynamics on surfactant effects in cloud droplet activation and found that different dynamic effects nearly cancel out at every time step. Noziere et al. (2014) assumed that both the bulk and surface reach a state of reasonable equilibrium with respect to organic adsorption at the aqueous surface within approximately 495 s. Therefore, we consider this assumption to be a reasonable first approximation.
Table 3Properties of the organic acids used in calculations of acid dissociation, including molecular properties (MW and ρ), well-known bulk solution acidity (pKa1, pKa2, and p), and surface-modulated dissociation properties (implemented as p and p).
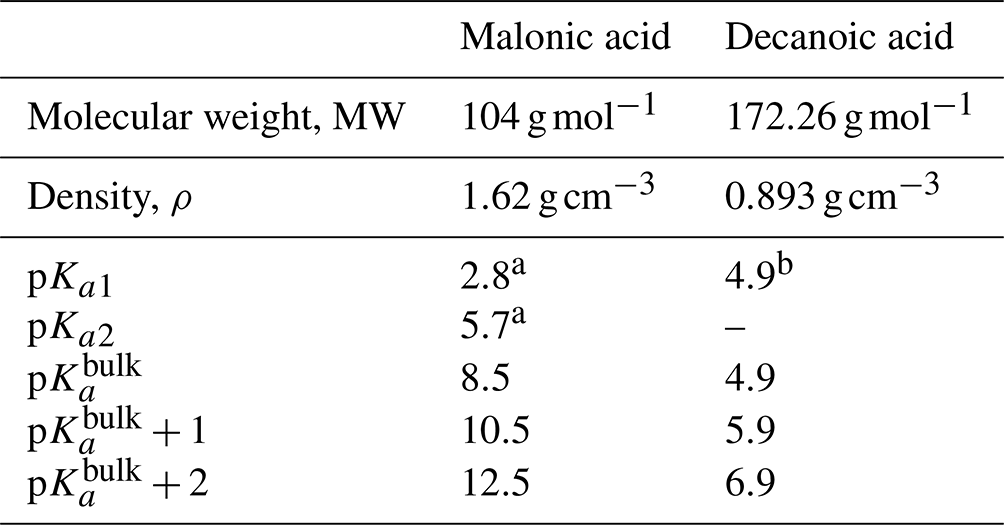
a Stahl and Wermuth (2002). b Martell and Smith (1974).
2.4.4 The van't Hoff factor for organic dissociation
Aqueous-phase dissociation also influences the available amount of solute species in aerosol particles and droplets, affecting the calculations of water activity and critical supersaturation (Sect. 2.2). The molar amount of available solute ns is calculated in SALSA2.0 from the molar amounts of all the internally mixed soluble species as
where the nSU, nOA, and nSS are the molar amounts of sulfate, organic aerosol, and sea salt, respectively, derived from the initial aerosol mass fractions given in Table 2, and iSU, iOA, and iSS are the corresponding van't Hoff factors for each soluble species. In SALSA2.0, the sulfate and sea salt are considered to be fully dissociated, such that iSU=3 and iSS=2. By default, organic aerosol is not considered as dissociated and iOA=1.
To include effects of organic acid dissociation, iOA is calculated with consideration of the acid dissociation degree α (from Eqs. 26 and 30) as
where nions=2 for the monoprotic acid and nions=3 for the diprotic acid is the number of ions formed from one formula unit of the organic acid. The total available molar amount of solute (ns, Eq. 33) is thus modified by organic acid dissociation according to iOA from Eq. (34) and reflected in the Raoult term B (Eq. 4), which changes the critical supersaturation Si for each aerosol size bin. The van't Hoff factor is calculated for each pKa using Eq. (34). The dissociation degrees and van't Hoff factors for the no organic dissociation (no diss), organic acid dissociation according to bulk acidity (p), and surface-modulated suppressed organic acid dissociation (p and p), for all the OA mass fractions considered here, are given in the Supplement (Table S1).
We present the results of HAMBOX simulations for sulfur chemistry, cloud microphysics, and aerosol–cloud–climate effects, considering organic acid bulk acidity (p), surface-modulated suppressed organic acid dissociation (p and 2), and no organic acid dissociation (no diss). Simulations were carried out assuming the entire OA fraction (Table 2) is composed of either malonic acid or decanoic acid. Results of sulfur chemistry calculations are presented in terms of total hydrogen ion concentration [H+]tot and secondary sulfate concentration in the aerosol population. The consecutive effect on cloud-activating properties is then presented in terms of change in cloud droplet number concentration ΔCDNC and cloud radiative effect RE predicted for bulk and surface-modulated suppressed dissociation of the organic acids, compared to no organic dissociation.
3.1 Aqueous aerosol hydrogen ion concentration
Figure 1 shows the total hydrogen ion concentration [H+]tot calculated with HAMBOX (Eq. 20) in the aqueous aerosol population with sizes between Dwet=0.317–40 µm after 1 h of simulation time, as a function of varying pKa, corresponding to representations of bulk (p) and surface-modulated (p and 2) organic acid dissociation, considering the OA fraction as malonic acid (panels a, b) or decanoic acid (panels c, d), for varying initial mass fraction of organic aerosol ( in blue, purple, pink, orange, and yellow, respectively). The hydrogen ion concentration for no organic dissociation (no diss, Eq. 19) is also shown as a black line. As expected, [H+]tot does not change with pKa when organic acid dissociation is not accounted for, whereas a significant increase is observed for both malonic and decanoic acids when organic acid dissociation is considered. The total hydrogen ion concentration is highest when organic acid dissociation is considered according to p and decreases for all χOA values as acid dissociation is increasingly suppressed according to the surface-modulated apparent p and p.
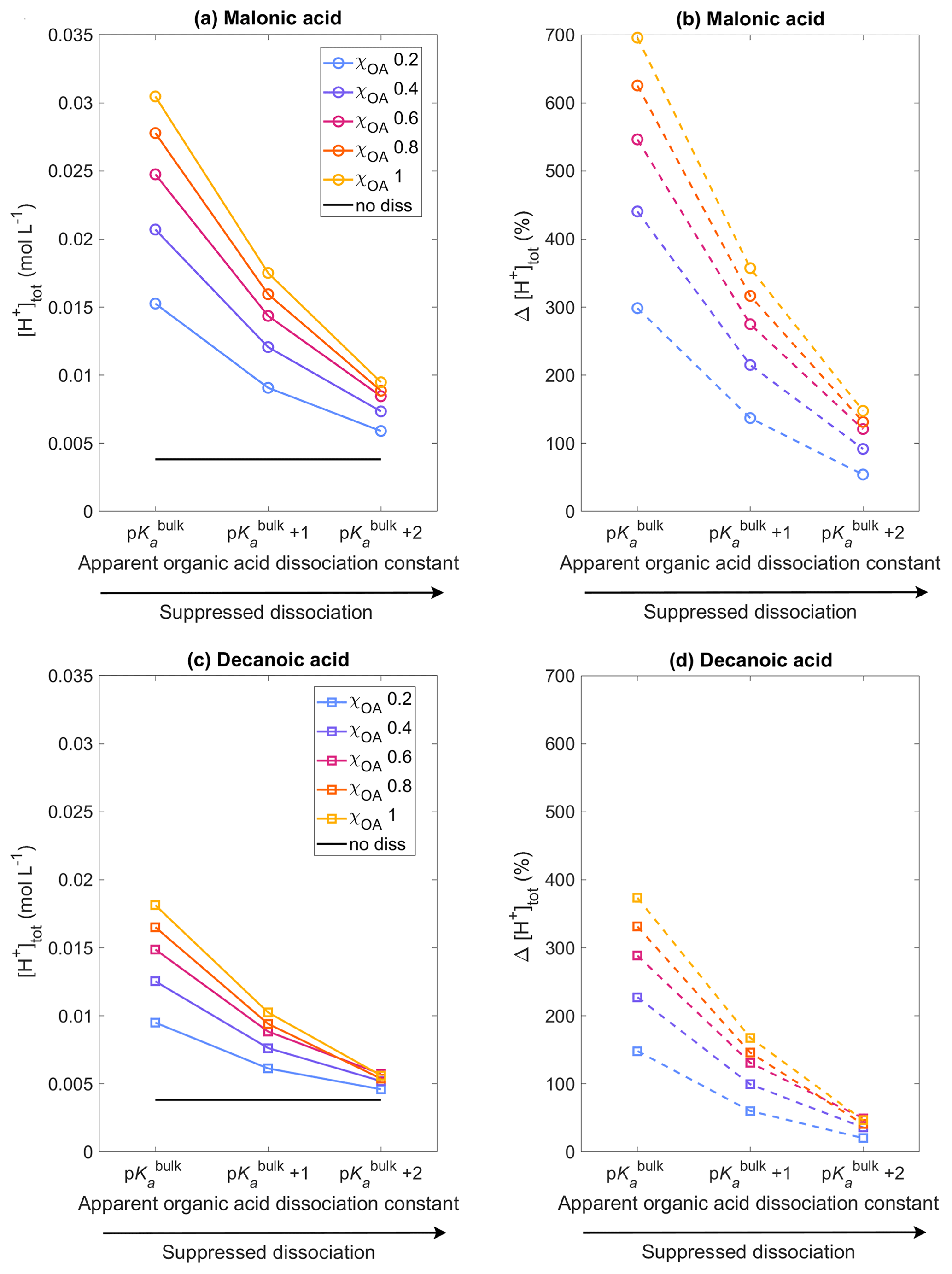
Figure 1Aqueous aerosol total hydrogen ion concentration, [H+]tot, calculated from Eq. (20) with the sulfur chemistry module of HAMBOX in the aqueous aerosol population with sizes between Dwet=0.317–40 µm, after 1 h of simulation time, assuming five different initial organic mass fractions , denoted by blue, purple, pink, orange, and yellow, respectively (see also Table 2), with varying pKa corresponding to representations of bulk (p) and surface-modulated (p and 2) organic acid dissociation. (a, b) All OA is assumed to be malonic acid, and (c, d) all OA is assumed to be decanoic acid. Simulations without accounting for organic acid dissociation are represented by no diss and (b) and (d) show the relative change in total hydrogen ion concentration Δ[H+]tot calculated from Eq. (35) for each of the acid dissociation conditions with respect to no diss.
The relative change in total hydrogen ion concentration with respect to the no diss condition
is shown for malonic and decanoic acids in panels (b) and (d), respectively, of Fig. 1. For malonic acid with p, % at the lowest OA mass fraction χOA=0.2 % and 696 % for the highest OA mass fraction χOA=1. Under the surface-modulated suppressed organic acid dissociation condition p, Δ[H+]tot decreases to 137 % and 357 % for χOA=0.2 and χOA=1, respectively. On further suppression of organic acid dissociation according to p, Δ[H+]tot further decreases to 54 % and 148 %, respectively, at these OA mass fractions. For decanoic acid organic aerosol, the total hydrogen ion concentration at p increases by 148% with respect to no diss for χOA=0.2 and by 374 % for χOA=1. At p, Δ[H+]tot decreases to 61 % and 168 %, respectively, and at p further to 20 % and 47 % for χOA=0.2 and 1, respectively. Therefore, even considering the stronger surface-modulated suppression of organic acid dissociation with p, the total hydrogen ion concentration in the aqueous aerosol is still 20 %–47 % higher than no diss for decanoic acid and 54 %–148 % higher for malonic acid, depending on the initial organic aerosol mass fraction.
The results shown in Fig. 1 and in the following are obtained for a constant ionic strength of I=0.5 mol kg−1. Ionic strength is a bulk solution phenomenon and not expected to affect surface adsorbed organic acids, which can be considered a (partially) liquid–liquid separated phase (Prisle et al., 2010a), to the same degree as in the bulk solution. Therefore, the number of hydrogen ions dissociated by the organic acid ([H+]HA, Eq. 20) is expected to depend on I mainly for the bulk solution condition and potentially to some extent for the surface-modulated conditions. The total hydrogen ion concentration ([H+]tot, Eq. 20) in the aerosol population is shown in Fig. S1 of the Supplement for malonic acid as the OA, considering χOA=0.4 and 0.6, and for varying ionic strengths mol kg−1.
The total hydrogen ion concentration decreases with increasing ionic strength, as expected. For χOA=0.4, [H+]tot is approximately 270 % greater for p than without consideration of dissociation (no diss) at I=5 mol kg−1. For the surface-modulated dissociation condition at the same ionic strength and organic aerosol mass fraction, [H+]tot is approximately 120 % greater for p compared to no diss. Even for the more strongly suppressed dissociation corresponding to p, [H+]tot is approximately 45 % higher than without dissociation. Therefore, for mol kg−1, the total hydrogen ion concentration in the aqueous aerosol has significant contribution from organic acid dissociation. Similar analysis for varying ionic strength considering the OA as decanoic acid was not immediately possible, due to lack of data on the variation of pKa with I for aqueous decanoic acid solutions. However, measurements of pKa for acetic acid in aqueous solutions with varying ionic strength were reported by Cohn et al. (1928). The total hydrogen ion concentration for varying ionic strengths in the aqueous aerosol is shown for acetic acid as the OA in Fig. S2 of the Supplement. For acetic acid as the OA, organic acid dissociation considering mol kg−1 results in approximately 270 %–380 % higher [H+]tot than without acid dissociation. Both acetic acid (pKa=4.76, Goldberg et al., 2002) and decanoic acid (pKa=4.9, Martell and Smith, 1974) are straight-chain monocarboxylic acids with comparable bulk acidity, and their aqueous dissociation properties are expected to be similar. Therefore, variation in I is expected to result in similar [H+]tot in the aqueous aerosol when the OA is decanoic acid as for acetic acid.
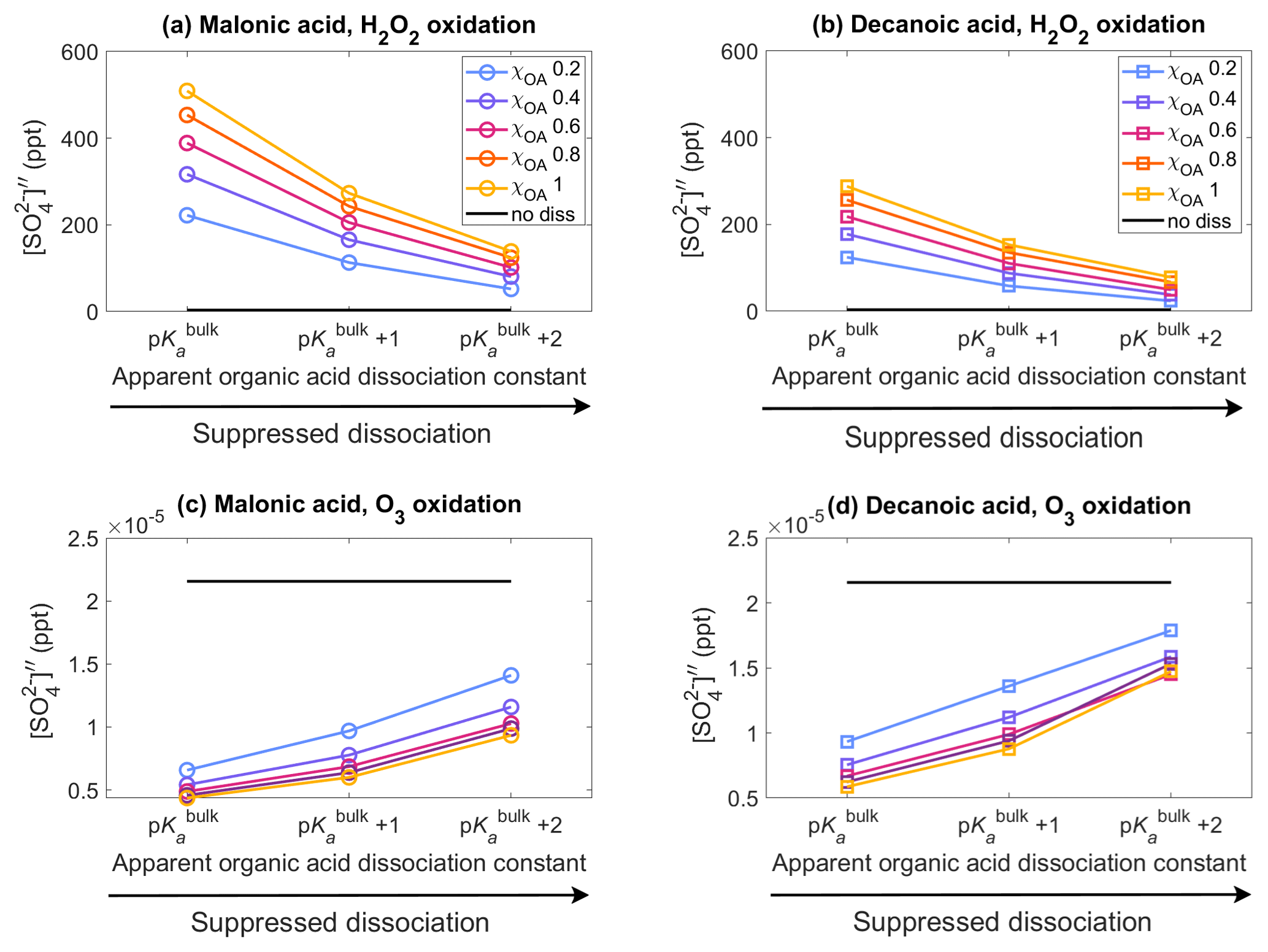
Figure 2Aqueous aerosol secondary sulfate concentrations from oxidation of SO2 by H2O2 (panels a, b, Eqs. 15 and 13) and O3 (panels c, d, Eq. 16), for malonic acid (panels a, c) and decanoic acid (panels b, d), with varying pKa corresponding to representations of bulk (p) and surface-modulated (p and 2) organic acid dissociation, in the aqueous aerosol population with sizes between Dwet=0.317–40 µm, calculated in the sulfur chemistry module of HAMBOX with five different initial organic mass fractions , denoted by blue, purple, pink, orange, and yellow, respectively (Table 2), after 1 h of simulation time. Simulations without accounting for organic acid dissociation are represented by no diss (black curves).
3.2 Aqueous aerosol sulfate concentration
Figure 2 shows the aqueous-phase secondary sulfate concentrations from oxidation of SO2 by H2O2 and O3 (Sect. 2.3) in the aqueous aerosol population with sizes between Dwet=0.317–40 µm, after 1 h of simulation time, for varying acid pKa corresponding to representations of bulk (p) and surface-modulated (p and p) organic dissociation, assuming five different initial organic mass fractions , denoted by blue, purple, pink, orange, and yellow, respectively, and considering the OA fraction as malonic acid (panels a, c) or decanoic acid (panels b, d). The secondary sulfate concentration obtained from simulations without consideration of organic acid dissociation (no diss, black line) is shown for reference. The relative changes in the sulfate concentration compared to no diss () for both oxidation pathways are given in Fig. S3 in the Supplement.
The sulfate concentration from H2O2 oxidation (panels a, b) increases drastically for both organic acids when organic acid dissociation is accounted for. From Eq. (15), it may seem that sulfate concentration should decrease with increasing [H+]tot, but the reverse is observed in Fig. 2. This is a property of the general acid-catalyzed mechanism, where the pKa-dependent rate constant kHA offsets the decrease in caused by increased [H+]tot concentration. These results are in line with Liu et al. (2020), who suggested the general acid-catalyzed H2O2 oxidation to explain “missing” sulfate during severe haze episodes. The oxidation of SO2 by O3 (panels c, d) follows a straightforward dependence on [H+]tot (Eq. 16), where increased hydrogen ion concentration results in decreased , compared to the no diss condition.
For malonic acid, H2O2 oxidation shows an increase in aqueous-phase secondary sulfate concentration compared to no diss (Fig. 2a), with ranging from 6434 %–14876 % at p with increasing χOA (Supplement Fig. S3a). With surface-modulated suppressed organic acid dissociation, decreases compared to p. The lowest is predicted for p at χOA=0.2. But even here, %, which is a strong increase compared to no diss. Similar trends are found for the H2O2 oxidation with decanoic acid (Fig. 2b), where the highest is obtained for p, with ranging from 3557 %–8367 % with increasing χOA (Supplement Fig. S3b). The lowest predicted for decanoic acid as the OA is seen for the stronger surface-modulated suppression of organic acid dissociation at p and χOA=0.2, as expected, with %. The aqueous-phase secondary sulfate concentration from O3 oxidation of SO2 (Fig. 2c) decreases by 70 %–80 % compared to no diss for malonic acid as the OA at p, with increasing χOA (Supplement Fig. S3c). The decrease is smaller for surface-modulated suppressed acid dissociation, as expected, with %–55 % for the more strongly suppressed dissociation at p. For decanoic acid as the OA (Fig. 2d), a similar trend is seen for from O3 oxidation, where the decrease in aqueous-phase secondary sulfate concentration is in the range of %–75 % for bulk and surface-modulated suppressed dissociation (Supplement Fig. S3d). Therefore, H2O2 oxidation of SO2 results in a far greater increase in in the aerosol population than the decrease in from the O3 oxidation of SO2, compared to “no diss”.
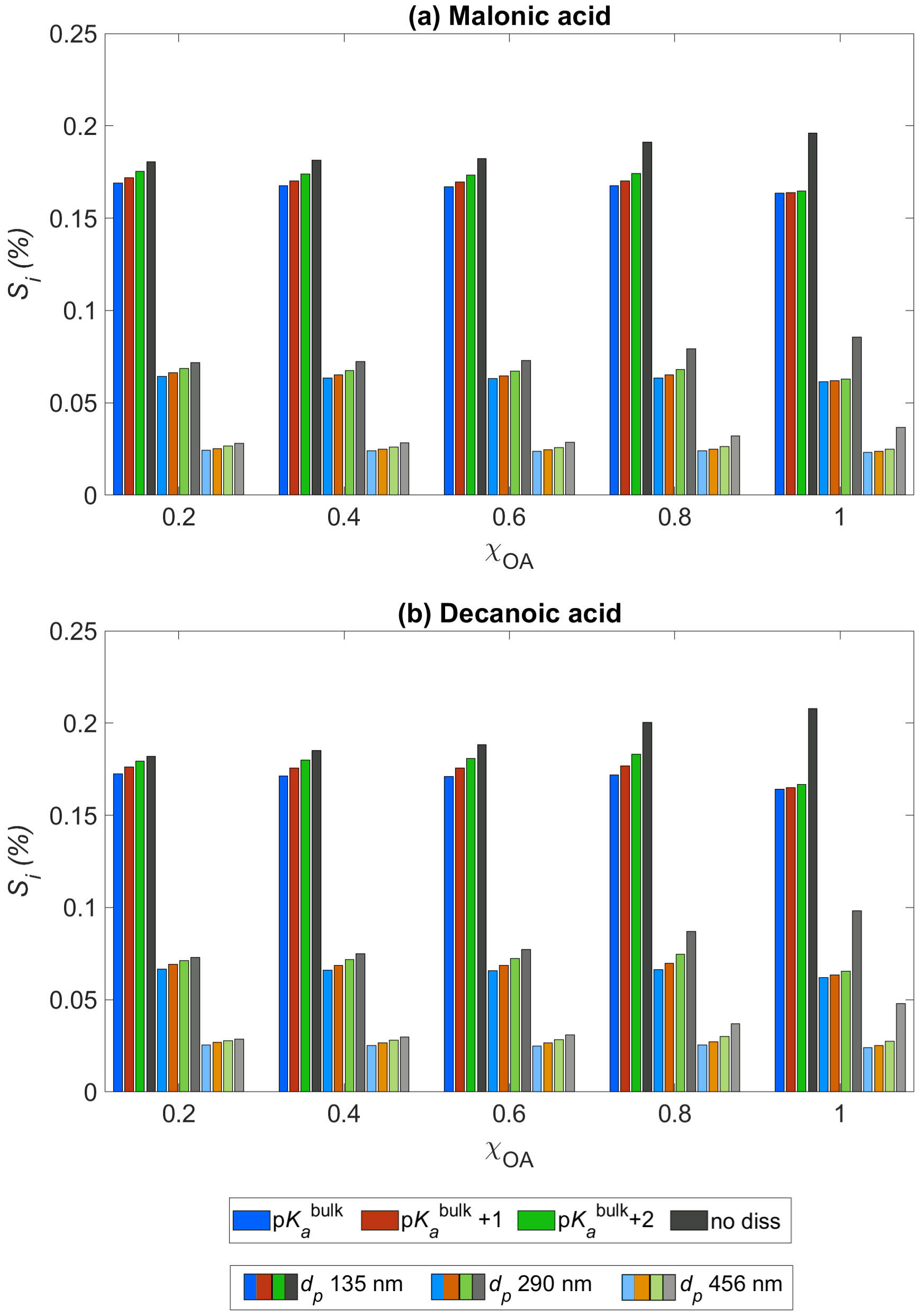
Figure 3Critical supersaturation Si as a function of the initial organic aerosol mass fraction χOA for (a) malonic acid and (b) decanoic acid, for three initial dry particle sizes, dp = 135 nm (darkest shade), 290 nm (lighter shade), and 456 nm (lightest shade), and pKa representing bulk (p, in blue) and surface (p, in orange, and p, in green) organic acid dissociation. Simulations without accounting for organic acid dissociation are represented by no diss (black and grey bars).
These results show how the increase in [H+]tot from organic acid dissociation in terms of [H+]HA results in significant increases in predicted in the aqueous aerosol, compared to when organic acid dissociation is not accounted for. As expected, the effect is smaller when organic dissociation is suppressed according to surface-modulated pKa, but even for the stronger suppression considered here, the effect is 1432 %–4000 % and 598 %–2500% with increasing χOA, for malonic and decanoic acid organic aerosol, respectively. We see that the effect of acid dissociation is larger for H2O2 oxidation, suggesting that this pathway is more sensitive to inclusion of organic aerosol acidity and dissociation effects.
3.3 Activation of droplets
Figure 3 shows the critical supersaturation Si (Eq. 3) predicted with HAMBOX for initial organic mass fractions , obtained for three dry particle sizes, dp = 135 nm (darkest shade), 290 nm (lighter shade), and 456 nm (lightest shade), with pKa represented by bulk (p) and surface-modulated (p and p) organic acid dissociation, considering the OA fraction as malonic acid (panel a) or decanoic acid (panel b). Results from simulations with the no diss condition (black bars) are also shown in both panels for each dry particle size. The dry particle sizes chosen here fall in the accumulation sub-range of size bins. We choose these dry particle sizes to investigate the critical supersaturation as particles in the accumulation size range have been shown to be the most effective in CDNC production (Patel and Jiang, 2021).
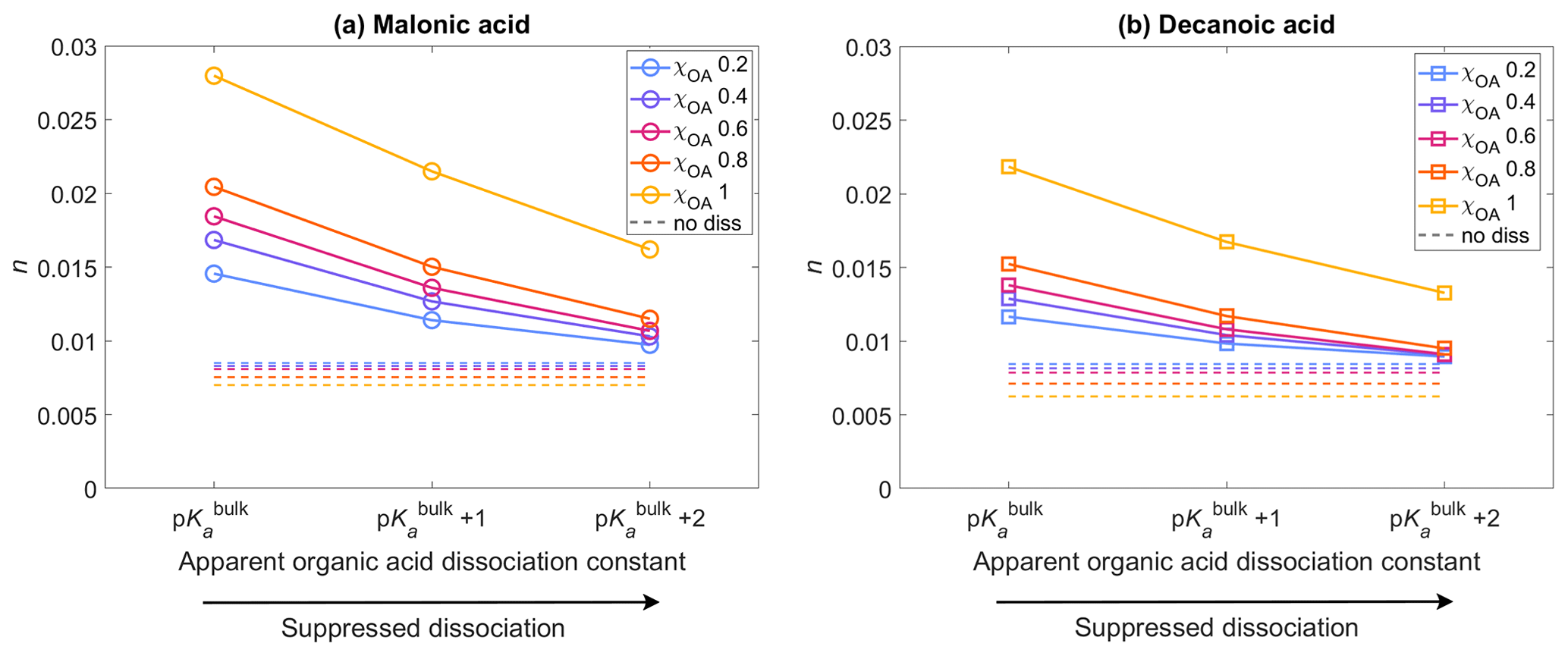
Figure 4The average activated fraction, n, of the aerosol population with droplet sizes between Dwet=0.317–40 µm, after 1 h of simulation time, for five different initial organic mass fractions , denoted by blue, purple, pink, orange, and yellow, respectively (see also Table 2), calculated for varying pKa corresponding to representations of bulk (p) and surface-modulated (p and 2) organic acid dissociation, and considering the entire OA fraction as (a) malonic acid and (b) decanoic acid. The average activated fraction in the no diss condition is shown as dashed lines in corresponding colors for each χOA.
The redistribution of aerosol sizes caused by the increased sulfate concentrations affects the calculation of the droplet size Dwet (Eq. 3). Therefore, Si is affected by bulk (p) and surface-modulated (p and p) acid dissociation of the OA. We see that for both organic acids considered, the increased secondary sulfate concentrations in the aqueous aerosols, compared to no diss, are sufficient to significantly decrease Si for all χOA at each of the three dry particle sizes considered. As expected, the decrease in Si is smaller when surface-modulated suppressed acid dissociation is considered, compared to simulations considering bulk acidity of OA. For decanoic acid as the OA (panel b), the difference between Si calculated for bulk and surface-modulated pKa is larger than for malonic acid as the OA (panel a), especially for the larger dry particle sizes (lightest shade) and higher organic mass fractions, χOA=0.6–1. This suggests that Si from consideration of decanoic acid dissociation is more susceptible to changes in the surface-modulated pKa than for malonic acid, especially at higher dp and χOA. While decanoic acid is the more surface-active of the two organic acids considered, because our simple empirical representation does not explicitly account for surface adsorption, this effect is caused here by the differences in bulk and apparent surface-modulated pKa with respect to the aerosol pH. As the organic mass fraction increases, the difference in Si between all pKa conditions and no diss increases for both organic acids.
The calculated Si in each size bin also includes any changes in aerosol water activity due to increased organic van't Hoff factor from organic acid dissociation (Eq. 34, Sect. 2.4.4). We see that for both organic acids, the water activity is sufficiently reduced, even for the surface-modulated suppressed acid dissociation, to significantly decrease Si, compared to no diss. It is well established that aerosol critical supersaturation typically increases with increasing OA mass fraction, due to the higher hygroscopicity of organic aerosol components compared to other soluble species, such as inorganic salts (Svenningsson et al., 2006). However, Fig. 3 shows that organic acid dissociation can partially counter this increase in Si. This effect is smaller if surface-modulated suppression of organic acid dissociation is considered, which is expected to be more relevant for smaller particles and droplets, due to their high ratio of surface area to bulk volume, and for aerosol populations with higher fractions of surface-active OA (Prisle, 2021).
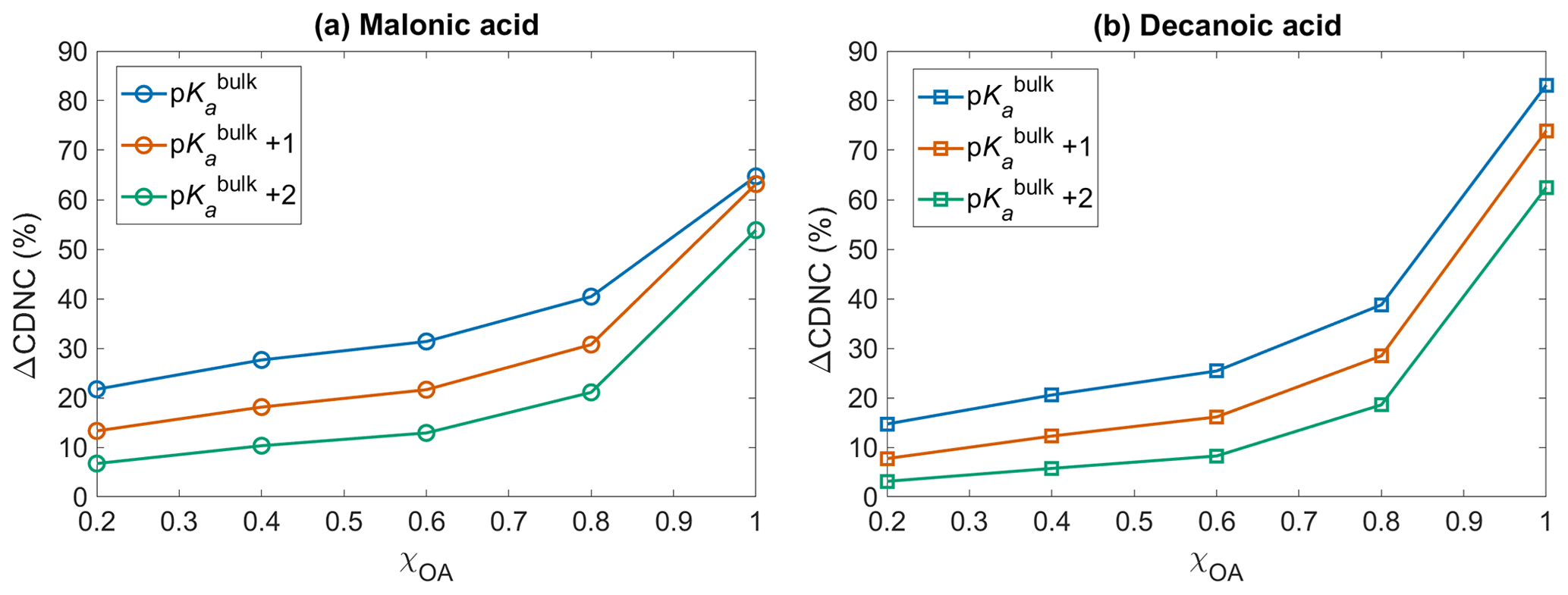
Figure 5The change in cloud droplet number concentration ΔCDNC (Eq. 10) with respect to no diss, as a function of initial organic mass fractions χOA, considering the OA fraction as (a) malonic acid OA and (b) decanoic acid OA, assuming organic acid dissociation according to bulk (p, blue) and surface-modulated properties (p, orange, and p, green).
Figure 4 shows the activated fraction n (Eq. 8) of the aerosol population, with droplet sizes between Dwet=0.317–40 µm, averaged for all aerosol size bins, calculated for initial organic mass fractions , denoted by blue, purple, pink, orange, and yellow, respectively (Table 2), with pKa representing bulk (p) and surface (p and p) organic acid dissociation, considering the OA fraction as malonic acid (panel a) or decanoic acid (panel b). The average activated fractions for simulations not considering organic acid dissociation (no diss) are shown as dashed lines in corresponding colors for each χOA, approximately 0.006–0.008 for both organic acids. The inclusion of organic acid dissociation effects results in higher activated fractions than no diss for both organic acids, with malonic acid dissociation resulting in a greater n than decanoic acid, for the same χOA and pKa. This is expected, as malonic acid is a stronger acid with lower p and [H+]tot from malonic acid dissociation under the same conditions is higher than for decanoic acid. The maximum activated fraction is observed for p with n=0.014–0.027 for malonic acid as the OA and n=0.012–0.021 for decanoic acid as the OA. For surface-modulated suppressed organic acid dissociation according to p, the activated fraction decreases to 0.011–0.021 and 0.010–0.016 for malonic and decanoic acid OA, respectively. As expected, the stronger surface-modulated acid dissociation suppression according to p further decreases the activated fraction, but n is still higher than for no diss (0.009–0.016 and 0.009–0.013 for malonic and decanoic acid OA, respectively). For both organic acids, the activated fraction also increases with increasing χOA, which is expected as the [H+]tot increases with increasing χOA (Fig. 1). For both organic acids, [H+]tot is sufficiently high to lead to a decrease in Si that translates into an increased activated fraction for both bulk and surface-modulated suppressed organic acid dissociation conditions, with a smaller effect for the suppressed acid dissociation, as expected.
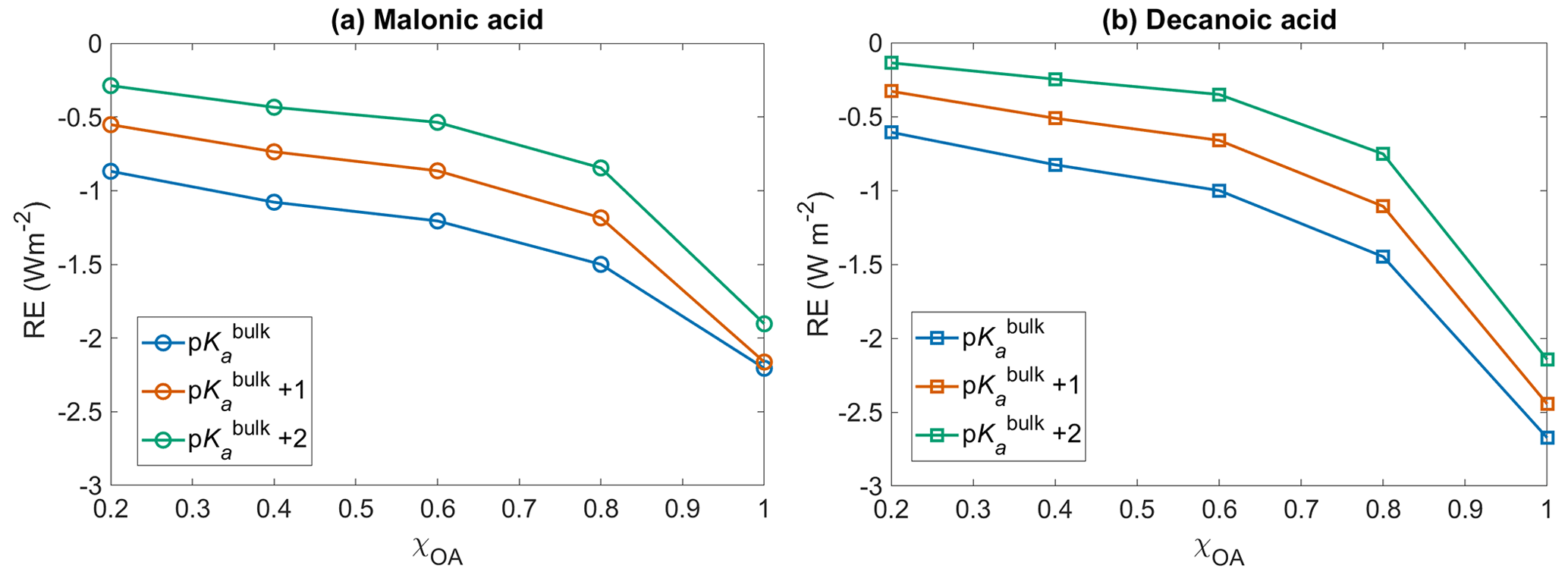
Figure 6Short wave radiative effect (RE, Eq. 12) with respect to no diss for (a) malonic acid and (b) decanoic acid OA, for varying initial organic mass fractions χOA, assuming organic dissociation according to bulk acidity (p, blue) and surface-modulated organic acid dissociation (p, orange, and p, green).
3.4 Cloud droplet number concentration
Figure 5 shows the change in cloud droplet number concentration ΔCDNC (Eq. 10) with respect to no diss, as a function of the initial organic mass fraction χOA, considering the entire OA fraction as malonic acid (panel a) and decanoic acid (panel b), for varying pKa corresponding to representations of bulk (p, in blue), and surface-modulated (p, in orange, and p, in green) organic acid dissociation. A significant enhancement in CDNC is seen for both malonic and decanoic acid, compared to when no organic acid dissociation is considered (no diss). Similar trends are seen for both acids, where p shows the highest CDNC enhancement compared to no diss. This is expected based on the calculated [H+]tot (Fig. 1) and (Fig. 2) from the bulk organic acidity and surface-modulated suppressed organic acid dissociation and consequent critical supersaturation (Si, Fig. 3) from both organic acids. For p and decanoic acid as the OA, ΔCDNC ranges from 14.7 % to 83.1 % with increasing χOA. For malonic acid as the OA, the ΔCDNC is smaller, ranging from 21.7 % to 64.7 % for corresponding χOA considering bulk acidity p. Under surface-modulated suppressed organic acid dissociation p, the CDNC enhancement is less than that obtained from p, ranging from 7.7 % to 73.9 % for decanoic acid and 13.3 % to 63.1 % for malonic acid. For the stronger surface-modulated acid dissociation suppression p, ΔCDNC is 3.1 % to 62.5 % for decanoic acid as the OA and 6.7 % to 53.9 % for malonic acid as the OA, with increasing χOA.
The CDNC enhancement upon including OA acid dissociation is caused by the change in aerosol size distribution due to the increased sulfate concentrations, which shifts the distribution towards larger particles, which are more effective in CDNC production (Hudson and Da, 1996; McFiggans et al., 2006). Since size plays a significant role in cloud droplet nucleating ability of aerosol particles (Dusek et al., 2006), the effect of organic acid dissociation on cloud response will be different depending on whether bulk or surface-modulated properties are used to describe the organic aerosol, and from Fig. 5 we see that this difference is significant for both malonic and decanoic acid OA under the simulation conditions. The aerosol size distribution after 1 h of simulation time, with and without including the sulfur chemistry module, is given in the Supplement (Figs. S4 and S5) for both organic acids, assuming initial organic mass fraction χOA=0.8. The aerosol size distribution is shown for varying pKa values, corresponding to representations of bulk (p) and surface-modulated (p and 2) organic acid dissociation, together with the no acid dissociation condition. Without organic acid dissociation, the size distribution is almost the same after 1 h for simulations with and without activating the sulfur chemistry module, for both organic acids. For bulk acidity p, the aerosol size distribution at 1 h is significantly different from the no diss size distribution. The change is smaller for the suppressed organic acid dissociation conditions p and p. However, even for the stronger suppressed dissociation the change in aerosol size distribution is sufficient to yield ΔCDNC by 6.7 %–53.8 % and 3.1 %–62.4 % for malonic acid and decanoic acid as the OA, respectively, compared to no diss.
3.5 Shortwave radiative effect
Figure 6 shows the shortwave radiative effect RE (Eq. 12) with respect to no diss, as a function of initial organic mass fractions χOA=0.2–1, considering the entire OA fraction as malonic acid (panel a) or decanoic acid (panel b), for varying pKa corresponding to representations of bulk (p, in blue), and surface-modulated (p, in orange, and p, in green) organic acid dissociation. The inclusion of organic acid dissociation leads to a cooling effect for both organic acids, compared to no diss. Considering organic acid dissociation with bulk properties (p), a larger cooling effect is observed for both acids, ranging from −0.6 to −2.7 W m−2 for decanoic acid and −0.86 to −2.2 W m−2 for malonic acid, as χOA increases from 0.2 to 1. The effect is smaller when surface-modulated suppression of acid dissociation is considered but still significant compared to no diss. For malonic acid as the OA, the range of RE extends from −0.5 to −2.2 W m−2 for p and from −0.3 to −1.6 W m−2 for the more strongly suppressed organic acid dissociation p. For decanoic acid as the OA, the range of RE varies from −0.3 to −2.4 W m−2 for p and from −0.1 to −2.1 W m−2 for p.
The effects of OA acid dissociation on cloud droplet number concentrations and radiative effect without considering the changes in the aqueous aerosol sulfur chemistry are shown in Fig. S6 of the Supplement. Here, the effects of organic acid dissociation arise from modification of the van't Hoff factor iOA (Eq. 34), reflected as changes in the Raoult term B (Eq. 4) and consequently Si (Eq. 3). The ΔCDNC and RE in Fig. S6 are therefore independent of the increased [H+]-driven sulfate concentrations in the aqueous aerosol and instead reflect the change in water activity due to the dissociation of the organic acid and consequent increase in the number of available moles of solute ns (Eq. 33). Considering OA acid dissociation effects in the water activity exclusively, malonic acid OA shows <0.5 % ΔCDNC with respect to no diss for all representations of organic acid dissociation. For decanoic acid OA, ΔCDNC with respect to no diss is slightly higher (≈1%), especially for bulk acidity p and higher χOA. The resulting RE with respect to no diss is within a range between 0 and −0.01 W m−2 for malonic acid as the OA and 0 to −0.05 W m−2 for decanoic acid as the OA.
3.6 Discussion
Our results show that acid dissociation of organic aerosols, exemplified with malonic and decanoic acid as common atmospheric moderately strong acids, can influence aqueous-phase sulfur chemistry to significantly impact the cloud shortwave radiative effect. Surface modulated suppressed acid dissociation of OA can further change the concentration of hydrogen ions in aqueous aerosol from what is immediately expected from the bulk acidity and aerosol pH. Under these surface-modulated conditions, the effects of organic acid dissociation on cloud properties are reduced but still significant. Since many components in atmospheric OA are both acidic and surface-active, this may be important to represent in large-scale models. Furthermore, the impact of OA acid dissociation on cloud-activating properties via aqueous-phase aerosol sulfur chemistry is significantly stronger than by changing the aerosol water activity and will be strongly underestimated if only effects on water activity are considered.
The increased hydrogen ion concentration in aqueous aerosols as a result of OA acid dissociation leads to enhanced sulfate mass from oxidation of SO2 by H2O2. Increased sulfate concentrations could potentially lead to enhanced formation of organosulfur compounds within the aerosols. Organosulfur compounds form in the atmosphere through heterogeneous reactions between volatile organic compounds and inorganic aerosol sulfate and can comprise over 15 % of the secondary organic aerosol mass (Hettiyadura et al., 2019; Chen et al., 2021a; Bruggemann et al., 2020; Riva et al., 2019). These organosulfur compounds could further increase OA mass and affect the resulting cloud droplet number concentrations. Organosulfates have been shown to exhibit acidic properties and primarily exist in the deprotonated form under atmospheric pH conditions (Fankhauser et al., 2022). Organosulfates are also known to be surface-active in aqueous solutions (Hansen et al., 2015; Prisle et al., 2010b, 2011; Lin et al., 2018; Malila and Prisle, 2018). Therefore, the mechanisms of both organic acid dissociation and its surface modulation studied here for atmospheric carboxylic acids could also apply to organosulfate aerosols, potentially affecting cloud droplet activation in a similar manner.
Bulk–surface partitioning in aqueous aerosols can be seen as a form of (potentially second order) liquid–liquid phase separation (Prisle, 2024). Phase separation of organic aerosols and its impact on cloud-activating properties of aerosol particles have been widely studied (Reid et al., 2018; Freedman, 2017; You et al., 2014). The partitioning of surface-active aerosol components occurs between the bulk and surface phases due to differences in composition and affinity for each phase. The suppression of organic acid dissociation considered here is exactly a consequence of the increased concentration of surface-active organic acid in the surface phase. Liquid–liquid phase separation in the bulk phase would effectively create two separate solutions with different compositions and ensuing properties. The modulation of organic acid dissociation could be taken into account separately for these phases, based on their individual concentrations, following analogous schemes as described by Prisle et al. (2010a).
Our results suggest that organic acid dissociation should be considered for accurate predictions of OA chemistry and cloud microphysics in the atmosphere. The specific magnitude of predictions with the present box model implementation may not be immediately representative of analogous simulations with full 3D aerosol–chemistry–climate models, due to their greater complexity and numerous coupled processes. For example, the effect of potential surface-modulated suppressed dissociation will further depend on aerosol surface activity and is expected to be especially relevant for smaller droplet sizes. However, as we have used a box model version of ECHAM–HAMMOZ, implementation of the OA acid dissociation mechanisms considered here will follow analogous strategies for the full model. The box model simulations contribute insights into the detailed mechanisms of OA acid dissociation and its impact on aerosol chemistry and cloud formation, and our present results provide a first assessment of the potential significance for resulting aerosol–cloud–climate parameters under conditions similar to those examined here.
We investigated the effects of organic aerosol acid dissociation on total hydrogen ion concentration in aqueous aerosols and the impact on resulting secondary sulfate aerosol mass, cloud droplet number concentration, and aerosol shortwave radiative effect, using the aerosol–chemistry–climate box model ECHAM6.3–HAM2.3 (HAMBOX). Simulations were carried out considering the entire OA to comprise organic acid and we used malonic and decanoic acid as proxies for atmospheric OA with different aqueous acidity and surface activity. Dissociation of organic acids was considered in three scenarios: (1) the current standard of no dissociation, (2) following well-known bulk solution acidity given by the reported acid constant p, and (3) accounting for a surface-modulated suppression of dissociation as observed in recent laboratory experiments.
Our results show that organic acid dissociation increases hydrogen ion concentrations in the aqueous aerosol phase, as expected. This leads to strongly increased secondary sulfate aerosol mass, which in turn decreases the critical supersaturation for cloud droplet activation and yields a higher activated aerosol fraction than if OA acid dissociation is not considered. The cloud response is observed as enhanced cloud droplet number concentration and a strong shortwave radiative effect of clouds. The effects of organic acid dissociation are greatest when considering the bulk acidity of OA but still significant even when potential surface-modulated suppression of dissociation is also included.
As many atmospheric organic aerosol components are acidic (Pye et al., 2020), their dissociation can have significant impacts on cloud properties. This work highlights the importance of including such organic acid dissociation effects in large scale atmospheric models. We suspect that, combined with the high surface area to bulk volume ratio and bulk–surface partitioning in small droplets (Bzdek et al., 2020; Prisle, 2021), the effects of organic dissociation and potential size-dependent surface-modulated acid dissociation could be significant in explaining some knowledge gaps about organic aerosol formation and acidity in atmospheric aerosols. OA acid dissociation could be particularly relevant in explaining discrepancies of atmospheric models with observations for polluted environments (Lee et al., 2013), where organic mass fraction is usually high, and the organic acid dissociation effects could become very significant. Many of these organic aerosol acids may also be surface-active in aqueous solutions (Gérard et al., 2019b), such as haze and activating cloud droplets, and therefore corrections to account for surface-modulated suppression of organic acid dissociation may also be necessary.
The HAMBOX code is available from the HAMMOZ Redmine at https://svn.iac.ethz.ch/external/echam-hammoz/echam6-hammoz/branches/fmi/HAM_box_OpenIFS/ (HAMMOZ, 2021). All simulation data and scripts underlying the figures are available from https://doi.org/10.5281/zenodo.8403178 (Sengupta et al., 2023).
GS did the model implementations and performed the calculations with contributions from MZ. GS and NLP analyzed the results and wrote the original and revised manuscripts and response to reviewers. NLP conceived, planned, supervised, and secured funding for the project. All authors approved the final text.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors warmly thank Kunal Ghosh and Harri Kokkola for valuable support with HAMBOX.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation program, project SURFACE (grant agreement no. 717022). The authors also gratefully acknowledge the financial contribution from the Academy of Finland, including grant nos. 308238, 314175, and 335649.
This paper was edited by Jason Surratt and reviewed by two anonymous referees.
Abdul-Razzak, H.: A parameterization of aerosol activation 3. Sectional representation, J. Geophys. Res., 107, 4026, https://doi.org/10.1029/2001jd000483, 2002. a
Abdul-Razzak, H. and Ghan, S. J.: A parameterization of aerosol activation 3. Sectional representation, J. Geophys. Res.-Atmos., 107, AAC–1, 2002. a
Abdul-Razzak, H., Ghan, S. J., and Rivera-Carpio, C.: A parameterization of aerosol activation: 1. Single aerosol type, J. Geophys. Res.-Atmos., 103, 6123–6131, https://doi.org/10.1029/97jd03735, 1998. a
Äijälä, M., Daellenbach, K. R., Canonaco, F., Heikkinen, L., Junninen, H., Petäjä, T., Kulmala, M., Prévôt, A. S. H., and Ehn, M.: Constructing a data-driven receptor model for organic and inorganic aerosol – a synthesis analysis of eight mass spectrometric data sets from a boreal forest site, Atmos. Chem. Phys., 19, 3645–3672, https://doi.org/10.5194/acp-19-3645-2019, 2019. a
Andreae, M. O. and Crutzen, P. J.: Atmospheric aerosols: Biogeochemical sources and role in atmospheric chemistry, Science, 276, 1052–1058, 1997. a
Angelis, M. D., Traversi, R., and Udisti, R.: Long-term trends of mono-carboxylic acids in Antarctica: Comparison of changes in sources and transport processes at the two EPICA deep drilling sites, Tellus B, 64, 17331, https://doi.org/10.3402/TELLUSB.V64I0.17331, 2012. a
Angle, K. J., Crocker, D. R., Simpson, R. M. C., Mayer, K. J., Garofalo, L. A., Moore, A. N., Mora Garcia, S. L., Or, V. W., Srinivasan, S., Farhan, M., Sauer, J. S., Lee, C., Pothier, M. A., Farmer, D. K., Martz, T. R., Bertram, T. H., Cappa, C. D., Prather, K. A., and Grassian, V. H.: Acidity across the interface from the ocean surface to sea spray aerosol, P. Natl. Acad. Sci. USA, 118, e2018397118, https://doi.org/10.1073/pnas.2018397118, 2021. a, b, c
Ault, A. P.: Aerosol Acidity: Novel measurements and implications for atmospheric chemistry, Accounts Chem. Res., 53, 1703–1714, 2020. a
Bruggemann, M., Xu, R., Tilgner, A., Kwong, K. C., Mutzel, A., Poon, H. Y., Otto, T., Schaefer, T., Poulain, L., Chan, M. N., and Herrmann, H.: Organosulfates in ambient aerosol: state of knowledge and future research directions on formation, abundance, fate, and importance, Environ. Sci. Technol., 54, 3767–3782, https://doi.org/10.1021/acs.est.9b06751, 2020. a
Bzdek, B. R., Reid, J. P., Malila, J., and Prisle, N. L.: The surface tension of surfactant-containing, finite volume droplets, P. Natl. Acad. Sci. USA, 7, 8335–8343, 2020. a, b, c
Chebbi, A. and Carlier, P.: Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review, Atmos. Environ., 30, 4233–4249, https://doi.org/10.1016/1352-2310(96)00102-1, 1996. a
Chen, Y., Dombek, T., Hand, J., Zhang, Z., Gold, A., Ault, A. P., Levine, K. E., and Surratt, J. D.: Seasonal Contribution of Isoprene-Derived Organosulfates to Total Water-Soluble Fine Particulate Organic Sulfur in the United States, ACS Earth Space Chem., 5, 2419–2432, 2021a. a
Chen, Y., Guo, H., Nah, T., Tanner, D. J., Sullivan, A. P., Takeuchi, M., Gao, Z., Vasilakos, P., Russell, A. G., Baumann, K., Huey, L. G., Weber, R. J., and Ng, N. L.: Low-Molecular-Weight Carboxylic Acids in the Southeastern U.S.: Formation, Partitioning, and Implications for Organic Aerosol Aging, Environ. Sci. Technol., 55, 6688–6699, http://10.1021/ACS.EST.1C01413/ASSET/, 2021b. a
{kind=link}
Cheng, Y., Li, S.-M., Leithead, A., Brickell, P. C., and Leaitch, W. R.: Characterizations of cis-pinonic acid and n-fatty acids on fine aerosols in the Lower Fraser Valley during Pacific 2001 Air Quality Study, Atmos. Environ., 38, 5789–5800, 2004. a
Cohn, E. J., Heyroth, F. F., and Menkin, M. F.: The Dissociation Constant of Acetic Acid and the Activity Coefficients of the Ions in Certain Acetate Solutions, J. Am. Chem. Soc., 50, 696–714, https://doi.org/10.1021/ja01390a012, 1928. a
Creux, P., Lachaise, J., Graciaa, A., and Beattie, J. K.: Specific cation effects at the hydroxide-charged air/water interface, The J. Phys. Chem. C, 111, 3753–3755, 2007. a
Drexler, C., Elias, H., Fecher, B., and Wannowius, K.: Kinetic investigation of sulfur (IV) oxidation by peroxo compounds R-OOH in aqueous solution, Fresen. J. Anal. Chem., 340, 605–615, 1991. a
Dusek, U., Frank, G., Hildebrandt, L., Curtius, J., Schneider, J., Walter, S., Chand, D., Drewnick, F., Hings, S., Jung, D., Borrmann, S., and Andreae, M. O.: Size matters more than chemistry for cloud-nucleating ability of aerosol particles, Science, 312, 1375–1378, 2006. a
Enami, S., Hoffmann, M. R., and Colussi, A. J.: Proton availability at the air/water interface, The J. Phys. Chem. Lett., 1, 1599–1604, 2010. a
Fankhauser, A. M., Lei, Z., Daley, K. R., Xiao, Y., Zhang, Z., Gold, A., Ault, B. S., Surratt, J. D., and Ault, A. P.: Acidity-Dependent Atmospheric Organosulfate Structures and Spectra: Exploration of Protonation State Effects via Raman and Infrared Spectroscopies Combined with Density Functional Theory, The J. Phys. Chem. A, 126, 5974–5984, 2022. a
Feichter, J., Kjellström, E., Rodhe, H., Dentener, F., Lelieveldi, J., and Roelofs, G.-J.: Simulation of the tropospheric sulfur cycle in a global climate model, Atmos. Environ., 30, 1693–1707, 1996a. a
Feichter, J., Kjellström, E., Rodhe, H., Dentener, F., Lelieveldi, J., and Roelofs, G.-J.: Simulation of the tropospheric sulfur cycle in a global climate model, Atmos. Environ., 30, 1693–1707, https://doi.org/10.1016/1352-2310(95)00394-0, 1996b. a
Freedman, M. A.: Phase separation in organic aerosol, Chem. Soc. Rev., 46, 7694–7705, 2017. a
Freedman, M. A., Ott, E.-J. E., and Marak, K. E.: Role of pH in aerosol processes and measurement challenges, The J. Phys. Chem. A, 123, 1275–1284, 2018. a
Frosch, M., Prisle, N. L., Bilde, M., Varga, Z., and Kiss, G.: Joint effect of organic acids and inorganic salts on cloud droplet activation, Atmos. Chem. Phys., 11, 3895–3911, https://doi.org/10.5194/acp-11-3895-2011, 2011. a
Gérard, V., Noziere, B., Baduel, C., Fine, L., Frossard, A. A., and Cohen, R. C.: Anionic, cationic, and nonionic surfactants in atmospheric aerosols from the baltic coast at askö, sweden: Implications for cloud droplet activation, Environ. Sci. Technol., 50, 2974–2982, 2016. a
Gérard, V., Noziere, B., Fine, L., Ferronato, C., Singh, D. K., Frossard, A. A., Cohen, R. C., Asmi, E., Lihavainen, H., Kivekäs, N., Aurela, M., Brus, D., Frka, S., and Cvitešić Kušan, A.: Concentrations and adsorption isotherms for amphiphilic surfactants in PM1 aerosols from different regions of Europe, Environ. Sci. Technol., 53, 12379–12388, 2019a. a
Gerard, V., Noziere, B., Fine, L., Ferronato, C., Singh, D. K., Frossard, A. A., Cohen, R. C., Asmi, E., Lihavainen, H., Kivekäs, N., Aurela, M., Brus, D., Frka, S., and Kušan, A. C.: Concentrations and adsorption isotherms for amphiphilic surfactants in PM1 aerosols from different regions of Europe, Environ. Sci. Technol., 53, 12379–12388, https://doi.org/10.1021/acs.est.9b03386, 2019b. a
Goldberg, R. N., Kishore, N., and Lennen, R. M.: Thermodynamic quantities for the ionization reactions of buffers, J. Phys. Chem. Refer. data, 31, 231–370, 2002. a
Gong, K., Ao, J., Li, K., Liu, L., Liu, Y., Xu, G., Wang, T., Cheng, H., Wang, Z., Zhang, X., et al.: Imaging of pH distribution inside individual microdroplet by stimulated Raman microscopy, P. Natl. Acad. Sci. USA, 120, e2219588120, https://doi.org/10.1073/pnas.2219588120, 2023. a
Graciaa, A., Morel, G., Saulner, P., Lachaise, J., and Schechter, R.: The ζ-potential of gas bubbles, J. Colloid Interf. Sci., 172, 131–136, 1995. a
Graham, B., Guyon, P., Taylor, P. E., Artaxo, P., Maenhaut, W., Glovsky, M. M., Flagan, R. C., and Andreae, M. O.: Organic compounds present in the natural Amazonian aerosol: Characterization by gas chromatography–mass spectrometry, J. Geophys. Res.-Atmos., 108, 4766, https://doi.org/10.1029/2003JD003990, 2003. a
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009. a
HAMMOZ consortium: HAMMOZ redmine repository, https://svn.iac.ethz.ch/external/echam-hammoz/echam6-hammoz/branches/fmi/HAM_box_OpenIFS/ (last access: 15 October 2021), 2021. a
Hansen, A. M. K., Hong, J., Raatikainen, T., Kristensen, K., Ylisirniö, A., Virtanen, A., Petäjä, T., Glasius, M., and Prisle, N. L.: Hygroscopic properties and cloud condensation nuclei activation of limonene-derived organosulfates and their mixtures with ammonium sulfate, Atmos. Chem. Phys., 15, 14071–14089, https://doi.org/10.5194/acp-15-14071-2015, 2015. a, b
Hettiyadura, A. P. S., Al-Naiema, I. M., Hughes, D. D., Fang, T., and Stone, E. A.: Organosulfates in Atlanta, Georgia: anthropogenic influences on biogenic secondary organic aerosol formation, Atmos. Chem. Phys., 19, 3191–3206, https://doi.org/10.5194/acp-19-3191-2019, 2019. a
Hudson, J. G. and Da, X.: Volatility and size of cloud condensation nuclei, J. Geophys. Res.-Atmos., 101, 4435–4442, 1996. a
Huebert, B., Bertram, T., Kline, J., Howell, S., Eatough, D., and Blomquist, B.: Measurements of organic and elemental carbon in Asian outflow during ACE-Asia from the NSF/NCAR C-130, J. Geophys. Res.-Atmos., 109, D19S11, https://doi.org/10.1029/2004JD004700, 2004. a
Hung, H.-M., Hsu, M.-N., and Hoffmann, M. R.: Quantification of SO2 oxidation on interfacial surfaces of acidic micro-droplets: Implication for ambient sulfate formation, Environ. Sci. Technol., 52, 9079–9086, 2018. a
IPCC: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 1535 pp., 2013. a, b
Jacob, D. J.: Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate, J. Geophys. Res.-Atmos., 91, 9807–9826, https://doi.org/10.1029/JD091ID09P09807, 1986. a
Jacobson, M. Z.: Fundamentals of Atmospheric Modeling, Cambridge University Press, Cambridge, 2 Edn., ISBN 9780521548656, https://doi.org/10.1017/CBO9781139165389, 2005. a
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005. a, b
Karraker, K. and Radke, C.: Disjoining pressures, zeta potentials and surface tensions of aqueous non-ionic surfactant/electrolyte solutions: theory and comparison to experiment, Adv. Coll. Interf. Sci., 96, 231–264, 2002. a
Kawamura, K., Ng, L. L., and Kaplan, I. R.: Determination of Organic Acids (C1-C10) in the Atmosphere, Motor Exhausts, and Engine Oils, Environ. Sci. Technol., 19, 1082–1086, http://doi.org/10.1021/ES00141A010/ASSET/, 1985. a
Keene, W. C. and Galloway, J. N.: Organic acidity in precipitation of North America, Atmos. Environ. (1967), 18, 2491–2497, https://doi.org/10.1016/0004-6981(84)90020-9, 1984. a
Kokkola, H., Kühn, T., Laakso, A., Bergman, T., Lehtinen, K. E. J., Mielonen, T., Arola, A., Stadtler, S., Korhonen, H., Ferrachat, S., Lohmann, U., Neubauer, D., Tegen, I., Siegenthaler-Le Drian, C., Schultz, M. G., Bey, I., Stier, P., Daskalakis, N., Heald, C. L., and Romakkaniemi, S.: SALSA2.0: The sectional aerosol module of the aerosol–chemistry–climate model ECHAM6.3.0-HAM2.3-MOZ1.0, Geosci. Model Dev., 11, 3833–3863, https://doi.org/10.5194/gmd-11-3833-2018, 2018. a, b
Kroflič, A., Frka, S., Simmel, M., Wex, H., and Grgić, I.: Size-Resolved Surface-Active Substances of Atmospheric Aerosol: Reconsideration of the Impact on Cloud Droplet Formation, Environ. Sci. Technol., 52, 9179–9187, https://doi.org/10.1021/acs.est.8b02381, 2018. a
Lee, L. A., Pringle, K. J., Reddington, C. L., Mann, G. W., Stier, P., Spracklen, D. V., Pierce, J. R., and Carslaw, K. S.: The magnitude and causes of uncertainty in global model simulations of cloud condensation nuclei, Atmos. Chem. Phys., 13, 8879–8914, https://doi.org/10.5194/acp-13-8879-2013, 2013. a
Legg, S.: IPCC, 2021: Climate change 2021-the physical science basis, Interaction, 49, 44–45, 2021. a
Li, Y.-C. and Yu, J. Z.: Simultaneous determination of mono-and dicarboxylic acids, ω-oxo-carboxylic acids, midchain ketocarboxylic acids, and aldehydes in atmospheric aerosol samples, Environ. Sci. Technol., 39, 7616–7624, 2005. a
Lin, J. J., Malila, J., and Prisle, N. L.: Cloud droplet activation of organic–salt mixtures predicted from two model treatments of the droplet surface, Environ. Sci.-Process. Impact., 20, 1611–1629, 2018. a, b
Lin, J. J., Kristensen, T. B., Calderón, S. M., Malila, J., and Prisle, N. L.: Effects of surface tension time-evolution for CCN activation of a complex organic surfactant, Environ. Sci.-Process. Impact., 22, 271–284, 2020. a, b
Liu, T., Clegg, S. L., and Abbatt, J. P.: Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles, P. Natl. Acad. Sci. USA, 117, 1354–1359, 2020. a, b, c, d, e, f
Liu, Y., Wu, Z., Huang, X., Shen, H., Bai, Y., Qiao, K., Meng, X., Hu, W., Tang, M., and He, L.: Aerosol phase state and its link to chemical composition and liquid water content in a subtropical coastal megacity, Environ. Sci. Technol., 53, 5027–5033, 2019. a
Lohmann, U. and Lesins, G.: Stronger constraints on the anthropogenic indirect aerosol effect, Science, 298, 1012–1015, 2002. a
Maaß, F., Elias, H., and Wannowius, K. J.: Kinetics of the oxidation of hydrogen sulfite by hydrogen peroxide in aqueous solution:: ionic strength effects and temperature dependence, Atmos. Environ., 33, 4413–4419, 1999. a
Malila, J. and Prisle, N.: A monolayer partitioning scheme for droplets of surfactant solutions, J. Adv. Model. Earth Syst., 10, 3233–3251, 2018. a, b
Margarella, A. M., Perrine, K. A., Lewis, T., Faubel, M., Winter, B., and Hemminger, J. C.: Dissociation of sulfuric acid in aqueous solution: determination of the photoelectron spectral fingerprints of H2SO4, HSO4–, and SO42–in water, The J. Phys. Chem. C, 117, 8131–8137, 2013. a
Martell, A. E. and Smith, R. M.: Critical stability constants, Vol. 1, Springer, New York: Plenum press, 1974. a, b
Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R., and Zhou, B.: Climate change 2021: the physical science basis, Contribution of working group I to the sixth assessment report of the intergovernmental panel on climate change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 33−144, https://doi.org/10.1017/9781009157896.002, 2021. a
McArdle, J. V. and Hoffmann, M. R.: Kinetics and mechanism of the oxidation of aquated sulfur dioxide by hydrogen peroxide at low pH, The J. Phys. Chem., 87, 5425–5429, 1983. a
McFiggans, G., Artaxo, P., Baltensperger, U., Coe, H., Facchini, M. C., Feingold, G., Fuzzi, S., Gysel, M., Laaksonen, A., Lohmann, U., Mentel, T. F., Murphy, D. M., O'Dowd, C. D., Snider, J. R., and Weingartner, E.: The effect of physical and chemical aerosol properties on warm cloud droplet activation, Atmos. Chem. Phys., 6, 2593–2649, https://doi.org/10.5194/acp-6-2593-2006, 2006. a, b
Michailoudi, G., Hyttinen, N., Kurten, T., and Prisle, N. L.: Solubility and Activity Coefficients of Atmospheric Surfactants in Aqueous Solution Evaluated Using COSMO therm, The J. Phys. Chem. A, 124, 430–443, 2019. a
Millet, D. B., Baasandorj, M., Farmer, D. K., Thornton, J. A., Baumann, K., Brophy, P., Chaliyakunnel, S., de Gouw, J. A., Graus, M., Hu, L., Koss, A., Lee, B. H., Lopez-Hilfiker, F. D., Neuman, J. A., Paulot, F., Peischl, J., Pollack, I. B., Ryerson, T. B., Warneke, C., Williams, B. J., and Xu, J.: A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 15, 6283–6304, https://doi.org/10.5194/acp-15-6283-2015, 2015. a
Mochida, M., Kawabata, A., Kawamura, K., Hatsushika, H., and Yamazaki, K.: Seasonal variation and origins of dicarboxylic acids in the marine atmosphere over the western North Pacific, J. Geophys. Res.-Atmos., 108, 4193, https://doi.org/10.1029/2002JD002355, 2003. a
Mochizuki, T., Kawamura, K., Aoki, K., and Sugimoto, N.: Long-range atmospheric transport of volatile monocarboxylic acids with Asian dust over a high mountain snow site, central Japan, Atmos. Chem. Phys., 16, 14621–14633, https://doi.org/10.5194/acp-16-14621-2016, 2016. a
Murphy, D., Cziczo, D., Froyd, K., Hudson, P., Matthew, B., Middlebrook, A., Peltier, R., Sullivan, A., Thomson, D., and Weber, R.: Single-particle mass spectrometry of tropospheric aerosol particles, J. Geophys. Res.-Atmos., 111, D23S32, https://doi.org/10.1029/2006JD007340, 2006. a
Narukawa, M., Kawamura, K., Li, S.-M., and Bottenheim, J.: Dicarboxylic acids in the Arctic aerosols and snowpacks collected during ALERT 2000, Atmosp. Environ., 36, 2491–2499, 2002. a
Noziere, B., Baduel, C., and Jaffrezo, J.-L.: The dynamic surface tension of atmospheric aerosol surfactants reveals new aspects of cloud activation, Nat. Commun., 5, 3335, https://doi.org/10.1038/ncomms4335, 2014. a
Nozière, B., Gérard, V., Baduel, C., and Ferronato, C.: Extraction and characterization of surfactants from atmospheric aerosols, JoVE-J. Vis. Exp., 122, e55622, https://doi.org/10.3791/55622, 2017. a
O'Dowd, C. D., Facchini, M. C., Cavalli, F., Ceburnis, D., Mircea, M., Decesari, S., Fuzzi, S., Yoon, Y. J., and Putaud, J.-P.: Biogenically driven organic contribution to marine aerosol, Nature, 431, 676–680, 2004. a, b
Öhrwall, G., Prisle, N. L., Ottosson, N., Werner, J., Ekholm, V., Walz, M.-M., and Björneholm, O.: Acid–base speciation of carboxylate ions in the surface region of aqueous solutions in the presence of ammonium and aminium ions, The J. Phys. Chem. B, 119, 4033–4040, 2015. a, b, c
Patel, P. N. and Jiang, J. H.: Cloud condensation nuclei characteristics at the Southern Great Plains site: role of particle size distribution and aerosol hygroscopicity, Environ. Res. Commun., 3, 075002, https://doi.org/10.1088/2515-7620/ac0e0b, 2021. a
Petersen, P. B. and Saykally, R. J.: Evidence for an enhanced hydronium concentration at the liquid water surface, The J. Phys. Chem. B, 109, 7976–7980, 2005. a
Petersen, P. B. and Saykally, R. J.: Is the liquid water surface basic or acidic? Macroscopic vs. molecular-scale investigations, Chem. Phys. Lett., 458, 255–261, 2008. a
Petters, S. S. and Petters, M. D.: Surfactant effect on cloud condensation nuclei for two-component internally mixed aerosols, J. Geophys. Res.-Atmos., 121, 1878–1895, 2016. a
Prisle, N.: Cloud Condensation Nuclei Properties of Organic Aerosol Particles: Effects of Acid Dissociation and Surfactant Partitioning, University of Copenhagen, 52, 183, https://doi.org/10.5281/zenodo.4650303, 2006. a
Prisle, N. L.: A predictive thermodynamic framework of cloud droplet activation for chemically unresolved aerosol mixtures, including surface tension, non-ideality, and bulk–surface partitioning, Atmos. Chem. Phys., 21, 16387–16411, https://doi.org/10.5194/acp-21-16387-2021, 2021. a, b, c, d, e, f, g
Prisle, N. L.: Surfaces of Atmospheric Droplet Models Probed with Synchrotron XPS on a Liquid Microjet, Accounts Chem. Res., 57, 177–187, https://doi.org/10.1021/acs.accounts.3c00201, 2024. a, b, c, d, e, f, g, h, i
Prisle, N. L., Raatikainen, T., Sorjamaa, R., Svenningsson, B., Laaksonen, A., and Bilde, M.: Surfactant partitioning in cloud droplet activation: a study of C8, C10, C12 and C14 normal fatty acid sodium salts, Tellus B, 60, 416–431, 2008. a, b, c
Prisle, N. L., Engelhart, G., Bilde, M., and Donahue, N.: Humidity influence on gas-particle phase partitioning of α-pinene+ O3 secondary organic aerosol, Geophys. Res. Lett., 37, L01802, https://doi.org/10.1029/2009GL041402, 2010a. a, b
Prisle, N. L., Raatikainen, T., Laaksonen, A., and Bilde, M.: Surfactants in cloud droplet activation: mixed organic-inorganic particles, Atmos. Chem. Phys., 10, 5663–5683, https://doi.org/10.5194/acp-10-5663-2010, 2010b. a, b, c
Prisle, N. L., Dal Maso, M., and Kokkola, H.: A simple representation of surface active organic aerosol in cloud droplet formation, Atmos. Chem. Phys., 11, 4073–4083, https://doi.org/10.5194/acp-11-4073-2011, 2011. a, b
Prisle, N. L., Asmi, A., Topping, D., Partanen, A. I., Romakkaniemi, S., Dal Maso, M., Kulmala, M., Laaksonen, A., Lehtinen, K. E. J., McFiggans, G., and Kokkola, H.: Surfactant effects in global simulations of cloud droplet activation, Geophys. Res. Lett., 39, L05802, https://doi.org/10.1029/2003JD004485, 2012a. a
Prisle, N. L., Ottosson, N., Öhrwall, G., Söderström, J., Dal Maso, M., and Björneholm, O.: Surface/bulk partitioning and acid/base speciation of aqueous decanoate: direct observations and atmospheric implications, Atmos. Chem. Phys., 12, 12227–12242, https://doi.org/10.5194/acp-12-12227-2012, 2012b. a, b, c, d
Putaud, J.-P., Raes, F., Van Dingenen, R., Brüggemann, E., Facchini, M.-C., Decesari, S., Fuzzi, S., Gehrig, R., Hüglin, C., Laj, P., Lorbeer, G., Maenhout, W., Mihalopoulos, N., Mueller, K., Querol, X., Rodriguez, S., Schneider, J., Spindler, G., Ten Brink, H., Toerseth, K., and Wiedensohler, A.: A European aerosol phenomenology—2: chemical characteristics of particulate matter at kerbside, urban, rural and background sites in Europe, Atmos. Environ., 38, 2579–2595, 2004. a
Putaud, J.-P., Van Dingenen, R., Alastuey, A., Bauer, H., Birmili, W., Cyrys, J., Flentje, H., Fuzzi, S., Gehrig, R., Hansson, H.-C., Harrison, R. M., Herrmann, H., Hitzenberger, R., Hüglin, C., Jones, A. M., Kasper-Giebl, A., Kiss, G., Kousa, A., Kuhlbusch, T. A. J., Löschau, G., Maenhaut, W., Molnar, A., Moreno, T., Pekkanen, J., Perrino, C., Pitz, M., Puxbaum, H., Querol, X., Rodriguez, S., Salma, I., Schwarz, J., Smolik, J., Schneider, J., Spindler, G., Ten Brink, H., Tursic, J., Viana, M., Wiedensohler, A., and Raes, F.: A European aerosol phenomenology–3: Physical and chemical characteristics of particulate matter from 60 rural, urban, and kerbside sites across Europe, Atmos. Environ., 44, 1308–1320, 2010. a
Pye, H. O. T., Nenes, A., Alexander, B., Ault, A. P., Barth, M. C., Clegg, S. L., Collett Jr., J. L., Fahey, K. M., Hennigan, C. J., Herrmann, H., Kanakidou, M., Kelly, J. T., Ku, I.-T., McNeill, V. F., Riemer, N., Schaefer, T., Shi, G., Tilgner, A., Walker, J. T., Wang, T., Weber, R., Xing, J., Zaveri, R. A., and Zuend, A.: The acidity of atmospheric particles and clouds, Atmos. Chem. Phys., 20, 4809–4888, https://doi.org/10.5194/acp-20-4809-2020, 2020. a, b, c, d
Reid, J. P., Bertram, A. K., Topping, D. O., Laskin, A., Martin, S. T., Petters, M. D., Pope, F. D., and Rovelli, G.: The viscosity of atmospherically relevant organic particles, Nat. Commun., 9, 956, https://doi.org/10.1038/s41467-018-03027-z, 2018. a
Riva, M., Chen, Y., Zhang, Y., Lei, Z., Olson, N. E., Boyer, H. C., Narayan, S., Yee, L. D., Green, H. S., Cui, T., Zhang, Z., Baumann, K., Fort, M., Edgerton, E., Budisulistiorini, S. H., Rose, C. A., Ribeiro, I. O., e Oliveira, R. L., dos Santos, E. O., Machado, C. M. D., Szopa, S., Zhao, Y., Alves, E. G., de Sá, S. S., Hu, W., Knipping, E. M., Shaw, S. L., Duvoisin Junior, S., de Souza, R. A. F., Palm, B. B., Jimenez, J.-L., Glasius, M., Goldstein, A. H., Pye, H. O. T., Gold, A., Turpin, B. J., Vizuete, W., Martin, S. T., Thornton, J. A., Dutcher, C. S., Ault, A. P., and Surratt, J. D.: Increasing isoprene epoxydiol-to-inorganic sulfate aerosol ratio results in extensive conversion of inorganic sulfate to organosulfur forms: implications for aerosol physicochemical properties, Environ. Sci. Technol., 53, 8682–8694, https://doi.org/10.1021/acs.est.9b01019, 2019. a
Roberts, G. C., Andreae, M. O., Zhou, J., and Artaxo, P.: Cloud condensation nuclei in the Amazon Basin: “Marine” conditions over a continent?, Geophys. Res. Lett., 28, 2807–2810, 2001. a
Saxena, P. and Hildemann, L. M.: Water-soluble organics in atmospheric particles: A critical review of the literature and application of thermodynamics to identify candidate compounds, J. Atmos. Chem., 24, 57–109, 1996. a
Saykally, R. J.: Two sides of the acid–base story, Nat. Chem., 5, 82–84, 2013. a
Sengupta, G., Zheng, M., and Prisle, N. L.: Simulation dataset and plotting scripts used for journal article “Impact of acidity and surface modulated acid dissociation on cloud response to organic aerosol” by Sengupta et al. (2023) [data set], Zenodo, https://doi.org/10.5281/zenodo.8403178, 2023. a
Seinfeld, J. H., Bretherton, C., Carslaw, K. S., Coe, H., DeMott, P. J., Dunlea, E. J., Feingold, G., Ghan, S., Guenther, A. B., Kahn, R., Kraucunas, I., Kreidenweis, S. M., Molina, M. J., Nenes, A., Penner, J. E., Prather, K. A., Ramanathan, V., Ramaswamy, V., Rasch, P. J., Ravishankara, A. R., Rosenfeld, D., Stephens, G., and Wood, R.: Improving our fundamental understanding of the role of aerosol- cloud interactions in the climate system, P. Natl. Acad. Sci. USA, 113, 5781–5790, 2016. a
Stahl, P. H. and Wermuth, C. G.: Handbook of pharmaceutical salts: properties, selection and use, Chem. Int., 24, 21, https://doi.org/10.1021/jm030019n, 2002. a
Svenningsson, B., Rissler, J., Swietlicki, E., Mircea, M., Bilde, M., Facchini, M. C., Decesari, S., Fuzzi, S., Zhou, J., Mønster, J., and Rosenørn, T.: Hygroscopic growth and critical supersaturations for mixed aerosol particles of inorganic and organic compounds of atmospheric relevance, Atmos. Chem. Phys., 6, 1937–1952, https://doi.org/10.5194/acp-6-1937-2006, 2006. a
Takahashi, M.: ζ potential of microbubbles in aqueous solutions: electrical properties of the gas- water interface, The J. Phys. Chem. B, 109, 21858–21864, 2005. a
Tedetti, M., Kawamura, K., Charrière, B., Chevalier, N., and Sempéré, R.: Determination of low molecular weight dicarboxylic and ketocarboxylic acids in seawater samples, Anal. Chem., 78, 6012–6018, 2006. a
Tegen, I., Neubauer, D., Ferrachat, S., Siegenthaler-Le Drian, C., Bey, I., Schutgens, N., Stier, P., Watson-Parris, D., Stanelle, T., Schmidt, H., Rast, S., Kokkola, H., Schultz, M., Schroeder, S., Daskalakis, N., Barthel, S., Heinold, B., and Lohmann, U.: The global aerosol–climate model ECHAM6.3–HAM2.3 – Part 1: Aerosol evaluation, Geosci. Model Dev., 12, 1643–1677, https://doi.org/10.5194/gmd-12-1643-2019, 2019. a, b
Zheng, Y. and Ming, Y.: Low-Level Cloud Budgets across Sea Ice Edges, J. Climate, 36, 3–18, https://doi.org/10.1175/JCLI-D-22-0301.1, 2022.
Tilgner, A., Schaefer, T., Alexander, B., Barth, M., Collett Jr., J. L., Fahey, K. M., Nenes, A., Pye, H. O. T., Herrmann, H., and McNeill, V. F.: Acidity and the multiphase chemistry of atmospheric aqueous particles and clouds, Atmos. Chem. Phys., 21, 13483–13536, https://doi.org/10.5194/acp-21-13483-2021, 2021a. a, b
Tilgner, A., Schaefer, T., Alexander, B., Barth, M., Collett Jr., J. L., Fahey, K. M., Nenes, A., Pye, H. O. T., Herrmann, H., and McNeill, V. F.: Acidity and the multiphase chemistry of atmospheric aqueous particles and clouds, Atmos. Chem. Phys., 21, 13483–13536, https://doi.org/10.5194/acp-21-13483-2021, 2021b. a
Tunved, P., Nilsson, E., Hansson, H.-C., Ström, J., Kulmala, M., Aalto, P., and Viisanen, Y.: Aerosol characteristics of air masses in northern Europe: Influences of location, transport, sinks, and sources, J. Geophys. Res.-Atmos., 110, D07201, https://doi.org/10.1029/2004JD005085, 2005. a, b
Tunved, P., Stroöm, J., Kulmala, M., Kerminen, V.-M., Dal Maso, M., Svenningson, B., Lunder, C., and Hansson, H.-C.: The natural aerosol over Northern Europe and its relation to anthropogenic emissions—implications of important climate feedbacks, Tellus B, 60, 473–484, 2008. a, b
Turnock, S., Mann, G., Woodhouse, M., Dalvi, M., O'Connor, F., Carslaw, K., and Spracklen, D.: The impact of changes in cloud water pH on aerosol radiative forcing, Geophys. Res. Lett., 46, 4039–4048, 2019. a
Twomey, S.: The influence of pollution on the shortwave albedo of clouds, J. Atmos. Sci., 34, 1149–1152, 1977. a
Vepsäläinen, S., Calderón, S. M., Malila, J., and Prisle, N. L.: Comparison of six approaches to predicting droplet activation of surface active aerosol – Part 1: moderately surface active organics, Atmos. Chem. Phys., 22, 2669–2687, https://doi.org/10.5194/acp-22-2669-2022, 2022. a, b
Vepsäläinen, S., Calderón, S. M., and Prisle, N. L.: Comparison of six approaches to predicting droplet activation of surface active aerosol – Part 2: Strong surfactants, Atmos. Chem. Phys., 23, 15149–15164, https://doi.org/10.5194/acp-23-15149-2023, 2023. a, b
Wang, G., Zhang, F., Peng, J., Duan, L., Ji, Y., Marrero-Ortiz, W., Wang, J., Li, J., Wu, C., Cao, C., Wang, Y., Zheng, J., Secrest, J., Li, Y., Wang, Y., Li, H., Li, N., and Zhang, R.: Particle acidity and sulfate production during severe haze events in China cannot be reliably inferred by assuming a mixture of inorganic salts, Atmos. Chem. Phys., 18, 10123–10132, https://doi.org/10.5194/acp-18-10123-2018, 2018. a
Wellen, B. A., Lach, E. A., and Allen, H. C.: Surface p K a of octanoic, nonanoic, and decanoic fatty acids at the air–water interface: Applications to atmospheric aerosol chemistry, Phys. Chem. Chem. Phys., 19, 26551–26558, 2017. a
Werner, J., Julin, J., Dalirian, M., Prisle, N. L., Öhrwall, G., Persson, I., Björneholm, O., and Riipinen, I.: Succinic acid in aqueous solution: connecting microscopic surface composition and macroscopic surface tension, Phys. Chem. Chem. Phys., 16, 21486–21495, 2014a. a
Werner, J., Julin, J., Dalirian, M., Prisle, N. L., Öhrwall, G., Persson, I., Björneholm, O., and Riipinen, I.: Succinic acid in aqueous solution: connecting microscopic surface composition and macroscopic surface tension, Phys. Chem. Chem. Phys., 16, 21486–21495, https://doi.org/10.1039/C4CP02776K, 2014b. a
Werner, J., Persson, I., Björneholm, O., Kawecki, D., Saak, C.-M., Walz, M.-M., Ekholm, V., Unger, I., Valtl, C., Caleman, C., et al.: Shifted equilibria of organic acids and bases in the aqueous surface region, Phys. Chem. Chem. Phys., 20, 23281–23293, 2018. a, b, c, d, e
Wu, L., Wei, L., Wang, G., and Zhao, J.: Comparison of Atmospheric Monocarboxylic and Dicarboxylic Acids in Xi’ an, China, for Source Apportionment of Organic Aerosols, Water Air, Soil Pollut., 231, 1–11, https://doi.org/10.1007/s11270-020-04675-y, 2020. a
Yassaa, N., Meklati, B. Y., Cecinato, A., and Marino, F.: Particulate n-alkanes, n-alkanoic acids and polycyclic aromatic hydrocarbons in the atmosphere of Algiers City Area, Atmos. Environ., 35, 1843–1851, 2001. a
Yli-Juuti, T., Barsanti, K., Hildebrandt Ruiz, L., Kieloaho, A.-J., Makkonen, U., Petäjä, T., Ruuskanen, T., Kulmala, M., and Riipinen, I.: Model for acid-base chemistry in nanoparticle growth (MABNAG), Atmos. Chem. Phys., 13, 12507–12524, https://doi.org/10.5194/acp-13-12507-2013, 2013. a
You, Y., Smith, M. L., Song, M., Martin, S. T., and Bertram, A. K.: Liquid–liquid phase separation in atmospherically relevant particles consisting of organic species and inorganic salts, Int. Rev. Phys. Chem., 33, 43–77, 2014. a
Zheng, Y. and Ming, Y.: Low-Level Cloud Budgets across Sea Ice Edges, J. Climate, 36, 3–18, https://doi.org/10.1175/JCLI-D-22-0301.1, 2022. a