the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 10 Jul 2023
| 10 Jul 2023
Chemical evolution of primary and secondary biomass burning aerosols during daytime and nighttime
Amir Yazdani
Satoshi Takahama
John K. Kodros
Marco Paglione
Mauro Masiol
Stefania Squizzato
Kalliopi Florou
Christos Kaltsonoudis
Spiro D. Jorga
Spyros N. Pandis
Athanasios Nenes
Primary emissions from wood and pellet stoves were aged in an atmospheric simulation chamber under daytime and nighttime conditions. The aerosol was analyzed with online aerosol mass spectrometry and offline Fourier transform infrared spectroscopy (FTIR). Measurements using the two techniques agreed reasonably well in terms of the organic aerosol (OA) mass concentration, OA:OC trends, and concentrations of biomass burning markers – lignin-like compounds and anhydrosugars. Based on aerosol mass spectrometry, around 15 % of the primary organic aerosol (POA) mass underwent some form of transformation during daytime oxidation conditions after 6–10 h of atmospheric exposure. A lesser extent of transformation was observed during the nighttime oxidation. The decay of certain semi-volatile (e.g., levoglucosan) and less volatile (e.g., lignin-like) POA components was substantial during aging, highlighting the role of heterogeneous reactions and gas–particle partitioning. Lignin-like compounds were observed to degrade under both daytime and nighttime conditions, whereas anhydrosugars degraded only under daytime conditions. Among the marker mass fragments of primary biomass burning OA (bbPOA), heavy ones (higher ) were relatively more stable during aging. The biomass burning secondary OA (bbSOA) became more oxidized with continued aging and resembled that of aged atmospheric organic aerosols. The bbSOA formed during daytime oxidation was dominated by acids. Organonitrates were an important product of nighttime reactions in both humid and dry conditions. Our results underline the importance of changes to both the primary and secondary biomass burning aerosols during their atmospheric aging. Heavier fragments from aerosol mass spectrometry seldom used in atmospheric chemistry can be used as more stable tracers of bbPOA and, in combination with the established levoglucosan marker, can provide an indication of the extent of bbPOA aging.
- Article
(2846 KB) - Full-text XML
-
Supplement
(1142 KB) - BibTeX
- EndNote





Fine particulate matter (PM) in the atmosphere impacts climate and visibility (McFiggans et al., 2004; Hallquist et al., 2009) and is known to cause respiratory and cardiovascular diseases, leading to premature mortality (Pope et al., 2009; Shiraiwa et al., 2017; Burnett et al., 2018). A major fraction (up to 90 %) of fine PM is organic. Organic aerosol (OA) has various sources and formation mechanisms in the atmosphere, resulting in its complex chemical composition (Russell, 2003; Kanakidou et al., 2005; Hallquist et al., 2009). Primary organic aerosols (POAs) are emitted directly from their sources, whereas secondary organic aerosols (SOAs) are formed through chemical reactions of organic vapors that produce lower-volatility compounds (Seinfeld and Pandis, 2016). Oxygenated OA often dominates atmospheric OA, highlighting the importance of atmospheric chemistry and aging for OA (Zhang et al., 2007) as particles and gases are exposed to oxidants for days in the atmosphere (Wang et al., 2018). The major types of aging include homogeneous gas-phase oxidation and condensation (Donahue et al., 2012), oligomerization (Kalberer et al., 2006), heterogeneous reactions with oxidants (Robinson et al., 2006; George et al., 2008), and photolysis (Bateman et al., 2011; Henry and Donahue, 2012). Oxidation reactions by excited state photosensitizers can also lead to SOA formation and composition changes in OA (Tsui and McNeill, 2018; Mabato et al., 2022; Zhang et al., 2022). Homogeneous gas-phase reactions are generally believed to dominate (Henry and Donahue, 2012), while other mechanisms may be important under different conditions (e.g., Hearn et al., 2005; Hung et al., 2005; Nah et al., 2014).
Biomass burning (BB) contributes significantly to atmospheric primary and secondary OA (POA and SOA) (Puxbaum et al., 2007; Qi et al., 2019; Lanz et al., 2010) and brown and black carbon (BrC and BC, respectively) (Bond et al., 2013). BB is expected to have an increasing contribution to PM2.5 in the foreseeable future (Ford et al., 2018). Primary (bbPOA) and secondary bbOA (bbSOA) formed during reactions with hydroxyl and nitrate radicals have been investigated in several environmental chamber and field studies (Johansson et al., 2004; Bäfver et al., 2011; Alves et al., 2011; Hennigan et al., 2011; Bruns et al., 2015; Tiitta et al., 2016; Bertrand et al., 2017, 2018a; Kodros et al., 2020; Jorga et al., 2021; Yazdani et al., 2021). These studies have suggested a net enhancement in the OA concentration with aging due to bbSOA formation. To estimate the contributions of bbPOA and bbSOA after aging in chambers or in field measurements, bbPOA implicitly (e.g., when using positive matrix factorization) or explicitly is assumed to be stable during the aging (Robinson et al., 2007; Grieshop et al., 2009; Tiitta et al., 2016; Kodros et al., 2020), neglecting the effects of heterogeneous reactions, photolysis, and the changing gas–particle partitioning of its components. This is an approximation, given that several studies report significant degradation of bbPOA markers like anhydrosugars and methoxyphenols during aging (Hennigan et al., 2010, 2011; Slade and Knopf, 2013; Bertrand et al., 2018a; Yazdani et al., 2021). These compounds constitute a significant fraction (up to 50 %) of the bbPOA mass (Fine et al., 2002; Bertrand et al., 2018a; Yazdani et al., 2021). This chemical processing can impact the bbPOA mass and composition significantly but has not been well-characterized to date.
Recent efforts using the volatility basis set (VBS; Donahue et al., 2006) address the volatility and gas–particle partitioning of POA including primary bbOA in simulations (Robinson et al., 2007; Theodoritsi et al., 2020). However, the chemical processing of bbPOA remains uncertain, and heterogeneous reactions of bbOA compounds are not included in most models.
Aerosol mass spectrometry and Fourier transform infrared spectroscopy (FTIR) are two methods used in this work to study the bbOA composition and its evolution with aging. The aerosol mass spectrometer (AMS; Aerodyne, Inc.), while being capable of analyzing most of the OA mass (Hallquist et al., 2009), is limited by the extensive molecule fragmentation and the variability of particle collection efficiency (CE) (Canagaratna et al., 2007; Faber et al., 2017; Kumar et al., 2018). FTIR is a non-destructive method that measures the abundance of certain functional groups but with a limited temporal resolution. Functional group abundances are then used to estimate the OA mass concentration and elemental ratios (Coury and Dillner, 2008; Ruthenburg et al., 2014; Reggente et al., 2016; Boris et al., 2019). A recent study also shows the ability of FTIR to quantify bbOA marker molecules (Yazdani et al., 2021).
In this work, primary biomass burning emissions generated from pellet and wood stoves are injected into an environmental simulation chamber and aged with hydroxyl and nitrate radicals. The AMS and FTIR are used in tandem to better understand and quantify the evolution of primary and the formation of secondary bbOA with aging. We adopt a particle wall loss correction method based on AMS organic measurements and develop a procedure to quantify the overall changes in the composition of bbPOA during the coarse of aging with the AMS and FTIR. The results of this study allow us to evaluate the stability of bbPOA under different conditions, the usefulness of different markers to identify aged atmospheric bbOA, and the importance of aging mechanisms other than the homogeneous gas-phase oxidation under atmospherically relevant conditions.
2.1 Experimental setup and procedure
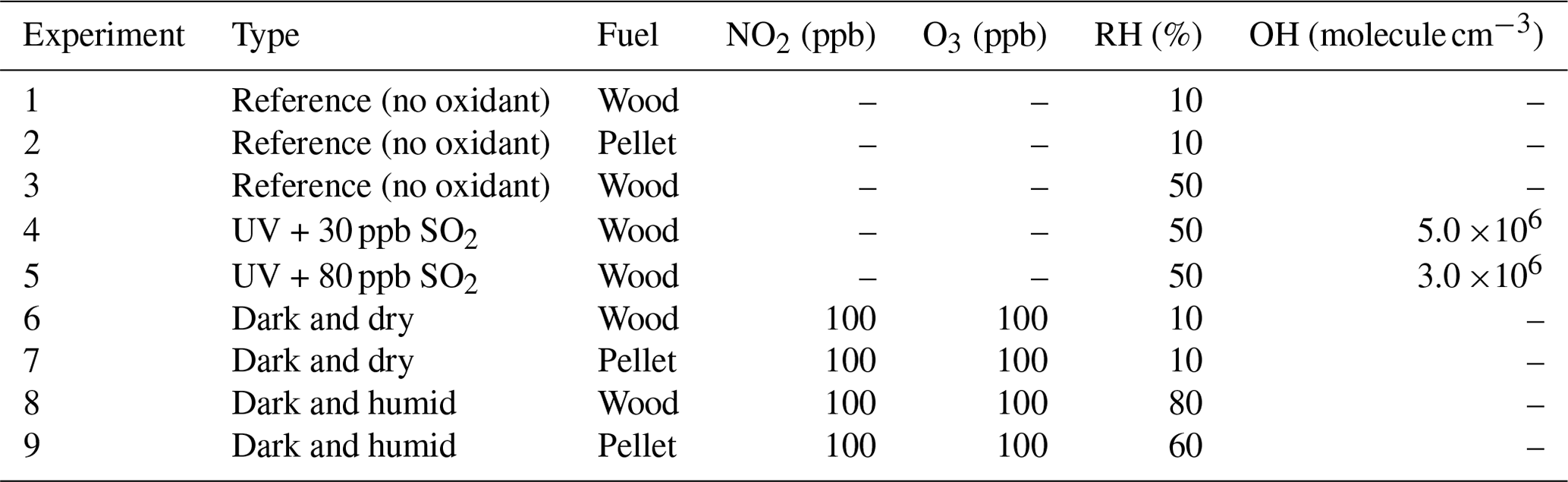
Primary emissions from common wood and pellet stoves (primarily flaming phase) were diluted and injected for 30–40 min into a 10 m3 Teflon atmospheric simulation chamber located at the Foundation for Research and Technology-Hellas (FORTH), Greece. Olive wood logs with bark and ENplus® A1 pellets were used as fuel. Details of the chamber and combustion facilities have been discussed elsewhere (Kaltsonoudis et al., 2017; Kodros et al., 2020). The fuels and stoves used in this work are commonly used in the region. Primary emissions were left in the chamber after the injection for around 2 h to ensure proper mixing and to characterize particle wall losses in the chamber.
Nine experiments were conducted in this work. For the three reference experiments (Table 1), emissions were left in the dark chamber without addition of any oxidants. For the two experiments simulating daytime aging, reactions were initiated by turning on UV fluorescent lamps (Osram, L 36W/73) ( min−1), and the hydroxyl radical was subsequently generated via ozone photolysis in the presence of water vapor for around 2 h. For these experiments the average RH was roughly 50 %, and the average OH concentration was (3–5) ×106 molecule cm−3. This corresponds to 6–10 h of aging in the atmosphere, assuming an average OH concentration of 106 molecule cm−3 (Seinfeld and Pandis, 2016). The OH concentration was estimated using a proton-transfer-reaction mass spectrometer (PTRMS; Ionicon Analytik) monitoring the concentration of 1-butanol-d9 injected initially into the chamber (Barmet et al., 2012). The four nocturnal aging experiments were conducted under two different RH regimes: dry (10 %) and humid (60 %–80 %). For these experiments, roughly 100 ppb of NO2 was injected into the chamber before the primary biomass burning emissions. Around 2 h after the injection of primary emissions, aging was initiated by injection of O3 (roughly 100 ppb), and the NO3 radical was produced via the reaction of O3 with NO2. Both NO2 and O3 concentrations were measured by Teledyne gas monitors (models T201 and 400E, respectively). NO3 radical concentrations were not measured during these experiments but are estimated to be 0–400 ppt from experiments with similar concentrations of NO2 and O3 (Kodros et al., 2023).
Table 1Description of experiments and initial O3 and NO2 concentrations.
2.2 On- and offline PM measurements
The composition of non-refractory aerosols in the chamber was measured by a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; Aerodyne Research Inc., Billerica, USA) (Jayne et al., 2000; Drewnick et al., 2005) sampling in V mode every 3 min. The sampling inlet was shared by the HR-ToF-AMS and a scanning mobility particle sizer (classifier model 3080, DMA model 3081, CPC model 3787, TSI), which was used to measure the particle number size distribution in the 15–700 nm range. No dryer was placed before the inlet of the HR-ToF-AMS and the SMPS. Ionization efficiency (IE) calibrations were performed twice a month using size-selected ammonium nitrate particles (300 nm in mobility diameter, Dm). For certain experiments, the HR-ToF-AMS also measured the composition of chamber aerosols after being passed through a thermodenuder (TD) to study their volatility in the 25–400 ∘C temperature range. The TD consisted of an insulated heating section for evaporation of the more volatile compounds and by a cooling section with activated carbon, which was used to avoid vapor condensation during the cooling stage (An et al., 2007; Louvaris et al., 2017).
Primary and aged PM1 was collected on two separate 47 mm PTFE filters (Pall corporation, 1 cm diameter of the collection surface) for each experiment. Sampling duration was 30 min for each filter at a flow rate of 8 L min−1 for 20 min, using a flow system composed of a silica gel denuder to minimize aerosol water and a sharp-cut-off cyclone. For the fresh bbOA emissions, the two filters (one quartz and one FTIR filter) were collected sequentially during this stabilized fresh emission period. The aged filters were collected 3.5 h after the O3 addition in order to allow sufficient time for the oxidation process to take place, as indicated by the O:C trend reaching is maximum value. Filters were immediately stored in petri dishes at 253 K to minimize volatilization of aerosols and chemical reactions. Filter samples were analyzed using a Bruker Vertex 80 FTIR instrument equipped with a α deuterated lanthanum alanine doped triglycine sulfate (DLaTGS) detector and a custom-made filter mini-chamber to minimize water vapor and CO2 interferences. The spectra (Yazdani et al., 2023) were obtained at a resolution of 4 cm−1 and were averaged over 128 scans.
2.3 Data analysis
2.3.1 Initial spectral post-processing
The AMS raw signal was post-processed using the AMS software toolkits SeQUential Igor data RetRiEvaL (SQUIRREL) v1.57 and the Peak Integration by Key Analysis (PIKA) v1.16. The elemental and OA:OC ratios were calculated using the approach of Canagaratna et al. (2015). The uncertainty deriving from the Improved-Ambient method for O:C is 28 %, while the Aiken-Explicit method yielded better results (20 %) when used with specific calibrations (Chen et al., 2011). The organic nitrate concentration was calculated based on NO+ and peak ratios in the AMS mass spectra following the approach of Farmer et al. (2010).
Baseline correction was performed on the FTIR spectra to eliminate the contribution of light scattering from the spectra (Russo et al., 2014; Parks et al., 2019) using smoothing splines. After baseline correction, blank subtraction was performed to recover some of the overlapping features with PTFE peaks (e.g., levoglucosan fingerprint bands 860–1050 cm−1). A multiple peak-fitting algorithm was applied on the FTIR spectra to estimate the contribution of absorption related to alcohol (referred to as aCOH), carboxylic acid (COOH), alkane (aCH), and non-acid carbonyl (naCO) groups (Takahama et al., 2013). The area under each peak was related to the abundance of the corresponding functional group using the measured absorption coefficient by Reggente et al. (2019).
The ratio of fingerprint absorbances related to levoglucosan (multiple peaks in the 860–1050 cm−1 range; Yazdani et al., 2021) in the FTIR spectra was compared between primary and aged aerosols to estimate the change of this biomass burning marker with aging. Absolute levoglucosan concentrations were estimated by averaging the heights of each of the three peaks in the fingerprint region and applying absorption coefficients estimated from measurements of Ruthenburg et al. (2014). The peak heights were defined relative to a local baseline defined by a linear interpolation among peak valleys (Yazdani, 2022).
The single sharp peak at 1515 cm−1 related to lignin-like compounds was used to estimate and compare the concentration of this group of compounds in primary and aged aerosols. Due to the lack of absorption coefficient for the 1515 cm−1 peak, the following approach was taken to estimate the concentration of lignin-like compounds: Fine et al. (2002) found syringaldehyde to be one of the most abundant lignin-like compounds in bbPOA. The total concentration of lignin-like compounds was estimated using the absorption coefficient of the 1515 cm−1 peak for syringaldehyde. As this coefficient was not available in the literature, it was calculated by scaling the measured coefficient of carbonyl (Reggente et al., 2019) by the ratio of absorbances at 1515 and 1680 cm−1 from a spectrum of the compound prepared in potassium bromide wafer taken from a reference database (John Wiley & Sons, Inc., 2022).
2.3.2 Quantifying the bbPOA transformation
Particle wall loss correction for AMS and FTIR measurements was carried out assuming a first-order, time-independent wall loss rate constant for the OA mass concentration (Pathak et al., 2007):
where kOA is the particle wall loss rate constant, and COA is the AMS OA mass concentration. The rate constant for each experiment was calculated using OA concentrations from 0.5–1 h after the injection of primary emissions into the chamber (to ensure proper mixing) up to the start of chemical aging or until the end for reference experiments. Wall-loss-corrected concentrations of individual AMS fragments were calculated by
where is the measured concentration of fragment i at time t, and is its wall-loss-corrected concentration at time t. In this process, it is assumed that particles are internally mixed, and there is a minor size dependence for the chemical composition of bbOA. These assumptions are supported by the measurements of Grieshop et al. (2009). The difference between the wall-loss-corrected and initial concentrations of diminishing fragments (those with ) was used to quantify the extent of bbPOA transformation at each time after the start of aging with the AMS.
2.3.3 Residual spectra
The apparent contribution of the fresh bbPOA to aged bbOA spectra (FTIR or AMS) was calculated using Eq. (3) assuming no evaporation, condensation, or heterogeneous reactions:
where sbbPOA(t) is the apparent bbPOA spectrum at time t, and sbbPOA(0) is the bbPOA spectrum at the start of aging. The residual spectrum at time t, sres(t), was defined as the result of subtraction of the apparent bbPOA spectrum at time t from that of the aged bbOA:
The FTIR and AMS residual spectra are composed of positive and negative elements. The positive elements, which indicate the formation of bbSOA species or oxidation products of fresh bbPOA, were studied in both AMS and FTIR residual spectra. The negative elements, on the other hand, indicate the loss of fresh bbPOA species. The negative peaks in the FTIR residual spectra were used to obtain another estimate of bbPOA transformation with aging that does not suffer from the limitations of the AMS.
2.3.4 Dimension reduction of AMS spectra
We used principal component analysis (PCA; Hotelling, 1933) to simplify the high-dimensional, inter-correlated AMS spectra while considering variations in the majority of mass fragments. This allows the better understanding of the evolution of bbOA during the course of aging in the chamber and to compare it to atmospheric aerosols. Before applying PCA, ion fragment data at 6 min intervals were normalized by total OA concentration to exclude the variation of ion fragment signals due to change in total OA mass (e.g., by SOA production or particle wall loss). PCA calculations were performed on the normalized and uncentered (not subtraction average value at each mass fragment) AMS spectra from the chamber experiments using singular value decomposition (Abdi and Williams, 2010). Different spectra such as positive matrix factorization (PMF) factors of atmospheric OA from previous studies were projected onto the obtained PC space for comparison. Specifically, moderately oxygenated OA (M-OOA) and very oxygenated OA factors from Athens and Patras in summer (Kostenidou et al., 2015) and fresh and aged bbOA factors from Patras (Florou et al., 2017) were used for comparison.
The composition of bbPOA and extent of aging in these experiments are described in Sect. 3.1. We discuss the range of estimated particle wall loss correction factors and uncertainties in Sect. 3.2 and, taking them into consideration, describe the results of each type of experiment in Sects. 3.3–3.5.
3.1 General description of primary biomass burning organic aerosols and extent of aging
The FTIR spectral profiles of primary wood- and pellet-burning aerosols are generally similar (Fig. 1). However, the higher relative absorbances of levoglucosan and the alcohol group and the lower absorbance of lignin-like compounds stand out for the pellet-burning aerosols. Primary pellet-burning aerosols also have similar FTIR spectra to that of wood (Pandey, 1999) but have higher relative concentrations of aCOH and levoglucosan compared to wood-burning aerosols (Fig. 1) that can be attributed to different combustion conditions and OA concentrations between the two fuels. For wood and pellet fuels, different ratios of lignin-related fragments (e.g., , , ; Li et al., 2012; Tolbert and Ragauskas, 2017; 2017) are observed, reflecting different composition of the fuels and combustion conditions (e.g., combustion efficiency). We also observe that the relative abundance of lignin- and levoglucosan-related fragments and their corresponding FTIR absorbances vary among different experiments with the same fuel probably due to slightly different combustion conditions.
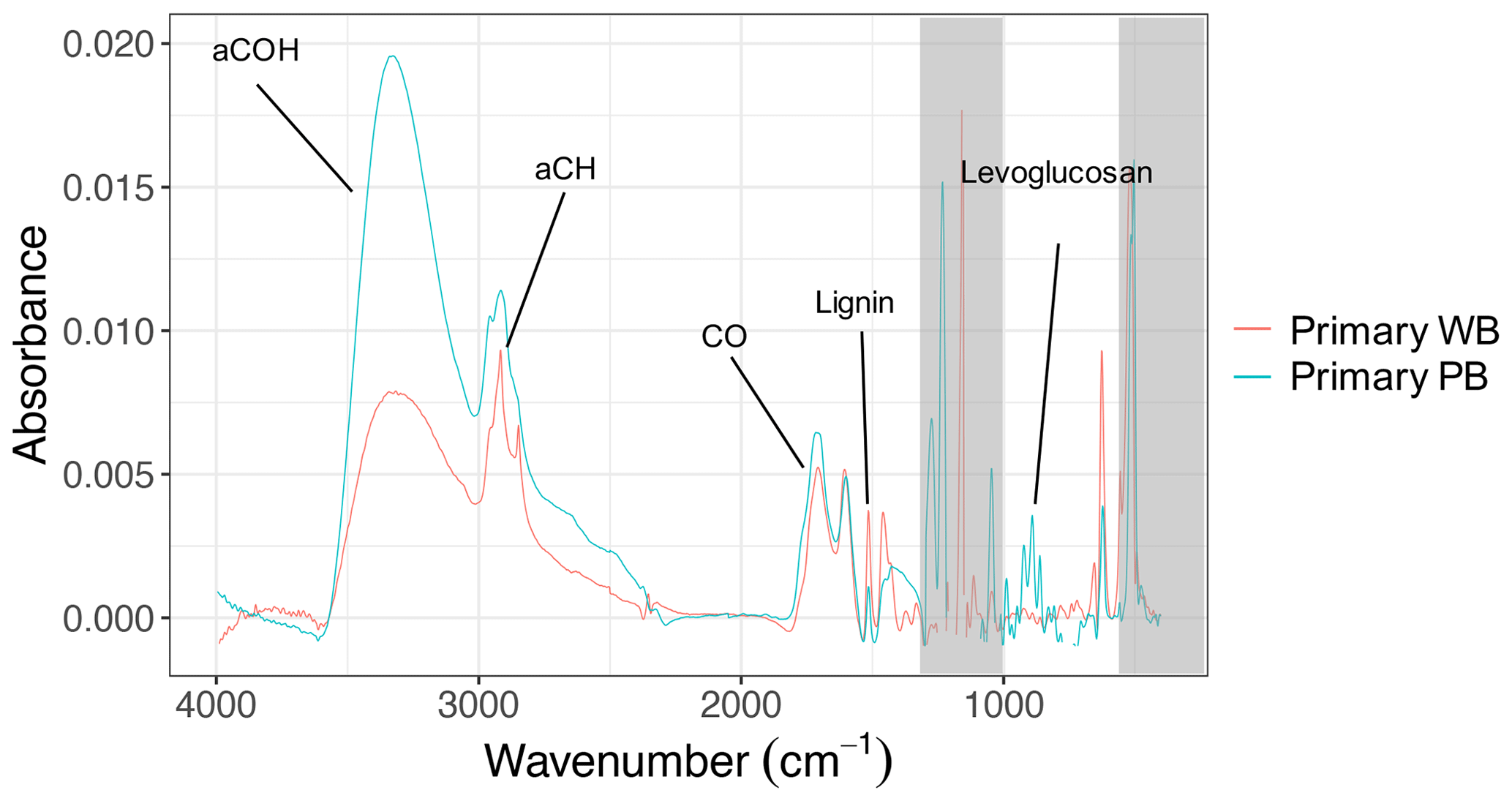
Figure 1FTIR spectra of primary wood-burning (WB) and pellet-burning (PB) aerosols of experiment 6 and 7, respectively. Important functional groups or biomass burning markers are indicated. Regions with strong interferences from the collection substrate are shaded in gray.
OA concentrations of the AMS (without CE correction) and FTIR were correlated with R2=0.75. The OM:OC estimated using these two also showed similar trends upon aging (Sect. S3 in the Supplement). The spectral features and chemical composition of primary wood-burning aerosols from FTIR – and mass fragment profiles from the AMS – in this work are consistent with those previously reported (e.g., Tiitta et al., 2016; Bertrand et al., 2017, 2018a; Yazdani et al., 2021). The FTIR spectra of these primary aerosols largely resemble those of wood constituents such as lignin (Yazdani et al., 2021). However, The aCH peaks are more prominent in the FTIR spectra of wood-burning aerosols of this work compared to those of Yazdani et al. (2021). While higher OA loadings can increase partitioning more volatile hydrocarbon compounds into the aerosol phase (Shilling et al., 2009), the primary OA concentrations in this work are comparable, and the prominent peaks are likely due to the combustion of wood with bark – which was not present in the experiments of Yazdani et al. (2021).
Visualizing the evolution of f44–f43 (–) and the first three principal components compares the extent to which primary and residual bbOA in these chamber experiments are similar to the atmospheric bbOA and OOA factors, respectively. As can be seen from Fig. 2, the trajectory for the total bbOA spans a small range of the plot and is located in the lower section of the triangle (Ng et al., 2011). For the residual OA formed using UV lights, however, the trajectories start in the lower section of the triangle and continue upward until they end close to the upper vertex, consistent with previously reported trajectories for wood burning in chamber experiments (Yazdani et al., 2021). The end points are close to the location of the very oxygenated OOA factor for atmospheric aerosols collected during summer in Athens and Patras (Kostenidou et al., 2015). The latter suggests that the extent of aging by UV in the chamber is comparable to that observed in the atmosphere.
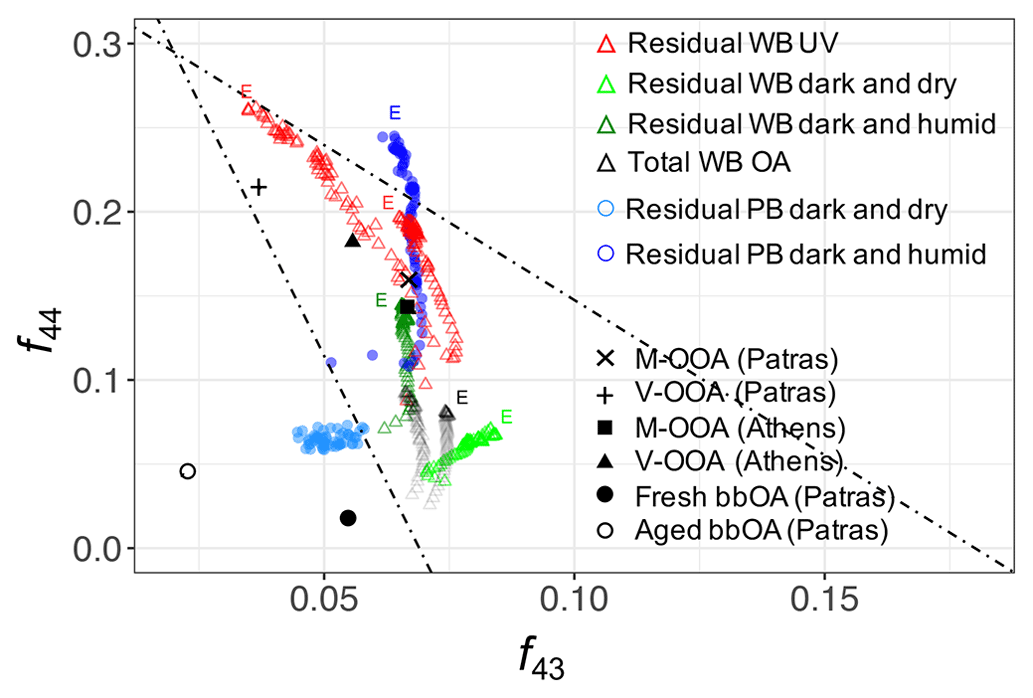
Figure 2Aging trajectories of wood-burning (WB) and pellet-burning (PB) aerosols in the chamber with UV lights and dark aging shown in the f44–f43 (–) plot. Atmospheric OA factors were obtained from Kostenidou et al. (2015) and Florou et al. (2017): V-OOA (very oxygenated OA), M-OOA (moderately oxygenated OA), and fresh and aged bbOA. The end of aging trajectories is denoted with the letter E.
The residual OA from wood and pellet-burning emissions aged in dark and dry conditions occupies the lower part of the triangle plot with a relatively less significant change in f44 and f43 with aging (Fig. 2), implying only mild oxidation. Contrary to dark and dry conditions, in dark and humid conditions, the trajectories resemble more those of UV experiments. The f44 and f43 increase more significantly, suggesting a stronger oxidation due to aqueous (nitrate radical) reactions. f43 is more elevated for aged bbOA in dark and humid conditions compared to that aged using UV (Fig. 2). The latter has also been observed by Kodros et al. (2020) and Kodros et al. (2023). Although the general oxidation trend is observed to be similar between PB and WB OA in dark conditions, the length of oxidation trajectories and their starting point appear to be somewhat different. These differences are believed to be related to different levels of aging in different experiments and slightly different composition of fresh PB and WB OA, respectively.
PCA extends the analysis of AMS spectra to additional fragments besides and C2H3O+. Principal components (PCs) were derived from chamber AMS spectra to highlight mass fragments describing most of variation in the chamber AMS spectra. Thereafter, the residual chamber spectra and atmospheric PMF factors from Kostenidou et al. (2015) and Florou et al. (2017) were projected onto the PC space including the first three PCs (Fig. 3). Since the first three principal components (PCs) in Fig. 3 describe the majority (87 %) of the total variance in AMS spectra, similar PC scores (closeness of points) imply their spectral similarity. These PCs have high loadings only for few fragments, making their interpretation straightforward (Sect. S4). The first PC has high negative loadings of oxygenated fragments ions such as CO+, , CHO+, and C2H3O+, showing the direction of aging. Similar results were found by Yazdani et al. (2021) for wood-burning OA in previous chamber experiments (with OA enhancement due to SOA condensation was approximately twice greater than those in this study). The CHO+ fragment has a high positive loading, and CO+ and have high negative loadings for PC2, suggesting that this PC distinguishes between carboxylic and dicarboxylic acids which are detected by and alcohols, which produce strong signals of CHO+. PC3 captures the degradation of biomass burning markers (e.g., levoglucosan) with aging through high negative loadings of and (Sect. S4 in the Supplement). As can be seen in Fig. 3, the atmospheric primary and aged bbOA factors from Florou et al. (2017) are located close to the primary wood-burning aerosols in both plots, suggesting their similar composition. The residual OA formed in dark and dry conditions appears to be the least aged among all residual spectra with higher PC1 loading and is located closer to aged chamber OA. The residual OA aged in dark and humid conditions is, however, to be more oxidized than that in dry conditions, and it is located closer to the atmospheric moderately and very oxygenated OA factors (M-OOA and V-OOA) from Kostenidou et al. (2015). The residual OA in the UV experiment has the most aged spectrum and is located the closest to the atmospheric V-OOA factors in the PC1–PC2 and PC1–PC3 biplots (Fig. 3). However, there is a noticeable difference in PC2 scores between atmospheric and chamber aerosols that is believed to be related to the general higher concentration of the CHO+ fragment for the chamber bbOA. The results of the PCA support those of the f44–f43 plot in terms of similarity of aged chamber aerosols to field measurements while considering a wider range of mass fragments. Additionally, the PCA characterizes the functional group content of aerosols discussed in this and following sections using fewer variables. For example, as can be seen in Fig. 3, pellet-burning aerosols have higher PC2 scores compared to wood burning, consistent with the higher alcohol content observed in their FTIR spectra (Fig. 1).
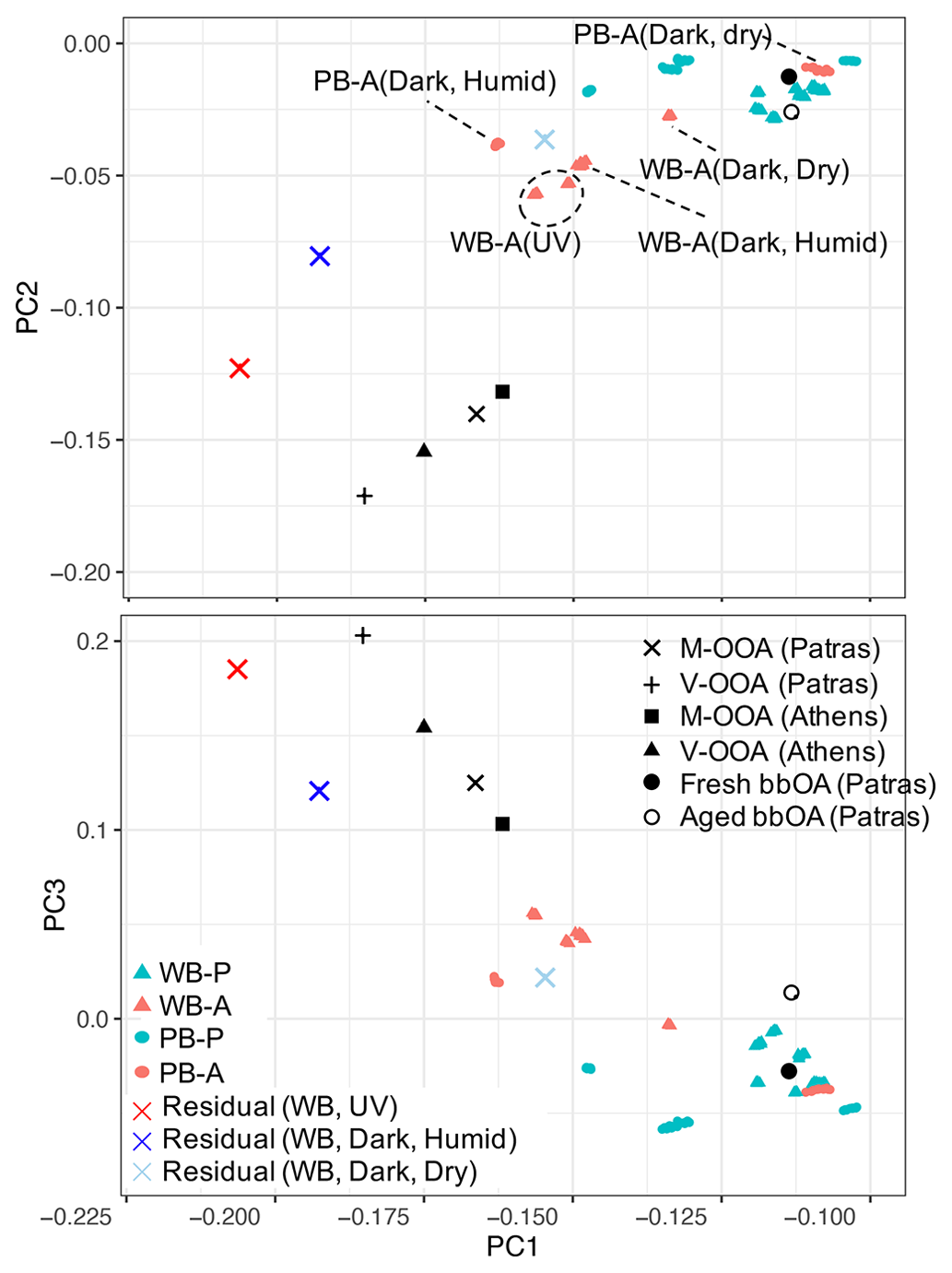
Figure 3PC1–PC2 and PC1–PC3 biplots for primary, aged, and residual bbOA in the chamber calculated for the period of PTFE filter sampling: WB-P (wood burning, primary), WB-A (wood burning, primary), PB-P (pellet burning, primary), and PB-A (pellet burning, aged). Atmospheric OA factors were obtained from Kostenidou et al. (2015) and Florou et al. (2017): V-OOA (very oxygenated OA), M-OOA (moderately oxygenated OA), and fresh and aged bbOA.
3.2 Particle wall loss corrections
The procedure for obtaining particle wall loss coefficients for correction of mass fragment profiles and residual spectra of the AMS and FTIR is illustrated here for a UV experiment (experiment 5). First, a first-order loss is fitted to the AMS OA concentration before the start of aging (in the −1.5–0 h range). The first-order model explains the OA trend in this experiment with R2=0.95. The fitted curve is shown by a dashed curve in Fig. 4.
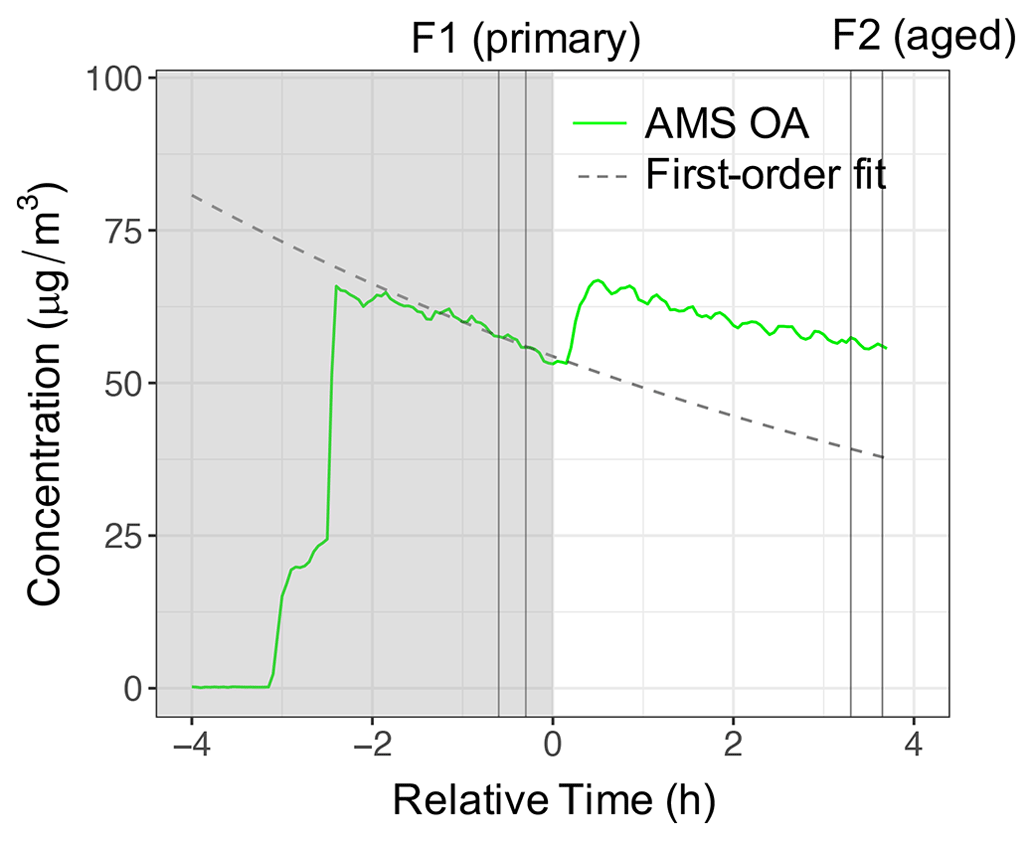
Figure 4The AMS OA and apparent POA concentrations using a first-order wall loss for experiment 5 (wood burning, aged with UV). Dark and light regions of the figure indicate periods before and after switching on UV lights. Vertical lines indicate the periods of filter sampling for primary and aged aerosols.
The estimated particle wall loss correction coefficients range from 0.06–0.11 with an estimated uncertainty of 5 %–12 % (Table 2). The first-order wall loss model is able to explain (R2>0.8) the overall variations in the AMS OA concentration for the reference experiments and the period prior to initiation of aging for other experiments well (Sect. S1). In addition, the decrease in the OA concentration estimated with FTIR for the reference experiments closely matches (less than 5 % difference) that estimated with the AMS (bbPOA ratios shown in Table 2). The latter supports the particle wall loss rate constants derived from AMS OA. Systematic differences up to 15 % are observed between the diminution of different fragments in reference experiments (Fig. 5a), which are investigated further in Sect. 3.3. These differences are relatively small compared to those in experiments with oxidants (Sects. 3.4–3.5).
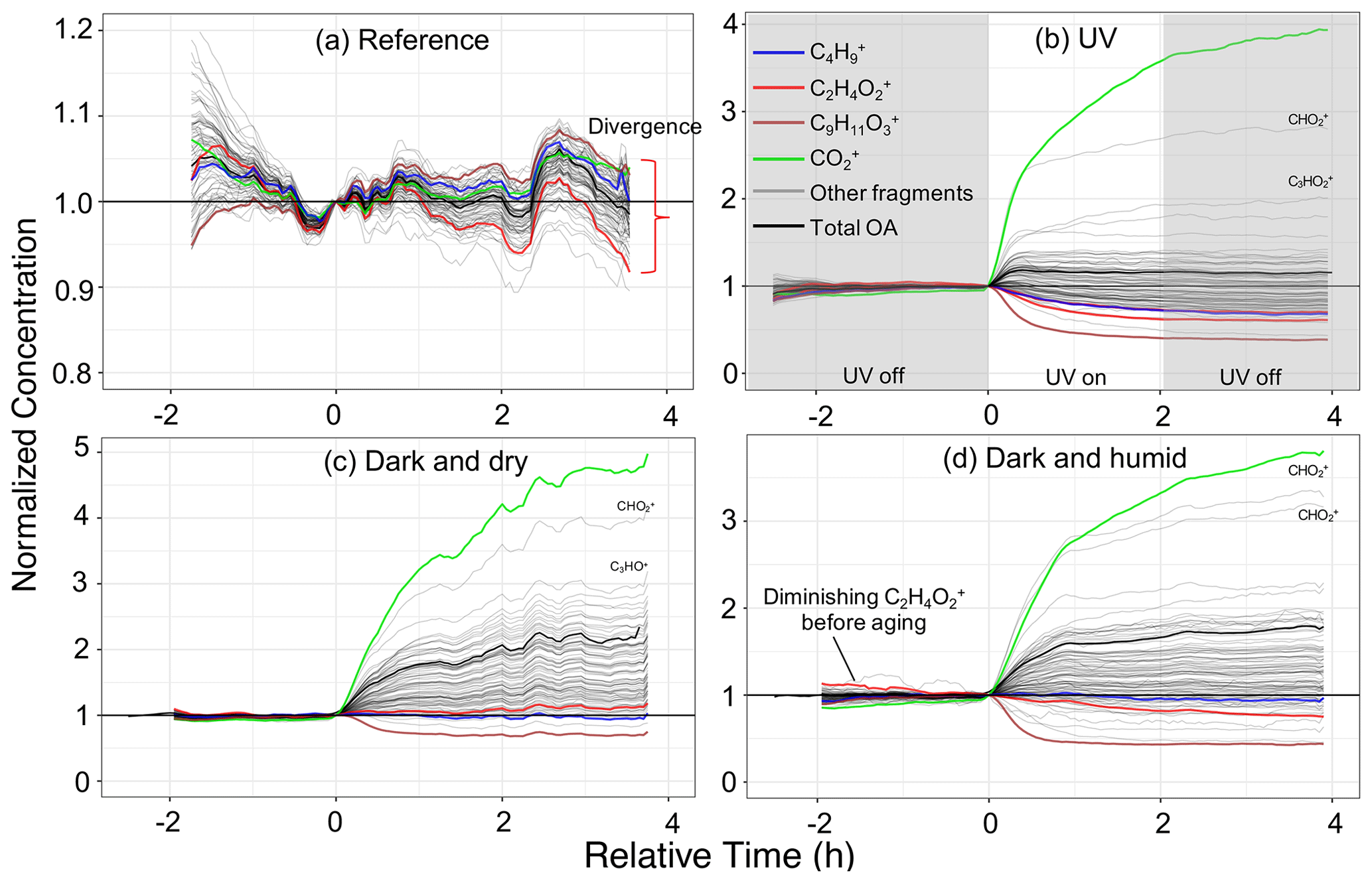
Figure 5Time series of wall-loss-corrected and normalized concentrations of individual mass fragments in different aging scenarios. Only mass fragments contributing more than 0.25 % to the total OA mass concentration are shown. Fragments often attributed specific species (e.g., hydrocarbons, acids) or biomass burning tracers (levoglucosan and lignin) are shown in color. Panels (a)–(d) represent experiments 1 (wood burning), 4 (wood burning), 6 (wood burning), and 8 (wood burning), respectively. Time zero indicates the initiation of aging.
3.3 Reference experiment
Interpretation of experiments in this and remaining sections is primarily done on wall-loss-corrected AMS fragments normalized by their concentrations at the start of aging. The trends were observed to be similar within each aging scenario regardless of the fuel burned, so an experiment representing each category is shown in Fig. 5. The increase in the normalized concentration of fragments (usually oxygenated fragments) indicates the appearance of new species, for example, through SOA condensation. A decrease in the normalized concentrations suggests that certain species diminish with aging either due to heterogeneous reactions or evaporation. Residual AMS and FTIR spectra for each category (Figs. 6 and 7) provide further support of conclusions drawn from the wall-loss-corrected time series profiles of AMS fragments.
As can be seen from Fig. 5a, in reference experiment 1, wall-loss-corrected fragments follow the general temporal trend of the total OA, with deviations within 10 %. Mass fragment concentrations in the residual AMS and absorbances in the residual FTIR spectra are noisy and close to zero (Figs. 6a and 7a), implying negligible emergence of new species. The loss of levoglucosan in experiment 2 with RH around 50 %, however, results in high uncertainty for its corresponding fragment, , in Fig. 6a. This observation suggests that the composition of OA does not change noticeably in the absence of oxidants; less than 2 % of bbPOA mass is transformed after leaving the emissions in the chamber for around 4 h. This value can be considered a baseline for other aging scenarios.
Some species-specific changes can be detected in these experiments. For instance, the systematic negative trend in the fragment attributed to levoglucosan (and anhydrosugars), , and the positive trend in a fragment related to lignin-like compounds, , that can be viewed in Fig. 5a indicate that their rate of loss in the chamber is greater than and less than total OA, respectively, since the wall loss correction is based on the OA. and small CxHy+ fragments (e.g., , , ) appear to have higher-than-average loss rates, while the fragments attributed to lignin-related compounds appear to diminish less. Similar trends are observed for all wood-burning experiments during the period before the start of aging, implying slight but systematic differences between the loss rates of different OA species. These differences are likely related to different volatility or size distribution of different species. Grieshop et al. (2009) reported bbPOA composition to be internally mixed and size-independent, but the range in volatility of compounds in bbPOA may be responsible (Bertrand et al., 2018b and Sect. S2).
Table 2Summary of wall loss rates and the ratio of the bbPOA concentration and certain markers in the aged bbOA to those in fresh bbOA.
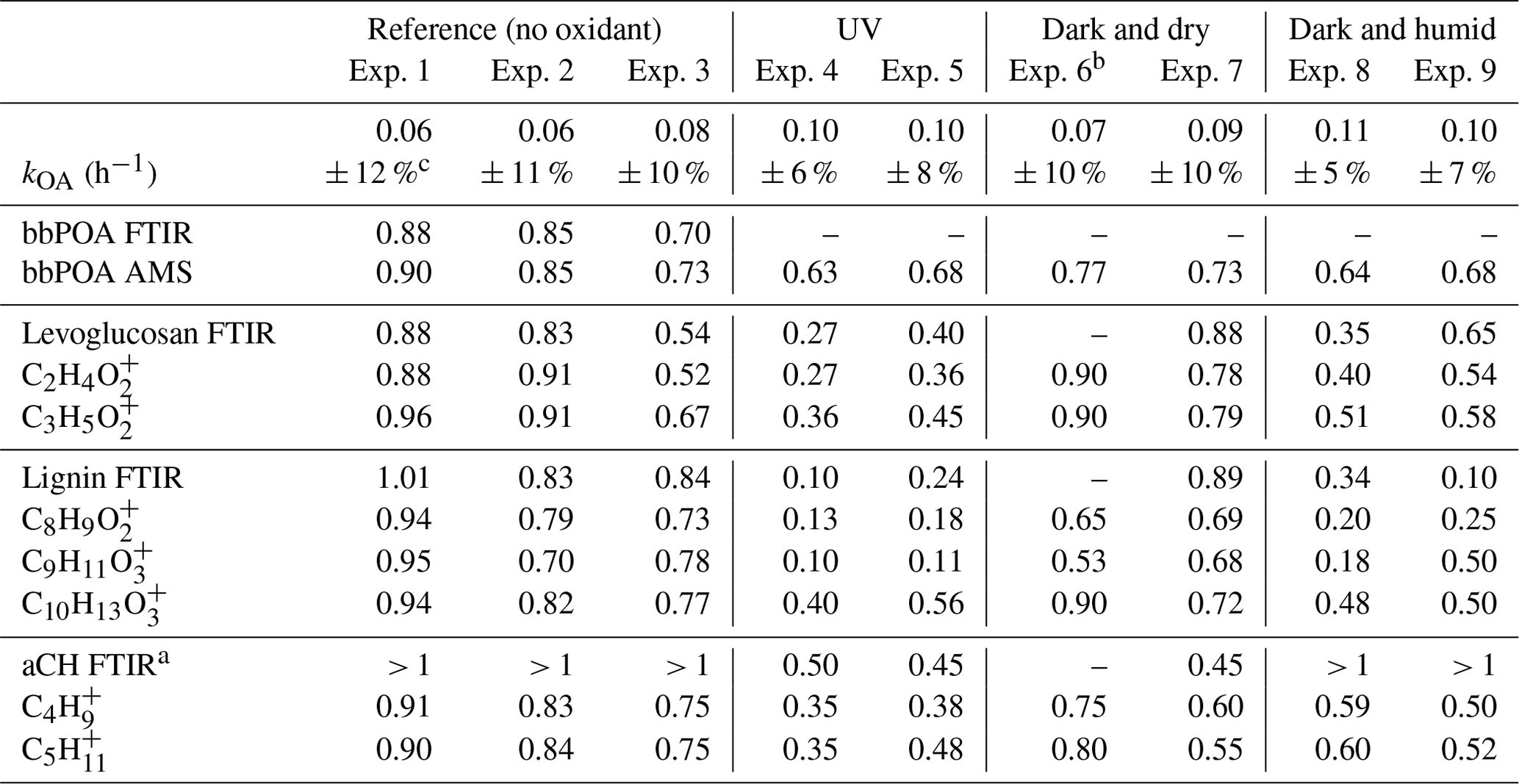
a Approximate values by which FTIR bbPOA spectra should be scaled to avoid inverted local CH peaks when subtracting from aged OA spectra. b FTIR measurements were discarded for experiment 6 due to the unusually low bbOA mass sampled on the filter. c Uncertainties are based on a 90 % confidence interval.
3.4 Aging by UV
In a UV experiment (experiment 4; Fig. 5b), as soon as the UV lights are turned on, several oxygenated fragment ions increase in concentration, as expected based on around 20 % mass formation observed; among these, has the most prominent growth. When emissions are aged with UV lights, we observe positive values for the majority of AMS fragment concentrations and FTIR absorbances in the residual spectra, suggesting a significant formation of new oxidized species. Since bbPOA transformation was discussed in the previous section, we only consider the positive elements of the residual spectra here as an approximate representation of bbSOA and aged bbPOA. As can be seen from Fig. 6b, the normalized residual spectrum in the UV experiments is mainly composed of (and the fragments directly estimated from it: CO+, H2O+, and OH+), suggesting the abundance of carboxylic acids (Aiken et al., 2007). A few heavier mass fragments with two or more oxygen atoms (e.g., , , , and ) statistically have non-zero values and can also be indicators of acids (Lambe et al., 2012) and polyfunctional organics (Fig. 6b). The sharp low-frequency carbonyl peak in the residual FTIR spectrum (1700 cm−1) besides the broad OH peak of dimerized acids (2400–3400 cm−1) is another indicator of carboxylic acids in the residual OA in the UV experiments (Fig. 7b). The latter, which is partially masked by the ammonium NH stretching peaks, becomes more prominent upon ammonium peak subtraction (Sect. S5). Although the relative contributions of bbSOA and oxidized bbPOA to the residual spectra are not clear, the formation of carboxylic acids via the oxidation of major wood-burning volatile organic compounds (VOCs) and the abundance of carboxylic acids in the bbSOA are consistent with previous reports (Yazdani et al., 2021; George et al., 2015; Chhabra et al., 2011). For certain atmospheric biomass burning samples, a unique FTIR spectral profile with high abundance of carboxylic acids has been observed, resembling the aged bbOA (with UV) of this work (Sect. S6).
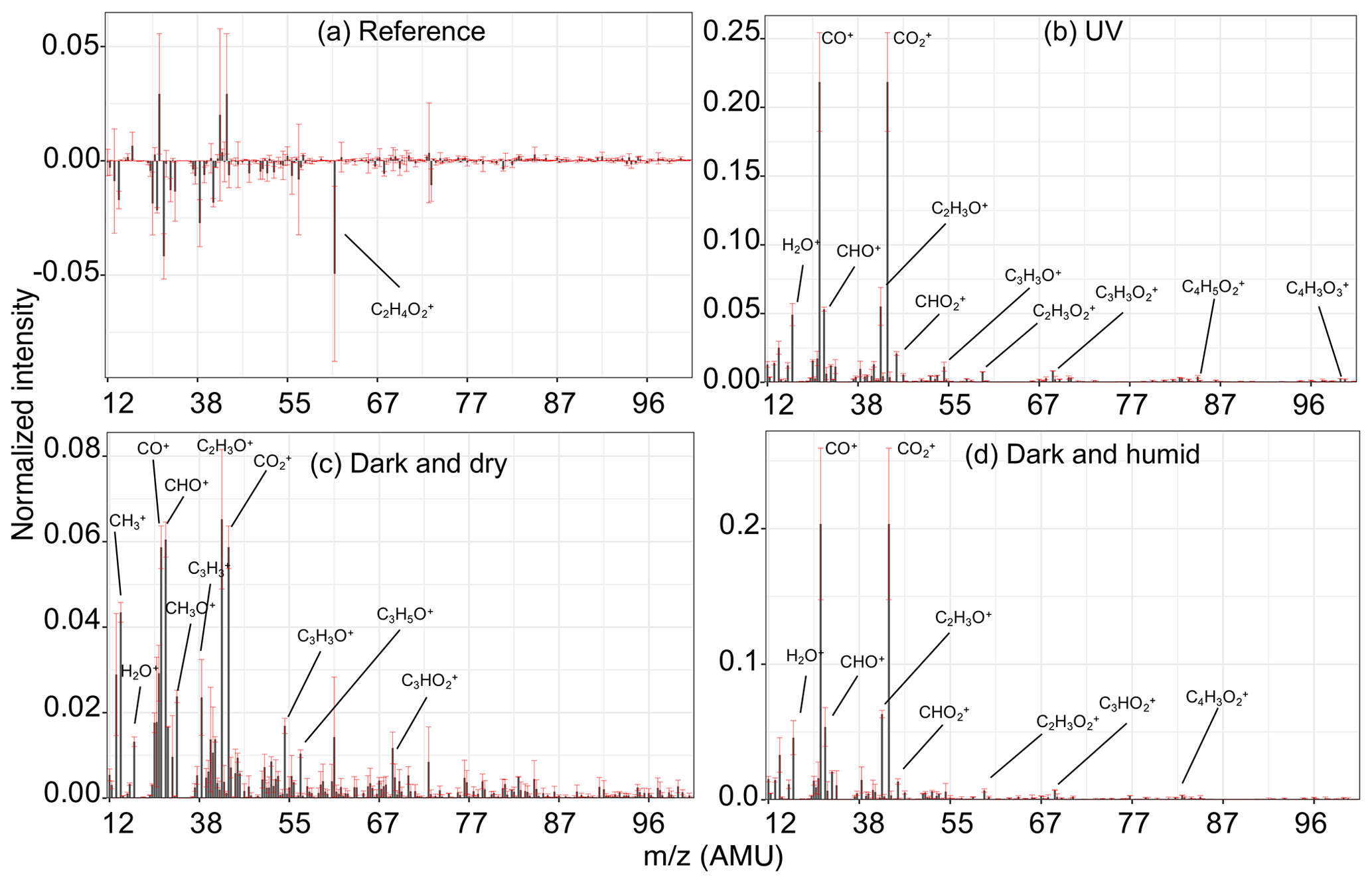
Figure 6Normalized residual AMS spectra of bbOA for different aging experiments. Residual spectra were calculated and averaged for the period of sampling the second PTFE filter for each experiment and then averaged over the experiments of the same aging category including both wood burning and pellet burning. Error bars show the range of variation for each fragment within each category.
The wall-loss-corrected concentrations of some mass fragments decrease by more than 50 % (Fig. 5b). Among the diminishing fragments, , attributed to lignin-like compounds, and , related to levoglucosan, decrease the most. Several non-oxygenated fragments with the CxHy+ formula, related to hydrocarbons and aromatics, are also lost with aging (e.g., , , , , and ; Table 2). The extent of the decay for these species is significantly higher than what can be attributed to particle wall loss rate uncertainties shown in Table 2. There are also fragments (with single oxygen like C3H4O+ or related to aromatics like ) that increase in concentration briefly after the start of aging (around 30 min) and fall below their initial concentration with continued aging. These fragments might indicate the moderately oxygenated intermediate species or those that partition to the particle phase with increased OA loading but with a decay that becomes more apparent with continued aging. The FTIR signatures of levoglucosan and lignin-like compounds also decrease significantly with the UV aging (Table 2), causing inverted peaks in the residual spectra (Fig. 7b). Locally inverted aCH peaks in the FTIR residual spectra of experiment 4 (Fig. 7b) support the hypothesis that hydrocarbons are lost with aging. The detectable decay of the aforementioned mentioned species is captured in both UV experiments 4 and 5 by both methods (use of the AMS and FTIR).
Gas–particle partitioning, heterogeneous oxidation reactions, and photolysis can play a role in the diminution of the mentioned species in the particle phase. For semivolatile compounds like levoglucosan, particle-phase depletion can result from gas-phase losses and subsequent mass transfer from particles to gas. Bertrand et al. (2018b) proposed the prominent role of gas–particle partitioning and the subsequent vapor loss to the chamber walls in the depletion of levoglucosan in the particle phase. Comparing the trends of AMS levoglucosan-related fragments (particle phase) between reference and UV experiments of this work, however, the reaction of levoglucosan with the hydroxyl radical appears to be the dominant loss pathway. In contrast, the fast diminution of AMS fragments from less volatile species (e.g., heavy lignin-like compounds) highlights the role of particle-phase photolysis and heterogeneous reactions. In addition, a similar fast depletion of fragments in the particle phase merely due to heterogeneous reactions with the hydroxyl radical was also reported by George et al. (2008).
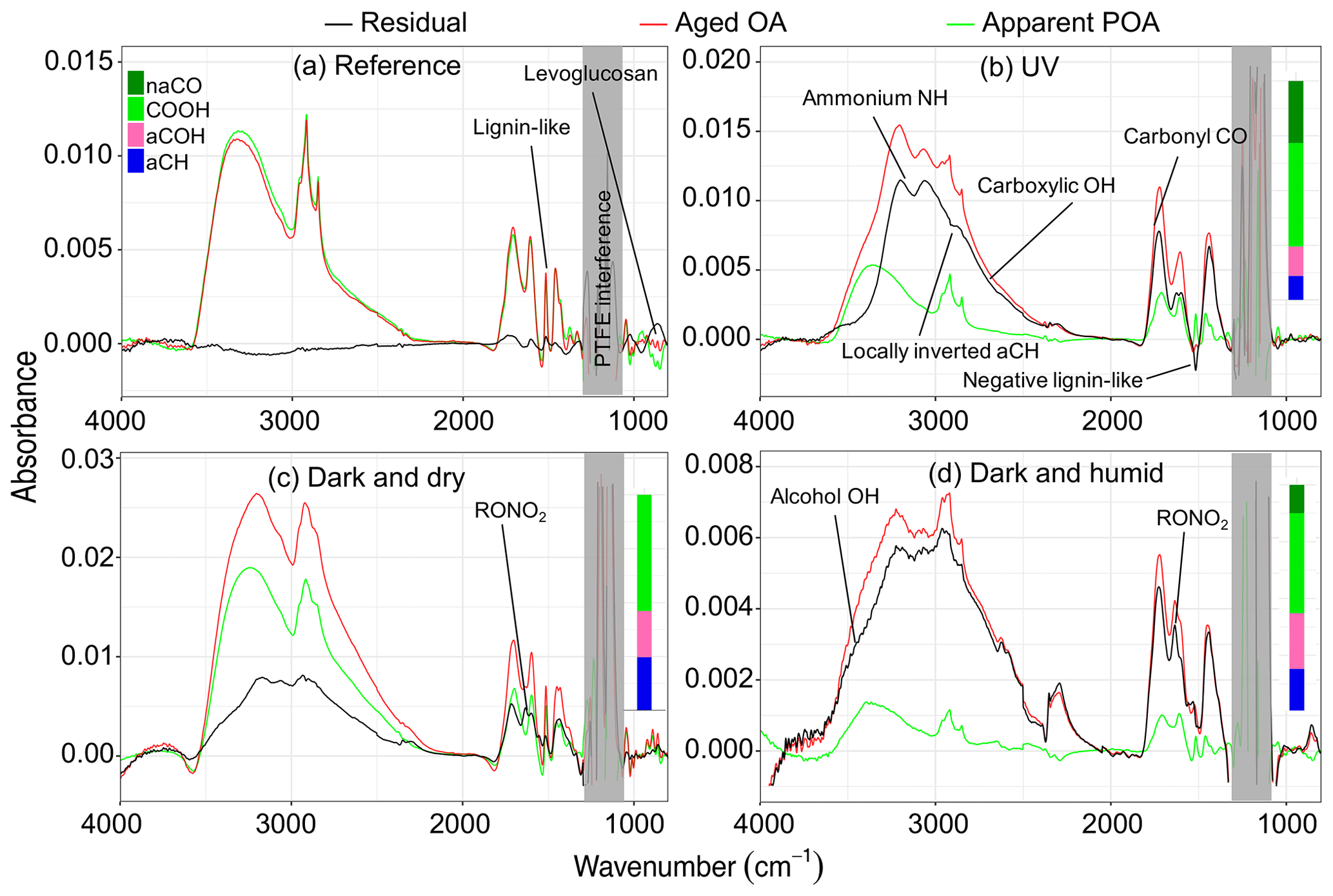
Figure 7FTIR spectra of aged aerosols (red), their apparent primary fraction (green), and the residual aerosols (black) for different aging experiments. Panels (a)–(d) represent Exp. 1 (wood burning), 4 (wood burning), 7 (pellet burning), and 8 (wood burning), respectively. Normalized functional group compositions from peak fitting are shown for the residual spectra. Ammonium-subtracted spectra for panels (b) and (c) are shown in Fig. S6.
The FTIR signature used for identification of lignin-like compounds in this work (sharp peak at 1515 cm−1) has also been observed in the FTIR spectra of the humic-like substance (HULIS) isolated from aqueous extracts of atmospheric aerosols (Graber and Rudich, 2006). As discussed by Yazdani et al. (2021), this peak can also be produced by small and relatively volatile molecules with a similar aromatic ring substitution to lignin (e.g., methoxyphenols and substituted syringols). The and fragments in the AMS spectra might not be exclusive to non-volatile lignin and can be produced by smaller, more volatile molecules in bbOA resulting from lignin pyrolysis. There are, however, two observations that suggest the lignin-related fragments in this work can be attributed to compounds with a lower volatility than levoglucosan that exist predominantly in the particle phase under the conditions of the experiments (e.g., OA loading and temperature). First, the thermodenuder data suggest the lower volatility of compounds producing and compared to species producing (Sect. S2). Second, the lower loss rate of in the absence of oxidants compared to other fragments including (Sect. S2) suggests the lower volatility of the corresponding species in that their concentration is mainly affected by particle wall losses compared to losses stemming from gas–particle partitioning for levoglucosan.
Between the two marker fragments attributed to lignin-like compounds, and , the former is a heavier and less abundant fragment (Li et al., 2012) and appears to be more stable with regard to aging with its loss rate being one-third of that of the latter (Sect. S7). Because of this difference in decay rates, the ratio of to can potentially be used as an aging clock of atmospheric bbOA and warrants further study.
In these UV aging experiments, transformation of 10 %–17 % of total bbPOA mass is estimated from the negative AMS residuals. The bbPOA transformation is estimated to be around 40 % more intense for experiment 4 compared to experiment 5, consistent with the higher average OH concentration for the former (Table 1). The values of bbPOA transformation obtained with the AMS should be considered as a lower bound estimate for the chemical processing of primary bbPOA. This is because the increase in the concentration of some mass fragments owing to SOA condensation might outweigh the decay for the same fragments generated by bbPOA oxidation or evaporation. This can especially render the loss of small oxygenated fragments that are common to several species (Yazdani et al., 2022) undetectable by the method. For instance in experiment 4, levoglucosan-marker fragments, and , decrease the most with aging. Unlike these fragments, CHO+, which is also produced in comparable amounts by levoglucosan fragmentation (Schneider et al., 2006), increases in concentration during the course of aging due to SOA condensation. By quantifying the inverted aliphatic CH, lignin, and levoglucosan peaks in the FTIR residual spectrum of this experiment (Fig. 7b) (representing the molecular structure of lignin by syringaldehyde), we estimate around 35 % (against 15 % estimated with the AMS) of the POA mass to transform with aging.
We note that the change in composition with aging is not necessarily accompanied by a change in overall OA mass. For instance, in experiment 4, the wall-loss-corrected OA mass concentration reaches a constant level about 1 h after the start of aging, while f44–f43 ratios for the total and residual OAs gradually increase and the particle-wall-loss-corrected organic carbon (OC) gradually decreases until the end of experiment (Sect. S8). The net loss of carbon from the particle phase and increased oxidation of the OA without an increase in its mass suggests that heterogeneous OH reactions and fragmentation are important mechanisms.
3.5 Aging under dark conditions
Emissions aged in dark conditions in both dry and humid conditions (experiments 6 and 8; Fig. 5c and d), and OA enhancement is similar to that reported by Kodros et al. (2020). We observe the highest increase in the fragment abundance. The average of the difference in residual spectra of wood and pellet burning is reported in Fig. 6c and d (differences among aging trajectories between fuel types are not substantial, as previously stated). In the residual FTIR spectra, higher abundance of the aCOH group relative to UV experiments is observed (Fig. 7c–d). The formation of organic nitrates is also confirmed in the residual FTIR spectra (Fig. 7c–d) as well as with the AMS. When emissions are aged in dry conditions, CHO+ and C2H3O+ are among the most prominent fragments in the residual spectra (besides ) (Fig. 6c). This observation suggests the abundance of non-acid oxygenated species in the SOA formed by the nitrate radical. Some heavier mass fragments with a single oxygen atom (e.g., C3H3O+ and C3H5O+) are also observed in the average residual spectrum, supporting this hypothesis (Fig. 6c). Apart from the oxygenated mass fragments, high abundances of light CxHy+ fragments (, and ) are observed in the residual AMS spectrum. When aged in humid conditions, the average residual AMS mass spectrum (Fig. 7d) is fairly similar to that of UV experiments (Fig. 7b).
Similar to the UV experiments, several mass fragments decrease more than what can be attributed to wall losses, but the mass fragments related to levoglucosan fragmentation do not diminish beyond that observed for the reference case. Under dry conditions, the majority of the diminishing fragments are in the form or those related to lignin-like compounds (Fig. 5d). Under humid conditions, we observe that lignin-related fragments decay more prominently in the humid compared to the dry conditions (Table 2). The decreasing trend for the levoglucosan-related fragments starts before the initiation of aging (injection of ozone into the chamber), and it is not affected by it (Fig. 5d). This observation suggests that the dark aging is not responsible for the decay of levoglucosan. In this case, other factors such as acid-catalyzed levoglucosan reactions in the aqueous phase (Holmes and Petrucci, 2006) or a more efficient removal of gas-phase levoglucosan by chamber walls might play a role. Similar diminution of hydrocarbons, anhydrosugars, and lignin-like compounds is observed in the FTIR spectra of experiment 8 (Table 2 and Fig. 5d). Nighttime aging under dry conditions of our experiments results in a negligible bbPOA transformation, close to what is observed in the reference experiments (approximate 2 %). Nighttime aging in humid conditions of our experiments, however, results in a slightly higher bbPOA transformation (up to 5 %).
3.6 Gas-phase wall losses
In this work, the changes in apparent mass concentrations reported are not corrected for gas-phase losses of semivolatile organic compounds (SVOCs) to the chamber walls. SVOCs in bbPOA evaporating during the course of the experiment can be adsorbed and absorbed to the walls (Huang et al., 2018), increasing the loss rate of molecules (in addition to degradation due to gas-phase reactions) from the system and driving the partitioning toward the gas phase compared to the scenario where wall losses are not present (Loza et al., 2010). The reported magnitude of these wall losses and effects on organic aerosol evolution in chamber experiments varies substantially according to different studies (Grosjean, 1985; Huang et al., 2018; Bian et al., 2015; Jiang et al., 2021; Ernle et al., 2023; Krechmer et al., 2020; Zhang et al., 2014; Pagonis et al., 2017; Pratap et al., 2020). However, unlike for particle wall losses, a generalizable result and correction remains elusive to date.
Given the above, Florou (2023) carried out two experiments (in the same chamber facility used in our study) where BB emissions from a pellet stove were allowed to reside in the chamber for 12 h (in the dark, no active chemistry). After particle corrections were applied, no significant changes in mass, AMS spectrum, and O:C ratio were observed in the BB aerosol, suggesting that vapor wall losses are not a dominating factor. However, in our present study, we also identify hydrocarbon fragments and aliphatic CH bonds that decrease over time, though the responsible compounds remain unidentified. The gas-phase wall loss rates are reported to be greater for those substances where appreciable fraction of mass resides in the vapor phase (Ye et al., 2016), but we do not anticipate the losses to be as significant to change the conclusions of our study. The results of this work establish the potential importance of bbPOA aging and provide a basis for estimation of rate constants in future studies, while identifying more stable markers that may be suitable for use in field measurements.
In this work, we characterized the evolution of bbPOA with aging using the AMS and FTIR. The similarity of AMS and FTIR spectra of oxidized bbOA in the chamber – especially when aged with UV or in dark and humid conditions – to those of oxidized atmospheric OA contextualizes the extent of aging in these chamber experiments and underscores the relevance of these conclusions to aging under ambient conditions. Similarity was assessed by f43:f44 plots, PCA, and functional group composition; e.g., the high concentration of carboxylic acids that was observed in the UV-aged chamber bbOA is similar to what is observed in atmospheric bbOA samples. The aged fraction of bbOA in the chamber became more oxidized with continued aging. In certain cases, oxidation happened without a substantial increase in the bbOA mass concentration (and with a decrease in the OC mass concentration), highlighting the role of heterogeneous reactions and photolysis.
Based on AMS fragments exhibiting a net negative change, we conservatively estimated the transformation (due to oxidation and evaporation) of bbPOA mass to be 2 %–5 % under nighttime aging conditions and up to 17 % of bbPOA with less than a day of atmospheric aging with UV. This latter amount was calculated to be twice as high with FTIR, based on absorptivities for aliphatic CH functional groups and signature peaks of levoglucosan and syringaldehyde (representing lignin-like compounds). The absorptivity-based approach considers mass transformations on a molecular rather than molecular fragment basis and may be more relevant according to the representation of OA used in numerical models.
Biomass burning markers such as lignin-like compounds and anhydrosugars, as well as hydrocarbons, were among the bbPOA compounds that degraded the most in the particle phase with aging (from 10 % to 50 % of original mass concentrations, up to 6 times greater than in reference experiments) according to both the AMS and FTIR. Unlike hydrocarbons and lignin-like compounds that degraded during both day- and nighttime oxidation, anhydrosugars were observed to degrade effectively only during the daytime oxidation. In addition, high humidity was observed to increase the loss rate of anhydrosugars in the particle phase. Since the degradation occurs for semi-volatile (levoglucosan) and less volatile (heavy lignin-like) compounds, gas–particle partitioning and heterogeneous reactions likely play a role. Different biomass burning-related fragments in the AMS spectra were observed to have different decay rates. These rate differences can potentially be used to identify the age of atmospheric bbOA, as well as the relative contribution of different aging mechanisms, provided that fragment ratios of anhydrosugar and lignin-like compound in various emission sources can be better constrained.
The infrared spectra of samples collected for this work can be found at https://doi.org/10.5281/zenodo.7982350 (Yazdani et al., 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-7461-2023-supplement.
AN, SNP, ST, and AY conceived the project. AY prepared the filter sampling setup. JKK, MP, KF, MM, and SS performed the chamber experiments. KF processed AMS row signals. AY processed FTIR spectra, performed the data analysis, and wrote the manuscript. ST, AN, and SNP provided regular input on the analysis and further editing of the manuscript. ST and AN provided overall supervision of the project.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work was supported by the project PyroTRACH (ERC-2016-COG) funded by H2020-EU.1.1. – Excellent Science – European Research Council (ERC), project ID 726165, and funding from the Swiss National Science Foundation (200021_172923). Spyros N. Pandis also acknowledges funding from the CHEVOPIN project of the Hellenic Foundation for Research & Innovation.
This research has been supported by the European Research Council, H2020 European Research Council (PyroTRACH (grant no. 726165)), the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (grant no. 200021_172923), and the Hellenic Foundation for Research and Innovation (CHEVOPIN grant).
This paper was edited by Qi Chen and reviewed by two anonymous referees.
Abdi, H. and Williams, L. J.: Principal Component Analysis, Wiley Interdiscip. Rev. Comput. Stat., 2, 433–459, https://doi.org/10.1002/wics.101, 2010. a
Aiken, A. C., DeCarlo, P. F., and Jimenez, J. L.: Elemental Analysis of Organic Species with Electron Ionization High-Resolution Mass Spectrometry, Anal. Chem., 79, 8350–8358, https://doi.org/10.1021/ac071150w, 2007. a
Alves, C., Gonçalves, C., Fernandes, A. P., Tarelho, L., and Pio, C.: Fireplace and Woodstove Fine Particle Emissions from Combustion of Western Mediterranean Wood Types, Atmos. Res., 101, 692–700, https://doi.org/10.1016/j.atmosres.2011.04.015, 2011. a
An, W. J., Pathak, R. K., Lee, B.-H., and Pandis, S. N.: Aerosol volatility measurement using an improved thermodenuder: Application to secondary organic aerosol, J. Aerosol Sci., 38, 305–314, https://doi.org/10.1016/j.jaerosci.2006.12.002, 2007. a
Bäfver, L. S., Leckner, B., Tullin, C., and Berntsen, M.: Particle Emissions from Pellets Stoves and Modern and Old-Type Wood Stoves, Biomass Bioenerg., 35, 3648–3655, https://doi.org/10.1016/j.biombioe.2011.05.027, 2011. a
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012. a
Bateman, A. P., Nizkorodov, S. A., Laskin, J., and Laskin, A.: Photolytic Processing of Secondary Organic Aerosols Dissolved in Cloud Droplets, Phys. Chem. Chem. Phys., 13, 12199–12212, https://doi.org/10.1039/C1CP20526A, 2011. a
Bertrand, A., Stefenelli, G., Bruns, E. A., Pieber, S. M., Temime-Roussel, B., Slowik, J. G., Prévôt, A. S. H., Wortham, H., El Haddad, I., and Marchand, N.: Primary Emissions and Secondary Aerosol Production Potential from Woodstoves for Residential Heating: Influence of the Stove Technology and Combustion Efficiency, Atmos. Environ., 169, 65–79, https://doi.org/10.1016/j.atmosenv.2017.09.005, 2017. a, b, c
Bertrand, A., Stefenelli, G., Jen, C. N., Pieber, S. M., Bruns, E. A., Ni, H., Temime-Roussel, B., Slowik, J. G., Goldstein, A. H., El Haddad, I., Baltensperger, U., Prévôt, A. S. H., Wortham, H., and Marchand, N.: Evolution of the chemical fingerprint of biomass burning organic aerosol during aging, Atmos. Chem. Phys., 18, 7607–7624, https://doi.org/10.5194/acp-18-7607-2018, 2018a. a, b, c, d
Bertrand, A., Stefenelli, G., Pieber, S. M., Bruns, E. A., Temime-Roussel, B., Slowik, J. G., Wortham, H., Prévôt, A. S. H., El Haddad, I., and Marchand, N.: Influence of the vapor wall loss on the degradation rate constants in chamber experiments of levoglucosan and other biomass burning markers, Atmos. Chem. Phys., 18, 10915–10930, https://doi.org/10.5194/acp-18-10915-2018, 2018b. a, b
Bian, Q., May, A. A., Kreidenweis, S. M., and Pierce, J. R.: Investigation of particle and vapor wall-loss effects on controlled wood-smoke smog-chamber experiments, Atmos. Chem. Phys., 15, 11027–11045, https://doi.org/10.5194/acp-15-11027-2015, 2015. a
Bond, T. C., Doherty, S. J., Fahey, D. W., Forster, P. M., Berntsen, T., DeAngelo, B. J., Flanner, M. G., Ghan, S., Kärcher, B., Koch, D., Kinne, S., Kondo, Y., Quinn, P. K., Sarofim, M. C., Schultz, M. G., Schulz, M., Venkataraman, C., Zhang, H., Zhang, S., Bellouin, N., Guttikunda, S. K., Hopke, P. K., Jacobson, M. Z., Kaiser, J. W., Klimont, Z., Lohmann, U., Schwarz, J. P., Shindell, D., Storelvmo, T., Warren, S. G., and Zender, C. S.: Bounding the Role of Black Carbon in the Climate System: A Scientific Assessment, J. Geophys. Res.-Atmos., 118, 5380–5552, https://doi.org/10.1002/jgrd.50171, 2013. a
Boris, A. J., Takahama, S., Weakley, A. T., Debus, B. M., Fredrickson, C. D., Esparza-Sanchez, M., Burki, C., Reggente, M., Shaw, S. L., Edgerton, E. S., and Dillner, A. M.: Quantifying organic matter and functional groups in particulate matter filter samples from the southeastern United States – Part 1: Methods, Atmos. Meas. Tech., 12, 5391–5415, https://doi.org/10.5194/amt-12-5391-2019, 2019. a
Bruns, E. A., Krapf, M., Orasche, J., Huang, Y., Zimmermann, R., Drinovec, L., Močnik, G., El-Haddad, I., Slowik, J. G., Dommen, J., Baltensperger, U., and Prévôt, A. S. H.: Characterization of primary and secondary wood combustion products generated under different burner loads, Atmos. Chem. Phys., 15, 2825–2841, https://doi.org/10.5194/acp-15-2825-2015, 2015. a
Burnett, R., Chen, H., Szyszkowicz, M., Fann, N., Hubbell, B., Pope, C. A., Apte, J. S., Brauer, M., Cohen, A., Weichenthal, S., Coggins, J., Di, Q., Brunekreef, B., Frostad, J., Lim, S. S., Kan, H., Walker, K. D., Thurston, G. D., Hayes, R. B., Lim, C. C., Turner, M. C., Jerrett, M., Krewski, D., Gapstur, S. M., Diver, W. R., Ostro, B., Goldberg, D., Crouse, D. L., Martin, R. V., Peters, P., Pinault, L., Tjepkema, M., van Donkelaar, A., Villeneuve, P. J., Miller, A. B., Yin, P., Zhou, M., Wang, L., Janssen, N. A. H., Marra, M., Atkinson, R. W., Tsang, H., Thach, T. Q., Cannon, J. B., Allen, R. T., Hart, J. E., Laden, F., Cesaroni, G., Forastiere, F., Weinmayr, G., Jaensch, A., Nagel, G., Concin, H., and Spadaro, J. V.: Global Estimates of Mortality Associated with Long-Term Exposure to Outdoor Fine Particulate Matter, P. Natl. Acad. Ssi. USA, 115, 9592–9597, https://doi.org/10.1073/pnas.1803222115, 2018. a
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007. a
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015. a
Chen, Q., Liu, Y., Donahue, N. M., Shilling, J. E., and Martin, S. T.: Particle-Phase Chemistry of Secondary Organic Material: Modeled Compared to Measured O:C and H:C Elemental Ratios Provide Constraints, Environ. Sci. Technol., 45, 4763–4770, https://doi.org/10.1021/es104398s, 2011. a
Chhabra, P. S., Ng, N. L., Canagaratna, M. R., Corrigan, A. L., Russell, L. M., Worsnop, D. R., Flagan, R. C., and Seinfeld, J. H.: Elemental composition and oxidation of chamber organic aerosol, Atmos. Chem. Phys., 11, 8827–8845, https://doi.org/10.5194/acp-11-8827-2011, 2011. a
Coury, C. and Dillner, A. M.: A Method to Quantify Organic Functional Groups and Inorganic Compounds in Ambient Aerosols Using Attenuated Total Reflectance FTIR Spectroscopy and Multivariate Chemometric Techniques, Atmos. Environ., 42, 5923–5932, https://doi.org/10.1016/j.atmosenv.2008.03.026, 2008. a
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006. a
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K.-H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of Biogenic Secondary Organic Aerosol via Gas-Phase OH Radical Reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012. a
Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R.: A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS)—Instrument Description and First Field Deployment, Aerosol Sci. Technol., 39, 637–658, https://doi.org/10.1080/02786820500182040, 2005. a
Ernle, L., Ringsdorf, M. A., and Williams, J.: Influence of ozone and humidity on PTR-MS and GC-MS VOC measurements with and without a Na2S2O3 ozone scrubber, Atmos. Meas. Tech., 16, 1179–1194, https://doi.org/10.5194/amt-16-1179-2023, 2023. a
Faber, P., Drewnick, F., Bierl, R., and Borrmann, S.: Complementary Online Aerosol Mass Spectrometry and Offline FT-IR Spectroscopy Measurements: Prospects and Challenges for the Analysis of Anthropogenic Aerosol Particle Emissions, Atmos. Environ., 166, 92–98, https://doi.org/10.1016/j.atmosenv.2017.07.014, 2017. a
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an Aerosol Mass Spectrometer to Organonitrates and Organosulfates and Implications for Atmospheric Chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010. a
Fine, P. M., Cass, G. R., and Simoneit, B. R. T.: Chemical Characterization of Fine Particle Emissions from the Fireplace Combustion of Woods Grown in the Southern United States, Environ. Sci. Technol., 36, 1442–1451, https://doi.org/10.1021/es0108988, 2002. a, b
Florou, K., Papanastasiou, D. K., Pikridas, M., Kaltsonoudis, C., Louvaris, E., Gkatzelis, G. I., Patoulias, D., Mihalopoulos, N., and Pandis, S. N.: The contribution of wood burning and other pollution sources to wintertime organic aerosol levels in two Greek cities, Atmos. Chem. Phys., 17, 3145–3163, https://doi.org/10.5194/acp-17-3145-2017, 2017. a, b, c, d, e
Floroum, K., Kodros, J. K., Paglione, M., Jorga, S., Squizzato, S., Masiol, M., Uruci, P., Nenes, A., and Pandis, S. N.: Characterization and dark oxidation of the emissions of a pellet stove, Environ. Sci.-Atmos., in review, 2023. a
Ford, B., Martin, M. V., Zelasky, S. E., Fischer, E. V., Anenberg, S. C., Heald, C. L., and Pierce, J. R.: Future Fire Impacts on Smoke Concentrations, Visibility, and Health in the Contiguous United States, GeoHealth, 2, 229–247, https://doi.org/10.1029/2018GH000144, 2018. a
George, I. J., Slowik, J., and Abbatt, J. P. D.: Chemical Aging of Ambient Organic Aerosol from Heterogeneous Reaction with Hydroxyl Radicals, Geophys. Res. Lett., 35, L13811, https://doi.org/10.1029/2008GL033884, 2008. a, b
George, K. M., Ruthenburg, T. C., Smith, J., Yu, L., Zhang, Q., Anastasio, C., and Dillner, A. M.: FT-IR Quantification of the Carbonyl Functional Group in Aqueous-Phase Secondary Organic Aerosol from Phenols, Atmos. Environ., 100, 230–237, https://doi.org/10.1016/j.atmosenv.2014.11.011, 2015. a
Graber, E. R. and Rudich, Y.: Atmospheric HULIS: How humic-like are they? A comprehensive and critical review, Atmos. Chem. Phys., 6, 729–753, https://doi.org/10.5194/acp-6-729-2006, 2006. a
Grieshop, A. P., Donahue, N. M., and Robinson, A. L.: Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 2: analysis of aerosol mass spectrometer data, Atmos. Chem. Phys., 9, 2227–2240, https://doi.org/10.5194/acp-9-2227-2009, 2009. a, b, c
Grosjean, D.: Wall loss of gaseous pollutants in outdoor Teflon chambers, Environ. Sci. Technol., 19, 1059–1065, https://doi.org/10.1021/es00141a006, 1985. a
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009. a, b, c
Hearn, J. D., Lovett, A. J., and Smith, G. D.: Ozonolysis of Oleic Acid Particles: Evidence for a Surface Reaction and Secondary Reactions Involving Criegee Intermediates, Phys. Chem. Chem. Phys., 7, 501–511, https://doi.org/10.1039/B414472D, 2005. a
Hennigan, C. J., Sullivan, A. P., Collett, J. L., and Robinson, A. L.: Levoglucosan Stability in Biomass Burning Particles Exposed to Hydroxyl Radicals, Geophys. Res. Lett., 37, L09806, https://doi.org/10.1029/2010GL043088, 2010. a
Hennigan, C. J., Miracolo, M. A., Engelhart, G. J., May, A. A., Presto, A. A., Lee, T., Sullivan, A. P., McMeeking, G. R., Coe, H., Wold, C. E., Hao, W.-M., Gilman, J. B., Kuster, W. C., de Gouw, J., Schichtel, B. A., Collett Jr., J. L., Kreidenweis, S. M., and Robinson, A. L.: Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber, Atmos. Chem. Phys., 11, 7669–7686, https://doi.org/10.5194/acp-11-7669-2011, 2011. a, b
Henry, K. M. and Donahue, N. M.: Photochemical Aging of Alpha-Pinene Secondary Organic Aerosol: Effects of OH Radical Sources and Photolysis, J. Phys. Chem. A, 116, 5932–5940, https://doi.org/10.1021/jp210288s, 2012. a, b
Holmes, B. J. and Petrucci, G. A.: Water-Soluble Oligomer Formation from Acid-Catalyzed Reactions of Levoglucosan in Proxies of Atmospheric Aqueous Aerosols, Environ. Sci. Technol., 40, 4983–4989, https://doi.org/10.1021/es060646c, 2006. a
Hotelling, H.: Analysis of a Complex of Statistical Variables into Principal Components, J. Educ. Psychol., 24, 417–441, https://doi.org/10.1037/h0071325, 1933. a
Huang, Y., Zhao, R., Charan, S. M., Kenseth, C. M., Zhang, X., and Seinfeld, J. H.: Unified Theory of Vapor–Wall Mass Transport in Teflon-Walled Environmental Chambers, Environ. Sci. Technol., 52, 2134–2142, https://doi.org/10.1021/acs.est.7b05575, 2018. a, b
Hung, H.-M., Katrib, Y., and Martin, S. T.: Products and Mechanisms of the Reaction of Oleic Acid with Ozone and Nitrate Radical, The J. Phys. Chem. A, 109, 4517–4530, https://doi.org/10.1021/jp0500900, 2005. a
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles, Aerosol Sci. Technol., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000. a
Jiang, J., El Haddad, I., Aksoyoglu, S., Stefenelli, G., Bertrand, A., Marchand, N., Canonaco, F., Petit, J.-E., Favez, O., Gilardoni, S., Baltensperger, U., and Prévôt, A. S. H.: Influence of biomass burning vapor wall loss correction on modeling organic aerosols in Europe by CAMx v6.50, Geosci. Model Dev., 14, 1681–1697, https://doi.org/10.5194/gmd-14-1681-2021, 2021. a
Johansson, L. S., Leckner, B., Gustavsson, L., Cooper, D., Tullin, C., and Potter, A.: Emission Characteristics of Modern and Old-Type Residential Boilers Fired with Wood Logs and Wood Pellets, Atmos. Environ., 38, 4183–4195, https://doi.org/10.1016/j.atmosenv.2004.04.020, 2004. a
John Wiley & Sons, Inc.: SpectraBase, https://spectrabase.com/, last access: 26 August, 2022. a
Jorga, S. D., Florou, K., Kaltsonoudis, C., Kodros, J. K., Vasilakopoulou, C., Cirtog, M., Fouqueau, A., Picquet-Varrault, B., Nenes, A., and Pandis, S. N.: Nighttime chemistry of biomass burning emissions in urban areas: A dual mobile chamber study, Atmos. Chem. Phys., 21, 15337–15349, https://doi.org/10.5194/acp-21-15337-2021, 2021. a
Kalberer, M., Sax, M., and Samburova, V.: Molecular Size Evolution of Oligomers in Organic Aerosols Collected in Urban Atmospheres and Generated in a Smog Chamber, Environ. Sci. Technol., 40, 5917–5922, https://doi.org/10.1021/es0525760, 2006. a
Kaltsonoudis, C., Kostenidou, E., Louvaris, E., Psichoudaki, M., Tsiligiannis, E., Florou, K., Liangou, A., and Pandis, S. N.: Characterization of fresh and aged organic aerosol emissions from meat charbroiling, Atmos. Chem. Phys., 17, 7143–7155, https://doi.org/10.5194/acp-17-7143-2017, 2017. a
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005. a
Kodros, J. K., Papanastasiou, D. K., Paglione, M., Masiol, M., Squizzato, S., Florou, K., Skyllakou, K., Kaltsonoudis, C., Nenes, A., and Pandis, S. N.: Rapid Dark Aging of Biomass Burning as an Overlooked Source of Oxidized Organic Aerosol, P. Natl. Acad. Sci. USA, 117, 33028–33033, https://doi.org/10.1073/pnas.2010365117, 2020. a, b, c, d, e
Kodros, J. K., Kaltsonoudis, C., Paglione, M., Florou, K., Jorga, S., Vasilakopoulou, C., Cirtog, M., Cazaunau, M., Picquet-Varrault, B., Nenes, A., and N. Pandis, S.: Secondary aerosol formation during the dark oxidation of residential biomass burning emissions, Environ. Sci.-Atmos., 2, 12211236, https://doi.org/10.1039/D2EA00031H, 2022. a, b
Kostenidou, E., Florou, K., Kaltsonoudis, C., Tsiflikiotou, M., Vratolis, S., Eleftheriadis, K., and Pandis, S. N.: Sources and chemical characterization of organic aerosol during the summer in the eastern Mediterranean, Atmos. Chem. Phys., 15, 11355–11371, https://doi.org/10.5194/acp-15-11355-2015, 2015. a, b, c, d, e, f
Krechmer, J. E., Day, D. A., and Jimenez, J. L.: Always Lost but Never Forgotten: Gas-Phase Wall Losses Are Important in All Teflon Environmental Chambers, Environ. Sci. Technol., 54, 12890–12897, https://doi.org/10.1021/acs.est.0c03381, 2020. a
Kumar, N. K., Corbin, J. C., Bruns, E. A., Massabó, D., Slowik, J. G., Drinovec, L., Močnik, G., Prati, P., Vlachou, A., Baltensperger, U., Gysel, M., El-Haddad, I., and Prévôt, A. S. H.: Production of particulate brown carbon during atmospheric aging of residential wood-burning emissions, Atmos. Chem. Phys., 18, 17843–17861, https://doi.org/10.5194/acp-18-17843-2018, 2018. a
Lambe, A. T., Onasch, T. B., Croasdale, D. R., Wright, J. P., Martin, A. T., Franklin, J. P., Massoli, P., Kroll, J. H., Canagaratna, M. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Transitions from Functionalization to Fragmentation Reactions of Laboratory Secondary Organic Aerosol (SOA) Generated from the OH Oxidation of Alkane Precursors, Environ. Sci. Technol., 46, 5430–5437, https://doi.org/10.1021/es300274t, 2012. a
Lanz, V. A., Prévôt, A. S. H., Alfarra, M. R., Weimer, S., Mohr, C., DeCarlo, P. F., Gianini, M. F. D., Hueglin, C., Schneider, J., Favez, O., D'Anna, B., George, C., and Baltensperger, U.: Characterization of aerosol chemical composition with aerosol mass spectrometry in Central Europe: an overview, Atmos. Chem. Phys., 10, 10453–10471, https://doi.org/10.5194/acp-10-10453-2010, 2010. a
Li, Y. J., Yeung, J. W. T., Leung, T. P. I., Lau, A. P. S., and Chan, C. K.: Characterization of Organic Particles from Incense Burning Using an Aerodyne High-Resolution Time-of-Flight Aerosol Mass Spectrometer, Aerosol Sci. Tech., 46, 654–665, https://doi.org/10.1080/02786826.2011.653017, 2012. a, b
Louvaris, E. E., Karnezi, E., Kostenidou, E., Kaltsonoudis, C., and Pandis, S. N.: Estimation of the volatility distribution of organic aerosol combining thermodenuder and isothermal dilution measurements, Atmos. Meas. Tech., 10, 3909–3918, https://doi.org/10.5194/amt-10-3909-2017, 2017. a
Loza, C. L., Chan, A. W. H., Galloway, M. M., Keutsch, F. N., Flagan, R. C., and Seinfeld, J. H.: Characterization of Vapor Wall Loss in Laboratory Chambers, Environ. Sci. Technol., 44, 5074–5078, https://doi.org/10.1021/es100727v, 2010. a
Mabato, B. R. G., Lyu, Y., Ji, Y., Li, Y. J., Huang, D. D., Li, X., Nah, T., Lam, C. H., and Chan, C. K.: Aqueous secondary organic aerosol formation from the direct photosensitized oxidation of vanillin in the absence and presence of ammonium nitrate, Atmos. Chem. Phys., 22, 273–293, https://doi.org/10.5194/acp-22-273-2022, 2022. a
McFiggans, G., Coe, H., Burgess, R., Allan, J., Cubison, M., Alfarra, M. R., Saunders, R., Saiz-Lopez, A., Plane, J. M. C., Wevill, D., Carpenter, L., Rickard, A. R., and Monks, P. S.: Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine, Atmos. Chem. Phys., 4, 701–713, https://doi.org/10.5194/acp-4-701-2004, 2004. a
Nah, T., Kessler, S. H., Daumit, K. E., Kroll, J. H., Leone, S. R., and Wilson, K. R.: Influence of Molecular Structure and Chemical Functionality on the Heterogeneous OH-Initiated Oxidation of Unsaturated Organic Particles, J. Phys. Chem. A, 118, 4106–4119, https://doi.org/10.1021/jp502666g, 2014. a
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011. a
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017. a
Pandey, K. K.: A Study of Chemical Structure of Soft and Hardwood and Wood Polymers by FTIR Spectroscopy, J. Appl. Polymer Sci.e, 71, 1969–1975, https://doi.org/10.1002/(SICI)1097-4628(19990321)71:12<1969::AID-APP6>3.0.CO;2-D, 1999. a
Parks, D. A., Raj, K. V., Berry, C. A., Weakley, A. T., Griffiths, P. R., and Miller, A. L.: Towards a Field-Portable Real-Time Organic and Elemental Carbon Monitor, Mining, Metall. Explor., 36, 765–772, https://doi.org/10.1007/s42461-019-0064-8, 2019. a
Pathak, R. K., Stanier, C. O., Donahue, N. M., and Pandis, S. N.: Ozonolysis of Alpha-Pinene at Atmospherically Relevant Concentrations: Temperature Dependence of Aerosol Mass Fractions (Yields), J. Geophys. Res.-Atmos., 112, D3, https://doi.org/10.1029/2006JD007436, 2007. a
Pope, C. A., Ezzati, M., and Dockery, D. W.: Fine-Particulate Air Pollution and Life Expectancy in the United States, N. Engl. J. Med., 360, 376–386, https://doi.org/10.1056/NEJMsa0805646, 2009. a
Pratap, V., Kiran, S. A., Bian, Q., Pierce, J. R., Hopke, P. K., and Nakao, S.: Observation of Vapor Wall Deposition in a Smog Chamber Using Size Evolution of Pure Organic Particles, Aerosol Air Qual. Res., 20, 2705–2714, https://doi.org/10.4209/aaqr.2020.05.0268, 2020. a
Puxbaum, H., Caseiro, A., Sánchez-Ochoa, A., Kasper-Giebl, A., Claeys, M., Gelencsér, A., Legrand, M., Preunkert, S., and Pio, C.: Levoglucosan Levels at Background Sites in Europe for Assessing the Impact of Biomass Combustion on the European Aerosol Background, J. Geophys. Res.-Atmos., 112, D23S05, https://doi.org/10.1029/2006JD008114, 2007. a
Qi, L., Chen, M., Stefenelli, G., Pospisilova, V., Tong, Y., Bertrand, A., Hueglin, C., Ge, X., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 2: Biomass burning influences in winter, Atmos. Chem. Phys., 19, 8037–8062, https://doi.org/10.5194/acp-19-8037-2019, 2019. a
Reggente, M., Dillner, A. M., and Takahama, S.: Predicting ambient aerosol thermal–optical reflectance (TOR) measurements from infrared spectra: extending the predictions to different years and different sites, Atmos. Meas. Tech., 9, 441–454, https://doi.org/10.5194/amt-9-441-2016, 2016. a
Reggente, M., Dillner, A. M., and Takahama, S.: Analysis of functional groups in atmospheric aerosols by infrared spectroscopy: systematic intercomparison of calibration methods for US measurement network samples, Atmos. Meas. Tech., 12, 2287–2312, https://doi.org/10.5194/amt-12-2287-2019, 2019. a, b
Robinson, A. L., Donahue, N. M., and Rogge, W. F.: Photochemical Oxidation and Changes in Molecular Composition of Organic Aerosol in the Regional Context, J. Geophys. Res.-Atmos., 111, D3, https://doi.org/10.1029/2005JD006265, 2006. a
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007. a, b
Russell, L. M.: Aerosol Organic-Mass-to-Organic- Carbon Ratio Measurements, Environ. Sci. Technol., 37, 2982–2987, https://doi.org/10.1021/es026123w, 2003. a
Russo, C., Stanzione, F., Tregrossi, A., and Ciajolo, A.: Infrared Spectroscopy of Some Carbon-Based Materials Relevant in Combustion: Qualitative and Quantitative Analysis of Hydrogen, Carbon, 74, 127–138, https://doi.org/10.1016/j.carbon.2014.03.014, 2014. a
Ruthenburg, T. C., Perlin, P. C., Liu, V., McDade, C. E., and Dillner, A. M.: Determination of Organic Matter and Organic Matter to Organic Carbon Ratios by Infrared Spectroscopy with Application to Selected Sites in the IMPROVE Network, Atmos. Environ., 86, 47–57, https://doi.org/10.1016/j.atmosenv.2013.12.034, 2014. a, b
Schneider, J., Weimer, S., Drewnick, F., Borrmann, S., Helas, G., Gwaze, P., Schmid, O., Andreae, M. O., and Kirchner, U.: Mass Spectrometric Analysis and Aerodynamic Properties of Various Types of Combustion-Related Aerosol Particles, Int. J. Mass Spectrom., 258, 37–49, https://doi.org/10.1016/j.ijms.2006.07.008, 2006. a
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Hoboken, NJ, 3rd Edn., 1152 pp., ISBN 978-1-118-94740-1, 2016. a, b
Shilling, J. E., Chen, Q., King, S. M., Rosenoern, T., Kroll, J. H., Worsnop, D. R., DeCarlo, P. F., Aiken, A. C., Sueper, D., Jimenez, J. L., and Martin, S. T.: Loading-dependent elemental composition of α-pinene SOA particles, Atmos. Chem. Phys., 9, 771–782, https://doi.org/10.5194/acp-9-771-2009, 2009. a
Shiraiwa, M., Ueda, K., Pozzer, A., Lammel, G., Kampf, C. J., Fushimi, A., Enami, S., Arangio, A. M., Fröhlich-Nowoisky, J., Fujitani, Y., Furuyama, A., Lakey, P. S. J., Lelieveld, J., Lucas, K., Morino, Y., Pöschl, U., Takahama, S., Takami, A., Tong, H., Weber, B., Yoshino, A., and Sato, K.: Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 51, 13545–13567, https://doi.org/10.1021/acs.est.7b04417, 2017. a
Slade, J. H. and Knopf, D. A.: Heterogeneous OH Oxidation of Biomass Burning Organic Aerosol Surrogate Compounds: Assessment of Volatilisation Products and the Role of OH Concentration on the Reactive Uptake Kinetics, Phys. Chem. Chem. Phys., 15, 5898–5915, https://doi.org/10.1039/C3CP44695F, 2013. a
Takahama, S., Johnson, A., and Russell, L. M.: Quantification of Carboxylic and Carbonyl Functional Groups in Organic Aerosol Infrared Absorbance Spectra, Aerosol Sci. Tech., 47, 310–325, https://doi.org/10.1080/02786826.2012.752065, 2013. a
Theodoritsi, G. N., Ciarelli, G., and Pandis, S. N.: Simulation of the evolution of biomass burning organic aerosol with different volatility basis set schemes in PMCAMx-SRv1.0, Geosci. Model Dev., 14, 2041–2055, https://doi.org/10.5194/gmd-14-2041-2021, 2021. a
Tiitta, P., Leskinen, A., Hao, L., Yli-Pirilä, P., Kortelainen, M., Grigonyte, J., Tissari, J., Lamberg, H., Hartikainen, A., Kuuspalo, K., Kortelainen, A.-M., Virtanen, A., Lehtinen, K. E. J., Komppula, M., Pieber, S., Prévôt, A. S. H., Onasch, T. B., Worsnop, D. R., Czech, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Transformation of logwood combustion emissions in a smog chamber: formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging, Atmos. Chem. Phys., 16, 13251–13269, https://doi.org/10.5194/acp-16-13251-2016, 2016. a, b, c
Tolbert, A. and Ragauskas, A. J.: Advances in Understanding the Surface Chemistry of Lignocellulosic Biomass via Time-of-Flight Secondary Ion Mass Spectrometry, Energy Sci. Eng., 5, 5–20, https://doi.org/10.1002/ese3.144, 2017. a
Tsui, W. G. and McNeill, V. F.: Modeling Secondary Organic Aerosol Production from Photosensitized Humic-like Substances (HULIS), American Chemical Society, Environ. Sci. Technol. Lett., 5, 255–259, https://doi.org/10.1021/acs.estlett.8b00101, 2018. a
Wang, N., Kostenidou, E., Donahue, N. M., and Pandis, S. N.: Multi-generation chemical aging of α-pinene ozonolysis products by reactions with OH, Atmos. Chem. Phys., 18, 3589–3601, https://doi.org/10.5194/acp-18-3589-2018, 2018. a
Yazdani, A.: Chemical characterization of organic aerosols with a focus on biomass burning and mid-infrared spectroscopy, Ph.D. thesis, École Polytechnique Fédérale de Lausanne, 371 pp., https://doi.org/10.5075/epfl-thesis-9382, 2022. a
Yazdani, A., Dudani, N., Takahama, S., Bertrand, A., Prévôt, A. S. H., El Haddad, I., and Dillner, A. M.: Characterization of primary and aged wood burning and coal combustion organic aerosols in an environmental chamber and its implications for atmospheric aerosols, Atmos. Chem. Phys., 21, 10273–10293, https://doi.org/10.5194/acp-21-10273-2021, 2021. a, b, c, d, e, f, g, h, i, j, k, l, m
Yazdani, A., Dudani, N., Takahama, S., Bertrand, A., Prévôt, A. S. H., El Haddad, I., and Dillner, A. M.: Fragment ion–functional group relationships in organic aerosols using aerosol mass spectrometry and mid-infrared spectroscopy, Atmos. Meas. Tech., 15, 2857–2874, https://doi.org/10.5194/amt-15-2857-2022, 2022. a
Yazdani, A., Takahama, S., Kodros, J. K., Paglione M., Masiol, M., Squizzato, S., Florou, K., Kaltsonoudis, C., Jorga, S. D., Pandis, S. N., and Nenes, A.: Infrared spectra of chamber aerosols, Zenodo [data set], https://doi.org/10.5281/zenodo.7982350, 2023. a, b
Ye, P., Ding, X., Hakala, J., Hofbauer, V., Robinson, E. S., and Donahue, N. M.: Vapor wall loss of semi-volatile organic compounds in a Teflon chamber, Aerosol Sci. Technol., 50, 822–834, https://doi.org/10.1080/02786826.2016.1195905, Taylor & Francis_eprint, 2016. a
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes, Geophys. Res. Lett., 34, 13, https://doi.org/10.1029/2007GL029979, 2007. a
Zhang, R., Gen, M., Liang, Z., Li, Y. J., and Chan, C. K.: Photochemical Reactions of Glyoxal during Particulate Ammonium Nitrate Photolysis: Brown Carbon Formation, Enhanced Glyoxal Decay, and Organic Phase Formation, Environ. Sci. Technol., 56, 1605–1614, https://doi.org/10.1021/acs.est.1c07211, 2022. a
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014. a