the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Jul 2023
| 05 Jul 2023
Volatility of aerosol particles from NO3 oxidation of various biogenic organic precursors
Emelie L. Graham
David M. Bell
Amelie Bertrand
Sophie L. Haslett
Urs Baltensperger
Imad El Haddad
Radovan Krejci
Ilona Riipinen
Secondary organic aerosol (SOA) is formed through the oxidation of volatile organic compounds (VOCs), which can be of both natural and anthropogenic origin. While the hydroxyl radical (OH) and ozone (O3) are the main atmospheric oxidants during the day, the nitrate radical (NO3) becomes more important during the nighttime. Yet, atmospheric nitrate chemistry has received less attention compared to OH and O3.
The Nitrate Aerosol and Volatility Experiment (NArVE) aimed to study the NO3-induced SOA formation and evolution from three biogenic VOCs (BVOCs), namely isoprene, α-pinene, and β-caryophyllene. The volatility of aerosol particles was studied using isothermal evaporation chambers, temperature-dependent evaporation in a volatility tandem differential mobility analyzer (VTDMA), and thermal desorption in a filter inlet for gases and aerosols coupled to a chemical ionization mass spectrometer (FIGAERO-CIMS). Data from these three setups present a cohesive picture of the volatility of the SOA formed in the dark from the three biogenic precursors. Under our experimental conditions, the SOA formed from NO3 + α-pinene was generally more volatile than SOA from α-pinene ozonolysis, while the NO3 oxidation of isoprene produced similar although slightly less volatile SOA than α-pinene under our experimental conditions. β-Caryophyllene reactions with NO3 resulted in the least volatile species.
Four different parameterizations for estimating the saturation vapor pressure of the oxidation products were tested for reproducing the observed evaporation in a kinetic modeling framework. Our results show that the SOA from nitrate oxidation of α-pinene or isoprene is dominated by low-volatility organic compounds (LVOCs) and semi-volatile organic compounds (SVOCs), while the corresponding SOA from β-caryophyllene consists primarily of extremely low-volatility organic compounds (ELVOCs) and LVOCs. The parameterizations yielded variable results in terms of reproducing the observed evaporation, and generally the comparisons pointed to a need for re-evaluating the treatment of the nitrate group in such parameterizations. Strategies for improving the predictive power of the volatility parameterizations, particularly in relation to the contribution from the nitrate group, are discussed.
- Article
(3271 KB) - Full-text XML
-
Supplement
(1584 KB) - BibTeX
- EndNote




Atmospheric aerosols, i.e., liquid or solid particles suspended in air, are important for the climate and human health. A large fraction of atmospheric particulate matter (PM) is secondary, formed from the condensation of atmospheric vapors after chemical reactions. Oxidation typically lowers the volatility (saturation vapor pressure) of the gas-phase precursors, resulting in increased atmospheric particle mass through condensation and possibly new particle formation (NPF). Secondary organic aerosol (SOA) is formed from the oxidation of volatile organic compounds (VOCs), which can be of both anthropogenic and natural origin. The atmosphere contains a plethora of VOCs, making it a complex system to understand and describe.
A way to describe the volatility of complex SOA is to use the volatility basis set (VBS), where the saturation vapor pressures of individual compounds are approximated as effective saturation vapor concentrations (C*; Donahue et al., 2006). In most VBS approaches, the C* values of individual molecular compounds are grouped into discrete bins. In this way, a complex mass spectrum containing thousands of compounds can be reduced to considerably fewer data points of volatilities, simplifying the description of, e.g., atmospheric aging processes in model frameworks. Volatility distributions of various SOA mixtures can be estimated experimentally based on the formation and growth or evaporation of the SOA (Berkemeier et al., 2020; Boyd et al., 2015; Karnezi et al., 2014; Lee et al., 2011; Tröstl et al., 2016) or through converting the molecular composition of SOA observed with mass spectrometric techniques to estimates of the volatilities present in the mixture (Donahue et al., 2011; Li et al., 2016; Pankow and Asher, 2008). Direct measurements of the volatility distributions are challenging, and typically a combination of techniques is needed to probe the full range of volatilities present in atmospheric mixtures and avoid misinterpretations of data.
While the hydroxyl radical (OH) and ozone are the two dominant oxidants in the atmosphere during daytime, the nitrate radical (NO3) is the major nocturnal oxidant. It is generated in the dark by the reaction of nitrogen dioxide (NO2) and O3, and its reaction rates with monoterpenes are similar to OH (Ng et al., 2017). There are much fewer studies on NO3 oxidation of biogenic VOCs (BVOCs) than those for ozonolysis and oxidation by OH, but they generally show more variable SOA yields (Ng et al., 2017). Specifically, reported SOA yields for nitrate oxidation are 0 %–82 % for α-pinene (Bates et al., 2022; Berkemeier et al., 2020; Fry et al., 2014; Hallquist et al., 2009; Nah et al., 2016; C. Wu et al., 2021), 2 %–24 % for isoprene (Ng et al., 2008; Rollins et al., 2009; C. Wu et al., 2021), and 86 %–146 % for β-caryophyllene (Fry et al., 2014; Jaoui et al., 2013; C. Wu et al., 2021). Nitrate oxidation of BVOCs is also a major pathway for forming atmospheric organic nitrates (ONs), which have been shown to be an important component of ambient SOA (Häkkinen et al., 2012; Holzinger et al., 2010; Kiendler-Scharr et al., 2016; Lee et al., 2016; O'Brian et al., 2014). Early modeling studies of the atmospheric fate of ONs assumed relatively high volatility, classifying all SOAs (including ON) as semi-volatile organic compounds (SVOCs) with C* > 10−0.5 µg m−3 (Chung and Seinfeld, 2002). ON volatility in the semi-volatility range was also established experimentally, based on equilibrium partitioning fits (Rollins et al., 2009). However, more recent studies, from both field and laboratory, suggest much lower volatilities to be associated with ON (e.g., Berkemeier et al., 2020; Häkkinen et al., 2012), with some ON even being characterized as highly oxygenated organic molecules (HOMs) and falling within the category of extremely low-volatility organic compounds (ELVOCs < 10−4.5 µg m−3; Pullinen et al., 2020; Zhao et al., 2021). These later results are in line with a rather large decrease in volatility associated with NO3 addition of about 2.5 orders of magnitude based on the group contribution method (see, e.g., Pankow and Asher, 2008; Donahue et al., 2011). Apart from a handful of pioneering studies (e.g., Berkemeier et al., 2020; C. Wu et al., 2021; R. Wu et al., 2021), research focusing specifically on the volatility of the nitrate-induced biogenic SOA is scarce. In particular, work combining multiple different techniques for studying the volatility of different chemical systems in a consistent manner is largely lacking.
This study is part of the Nitrate Aerosol Volatility Experiment (NArVE; see also Bell et al., 2022 and C. Wu et al., 2021), aimed to improve our understanding of nitrate-induced biogenic SOA formation. Bell et al. (2022) showed that the dark aging of α-pinene + NO3 was driven primarily by further oxidation reactions, in addition to fragmentation and dimer degradation reactions. C. Wu et al. (2021), on the other hand, studied the transition from dark to light conditions, revealing substantial changes in the chemical composition but relatively low evaporation. In this paper, the focus is on the volatility of biogenic SOA formed from the nitrate oxidation of α-pinene, isoprene, and β-caryophyllene. This study combines three experimental methods, namely isothermal evaporation chambers, temperature-dependent evaporation in the volatility tandem differential mobility analyzer (VTDMA), and a chemical ionization mass spectrometer with a filter inlet for gases and aerosols (FIGAERO-CIMS). The chemical information obtained from the FIGAERO-CIMS is used to estimate the volatility distributions for the SOA systems, which is then used as input in a kinetic model simulating the evaporation of aerosol particles in the isothermal evaporation chamber and the thermodenuder (TD) of the VTDMA. Additionally, experiments of α-pinene ozonolysis were performed for comparison and to benchmark our methods against the literature on the volatility of this well-studied system. The performance of the composition-based volatility predictions is evaluated, and the observations from the three systems are compared to available literature data. Finally, implications for the volatility distributions of the studied systems are discussed, together with strategies for improving the predictive power of the volatility parameterizations especially for ONs.
2.1 Chamber conditions and experimental overview of the NArVE campaign
Atmospheric simulation chamber experiments of the oxidation of α-pinene, isoprene, and β-caryophyllene were performed at the Paul Scherrer Institute in an 8 m3 Teflon chamber (see Platt et al., 2013), operating at 18–23 ∘C and 58 %–65 % relative humidity (RH) (see Table 1 for an overview of the experiments). Gas-phase instruments included a quadrupole proton transfer reaction mass spectrometer (Q-PTR-MS; Ionicon), an ozone monitor (Thermo 49C), and a NOx monitor (Thermo 42C). Particle-phase instruments included a scanning mobility particle sizer (SMPS; TSI model 3938), a custom-built VTDMA, isothermal evaporation chambers coupled to a custom-built SMPS, and a FIGAERO-CIMS (Aerodyne). Clean air was supplied by a zero-air generator (AADCO) and was used to flush the chamber overnight following every experiment at 50 L min−1 with the chamber volume heated to 30 ∘C.
In the NO3 experiments, the chamber was humidified with milliQ water (resistivity > 18 MΩ cm) until it reached the desired humidity, then the VOC of choice (α-pinene – Sigma Aldrich, 99+ %; isoprene – Sigma Aldrich, 95+ %; or β-caryophyllene – Sigma Aldrich, 99+ %) was injected into the chamber, and finally N2O5 (sampling from the head space above solid N2O5, which was kept chilled in an ice bath) was injected in a short burst (∼ 5 s at ∼ 1 L min−1), and the chamber was stirred by injecting 100 L min−1 of air for 10 s. N2O5 was generated by reacting liquid NO2 with high concentrations (∼ 1 %) of O3 and collecting the N2O5 crystals in a dry ice acetone bath. In the O3 experiments, O3 was generated by photolysis of air. Similar to the NO3 experiments, the chamber was first humidified and then O3 and α-pinene with the desired levels were added to the chamber volumetrically.
To determine the initial N2O5 concentrations, the decay rates of the VOC precursors were modeled with the Master Chemical Mechanism (MCM) using a simple 0-D box model (F0AM; Wolfe et al., 2016). For the β-caryophyllene experiment, its concentration could not be monitored with the PTR-MS because its mass-to-charge ratio () of 204 Th is outside of the mass transmission range for quantitative measurements; thus we estimated the concentration from the average N2O5 concentrations in the other experiments.
The particle mass concentrations (in µg m−3) were derived from integrated number size distributions from the SMPS and their conversions to mass using an assumed organic aerosol density (1.19 g cm−3; Vaden et al., 2011). They were further corrected for wall losses using a uniform dynamic wall loss rate kwall for the whole size range. The wall loss rate (kwall) was calculated from the exponential decay of the total particle number concentration (cm−3) measured by the SMPS, corrected for coagulation. The particulate wall loss is defined by
where N is the particle number concentration and kcoag. corresponds to the coagulation coefficient (5 × 10−10 s−1; Pospisilova et al., 2020). The wall-loss-corrected particle mass was divided by the uncorrected mass to obtain a wall loss correction factor for correction of the signals from the FIGAERO-CIMS.
Table 1Overview of the experimental conditions, and maximal OA load and SOA yield together with initial particle diameter (Dsel.) and concentration (Csel.) used as input in the kinetic model for the thermodenuder and isothermal evaporation (experiments marked with hyphens are not simulated due to either missing data about the molecular composition or experimental evaporation data). All parameters are derived from the experimental data.
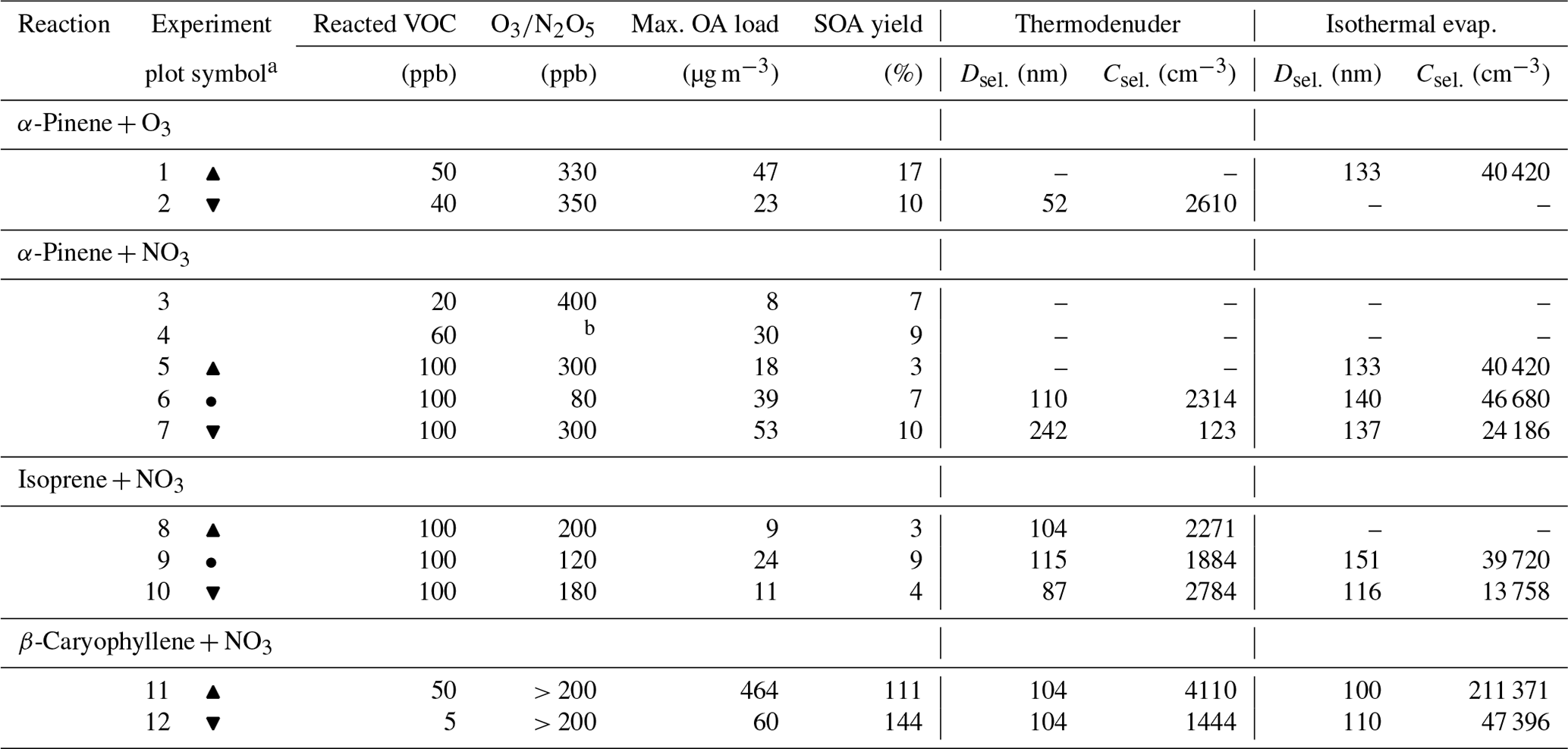
a Symbols used in Figs. 4, 5, and S7 to mark each experiment.
b N2O5 concentration was not available.
2.2 Volatility measurements
We assessed the SOA volatility using three methods: (1) a VTDMA (Sect. 2.2.1), (2) an isothermal evaporation chamber (Sect. 2.2.2), and (3) a FIGAERO-CIMS (Sect. 2.2.4). From the first two methods, we observed reduced particle diameters due to heating and dilution, respectively, and calculated the volume fraction remaining (VFR) (see Sect. 2.2.3). With the FIGAERO-CIMS, we obtained thermal desorption profiles of individual compounds as well as the bulk sample. In addition, we also estimated the volatility based on the chemical composition measured by the FIGAERO-CIMS and modeled the temperature- and time-dependent VFR using a kinetic evaporation model (Sect. 2.2.5) for comparison with the VTDMA and the isothermal evaporation chamber, respectively.
2.2.1 Volatility tandem differential mobility analyzer (VTDMA)
A custom-built VTDMA was used to measure the change in particle diameter upon heating. The VTDMA (see, e.g., Tritscher et al., 2011) sampled aerosol particles through a silica gel diffusion drier connected to an approximately 5.5 m long stainless steel tube reaching to the chamber. The VTDMA combines two differential mobility particle sizer (DMPS) systems, coupled in series with a thermodenuder (TD) in between. Both DMPS systems consisted of a custom-made short differential mobility analyzer (DMA; Stockholm University, operated with closed-loop sheath air and 1 L min−1 sample flow) coupled to a condensation particle counter (CPC; TSI 3010). Before entering each DMA system, the particles were brought into charge equilibrium using a Ni-63 neutralizer.
The first DMA selected a nearly monodisperse aerosol with the diameter (Dsel.) set to the geometric mean diameter of the aerosol population (varying from 52 to 242 nm between experiments) measured by the SMPS upstream. The sample flow was split in two, with half going to the CPC measuring the selected particle concentration (Csel.) and the other half entering a 35 cm long custom-built TD, with a residence time of 1.9 s. For each experiment, a set of temperature ramps from 50 to 225 ∘C in increments of 25 ∘C were performed with the TD. The size distribution of the heated aerosol was measured in the second DMPS system with a time resolution of approximately 5 min.
2.2.2 Isothermal evaporation chamber
Samples were size selected with a DMA (TSI Inc. Model 3082) and placed into a 20 L stainless steel chamber, which operated at room temperature (20 ∘C, set by the air conditioning in the instrument container). The chamber was equipped with a layer of charcoal at the bottom of the chamber. The charcoal continuously removed the gas-phase species from the chamber, ensuring the particles never fully reached equilibrium with the surrounding gases, promoting evaporation. Changes in particle sizes due to isothermal evaporation were monitored using an SMPS (TSI Inc., model 3938). The experimental setup has been described in detail elsewhere (Bell et al., 2017; Vaden et al., 2011; Wilson et al., 2015).
2.2.3 Volume fraction remaining (VFR)
As particles evaporate in the VTDMA and isothermal evaporation chamber, they shrink in size, and the resulting volume fraction remaining (VFR) can be used as proxy for the aerosol population volatility. We calculated the VFR values for the VTDMA and evaporation chamber experiments from the particle diameters as follows:
where Dsel. is the initial geometric mean mobility particle diameter, and Devap. is the size observed after evaporation took place. In the VTDMA, the Devap. values are the heated diameters, represented by the geometric mean diameter estimated using a Gaussian fit over the whole heated size distribution. The uncertainty in the temperature-dependent VFR is the sum of the standard deviation of the VFR and the error of the two DMPS systems within the VTDMA, estimated using error propagation law and polystyrene latex (PSL) spheres for calibration. For the evaporation chamber experiments, Devap. was directly reported by the SMPS coupled to the chamber.
2.2.4 Deriving the volatility distribution from the FIGAERO-CIMS data
The molecular composition of the generated SOA was measured using the FIGAERO-CIMS, whose design, operation, and related data analysis are described in C. Wu et al. (2021). Briefly, the FIGAERO-CIMS was set up using iodide (I−) as reagent ion and an X-ray generator as ion source. Particles were collected on a 25 mm Zefluor® PTFE filter (Pall Corp.) with 2 µm pore size via a sampling port directly connected to the chamber (flow rate of 5 L min−1). The duration of sampling was 10–20 min for most of the experiments. When the sampling was done, particles on the filter were desorbed by a flow of gradually heated ultra-high-purity (99.999 %) nitrogen. A FIGAERO-CIMS desorption round lasted 40 min: 20 min of ramping the temperature of the nitrogen flow up to 200 ∘C, followed by a 20 min “soak period” at a constant 200 ∘C to allow signal to go back to background values. After that, it was cooled down to room temperature (for 15 min). Artefacts from thermal fragmentation (< 30 % of total signal for all experiments; for details, see C. Wu et al., 2021) were subtracted for further analysis. The potential uncertainties induced by thermal fragmentation are discussed in Sect. 3.4.
For each individual filter sample and the ensemble of deposited particles, we estimated the volatility of individual compounds based on their molecular composition measured by the FIGAERO-CIMS and further binned their concentrations into a volatility distribution. The effective saturation vapor concentration (C*) of individual compounds was calculated using four different, previously published parameterizations, which have explicit formulations for nitrogen or nitrate groups. These are as follows.
-
An updated version of the parameterization by Donahue et al. (2011), modified based on the saturation vapor concentrations of highly oxygenated molecules (HOMs) detected by Tröstl et al. (2016) and published by Mohr et al. (2019).
Here, n0= 25, bC= 0.475, bO= 0.2, bCO= 0.9, and bN= 2.5. nC, nO, and nN are the number of carbon, oxygen, and nitrogen atoms in the compound, respectively. This parameterization hence assumes that a nitrate group (one nitrogen atom and three oxygen atoms) reduces a compound's saturation vapor pressure by 2.5 orders of magnitude (Donahue et al., 2011; Pankow and Asher, 2008).
-
An updated version of Daumit et al. (2013) with a nitrate group contribution term.
Here, (conversion factor, where MW is the molecular weight in grams per mol, T is the temperature in Kelvin, and R is the gas constant) and b terms are the different group contribution terms for quantifying the contribution of each chemical moiety to the saturation vapor pressure: b0 is the zero order term, bC is the carbon number term, b=O is the carbonyl group term, b−OH is the hydroxyl group term, and is the nitrate group term (for MW = 200 g mol−1 and T= 293 K, it is equal to 1.79, −0.438, −0.935, −2.23, and −2.23, respectively). Here, we assume all nitrogen atoms are from nitrate groups; i.e., .
We modified the parameterization from Daumit et al. (2013) by including nitrate groups. Functional groups in the molecules are thus limited to carbonyls, hydroxyls, nitrates, or some combination of the three. A carbonyl or a hydroxyl group contains one oxygen atom, and a nitrate group contains three oxygen atoms; thus,
The level of unsaturation (double bound equivalent (DBE)) arises from carbonyl groups and nitrate groups (assuming the double bonds in the carbon chain of the precursor have reacted). Since a nitrate group has a degree of unsaturation of 1,
We note that including the nitrate group in the calculation of n=O can be critically discussed, as the nitrate group does not yield any information on double-bonded carbons. However, treating the N of the nitrate group as a hydrogen in the DBE formulation yields the same final formula for n=O (Eq. 6). Based on Eqs. (5) and (6), the number of hydroxyl groups could be calculated as
from which the modified Daumit parameterization follows as
-
An updated version of Li et al. (2016) with a modified nitrogen coefficient for organic nitrates (Isaacman-VanWertz and Aumont, 2020).
Here, n0= 22.66, bC= 0.4481, bO= 1.656, and bCO= −0.7790 for CHO compounds; and n0= 24.13, bC= 0.3667, bO= 0.7732, bCO= −0.07790, and bN= 0.7732 for CHON compounds. The modification of the parameterization is based on the observation that a nitrate group has a similar impact on the saturation vapor pressure as a hydroxyl group (SIMPOL; Pankow and Asher, 2008).
-
A simple parameterization based on highly oxygenated organic molecules (HOMs) from α-pinene ozonolysis (Peräkylä et al., 2020).
Here, nH is the number of hydrogen atoms in the compound. With this parameterization, a nitrate group reduces a compound's saturation vapor pressure by 0.34 orders of magnitude.
The temperature dependence of C* is considered as follows:
where C*(300 K) is the saturation vapor concentration at 300 K, and ΔHVAP is the vaporization enthalpy (Epstein et al., 2010):
Qualitative volatility information was also derived from the FIGAERO-CIMS desorption profiles, referred to as thermograms. We calculated the mass-weighted average thermogram and corresponding Tmax of the entire particle population deposited on the filters from the FIGAERO-CIMS. The desorption temperature at which a compound's signal exhibits a maximum in the FIGAERO-CIMS (referred to as Tmax) has earlier been shown to be connected qualitatively to the volatility of the compound (log10C* or enthalpy of vaporization; Bannan et al., 2019; Lopez-Hilfiker et al., 2014; Thornton et al., 2020).
2.2.5 Kinetic evaporation modeling
We used a kinetic evaporation model (Riipinen et al., 2010) to predict the evaporation of the aerosol populations in the TD and the isothermal evaporation chamber; i.e., temperature- and time-dependent VFR, respectively. The model solves the equations describing the mass transfer between the particulate and the gas phase due to condensation and evaporation. The model is applicable over a wide range of particle sizes, as it describes the transport over kinetic, transition and continuum regimes using the Fuchs and Sutugin transition regime correction factor (see Riipinen et al., 2010 and the references therein for details). The particle size distribution and volatility distribution, initial gas-phase concentrations of the relevant species, and the temperature evolution are given as inputs to the model – together with the molecular properties of the chemical components of the system (see Table S1 in the Supplement). The initial particle diameter was set to the same sizes as the monodisperse size selection prior to each TD and isothermal evaporation measurement (see Table 1). Additionally, the initial particle concentration for each Dsel. was set to the corresponding concentration measured by the CPC within the VTDMA and using the SMPS for the TD and isothermal evaporation chamber, respectively (see Table 1). For the VTDMA setup, an initial temperature corresponding to room temperature (20 ∘C) and a flat temperature profile (constant temperature throughout the TD) with final temperatures varying between 25 and 225 ∘C together with a residence time of 1.9 s were used for the TD section. The gas phase was assumed to be initially in equilibrium with the particle phase for the VTDMA setup. For the isothermal evaporation chamber setup, the gas phase was assumed to be effectively stripped from the condensable vapors throughout the evaporation, and the evaporation took place at room temperature (20 ∘C) over the residence time of 240 min.
In addition to the physical and chemical properties presented in Table S1, the model inputs included saturation vapor concentration estimates based on the chemical composition measured by the FIGAERO-CIMS. The three volatility parameterizations (see previous section) were used as model inputs for the C* values of the SOA compounds, and the corresponding ΔHVAP was calculated using Eq. (12). The sensitivity of the model setup was evaluated (see Figs. S3–S6) also for a fixed vaporization enthalpy, ΔHVAP= 70 kJ mol−1 (see, e.g., Hong et al., 2017), reduced accommodation (αm, set to be either 0.1 or 0.01; see, e.g., Riipinen et al., 2010), and a mass-dependent diffusion coefficient (see, e.g., Poling et al., 2001, for more details) from the measured compositions:
P is the pressure (1 bar), and M is the molecular weight (g mol−1) for gases A and B, the latter being air and the former the organic vapor. Σv is the sum of the atomic diffusion volumes of the two components (see Table S1). Particle-phase diffusion or any chemical reactions during the evaporation were ignored as a first-order approximation.
3.1 SOA formation and yields
An overview of the initial precursor and oxidant concentrations is presented in Table 1, together with the resulting maximal mass load in the particle phase. Prompt particle formation, due to rapid reaction with NO3 or O3, followed by steady particle mass decay due to evaporation, was observed for all three precursors. For the NO3 experiments, the initial particle composition followed primarily from the reactions between peroxy radicals (RO2–RO2) and between RO2 and NO3, resulting in a substantial fraction of oligomers with the dimer-to-monomer ratio increasing with decreasing precursor carbon number (Bates et al., 2022; Bell et al., 2022; C. Wu et al., 2021). According to the FIGAERO-CIMS, the mass fractions of oligomers were 86 %–88 %, ∼ 100 %, and 60 %–63 % for the α-pinene, isoprene, and β-caryophyllene SOA, respectively. Compared to the nitrate systems, the oligomer mass fraction for α-pinene + O3 was lower and in the range of 45 %–60 %. The aerosol particles formed from nitrate oxidation of α-pinene and isoprene were dominated by oligomers with two and three to four nitrate groups, respectively. The β-caryophyllene SOA was dominated by both monomers with one to two nitrate groups and dimers with two to three nitrate groups (C. Wu et al., 2021). The wall-loss-corrected maximum mass concentrations formed were 23–47 µg m−3 for α-pinene + O3, 8–53 µg m−3 for α-pinene + NO3, 9–24 µg m−3 for isoprene + NO3, and 60–464 µg m−3 for β-caryophyllene + NO3. The corresponding SOA yields of 10 %–17 % (α-pinene + O3), 3 %–10 % (α-pinene + NO3), 3 %–9 % (isoprene + NO3), and 111 %–144 % (β-caryophyllene + NO3) are generally in line with the literature (Berkemeier et al., 2020; Ng et al., 2017; R. Wu et al., 2021).
3.2 Measured evaporation of SOA from α-pinene, isoprene, and β-caryophyllene oxidation
Figure 1 presents the experimental data on the evaporation of α-pinene SOA from oxidation with O3 and NO3, measured as temperature-dependent VFR in the VTDMA (Fig. 1a), time-dependent VFR in the isothermal evaporation chamber (Fig. 1b), and temperature-dependent normalized signal (thermograms) during heating rounds from the FIGAERO-CIMS (Fig. 1c). The VTDMA plots (Fig. 1a) show that particles formed through nitrate oxidation consistently display a lower VRF at given temperatures than the particles from ozonolysis, and hence they have higher bulk volatility. We also compared the VFR data from the α-pinene ozonolysis experiments to available literature data (see, e.g., Tritscher et al., 2011, and references therein) in Fig. S1. The comparison shows general agreement with the previous measurements, with some variation arising from the different experimental conditions such as the residence time within the TD, which ranges from < 1 up to 31.6 s in the data sets included in Fig. S1. With the residence time of 1.9 s, our measurements are at the short end of the range, with correspondingly higher VFR to be expected. The lower VFRs during isothermal evaporation for NO3 oxidation as compared with ozonolysis agree with the VTDMA data, indicating that nitrate oxidation of α-pinene SOA produces more volatile compounds (Fig. 1b). The FIGAERO-CIMS average thermogram also reveals lower maximum desorption temperature, indicating higher bulk volatility for α-pinene + NO3 than for α-pinene ozonolysis (Fig. 1c). The three methods are therefore overall consistent in this regard. The time-dependent VFR for nitrate SOA with different loadings in the isothermal evaporation exhibits a tendency for lower VFR for higher initial mass loadings. This may be related to, e.g., variation in the volatility distributions depending on the precursor and oxidant loadings. However, no significant dependence of the evaporation on the mass loading was observed for the VTDMA results (see Fig. S2).
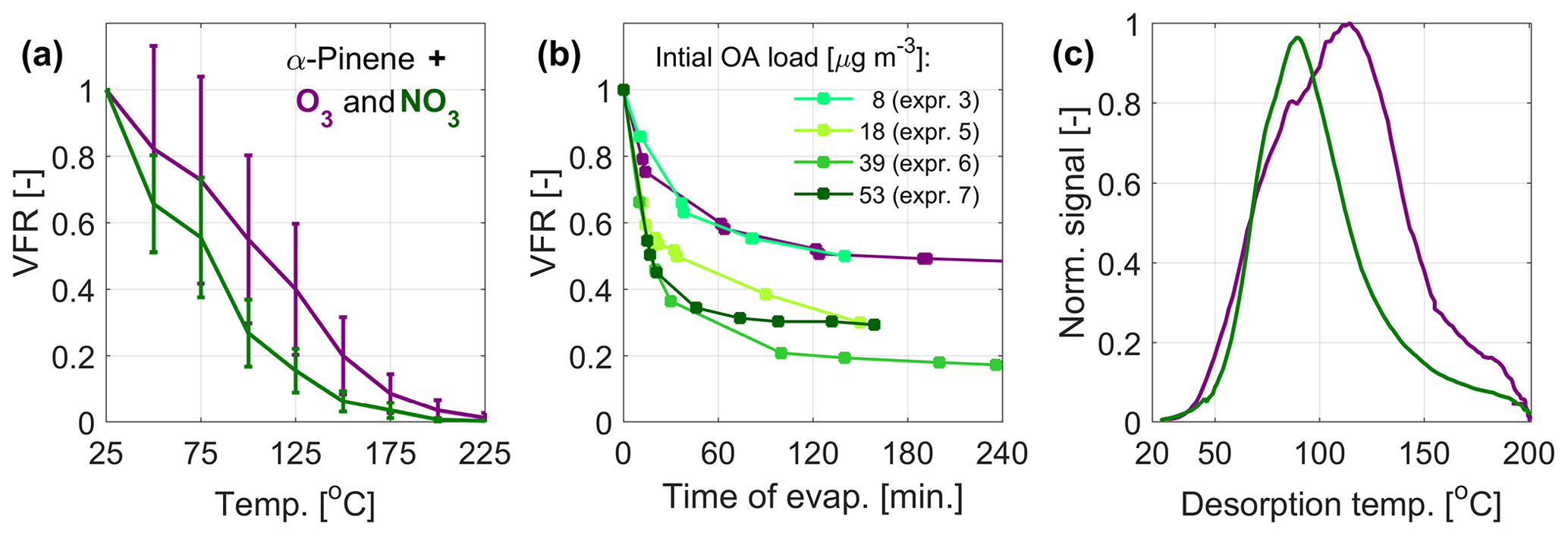
Figure 1Measured volatility of SOA particles from α-pinene oxidation by O3 (purple) and NO3 (green). (a) Mean VFR evolution during the VTDMA temperature ramps, (b) the isothermal evaporation, and (c) mean thermograms measured by the FIGAERO-CIMS. The VFR at room temperature (a) is estimated based on predicted evaporation at room temperature (b). For the VTDMA experiments, the VFR values correspond to the mean of two consecutive recordings during one ozonolysis experiment and the mean of the four individual nitrate oxidation experiments (see Table 1), where the uncertainty bars correspond to the sum of the standard deviation of the VFR and the error of the two DMPS systems within the VTDMA (see Sect. 2.2.3).
Figure 2 presents the volatility data from the VTDMA, isothermal evaporation chamber, and FIGAERO-CIMS for the nitrate oxidation of isoprene (Fig. 2a–c) and β-caryophyllene (Fig. 2d–f) in comparison with α-pinene. The isoprene + NO3 SOA has similar bulk volatility as the α-pinene + NO3 SOA. Both the temperature ramp in the VTDMA and residence time in the isothermal evaporation chamber result in slightly higher VFR and hence somewhat lower volatility than for the corresponding SOA from α-pinene oxidation. The effect is not as clear in the FIGAERO-CIMS thermogram, where the peak of the bulk thermogram is observed at slightly lower temperatures than for α-pinene, although with a prominent shoulder at higher temperatures. The temperature-dependent VFRs for the β-caryophyllene system are even higher, and in the isothermal chamber no significant evaporation could be observed. In the FIGAERO-CIMS analysis, the β-caryophyllene data display a bimodal thermogram with one peak (from monomers) at the same temperature as α-pinene, while the dominant peak (from dimers) is located at higher temperatures, in agreement with the higher measured VFRs.
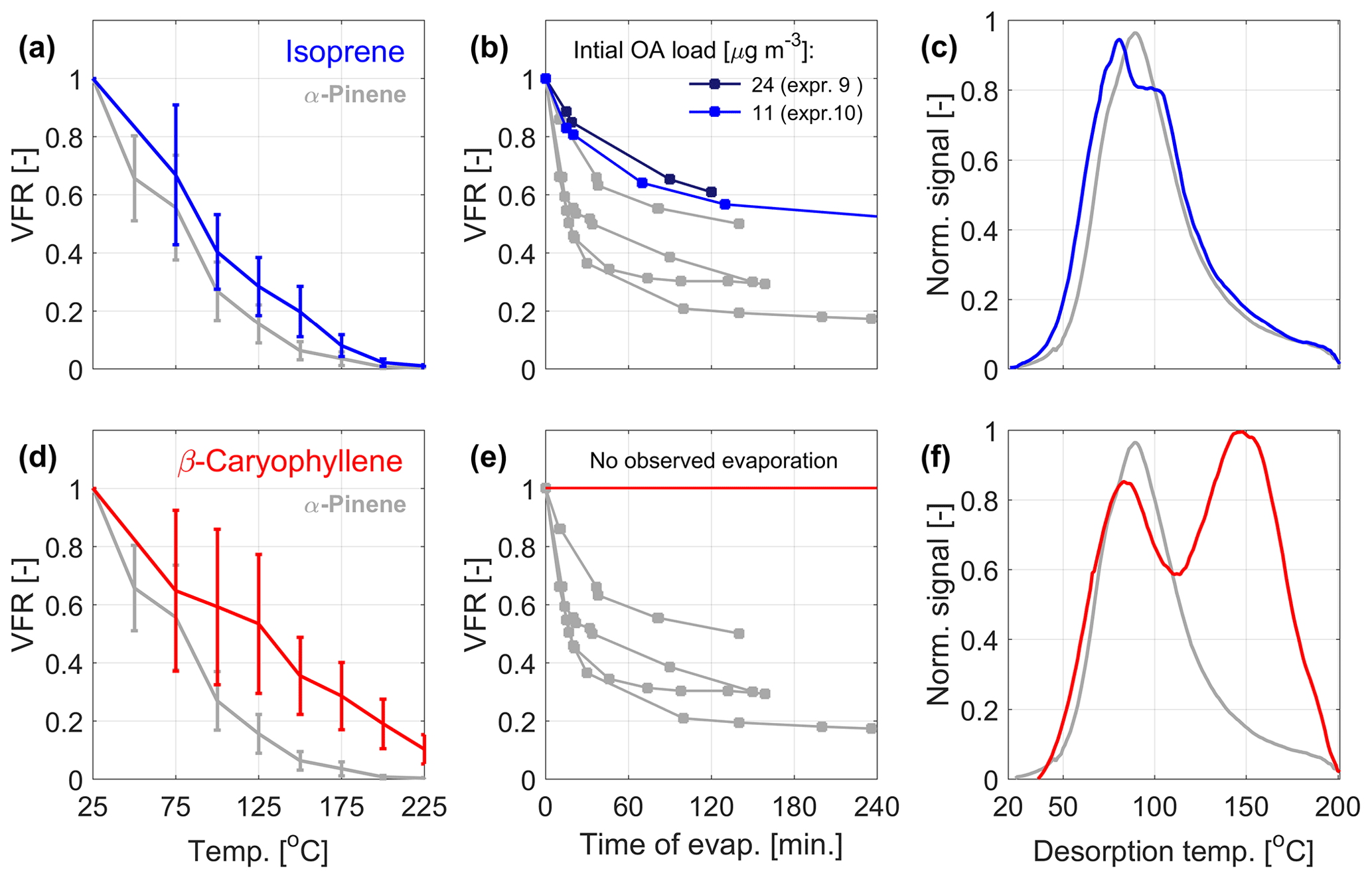
Figure 2Measured volatility of SOA from isoprene (a–c; blue curves) and β-caryophyllene (d–f; red curves) oxidation by NO3. The corresponding data from α-pinene oxidation are marked in gray. (a, d) The mean VFR evolution during the VTDMA temperature ramps, (b, e) the isothermal evaporation, and (c, f) the mean thermograms measured by the FIGAERO-CIMS. The VFR at room temperature (a, d) is estimated based on predicted evaporation at room temperature (b, e). For the VTDMA experiments, the VFR values correspond to the mean of three and two individual experiments for isoprene and β-caryophyllene, respectively (see Table 1), where the uncertainty bars correspond to the sum of the standard deviation of the VFR and the error of the two DMPS systems within the VTDMA (see Sect. 2.2.3).
3.3 Volatility distributions derived from the molecular composition
The volatility distributions in the form of VBS resulting from the parameterizations by Mohr et al. (2019), modified Daumit et al. (2013), modified Li et al. (2016), and Peräkylä et al. (2020) (hereafter referred to as MHR, DMT, Li, and PRK, respectively) are presented in Fig. 3 for all the studied chemical systems, including α-pinene ozonolysis. PRK produces the highest volatilities, followed by Li, with MHR resulting in the least volatile compounds for all nitrate systems and DMT resulting in the least volatile compounds for the α-pinene ozonolysis system. The volatility distributions from MHR, DMT, and Li tend to contain a wider range of C* values, while the volatility distributions by PRK are narrower and generally unimodal; the same trend can be observed for all the four systems.
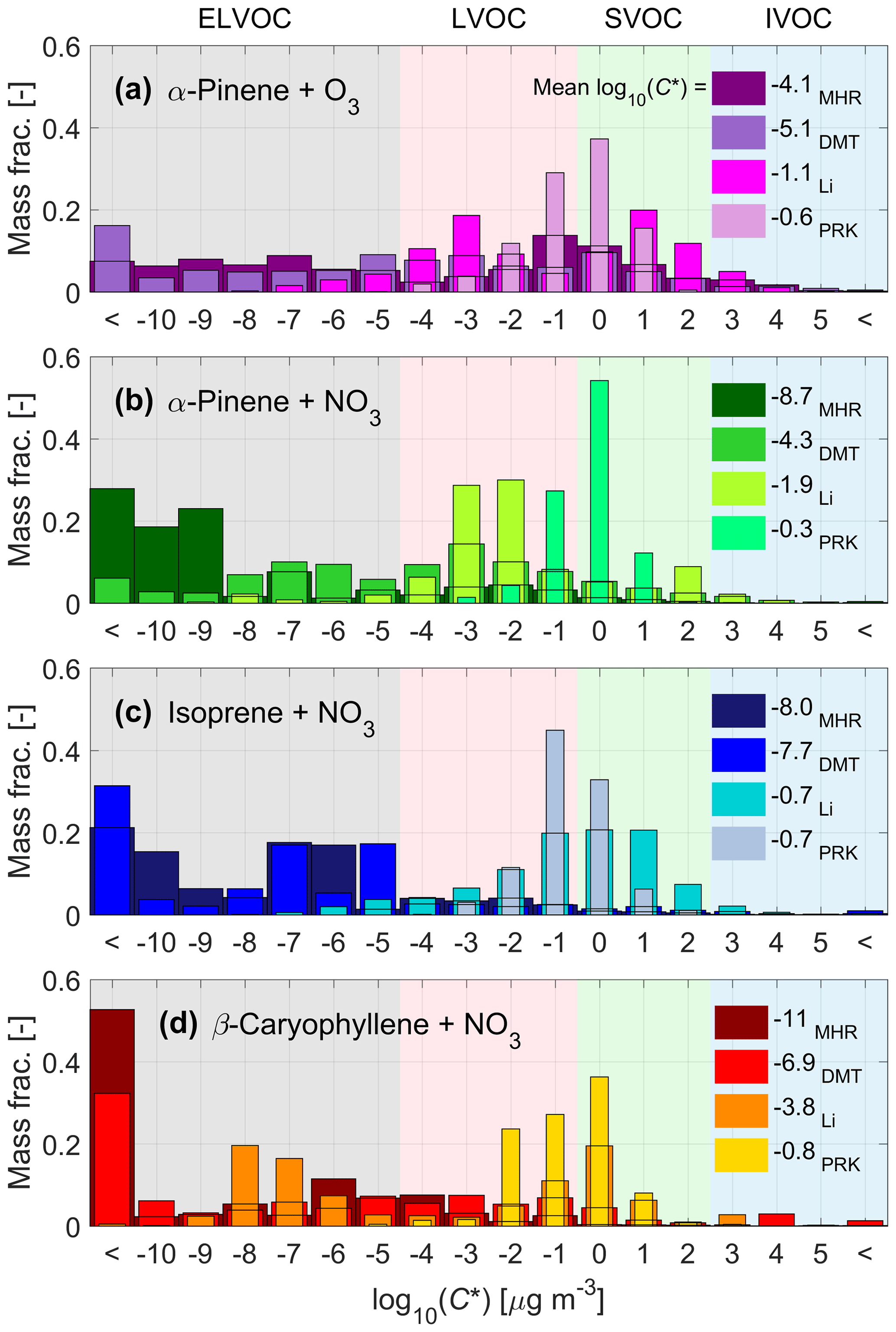
Figure 3Volatility distributions in the form of VBS as derived from the molecular composition for α-pinene ozonolysis (a) and nitrate oxidation of α-pinene (b), isoprene (c), and β-caryophyllene (d). The four volatility parameterizations used (MHR: Mohr et al., 2019; DMT: modified Daumit et al., 2013; Li: modified Li et al., 2016; PRK: Peräkylä et al., 2020) are depicted with different shades of the main color for each of the precursors. The averaged log10(C*) for each parameterization is also listed in the legend.
The MHR parameterization predicts the presence of extremely low volatilities for a major part of the nitrate oxidation products of all three different precursors, with β-caryophyllene (Fig. 3d) producing the least volatile compounds, which is qualitatively in line with the observed VFRs (see Fig. 2). For α-pinene and isoprene, MHR predicts the majority of the compounds to fall into the volatility bins located below log10(C*) ∼ −6 and −5, respectively. For β-caryophyllene, MHR predicts two main classes of volatilities: one below log10(C*) ∼ −10 (oligomers) and another, broader peak around log10(C*) ∼ −5.5 (monomers). Comparing the nitrate system with the ozonolysis system, MHR predicts that α-pinene nitrate (Fig. 3b) SOA has lower volatility than α-pinene ozonolysis SOA (Fig. 3a). This result is in contrast with the direct volatility measurements displaying lower VFR, indicating higher volatility, for nitrate oxidation compared to α-pinene ozonolysis (see Fig. 1a). All in all, the MHR parameterization predicts ELVOCs to dominate the composition of the SOA from nitrate oxidation of BVOCs.
The DMT parameterization also predicts a broad range of volatility, similar to MHR. It predicts more ELVOC for the ozonolysis system, with a mean log10(C*) of −5.2, which is lower than the mean log10(C*) of −4.2 by MHR. However, for all nitrate systems, it predicts significantly less ELVOC than MHR, and the mean log10(C*) is also higher than that by MHR.
The other two parameterizations yield narrower distributions that are generally shifted towards higher volatility. PRK results in higher volatilities than Li for all three precursors, with the majority of the species being in the LVOC and SVOC range. With the Li parameterization, the isoprene nitrate products are more volatile than the α-pinene nitrate products, with isoprene peaking around log10(C*) ∼ 0.5 compared to log10(C*) ∼ −2.5 for α-pinene. For the SOA from β-caryophyllene, Li also produces a bimodal distribution, overlapping both the MHR and PRK distributions. The least volatile peak for Li overlaps the most volatile peak in the MHR distribution, at log10(C*) ∼ −7.5, while the more volatile peak appears at log10(C*) ∼ −0.5, instead overlapping with the whole distribution by PRK for β-caryophyllene. Finally, with the PRK parameterization, the VBS distributions of all systems are similar and in a narrow range between −3 to 1, which suggests that this parameterization is the least sensitive. Although the differences in the VBS for the α-pinene nitrate and ozonolysis system are smaller using these two parameterizations, they still predict lower-volatility products of the nitrate system compared to the α-pinene ozonolysis SOA, which is in contrast with the observations. Clearly, the four parameterizations yield different volatility distributions, which are not always in line with the measured evaporation described in Sect. 3.2.
3.4 Comparison to evaporation calculated with a kinetic model
In order to better compare the volatility derived from the chemical composition to the evaporation observed in the VTDMA and isothermal evaporation chamber, the volatility distributions derived from the four investigated parameterizations (Sect. 3.3, Fig. 3) were entered into a kinetic model describing the mass transport to and from the aerosol surface (Riipinen et al., 2010, Sect. 2.2.5). The model results are compared to the experiments in Fig. 4.
The calculated evaporation of SOA from α-pinene ozonolysis in the VTDMA (Fig. 4a) using MHR and DMT reproduces the evaporation well, while the other two parameterizations, Li and PRK, result in lower VFR compared to the measurements. The overall performance is comparable for the isothermal evaporation data (Fig. 4e): i.e., MHR reproduces the evaporation best, while Li and PRK underestimate VFR; however, DMT overestimates the evaporation over time. The α-pinene ozonolysis data set was used to demonstrate the sensitivity of the model to the key input parameters (see Table S1). In this regard, we tested the values of 0.1 and 0.01 for the accommodation coefficient (see, e.g., Riipinen et al., 2010), a fixed value of 70 kJ mol−1 for the vaporization enthalpy (see, e.g., Hong et al., 2017), as well as the effect of estimating the diffusion coefficient based on the molecular composition, which was now directly available from the FIGAERO-CIMS data (see Eq. 13). The results are presented in Fig. S3. While modifying the accommodation coefficient or the vaporization enthalpy can bring the other parameterizations somewhat closer to the observations, it is clear that the MHR parameterization captures the evaporation of this system exceptionally well with the base case assumptions in the model. Including a detailed description of the diffusion coefficient based on the molecular structure had no significant effect on the output of the data for any of the volatility parameterizations.
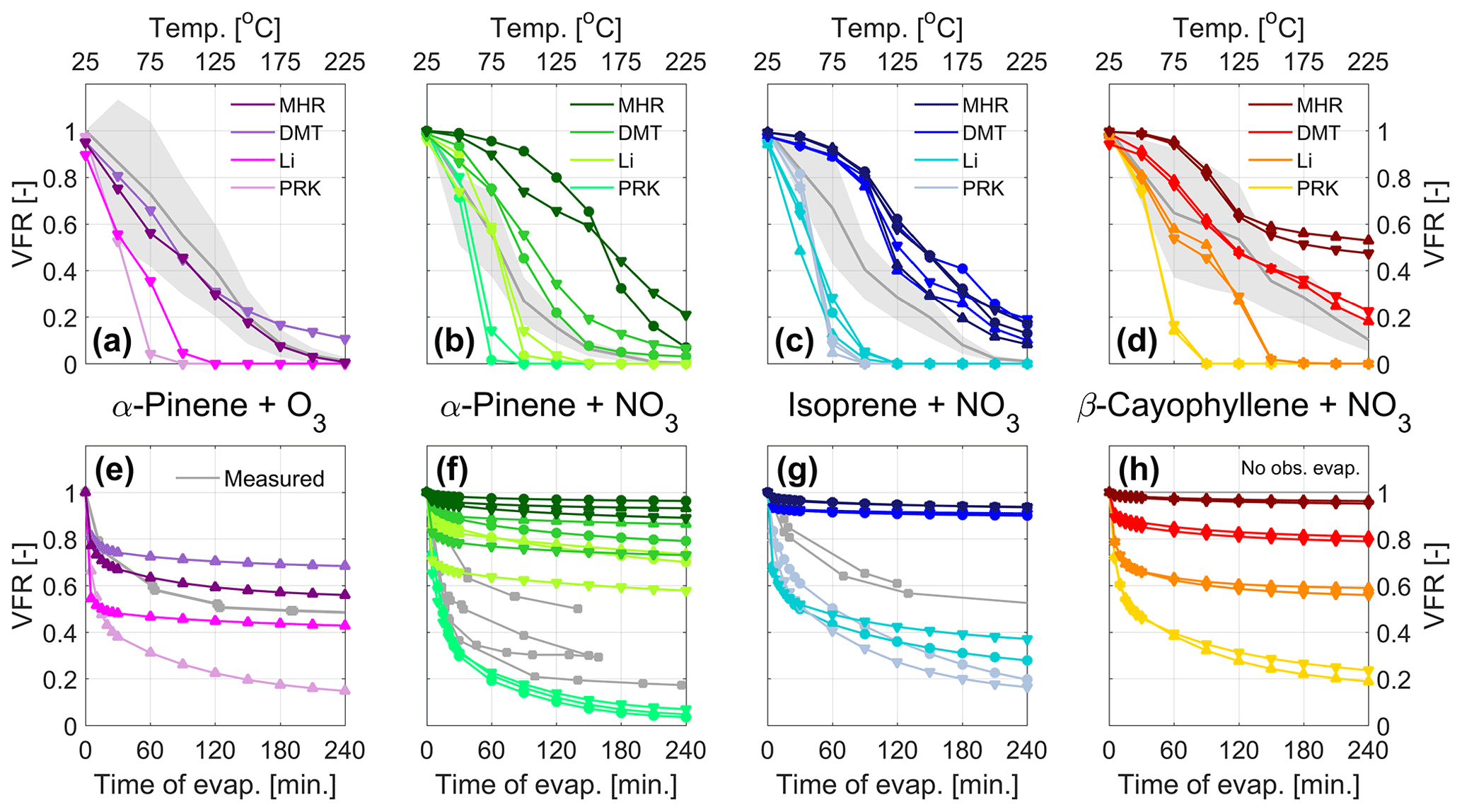
Figure 4Evaporation in the TD (a–d) and isothermal evaporation chamber (e–h) predicted by the kinetic model using the VBS from Fig. 3 as input for α-pinene ozonolysis and nitrate oxidation of α-pinene , isoprene, and β-caryophyllene. The symbols (▴ • ▾) mark the experiments in chronological order, as indicated in Table 1, during which the VBS data were collected. For comparison, the experimental VFR data are displayed in gray, with the shaded areas displaying the uncertainty of the VTMDA measurement.
For the nitrate systems, MHR, in contrast to its good performance for the ozonolysis system, results in too high VFR compared to what is measured with the VTDMA (Fig. 4b, c, and d) and the isothermal evaporation chamber (Fig. 4f and g). PRK, which results in highest volatility (Fig. 3), underestimates the VFR for all cases. The VFR estimates using Li and DMT fall in between. DMT tends to overestimate, and Li tends to underestimate the VFR with the VTDMA (Fig. 4b, e, and d). We simulated the VFR using averaged Li and DMT values in Fig. S7 and it works well for the α-pinene and isoprene systems but not the β-caryophyllene system. For simulating the VFR with the isothermal evaporation chamber, their performance is different and depends on individual systems: DMT and Li both overestimate the VFR for the α-pinene system, DMT overestimates the VFR and Li underestimates the VFR for the isoprene system, and both underestimate the VFR for the β-caryophyllene system.
Based on these results, no parameterization can be deemed to be systematically the best for reproducing both the VTDMA and the isothermal evaporation chamber data. This is understandable, since these experimental techniques are sensitive to different volatility ranges given the different operating temperatures. Combining these results with the volatility distributions (Fig. 4) suggests that the material contributing to the VFRs from the VTDMA setup consists of species with volatilities at the low end of the spectrum (log10(C*) values of about −5 and lower), while the species that remain in the particle phase in the isothermal evaporation chamber seem to have log10(C*) values below about −2. This is especially clear for the isoprene simulations, with both the TD and isothermal evaporation simulations being very sensitive in the volatility range between log10(C*) ∼ −5 to −2 (see Figs. 3 and 4).
It is worth noting that with the FIGAERO-CIMS, subtraction of the thermal decomposition compounds may also induce uncertainties. Thermal fragmentation contributed 5 %–27 %, 1 %–4 %, and 10 %–23 % of the total organic signal of the isoprene, α-pinene, and β-caryophyllene nitrate SOA, respectively, and 3 %–13 % for α-pinene ozonolysis SOA. Since thermal decomposition due to dehydration or decarboxylation reactions normally occurs at temperatures higher than 120 ∘C (Buchholz et al., 2020; Stark et al., 2017) and the parent compounds of the decomposing molecules have an even higher thermal desorption temperature, we can conclude that the compounds we remove are of very low volatility. Unfortunately, their volatility could not be identified with the parameterization based on the formula of the thermal decomposition compounds, as they are much smaller than the parent compounds and thus have much higher volatilities. We subtracted these compounds for the further calculation of the bulk volatility and the simulation of evaporation, but we also note that it may lead to decreases in the simulated VFR in the higher temperature range (> 120 ∘C) in the VTDMA; thus it could be a potential reason for the discrepancies between the calculated VFR and the measured VFR with the VTDMA. In addition, as α-pinene nitrate SOA exhibits much less thermal fragmentation compared to isoprene and β-caryophyllene, it could also explain partly the different performance of parameterization in simulating the evaporation in the VTDMA in different systems.
Taken together, the results from Fig. 4 suggest that the volatility reduction by the nitrate group is considerably overestimated by the MHR parameterization, while PRK tends to systematically overestimate the volatility of the studied systems in the TD and isothermal evaporation setups. This is generally in line with previous findings, particularly the conclusion by R. Wu et al. (2021) for the isoprene + NO3 system, who find that the MHR parameterization gives clearly lower volatilities than that estimated by other parameterizations, e.g., SIMPOL (Pankow and Asher, 2008) and PRK, as well as that obtained by experimental approaches. Furthermore, these results show a substantial contribution of species in the LVOC range in the SOA from both α-pinene + NO3 and isoprene + NO3, and probably the presence of SVOCs, which show considerable evaporation even at room temperature (since the isothermal evaporation is mainly sensitive in this range, as described above). The SOA from β-caryophyllene + NO3 on the other hand seems to consist primarily of species in the ELVOC and LVOC range. Our data present overall lower volatilities of SOA formed from α-pinene + NO3 and isoprene + NO3 compared with the few other existing studies (Berkemeier et al., 2020; Boyd et al., 2015; Ng et al., 2017; R. Wu et al., 2021), while for the β-caryophyllene + NO3 system, no previous data exist. When interpreting these comparisons, however, one should be aware of the somewhat different experimental conditions in the existing studies, remembering, e.g., the fact that our experiments show a substantial contribution from oligomers.
The MHR and Li parameterizations are of similar mathematical form, with the exception that the former subtracts the oxygen atoms from the nitrate groups in the carbon–oxygen non-ideality, while the latter includes all oxygen atoms in the carbon–oxygen non-ideality. In MHR, a nitrate group reduces the saturation vapor pressure of a compound by 2.5 orders of magnitude, instead of 0.7732 orders of magnitude in Li (which is similar to a hydroxyl group). The difference is due to the different training data sets used to create the parameterizations. The MHR and DMT parameterizations are similar in the bN values but different in the way they treat other functional groups containing oxygen atoms. DMT assumes all oxygen atoms except those from −NO3 are from −OH and =O functional groups, while MHR was tuned for −OOH functional groups, and the bO (= 0.2) is much smaller than b−OH (= −2.23) and b=O (= −0.935) in DMT. As MHR works better for the α-pinene + O3 system and DMT works better for the α-pinene + NO3 system, it is possible that the α-pinene + O3 system has more −OOH and the α-pinene + NO3 system has more −OH and =O. However, it is worth noting that, with the assumption in DMT, the level of unsaturation (DBE) arises from carbonyl and nitrate groups. Ring structures (which are highly possible in many α-pinene and β-caryophyllene oxidation products) are treated as the former. This could lead to underestimation of the volatility. The PRK parameterization, on the other hand, is based on experimental methods, specifically on HOMs from α-pinene ozonolysis, and displays a simple dependence on the individual atoms instead of specific functional groups (like the nitrate group). With this in mind, it is not surprising that PRK best captures α-pinene. Finally, the uncertainty of the effect of nitrogen groups on the saturation vapor pressures of organic species is also to be expected, given the lack of quantitative experimental data to use to train the parameterizations (see, e.g., Pankow and Asher, 2008).
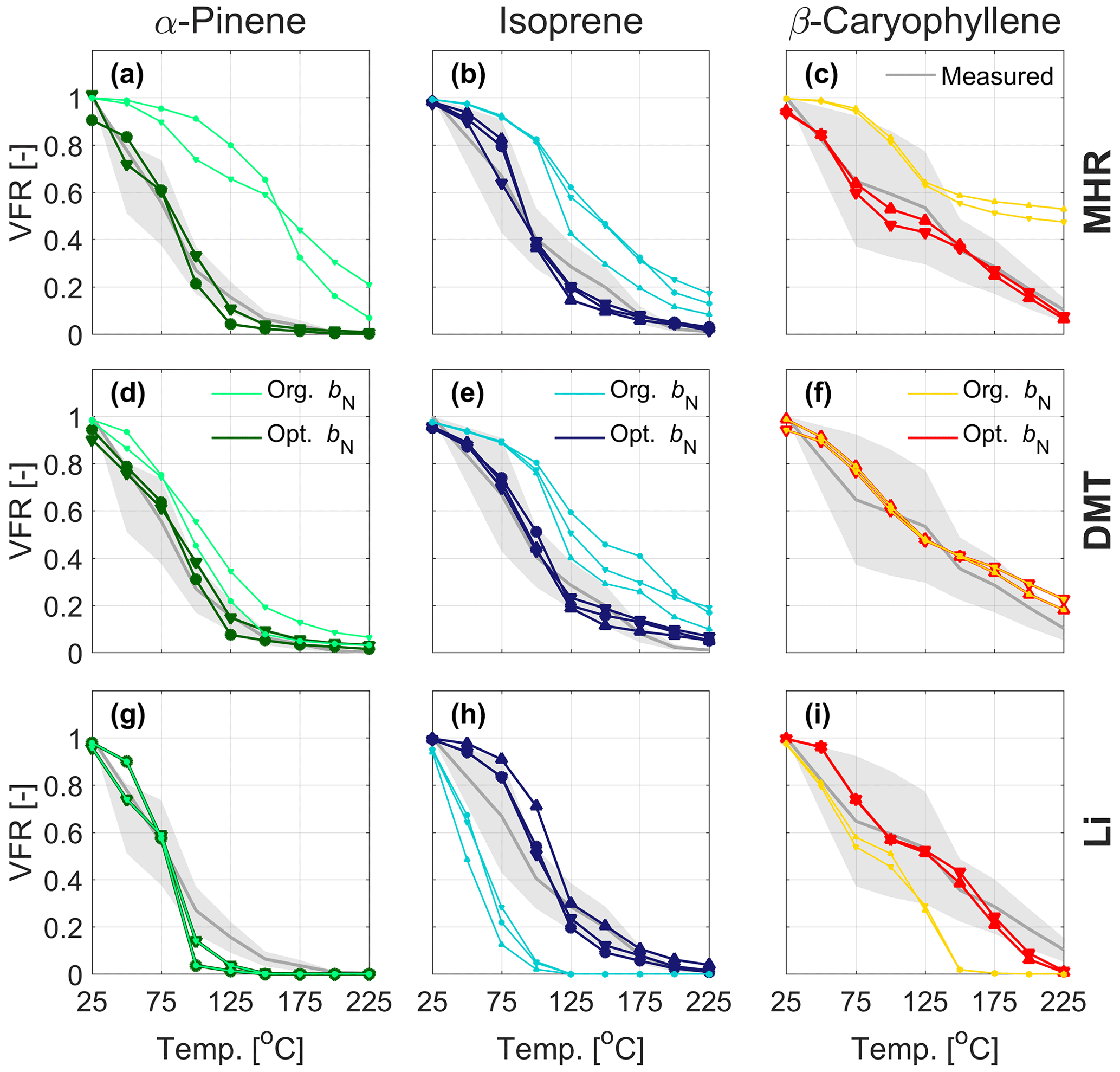
Figure 5Evaporation in the TD predicted by the kinetic model using original parameterizations (MHR, DMT, and Li) and after optimizing the nitrate influence for the nitrate oxidation of α-pinene (a, d, g), isoprene (b, e, h), and β-caryophyllene (c, f, i). The symbols () mark the experiments in chronological order, as indicated in Table 1, during which the VBS data were collected. For comparison, the experimental evaporation (in gray with the shaded areas denoting the uncertainty of the VTDMA measurement) are displayed.
Therefore, we investigated the effect of modifying the parameter bN in steps for MHR, DMT, and Li for better reproducing the observations (see Table 2 and Fig. 5). When applying the MHR parameterization, the nitrate-induced reduction in saturation vapor pressure needed to be substantially reduced for all three systems to get predictions closer to the observations. For the isoprene system, bN values between 1.0 and 1.5 provided the best fit to the observations (Fig. 5b), still however indicating a saturation vapor pressure reducing effect of the nitrate group. For the α-pinene and β-caryophyllene systems, however, the bN values needed to be reduced to negative values (from −0.75 to −0.5) to fit the observations (Fig. 5a, c). The negative values would indicate that the nitrate group would increase the volatility. With DMT, a reduction of bN from 2.23 (original) to 1–1.5 would lead a better simulation for the α-pinene and isoprene systems. For the β-caryophyllene system, the original value did the best job. In the case of Li, the original value of bN= 0.7732 already performed exceptionally well for the nitrate oxidation of α-pinene, and hence no adjustment was warranted here (Fig. 5g). For isoprene and β-caryophyllene, the saturation vapor pressure reduction by the nitrate group needed to be increased to 1.8–2.8 orders of magnitude, indicating similar nitrate influence as previously estimated by Pankow and Asher (2008). Since none of the bN values were able to reproduce the data from different precursors for MHR, DMT, or Li, it is clear that simply adjusting the bN value would not resolve the discrepancy we observed between the measured and simulated VFR based on the chemical composition. The logC* is closely related to the other interaction terms, e.g., the carbon–carbon interaction term and the carbon–oxygen non-ideality. This finding indicates complexity of the interactions between different functional groups and warrants further and more detailed studies on nitrate-containing systems.
Table 2Optimal nitrate influence (bN) for MHR, DMT, and Li tuned for the α-pinene, isoprene, and β-caryophyllene + NO2 systems.
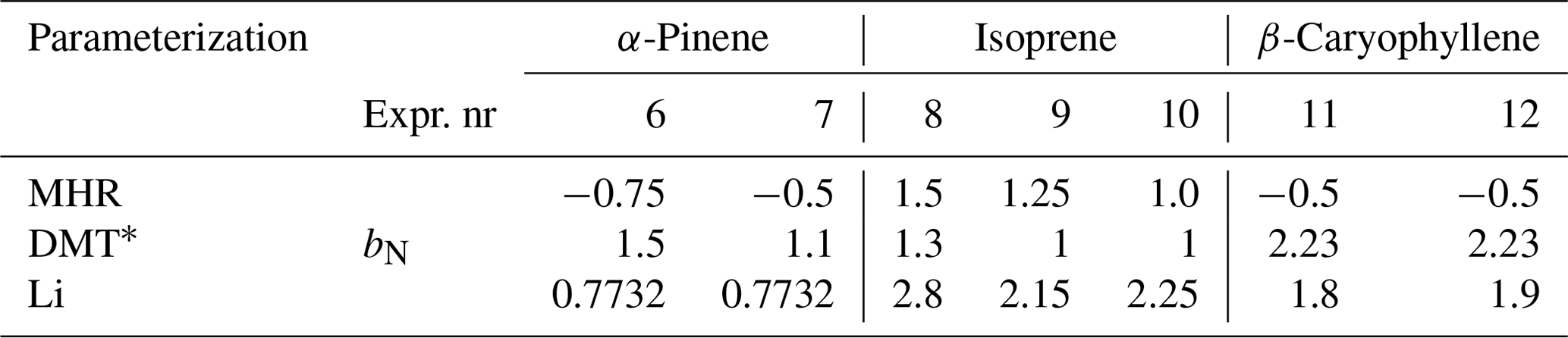
* All b values for DMT were multiplied by −1, in order to keep the same form as for the other parameterizations.
Here we studied the volatility of SOA particles from nitrate oxidation of α-pinene, isoprene, and β-caryophyllene using a set of experimental techniques, presenting a cohesive picture of the three studied precursors. The experimental data from this work as well as the comparison to current literature reveal that under our experimental conditions, the SOA formed from nitrate oxidation of α-pinene is more volatile than the corresponding α-pinene ozonolysis products. Further, the nitrate oxidation of β-caryophyllene produces less volatile SOA than α-pinene and isoprene.
Besides observing the volatility directly through following the evaporation of the particles, the particle-phase composition observed by the FIGAERO-CIMS was used to predict the volatilities present in the SOA mixtures. Four different parameterizations (modified Daumit et al., 2013; modified Li et al., 2016; Mohr et al., 2019; and Peräkylä et al., 2020) were used to convert the SOA mass spectra to volatility distributions (VBS). The typical bulk volatilities of SOA from individual systems varied over several orders of magnitude with the four parameterizations, as also reported by R. Wu et al. (2021) for the isoprene + NO3 system. For all systems, the parameterization by Mohr et al. (2019) produced volatilities mainly in the ELVOC range, while Peräkylä et al. (2020) produced narrow volatility distributions mainly in the SVOC range. Modified Daumit et al. (2013) and modified Li et al. (2016) produced volatilities between the two other methods.
When used as input to a kinetic model, the VBS predicted by Mohr et al. (2019) reproduced the evaporation of α-pinene ozonolysis generated SOA most accurately out of the four parameterizations. However, when simulating the evaporation of the nitrate-initiated SOA, no parameterization performed perfectly for all three systems. The Mohr et al. (2019) parameterization was found to generally substantially underpredict the volatility and evaporation for all three precursors, while the Peräkylä et al. (2020) parameterization generally overpredicted the volatility and evaporation for all the systems, and the modified Li et al. (2016) and modified Daumit et al. (2013) were in between. This warrants a thorough re-evaluation of the parameter describing the magnitude of the saturation vapor pressure reduction due to nitrogen-containing functional groups.
By comparing the volatility information from different parameterizations with that derived from temperature-dependent evaporation in VTDMA and isothermal evaporation chambers, our study suggests that the SOA from the nitrate oxidation of α-pinene and isoprene contains a wide range of volatilities from the ELVOC to the SVOC range, with LVOCs dominating the composition. The SOA from the nitrate oxidation of β-caryophyllene, on the other hand, seems to be dominated by volatilities in the ELVOC and LVOC range. Our nitrate system represents SOA formation conditions where reactions between peroxy radicals and between peroxy radical and NO3 are favored and oligomers dominate. Further investigations to probe different ambient conditions are warranted to complete the picture.
The data sets are available upon request from the corresponding authors. The data sets will also be published on 31 August 2023 in the open-access database of the Bolin Centre for Climate Research (https://doi.org/10.17043/graham-2023-no3-1, Graham et al., 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-7347-2023-supplement.
ELG, CW, DMB, IR, and CM designed the study. Chamber experiments were carried out by ELG, CW, DMB, SH, AB, and CM. Data analysis and interpretation was performed by ELG, CW, DMB, IR, and CM. ELG and CW wrote the paper, with input from all the co-authors. All co-authors read and commented on the paper.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We would also like to thank Rene Richter for his assistance with installing and assembling the experimental setup.
Financial support from the European
Union’s Horizon 2020 research and innovation program (project FORCeS under grant agreement no. 821205), the European Research Council (Consolidator grant INTEGRATE no. 865799), and the Knut and Alice Wallenberg Foundation (Wallenberg Academy Fellowship projects AtmoRemove no. 2015.0162, AtmoCLOUD no. 2021.0169, and CLOUDFORM no. 2017.0165) is gratefully acknowledged. Financial support from the Max Planck Society is also gratefully acknowledged. We would also like to thank the European Union’s Horizon 2020 research and innovation program through the EUROCHAMP-2020 Infrastructure Activity under grant agreement no. 730997.
The article processing charges for this open-access publication were covered by Stockholm University.
This paper was edited by Thomas Berkemeier and reviewed by Gabriel Isaacman-VanWertz and one anonymous referee.
Bannan, T. J., Le Breton, M., Priestley, M., Worrall, S. D., Bacak, A., Marsden, N. A., Mehra, A., Hammes, J., Hallquist, M., Alfarra, M. R., Krieger, U. K., Reid, J. P., Jayne, J., Robinson, W., McFiggans, G., Coe, H., Percival, C. J., and Topping, D.: A method for extracting calibrated volatility information from the FIGAERO-HR-ToF-CIMS and its experimental application, Atmos. Meas. Tech., 12, 1429–1439, https://doi.org/10.5194/amt-12-1429-2019, 2019.
Bates, K. H., Burke, G. J. P., Cope, J. D., and Nguyen, T. B.: Secondary organic aerosol and organic nitrogen yields from the nitrate radical (NO3) oxidation of alpha-pinene from various RO2 fates, Atmos. Chem. Phys., 22, 1467–1482, https://doi.org/10.5194/acp-22-1467-2022, 2022.
Bell, D. M., Imre, D., T. Martin, S., and Zelenyuk, A.: The properties and behavior of α-pinene secondary organic aerosol particles exposed to ammonia under dry conditions, Phys. Chem. Chem. Phys., 19, 6497–6507, https://doi.org/10.1039/C6CP08839B, 2017.
Bell, D. M., Wu, C., Bertrand, A., Graham, E., Schoonbaert, J., Giannoukos, S., Baltensperger, U., Prevot, A. S. H., Riipinen, I., El Haddad, I., and Mohr, C.: Particle-phase processing of α-pinene NO3 secondary organic aerosol in the dark, Atmos. Chem. Phys., 22, 13167–13182, https://doi.org/10.5194/acp-22-13167-2022, 2022.
Berkemeier, T., Takeuchi, M., Eris, G., and Ng, N. L.: Kinetic modeling of formation and evaporation of secondary organic aerosol from NO3 oxidation of pure and mixed monoterpenes, Atmos. Chem. Phys., 20, 15513–15535, https://doi.org/10.5194/acp-20-15513-2020, 2020.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Buchholz, A., Ylisirniö, A., Huang, W., Mohr, C., Canagaratna, M., Worsnop, D. R., Schobesberger, S., and Virtanen, A.: Deconvolution of FIGAERO–CIMS thermal desorption profiles using positive matrix factorisation to identify chemical and physical processes during particle evaporation, Atmos. Chem. Phys., 20, 7693–7716, https://doi.org/10.5194/acp-20-7693-2020, 2020.
Chung, S. H. and Seinfeld, J. H.: Global distribution and climate forcing of carbonaceous aerosols, J. Geophys. Res., 107, 4407, https://doi.org/10.1029/2001JD001397, 2002.
Daumit, K. E., Kessler, S. H., and Kroll, J. H.: Average chemical properties and potential formation pathways of highly oxidized organic aerosol, Faraday Discuss., 165, 181–202, https://doi.org/10.1039/C3FD00045A, 2013.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Epstein, S. A., Riipinen, I., and Donahue, N. M.: A Semiempirical Correlation between Enthalpy of Vaporization and Saturation Concentration for Organic Aerosol, Environ. Sci. Technol., 44, 743–748, https://doi.org/10.1021/es902497z, 2010.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Graham, E. L., Wu, C., Bell, D. M., Bertrand, A., Haslett, S. L., Baltensperger, U., El Haddad, I., Krejci, R., Riipinen, I., and Mohr, C.: Volatility of aerosol particles from NO3 oxidation of α-pinene, isoprene and β-caryophyllene, Bolin [data set], https://doi.org/10.17043/graham-2023-no3-1, 2023.
Häkkinen, S. A. K., Äijälä, M., Lehtipalo, K., Junninen, H., Backman, J., Virkkula, A., Nieminen, T., Vestenius, M., Hakola, H., Ehn, M., Worsnop, D. R., Kulmala, M., Petäjä, T., and Riipinen, I.: Long-term volatility measurements of submicron atmospheric aerosol in Hyytiälä, Finland, Atmos. Chem. Phys., 12, 10771–10786, https://doi.org/10.5194/acp-12-10771-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Holzinger, R., Kasper-Giebl, A., Staudinger, M., Schauer, G., and Röckmann, T.: Analysis of the chemical composition of organic aerosol at the Mt. Sonnblick observatory using a novel high mass resolution thermal-desorption proton-transfer-reaction mass-spectrometer (hr-TD-PTR-MS), Atmos. Chem. Phys., 10, 10111–10128, https://doi.org/10.5194/acp-10-10111-2010, 2010.
Hong, J., Äijälä, M., Häme, S. A. K., Hao, L., Duplissy, J., Heikkinen, L. M., Nie, W., Mikkilä, J., Kulmala, M., Prisle, N. L., Virtanen, A., Ehn, M., Paasonen, P., Worsnop, D. R., Riipinen, I., Petäjä, T., and Kerminen, V.-M.: Estimates of the organic aerosol volatility in a boreal forest using two independent methods, Atmos. Chem. Phys., 17, 4387–4399, https://doi.org/10.5194/acp-17-4387-2017, 2017.
Isaacman-VanWertz, G. and Aumont, B.: Impact of organic molecular structure on the estimation of atmospherically relevant physicochemical parameters, Atmos. Chem. Phys., 21, 6541–6563, https://doi.org/10.5194/acp-21-6541-2021, 2021.
Jaoui, M., Kleindienst, T. E., Docherty, K. S., Lewandowski, M., and Offenberg, J. H.: Secondary organic aerosol formation from the oxidation of a series of sesquiterpenes: α-cedrene, β-caryophyllene, α-humulene and á-farnesene with O3, OH and NO3 radicals, Environ. Chem., 10, 178–193, https://doi.org/10.1071/EN13025, 2013.
Karnezi, E., Riipinen, I., and Pandis, S. N.: Measuring the atmospheric organic aerosol volatility distribution: a theoretical analysis, Atmos. Meas. Tech., 7, 2953–2965, https://doi.org/10.5194/amt-7-2953-2014, 2014.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett, 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Lee, B.-H., Pierce, J. R., Engelhart, G. J., and Pandis, S. N.: Volatility of secondary organic aerosol from the ozonolysis of monoterpenes, Atmos. Environ., 45, 2443–2452, https://doi.org/10.1016/j.atmosenv.2011.02.004, 2011.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Mohr, C., Thornton, J. A., Heitto, A., Lopez-Hilfiker, F. D., Lutz, A., Riipinen, I., Hong, J., Donahue, N. M., Hallquist, M., Petäjä, T., Kulmala, M., and Yli-Juuti, T.: Molecular identification of organic vapors driving atmospheric nanoparticle growth, Nat. Commun., 10, 4442, https://doi.org/10.1038/s41467-019-12473-2, 2019.
Nah, T., Sanchez, J., Boyd, C., and Ng, N. L.: Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
O'Brian, R. E., Laskin, A., Laskin, J., Rubitschun, C. L., Surratt, J. D., and Goldstein, A. H.: Molecular characterization of S- and N-containing organic constituents in ambient aerosols by negative ion mode high-resolution Nanospray Desorption Electrospray Ionization Mass Spectrometry: CalNex 2010 field study, J. Geophys. Res.-Atmos., 119, 706–720, https://doi.org/10.1002/2014JD021955, 2014.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Peräkylä, O., Riva, M., Heikkinen, L., Quéléver, L., Roldin, P., and Ehn, M.: Experimental investigation into the volatilities of highly oxygenated organic molecules (HOMs), Atmos. Chem. Phys., 20, 649–669, https://doi.org/10.5194/acp-20-649-2020, 2020.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Moènik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Poling, B. E., Prausnitz, J. M., and O'Connell, J. P.: The properties of gases and liquids, 5th edn., McGra-Hill, ISBN 0070116822, 2001.
Pospisilova, V., Lopez-Hilfiker, F. D., Bell, D. M., El Haddad, I., Mohr, C., Huang, W., Heikkinen, L., Xiao, M., Dommen, J., Prevot, A. S. H., Baltensperger, U., and Slowik, J. G.: On the fate of oxygenated organic molecules in atmospheric aerosol particles, Sci. Adv., 6, eaax8922, https://doi.org/10.1126/sciadv.aax8922, 2020.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Riipinen, I., Pierce, J. R., Donahue, N. M., and Pandis, S. N.: Equilibration time scales of organic aerosol inside thermodenuders: Evaporation kinetics versus thermodynamics, Atmos. Environ., 44, 597–607, https://doi.org/10.1016/j.atmosenv.2009.11.022, 2010.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Thornton, J. A., Mohr, C., Schobesberger, S., D'Ambro, E. L., Lee, B. H., and Lopez-Hilfiker, F. D.: Evaluating Organic Aerosol Sources and Evolution with a Combined Molecular Composition and Volatility Framework Using the Filter Inlet for Gases and Aerosols (FIGAERO), Acc. Chem. Res., 53, 1415–1426, https://doi.org/10.1021/acs.accounts.0c00259, 2020.
Tritscher, T., Dommen, J., DeCarlo, P. F., Gysel, M., Barmet, P. B., Praplan, A. P., Weingartner, E., Prévôt, A. S. H., Riipinen, I., Donahue, N. M., and Baltensperger, U.: Volatility and hygroscopicity of aging secondary organic aerosol in a smog chamber, Atmos. Chem. Phys., 11, 11477–11496, https://doi.org/10.5194/acp-11-11477-2011, 2011.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Vaden, T. D., Imre, D., Beranek, J., Shrivastava, M., and Zelenyuk, A.: Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol, P. Natl. Acad. Sci., 108, 2190–2195, https://doi.org/10.1073/pnas.1013391108, 2011.
Wilson, J., Imre, D., Beránek, J., Shrivastava, M., and Zelenyuk, A.: Evaporation Kinetics of Laboratory-Generated Secondary Organic Aerosols at Elevated Relative Humidity, Environ. Sci. Technol., 49, 243–249, https://doi.org/10.1021/es505331d, 2015.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wu, C., Bell, D. M., Graham, E. L., Haslett, S., Riipinen, I., Baltensperger, U., Bertrand, A., Giannoukos, S., Schoonbaert, J., El Haddad, I., Prevot, A. S. H., Huang, W., and Mohr, C.: Photolytically induced changes in composition and volatility of biogenic secondary organic aerosol from nitrate radical oxidation during night-to-day transition, Atmos. Chem. Phys., 21, 14907–14925, https://doi.org/10.5194/acp-21-14907-2021, 2021.
Wu, R., Vereecken, L., Tsiligiannis, E., Kang, S., Albrecht, S. R., Hantschke, L., Zhao, D., Novelli, A., Fuchs, H., Tillmann, R., Hohaus, T., Carlsson, P. T. M., Shenolikar, J., Bernard, F., Crowley, J. N., Fry, J. L., Brownwood, B., Thornton, J. A., Brown, S. S., Kiendler-Scharr, A., Wahner, A., Hallquist, M., and Mentel, T. F.: Molecular composition and volatility of multi-generation products formed from isoprene oxidation by nitrate radical, Atmos. Chem. Phys., 21, 10799–10824, https://doi.org/10.5194/acp-21-10799-2021, 2021.
Zhao, D., Pullinen, I., Fuchs, H., Schrade, S., Wu, R., Acir, I.-H., Tillmann, R., Rohrer, F., Wildt, J., Guo, Y., Kiendler-Scharr, A., Wahner, A., Kang, S., Vereecken, L., and Mentel, T. F.: Highly oxygenated organic molecule (HOM) formation in the isoprene oxidation by NO3 radical, Atmos. Chem. Phys., 21, 9681–9704, https://doi.org/10.5194/acp-21-9681-2021, 2021.