the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Mar 2023
| 06 Mar 2023
Characterization of volatile organic compounds and submicron organic aerosol in a traffic environment
Sanna Saarikoski
Heidi Hellén
Arnaud P. Praplan
Simon Schallhart
Petri Clusius
Jarkko V. Niemi
Anu Kousa
Toni Tykkä
Rostislav Kouznetsov
Minna Aurela
Laura Salo
Topi Rönkkö
Luis M. F. Barreira
Liisa Pirjola
Hilkka Timonen
Urban air consists of a complex mixture of gaseous and particulate species from anthropogenic and biogenic sources that are further processed in the atmosphere. This study investigated the characteristics and sources of volatile organic compounds (VOCs) and submicron organic aerosol (OA) in a traffic environment in Helsinki, Finland, in late summer. The anthropogenic VOCs (aVOCs; aromatic hydrocarbons) and biogenic VOCs (bVOCs; terpenoids) relevant for secondary-organic-aerosol formation were analyzed with an online gas chromatograph mass spectrometer, whereas the composition and size distribution of submicron particles was measured with a soot particle aerosol mass spectrometer.
This study showed that aVOC concentrations were significantly higher than bVOC concentrations in the traffic environment. The largest aVOC concentrations were measured for toluene (campaign average of 1630 ng m−3) and xylene (campaign average of 1070 ng m−3), while the dominating bVOC was α-pinene (campaign average of 200 ng m−3). For particle-phase organics, the campaign-average OA concentration was 2.4 µg m−3. The source apportionment analysis extracted six factors for OA. Three OA factors were related to primary OA sources – traffic (24 % of OA, two OA types) and a coffee roastery (7 % of OA) – whereas the largest fraction of OA (69 %) consisted of oxygenated OA (OOA). OOA was divided into less oxidized semi-volatile OA (SV-OOA; 40 % of OA) and two types of low-volatility OA (LV-OOA; 30 %).
The focus of this research was also on the oxidation potential of the measured VOCs and the association between VOCs and OA in ambient air. Production rates of the oxidized compounds (OxPR) from the VOC reactions revealed that the main local sources of the oxidation products were O3 oxidation of bVOCs (66 % of total OxPR) and OH radical oxidation of aVOCs and bVOCs (25 % of total OxPR). Overall, aVOCs produced a much smaller portion of the oxidation products (18 %) than bVOCs (82 %). In terms of OA factors, SV-OOA was likely to originate from biogenic sources since it correlated with an oxidation product of monoterpene, nopinone. LV-OOA consisted of highly oxygenated long-range or regionally transported OA that had no correlation with local oxidant concentrations as it had already spent several days in the atmosphere before reaching the measurement site.
In general, the main sources were different for VOCs and OA in the traffic environment. Vehicle emissions impacted both VOC and OA concentrations. Due to the specific VOCs attributed to biogenic emissions, the influence of biogenic emissions was more clearly detected in the VOC concentrations than in OA. In contrast, the emissions from the local coffee roastery had a distinctive mass spectrum for OA, but they could not be seen in the VOC measurements due to the measurement limitations for the large VOC compounds. Long-range transport increased the OA concentration and oxidation state considerably, while its effect was observed less clearly in the VOC measurements due to the oxidation of most VOC in the atmosphere during the transport. Overall, this study revealed that in order to properly characterize the impact of different emission sources on air quality, health, and climate, it is of importance to describe both gaseous and particulate emissions and understand how they interact as well as their phase transfers in the atmosphere during the aging process.
- Article
(4562 KB) - Full-text XML
-
Supplement
(4111 KB) - BibTeX
- EndNote




Anthropogenic air pollution is one of the greatest environmental issues, with broad impacts on air quality, climate, and health (Lelieveld et al., 2015; Schraufnagel, 2020; IPCC, 2021). Impaired air quality has been estimated to be responsible for a large portion of annual morbidity and mortality around the world due to several respiratory, cardiovascular, immune, and nervous system diseases (e.g., Dominici et al., 2006; Genc et al., 2012; Glencross et al., 2020). In 2019, air pollution accounted for an estimated 6.7 million deaths, about 12 % of all deaths registered in the same year (Brauer et al., 2021). The spatial and temporal variation in gaseous and particulate pollutants from anthropogenic sources depends largely on source type, atmospheric lifetime of pollutants, and local meteorology. Biogenic emissions also play an important role in atmospheric pollution. For example, it has been shown that biogenic precursors can form secondary organic aerosol (SOA), which can be enhanced by the presence of anthropogenic pollutants such as nitrogen oxides (NOx) and sulfur dioxide (SO2) (Edney et al., 2005; Kroll et al., 2006; Budisulistiorini et al., 2015).
Volatile organic compounds (VOCs) are particularly important atmospheric gaseous pollutants. Globally, anthropogenic VOCs (aVOCs) contribute approximately 14 % to the total VOC emission, while the contribution of biogenic VOCs (bVOCs) is over 80 % (Guenther et al., 2012; Crippa et al., 2020). In urban environments, the contribution of aVOCs is much larger. While traffic has been historically the main contributor to aVOCs, its significance is decreasing in developed countries due to the imposed regulations (e.g., EEA, 2019). Therefore, other VOC sources, like the use of volatile chemical products (VCPs), are becoming more important and are suspected to be responsible for already half of the aVOC emissions in urban areas (McDonald et al., 2018; Coggon et al., 2021; Gkatzelis et al., 2021). This can be seen in the study of Karl et al. (2018), where VOC fluxes were measured above a city. From their measurements, several sources of VCPs were identified, e.g., cleaning agents, paint, human emissions (skin), healthcare products, and disinfectants. The main source of bVOCs in urban environments is green urban infrastructure (e.g., parks, green roofs, forests), which is used in many cities not only as recreation zones but also for heat and air pollution mitigation and water interception (Livesley et al., 2016; Fitzky et al., 2019). However, it should be noted that also some VCPs can be included in bVOCs as, for example, cleaning agents and personal care products contain the same compounds as naturally emitted bVOCs. Once emitted into the atmosphere, VOCs get oxidized by either the hydroxyl radical (OH), ozone (O3), or the nitrate radical (NO3). The oxidation products vary greatly depending on the VOC composition and atmospheric conditions, but often they have a lower volatility than their precursors and are potential contributors to the formation and/or growth of SOA.
SOA can be produced from both aVOCs and bVOCs via new particle formation or the condensation of oxidation products on existing particles. The volatility and oxidation of organics continues further in the particle phase with photochemical processing. For example, in a study performed in Mexico City, semi-volatile oxygenated organic aerosol (SV-OOA) was the dominant organic aerosol (OA) type, but the oxygen-to-carbon ratio (O:C) and the contribution of low-volatility oxygenated organic aerosol (LV-OOA) increased with OA aging when measured with the aerosol mass spectrometer (AMS; Jimenez et al., 2009). A similar transformation has also been observed in the laboratory studies. SOA formed from the oxidation of α-pinene became more similar to ambient SV-OOA after some aging, and then with continued oxidation, it evolved to be similar to ambient LV-OOA (Jimenez et al., 2009). These results suggest that oxygenated organic aerosol (OOA) components become more chemically similar with photochemical aging regardless of the original source of OOA; however, this finding could be partially caused by the limitations of the AMS detection due to the substantial fragmentation of the chemical species. In addition to secondary production, OA can also be emitted directly from the sources (primary OA, POA). The main sources of POA in urban areas are traffic, residential biomass combustion, industry, and energy production (e.g., Crippa et al., 2014; Timonen et al., 2013; Zhang et al., 2019). However, it should be noted that these sources also emit gaseous precursors for SOA.
To understand the secondary-aerosol-formation processes in urban areas, detailed information about the chemistry of both gaseous compounds and primary and secondary particulate species as well as local meteorology is needed. Harrison (2018) underlined that urban environments usually have high levels of primary emissions with strong concentration gradients as mixing processes are heavily influenced by the presence of buildings and potentially by the urban heat island. Reaction timescales are therefore shorter in urban areas compared to the well-mixed regional atmosphere. Kim et al. (2018) found that in Seoul, Korea, the formation of LV-OOA and sulfate was mainly promoted by elevated ozone concentrations and photochemical reactions during daytime, whereas SV-OOA and nitrate formation were attributed to both nocturnal processing and daytime photochemical reactions. Yu et al. (2019) identified three bVOCs (α-pinene, limonene, and camphene) and one aVOC (styrene) as the possible key VOC precursors to particulate organic nitrates in the megacity of Shenzhen, China. Sjostedt et al. (2011) concluded that biogenic precursors contribute significantly to the total amount of SOA formation, even during periods of urban outflow. They found that the importance of aromatic precursors was more difficult to assess given that their sources are likely to be localized and thus of variable impact at the sampling location.
The aim of this study was to investigate the characteristics and sources of VOCs and particulate submicron (<1 µm in diameter) OA in a traffic environment in late summer. For the first time in Helsinki, a wide range of aVOCs and bVOCs were analyzed with an online gas chromatograph–mass spectrometer (GC–MS). The 1 h time resolution for the VOC data enabled the study of short-term variability in the concentrations and allowed for comparisons with particle measurements conducted with a real-time aerosol mass spectrometer. The OA mass spectra from the AMS were further analyzed by positive matrix factorization (PMF) for the sources and properties of OA. Moreover, the oxidation of VOCs was investigated thoroughly by calculating the production rates of the VOC oxidation products, and their contribution to SOA formation was assessed. This study provides novel information on the sources of anthropogenic and biogenic VOCs and OA in an urban environment and elucidates atmospheric oxidation processes and SOA formation in a traffic environment. This information is currently highly needed by air quality authorities and modelers all over the world to improve urban air quality as well as the models for aerosol dynamics and atmospheric chemistry.
2.1 Measurement site
The measurement campaign was conducted from 14 August to 13 September 2019 at the Helsinki supersite measurement station (street address: Mäkelänkatu 50; Fig. S1 in the Supplement), Finland. The station is located at the curbside of the street and is maintained by the Helsinki Region Environmental Services Authority (HSY). The street consists of six lanes for motorized traffic, two rows of trees, two tram lanes, and two sidewalks, for a total width of 42 m (Hietikko et al., 2018). Mäkelänkatu is one of the busiest traffic sites in the Helsinki city center, with a traffic density of about 28 000 vehicles per weekday and a heavy-duty vehicle share of 10 % (statistics from the City of Helsinki). Long-term concentrations, composition, and trends of submicron particulate matter (PM1) at the Helsinki supersite have been presented in Barreira et al. (2021), and the spatial variability in air pollutant concentrations at the measurement site has been investigated previously in Järvi et al. (2023).
2.2 Instruments
2.2.1 Online TD–GC–MS
The concentrations of VOCs were measured with an in situ thermal desorption–gas chromatograph–mass spectrometer (TD–GC–MS; Perkin Elmer Inc., Waltham, US). Studied compounds were hydrocarbons with 5 to 15 carbon atoms, which are known to be important SOA precursors. Based on their most probable origin, compounds were classified as aVOCs and bVOCs even though some bVOCs are also known to have anthropogenic sources. The studied aVOCs consisted of aromatic hydrocarbons (benzene, toluene, ethylbenzene, p/m-xylene, styrene, o-xylene, 3-ethyltoluene, 4-ethyltoluene, 1,3,5-trimethylbenzene, 2-ethyltoluene, 1,2,4-trimethylbenzene, and 1,2,3-trimethylbenzene). The analyzed bVOCs were isoprene, monoterpenoids (α-pinene, camphene, β-pinene, Δ3-carene, p-cymene, 1,8-cineol, and limonene), sesquiterpenes (longicyclene, iso-longifolene, β-caryophyllene, and α-humulene), and an oxidation product of β-pinene (nopinone).
VOCs were collected into the cold trap (Tenax TA 60–80/Carbopack B 60–80) of the thermal desorption unit (TurboMatrix 350, Perkin-Elmer Inc., Waltham, US) connected to a gas chromatograph (Clarus 680, Perkin-Elmer Inc., Waltham, US) coupled to a mass spectrometer (Clarus SQ 8 T, Perkin-Elmer Inc., Waltham, US). The hydrophobic cold trap was kept at 25 ∘C for the removal of humidity; 30 min samples were taken with a 1 h time resolution and a flow rate of 40 mL min−1. The main flow going to the instruments through fluorinated ethylene propylene (FEP) tubing (ca. 5 m length, i.d. ) was approximately 0.8 L min−1. To also enable the measurements of highly ozone-reactive terpenes, a heated stainless-steel tube was connected to the main flow path to remove ozone before sampling (see Hellén et al., 2012a). For calibration, standards were injected as methanol solutions into sorbent tubes (Tenax TA 60–80/Carbopack B 60–80), methanol was flushed away in nitrogen (6.0) flow, and the tubes were thermally desorbed and analyzed as samples. Five-point calibration curves were used. For isoprene calibration, a gas standard from National Physical Laboratories (UK) was used. The method has been described in detail by Helin et al. (2020).
2.2.2 SP-AMS
Size-resolved chemical composition of submicron particles, i.e., organics, sulfate, nitrate, ammonium, chloride, and refractory black carbon (rBC), was determined with a soot particle aerosol mass spectrometer (SP-AMS; Aerodyne Research Inc., Billerica, US; Onasch et al., 2012). The SP-AMS collected data with 2 min time resolution, of which half of the time the instrument operated in a mass spectra mode (mass concentrations) and half of the time in a particle time-of-flight (PToF) mode (mass size distributions). The measured particle size range of the SP-AMS is roughly from 40 nm to 1 µm. A collection efficiency (CE) of 1 was applied to the data as with this value total PM1 from the aethalometer (equivalent black carbon, eBC) and the SP-AMS (excluding rBC) was comparable with that from the differential mobility particle sizer (DMPS) operating at the site. The CE of 1 was larger than that usually calculated for the AMS (Middlebrook et al., 2012) or SP-AMS (Onasch et al., 2012), which could be due to the inaccuracy in the ammonium nitrate calibration. A relative ionization efficiency (RIE) of 0.1 was used for rBC based on the calibration with REGAL black (REGAL 400R black pigment, Cabot Corp.). However, due to the considerable uncertainties related to the quantification of rBC with the SP-AMS (e.g., imperfect laser beam alignment), black carbon (BC) concentrations presented in this paper are taken from the aethalometer. The SP-AMS data were analyzed with IGOR 6.37, SQRL 1.62A, and PIKA 1.22A software.
2.2.3 Aethalometer, DMPS, and auxiliary measurements
Equivalent black carbon measurements were conducted using a dual-spot aethalometer (AE33, Aerosol d.o.o., Ljubljana, Slovenia), which allows real-time measurement of aerosol light absorption at seven wavelengths (370–950 nm; Drinovec et al., 2015). The sampling flow rate was set to 5 L min−1, and the inlet cut-off size was 1 µm (sharp-cut cyclone, BGI model SCC1.197). The time resolution was 1 min. The filter tape was a M8060 and consisted of tetrafluoroethylene (TFE)-coated glass fiber filters.
The sources of eBC can be examined by analyzing the absorption spectra of light-absorbing material in particles as particles from fossil fuel and biomass combustion are characterized by different spectral dependencies. The source apportionment method based on the light absorption at different wavelengths is usually called an aethalometer model (Sandradewi et al., 2008) based on the multi-wavelength optical instrument typically used in the measurements. In the aethalometer model, BC concentrations from wood burning (BCwb) and fossil fuel combustion (BCff) are estimated by the following equations:
and
where babs is an aerosol light absorption coefficient given by the AE33 at the wavelengths of 470 and 950 nm. Absorption Ångström exponents (α) of 1.1 and 1.6 were applied to fossil fuel (αff) and wood burning (αwb), respectively, as those values have been previously optimized for the measurement site (Helin et al., 2018).
Submicron particle number size distributions were measured using a DMPS (Knutson and Whitby, 1975). The DMPS includes a differential mobility analyzer (DMA; Vienna type), used for particle sizing, and a condensation particle counter (CPC; A20 Airmodus, Helsinki, Finland) for obtaining particle number concentrations for each size bin. The time resolution of the DMPS was 9 min, and the scanned particle size range was 6 to 1000 nm (mobility diameter, Dm), but due to a power source issue, the three smallest stages of the DMPS were excluded from data, and the size distribution was calculated only for the size range of 10–1000 nm. The DMPS size distribution was compared to that of the electrical low-pressure impactor (ELPI+, Dekati Ltd., Tampere, Finland; Järvinen et al., 2014), which operated at the site during the last week of the measurement campaign (5–12 September 2019). The number size distributions from the DMPS and ELPI+ were similar, indicating that the DMPS data were reliable after excluding the smallest stages of the DMPS. The DMPS number size distribution was converted to the mass size distribution by assuming spherical particles and a particle density of 1.42 g cm−3, which has been shown to be the average density of submicron particles at the site (Barreira et al., 2021).
Basic air quality parameters were also measured at the site. The concentration of NOx was measured by an APNA-370 analyzer (Horiba, Kyoto, Japan), O3 by using an ambient O3 monitor (APOA-370, Horiba, Kyoto, Japan), CO by APMA-360 (Horiba, Kyoto, Japan), and PM2.5 and PM10 concentrations by a tapered element oscillating microbalance (1405 TEOMTM, Thermo Fischer Scientific, Waltham, US) with a time resolution of 1 min. The mass concentration of coarse particles (PM2.5–10) was calculated by subtracting PM2.5 from PM10. Of meteorological parameters, temperature (T), relative humidity (RH), and precipitation were measured at the Helsinki supersite, while wind speed and wind direction were measured at a meteorological station above the roof level (53 m above the land surface) located approximately 900 m northwest of the measurement site. The mixing height was calculated using the model (MPP-FMI) presented by Karppinen et al. (2000). In a previous study of Järvi et al. (2023), they have found that the concentration levels at the street canyon are more affected by traffic rates, whereas in surrounding areas meteorological conditions dominate pollutant levels.
In order to investigate a long-range transport (LRT) episode detected at the site on 9–11 September 2019, the origins of the air masses were calculated using the NOAA HYSPLIT model (Stein et al., 2015; Rolph et al., 2017); 96 h back trajectories were calculated for every 6 h at a height of 100 m a.s.l. (above sea level).
2.3 Data analysis
2.3.1 Calculation of the production rates of oxidized compounds
Production rates of oxidized compounds (OxPRs) from VOCsi were calculated from their concentration, the concentration of the oxidant, and their respective reaction rate:
where ki is the reaction rate coefficient of a VOC with an oxidant (OH, O3, or NO3), and [VOCi] is the concentration of the corresponding VOC or oxidant. Details of the reaction rate coefficients used in this study can be found in Table S1 in the Supplement. Concentrations of O3 were from the local measurements, while OH and NO3 radical concentrations were modeled using the ARCA box model as described in Sect. 2.4.
2.3.2 PMF for the SP-AMS data
The SP-AMS data set was analyzed for the sources and types of OA with a positive matrix factorization algorithm (CU AMS PMF tool v2.08D; Paatero and Tapper, 1994; Ulbrich et al., 2009). The number of factors was varied from two to eight (Fig. S2), and the solution obtained with six factors provided the most reasonable results. The factors were identified as two hydrocarbon-like OA (HOA) factors referred to as HOA-1 and HOA-2, one semi-volatile oxygenated OA factor (SV-OOA), one low-volatility oxygenated OA factor (LV-OOA), one LV-OOA factor from long-range transport (LV-OOA-LRT), and a coffee roastery OA factor (CoOA). HOA-1 and HOA-2 correlated only moderately in terms of time series (Pearson R=0.42) and mass spectra (R=0.69), and therefore, they were supposed to represent different types of OA and were not combined. The results for seven and eight factors did not provide any additional information; in the seven-factor solution, HOA-2 was split further into two factors, whereas in the eight-factor solution LV-OOA-LRT was also divided into two identical factors. Two periods of high OA concentrations were excluded from the PMF data matrix. Those periods were (1) from 02:00 to 04:15 LT on 31 August 2019 (Saturday) and (2) from 23:00 LT on 31 August 2019 to 01:20 LT on 1 September 2019 (Saturday–Sunday night). The average OA and PM1 concentrations during period (1) were 65.3 and 67.1 µg m−3 and during period (2) 47.5 and 52.6 µg m−3, respectively. During those periods OA consisted purely of hydrocarbon fragments (similar to the HOA-1 factor), but when those cases were included in the data set, they distorted the calculation of the campaign and diurnal averages as well as the PMF analysis. The source for the high HOA-1 concentrations was not found, but since CO, CO2, and NOx concentrations did not increase during those periods, the source was not likely to be any typical combustion process.
A PMF solution with six factors was investigated for the rotational freedom by varying fpeak, a tool that allows a single one-dimensional transect through the multidimensional solution space to be explored (Ulbrich et al., 2009), and for the accuracy with bootstrapping and multiple seeds (Figs. S3 and S4). These validation tests showed that the six-factor solution was very stable. Also detailed figures on the residuals for the six-factor solution are given in Fig. S5. Residuals show that there was a small amount of unexplained mass during early morning, afternoon, and evening, and in terms of the mass spectra, the largest relative residuals were noticed for larger 's that had the smallest absolute signal values.
Besides OA, PMF was also applied to the mass spectra of organics accompanied by NO+ and ions to explore the presence of organonitrates in the mass spectra of the PMF factors. As in the OA PMF analysis, PMF was run with up to eight factors with NO+ and ions (hereafter called the solution). The seven-factor solution corresponded closely to the six-factor solution with OA since the seventh factor in the solution represented inorganic ammonium nitrate, consisting mostly of NO+ and ions and contributing only 1 % to the total OA signal. The comparison of the PMF solutions for OA (six-factor solution) and (seven-factor solution) in terms of high-resolution mass spectra and mass concentrations is presented in Figs. S6 and S7. For the POA factors (HOA-1, HOA-2, CoOA), the correlation was good for both mass spectra and mass concentrations (R=0.994–1.00), while for the oxygenated OA factors (especially for LV-OOA and LV-OOA-LRT) there were small differences between the solutions. The solution was utilized only to assess the contribution of organonitrates to the PMF factors (Sect. 3.5.2), and all the other data shown in this paper were obtained from the PMF solution for OA (OA solution with six factors).
2.4 Air chemistry modeling with ARCA box
The Atmospherically Relevant Chemistry and Aerosol box model (ARCA box; Clusius et al., 2022) was used to estimate the concentrations of OH and NO3. ARCA box combines the most recent development in terms of atmospheric modeling, including the latest master chemical mechanism (MCM) version (http://mcm.york.ac.uk/, last access: 6 July 2022), complemented by the peroxy radical autoxidation mechanism (PRAM; Roldin et al., 2019), as well as atmospheric cluster dynamics code (ACDC; McGrath et al., 2012) for molecular clustering and representation of aerosol particle size distribution and its evolution.
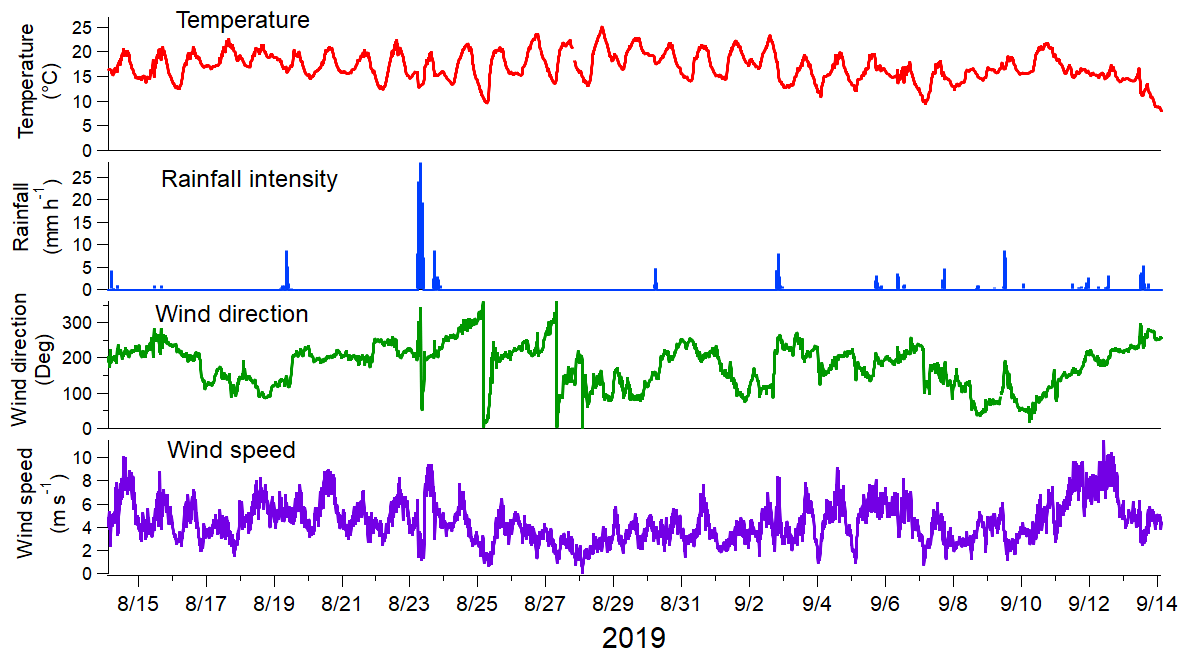
Figure 1Meteorological parameters during the measurement period. Observations were done every 10 min.
Six periods from the campaign period during which VOC measurements were available were simulated in ARCA box (v1.2.0) with a 1 h time resolution. The input for the model consisted of in situ measurements of meteorological parameters (temperature, pressure, relative humidity), trace gas concentrations (NO, NO2, O3, CO), and VOC concentrations (benzene, toluene, xylenes, ethylbenzene, ethyltoluenes, trimethylbenzenes, styrene, isoprene, pinenes, limonene, carene, and β-caryophyllene). In addition, global irradiance from the SMEAR III station located ca. 940 m to the northeast was used (https://smear.avaa.csc.fi/, last access: 6 July 2022), as well as SO2 concentrations from the urban site Kallio, about 1.0 km south of the Helsinki supersite. The surface albedo, used in calculating the actinic flux from the measured irradiance, was set to 0.2. The modeled concentrations were linearly interpolated to match the times of the VOC measurements for the calculation of OxPRs.
In the present study, new particle formation and coagulation were not simulated. The particle size distribution measured at the site was used to calculate the condensation sink and oxidation products, which were allowed to condense on the particles. The sensitivity of the model was tested by varying VOC concentrations by 20 % (uncertainty in our method), and it was found that [OH] varies by 23 % at most and [NO3] by 11 % at most.
3.1 Meteorology and inorganic gases
The measurement period from 14 August to 13 September 2019 was characterized by warm late-summer and early-autumn weather. The temperature was on average 17 ∘C with a clear variation between daytime (maximum of 25 ∘C, minimum 16.5 ∘C) and nighttime (maximum of 17.5 ∘C, minimum 9.3 ∘C; Fig. 1). There was rain on a total of 15 d, with the maximum rainfall observed on 23 August. Wind speed varied from 0 to 10.5 m s−1, with an average of 4.4 m s−1. The dominant wind direction was from the south to the southwest sector (Fig. S8), and consequently, the measured concentrations were likely to be impacted by the emissions from central Europe. Moreover, there was a distinctive LRT pollution episode between 9 and 11 September, and based on the air mass trajectories, the air masses originated from eastern Europe and Russia during that period (Fig. S9). This LRT period has been studied earlier in Salo et al. (2021) in terms of the lung-deposited surface area (LDSA) of particles.
For the inorganic gases, the campaign-average NO, NO2, NOx, O3, and CO concentrations were 18.9 (±26.7), 30.3 (±20.2), 59.4 (±57.9), 45.1 (±18.0), and 200 (±87.2) µg m−3, respectively. As expected for a traffic environment, NO, NO2, and NOx had a clear daily variation displaying a maximum during morning traffic (07:00–09:00 LT) and a second, but less pronounced, peak in the afternoon (15:00–17:00 LT) (Fig. 2). O3 had an opposite diurnal trend to NO, NO2, and NOx, with a minimum in the morning (07:00–09:00 LT). CO was slightly elevated in daytime, with an increase of ∼50 µg m−3 compared to the nighttime concentrations. The time series of NO, NO2, O3, and CO during the measurement campaign can be found in the Supplement (Fig. S10).
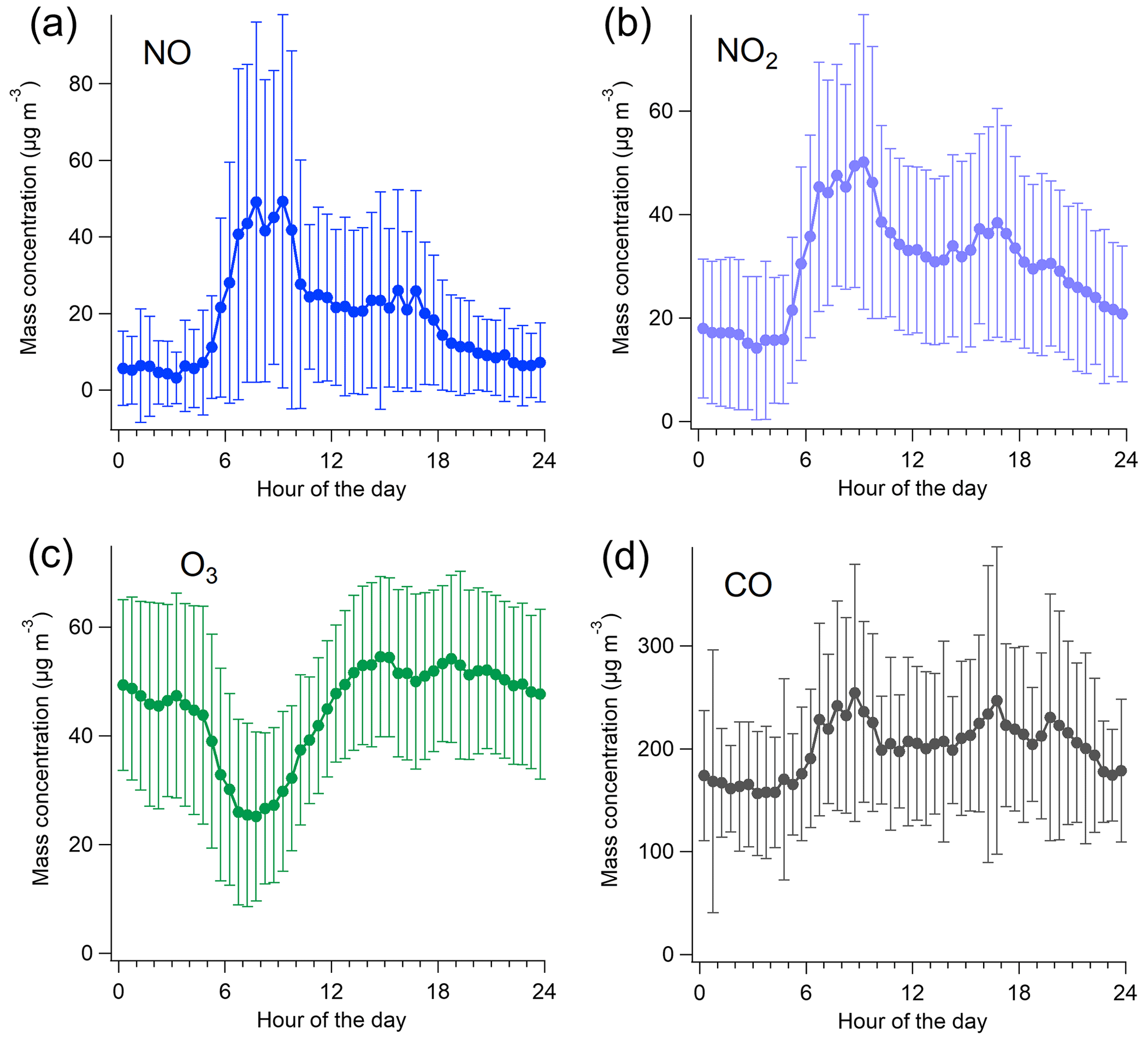
Figure 2Campaign-average concentrations of NO (a), NO2 (b), O3 (c), and CO (d) with standard deviations.
3.2 Volatile organic compounds
Studied anthropogenic VOCs had clearly higher concentrations (campaign average of 4.8 µg m−3) than biogenic VOCs (campaign average of 0.57 µg m−3), with toluene and -xylene being the most abundant aVOCs (Table S1). Previous source apportionment studies conducted in Helsinki in the early 2000s indicated that traffic was clearly the most important source of aVOCs (Hellén et al., 2006). However, since then, the traffic emissions have decreased due to the emission regulations (e.g., EEA, 2019), and therefore, the relative importance of other sources (e.g., VCPs) might have increased (McDonald et al., 2018).
Concentrations of aVOCs were highest during the rush hours, with the morning peak being more intense than the evening peak (Fig. 3a). This is possibly due to a lower mixing layer height and therefore less dilution in the morning (Fig. 3b). In the previous study conducted at the same site in summer (Järvi et al., 2023), they found a stable atmosphere mostly at nighttime, indicating limited vertical mixing, whereas in daytime (between 11:00–14:00 LT) unstable conditions took place, indicating a well-mixed lower atmosphere. Also the direction of vehicles depends on the time of day since in the morning there is more traffic in the lanes close to the site (southbound, towards the city center), whereas in the evening there is more traffic towards the north using the lanes farther from the site. Styrene had a different diurnal variation from all the other VOCs as it is only aVOC having significant reactions with ozone (Fig. 3c). The measured average benzene concentration (0.34±0.220 µg m−3) was well below the lowest annual average concentration threshold (2 µg m−3) given by EU (2008). Usually, the highest concentrations of aromatic hydrocarbons are measured in the winter due to longer lifetimes and higher emissions in the winter (Hellén et al., 2012b).
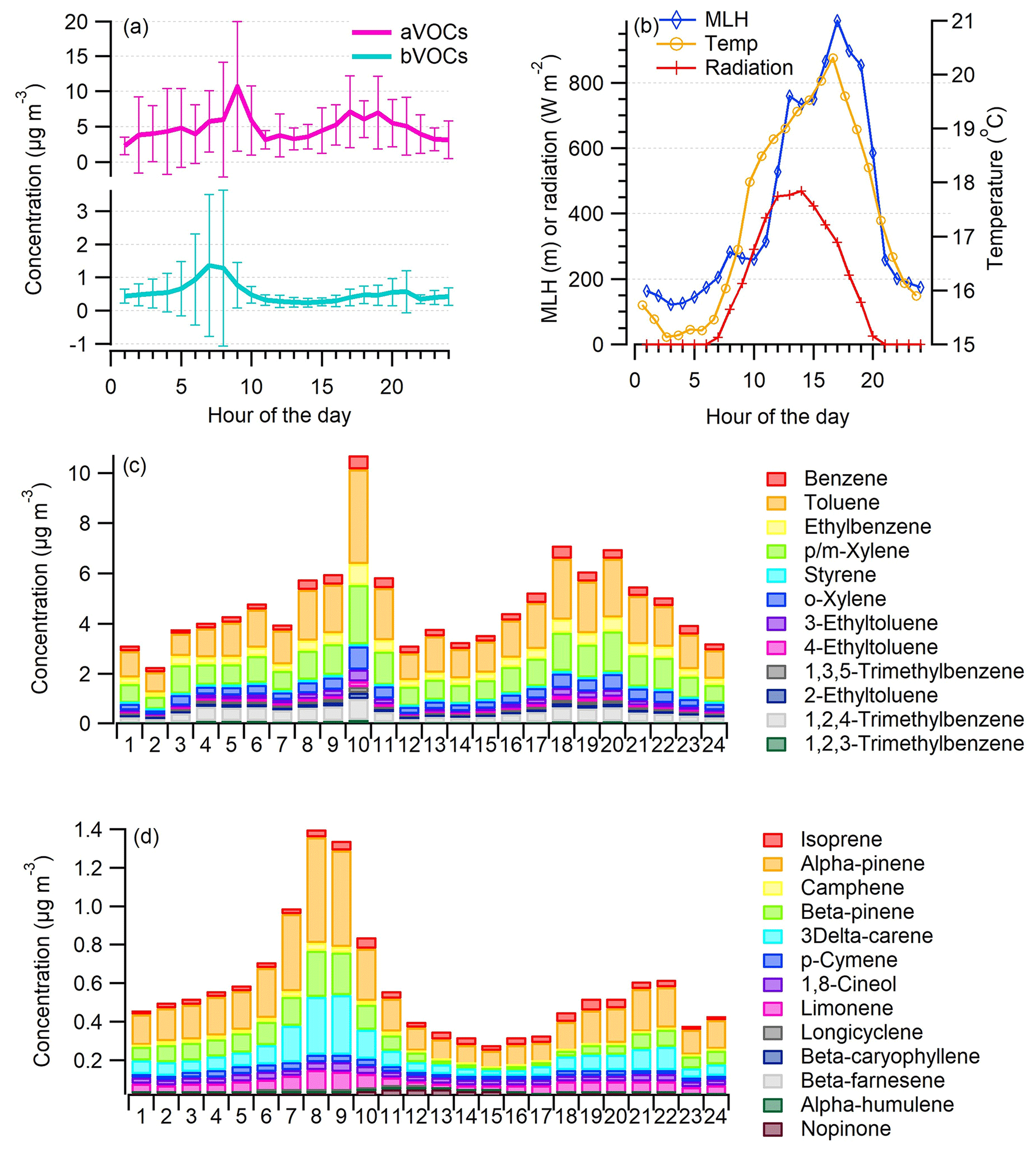
Figure 3Campaign-average diurnal variation in aVOC and bVOC concentrations with standard deviation (a) and the average mixing layer height (MLH), ambient temperature (Temp), and solar radiation (Radiation) (b) as well as concentrations of specific aVOCs (c) and concentrations of specific bVOCs (d) during the VOC measurements.
Most bVOC concentrations (Table S1) were well above the detection limits during these measurements in late August/early September even though generally bVOC emissions and concentrations are known to be highest during the main growing season in July/early August (Tarvainen et al., 2007; Hellén et al., 2018). Since the emissions from vegetation are known to be temperature- and light-dependent, the relatively high ambient temperature during the measurements at least partly explains the high bVOC concentrations (Fig. 3d). Of bVOCs, monoterpenes had the highest concentrations, α-pinene being the most abundant. Isoprene concentrations were clearly lower than those of monoterpenes. This was expected since the most common trees in Finland are known to be mainly mono- and sesquiterpene emitters (e.g., Scots pine, Norway spruce, silver/downy birch; Tarvainen et al., 2007). Also some sesquiterpenes were detected with the concentrations close to their quantification limits. Even with relatively high emissions, sesquiterpene concentrations in ambient air remain low due to their high reactivity and short lifetimes in the atmosphere (Hellén et al., 2018). The main sesquiterpene was β-caryophyllene, which has been detected previously in the emissions of the main tree species in Finland (Scots pine, Norway spruce, silver/downy birch; Hakola et al., 2001, 2006, 2017; Hellen et al., 2021).
Both mono- and sesquiterpenes had similar diurnal variation, with the highest concentrations measured during the early morning hours. In general, the emissions of bVOCs from the vegetation follow the variations in temperature and light, which are the highest in the afternoon (e.g., Hakola et al., 2017; Hellén et al., 2021); however, for these highly reactive compounds with short atmospheric lifetimes, mixing has a strong effect on the local concentration levels. Due to a much lower mixing layer with lower dilution during nighttime, higher nighttime concentrations have been observed for bVOCs (Mogensen et al., 2011; Hellén et al., 2018). In this study, the morning peak of bVOCs is expected to be a balance between the emissions and mixing. In addition to biogenic emissions, terpenes have some anthropogenic sources. Personal care products and cleaning agents are known to be a source of especially limonene (Claflin et al., 2021), which was also detected here with an average concentration of 0.054 (±0.063) µg m−3.
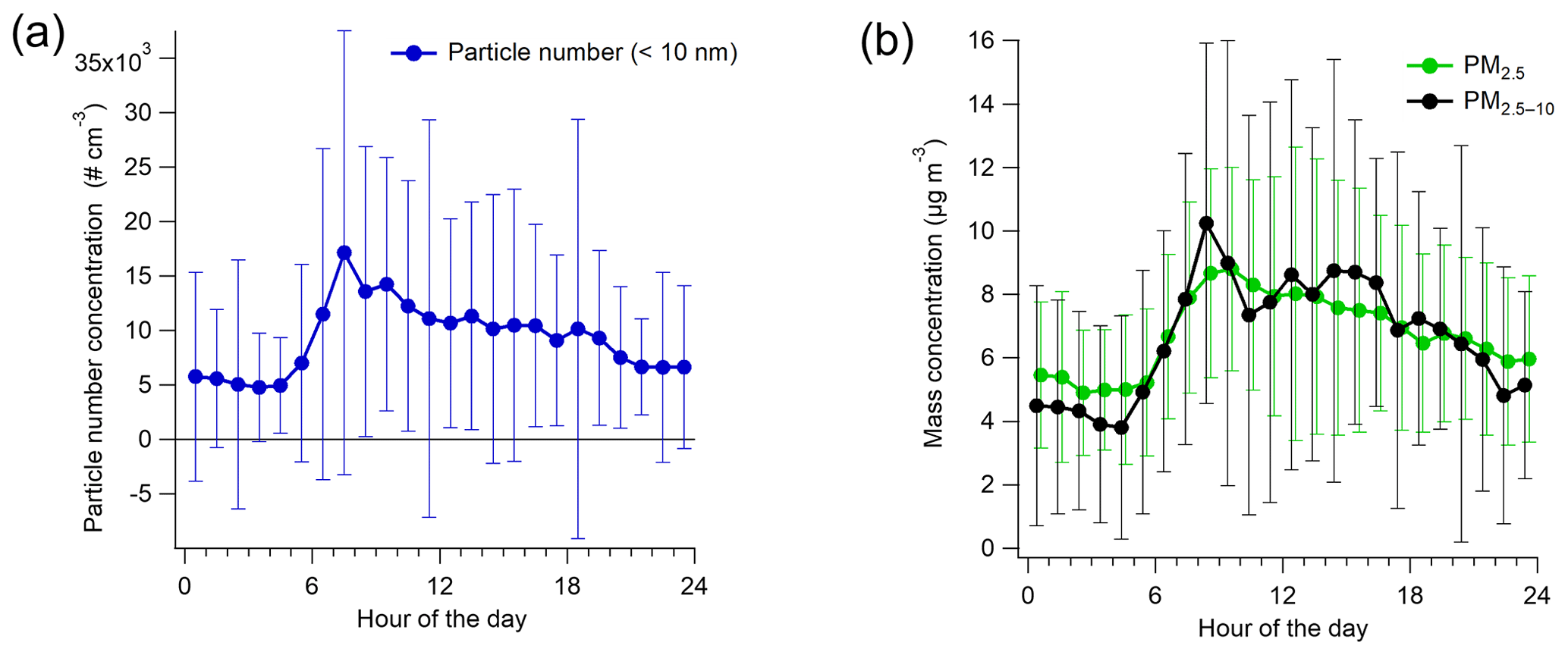
Figure 4Campaign-average diurnal variation in particle number (>10 nm) (a) and PM2.5 and PM2.5–10 concentrations (b) with standard deviations.
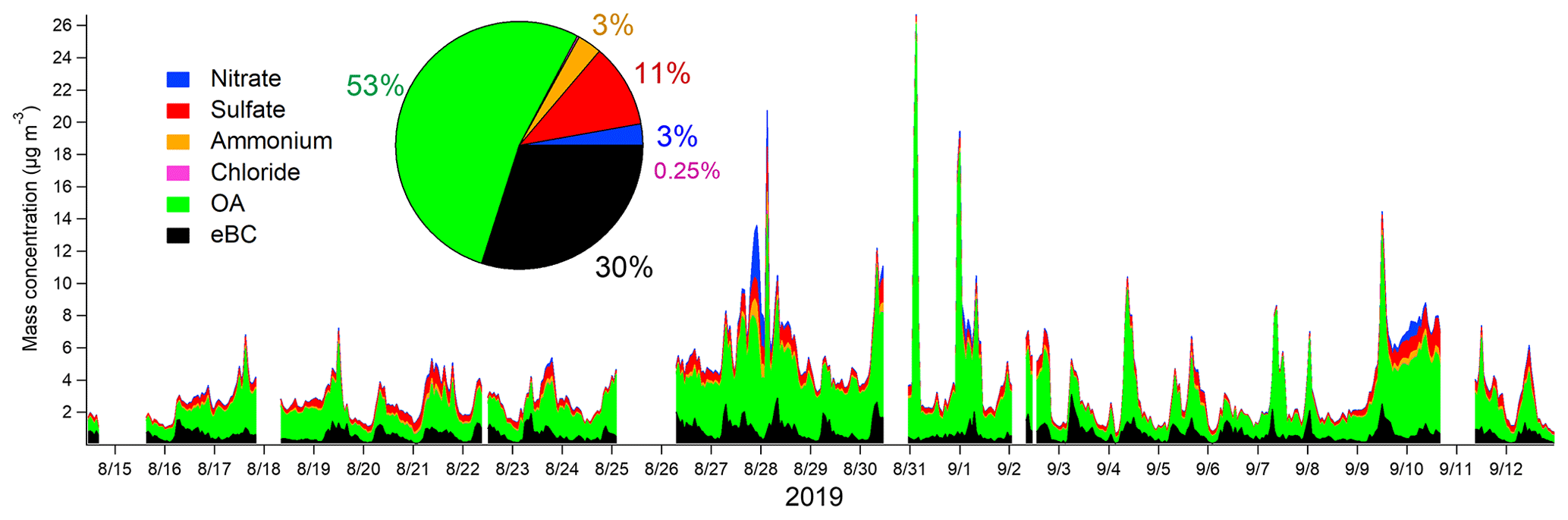
Figure 5Time series of the mass concentrations and the mass fractions of nitrate, sulfate, ammonium, chloride, OA, and eBC calculated with 1 h averages.
3.3 Particle number and mass concentrations
The average particle number concentration for >10 nm particles was 9200 (±11 800) particles per cubic centimeter during the measurement campaign. Particle number concentration followed the traffic pattern with the largest concentrations during the morning rush hour (∼17 000 particles per cubic centimeter) and the smallest concentrations (∼4800 particles per cubic centimeter) during the early morning hours (∼ 03:00–05:00 LT) (Fig. 4a). In terms of the smallest size fraction, 51 % of the particles were in the size range of 10–25 nm, with the 10–25 nm fraction being the smallest in the early morning (38 %; Fig. S11). In general, the number size distribution compared well with the previous studies carried out at the site (e.g., Barreira et al., 2021).
The average mass concentrations of fine (PM2.5) and coarse (PM2.5–10) particles were virtually equal (6.8±3.3 and 6.7±5.7 µg m−3, respectively), but there was more variation in the PM2.5–10 than in the PM2.5 concentration during the campaign (Fig. S12b). Both PM2.5 and PM2.5−10 had elevated concentrations during the day and the smallest concentrations in the early morning hours (Fig. 4b), similar to the number concentrations. Based on the DMPS data, the average mass concentration of PM1 was 3.7±3.5 µg m−3 (calculated with the density of 1.42 g cm−3), while the sum of the SP-AMS species and eBC from the aethalometer was slightly larger, on average 4.5±6.1 µg m−3.
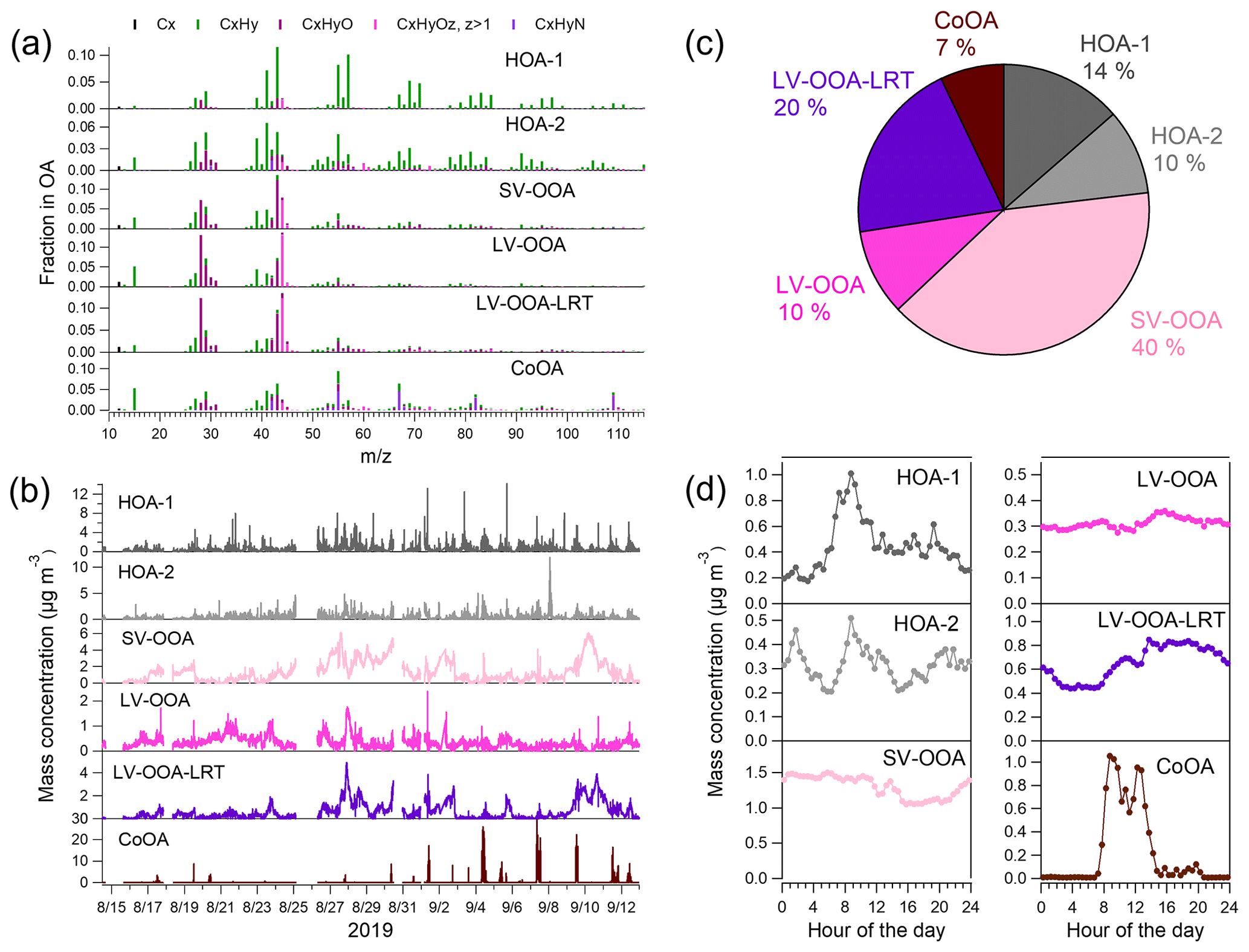
Figure 6Mass spectra (a), time series (b), campaign-average mass fractions (c), and campaign-average diurnal trends (d) for six OA PMF factors in the traffic environment.
3.4 PM1 chemical composition
PM1 particles consisted mostly of OA (53 %), followed by eBC (30 %) (Fig. 5). The average contributions of inorganic species were small, 11 %, 2.8 %, 3.2 %, and 0.25 % for sulfate, nitrate, ammonium, and chloride, respectively. Compared to the average composition at the site presented in Barreira et al. (2021), the contribution of eBC was larger, and the contributions of inorganic species were smaller in this study. Larger eBC can be explained to some extent by the use of a multi-angle absorption photometer (MAAP) in Barreira et al. (2021), as the MAAP gave approximately 72 % of the AE33 values at the Helsinki supersite (Helin et al., 2018). The eBC concentrations followed the traffic pattern with a maximum in the morning and a smaller concentration peak in the afternoon during the evening traffic. OA had slightly larger concentrations before noon, but besides that, there was no clear diurnal trend for OA. In terms of inorganic species, nitrate had smaller concentrations in daytime, indicating its semi-volatile characteristic. Sulfate, ammonium, and chloride did not display any diurnal pattern.
3.4.1 Primary OA
The sources of OA were investigated by the PMF analysis. PMF extracted six different types of OA at the traffic environment (Fig. 6), of which three can be considered to be POA (HOA-1, HOA-2, and CoOA). HOA-1 had a contribution of 14 % to OA. The mass spectrum of HOA-1 was similar to that from engine emissions, which have the largest signal for the hydrocarbon ions , , , , , and at mass-to-charge ratios ('s) of 57, 43, 55, 41, 69, and 71, respectively (Canagaratna et al., 2004). HOA-1 also displayed a similar diurnal trend with the traffic-related components BCff and nitrogen oxides (Fig. 7a) having the maximum in the morning; however, BCff increased more sharply in the morning, whereas HOA peaked later at ∼ 09:00 LT. Also, the evening rush hour peak detected for BCff and NOx (15:00–17:00 LT), was not apparent for HOA-1. Overall, HOA-1 correlated only moderately with BCff (R=0.52) and NOx (R=0.53). Also the total concentration of aVOCs correlated with HOA-1 (R=0.67). Both species peaked during morning rush hour and in the evening, but relative to the morning peak, aVOCs had larger concentrations in the evening than HOA-1 (Fig. 7b). In terms of mass size distributions, HOA-1 was likely to be found in a relatively small particle size. Unit-mass-resolution of 57, used as a surrogate for HOA-1, peaked at ∼100–150 nm during the period of high traffic emissions, but there was also a second mode at larger particle size (250–450 nm; Fig. S13).
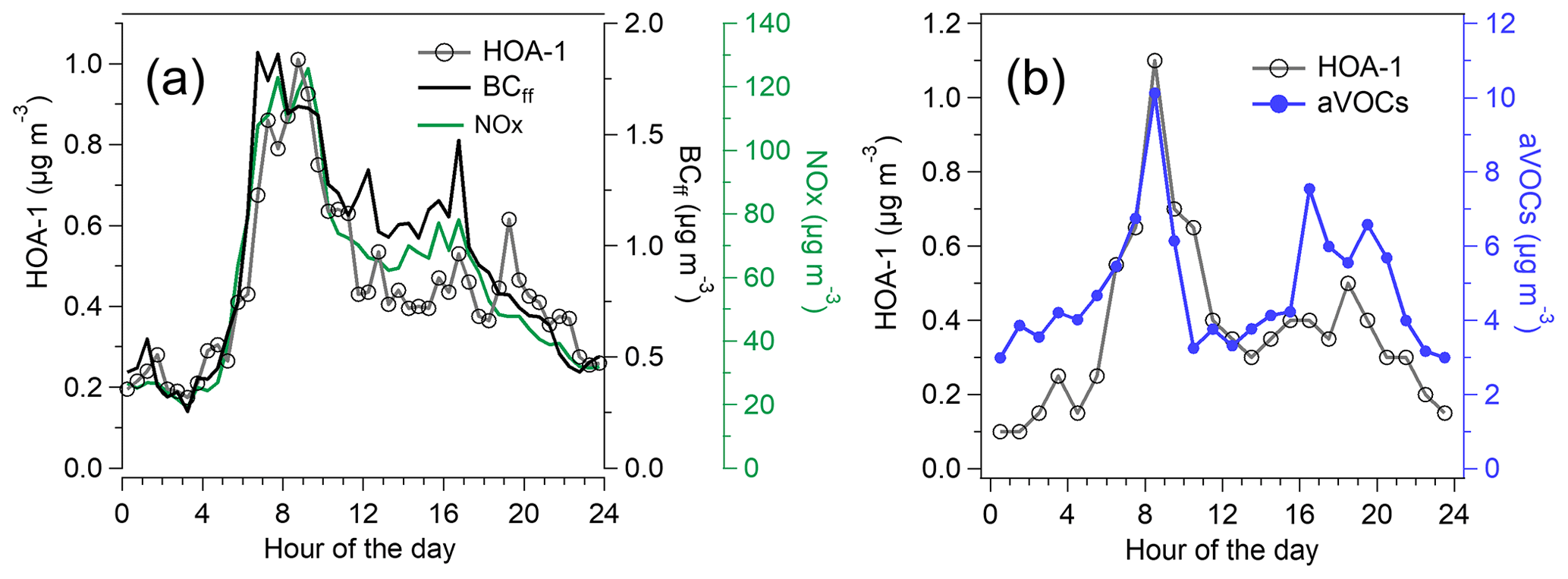
Figure 7Campaign-average diurnal trend of HOA-1, BCff, and NOx (a) and the diurnal trends of HOA-1 and aVOCs averaged over the VOC measurement periods (b).
HOA-2 had a smaller contribution to OA than HOA-1 (10 %), and it was more oxygenated than HOA-1. Also HOA-2 had a clear pattern for the hydrocarbon ions, but it was different from HOA-1. HOA-2 had more signal at lower 's, with the largest signal being for , , , and at 's of 41, 39, 55, and 43, respectively, indicating that HOA-2 had more double bonds in the hydrocarbon ions, and therefore they were less saturated than in HOA-1. The mass spectrum of HOA-2 also had a distinct signal for the oxygenated ions and at 's of 60 and 73, respectively, that are usually considered to be typical ions for biomass burning OA (BBOA; Alfarra et al., 2007). In this study, these fragments are unlikely to be related to biomass combustion, as similar to HOA-1, HOA-2 peaked in the morning between 08:00 and 09:00 LT; however, the morning peak was smaller for HOA-2 than for HOA-1. At nighttime, the concentrations of HOA-1 and HOA-2 were almost similar. HOA-2 correlated moderately with BCwb (R=0.41), but the correlation was stronger for HOA-1 and BCwb (R=0.59) since BCwb also had a clear morning peak. This is possibly explained by the fact that the aethalometer model cannot resolve BCwb and BCff completely. Furthermore, traffic also emits carbon, which absorbs at near-ultraviolet and lower-visible wavelengths (brown carbon), which can be attributed to BCwb in the aethalometer model, regardless of its original source. The contribution of biomass burning to OA and eBC was likely to be small since the measurements were carried out in late summer, when ambient temperature was still quite high, and the site was located in the area with apartment buildings with no wood stoves or wood-heated saunas.
It can be speculated that HOA-2 and HOA-1 were related to the emissions from different types of vehicles. In the previous studies, it has been shown that, for example, the exhaust emission of diesel–electric hybrid and ethanol buses equipped with exhaust aftertreatment systems can contain ( of 60) and ( of 73) ions in their mass spectra (Saarikoski et al., 2017). It should be noted that SOA produced from the vehicle emissions is also likely to include these oxygenated ions (Timonen et al., 2017); however, it has been shown that modern exhaust aftertreatment systems reduce SOA emissions in general (Karjalainen et al., 2019).
Another source for POA at the traffic site was the local coffee roastery. The mass spectrum of CoOA had pronounced peaks at 's of 55, 67, 82, and 109, corresponding to the ions of C3H5N+, , , and , respectively, those ions being characteristic for caffeine in the AMS mass spectra (Timonen et al., 2013). As can be seen from the time series, CoOA was detected sporadically, while most of the time its concentration was near zero. On average, CoOA comprised 7 % of total OA, but during its maximum concentrations, its contribution to OA was as large as 80 %. Regarding diurnal trends, OA from the coffee roastery was detected mostly between 07:00 and 14:00 LT, which agreed with the operation hours of the roastery. CoOA did not correlate with any of the inorganic SP-AMS species. Based on the wind direction data, CoOA was clearly associated with the south sector from the measurement site (Fig. S14), which is the direction of the coffee roastery (Fig. S1). CoOA has been observed earlier in Helsinki at the SMEAR III station (1 % of OA; Timonen et al., 2013), but compared to SMEAR III, CoOA concentration and contribution was much larger at the Helsinki supersite because it was much closer to the coffee roastery (the Helsinki supersite is ∼600 m north of the roastery, while SMEAR III is ∼1.5 km northeast of the roastery). Regarding the mass size distributions, of 109, a characteristic unit-mass-resolution for CoOA, peaked at ∼300 nm during an intense coffee roastery emission event (Fig. S13).
The concentrations of CoOA might have been overestimated in this study. Compared to the PM1 mass from the DMPS, the sum of eBC from the aethalometer and the SP-AMS species (excluding rBC) was clearly larger when the coffee roastery emissions dominated OA (Fig. S15). This is likely due to the larger relative ionization efficiency for organics in the coffee roastery emissions since a constant RIE value (default 1.4) was used for organics regardless of the composition. For LV-OOA, the impact of RIE was opposite to CoOA as PM1 from the SP-AMS and aethalometer was smaller than that from the DMPS when the LV-OOA fraction had the largest values. For the other PMF factors, the impact of RIE was less clear. Another reason for higher PM1 from the SP-AMS and aethalometer could be the enhanced collection efficiency in the SP-AMS. A constant CE of 1 was used in this study, but the collection efficiency calculated by Middlebrook et al. (2012) resulted in a CE varying in the range of 0.45–0.65. Even so, the CE did not seem to explain the difference in PM1 between the DMPS and the sum of the SP-AMS and aethalometer (Fig. S16).
Although there were several restaurants near the measurement site (Fig. S1), cooking-related OA was not found at the site. That can be explained by the fact that there were no street kitchens or outdoor dining places near the site. However, based on the method presented in Mohr et al. (2012) to estimate cooking OA in the ambient data set, the ratios of f55 to f57 and to for CoOA were close to those of cooking OA (Fig. S17). Therefore, it is possible to explain CoOA as cooking OA if meteorological data, prior knowledge of the local sources, and reference mass spectra are not available.
3.4.2 Secondary OA
The largest fraction of OA (78 %) consisted of three types of oxygenated OAs that were likely to be related to SOA. Two of the OOA factors had similar mass spectra, with the largest signal for the CO2-related ions and CO+ at 's of 44 and 28, respectively, and the ion C2H3O+ at a of 43. Based on their mass spectra, these OOAs were classified as LV-OOAs. However, the time series of two LV-OOAs differed clearly as one of the factors had more stable concentrations throughout the measurement period, whereas the other one increased clearly at the end of the measurement campaign, when the air masses came from eastern Europe and Russia (9–11 September 2019; Fig. S9). Therefore, this LV-OOA is called LV-OOA-LRT. The LRT episode was defined by large concentrations of inorganic species, namely sulfate, nitrate, and ammonium, and LV-OOA-LRT had a strong correlation with sulfate (R=0.87) and ammonium (R=0.81), while the corresponding correlations with LV-OOA were less significant (R=0.35 and R=0.53, respectively.) LV-OOA had a rather flat diurnal trend, while LV-OOA-LRT had smaller concentrations from 02:00 to 10:00 LT than at the other times of the day. The contributions of LV-OOA and LV-OOA-LRT to OA were 10 % and 20 %, respectively. According to wind direction and speed data, LV-OOA-LRT was mostly related to western winds, whereas LV-OOA was associated with the south and southwest direction (Fig. S14).
During the LRT episode, the mass size distributions of unit-mass-resolution of 44, representative of LV-OOA, peaked at ∼450 nm (Fig. S13). The large size was probably due to the condensation of gaseous species on particles during transport and aging, increasing their size. Previous studies have also shown that the largest average particle sizes are observed for atmospherically processed particles that have grown, for instance, during the long-range transport of the air mass (Niemi et al., 2005; Timonen et al., 2008).
The third OOA factor was classified as semi-volatile OOA as it had the largest signal for the ion C2H3O+ at a of 43 followed by the CO2-related ions. Of all six PMF factors, SV-OOA had the largest campaign-average contribution to OA with a fraction of 40 %. The SV-OOA concentration was smaller from 11:00 LT to midnight than at the other times of the day, similar to nitrate, suggesting its semi-volatile character. Additionally, SV-OOA had a small increase around 14:00 LT in the afternoon that could be due to the SOA formation in the afternoon. SV-OOA was at least partly related to biogenic SOA (discussed later in detail). During large biogenic emissions (see explanation in Fig. S13), unit-mass-resolution of 43 peaked at ∼350 nm. Compared to the other sources, the size for a of 43 was larger than the of 57 for traffic emissions and the of 109 for coffee roastery emissions but smaller than the of 44 for the LRT episode.
Of the six PMF factors, LV-OOA-LRT was clearly the most oxygenated factor and had the largest oxidation state (Table 1). That was expected as long-range-transported OA had already spent several days in the atmosphere and was exposed to the oxidants before arriving in Helsinki. Also LV-OOA was highly oxygenated, whereas SV-OOA was much less oxygenated. As anticipated, primary OA sources were the least oxygenated factors. The same pattern was observed when the PMF factors were placed in the O:C–H:C space (Van Krevelen diagram in Fig. 8); POA factors were located at smaller O:C and larger H:C values than the OOA factors.
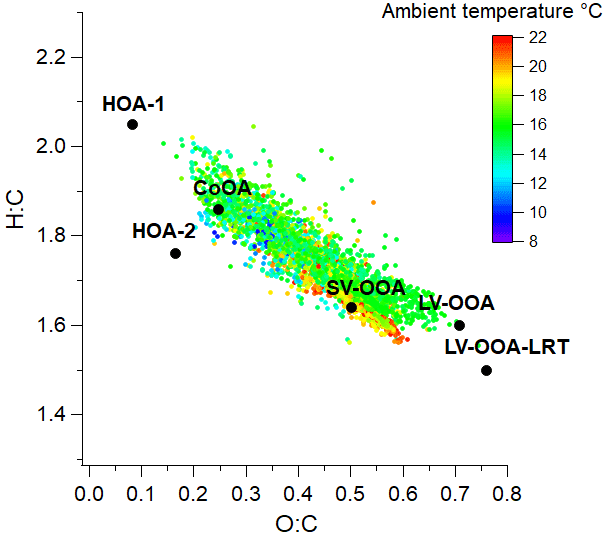
Figure 8Location of OA and PMF factors in O:C and H:C spaces; 1 h average OA values were colored according to ambient temperature.
Table 1Elemental ratios, oxidation states, and NO+-to- ratios for the PMF factors.
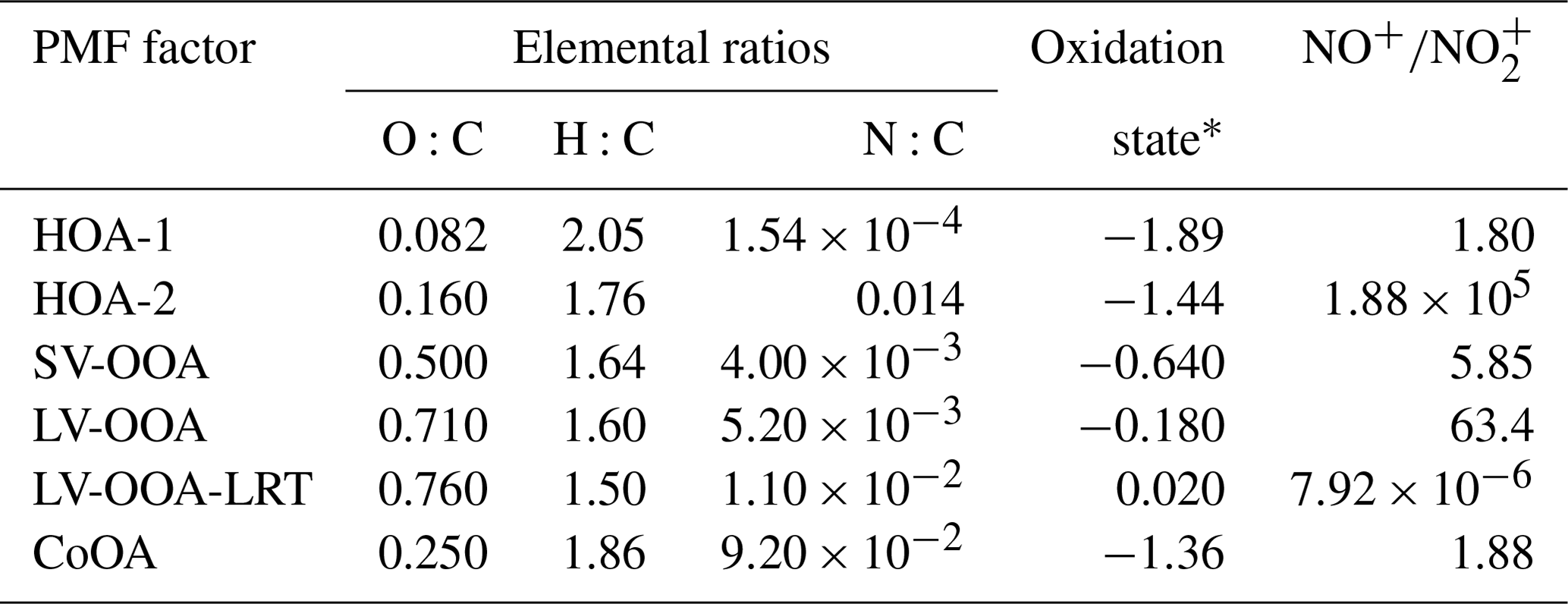
* calculated as .
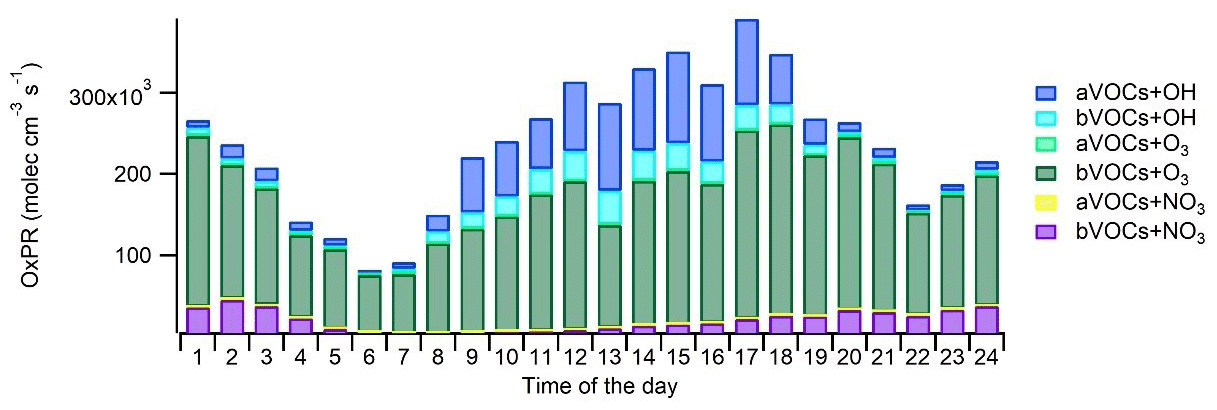
Figure 9Diurnal variation in the production of oxidation products (OxPRs) from the oxidation of anthropogenic VOCs (aVOC) and biogenic VOCs (bVOCs) with hydroxyl radicals (OH), nitrate radicals (NO3), and ozone (O3).
3.5 Oxidation of VOCs and related SOA formation
3.5.1 Oxidation of measured VOCs
Oxidation of VOCs under various environmental conditions produces a variety of gas- and particle-phase products that are relevant for atmospheric chemistry and SOA production. To describe this, the production rates of oxidized compounds were calculated for studied VOCs from the VOC reactions as described in Sect. 2.3. The main local sources of oxidation products were O3 oxidation (OxPR) of bVOCs (66 % of total OxPR) and OH radical oxidation (OxPROH) of aVOCs and bVOCs (25 % of total OxPR; Fig. 9). OxPR stayed relatively constant over the day, while OxPROH was significant only during daytime. This is expected since OH radicals are produced in photochemical reactions only during light hours. NO3 radical oxidation had only 8 % contribution to total OxPR. In an earlier study in a forest environment in Finland, O3 oxidation was found to be a major oxidation pathway (Hellén et al., 2018), and even with much higher mixing ratios of aVOCs, it was also the case here. However, at least part of the bVOCs detected here are emitted from anthropogenic sources, such as personal care and cleaning products. Nonetheless, this describes only the local situation in the traffic environment, and with a bit more regional perspective, the situation may change.
On average, aVOCs produced 18 % of the oxidation products at the site and had a major contribution to the OH radical oxidation (72 % of OxPROH). Oxidation of aVOCs with NO3 radicals had a low contribution (0.3 %) and no contribution to O3 oxidation. The major aVOC for OxPR was -xylene (36 % of aVOC OxPROH), followed by toluene (10 % of the aVOC OxPROH). Even though the contribution of trimethylbenzenes was only 13 % of the aVOC OxPROH, their impact on SOA formation may still be significant due to their higher SOA formation potentials.
The contribution of bVOCs to total OxPR was 82 %. O3 oxidation was the main oxidation pathway for bVOCs, with 82 % contribution. Contributions of OH and NO3 radicals were 9 % and 10 %, respectively. OxPR was driven by three monoterpenes, (α-pinene, limonene, and terpinolene) and a sesquiterpene (β-caryophyllene), with 20 %, 13 %, 33 %, and 26 % contributions to OxPR, respectively. For OxPROH, isoprene and α-pinene were the most significant bVOCs. NO3 radical oxidation was driven by four monoterpenes (α-pinene, 3Δ-carene, limonene, and terpinolene), which had a 92 % contribution to total OxPR. It is clear that bVOCs with lower concentrations and sesquiterpenes with very low concentrations (∼0.004 µg m−3) also have a significant effect on local chemistry and, due to their high SOA yields (e.g., Lee et al., 2006), possibly also on the SOA formation. During summertime, when bVOC emissions from vegetation as well as their ambient air concentrations are higher (Hellén et al., 2012b), their contribution is expected to be even more significant.
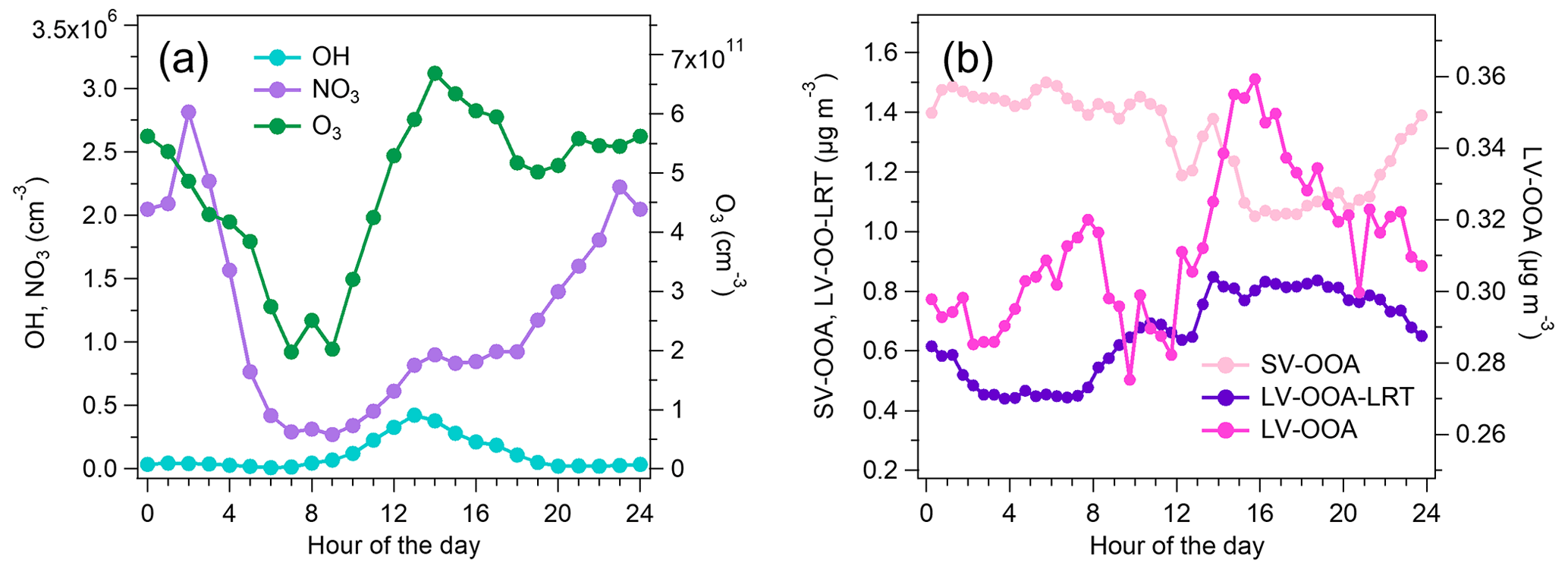
Figure 10Diurnal trends of OH and NO3 and O3 concentrations (a) and the PMF factors related to SOA (SV-OOA, LV-OOA-LRT, and LV-OOA) (b).
There are also other VOCs, e.g., non-methane hydrocarbons and oxygenated volatile organic compounds (OVOCs) including ethanol, acetone, formaldehyde, and acetaldehyde, in urban air with possibly even higher concentrations (e.g., Hellén et al., 2006) than VOCs measured in this study. Most of them are more volatile and less reactive, and even with high concentrations, their oxidation products are not expected to have significant impacts on local SOA formation. As recent studies on volatile chemical products (McDonald et al., 2018; Coggon et al., 2021; Pennington et al., 2021) show, it is highly probable that there are also other VOCs (e.g., siloxanes and intermediate volatile organic compounds, IVOCs) which could contribute to SOA production. They estimate that volatile chemical product (VCP) emissions, which are not traditionally considered to be a significant VOC source, may be as high as traffic emissions. However, the total OH reactivity measurements in urban ambient air indicate that missing OH reactivity in urban areas has not been this high (see the review by Yang et al., 2016). VCP emissions include lots of different compounds, but part of the VCP emissions are aromatics and terpenes, which were also measured in this study and are measured in most total OH reactivity studies. This could also explain why actual missing reactivity in urban air has not been as high as missing VCP emissions.
3.5.2 SOA formation
SOA formation was studied by comparing the diurnal trends of the OOA factors with the diurnal trends of the modeled OH and NO3 radical concentrations (Fig. 10). LV-OOA had only a small variation throughout the day; however, the largest concentrations were measured in daytime, suggesting that its source is likely to be OH radical reactions with aVOCs. LV-OOA also had a peak during the morning rush hour from 06:30 to 09:00 LT that was not seen for OH; however, the relative contribution of LV-OOA to OA was smallest in the morning, suggesting that the increase was probably due to the low mixing layer height in the morning. Also the advanced exhaust aftertreatment in vehicles can possibly increase the direct emissions of LV-OOA. For instance, the study of Arnold et al. (2012) indicated elevated exhaust concentrations of organic acids as a consequence of oxidation processes in the exhaust aftertreatment devices. Thus, the distinctive peak during the morning rush hours can also be caused partly by direct emissions of low-volatility organic compounds and their condensation to the particulate phase immediately after the emission. The diurnal trend of SV-OOA differed from that of radical concentrations, being smaller in daytime. This indicates that the main factor behind the diurnal trend of SV-OOA was ambient temperature as SV-OOA is likely to be semi-volatile. LV-OOA-LRT had a slightly larger concentration in daytime than in the early morning hours, but this was likely to be due to meteorological parameters such as wind direction as LV-OOA-LRT was already highly oxygenated when it arrived in Helsinki. On the other hand, the diurnal variation in LV-OOA-LRT had a correlation with OxPR (R=0.71; Fig. S18). Ozone is also strongly related to long-range transport in Finland, but due to the short lifetime of terpenes, this production would be quite local.
Nopinone, an oxidation product of the monoterpene β-pinene, correlated with SV-OOA when the intense long-range-transported episode at the end of the measurement period was excluded (R=0.71). Nopinone is produced in the air through the oxidation of β-pinene by OH (Calvert et al., 2011; Kaminski et al., 2017) and ozone (Grosjean et al., 1993; Hakola et al., 1994; Winterhalter et al., 2000). The concentration of nopinone is a balance between the production from these reactions and its own oxidation by OH (Hellén et al., 2018). The main source of β-pinene is expected to be vegetation, but also some anthropogenic sources are possible (e.g., personal care products or cleaning agents).
The high correlation of the nopinone concentration with the SV-OOA factor supports the fact that SV-OOA is at least partly related to biogenic emissions. Both nopinone and SV-OOA had maximum concentrations just before midday (Fig. 11). This can be at least partly explained by the mixing layer height and oxidation rates. During the night, monoterpenes (and other VOCs) often accumulate in the air due to low mixing layer and lower reaction sinks (Hellén et al., 2018), as is also seen here by high early-morning concentrations including nopinone precursor β-pinene (Fig. 3d). After sunrise, when OH radical production starts, oxidation rates increase, and more oxidation products (including nopinone) are formed. The mixing layer height was still relatively low during the morning hours, and nopinone and SV-OOA concentrations increased. Later during the day, when high production of semi-volatile compounds continued, dilution started to play a role due to the higher mixing layer, and the concentrations decreased. This also indicated the local origin of SV-OOA. Nonetheless, the diurnal variation in SV-OOA and nopinone was relatively small. This could be explained by the production of semi-volatile compounds from O3 and NO3 reactions of primary VOCs also during the night. In terms of ambient temperature, OA was located close to the SV-OOA factor in the Van Krevelen diagram when the temperature was high (Fig. 8), which agrees with the larger VOC emissions from vegetation at higher temperatures.
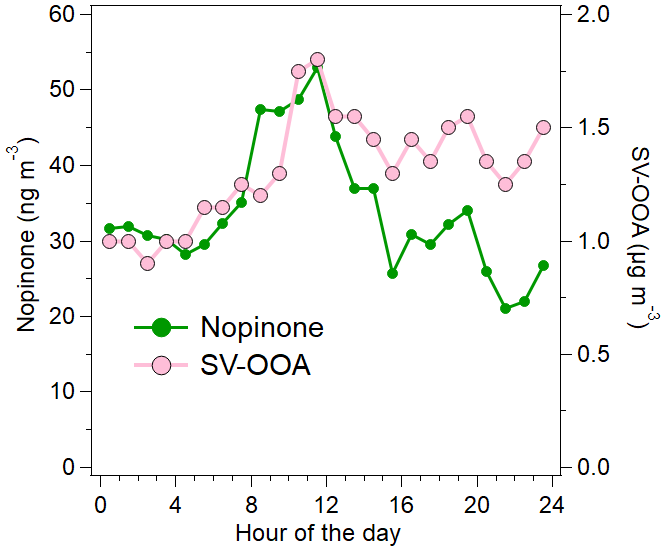
Figure 11Diurnal trends for nopinone and SV-OOA. The intense long-range-transported episode at the end of the measurement period has been excluded from the data.
SOA formation was also studied in terms of organonitrates. In order to investigate secondary organonitrates in the mass spectra of the PMF factors, NO+ and ions were added to the PMF input data matrix. Both inorganic nitrate and organic nitrate consist dominantly of the ions NO+ and ; however, the ratio of NO+ to () is different for organonitrates and ammonium nitrate and therefore allows the determination of organonitrates in OA (Farmer et al., 2010). Secondary organonitrates are formed mainly during dark aging, via gas-phase NO3 reactions and RO2−NO reactions (Atkinson, 2000; Kiendler-Scharr et al., 2016). Fry et al. (2009) have published a value of ∼10 for the particles produced in the reaction of α-pinene with nitrate radicals, whereas Bruns et al. (2010) have measured values of 10–15 and ∼5 for the reactions of nitrate radicals with monoterpenes and isoprene, respectively. In this study, SV-OOA and LV-OOA had ratios of ∼6 and ∼63 (Table 1), suggesting the presence of organonitrates in the SV-OOA and LV-OOA factors. For comparison, for pure ammonium nitrate salt and PMF factors consisting almost solely of ammonium nitrate fragments the ratio was much lower, 0.8–1.0 and 1.2, respectively. Of the local production of the oxidation products, 10 % was estimated to be from nitrate radical reactions of bVOCs (Fig. 9).
LV-OOA-LRT had an extremely low , which can be explained by the fact that the oxidation products of nitrate radicals can be further oxidized with daytime oxidants (Tiitta et al., 2016). In terms of the POA factors, HOA-2 had a large , so it can be speculated that HOA-2 has been oxidized with a nitrate radical during the night; however, the contributions of NO+ and were small in the HOA-2 factor, causing a high uncertainty in the ratio. Furthermore, HOA-2 also had the second-largest nitrogen-to-carbon ratio (N:C; 0.014) after the CoOA factor (Table 1), which included several N-containing ions related to the caffeine (e.g., C3H5N+, , , and ).
In this study, state-of-the-art instrumentation was used to measure the concentrations of the main anthropogenic and biogenic VOCs as well as the chemical characteristics of submicron OA in a traffic environment to elucidate the sources and features of particulate and gaseous pollutants in urban air. Furthermore, the production rates of the oxidation products were studied to reveal the main oxidation products and pathways for aVOCs and bVOC in the traffic environment.
The concentrations of aVOCs were clearly higher than bVOC concentrations at the traffic site. Although the concentrations were lower, the oxidation of bVOCs with ozone was a greater source of oxidation products than oxidation of studied aVOCs; bVOCs produced a much larger portion of the oxidation products (82 %) than aVOCs (18 %) even though the site was one of the busiest traffic sites in the Helsinki area. Generally bVOC emissions and ambient concentrations are known to be highest during the main growing season, in July/early August, but the relatively high ambient temperature during the measurements can at least partly explain the high influence of bVOCs. However, during light hours, OH radical oxidation with aVOCs was also a significant local pathway producing oxidation products. Based on the earlier literature (e.g., McDonald et al., 2018; Coggon et al., 2021; Pennington et al., 2021), it is highly probable that there are additional anthropogenic VOCs relevant for SOA formation which were not quantified here, and they may have as high an impact on local chemistry as the measured aVOCs. However, even with this much higher aVOC contribution, bVOCs would still be the main source of these oxidation products.
Roughly one-third of submicron OA consisted of primary OA, its main sources being traffic and a local coffee roastery. Biomass-burning-related OA was not observed since ambient temperature was still quite high, and the site was in an area with apartment buildings with no wood stoves or wood-heated saunas. On the other hand, secondary organic aerosol, especially from biogenic VOCs as well as from the long-range transport, significantly influenced the OA concentrations.
Both VOC and OA data indicated that the dominating sources at the site were traffic and biogenic emissions. The sum of aVOCs correlated with HOA (R=0.67), both having a clear maximum during the morning rush hour. The oxidation product of a bVOC, nopinone, correlated with SV-OOA, both having maximum concentrations just before midday, representing particles originating from biogenic sources. The maximum concentration caused by the biogenic sources was observed later than that for traffic-related emissions due to different diurnal behavior of biogenic emissions and local meteorology. During the night, bVOCs often accumulate in the air due to the low mixing layer height and lower sink reactions, as was also seen here by high early-morning concentrations. After sunrise, when OH radical production starts, oxidation rates of primary compounds increase, and more oxidation products (including nopinone) are formed.
For the biogenic sources, VOCs are important for source classification as the separation and identification of biogenic compounds from the AMS data are challenging due to their extensive fragmentation and similarity to the other SOA sources. In contrast, primary OA sources, e.g., traffic and biomass combustion, can be separated from the AMS data by using PMF. As shown in this paper, coffee roastery emissions can also be identified from the AMS data due to the unique mass spectrum for caffeine. The gaseous compounds affiliated with coffee roastery activities, for example furfurylthiol (Cerny et al., 2021), could also be specific for the coffee roastery emissions; however, they could not be extracted with the GC–MS method selected for this study. This highlights the need for a wider range of VOC measurements as cooking emissions could also be identified with the specific VOCs such as unsaturated aldehydes (Klein et al., 2019).
Long-range-transported aerosol was easily separated from OA, the identification supported by inorganic species and air mass trajectories, while its effect was observed less clearly in the VOC measurements due to the oxidation of most VOCs in the atmosphere during the transport. Instead, the VOC measurements identified elevated limonene concentrations, associated with cleaning agents and personal care products. This source could not be separated from the OA data though. Overall, the results of this study indicate that the use of volatile organic markers complement the source apportionment of OA. However, proper markers for both gas and particle phases still need to be identified to achieve a comprehensive source analysis for gas- and particle-phase organics. Another approach could be combining VOC and OA data in the same statistical data analysis method, but the interpretation of the results can be challenging due to, for example, different rates for the atmospheric processes.
The data shown in the paper are available on request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-2963-2023-supplement.
SaS, JVN, HH, TR, and HT designed the experiments, and SaS, TT, LMFB, and MA performed the measurements. SaS, LP, APP, SiS, PC, RK, LS, and AK performed the data analysis. SaS and HH wrote the first version of the manuscript, but all authors participated in the writing process. SaS, TR, HT, HH, APP, SiS, LP, and JVN contributed to the acquisition of funding for the study.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors gratefully acknowledge the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport and dispersion model and/or READY website (https://www.ready.noaa.gov, last access: 7 November 2022) used in this publication.
This research has been supported by the European Commission, Horizon 2020 framework program (TUBE; grant no. 814978), the Academy of Finland (grant nos. 316151, 307797, 323255, 337552, and 337551), Business Finland (grant nos. 528/31/2019, 530/31/2019, and 7517/31/2018), and the European Cooperation in Science and Technology (grant no. CA16109).
This paper was edited by Harald Saathoff and reviewed by two anonymous referees.
Alfarra, M. R., Prevot, A. S. H., Szidat, S., Sandradewi, J., Weimer, S., Lanz, V. A., Schreiber, D., Mohr, M., and Baltensperger, U.: Identification of the mass spectral signature of organic aerosols from wood burning emissions, Environ. Sci. Technol., 41, 5770–5777, https://doi.org/10.1021/es062289b, 2007.
Arnold, F., Pirjola, L., Rönkkö, T., Reichl, U., Schlager, H., Lähde, T., Heikkilä, J., and Keskinen, J.: First on-line measurements of sulphuric acid gas in modern heavy duty diesel engine exhaust: Implications for nanoparticle formation, Environ. Sci. Technol., 46, 11227–11234, https://doi.org/10.1021/es302432s, 2012.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/S1352-2310(99)00460-4, 2000.
Barreira, L. M. F., Helin, A., Aurela, M., Teinilä, K., Friman, M., Kangas, L., Niemi, J. V., Portin, H., Kousa, A., Pirjola, L., Rönkkö, T., Saarikoski, S., and Timonen, H.: In-depth characterization of submicron particulate matter inter-annual variations at a street canyon site in Northern Europe, Atmos. Chem. Phys., 21, 6297–6314, https://doi.org/10.5194/acp-21-6297-2021, 2021.
Brauer, M., Casadei, B., Harrington, R. A., Kovacs, R., Sliwa, K., and the WHF Air Pollution Expert Group: Taking a Stand Against Air Pollution – The Impact on Cardiovascular Disease, Circulation, 143, e800–e804, https://doi.org/10.1161/CIRCULATIONAHA.120.052666, 2021.
Bruns, E. A., Perraud, V., Zelenyuk, A., Ezell, M. J., Johnson, S. N., Yu, Y., Imre, D., Finlayson-Pitts, B. J., and Alexander, M. L.: Comparison of FTIR and Particle Mass Spectrometry for the Measurement of Particulate Organic Nitrates, Environ. Sci. Technol., 44, 1056–1061, https://doi.org/10.1021/es9029864, 2010.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015.
Calvert, J. G., Mellouki, A., and Orlando, J. J.: The mechanism of atmospheric oxidation of the oxygenates, Oxford University Press, New York, 2011.
Canagaratna, M. R., Jayne, J. T., Ghertner, D. A., Herndon, S., Shi, Q., Jimenez, J. L., Silva, P. J., Williams, P., Lanni, T., Drewnick, F., Demerjian, K. L., Kolb, C. E., and Worsnop, D. R.: Chase Studies of Particulate Emissions from in-use New York City Vehicles, Aerosol Sci. Tech., 38, 555-537, https://doi.org/10.1080/02786820490465504, 2004.
Cerny, C., Schlichtherle-Cerny, H., Gibe, R., and Yuan, Y.: Furfuryl alcohol is a precursor for furfurylthiol in coffee, Food Chem., 337, 128008, https://doi.org/10.1016/j.foodchem.2020.128008, 2021.
Claflin, M. S., Pagonis, D., Finewax, Z., Handschy, A. V., Day, D. A., Brown, W. L., Jayne, J. T., Worsnop, D. R., Jimenez, J. L., Ziemann, P. J., de Gouw, J., and Lerner, B. M.: An in situ gas chromatograph with automatic detector switching between PTR- and EI-TOF-MS: isomer-resolved measurements of indoor air, Atmos. Meas. Tech., 14, 133–152, https://doi.org/10.5194/amt-14-133-2021, 2021.
Clusius, P., Xavier, C., Pichelstorfer, L., Zhou, P., Olenius, T., Roldin, P., and Boy, M.: Atmospherically Relevant Chemistry and Aerosol box model – ARCA box (version 1.2), Geosci. Model Dev., 15, 7257–7286, https://doi.org/10.5194/gmd-15-7257-2022, 2022.
Coggon, M. M., Gkatzelis, G. I., McDonald, B. C., Gilman, J. B., Schwantes, R. H., Abuhassan, N., Aikin, K. C., Arend, M. F., Berkoff, T. A., Brown, S. S., Campos, T. L., Dickerson, R. R., Gronoff, G., Hurley, J. F., Isaacman-VanWertz, G., Koss, A. R., Li, M., McKeen, S. A., Moshary, F., Peischl, J., Pospisilova, V., Ren, X., Wilson, A., Wu, Y., Trainer, M., and Warneke, C.: Volatile chemical product emissions enhance ozone and modulate urban chemistry, P. Natl. Acad. Sci. USA, 118, e2026653118, https://doi.org/10.1073/pnas.2026653118, 2021.
Crippa, M., Canonaco, F., Lanz, V. A., Äijälä, M., Allan, J. D., Carbone, S., Capes, G., Dall'Osto, M., Day, D. A., DeCarlo, P. F., Di Marco, C. F., Ehn, M., Eriksson, A., Freney, E., Hildebrandt Ruiz, L., Hillamo, R., Jimenez, J.-L., Junninen, H., Kiendler-Scharr, A., Kortelainen, A.-M., Kulmala, M., Mensah, A. A., Mohr, C., Nemitz, E., O'Dowd, C., Ovadnevaite, J., Pandis, S.N., Petäjä, T., Poulain, L., Saarikoski, S., Sellegri, K., Swietlicki, E., Tiitta, P., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S.: Organic aerosol components derived from 25 AMS datasets across Europe using a newly developed ME-2 based source apportionment strategy, Atmos. Chem. Phys. 14, 6159–6176, https://doi.org/10.5194/acp-14-6159-2014, 2014.
Crippa, M., Solazzo, E., Huang, G., Guizzardi, D., Koffi, E., Muntean, M., Schieberle, C., Friedrich, R., and Janssens-Maenhout, G.: High resolution temporal profiles in the Emissions Database for Global Atmospheric Research, Sci. Data, 7, 121, https://doi.org/10.1038/s41597-020-0462-2, 2020.
Dominici, F., Peng, R. D., Bell, M. L., Pham, L., McDermott, A., Zeger, S. L., and Samet, J. M.: Fine Particulate Air Pollution and Hospital Admission for Cardiovascular and Respiratory Diseases, J. Am. Med. Assoc., 295, 1127–1134, https://doi.org/10.1001/jama.295.10.1127, 2006.
Drinovec, L., Močnik, G., Zotter, P., Prévôt, A. S. H., Ruckstuhl, C., Coz, E., Rupakheti, M., Sciare, J., Müller, T., Wiedensohler, A., and Hansen, A. D. A.: The “dual-spot” Aethalometer: an improved measurement of aerosol black carbon with real-time loading compensation, Atmos. Meas. Tech., 8, 1965–1979, https://doi.org/10.5194/amt-8-1965-2015, 2015.
Edney, E. O., Kleindienst, T. E., Jaoui, M., Lewandowski, M.,Offenberg, J. H., Wang, W., and Claeys, M.: Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States, Atmos. Environ., 39, 5281–5289, https://doi.org/10.1016/j.atmosenv.2005.05.031, 2005.
EEA: Air quality in Europe – 2019 report, Report 10/2019, Publications Office of the European Union, European Environment Agengy, https://doi.org/10.2800/822355, 2019.
EU: Directive 2008/50/EC of the European Parliament and of the Council of 21 May 2008 on ambient air quality and cleaner air for Europe, 1–44, http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32008L0050&from=EN (last access: September 2021), 2008.
Farmer, D .K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Fitzky, A. C., Sandén, H., Karl, T., Fares, S., Calfapietra, C., Grote, R., Saunier, A., and Rewald, B.: The interplay betweenozone and urban vegetation BVOC emissions, ozone deposi-tion, and tree ecophysiology, Front. Forest. Global Change, 2, 50, https://doi.org/10.3389/ffgc.2019.00050, 2019.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Genc, S., Zadeoglulari, Z., Fuss, S. H., and Genc, K.: The Adverse Effects of Air Pollution on the Nervous System, J. Toxicol., 2012, 782462, https://doi.org/10.1155/2012/782462, 2012.
Gkatzelis, G. I., Coggon, M. M., McDonald, B. C., Peischl, J., Aikin, K. C., Gilman, J. B., Trainer, M., and Warneke, C.: Identifying Volatile Chemical Product Tracer Compounds in U.S. Cities, Environ. Sci. Technol., 55, 188–199, https://doi.org/10.1021/acs.est.0c05467, 2021.
Glencross, D. A., Ho, T.-R., Camiña, N., Hawrylowicz, C. M., and Pfeffer, P E.: Air pollution and its effects on the immune system, Free Radic. Biol. Med., 151, 56–68, https://doi.org/10.1016/j.freeradbiomed.2020.01.179, 2020.
Grosjean, D., Williams, E. L., Grosjean, E., Andino, J. M., and Seinfeld, J. H.: Atmospheric oxidation of biogenic hydrocarbons: Reaction of ozone with β-pinene, D-limonene and trans-caryophyllene, Environ. Sci. Technol., 27, 2754–2758, https://doi.org/10.1021/es00049a014, 1993.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hakola, H., Arey, J., Aschmann, S. M., and Atkinson, R.: Product formation from the gas-phase reactions of OH radicals and O3 with a series of monoterpenes, J. Atmos. Chem., 18, 75–102, 1994.
Hakola, H., Laurila, T., Lindfors, V., Hellén, H., Gaman, A., and Rinne, J.: Variation of the VOC emission rates of birch species during the growing season, Boreal. Env. Res., 6, 237–249, 2001.
Hakola, H., Tarvainen, V., Bäck, J., Ranta, H., Bonn, B., Rinne, J., and Kulmala, M.: Seasonal variation of mono- and sesquiterpene emission rates of Scots pine, Biogeosciences, 3, 93–101, https://doi.org/10.5194/bg-3-93-2006, 2006.
Hakola, H., Tarvainen, V., Praplan, A. P., Jaars, K., Hemmilä, M., Kulmala, M., Bäck, J., and Hellén, H.: Terpenoid and carbonyl emissions from Norway spruce in Finland during the growing season, Atmos. Chem. Phys., 17, 3357–3370, https://doi.org/10.5194/acp-17-3357-2017, 2017.
Harrison, R.: Urban Atmospheric Chemistry: A Very Special Case for Study, Clim. Atmos. Sci., 5, 20175, https://doi.org/10.1038/s41612-017-0010-8, 2018.
Helin, A., Niemi, J. V., Virkkula, A., Pirjola, L., Teinilä, K., Backman, J., Aurela, M., Saarikoski, S., Rönkkö, T., Asmi, E., and Timonen, H.: Characteristics and source apportionment of black carbon in the Helsinki metropolitan area, Finland, Atmos. Environ., 190, 87–98, https://doi.org/10.1016/j.atmosenv.2018.07.022, 2018.
Helin, A., Hakola, H., and Hellén, H.: Optimisation of a thermal desorption–gas chromatography–mass spectrometry method for the analysis of monoterpenes, sesquiterpenes and diterpenes, Atmos. Meas. Tech., 13, 3543–3560, https://doi.org/10.5194/amt-13-3543-2020, 2020.
Hellén, H., Hakola, H., Pystynen, K.-H., Rinne, J. and Haapanala, S.: C2–C10 hydrocarbon emissions from a boreal wetland and forest floor, Biogeosciences, 3, 167–174, https://doi.org/10.5194/bg-3-167-2006, 2006.
Hellén, H., Kuronen, P., and Hakola, H.: Heated stainless steel tube for ozone removal in the ambient air measurements of mono- and sesquiterpenes, Atmos. Environ., 57, 35-40, https://doi.org/10.1016/j.atmosenv.2012.04.019, 2012a.
Hellén, H., Tykkä, T., and Hakola, H.: Importance of isoprene and monoterpenes in urban air in Northern Europe, Atmos. Environ., 59, 59–66, https://doi.org/10.1016/j.atmosenv.2012.04.049, 2012b.
Hellén, H., Praplan, A. P., Tykkä, T., Helin, A., Schallhart, S., Schiestl-Aalto, P. P., Back, J., and Hakola, H.: Sesquiterpenes and oxygenated sesquiterpenes dominate the VOC (C5–C20) emissions of downy birches, Atmos. Chem. Phys., 21, 8045–8066, https://doi.org/10.5194/acp-21-8045-2021, 2021.
Hellén, H., Praplan, A. P., Tykkä, T., Ylivinkka, I., Vakkari, V., Bäck, J., Petäjä, T., Kulmala, M., and Hakola, H.: Long-term measurements of volatile organic compounds highlight the importance of sesquiterpenes for the atmospheric chemistry of a boreal forest, Atmos. Chem. Phys., 18, 13839–13863, https://doi.org/10.5194/acp-18-13839-2018, 2018.
Hietikko, R., Kuuluvainen, H., Harrison, R. M., Portin, H., Timonen, H., Niemi, J. V., and Rönkkö, T.: Diurnal variation of nanocluster aerosol concentrations and emission factors in a street canyon, Atmos. Environ., 189, 98–106, https://doi.org/10.1016/j.atmosenv.2018.06.031, 2018.
IPCC: Climate Change 2021: The Physical Science Basis, in: Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M. I., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R., and Zhou, B., Cambridge University Press, Cambridge, UK and New York, NY, USA, https://doi.org/10.1017/9781009157896, in press, 2021.
Järvi, L., Kurppa, M., Kuuluvainen, H., Rönkkö, T., Karttunen, S., Balling, A., Timonen, H., Niemi, J. V., and Pirjola, L.: Determinants of spatial variability of air pollutant concentrations in a street canyon network measured using a mobile laboratory and a drone, Sci. Total Environ., 856, 158974, https://doi.org/10.1016/j.scitotenv.2022.158974, 2023.
Järvinen, A., Aitomaa, M., Rostedt, A., Keskinen, J., and Yli-Ojanperä, J.: Calibration of the new electrical low pressure impactor (ELPI+), J. Aerosol Sci., 69, 150–159, https://doi.org/10.1016/j.jaerosci.2013.12.006, 2014.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo. P. F., Allan. J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. I., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J.,Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, Borrmann, F. S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and D. R. Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kaminski, M., Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., and Wahner, A.: Investigation of the β-pinene photooxidation by OH in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 17, 6631–6650, https://doi.org/10.5194/acp-17-6631-2017, 2017.
Karjalainen, P., Rönkkö, T., Simonen, P., Ntziachristos, L., Juuti, P., Timonen, H., Teinilä, K., Saarikoski, S., Saveljeff, H., Lauren, M., Happonen, M., Matilainen, P., Maunula, T., Nuottimäki, J., and Keskinen, J.: On the strategies to diminish the emissions of particles and secondary aerosol formation from diesel engines, Environ. Sci. Technol., 53, 10408–10416, https://doi.org/10.1021/acs.est.9b04073, 2019.
Karl, T., Striednig, M., Graus, M., Hammerle, A., and Wohlfahrt, G.: Urban flux measurements reveal a large pool of oxygenated volatile organic compound emissions, P. Natl. Acad. Sci. USA, 115, 1186–1191, https://doi.org/10.1073/pnas.1714715115, 2018.
Karppinen, A., Joffre, S. M., and Kukkonen, J.: The refinement of a meteorological pre-processor for the urban environment, Int. J. Environ. Pollut., 14, 565–572, https://doi.org/10.1504/IJEP.2000.000580, 2000.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prévôt, A. S., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Ostoc, M., Day, D. A., DeCarlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kim, H., Zhang, Q., and Heo, J.: Influence of intense secondary aerosol formation and long-range transport on aerosol chemistry and properties in the Seoul Metropolitan Area during spring time: results from KORUS-AQ, Atmos. Chem. Phys., 18, 7149–7168, https://doi.org/10.5194/acp-18-7149-2018, 2018.
Klein, F., Baltensperger, U., Prévôt, A. S. H., and El Haddad, I.: Quantification of the impact of cooking processes on indoor concentrations of volatile organic species and primary and secondary organic aerosols, Indoor Air, 29, 926–942, https://doi.org/10.1111/ina.12597, 2019.
Knutson, E. O. and Whitby, K. T.: Accurate measurement of aerosol electric mobility moments, J. Aerosol Sci., 6, 453–460, https://doi.org/10.1016/0021-8502(75)90061-0, 1975
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary Organic Aerosol Formation from Isoprene Photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Lee, A., Goldstein, A. H., Kroll, J. H., Ng, N. L., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes, J. Geophys. Res., 111, D17305, https://doi.org/10.1029/2006JD007050, 2006.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367-371, https://doi.org/10.1038/nature15371, 2015.
Livesley, S. J., McPherson, G. M., and Calfapietra, C.: The urban forest and ecosystem services: impacts on urban water, heat, and pollution cycles at the tree, street, and city scale, J. Environ. Qual., 45, 119–124, https://doi.org/10.2134/jeq2015.11.0567, 2016.
McDonald, B. C., de Gouw, J. A., Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L., Lee-Taylor, J., Hayes, P. L., McKeen, S. A., Cui, Y. Y., Kim, S. W., Gen-tner, D. R., Isaacman-VanWertz, G., Goldstein, A. H., Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochem-ical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018.
McGrath, M. J., Olenius, T., Ortega, I. K., Loukonen, V., Paasonen, P., Kurtén, T., Kulmala, M., and Vehkamäki, H.: Atmospheric Cluster Dynamics Code: a flexible method for solution of the birth-death equations, Atmos. Chem. Phys., 12, 2345–2355, https://doi.org/10.5194/acp-12-2345-2012, 2012.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of composition-dependent collection efficiencies for the aerodyne aerosol mass spectrometer using field data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Mogensen, D., Smolander, S., Sogachev, A., Zhou, L., Sinha, V., Guenther, A., Williams, J., Nieminen, T., Kajos, M. K., Rinne, J., Kulmala, M., and Boy, M.: Modelling atmospheric OH-reactivity in a boreal forest ecosystem, Atmos. Chem. Phys., 11, 9709–9719, https://doi.org/10.5194/acp-11-9709-2011, 2011.
Mohr, C., DeCarlo, P. F., Heringa, M. F., Chirico, R., Slowik, J. G., Richter, R., Reche, C., Alastuey, A., Querol, X., Seco, R., Peñuelas, J., Jiménez, J. L., Crippa, M., Zimmermann, R., Baltensperger, U., and Prévôt, A. S. H.: Identification and quantification of organic aerosol from cooking and other sources in Barcelona using aerosol mass spectrometer data, Atmos. Chem. Phys., 12, 1649–1665, https://doi.org/10.5194/acp-12-1649-2012, 2012.
Niemi, J. V., Tervahattu, H., Vehkamaki, H., Martikainen, J., Laakso, L., Kulmala, M., Aarnio, P., Koskentalo, T., Sillanpaa, M., and Makkonen, U.: Characterization of aerosol particle episodes in Finland caused by wildfires in Eastern Europe, Atmos. Chem. Phys., 5, 2299–2310, https://doi.org/10.5194/acp-5-2299-2005, 2005.
Onasch, T. B., Trimborn, A., Fortner, E. C., Jayne, J. T., Kok, G. L., Williams, L. R., Davidovits, P., and Worsnop, D. R.: Soot particle aerosol mass spectrometer: Development, validation, and initial application, Aerosol Sci. Tech., 46, 804–817, https://doi.org/10.1080/02786826.2012.663948, 2012.
Paatero, P. and Tapper, U.: Positive matrix factorization – A nonnegative factor model with optimal utilization of error-estimates of data values, Environmetrics, 5, 111–126, https://doi.org/10.1002/env.3170050203, 1994.
Pennington, E. A., Seltzer, K. M., Murphy, B. N., Qin, M., Seinfeld, J. H., and Pye, H. O. T.: Modeling secondary organic aerosol formation from volatile chemical products, Atmos. Chem. Phys., 21, 18247–18261, https://doi.org/10.5194/acp-21-18247-2021, 2021.
Roldin, P., Ehn, M., Kurtén, T., Olenius, T., Rissanen, M. P., Sarnela, N., Elm, J., Rantala, P., Hao, L., Hyttinen, N., Heikkinen, L., Worsnop, D. R., Pichelstorfer, L., Xavier, C., Clusius, P., Öström, E., Petäjä, T., Kulmala, M., Vehkamäki, H., Virtanen, A., Riipinen, I., and Boy, M.: The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system, Nat. Commun., 10, 4370, https://doi.org/10.1038/s41467-019-12338-8, 2019.
Rolph, G., Stein, A., and Stunder, B.: Real-time Environmental Applications and Display sYstem: READY, Environ. Model. Softw., 95, 210–228, https://doi.org/10.1016/j.envsoft.2017.06.025, 2017.
Saarikoski, S., Timonen, H., Carbone, S., Kuuluvainen, H., Niemi, J. V., Kousa, A., Rönkkö, T., Worsnop, D., Hillamo, R., and Pirjola, L.: Investigating the chemical species in submicron particles emitted by city buses, Aerosol Sci. Tech., 51, 317–329, https://doi.org/10.1080/02786826.2016.1261992, 2017.
Salo, L., Hyvärinen, A., Jalava, P., Teinilä, K., Hooda, R. K., Datta, A., Saarikoski, S., Lintusaari, H., Lepistö, T., Martikainen, S., Rostedt, A., Sharma, V. P., Rähmään, M. H. Subudhi, S., Asmi, E., Niemi, J. V., Lihavainen, H., Lal, B., Keskinen, J., Kuuluvainen, H., Timonen, H., and Rönkkö, T.: The characteristics and size of lung-depositing particles vary significantly between high and low pollution Ftraffic environments, Atmos. Environ., 255, 118421, https://doi.org/10.1016/j.atmosenv.2021.118421, 2021.
Sandradewi, J., Prévôt, A. S., Szidat, S., Perron, N., Alfarra, M. R., Lanz, V. A., Weingartner, E., and Baltensperger, U.: Using aerosol light absorption measurements for the quantitative determination of wood burning and traffic emission contributions to particulate matter, Environ. Sci. Technol., 42, 3316–3323, https://doi.org/10.1021/es702253m, 2008.
Schraufnagel, D. E.: The health effects of ultrafine particles, Exp. Mol. Med., 52, 311–317, https://doi.org/10.1038/s12276-020-0403-3, 2020.
Sjostedt, S. J., Slowik, J. G., Brook, J. R., R. Chang, R. Y.-W., C. Mihele, C., Stroud, C. A., Vlasenko, A., and Abbatt, J. P. D. :Diurnally Resolved Particulate and VOC Measurements at a Rural Site: Indication of Significant Biogenic Secondary Organic Aerosol Formation, Atmos. Chem. Phys., 11, 5745–5760, https://doi.org/10.5194/acp-11-5745-2011, 2011.
Stein, A. F., Draxler, R. R., Rolph, G. D., Stunder, B. J. B., Cohen, M. D., and Ngan, F.: NOAA's HYSPLIT atmospheric transport and dispersion modeling system, B. Am. Meteorol. Soc., 96, 2059–2077, https://doi.org/10.1175/BAMS-D-14-00110.1, 2015.
Tarvainen, V., Hakola, H., Rinne, J., Hellén, H., and Haapanala, S.: Towards a comprehensive emission inventory of terpenoids from boreal ecosystems, Tellus B, 59, 526–534, https://doi.org/10.1111/j.1600-0889.2007.00263.x, 2007.
Tiitta, P., Leskinen, A., Hao, L., Yli-Pirilä, P., Kortelainen, M., Grigonyte, J., Tissari, J., Lamberg, H., Hartikainen, A., Kuuspalo, K., Kortelainen, A.-M., Virtanen, A., Lehtinen, K. E. J., Komppula, M., Pieber, S., Prévôt, A. S. H., Onasch, T. B., Worsnop, D. R., Czech, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Transformation of logwood combustion emissions in a smog chamber: formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging, Atmos. Chem. Phys., 16, 13251–13269, https://doi.org/10.5194/acp-16-13251-2016, 2016.
Timonen, H., Saarikoski, S., Tolonen-Kivimäki, O., Aurela, M., Saarnio, K., Petäjä, T., Aalto, P. P., Kulmala, M., Pakkanen, T., and Hillamo, R.: Size distributions, sources and source areas of water-soluble organic carbon in urban background air, Atmos. Chem. Phys., 8, 5635–5647, https://doi.org/10.5194/acp-8-5635-2008, 2008.
Timonen, H., Carbone, S., Aurela, M., Saarnio, K., Saarikoski, S., Ng, N. L., Canagaratna, M. R., Kulmala, M., Kerminen, V.-M., Worsnop, D. R., and Hillamo, R.: Characteristics, sources and water-solubility of ambient submicron organic aerosol in springtime in Helsinki, Finland, J. Aerosol Sci., 56, 61–77, https://doi.org/10.1016/j.jaerosci.2012.06.005, 2013.
Timonen, H., Karjalainen, P., Saukko, E., Saarikoski, S., Aakko-Saksa, P., Simonen, P., Murtonen, T., Dal Maso, M., Kuuluvainen, H., Bloss, M., Ahlberg, E., Svenningsson, B., Pagels, J., Brune, W. H., Keskinen, J., Worsnop, D. R., Hillamo, R., and Rönkkö, T.: Influence of fuel ethanol content on primary emissions and secondary aerosol formation potential for a modern flex-fuel gasoline vehicle, Atmos. Chem. Phys., 17, 5311–5329, https://doi.org/10.5194/acp-17-5311-2017, 2017.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Winterhalter, R., Neeb, P., Grossmann, D., Kolloff, A., Horie, O., and Moortgat, G.: Products and Mechanism of the Gas Phase Reaction of Ozone with β-Pinene, J. Atmos. Chem., 35, 165–197, https://doi.org/10.1023/A:1006257800929, 2000.
Yang, Y, Shao, M., Wang, X., Noelscher, A. C., Kessel, S., Guenther, A., and Williams, J.: Towards a quantitative understanding of total OH reactivity: A review, Atmos. Environ., 143, 147–161, 2016.
Yu, K., Zhu, Q., Du, K., and Huang, X.-F.: Characterization of nighttime formation of particulate organic nitrates based on high-resolution aerosol mass spectrometry in an urban atmosphere in China, Atmos. Chem. Phys., 19, 5235–5249, https://doi.org/10.5194/acp-19-5235-2019, 2019.
Zhang, Y., Favez, O., Petit, J.-E., Canonaco, F., Truong, F., Bonnaire, N., Crenn, V., Amodeo, T., Prévôt, A. S. H., Sciare, J., Gros, V., and Albinet, A.: Six-year source apportionment of submicron organic aerosols from near-continuous highly time-resolved measurements at SIRTA (Paris area, France), Atmos. Chem. Phys., 19, 14755–14776, https://doi.org/10.5194/acp-19-14755-2019, 2019.