the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Feb 2023
| 08 Feb 2023
Experimental chemical budgets of OH, HO2, and RO2 radicals in rural air in western Germany during the JULIAC campaign 2019
Changmin Cho
Andreas Hofzumahaus
Frank Holland
William J. Bloss
Birger Bohn
Hans-Peter Dorn
Marvin Glowania
Thorsten Hohaus
Paul S. Monks
Doreen Niether
Franz Rohrer
Roberto Sommariva
Zhaofeng Tan
Ralf Tillmann
Astrid Kiendler-Scharr
Andreas Wahner
Photochemical processes in ambient air were studied using the atmospheric simulation chamber SAPHIR at Forschungszentrum Jülich, Germany. Ambient air was continuously drawn into the chamber through a 50 m high inlet line and passed through the chamber for 1 month in each season throughout 2019. The residence time of the air inside the chamber was about 1 h. As the research center is surrounded by a mixed deciduous forest and is located close to the city Jülich, the sampled air was influenced by both anthropogenic and biogenic emissions. Measurements of hydroxyl (OH), hydroperoxyl (HO2), and organic peroxy (RO2) radicals were achieved by a laser-induced fluorescence instrument. The radical measurements together with measurements of OH reactivity (kOH, the inverse of the OH lifetime) and a comprehensive set of trace gas concentrations and aerosol properties allowed for the investigation of the seasonal and diurnal variation of radical production and destruction pathways. In spring and summer periods, median OH concentrations reached 6 × 106 cm−3 at noon, and median concentrations of both HO2 and RO2 radicals were 3 × 108 cm−3. The measured OH reactivity was between 4 and 18 s−1 in both seasons. The total reaction rate of peroxy radicals with NO was found to be consistent with production rates of odd oxygen (Ox= NO2 + O3) determined from NO2 and O3 concentration measurements. The chemical budgets of radicals were analyzed for the spring and summer seasons, when peroxy radical concentrations were above the detection limit. For most conditions, the concentrations of radicals were mainly sustained by the regeneration of OH via reactions of HO2 and RO2 radicals with nitric oxide (NO). The median diurnal profiles of the total radical production and destruction rates showed maxima between 3 and 6 ppbv h−1 for OH, HO2, and RO2. Total ROX (OH, HO2, and RO2) initiation and termination rates were below 3 ppbv h−1. The highest OH radical turnover rate of 13 ppbv h−1 was observed during a high-temperature (max. 40 ∘C) period in August. In this period, the highest HO2, RO2, and ROX turnover rates were around 11, 10, and 4 ppbv h−1, respectively. When NO mixing ratios were between 1 and 3 ppbv, OH and HO2 production and destruction rates were balanced, but unexplained RO2 and ROX production reactions with median rates of 2 and 0.4 ppbv h−1, respectively, were required to balance their destruction. For NO mixing ratios above 3 ppbv, the peroxy radical reaction rates with NO were highly uncertain due to the low peroxy radical concentrations close to the limit of NO interferences in the HO2 and RO2 measurements. For NO mixing ratios below 1 ppbv, a missing source for OH and a missing sink for HO2 were found with maximum rates of 3.0 and 2.0 ppbv h−1, respectively. The missing OH source likely consisted of a combination of a missing inter-radical HO2 to OH conversion reaction (up to 2 ppbv h−1) and a missing primary radical source (0.5–1.4 ppbv h−1). The dataset collected in this campaign allowed analyzing the potential impact of OH regeneration from RO2 isomerization reactions from isoprene, HO2 uptake on aerosol, and RO2 production from chlorine chemistry on radical production and destruction rates. These processes were negligible for the chemical conditions encountered in this study.
- Article
(9074 KB) - Full-text XML
-
Supplement
(2265 KB) - BibTeX
- EndNote





The hydroxyl (OH) radical is the dominant daytime atmospheric oxidant. It reacts with most trace gases in the troposphere and thereby controls the rate of their removal and chemical transformation. In the lower troposphere, OH is primarily produced by solar photolysis of ozone (O3) and nitrous acid (HONO). The reaction of OH with trace gases leads to the formation of hydroperoxy (HO2) or organic peroxy (RO2, with R= organic group) radicals, which undergo further radical reactions. Generally, these reactions are cyclic chain reactions, in which OH, HO2, and RO2 are converted into each other, while at the same time emitted pollutants are oxidized and converted into secondary pollutants such as ozone and oxygenated volatile organic compounds (OVOCs). Because the conversion of radicals occurs on a timescale of seconds to minutes, they are often referred to as the ROX family (OH + HO2+ RO2). The most important radical reactions in the lower atmosphere are summarized in Table 1. Understanding radical chemistry is the basis for reliable predictions of the atmospheric lifetime and chemical transformation of air pollutants and climate-relevant gases by atmospheric chemistry models (Stone et al., 2012).
The level of agreement between simulated and observed radical concentrations in various environments shows the degree of understanding of the underlying radical chemical mechanism. Even though good agreement is found in some cases (Tan et al., 2001; Konrad et al., 2003; Mihelcic et al., 2003; Lelieveld et al., 2008; Kubistin et al., 2010; Whalley et al., 2011), there are significant unexplained discrepancies between modeled and measured OH in forested regions (Wolfe et al., 2011; Griffith et al., 2013; Kim et al., 2013; Hens et al., 2013; Wolfe et al., 2014) and of HO2 and RO2 in polluted areas (Ren et al., 2003, 2006; Kanaya et al., 2007; Dusanter et al., 2009; Chen et al., 2010; Ren et al., 2013; Brune et al., 2016; Tan et al., 2018; Slater et al., 2020; Whalley et al., 2021), while different results are found depending on the abundance of nitric oxide (NO) in rural environments (Hofzumahaus et al., 2009; Lou et al., 2010; Elshorbany et al., 2012; Kanaya et al., 2012; Tan et al., 2017).
A chemical budget analysis using measured OH, HO2, and RO2 radical concentrations can help assess the strength of different radical production and loss paths. This allows identifying possible missing chemical processes by comparing the total production and destruction rates for the different radicals as concentrations are expected to be in steady state due to their short chemical lifetime. A large number of measurements needs to be available (e.g., OH reactivity, OH, peroxy radicals); therefore, there have only been a few studies focusing on the analysis of the chemical budget for OH radicals so far (Handisides et al., 2003; Hofzumahaus et al., 2009; Brune et al., 2016; Whalley et al., 2018; Tan et al., 2019; Whalley et al., 2021).
Results from field campaigns in China showed a larger OH radical destruction rate compared to its production rate in the afternoon, which points to an OH radical source that is unaccounted for. Discrepancies were highest when NO mixing ratios were lower than 2 ppbv (Hofzumahaus et al., 2009; Tan et al., 2019; Whalley et al., 2021). On the other hand, studies in urban areas in California (Brune et al., 2016) and in London (Whalley et al., 2018) as well as in a rural area in Hohenpeissenberg (Handisides et al., 2003) showed no significant gap between the OH production and destruction rates. Recently, radical measurements including RO2 enabled the investigation of HO2, RO2, and ROX production and destruction rates in field campaigns in China (Tan et al., 2019; Whalley et al., 2021). Tan et al. (2019) showed that an RO2 loss process was required in a campaign in Wangdu in summer, while HO2 production and destruction rates were balanced. This suggests a missing conversion of RO2 to OH in addition to the reaction of peroxy radicals with NO. Furthermore, Whalley et al. (2021) found large imbalances between peroxy radical production and destruction rates in Beijing, indicating substantially slower propagation of RO2 to HO2 radicals than anticipated.
In this study, OH, HO2, and RO2 radical concentrations as well as OH reactivity, the inverse of the OH radical lifetime, were measured in the atmospheric simulation chamber SAPHIR on the campus of Forschungszentrum Jülich (FZJ), Germany, in the Jülich Atmospheric Chemistry project (JULIAC) campaign. Ambient air was sampled from 50 m height into the SAPHIR chamber. From this dataset, a chemical budget analysis of OH, HO2, RO2 radicals, and their sum (ROX) was done using measured concentrations allowing the investigation of whether all radical production and destruction processes were accounted for during spring and summer.
2.1 JULIAC
The Jülich Atmospheric Chemistry (JULIAC) project campaign was conducted at Forschungszentrum Jülich (FZJ, 50.9∘ N, 6.4∘ E), Germany. The project consisted of four 1-month-long intensive campaigns studying atmospheric chemistry in ambient air in each season throughout 2019. The location is surrounded by a deciduous forest and is located in a rural environment near a town, Jülich (33 000 inhabitants), 25 km northeast, 40 km west, and 43 km southwest from the three large cities of Aachen, Cologne, and Düsseldorf, respectively. Therefore, ambient air is influenced by both biogenic and anthropogenic emission sources.
Table 1Chemical reactions and rate constants used for the analysis of the chemical budgets of radicals. Values of reaction rate constants are given for standard conditions (298 K, 1 atm). Actual numbers are used for the calculations.
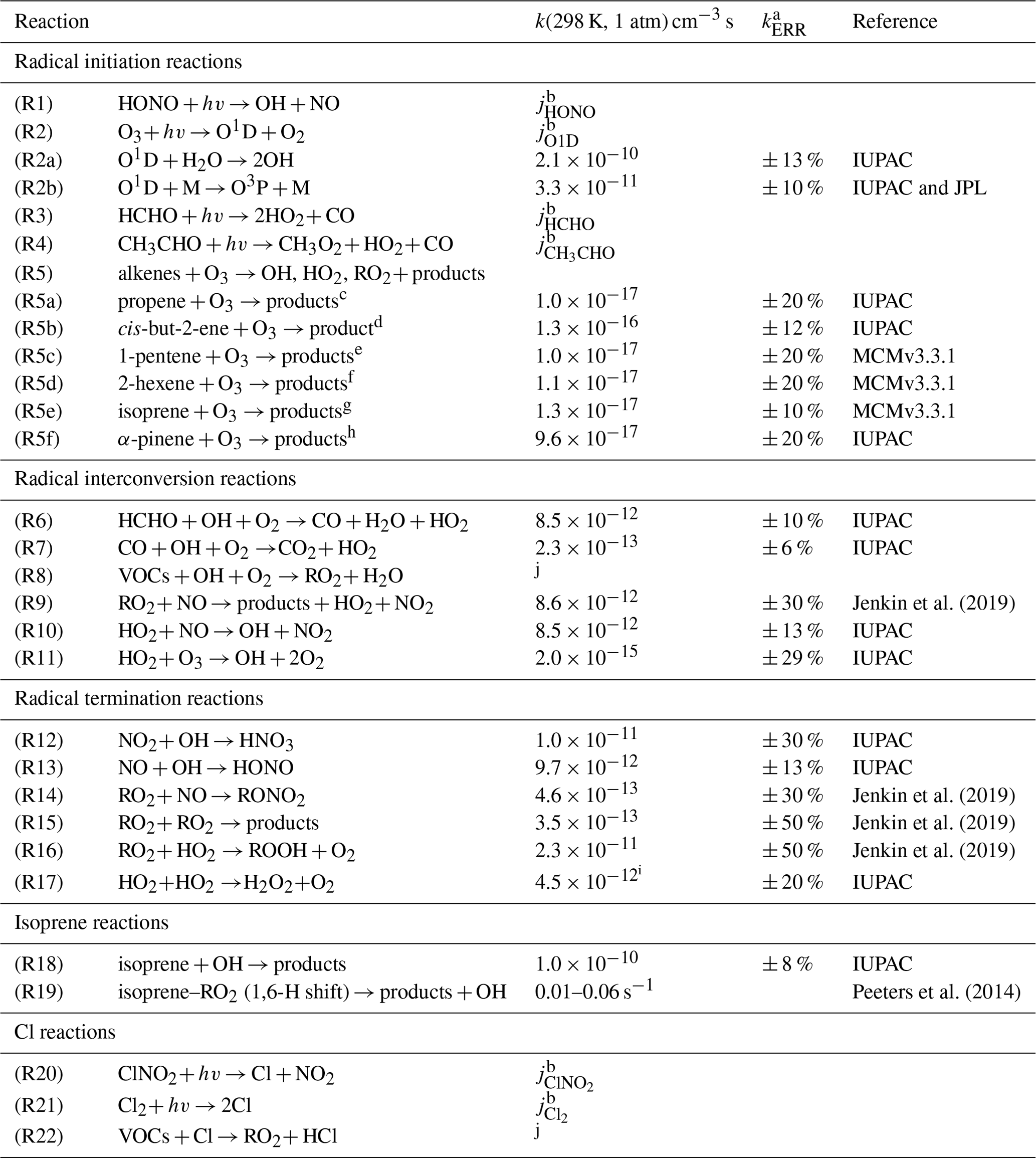
a 1σ uncertainty. b Measured photolysis frequencies. c Yield for OH: 0.36, HO2: 0.10, RO2: 0.42 from Novelli et al. (2021). d Yield for OH: 0.36, HO2: 0.15, RO2: 0.51 from Novelli et al. (2021). e Yield for OH: 0.32, HO2: 0.09, RO2: 0.37 from Novelli et al. (2021). f Yield for OH: 0.48, HO2: 0.11, RO2: 0.59 from Novelli et al. (2021). g Yield for OH: 0.26, HO2: 0.26 from Malkin et al. (2010). h Yield for OH: 0.8 from Cox et al. (2020). i At 1 % water vapor mixing ratio. j Highly variable depending on the specific VOC.
Table 2Specification of instruments used in the JULIAC campaign for the analysis in this work.
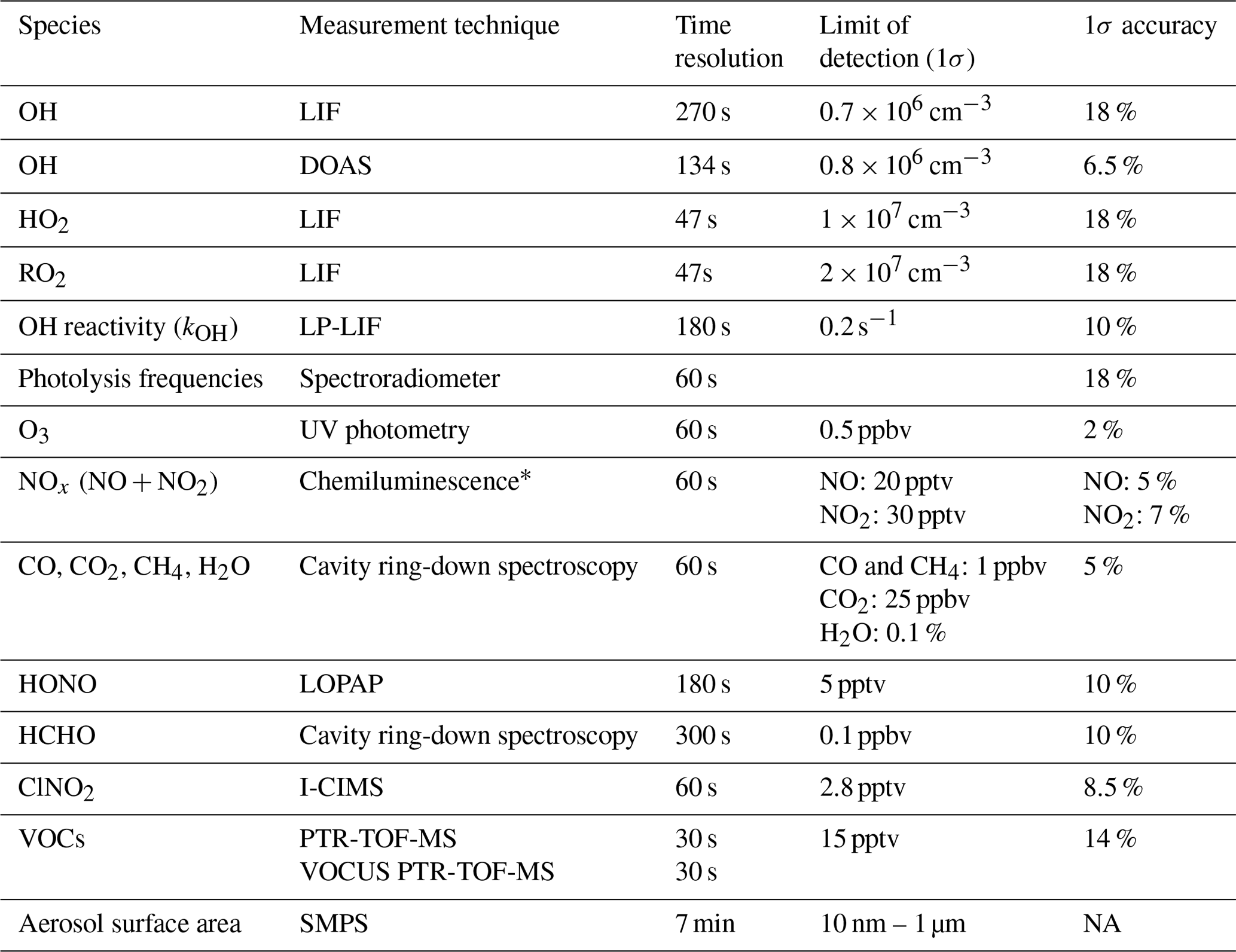
* NO2 was converted to NO before detection using a custom-built photolytic converter. NA: not available.
The investigation of the photochemistry was performed in the SAPHIR chamber, which was equipped with a large set of instruments measuring radicals, trace gases, and aerosol (Table 2). The SAPHIR chamber has a cylindrical shape and is made of a double-wall Teflon (FEP) film. A slight overpressure (35 Pa) is maintained in the chamber, and the space between the two films is permanently flushed with pure nitrogen (Linde, purity: > 99:99990 %) to prevent outside air from penetrating the inner chamber. The chamber is equipped with a shutter system allowing the air to be either shielded from or exposed to solar radiation.
In the JULIAC campaign, ambient air was sampled at a high flow rate of 660 m3 h−1 from a 50 m high inlet line (104 mm inner diameter, SilcoNert® coated stainless steel) by means of an oil-free turbo blower (Aerzener Maschinenfabrik, AERZEN Turbo G3, type TB 50–0.6 S). Large particles (> 10 µm diameter) were removed by a SilcoNert®-coated cyclone (LTG, ZSB-6). The temperatures in the inlet line and cyclone were controlled to be slightly higher than ambient temperature (+1 to 2 ∘C) to avoid water vapor condensation in the inlet system. A -way valve directed some of the air (flow rate of 250 m3 h−1) into the chamber. Two fans inside the chamber ensured fast mixing on a timescale of a few minutes. As a result, the chamber behaved as a continuously stirred photochemical flow reactor with a mean residence time of air of 1.1 h. During the transition time of 3.5 s from the tip of the inlet to the SAPHIR chamber, atmospheric ROX radicals are lost on walls, but concentrations are rapidly re-established in the sampled ambient air inside the sunlit chamber.
The use of the chamber as a flow reactor has advantages compared to field measurements in the open air. Perturbations of the studied chemistry due to local emissions of VOCs or NOx can be avoided. Transient fluctuations of reactants in the sampled air, for example due to spikes of NO from passing cars, are smoothed out in the chamber. Due to the homogeneous mixing, instruments connected to the chamber measure the same air composition, and segregation effects on reaction rates are insignificant.
The air composition could be influenced by the inlet line and chamber surfaces. As the whole inlet line is heated and chemically inert due to the SilcoNert® coating, no relevant wall loss or desorption of trace gases is expected from the inlet. This assumption was confirmed by comparing OH reactivity measured at several positions of the inlet line. No significant differences were found between measurements whether the air was sampled upstream of the cyclone or downstream of the blower. Wall losses of trace gases (VOCs, NOx, O3) inside the SAPHIR chamber were found to be negligible in previous experiments (e.g., Kaminski et al., 2017, Rolletter et al., 2020).
Nitrous acid (HONO) and formaldehyde (HCHO) are known to be emitted from the chamber film when it is exposed to solar radiation (Rohrer et al., 2005). These emissions significantly increase the concentrations of HONO and HCHO in the chamber. Due to the transmission through the Teflon film and shading from construction elements of the chamber, the absolute actinic flux density is reduced by 20 % to 40 % compared to outside the chamber. It is worth noting, however, that the relative spectral distribution of the solar radiation is not changed by the transmission through the chamber film (Bohn and Zilken, 2005).
The floor underneath the chamber is heated by the solar radiation. Although it is not in direct contact with the foil, the air temperature in the chamber was on average 0.7 ∘C higher during winter and autumn and 1.9 ∘C higher during spring and summer than the temperature outside the chamber in daytime. Since photochemistry was studied in the chamber, all data for chemical and physical conditions shown in this work refer to conditions inside the chamber.
The measurements in the campaign were interrupted at least once a week for calibration and maintenance of instruments. Some days were also excluded from the analysis in this work because the chamber shutter system was kept closed to protect the chamber film during bad weather from strong wind gusts and/or precipitation. Reference experiments with clean synthetic air were performed to investigate possible changes in the strength of chamber emissions and to check for instrumental backgrounds. In addition, chemical actinometry experiments, in which NO2 was photolyzed in synthetic air, were performed before and after each intensive period. The comparison of actinometric and spectroradiometric values was used to track and correct for changes in light transmission due to aging of the chamber wall (Bohn et al., 2005).
2.2 Instrumentation
2.2.1 OH, HO2, and RO2 radical and OH reactivity (kOH) measurements
OH, HO2, and RO2 radicals were measured by the FZJ laser-induced fluorescence (LIF) instrument, which included a newly developed chemical modulation reactor (CMR) for interference-corrected measurements of OH radicals (Cho et al., 2021). The signals of the instrument were calibrated against well-defined radical concentrations that were produced from water photolysis in synthetic air at a wavelength of 185 nm using radiation from a mercury lamp. A detailed description of the LIF instrument and its calibration can be found in previous publications (Holland et al., 2003; Fuchs et al., 2008, 2011, 2012).
The OH radical is sampled through a nozzle with a 0.4 mm diameter pinhole and is excited by a pulsed laser at a wavelength of 308 nm in a low-pressure (4 hPa) fluorescence cell. The emitted resonant fluorescence is detected with a time delay by a time-gated micro-channel plate detector (MCP). In the JULIAC campaign, a chemical modulation reactor (CMR) was implemented on top of the OH cell to quantify potential interferences. This is achieved by periodically removing ambient OH by an OH scavenger that is injected in the reactor (propane, Air Liquide, purity > 99.95 %, (5.0 ± 0.1) % mixture in nitrogen) before the air enters the fluorescence cell. During the campaign, the observed interference could be fully explained by the well-characterized interference from the photolysis of ozone in humid air inside the detection cell. No evidence for unexplained interference was found (Cho et al., 2021). The limit of detection for OH was 0.7 × 106 cm−3 and the accuracy was 18 % (1σ).
OH radical concentrations were also measured by differential optical absorption spectroscopy (DOAS) using a multiple folded light path for absorption inside along the chamber. The DOAS technique is a calibration-free technique (Hausmann et al., 1997; Schlosser et al., 2007, 2009). The limit of detection was 0.8 × 106 cm−3 and the 1σ accuracy was 6.5 %. Due to a technical laser problem, the DOAS instrument was not available in spring.
HO2 radicals were detected by the LIF instrument in a separate detection cell, where HO2 is chemically converted to OH radicals in the reaction with NO (Air Liquide, 1 % NO in N2, purity > 99.5 %) that is injected in the fluorescence cell (Fuchs et al., 2011). During the JULIAC campaign, two different concentrations (2.5×1013 and 1.0×1014 cm−3) of NO in the fluorescence cell were used to observe possible interference from specific RO2 radicals as highlighted by Fuchs et al. (2011). No difference between HO2 measurements at high and low NO concentrations was found, suggesting that there was no significant interference from RO2.
In addition, the sum of OH, HO2, and RO2 (ROX) was measured by the ROX–LIF system. Air is sampled into a chemical converter (pressure of ∼ 25 hPa), where a mixture of NO (Air Liquide, 500 ppmv NO in N2, purity > 99.5 %) and CO (Air Liquide, 10 % CO in N2, purity > 99.997 %) is injected. The NO converts RO2 radicals to HO2 radicals, and CO converts OH radicals formed from the reaction of HO2 radicals with NO back to HO2. Therefore, an equilibrium between OH and HO2 is established. Concentrations are chosen so that the equilibrium is on the side of HO2. In a low-pressure cell downstream of the converter HO2 radicals are converted to OH radicals by injecting excess NO (Air Liquide, pure NO, purity > 99.5 %) (Fuchs et al., 2008) that shifts the equilibrium between OH and HO2 to OH. The RO2 concentration is obtained from the difference between the sum measurement of ROX and measurements of OH and HO2 concentrations in the other two detection cells. The RO2 detection sensitivity was calibrated for methyl peroxy radicals (CH3O2), which are produced from the reaction of OH with methane (CH4) in the calibration system. The resulting calibration is also applicable to the majority of other atmospheric alkyl peroxy radicals (Fuchs et al., 2008, 2011), and recent laboratory tests performed with a variety of VOCs including monoterpenes and chained alkanes for the CO and NO mixing ratios applied in the ROX converter during the JULIAC campaign showed a decrease of less than 15 % sensitivity compared to methyl peroxy radicals, which is within the accuracy of the instrument.
The signals in the HO2 and RO2 detection systems contain a background signal observed when NO is injected into the detection cells, even if no radicals are present in the air sampled. The background signal can be characterized when the inlet of the detection system is overflown with synthetic air, which is part of the calibration procedures. During the JULIAC campaign the background varied from calibration to calibration and was often larger than the smallest signals measured in ambient air from the chamber (Table S1). The highest background signals obtained from calibrations are therefore regarded as an upper limit, and the variability is considered to be an additional uncertainty in the measured HO2 and RO2 concentrations. HO2 and RO2 background signals, which are subtracted in the evaluation of HO2 and RO2 measurements, were taken from reference experiments in the dark clean chamber, when no HO2 or RO2 radicals are expected. The subtracted signals for each period are available in Table S1 and in most cases were equivalent to concentrations lower than 1 × 107 cm−3 for both HO2 and RO2 measurements.
The total OH reactivity (kOH), the inverse of the chemical lifetime of OH radicals, was measured in ambient air by a laser-flash photolysis LIF instrument (Lou et al., 2010; Fuchs et al., 2017). A high concentration of OH radicals is produced by flash photolysis (266 nm, 1 Hz repetition rate) of ozone in humid air (Reaction R2 in Table 1) in a flow tube that is on top of an OH fluorescence cell. The pseudo-first-order decay of OH in the chemical reactions with atmospheric reactants is measured, directly giving the OH reactivity.
2.2.2 Other trace gas, aerosol property, and photolysis frequency measurements
A comprehensive set of instruments operated during the JULIAC campaign (Table 2) analyzed the air composition inside the chamber. Photolysis frequencies inside the chamber were derived from solar actinic flux densities measured by a spectroradiometer mounted on the roof of the nearby institute building. Chamber values were calculated using a model approach considering shading effects and the influence of the chamber film (Bohn et al., 2005; Bohn and Zilken, 2005). Formaldehyde (HCHO) was detected by cavity ring-down spectroscopy (Picarro, G2307, Glowania et al., 2021). NO and NO2 were measured by chemiluminescence (Eco Physics, TR780, NO2 conversion by a custom-built photolytic converter). In addition, HONO was measured by long-path absorption photometry (LOPAP, Kleffmann et al., 2006; Häseler et al., 2009), CO, CO2, CH4, and H2O by cavity ring-down spectroscopy (Picarro, G2401), and O3 by UV absorption (Ansyco-41M and Thermo scientific-49I). Volatile organic compounds (VOCs) were detected by a proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS, Ionicon) (Jordan et al., 2009) and a VOCUS PTR-TOF-MS instrument (Aerodyne). The VOCs included in this study are listed in Table S2 and include isoprene and some carbonyl compounds. Total aerosol surface area was determined from measurements by a scanning mobility particle sizer (SMPS). In the summer and autumn periods, nitryl chloride (ClNO2) was detected by a chemical ionization mass spectrometer using iodine as the reagent ion (I-CIMS) (Sommariva et al., 2018; Tan et al., 2022).
In addition to measurements in the chamber, concentrations of O3 and NOx were also measured in the inlet system before the air flowed into the SAPHIR chamber. For these measurements, a combined system (Eco Physics, CraNOX) consisting of an ozone photometer and a chemiluminescence instrument for NOx was deployed. Measurements were used to determine the photochemical ozone production in the JULIAC campaign. The measurement setup and concept of the evaluation will be discussed in detail in a further publication.
2.3 Chemical budget calculations
A chemical budget analysis, similar to that in Tan et al. (2019) and Whalley et al. (2021), was applied for OH, HO2, RO2, and the sum of all three radicals (ROX) to the dataset from the JULIAC campaign. All reactions typically considered to be relevant for the generation and destruction of these radicals are considered (Table 1). Rate constants and their uncertainties were mainly taken from IUPAC recommendations (Atkinson et al., 2004, 2006; Cox et al., 2020) or more recent studies. If not otherwise specified, radical production and destruction rates are calculated from measured concentrations of reactants.
2.3.1 Chemical budget of OH radicals
The production rate of OH radicals includes primary production reactions (Reactions R1, R2, and R5) and radical interconversion reactions (Reactions R10 and R11):
Here, φOH is the effective OH yield of the ozone photolysis including the reaction of excited oxygen atoms O(1D) with H2O, producing two OH radicals. is the OH yield of the ozonolysis reaction of alkenes, and represents the rate constants of the corresponding reactions.
POH,Isop is the effective production of OH radicals from unimolecular reactions (1,6-hydrogen shift reactions) of isoprene–RO2 radicals (Z-δ-RO2-I and II, Peeters et al., 2014) and the subsequent chemistry of products. As there was no measurement of speciated RO2 radicals, isoprene–RO2 radical concentrations are estimated from steady-state conditions considering their production from the reaction of isoprene with OH and their destruction in bimolecular reaction (reaction rate kbi) as well as unimolecular reactions (bulk reaction rate kbulk 1,6-H as defined in Peeters et al., 2014).
Bimolecular loss reactions include reactions with NO (Reactions R9 and R14), RO2 (Reaction R15), and HO2 (Reaction R16). The OH production from isoprene–RO2 isomerization reactions is simplified in the calculation of the total OH production in this work by assuming that each isomerization reaction rapidly produces one OH radical from the subsequent reactions of products such as photolysis of hydroxy-peroxy aldehyde (HPALD). In this case, the radical production rate is equal to the loss rate of the isoprene–RO2 due to isomerization reactions ().
The total loss rate of OH radicals for the chemical budget analysis is determined by the product of the total OH reactivity (kOH) and the OH radical concentration.
2.3.2 Chemical budget of HO2 radicals
The production rate of HO2 radicals includes primary reactions (Reactions R3, R4, and R5) and interconversion reactions (Reactions R6, R7, and R9, Table 1).
Here, the photolysis frequency of HCHO (jHCHO) includes only paths generating radicals. is the HO2 yield from the ozonolysis of alkenes. The reactions of OH with H2 and O3 are not considered due to their negligible contributions to the HO2 production.
The loss rate of HO2 is determined by the reactions with NO (Reaction R10), O3 (Reaction R11), RO2 (Reaction R16), and HO2 (Reaction R17).
Here, the humidity dependence of k17 was taken into account. The reaction of HO2 radicals with NO2 is not included as the thermal decomposition of peroxynitric acid (HO2NO2) forming back HO2 radicals and NO2 is instantaneous for the temperatures experienced during the JULIAC campaign.
In a sensitivity calculation (Sect. 4.2.3), potential loss of HO2 due to heterogeneous uptake on aerosol is investigated. The first-order loss rate (khet.) can be described as
where is the mean molecular velocity of HO2 (4.44 × 105 cm s−1 at 298 K), [AS] is the measured aerosol surface area concentration, and γeff. is the effective uptake coefficient.
2.3.3 Chemical budget of RO2 radicals
Primary sources of RO2 radicals include all oxidation reactions of VOCs with OH, Cl, NO3 radicals, and O3. As the number of measured VOC species in this study was limited (Table S2) and because it is generally difficult to capture the entire spectrum of atmospheric VOCs (Goldstein and Galbally, 2007; Lou et al., 2010), the measured total OH reactivity (kOH) can be used to calculate the RO2 radical production from the reactions of VOCs with OH. First, the contributions from CO, NO, NO2, HCHO, and O3 are removed from the measured OH reactivity as these species do not form RO2 radicals in the reaction with OH. It is then assumed that the remaining fraction can be attributed to organic compounds (VOC reactivity (kVOC)) including measured and unmeasured VOCs, which produce RO2 radicals in their reaction with OH.
For some aromatics, such as toluene, benzene, and xylene, the prompt formation of HO2 is expected from their reaction with OH (Nehr et al., 2011, 2014; Jenkin et al., 2019). However, in this campaign, their concentrations were small and their average contributions to the OH reactivity from VOCs were only 2.8 %. Therefore, their potential impacts on the RO2 production are negligible (less than 1 %).
In addition, RO2 production from ozonolysis needs to be included. In this work, only the reactions of measured organic compounds are considered. The contribution to the RO2 production from the oxidation of VOCs by the NO3 radical was negligible during daytime due to the low VOC load (low OH reactivity), so NO3 destruction by photolysis and reaction with NO dominated.
Reactions of chloride (Cl) also produce RO2 radicals, but the concentration was not measured in the JULIAC campaign. However, one of the most important precursor species, nitryl chloride (ClNO2), was detected during the campaign (except in spring, Tan et al., 2022). ClNO2 can accumulate during nighttime, but it is photolyzed after sunrise, yielding NO2 and Cl atoms (Reaction R20). Assuming as an upper limit that each Cl atom reacts with a VOC (Tanaka et al., 2003), the RO2 production rate from Cl radicals can be calculated as
The total RO2 production rate is then calculated as
Here, is the RO2 yield from the ozonolysis of alkenes species (Table 1).
With respect to the destruction rate of RO2, its reactions with NO, HO2, and other RO2 as well as unimolecular reactions of specific isoprene–RO2 radicals () (Eq. 4) are considered in this work:
2.3.4 Chemical budget of ROX radicals
In the chemical budget of the sum of OH, HO2, and RO2 (ROX), inter-radical conversion reactions cancel out, and only initiation and termination reactions are included. Therefore, the ROX radical budget analysis allows investigating whether primary radical source reactions or termination processes are missing in the chemical mechanism used (Table 1).
The production rate of the ROX radicals is given by the sum of rates from radical initiation reactions (Reactions R1–R5, R20–R22, Table 1).
Radicals can additionally be produced from the photolysis of other oxygenated organic compounds (OVOCs, e.g., Reaction R4) not included in Eq. (12). Their potential impact is further discussed in Sect. 4.2.2.
The loss rate of the ROX radical is calculated by the sum of rates from radical termination reactions (Reactions R12–R17).
2.3.5 Uncertainties in the calculated production and destruction rates
The uncertainty of each production or loss rate is calculated by Gaussian summation of the 1σ errors of the measured quantities (Table 2) and the uncertainties of the reaction rate constants (Table 1).
For reactions of RO2 with NO (Reactions R9, R14), HO2 (Reaction R16), and RO2 (Reaction R15), generic rate constants are used for the sum of RO2 radicals (Table 1, Jenkin et al., 2019). Rate constants of the NO reaction with RO2 derived from hydrocarbons (< C5) and with oxygenated peroxy radicals range from to cm3 s−1 (Jenkin et al., 2019). The 1σ uncertainty of the rate constants varies from 6 % to 30 %. In the error calculations here, an upper limit value of 30 % is applied. However, for reactions of RO2 with HO2 and with RO2, the range of rate constants varies by more than an order of magnitude. In the calculations, an uncertainty of 50 % is used for the reaction rate constants of RO2 with HO2 and with RO2.
As there are no measurements of speciated RO2 radicals, a yield of 5 % for the formation of organic nitrates is assumed for all RO2, but the yield can vary between 1 % for methyl peroxy radicals (CH3O2) and more than 20 % for RO2 from monoterpene species. This simplification can introduce systematic errors in the calculations (Sect. 4.2.1).
2.4 Odd oxygen production rate
In the troposphere, ozone is formed exclusively by the oxidation of NO to NO2 through reaction with RO2 (Reaction R9) and HO2 (Reaction R10), followed by NO2 photolysis (Fishman and Carney, 1984; Sillman et al., 1990; Kleinman et al., 2002).
During the day, the photolysis of NO2 and the back reaction of NO with O3 form a rapid photochemical equilibrium between O3 and NO2. The sum of O3 and NO2 is therefore defined as odd oxygen (Ox) (Han et al., 2011; Goldberg et al., 2015). The relative composition of Ox depends on the NO2 photolysis frequency and the NO concentration. For the conditions of the spring and summer periods in the JULIAC campaign, Ox consisted predominantly (> 85 %) of O3.
In this work, the net production rate of Ox () was determined experimentally from the increase in Ox in the sunlit SAPHIR chamber. Furthermore, measurements of radicals and NOx were used to calculate from the rate of Ox formation reactions (Reactions R9, R10) and Ox loss by the reaction of NO2 with OH (Reaction R12) (Mihelcic et al., 2003; Cazorla et al., 2012).
This calculation neglects minor Ox destruction processes such as the reaction of O3 with NO2, OH, HO2, Cl, or alkenes since they did not play a notable role during the day in this campaign.
3.1 Data quality of radical measurements
Performing measurements in the SAPHIR chamber allowed testing the accuracy of radical measurements in different ways that are typically not available in field experiments. First, OH radicals was measured by two independent instruments, the OH DOAS and LIF instruments (Cho et al., 2021). Second, the Ox production rate calculated from measured concentrations of HO2 and RO2 could be compared to the observed increase in Ox concentrations in the chamber, which can be solely attributed to chemical reactions. This is possible because other factors typically impacting the Ox concentration in field experiments such as transportation processes are not effective.
OH concentrations were measured by the LIF instrument by applying the chemical modulation scheme and the DOAS in the winter, summer, and autumn periods of the campaign. As OH concentrations were close to the limit of detection in autumn and winter, a meaningful comparison of measurements was only possible for the summer period. A detailed comparison of measurements can be found in Cho et al. (2021). In general, the OH measurements of the two instruments agreed within their measurement errors (Table 1), giving a slope of 1.1 ± 0.02 in a linear regression analysis. The good agreement confirms that the newly developed chemical modulation system of the LIF instrument allowed for interference-free OH concentration measurements for conditions of the campaign. Only in the period from 22 to 26 August, which was characterized by exceptionally high temperatures (30 to 40 ∘C), were OH concentrations measured by the LIF instrument systematically higher by 25 % than those measured by the DOAS instrument for unknown reasons (Cho et al., 2021). OH concentrations measured by the DOAS instrument were used for the analysis of the radical budgets in this period.
Net Ox production rates were determined from the measured increase in Ox concentrations in the chamber and compared to calculations from the turnover rates of HO2 and RO2 reactions with NO. This calculation also takes the NO2 loss due to its reaction with OH into account (Eq. 14). The odd oxygen production rate did not exceed 1 ppbv h−1 in winter and autumn due to the generally low photochemical activity in these seasons. In spring and summer, the Ox production rate showed clear diurnal variations with noontime maxima that reached up to 16 ppbv h−1. In these seasons, both methods for determining the Ox production rate agreed within ± 15 % (1σ). Observed discrepancies were less than 1 ppbv h−1 when NO mixing ratios were lower than 1 ppbv but reached values of 3 ppbv h−1 for NO mixing ratios of 3–4 ppbv NO. The largest discrepancy of 8.5 ppbv h−1 was found in the morning on 29 April, when the NO mixing ratio exceeded 9 ppbv. High NO values suppressed HO2 and RO2 concentrations to values below 2.0 ×107 cm−3, which is within the range of the background corrections for the HO2 and RO2 measurements (Table S1). Under these conditions, an erroneous background subtraction may have caused the observed discrepancies.
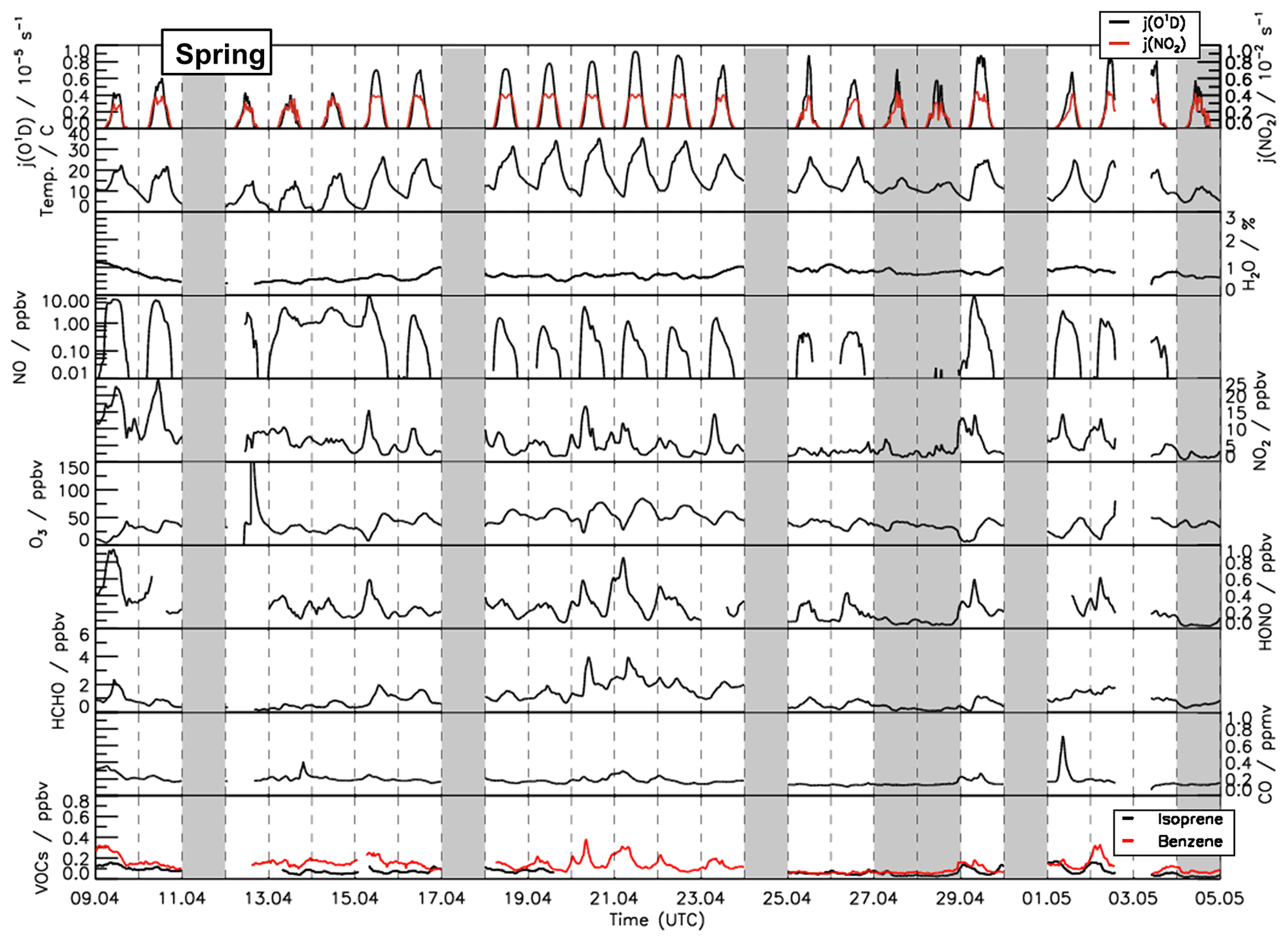
Figure 1Time series of temperature and trace gas concentrations during the spring period of the JULIAC campaign. Vertical dashed lines denote midnight. Grey shaded areas indicate calibration days when no measurements were done and days when the chamber roof was closed due to bad weather conditions.
3.2 Meteorological and chemical conditions during the JULIAC campaign
A broad range of meteorological and chemical conditions was encountered during the JULIAC campaign. During the winter and autumn periods (Figs. S1 and S2), the sky was often overcast and it rained frequently. Temperatures were generally below 10 ∘C and the photolysis frequencies of ozone () and nitrogen dioxide () mostly remained below and s−1, respectively. During spring and summer, temperatures in the chamber were up to 35 ∘C in mid-April and 40 ∘C between 24 and 31 August (Figs. 1 and 2). Photolysis frequencies in the chamber were s−1 () and s−1 ().
The air was sampled at all times from 50 m above the ground. The temperature at different heights measured between 5 m and 120 m at a meteorological tower near the SAPHIR chamber showed that the air was well mixed within this height range during the day. Therefore, it can be assumed that the chemical composition of the air sampled into the chamber is representative of the air within the atmospheric boundary layer. At night, vertical temperature profiles showed atmospheric stratification below 100 m. The air at 50 m can be assumed to be isolated from the ground and therefore not affected by surface emissions or deposition on surfaces at the ground level.
Overall, relatively clean air was sampled during the whole of the JULIAC campaign, as indicated by CO and NO mixing ratios below 0.3 ppmv and 2 ppbv, respectively. Concentrations of anthropogenic organic compounds (e.g., benzene and toluene) were low with mixing ratios of less than 0.5 ppbv. Even though the measurement site is surrounded by a deciduous forest, the concentrations of biogenic organic compounds such as isoprene and monoterpenes were also low (median 0.8 ppbv and 0.15 ppbv, respectively) compared to previously reported values measured on the campus of FZJ in summer, when isoprene concentrations ranged between 0.5 and 4 ppbv (Komenda et al., 2003; Spirig et al., 2005; Kanaya et al., 2012). A possible reason for the low values could be damage to trees from severe droughts in the previous year (BMEL, 2021).
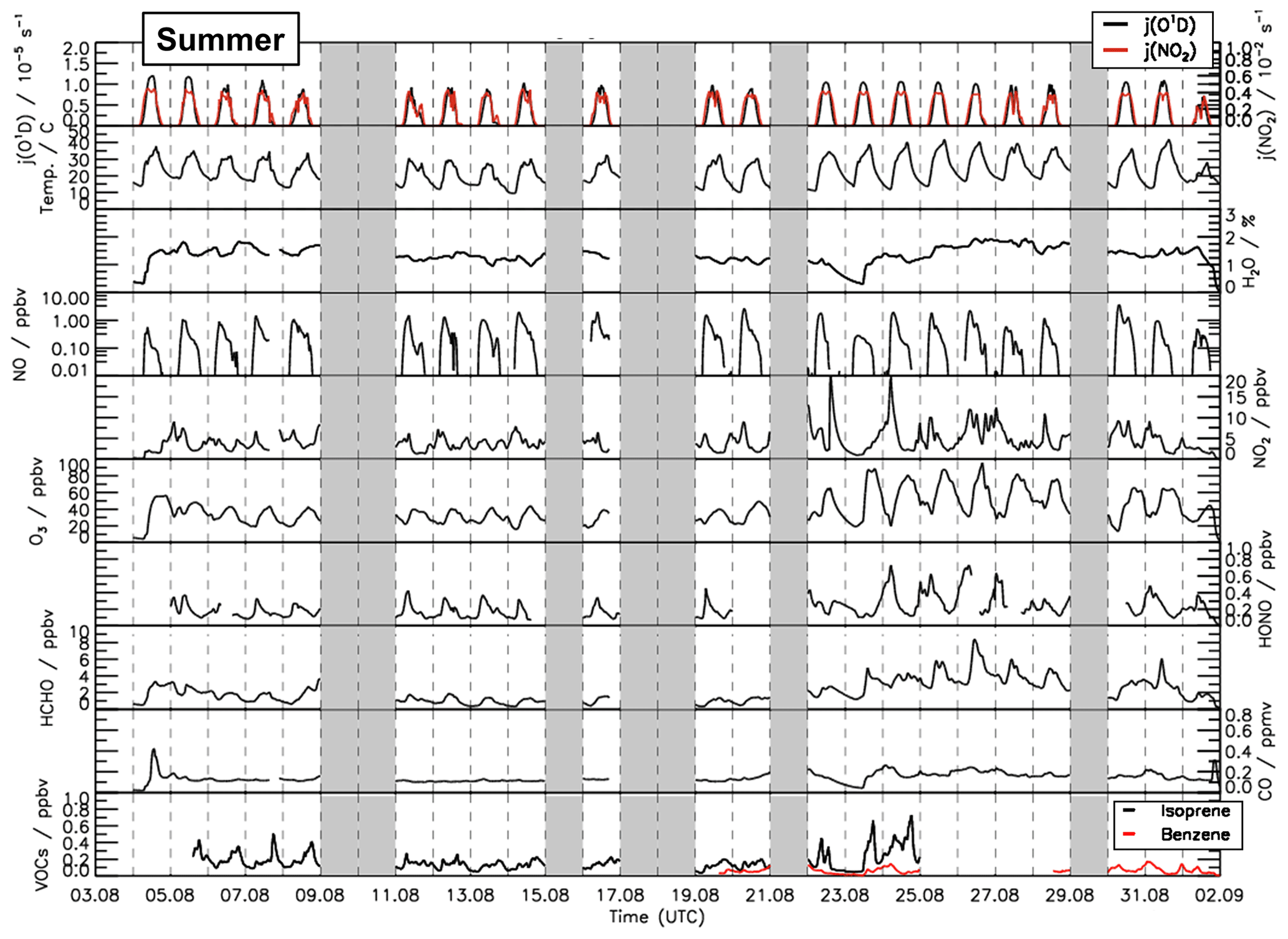
Figure 2Time series of temperature and trace gas concentrations during the summer period of the JULIAC campaign. Vertical dashed lines denote midnight. Grey shaded areas indicate calibration days when no measurements were done and days when the chamber roof was closed due to bad weather conditions.
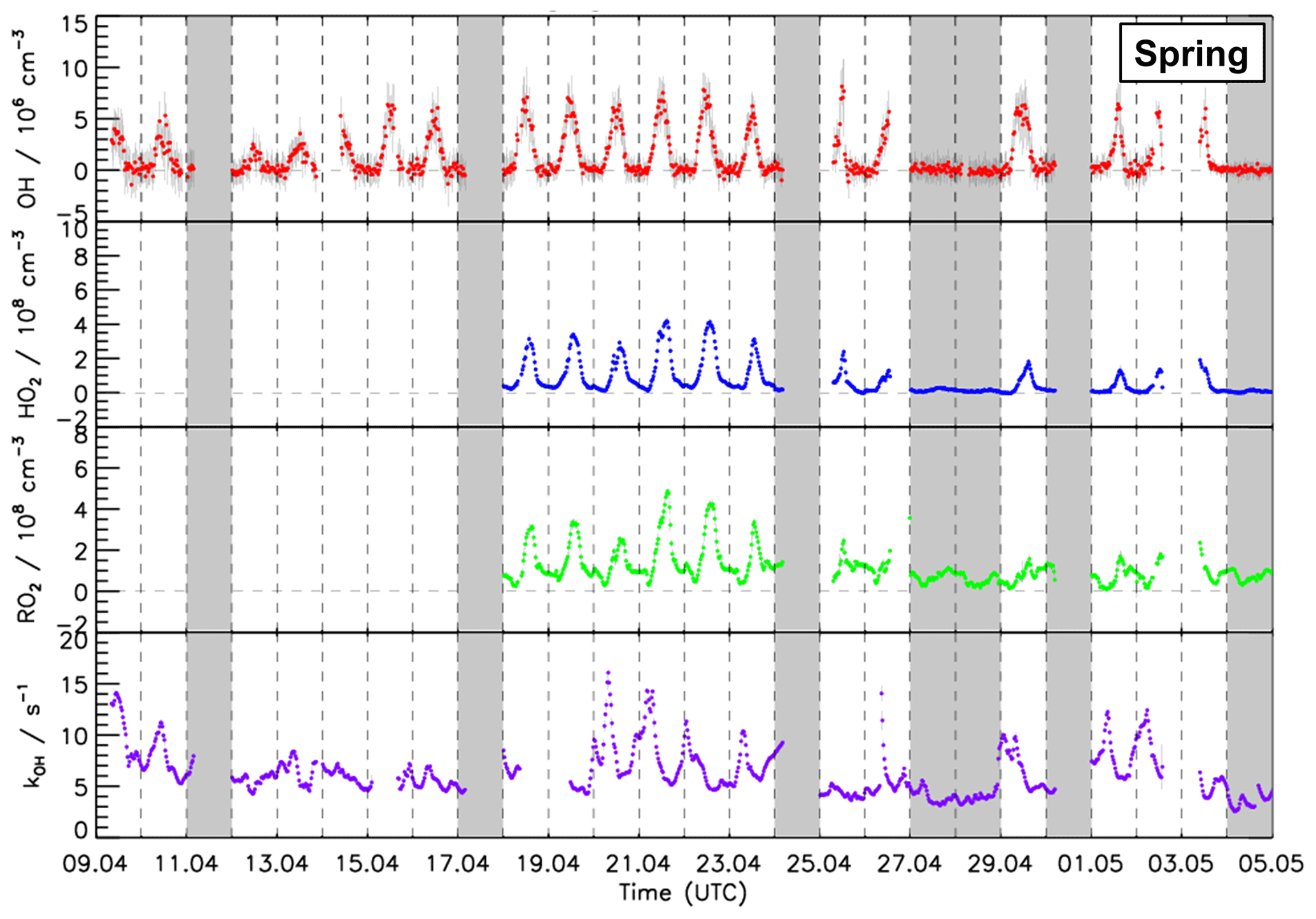
Figure 3Time series of OH, HO2, and RO2 radical concentration measured by the FZJ LIF–CMR instrument and measurements of the OH reactivity (kOH) measured in the spring period of the JULIAC campaign (Cho et al., 2022). Vertical bars represent 1σ statistical errors. Vertical dashed lines denote midnight. Grey shaded areas indicate calibration days when no measurements were done and days when the chamber roof was closed due to bad weather conditions.
3.3 OH, HO2, and RO2 radical concentrations and OH reactivity during the winter and autumn periods of the JULIAC campaign
During winter (Fig. S3) and autumn (Fig. S4), daytime OH radical concentrations were below 1×106 cm−3, mainly due to low primary radical production. Daytime peroxy radical (HO2 and RO2) concentrations during these periods were also very low with average values below 2×107 cm−3 (Fig. S5), which is close to the limit of detection of RO2 radicals (Table 2) and within the uncertainty of the background corrections for HO2 and RO2 (Table S1). During winter and autumn, HO2 concentrations typically increased in the morning and reached peak concentrations of 2×107 cm−3 at noon. Concentrations decreased in the evening and night with minimum values right before sunrise. In contrast, nighttime RO2 concentrations increased to values between 3 and 4×107 cm−3 after sunset, when the chemical loss due to their reaction with NO became negligible, while RO2 radicals were still produced from reactions of VOCs with NO3 and O3. NO concentrations were essentially zero at that time because NO production by the photolysis of NO2 stopped and NO rapidly reacted with ozone. RO2 radical concentrations decreased in the morning to values that were similar to those of HO2 radicals as can be expected for conditions with high NO mixing ratios, which led to a fast loss of RO2 and HO2 in their reactions with NO.
The measured OH reactivity (kOH) ranged between 4 and 33 s−1 during winter and autumn periods. The highest value was observed on 21 January, when a highly polluted plume containing 50 ppbv of NO was sampled.
The measured OH reactivity can be compared to OH reactivity calculated by summing up the product between measured OH reactant concentrations and their reaction rate constants with the OH radical. On average, 1.3 s−1 (18 %) of the measured OH reactivity could not be explained by the measured OH reactants during the winter and autumn periods (Fig. S5). NOx, CH4, CO, and VOCs contributed approximately 43 %, 3 %, 20 %, and 13 %, respectively, to the measured OH reactivity.
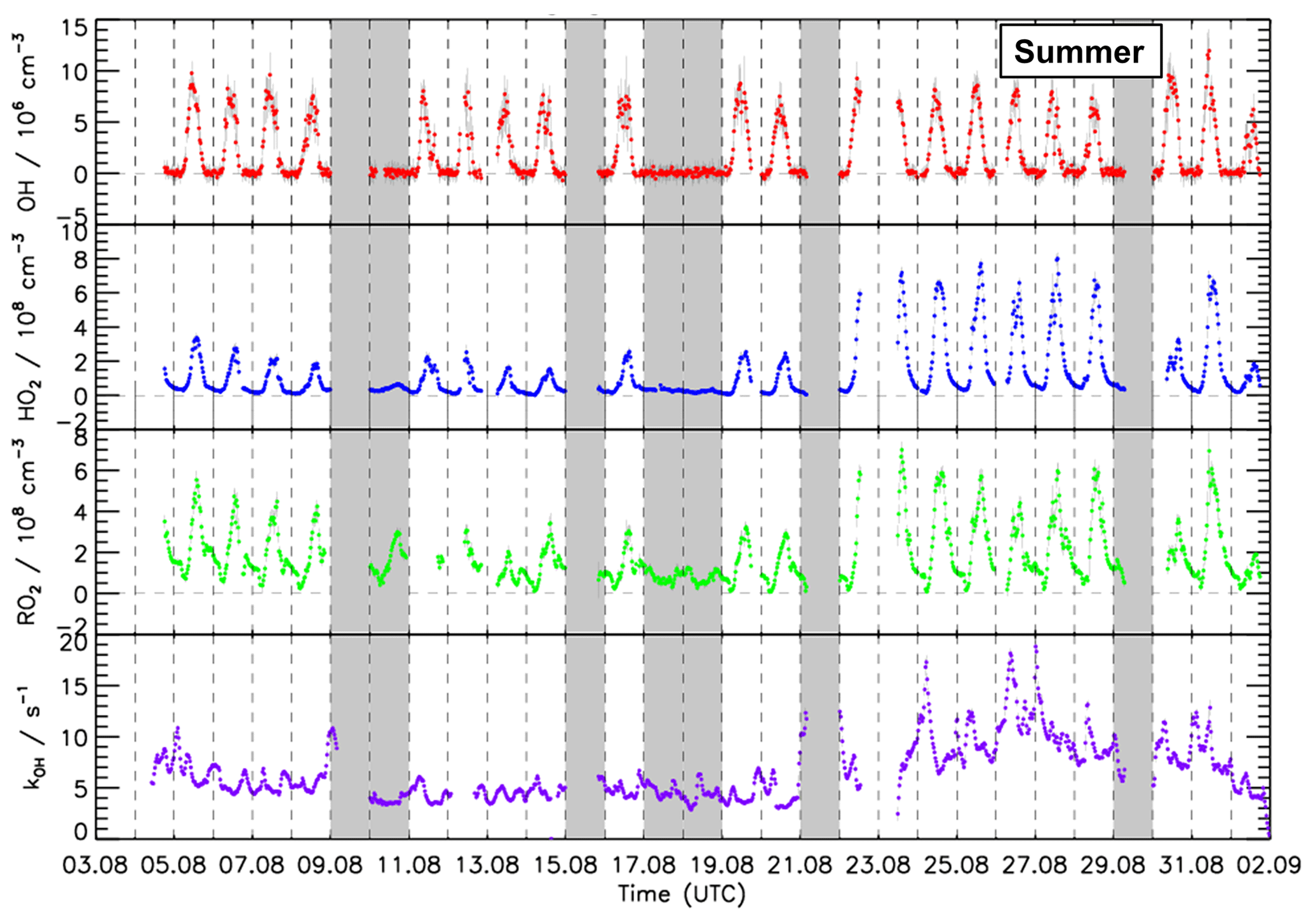
Figure 4Time series of OH, HO2, and RO2 concentration measured by the FZJ LIF–CMR instrument and measurements of the OH reactivity (kOH) in the summer period of the JULIAC campaign (Cho et al., 2022). Vertical bars represent 1σ statistical errors. Vertical dashed lines denote midnight. Grey shaded areas indicate calibration days when no measurements were done and days when the chamber roof was closed due to bad weather conditions.
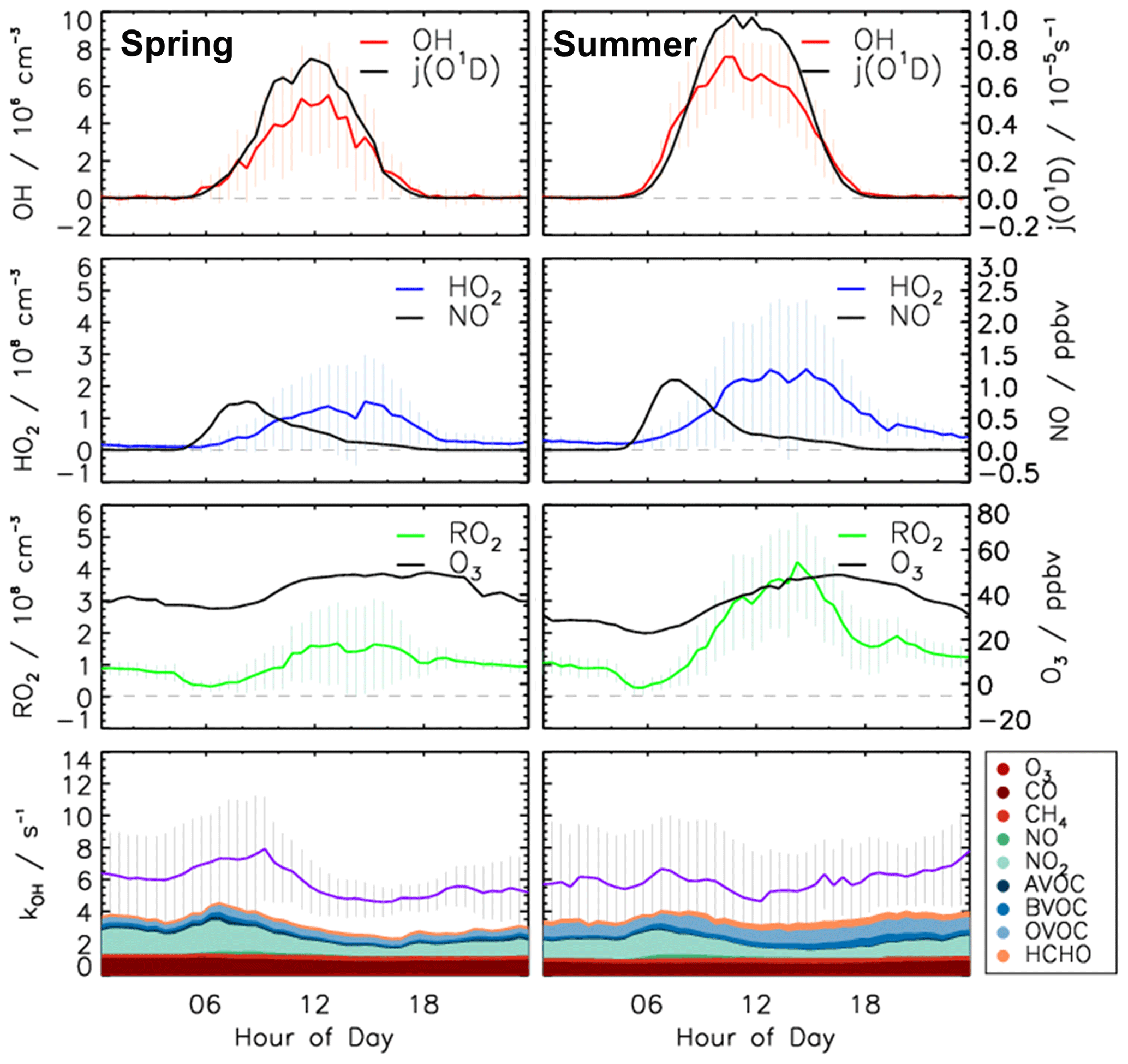
Figure 5Median values of the diurnal profiles of OH, HO2, RO2, kOH, , NO, and O3 measured in the spring and summer periods of the JULIAC campaign. Colored areas represent the contributions of measured reactants to the total OH reactivity. Vertical lines give 25th and 75th percentile values.
3.4 OH, HO2, and RO2 radical concentrations and OH reactivity during the spring and summer periods of the JULIAC campaign
During spring and summer (Figs. 3, 4 and 5), maximum daytime OH concentrations were between 6 and 8 × 106 cm−3. The highest OH concentration (1.2 ×107 cm−3) occurred on 31 August. The diurnal OH concentration profile shows a high correlation with the ozone photolysis frequency () as observed in previous field campaigns (e.g., Ehhalt and Rohrer, 2000; Handisides et al., 2003; Holland et al., 2003).
Unfortunately, the measurements of HO2 and RO2 radicals were not available for the first 2 weeks of the spring campaign due to a malfunction of the instrument. Daily maximum HO2 and RO2 concentrations were in the range of 2 to 4 ×108 cm−3 during the spring period and the first half of the summer period. Maximum HO2 and RO2 concentrations were 8.0 ×108 and 7.0 ×108 cm−3, respectively, during the second half of summer period. In spring and summer, peroxy radical concentrations showed a distinct diurnal pattern. Both HO2 and RO2 radical concentrations were suppressed in the early morning (between 04:00 and 07:00 UTC) due to the reaction with elevated NO mixing ratios of up to 1.5 ppbv. Maximum peroxy radical concentrations were usually reached in the afternoon (∼ 14:00), when NO concentrations were lowest.
The measured OH reactivity values were in the range of 4 to 18 s−1. High values were observed between 23 and 31 August due to high emissions of biogenic volatile organic compounds (BVOCs) from plants at high ambient temperatures. The OH reactivity that cannot be attributed to the measured OH reactants was on average 2.5 s−1 (40 %), which is much higher than observed in the winter and autumn periods (Fig. S5). CO and CH4 contributed 10 % and 4 %, respectively. Due to the high emissions of biogenic organic compounds in spring and summer, the attributed contribution of organic compounds to the total measured OH reactivity was 20 % and the contribution of NOx was only 19 %, which is much less compared to the winter and autumn periods. Isoprene had the largest contribution among all VOCs, accounting for up to 5 % of the total measured OH reactivity. Unfortunately, the number of detected VOC species in the JULIAC campaign was small (Table S2).
In the JULIAC campaign, nighttime OH concentrations were clearly below the limit of detection of the FZJ CMR–LIF instrument (0.7 ×106 cm−3). When all nighttime data are averaged, mean OH concentrations with 1σ standard errors of and cm−3 are obtained for the spring and summer periods, respectively. These low values support the absence of instrumentally produced OH and indicate very low nocturnal OH production at 50 m height in the absence of NO and solar UV.
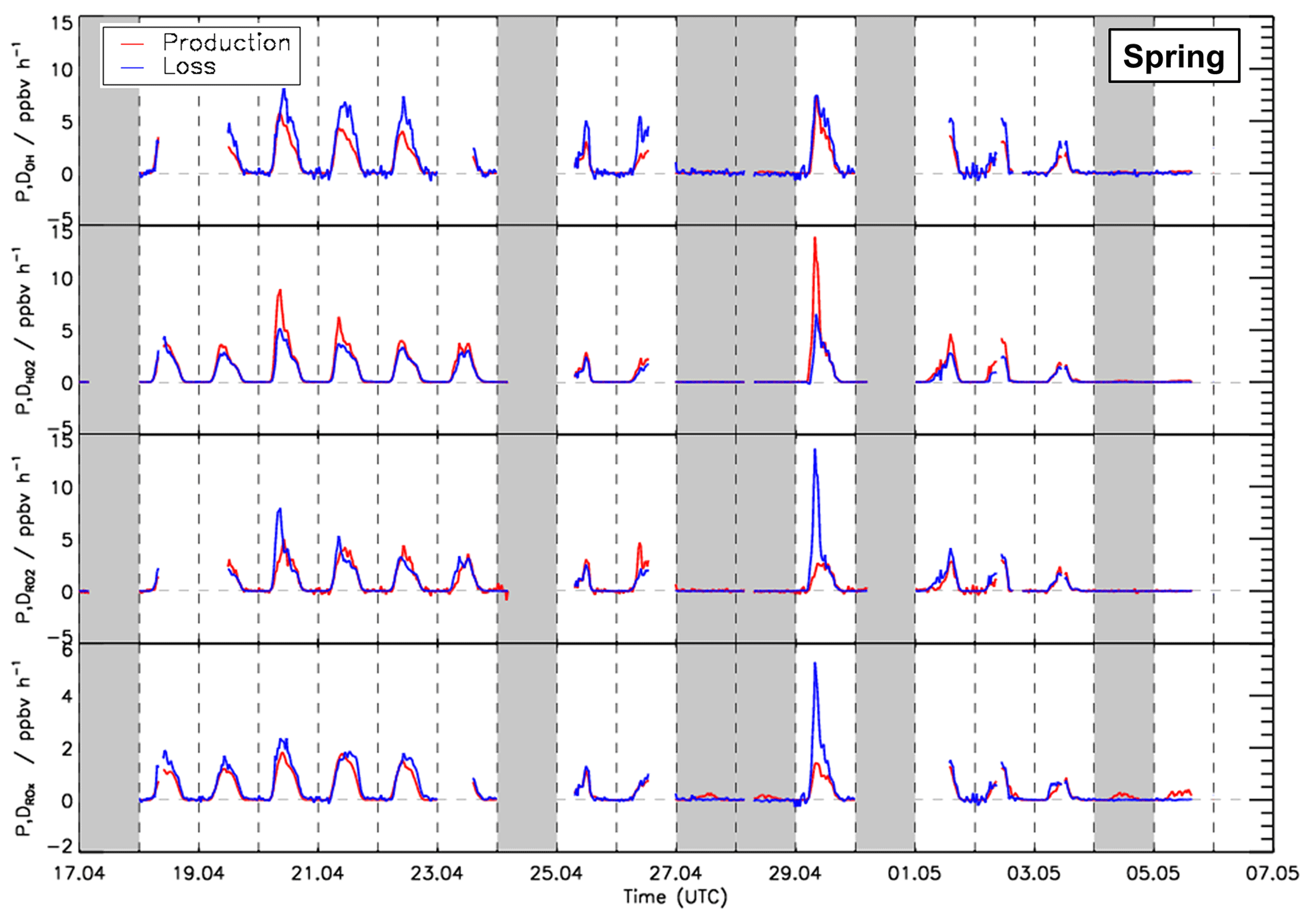
Figure 6Time series of total production and destruction rates of OH, HO2, RO2, and ROX radicals in the spring period of the JULIAC campaign. Vertical dashed lines denote midnight. Grey areas indicate calibration days and days when the chamber roof was closed.
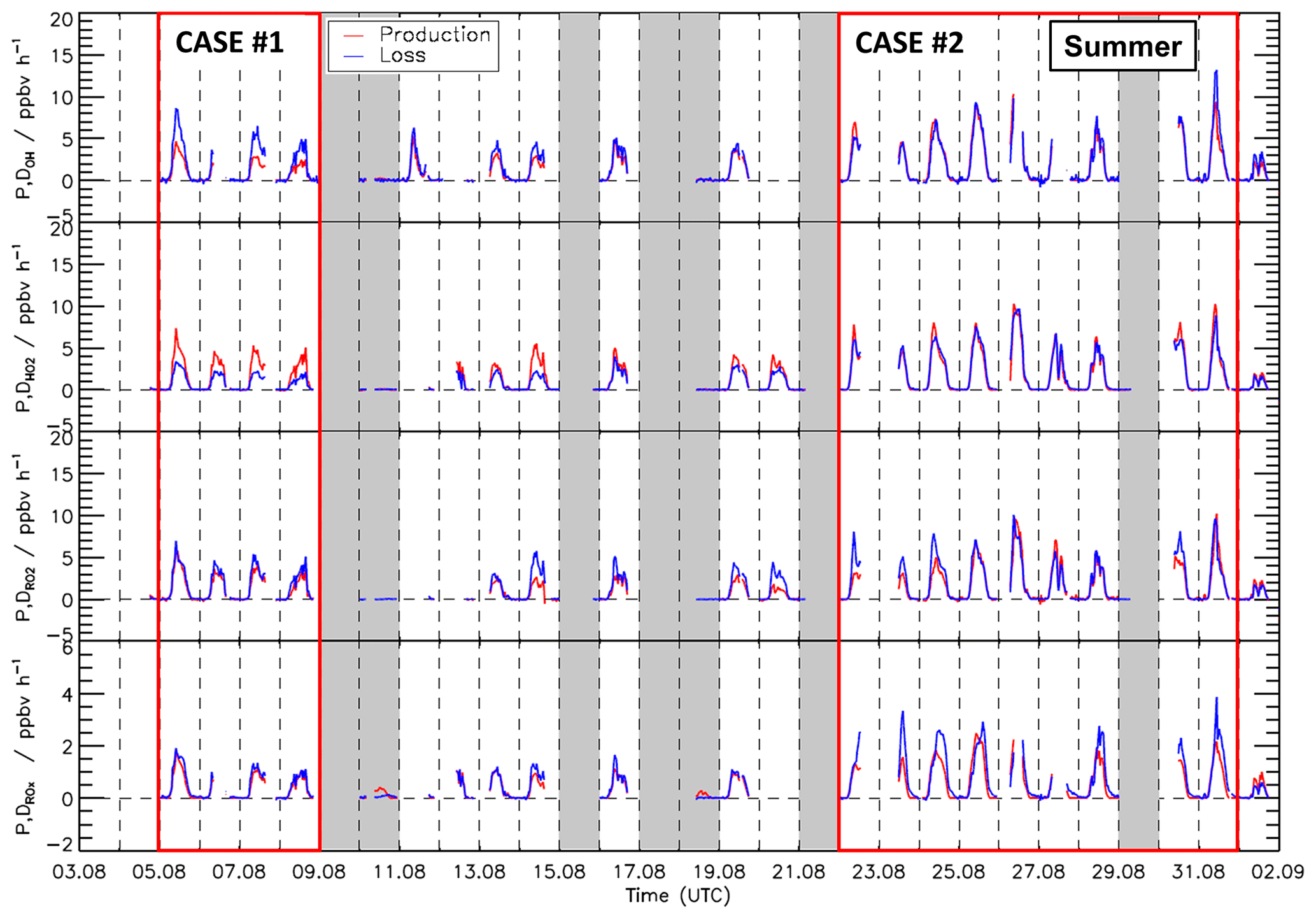
Figure 7Time series of total production and destruction rates of OH, HO2, RO2, and ROX radicals in the summer period of the JULIAC campaign. Vertical dashed lines denote midnight. Grey areas indicate calibration days and days when the chamber roof was closed. The red boxes denote periods that are discussed in more detail (Case 1 and Case 2).
3.5 Chemical budgets of OH, HO2, RO2, and ROX radicals in the spring and summer periods
Due to the very low photochemical activity observed in autumn and winter, which resulted in radical concentrations close to the detection limit of the instrument, the chemical budget analysis is only discussed for data from the spring and summer periods. It focuses on daytime conditions.
Time series of turnover rates of reactions involving OH, HO2, RO2, and ROX radicals in the spring and summer periods are presented in Figs. 6 and 7, respectively, and median diurnal profiles in Fig. 8. Typical daytime turnover rates of OH, HO2, and RO2 radicals were between 3 and 10 ppbv h−1. The rates of ROX production and destruction ranged from 1 to 3 ppbv h−1, which is 2 to 4 times lower than those of OH, HO2, and RO2 because radical conversion reactions cancel out. The highest OH turnover rate of 13 ppbv h−1 was observed on 31 August, when the air temperature in the chamber reached up to 40 ∘C. Unusually high turnover rates for HO2, RO2, and ROX radicals occurred on 29 April with values of 14, 15, and 4 ppbv h−1, respectively, when the NO mixing ratio exceeded 9 ppbv. For the reasons stated in Sect. 3.1, the HO2 and RO2 data on this date are considered highly uncertain and were excluded from further analysis of the chemical budgets.
Diurnal variations of total radical production and destruction rates, as well as of the contributions of the most important reactions, are shown as median values for the entire spring and summer period in Fig. 8. For OH, the reaction of HO2 with NO (Reaction R10) was the dominant production pathway, contributing more than 70 % to the total production rate in both spring and summer periods. The photolysis of HONO (Reaction R1) was the most important primary OH source during daytime, contributing approximately 20 % to the total OH production. The reaction of HO2 with ozone (Reaction R11), the photolysis of ozone (Reaction R2), and the ozonolysis of alkenes (Reaction R5) contributed less than 3 % to the total OH production. The maximum median total OH production rate of 3.5 ppbv h−1 was observed in the morning shortly after the peak NO concentration in both spring and summer (Fig. 5). Values gradually decreased until sunset. Median total OH destruction rates were higher than production rates and reached up to 5 and 6 ppbv h−1 at noon in spring and summer, respectively. The contributions of different reactions to the total OH destruction rate are described by the contribution of OH reactants to the OH reactivity (Sect. 3.4, Fig. 5).
Short-lived radicals are expected to be in a steady state, and therefore radical production and destruction rates must be balanced. An imbalance between the calculated rates indicates inaccurate data or a missing radical production or destruction process. The daily peak of the OH production rates was typically lower than the destruction rate by approximately 1.8 ppbv h−1 in the spring and 2.5 ppbv h−1 in the summer period (36 % and 43 % of the total OH destruction rate). These discrepancies are higher than the uncertainty of the calculation (Fig. 8).
80 % of the HO2 production rate consisted of the reaction of RO2 with NO (Reaction R9). The remaining part of the HO2 production rate was due to the photolysis of formaldehyde (9 %) and the reaction of formaldehyde with OH (10 %). Other reactions producing HO2 played a minor role (<1 %). The HO2 destruction was mostly due to the reaction of HO2 with NO (Reaction R10), contributing on average 88 % to the total production rate. The loss due to reaction of HO2 with RO2 radicals (Reaction R16) contributed on average 9 % to the total loss.
Median values of the total HO2 destruction and production rates were well balanced in the spring period, with the production rate being slightly higher than the destruction rate. The maximum difference of 1 ppbv h−1, however, was insignificant compared to the uncertainty of the calculation. A similar tendency but more pronounced feature was observed in summer. Here, the median value of the production rate was higher than that of the destruction rate by 1.8 ppbv h−1 (38 % of the total HO2 production rate), but differences were variable (Fig. 7). This aspect is discussed in more detail for two periods (Sect. 3.7 and 3.8), which exhibited different degrees of imbalance in the radical budgets.
The RO2 production rate was dominated by the reaction of VOCs with OH (Reaction R8). The contributions of ozonolysis of measured alkenes to the RO2 production were very small (less than 1 %). The reaction of RO2 with NO (Reaction R9) dominated the RO2 destruction and contributed more than 90 % to the total loss rate. In the late afternoon, the RO2 termination reaction with HO2 gained importance with contributions of up to 10 %. Although slight imbalances of up to 1 ppbv were observed in the early morning, the RO2 production and destruction rates were generally balanced within the uncertainty of calculations in both spring and summer.
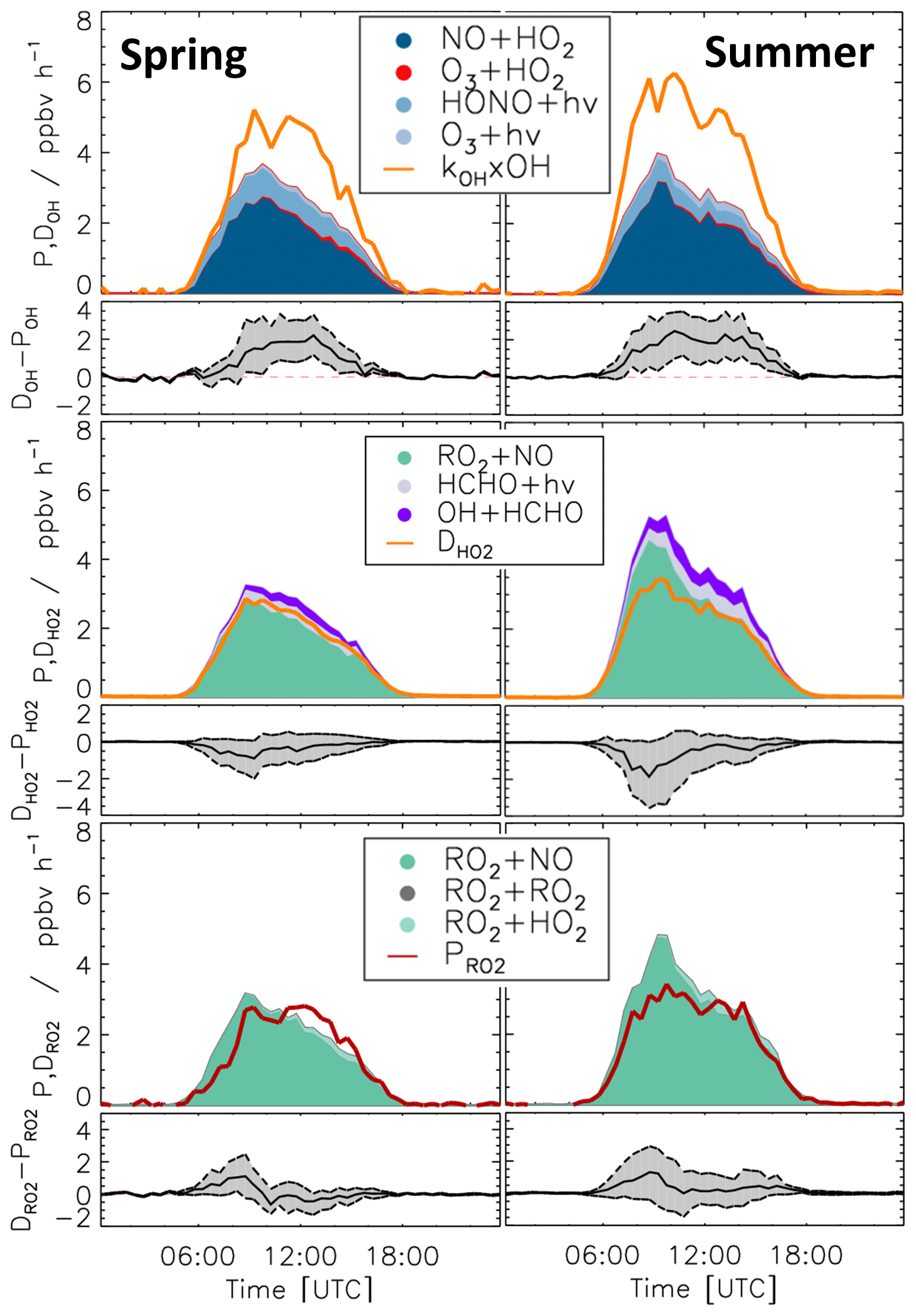
Figure 8Median values of production and destruction rates of OH, HO2, and RO2 radicals in the spring and summer periods of the JULIAC campaign, with data from 29 April excluded. In addition, the differences between the destruction and production rates are shown. Grey areas indicate the 1σ uncertainty derived from experimental errors of the measured quantities (Table 2) and of the reaction rate constants (Table 1). The reactions that have insignificant contributions to the production or destruction rates are not shown.
Figure 9 shows the calculated ROX production and destruction rates. The photolysis of HONO (Reaction R1), HCHO (Reaction R3), and O3 (Reaction R2) was the dominant process initiating radical chemistry and contributed 45 %, 38 %, and 15 %, respectively, to the total ROX production rate on average in both periods. In the morning, the reaction of OH with NO2 (Reaction R12) was the most important radical termination process, contributing up to 65 % to the total ROX destruction rate. In addition, due to relatively high NO mixing ratios in the early morning, the reactions of OH with NO (Reaction R13) and RO2 with NO, which yields organic nitrate (Reaction R14), were also significant radical termination processes, contributing 13 % and 17 % to the total ROX destruction rate, respectively. In the afternoon, radical self-reactions (Reactions R15–R17) and, in particular, the reaction of RO2 with HO2 (Reaction R16) dominated the ROX destruction due to the low NO and NO2 mixing ratios. In both the spring and summer periods , the total ROX destruction rate was slightly higher than the production rate, in particular in the afternoon. The imbalance was up to 0.5 ppbv h−1, which is higher than the uncertainty of the calculations.
Meteorological and chemical conditions were variable, especially in the summer period, causing variations in the balance between radical production and destruction rates (Fig. 7 and Table S3). In the following, the chemical budgets with the largest and smallest observed imbalances are discussed: 5–8 August (Case 1) and 22–31 August (Case 2).
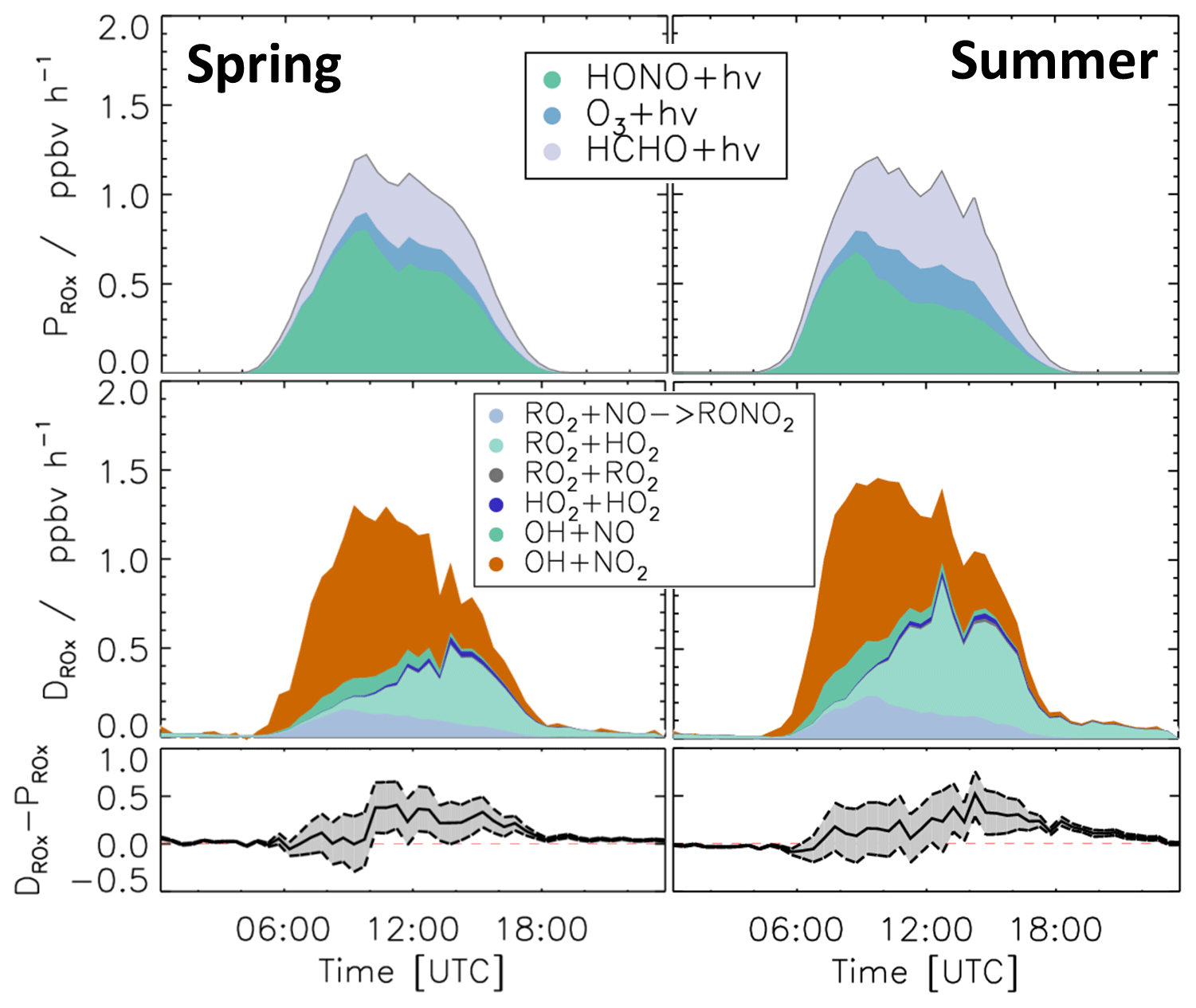
Figure 9Median values of production and destruction rates of ROX radicals during the spring and summer periods of the JULIAC campaign. In addition, the differences between the destruction and production rates are shown. Grey areas indicate the 1σ uncertainty derived from experimental errors of the measured quantities (Table 2) and of the reaction rate constants (Table 1). The reactions that have insignificant contributions to the production or destruction rates are not shown.
3.5.1 Case 1: 5–8 August 2019
For the period between 5 and 8 August, relatively low NO mixing ratios (maximum: 1 ppbv, median: 0.26 ppbv) and typical summer temperatures for this region (median: 27 ∘C) were observed (Fig. 10 and Table S3).
As for the whole summer period (Fig. 8), the reactions of peroxy radicals with NO (Reactions R9, R10) dominated the inter-radical conversion reactions of OH, HO2, and RO2 in this period (Fig. 10). A significant imbalance between the OH production and destruction rates of up to 3.0 ppbv h−1 (51 % of the total OH destruction rate) is found, which cannot be explained by the uncertainty of the calculations. The total HO2 production rate was 2.0 ppbv h−1 higher than the destruction rate (48 % of the total HO2 production rate), whilst the RO2 production and destruction rates were well balanced. Relatively small but nevertheless significant differences between ROX production and destruction rates (0.5 ppbv h−1) were observed during daytime (Fig. 11).
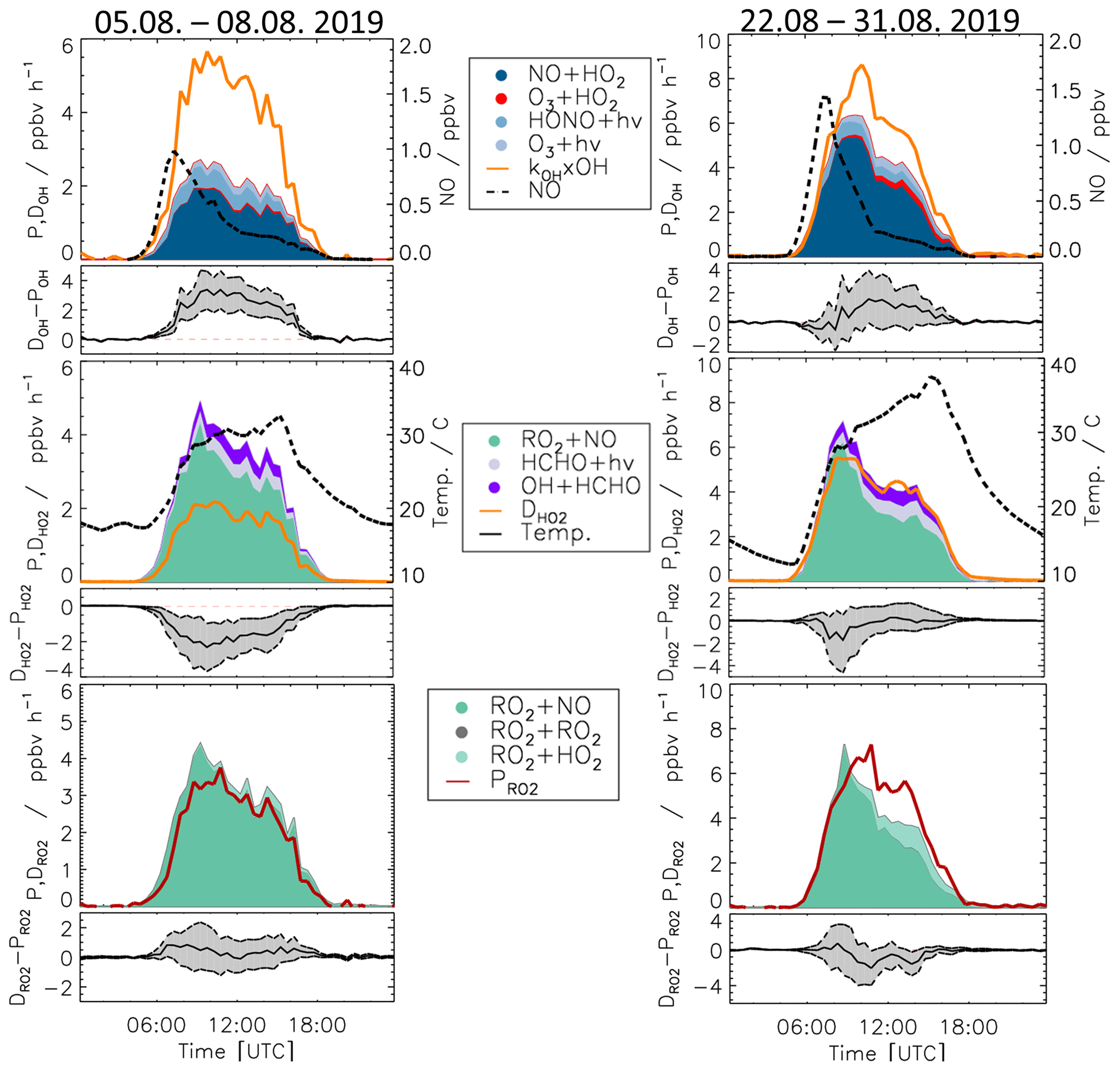
Figure 10Production and destruction rates of OH, HO2, and RO2 radicals for Case 1 (5–8 August 2019) and Case 2 (22–31 August 2019). In addition, the differences between the destruction and production rates are shown. Grey areas give the 1σ uncertainty derived from experimental errors of the measured quantities (Table 2) and of the reaction rate constants (Table 1). The reactions that have insignificant contributions to the production or destruction rates are not shown.
3.5.2 Case 2: 22–31 August 2019
During the period from 22 to 31 August, the temperature was generally high and reached a maximum value of 42 ∘C inside the chamber. The concentrations of the radical precursors (HONO, HCHO, and O3) were higher than those observed in Case 1 (Table S3). Ozone mixing ratios reached values up to 100 ppbv, while daytime NO mixing ratios were similar as in Case 1 (<1.5 ppbv, median value of 0.22 ppbv). The conditions outside the chamber were characterized by stagnant air (wind speed <4 m s−1 at 50 m height) with no precipitation. At these conditions, vigorous biogenic emissions can be expected (Vilà-Guerau de Arellano et al., 2009; Sarkar et al., 2020). Enhanced biogenic VOC emissions and their photochemical degradation can therefore explain the higher VOC and HCHO concentrations in Case 2 compared to the cooler period at the beginning of the month (Table S3). The larger VOC reactivity and comparable OH concentrations resulted in HO2 and RO2 concentrations that were approximately 2 to 3 times higher than in Case 1 (Table S3).
Imbalances between the radical production and destruction rates were a factor of 2 smaller in the warmer and more photochemically active period of Case 2 compared to Case 1. OH destruction rates were up to 1.5 ppbv h−1 (25 % of the total OH destruction rate) higher than the total production rate (Fig. 10). The HO2 production and destruction rates agree within ± 1 ppbv h−1. The contributions from photolysis of HCHO and the reaction of HCHO with OH to the HO2 production rate were larger compared to other periods with values of up to 15 % and 13 %, respectively, due to high HCHO mixing ratios of up to 8 ppbv (Fig. 2). The RO2 production and destruction rates showed imbalances up to 1.5 ppbv h−1 in the late afternoon.
While HONO photolysis was the dominating ROX source most of the time in spring and summer (Fig. 9), HO2 production from the photolysis of HCHO was the most important primary radical source in Case 2 due to the high concentration of HCHO (Fig. 11). Although the chemical budgets for each radical species were essentially closed within the experimental uncertainty, the total loss rate of ROX was consistently higher than the production rate during daytime. The deviation was higher than the experimental uncertainty and reached a maximum value of 1.4 ppbv h−1 at noontime.
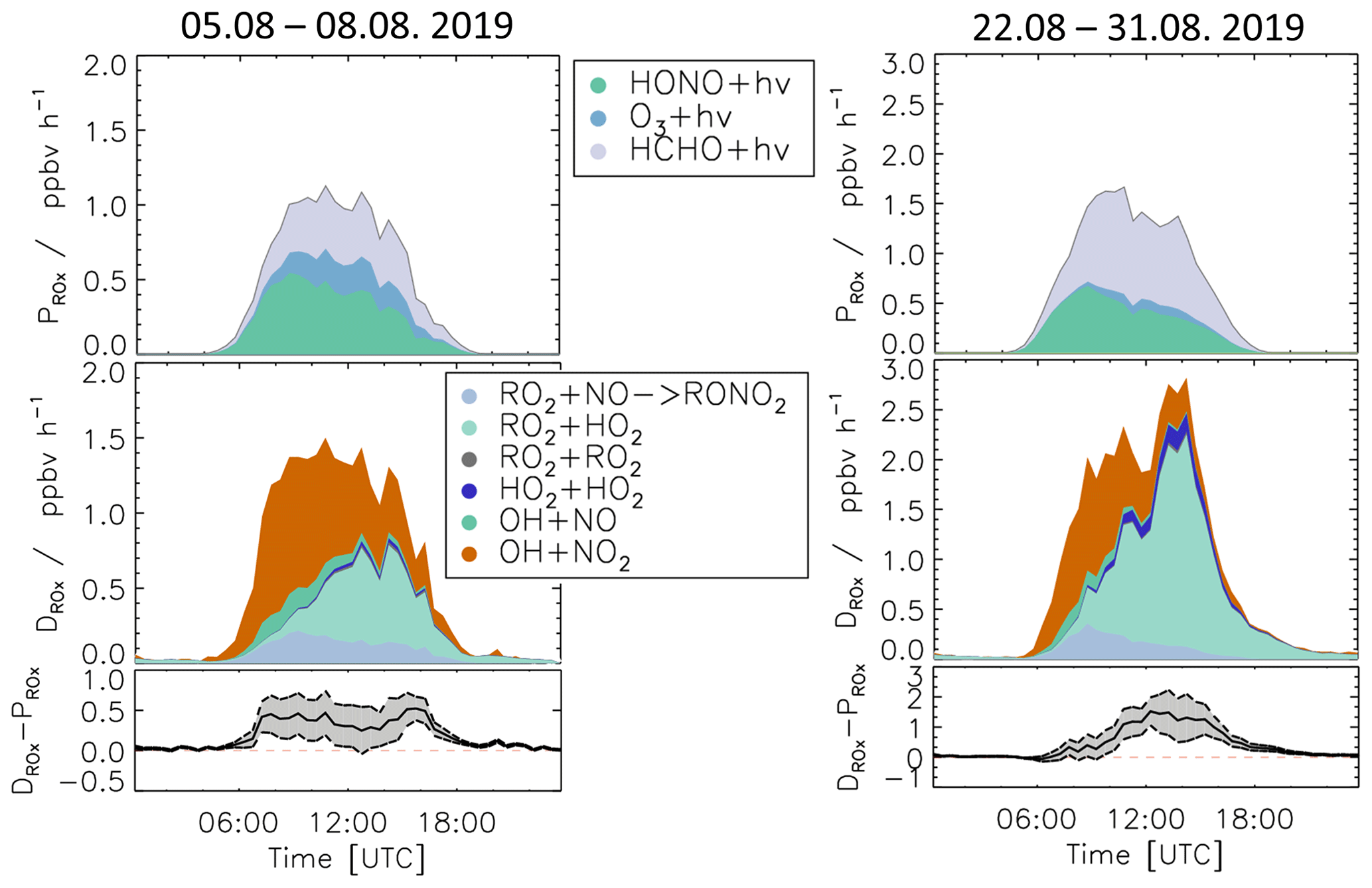
Figure 11Production and destruction rates of ROX for the periods of the case studies (Case 1 and Case 2). In addition, the differences between the destruction and production rates are shown. Grey areas indicate the 1σ uncertainty derived from experimental errors of the measured quantities (Table 2) and of the reaction rate constants (Table 1). The reactions that have insignificant contributions to the production or destruction rates are not shown.
3.5.3 NO dependence of radical production and destruction rates
One of the most influential parameters for the radical chemistry is the concentration of NO, since the reaction with NO dominates the conversion rate of RO2 to HO2 (Reaction R10) and HO2 to OH (Reaction R9) (Fig. 10). Figure 12 shows the NO dependence of median values of the calculated production and destruction rates for the different radicals for the spring and summer period.
For OH, the production rates are consistently lower than the destruction rates by about 1.5 ppbv h−1 for NO mixing ratios lower than 1 ppbv NO. At higher NO, the OH budget is balanced within the experimental uncertainty. For HO2, an inverse pattern is observed. Below 1 ppbv NO, the production rate is higher than the destruction rate by about 1 ppbv h−1. Only for lowest NO mixing ratios are the production and destruction rates balanced. For NO mixing ratios above 1 ppbv, the chemical budget of HO2 is essentially closed. For NO mixing ratios of 3.5 ppbv, the difference between the production and destruction rate is noticeably high (more than 4 ppbv h−1) but also has large uncertainty. For RO2 radicals, the chemical budget is closed for NO mixing ratios below 1 ppbv, but an increasing discrepancy between the loss and production rates is observed with increasing NO mixing ratios. While the production rate is relatively constant at a value of 2.5 ppbv h−1, the loss rate increases to values of up to 7.5 ppbv h−1 at 3.5 ppbv NO. The budget of ROX, in which radical interconversion reactions cancel out, is mostly balanced over the whole range of NO. Only for the lowest and highest NO mixing ratios is the destruction rate 0.6 ppbv h−1 higher than the production rate.
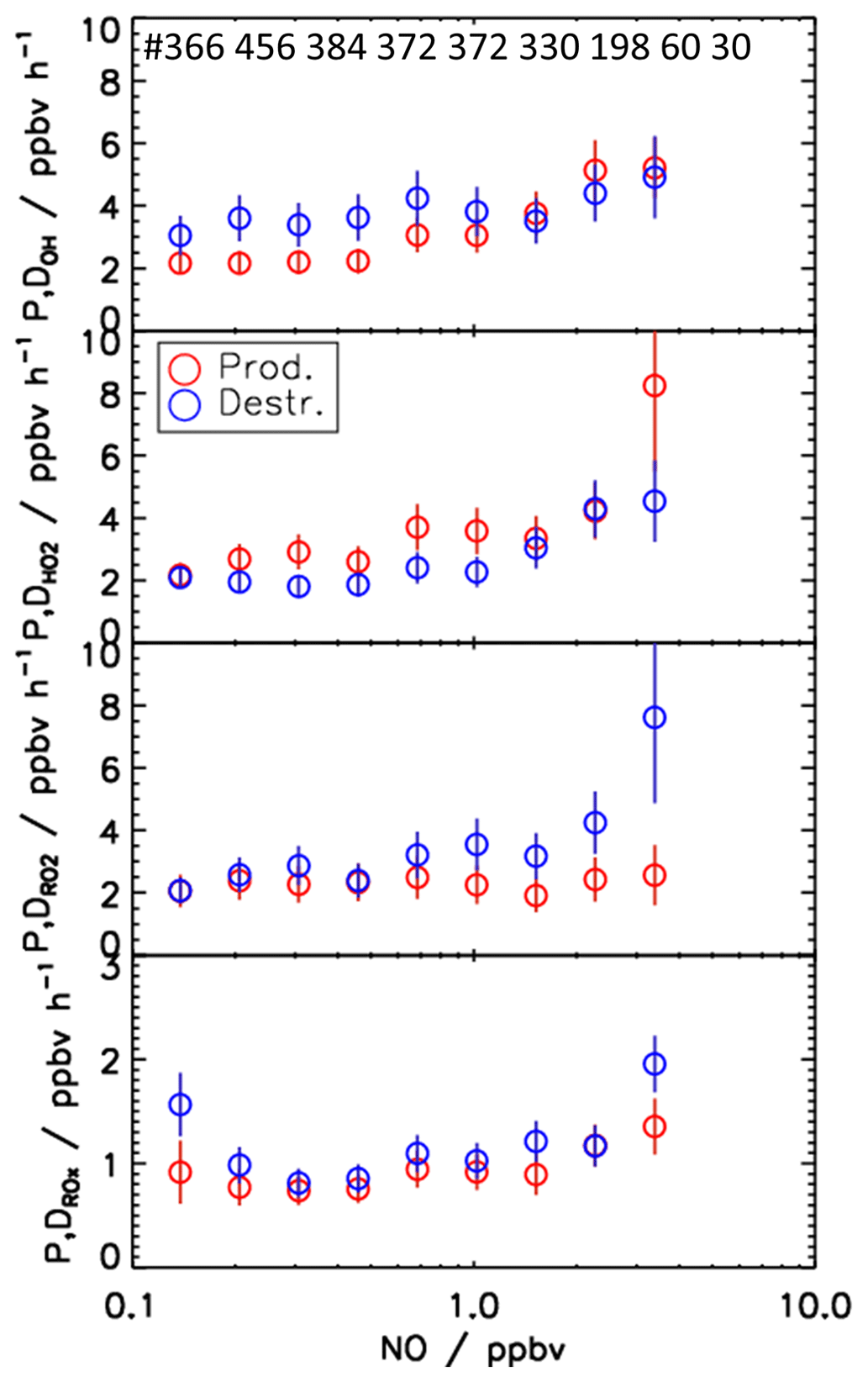
Figure 12NO dependence of median production and destruction rates of OH, HO2, RO2, and ROX radicals. Median values include all data from the spring and summer periods of the JULIAC campaign (NO intervals: ln(NO) = 0.4 ppbv). Vertical bars represent the 1σ uncertainty from experimental errors of the measured quantities (Table 2) and of the reaction rate constants (Table 1). The number of data points in each NO bin is represented on the top panel.
4.1 Discrepancies in the chemical budgets of radicals
The highest imbalances in the chemical budgets of radicals are found for OH radicals. In spring and summer, their production rate was consistently lower than the loss rate (Fig. 8). This deficit was largest at the beginning of August (Case 1, Fig. 10) when the discrepancy reached (3.0 ± 1) ppbv h−1.
Imbalances in the radical budgets can be observed for different reasons. They can be caused by missing processes or incorrect rate constants in the calculations of the production or destruction rates (Sect. 4.2). It is also possible that measured concentrations that are used for the calculation contain unknown errors. The technically difficult radical measurements have a large potential for artifacts (Hofzumahaus and Heard, 2016). Precautions were taken to minimize measurement interferences for OH and HO2 in this campaign.
-
The measurements of OH by the LIF instrument were interference-corrected using chemical modulation and agreed with simultaneous OH measurements by the DOAS instrument within the experimental uncertainties. The measured OH reactivity quantifies the total chemical loss rate of OH caused by atmospheric reactants and has a total accuracy of 10 %. Thus, the destruction rate of OH, which is the product of the concentration and reactivity of OH, is known within 20 % and is unlikely to be biased by unknown OH interferences or unknown atmospheric reactants.
-
The Ox production rate calculated from the reaction of peroxy radicals with NO agrees with the measured increase in Ox concentrations within ± 1 ppbv h−1 for most conditions (Sect. 3.1). As more than 70 % of the OH production is due to the reaction of HO2 with NO (Reaction R10), a bias of more than 1 ppbv h−1 due to an HO2 measurement error that is unaccounted for seems unlikely.
-
The analysis of the chemical budget of OH in previous chamber experiments performed at various chemical conditions showed no evidence for a missing OH source originating from chamber wall effects (Kaminski et al., 2017; Fuchs et al., 2018; Novelli et al., 2018; Rolletter et al., 2019; Rolletter et al., 2020).
Thus, there is no evidence for instrumental errors that are not included in the estimated errors of the calculated turnover rates. The observed imbalances in the OH budget of up to 3 ppbv h−1 are therefore most likely due to a missing OH source.
The missing OH production is correlated with the imbalance in the HO2 budget, for which the production rate is larger than the loss rate at low NO mixing ratios (Fig. 12). This is most clearly seen in the period of Case 1, when the discrepancy reaches (2.0± 1) ppbv h−1 (Fig. 10). The production rate of HO2 is nearly equal to the RO2 loss rate () because both are controlled by the reaction of RO2 with NO (Reaction R9). Furthermore, the RO2 loss rate is well balanced by the RO2 production rate within the experimental uncertainty of ± 1 ppbv h−1 (Figs. 8 and 10). Thus, there is no hint that the calculated turnover rate of the RO2+ NO reaction had a bias higher than 1 ppbv h−1. In addition, turnover rates of the reactions of HO2 and RO2 with NO, producing ozone, are consistent with the observed Ox increase in the chamber (Sect. 3.1). This suggests that these rates are correct in the chemical budget analysis. For the above reasons, the discrepancy between HO2 production and destruction rates is most likely due to a missing HO2 loss process and not measurement errors of HO2, RO2, or NO.
ROX destruction rates are generally higher than the production rates, but differences are on average lower than 0.5 ppbv h−1 (Fig. 9). In the periods of Case 1 and Case 2, the corresponding discrepancies reach 0.5 and 1.4 ppbv h−1, respectively (Fig. 10). If these discrepancies were due to a missing primary OH source, they could also explain a small part (17 %) of the imbalance in the chemical OH budget in Case 1 and the complete imbalance in the OH budget in Case 2.
It is difficult to identify the exact cause of the differences in OH and HO2 budgets observed for Case 1 and 2 with only the available data. Case 2 was characterized by high temperature with increased BVOC emissions and high levels of HCHO (Table S3). No clear correlation was found between the ratio of the production and destruction rates of the radicals and the concentration of chemical species such as NO, NO2, O3, and HCHO. A weak correlation was observed with temperature with an improved balance in the budgets the higher the temperature was. This could indicate that the processes that are unaccounted for become less competitive for high radical turnover rates with chemical conditions being dominated by organic compounds from biogenic emissions.
In conclusion, the radical budget analysis suggests the presence of a missing OH source and a missing HO2 loss process with a similar turnover rate at NO mixing ratios below 1 ppbv for typical temperatures in summer. The opposing imbalances in the OH and HO2 budgets could be due to an unknown mechanism that converts HO2 to OH, or they could indicate a missing primary OH source and a similar fast, but independent, termination reaction removing HO2. The remaining imbalance in the ROX budget would be consistent with a primary OH source that is unaccounted for. This fits the observations best in Case 2, which is characterized by high temperatures and VOC emissions.
For NO mixing ratios that are higher than 1 ppbv, production and destruction rates of OH and HO2 radicals are generally balanced (Fig. 12). An exception is observed for HO2 for the highest NO mixing ratios of 3.5 ppbv, for which the production rate is 3.5 ppbv h−1 higher than the loss rate.
For RO2, the radical budget is not closed, but the loss rate increases with NO in contrast to the production rate. The difference reaches a value of 5 ppbv h−1 at 3.5 ppbv NO. In the same range of NO mixing ratios, the odd oxygen production rate () calculated by peroxy radicals (Eq. 14) overestimates the observed increase in the Ox mixing ratio by about 3 ppbv h−1. This difference points to a systematic error in the peroxy radical measurements, explaining a considerable part of the imbalance in the RO2 budget. A reduction of the RO2 concentration by 3 ×107 cm−3 would reduce the HO2 production rate by 3 ppbv h−1 and resolve the discrepancy in the odd oxygen production calculations for the highest NO mixing ratio. The presumed bias in the RO2 measurement may be caused by an incorrect background subtraction that becomes most relevant at high NO concentrations (Sect. 3.1). However, even after correction of this bias a discrepancy in the RO2 budget would remain, requiring an additional RO2 source of approximately 2 ppbv h−1 to be balanced.
Further information on the nature of the missing RO2 source can be obtained from the chemical budget of ROX, for which the production rate is 0.5 ppbv h−1 smaller than the loss rate at 3.5 ppbv NO (Fig. 12). This discrepancy cannot be explained by the instrumental uncertainties in HO2 and RO2 measurements because the ROX budget at high NO in the morning was dominated by OH reactions with NO2 and (Fig. 9). Thus, the imbalance in the ROX budget at high NO indicates a missing primary radical source, which on a single day (29 April) even reached 3 ppbv h−1 (Fig. 6). As the OH budget is balanced most of the time and the corresponding HO2 budget does not require an additional HO2 source, a missing primary RO2 source is a likely explanation for the discrepancy in the ROX budget. This would also explain part of the imbalance in the RO2 budget at high NO concentrations.
4.2 Potentially missing chemical processes
The above discussion shows that imbalances between calculated production and destruction rates are highly variable over time and change with chemical conditions. As main general features in spring and summer, the radical budget analysis indicates OH production processes that are unaccounted for with a typical strength of 1.5–3 ppbv h−1 at low NO concentrations, which coincides with a missing HO2 sink of 1–2 ppbv h−1. At high NO mixing ratios (> 1 ppbv), the radical budgets for OH and HO2 radicals are relatively well balanced, but RO2 production processes of about 2 ppbv h−1 appear to be missing in the RO2 radical budget. In the following, potential reasons for the observed discrepancies in the radical budgets are discussed.
4.2.1 Differences in the chemical behavior of specific RO2 radicals
As no speciated RO2 radicals were detected but the sum of all RO2 species, effective rate coefficients for the reaction of all RO2 species with NO (Reactions R9, R14), RO2 (Reaction R15), and HO2 (Reaction R16) are used from the structure–activity relationship (SAR) by Jenkin et al. (2019) for the calculations of turnover rates. Potential systematic errors due to this simplification for reactions of RO2 with RO2 and HO2 are expected to be negligible due to their small contributions to the total turnover rates.
In contrast, the reaction of RO2 with NO plays an important role in the chemical budgets of HO2 and RO2. The reaction has one channel that converts RO2 to HO2 (Reaction R9) and one radical termination channel that produces organic nitrates (RONO2) (Reaction R14). The unknown speciation of RO2 causes uncertainty with respect to the total rate constant of the RO2+ NO reaction (k9+k14). An effective value of cm−3 s−1 was taken from Jenkin et al. (2019). A high limit for the total rate coefficient of RO2+ NO (for example, cm−3 s−1 at 298 K for c-C5H9O2) would slightly increase the imbalances between production and destruction rates for HO2 and RO2 radicals by 13 % for both spring and summer. A lower limit would be the rate constant of the reaction of methyl peroxy radicals (CH3O2) with NO having a value of cm−3 s−1 (298 K). Applying this number in the calculations for HO2 production and RO2 destruction rates (Fig. S6) for the period when observed discrepancies in the HO2 budget were highest (Case 1) further improves the already well-balanced budget of RO2 radicals. This also reduces the imbalance between HO2 destruction and destruction rates, but the effect is rather small (approximately 10 %) and not sufficient to explain the total difference. For the other periods such as the spring period and the period of Case 2, a reduced reaction rate would worsen the observed imbalances.
An additional uncertainty in the HO2 production rate comes from the assumed yield of organic nitrates in the reaction of RO2 with NO. Typical organic nitrate yields range from 5 % to 20 % (Jenkin et al., 2019). The low-limit value is applied in the calculations above. Using a value of 20 % decreases the discrepancy between HO2 production and destruction rates from 2.0 to 1.5 ppbv h−1 for the period of Case 1.
It is worth noting that the organic nitrate yield is generally higher for larger hydrocarbons, but the rate constant for the RO2+ NO reaction is also often higher so that there are compensating effects in the production efficiency of HO2. In addition, it is expected that only a fraction of RO2 radicals produced from large hydrocarbons due to the major composition of RO2 would be methyl peroxy radicals.
For the above reasons, the unknown speciation of RO2 is unlikely to be the reason for the observed imbalances in the HO2 budget that are most prominent in the period of Case 1.
4.2.2 Missing primary radical sources
Primary ROX radical production that may not be appropriately accounted for in the calculations could be OH, HO2, and RO2 production from the ozonolysis of alkenes. Only a few alkene compounds were measured in the JULIAC campaign. The contribution from the ozonolysis of these alkenes to the radical production was very small with values in the range of 0.005 to 0.03 ppbv h−1 (Sect. 3.5). The ozonolysis of small alkenes such as propene and cis-2-butene that were not measured but are often abundant, for example, in forested areas (Goldstein et al., 1996; Rhew et al., 2017) may have significantly contributed to the radical production.
The potential impact of unmeasured alkenes on the primary radical production is tested by assuming that the OH reactivity that cannot be explained by measured OH reactants (on average, 2.5 s−1) originates from 1.5 ppbv propene and 1.0 ppbv cis-2-butene. The radical production by ozonolysis of the additional propene and cis-2-butene increases the production from ozonolysis of measured species by more than an order of magnitude in both the spring and summer periods of the JULIAC campaign (Fig. S7). The discrepancies between the total ROX production and destruction rates are significantly decreased for the period of 2 Case by approximately 0.2 ppbv h−1. However, the additional OH production is by far insufficient to explain the missing OH source that was generally found during the JULIAC campaign. In addition, the corresponding OH and O3 reactivity from the additional alkene compounds is about a factor of 6 larger than of alkenes (e.g., ethene, propene, trans-2-butene, cis-2-pentene) that were measured in ambient air next to the SAPHIR chamber in the HOxComp campaign in July 2005 (Elshorbany et al., 2012; Kanaya et al., 2012).
The photolysis of oxygenated organic compounds is another source for radicals that could be underestimated in the calculations. Only the photolysis of HCHO is included in the production rate of HO2 and ROX at all times of the campaign. In addition, acetaldehyde (CH3CHO), methyl vinyl ketone (MVK), methacrolein (MACR), and methylglyoxal were measured during part of the campaign and were not included in the analysis in Sect. 3. Calculations show that the radical production rate from their photolysis was less than 0.1 ppbv h−1. Thus, photolysis of unmeasured OVOCs was very likely unimportant in the present study. This is consistent with similar small contributions from photolysis of OVOCs other than HCHO found in the HOxComp campaign (Kanaya et al., 2012). In addition, during the HOxComp campaign the modeled OH reactivity could be matched with the measured reactivity by including either additional primary emissions (Kanaya et al., 2012) or model-produced oxygenated secondary products (Elshorbany et al., 2012). Neither of the additional species contributed enough to close the radical budgets. If it is assumed that the missing OH reactivity (2.5 s−1) is all due to glyoxal (9 ppb), additional OH production of 0.3 ppb h−1 could be expected. This would still not be enough to close the radical budget, suggesting that unmeasured OVOCs do not play a large role.
The photolysis of ClNO2 constitutes a primary radical source (Reactions R20, R22) that can be found in coastal environments (e.g., Osthoff et al., 2008) and mid-continental regions (e.g., Thornton et al., 2010). The availability of ClNO2 data during the summer period allowed assessing the potential impact of its photolysis on the RO2 radical production (Eq. 9). Due to the low mixing ratio of ClNO2 of less than 0.4 ppbv (Tan et al., 2022), the RO2 production from Cl oxidation processes was insignificant (<0.1 ppbv h−1) and cannot explain the observed discrepancies in the primary production and destruction rates of radicals in the summer period and in the case studies. The instrument detecting ClNO2 was not available in the spring period of the campaign. Therefore, the extent to which ClNO2 photolysis contributed in spring, for example to the large missing ROx source (up to 3 ppbv h−1) on 29 April, remains unknown.
4.2.3 Missing radical termination reactions
Heterogeneous uptake of HO2 on aerosol is a potential termination reaction that is not included in the HO2 and ROX destruction rates above. However, the impact of including the heterogeneous HO2 loss on an aerosol surface (Reaction 8) on the total loss rate is insignificant (less than 1 %), even if a high effective uptake coefficient of 0.2 is assumed (Fig. S7).
As HO2 uptake is a radical termination process, its relative contribution to the total ROX loss rate can be higher compared to the relative contribution to the total HO2 loss rate. However, the only notable influence would be for the period of Case 2 (8 % of total ROX loss rate), when the aerosol surface area concentration was high with values of up to 3.0 ×102 µm2 cm−3.
The estimate for the heterogeneous HO2 loss rate has high uncertainty because the uptake coefficient highly depends on the aerosol properties that were not fully characterized in this campaign. Previous laboratory investigations showed large variability for the uptake coefficient with values ranging from 0.08 to 0.6 depending on the aerosol chemical composition and the physical state (George et al., 2007; Taketani et al., 2008, 2009; George et al., 2013; Lakey et al., 2015; Song et al., 2020; Tan et al., 2020). Even the largest reported HO2 uptake coefficients cannot explain the observed differences in the chemical budget of HO2 radicals. Therefore, heterogeneous HO2 reactions can be ruled out as an explanation for the unexplained HO2 loss rate.
4.2.4 Missing radical interconversion reactions
In the last decade, it has been discovered that unimolecular reactions of RO2 can significantly increase atmospheric OH concentrations in low-NO environments where they can compete with the reaction of RO2 with NO. The most important, atmospherically relevant example is the production of OH from the isomerization of isoprene–RO2 radicals (Peeters et al., 2009; da Silva et al., 2010; Peeters and Müller, 2010; Crounse et al., 2011; Fuchs et al., 2013; Peeters et al., 2014; Teng et al., 2017; Novelli et al., 2020). The SAPHIR chamber is surrounded by a deciduous forest that emits isoprene, especially in summer. Compared to previous campaigns on the campus wherein up to several parts per billion by volume of isoprene were measured (Komenda et al., 2003; Spirig et al., 2005; Kanaya et al., 2012), concentrations were relatively low during the JULIAC campaign (<0.4 ppbv on average).
The effect of the conversion of RO2 to OH by the isomerization of isoprene–RO2 (Eq. 4) is tested in the analysis of the OH and RO2 budgets. In the afternoon of days in the spring period and the period of Case 2, the total OH production increases only 1 % due to the low isoprene mixing ratios (<0.2 ppbv) and the competition of unimolecular reactions with bimolecular reactions of RO2 with NO. Even in the summer period, when isoprene mixing ratios were up to 0.8 ppbv, the contribution of isomerization reactions from isoprene–RO2 radicals to the total turnover rate of RO2 is still small with values of less than 4 %. This implies that unimolecular decomposition reactions of isoprene–RO2 radicals made a minor contribution to the RO2 destruction and OH production rates.
Another known isomerization process that produces OH applies to RO2 formed by OH oxidation of methacrolein (MACR) (Crounse et al., 2012; Fuchs et al., 2014), which is an oxidation product of isoprene. MACR mixing ratios were up to 0.5 ppbv in the JULIAC campaign. Because the rate constant for the OH reaction of MACR is smaller than for isoprene, OH regeneration from MACR-RO2 radicals is even less important than from isoprene–RO2.
For acyl and carbonyl peroxy radicals it was shown that the reaction of RO2 with HO2, which mainly forms hydroperoxides (ROOHs) (Reaction R16), can produce OH with yields up to 80 % (Hasson et al., 2004; Dillon and Crowley, 2008; Groß et al., 2014; Praske et al., 2015; Winiberg et al., 2016; Fuchs et al., 2018; Jenkin et al., 2019). It is also noteworthy that the rate constant for the reaction of HO2 with this class of RO2 species is almost a factor of 2 higher than for other RO2 species (Jenkin et al., 2019). However, even if it is assumed that all the measured RO2 constitutes acyl and carbonyl peroxy radicals, the formation of OH from their reaction with NO could only explain up to 0.5 ppbv h−1 of the imbalances in both OH and HO2 budgets.
Studies in the remote marine boundary layer show that HO2 to OH conversion mediated by halogen oxides (XO, X= Cl, Br, I) (e.g., Bloss et al., 2005; Sommariva et al., 2006; Kanaya et al., 2007; Stone et al., 2018; Fan and Li, 2022) can significantly contribute to the interconversion of radicals and destroy ozone.
This conversion mechanism would only be effective at low NO, when the consumption of XO by NO (Reaction R25) is comparatively slow and when X is not depleted by other reactions as in the case of Cl by reactions with VOCs (Reaction R22).
For BrO, the rate constants for Reactions (R23) and (R25) are about the same ( cm−3 s−1 at 298 K; Burkholder et al., 2019). Thus, the reaction of BrO with HO2 would only be dominant if the NO concentration were smaller than the concentration of HO2, i.e., less than 10 pptv in this campaign. For IO, the situation is similar and NO mixing ratios would need to be less than 40 pptv. Such low NO mixing ratios were not observed during daytime and rule out significant halogen-oxide-mediated HO2 to OH conversion. The required XO concentrations to achieve an HO2 loss rate of 1 ppbv h−1 at an HO2 concentration of 2 ×108 cm3 would be 66 pptv of BrO or 16 pptv of IO, which exceeds the abundances reported for marine environments, where halogen sources are known to exist, by more than an order of magnitude. For these reasons, halogen oxide chemistry cannot explain the missing HO2 sink and missing OH source in this study.
4.3 Comparison with results from other field campaigns
Although the chemical and physical conditions were partly influenced by the chamber properties (Sect. 2.1), the radical concentrations observed during spring and summer were within the range of values that have been observed in other field studies in summertime in urban and suburban areas (Tan et al., 2001; Ren et al., 2003; Kanaya et al., 2007; Mao et al., 2010; Lu et al., 2013; Brune et al., 2016; Tan et al., 2017; Whalley et al., 2018; Tan et al., 2019). The impact of the decreased solar radiation by the chamber transmission on the radical production was compensated for by the radical production from the photolysis of HONO and HCHO emitted from the chamber film.
This effect is also shown in the relationship between the OH concentration and the photolysis frequencies of ozone, (Sect. 3.4). The slope (8.0 ×1011 cm−3 s−1) of the correlation for the data from the JULIAC campaign is much higher than obtained for data in other field campaigns in similar environments (Ehhalt and Rohrer, 2000; Handisides et al., 2003; Holland et al., 2003; Tan et al., 2017) due to the high OH production by the photolysis of chamber-produced HONO (Reaction R1). This is further confirmed by the similarity in OH and HO2 radical concentrations between this campaign and what was observed in the HOxComp campaign when measurements were performed in front of the SAPHIR chamber for 3 d in July 2005 (Elshorbany et al., 2012).
In contrast, daytime OH concentrations observed during winter and autumn in the JULIAC campaign were lower than OH concentrations observed in previous wintertime field campaigns (Heard et al., 2004; Ren et al., 2006; Kanaya et al., 2007; Tan et al., 2018; Ma et al., 2019). This is due to the lower photolysis frequencies in the chamber compared to outside, which is not compensated for by chamber-produced HONO in wintertime because the emission strength is low at low temperature and low solar radiation.
The very low nighttime OH concentration in all seasons of the JULIAC campaign (Sect. 3.4) is consistent with observations in previous field campaigns in rural areas in Germany (Ehhalt and Rohrer, 2000; Handisides et al., 2003; Holland et al., 2003), in which nighttime OH concentrations were less than 1 ×105 cm−3. However, in several other field studies performed in urban areas, nighttime OH concentrations were in the range of 0.2 to 3 ×106 cm−3, for example in China (Lu et al., 2014; Rohrer et al., 2014; Tan et al., 2017, 2018; Ma et al., 2019; Tan et al., 2019; Wang et al., 2019; Whalley et al., 2021), in the US (Martinez et al., 2003; Brune et al., 2016; Griffith et al., 2016), and in the UK (Ren et al., 2003; Vaughan et al., 2012). In these studies, the high nighttime OH concentrations could not be explained by model predictions and raised questions about the presence of potential interference in nighttime OH signals measured by LIF instruments (Mao et al., 2012; Lu et al., 2014; Novelli et al., 2014).
Similar studies investigating the chemical budgets of OH, HO2, RO2, and ROX radicals like in this study were performed for data from field campaigns in a suburban area in the Pearl River Delta (PRD), China, in autumn 2014 (Tan et al., 2019), and in central Beijing, China (Whalley et al., 2021), in summer 2017.
Tan et al. (2019) observed median values of turnover rates of OH, HO2, and RO2 radicals ranging from 10 to 15 ppbv h−1, while rates for ROX initiation and termination were on the order of 3 to 4 ppbv h−1 during daytime for chemical conditions affected by anthropogenic emissions. From the comparison between the radical production and destruction rates, a missing OH source and a missing RO2 sink with a similar rate up to 7 ppbv h−1 (45 % of the total OH turnover) were found at low NO mixing ratios below 1 ppbv, while HO2 production and destruction rates were balanced. The authors suggested that an additional chemical mechanism is required that efficiently converts RO2 to OH without the involvement of NO. One possibility proposed by Tan et al. (2019) is that HOX radicals are formed from the auto-oxidation of specific RO2 species which include multifunctional groups such as -OH, -OOH, or -CHO groups.
The analysis of the chemical budget of OH radicals in the JULIAC campaign shows that an OH source that is unaccounted for with a rate ranging between 2 and 3 ppbv h−1 (about 50 % of the total OH destruction rate) is required at low NO mixing ratios to balance OH production and destruction rates. This rate is smaller than the rate determined in Tan et al. (2019). However, considering that the OH radical turnover rates in the JULIAC campaign were about half compared to values in the campaign in the PRD area, the relative importance of the unaccounted for OH source was comparable in both campaigns. However, the mechanism suggested by Tan et al. (2019) is likely not the only explanation for discrepancies in the radical budgets observed in this study. In the JULIAC campaign, to balance the budget of RO2 radicals requires an additional radical source rather than additional loss processes, particularly at high NO mixing ratios above 1 ppbv, and the missing OH sources likely originate from an HO2 to OH conversion process and/or a missing primary OH source.
Whalley et al. (2021) also investigated the chemical budgets for radicals over a wide range of NO mixing ratios (0.1 to 104 ppbv) from measurements performed in central Beijing, China. Compared to the results in Tan et al. (2019) and results in this study, the rates of ROX initiation and termination reactions were 2 to 4 times higher. Also, the rates of radical propagation reactions for OH, HO2, and RO2 radicals were 5 to 10 times higher due to fast inter-radical conversion reactions at conditions with high concentrations of NO. Similar to the results in this study, an OH source with a high rate of up to 15 ppbv h−1 (50 % of the total OH destruction) was required to balance OH production and destruction rates for low NO mixing ratios. This unaccounted for OH source is more than 3 times higher than that determined in the JULIAC campaign and in the campaign in China reported by Tan et al. (2019). The HO2 production rate observed in Beijing largely exceeded the destruction rate by 3 to 5 times for low NO mixing ratios. In contrast, production and destruction of RO2 and ROX radicals were well balanced. On the other hand, for results in conditions of low NO concentrations, production and destruction of OH radicals were balanced at high NO mixing ratios, while very high imbalances of up to 50 ppbv h−1 were observed for HO2 and RO2 radicals. Whalley et al. (2021) showed that reducing the rate constant of the reaction between RO2 and NO by a factor of 10 could close the gaps between production and destruction rates. The authors suggested that the presence of a significant fraction of RO2 radicals from the oxidation of large and multifunctional VOCs such as monoterpenes and long-chain alkanes could explain observations. These radicals can undergo multiple RO2 to RO2 conversion reactions by unimolecular isomerization of alkoxy radicals (RO), which are formed from the reaction of RO2 with NO so that no HO2 is produced. Such an RO2 radical reaction chain would be equivalent to an increased chemical lifetime of RO2 radicals if RO2 species cannot be distinguished by instruments like in the sum measurements performed by ROX–LIF instruments. Whalley et al. (2021) showed that RO2 production by this mechanism would largely reconcile discrepancies between modeled and measured RO2 concentrations (the model–measurement ratio decreases from 6.2 to 1.8) if the OH reactivity that could not be accounted for by measured OH reactants is attributed to α-pinene.
Applying a reduced rate constant for RO2 to HO2 propagation reactions as suggested in Whalley et al. (2021) in the calculations in this study could help explain the observed discrepancies between HO2 and RO2 production and destruction rates. The largest effect is expected when high NO mixing ratios up to 10 ppbv like on 29 April are experienced. In this case, a large reduction of the rate constant by a factor of 2 for all measured RO2 would be required to close the observed gaps between production and destruction rates. Reduced reaction rate constants of the RO2+ NO reaction could be expected for RO2 from large VOCs. However, the fraction of these RO2 species is expected to be small for conditions of this campaign, even if OH reactivity that is not explained by measured OH reactants is attributed to large VOCs. Therefore, it seems unlikely that the mechanism suggested by Whalley et al. (2021) affects the observed discrepancies in the radical budgets in this study.
It is interesting to point out that similar discrepancies in the OH and HO2 budgets were observed during the HOxComp campaign in July 2005 (Elshorbany et al., 2012). Although measurements were only done for 3 d and despite the fact that these were 14 years earlier than measurements in this work, the chemical composition was similar with comparable values of NOx, O3, isoprene concentrations, and OH reactivity. As observed in this study, a missing OH radical source in the range of 2 to 4 ppbv h−1 was needed to close the OH budget for low-NO chemical regimes. The lack of measured RO2 radicals did not allow performing a measurement-only budget for HO2 radicals. Nevertheless, model calculations overestimated measured HO2 radicals after the correction for RO2 radical interference (Fuchs et al., 2011) by up to 30 % at low NO (Elshorbany et al., 2012; Kanaya et al., 2012). Like in this study, good agreement was found between modeled and measured OH and HO2 radical concentrations only if an unknown loss process for HO2 radicals that would recycle OH was introduced.
4.4 Potential role of the missing radical processes in the evaluation of the ozone production rate
The good agreement of the odd oxygen production rates calculated by the two different methods (Sect. 3.1) not only gives high confidence in the measured peroxy radical concentrations but also confirms the current chemical understanding of tropospheric ozone formation from the reaction of peroxy radicals with NO. Therefore, results demonstrate that accurate predictions of radical concentrations in atmospheric models are crucial to accurately predict the surface ozone level.
However, the significant level of missing radical processes found in this study implies difficulties in the prediction of radical concentrations by models without constraining radicals by their measurements. In low NO mixing ratios, there are two opposing effects of the missing radical processes on the O3 formation. At first, a missing OH source and therefore an underestimation of OH concentrations by the models would lower the loss of NO2 by the reduced reaction rate with OH and essentially produce more O3 by its photolysis. Furthermore, the production of RO2 would be underpredicted due to the lower OH concentrations in the models. At the same time, an unexplained HO2 sink would result in the overprediction of HO2 concentrations and thus O3 production. In high-NO environments, missing RO2 and ROX production processes would result in an underestimation of the O3 production.
Ambient measurements of atmospheric radicals, trace gases, and aerosol properties were performed during the Jülich Atmospheric Chemistry (JULIAC) project campaign using the atmospheric simulation chamber SAPHIR at Forschungszentrum Jülich, Germany. Ambient air was continuously drawn at a high rate into the chamber (1 h residence time) through a 50 m high inlet line for 1 month in each season throughout 2019.
For parts of the campaign, measurements of OH concentrations were achieved by two different methods, laser-induced fluorescence with a chemical modulation system for zeroing (FZJ LIF–CMR) and differential optical absorption spectroscopy (FZJ DOAS). Measurements of both instruments agreed within 11 % (Cho et al., 2021).
The production rate of odd oxygen (Ox) was determined by using either measured HO2 and RO2 concentrations or O3 and NO2 concentrations measured in the chamber and in the incoming flow. Results showed excellent agreement between the two different methods, confirming that HO2 and RO2 are responsible for the formation of tropospheric O3 and giving additional confidence to the reliability of peroxy radical concentration measurements performed in the JULIAC campaign.
An analysis of the chemical budgets of OH, HO2, RO2, and ROX radicals was performed for data obtained in the spring and summer periods of the campaign. On average, daytime radical turnover rates ranged between 3 and 6 ppbv h−1 and between 4 and 10 ppbv h−1 in spring and summer, respectively, for OH, HO2, and RO2 radicals, while total rates of ROX initiation and termination reactions were below 2.0 ppbv h−1. For most conditions, radical production and destruction rates highly depended on the turnover rate of the reaction of peroxy radicals with NO. For the total turnover rate of the sum of all radicals (ROX), the photolysis of HONO and HCHO contributed most to the primary radical production, and the reactions of OH with NO2 and RO2 with HO2 dominated the radical termination processes.
Differences between radical production and destruction rates were often small and below the accuracy of the calculations in the JULIAC campaign in winter and autumn. However, for both spring and summer, an additional OH source is required to explain the observed discrepancy between production and destruction rates. The OH production rate of this source would need be on average 2 and 3 ppbv h−1 in the spring and summer period, respectively. This discrepancy is in the same range as observed for measurements at the same location during the HOxComp campaign in July 2005 (Elshorbany et al., 2012).
Discrepancies between production and destruction rates of OH radicals were highest for conditions with low NO mixing ratios in this study. This is similar to findings in other field campaigns in China (Tan et al., 2017, 2019; Whalley et al., 2021). The high reliability of radical data in this study gives further confidence that the discrepancies arise from chemical processes that are unaccounted for rather than from instrumental artifacts.
The highest unaccounted for OH source with a rate of 3.0 ppbv h−1 (51 % of the observed total OH destruction rate) is observed in the period from 5 to 8 August (Case 1), when NO mixing ratios were less than 1 ppbv and median maximum temperatures in the chamber were 31 ∘C. At the same time, an additional HO2 destruction process with a rate of up to 2.0 ppbv h−1 is required to balance the HO2 production rate, while production and destruction rates for RO2 radicals are well balanced. The opposing imbalances in the OH and HO2 budgets could be due to an unknown mechanism that converts HO2 to OH, or this could indicate a missing primary OH source and a similar fast, but independent, termination reaction removing HO2. If an unknown HO2 to OH conversion mechanism played a major role, it would not explain the complete rate of the missing OH source. Since the missing OH source is slightly larger than the rate of the missing HO2 sink, part of the missing OH source could have originated from a missing primary OH production process because a small difference between the total ROX production and destruction rates is also observed. The missing ROX source was up to 0.5 ppbv h−1 for Case 1 but was even higher with a rate of 1.4 ppbv h−1 in the summer when temperature was highest (Case 2). Since the calculated reaction rates of the HO2 and RO2 radicals with NO were able to reproduce the observed Ox production within 1 ppbv h−1, the unknown missing processes do not seem to have a direct impact on net ozone production.
For NO mixing ratios in the range of 1 to 3 ppbv, production and destruction rates for OH and HO2 radicals were balanced, while additional sources of RO2 and ROX having average rates of 1.6 and 0.4 ppbv h−1, respectively, were required to balance their production and destruction rates. Therefore, part of the missing RO2 source can be explained by a primary radical source, but the remaining RO2 source is still unresolved.
For high NO mixing ratios above 3 ppbv (4 to 5 ppbv h−1), large discrepancies between production and destruction rates of HO2 and RO2 radicals were found, but the calculations for these conditions have a higher uncertainty due to low HO2 and RO2 concentrations close to background signals. Whereas the imbalance in the budget for HO2 radicals is due to a loss process that is unaccounted for, an additional RO2 production process is required to close the chemical budget for RO2 radicals. For the same conditions, a primary ROX source with a rate of 0.5 ppbv h−1 was needed to balance the ROX destruction rate. Therefore, the missing primary ROX source is likely a primary RO2 source that is unaccounted for.
Production of radicals from the oxidation of organic compounds by chlorine could have been one additional source. Unfortunately, the potential impact of chlorine chemistry could not be examined in the spring periods, when these conditions were experienced, because ClNO2 measurements were not available. During times when ClNO2 concentrations were measured, chlorine chemistry initiated by the photolysis of ClNO2 did not significantly contribute to the radical production.
For chemical conditions when the contribution of the reaction of HO2 with NO to the OH production was reduced, i.e., at lower NO levels, other radical formation pathways such as isomerization reactions of RO2 radicals, OH formation from ozonolysis of alkenes, or photolysis of multifunctional organic compounds could gain importance and need to be properly accounted for. These processes remain relatively poorly constrained due to the lack of direct measurements of, e.g., multifunctional organic compounds.
Although the exact mechanism for the missing production or destruction processes for OH, HO2, and RO2 radicals could not be determined from measurements in this campaign, knowing the magnitudes of the missing radical processes gives indicative information about the disagreements of model simulations and observations for radicals and secondary air pollutants.
More investigations of the chemical budgets of radicals, for example in environments with high NO mixing ratios, including the determination of the impact of chlorine chemistry and with a detailed characterization of the chemical composition of air masses with respect to the presence of complex organic compounds, would be beneficial for the understanding of radical chemistry as well as the formation of secondary air pollution such as ozone.
Data from the JULIAC campaign analyzed in this work are available from the Jülich Data repository (https://doi.org/10.26165/JUELICH-DATA/3J80BW, Cho et al., 2022).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-2003-2023-supplement.
AH designed the JULIAC campaign and organized it together with HF and FH. CC performed the measurements of radicals, analyzed the data, and wrote the paper together with AN and HF. All co-authors contributed with data and helped the writing through intensive discussions of the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank all JULIAC team members for discussion as well as technical and logistical support.
This research has been supported by the H2020 European Research Council (SARLEP (grant no. 681529)) and the European Commission, Horizon 2020 Framework Programme (EUROCHAMP-2020 (grant no. 730997)).
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Christopher Cantrell and reviewed by two anonymous referees.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and Subcommittee, I.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Bloss, W. J., Lee, J. D., Johnson, G. P., Sommariva, R., Heard, D. E., Saiz-Lopez, A., Plane, J. M. C., McFiggans, G., Coe, H., Flynn, M., Williams, P., Rickard, A. R., and Fleming, Z. L.: Impact of halogen monoxide chemistry upon boundary layer OH and HO2 concentrations at a coastal site, Geophys. Res. Lett., 32, 1–4, https://doi.org/10.1029/2004GL022084, 2005.
BMEL (Federal Ministry of Food and Agriculture): Ergebnisse der Waldzustandserhebung 2020, Bonn, Germany, https://www.bmel.de/ (last access: January 2023), 2021.
Bohn, B., Rohrer, F., Brauers, T., and Wahner, A.: Actinometric measurements of NO2 photolysis frequencies in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 493–503, https://doi.org/10.5194/acp-5-493-2005, 2005.
Bohn, B. and Zilken, H.: Model-aided radiometric determination of photolysis frequencies in a sunlit atmosphere simulation chamber, Atmos. Chem. Phys., 5, 191–206, https://doi.org/10.5194/acp-5-191-2005, 2005.
Brune, W. H., Baier, B. C., Thomas, J., Ren, X., Cohen, R. C., Pusede, S. E., Browne, E. C., Goldstein, A. H., Gentner, D. R., Keutsch, F. N., Thornton, J. A., Harrold, S., Lopez-Hilfiker, F. D., and Wennberg, P. O.: Ozone production chemistry in the presence of urban plumes, Faraday Discuss., 189, 169–189, https://doi.org/10.1039/C5FD00204D, 2016.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory, Pasadena, https://jpldataeval.jpl.nasa.gov/ (last access: January 2023), 2019.
Cazorla, M., Brune, W. H., Ren, X., and Lefer, B.: Direct measurement of ozone production rates in Houston in 2009 and comparison with two estimation methods, Atmos. Chem. Phys., 12, 1203–1212, https://doi.org/10.5194/acp-12-1203-2012, 2012.
Chen, S., Ren, X., Mao, J., Chen, Z., Brune, W. H., Lefer, B., Rappenglück, B., Flynn, J., Olson, J., and Crawford, J. H.: A comparison of chemical mechanisms based on TRAMP-2006 field data, Atmos. Environ., 44, 4116–4125, https://doi.org/10.1016/j.atmosenv.2009.05.027, 2010.
Cho, C., Hofzumahaus, A., Fuchs, H., Dorn, H. P., Glowania, M., Holland, F., Rohrer, F., Vardhan, V., Kiendler-Scharr, A., Wahner, A., and Novelli, A.: Characterization of a chemical modulation reactor (CMR) for the measurement of atmospheric concentrations of hydroxyl radicals with a laser-induced fluorescence instrument, Atmos. Meas. Tech., 14, 1851–1877, https://doi.org/10.5194/amt-14-1851-2021, 2021.
Cox, R. A., Ammann, M., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 20, 13497–13519, https://doi.org/10.5194/acp-20-13497-2020, 2020.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/C1CP21330J, 2011.
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein, 1. Peroxy Radical Isomerization Following Addition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012.
da Silva, G., Graham, C., and Wang, Z.-F.: Unimolecular â-hydroxyperoxy radical decomposition with OH recycling in the photochemical oxidation of isoprene, Environ. Sci. Technol., 44, 250–256, https://doi.org/10.1021/es900924d, 2010.
Dillon, T. J. and Crowley, J. N.: Direct detection of OH formation in the reactions of HO2 with CH3C(O)O2 and other substituted peroxy radicals, Atmos. Chem. Phys., 8, 4877–4889, https://doi.org/10.5194/acp-8-4877-2008, 2008.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., Molina, L. T., Baker, A., Meinardi, S., Blake, D., Sheehy, P., Merten, A., Zhang, R., Zheng, J., Fortner, E. C., Junkermann, W., Dubey, M., Rahn, T., Eichinger, B., Lewandowski, P., Prueger, J., and Holder, H.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 9, 6655–6675, https://doi.org/10.5194/acp-9-6655-2009, 2009.
Ehhalt, D. H. and Rohrer, F.: Dependence of the OH concentration on solar UV, J. Geophys. Res.-Atmos., 105, 3565–3571, https://doi.org/10.1029/1999jd901070, 2000.
Elshorbany, Y. F., Kleffmann, J., Hofzumahaus, A., Kurtenbach, R., Wiesen, P., Brauers, T., Bohn, B., Dorn, H.-P., Fuchs, H., Holland, F., Rohrer, F., Tillmann, R., Wegener, R., Wahner, A., Kanaya, Y., Yoshino, A., Nishida, S., Kajii, Y., Martinez, M., Kubistin, D., Harder, H., Lelieveld, J., Elste, T., Plass-Dülmer, C., Stange, G., Berresheim, H., and Schurath, U.: HOx budgets during HOxComp: A case study of HOx chemistry under NOx-limited conditions, J. Geophys. Res.-Atmos., 117, D03307, https://doi.org/10.1029/2011JD017008, 2012.
Fan, S. and Li, Y.: The impacts of marine-emitted halogens on OH radicals in East Asia during summer, Atmos. Chem. Phys., 22, 7331–7351, https://doi.org/10.5194/acp-22-7331-2022, 2022.
Fishman, J. and Carney, T. A.: A one-dimensional photochemical model of the troposphere with planetary boundary-layer parameterization, J. Atmos. Chem., 1, 351–376, https://doi.org/10.1007/BF00053800, 1984.
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 79, 084104, https://doi.org/10.1063/1.2968712, 2008.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Fuchs, H., Dorn, H. P., Bachner, M., Bohn, B., Brauers, T., Gomm, S., Hofzumahaus, A., Holland, F., Nehr, S., Rohrer, F., Tillmann, R., and Wahner, A.: Comparison of OH concentration measurements by DOAS and LIF during SAPHIR chamber experiments at high OH reactivity and low NO concentration, Atmos. Meas. Tech., 5, 1611–1626, https://doi.org/10.5194/amt-5-1611-2012, 2012.
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Häseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/ngeo1964, 2013.
Fuchs, H., Acir, I. H., Bohn, B., Brauers, T., Dorn, H. P., Häseler, R., Hofzumahaus, A., Holland, F., Kaminski, M., Li, X., Lu, K., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., and Wahner, A.: OH regeneration from methacrolein oxidation investigated in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 14, 7895–7908, https://doi.org/10.5194/acp-14-7895-2014, 2014.
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017.
Fuchs, H., Albrecht, S., Acir, I., Bohn, B., Breitenlechner, M., Dorn, H. P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Kaminski, M., Keutsch, F. N., Novelli, A., Reimer, D., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., Zaytsev, A., Kiendler-Scharr, A., and Wahner, A.: Investigation of the oxidation of methyl vinyl ketone (MVK) by OH radicals in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 18, 8001–8016, https://doi.org/10.5194/acp-18-8001-2018, 2018.
George, I. J., Vlasenko, A., Slowik, J. G., Broekhuizen, K., and Abbatt, J. P. D.: Heterogeneous oxidation of saturated organic aerosols by hydroxyl radicals: uptake kinetics, condensed-phase products, and particle size change, Atmos. Chem. Phys., 7, 4187–4201, https://doi.org/10.5194/acp-7-4187-2007, 2007.
George, I. J., Matthews, P. S. J., Whalley, L. K., Brooks, B., Goddard, A., Baeza-Romero, M. T., and Heard, D. E.: Measurements of uptake coefficients for heterogeneous loss of HO2 onto submicron inorganic salt aerosols, Phys. Chem. Chem. Phys, 15, 12829–12845, https://doi.org/10.1039/C3CP51831K, 2013.
Glowania, M., Rohrer, F., Dorn, H.-P., Hofzumahaus, A., Holland, F., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Comparison of formaldehyde measurements by Hantzsch, CRDS and DOAS in the SAPHIR chamber, Atmos. Meas. Tech., 14, 4239–4253, https://doi.org/10.5194/amt-14-4239-2021, 2021.
Goldberg, D. L., Vinciguerra, T. P., Hosley, K. M., Loughner, C. P., Canty, T. P., Salawitch, R. J., and Dickerson, R. R.: Evidence for an increase in the ozone photochemical lifetime in the eastern United States using a regional air quality model, J. Geophys. Res.-Atmos., 120, 12778–12793, https://doi.org/10.1002/2015JD023930, 2015.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Goldstein, A. H., Fan, S. M., Goulden, M. L., Munger, J. W., and Wofsy, S. C.: Emissions of ethene, propene, and 1-butene by a midlatitude forest, J. Geophys. Res.-Atmos., 101, 9149–9157, https://doi.org/10.1029/96JD00334, 1996.
Griffith, S. M., Hansen, R. F., Dusanter, S., Stevens, P. S., Alaghmand, M., Bertman, S. B., Carroll, M. A., Erickson, M., Galloway, M., Grossberg, N., Hottle, J., Hou, J., Jobson, B. T., Kammrath, A., Keutsch, F. N., Lefer, B. L., Mielke, L. H., O'Brien, A., Shepson, P. B., Thurlow, M., Wallace, W., Zhang, N., and Zhou, X. L.: OH and HO2 radical chemistry during PROPHET 2008 and CABINEX 2009 – Part 1: Measurements and model comparison, Atmos. Chem. Phys., 13, 5403–5423, https://doi.org/10.5194/acp-13-5403-2013, 2013.
Griffith, S. M., Hansen, R. F., Dusanter, S., Michoud, V., Gilman, J. B., Kuster, W. C., Veres, P. R., Graus, M., de Gouw, J. A., Roberts, J., Young, C., Washenfelder, R., Brown, S. S., Thalman, R., Waxman, E., Volkamer, R., Tsai, C., Stutz, J., Flynn, J. H., Grossberg, N., Lefer, B., Alvarez, S. L., Rappenglueck, B., Mielke, L. H., Osthoff, H. D., and Stevens, P. S.: Measurements of hydroxyl and hydroperoxy radicals during CalNex-LA: Model comparisons and radical budgets, J. Geophys. Res.-Atmos., 121, 4211–4232, https://doi.org/10.1002/2015jd024358, 2016.
Groß, C. B. M., Dillon, T. J., Schuster, G., Lelieveld, J., and Crowley, J. N.: Direct Kinetic Study of OH and O3 Formation in the Reaction of CH3C(O)O2 with HO2, J. Phys. Chem. A, 118, 974–985, https://doi.org/10.1021/jp412380z, 2014.
Han, S., Bian, H., Feng, Y., Liu, A., Li, X., Zeng, F., and Zhang, X.: Analysis of the Relationship between O3, NO and NO2 in Tianjin, China, Aerosol Air Qual. Res., 11, 128–139, https://doi.org/10.4209/aaqr.2010.07.0055, 2011.
Handisides, G. M., Plass-Dülmer, C., Gilge, S., Bingemer, H., and Berresheim, H.: Hohenpeissenberg Photochemical Experiment (HOPE 2000): Measurements and photostationary state calculations of OH and peroxy radicals, Atmos. Chem. Phys., 3, 1565–1588, https://doi.org/10.5194/acp-3-1565-2003, 2003.
Häseler, R., Brauers, T., Holland, F., and Wahner, A.: Development and application of a new mobile LOPAP instrument for the measurement of HONO altitude profiles in the planetary boundary layer, Atmos. Meas. Tech. Discuss., 2, 2027–2054, https://doi.org/10.5194/amtd-2-2027-2009, 2009.
Hasson, A. S., Tyndall, G. S., and Orlando, J. J.: A Product Yield Study of the Reaction of HO2 Radicals with Ethyl Peroxy (C2H5O2), Acetyl Peroxy (CH3C(O)O2), and Acetonyl Peroxy (CH3C(O)CH2O2) Radicals, J. Phys. Chem. A, 108, 5979–5989, https://doi.org/10.1021/jp048873t, 2004.
Hausmann, M., Brandenburger, U., Brauers, T., and Dorn, H.-P.: Detection of tropospheric OH radicals by long-path differential optical-absorption spectroscopy: Experimental setup, accuracy, and precision, J. Geophys. Res., 102, 16011–16022, https://doi.org/10.1029/97jd00931, 1997.
Heard, D. E., Carpenter, L. J., Creasey, D. J., Hopkins, J. R., Lee, J. D., Lewis, A. C., Pilling, M. J., Seakins, P. W., Carslaw, N., and Emmerson, K. M.: High levels of the hydroxyl radical in the winter urban troposphere, Geophys. Res. Lett., 31, L18112, https://doi.org/10.1029/2004GL020544, 2004.
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified trace gas removal in the troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Hofzumahaus, A. and Heard, D. H.: Assessment of local HOx and ROx measurement techniques: achivements, challenges, and future directions – Outcomes of the 2015 international HOx workshop, Forschungszentrum Jülich, Jülich, 20–21, https://juser.fz-juelich.de/record/826803 (last access: January 2023), 2016.
Holland, F., Hofzumahaus, A., Schäfer, J., Kraus, A., and Pätz, H.-W.: Measurements of OH and HO2 radical concentrations and photolysis frequencies during BERLIOZ, J. Geophys. Res.-Atmos., 108, 8246, https://doi.org/10.1029/2001jd001393, 2003.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, https://doi.org/10.1016/j.ijms.2009.07.005, 2009.
Kaminski, M., Fuchs, H., Acir, I. H., Bohn, B., Brauers, T., Dorn, H. P., Häseler, R., Hofzumahaus, A., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., and Wahner, A.: Investigation of the â-pinene photooxidation by OH in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 17, 6631–6650, https://doi.org/10.5194/acp-17-6631-2017, 2017.
Kanaya, Y., Cao, R., Akimoto, H., Fukuda, M., Komazaki, Y., Yokouchi, Y., Koike, M., Tanimoto, H., Takegawa, N., and Kondo, Y.: Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004, J. Geophys. Res.-Atmos., 112, D21312, https://doi.org/10.1029/2007jd008670, 2007.
Kanaya, Y., Hofzumahaus, A., Dorn, H. P., Brauers, T., Fuchs, H., Holland, F., Rohrer, F., Bohn, B., Tillmann, R., Wegener, R., Wahner, A., Kajii, Y., Miyamoto, K., Nishida, S., Watanabe, K., Yoshino, A., Kubistin, D., Martinez, M., Rudolf, M., Harder, H., Berresheim, H., Elste, T., Plass-Dülmer, C., Stange, G., Kleffmann, J., Elshorbany, Y., and Schurath, U.: Comparisons of observed and modeled OH and HO2 concentrations during the ambient measurement period of the HOxComp field campaign, Atmos. Chem. Phys., 12, 2567–2585, https://doi.org/10.5194/acp-12-2567-2012, 2012.
Kim, S., Wolfe, G. M., Mauldin, L., Cantrell, C., Guenther, A., Karl, T., Turnipseed, A., Greenberg, J., Hall, S. R., Ullmann, K., Apel, E., Hornbrook, R., Kajii, Y., Nakashima, Y., Keutsch, F. N., DiGangi, J. P., Henry, S. B., Kaser, L., Schnitzhofer, R., Graus, M., Hansel, A., Zheng, W., and Flocke, F. F.: Evaluation of HOx sources and cycling using measurement-constrained model calculations in a 2-methyl-3-butene-2-ol (MBO) and monoterpene (MT) dominated ecosystem, Atmos. Chem. Phys., 13, 2031–2044, https://doi.org/10.5194/acp-13-2031-2013, 2013.
Kleffmann, J., Lörzer, J. C., Wiesen, P., Kern, C., Trick, S., Volkamer, R., Rodenas, M., and Wirtz, K.: Intercomparison of the DOAS and LOPAP techniques for the detection of nitrous acid (HONO), Atmos. Environ., 40, 3640–3652, https://doi.org/10.1016/j.atmosenv.2006.03.027, 2006.
Kleinman, L. I., Daum, P. H., Lee, Y.-N., Nunnermacker, L. J., Springston, S. R., Weinstein-Lloyd, J., and Rudolph, J.: Ozone production efficiency in an urban area, J. Geophys. Res.-Atmos., 107, 4733, https://doi.org/10.1029/2002JD002529, 2002.
Komenda, M., Schaub, A., and Koppmann, R.: Description and characterization of an on-line system for long-term measurements of isoprene, methyl vinyl ketone, and methacrolein in ambient air, J. Chromatogr. A, 995, 185–201, https://doi.org/10.1016/S0021-9673(03)00518-1, 2003.
Konrad, S., Schmitz, T., Buers, H.-J., Houben, N., Mannschreck, K., Mihelcic, D., Müsgen, P., Pätz, H.-W., Holland, F., Hofzumahaus, A., Schäfer, H.-J., Schröder, S., Volz-Thomas, A., Bächmann, K., Schlomski, S., Moortgat, G., and Großmann, D.: Hydrocarbon measurements at Pabstthum during the BERLIOZ campaign and modeling of free radicals, J. Geophys. Res.-Atmos., 108, 8251, https://doi.org/10.1029/2001JD000866, 2003.
Kubistin, D., Harder, H., Martinez, M., Rudolf, M., Sander, R., Bozem, H., Eerdekens, G., Fischer, H., Gurk, C., Klüpfel, T., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Taraborrelli, D., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 10, 9705–9728, https://doi.org/10.5194/acp-10-9705-2010, 2010.
Lakey, P. S. J., George, I. J., Whalley, L. K., Baeza-Romero, M. T., and Heard, D. E.: Measurements of the HO2 Uptake Coefficients onto Single Component Organic Aerosols, Environ. Sci. Technol., 49, 4878–4885, https://doi.org/10.1021/acs.est.5b00948, 2015.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008.
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010.
Lu, K. D., Hofzumahaus, A., Holland, F., Bohn, B., Brauers, T., Fuchs, H., Hu, M., Häseler, R., Kita, K., Kondo, Y., Li, X., Lou, S. R., Oebel, A., Shao, M., Zeng, L. M., Wahner, A., Zhu, T., Zhang, Y. H., and Rohrer, F.: Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006, Atmos. Chem. Phys., 13, 1057–1080, https://doi.org/10.5194/acp-13-1057-2013, 2013.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Brauers, T., Oebel, A., Dlugi, R., Hu, M., Li, X., Lou, S. R., Shao, M., Zhu, T., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Nighttime observation and chemistry of HOX in the Pearl River Delta and Beijing in summer 2006, Atmos. Chem. Phys., 14, 4979–4999, https://doi.org/10.5194/acp-14-4979-2014, 2014.
Ma, X., Tan, Z., Lu, K., Yang, X., Liu, Y., Li, S., Li, X., Chen, S., Novelli, A., Cho, C., Zeng, L., Wahner, A., and Zhang, Y.: Winter photochemistry in Beijing: Observation and model simulation of OH and HO2 radicals at an urban site, Sci. Tot. Environ., 685, 85–95, https://doi.org/10.1016/j.scitotenv.2019.05.329, 2019.
Malkin, T. L., Goddard, A., Heard, D. E., and Seakins, P. W.: Measurements of OH and HO2 yields from the gas phase ozonolysis of isoprene, Atmos. Chem. Phys., 10, 1441–1459, https://doi.org/10.5194/acp-10-1441-2010, 2010.
Mao, J., Ren, X., Chen, S., Brune, W. H., Chen, Z., Martinez, M., Harder, H., Lefer, B., Rappenglück, B., Flynn, J., and Leuchner, M.: Atmospheric oxidation capacity in the summer of Houston 2006: Comparison with summer measurements in other metropolitan studies, Atmos. Environ., 44, 4107–4115, https://doi.org/10.1016/j.atmosenv.2009.01.013, 2010.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J. H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Martinez, M., Harder, H., Kovacs, T. A., Simpas, J. B., Bassis, J., Lesher, R., Brune, W. H., Frost, G. J., Williams, E. J., Stroud, C. A., Jobson, B. T., Roberts, J. M., Hall, S. R., Shetter, R. E., Wert, B., Fried, A., Alicke, B., Stutz, J., Young, V. L., White, A. B., and Zamora, R. J.: OH and HO2 concentrations, sources, and loss rates during the Southern Oxidants Study in Nashville, Tennessee, summer 1999, J. Geophys. Res.-Atmos., 108, 4617, https://doi.org/10.1029/2003JD003551, 2003.
Mihelcic, D., Holland, F., Hofzumahaus, A., Hoppe, L., Konrad, S., Müsgen, P., Pätz, H.-W., Schäfer, H.-J., Schmitz, T., Volz-Thomas, A., Bächmann, K., Schlomski, S., Platt, U., Geyer, A., Alicke, B., and Moortgat, G. K.: Peroxy radicals during BERLIOZ at Pabstthum: Measurements, radical budgets and ozone production, J. Geophys. Res.-Atmos., 108, D4, https://doi.org/10.1029/2001JD001014, 2003.
Nehr, S., Bohn, B., Fuchs, H., Hofzumahaus, A., and Wahner, A.: HO2 formation from the OH+ benzene reaction in the presence of O2, Phys. Chem. Chem. Phys., 13, 10699–10708, https://doi.org/10.1039/C1CP20334G, 2011.
Nehr, S., Bohn, B., Dorn, H. P., Fuchs, H., Häseler, R., Hofzumahaus, A., Li, X., Rohrer, F., Tillmann, R., and Wahner, A.: Atmospheric photochemistry of aromatic hydrocarbons: OH budgets during SAPHIR chamber experiments, Atmos. Chem. Phys., 14, 6941–6952, https://doi.org/10.5194/acp-14-6941-2014, 2014.
Novelli, A., Hens, K., Tatum Ernest, C., Kubistin, D., Regelin, E., Elste, T., Plass-Dülmer, C., Martinez, M., Lelieveld, J., and Harder, H.: Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals, Atmos. Meas. Tech., 7, 3413–3430, https://doi.org/10.5194/amt-7-3413-2014, 2014.
Novelli, A., Kaminski, M., Rolletter, M., Acir, I. H., Bohn, B., Dorn, H. P., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Holland, F., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Evaluation of OH and HO2 concentrations and their budgets during photooxidation of 2-methyl-3-butene-2-ol (MBO) in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 18, 11409–11422, https://doi.org/10.5194/acp-18-11409-2018, 2018.
Novelli, A., Vereecken, L., Bohn, B., Dorn, H. P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Reimer, D., Rohrer, F., Rosanka, S., Taraborrelli, D., Tillmann, R., Wegener, R., Yu, Z., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 3333–3355, https://doi.org/10.5194/acp-20-3333-2020, 2020.
Novelli, A., Cho, C., Fuchs, H., Hofzumahaus, A., Rohrer, F., Tillmann, R., Kiendler-Scharr, A., Wahner, A., and Vereecken, L.: Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 23, 5474–5495, https://doi.org/10.1039/d0cp05555g, 2021.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/ngeo177, 2008.
Peeters, J. and Müller, J.-F.: HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations, II: experimental evidence and global impact, Phys. Chem. Chem. Phys, 12, 14227–14235, https://doi.org/10.1039/C0CP00811G, 2010.
Peeters, J., Nguyen, T., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys, 11, 5935–5939, https://doi.org/10.1039/b908511d, 2009.
Peeters, J., Müller, J. F., Stavrakou, T., and Vinh Son, N.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem.. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014.
Praske, E., Crounse, J. D., Bates, K. H., Kurtén, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015.
Ren, X., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Simpas, J. B., Brune, W. H., Schwab, J. J., Demerjian, K. L., He, Y., Zhou, X., and Gao, H.: OH and HO2 Chemistry in the urban atmosphere of New York City, Atmos. Environ., 37, 3639–3651, https://doi.org/10.1016/S1352-2310(03)00459-X, 2003.
Ren, X., Brune, W. H., Mao, J., Mitchell, M. J., Lesher, R. L., Simpas, J. B., Metcalf, A. R., Schwab, J. J., Cai, C., Li, Y., Demerjian, K. L., Felton, H. D., Boynton, G., Adams, A., Perry, J., He, Y., Zhou, X., and Hou, J.: Behavior of OH and HO2 in the winter atmosphere in New York City, Atmos. Environ., 40, 252–263, https://doi.org/10.1016/j.atmosenv.2005.11.073, 2006.
Ren, X., van Duin, D., Cazorla, M., Chen, S., Mao, J., Zhang, L., Brune, W. H., Flynn, J. H., Grossberg, N., Lefer, B. L., Rappenglück, B., Wong, K. W., Tsai, C., Stutz, J., Dibb, J. E., Thomas Jobson, B., Luke, W. T., and Kelley, P.: Atmospheric oxidation chemistry and ozone production: Results from SHARP 2009 in Houston, Texas, J. Geophys. Res.-Atmos., 118, 5770–5780, https://doi.org/10.1002/jgrd.50342, 2013.
Rhew, R. C., Deventer, M. J., Turnipseed, A. A., Warneke, C., Ortega, J., Shen, S., Martinez, L., Koss, A., Lerner, B. M., Gilman, J. B., Smith, J. N., Guenther, A. B., and de Gouw, J. A.: Ethene, propene, butene and isoprene emissions from a ponderosa pine forest measured by relaxed eddy accumulation, Atmos. Chem. Phys., 17, 13417–13438, https://doi.org/10.5194/acp-17-13417-2017, 2017.
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F. J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005.
Rohrer, F., Lu, K., Hofzumahaus, A., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Häseler, R., Holland, F., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S., Oebel, A., Shao, M., Zeng, L., Zhu, T., Zhang, Y., and Wahner, A.: Maximum efficiency in the hydroxyl-radical-based self-cleansing of the troposphere, Nat. Geosci., 7, 559–563, https://doi.org/10.1038/ngeo2199, 2014.
Rolletter, M., Kaminski, M., Acir, I. H., Bohn, B., Dorn, H. P., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Investigation of the á-pinene photooxidation by OH in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 19, 11635–11649, https://doi.org/10.5194/acp-19-11635-2019, 2019.
Rolletter, M., Blocquet, M., Kaminski, M., Bohn, B., Dorn, H. P., Hofzumahaus, A., Holland, F., Li, X., Rohrer, F., Tillmann, R., Wegener, R., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Photooxidation of pinonaldehyde at ambient conditions investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 13701–13719, https://doi.org/10.5194/acp-20-13701-2020, 2020.
Schlosser, E., Bohn, B., Brauers, T., Dorn, H.-P., Fuchs, H., Häseler, R., Hofzumahaus, A., Holland, F., Rohrer, F., Rupp, L. O., Siese, M., Tillmann, R., and Wahner, A.: Intercomparison of two hydroxyl radical measurement techniques at the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 56, 187–205, https://doi.org/10.1007/s10874-006-9049-3, 2007.
Schlosser, E., Brauers, T., Dorn, H. P., Fuchs, H., Häseler, R., Hofzumahaus, A., Holland, F., Wahner, A., Kanaya, Y., Kajii, Y., Miyamoto, K., Nishida, S., Watanabe, K., Yoshino, A., Kubistin, D., Martinez, M., Rudolf, M., Harder, H., Berresheim, H., Elste, T., Plass-Dülmer, C., Stange, G., and Schurath, U.: Technical Note: Formal blind intercomparison of OH measurements: results from the international campaign HOxComp, Atmos. Chem. Phys., 9, 7923–7948, https://doi.org/10.5194/acp-9-7923-2009, 2009.
Sarkar, C., Guenther, A. B., Park, J. H., Seco, R., Alves, E., Batalha, S., Santana, R., Kim, S., Smith, J., Tóta, J., and Vega, O.: PTR-TOF-MS eddy covariance measurements of isoprene and monoterpene fluxes from an eastern Amazonian rainforest, Atmos. Chem. Phys., 20, 7179–7191, https://doi.org/10.5194/acp-20-7179-2020, 2020.
Sillman, S., Logan, J. A., and Wofsy, S. C.: The sensitivity of ozone to nitrogen oxides and hydrocarbons in regional ozone episodes, J. Geophys. Res.-Atmos., 95, 1837–1851, https://doi.org/10.1029/JD095iD02p01837, 1990.
Slater, E. J., Whalley, L. K., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Crilley, L. R., Kramer, L., Bloss, W., Vu, T., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Elevated levels of OH observed in haze events during wintertime in central Beijing, Atmos. Chem. Phys., 20, 14847–14871, https://doi.org/10.5194/acp-20-14847-2020, 2020.
Sommariva, R., Bloss, W. J., Brough, N., Carslaw, N., Flynn, M., Haggerstone, A. L., Heard, D. E., Hopkins, J. R., Lee, J. D., Lewis, A. C., McFiggans, G., Monks, P. S., Penkett, S. A., Pilling, M. J., Plane, J. M. C., Read, K. A., Saiz-Lopez, A., Rickard, A. R., and Williams, P. I.: OH and HO2 chemistry during NAMBLEX: roles of oxygenates, halogen oxides and heterogeneous uptake, Atmos. Chem. Phys., 6, 1135–1153, https://doi.org/10.5194/acp-6-1135-2006, 2006.
Sommariva, R., Hollis, L. D. J., Sherwen, T., Baker, A. R., Ball, S. M., Bandy, B. J., Bell, T. G., Chowdhury, M. N., Cordell, R. L., Evans, M. J., Lee, J. D., Reed, C., Reeves, C. E., Roberts, J. M., Yang, M., and Monks, P. S.: Seasonal and geographical variability of nitryl chloride and its precursors in Northern Europe, Atmos. Sci. Lett., 19, UNSP e844, https://doi.org/10.1002/asl.844, 2018.
Song, H., Chen, X., Lu, K., Zou, Q., Tan, Z., Fuchs, H., Wiedensohler, A., Moon, D. R., Heard, D. E., Baeza-Romero, M. T., Zheng, M., Wahner, A., Kiendler-Scharr, A., and Zhang, Y.: Influence of aerosol copper on HO2 uptake: a novel parameterized equation, Atmos. Chem. Phys., 20, 15835–15850, https://doi.org/10.5194/acp-20-15835-2020, 2020.
Spirig, C., Neftel, A., Ammann, C., Dommen, J., Grabmer, W., Thielmann, A., Schaub, A., Beauchamp, J., Wisthaler, A., and Hansel, A.: Eddy covariance flux measurements of biogenic VOCs during ECHO 2003 using proton transfer reaction mass spectrometry, Atmos. Chem. Phys., 5, 465–481, https://doi.org/10.5194/acp-5-465-2005, 2005.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/C2CS35140D, 2012.
Stone, D., Sherwen, T., Evans, M. J., Vaughan, S., Ingham, T., Whalley, L. K., Edwards, P. M., Read, K. A., Lee, J. D., Moller, S. J., Carpenter, L. J., Lewis, A. C., and Heard, D. E.: Impacts of bromine and iodine chemistry on tropospheric OH and HO2: comparing observations with box and global model perspectives, Atmos. Chem. Phys., 18, 3541–3561, https://doi.org/10.5194/acp-18-3541-2018, 2018.
Taketani, F., Kanaya, Y., and Akimoto, H.: Kinetics of Heterogeneous Reactions of HO2 Radical at Ambient Concentration Levels with (NH4)2SO4 and NaCl Aerosol Particles, J. Phys. Chem.. A, 112, 2370–2377, https://doi.org/10.1021/jp0769936, 2008.
Taketani, F., Kanaya, Y., and Akimoto, H.: Heterogeneous loss of HO2 by KCl, synthetic sea salt, and natural seawater aerosol particles, Atmos. Environ., 43, 1660–1665, https://doi.org/10.1016/j.atmosenv.2008.12.010, 2009.
Tan, D., Faloona, I., Simpas, J. B., Brune, W., Shepson, P. B., Couch, T. L., Sumner, A. L., Carroll, M. A., Thornberry, T., Apel, E., Riemer, D., and Stockwell, W.: HOx budgets in a deciduous forest: Results from the PROPHET summer 1998 campaign, J. Geophys. Res.-Atmos., 106, 24407–24427, https://doi.org/10.1029/2001jd900016, 2001.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tan, Z., Lu, K., Hofzumahaus, A., Fuchs, H., Bohn, B., Holland, F., Liu, Y., Rohrer, F., Shao, M., Sun, K., Wu, Y., Zeng, L., Zhang, Y., Zou, Q., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 19, 7129–7150, https://doi.org/10.5194/acp-19-7129-2019, 2019.
Tan, Z., Hofzumahaus, A., Lu, K., Brown, S. S., Holland, F., Huey, L. G., Kiendler-Scharr, A., Li, X., Liu, X., Ma, N., Min, K.-E., Rohrer, F., Shao, M., Wahner, A., Wang, Y., Wiedensohler, A., Wu, Y., Wu, Z., Zeng, L., Zhang, Y., and Fuchs, H.: No Evidence for a Significant Impact of Heterogeneous Chemistry on Radical Concentrations in the North China Plain in Summer 2014, Environ. Sci. Technol., 54, 5973–5979, https://doi.org/10.1021/acs.est.0c00525, 2020.
Tan, Z., Fuchs, H., Hofzumahaus, A., Bloss, W. J., Bohn, B., Cho, C., Hohaus, T., Holland, F., Lakshmisha, C., Liu, L., Monks, P. S., Novelli, A., Niether, D., Rohrer, F., Tillmann, R., Valkenburg, T. S. E., Vardhan, V., Kiendler-Scharr, A., Wahner, A., and Sommariva, R.: Seasonal variation in nitryl chloride and its relation to gas-phase precursors during the JULIAC campaign in Germany, Atmos. Chem. Phys., 22, 13137–13152, https://doi.org/10.5194/acp-22-13137-2022, 2022.
Tanaka, P. L., Riemer, D. D., Chang, S., Yarwood, G., McDonald-Buller, E. C., Apel, E. C., Orlando, J. J., Silva, P. J., Jimenez, J. L., Canagaratna, M. R., Neece, J. D., Mullins, C. B., and Allen, D. T.: Direct evidence for chlorine-enhanced urban ozone formation in Houston, Texas, Atmos. Environ., 37, 1393–1400, https://doi.org/10.1016/S1352-2310(02)01007-5, 2003.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dubé, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/nature08905, 2010.
Vaughan, S., Ingham, T., Whalley, L. K., Stone, D., Evans, M. J., Read, K. A., Lee, J. D., Moller, S. J., Carpenter, L. J., Lewis, A. C., Fleming, Z. L., and Heard, D. E.: Seasonal observations of OH and HO2 in the remote tropical marine boundary layer, Atmos. Chem. Phys., 12, 2149–2172, https://doi.org/10.5194/acp-12-2149-2012, 2012.
Vilà-Guerau de Arellano, J., van den Dries, K., and Pino, D.: On inferring isoprene emission surface flux from atmospheric boundary layer concentration measurements, Atmos. Chem. Phys., 9, 3629–3640, https://doi.org/10.5194/acp-9-3629-2009, 2009.
Wang, F., Hu, R., Chen, H., Xie, P., Wang, Y., Li, Z., Jin, H., Liu, J., and Liu, W.: Development of a field system for measurement of tropospheric OH radical using laser-induced fluorescence technique, Optics Express, 27, 419–435, https://doi.org/10.1364/OE.27.00A419, 2019.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Whalley, L. K., Stone, D., Dunmore, R., Hamilton, J., Hopkins, J. R., Lee, J. D., Lewis, A. C., Williams, P., Kleffmann, J., Laufs, S., Woodward-Massey, R., and Heard, D. E.: Understanding in situ ozone production in the summertime through radical observations and modelling studies during the Clean air for London project (ClearfLo), Atmos. Chem. Phys., 18, 2547–2571, https://doi.org/10.5194/acp-18-2547-2018, 2018.
Whalley, L. K., Slater, E. J., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Mehra, A., Worrall, S. D., Bacak, A., Bannan, T. J., Coe, H., Percival, C. J., Ouyang, B., Jones, R. L., Crilley, L. R., Kramer, L. J., Bloss, W. J., Vu, T., Kotthaus, S., Grimmond, S., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Evaluating the sensitivity of radical chemistry and ozone formation to ambient VOCs and NOx in Beijing, Atmos. Chem. Phys., 21, 2125–2147, https://doi.org/10.5194/acp-21-2125-2021, 2021.
Winiberg, F. A. F., Dillon, T. J., Orr, S. C., Groß, C. B. M., Bejan, I., Brumby, C. A., Evans, M. J., Smith, S. C., Heard, D. E., and Seakins, P. W.: Direct measurements of OH and other product yields from the HO2 + CH3C(O)O2 reaction, Atmos. Chem. Phys., 16, 4023–4042, https://doi.org/10.5194/acp-16-4023-2016, 2016.
Wolfe, G. M., Thornton, J. A., Bouvier-Brown, N. C., Goldstein, A. H., Park, J. H., McKay, M., Matross, D. M., Mao, J., Brune, W. H., LaFranchi, B. W., Browne, E. C., Min, K. E., Wooldridge, P. J., Cohen, R. C., Crounse, J. D., Faloona, I. C., Gilman, J. B., Kuster, W. C., de Gouw, J. A., Huisman, A., and Keutsch, F. N.: The Chemistry of Atmosphere-Forest Exchange (CAFE) Model – Part 2: Application to BEARPEX-2007 observations, Atmos. Chem. Phys., 11, 1269–1294, https://doi.org/10.5194/acp-11-1269-2011, 2011.
Wolfe, G. M., Cantrell, C., Kim, S., Mauldin Iii, R. L., Karl, T., Harley, P., Turnipseed, A., Zheng, W., Flocke, F., Apel, E. C., Hornbrook, R. S., Hall, S. R., Ullmann, K., Henry, S. B., DiGangi, J. P., Boyle, E. S., Kaser, L., Schnitzhofer, R., Hansel, A., Graus, M., Nakashima, Y., Kajii, Y., Guenther, A., and Keutsch, F. N.: Missing peroxy radical sources within a summertime ponderosa pine forest, Atmos. Chem. Phys., 14, 4715–4732, https://doi.org/10.5194/acp-14-4715-2014, 2014.