the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Dec 2023
| 20 Dec 2023
Gas–particle partitioning of toluene oxidation products: an experimental and modeling study
Evangelia Kostenidou
Florian Couvidat
Alvaro Martinez-Valiente
Philipp Eichler
Armin Wisthaler
Markus Müller
Brice Temime-Roussel
Richard Valorso
Karine Sartelet
Toluene represents a large fraction of anthropogenic emissions and significantly contributes to tropospheric ozone and secondary organic aerosol (SOA) formation. Despite the fact that toluene is one of the most studied aromatic compounds, detailed chemical mechanisms still fail to correctly reproduce the speciation of toluene gaseous and condensed oxidation products. This study aims to elucidate the role of initial experimental conditions in toluene SOA mass loadings and to investigate gas–particle partitioning of its reaction products at different relevant temperatures. Gaseous and particulate reaction products were identified and quantified using a proton transfer reaction time-of-flight mass spectrometer (PTR-ToF-MS) coupled to a CHemical Analysis of aeRosol ONline (CHARON) inlet. The chemical system exhibited a volatility distribution mostly in the semi-volatile regime. Temperature decrease caused a shift of saturation concentration towards lower values. The CHARON–PTR-ToF-MS instrument identified and quantified approximately 60 %–80 % of the total organic mass measured by an aerosol mass spectrometer. A detailed mechanism for toluene gaseous oxidation was developed based on the Master Chemical Mechanism (MCM) and Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) deterministic mechanisms, modified following the literature. The new mechanism showed improvements in modeling oxidation product speciation with more observed species represented and more representative concentrations compared to the MCM–GECKO-A reference. Tests on partitioning processes, nonideality, and wall losses highlighted the high dependency of SOA formation on the considered processes. Our results underline the fact that volatility is not sufficient to explain the gas–particle partitioning: the organic and the aqueous phases need to be considered as well as the interactions between compounds in the particle phase.
- Article
(2331 KB) - Full-text XML
-
Supplement
(3393 KB) - BibTeX
- EndNote




Aromatic compounds represent an important fraction of hydrocarbons emitted in the urban atmosphere (Calvert et al., 2002). Their photochemical oxidation is a source of ozone and secondary organic gas-phase compounds which can lead to the formation of secondary organic aerosol (SOA) (Seigneur, 2019). SOA represents a significant fraction of the total fine particles (Gelencsér et al., 2007; Jimenez et al., 2009) which are key components of the Earth's atmospheric system, significantly impacting visibility (Han et al., 2012), human health (Lim et al., 2012, 2018; Malley et al., 2017), and the Earth's climate (Boucher et al., 2013).
Toluene is the most abundant alkyl-aromatic compound, especially in urban areas (Lee et al., 2002; Yan et al., 2019; Zhang et al., 2015a). It is emitted by vehicle exhaust but also from biomass burning, fuel evaporation, solvent usage, and industrial processes (Singh et al., 1992; Skorokhod et al., 2017). It is classified as a toxic compound as it affects the central nervous system (US Environmental Protection Agency, 2012). The gas-phase products from toluene–OH radical reaction have been widely studied in the last 20 years via both experimental and modeling approaches (Baltaretu et al., 2009; Bohn, 2001; Jang and Kamens, 2001; Klotz et al., 1998; Sato et al., 2007; Schwantes et al., 2017; Suh et al., 2002; Wagner et al., 2003; Xu et al., 2015; Zaytsev et al., 2019). Toluene photo-oxidation leads to the formation of gas-phase products of low volatility that can easily condense on the pre-existing aerosol particles to form SOA (Ng et al., 2007; Seigneur, 2019; White et al., 2014). Despite the amount of work devoted to the degradation of toluene, many uncertainties remain regarding the oxidation mechanisms (species and reactions involved) and the physicochemical processes leading to the formation of SOA from toluene (Calvert et al., 2002; Jang and Kamens, 2001; Schwantes et al., 2017; Zaytsev et al., 2019). Moreover, properties of the compounds formed (and thus their ability to condense and form SOA) strongly depend on environmental parameters such as temperature, NOX, humidity, pre-existing particle surface, toluene concentration, and OH radical concentration (Kamens et al., 2011; Kim et al., 2011; Lannuque et al., 2018; Ng et al., 2007; Sato et al., 2007; Takekawa et al., 2003; Xu et al., 2015). In order to reproduce the observed SOA variations well with modeling and to interpret experimental measurements, detailed and representative oxidation mechanisms and partitioning processes are required.
The reference explicit chemical mechanism for toluene oxidation is the Master Chemical Mechanism (MCM) (Bloss et al., 2005; Jenkin et al., 2003). The MCM is a detailed mechanism that describes the chemical evolution of organic matter from toluene through 270 secondary species and 814 reactions. The main reaction pathways of toluene are (i) addition of an OH function on the aromatic ring and the formation of an RO2 radical, leading to the formation of bicyclic compounds with an O–O bridge (65 %); (ii) addition of an OH function on the ring and the formation of cresol and HO2 (18 %); (iii) addition of an OH function on the aromatic ring, leading to the formation of epoxides and ring opening (10 %); and (iv) hydrogen abstraction from the methyl group (7 %). Based on recent experiments in atmospheric simulation chambers, the cresol formation pathway and the successive addition of alcohol function on the aromatic ring seem to be underestimated or neglected in the MCM (Schwantes et al., 2017), and branching ratios should be revised (Zaytsev et al., 2019). These changes necessarily affect SOA formation, which is favored by the formation of (i) low-volatility oxygenated compounds (Molteni et al., 2018; Ng et al., 2007; Schwantes et al., 2017; Wang et al., 2017, 2020) and (ii) volatile organic acids and dicarbonyls such as methylglyoxal and glyoxal, which oligomerize, hydrate, or oxidize in the aqueous phase (De Haan et al., 2018; Hu et al., 2022; Li et al., 2021a, b; Marrero-Ortiz et al., 2019). Nonetheless, partitioning processes toward an aqueous-phase and particle-phase chemistry (such as oligomerization or oxidation) remain poorly represented in models.
The recent development of soft-ionization analytical tools for particles as the CHemical Analysis of aeRosol ONline (CHARON) coupled to a proton transfer reaction time-of-flight mass spectrometer (PTR-ToF-MS) allows for direct characterization of organic aerosol on a molecular composition level (Eichler et al., 2015). At the same time the molecular gas-phase characterization is provided by a separate PTR-ToF-MS. The above instruments were deployed in an effort to better understand the chemistry of toluene and the formation of SOA at different temperatures using a thermostated oxidation flow reactor (OFR). Then these experiments were numerically reproduced using detailed chemical mechanisms based on a combination of the MCM and the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) (Aumont et al., 2005) modified according to recent studies.
The main goals of this study are (1) the chemical identification of gaseous- and particulate-phase products, (2) the determination of the partition coefficient of the individual SOA products at different temperatures, (3) the development of a new detailed oxidation mechanism for toluene to better reproduce the experimental speciation, and (4) the exploration and impact analysis of the different processes influencing the partitioning (hydrophilic partitioning, nonideality, wall losses, and methylglyoxal irreversible partitioning) on the modeling of experimental SOA. To our knowledge this is the first time that partitioning and volatility data on individual toluene SOA components are provided at relevant ambient temperatures.
2.1 Experimental conditions and instrumentation
2.1.1 Oxidation experiments
Toluene photo-oxidation experiments were carried out at atmospheric pressure in a horizontal cylindrical OFR made of Pyrex (12 cm internal diameter, 152 cm length) surrounded by seven fluorescent lamps (Philips CLEO) with a continuous emission spectrum in the 300–420 nm range and a total irradiance of 3 × 1016 photons cm−2 s−1. Temperature was controlled using a circulating water bath with the outer jacket (Huber CC 405). The experiments were conducted at 280, 285, and 295 K. The total flow varied from 1.3 to 1.8 L min−1, and the residence time was 10–13 min. The relative humidity (RH) was adjusted by bubbling pure nitrogen though ultrapure distilled and deionized water and ranged from 23 %–50 %. Monodisperse ammonium sulfate (AS) particles were nebulized using a TSI atomizer (model 3076), dried using a silica diffusion drier, and then passed through a differential mobility analyzer (DMA, TSI model 3081) to generate monodisperse aerosol ranging from 113 to 169 nm (mobility diameter). Toluene (99.8 % purity, Sigma Aldrich) was constantly introduced in the OFR via a permeation tube kept at 353 K and purged with pure nitrogen, and its concentration inside the OFR varied from 8 to 162 ppbv. Isopropyl nitrite (IPN) was used as a hydroxyl radical precursor and was prepared from the corresponding alcohol following the procedure reported by Noyes (1936). The IPN was constantly introduced in the OFR using a permeation tube kept at 308–313 K and flushed with pure nitrogen. During IPN photolysis NO and HO2 are produced and can further react, producing OH and NO2 radicals (Raff and Finlayson-Pitts, 2010). The average oxidant concentration during the experiment was calculated from the toluene decay (kOH≈6 × 10−12 cm3 molec.−1 s−1) and ranged from 7.4 to 13 × 107 molec. cm−3, corresponding approximately to 10 and 20 h of atmospheric OH radical exposure, taking into account a diurnal average hydroxyl radical concentration of 1.5 × 106 molec. cm−3 (Mao et al., 2009). High amounts of acetone, a side product of IPN photolysis, inhibit the detection of glyoxal, a major gas-phase product. A total of 24 photo-oxidation experiments have been carried out. A detailed list of the experimental conditions is provided in Table S1 in the Supplement. A subset of six experiences is reported here for detailed analysis at the molecular level of the gaseous and particulate phases; these experiments are indicated in red in Table S1.
2.1.2 Instrumentation
A high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Tofwerk AG, Aerodyne Inc., USA) (DeCarlo et al., 2006), a scanning mobility particle sizer (SMPS, TSI Classifier model 3081, DMA, TSI CPC 3776), and a CHemical Analysis of aeRosol ONline (CHARON) inlet coupled to a commercial PTR-ToF-MS 8000 (Ionicon Analytik GmbH, Innsbruck, Austria) were used to measure aerosol mass, aerosol chemical composition, and size distribution. The AMS operating principles, calibration procedures, and analysis protocols are described in Drewnick et al. (2005, 2009). Uncertainties from the HR-ToF-AMS are typically of the order of ± 30 %.
The PTR-ToF-MS instrument and methods of data analysis are described in Jordan et al. (2009), Graus et al. (2010), and Müller et al. (2013). Data analysis was performed using the standard PTR data analysis toolkits (Tofwerk Inc, Switzerland) version 2.5.10 with Igor Pro 6.37 version (Wavemetrics, Inc.) and PTR-ToF-MS data analyzer toolbox (version 4.46) (Müller et al., 2013). Calibration and quantification procedures are described in the Supplement of the article. A detailed description of the CHARON inlet has been provided elsewhere by Eichler et al. (2015), Müller et al. (2017), and Leglise et al. (2019), and therefore only a brief description is provided here. The CHARON inlet used in these experiments was a noncommercial prototype almost identical to that described by Eichler et al. (2015). The air sample is drawn through an activated carbon monolith denuder which removes gaseous organics. An aerodynamic lens is used for enriching the particle concentration by a factor of ∼ 8–25 depending on the particle size. Enrichment factor calibration was repeated several times during the campaign using ammonium nitrate monodispersed particles (see Fig. S1 in the Supplement). Finally, a thermodesorption unit vaporizes the particles at 413 K. The instrumental background is determined by periodically diverting the sampling flow through a high-efficiency particulate air filter (HC01-5N-B miniature HEPA capsule filter) placed upstream of the CHARON inlet. Operational and software details as well as routines are given in the Supplement. An additional NOX detector (Thermo 42C chemiluminescent analyzer) was also used at the exit of the OFR during some experiments.
2.1.3 Experimental partitioning and volatility distribution
The experimental partitioning coefficient Kp,i (in µg−1 m3) is calculated for the experimental data by applying the partitioning theory (Yamasaki et al., 1982):
where Cp,i and Cg,i are the particle- and gas-phase concentrations measured by CHARON–PTR-ToF-MS and PTR-ToF-MS of the species i (in µg m−3), and TSP is the total suspended particulate matter (in µg m−3). The saturation concentration (in µg m−3), commonly used to define the volatility of different chemical species (Donahue et al., 2011), is linked to the Kp,i through the equation
The saturation concentration of the major particle components is calculated for experiments at 295 and 280 K. For species having concentrations close to the detection limits, the was not evaluated because of high uncertainty associated with it.
2.2 Model setup and chemical mechanisms
2.2.1 Toluene oxidation mechanisms
Three versions of a detailed chemical mechanism are used in this study to reproduce the experimental oxidation of toluene and the formation of condensable species. All three are based on a detailed mechanism coupling MCM and GECKO-A to which additions and modifications are successively made. All the chemical mechanisms of toluene oxidation as well as the chemical compounds are presented in the Supplement with the details of the modifications made (Tables S4 to S8 in the Supplement), and the first oxidation step is illustrated in Fig. S11.
The MCM–GECKO base mechanism
The first oxidation mechanism is generated with GECKO-A (Aumont et al., 2005). The GECKO-A tool is a computer program that automatically generates explicit chemical mechanisms following an established writing protocol. It integrates the elementary kinetic and mechanistic data available in the literature and estimates the missing ones using structure–activity relationships (SARs). The version of GECKO-A used in this study is based on the protocol presented in Aumont et al. (2005) and includes updates described in Valorso et al. (2011), Aumont et al. (2012, 2013), and La et al. (2016). In this version, the MCM v3.3.1 is used to describe the chemistry of compounds possessing an aromatic ring (Bloss et al., 2005; Jenkin et al., 2003), and GECKO-A then generates the oxidation mechanism for nonaromatic compounds.
Because the explicit description of toluene oxidation involves thousands of species and reactions, simplifications are made during generation to limit the size of the mechanism. Gaseous compounds with a saturating vapor pressure (Psat) below 10−13 atm are considered nonreactive. Positional isomers are lumped together if their production yield is less than 10−3 (Valorso et al., 2011). RO2 from the stepwise oxidation of toluene can only react with NO, HO2, and CH3O2. The other RO2+ RO2 reactions which represent less than 10 % of RO2 reactivity under atmospheric conditions (Lannuque et al., 2018) are not generated. The oxidation mechanism stops after the formation of the sixth generation of stable compounds. The reaction pathways thereby simplified or eliminated concern a minor fraction of matter. These simplifications do not significantly impact the simulated SOA mass or the major simulated species.
Only the reaction pathways leading to the formation of the main gaseous and/or condensed compounds experimentally identified (i.e., at the measured mass–charge ratio ) as well as to the formation of the major contributors to SOA identified by the modeling (i.e., representing more than 1 % of the simulated SOA mass under the experimental conditions) are considered here. One of the major gaseous compounds formed during the toluene oxidation is methylglyoxal. It is produced by more than 100 reactions and belongs to all six generations of the chemical mechanism. The simplifications made reduced the number of these reactions to about 15 major reaction pathways that represent about 85 % of the simulated production of methylglyoxal. The resulting chemical mechanism is hereafter denoted MG (for MCM–GECKO-A) and is composed of 125 species and 366 reactions (277 reactions leading to the formation of new species and 89 loss reactions to avoid the accumulation of final products).
Modification of the cresol chemistry
The second chemical mechanism of toluene oxidation studied is based on the MG mechanism with the addition of the chemistry of cresols and, in particular, the successive addition of alcohol functions (-OH) on the aromatic ring, the fragmentation of the compounds formed into two aliphatic compounds, and the associated kinetics as described in Schwantes et al. (2017). Additional modifications were made to the mechanism proposed in Schwantes et al. (2017) based on MCM and GECKO-A. For example, the first two products of reaction B2-017 (see Table S5) are from MCM, while the others are from Schwantes et al. (2017), and the branching ratios are estimated using GECKO-A. The products of reactions B2-078, B2-084, and B2-090 (see Table S5) are those proposed in Schwantes et al. (2017), and the same branching ratios are taken for each pair of compounds formed due to the lack of more accurate estimations. Hydrogen abstraction of an -OH group and then organic nitrate or peroxyl formation reactions (by addition of NO2 or reaction with O3 and then HO2, respectively) at each oxidation step are added to the mechanism of Schwantes et al. (2017) following kinetics estimated by the SAR implemented in GECKO-A (Jenkin et al., 2019; Platz et al., 1998; Tao et al.,1999) and branching ratios similar to MCM cresol oxidation reaction (see reactions B2-059 to B2-074 in Table S5). Finally, loss reactions are added for all end products of a new reaction pathway. This chemical mechanism is hereafter denoted MG–Cr and has 168 species and 422 reactions (including 110 loss reactions).
Modification of the aliphatic chemistry and other modifications
The third chemical mechanism studied is similar to MG–Cr, with the addition of a new ring-opening reaction pathway for toluene and new pathways for the formation of furan-like compounds based on the work of Jang and Kamens (2001). The new aromatic-ring-opening pathway is added to the first oxidation step (reactions B5-001 to B5-024 in Table S5). It is different from the one leading to the formation of MG mechanism epoxides. In Jang and Kamens (2001), no kinetics and branching ratios are proposed. These are therefore estimated using the SAR implemented in GECKO-A (Jenkin et al., 2018, 2019; Vereecken and Peeters, 2009). Reaction pathways for the formation of 5-atom-ring compounds (4 carbons and one oxygen) are therefore added with kinetics estimated by GECKO-A. Some branching ratios are adjusted in order to reproduce the measurements as well as possible (for the B3-010 reaction in particular).
In parallel to the addition of this reaction pathway, other modifications are considered. A simplified formation of nitro-toluene, an abundant gaseous first-generation product, is added following the parameterization proposed by Atkinson et al. (1989). A formation pathway of highly oxygenated matter (HOM) is added following Wang et al. (2017) (reactions B3-004 to B3-009 in Table S5). The kinetics of the first reaction of the pathway (B3-004) are estimated by taking into account the fraction of biperoxy radicals leading to HOM formation (Wu et al., 2014). In the absence of experimental or theoretical values, the kinetics of the second successive autoxidation reaction (B3-005) are set to 0.5 s−1 (similar to the value proposed in Ehn et al., 2014, for autoxidation during α-pinene oxidation). These additions also lead to the modification of the branching ratios of the first oxidation step of toluene (B0-001). The branching ratios are modified to incorporate the new reaction pathways, to be closer to those proposed in Zaytsev et al. (2019), and to better reproduce the experimental observations. The latter chemical mechanism is denoted MG–Cr–Al hereafter and has 210 species and 493 reactions (including 136 loss reactions).
2.2.2 Partitioning modeling with SSH-aerosol
The chemical mechanisms are implemented in the SSH-aerosol box model (Sartelet et al., 2020). SSH-aerosol is a modular model that represents the formation and evolution of the mass, number, and mixing state of primary and secondary particles with different levels of complexity. It results from the coupling of three models: the Size-Composition Resolved Aerosol Model (SCRAM) (Zhu et al., 2015) for aerosol dynamics, the Secondary Organic Aerosol Processor (SOAP) (Couvidat and Sartelet, 2015) for organic gas–particle partitioning, and the hydrophobic–hydrophilic organics mechanism (H2O) (Couvidat et al., 2012) for the gas-phase chemical mechanism that represents the formation of condensable species. SSH-aerosol models the physicochemical processes driving the evolution of atmospheric particles such as (i) nucleation and coagulation as well as (ii) partitioning of inorganic and organic compounds dynamically or assuming thermodynamic equilibrium. For organics, compounds can partition towards the organic or aqueous phases. The influence of hygroscopicity, viscosity, and nonideality of condensed phases can be taken into account.
SSH-aerosol is used here to represent the partitioning of organic compounds formed following the different chemical mechanisms presented in Sect. 2.2.1. The partitioning is modeled for all stable organic compounds (i) considering an accommodation coefficient equal to 0.7 (close to the value reported by Liu et al., 2019), (ii) by estimating the Psat with the structure–property relationship of Nannoolal et al. (2008), and (iii) by estimating the effective Henry's law constants (Heff) following the universal functional group activity coefficient (UNIFAC) (Fredenslund et al., 1975) method from their Psat.
Nonideality of condensed phases can be taken into account considering (i) interactions between uncharged organic molecules in both organic and aqueous phases only (computed using UNIFAC) or (ii) the combination of interactions between uncharged organic molecules in both organic and aqueous phases as well as interactions of organic molecules with inorganic ions in the aqueous phase (computed using the Aerosol Inorganic–Organic Mixtures Functional groups Activity Coefficients model, AIOMFAC, Zuend et al., 2008). Each organic molecule is automatically associated with a list of UNIFAC functional groups. Due to the high functionalization of several compounds, an exact decomposition in UNIFAC functional groups is not always possible. In those cases, some approximations are made in order to use the decomposition of a similar molecule such as relocating a functional group on a free carbon or adding a carbon to the molecule to take into account every oxygenated group.
A wall loss parameterization of gaseous organic compounds was implemented in SSH-aerosol for the modeled partitioning study of this work. In the absence of wall loss studies in Pyrex OFR, this parameterization follows the approach developed for Teflon chamber studies (Huang et al., 2018; Krechmer et al., 2016; Zhang et al., 2015b). The parameterization represents an irreversible first-order process whose kinetics depend on the saturation concentration of each species. Two parameters are tuned to correctly reproduce the final SOA concentration of the studied case and allow the analysis of wall loss effects on the speciation and distribution of secondary organic compounds. The parameterization details are presented in the Supplement.
A simplified parameterization of the irreversible gas–particle partitioning (due to oxidation in the condensed phase) is tested only for methylglyoxal (as glyoxal was not measured in this study). This parameterization represents irreversible partitioning as dependent on RH. This empiric parameterization is based on effective uptake rates (keff,uptake) calculated with atmospheric observation values in Hu et al. (2022) for high-RH cases (RH > 40 %) and with experimental values in De Haan et al. (2018) for low-RH cases (RH < 5 %). A third-degree polynomial is fitted on experimental keff,uptake to establish the RH dependency used in this study. Polynomial, experimental keff,uptake, and details of the parameterization are presented in the Supplement.
Here modeled Kp is not directly compared to experimental Kp for several reasons: (i) the significant variations and uncertainties in the calculation of the modeled Kp depending on the processes considered, especially nonideality; (ii) the impossibility of calculating Kp for many major compounds of the modeled SOA due to their total transfer in the condensed phase; and (iii) the impossibility of calculating Kp for compounds with experimental Kp but present only in the gas phase in the simulations (i.e., light compounds).
2.2.3 Simulation setup
To model the experimental oxidation conditions, chemical equilibria, and radical concentrations, a modified version of the Regional Atmospheric Chemistry Mechanism version 2 (RACM2) (Goliff et al., 2013) is used. The modifications to RACM2 are described in detail in Lannuque et al. (2021). This new version aims at better representing the evolution of HOX radical concentrations during the oxidation of mono-aromatic species such as toluene and xylenes. The experimental formation of NOX and OH radical is represented by five reactions from the IPN photochemistry, which are added to the mechanism as described in Lannuque et al. (2021). The kinetics of these reactions are based on the work of Fittschen et al. (1999) and Raff and Finlayson-Pitts (2010). The photolysis rate of IPN is calculated considering a quantum yield of 1 based on the absorption spectrum of Raff and Finlayson-Pitts (2010). The heterogeneous reaction of NO2 on the OFR walls is also modeled to regulate the simulated NOX concentrations (Lannuque et al., 2021). The kinetics of this reaction take into account the gas diffusion and the uptake. It is estimated according to the method proposed in Fiorentino et al. (2021) considering an uptake coefficient of 10−5 as recommended in Jacob (2000).
The toluene oxidation mechanisms presented in Sect. 2.2.1 are added in SSH-aerosol to represent the formation of condensable species for SOA formation. The consumption and production of oxidants are neglected in these added mechanisms as they are computed by RACM2. All the photolysis rates (from RACM2, IPN chemistry and toluene oxidation mechanisms) are recalculated considering the irradiation spectrum of the lamps of the experiments.
The simulations were conducted using the SSH-aerosol box model. The temperature and RH were forced by the experimental conditions. The simulation duration was set to 13 min to match the residence time in the OFR. The simulated initial toluene and pre-existing aerosol concentrations corresponded to the experimental measured values at the inlet of the OFR. Since IPN concentration was not monitored, the initial simulated IPN concentration was set to reproduce experimental toluene decay.
Only one size bin was considered (no size distribution), and the nucleation and coagulation processes were ignored. The phase partitioning is represented dynamically considering the transfer to nonideal organic and aqueous phases with only interactions between uncharged organic molecules (computed with UNIFAC). As presented in Sect. 4.2, several tests on partitioning processes were performed considering the transfer (i) to an ideal organic phase only (without interactions between compounds within the phase), (ii) to ideal organic and aqueous phases, and (iii) to nonideal organic and aqueous phases with interactions between uncharged organic molecules only or also with interactions of organic molecules with inorganic ions. Tests taking into account wall losses of the gaseous organic compounds and irreversible partitioning of methylglyoxal are also performed.
The simulation results presented in this study focus on two experiments carried out at 280 and 295 K with 24 % RH, 112 ppbv of toluene, and 120 ppbv of IPN (experiment nos. 21 and 7, respectively, in Table S1). The pre-existing aerosol concentration is 9 µg m−3 of AS, and a small concentration of organic matter (0.01 µg m−3) is also added to the initial concentrations to allow for the formation of organic aerosols in the model.
Nitro-aliphatic and peroxy acyl nitrate (PAN) compounds are not correctly quantified by the PTR-ToF-MS due to fragmentation. A correction is therefore applied to simulated aliphatic nitrates and PAN concentrations to reflect this instrumental feature. Concretely, during post-processing of model results on speciation, we consider these simulated compounds to automatically fragment and lose their -ONO2 or -OONO2 groups (replaced by a hydrogen atom).
3.1 Temperature dependence of SOA formation and yield
SOA yield from toluene was investigated for 24 experiments using different initial conditions of temperature, toluene concentration, OH radical concentration, and ammonium sulfate seeds. Detailed information on experimental conditions and SOA derived by HR-ToF-AMS and CHARON–PTR-ToF-MS can be found in Table S1 in the Supplement. Around 40 % to 60 % of the toluene concentration was consumed during the experiments. Figure 1 compares the calculated SOA yield as a function of the produced SOA mass with previous published works using the data from the HR-ToF-AMS. All experiments were carried out under high-NOX conditions. The red squares refer to experiments carried out at 295 K, the green circles at 285 K, and the blue triangles at 280 K. The empty markers correspond to experiments with a low initial toluene concentration (below 23 ppbv) and the filled markers correspond to a higher initial toluene concentration (above 100 ppbv). Both the calculated SOA yield and measured aerosol concentrations are strongly dependent on the experimental conditions and are consistent with previous studies carried out in smog chambers using higher toluene (200–1000 ppbv) and seed concentrations (Hildebrandt et al., 2009; Ng et al., 2007; Takekawa et al., 2003). For toluene concentrations between 112–162 ppbv SOA yields are located at the lower edge of the Hildebrandt et al. (2009) gray area, determined using basis set parameterizations, and are closer to Ng et al. (2007) yields. One reason might be the yield correction for both organic vapor and particle wall losses performed by Hildebrandt et al. (2009), while Ng et al. (2007) applied wall loss corrections for particles only. Since the residence time into the OFR was below 13 min and seed particles (between 113–169 nm) did not show any significant depletion (below 5 %), we did not correct for particle losses, while between 5 %–8 % of toluene wall losses were measured depending on the flow tube temperature.
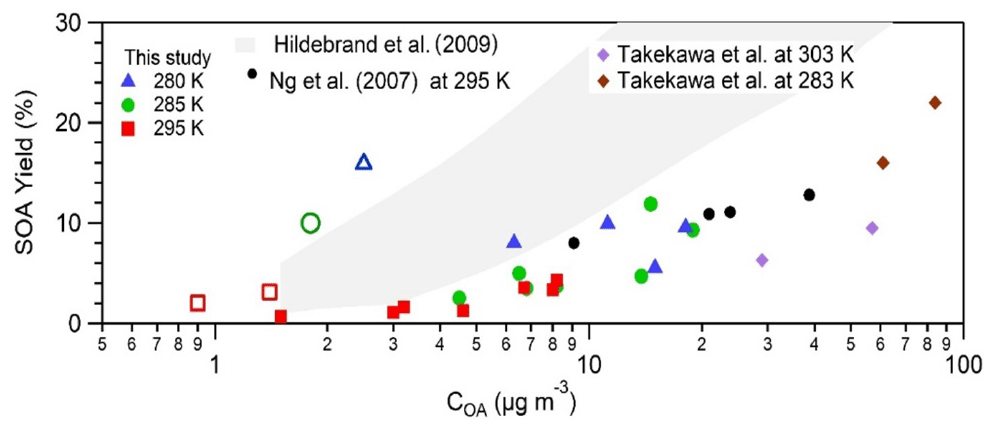
Figure 1SOA yield (measured by HR-ToF-AMS) vs. organic aerosol formed. Red squares refer to 295 K, green circles stand for 285 K, and blue triangles represent data points at 28 K. Empty markers represent experiments with a low toluene concentration (8 and 23 ppbv), and filled markers indicate high-toluene-concentration experiments (above 110 ppbv). Data points from this work are compared with toluene experiments from Ng et al. (2007), Hildebrandt et al. (2009), and Takekawa et al. (2003).
For the same initial reactant concentration, the SOA yield is linearly dependent on the temperature (in the range of temperatures investigated); for toluene concentration above 110 ppbv (filled symbols) a slope of 0.4 can be traced and for toluene concentration below 23 ppbv (empty symbols) a much steeper slope of about 11 can be drawn. The temperature dependence observed in this work is consistent with previous studies (Takekawa et al., 2003) that reported a toluene SOA yield twice as high for a temperature decrease from 303 to 283 K. Also, Hildebrandt et al. (2009) reported a significant increase in toluene SOA yield when the temperature decreased from 305 to 284 K (Fig. 1). Other experiments clearly show the influence of seed (ammonium sulfate) concentration on SOA yield, presented in Fig. S2. An increase in seed concentration from 5 to 10 µg m−3 led to an increment of the SOA yield from 0.8 % to almost 2.3 % at 295 K and from 4 %–10 % at 280 K. These findings are in agreement with previous studies (Ng et al., 2007). The observed behavior can be rationalized by an increased seed surface that favors condensation of toluene semi-volatile oxidation products at lower temperatures, with enhancement of aerosol concentration and SOA yield.
3.2 Experimental speciation of toluene oxidation products
In total 97 significant signals were detected in the gas phase (from 31 to 171), 103 signals were detected in the particle phase (from 47 to 189), and only 65 of them were in common among the two phases. Overall chemical characteristics of toluene reaction products in the gas (PTR-ToF-MS) and particle (CHARON–PTR-ToF-MS) phases are presented in Fig. 2a and b, respectively, for an experiment at 280 K with an initial toluene concentration of 112 ppbv (experiment no. 21, see Table S1). A similar plot for the same experiment at 295 K (experiment no. 7, see Table S1) is presented in Fig. S4 in the Supplement. The overall product distribution is comparable at the two temperatures. The mass fractions (in µg m−3) of the reaction products were further classified depending on their number of oxygen atoms and the molecular weight distribution, and they were divided into four groups: 30–50, 51–100, 101–150, and above 150. The detected gas-phase products account for 31 %–60 % of the toluene carbon balance depending on the oxidation conditions and considering that CO, CO2, and glyoxal were not measured in these experiments, as IPN degradation produces high amount of acetone that interferes with the glyoxal signal. The SOA formed accounted for 1 % and 3 % at 295 and 280 K of the toluene carbon balance, respectively. Additional information on gas and particle chemical composition for six experiments is available in the Supplement (Table S9).
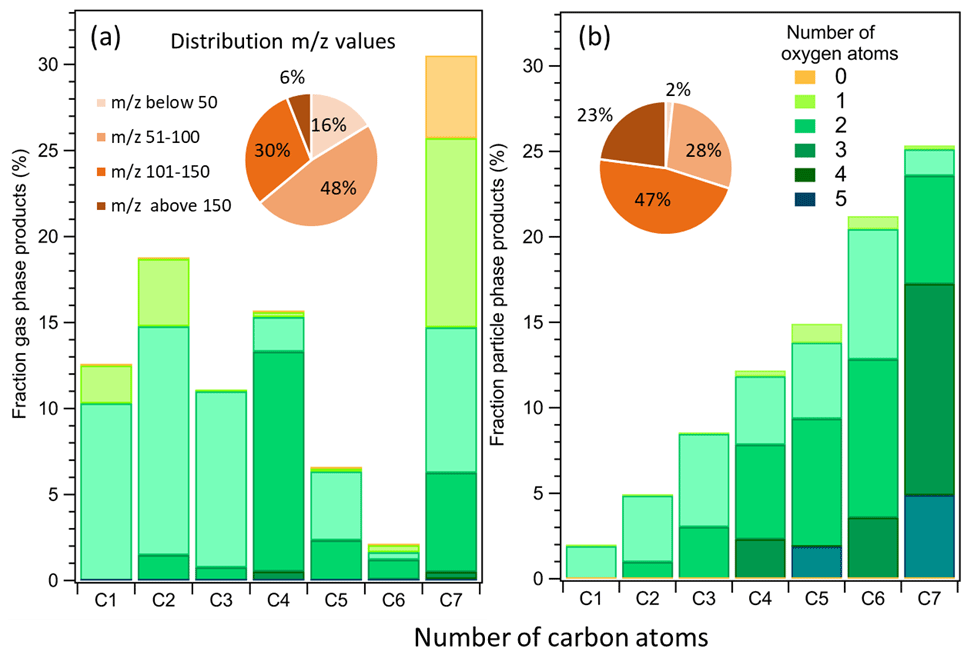
Figure 2Mass product fraction (y axis) distribution based on the number of carbon atoms (x axis) for an experiment with 112 ppbv of toluene at 280 K. Detected ions in the (a) gas phase and (b) particle phase. Pie charts correspond to the molecular weight contribution to the overall mass.
The identified gas-phase products shown in Fig. 2a are characterized by a high fraction of C7 compounds (31 %) followed by C2 (19 %), C4 (16 %), C1(12 %), C3 (11 %), C5 (6 %), and C6 (2 %). The product distribution in the particle phase (Fig. 2b) exhibits a different trend with increasing mass fraction from C1 (2 %), C2 (5 %), C3 (9 %), C4 (12 %), C5 (15 %), C6 (21 %), and finally C7 (26 %). The condensed phase is generally more oxygenated. More than half of the molecules contain 3 to 5 oxygen atoms and show higher molecular weight (pie chart in Fig. 2b) with respect to the gas-phase product distribution.
Table 1 lists the measured signals, the assigned ion formula, and a tentative assignment to the reaction products. The tentative chemical assignment is based on the results of a consistent number of dedicated studies (Bloss et al., 2005; Borrás and Tortajada-Genaro, 2012; Forstner et al., 1997; Hamilton et al., 2005; Jang and Kamens, 2001; Kleindienst et al., 2004; Schwantes et al., 2017; White et al., 2014; Wu et al., 2014; Zaytsev et al., 2019) and the CHARON assignment procedure recommended by Gkatzelis et al. (2018a), where average saturation mass concentration log(C) is used to discriminate between parent and fragment ions (see Fig. S3 in the Supplement). Furthermore, some reference compounds detected by CHARON–PTR-ToF-MS are listed in Table S2 in the Supplement.
Table 1List of ion sum formulas of the major toluene degradation products monitored during the experiments. Reaction products are given as a percent of the total carbon of toluene consumed (carbon yield), as a fraction of the gas-phase products (in % of µg m−3), and as a fraction of the SOA products (% of µg m−3).
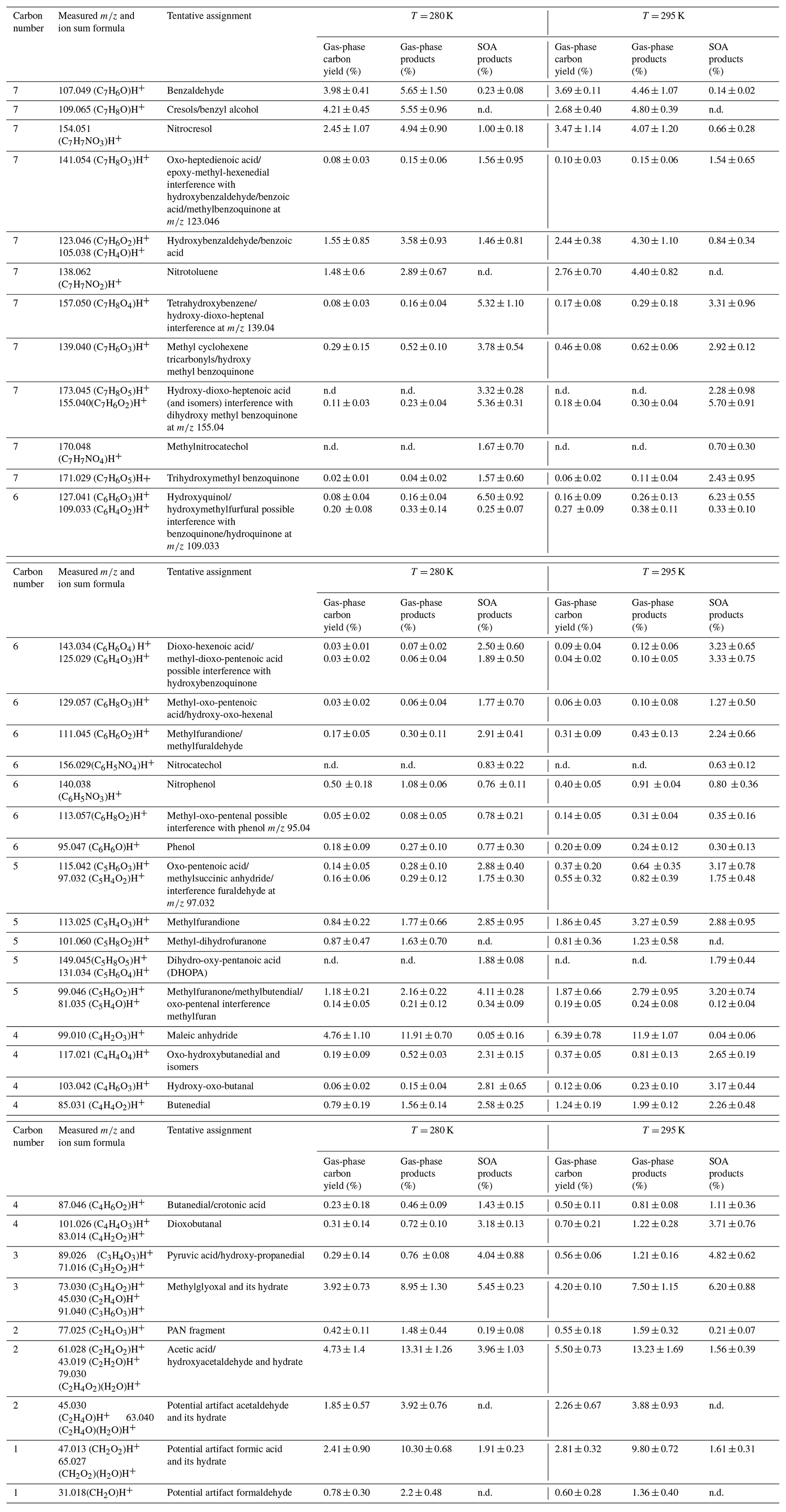
In Table 1 the compounds are classified with respect to the decreasing number of carbon atoms for two sets of experiments with an initial toluene concentration of 112 ppbv at 280 and 295 K. For each temperature three columns are presented: the first indicates the gas-phase carbon balance (% of ppbC with respect to the toluene consumed), the second one presents the fraction of gas-phase reaction products (% of µg m−3), and the third presents the fraction of the SOA products (% of µg m−3). The C7 compounds account for approximately 31 % of the total gas-phase products. The most abundant C7 ions are associated with ring-retaining compounds and could be tentatively assigned to benzaldehyde ( 107.049), cresols and isomers ( 109.065, 91.057), or benzoic acid and isomers ( 123.046), but also nitro-compounds such as nitrocresol ( 154.051 and 136.039) and nitrotoluene ( 138.057). These results are in very good agreement with previous works listed above. The C7 particle-phase compounds are the most abundant, accounting for 25 % of the SOA mass fraction. The major identified ions contain several oxygen atoms associated with the following ion formulas: (C7H6O4)H+ at 155.034, (C7H8O4)H+ at 157.050, (C7H6O3)H+ at 139.044, (C7H8O3)H+ at 141.054, (C7H6O5)H+ at 171.029, and (C7H8O5)H+ at 173.044. The highest number of carbon atoms observed in the particle phase is 7. Oligomers were therefore not observed as the operating conditions used in this study ( = 105 Td) might induce oligomer fragmentation upon protonation.
The C6 carbon compounds account for 2 % only of the gas-phase products but reach 21 % of the organic fraction in the condensed phase. Major C6 ions found are (C6H6O3)H+ at 127.041, (C6H6O4)H+ at 143.034, and (C6H4O3)H+ at 125.029, tentatively assigned to hydroxyquinol, hydroxymethylfurfural, and functionalized acids. Other less abundant C6 ions found are (C6H6O2)H+ at 111.04, (C6H8O2)H+ at 113.06, (C6H6O)H+ at 97.064, and (C6H8O3)H+ at 129.057 (Table 1). Among the C7 and C6 compounds several nitro-aromatic compounds have been identified (nitrotoluene, nitrocresol, methylnitrocatechol, and nitrophenol); these nitro-aromatic compounds account for 13 % and 4 % of the gas- and particle-phase products, respectively, in agreement with previous studies (Sato et al., 2007; Tuazon et al., 1984; Zaytsev et al., 2019).
The C1–C5 compounds correspond to products formed upon cleavage of the aromatic ring. Most of the C3–C5 products are associated with nonaromatic ring-retaining compounds as functionalized furans and anhydrides or ring-opening products (oxocarboxylic acids, dicarbonyl, or oxo-carbonyl compounds) (Hamilton et al., 2005; Jang and Kamens, 2001; Zaytsev et al., 2019). The C5 compounds represent 7 % and 15 % of the gas- and particle-phase reaction products, respectively. The most important ions in the particle phase are (C5H6O3)H+ at 115.042, (C5H4O2)H+ at 97.032, (C5H6O2)H+ at 99.046, and (C5H4O3)H at 113.025. See Table 1 for tentative assignment.
The C4 compounds represent 16 % and 12 % of the gaseous and particulate phase products, respectively. A far major ion fragment is detected at 99.010, assigned to maleic anhydride, that alone represents more than 12 % of the gas-phase products, in agreement with several previous studies (Hamilton et al., 2005; Jang and Kamens 2001). In the condensed phase other identified ions are (C4H6O3)H+at 103.042, (C4H4O2)H+at 85.031, (C4H6O2)H+at 87.046, (C4H4O3)H+at 101.026, and (C4H4O4)H+ at 117.021.
The C3 compounds represent approximately 11 % and 9 % of the gaseous- and particulate-phase products, respectively. Methylglyoxal and its hydrate are among the most important reaction products in both phases. Other ions at 89.026 and 71.016 assigned to pyruvic acid (White et al., 2014) are mostly present in the particle phase. Based on previous works, glyoxal molar yield under medium- and high-NOX conditions would vary between 8 %–20 % as a function of the NOX concentration (Nishino et al., 2010; Tuazon et al., 1984), but during our experiment it could not be detected due to interference with acetone produced upon photolysis of IPN.
Thermal decomposition such as the decarboxylation of organic acids does not occur at the temperatures used in this study (Piel et al., 2021). This is also confirmed by the ion distribution of formic and succinic acid in Table S2. Piel et al. (2021) found evidence of the thermal decomposition in the CHARON inlet of other compounds as hydroperoxides. But previous works (Gkatzelis et al., 2018a; Leglise et al., 2019) show that at the conditions used in this study ( = 105 Td), scission of the C–C bond may occur. Therefore, a minor fraction of the low-C compounds found in the particle phase could thus be measurement artifacts. Acetic acid–hydroxyacetaldehyde at 61.028 and formic acid at 47.013 are found both in the gas and particle phase; both can be produced during toluene degradation and be surface artifacts due to heterogeneous reactions. Small aldehydes such as acetaldehyde and formaldehyde can also be surface artifacts, as adsorbed molecules on OFR walls can undergo chemical reaction and produce such small molecules (Doussin et al., 2023). Aliphatic nitrogen-containing compounds are not easily detected due to fragmentation in the PTR-MS. Nevertheless the fragment ion at 77.024 (C2H3O)H+ previously assigned to the peroxy acetyl nitrate fragment has been detected (Müller et al., 2012).
The particle-phase mass loading recovered by CHARON–PTR-ToF-MS accounts for approximately 77 % of the mass measured by the HR-ToF-AMS (Fig. S5.a) when considering all 24 experiments (Table S1). Part of this discrepancy can be explained by the loss of water occurring in the PTR-ToF-MS technique (see Table S2) but also from the different size cut-off of the two instruments inlets. This hypothesis is confirmed in Fig. S5b, where CHARON–PTR-ToF-MS mass recovery decreases to 55 % of that observed by HR-ToF-AMS when only particles below 150 nm (mobility diameter) are considered.
3.3 Experimentally derived gas–particle phase partitioning
Figure 3 depicts the oxidation state (OSc) of the identified major ion fragments i versus the log at 295 K (magenta) and 280 K (light violet). The circles are proportional to the mass associated with each ion. The error bars indicate the standard deviation of repeated experiments. The 2D-VBS (Donahue et al., 2011; Murphy et al., 2012) framework was used to implement the results, with background colors corresponding to the different volatility classes, ranging from intermediate-volatility organic compounds (IVOCs, light blue) to semi-volatile organic compounds (SVOCs, light green). The OSc is derived from the Kroll et al. (2011) method for each individual species based on the number of carbon, hydrogen, and oxygen atoms. In total 103 ions were detected in the particulate phases; among them 29 were classified as parent ions and partitioned between the gas and particle phases. Table S3 (the Supplement) lists log calculated at both 280 and 295 K for the identified parent ions. A total of 20 other ions were detected in the particle phase, only accounting for 10 %–17 % of the SOA mass. The remaining 62 %–71 % of the particle mass occupied the SVOC area, and approximately 12 %–19 % were in the IVOC area. A shift of the experimental temperature from 295 to 280 K induced an important increase in the total organic aerosol mass (from 8 to 15.7 µg m−3) since a higher fraction of the oxidation products was able to condense onto the particulate phase. This can be observed in Fig. 3 where the volatility of the individual parent ions appreciably shifts toward lower log values with Δlog varying from 0.29 to 1.07 (Table S3).
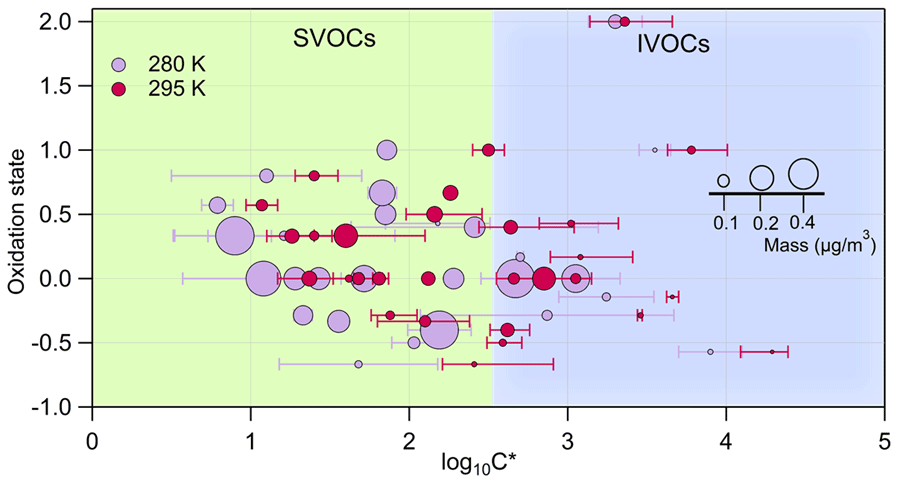
Figure 3The oxidation state for the detected parent ions (OSc) versus the averaged saturation concentration in terms of log. The area of the dots denotes the mass associated with each species. In light violet the ion fragments identified for an experiment carried out at 280 K are represented, and in magenta are those at 295 K. Errors bars correspond to ± 1σ of the experimental average.
The saturation concentration () values ranged between 3 and 31 000 µg m−3, exhibiting similar values with respect to those observed for other SOA systems (Gkatzelis et al., 2018b). Previous findings suggested a volatility decrease with increasing OSc and oxygen atom number (Gkatzelis et al., 2018b; Jimenez et al., 2009; Kroll et al., 2011). In this work, however, such a trend is not observed (see also Fig. S6 in the Supplement) as it was not for SOA formed from tree emissions (Gkatzelis et al., 2018b), suggesting that a higher oxidation state may not always be the key parameter for explaining volatility (Kostenidou et al., 2018). Additionally, in this study we report volatility properties for all ions detected, while previous works considered only compounds with more than 5 carbon atoms.
In Fig. 4, the volatility distribution of the toluene SOA is compared to a previous study (Hildebrandt et al., 2009). CHARON data are categorized in volatility bins in order to be comparable to the above work. Our results indicate that our products were generally in good agreement with those extracted by Hildebrandt et al. (2009) applying the volatility basis set parameterization for four products to chamber experiments at 293 K and in low-NOx conditions. However, our volatility distribution is shifted somehow to lower-volatility products as we observed a higher mass fraction of compounds with saturation concentration 100 µg m−3 (log) and a lower contribution of species with saturation concentration 1000 µg m−3 (log) in comparison to Hildebrandt et al. (2009). One explanation for this behavior could be the lower initial toluene concentrations (105 ± 49 ppb) used in this study compared to those used in Hildebrandt et al. (2009) (362 ± 176 ppb), resulting in a higher fraction of less volatile species. The sum of the volatility bins mass fraction in Fig. 4 is less than unity since approximately 12 %–15 % of the mass was detected in the particle phase only and it has not been included in these bins.
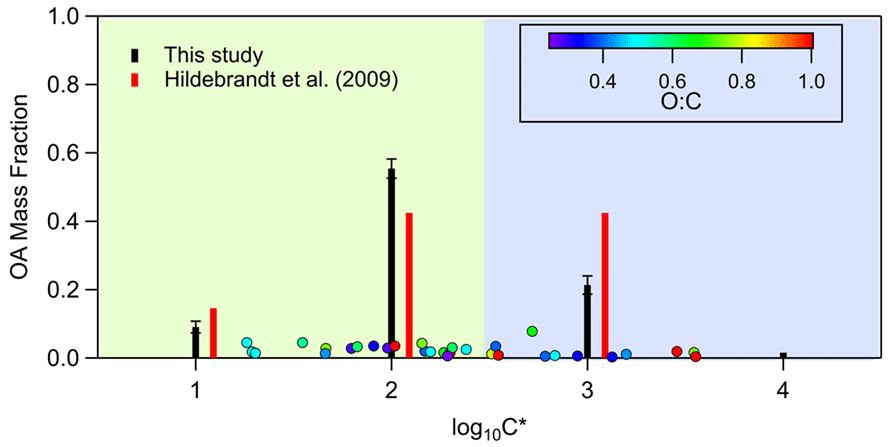
Figure 4Saturation concentration (shown as volatility distribution) of the toluene SOA species. The black bars correspond to the categorization in volatility bins of this work, while the red bars correspond to the volatility distribution derived from Hildebrandt et al. (2009) applying the volatility basis set parameterization for four products to chamber experiments (20 ∘C, low NOx).
4.1 Modeling of products chemical composition
Table 2 shows a comparison between the final experimental concentrations at the outlet of the OFR and the simulated concentrations estimated with the different gas-phase chemical mechanisms for the experiments with 24 % RH and 112 ppbv of toluene at 280 and 295 K. In these simulations partitioning to nonideal organic and aqueous condensed phases (with only interactions between uncharged organic molecules estimated with UNIFAC) is considered and no wall loss process is represented. The normalized root mean square error (NRMSE) for each mechanism is calculated as
where and are the mass concentrations of each simulated and experimental i, respectively, and n is the number of values for which the concentrations are nonzero according to both experiments and modeling. For comparison, experimental values are rounded to the unit. At 280 K (295 K), about 81 (88) rounded experimental values are measured. We observe an important variation of the total mass formed (gas + particle) with the temperature during the experiments (139.9 µg m−3 at 280 K and 185.4 µg m−3 at 295 K: +32.5 %) not reproduced by the model, which overestimates the mass at 280 K and underestimates it at 295 K. The mechanism modifications lead to a decrease in the simulated total mass of about 3 % and 11 % whatever the temperature with MG–Cr and MG–Cr–Al, respectively, which is mainly due to the increasing importance of fragmentation pathways compared to functionalization ones. The modifications of the chemical mechanism improved the number of represented experimental values: 31, 35, and 42 (33, 37, and 44) with MG, MG–Cr, and MG–Cr–Al, respectively. The better reproduction of the experimental by the modified mechanisms results in a decrease in the NRMSE, which is 2.74, 2.23, and 1.89 for MG, MG–Cr, and MG–Cr–Al, respectively, at 280 K (2.13, 1.81, and 1.62 at 295 K). However, this NRMSE remains very high.
Table 2Number of identified values, total mass, and NRMSE for the three mechanisms and experiments.
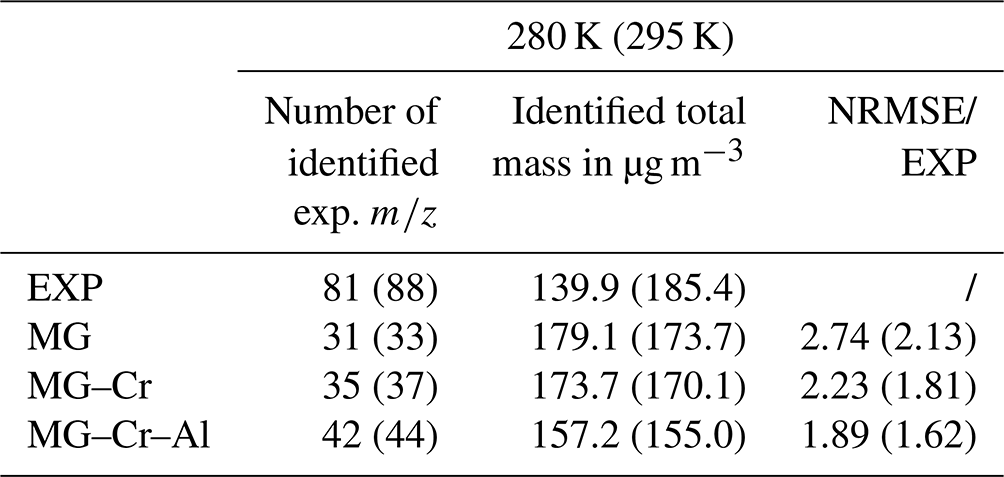
Figure 5 shows the chemical characteristics of simulated toluene reaction products at 280 K in both the gaseous and particulate phases. Simulated glyoxal is not considered, and the correction for nitro-products is applied. Similarly to the experimental results (see Fig. 2), the distributions of the simulated compounds in the gas phase is dominated by C7 compounds, but their mass fraction is higher (∼ 39 % for MG and MG–Cr and ∼ 43 % for MG–Cr–Al) than the measured fraction (31 %). Although the three mechanisms correctly reproduce the low fraction of C6 in the gas phase, they all underestimate the C2 gas-phase fraction (∼ 10 % for MG and MG–Cr and ∼ 13 % for MG–Cr–Al). With all three mechanisms, the gas-phase fraction of C5 is the second-most important (∼ 25 %), much higher than the measured value (7 %). The gas-phase fraction of C1 is underestimated. Although there is little difference in the gas-phase distribution of compounds between the MG and MG–Cr mechanisms, the MG–Cr–Al mechanism introduces some differences: (i) a more important C7 gas-phase fraction (+5 %), (ii) C1 is no longer negligible (∼ 3 % due to the higher formation of formaldehyde), and (iii) the gas-phase fraction of C2 is proportionally higher due to the higher formation of acetaldehyde. In terms of compound oxidation, the main difference between the experimental and simulated gas-phase distribution is the formation of more oxidized C7 compounds (∼ 40 % of the gaseous C7 has 4 or 5 oxygen atoms).
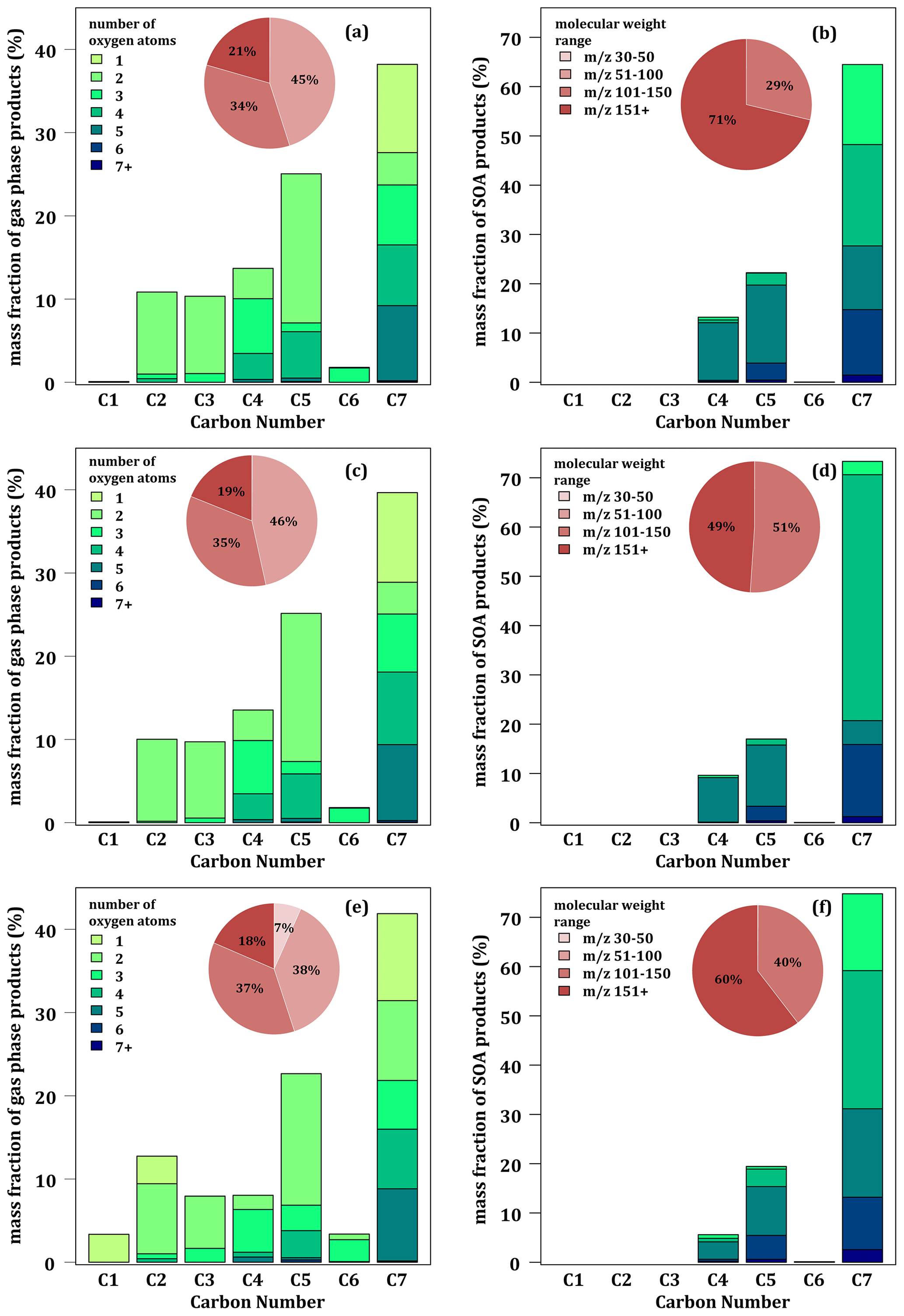
Figure 5Simulated mass product fraction (y axis) distribution based on the number of carbon atoms (x axis) for the oxidation of 112 ppbv of toluene at 24 % RH and 280 K with MG (a, b), MG–Cr (c, d), and MG–Cr–Al (e, f) mechanisms in the gas (a, c, e) and condensed phases (b, d, f). Pie charts correspond to the molecular weight contribution to the overall mass.
In the condensed phase, the simulated distribution of compounds by carbon number strongly differs from measured ones. First, the simulations do not reproduce the “staircase” distribution observed experimentally, regardless of the mechanism used. According to the simulations, the SOA is composed of a large majority of C7 (∼ 64 % with MG–Cr and ∼ 73 % with MG and MG–Cr–Al), followed by C5 (17 % and 22 %) and C4 (7 % and 14 %). The other compounds (C6, C3, C2, and C1) represent less than 1 % of the mass of SOA formed. The simulated SOA is more oxidized than the experimentally observed SOA. It is mostly composed of molecules with 4 or more oxygen atoms (taking into account the breakup of aliphatic nitro-compounds), and only a fraction of C7 has just 3 oxygens. The large fraction of C7 with 4 oxygen atoms simulated with the MG mechanism is due to the accumulation of methylnitrocatechol. The near absence of C6 in the simulated condensed phase can be explained by the low formation of these compounds in the gas phase, while the absence of short-carbon-chain compounds (C1 to C3) could be mainly explained by partitioning processes implemented in the model: these compounds have Psat and Heff too high and low, respectively, to condense according to Raoult's or Henry's laws as implemented in the SSH-aerosol model under the low RH conditions of simulation.
The simulated distributions indicate that (i) the MG and MG–Cr mechanisms overestimate the gaseous formation of heavy compounds and underestimate the formation of light ones (0 % of the total mass with ≤ 50 against 17 % experimentally), (ii) the MG–Cr–Al mechanism improves the representation of ≤ 50 but mainly at the expense of the fraction with between 50 and 100, and (iii) there is an overrepresentation of heavy compounds in the condensed phase ( > 150 representing between 48.9 % and 71.2 % of the SOA mass against 23 % experimentally) compared to the lighter ones ( < 100 representing less than 0.1 % against 30 % experimentally).
Figure 6 represents the experimental mass spectra for the gas and condensed phases (Fig. 6a) and the reconstructions of similar spectra from the simulated concentrations with the three mechanisms (Fig. 6b to d) at 280 K. Glyoxal is not represented in Fig. 6. Species formed according to the MG mechanism are less numerous and heavier than those observed in the experiment, with only three significant peaks of species with below 99 and none below 61 (Fig. 6b). In contrast, there is a strong accumulation of species with above 140 (up to > 250) in both the gas and the condensed phases. A very high peak in the gas phase is observed at 99. It consists of more than 50 % of aliphatic nitro-compounds (with one or two nitrate or PAN functions) of higher from which the nitro-groups have been subtracted to represent the effect of PTR-ToF-MS on these compounds. Similarly, the peak at 61, normally corresponding to acetic acid, consists almost exclusively of the 2-carbon PAN here, from which -OONO2 has been removed. Finally, with MG, an important accumulation of methylnitrocatechol at 170 (AR0128, see Table S4 in the Supplement) is observed in the condensed phase (23 µg m−3). The methylnitrocatechol strong formation originates from the MCM, and it has already been reported in other simulation studies, which estimated this species as the major contributor to the SOA of toluene (Kelly et al., 2010), in disagreement with our experimental observations.
The modification of the cresol chemistry in the MG–Cr mechanism corrects the overestimation of methylnitrocatechol whose formation is now competing with alternative oxidation pathways. Its concentration in the condensed phase is thus divided by about 10 (∼ 2.5 µg m−3). However, new compounds accumulate in the condensed phase, namely methylbenzenes with 3-, 4-, and 5-OH groups on the ring, corresponding to 141, 157, and 170 with concentrations of 6.5, 4.3, and 1.6 µg m−3, respectively. This version of the mechanism does not improve the representation of light compounds, which remain largely underestimated.
The latest modifications and the addition of a new ring-opening chemistry in the MG–Cr–Al mechanism lead to a clear improvement in the representation of light compounds with (i) enhanced formation, in particular of formaldehyde ( 31) and acetaldehyde ( 45), and (ii) reduction of 99 species. This mechanism leads to a decrease in large compounds able to partition to the condensed phase with a consequent decrease in the SOA mass formed. The modifications lead to a significant improvement in the representation of the gas-phase products with the MG–Cr and then MG–Cr–Al mechanisms. Despite some important improvement, all the three mechanisms show some limitation in reproducing experimental light compound concentrations, while they tend to overestimate the formation of heavy compounds (above 151) in the gas phase. Additionally, for all three mechanisms we observe the absence of compounds with < 130 in the simulated SOA, while they represent the largest fraction according to the experimental results of other recent studies on toluene-derived SOA (Zaytsev et al., 2019). This discrepancy between model and experimental results can be tentatively explained by (i) the absence of some reaction products in the gas-phase oxidation mechanisms or (ii) unaccounted for condensation processes of many gas-phase products in the common version of the model such as irreversible gas–particle partitioning processes of α-dicarbonyl compounds and small acids (Hu et al., 2022). Even though the MG–Cr–Al mechanism leads to a better representation of light compounds in the gas phase, it does not result in better agreement regarding the distribution by carbon number in the condensed phase, suggesting that the main problem is the representation of products partitioning in the model. Several tests on the partitioning are presented in the next section in an effort to provide better insight into the process.
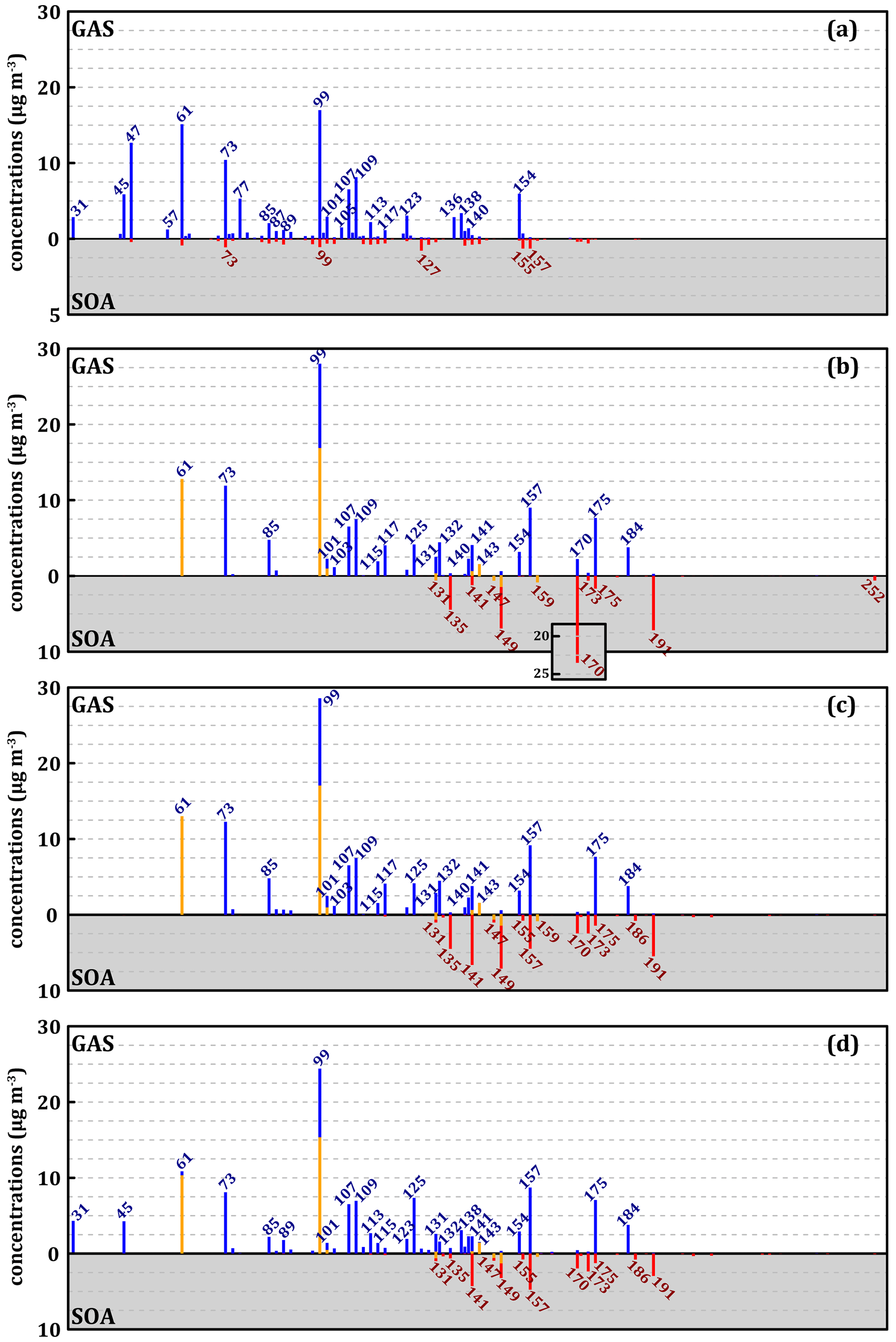
Figure 6Mass spectra of gaseous (blue) and condensed (red) secondary compounds during the oxidation of 112 ppbv of toluene at 280 K according to experiments with the (a), MG (b), MG–Cr (c), and MG–Cr–Al (d) mechanisms. The yellow fractions of spectra represent simulated mass after recalculation by removing functional groups with nitrogen for aliphatic compounds. The scale of the experimental SOA (a) is modified for better readability of concentrations.
4.2 Partitioning modeling and tests on partitioning processes
To explore the partitioning of organic compounds and to try to improve the representation of the experimental observations, different tests on the partitioning processes have been carried out. In this section the formation of SOA is studied using the MG–Cr–Al chemical mechanism to represent the formation of secondary condensable species. The partitioning of these species between the gaseous and condensed phases is simulated under the same experimental conditions as presented in Sect. 2.2.3 at 280 and 295 K by considering different physicochemical processes impacting it. The studied cases are the following.
-
T1: partitioning to an ideal organic phase
-
T2: T1 conditions + partitioning to an ideal aqueous phase
-
T3: partitioning to nonideal organic and aqueous phases, considering the interactions between the uncharged organic molecules in both condensed phases (according to the UNIFAC method)
-
T4: T3 conditions + consideration interactions of organic molecules with inorganic ions in the aqueous phase (according to the AIOMFAC method)
-
T5: T4 conditions + consideration of the loss at the walls of the OFR of gaseous organic compounds according to the parameterization presented in the Supplement
-
T6: T5 conditions + consideration of an irreversible pathway for methylglyoxal condensation according to the parameterization presented in the Supplement
Figure 7 represents the temporal evolution of the concentrations of SOA formed during 13 min of toluene oxidation for the six test cases as well as the final experimental concentration measured by the HR-ToF-AMS at 280 K. In addition, results at 295 K are presented in Fig. S10 in the Supplement, and reconstructions of mass spectra at 280 K with the simulated concentrations for the test cases are presented in Figs. S8 and S9. For partitioning to an ideal organic phase (T1), driven by Raoult's law only and thus depending on the Psat of the compounds, the SOA formation is very slow and reaches ∼ 3 µg m−3 after 13 min. This is due to the small amount of pre-existing particulate organic matter (0.01 µg m−3) onto which the compounds can condense (inorganic seeds). The addition of partitioning toward an aqueous phase, also ideal (T2), following Henry's law and thus depending on the Heff of the compounds accelerates SOA formation. In this case SOA mass concentration reaches ∼ 19 µg m−3 after 13 min, closer to the mass concentration measured experimentally (15.7 µg m−3). SOA formation is enhanced here by the presence of inorganic ions in the pre-existing condensed phase (NH and SO). Considering the interactions between organic compounds within the particulate phase according to the UNIFAC method (T3) leads to an overestimation of SOA formation (∼ 30 µg m−3) by a factor of 2 compared to the experimental value. Considering the interactions of organic molecules with inorganic ions in the aqueous phase (according to AIOMFAC) and the interactions between uncharged organic molecules in the both particulate phases (T4) leads to the formation of SOA mass concentration (∼ 21 µg m−3) closer to the experimental value. The interactions between organic compounds in the condensed phase tend to shift the equilibria towards the particulate phase (reflecting an affinity of the compounds for each other), while the interactions between organic and inorganics compounds result in the opposite effect. Even though these two processes compensate for each other, there is still a relatively high error (+31 %) between the simulated and the experimental total SOA mass concentration.
Even though T2 and T4 final SOA concentrations are close, the modeled mass spectra show significant differences in composition (see Fig. S8). The interactions between organic molecules favor the condensation of many compounds (, 155, 157, 170, 175, or 191, for example) and particularly the lighter ones (, 132, 135). This also reduces the accumulation of the 141 compounds. The interactions between organic and inorganic compounds in the aqueous phase modify the SOA speciation by limiting the partitioning of several chemical species (, 157, 170, 175, and 191 in particular) and favoring the uptake of others (, 186, or 202). The result is a condensed phase composed of a greater variety of compounds covering a wider range of values.
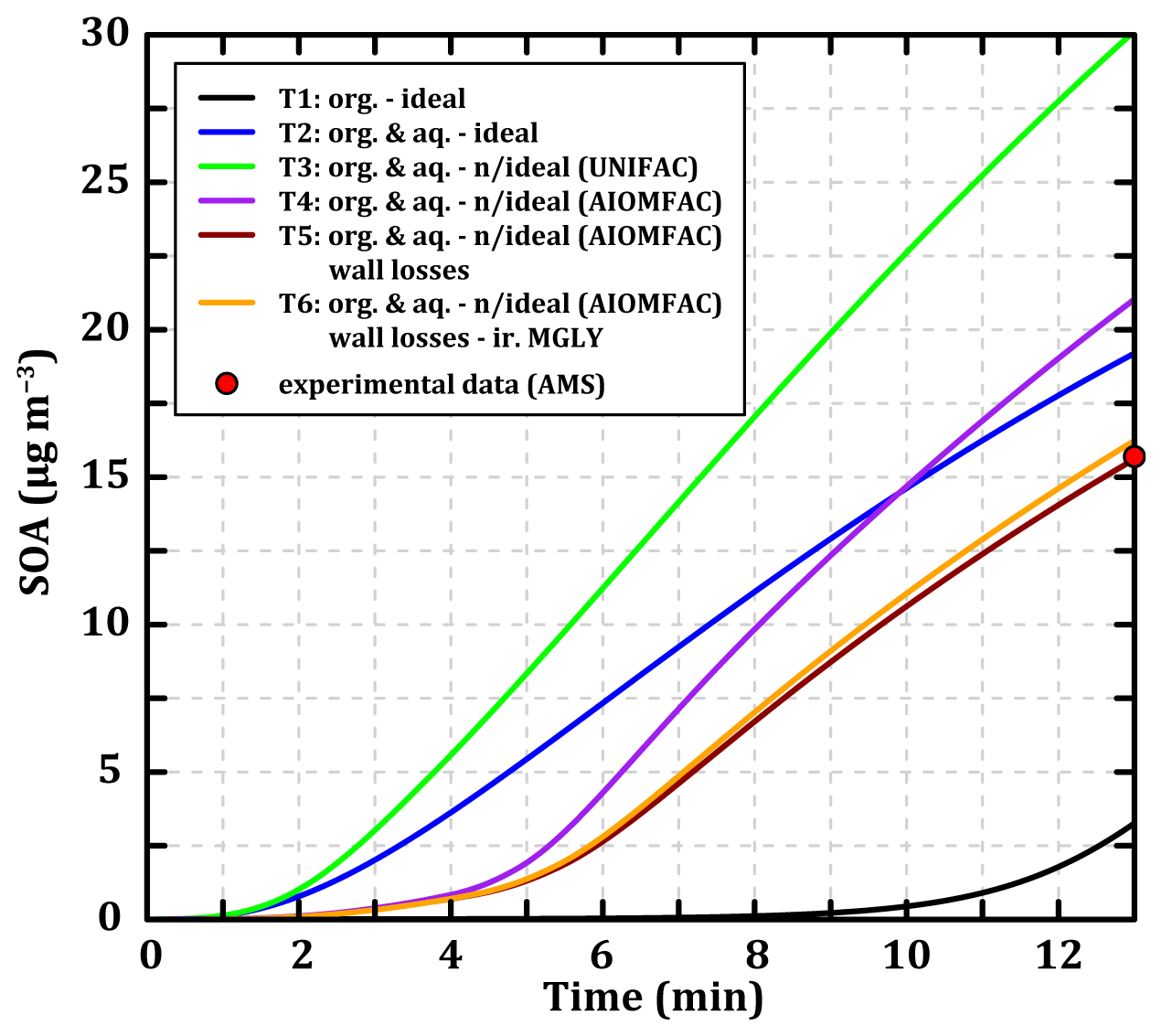
Figure 7Temporal evolution of the SOA concentrations during toluene oxidation under experimental conditions (see Sect. 2.2.3) simulated with MG–Cr–Al mechanism at 280 K considering mass transfer through an ideal organic phase (T1, black line), ideal organic and aqueous phases (T2, blue line), organic and aqueous phases with interactions between organic molecules (T3, green line), organic and aqueous phases with interactions between organic molecules and inorganic ions (T4, purple line), organic and aqueous phases with interactions between organic molecules and inorganic ions and wall losses (T5, dark red line), and organic and aqueous phases with interactions between organic molecules and inorganic ions, wall losses, and an irreversible pathway for methylglyoxal condensation (T6, orange line). The red point represents the experimental SOA concentration measured by AMS.
The T5 test involves adding a wall loss parameterization for gaseous organic compounds as described in Sect. 2.2.2. These -dependent wall losses logically lead to a better reproduction of the SOA concentration (∼ 15.7 µg m−3) as the parameters have been optimized. However, it is interesting to analyze the effect of the wall loss parameterization on the mass spectra of the secondary compounds (see Fig. S8). For the gas phase, we observe slight decreases in the concentrations of some heavy compounds (as , 175, 170, and 157, for example) and also of some lighter ones ( and 99) due to the loss of heavy compounds before their fragmentation in the PTR-ToF-MS (indicated in yellow in Fig. S8). Regarding the particulate phase, the parameterization reduces the concentrations of all the simulated values proportionally to their mass, meaning that all the condensed compounds have low enough volatility to undergo partial wall losses. Finally, the parameterization results in a better representation of the condensed fraction without significantly affecting the representation of the gas phase.
T1 to T5 simulations at 295 K show the same behavior for the SOA mass evolution depending on the considered processes (see Fig. S10). Even if wall loss parameterization was optimized on the 280 K case, The T5 test reproduces the experimental SOA mass well at 295 K (with 8.7 µg m−3 simulated versus 8.2 µg m−3 measured by the HR-Tof-AMS, see Fig. S10).
Regardless of the considered partitioning processes, the model fails to represent the experimentally observed partitioning of light compounds. In the T6 test, an irreversible gas–particle partitioning parameterization for methylglyoxal based on atmospheric observations by Hu et al. (2022) and chamber experiments by De Haan et al. (2018) is implemented to the SSH-aerosol model in an attempt to reproduce the experimentally observed partitioning of this compound (see Sect. 2.2.2). The mass spectrum of the simulation at 280 K considering all the processes tested in this section and adding this new parameterization is presented in Fig. S9 in the Supplement. The addition of the irreversible partitioning at 280 K leads to the condensation of about 0.49 µg m−3 of the simulated methylglyoxal, which is of the same order of magnitude as the experimental value (∼ 0.56 µg m−3) despite an underestimation of the gaseous methylglyoxal mass concentration (∼ 7.5 µg m−3 simulated versus ∼ 10.4 µg m−3 experimentally observed). This increase in the condensed mass concentration of methylglyoxal logically affects the simulated mass concentration of SOA (∼ 16.2 µg m−3). At 295 K, the irreversible partitioning leads to the condensation of about 0.47 µg m−3 of the simulated methylglyoxal, which is higher than the measured value (∼ 0.30 µg m−3). Experimental and simulated values are still of the same order of magnitude with a higher error at 295 K (+56 % vs. −12.5 % at 280 K). The low variation of the condensed methylglyoxal with the temperature variation shows the limits of the simplified parameterization only based on methylglyoxal concentration and RH (here fixed at 24 % for both temperatures).
The objective of the modeling exercise was to reproduce the experimentally observed SOA masses for the right reasons supported by molecular information. However, by making the chemistry representation more complex, it became necessary to increase the complexity of the partitioning processes as well. In this study, neglecting the aqueous partitioning leads to SOA underestimation because of the quasi-absence of a pre-existing organic phase and because compounds are too volatile to condense on an organic phase. Considering organic compound interactions in the particulate phase leads to a shift of equilibria towards the particulate phase and thus favors the condensation of molecules. In the simulation at 280 K, this increases the SOA mass formed by ∼ 50 % (+11 µg m−3) compared to the ideal case. Contrariwise, the interactions between inorganic ions and organic compounds in the aqueous phase limit the condensation of gaseous organic species, therefore reducing the estimated SOA mass concentration by ∼ 33 % (−9 µg m−3) compared to the case in which only organic interactions are considered. All these tests highlight the large uncertainties in SOA formation related to the representation of phase transfer processes independently of the gaseous oxidation mechanism. Whether or not the different processes are taken into account has a considerable effect on the simulated SOA mass. This work therefore raises questions about the significance of the results and the representativeness of (i) simplified models that do not include them, (ii) models that include only some of them with the risk of introducing biases in the results, or (iii) more complex models that include as many processes as possible, increasing the potential sources of error and the resources needed to resolve them. This study underlines the need to develop semi-explicit mechanisms, such as those presented by Wang et al. (2022), preserving the SOA properties in terms of solubility and interactions with organics and inorganics.
This work focuses on the experimental and modeling study of toluene SOA formation upon OH radical reaction. The calculated SOA yield and measured aerosol concentrations were strongly dependent on the experimental conditions (temperature, seed concentration, and toluene initial concentrations) and agreed with previous works. The CHARON inlet coupled to a PTR-ToF-MS successfully quantified approximately 77 % of the total organic mass measured from an aerosol mass spectrometer (HR-ToF-AMS). Despite possible fragmentation of the measured compounds, the major products both in the gas and particulate phases were confirmed through intercomparison with the literature. The partitioning properties of the identified reaction products were determined. Approximately 80 %–85 % of the organic aerosol fraction detected by CHARON showed volatility distributions, expressed as saturation mass concentration, logCi*, ranging from 0.65 to 5.30, and another 10 %–17 % was detected in the condensed phase only. Temperature variation from 295 to 280 K induced an appreciable decrease in the compound's volatility with a Δlog varying from 0.28 to 1.07. Parent ions were mapped onto the 2D-VBS framework, and results did not exhibit a clear correlation with increasing oxidation state or oxygen atom number.
To reproduce the observed SOA mass loading and composition, a new semi-detailed chemical mechanism for the gaseous oxidation of toluene has been developed to reproduce the experimentally observed speciation of secondary compounds in the gas phase and SOA. It is based on an explicit reference mechanism using MCM for the aromatic chemistry and GECKO-A for the aliphatic chemistry. This reference has been simplified and modified on the basis of the experimental data and previous works on cresol chemistry, furan formation and aromatic-ring-opening chemistry, formation of HOM, and SAR of GECKO-A to estimate missing kinetic data. All these modifications improved the representativeness of the chemical mechanism with +35 % of major secondary compounds identified across all phases (number of species) and an error in the total secondary organic mass reduced by ∼ 55 %. At the molecular scale, this translates into a better representation of the formation of light compounds (i.e., < 100) and lower concentration of larger compounds (i.e., ≥ 100). However, the mechanism still tends to underestimate the formation of light compounds and overestimate that of heavier ones compared to experimental observations.
The modeled reaction products are more oxidized and less fragmented than the experimentally observed ones, especially in the condensed phase. In the gas phase, the model favors the formation of C7 and C5 at the expense of the shorter carbon chains. The level of oxidation of the reaction products is generally similar to the observations except for the more oxidized C7 compounds. The underestimation of the fragmentation pathways compared to the functionalization ones and the risk of product fragmentation during the experimental measurement may lead to some discrepancy between simulated and observed concentrations in both phases. The larger errors in the condensed phase than in the gas phase highlight the remaining uncertainty in the partitioning processes and their modeling. Tests on partitioning reveal that taking into account some processes leads to large variation of the simulated SOA mass concentration, resulting in discrepancies with respect to the experimental values from +88 % to −81 %. The addition of an aqueous phase with few organic seeds favored the formation of SOA. Taking into account the interactions of the organic compounds among themselves shifted the equilibrium towards the condensed phase (error raised to +88 %), while the interaction between inorganic ions and organic compounds in the aqueous phase had the opposite effect (error reduced to +31 %). The introduction of wall loss parameterization also reduced the discrepancy. When including all these processes, the model reproduces the experimental SOA mass concentration and its temperature dependence much better, despite relevant discrepancies in the chemical speciation. The model, in its current state, does not reproduce the partitioning of light compounds, but a test of a simplified parameterization for methylglyoxal irreversible partitioning led to an improvement of the representation of this light compound in SOA, thus opening the way to further research and developments.
Experimental and modeling data analyzed in the article are available in the Supplement. The SSH-aerosol model is open-source (GNU GPL-3 license). The latest version of SSH-aerosol is available at https://github.com/sshaerosol/ssh-aerosol (last access: 8 June 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-15537-2023-supplement.
VL, BD, AW, and KS designed the research. PI, BTR, AMV, and MM performed the experiments and contributed to the acquisition and analysis of the data. VL, KS, and FC developed the software. VL and RV contributed to photolysis and mechanistic data for the model. VL, KS, BD, EK, and PI analyzed and interpreted the data. VL and BD drafted the article. VL, BD, KS, EK, MM, and AW revised the article. KS, BD, and VL were responsible for funding acquisition.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The France–Austria Hubert Curien Partnership 2016 with the Amadeus Program provided financial support for mobility of PhD students between the University of Innsbruck (Austria) and IRCELYON (France).
This work was funded by the POLEMICS project of the Agence Nationale de la Recherche (ANR) program (grant no. ANR-18-CE22-0011), DIM QI2 (Air Quality Research Network on air quality in the Ile-de-France region), and the MAESTRO-EU6 project (ADEME CORTEA no. 1866C0001). This work also benefited from the IPSL-CGS EUR and was partially supported by a government grant under the Programme d'Investissements d'avenir (reference ANR-11-IDEX-0004-17-EURE-0006), managed by the Agence Nationale de la Recherche.
This paper was edited by Qi Chen and reviewed by two anonymous referees.
Atkinson, R., Aschmann, S. M., Arey, J., and Carter, W. P. L.: Formation of ring-retaining products from the OH radical-initiated reactions of benzene and toluene, Int. J. Chem. Kinet., 21, 801–827, https://doi.org/10.1002/KIN.550210907, 1989.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Aumont, B., Valorso, R., Mouchel-Vallon, C., Camredon, M., Lee-Taylor, J., and Madronich, S.: Modeling SOA formation from the oxidation of intermediate volatility n-alkanes, Atmos. Chem. Phys., 12, 7577–7589, https://doi.org/10.5194/acp-12-7577-2012, 2012.
Aumont, B., Camredon, M., Mouchel-Vallon, C., La, S., Ouzebidour, F., Valorso, R., Lee-Taylor, J., and Madronich, S.: Modeling the influence of alkane molecular structure on secondary organic aerosol formation, Faraday Discuss., 165, 105, https://doi.org/10.1039/c3fd00029j, 2013.
Baltaretu, C. O., Lichtman, E. I., Hadler, A. B., and Elrod, M. J.: Primary atmospheric oxidation mechanism for toluene, J. Phys. Chem. A, 113, 221–230, https://doi.org/10.1021/jp806841t, 2009.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005.
Bohn, B.: Formation of peroxy radicals from OH-toluene adducts and O2, J. Phys. Chem. A, 105, 6092–6101, https://doi.org/10.1021/jp0033972, 2001.
Borrás, E. and Tortajada-Genaro, L. A.: Determination of oxygenated compounds in secondary organic aerosol from isoprene and toluene smog chamber experiments, Int. J. Environ. Anal. Chem., 92, 110–124, https://doi.org/10.1080/03067319.2011.572164, 2012.
Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G., Forster, P., Kerminen, V.-M., Kondo, Y., Liao, H., Lohmann, U., Rasch, P., Satheesh, S. K., Sherwood, S., Stevens, B., and Zhang, X. Y.: Clouds and Aerosols, in: Climate Change 2013 – The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 571–658, https://doi.org/10.1017/CBO9781107415324.016, 2013.
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. J., and Yarwood, G.: The mechanisms of atmospheric oxidation of aromatic hydrocarbons, Oxford University Press, Oxford, UK, ISBN: 9780195146288, 2002.
Couvidat, F. and Sartelet, K.: The Secondary Organic Aerosol Processor (SOAP v1.0) model: a unified model with different ranges of complexity based on the molecular surrogate approach, Geosci. Model Dev., 8, 1111–1138, https://doi.org/10.5194/gmd-8-1111-2015, 2015.
Couvidat, F., Debry, É., Sartelet, K., and Seigneur, C.: A hydrophilic/hydrophobic organic (H2O) aerosol model: Development, evaluation and sensitivity analysis, J. Geophys. Res., 117, D10304, https://doi.org/10.1029/2011JD017214, 2012.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
De Haan, D. O., Jimenez, N. G., de Loera, A., Cazaunau, M., Gratien, A., Pangui, E., and Doussin, J.-F.: Methylglyoxal Uptake Coefficients on Aqueous Aerosol Surfaces, J. Phys. Chem. A, 122, 4854–4860, https://doi.org/10.1021/acs.jpca.8b00533, 2018.
Donahue, N. M., Epstein, S. a., Pandis, S. N., and Robinson, a. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Doussin, J.-F., Fuchs, H., Kiendler-Scharr, A., Seakins, P., and Wenger, J. (Eds.): A Practical Guide to Atmospheric Simulation Chambers, Springer International Publishing, Cham, https://doi.org/10.1007/978-3-031-22277-1, 2023.
Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R.: A new time-of-flight aerosol mass spectrometer (TOF-AMS) – Instrument description and first field deployment, Aerosol Sci. Technol., 39, 637–658, https://doi.org/10.1080/02786820500182040, 2005.
Drewnick, F., Hings, S. S., Alfarra, M. R., Prevot, A. S. H., and Borrmann, S.: Aerosol quantification with the Aerodyne Aerosol Mass Spectrometer: detection limits and ionizer background effects, Atmos. Meas. Tech., 2, 33–46, https://doi.org/10.5194/amt-2-33-2009, 2009.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/Nature13032, 2014.
Eichler, P., Müller, M., D'Anna, B., and Wisthaler, A.: A novel inlet system for online chemical analysis of semi-volatile submicron particulate matter, Atmos. Meas. Tech., 8, 1353–1360, https://doi.org/10.5194/amt-8-1353-2015, 2015.
Fiorentino, E.-A., Wortham, H., and Sartelet, K.: Combining homogeneous and heterogeneous chemistry to model inorganic compound concentrations in indoor environments: the H2I model (v1.0), Geosci. Model Dev., 14, 2747–2780, https://doi.org/10.5194/gmd-14-2747-2021, 2021.
Fittschen, C., Frenzel, A., Imrik, K., and Devolder, P.: Rate constants for the reactions of C2H5O, i-C3H7O, and n-C3H7O with NO and O2 as a function of temperature, Int. J. Chem. Kinet., 31, 860–866, https://doi.org/10.1002/(SICI)1097-4601(1999)31:12<860::AID-KIN4>3.0.CO;2-E, 1999.
Forstner, H. J. L., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol from the photooxidation of aromatic hydrocarbons: Molecular composition, Environ. Sci. Technol., 31, 1345–1358, https://doi.org/10.1021/es9605376, 1997.
Fredenslund, A., Jones, R. L., and Prausnitz, J. M.: Group-contribution estimation of activity coefficients in nonideal liquid mixtures, AIChE J., 21, 1086–1099, https://doi.org/10.1002/aic.690210607, 1975.
Gelencsér, A., May, B., Simpson, D., Sánchez-Ochoa, A., Kasper-Giebl, A., Puxbaum, H., Caseiro, A., Pio, C., and Legrand, M.: Source apportionment of PM2.5 organic aerosol over Europe: Primary/secondary, natural/anthropogenic, and fossil/biogenic origin, J. Geophys. Res., 112, D23S04, https://doi.org/10.1029/2006JD008094, 2007.
Gkatzelis, G. I., Tillmann, R., Hohaus, T., Müller, M., Eichler, P., Xu, K.-M., Schlag, P., Schmitt, S. H., Wegener, R., Kaminski, M., Holzinger, R., Wisthaler, A., and Kiendler-Scharr, A.: Comparison of three aerosol chemical characterization techniques utilizing PTR-ToF-MS: a study on freshly formed and aged biogenic SOA, Atmos. Meas. Tech, 11, 1481–1500, https://doi.org/10.5194/amt-11-1481-2018, 2018a.
Gkatzelis, G. I., Hohaus, T., Tillmann, R., Gensch, I., Müller, M., Eichler, P., Xu, K.-M., Schlag, P., Schmitt, S. H., Yu, Z., Wegener, R., Kaminski, M., Holzinger, R., Wisthaler, A., and Kiendler-Scharr, A.: Gas-to-particle partitioning of major biogenic oxidation products: a study on freshly formed and aged biogenic SOA, Atmos. Chem. Phys, 18, 12969–12989, https://doi.org/10.5194/acp-18-12969-2018, 2018b.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013.
Graus, M., Müller, M., and Hansel, A.: High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time, J. Am. Soc. Mass. Spectrom., 21, 1037–1044, https://doi.org/10.1016/j.jasms.2010.02.006, 2010.
Hamilton, J. F., Webb, P. J., Lewis, A. C., and Reviejo, M. M.: Quantifying small molecules in secondary organic aerosol formed during the photo-oxidation of toluene with hydroxyl radicals, Atmos. Environ., 39, 7263–7275, https://doi.org/10.1016/J.ATMOSENV.2005.09.006, 2005.
Han, S., Bian, H., Zhang, Y., Wu, J., Wang, Y., Tie, X., Li, Y., Li, X., and Yao, Q.: Effect of Aerosols on Visibility and Radiation in Spring 2009 in Tianjin, China, Aerosol Air Qual. Res., 12, 211–217, https://doi.org/10.4209/aaqr.2011.05.0073, 2012.
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009.
Hu, J., Chen, Z., Qin, X., and Dong, P.: Reversible and irreversible gas-particle partitioning of dicarbonyl compounds observed in the real atmosphere, Atmos. Chem. Phys, 22, 6971–6987, https://doi.org/10.5194/acp-22-6971-2022, 2022.
Huang, Y., Zhao, R., Charan, S. M., Kenseth, C. M., Zhang, X., and Seinfeld, J. H.: Unified Theory of Vapor-Wall Mass Transport in Teflon-Walled Environmental Chambers, Environ. Sci. Technol., 52, 2134–2142, https://doi.org/10.1021/acs.est.7b05575, 2018.
Jacob, D.: Heterogeneous chemistry and tropospheric ozone, Atmos. Environ., 34, 2131–2159, https://doi.org/10.1016/S1352-2310(99)00462-8, 2000.
Jang, M. and Kamens, R. M.: Characterization of secondary aerosol from the photooxidation of toluene in the presence of NOx and 1-propene, Environ. Sci. Technol., 35, 3626–3639, https://doi.org/10.1021/es010676, 2001.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, https://doi.org/10.1016/J.IJMS.2009.07.005, 2009.
Kamens, R. M., Zhang, H., Chen, E. H., Zhou, Y., Parikh, H. M., Wilson, R. L., Galloway, K. E., and Rosen, E. P.: Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: Water and particle seed effects, Atmos. Environ., 45, 2324–2334, https://doi.org/10.1016/j.atmosenv.2010.11.007, 2011.
Kelly, J. L., Michelangeli, D. V., Makar, P. A., Hastie, D. R., Mozurkewich, M., and Auld, J.: Aerosol speciation and mass prediction from toluene oxidation under high NOx conditions, Atmos. Environ., 44, 361–369, https://doi.org/10.1016/J.ATMOSENV.2009.10.035, 2010.
Kim, Y., Couvidat, F., Sartelet, K., and Seigneur, C.: Comparison of Different Gas-Phase Mechanisms and Aerosol Modules for Simulating Particulate Matter Formation, J. Air Waste Manage. Assoc., 61, 1218–1226, https://doi.org/10.1080/10473289.2011.603999, 2011.
Kleindienst, T. E., Conver, T. S., McIver, C. D., and Edney, E. O.: Determination of secondary organic aerosol products from the photooxidation of toluene and their implications in ambient PM2.5, J. Atmos. Chem., 47, 79–100, https://doi.org/10.1023/B:JOCH.0000012305.94498.28, 2004.
Klotz, B., Sørensen, S., Barnes, I., Becker, K. H., Etzkorn, T., Volkamer, R., Platt, U., Wirtz, K., and Martin-Reviejo, M.: Atmospheric oxidation of toluene in a large-volume outdoor photoreactor: In situ determination of ring-retaining product yields, J. Phys. Chem. A, 102, 10289–10299, https://doi.org/10.1021/jp982719n, 1998.
Kostenidou, E., Karnezi, E., Hite, J., Bougiatioti, A., Cerully, K., Xu, L., Ng, N., Nenes, A., and Pandis, S.: Organic aerosol in the summertime southeastern United States: Components and their link to volatility distribution, oxidation state and hygroscopicity, Atmos. Chem. Phys., 18, 5799–5819, https://doi.org/10.5194/acp-18-5799-2018, 2018.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol., Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
La, Y. S., Camredon, M., Ziemann, P. J., Valorso, R., Matsunaga, A., Lannuque, V., Lee-Taylor, J., Hodzic, A., Madronich, S., and Aumont, B.: Impact of chamber wall loss of gaseous organic compounds on secondary organic aerosol formation: Explicit modeling of SOA formation from alkane and alkene oxidation, Atmos. Chem. Phys., 16, 1417–1431, https://doi.org/10.5194/acp-16-1417-2016, 2016.
Lannuque, V., Camredon, M., Couvidat, F., Hodzic, A., Valorso, R., Madronich, S., Bessagnet, B., and Aumont, B.: Exploration of the influence of environmental conditions on secondary organic aerosol formation and organic species properties using explicit simulations: development of the VBS-GECKO parameterization, Atmos. Chem. Phys., 18, 13411–13428, https://doi.org/10.5194/acp-18-13411-2018, 2018.
Lannuque, V., D'Anna, B., Couvidat, F., Valorso, R., and Sartelet, K.: Improvement in Modeling of OH and HO2 Radical Concentrations during Toluene and Xylene Oxidation with Racm2 using Mcm/Gecko-A, Atmosphere, 12, 732, https://doi.org/10.3390/atmos12060732, 2021.
Lee, S. C., Chiu, M. Y., Ho, K. F., Zou, S. C., and Wang, X.: Volatile organic compounds (VOCs) in urban atmosphere of Hong Kong, Chemosphere, 48, 375–382, https://doi.org/10.1016/S0045-6535(02)00040-1, 2002.
Leglise, J., Müller, M., Piel, F., Otto, T., and Wisthaler, A.: Bulk Organic Aerosol Analysis by Proton-Transfer-Reaction Mass Spectrometry: An Improved Methodology for the Determination of Total Organic Mass, O : C and H : C Elemental Ratios, and the Average Molecular Formula, Anal. Chem., 91, 12619–12624, https://doi.org/10.1021/acs.analchem.9b02949, 2019.
Li, Y., Zhao, J., Wang, Y., Seinfeld, J. H., and Zhang, R.: Multigeneration Production of Secondary Organic Aerosol from Toluene Photooxidation, Environ. Sci. Technol., 55, 8592–8603, https://doi.org/10.1021/acs.est.1c02026, 2021a.
Li, Y., Ji, Y., Zhao, J., Wang, Y., Shi, Q., Peng, J., Wang, Y., Wang, C., Zhang, F., Wang, Y., Seinfeld, J. H., and Zhang, R.: Unexpected Oligomerization of Small α-Dicarbonyls for Secondary Organic Aerosol and Brown Carbon Formation, Environ. Sci. Technol., 55, 4430–4439, https://doi.org/10.1021/acs.est.0c08066, 2021b.
Lim, C. C., Hayes, R. B., Ahn, J., Shao, Y., Silverman, D. T., Jones, R. R., Garcia, C., and Thurston, G. D.: Association between long-term exposure to ambient air pollution and diabetes mortality in the US, Environ. Res., 165, 330–336, https://doi.org/10.1016/J.ENVRES.2018.04.011, 2018.
Lim, S. S., Vos, T., Flaxman, A. D., Danaei, G., Shibuya, K., Adair-Rohani, H., AlMazroa, M. A., Amann, M., Anderson, H. R., Andrews, K. G., Aryee, M., Atkinson, C., Bacchus, L. J., Bahalim, A. N., Balakrishnan, K., Balmes, J., Barker-Collo, S., Baxter, A., Bell, M. L., Blore, J. D., Blyth, F., Bonner, C., Borges, G., Bourne, R., Boussinesq, M., Brauer, M., Brooks, P., Bruce, N. G., Brunekreef, B., Bryan-Hancock, C., Bucello, C., Buchbinder, R., Bull, F., Burnett, R. T., Byers, T. E., Calabria, B., Carapetis, J., Carnahan, E., Chafe, Z., Charlson, F., Chen, H., Chen, J. S., Cheng, A. T.-A., Child, J. C., Cohen, A., Colson, K. E., Cowie, B. C., Darby, S., Darling, S., Davis, A., Degenhardt, L., Dentener, F., Des Jarlais, D. C., Devries, K., Dherani, M., Ding, E. L., Dorsey, E. R., Driscoll, T., Edmond, K., Ali, S. E., Engell, R. E., Erwin, P. J., Fahimi, S., Falder, G., Farzadfar, F., Ferrari, A., Finucane, M. M., Flaxman, S., Fowkes, F. G. R., Freedman, G., Freeman, M. K., Gakidou, E., Ghosh, S., Giovannucci, E., Gmel, G., Graham, K., Grainger, R., Grant, B., Gunnell, D., Gutierrez, H. R., Hall, W., Hoek, H. W., Hogan, A., Hosgood, H. D., Hoy, D., Hu, H., Hubbell, B. J., Hutchings, S. J., Ibeanusi, S. E., Jacklyn, G. L., Jasrasaria, R., Jonas, J. B., Kan, H., Kanis, J. A., Kassebaum, N., Kawakami, N., Khang, Y.-H., Khatibzadeh, S., Khoo, J.-P., Kok, C., Laden, F., Lalloo, R., Lan, Q., Lathlean, T., Leasher, J., Leigh, J., Li, Y., Lin, J., Lipshultz, S., London, S., Lozano, R., Lu, Y., Mak, J., Malekzadeh, R., Mallinger, L., Marcenes, W., March, L., Marks, R., Martin, R., McGale, P., McGrath, J., Mehta, S., Mensah, G., Merriman, T., Micha, R., Michaud, C., Mishra, V., Hanafiah, K., Mokdad, A., Morawska, L., Mozaffarian, D., Murphy, T., Naghavi, M., Neal, B., Nelson, P., Nolla, J., Norman, R., Olives, C., Omer, S., Orchard, J., Osborne, R., Ostro, B., Page, A., Pandey, K., Parry, C., Passmore, E., Patra, J., Pearce, N., Pelizzari, P., Petzold, M., Phillips, M., Pope, D., Pope, C., Powles, J., Rao, M., Razavi, H., Rehfuess, E., Rehm, J., Ritz, B., Rivara, F., Roberts, T., Robinson, C., Rodriguez-Portales, J., Romieu, I., Room, R., Rosenfeld, L., Roy, A., Rushton, L., Salomon, J., Sampson, U., Sanchez-Riera, L., Sanman, E., Sapkota, A., Seedat, S., Shi, P., Shield, K., Shivakoti, R., Singh, G., Sleet, D., Smith, E., Smith, K., Stapelberg, N., Steenland, K., Stöckl, H., Stovner, L., Straif, K., Straney, L., Thurston, G., Tran, J., Van Dingenen, R., Van Donkelaar, A., Veerman, J., Vijayakumar, L., Weintraub, R., Weissman, M., White, R., Whiteford, H., Wiersma, S., Wilkinson, J., Williams, H., Williams, W., Wilson, N., Woolf, A., Yip, P., Zielinski, J., Lopez, A., Murray, C., and Ezzati, M.: A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010, Lancet, 380, 2224–2260, https://doi.org/10.1016/S0140-6736(12)61766-8, 2012.
Liu, X., Day, D. A., Krechmer, J. E., Brown, W., Peng, Z., Ziemann, P. J., and Jimenez, J. L.: Direct measurements of semi-volatile organic compound dynamics show near-unity mass accommodation coefficients for diverse aerosols, Commun. Chem., 21, 1–9, https://doi.org/10.1038/s42004-019-0200-x, 2019.
Malley, C. S., Kuylenstierna, J. C. I., Vallack, H. W., Henze, D. K., Blencowe, H., and Ashmore, M. R.: Preterm birth associated with maternal fine particulate matter exposure: A global, regional and national assessment, Environ. Int., 101, 173–182, https://doi.org/10.1016/j.envint.2017.01.023, 2017.
Mao, J., Ren, X., Brune, W. H., Olson, J. R., Crawford, J. H., Fried, A., Huey, L. G., Cohen, R. C., Heikes, B., Singh, H. B., Blake, D. R., Sachse, G. W., Diskin, G. S., Hall, S. R., and Shetter, R. E.: Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 9, 163–173, https://doi.org/10.5194/acp-9-163-2009, 2009.
Marrero-Ortiz, W., Hu, M., Du, Z., Ji, Y., Wang, Y., Guo, S., Lin, Y., Gomez-Hermandez, M., Peng, J., Li, Y., Secrest, J., Zamora, M. L., Wang, Y., An, T., and Zhang, R.: Formation and Optical Properties of Brown Carbon from Small α-Dicarbonyls and Amines, Environ. Sci. Technol., 53, 117–126, https://doi.org/10.1021/acs.est.8b03995, 2019.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Müller, M., Graus, M., Wisthaler, A., Hansel, A., Metzger, A., Dommen, J., and Baltensperger, U.: Analysis of high mass resolution PTR-TOF mass spectra from 1,3,5-trimethylbenzene (TMB) environmental chamber experiments, Atmos. Chem. Phys., 12, 829–843, https://doi.org/10.5194/acp-12-829-2012, 2012.
Müller, M., Mikoviny, T., Jud, W., D'Anna, B., and Wisthaler, A.: A new software tool for the analysis of high resolution PTR-TOF mass spectra, Chemom. Intell. Lab. Syst., 127, 158–165, https://doi.org/10.1016/J.CHEMOLAB.2013.06.011, 2013.
Müller, M., Eichler, P., D'Anna, B., Tan, W., and Wisthaler, A.: Direct Sampling and Analysis of Atmospheric Particulate Organic Matter by Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 89, 10889–10897, https://doi.org/10.1021/acs.analchem.7b02582, 2017.
Murphy, B. N., Donahue, N. M., Fountoukis, C., Dall'osto, M., O'dowd, C., Kiendler-Scharr, A., and Pandis, S. N.: Atmospheric Chemistry and Physics Functionalization and fragmentation during ambient organic aerosol aging: application of the 2-D volatility basis set to field studies, Atmos. Chem. Phys, 12, 10797–10816, https://doi.org/10.5194/acp-12-10797-2012, 2012.
Nannoolal, Y., Rarey, J., and Ramjugernath, D.: Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilib., 269, 117–133, https://doi.org/10.1016/j.fluid.2008.04.020, 2008.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Nishino, N., Arey, J., and Atkinson, R.: Formation yields of glyoxal and methylglyoxal from the gas-phase OH radical-initiated reactions of toluene, xylenes, and trimethylbenzenes as a function of NO2 concentration, J. Phys. Chem. A, 114, 10140–10147, https://doi.org/10.1021/jp105112h, 2010.
Noyes, W. A.: n-Butyl Nitrite, Org. Synth., 16, 7, https://doi.org/10.15227/orgsyn.016.0007, 1936.
Piel, F., Müller, M., Winkler, K., Skytte Af Sätra, J., and Wisthaler, A.: Introducing the extended volatility range proton-transfer-reaction mass spectrometer (EVR PTR-MS), Atmos. Meas. Tech, 14, 1355–1363, https://doi.org/10.5194/amt-14-1355-2021, 2021.
Platz, J., Nielsen, O. J., Wallington, T. J., Ball, J. C., Hurley, M. D., Straccia, A. M., Schneider, W. F., and Sehested, J.: Atmospheric Chemistry of the Phenoxy Radical, C6H5O(•): UV Spectrum and Kinetics of Its Reaction with NO, NO2 , and O2, J. Phys. Chem. A, 102, 7964–7974, https://doi.org/10.1021/jp982221l, 1998.
Raff, J. D. and Finlayson-Pitts, B. J.: Hydroxyl Radical Quantum Yields from Isopropyl Nitrite Photolysis in Air, Environ. Sci. Technol., 44, 8150–8155, https://doi.org/10.1021/es102218d, 2010.
Sartelet, K., Couvidat, F., Wang, Z., Flageul, C., and Kim, Y.: SSH-Aerosol v1.1: A Modular Box Model to Simulate the Evolution of Primary and Secondary Aerosols, Atmosphere, 11, 525, https://doi.org/10.3390/atmos11050525, 2020.
Sato, K., Hatakeyama, S., and Imamura, T.: Secondary Organic Aerosol Formation during the Photooxidation of Toluene: NOx Dependence of Chemical Composition, J. Phys. Chem. A, 111, 9796–9808, https://doi.org/10.1021/jp071419f, 2007.
Schwantes, R. H., Schilling, K. A., Mcvay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys, 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017.
Seigneur, C.: Air Pollution, Cambridge University Press, https://doi.org/10.1017/9781108674614, 2019.
Singh, H. B., Salas, L., Viezee, W., Sitton, B., and Ferek, R.: Measurement of volatile organic chemicals at selected sites in California, Atmos. Environ. Pt. A, 26, 2929–2946, https://doi.org/10.1016/0960-1686(92)90285-S, 1992.
Skorokhod, A. I., Berezina, E. V., Moiseenko, K. B., Elansky, N. F., and Belikov, I. B.: Benzene and toluene in the surface air of northern Eurasia from TROICA-12 campaign along the Trans-Siberian Railway, Atmos. Chem. Phys., 17, 5501–5514, https://doi.org/10.5194/acp-17-5501-2017, 2017.
Suh, I., Zhang, D., Zhang, R., Molina, L. T., and Molina, M. J.: Theoretical study of OH addition reaction to toluene, Chem. Phys. Lett., 364, 454–462, https://doi.org/10.1016/S0009-2614(02)01364-7, 2002.
Takekawa, H., Minoura, H., and Yamazaki, S.: Temperature dependence of secondary organic aerosol formation by photo-oxidation of hydrocarbons, Atmos. Environ., 37, 3413–3424, https://doi.org/10.1016/S1352-2310(03)00359-5, 2003.
Tao, Z. and Li, Z.: A kinetics study on reactions of C6H5O with C6H5O and O3 at 298 k, Int. J. Chem. Kinet., 31, 65–72, https://doi.org/10.1002/(SICI)1097-4601(1999)31:1<65::AID-KIN8>3.0.CO;2-J, 1999.
Tuazon, E. C., Atkinson, R., Leod, H. Mac, Biermann, H. W., Winer, A. M., Carter, W. P. L., and Pitts, J. N.: Yields of Glyoxal and Methylglyoxal from the NOx-Air Photooxidations of Toluene and m- and p-Xylene, Environ. Sci. Technol., 18, 981–984, https://doi.org/10.1021/es00130a017, 1984.
US Environmental Protection Agency: Health Effects Note-book for Hazardous Air Pollutants, https://www.epa.gov/sites/default/files/2016-09/documents/toluene.pdf, (last access: 13 March 2023), 2012.
Valorso, R., Aumont, B., Camredon, M., Raventos-Duran, T., Mouchel-Vallon, C., Ng, N. L., Seinfeld, J. H., Lee-Taylor, J., and Madronich, S.: Explicit modelling of SOA formation from α-pinene photooxidation: Sensitivity to vapour pressure estimation, Atmos. Chem. Phys., 11, 6895–6910, https://doi.org/10.5194/acp-11-6895-2011, 2011.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – Part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062, https://doi.org/10.1039/b909712k, 2009.
Wagner, V., Jenkin, M. E., Saunders, S. M., Stanton, J., Wirtz, K., and Pilling, M. J.: Modelling of the photooxidation of toluene: Conceptual ideas for validating detailed mechanisms, Atmos. Chem. Phys., 3, 89–106, https://doi.org/10.5194/acp-3-89-2003, 2003.
Wang, M., Chen, D., Xiao, M., Ye, Q., Stolzenburg, D., Hofbauer, V., Ye, P., Vogel, A. L., Mauldin, R. L., Amorim, A., Baccarini, A., Baumgartner, B., Brilke, S., Dada, L., Dias, A., Duplissy, J., Finkenzeller, H., Garmash, O., He, X.-C., Hoyle, C. R., Kim, C., Kvashnin, A., Lehtipalo, K., Fischer, L., Molteni, U., Petäjä, T., Pospisilova, V., Quéléver, L. L. J., Rissanen, M., Simon, M., Tauber, C., Tomé, A., Wagner, A. C., Weitz, L., Volkamer, R., Winkler, P. M., Kirkby, J., Worsnop, D. R., Kulmala, M., Baltensperger, U., Dommen, J., El-Haddad, I., and Donahue, N. M.: Photo-oxidation of Aromatic Hydrocarbons Produces Low-Volatility Organic Compounds, Environ. Sci. Technol., 54, 7911–7921, https://doi.org/10.1021/acs.est.0c02100, 2020.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, Z., Couvidat, F., and Sartelet, K.: GENerator of reduced Organic Aerosol mechanism (GENOA v1.0): an automatic generation tool of semi-explicit mechanisms, Geosci. Model Dev., 15, 8957–8982, https://doi.org/10.5194/gmd-15-8957-2022, 2022.
White, S. J., Jamie, I. M., and Angove, D. E.: Chemical characterisation of semi-volatile and aerosol compounds from the photooxidation of toluene and NOx, Atmos. Environ., 83, 237–244, https://doi.org/10.1016/J.ATMOSENV.2013.11.023, 2014.
Wu, R., Pan, S., Li, Y., and Wang, L.: Atmospheric Oxidation Mechanism of Toluene, J. Phys. Chem. A, 118, 4533–4547, https://doi.org/10.1021/JP500077F, 2014.
Xu, J., Griffin, R. J., Liu, Y., Nakao, S., and Cocker, D. R.: Simulated impact of NOx on SOA formation from oxidation of toluene and m-xylene, Atmos. Environ., 101, 217–225, https://doi.org/10.1016/J.ATMOSENV.2014.11.008, 2015.
Yamasaki, H., Kuwata, K., and Miyamoto, H.: Effects of ambient temperature on aspects of airborne polycyclic aromatic hydrocarbons, Environ. Sci. Technol., 16, 189–194, https://doi.org/10.1021/es00098a003, 1982.
Yan, Y., Cabrera-Perez, D., Lin, J., Pozzer, A., Hu, L., Millet, D. B., Porter, W. C., and Lelieveld, J.: Global tropospheric effects of aromatic chemistry with the SAPRC-11 mechanism implemented in GEOS-Chem version 9-02, Geosci. Model Dev., 12, 111–130, https://doi.org/10.5194/gmd-12-111-2019, 2019.
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., Keutsch, F. N., and Paulson, J. A.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys, 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019.
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of Urban Fine Particulate Matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015a.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015b.
Zhu, S., Sartelet, K. N., and Seigneur, C.: A size-composition resolved aerosol model for simulating the dynamics of externally mixed particles: SCRAM (v 1.0), Geosci. Model Dev., 8, 1595–1612, https://doi.org/10.5194/gmd-8-1595-2015, 2015.
Zuend, A., Marcolli, C., Luo, B. P., and Peter, T.: A thermodynamic model of mixed organic-inorganic aerosols to predict activity coefficients, Atmos. Chem. Phys., 8, 4559–4593, https://doi.org/10.5194/acp-8-4559-2008, 2008.