the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Aug 2022
| 03 Aug 2022
Functionality-based formation of secondary organic aerosol from m-xylene photooxidation
Yixin Li
Jiayun Zhao
Mario Gomez-Hernandez
Michael Lavallee
Natalie M. Johnson
Photooxidation of volatile organic compounds (VOCs) produces condensable oxidized organics (COOs) to yield secondary organic aerosol (SOA), but the fundamental chemical mechanism for gas-to-particle conversion remains uncertain. Here we elucidate the production of COOs and their roles in SOA and brown carbon (BrC) formation from m-xylene oxidation by simultaneously monitoring the evolution of gas-phase products and aerosol properties in an environmental chamber. Four COO types with the distinct functionalities of dicarbonyls, carboxylic acids, polyhydroxy aromatics/quinones, and nitrophenols are identified from early-generation oxidation, with the yields of 25 %, 37 %, 5 %, and 3 %, respectively. SOA formation occurs via several heterogeneous processes, including interfacial interaction, ionic dissociation/acid–base reaction, and oligomerization, with the yields of (20 ± 4) % and (32 ± 7) % at 10 % and 70 % relative humidity (RH), respectively. Chemical speciation shows the dominant presence of oligomers, nitrogen-containing organics, and carboxylates at high RH and carboxylates at low RH. The identified BrC includes N-heterocycles/N-heterochains and nitrophenols, as evident from reduced single scattering albedo. The measured uptake coefficient (γ) for COOs is dependent on the functionality, ranging from 3.7 × 10−4 to 1.3 × 10−2. A functionality-based kinetic framework is developed to predict SOA production from the observed concentrations and uptake coefficients for COOs, which reproduces the measurement from m-xylene oxidation. Our results reveal that photochemical oxidation of m-xylene represents a major source for SOA and BrC formation under urban environments, because of its large abundance, high reactivity with OH, and high yields for COOs.
- Article
(3376 KB) - Full-text XML
-
Supplement
(1052 KB) - BibTeX
- EndNote





Photooxidation of anthropogenic and biogenic volatile organic compounds (VOCs) produces tropospheric ozone, secondary organic aerosol (SOA), and brown carbon (BrC), with profound implications for air quality, human health, and climate (Pope et al., 2002; Li et al., 2007; IPCC, 2013; NASEM, 2016; Molina, 2021; Zhang et al., 2021). For example, SOA contributes to the Earth energy budget, directly by scattering solar radiation and indirectly by serving as cloud condensation nuclei to influence cloud formation and precipitation (IPCC, 2013; Wang et al., 2014; Zhu et al., 2017; Zhang et al., 2020). Also, light-absorbing BrC interferes with solar radiation transfer, contributing to positive radiative forcing (Wang et al., 2013; NASEM, 2016). VOC oxidation is initiated by various oxidants (e.g., OH, O3, and NO3) and proceeds via multiple pathways and stages (Atkinson, 2000; Suh et al., 2001; Zhang et al., 2002; Zhao et al., 2004; Wennberg et al., 2018), yielding condensable oxidized organics (COOs) to form SOA and BrC via gas-to-particle conversion (Finlayson-Pitts and Pitts, 2000; Zhang et al., 2009; Moise et al., 2015; Seinfeld and Pandis, 2016). Currently, the enormous chemical complexity for VOC oxidation and gas-to-particle conversion represents one of the greatest challenges in atmospheric chemistry research (Ravishankara, 1997; Zhao et al., 2005a; NASEM, 2016). Aromatic hydrocarbons (e.g., benzene, toluene, xylenes, and trimethylbenzene) account for 20 %–30 % of the total VOCs and are the major anthropogenic SOA precursors in the urban atmosphere (Calvert et al., 2002; Ng et al., 2007; Song et al., 2007; Guo et al., 2014; Seinfeld and Pandis, 2016). For example, m-xylene or C6H4(CH3)2 represents an important type of aromatic hydrocarbons, which is emitted primarily from industrial and traffic sources. The concentration of m-xylene ranges in a level from sub-parts per billion (sub-ppb) up to several tens of ppb under urban environments (Calvert et al., 2002; Fortner et al., 2009). Photooxidation of m-xylene is primarily initiated by OH with a rate constant of 2.4 × 10−11 cm3 molec.−1 s−1, which is nearly 4 times higher than that of toluene (Fan and Zhang, 2008; Ji et al., 2017). A laboratory study identified m-tolualdehyde, m-dimethylphenols, and dicarbonyls (i.e., glyoxal, methylglyoxal, unsaturated dicarbonyls, and epoxy carbonyls) as the main gas-phase products from OH-initiated oxidation of m-xylene (Zhao et al., 2005b).
Several types of COOs with distinct functionality and volatility are produced from m-xylene photooxidation, contributing to aerosol nucleation and growth (Zhang et al., 2004, 2015; Guo et al., 2020). SOA formation is conventionally considered to be mainly resulted from equilibrium gas–particle partitioning of semi-volatile, intermediate-volatility, or low-volatility products (Shrivastava et al., 2017). Aromatic oxidation by OH yields low-volatility polyhydroxy aromatics/quinones (denoted as PAQ hereafter), which condense to the aerosol phase (Schwantes et al., 2017). Also, chain autoxidation reactions of RO2 from aromatics photooxidation likely yield highly oxygenated molecules (HOMs) with low volatility (Molteni et al., 2018; Garmash et al., 2020; Wang et al., 2020), although the reported yield of HOMs from m-xylene oxidation is small (1.0 %–1.7 %) (Molteni et al., 2018). Several other oxidation products of aromatic oxidation have been shown to readily engage in multiphase reactions and contribute to SOA formation (Ji et al., 2020; Li et al., 2021a). A recent experimental study demonstrated that SOA formation from toluene photooxidation is mainly contributed by volatile dicarbonyls and organic acids (Li et al., 2021b). Moreover, the aqueous reactions between small α-dicarbonyls and base species (e.g., ammonia and amines) produce light-absorbing brown carbon (BrC) (De Haan et al., 2011, 2017; Marrero-Ortiz et al., 2019; Li et al., 2021a, b). Previous experimental studies also identified the presence of hydroxy (C–OH), carbonyl (C=O), and acetal (C–O–C) functional groups in aromatic-derived SOA formed via aqueous reactions, implying a role of oxygenated organics in SOA formation (Jia and Xu, 2014, 2018; Zhang et al., 2019). Also, laboratory experiments showed that a coating of m-xylene SOA on black carbon particles significantly impacts scattering, absorption, and single scattering albedo (SSA) (Guo et al., 2016).
Current atmospheric models mainly parameterize SOA formation based on equilibrium partitioning for semi-volatile to low-volatility COOs (Shrivastava et al., 2017). However, gas-to-particle conversion corresponds to decreasing entropy (i.e., ΔS≪0), representing highly non-equilibrium chemical processes (Peng et al., 2021). Moreover, the occurrence of particle-phase reactions significantly alters the physiochemical properties (including volatility, hygroscopicity, and optical properties) for COOs (Tan et al., 2012; Faust et al., 2017; Ji et al., 2020; Li et al., 2021a, b; Liu et al., 2021). Notably, the volatility-based approach consistently underpredicts SOA formation (Heald et al., 2005; Zhang et al., 2015; Hodzic et al., 2016), particularly during haze formation under polluted conditions (Guo et al., 2014; Peng et al., 2021). Here we investigate the production of COOs with different functionalities and assess their roles in SOA formation from m-xylene oxidation. A primary objective of this work is to establish a functionality-based framework to predict SOA formation from VOC photooxidation.
2.1 Chamber experiments
The production of COOs and their roles in SOA formation from m-xylene oxidation were investigated using a 1 m3 chamber (Teflon® PFA, Fig. S1 in the Supplement), similar to our previous studies (Li et al., 2021a, b). The environmental chamber was equipped with 18 black light lamps (18 × 30 W, F30T8/350BL, Sylvania). A water bubbler at a temperature of 30 ∘C was used to humidify the chamber to 10 %, 30 %, 50 %, or 70 % relative humidity (RH), and all experiments were performed at 298 K. Three types of seed particles were produced using a constant output atomizer (Model 3076, TSI) to represent various chemical compositions, i.e., NH4HSO4 – ammonium bisulfate (ABS), (NH4)2SO4 – ammonium sulfate (AS) in the presence and absence of NH3, and NaCl – sodium chloride. Seed particles were dried to RH of ∼2 % by a Nafion dryer (PD-070-18T-12SS, Perma Pure) and size-selected for an initial size of 100 nm by a differential mobility analyzer (DMA; Model 3081, TSI). Size-selected particles were injected into the chamber with an initial particle concentration of 1.5 × 104 cm−3 measured by a condensation particle counter (CPC; Model 3760A, TSI). The acidity of seed particles was estimated using a thermodynamic model (Fountoukis and Nenes, 2007; Wang et al., 2018), with the pH value of 3–5 in the presence of gaseous NH3 and 0–1 in the absence of gaseous NH3 for (NH4)2SO4, −0.5 for NH4HSO4, and 7 for NaCl. To investigate the effects of NH3 and NOx, commercially available gaseous NH3 (2000 ppm NH3 in N2) and NO2 (500 ppm NO2 in air) were injected into the chamber with a flow of 100 sccm. Quantities of 600 µL H2O2 (35 wt %, Sigma-Aldrich) and 10 µL m-xylene (99.5 %, Sigma-Aldrich) were injected into a glass reservoir and flushed into the chamber by a 10 slpm flow from a pure air generator (Aadco 737-11, Aadco Inc.) for 10 min. The gases were then mixed by a fan inside the chamber. Once desired concentrations were established, the black light lamps were turned on to initiate H2O2 photolysis to generate OH radicals. The initial concentration for m-xylene and the steady-state concentration for OH were estimated to be 1.9 ppm and 2.1 × 106 molec. cm−3, respectively (Fig. 1a). The experimental conditions are summarized in Table 1.
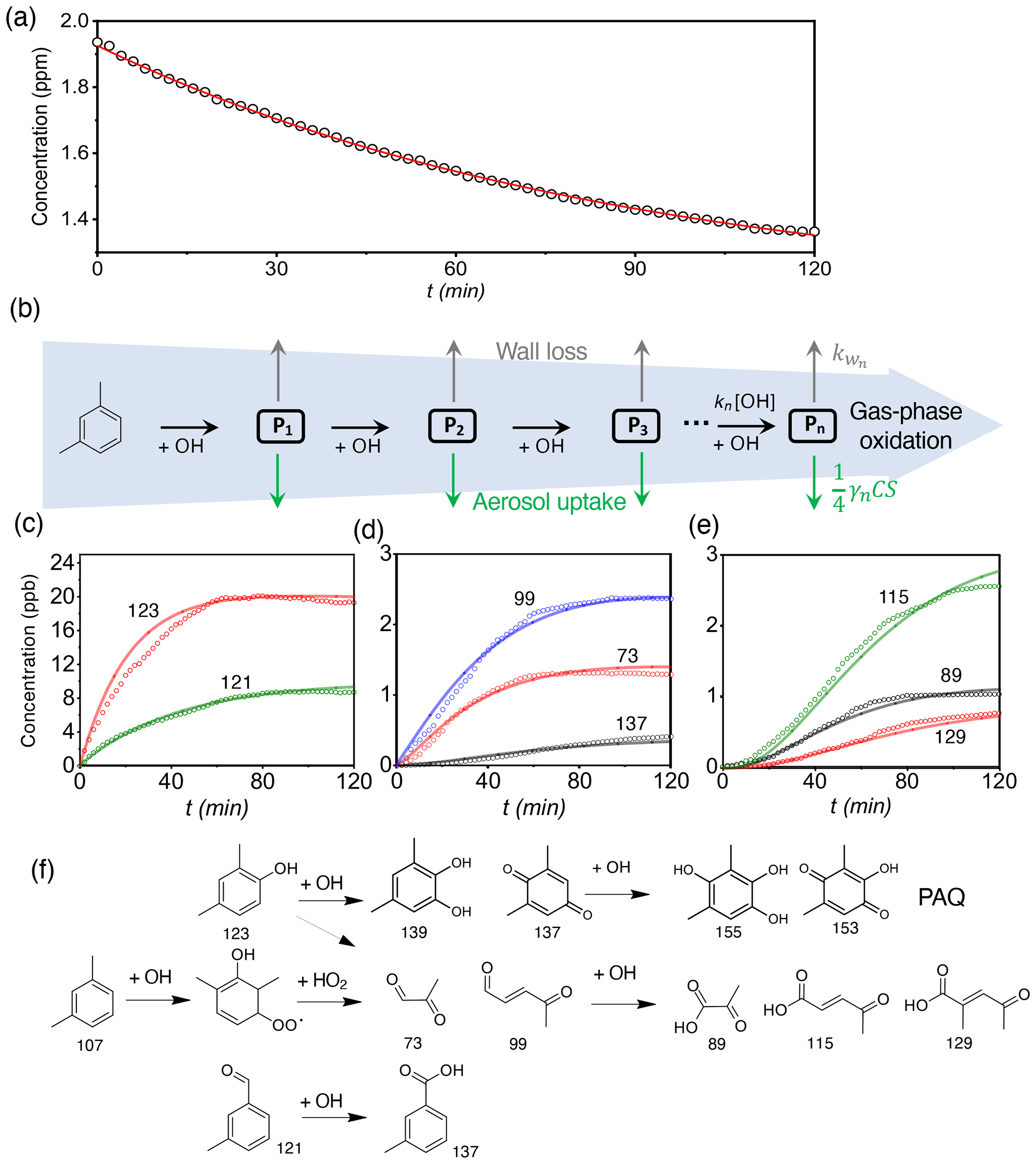
Figure 1OH-initiated photooxidation of m-xylene. (a) Time-dependent concentration of m-xylene. The symbols are from the measurements, and the solid red line corresponds to exponential fitting of the m-xylene concentration. The initial concentration for m-xylene and the estimated steady-state concentration for OH are 1940 ppb and 2.1 × 106 molec. cm−3, respectively, with a bimolecular rate constant of 2.4 × 10−11 cm3 molec.−1 s−1. (b) Schematic representation leading to the multi-generation product, Pn, where n denotes the sequence of OH-initiated oxidation. The black, gray, and green arrows denote the gas-phase oxidation, chamber wall loss, and aerosol uptake, respectively. kn and are the rate coefficients for the gas-phase oxidation and wall loss, respectively, γn represents the aerosol uptake coefficient, is the thermal velocity, and S is the aerosol surface area. (c–e) Time-dependent gas-phase concentrations of P1 (c), P2 (d), and P3 (e) products. The symbols are from measurements, and the solid curves are simulated according to the tri-exponential kinetics. (f) The gaseous oxidation pathways leading to the detected products. The top row corresponds to the pathways leading to polyhydroxy aromatics/quinones (PAQ). The numbers in (c) to (f) represent the mass-to-charge ratio (). All experiments are carried out for (NH4)2SO4 seed particles with 19 ppb NH3 at 298 K and RH = 70 %. Initiation of photooxidation by ultraviolet light occurs at t=0.
2.2 Analytical instruments
The particle size growth, density, and light scattering/absorption were simultaneously and continuously monitored, after monodispersed seed particles were exposed to the products of m-xylene–OH oxidation in the reaction chamber. A scanning mobility particle sizer (SMPS) consisting of a DMA and a CPC was used to continuously measure the particle size growth factor, GF = , where Dp is the diameter after exposure and D0=100 nm is the initial diameter. Note both Dp and D0 correspond to the dry particle diameter measured after passing through a Nafion dryer (∼ 2 % RH). An aerosol particle mass analyzer (APM; Model 3600, Kanomax) was used to derive the particle density throughout the experiments (Li et al., 2021b). The density for the SOA materials (ρSOA) is calculated by
where ρ and GF are measured particle density (g cm−3) and growth factor at t (min), respectively. ρ0 is the initial density of seed particles. The uncertainty in the density measurements was estimated to be ±0.03 g cm−3.
To measure the optical properties of exposed particles, a 1 slpm flow from the chamber was diluted by 4 slpm N2 and introduced into a commercial integrating nephelometer (TSI 3563) and a home-built cavity ring-down spectrometer (CRDS) throughout the experiment. Light scattering (bsca) and extinction (bext) at 532 nm were determined by the nephelometer and CRDS, respectively. The nephelometer was calibrated by comparing measured bsca with bext from CRDS when measuring ammonium sulfate particles with diameters of 100, 150, 200, 250, and 300 nm. The absorption coefficient (babs) was determined from (bext−bsca), and SSA was calculated from .
An ion drift–chemical ionization mass spectrometer (ID-CIMS) using the hydronium reagent ion (H3O+) was employed to analyze gaseous concentrations with a sample flow rate of 0.5 slpm from the chamber (Fortner et al., 2004). The sampling line was heated to 70 ∘C to reduce wall loss. Gas-phase concentrations of m-xylene and its oxidation products were quantified by the ID-CIMS. Briefly, the concentration of species A from the proton transfer reaction (H3O+ + A → H2O + HA+) is determined by
where SA and SRI are the mass spectrum intensities for A and reagent ions, respectively. kPTR is the proton transfer rate constant between A and H3O+ (Zhao and Zhang, 2004). is the retention time of ions, which is determined by the length of the drift tube l=8 cm and the ion drift velocity Ui. The ion drift velocity Ui is determined from
where u0=2 cm2 V−1 s−1 is the reduced ionic mobility (Dotan et al., 1976), p=3.0 Torr (where 1 Torr is equivalent to 133.3 Pa), T=298 K, and E=133 V cm−1 is the voltage gradient in the drift tube. Alternatively, the concentrations of m-xylene, dicarbonyls, and organic acids were also calibrated by preparing the vapors of m-xylene and methylglyoxal with known mixing ratios in N2 and using a permeation tube of propanoic acid, respectively (Li et al., 2021a). The detection limit (defined by the signal-to-noise ratio of 3) for the oxidation products from m-xylene–OH reactions was estimated to be 50 ppt by the ID-CIMS.
The steady-state OH concentration was determined by exponential fitting of the decay of m-xylene concentration (Fig. 1a):
where b is the slope of the exponential fitting, and cm3 molec.−1 s−1 is the second-order rate constant for m-xylene–OH oxidation (Fan and Zhang, 2008). The estimated concentration of OH was 2.1 × 106 molec. cm−3 in our experiments.
We also estimated loss of condensable vapors to the chamber wall using the first-order wall loss coefficient, kw (s−1), by considering gas-phase transport within the chamber (Zhang et al., 2014):
where is the surface-to-volume ratio of the chamber (equal to for a square chamber, where L=1 m is its dimension), αw is the mass accommodation coefficient of vapors onto Teflon chamber walls, is the mean thermal speed of the molecules, and ke is the coefficient of eddy diffusion, which is estimated to be 0.5 based on the loss rate of particles (McMurry and Rader, 1985):
where β0 is the fractional loss rate of particles:
where N is the particle number concentration, and m2 s−1 is the Brownian diffusion coefficient for particles. Dg is the gas-phase diffusion coefficient. For NH3, the mass accommodation coefficient of vapors onto the chamber wall is 0.05 (Zhang et al., 2014). The gas-phase diffusion constant Dg is 1.98 × 10−5 m2 s−1. The mean thermal speed is 603 m s−1. The first-order wall loss coefficient of NH3 was calculated to be 1.2 × 10−2 s−1.
The average concentration of NH3 or NOx (denoted as X) is estimated by
For initial injection of 400 ppb NH3, the average concentration of NH3 was estimated to be 19 ppb. The first-order wall loss rate of NOx was measured to be 2.5 × 10−3 h−1, leading to negligible wall loss (Qi et al., 2020).
The particle-phase chemical composition was analyzed by a thermal desorption (TD) ID-CIMS (TD-ID-CIMS) (Wang et al., 2010). Seed particles after 20 min of exposure to photooxidation were collected for 2 h by a platinum filament (with a collection voltage of about 3000 V) in a 2.5 slpm flow from the reaction chamber. The analytes were evaporated by heating the filament to 350 ∘C for 2 s and detected by the ID-CIMS using H3O+ as the reagent ions. The mass resolution of the TD-ID-CIMS was about 0.5 amu. The desorption signal was represented by the relative intensity (RI) of the integrated peak area during heating. The uncertainties of TD-ID-CIMS measurements arose from the flow rate, voltage, collection time, evaporation voltage, and mass spectrometer ionization/detection efficiencies during particle collection and were represented by the standard deviation of three repeated measurements. In both ID-CIMS and TD-ID-CIMS configurations, an ion drift tube was used. An electric field of Td was applied for the ID-CIMS at high RH (i.e., 70 %), and Td was used for the TD-ID-CIMS at low RH (<1 %). Formation of cluster ions was effectively suppressed, since a uniform electrical field in the drift tube decomposed weakly bonded cluster ions. Fragmentation using the ID-CIMS or TD-ID-CIMS was minimal, and all gaseous and particle-phase products were detected at their respective parent peaks, indicating little dissociation effect from either thermal desorption or ionization by H3O+ (Yuan et al., 2017). For example, plausible fragment ion peaks for oligomers and organic acids are the monomeric ion peaks and [M-17]+ or [M-45]+ from α-cleavage, respectively. From our detected particle-phase products (Fig. 2c), non-fragmental oligomers/nitrogen-containing organics (NCOs) and organic acids represented the dominant peaks, while negligible monomers (e.g., ) and fragments for organic acids (e.g., [M-17]+ or [M-45]+) were detected, confirming little fragmentation by TD-ID-CIMS measurement. Also, the major acetal/hemiacetal oligomers detected by the TD-ID-CIMS at high RH from m-xylene oxidation were consistent with a previous study showing intact oligomers with thermal desorption (Claflin and Ziemann, 2019).
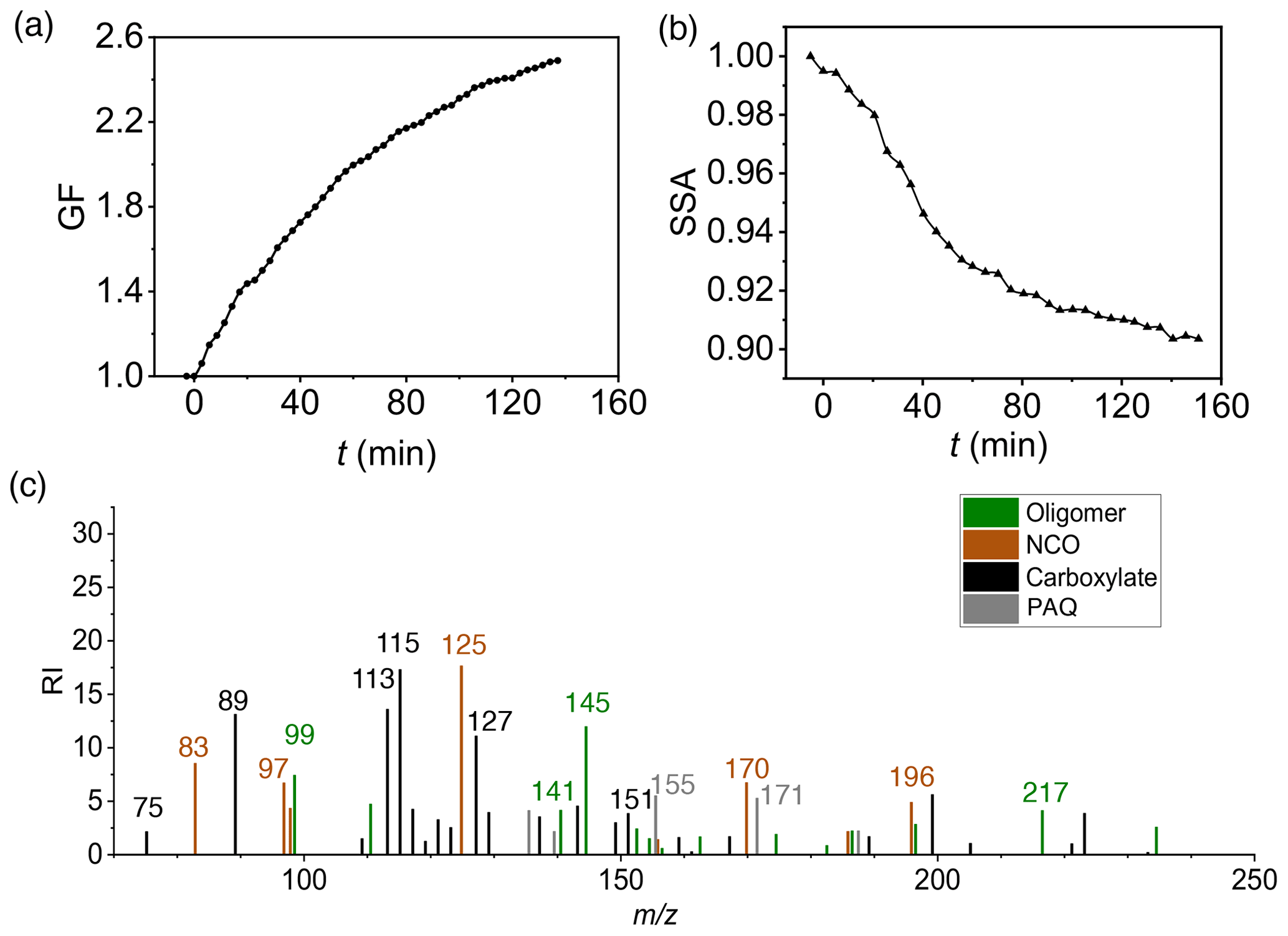
Figure 2SOA and BrC formation from m-xylene photooxidation. Time-dependent growth factor (GF = , a) and single scattering albedo (SSA, b) of seed particles exposed to m-xylene oxidation products. (c) Chemical speciation of aerosol-phase products by the TD-ID-CIMS. Initiation of photooxidation by ultraviolet light occurred at t=0. RI denotes the relative intensity for the integrated peak area during thermal desorption. All experiments are carried out for (NH4)2SO4 seed particles with 19 ppb NH3 at 298 K and RH = 70 %.
While the gaseous concentrations of the oxidation products, GF, SSA, and density were temporally resolved in our experiments, the particle chemical composition reflected an overall aerosol chemical makeup during an experiment. To assess the effects of different seed particles, RH, and NH3 on SOA and BrC formation, we compared the measured GF and SSA values after 120 min exposure to the m-xylene–OH oxidation.
2.3 Uptake coefficient (γ) determination
The uptake coefficient (γ) for COOs is calculated by
where FR is the percent contributions of the COO species from m-xylene–OH oxidation to the particle-phase mass intensity measured by the TD-ID-CIMS. kp is the measured first-order particle uptake rate constant for COOs (i.e., glyoxal, methylglyoxal, γ-dicarbonyls, organic acids, PAQ, and nitrophenols), which is calculated from the measured growth rate of seed particles:
where Dp and D0 are the final and initial diameters of the particles during the time period of 120 min, respectively, ρ is the density of SOA, = 11 250 cm−3 is the average particle number concentration during 120 min, Δt=120 min is the exposure time, NA is the Avogadro constant, MW is the molar weight of species A, and [A] is the average gas-phase concentration of species A. S is the average surface area of aerosols (square centimeter of surface per cubic centimeter of air) during the period of Δt=120 min:
where D is the time-dependent particle diameter, HGf is the hygroscopic growth factor of particles (Seinfeld and Pandis, 2016). The error bars of reactive coefficients are derived from the propagation of uncertainties for the parameters in Eqs. (9)–(11) (i.e., 1σ of at least three repeated measurements for each parameter).
2.4 Simulation of gaseous oxidation products
The gas-phase concentration of m-xylene–OH oxidation products was analyzed by a tri-exponential kinetic model to account for the gas-phase oxidation, chamber wall loss, and particle uptake. The precursor A with an initial concentration of [A]0 undergoes multiple steps of oxidation to yield multi-generation products, Pn (where n=1, 2, 3, …). Once formed, each product engages in further oxidation with a pseudo-first-order rate constant of , particle uptake with a rate constant of , and wall loss with a rate of . The forward rate constants, kn+1, were taken from MCM 3.3.1 (Jenkin et al., 2003). The rate constant of uptake is expressed by
and the first-order wall loss rate constants were estimated by Eq. (5) with the mass accommodation coefficients αw constrained by the measured time-dependent concentration. The gas-phase concentrations of P1 to P3 are calculated by
where C1 to C7 are constants, and denotes the pseudo-first-order rate constant:
The rate constants and values for C1 to C7 are summarized in Table S1 in the Supplement.
2.5 COO yield
The COO yield (YCOO) from m-xylene oxidation is determined from
where i represents the type of COO species, and Δ[C6H4(CH3)2] denotes the concentration of m-xylene consumed due to OH oxidation. There were four types of COOs identified from our experiments, i.e., dicarbonyls for i=1, carboxylic acids for i=2, PAQ for i=3, and nitrophenols for i=4. The measured gaseous concentration for each COO is corrected for wall loss and unreacted earlier-generation products:
where [COOi,f] and are the final and time-averaged gas-phase concentrations of COOi at 120 min, respectively. [Af] and [At] are the unreacted and the total production of the earlier-generation species A, respectively.
2.6 SOA mass concentration and yield
The measured SOA mass concentration is calculated based on the aerosol size growth and density,
where ρt and ρ0 are the final and initial particle density during 120 min, respectively. Alternatively, by categorizing COOs in accordance with the functional groups, we established a framework to predict SOA formation from their measured production and reactivity. The SOA mass concentration (MSOA) is predicted from the measured uptake coefficients and average gas-phase concentrations of COOs,
where MWi is the molar weight of species i. γi is averaged over all identified species for each type (i) of COOs, and [Ai] is the sum of the measured concentrations of all identified species for type i (time-averaged). The SOA mass yield is calculated by
where MWm is the molar weight of m-xylene.
To compare the measured and predicted SOA formation at RH = 10 % and 70 %, we corrected the vapor wall loss and particle surface area at RH = 70 % compared to 10 % RH. A correction factor is determined from the average measured gas concentration ratios (Rc) between 10 % and 70 % RH for each type of COOs. Additionally, a hygroscopic volume enhancement factor (HVE) of 1.2 between 10 % and 70 % RH is adopted. The RH-corrected SOA mass for each COO is estimated by
where S1 and S2 are the particle surface areas at 10 % and 70 % RH, respectively.
3.1 Evolution of oxidation products
To elucidate the production of COOs, we measured time-dependent gaseous concentrations of m-xylene oxidation products. The OH–m-xylene reactions occur via dominant OH addition to the aromatic ring to yield m-xylene–OH adducts and minor H abstraction from the methyl group to form methylbenzyl radicals (about 4 %) (Fan and Zhang, 2008). The m-xylene–OH adducts then react with O2 to form dimethylphenol via H extraction or OH–m-xylene–O2 peroxy radicals (RO2) via O2 addition (Fig. S2a). While the m-xylene mixing ratio exhibits an exponential decay throughout the experiments (Fig. 1a), the formation of the gas-phase oxidation products follows the rate-determining steps involving successive OH oxidation with three major generations (Fig. 1b–e). The first-generation products (P1) include dimethylphenol () and methylbenzaldehyde (), corresponding to OH addition to the aromatic ring (∼ 96 %) and hydrogen extraction (∼ 4 %) from the methyl group, respectively (Fan and Zhang, 2008; Li et al., 2021b). The second-generation products (P2) mainly consist of methylglyoxal (), methylbutenedial (), toluic acid (), and dimethylresorcinol (Figs. 1f and S2b in the Supplement), which are produced from the P1 reactions with OH or HO2. For example, methylglyoxal and methylbutenedial are formed from OH oxidation of dimethylphenol and subsequent ring-opening or from the primary peroxy radical. The latter undergoes cyclization to form the bicyclic radical, which then reacts with O2 to form the secondary peroxy radical, followed by reactions with HO2 and subsequent ring-cleavage (Fan and Zhang, 2008). The third-generation products (P3) contain mainly multi-functional organic acids, including pyruvic acid (), 4-oxo-2-pentenoic acid (), and 3-methyl-4-oxo-2-pentenoic acid () produced from the subsequent reactions of dicarbonyls with OH. Note that Pn in Fig. 1b denotes the lumped product of the nth generation, which is related the sequence of OH addition for m-xylene oxidation and accounts for the various species detected by the ID-CIMS (Fig. S2b).
The P1 concentrations rise immediately upon initiating photooxidation and reach the peak mixing ratios of about 20 ppb for dimethylphenol and 9 ppb for methylbenzaldehyde. There is a slight decline in P1 concentrations after 70 to 100 min, reflecting their consumption from further OH oxidation (Fig. 1c). The P2 concentrations follow those of P1 (Figs. 1d and S3a) and reach the peak mixing ratios ranging from 0.4 to 2.4 ppb, dependent on their subsequent oxidation by OH as well as loss to the chamber wall and aerosols. Note that the concentration of toluic acid rises later than those of the dicarbonyls, attributable to the slower rates for H-abstraction by OH from the methyl group than OH addition to the aromatic ring (Fan and Zhang, 2008). The P3 concentrations increase monotonically after a delay of 10–20 min and reach a mixing ratio from sub-ppb to 2.5 ppb at 120 min (Figs. 1e and S3b). The initial concentration of m-xylene (e.g., 1940 ppb) in our experiment is higher than that in the atmosphere, potentially inducing self- and cross-reactions of RO2 to form alkoxy radicals (RO) or dialkyl peroxides (ROOR′) leading to HOMs. However, negligible products relevant to HOMs were detected in our experiments, indicating a minor importance for the self- and cross-reactions of RO2 compared to the competing reactions between RO2 and HO2, NO, or RO2 to form ring-opening products. Our results for insignificant contribution of HOMs to SOA formation from m-xylene oxidation are consistent with a small yield of HOMs reported in a previous study (Molteni et al., 2018).
We simulated the time-dependent evolution of the gaseous products by employing a tri-exponential kinetic model (Fig. 1b and Table S1), according to Eqs. (13)–(15). Multi-generation oxidation of m-xylene occurs via consecutive reaction steps to produce a multi-generation product, Pn (where n denotes the sequence of OH oxidation), which subsequently engages in additional OH oxidation, wall loss, and aerosol loss. Figure 1b–d indicate that the simulated concentrations reproduce those measured for P1 to P3 by adopting the measured γ (to be discussed below) and estimated wall loss rates (see Methods and Table S1).
3.2 Particle size growth, SSA, and chemical speciation
To evaluate SOA and BrC formation from COOs, we measured the time-dependent GF, SSA, and density, after (NH4)2SO4 seed particles are exposed to the m-xylene oxidation products in the presence of 19 ppb NH3 and at 70 % RH (Fig. 2a–c, Exp. 1). The GF increases monotonically and reaches a value of (2.41 ± 0.03) at 120 min (Fig. 2a), while the SSA declines steadily throughout the exposure and reaches a value of (0.91 ± 0.01) at 120 min (Fig. 2b). The latter is indicative of the production of light-absorbing BrC. The measured SSA for m-xylene is lower than that previously reported for toluene under comparable experimental conditions (Li et al., 2021b). The difference in SSA/BrC formation between toluene and m-xylene is explained by a higher yield of methylglyoxal from m-xylene than from toluene (Jenkin et al., 2003; Nishino et al., 2010), since methylglyoxal produces BrC more efficiently via aqueous reaction than glyoxal (Li et al., 2021a). Analysis of particle chemical composition using the TD-ID-CIMS reveals high abundances of oligomers, NCOs consisting of N-heterocycles/N-heterochains and carboxylates, along with a small amount of PAQ (Fig. 2c). The identified mass peaks are summarized in Tables S2 to S4. Assuming similar sensitivity to proton transfer reactions for the aerosol-phase organics, i.e., with the proton transfer reaction rate constants of (2 to 4) × 10−9 cm3 molec.−1 s−1 (Zhao and Zhang, 2004), the contributions of oligomers, NCOs, carboxylates, and PAQ to the total SOA formation are estimated to be 22 %, 23 %, 47 %, and 8 %, respectively, at 70 % RH (Table S5). Note that the TD-ID-CIMS method preserves the identity of all organic species without fragmentation, providing unambiguous chemical speciation for the aerosol-phase products.
3.3 Effects of seed particles, NH3, RH, and NOx
We assessed the dependence of SOA/BrC formation from m-xylene–OH oxidation on different seed particles (Exp. 1–4), NH3 concentration (Exp. 1–12), RH (Exp. 1, 13–15), and NOx concentration (Exp. 1, 16–18). Figure 3a shows that (NH4)2SO4 seed particles in the presence of 19 ppb NH3 exhibit the largest GF and lowest SSA at 120 min, which are explained by the chemical mechanisms leading to the formation of the observed aerosol-phase products. While dicarbonyls undergo aqueous-phase reactions to form oligomers (Fig. S4) (Ji et al., 2020; Li et al., 2021a), organic acids engage in ionic dissociation or acid–base reaction to yield carboxylates (Fig. 3b). In addition, the reaction of dicarbonyls with NH3 produces NCOs (Fig. S5), which are light-absorbing (Marrero-Ortiz et al., 2019; Li et al., 2021a). Oligomerization of dicarbonyls involves protonation, hydration, and deprotonation, which are mediated by carbenium ions via nucleophilic addition (Ji et al., 2020; Li et al., 2021a). While protonation of dicarbonyls to yield carbenium ions is promoted by hydrogen ion activity, high acidity hinders hydration and deprotonation, explaining the largest GF and the smallest SSA on weakly acidic (NH4)2SO4 particles in the presence of NH3 (Fig. 3a). In addition, uptake of dicarbonyls, organic acids, and PAQ is likely facilitated on aqueous (NH4)2SO4 particles, because of surface propensity of charge separation at the interface (Hua et al., 2011; Shi et al., 2020). Specifically, surface-abundant NH cations arising from interfacial charge separation of (NH4)2SO4 likely exert electrostatic attraction to gaseous oxygenated species (with a negative charge character) to enhance uptake for dicarbonyls, organic acids, and PAQ (Li et al., 2021a, b). The most efficient formation of SOA and BrC on (NH4)2SO4 particles in the presence of NH3 is also consistent with the measured highest intensities for oligomers and NCOs (Fig. 3b). On the other hand, ionic dissociation and acid–base reaction to yield carboxylates occur efficiently in the presence of NH3/NaCl, as evident from the increasing carboxylate intensity from NH4HSO4 to NaCl (Fig. 3b). The GF and SSA increase and decrease, respectively, with NH3 from 0 to 27 ppb for all three types of seed particles (Fig. 4a, b), relevant to the reactions of NH3 with dicarbonyls to form NCOs and with organic acids to form ammonium carboxylates. The analysis of the particle composition corroborates that the intensities of oligomers, NCOs, and carboxylates increase with the NH3 concentration (Fig. 4c–e).

Figure 3Dependence of SOA/BrC formation on seed particles. (a) GF (black) and SSA (red) at 120 min of exposure for (NH4)2SO4 particles in the presence (AS w/ NH3) and absence (AS w/o NH3) of 19 ppb NH3 and for NH4HSO4 (ABS) and NaCl particles in the absence of NH3. (b) Aerosol-phase relative intensity (RI) for carboxylates (black), oligomers (green), and NCOs (brown) on different seed particles. The error bar denotes 1σ of three replicated measurements. All particle properties were measured relevant to dry conditions (less than 5 % RH) and were dominantly contributed by non-volatile aerosol-phase products.

Figure 4Variation of SOA formation with seed particles and NH3 concentration. (a, b) Growth factor (a) and SSA (b) at t=120 min with varying NH3 concentration between 0 and 27 ppb for ammonium sulfate (AS, red), ammonium bisulfate (ABS, green), and sodium chloride (NaCl, blue) seed particles. (c–e) Aerosol-phase mass relative intensity (RI) for NCOs (red), oligomers (green), and carboxylates (blue) on AS (c), ABS (d), and NaCl (e) seed particles. The error bar denotes 1σ of three replicated measurements.
We carried out additional experiments by varying RH from 10 % to 70 % in the presence of 19 ppb NH3 (Fig. 5). The GF decreases with increasing RH from (3.10 ± 0.06) at RH = 10 % to (2.41 ± 0.03) at RH = 70 % (Fig. 5a). The measured SSA at 120 min is close to unity at 10 % and 30 % RH and then decreases with increasing RH (Fig. 5a), indicating negligible NCO formation at low RH but significant NCO formation at RH above 50 %. Carboxylates represent the dominant constituent throughout the RH range (i.e., 85 % at 10 % RH to 47 % at 70 % RH), while the contributions of oligomers and NCOs are small at low RH (2 %–5 % at RH < 40 %) and become increasingly significant at high RH (15 %–23 % at RH > 50 %) (Fig. 5b). The fraction for PAQ (8 %) is nearly invariant with RH. For (NH4)2SO4 particles, the deliquescent and efflorescent points are at 80 % and 36 % RH, respectively (Li et al., 2021b). At low RH (10 % and 30 %), aqueous reactions to yield oligomers/NCOs are minimal because of small water activity and significantly suppressed protonation and oligomerization. On the other hand, carboxylic acids readily undergo ionic dissociation or acid–base reaction, since organic acids efficiently retain water, even at low RH (Xue et al., 2009). The equilibrium vapor pressures for PAQ are much lower than those for organic acids (Table S6), facilitating more efficient condensation. Additionally, wall loss of COOs is more pronounced at high RH (Li et al., 2021b), explaining the decreased GF with increasing RH. Measurement of gaseous concentrations for COOs shows that wall loss is 1.5 to 4.6 times more efficient at 70 % RH than at 10 % RH, while wall loss of m-xylene is negligible at both RH levels (Table S7).
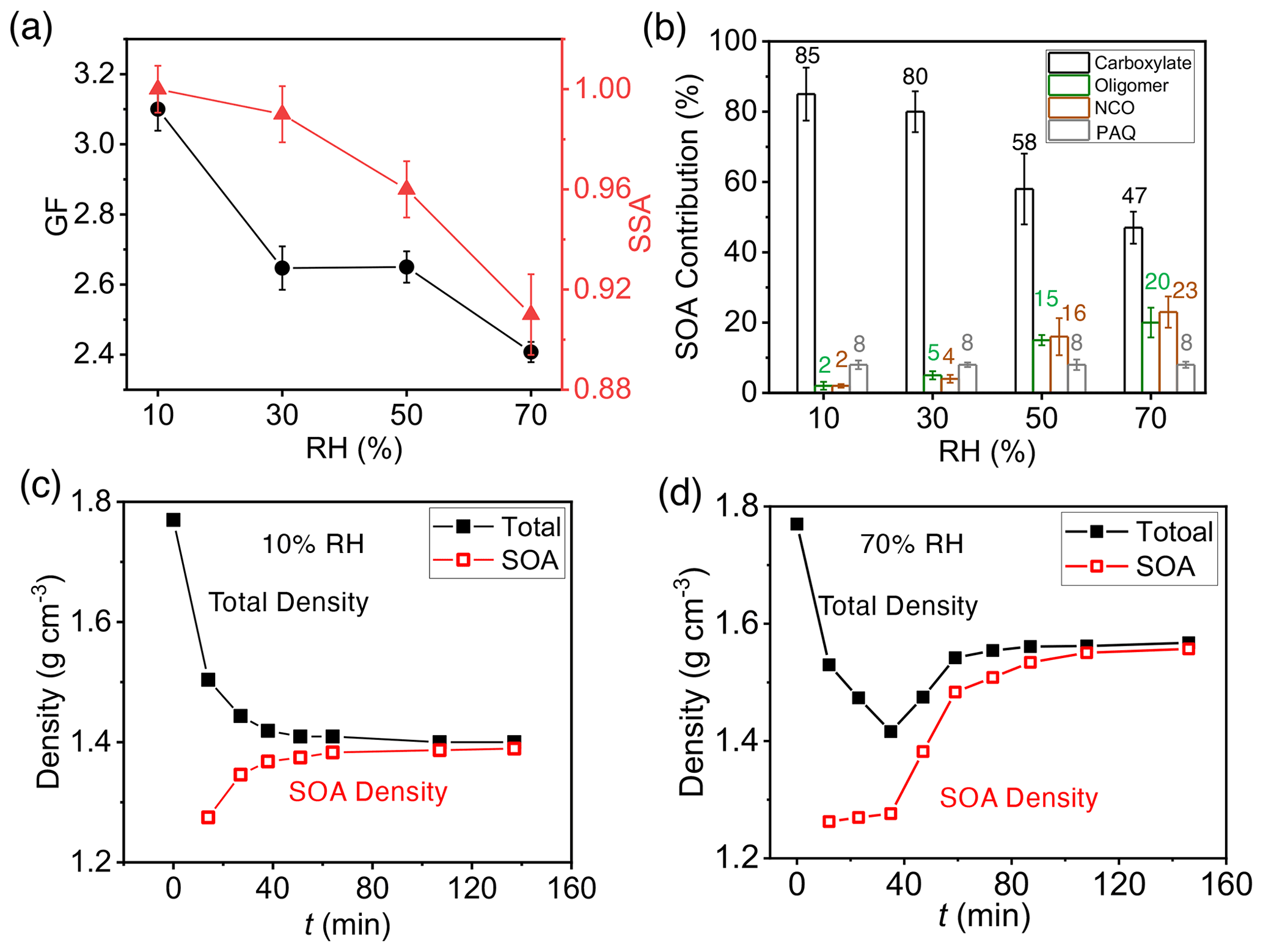
Figure 5Dependence of SOA/BrC formation on RH. (a) GF (black) and SSA (red) at 120 min with varying RH from 10 % to 70 %. (b) Contribution of COOs to SOA formation with varying RH from 10 % to 70 %: carboxylate (black), oligomers (green), NCOs (brown), and PAQ (gray) to SOA formation. The number denotes the percentage contribution ( %) of each type of aerosol-phase products. (c, d) Time-dependent particle (black) and SOA (red) densities of seed particles exposed to m-xylene oxidation products at RH = 10 % (c) and 70 % (d), respectively. The error bar denotes 1σ of three replicated measurements.
We determined the total density and the density of the SOA fraction for (NH4)2SO4 seed particles exposed to m-xylene oxidation (Fig. 5c, d). The measured densities are distinct between 10 and 70 % RH. At 10 % RH, the total density decreases monotonically, while the SOA density increases slightly with reaction time, i.e., from 1.27 to 1.39 g cm−3 (Fig. 5c), indicating minor oligomers and dominant carboxylic acids at low RH. At 70 % RH, the total particle density initially decreases from 1.77 to 1.41 g cm−3 (at a GF of 1.24) and subsequently increases steadily to 1.56 g cm−3 at 120 min. The SOA density on (NH4)2SO4 particles increases from 1.26 g cm−3 at 10 min to 1.55 g cm−3 at 120 min (Fig. 5d). The evolution in the densities reflects the variation in the chemical composition. The initial particle growth is dominated by small oligomers, imidazoles from methylglyoxal/methylbutenedial, and early-generation organic acids (e.g., toluic acid), with densities from 0.98 to 1.27 g cm−3 (Table S8). Subsequent particle growth from methylglyoxal/methylbutenedial yields large oligomers with the densities of 1.71–1.90 g cm−3 (Table S8).
To survey the NOx effects, we performed experiments on (NH4)2SO4 seed particles by varying the initial NOx concentration from 0 to 500 ppb (Fig. 6). Three major nitrophenols are identified from NO2 addition to the OH–m-xylene adduct, i.e., 4-methyl-2-nitrophenol (), dimethyl nitrophenol (), and dimethyl-4-nitrocatechol () (Fig. 6a). The production of nitrophenols is much smaller than that of dicarbonyls and organic acids, consistent with a smaller yield (less than 7 %) for nitrophenols (Fan and Zhang, 2008). The GF on (NH4)2SO4 seed particles with NH3 decreases from 2.41 ± 0.03 to 2.18 ± 0.03 with 0 to 500 ppb NOx (Fig. 6b). The SSA decreases significantly from 0.911 ± 0.006 to 0.839 ± 0.003 with increasing NOx (Fig. 6b), because of the formation of light-absorbing nitrophenols (Hems and Abbatt, 2018; Li et al., 2021b). Analysis of particle composition reveals that the decreasing GF with increasing NOx correlates with simultaneous decreases of carboxylates, oligomers, and NCOs and with increasing nitrophenols in the aerosol phase (Fig. 6c). Overall, nitrophenols contribute 2 % to 4 % to the SOA formation. Addition of NOx not only produces nitrophenols, but also alters the cycling between RO2 and alkoxy (RO) radicals, leading to redistributions of COOs. The presence of NOx decreases dicarbonyls and organic acids, as evident from the decreased intensities for oligomers, NCOs, and carboxylates in the aerosol phase with increasing NOx (Fig. 2h).

Figure 6The effects of NOx. (a) Time-dependent gas-phase concentrations of 5-methyl-2-nitrophenol (black, ), dimethyl nitrophenol (red, ), and dihydroxy nitrotoluene (blue, ). The numbers denote the mass-to-charge ratio (). Initiation of photooxidation by ultraviolet light occurred at t=0. (b) GF (black) and SSA (red) at 120 min with varying NOx concentration from 0 to 500 ppb. (h) Dependence of aerosol-phase relative mass intensities (RI) for carboxylates (black), oligomers (green), NCOs (brown), and nitrophenols (red) on NOx concentration for (NH4)2SO4 seed particles with 19 ppb NH3 at RH = 70 %. The error bar denotes 1σ of three replicated measurements.
3.4 COO and SOA yields
From the measured GF and COO concentrations, we derived the γ, which is widely employed to represent aerosol formation in atmospheric models (Zhang et al., 2015). The measured γ varies with the functionality of COOs and RH on (NH4)2SO4 seed particles (Fig. 7a). Results of the γ values for COOs are also summarized in Table S9. The γ for dicarbonyls is the highest (from 3 × 10−3 to 1.3 × 10−2) at 70 % RH but is significantly reduced (from 3.7 × 10−4 to 1.0 × 10−3) at 10 % RH, while the γ's for organic acids, PAQ, and nitrophenols are slightly higher at 70 % RH. Among the organic acids, the γ correlates with the acid dissociation constant and solubility, which are the highest for pyruvic acid and the lowest for toluic acid. The standard deviation in γ (within 1σ) among each COO type is within 50 %, 40 %, 30 %, and 15 % for dicarbonyls, organic acids, PAQ, and nitrophenols, respectively. From the measured concentrations of COOs and m-xylene, we determined the COO yields (YCOO) using Eq. (17), by considering four COO species, i.e., dicarbonyls for i=1, carboxylic acids for i=2, PAQ for i=3, and nitrophenols for i=4 (Table S7). The YCOO values for dicarbonyls, organic acids, PAQ, and nitrophenols are 25 %, 37 %, 5 %, and 3 %, respectively (Fig. 7b), indicating significant production of COOs from m-xylene oxidation.
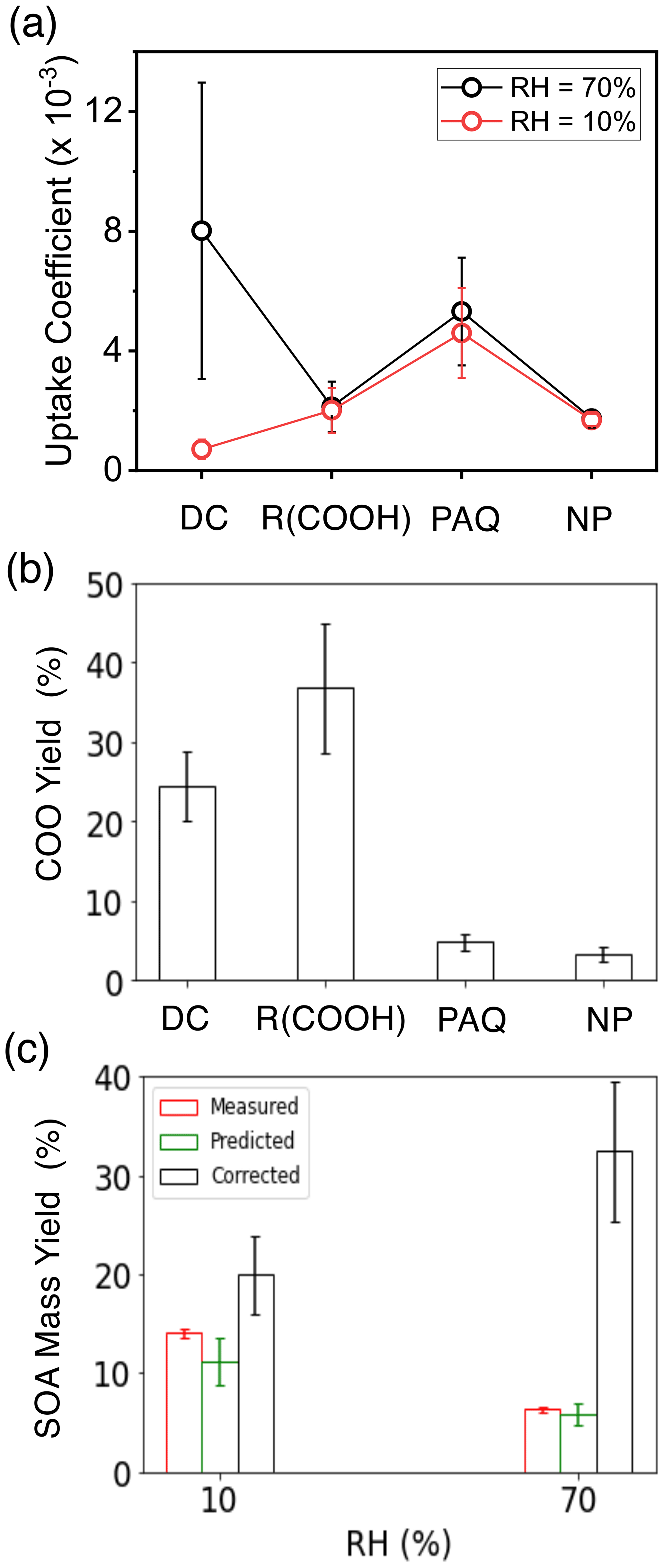
Figure 7Uptake coefficient, COO yield, and SOA mass yield. (a) Average uptake coefficients (γ) for different types of COOs at 10 % (red) and 70 % (black) RH for (NH4)2SO4 seed particles with 19 ppb NH3 at 298 K. (b) COO yields: dicarbonyls, organic acids, polyhydroxy aromatics/quinones, and nitrophenols are represented by DC, R(COOH)n, PAQ, and NP, respectively. (c) SOA mass yields at 10 % (left columns) and 70 % RH (right). The red, green, and black columns represent the measured, predicted, and corrected (for wall loss) SOA mass yields according to Eqs. (19)–(22). All experiments are carried out for 14 µg m−3 (NH4)2SO4 seed particles with 19 ppb NH3 at 298 K. The error bars denote the 1σ of three replicated measurements or by accounting for error propagation of the measured parameters.
Our work shows a significant increase in particle size (Fig. 2a) and constantly varying particle properties, i.e., SSA (Fig. 2b) and density (Fig. 5c, d), which correlate closely with the gaseous COO production (Fig. 1). These results imply a highly nonequilibrium kinetic process leading to SOA formation from m-xylene oxidation, which cannot be described only by equilibrium partitioning. The gas-to-particle conversion from m-xylene oxidation is dominated by several volatile COO species (i.e., organic acids and dicarbonyls) and with minor contribution from condensation of low-volatility COO (i.e., PAQs and nitrophenols) (Fig. 5b). In our study, the vapor pressures of the detected gaseous oxidation products for organic acids and dicarbonyls are too large to explain the measured particle growth via equilibrium partitioning (Table S6). Specifically, the saturation vapor pressures of organic acids detected in the particle phase range from 1.9 × 10−3 to 6.6 × 10−6 atm, while their gas-phase concentrations range from 0.5 to 2.5 ppb. Similarly, the saturation vapor pressures of dicarbonyls detected in the particle phase range from 1.6 × 10−1 to 3.9 × 10−4 atm, while their gas-phase concentrations range from 0.5 to 2.2 ppb. The high saturation vapor pressures and low gas-phase concentrations for dicarbonyls and organic acids render equilibrium partitioning implausible for these COOs in our experiments. Also, the detection of the particle-phase products confirms that organic acids and dicarbonyls directly participate in heterogeneous reactions to yield low-volatility products. Moreover, volatility-based equilibrium partitioning cannot explain the variations in the particle size growth, SSA, and chemical compositions on different seed particles (Fig. 3). Clearly, the gas-to-particle conversion from m-xylene oxidation involves several distinct heterogeneous processes, including the interfacial interaction, ionic dissociation/acid–base reaction, and nucleophilic oligomerization (Li et al., 2021a, b).
We establish a functionality-based SOA formation to predict the aerosol mass concentration (MSOA) from the gaseous concentrations and γ's for COOs using Eq. (20). The MSOA is calculated from the measurements of the averaged γ (Fig. 7a and Table S10) and the time-averaged concentrations for each COO type (Table S7). While the production of COOs is dependent on the VOC types and photooxidation, the aerosol-phase reactivity of COOs, as represented by γ, is dependent on the functionality for a given aerosol type. Thus, SOA formation from various VOC types can be predicted from the production and reactivity for COOs based on the experimental measurements, by categorizing COOs in accordance with their functional groups. The overall gas uptake onto liquid or solid particles is commonly described by an empirical framework on the basis of an effective γ, which accounts for the gas-phase diffusion, the interfacial process (i.e., accommodation), and the aerosol-phase processes (Zhang et al., 1994; Ravishankara, 1997). The aerosol-phase processes include thermodynamically driven surface- or volume-limited partitioning (including condensation for low-volatility species or solubility for volatile species) and chemically driven reactivity relevant to the functionality (Gomez et al., 2015). The SOA growth is ultimately dependent on the volatility of the aerosol-phase products. Evidently, there are two pathways to produce low-volatility compounds, i.e., gas-phase oxidation to yield low-volatility COOs that subsequently undergo condensation and multiphase reactions of volatile COOs to form low-volatility products in the aerosol phase. In our study, the gas-to-particle conversion from m-xylene oxidation occurs dominantly via the latter, i.e., reactive uptake for dicarbonyls and organic acids, with a relatively small contribution from the former, i.e., condensation by low-volatility PAQ and nitrophenols. Also, our measured uptake coefficient for each COO type (γi) is derived from the measured particle growth (Eqs. 9 and 10), which implicitly accounts for non-continuum diffusion, imperfect accommodation, and evaporation.
To assess the consistency of our approach, we compared the predicted and measured SOA mass yields (YSOA), according to Eq. (21). The predicted YSOA is (11 ± 3) % and (6 ± 1) %, respectively, at 10 % and 70 % RH, comparable to the measured values of (14 ± 0.4) % and (6.3 ± 0.2) % on (NH4)2SO4 seed particles (Fig. 7c). The slight differences between the predicted and measured YSOA's are explainable by the uncertainties related to lumping each COO type (i.e., averaging the γ values) as well as unaccounted for low-abundant COO species. To account for the RH-dependent wall loss for COOs, we corrected YSOA from the measured gaseous concentrations at varying RH (see Methods and Table S7). Such correction results in an increase of 1 to 4 times for YSOA at 10 % and 70 % RH, yielding the values of (20 ± 4) % and (32 ± 7) %, respectively (Fig. 7c). Under atmospheric conditions, SOA/BrC formation from m-xylene is expected to be enhanced at high RH, because of significantly enhanced aqueous reactions, larger aerosol surface area, and higher water activity. Field measurements showed significantly increased SOA formation with increasing RH during severe haze evolution (Guo et al., 2014; Peng et al., 2021).
In this work we have elucidated the production of COOs and their roles in SOA/BrC formation from m-xylene oxidation. The major advantages of our experimental methodologies lie in simultaneous monitoring of the evolutions of gaseous oxidation products and aerosol properties when seed particles are exposed to m-xylene oxidation, which allows for assessment of multi-generation production of SOA and BrC. Hence, our approach remedies the deficiency of wall loss of reactive gases and particles, which represents a main obstacle in experimental investigation using environmental chambers (Zhang et al., 2014; Huang et al., 2018). OH-initiated oxidation of m-xylene produces four distinct COO types consisting of dicarbonyls, carboxylic acids, polyhydroxy aromatics/quinones, and nitrophenols from early-generation products (P2 and P3), with the yields of 25 %, 37 %, 5 %, and 3 %, respectively. SOA formation occurs via several heterogeneous processes, including interfacial interaction, ionic dissociation/acid–base reaction, and oligomerization, with the yields of (20 ± 4) % at 10 % RH and (32 ± 7) % at 70 % RH. The identified aerosol-phase products include dominant oligomers, N-heterocycles/N-heterochains, and carboxylates at high RH and dominant carboxylates at low RH, along with small amounts of PAQ and nitrophenols (in the presence of NOx). The nitrogen-containing organics consisting of N-heterocycles, N-heterochains, and nitrophenols are light-absorbing, characterized by low SSA. The measured γ for COOs is dependent on the functionality, ranging from 3.7 × 10−4 to 1.3 × 10−2. A kinetic framework is developed to predict SOA formation from the concentration and γ for COOs. Our results illustrate that SOA formation from m-xylene oxidation is accurately quantified from the abundance and reactivity for COOs (i.e., Eq. 20). Notably, this kinetic framework accounts for the interfacial process (i.e., mass accommodation) as well as aqueous reactions (i.e., oligomerization and acid–base reactions) without the assumption of gas–particle equilibrium. We envisage that this functionality-based approach is broadly applicable to predict SOA formation from VOC photooxidation using experiment/field-measured or model-simulated COOs, aerosol surface area, and reactivity. We conclude that photochemical oxidation of m-xylene represents a major source for SOA and BrC formation under urban conditions, because of its large abundance, high reactivity with OH, and high yields for COOs.
All data can be provided by the corresponding authors upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-9843-2022-supplement.
YL and RZ designed the research. YL and JZ performed the research. YL, JZ, MGH, ML, NMJ, and RZ analyzed data. YL and RZ wrote the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This research was supported by the Robert A. Welch Foundation (grant no. A-1417). Yixin Li was supported by a dissertation fellowship at Texas A&M University.
This paper was edited by Ivan Kourtchev and reviewed by three anonymous referees.
Atkinson, R.: Atmospheric Chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, 2000.
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. H., and Yarwood, G.: The Mechanisms of Atmospheric Oxidation of Aromatic Hydrocarbons, Oxford University Press, New York, ISBN 9780195146288, 2002.
Claflin, M. S. and Ziemann, P. J.: Thermal desorption behavior of hemiacetal, acetal, ether, and ester oligomers, Aerosol Sci. Tech., 53, 473–484, https://doi.org/10.1080/02786826.2019.1576853, 2019.
De Haan, D. O., Hawkins, L. N., Kononenko, J. A., Turley, J. J., Corrigan, A. L., Tolbert, M. A., and Jimenez, J. L.: Formation of Nitrogen-Containing Oligomers by Methylglyoxal and Amines in Simulated Evaporating Cloud Droplets, Environ. Sci. Technol., 45, 984–991, 2011.
De Haan, D. O., Hawkins, L. N., Welsh, H. G., Pednekar, R., Casar, J. R., Pennington, E. A., de Loera, A., Jimenez, N. G., Symons, M. A., Zauscher, M., Pajunoja, A., Caponi, L., Cazaunau, M., Formenti, P., Gratien, A., Pangui, E., and Doussin, J.-F.: Brown Carbon Production in Ammonium- or Amine-Containing Aerosol Particles by Reactive Uptake of Methylglyoxal and Photolytic Cloud Cycling, Environ. Sci. Technol., 51, 7458–7466, https://doi.org/10.1021/acs.est.7b00159, 2017.
Dotan, I., Albritton, D. L., Lindinger, W., and Pahl, M.: Mobilities of CO, N2H+, H3O+, H3O, and H3O ions in N2, J. Chem. Phys., 65, 5028–5030, https://doi.org/10.1063/1.432943, 1976.
Fan, J. and Zhang, R.: Density Functional Theory Study on OH-Initiated Atmospheric Oxidation of m-Xylene, J. Phys. Chem. A, 112, 4314–4323, https://doi.org/10.1021/jp077648j, 2008.
Faust, J. A., Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Role of Aerosol Liquid Water in Secondary Organic Aerosol Formation from Volatile Organic Compounds, Environ. Sci. Technol., 51, 1405–1413, 2017.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the Upper and Lower Atmosphere: Theory, Experiments and Applications, Academic Press, San Diego, ISBN 978-0-12-257060-5, 2000.
Fortner, E. C., Zhao, J., and Zhang, R.: Development of ion drift-chemical ionization mass spectrometry, Anal. Chem., 76, 5436–5440, 2004.
Fortner, E. C., Zheng, J., Zhang, R., Berk Knighton, W., Volkamer, R. M., Sheehy, P., Molina, L., and André, M.: Measurements of Volatile Organic Compounds Using Proton Transfer Reaction – Mass Spectrometry during the MILAGRO 2006 Campaign, Atmos. Chem. Phys., 9, 467–481, https://doi.org/10.5194/acp-9-467-2009, 2009.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+--Na+---Cl−-H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Gomez, M. E., Lin, Y., Guo, S., and Zhang, R.: Heterogeneous chemistry of glyoxal on acidic solutions – An oligomerization pathway for secondary organic aerosol formation, J. Phys. Chem., 118, 4457–4463, https://doi.org/10.1021/jp509916r, 2015.
Guo, S., Hu, M., Zamora, M. L., Peng, J., Shang, D., Zheng, J., Du, Z., Wu, Z., Shao, M., Zeng, L., Molina, M. J., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, https://doi.org/10.1073/pnas.1419604111, 2014.
Guo, S., Hu, M., Lin, Y., Gomez-Hernandez, M., Zamora, M. L., Peng, J., Collins, D. R., and Zhang, R.: OH-Initiated Oxidation of m-Xylene on Black Carbon Aging, Environ. Sci. Technol., 50, 8605–8612, https://doi.org/10.1021/acs.est.6b01272, 2016.
Guo, S., Hu, M., Peng, J., Wu, Z., Zamora, M. L., Shang, D., Du, Z., Zheng, J., Fang, X., Tang, R., Wu, Y., Zeng, L., Shuai, S., Zhang, W., Wang, Y., Ji, Y., Li, Y., Zhang, A. L., Wang, W., Zhang, F., Zhao, J., Gong, X., Wang, C., Molina, M. J., and Zhang, R.: Remarkable nucleation and growth of ultrafine particles from vehicular exhaust, P. Natl. Acad. Sci. USA, 117, 3427–3432, https://doi.org/10.1073/pnas.1916366117, 2020.
Heald, C. L., Jacob, D. J., Park, R. J., Russell, L. M., Huebert, B. J., Seinfeld, J. H., Liao, H., and Weber, R. J.: A Large Organic Aerosol Source in the Free Troposphere Missing from Current Models, Geophys. Res. Lett., 32, 1–4, 2005.
Hems, R. F. and Abbatt, J. P. D.: Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Sp. Chem., 2, 225–234, https://doi.org/10.1021/acsearthspacechem.7b00123, 2018.
Hodzic, A., Kasibhatla, P. S., Jo, D. S., Cappa, C. D., Jimenez, J. L., Madronich, S., and Park, R. J.: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 16, 7917–7941, https://doi.org/10.5194/acp-16-7917-2016, 2016.
Hua, W., Jubb, A. M., and Allen, H. C.: Electric Field Reversal of Na2SO4, (NH4)2SO4, and Na2CO3 Relative to CaCl2 and NaCl at the Air/Aqueous Interface Revealed by Heterodyne Detected Phase-Sensitive Sum Frequency, J. Phys. Chem. Lett., 2, 2515–2520, https://doi.org/10.1021/jz200888t, 2011.
Huang, Y., Zhao, R., Charan, S. M., Kenseth, C. M., Zhang, X., and Seinfeld, J. H.: Unified Theory of Vapor–Wall Mass Transport in Teflon-Walled Environmental Chambers, Environ. Sci. Technol., 52, 2134–2142, https://doi.org/10.1021/acs.est.7b05575, 2018.
IPCC (Intergovernmental Panel on Climate Change): Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, 2013.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, P. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1705463114, 2017.
Ji, Y., Shi, Q., Li, Y., An, T., Zheng, J., Peng, J., Gao, Y., Chen, J., Li, G., Wang, Y., Zhang, F., Zhang, A. L., Zhao, J., Molina, M. J., and Zhang, R.: Carbenium ion-mediated oligomerization of methylglyoxal for secondary organic aerosol formation, P. Natl. Acad. Sci. USA, 117, 13294–13299, https://doi.org/10.1073/pnas.1912235117, 2020.
Jia, L. and Xu, Y.: Effects of Relative Humidity on Ozone and Secondary Organic Aerosol Formation from the Photooxidation of Benzene and Ethylbenzene, Aerosol Sci. Tech., 48, 1–12, https://doi.org/10.1080/02786826.2013.847269, 2014.
Jia, L. and Xu, Y.: Different roles of water in secondary organic aerosol formation from toluene and isoprene, Atmos. Chem. Phys., 18, 8137–8154, https://doi.org/10.5194/acp-18-8137-2018, 2018.
Li, G., Zhang, R., Fan, J., and Tie, X.: Impacts of biogenic emissions on photochemical ozone production in Houston, Texas, J. Geophys. Res., 112, D10309, https://doi.org/10.1029/2006JD007924, 2007.
Li, Y., Ji, Y., Zhao, J., Wang, Y., Shi, Q., Peng, J., Wang, Y., Wang, C., Zhang, F., Wang, Y., Seinfeld, J. H., and Zhang, R.: Unexpected Oligomerization of Small α-Dicarbonyls for Secondary Organic Aerosol and Brown Carbon Formation, Environ. Sci. Technol., 55, 4430–4439, https://doi.org/10.1021/acs.est.0c08066, 2021a.
Li, Y., Zhao, J., Wang, Y., Seinfeld, J. H., and Zhang, R.: Multigeneration Production of Secondary Organic Aerosol from Toluene Photooxidation, Environ. Sci. Technol., 55, 8592–8603, https://doi.org/10.1021/acs.est.1c02026, 2021b.
Liu, J., Zhang, F., Xu, W., Sun, Y., Chen, L., Li, S., Ren, J., Hu, B., Wu, H., and Zhang, R.: Hygroscopicity of Organic Aerosols Linked to Formation Mechanisms, Geophys. Res. Lett., 48, e2020GL091683, https://doi.org/10.1029/2020GL091683, 2021.
Marrero-Ortiz, W., Hu, M., Du, Z., Ji, Y., Wang, Y. Y., Guo, S., Lin, Y., Gomez-Hermandez, M., Peng, J., Li, Y., Secrest, J., Zamora, M. L., Wang, Y. Y., An, T., and Zhang, R.: Formation and Optical Properties of Brown Carbon from Small α-Dicarbonyls and Amines, Environ. Sci. Technol., 53, 117–126, https://doi.org/10.1021/acs.est.8b03995, 2019.
McMurry, P. H. and Rader, D. J.: Aerosol Wall Losses in Electrically Charged Chambers, Aerosol Sci. Tech., 4, 249–268, https://doi.org/10.1080/02786828508959054, 1985.
Molina, L. T.: Introductory lecture: air quality in megacities, Faraday Discuss., 226, 9–52, https://doi.org/10.1039/d0fd00123f, 2021.
Moise, T., Flores, J. M., and Rudich, Y.: Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes, Chem. Rev., 115, 4400–4439, 2015.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
NASEM (National Academies of Sciences Engineering and Medicine): The Future of Atmospheric Chemistry Research: Remembering Yesterday, Understanding Today, Anticipating Tomorrow, The National Academies Press, Washington, DC, https://doi.org/10.17226/23573, 2016.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
Nishino, N., Arey, J., and Atkinson, R.: Formation Yields of Glyoxal and Methylglyoxal from the Gas-Phase OH Radical-Initiated Reactions of Toluene, Xylenes, and Trimethylbenzenes as a Function of NO2 Concentration, J. Phys. Chem. A, 114, 10140–10147, https://doi.org/10.1021/jp105112h, 2010.
Peng, J., Hu, M., Shang, D., Wu, Z., Du, Z., Tan, T., Wang, Y., Zhang, F., and Zhang, R.: Explosive secondary aerosol formation during severe haze in the North China Plain, Environ. Sci. Technol., 55, 2189–2207, 2021.
Pope III, C. A., Burnett, R. T., Thun, M. J., Calle, E. E., Krewski, D., Ito, K., and Thurston, G. D.: Lung Cancer, Cardiopulmonary Mortality, and Long-Term Exposure to Fine Particulate Air Pollution, J. Am. Med. Assoc., 287, 1132–1141, 2002.
Qi, X., Zhu, S., Zhu, C., Hu, J., Lou, S., Xu, L., Dong, J., and Cheng, P.: Smog chamber study of the effects of NOx and NH3 on the formation of secondary organic aerosols and optical properties from photo-oxidation of toluene, Sci. Total Environ., 727, 138632, https://doi.org/10.1016/j.scitotenv.2020.138632, 2020.
Ravishankara, A. R.: Heterogeneous and multiphase chemistry in the troposphere, Science, 276, 1058–1065, 1997.
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, ISBN 978-1-118-94740-1, 2016.
Shi, Q., Zhang, W., Ji, Y., Wang, J., Qin, D., Chen, J., Gao, Y., Li, G., and An, T.: Enhanced uptake of glyoxal at the acidic nanoparticle interface: implications for secondary organic aerosol formation, Environ. Sci. Nano, 7, 1126–1135, 2020.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Song, C., Na, K., Warren, B., Malloy, Q., and Cocker, D. R.: Secondary Organic Aerosol Formation from m-Xylene in the Absence of NOx, Environ. Sci. Technol., 41, 7409–7416, https://doi.org/10.1021/es070429r, 2007.
Suh, I., Lei, W., and Zhang, R.: Experimental and theoretical studies of isoprene reaction with NO3, J. Phys. Chem., 105, 6471–6478, 2001.
Tan, Y., Lim, Y. B., Altieri, K. E., Seitzinger, S. P., and Turpin, B. J.: Mechanisms leading to oligomers and SOA through aqueous photooxidation: insights from OH radical oxidation of acetic acid and methylglyoxal, Atmos. Chem. Phys., 12, 801–813, https://doi.org/10.5194/acp-12-801-2012, 2012.
Wang, G., Zhang, F., Peng, J., Duan, L., Ji, Y., Marrero-Ortiz, W., Wang, J., Li, J., Wu, C., Cao, C., Wang, Y., Zheng, J., Secrest, J., Li, Y., Wang, Y., Li, H., Li, N., and Zhang, R.: Particle acidity and sulfate production during severe haze events in China cannot be reliably inferred by assuming a mixture of inorganic salts, Atmos. Chem. Phys., 18, 10123–10132, https://doi.org/10.5194/acp-18-10123-2018, 2018.
Wang, L., Khalizov, A. F., Zheng, J., Xu, W., Ma, Y., Lal, V., and Zhang, R.: Atmospheric nanoparticles formed from heterogeneous reactions of organics, Nat. Geosci., 3, 238–242, https://doi.org/10.1038/ngeo778, 2010.
Wang, M., Chen, D., Xiao, M., Ye, Q., Stolzenburg, D., Hofbauer, V., Ye, P., Vogel, A. L., Mauldin, R. L., Amorim, A., Baccarini, A., Baumgartner, B., Brilke, S., Dada, L., Dias, A., Duplissy, J., Finkenzeller, H., Garmash, O., He, X.-C., Hoyle, C. R., Kim, C., Kvashnin, A., Lehtipalo, K., Fischer, L., Molteni, U., Petäjä, T., Pospisilova, V., Quéléver, L. L. J., Rissanen, M., Simon, M., Tauber, C., Tomé, A., Wagner, A. C., Weitz, L., Volkamer, R., Winkler, P. M., Kirkby, J., Worsnop, D. R., Kulmala, M., Baltensperger, U., Dommen, J., El-Haddad, I., and Donahue, N. M.: Photo-oxidation of Aromatic Hydrocarbons Produces Low-Volatility Organic Compounds, Environ. Sci. Technol., 54, 7911–7921, https://doi.org/10.1021/acs.est.0c02100, 2020.
Wang, Y., Khalizov, A., Levy, M., and Zhang, R.: Light absorbing aerosols and their atmospheric impacts, Atmos. Environ., 81, 713–715, https://doi.org/10.1016/j.atmosenv.2013.09.034, 2013.
Wang, Y., Lee, K.-H., Lin, Y., Levy, M., and Zhang, R.: Distinct effects of anthropogenic aerosols on tropical cyclones, Nat. Clim. Change, 4, 368–373, https://doi.org/10.1038/nclimate2144, 2014.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, 2018.
Xue, H., Khalizov, A. F., Wang, L., Zheng, J., and Zhang, R.: Effects of Coating of Dicarboxylic Acids on the Mass–Mobility Relationship of Soot Particles, Environ. Sci. Technol., 43, 2787–2792, https://doi.org/10.1021/es803287v, 2009.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Zhang, D., Lei, W., and Zhang, R.: Mechanism of OH formation from ozonolysis of isoprene: Kinetics and product yields, Chem. Phys. Lett., 358, 171–179, 2002.
Zhang, F., Wang, Y., Peng, J., Chen, L., Sun, Y., Duan, L., Ge, X., Li, Y., Zhao, J., Liu, C., Zhang, X., Zhang, G., Pan, Y., Wang, Y., Zhang, A. L., Ji, Y., Wang, G., Hu, M., Molina, M. J., and Zhang, R.: An unexpected catalyst dominates formation and radiative forcing of regional haze, P. Natl. Acad. Sci. USA, 117, 3960–3966, https://doi.org/10.1073/pnas.1919343117, 2020.
Zhang, Q., Xu, Y., and Jia, L.: Secondary organic aerosol formation from OH-initiated oxidation of m-xylene: effects of relative humidity on yield and chemical composition, Atmos. Chem. Phys., 19, 15007–15021, https://doi.org/10.5194/acp-19-15007-2019, 2019.
Zhang, R., Leu, M. T., and Keyser, L. F.: Heterogeneous reactions involving ClONO2, HCl, and HOCl on liquid sulfuric acid surfaces, J. Phys. Chem., 98, 13563–13574, 1994.
Zhang, R., Suh, I., Zhao, J., Zhang, D., Fortner, E. C., Tie, X., Molina, L. T., and Molina, M. J.: Atmospheric New Particle Formation Enhanced by Organic Acids, Science, 304, 1487–1490, https://doi.org/10.1126/science.1095139, 2004.
Zhang, R., Wang, L., Khalizov, A. F., Zhao, J., Zheng, J., McGraw, R. L., and Molina, L. T.: Formation of nanoparticles of blue haze enhanced by anthropogenic pollution, P. Natl. Acad. Sci. USA, 106, 17650–17654, https://doi.org/10.1073/pnas.0910125106, 2009.
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of Urban Fine Particulate Matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015.
Zhang, R., Johnson, N. M., and Li, Y.: Establishing the exposure-outcome relation between airborne particulate matter and children's health, Thorax, 77, 322–323, https://doi.org/10.1136/thoraxjnl-2021-217915, 2021.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of Vapor Wall Loss in Laboratory Chambers on Yields of Secondary Organic Aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, 2014.
Zhao, J. and Zhang, R.: Proton transfer reaction rate constants between hydronium ion (H3O+) and volatile organic compounds, Atmos. Environ., 38, 2177–2185, https://doi.org/10.1016/j.atmosenv.2004.01.019, 2004.
Zhao, J., Zhang, R., Fortner, E. C., and North, S. W.: Quantification of hydroxycarbonyls from OH-isoprene reactions, J. Am. Chem. Soc., 126, 2686–2687, 2004.
Zhao, J., Levitt, N. P., and Zhang, R.: Heterogeneous chemistry of octanal and 2,4-hexadienal with sulfuric acid, Geophys. Res. Lett., 32, L09802, https://doi.org/10.1029/2004GL022200, 2005a.
Zhao, J., Zhang, R., Misawa, K., and Shibuya, K.: Experimental product study of the OH-initiated oxidation of m-xylene, J. Photochem. Photobio. A, 176, 199–207, https://doi.org/10.1016/j.jphotochem.2005.07.013, 2005b.
Zhu, J., Penner, J. E., Lin, G., Zhou, C., Xu, L., and Zhuang, B.: Mechanism of SOA formation determines magnitude of radiative effects, P. Natl. Acad. Sci. USA, 114, 12685–12690, https://doi.org/10.1073/pnas.1712273114, 2017.