the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Jan 2022
| 20 Jan 2022
An assessment of the tropospherically accessible photo-initiated ground state chemistry of organic carbonyls
Keiran N. Rowell
Scott H. Kable
Meredith J. T. Jordan
Carbonyls are among the most abundant volatile organic compounds in the atmosphere. They are central to atmospheric photochemistry as absorption of near-UV radiation by the C=O chromophore can lead to photolysis. If photolysis does not occur on electronic excited states, non-radiative relaxation to the ground state will form carbonyls with extremely high internal energy. These “hot” molecules can access a range of ground state reactions. Up to nine potential ground state reactions are investigated at the B2GP-PLYP-D3/def2-TZVP level of theory for a test set of 20 representative carbonyls. Almost all are energetically accessible under tropospheric conditions. Comparison with experiment suggests the most significant ground state dissociation pathways will be concerted triple fragmentation in saturated aldehydes, Norrish type III dissociation to form another carbonyl, and H2 loss involving the formyl H atom in aldehydes. Tautomerisation, leading to more reactive unsaturated species, is also predicted to be energetically accessible and is likely to be important when there is no low-energy ground state dissociation pathway, for example in α,β-unsaturated carbonyls and some ketones. The concerted triple fragmentation and H2-loss pathways have immediate atmospheric implications for global H2 production, and tautomerisation has implications for the atmospheric production of organic acids.
- Article
(2642 KB) - Full-text XML
-
Supplement
(4243 KB) - BibTeX
- EndNote





Carbonyls are a class of volatile organic compounds (VOCs) central to atmospheric chemistry. They arise, in large quantities, from primary anthropogenic and biogenic emissions and via secondary atmospheric processes (Kesselmeier and Staudt, 1999; Millet et al., 2010; Chen et al., 2014). For example, small organic carbonyls, such as acetone, formaldehyde and acetaldehyde, which are ranked in the top 25 of all anthropogenically emitted molecules by mass (Simon et al., 2010), are emitted as pollutants (Chen et al., 2014). Up to 10 % of carbon initially fixed by plants is also subsequently emitted as biological volatile organic compounds (BVOCs), which include directly emitted carbonyls, as well as other volatile species that are subsequently oxidised to carbonyls (Seco et al., 2007). Indeed, carbonyls are generated throughout the oxidative degradation pathways of all VOCs (Carlier et al., 1986; Atkinson and Arey, 2003). Atmospheric concentrations of organic carbonyls are in the pptV–ppbV range (Tanner et al., 1996), with high concentrations found at low altitudes and in polluted environments (Lee et al., 1998; Pal et al., 2008; Guo et al., 2014; Menchaca-Torre et al., 2015).
There are of the order of 106 known or suspected VOCs in the atmosphere (Goldstein and Galbally, 2007). As a category, BVOCs are structurally complex, and carbonyls formed by their subsequent oxidation will reflect this structural diversity. Consequently, atmospheric carbonyls can have varied chemical structures and reactivities (Kawamura et al., 2000; Atkinson et al., 2006). Structural complexities include double bonds, branched carbon chains and varied functionalisation.
The atmospheric importance of carbonyls arises because they are one of the few classes of VOC that can efficiently absorb solar radiation in the troposphere. In the atmosphere, carbonyl photochemistry is most notable for its generation of radicals following excited state bond cleavage, the Norrish type I reaction (NTI) (Norrish and Appleyard, 1934), and these radicals driving key atmospheric reactions (Lary and Shallcross, 2000; Yang et al., 2018). However, many other reactions, including reactions on the ground electronic state, S0, are possible.
The intensity of solar radiation reaching the troposphere is shown in grey on the right axis of Fig. 1. Most UV-B radiation (280–315 nm) is absorbed by the ozone layer, and the intensity of UV-A radiation (315–400 nm) increases with increasing wavelength. The maximum photon energy available in the troposphere is ∼400 kJ/mol (300 nm) and is shown in violet on relevant energetic plots below. The overlap of a given carbonyl absorption spectrum and the solar spectrum defines the actinic energy window in which photo-initiated chemistry can occur. In carbonyls, a near-UV photon excites the S1←S0 () transition of the C=O chromophore. The absorption wavelength depends on the structural class of the carbonyl and is determined by the substituents attached to the C=O. UV absorption spectra for representative carbonyls are shown in Fig. 1.
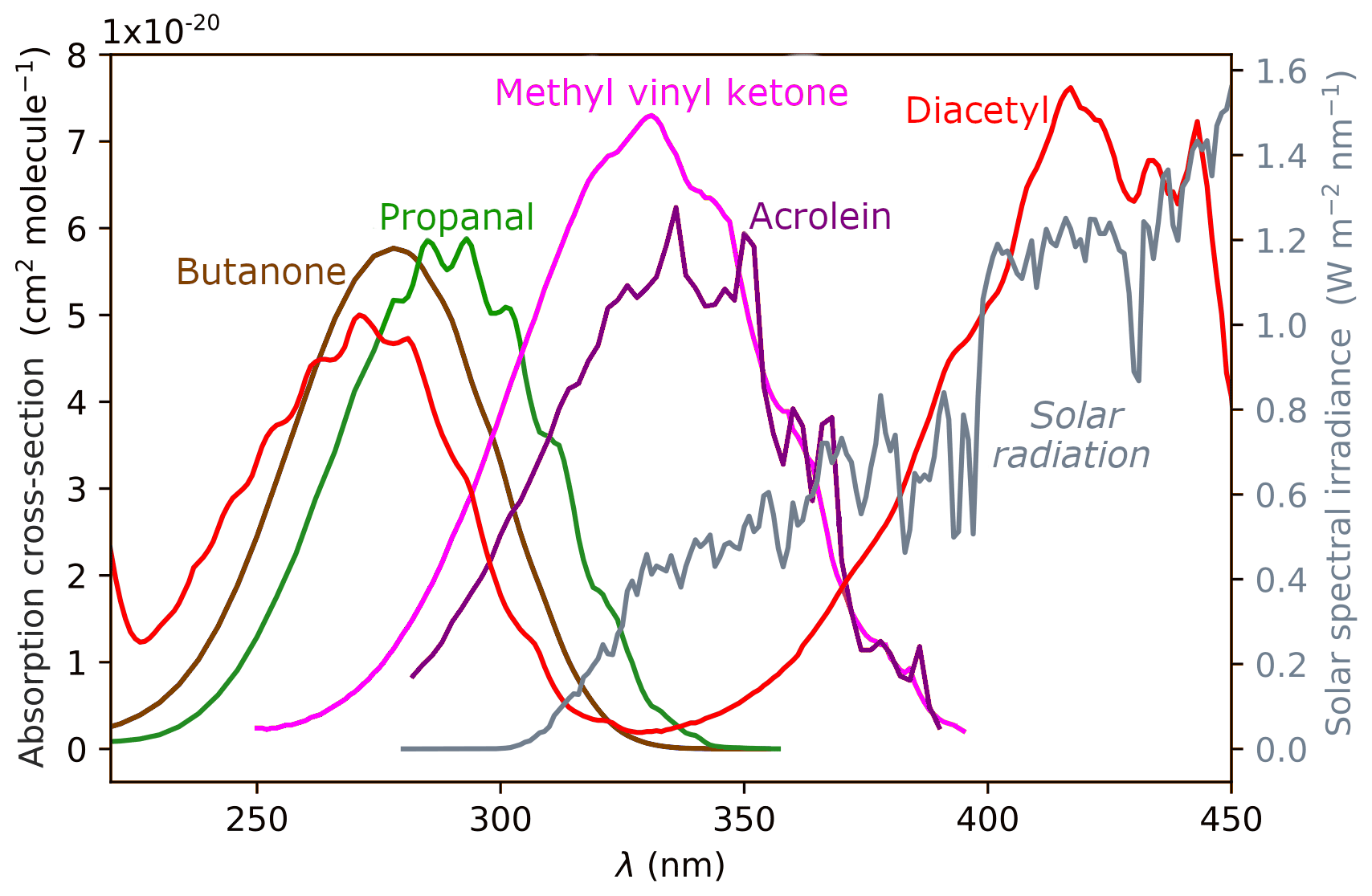
Figure 1UV absorption spectra of representative carbonyls: aldehydes (represented by propanal), ketones (butanone), enals (acrolein), enones (methyl vinyl ketone) and α-dicarbonyls (diacetyl). The solar spectrum is shown in grey, plotted against the right ordinate.
Over the past couple of decades, it has become apparent that there are carbonyl “photochemical” pathways on the ground electronic state (S0). These photo-initiated reactions on S0 include the novel class of “roaming” reactions (Townsend et al., 2004; Bowman and Houston, 2017), in which partly dissociated radical fragments become trapped in each other's van der Waals well and recombine to generate molecular products. However, more conventional S0 transition state (TS) reactions, which occur over a barrier, as well as barrierless S0 reactions, are also being discovered (Heazlewood et al., 2011; So et al., 2018; Toulson et al., 2018). Despite the growing evidence of carbonyl ground state reactions, following excitation at energies accessible in the troposphere (λ>300 nm), these reactions are almost entirely absent from the reaction schemes of contemporary atmospheric models (Wild et al., 2000, 1997, 2002; Bey et al., 2001).
1.1 The tropospheric fate of carbonyls
The tropospheric fate of an illustrative carbonyl, acetaldehyde (CH3CHO), is shown schematically in Fig. 2. A near-UV photon excites the S1←S0 () transition. The actinic absorption window, indicated in Fig. 2 by violet shading, is bounded by the lowest energy excitation to S1 and the highest energy photon available in the troposphere. For most carbonyls the S1 state is bound at actinic energies, as shown in Fig. 2, and S1 reactions are inaccessible (Haas, 2004; Rowell et al., 2019). Intersystem crossing (ISC) from S1 to the lowest energy triplet state, T1, is formally spin-forbidden; however, as shown schematically in Fig. 2, carbonyl S1 and T1 minimum energy geometries are almost identical, and the energetic separation between them is small. This allows relatively fast S1→T1 ISC (Hansen and Lee, 1975), indicated in Fig. 2 by a thick ISC arrow. On T1, NTI α-bond cleavage is the most common photolysis reaction (Kirkbride and Norrish, 1931; Zhu et al., 2009), and we define NTIa and NTIb as cleavage of the α-bond to the larger and smaller substituents, respectively. In CH3CHO NTIa results in the formation of •CH3 + •HCO and NTIb yields CH3CO•+•H. On T1 both NTIa and NTIb involve an energetic barrier, shown in orange in Fig. 2.
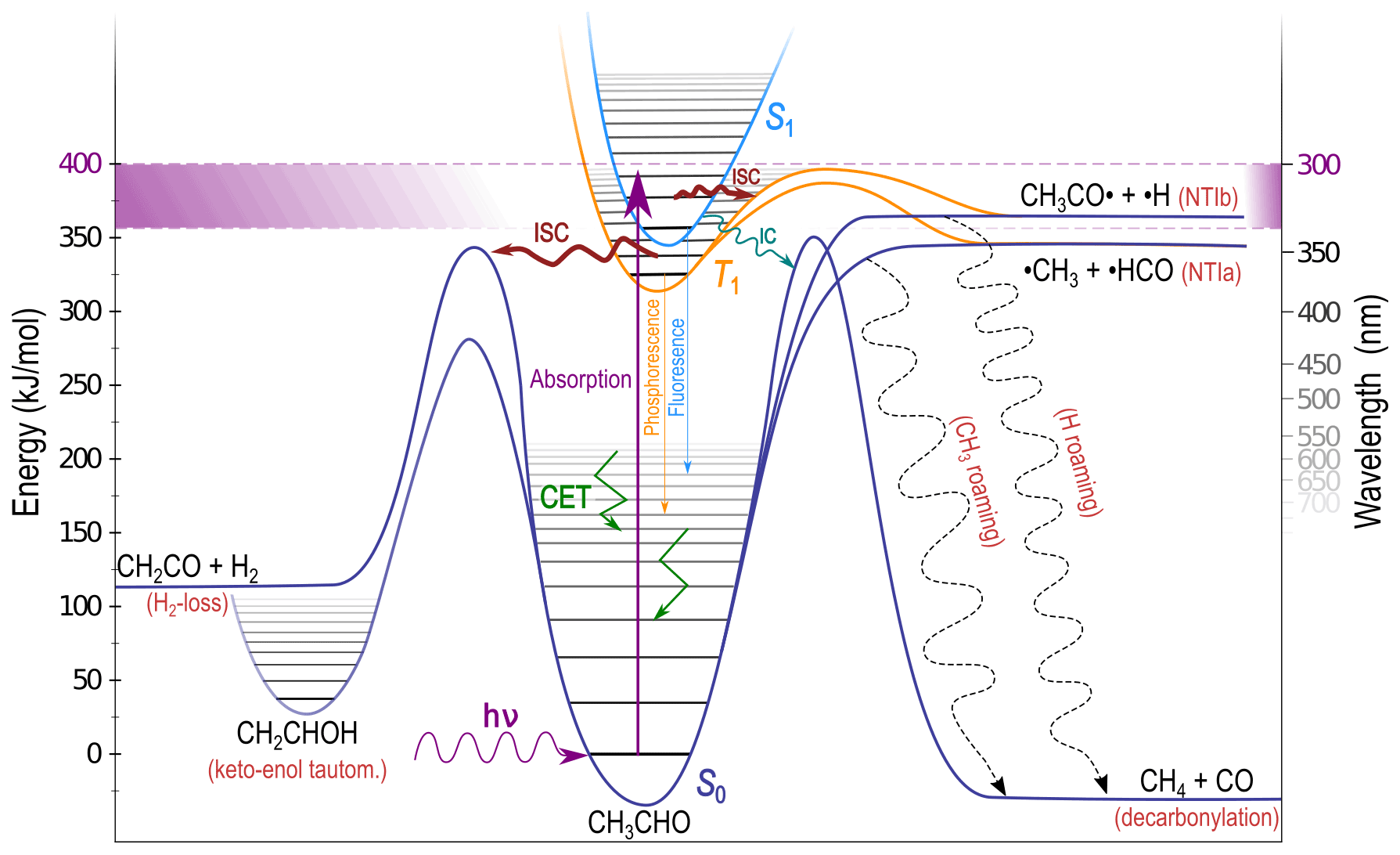
Figure 2Schematic of the atmospheric fate of acetaldehyde (CH3CHO) following absorption of a near-UV photon. Possible outcomes depend on excitation energy and include electronic surface crossing, collisional energy transfer (CET), unimolecular dissociation including roaming (dashed arrows) and isomerisation.
For larger carbonyls, the Norrish type II (NTII) intramolecular hydrogen-transfer reaction, forming an alkene and an enol, also occurs and is initiated in an electronic excited state (Tadić et al., 2002, 2012). Although NTI and NTII are typically the major contributors to the photolysis quantum yield (QY) of carbonyls (Wagner and Zepp, 1972; Wagner and Klán, 2004; Zhu et al., 2009), if excitation is to energies below excited state photolysis thresholds, or if the electronically excited carbonyl is collisionally cooled below these thresholds, non-radiative relaxation to S0 must occur; fluorescence and phosphorescence QYs in carbonyls are typically <1 % (Heicklen and Noyes, 1959; Copeland and Crosley, 1985). The large energy separation between S1 and S0 means S1→S0 internal conversion (IC) will be slow and less important than ISC (Lee and Chen, 1997). Indeed, rapid IC is usually associated with conical intersections: geometric configurations in which two electronic surfaces are degenerate (Schuurman and Stolow, 2018). In the main, these lie above the actinic energy range in carbonyls (Diau et al., 2001; Chen and Fang, 2009; Toulson et al., 2017).
The carbonyl RC=O group is planar in the S0 minimum energy geometry but pyramidalised in the S1 and T1 minima (Godunov et al., 1995). The change in electron spin, geometric dissimilarity and energetic separation between the T1 and S0 minima suggest T1→S0 ISC may be slow. However, the non-bonding oxygen lone pair orbital, n, in C=O is approximately orthogonal to the anti-bonding π* excited state orbital. Thus the change in electron spin angular momentum in T1→S0 relaxation is accompanied by a change in electron orbital angular momentum. This results in conservation of total electron angular momentum and a high T1→S0 ISC rate, as dictated by El-Sayed's rule (El-Sayed, 1961). Indeed, ISC to S0 can be kinetically competitive even when NTI reactions are accessible (Heazlewood et al., 2009; Amaral et al., 2010).
The most likely carbonyl relaxation route to S0 is therefore via two sequential ISC steps (Toulson et al., 2017). This generates vibrationally “hot” photoexcited S0 molecules that retain much of the initial UV photon energy as internal energy. This “hot” carbonyl can collide with other atmospheric molecules, and collisional energy transfer (CET) removes excess vibrational energy, returning the molecule to thermal equilibrium. This is indicated in Fig. 2 by angular green arrows in the S0 well, although CET will also occur in S1, T1 and isomer wells (Heazlewood et al., 2011; Andrews et al., 2012). The photoexcited S0 carbonyls are conformationally flexible, and S0 dissociation and isomerisation will compete with CET.
This paper explores, using computational chemistry, the possible S0 dissociation and isomerisation pathways and how they depend on the structural class of the carbonyl.
1.2 The test set
There are a vast number of carbonyls in the atmosphere with little experimental data in comparison to this diversity (Atkinson et al., 1992). Here we consider a test set of 20 carbonyls, which includes the 12 species that have explicit photolysis reactions within the Master Chemical Mechanism (Jenkin et al., 1997) and 5 species in the GEOS-Chem (Bey et al., 2001) atmospheric chemistry models. We also include carbonyls with atmospherically representative structural features based on bond order or substitution around the C=O chromophore. The 20 carbonyls in the test set are shown in Fig. 3 and were previously used in a study of excited state NTI photolysis (Rowell et al., 2019). They are colour-coded into seven structurally distinct classes, which determine their absorption spectra (Fig. 1) and chemical behaviour (Atkinson et al., 1992).

Quantum chemistry methods are used to calculate reaction thresholds for up to nine S0 unimolecular reactions that may be accessible under tropospheric conditions. In our quantum chemistry calculations, the threshold is defined as the limiting zero-point-corrected electronic energy for the reaction to occur. Experimental thresholds are the lowest energies for which reaction has been observed to occur and hence include tunnelling through any barrier to reaction. In the figures below, the nine possible unimolecular reactions are colour-coded according to Fig. 3. Explicitly, the reactions are as follows:
-
decarbonylation (CO loss)
-
concerted triple fragmentation (TF)
-
Norrish type III β-H transfer (NTIII)
-
H2 loss from hydrogens at the formyl and α positions (formyl + α)
-
H2 loss from hydrogens at the α and β positions (α+β)
-
H2 loss from hydrogens at the β and γ positions (β+γ)
-
alkane/alkene elimination (AE) in saturated/unsaturated ketones
-
keto–enol tautomerisation
-
enal–ketene tautomerisation.
Ground state NTI reactions are also possible for all carbonyls in the test set. These reactions are barrierless, and their asymptotic energies have been previously reported (Rowell et al., 2019). Other isomerisations are possible. For example, acetaldehyde can isomerise to oxirane or methylhydroxycarbene. Although these isomers are theoretically accessible at actinic energies, their formation barriers are very high with low barriers for the reverse isomerisation (Heazlewood et al., 2011). Thus they are unlikely to be collisionally stabilised before isomerising back to the parent carbonyl. Norrish type II reactions are possible for butanal, pentanal and pentan-2-one and have not been considered here as they involve excited electronic states (Tadić et al., 2002, 2012).
Seven of the S0 reactions listed above are illustrated for butanal in Fig. S1 of the Supplement; alkane elimination is only available to ketones and enal–ketene tautomerisation is only available to acrolein, methacrolein and crotonaldehyde. The figure also includes schematic representations of the concerted TF and NTIII mechanisms.
The calculated S0 thresholds are used to determine general energetic trends that can be applied to larger carbonyls and to identify the most likely S0 reactions for each class of carbonyl under tropospheric conditions. These reactions are then assessed in terms of their tropospheric significance. Ultimately, however, the most important reactions will be those with the highest reaction rate coefficients. Although the threshold energy is, in general, the largest contributor to the magnitude of a reaction rate coefficient, entropy is also important. For similar thresholds, reactions with loose transition states, for example, the variational transition states associated with barrierless reactions like the S0 NTI reactions, will have higher rate coefficients. By identifying reactions that may be important we will be able to focus future work on calculating their reaction rate coefficients and incorporating them into tropospheric master equation models.
S0 calculations were performed with the B2GP-PLYP double-hybrid density functional (Karton et al., 2008), using the def2-TZVP canonical basis set (Weigend and Ahlrichs, 2005) and the RIJK resolution of the identity approximation with the def2/JK and def2-TZVP/C auxiliary basis sets. The use of the RIJK approximation, and the “RI-” prefix, is taken as implicit. All calculations were dispersion-corrected using the D3(BJ) scheme (Grimme et al., 2010, 2011), abbreviated D3 below. Geometry optimisations and frequency calculations were performed using the ORCA electronic structure programme (Neese, 2017). All zero-point energies were scaled with the literature scaling factor of 0.9752 (Kesharwani et al., 2015).
The B2GP-PLYP functional has been found to give errors <8 kJ/mol across a diverse range of benchmark data (Karton et al., 2008). B2GP-PLYP-D3/def2-TZVP was also validated by the authors against experimental carbonyl T1 photolysis data, with mean absolute deviations of ∼6 kJ/mol (Rowell et al., 2019). Comparisons of our B2GP-PLYP-D3/def2-TZVP thresholds with previous literature are provided in Tables S1–S6 of the Supplement and indicate excellent agreement with high-level correlated methods using accurate geometries. Note that, for S0 energy differences between structurally similar molecules, as studied here, relative errors would be expected to be lower, approaching 4 kJ/mol “chemical accuracy” (Pople, 1999).
Transition states were confirmed as first-order saddle points with a single imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were performed to ensure that the normal mode corresponding to the imaginary frequency connected the desired reactant and product minima. In order to reduce computational burden, IRC calculations were not performed where a TS had a structure and reaction coordinate directly analogous to a homologous molecule for which the reaction mechanism had been verified.
Schematic representation of the optimised saddle point structures are shown in the figures below. They are also represented in Figs. S2–S7, and their Cartesian coordinates are provided in the Supplement. In general, only the lowest energy thresholds for each reaction class are discussed below, and higher energy thresholds are reported in the Supplement.
The lowest energy calculated reaction thresholds for the up to nine S0 reactions considered are reported in Table 1 for carbonyls in the test set. Additional, higher energy thresholds are reported in the Supplement. For some of the carbonyls considered, asymptotic energies for NTI reactions can be obtained from (effectively) experimental enthalpies of formation at 0 K, tabulated in the Active Thermochemical Tables (Ruscic et al., 2005; Ruscic and Bross, 2020). The results in Table 1 with uncertainties are based on these experimental enthalpies of formation. The remaining NTI asymptotic energies shown in Table 1 have been previously calculated and are reproduced from Rowell et al. (2019).
Table 1Lowest energy zero-point vibrational-energy-corrected B2GP-PLYP-D3/def2-TZVP S0 thresholds (kJ/mol) for the indicated unimolecular reactions of the carbonyls in the test set (see text).
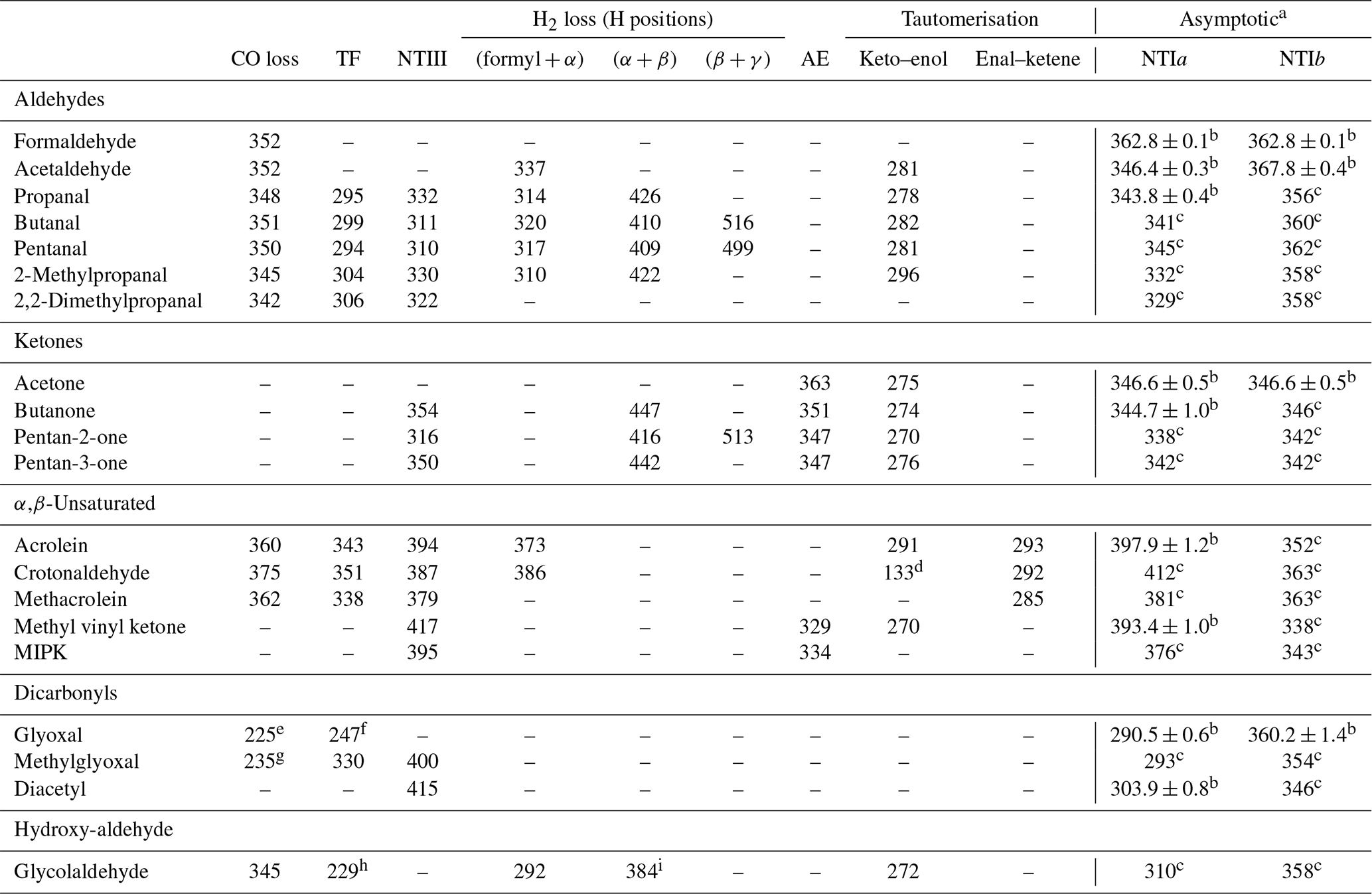
a NTIa and NTIb are cleavages to the larger and smaller fragments, respectively. b Values with uncertainties are determined from experimental enthalpies of formation from the Active Thermochemical Tables (v. 1.22p) (Ruscic and Bross, 2020; Ruscic et al., 2005). c Reproduced from Rowell et al. (2019). d For a [1,5]-H atom shift to form but-1,3-dien-1-ol. e For formation of formaldehyde + CO. f For a four-centre TS involving the two formyl H atoms. g For formation of acetaldehyde + CO. h The β-H for the TF transition state is from the OH moiety. i The H2-loss TS involves the OH hydrogen.
Each reaction type is discussed separately below, with the three H2-loss reactions discussed together. These data and available literature and experimental results are then analysed to determine chemically rationalised trends that will be applicable to larger carbonyls.
3.1 Decarbonylation (CO loss)
Decarbonylation, the unimolecular loss of a CO molecule, is uncommon in gas-phase ketones. It has been observed in acetone, although the mechanism is unclear; Skorobogatov et al. (2002) postulated a direct mechanism, presumably via a TS, and roaming reactions have also been suggested in acetone (Goncharov et al., 2008; Saheb and Zokaie, 2018). Here, like previous studies (Saheb and Zokaie, 2018; So et al., 2018), we find no computational evidence for direct decarbonylation TSs in ketones.
In aldehydes, the formyl hydrogen is transferred to the main alkyl chain, forming CO and an alkane: R–(C=O)–H → CO + RH. Both roaming and TS decarbonylation mechanisms have been observed in gas-phase reactions of small aldehydes (Townsend et al., 2004; Houston and Kable, 2006; Heazlewood et al., 2008; Rubio-Lago et al., 2012; Yang et al., 2020). Figure 4 shows TS decarbonylation thresholds for the aldehydes in the test set.
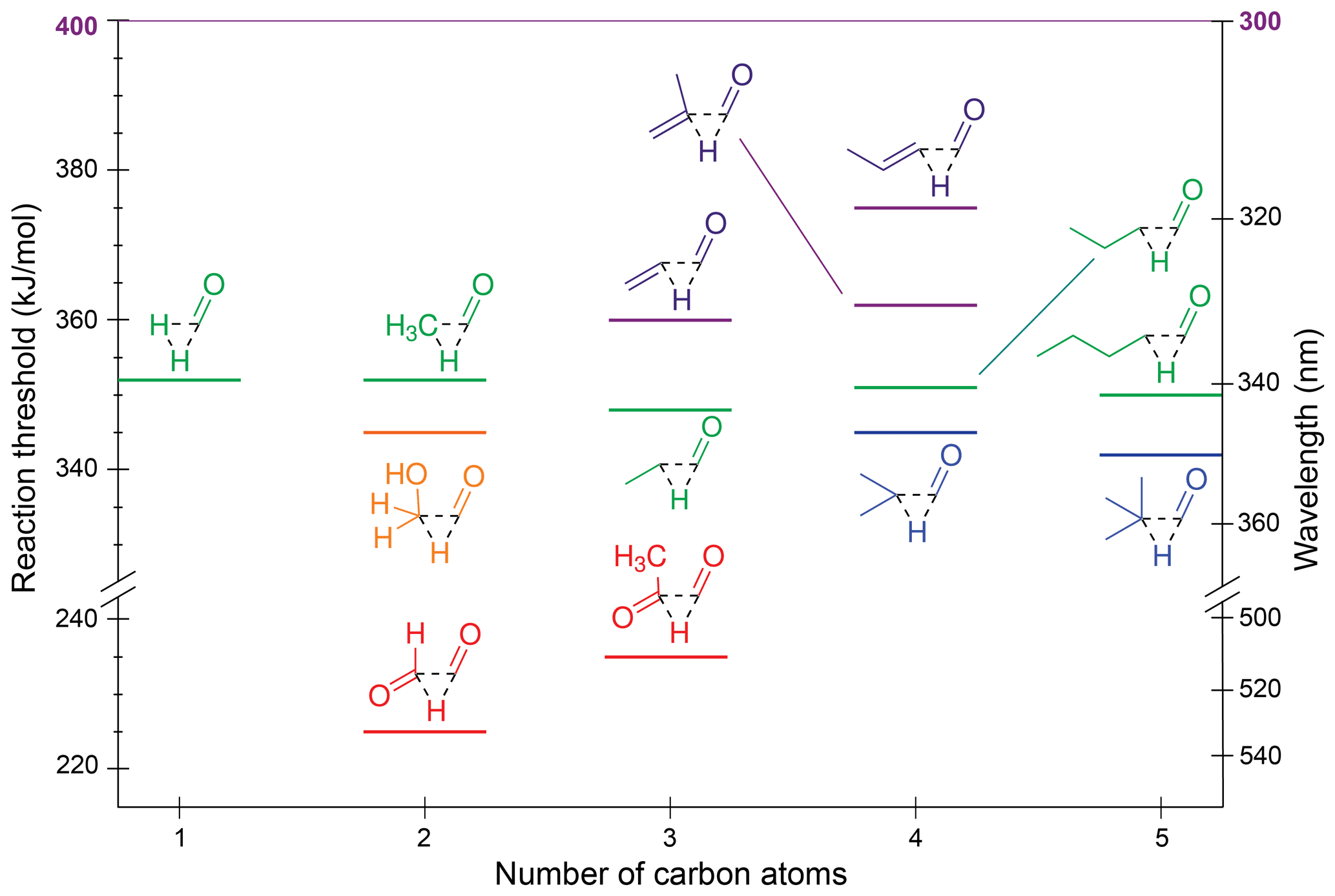
Figure 4Zero-point vibrational-energy-corrected B2GP-PLYP-D3/def2-TZVP S0 decarbonylation thresholds.
One trend apparent in Fig. 4 is that extension of the alkyl chain has little effect on the decarbonylation threshold, with formaldehyde through pentanal all predicted to have ∼350 kJ/mol thresholds. The predicted decrease in threshold upon branching at the α position is <5 kJ/mol.
The decarbonylation thresholds in the α,β-unsaturated enals, acrolein and methacrolein, are ∼10 kJ/mol higher than those for saturated aldehydes as decarbonylation involves breaking a resonance-stabilised C–C bond. Further delocalisation of the π system increases the threshold to 375 kJ/mol for crotonaldehyde.
The α-dicarbonyls are predicted to have the lowest decarbonylation thresholds, which is consistent with the two electron-withdrawing oxygen substituents reducing the electron density between the carbonyl moieties and weakening the α-C–C bond. Four decarbonylation thresholds were found for glyoxal (Table S1 and Fig. S3 in Sect. S2 of the Supplement), with the lowest energy threshold of 225 kJ/mol corresponding to a non-planar TS and formation of formaldehyde and CO. This threshold is lower than the 251 kJ/mol threshold to form hydroxymethylene, HOCH and CO. A similar non-planar TS is found for decarbonylation in methylglyoxal to form acetaldehyde, which has a slightly higher threshold of 235 kJ/mol. Here, substitution strengthens the central C–C bond due to electron donation from the CH3 substituent, and there is also a steric penalty at the TS (Fig. S3a vs. S3e).
All calculated decarbonylation thresholds are accessible at the maximum tropospheric photon energy of 400 kJ/mol, although, with the exception of formaldehyde (Moortgat et al., 1983), CO-loss QYs are low. For example, they are measured to be ∼0.02 in acetaldehyde (Blacet et al., 1942) and ∼0.01 for butanal (Blacet and Calvert, 1951).
The thresholds for roaming decarbonylation reactions are linked to the S0 NTI asymptotic energies (Andrews et al., 2013; Mauguière et al., 2015; Bowman and Houston, 2017). For example, the roaming threshold in formaldehyde is ∼ 2 kJ/mol below the radical dissociation threshold (Townsend et al., 2004; Quinn et al., 2017), and both H and CH3 roaming has been observed in acetaldehyde (Lee et al., 2014). As such, the majority of aldehydes in the test set have roaming thresholds that are similar to or lower in energy than the TS decarbonylation threshold, with formaldehyde a notable exception. Roaming pathways associated with NTI may also be present in ketones (Goncharov et al., 2008; Saheb and Zokaie, 2018). This suggests decarbonylation will be energetically accessible under tropospheric conditions for all carbonyls.
3.2 Triple fragmentation (TF)
We define the TF reaction as a concerted S0 reaction forming three products via a single TS, in which one of these products is H2. For example, the TF reaction in the saturated aldehyde, propanal, is CH3–CH2–(C=O)H → H2C=CH2 + H2 + CO. For enals, the hydrocarbon product will be an alkyne. In dicarbonyls, TF forms H2, CO, and either a second CO or ketene. Predicted S0 TF thresholds are shown in Fig. 5. All of the TF reactions shown, except that for glyoxal, involve a five-centre TS and H loss from a β-hydrogen. In glyoxal, the four-centre TS involves the two formyl hydrogens. The TSs are shown explicitly in Fig. S4 of the Supplement and are “late”, resembling the photolysis products. As a result, TF threshold energies are strongly influenced by the stability of the products.
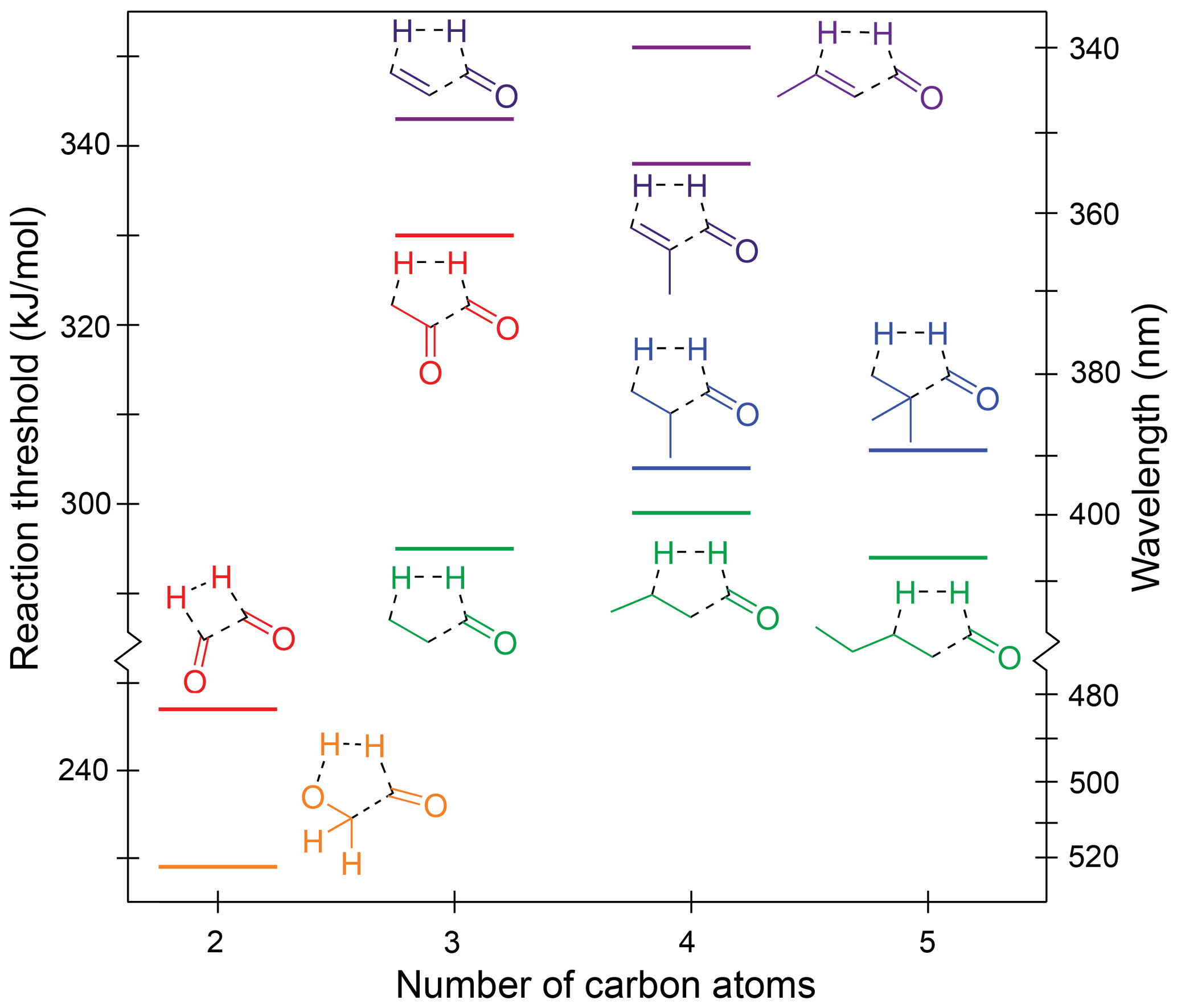
Figure 5Zero-point vibrational-energy-corrected B2GP-PLYP-D3/def2-TZVP S0 concerted triple fragmentation thresholds.
The TF thresholds for the saturated aldehydes are not significantly affected by chain extension, with thresholds of 295, 299 and 294 kJ/mol for propanal, butanal and pentanal, respectively. The effect of alkyl branching at the α position is inconsistent and small. It raises the thresholds to 304 kJ/mol for 2-methylpropanal and 306 kJ/mol for 2,2-dimethylpropanal but lowers it for methacrolein (338 kJ/mol) compared to acrolein (343 kJ/mol). These differences are close to the likely accuracy of the B2GP-PLYP-D3 calculations.
The TF thresholds of the enals are significantly higher than those of the saturated aldehydes at ∼340–350 kJ/mol. This increase is larger than the ∼10 kJ/mol increase seen in decarbonylation thresholds for breaking a delocalised C–C bond and arises because of bond angle strain in the five-centre TF TSs of the α,β-unsaturated species. As shown in Fig. S4, the C–C–C backbone angles in the TSs deviate significantly from their equilibrium values, and the energetic penalty is increased, relative to saturated species, because of the rigidity of the delocalised C–C bonds.
The predicted S0 TF thresholds of the two α-dicarbonyls, glyoxal and methylglyoxal, differ markedly by 83 kJ/mol. This arises because their TS structures and reaction products are qualitatively different (Fig. S4j and k). Methylglyoxal has a five-centre TS that involves strain across the O=C–C=O backbone and also forms a relatively high-energy ketene product. Glyoxal, in contrast, forms exothermically favourable products: H2 and two CO molecules, resulting in a significantly lower S0 TF threshold.
Glycolaldehyde is an atypical carbonyl and has the lowest energy TF pathway, calculated at 229 kJ/mol. Here, the H2-loss channel arises from the combination of the formyl hydrogen and the OH hydrogen, which is more labile than the backbone C–H hydrogens. The C–OH angle in the glycolaldehyde TS also involves minimal ring strain (Fig. S4l). This pathway has not been previously proposed for glycolaldehyde (Wallington et al., 2018; Bacher et al., 2001; Zhu and Zhu, 2010; Cui and Fang, 2011; So et al., 2015) and occurs via a five-centre TS, which is lower in energy than the four-centre TF TS for H2 formation from the formyl and a β CH2 hydrogen. Indeed it is the lowest energy dissociation pathway we have found in glycolaldehyde.
3.3 Norrish type III β-H transfer reaction (NTIII)
Alongside the Norrish type I and II reactions, the lesser known “Norrish type III” reaction (NTIII) was proposed by Zahra and Noyes (1965) to explain the observation of acetaldehyde and propene as products of 3-methylbutan-2-one photoexcitation. NTIII involves a four-centre TS, with β-hydrogen transfer from the backbone to the carbonyl moiety leading to the formation of an aldehyde and an alkene; for example, RCH2CH2(C=O)R′ → R–CH=CH2 + H(C=O)R′. Experimentally, NTIII has been found to be a minor channel (Zahra and Noyes, 1965). However, since the alkene product from NTIII can also be formed in other S0 reactions (TF, H2 loss), NTIII needs to be well understood to disambiguate these mechanisms.
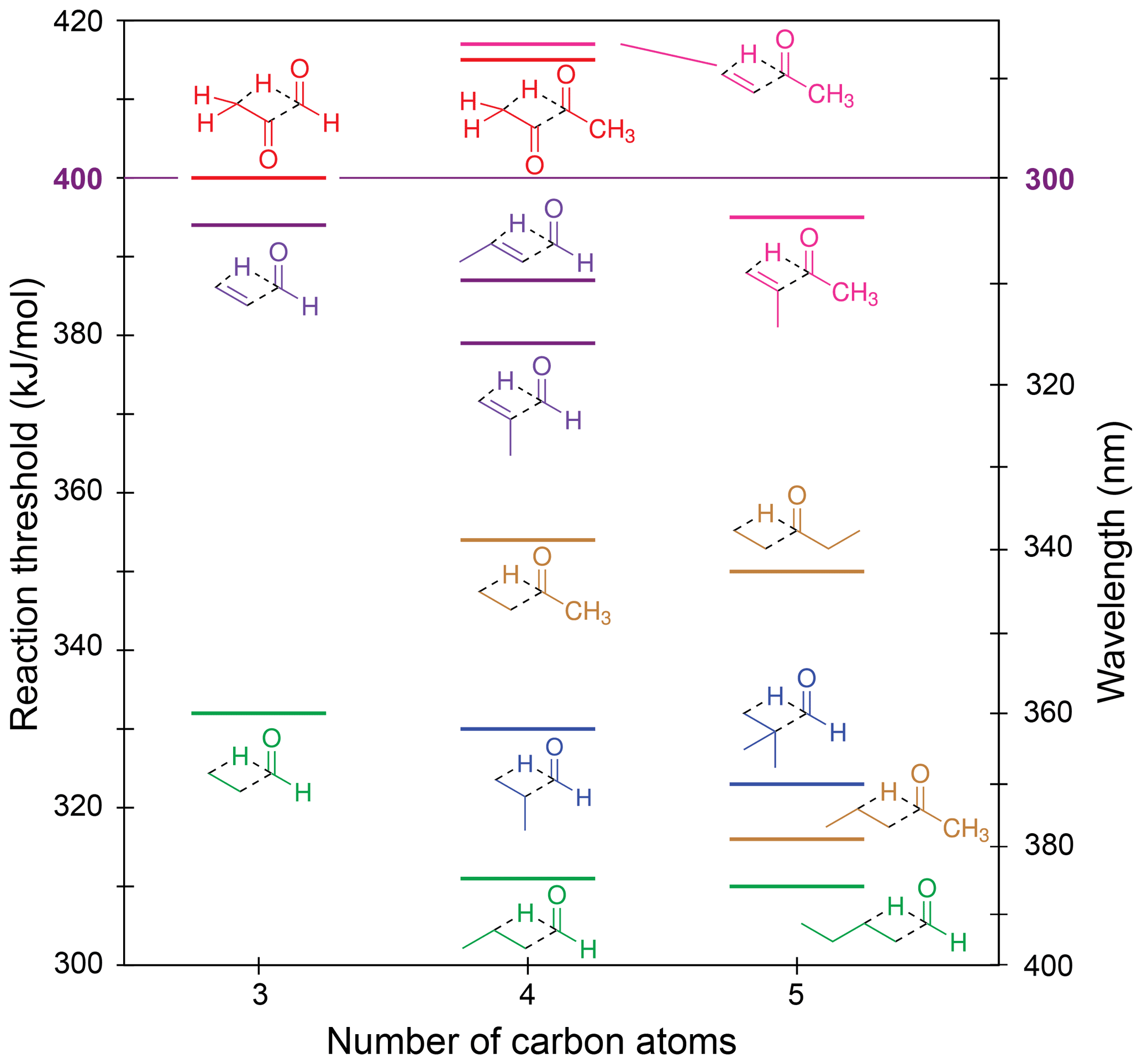
Figure 6Zero-point vibrational-energy-corrected B2GP-PLYP-D3/def2-TZVP S0 Norrish type III thresholds.
Figure 6 shows our calculated NTIII thresholds. These vary across a large energy range from 310 to 417 kJ/mol. In glyoxal, methylglyoxal and methyl vinyl ketone the NTIII thresholds are at or above the actinic maximum energy of 400 kJ/mol. The NTIII thresholds of the other α,β-unsaturated carbonyls lie between 380 and 400 kJ/mol. The high thresholds arise from resonance stabilisation of the breaking C–C bond and suggest NTIII is unlikely to be important in these species under tropospheric conditions.
The saturated carbonyls in Fig. 6 have NTIII thresholds in the range of 310–355 kJ/mol. The NTIII thresholds decrease significantly when the main alkyl chain is lengthened past the β position: there is a 38 kJ/mol lowering of the NTIII threshold from butanone to pentan-2-one, and a 21 kJ/mol lowering from propanal to butanal. The effect of alkyl chain lengthening beyond the β position is also present in α,β-unsaturated carbonyls, although it is smaller, with only a 7 kJ/mol decrease in threshold from acrolein to crotonaldehyde. The NTIII threshold, however, is unchanged with further alkyl chain lengthening. For example, butanal and pentanal have almost the same NTIII threshold (311 and 310 kJ/mol, respectively), and there is only a 4 kJ/mol difference in threshold between butanone and pentan-3-one. This suggests chain lengthening past the γ position will not result in any change to the reaction threshold. The NTIII threshold also appears to be independent of the “spectator” alkyl substituent in the ketones.
Branching at the α position decreases the NTIII threshold, though to a lesser extent than the addition of a γ-carbon. The decreases are as follows: 2 kJ/mol from propanal to 2-methylpropanal, 7 kJ/mol from 2-methylpropanal to 2,2-dimethylpropanal, 15 kJ/mol from acrolein to methacrolein, and 23 kJ/mol ketone to methyl isopropenyl ketone. These decreases in threshold energy exceed the expected uncertainty of our B2GP-PLYP-D3 calculations, except for the smallest 2 kJ/mol energy difference. The trends in NTIII threshold with both alkyl chain length and branching can be rationalised in terms of the alkene product formed: the relative stability of the alkene increases with increasing substitution about the double bond (Whangbo and Stewart, 1982).
3.4 Concerted four-centre H2 loss
H2 can be formed on S0 via a four-centre TS where adjacent hydrogen atoms form an H–H bond and dissociate as H2, leaving an unsaturated carbonyl product. In aldehydes these adjacent hydrogens can be the formyl and α-hydrogens, but as the alkyl chain lengthens possibilities include hydrogens in the α+β, β+γ, etc. positions. These H2-loss mechanisms can be distinguished by the point of unsaturation in the co-product.
The S0 reaction thresholds for the possible H2-loss channels are given in Table 1 and are shown in Fig. 7, where solid lines denote H2 loss from the formyl and α positions, dashed lines H2 loss from the α and β positions, and dot-dashed lines H2 loss from the β and γ positions. Note that the y axis of Fig. 7 is broken into three energy sections to indicate energetic separation between the different mechanisms.
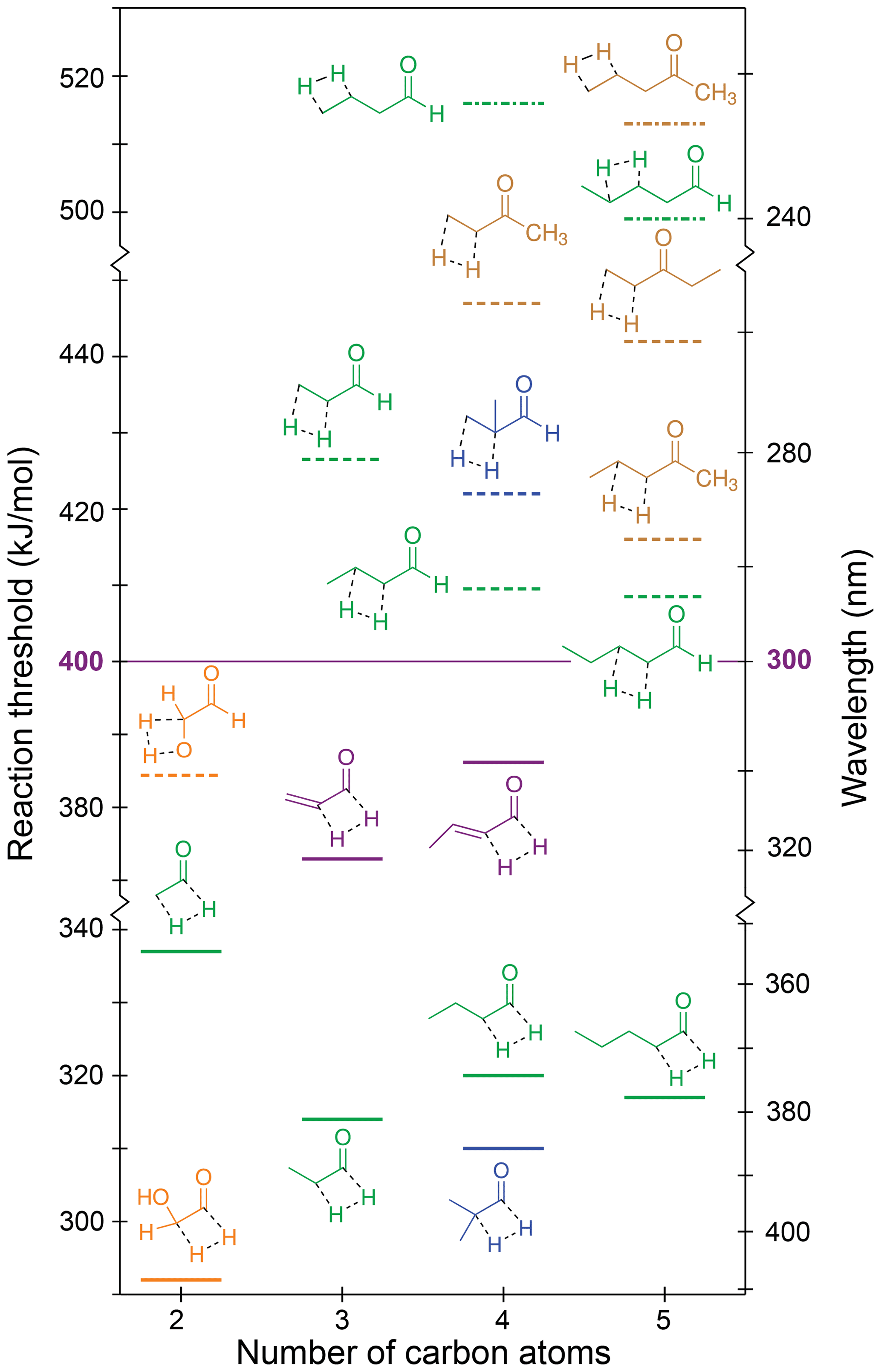
Figure 7Zero-point vibrational-energy-corrected B2GP-PLYP-D3/def2-TZVP S0 H2-loss thresholds. Solid lines: formyl H + α-H; dashed lines: α-H + β-H; dot-dashed lines: β-H + γ-H.
Figure 7 indicates that thresholds for H2 loss from the α and β positions are higher than from the formyl and α positions, with H2-loss thresholds from the β and γ positions higher still. For a given carbonyl, the highest H2-loss threshold is predicted for removal of a hydrogen from the terminal carbon. Like the NTIII reaction, the TSs for four-centre H2 loss are “late”, and the threshold energies are related to the stability of the forming alkene product. Increasing substitution around the double bond increases alkene stability (Whangbo and Stewart, 1982), and so products with a terminal C=C bond are comparatively less stable than products from H2 loss at other sites. For example, there is an approximately 20 kJ/mol decrease in threshold for formyl + α H2 loss from acetaldehyde to propanal. Like NTIII, there is little effect on the formyl and α H2-loss threshold upon further chain extension. For example, the formyl and α H2-loss thresholds for propanal, butanal and pentanal are all ∼315 kJ/mol. Branching at the α position also has little effect on the formyl and α H2-loss threshold (cf. propanal and 2-methylpropanal). Similar trends are seen for H2 loss from the α and β and β and γ positions.
The results in Table 1 and Fig. 7 reinforce, for multiple carbonyl species, that only H2 loss from the formyl and α positions is energetically accessible in the actinic energy range. Indeed, the four-centre H2-loss channel in acetaldehyde has recently been observed experimentally under tropospherically relevant conditions (Harrison et al., 2019). In the absence of a formyl hydrogen, none of the ketone H2-loss channels are accessible in the troposphere. Similarly, H2 loss is not accessible in species lacking an α-hydrogen, like 2,2-dimethylbutanal and methacrolein.
The thresholds for H2 loss from the formyl and α positions in acrolein and crotonaldehyde are also in the actinic range at 373 and 386 kJ/mol, respectively. These thresholds are significantly higher than those for the saturated aldehydes because of the high energy of the product propadienone and 1,2-butadienone species, and they are close to the maximum actinic energy, suggesting S0 H2 loss is unlikely to be important in α,β-unsaturated aldehydes.
Glycolaldehyde is calculated to have the lowest H2-loss threshold, 292 kJ/mol, for loss of the formyl and α-hydrogens. This can be rationalised in terms of the electron-withdrawing nature of the OH stabilising the four-centre TS and the hydroxyketene product. Glycolaldehdye is also the only carbonyl with an energetically accessible β-H-loss channel. Loss of the OH and β-hydrogen forms glyoxal and H2 with a 384 kJ/mol threshold, close to the actinic maximum energy.
3.5 Alkane/alkene elimination (AE)
In ketones, migration of an α-H atom to the “other” α-carbon via a four-centre TS can form an alkane and a ketene. For example, acetone can dissociate to methane and ketene. In methyl vinyl ketone, the unsaturation leads to alkene elimination and formation of ethene and ketene.
The S0 reaction thresholds for the lowest energy AE channels are given in Table 1 and are shown in Fig. 8. In asymmetric ketones two AE channels are possible, and these are described in Sect. S6 of the Supplement.

Figure 8Zero-point vibrational-energy-corrected B2GP-PLYP-D3/def2-TZVP S0 alkane/alkene elimination thresholds.
Figure 8 shows our calculated AE thresholds vary from 329 to 363 kJ/mol, with all accessible under tropospheric conditions. The TSs for AE are shown in Fig. S6.
In linear unsaturated ketones, the AE TSs are “late”, with C–C breaking bond lengths over 1.8 Å. Naïvely, we would therefore infer AE thresholds for these species will reflect the relative stability of the forming products. From tabulated 0 K enthalpies of formation (Ruscic et al., 2005; Ruscic and Bross, 2020), although methylketene is 9 kJ/mol more stable than ketene, butanone is 17 kJ/mol more stable than acetone. This would imply a lower AE threshold in acetone. The AE threshold in butanone, however, is calculated to be 12 kJ/mol lower than that for acetone. The TS for AE in butanone (Fig. S6b) is marginally earlier than in acetone (Fig. S6a), suggesting electronic effects are responsible for the lower threshold. Only a 4 kJ/mol decrease in threshold, to 347 kJ/mol, is seen for production of ethylketene in pentan-2-one, indicating further chain lengthening has little effect. This threshold is the same as that calculated for pentan-3-one forming ethane and methylketene; that is, there is a small reduction in threshold energy on formation of a larger alkane. This is also seen in the alternate AE pathways (Sect. S6). As shown in Fig. S6, in linear unsaturated ketones, thresholds for producing ketene (via the alternate AE channels) are ∼360 kJ/mol, and thresholds for producing methylketene are ∼350 kJ/mol. We expect these thresholds to be generalisable to larger linear unsaturated ketones.
The lowest AE thresholds are for methyl vinyl ketone and methyl isopropenyl ketone (MIPK), both of which yield ketene. The AE TSs in these molecules are much “tighter” than in the linear unsaturated ketones (Fig. S6). Here there is resonance stabilisation of the saddle points, leading to thresholds of 329 and 334 kJ/mol, respectively.
3.6 Keto–enol tautomerisation
Carbonyls can exist in two tautomeric forms: a keto form (encompassing, here, both ketones and aldehydes) and an enol form in which an H atom has transferred to the carbonyl oxygen, forming an OH substituent and a point of unsaturation. Keto–enol tautomerisation is known to occur as a dynamic equilibrium in S0 carbonyls in aqueous solution at room temperature, although the keto tautomer is thermodynamically favoured (Keeffe et al., 1988). Keto–enol tautomerisation has been observed in gas-phase photolysis experiments on acetaldehyde (Clubb et al., 2012; Shaw et al., 2018), and the authors suggest it may occur in many other carbonyls under tropospheric conditions. In solution or at higher gas pressures, keto–enol isomerisation is also readily catalysed by species containing acidic hydrogens – including water, alcohols, and organic and inorganic acids.
The lowest energy calculated keto–enol tautomerisation thresholds for the relevant carbonyls in the test set are given in Table 1 and shown in Fig. 9. A keto–enol TS involving an α-hydrogen, that is, a [1,2]-H atom shift, was found in all of these species. In crotonaldehyde, however, the lowest energy threshold, 133 kJ/mol, was for a [1,5]-H atom shift involving a γ-hydrogen, and this is less than half the 294 kJ/mol threshold we predict for a [1,2]-H atom shift. This mechanism is analogous to the [1,5]-H atom shift in the Norrish type II reaction. Here, however, because it occurs in an enal, conjugation prevents the bond between the α- and β-carbons from breaking, and, instead, but-1,3-diene-1-ol is formed.

Figure 9Zero-point vibrational-energy-corrected B2GP-PLYP-D3/def2-TZVP S0 keto–enol tautomerisation thresholds.
There may be multiple possible [1,2]-H atom shift keto–enol tautomerisation pathways for a given carbonyl. These correspond to the formation of geometric isomers (e.g. cis- or trans-enols) or, in asymmetric ketones, tautomerisation involving hydrogens from either alkyl substituent. These additional keto–enol tautomerisation thresholds are reported in Table S7, and all optimised saddle point geometries are shown in Fig. S7 of the Supplement. As described in more detail in Sect. S7, due to steric factors, thresholds for tautomerisation to trans-enols are typically ∼20 kJ/mol lower than those to the corresponding cis-enol.
As shown in Fig. 9, the keto–enol tautomerisation thresholds for linear aldehydes lie in a narrow energy range (278–281 kJ/mol), indicating chain extension has no effect on threshold as long as the bulky alkyl group can be oriented trans to the enol OH group. This is not the case for the α-branched 2-methylpropanal, and the steric penalty leads to the highest keto–enol tautomerisation threshold calculated here (296 kJ/mol).
The tautomerisation thresholds for ketones are also in a narrow range (270–276 kJ/mol), ∼5 kJ/mol lower than the corresponding aldehydes. As shown in Table S7 this extends to the alternate pathway in asymmetrically substituted ketones, which have thresholds for the formation of the alternate trans-enol within ∼1 kJ/mol of those shown in Fig. 9.
In α,β-unsaturated carbonyls, the keto–enol tautomerisation thresholds are low when tautomerisation involves an H atom from an aliphatic group, for example, the CH3 group in methyl vinyl ketone and crotonaldehyde. Tautomerisation thresholds involving olefinic H atoms, however, are significantly higher, for example 292 kJ/mol in acrolein, reflecting the relatively unstable propadienol product.
Glycolaldehyde has an –OH electron-withdrawing functional group. For this molecule the tautomerisation threshold is predicted to be lowered by 8 kJ/mol compared to acetaldehyde; that is, the electron-withdrawing group stabilises the TS to the forming enol.
Notably, all calculated keto–enol tautomerisation thresholds are significantly below the maximum actinic photon energy. We expect this pathway to be energetically accessible in all carbonyls with an α-hydrogen and appropriate unsaturated species with a γ-hydrogen. Moreover, all linear aldehydes and ketones are calculated to have keto–enol tautomerisation thresholds close to, or below, that of acetaldehyde. Given the experimental observation of keto–enol tautomerisation in acetaldehyde (Clubb et al., 2012; Shaw et al., 2018) and in acetone and methyl vinyl ketone (Couch et al., 2021), this tautomerisation may be important under tropospheric conditions in these species.
3.7 Enal–ketene tautomerisation
There has been recent interest in the formation of ketenes as atypical and relatively uncharacterised products of carbonyl photolysis (Harrison et al., 2019; Toulson et al., 2018). As seen above, the formyl + α H2-loss mechanism forms ketenes in aldehydes, and the AE mechanism forms ketenes in ketones. In enals there is also an S0 tautomerisation mechanism involving a [1,3]-H shift of the formyl hydrogen to the β-carbon that can form ketenes. First order saddle points have been optimised for enal–ketene tautomerisation in acrolein, crotonaldehyde and methacrolein. These are shown in Fig. 10, together with the calculated B2GP-PLYP-D3 threshold energies. The enal–ketene tautomerisation threshold in methacrolein, 285 kJ/mol, is ∼ 14 kJ/mol lower in energy than the 299 kJ/mol G3X-K//M06-2X/6-31G(2df,p) threshold previously calculated by So et al. (2018). This difference is consistent with the variation between B2GP-PLYP-D3 and G3X-K thresholds for other reactions considered in methacrolein and methyl vinyl ketone (see Supplement) and may be due to the treatment of dispersion in the saddle point geometries.
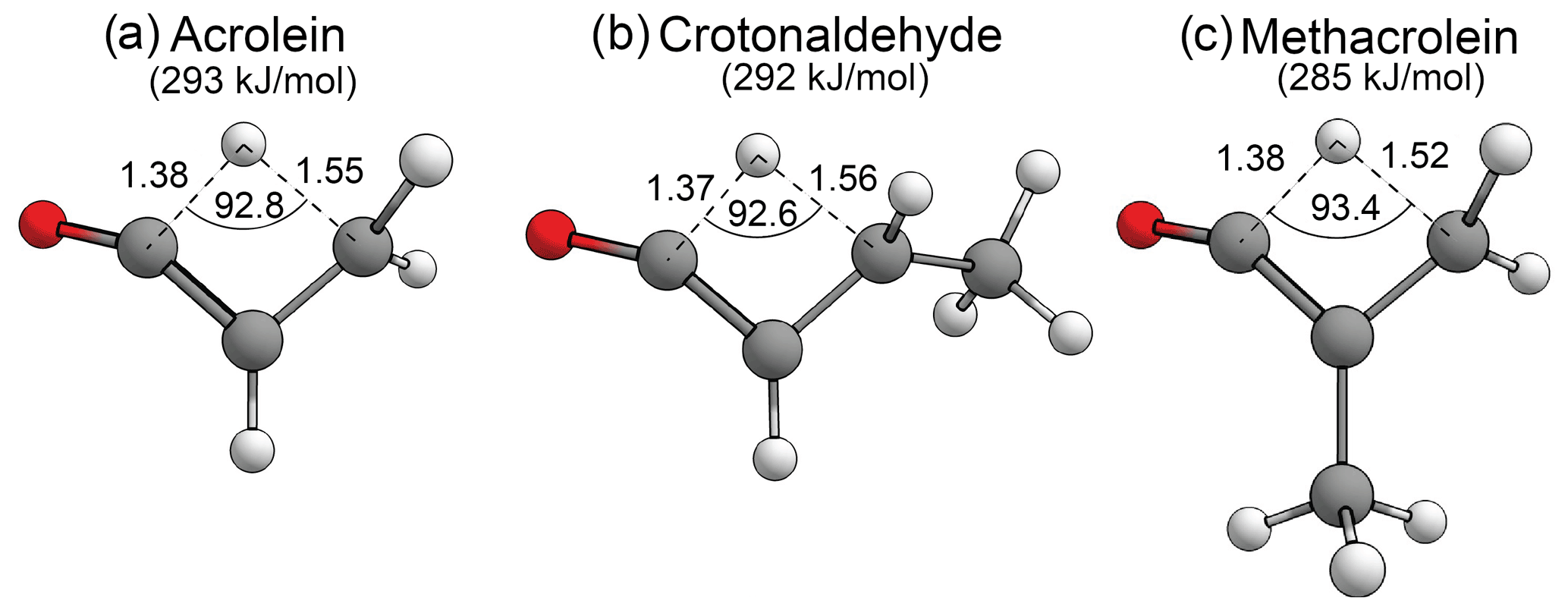
Figure 10Optimised B2GP-PLYP-D3/def2-TZVP S0 TSs and zero-point vibrational-energy-corrected threshold energies for enal–ketene tautomerisation, as shown. Key structural parameters in angstroms (Å) and degrees.
Figure 10 shows that the key structural parameters describing the TSs for S0 enal–ketene tautomerisation are almost identical for the three enals in the test set. This indicates that α-branching and main alkyl chain extension have a negligible effect on the TS geometry and little effect on the threshold energy. The predicted enal–ketene tautomerisation thresholds are all well below the maximum actinic energy of 400 kJ/mol and suggest that, in appropriate species, enal–ketene tautomerisation may be competitive in the troposphere.
Our calculated S0 thresholds can be compared to relevant experimental results to validate the calculations, to generalise our results and to predict the most important S0 reactions for each class of carbonyl. Indeed, the seven reaction thresholds depicted for butanal in Fig. S1 are illustrative of the energetic relevance of these channels in other saturated aldehydes.
4.1 Saturated aldehydes
For saturated aldehydes, the lowest energy dissociation pathway on S0 is concerted TF with a threshold of ∼300 kJ/mol. At wavelengths and internal energies where excited state chemistry is energetically inaccessible, TF may therefore be a significant photo-induced mechanism. Triple fragmentation has been observed as a primary photolysis mechanism in propanal and 2-methylpropanal, with QYs of 4 % and 9 %, respectively, at 1 atm pressure of N2 (Kharazmi, 2019). On the basis of kinetic modelling and in the absence of excited state primary pathways, both sets of TF products were found to be consistent with S0 reaction. The increased QY for 2-methylpropanal over propanal was attributed to increased reaction path degeneracy rather than a lower reaction threshold. This suggests TF QYs will be higher in branched aldehydes in proportion to the ratio of β-hydrogens. For example, we predict 2,2-dimethylpropanal, with nine β-hydrogens, to have a higher TF QY than 2-methylpropanal, with six β-hydrogens, and propanal, with three β-hydrogens.
The next lowest energy S0 dissociation pathway is either H2 loss from the formyl and α-H atoms in the smaller species (acetaldehyde, propanal, methylpropanal) or NTIII in the larger species (butanal, pentanal, 2,2-dimethylpropanal). For formyl + α H2 loss, acetaldehyde has three α-hydrogens and the highest threshold at 337 kJ/mol. This channel has been observed experimentally, with a QY of ∼1 % at 305 nm (392 kJ/mol) and 1 atm of N2, and is demonstrated to be a primary process by single molecule velocity map ion imaging (VMI) experiments (Harrison et al., 2019). Although classical trajectory simulations initiated at the H2-loss TS could not reproduce the VMI product state distributions, they are consistent with an S0 roaming mechanism (Harrison et al., 2019). The equivalent H2-loss thresholds for the other saturated aldehydes, which have two rather than three α-hydrogen atoms, are significantly lower at ∼315 kJ/mol. Thus we expect H2-loss reactions to be present in other saturated aldehydes with similar or slightly higher QYs than in acetaldehyde. Indeed, under tropospheric conditions, methylketene and dimethylketene have been observed as minor products following the photolysis of propanal and 2-methylpropanal, respectively, with kinetic modelling consistent with S0 reaction (Kharazmi, 2019). In saturated aldehydes, the NTIII thresholds are similar to those for H2 loss. Experimental QYs for this pathway, however, have not been reported. The NTIII mechanism shares common products with TF (alkene) and decarbonylation (CO), which may complicate its experimental identification. Our calculations suggest NTIII is energetically competitive, and it may be important in interpreting photolysis QYs of saturated aldehydes.
With the exception of formaldehyde, the next lowest energy S0 dissociation threshold is for ground state NTIa, that is, α-bond cleavage to the alkyl substituent. This reaction is typically assumed to occur only on T1, but the S0 reaction has been identified in formaldehyde (Quinn et al., 2017), acetaldehyde (Heazlewood et al., 2009; Amaral et al., 2010) and acetone (Lee et al., 2017). Because it is barrierless we expect S0 NTIa to dominate as internal energy increases above its threshold. Thus, we expect to observe NTIa dissociation products at energies well below the T1 threshold.
Decarbonylation (CO loss) has the highest calculated S0 dissociation thresholds for all the saturated carbonyls in the test set, bar formaldehyde, for which the decarbonylation threshold is slightly below that of NTI. All decarbonylation thresholds are predicted to be 340–350 kJ/mol, and experimental CO-loss QYs are correspondingly low: in the actinic energy range, the atmospheric pressure decarbonylation QY for acetaldehyde is ∼0.5 % (Moortgat et al., 2010; Warneck and Moortgat, 2012) and ∼0.2 % for 2-methylbutanal (Gruver and Calvert, 1958). Although two S0 decarbonylation mechanisms are known, roaming reactions and reaction via a TS, there is no known excited state decarbonylation mechanism. Given the relatively high threshold energies, we predict other S0 pathways will be favoured in larger saturated aldehydes over decarbonylation, which will be at most a minor channel.
Our calculations predict keto–enol tautomerisation to be the lowest energy pathway on S0 for all saturated aldehydes, lying about 20 kJ/mol below TF. Unlike dissociation, however, tautomerisation is reversible. Collisional cooling into the enol well therefore competes with tautomerisation back to the parent aldehyde (Andrews et al., 2012). Dissociation of the aldehyde reduces its concentration, and hence, while the rate coefficient is constant, the rate of enol formation is reduced. Although experimentally observed in ketones, the only aldehyde for which photo-initiated keto–enol tautomerisation has been observed is acetaldehyde (Clubb et al., 2012; Shaw et al., 2018), in which TF is not available, and master equation modelling confirms that it is an S0 process (Shaw et al., 2018). Whilst it is likely keto–enol tautomerisation is occurring in other saturated aldehydes, QYs are likely small, and the reactivity of the enol will make characterisation difficult.
4.2 Saturated ketones
Following excitation by an actinic photon, the relevant S0 chemistry of ketones is much simpler than that of aldehydes. No TS for direct decarbonylation was found, and the absence of a formyl H atom in ketones removes the TF and formyl + α H2-loss pathways. The only S0 dissociation pathways in ketones are NTIII, AE and NTI. The thresholds for AE and NTI are similar in the ketones considered at ∼340 kJ/mol. Given NTI is barrierless, it is likely to dominate over AE. We therefore expect NTI to be the dominant dissociation mechanism in acetone, butanone and pentan-2-one, where it has a lower threshold than NTIII, although NTIII may be important in pentan-2-one and in larger saturated ketones, where there is alkyl chain extension past the β-hydrogen involved in the reaction. For example, the production of acetaldehyde from 3-methylbutan-2-one is one of the few experimental examples of the – assumed S0 – NTIII mechanism in the literature (Zahra and Noyes, 1965).
The keto–enol tautomerisation thresholds for saturated ketones are slightly lower than in saturated aldehydes. This, combined with the absence of low-energy S0 dissociation pathways and the experimental observation of keto–enol tautomerisation in acetone under thermal conditions, where reaction must occur on S0 (Couch et al., 2021), suggests photo-initiated tautomerisation may be important in ketones.
4.3 α,β-unsaturated carbonyls
In general, because of resonance stabilisation of the bond between α-carbons and formyl carbons, we calculate higher S0 thresholds in α,β-unsaturated carbonyls than in equivalent saturated carbonyls. However, excited state NTIa thresholds are also elevated, and all excited state NTI thresholds are close to, or above, the maximum available actinic energy (Rowell et al., 2019). Thus any photolysis must occur on S0. Further, unlike many other carbonyls, the electronic structure of the α,β-unsaturated carbonyls promotes rapid electronic relaxation from S1 to S0 (Lee et al., 2007; Schalk et al., 2014; Cao and Xie, 2016), suggesting high S0 internal energies and therefore a relatively high probability of S0 reaction.
Triple fragmentation is the lowest energy S0 dissociation pathway for the unsaturated aldehydes in the test set. The TF thresholds are ∼40 kJ/mol higher than in saturated aldehydes and are slightly lower than the S0 NTIb asymptotic energies and decarbonylation thresholds. The other possible S0 dissociation reactions (H2 loss, NTIII and S0 NTIa) have thresholds >370 kJ/mol and are unlikely to be significant under tropospheric conditions.
Unsaturated ketones, like their saturated counterparts, have fewer S0 dissociation pathways, with only NTIII, AE, NTIa and NTIb possible. In methyl vinyl ketone, AE has the lowest threshold at 329 kJ/mol, 9 kJ/mol lower than the NTIb threshold. NTIb dissociation, however, is barrierless. The 334 kJ/mol threshold for AE is the lowest energy S0 pathway in methyl isopropenyl ketone, 10 kJ/mol lower than the threshold for NTIb dissociation. Although NTIb will dominate at higher energies, there may be a small energy window where AE may be important, although it has not been experimentally observed. Similarly, there may be an energy window for AE in methyl vinyl ketone. The other possible dissociations in methyl vinyl ketone, NTIa and NTIII, have thresholds >375 kJ/mol and are unlikely to be important under tropospheric conditions.
Given the lack of low-energy ground or excited state dissociation pathways, we expect photolysis QYs for α,β-unsaturated carbonyls to be small. Indeed the total photolysis QY of methacrolein is around 1 % at atmospheric pressure (Raber and Moortgat, 1996). Photo-induced keto–enol and enal–ketene tautomerisations, however, have thresholds under 300 kJ/mol, with crotonaldehyde having one of the lowest energy keto–enol tautomerisation thresholds of 133 kJ/mol. Tautomerisation has been observed in methyl vinyl ketone under thermal conditions (Couch et al., 2021), and S0 tautomerisation may therefore be competitive for appropriate α,β-unsaturated species (So et al., 2018). The higher energy enol and ketene isomers may be collisionally stabilised under tropospheric conditions, although they are significantly more reactive and will be difficult to experimentally isolate.
4.4 Other carbonyls
The α-dicarbonyls have low-energy excited states (Arnett et al., 1974; Dykstra and Schaefer, 1976), red-shifted absorption spectra compared to other carbonyls (Fig. 1) and weakened α-C–C bonds due to the electron-withdrawing nature of the two C=O chromophores. As a result, the excited state NTIa thresholds in dicarbonyls are lowered to ∼390 kJ/mol on S1 and ∼300 kJ/mol on T1 (Rowell et al., 2019), with both accessible in the troposphere. The S0 asymptotic energies for NTIa are slightly lower than the triplet thresholds, giving an energetic window for S0 radical dissociation. Indeed, in glyoxal, two distinct, wavelength-dependent mechanisms of HCO• formation have been observed and attributed to dissociation on two electronic states (Chen and Zhu, 2003; Kao et al., 2004; Salter et al., 2013). Decarbonylation to form H2CO + CO and TF to form H2 + 2 CO have the lowest energy thresholds in glyoxal, at 225 and 247 kJ/mol, respectively. Indeed, CO is known to form following irradiation of glyoxal at energies below 272 kJ/mol (Loge and Parmenter, 1981; Hepburn et al., 1983; Burak et al., 1987; Dobeck et al., 1999). Decarbonylation in methylglyoxal also has a low threshold energy, 235 kJ/mol, and we expect it to be the dominant S0 dissociation mechanism. In diacetyl, without a formyl hydrogen, NTIa has the lowest dissociation threshold. We therefore predict, for larger α-dicarbonyls, there will be an actinic window from ∼300 to 266 kJ/mol with no accessible dissociation pathways. In this case, collisional cooling and thermalisation to the parent carbonyl are the likely fate.
The remaining carbonyl in the test set is glycolaldehyde, with an actinic range between 352 and 400 kJ/mol (Bacher et al., 2001). The NTIa thresholds on S1 and T1 for glycolaldehyde are 379 and 334 kJ/mol, respectively (Rowell et al., 2019), and this channel dominates at all actinic energies (Bacher et al., 2001; Zhu and Zhu, 2010), with cleavage of the C–OH bond to form •OH also reported (Zhu and Zhu, 2010). Thus, despite having the lowest calculated TF and H2-loss thresholds, these reactions have not been experimentally observed in glycolaldehyde. Keto–enol tautomerisation to 1,2-ethenediol, for which we calculate a threshold of 272 kJ/mol, has been postulated as an atmospheric route to the formation of HO and formic acid (So et al., 2015), although it has not been experimentally observed.
A few guiding principles can be used to determine which of the possible S0 pathways may be tropospherically relevant in a given carbonyl following absorption of an actinic photon.
-
Reactions with thresholds greater than the actinic maximum energy of 400 kJ/mol are inaccessible at tropospheric photon energies. This rules out most S1 reactions, with the exceptions of NTIa dissociation in glycolaldehyde and NTIb in methyl vinyl ketone and methyl isopropenyl ketone (Haas, 2004; Rowell et al., 2019). T1 NTI dissociations are accessible. T1 NTIb thresholds are close to the maximum available energy of 400 kJ/mol and have negligible contributions to actinic photolysis (Zhu et al., 2009), T1 NTIa thresholds are generally lower (Rowell et al., 2019), and this reaction dominates the photolysis QY of small carbonyls (Kirkbride and Norrish, 1931; Zhu et al., 2009). In larger carbonyls (alkyl chain lengths ≥4), excited state NTII intramolecular γ-H abstraction is also accessible (Wagner and Zepp, 1972; Wagner and Klán, 2004; Zhu et al., 2009).
-
For photon energies above their thresholds, T1 reactions are fast and dominate photolysis (Kirkbride and Norrish, 1931; Zhu et al., 2009). If the photon energy is near threshold, non-radiative transitions and S0 reaction may be competitive with excited state reaction. For photon energies (or collisional cooling on S1 or T1) below any excited state threshold, any photolysis must occur on S0. There is significant overlap of the absorption spectrum of most carbonyls with these lower energy photons (Fig. 1), and S0 reactions have been observed in saturated carbonyls under these conditions (Heazlewood et al., 2009; Amaral et al., 2010; Andrews et al., 2012; Tsai et al., 2015; Quinn et al., 2017; Toulson et al., 2018).
-
For an S0 process to be tropospherically important, its rate must be competitive with collisional cooling and thermal equilibrium. Only S0 dissociations with thresholds <350 kJ/mol have been experimentally observed following photoexcitation of aldehydes in 1 atm of N2 (Clubb et al., 2012; Shaw et al., 2018; Harrison et al., 2019). Because of the efficiency of collisional cooling, the most important S0 dissociations are likely to be those with the lowest thresholds. These include TF if available, NTI in ketones, and selected AE, NTIII and formyl + α H2-loss reactions.
-
Although S0 keto–enol and enal–ketene tautomerisations have the lowest calculated thresholds, these are reversible reactions in which collisional stabilisation of the tautomer competes with isomerisation back to the parent carbonyl. Tautomerisation, per se, is therefore unlikely to be important if there are low-energy S0 dissociation pathways. It will be important in the absence of such a channel, for example, in acetaldehyde (Clubb et al., 2012; Shaw et al., 2018), some ketones and some α,β-unsaturated carbonyls. Tautomerisation may also have an indirect effect on other S0 reactions. Rapid interconversion of tautomers will reduce the effective concentration of the “keto” form and hence reduce the probability of other possible S0 reactions.
The atmospheric importance of photo-initiated S0 reactions is as yet unknown; however, S0 reactions are likely to be broadly accessible under tropospheric conditions for all carbonyls. They are therefore likely in regions of the troposphere with high carbonyl concentrations: in highly polluted environments, where carbonyls are both directly emitted and are oxidation products of other VOCs, and in unpolluted, forested environments, where oxidation of BVOCs leads to unsaturated carbonyls like methacrolein and methyl vinyl ketone (Kesselmeier and Staudt, 1999; Millet et al., 2010; Chen et al., 2014). Carbonyls are also important species in the marine boundary layer, although there are significant discrepancies between observed and modelled concentrations (Vigouroux et al., 2009; Anderson et al., 2017; Ahn et al., 2019).
Photo-initiated S0 reactions of carbonyls have a number of atmospheric consequences. The majority of reactions lead to molecular, rather than radical, products, and this may effect modelled radical quantum yields and therefore radical propagation reactions. Many of the S0 reaction products are also “unexpected” and form unsaturated species such as alkenes, alkynes, enols and ketenes that are more reactive than their parent carbonyl, particularly to addition reactions, for example, with •OH and NOx radicals and even atmospheric H2O (Atkinson et al., 2006; So et al., 2014; Kahan et al., 2013). We suggest ground state reactions should be considered whenever unexpected products are found in the laboratory photolysis of carbonyls and when products are observed following photolysis below excited state thresholds.
The experimental observations of photo-initiated S0 reactions in carbonyls described in this paper indicate the parent carbonyl is returning to the ground electronic state with energies close to the original photon energy. In addition to dissociation and isomerisation, these vibrationally “hot” S0 molecules could also react with other atmospheric species: their high internal energy makes otherwise inaccessible reactions energetically feasible. For example, internally excited carbonyls could undergo rapid bimolecular reaction with species such as O2, H2O, •OH, NOx and NH3. The possibility of a reaction between O2 and internally hot acetaldehyde, formed following excitation at 248 nm, considerably above the actinic range, was speculated by Morajkar et al. (2014), although this was not further investigated or resolved. Such bimolecular reactions may lead to radical species, and we suggest they may be responsible for radical QYs observed following photolysis at energies below the NTI thresholds in formaldehyde (Horowitz and Calvert, 1978; Moortgat and Warneck, 1979; Valachovic et al., 2000). In this sense, whilst the total experimental radical QY may be correct, the reaction mechanism is not simply unimolecular dissociation, and radical QYs from bimolecular reaction will have different pressure dependence to those from unimolecular dissociation.
The molecular products of some of the S0 reactions considered are also formed with extremely high internal energy. Experimentally, the roaming pathway in formaldehyde forms H2 with up to 12 quanta of vibrational excitation (Quinn et al., 2017). Similarly, CH4 from roaming in acetaldehyde is formed with internal energies up to 95 % of the CH bond dissociation energy (Heazlewood et al., 2008). Although its mechanism has not been elucidated, the formyl + α H2-loss channel in acetaldehyde has been shown experimentally to yield internally hot ketene with energy >150 kJ/mol (Harrison et al., 2019). These “hot” molecular products could also potentially undergo bimolecular reactions in the atmosphere and contribute toward radical QYs.
There are two cases when photo-initiated S0 reactions of carbonyls have immediate tropospheric implications and missing or underestimated photochemical sources have previously been speculated. These are the formation of organic acids and the photolytic generation of molecular hydrogen, H2. In both polluted and pristine regions there are discrepancies between the predictions of atmospheric models and field measurements of the concentrations of organic acids (Cady-Pereira et al., 2014; Yuan et al., 2015; Millet et al., 2015), although a recent paper by Franco et al. (2021) proposes a pathway that may close the gap. Depending on the type of model and the estimation of soil uptake, there are also large differences in estimates of global photochemical production of H2 (Novelli, 1999; Rhee et al., 2006; Price et al., 2007; Xiao et al., 2007; Ehhalt and Rohrer, 2009; Yashiro et al., 2011; Patterson et al., 2020). Notably, in “top-down” models, Rhee et al. (2006) and Xiao et al. (2007) propose increased H2 production from photolysis of oxidised non-methane VOCs.
The tautomerisation of acetaldehyde to vinyl alcohol, the addition of atmospheric •OH and subsequent oxidation have been modelled and shown to produce significant global tropospheric formic acid and were found to be the dominant mechanism for formic acid formation in the marine boundary layer (Shaw et al., 2018). This one reaction, however, is not sufficient to explain the factor of 2 discrepancy between modelled and measured global organic acid concentrations (Shaw et al., 2018). Keto–enol isomerisation is present in almost all carbonyls we have considered, having amongst the lowest S0 reaction thresholds. Similar reactions involving other unsaturated products, for example alkenes and ketenes (Atkinson et al., 2006; Kahan et al., 2013), will also lead to organic acid formation. We expect these reactions to be energetically accessible in most atmospheric carbonyls, and their cumulative effect may address the modelled deficit (Shaw et al., 2018; So et al., 2018). Understanding the mechanism of formation of organic acids in the troposphere has further application to secondary aerosol formation; the higher oxygen content of organic acids reduces their volatility, and, as proton donors, they are believed to be key species in the nucleation and growth of atmospheric particles (Zhang et al., 2004; Bianchi et al., 2016; Tröstl et al., 2016; Liu et al., 2021).
Molecular hydrogen is an important atmospheric reducing agent and is an indirect greenhouse gas because it increases the atmospheric lifetime of CH4 (Ehhalt and Rohrer, 2009). Current understanding indicates the major photochemical source of H2 in the atmosphere is photolysis of formaldehyde, which accounts for at least half of the photochemically generated H2 (Hauglustaine, 2002). The mechanisms generating the other half are unknown (Grant et al., 2010). The energetically accessible S0 TF and/or H2-loss reactions present in all aldehydes in our test set, and expected in all atmospheric aldehydes, provide primary photolysis routes to H2 that have not previously been considered. By better understanding the current atmospheric H2 budget, we are better placed to model any future increase in atmospheric H2, for example, due to leakage of H2 in any transition to a hydrogen economy.
Challenges to address
To definitively answer the question “how important is the photo-initiated ground state chemistry of carbonyls in the atmosphere”, a number of challenges need to be addressed. Experimentally these include observation of primary products formed on S0 and characterisation of S0 QYs. An understanding of the competition between collisional cooling and reaction under tropospheric conditions is also required.
Primary photolysis products can be observed in single molecule experiments; for example, Harrison et al. (2019) recently observed the formyl + α H2-loss channel in acetaldehyde using velocity map ion imaging. These experiments, however, do not provide absolute QYs. An additional challenge is that many of the products formed are either transient and difficult to observe under atmospheric conditions or can be formed from multiple reaction pathways. Determination of S0 QYs therefore relies on master equation modelling and hence on an accurate characterisation of reaction mechanisms. The results in this paper are a necessary step toward such modelling and will inform the interpretation of photolysis experiments on individual carbonyls, box models of specific locations and ultimately global chemical transport models.
Very little is known about collisional cooling of highly excited ground state molecules. Modelling of experimental photolysis QYs of acetaldehyde, using a simple exponential-down model, suggests an average loss of 150 cm−1 (∼ 1.8 kJ/mol) internal energy per collision with N2 (Andrews et al., 2012). The accuracy of this model and the generality of this result to other carbonyls and their isomers are yet to be tested. Nevertheless, it suggests approximately 200 collisions are required to thermalise carbonyls after absorption of an actinic photon. Given collision frequencies of the order of 109 s−1 at atmospheric pressure, unimolecular reactions with rate coefficients of magnitude 107 or more are likely to be competitive under tropospheric conditions. For example, for deuterated acetaldehyde following excitation by 322.9 nm light, Heazlewood et al. (2011) calculated unimolecular rate coefficients for S0 NTI dissociation, decarbonylation, formyl + α H2 loss and keto–enol tautomerisation of approximately 1×107, 1×106, 2×106 and 2×107 s−1, respectively. There is experimental evidence for all of these S0 reactions under tropospheric conditions (Heazlewood et al., 2009; Horowitz and Calvert, 1982; Heazlewood et al., 2008; Moortgat et al., 2010; Harrison et al., 2019; Clubb et al., 2012; Shaw et al., 2018), although decarbonylation and formyl + α H2 loss are minor channels. Many of the S0 thresholds in Table 1 are lower than those for acetaldehyde, implying higher reaction rate coefficients, although these are yet to be calculated.
Meeting these challenges will enable the tropospheric importance of photo-initiated S0 reactions in individual carbonyls to be determined. Even if these individual reactions have a small QY, their presence in all atmospheric carbonyls may lead to cumulative products that may be atmospherically significant.
We have calculated S0 reaction thresholds for nine reaction types in seven classes of carbonyl within a “small” carbonyl test set. In general, the S0 transition states are “late” and resemble the products. Reaction threshold energies typically correlate with the stability of the product molecules, enabling our results to be generalised to larger carbonyls.
In the smallest carbonyls and dicarbonyls, formaldehyde, glyoxal and methylglyoxal, the lowest energy threshold is for direct decarbonylation, and this mechanism will compete with NTI dissociation. In larger aldehydes direct decarbonylation via a TS mechanism has a threshold of ∼350 kJ/mol in saturated species and ∼360–375 in α,β-unsaturated species. This reaction is therefore likely unimportant in larger aldehydes. However, alternate roaming pathways may be viable if there are low-energy barrierless S0 NTI pathways.
In larger carbonyls, the lowest energy S0 dissociation thresholds are for triple fragmentation (TF) of both saturated (∼300 kJ/mol) and α,β-unsaturated (∼340 kJ/mol) aldehydes, with a negligible impact from the extension of the main alkyl chain. Branching at the α position only increases TF thresholds by ∼5 kJ/mol but increases the reaction path degeneracy and hence, likely, the QY.
The only four-centre H2-loss mechanism relevant at actinic energies involves the formyl and α-hydrogens, and so it needs only to be considered for aldehydes. This threshold is highest, at 337 kJ/mol, for acetaldehyde, and we expect it to be ∼315 in other aldehydes since a terminal double bond is not being formed.
The NTIII β-hydrogen transfer reaction is energetically accessible in saturated carbonyls but largely inaccessible for α,β-unsaturated species. This pathway is often overlooked in the interpretation of photolysis experiments but should be considered when the production of alkenes and aldehydes shows little-to-no pressure dependence.
We expect alkane elimination to have thresholds of ∼ 350 kJ/mol in linear unsaturated ketones. Whilst energetically accessible under tropospheric conditions, these thresholds are above those for NTI dissociation, and we do not expect alkane elimination to be significant. The AE threshold, however, is reduced to ∼330 kJ/mol by branching or unsaturation at the α position, and alkene elimination may be important in these species.
Finally, when present, tautomerisation pathways have the overall lowest S0 thresholds and yield highly reactive unsaturated species: enols or ketenes. The keto–enol tautomerisation of acetaldehyde to vinyl alcohol leads to significant formation of formic acid in the troposphere. This process may be relevant to other atmospheric carbonyls. In particular, the α,β-unsaturated carbonyls have UV absorption spectra that extend to low energies, high excited state reaction thresholds and relatively high S0 dissociation thresholds.
The calculations in this paper demonstrate a range of ground state reactions are energetically accessible within the tropospheric “photochemistry” of carbonyls. The energetic thresholds for these S0 reactions are some of the lowest calculated for any carbonyl reaction on any electronic state. Many of the TF reactions, as well as the keto–enol and enal–ketene tautomerisation, are predicted to have reaction thresholds ≲300 kJ/mol. These reactions are likely to be important following photoexcitation at energies below any excited state reaction threshold or following collisional cooling of excited state carbonyl molecules below such thresholds. An assessment of the likelihood of these S0 reactions and determination of their product yields, however, require the calculation of the respective reaction rate coefficients and master equation modelling. Our results will help target such calculations, as well as future experimental efforts, to reactions most likely to have tropospheric consequences. It may also be that the QY for the S0 reactions of an individual carbonyl is relatively small. The fact that one or more of these reactions are expected to occur in all atmospheric carbonyls suggests that, cumulatively, they may be significant in the troposphere and may have atmospheric consequences.
The code is all available in the ORCA suite of programmes, which is referred to in the computational methods, and as referenced by Neese (2017).
No data sets were used in this article.
The Supplement related to this article contains a figure and discussion of the energetic thresholds for relevant S0 reactions in butanal: for the reaction classes considered, excepting enal–ketene tautomerisation, there is a review of the previous computational literature, additional higher energy thresholds, as applicable, and representations of the optimised S0 first-order saddle points (excepting those for H2 loss). Cartesian coordinates for all optimised saddle points are provided as text filed in a ZIP folder. The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-929-2022-supplement.
KNR performed all calculations and data analysis. MJTJ and SHK conceived and directed the project, and MJTJ supervised the calculations. All authors contributed to data interpretation and the drafting of the manuscript.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This research was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government, as well as computer time on the computational cluster Katana supported by the Faculty of Science, UNSW, Australia, and the computational cluster Artemis supported by the Sydney Informatics Hub at the University of Sydney.
This work was supported by the Australian Research Council (grant nos. DP160101792 and DP190102013). Keiran N. Rowell was supported by an Australian Government Research Training Program (RTP) scholarship.
This paper was edited by Andreas Hofzumahaus and reviewed by two anonymous referees.
Ahn, D. H., Choi, T., Kim, J., Park, S. S., Lee, Y. G., Kim, S.-J., and Koo, J.-H.: Southern Hemisphere mid- and high-latitudinal AOD, CO, NO2, and HCHO: Spatiotemporal patterns revealed by satellite observations, Prog. Earth Planet. Sci., 6, 34, https://doi.org/10.1186/s40645-019-0277-y, 2019. a
Amaral, G. A., Arregui, A., Rubio-Lago, L., Rodríguez, J. D., and Baares, L.: Imaging the radical channel in acetaldehyde photodissociation: Competing mechanisms at energies close to the triplet exit barrier, J. Chem. Phys., 133, 064 303, https://doi.org/10.1063/1.3474993, 2010. a, b, c
Anderson, D. C., Nicely, J. M., Wolfe, G. M., Hanisco, T. F., Salawitch, R. J., Canty, T. P., Dickerson, R. R., Apel, E. C., Baidar, S., Bannan, T. J., Blake, N. J., Chen, D., Dix, B., Fernandez, R. P., Hall, S. R., Hornbrook, R. S., Gregory Huey, L., Josse, B., Jöckel, P., Kinnison, D. E., Koenig, T. K., Le Breton, M., Marécal, V., Morgenstern, O., Oman, L. D., Pan, L. L., Percival, C., Plummer, D., Revell, L. E., Rozanov, E., Saiz-Lopez, A., Stenke, A., Sudo, K., Tilmes, S., Ullmann, K., Volkamer, R., Weinheimer, A. J., and Zeng, G.: Formaldehyde in the tropical Western Pacific: Chemical sources and sinks, convective transport, and representation in CAM-Chem and the CCMI models, J. Geophys. Res.-Atmos., 122, 11201–11226, https://doi.org/10.1002/2016JD026121, 2017. a
Andrews, D. U., Heazlewood, B. R., Maccarone, A. T., Conroy, T., Payne, R. J., Jordan, M. J. T., and Kable, S. H.: Photo-Tautomerization of acetaldehyde to vinyl alcohol: A potential route to tropospheric acids, Science, 337, 1203–1206, https://doi.org/10.1126/science.1220712, 2012. a, b, c, d
Andrews, D. U., Kable, S. H., and Jordan, M. J.: A phase space theory for roaming reactions, J. Phys. Chem. A, 117, 7631–7642, https://doi.org/10.1021/jp405582z, 2013. a
Arnett, J. F., Newkome, G., Mattice, W. L., and McGlynn, S. P.: Excited electronic states of the α-dicarbonyls, J. Am. Chem. Soc., 96, 4385–4392, https://doi.org/10.1021/ja00821a007, 1974. a
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003. a
Atkinson, R., Baulch, D., Cox, R., Hampson, R., Kerr, J., Troe, J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Supplement IV: IUPAC subcommittee on gas kinetic data evaluation for atmospheric chemistry, Atmos. Environ. A, 26, 1187–1230, https://doi.org/10.1016/0960-1686(92)90383-V, 1992. a, b
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a, b, c
Bacher, C., Tyndall, G. S., and Orlando, J. J.: The atmospheric chemistry of glycolaldehyde, J. Atmos. Chem., 39, 171–189, https://doi.org/10.1023/A:1010689706869, 2001. a, b, c
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q., Liu, H. Y., Mickley, L. J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res.-Atmos., 106, 23073–23095, https://doi.org/10.1029/2001JD000807, 2001. a, b
Bianchi, F., Tröstl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kürten, A., Manninen, H. E., Münch, S., Peräkylä, O., Petäjä, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, https://doi.org/10.1126/science.aad5456, 2016. a
Blacet, B. F. E., Heldman, J. D., and Heldman, J. D.: The photolysis of the aliphatic aldehydes. Acetaldehyde and iodine mixtures, J. Am. Chem. Soc., 64, 889–893, https://doi.org/10.1021/ja01256a044, 1942. a
Blacet, F. E. and Calvert, J. G.: The photolysis of aliphatic aldehydes. XV. The butyraldehydes with iodine vapor, J. Am. Chem. Soc., 73, 667–674, https://doi.org/10.1021/ja01146a049, 1951. a
Bowman, J. M. and Houston, P. L.: Theories and simulations of roaming, Chem. Soc. Rev., 46, 7615–7624, https://doi.org/10.1039/C7CS00578D, 2017. a, b
Burak, I., Hepburn, J. W., Sivakumar, N., Hall, G. E., Chawla, G., and Houston, P. L.: State‐to‐state photodissociation dynamics of trans‐glyoxal, J. Chem. Phys., 86, 1258–1268, https://doi.org/10.1063/1.452215, 1987. a
Cady-Pereira, K. E., Chaliyakunnel, S., Shephard, M. W., Millet, D. B., Luo, M., and Wells, K. C.: HCOOH measurements from space: TES retrieval algorithm and observed global distribution, Atmos. Meas. Tech., 7, 2297–2311, https://doi.org/10.5194/amt-7-2297-2014, 2014. a
Cao, J. and Xie, Z.-Z.: Internal conversion and intersystem crossing in α,β-enones: a combination of electronic structure calculations and dynamics simulations, Phys. Chem. Chem. Phys., 18, 6931–6945, https://doi.org/10.1039/c5cp06833a, 2016. a
Carlier, P., Hannachi, H., and Mouvier, G.: The chemistry of carbonyl compounds in the atmosphere-A review, Atmos. Environ., 20, 2079–2099, https://doi.org/10.1016/0004-6981(86)90304-5, 1986. a
Chen, S. L. and Fang, W. H.: Insights into photodissociation dynamics of acetaldehyde from ab initio calculations and molecular dynamics simulations, J. Chem. Phys., 131, 054306, https://doi.org/10.1021/jp073875+, 2009. a
Chen, W. T., Shao, M., Lu, S. H., Wang, M., Zeng, L. M., Yuan, B., and Liu, Y.: Understanding primary and secondary sources of ambient carbonyl compounds in Beijing using the PMF model, Atmos. Chem. Phys., 14, 3047–3062, https://doi.org/10.5194/acp-14-3047-2014, 2014. a, b, c
Chen, Y. and Zhu, L.: Wavelength-dependent photolysis of glyoxal in the 290–420 nm region, J. Phys. Chem. A, 107, 4643–4651, https://doi.org/10.1021/jp022440d, 2003. a
Clubb, A. E., Jordan, M. J. T., Kable, S. H., and Osborn, D. L.: Phototautomerization of acetaldehyde to vinyl alcohol: A primary process in UV-irradiated acetaldehyde from 295 to 335 nm, J. Phys. Chem. Lett., 3, 3522–3526, https://doi.org/10.1021/jz301701x, 2012. a, b, c, d, e, f
Copeland, R. A. and Crosley, D. R.: Radiative, collisional and dissociative processes in triplet acetone, Chem. Phys. Lett., 115, 362–368, https://doi.org/10.1016/0009-2614(85)85149-6, 1985. a
Couch, D. E., Nguyen, Q. L., Liu, A., Hickstein, D. D., Kapteyn, H. C., Murnane, M. M., and Labbe, N. J.: Detection of the keto-enol tautomerization in acetaldehyde, acetone, cyclohexanone, and methyl vinyl ketone with a novel VUV light source, Proc. Combust. Inst., 38, 1737–1744, https://doi.org/10.1016/j.proci.2020.06.139, 2021. a, b, c
Cui, G. and Fang, W.: Mechanistic photodissociation of glycolaldehyde: Insights from ab initio and RRKM calculations, ChemPhysChem, 12, 1351–1357, https://doi.org/10.1002/cphc.201000968, 2011. a
Diau, E. W.-G., Kötting, C., and Zewail, A. H.: Femtochemistry of Norrish Type-I reactions: I. Experimental and theoretical studies of acetone and related ketones on the S1 surface, Chem. Phys. Chem., 2, 273–293, https://doi.org/10.1002/1439-7641(20010518)2:5<273::AID-CPHC273>3.0.CO;2-H, 2001. a
Dobeck, L. M., Lambert, H. M., Kong, W., Pisano, P. J., and Houston, P. L.: H2 production in the 440-nm photodissociation of glyoxal, System, 103, 10312–10323, https://doi.org/10.1021/jp992333s, 1999. a
Dykstra, C. E. and Schaefer, H. F.: Electronic structure of dicarbonyls. Glyoxal excited states, J. Am. Chem. Soc., 98, 401–406, https://doi.org/10.1021/ja00418a014, 1976. a
Ehhalt, D. H. and Rohrer, F.: The tropospheric cycle of H2: A critical review, Tellus B Chem. Phys. Meteorol., 61B, 500–535, https://doi.org/10.1111/j.1600-0889.2009.00416.x, 2009. a, b
El-Sayed, M. A.: Spin-orbit coupling and the radiationless processes in nitrogen heterocyclics, Chem. Phys., 38, 2834–2838, 1961. a
Franco, B., Blumenstock, T., Cho, C., Clarisse, L., Clerbaux, C., Coheur, P.-F., De Mazière, M., De Smedt, I., Dorn, H.-P., Emmerichs, T., Fuchs, H., Gkatzelis, G., Griffith, D. W. T., Gromov, S., Hannigan, J. W., Hase, F., Hohaus, T., Jones, N., Kerkweg, A., Kiendler-Scharr, A., Lutsch, E., Mahieu1, E., Novelli, A., Ortega, I., Paton-Walsh, C., Pommier, M., Pozzer, A., Reimer, D., Rosanka, S., Sander, R., Schneider, M., Strong, K., Tillmann, R., Van Roozendael, M., Vereecken, L., Vigouroux, C., Wahner, A., and Taraborrelli, D.: Ubiquitous atmospheric production of organic acids mediated by cloud droplets, Nature, 593, 233–237, https://doi.org/10.1038/s41586-021-03462-x, 2021. a
Godunov, I. A., Yakovlev, N. N., and Khimii, Z. S.: Experimental structural and conformational studies of carbonyl molecules in the ground and lower excited states, J. Struct. Chem., 36, 238–253, https://doi.org/10.1007/BF02578062, 1995. a
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007. a
Goncharov, V., Herath, N., and Suits, A. G.: Roaming dynamics in acetone dissociation, J. Phys. Chem. A, 112, 9423–9428, https://doi.org/10.1021/jp802534r, 2008. a, b
Grant, A., Witham, C. S., Simmonds, P. G., Manning, A. J., and O'Doherty, S.: A 15 year record of high-frequency, in situ measurements of hydrogen at Mace Head, Ireland, Atmos. Chem. Phys., 10, 1203–1214, https://doi.org/10.5194/acp-10-1203-2010, 2010. a
Grimme, S., Antony, J., Ehrlich, S., Krieg, H., Grimme, S., Antony, J., Ehrlich, S., and Krieg, H.: A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 132, 154104, https://doi.org/10.1063/1.3382344, 2010. a
Grimme, S., Ehrlich, S., and Goerigk, L.: Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 32, 1456–1465, https://doi.org/10.1002/jcc.21759, 2011. a
Gruver, J. T. and Calvert, J. G.: The vapor phase photolysis of (+)2-methylbutanal-iodine mixtures at wave length 3130 Å, J. Am. Chem. Soc., 80, 3524–3527, https://doi.org/10.1021/ja01547a009, 1958. a
Guo, S. J., Chen, M., He, X. L., Yang, W. W., and Tan, J. H.: Seasonal and diurnal characteristics of carbonyls in urban air in Qinzhou, China, Aerosol Air Qual. Res., 14, 1653–1664, https://doi.org/10.4209/aaqr.2013.12.0351, 2014. a
Haas, Y.: Photochemical α-cleavage of ketones: Revisiting acetone, Photochem. Photobiol. Sci., 3, 6–16, https://doi.org/10.1039/b307997j, 2004. a, b
Hansen, D. A. and Lee, E. K.: Radiative and nonradiative transitions in the first excited singlet state of simple linear aldehydes, J. Chem. Phys., 63, 3272–3277, https://doi.org/10.1063/1.431802, 1975. a
Harrison, A. W., Kharazmi, A., Shaw, M. F., Quinn, M. S., Lee, K. L. K., Nauta, K., Rowell, K. N., Jordan, M. J. T., and Kable, S. H.: Dynamics and quantum yields of H2 + CH2CO as a primary photolysis channel in CH3CHO, Phys. Chem. Chem. Phys., 21, 14284–14295, https://doi.org/10.1039/c8cp06412a, 2019. a, b, c, d, e, f, g, h
Hauglustaine, D. A.: A three-dimensional model of molecular hydrogen in the troposphere, J. Geophys. Res.-Atmos., 107, 4330, https://doi.org/10.1029/2001JD001156, 2002. a
Heazlewood, B. R., Jordan, M. J. T., Kable, S. H., Selby, T. M., Osborn, D. L., Shepler, B. C., Braams, B. J., and Bowman, J. M.: Roaming is the dominant mechanism for molecular products in acetaldehyde photodissociation, P. Natl. Acad. Sci. USA, 105, 12719–12724, https://doi.org/10.1073/pnas.0802769105, 2008. a, b, c
Heazlewood, B. R., Rowling, S. J., Maccarone, A. T., Jordan, M. J., and Kable, S. H.: Photochemical formation of HCO and CH3 on the ground S0 (1A′) state of CH3CHO, J. Chem. Phys., 130, 054310, https://doi.org/10.1063/1.3070517, 2009. a, b, c, d
Heazlewood, B. R., Maccarone, A. T., Andrews, D. U., Osborn, D. L., Harding, L. B., Klippenstein, S. J., Jordan, M. J. T., and Kable, S. H.: Near-threshold H/D exchange in CD3CHO photodissociation, Nat. Chem., 3, 443–448, https://doi.org/10.1038/nchem.1052, 2011. a, b, c, d
Heicklen, J. and Noyes, W. A.: The photolysis and fluorescence of acetone and acetone-biacetyl mixtures, J. Am. Chem. Soc., 81, 3858–3863, https://doi.org/10.1021/ja01524a016, 1959. a
Hepburn, J. W., Buss, R. J., Butler, L. J., and Lee, Y. T.: Molecular beam study of the photochemistry of S1 glyoxal, J. Phys. Chem., 87, 3638–3641, https://doi.org/10.1021/j100242a014, 1983. a
Horowitz, A. and Calvert, J. G.: Wavelength dependence of the quantum efficiencies of the primary processes in formaldehyde photolysis at 25 ∘C, Int. J. Chem. Kinet., 10, 805–819, https://doi.org/10.1002/kin.550100803, 1978. a
Horowitz, A. and Calvert, J. G.: Wavelength dependence of the primary processes in acetaldehyde photolysis, J. Phys. Chem., 86, 3105–3114, https://doi.org/10.1021/j100213a011, 1982. a
Houston, P. L. and Kable, S. H.: Photodissociation of acetaldehyde as a second example of the roaming mechanism, P. Natl. Acad. Sci. USA, 103, 16079–16082, https://doi.org/10.1073/pnas.0604441103, 2006. a
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a, b
Jenkin, M. E., Saunders, S. M., Derwent, R. G., and Pilling, M. J.: Development of a reduced speciated VOC degradation mechanism for use in ozone models, Atmos. Environ., 36, 4715–4734, https://doi.org/10.1016/S1352-2310(02)00563-0, 2002. a
Kahan, T. F., Ormond, T. K., Ellison, G. B., and Vaida, V.: Acetic acid formation via the hydration of gas-phase ketene under ambient conditions, Chem. Phys. Lett., 565, 1–4, https://doi.org/10.1016/j.cplett.2013.02.030, 2013. a, b
Kao, C. C., Ho, M.-L., Chen, M.-W., Lee, S.-J., and Chen, I.-C.: Internal state distributions of fragment HCO via S0 and T1 pathways of glyoxal after photolysis in the ultraviolet region, J. Chem. Phys, 120, 5087–5095, https://doi.org/10.1063/1.1647538, 2004. a
Karton, A., Tarnopolsky, A., Lamère, J.-F., Schatz, G. C., and Martin, J. M. L.: Highly accurate first-principles benchmark data sets for the parametrization and validation of density functional and other approximate methods. Derivation of a robust, generally applicable, double-hybrid functional for thermochemistry and thermochemical kinetics, J. Phys. Chem. A, 112, 12868, https://doi.org/10.1021/jp801805p, 2008. a, b
Kawamura, K., Steinberg, S., and Kaplan, I. R.: Homologous series of C1-C10 monocarboxylic acids and C1-C6 carbonyls in Los Angeles air and motor vehicle exhausts, Atmos. Environ., 34, 4175–4191, https://doi.org/10.1016/S1352-2310(00)00212-0, 2000. a
Keeffe, J. R., Kresge, A. J., and Schepp, N. P.: Generation of simple enols by photooxidation. Keto-enol equilibrium constants of some aliphatic systems in aqueous solution, J. Am. Chem. Soc., 110, 1993–1995, https://doi.org/10.1021/ja00214a069, 1988. a
Kesharwani, M. K., Brauer, B., and Martin, J. M.: Frequency and zero-point vibrational energy scale factors for double-hybrid density functionals (and other selected methods): Can anharmonic force fields be avoided?, J. Phys. Chem. A, 119, 1701–1714, https://doi.org/10.1021/jp508422u, 2015. a
Kesselmeier, J. and Staudt, M.: Biogenic volatile organic compunds (VOC): An overview on emission, physiology and ecology., J. Atmos. Chem., 33, 23–88, https://doi.org/10.1023/A:1006127516791, 1999. a, b
Kharazmi, A.: Investigating the complex photochemistry of atmospheric carbonyls, PhD thesis, The University of New South Wales, Sydney, Australia, 2019. a, b
Kirkbride, F. W. and Norrish, R. G. W.: The photochemical properties of the carbonyl group, Trans. Faraday Soc., 27, 404–408, https://doi.org/10.1039/TF9312700404, 1931. a, b, c
Lary, D. J. and Shallcross, D. E.: Central role of carbonyl compounds in atmospheric chemistry, J. Geophys. Res.-Atmos., 105, 19771–19778, https://doi.org/10.1029/1999JD901184, 2000. a
Lee, A. M. D., Coe, J. D., Ullrich, S., Ho, M. L., Lee, S. J., Cheng, B. M., Zgierski, M. Z., Chen, I. C., Martinez, T. J., and Stolow, A.: Substituent effects on dynamics at conical intersections: α,β-enones, J. Phys. Chem. A, 111, 11948–11960, https://doi.org/10.1021/jp074622j, 2007. a
Lee, K. L. K., Quinn, M. S., Maccarone, A. T., Nauta, K., Houston, P. L., Reid, S. A., Jordan, M. J., and Kable, S. H.: Two roaming pathways in the photolysis of CH3CHO between 328 and 308 nm, Chem. Sci., 5, 4633–4638, https://doi.org/10.1039/c4sc02266a, 2014. a
Lee, K. L. K., Nauta, K., and Kable, S. H.: Photodissociation of acetone from 266 to 312 nm: Dynamics of CH3 + CH3CO channels on the S0 and T1 states, J. Chem. Phys., 146, 044304, https://doi.org/10.1063/1.4974035, 2017. a
Lee, S. H. and Chen, I. C.: Non-exponential decays of the S1 vibronic levels of acetaldehyde, Chem. Phys., 220, 175–189, https://doi.org/10.1016/S0301-0104(97)00148-1, 1997. a
Lee, Y., Zhou, X., Kleinman, L. I., Nunnermacker, L. J., Springston, S. R., Daum, P. H., Newman, L., Keigley, W. G., Holdren, M. W., Spicer, C. W., Young, V., Fu, B., Parrish, D. D., Holloway, J., Williams, J., Roberts, J. M., Ryerson, T. B., and Fehsenfeld, F. C.: Atmospheric chemistry and distribution of formaldehyde and several multioxygenated carbonyl compounds during the 1995 Nashville/Middle Tennessee Ozone Study, J. Geophys. Res.-Atmos., 103, 22449–22462, https://doi.org/10.1029/98JD01251, 1998. a
Liu, J.-Y., Long, Z.-W., Mitchell, E., and Long, B.: New mechanistic pathways for the reactions of formaldehyde with formic acid catalyzed by sulfuric acid and formaldehyde with sulfuric acid catalyzed by formic acid: Formation of potential secondary organic aerosol precursors, ACS Earth Space Chem. Article ASAP, 5, 1363–1372, https://doi.org/10.1021/acsearthspacechem.1c00002, 2021. a
Loge, G. W. and Parmenter, C. S.: Collision-free dissociation after excitation of single rotational levels in S1 glyoxal, J. Phys. Chem., 85, 1653–1662, https://doi.org/10.1021/j150612a011, 1981. a
Mauguière, F. A. L., Collins, P., Kramer, Z. C., Carpenter, B. K., Ezra, G. S., Farantos, S. C., and Wiggins, S.: Phase space structures explain hydrogen atom roaming in formaldehyde decomposition, J. Phys. Chem. Lett., 6, 4123–4128, https://doi.org/10.1021/acs.jpclett.5b01930, 2015. a
Menchaca-Torre, H. L., Mercado-Hernández, R., Rodríguez-Rodríguez, J., and Mendoza-Domínguez, A.: Diurnal and seasonal variations of carbonyls and their effect on ozone concentrations in the atmosphere of Monterrey, Mexico, J. Air Waste Manag. Assoc., 65, 500–510, https://doi.org/10.1080/10962247.2015.1005849, 2015. a
Millet, D. B., Guenther, A., Siegel, D. A., Nelson, N. B., Singh, H. B., de Gouw, J. A., Warneke, C., Williams, J., Eerdekens, G., Sinha, V., Karl, T., Flocke, F., Apel, E., Riemer, D. D., Palmer, P. I., and Barkley, M.: Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys., 10, 3405–3425, https://doi.org/10.5194/acp-10-3405-2010, 2010. a, b
Millet, D. B., Baasandorj, M., Farmer, D. K., Thornton, J. A., Baumann, K., Brophy, P., Chaliyakunnel, S., de Gouw, J. A., Graus, M., Hu, L., Koss, A., Lee, B. H., Lopez-Hilfiker, F. D., Neuman, J. A., Paulot, F., Peischl, J., Pollack, I. B., Ryerson, T. B., Warneke, C., Williams, B. J., and Xu, J.: A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 15, 6283–6304, https://doi.org/10.5194/acp-15-6283-2015, 2015. a
Moortgat, G. K. and Warneck, P.: CO and H2 quantum yields in the photodecomposition of formaldehyde in air, J. Chem. Phys., 70, 3639–3651, https://doi.org/10.1063/1.437956, 1979. a
Moortgat, G. K., Seiler, W., and Warneck, P.: Photodissociation of HCHO in air: CO and H2 quantum yields at 220 and 300 K, J. Chem. Phys., 78, 1185–1190, https://doi.org/10.1063/1.444911, 1983. a
Moortgat, G. K., Meyrahn, H., and Warneck, P.: Photolysis of acetaldehyde in air: CH4, CO and CO2 quantum yields, Chem. Phys. Chem., 11, 3896–3908, https://doi.org/10.1002/cphc.201000757, 2010. a, b
Morajkar, P., Bossolasco, A., Schoemaecker, C., and Fittschen, C.: Photolysis of CH3CHO at 248 nm: Evidence of triple fragmentation from primary quantum yield of CH3 and HCO radicals and H atoms, J. Chem. Phys., 140, 214 308, https://doi.org/10.1063/1.4878668, 2014. a
Neese, F.: Software update: The ORCA program system, version 4.0, Wiley Interdiscip. Rev. Comput. Mol. Sci. [code], 8, 4–9, https://doi.org/10.1002/wcms.1327, 2017. a
Norrish, R. G. W. and Appleyard, M. E. S.: 191. Primary photochemical reactions. Part IV. Decomposition of methyl ethyl ketone and methyl butyl ketone, J. Chem. Soc., 874–880, https://doi.org/10.1039/JR9340000874, 1934. a
Novelli, P. C.: Molecular hydrogen in the troposphere: Global distribution and budget, J. Geophys. Res.-Atmos., 104, 30427–30444, https://doi.org/10.1029/1999JD900788, 1999. a
Pal, R., Kim, K. H., Hong, Y. J., and Jeon, E. C.: The pollution status of atmospheric carbonyls in a highly industrialized area, J. Hazard. Mater., 153, 1122–1135, https://doi.org/10.1016/j.jhazmat.2007.09.068, 2008. a
Patterson, J. D., Aydin, M., Crotwell, A. M., Petron, G., Severinghaus, J. P., and Saltzman, E. S.: Atmospheric history of H2 over the past century reconstructed from South Pole firn air, Geophys. Res. Lett., 47, e2020GL087787, https://doi.org/10.1029/2020GL087787, 2020. a
Pople, J. A.: Quantum chemical models (Nobel lecture), Angew. Chem. Int. Ed., 38, 1894–1902, https://doi.org/10.1002/(SICI)1521-3773(19990712)38:13/14<1894::AID-ANIE1894>3.0.CO;2-H, 1999. a
Price, H., Jaeglé, L., Rice, A., Quay, P., Novelli, P. C., and Gammon, R.: Global budget of molecular hydrogen and its deuterium content: Constraints from ground station, cruise, and aircraft observations, J. Geophys. Res.-Atmos., 112, D22108, https://doi.org/10.1029/2006JD008152, 2007. a
Quinn, M. S., Andrews, D. U., Nauta, K., Jordan, M. J., and Kable, S. H.: The energy dependence of CO(v,J) produced from H2CO via the transition state, roaming, and triple fragmentation channels, J. Chem. Phys., 147, 013935, https://doi.org/10.1063/1.4983138, 2017. a, b, c, d
Raber, W. H. and Moortgat, G. K.: Photooxidation of selected carbonyl compounds in air: methyl ethyl ketone, methyl vinyl ketone, methacrolein and methylglyoxal, in: Progress and Problems in Atmospheric Chemistry, edited by: Barker, R. J., World Scientific Publishing Company, Singapore, 318–373, 1996. a
Rhee, T. S., Brenninkmeijer, C. A. M., and Röckmann, T.: The overwhelming role of soils in the global atmospheric hydrogen cycle, Atmos. Chem. Phys., 6, 1611–1625, https://doi.org/10.5194/acp-6-1611-2006, 2006. a, b
Rickard, A. and Young, J.: The Master Chemical Mechanism (MCM) v3.2, available at: http://mcm.york.ac.uk, last access: 13 December 2021. a, b
Rowell, K. N., Kable, S. H., and Jordan, M. J. T.: Structural effects on the Norrish Type I α-bond cleavage of tropospherically important carbonyls, J. Phys. Chem. A, 123, 10381–10396, https://doi.org/10.1021/acs.jpca.9b05534, 2019. a, b, c, d, e, f, g, h, i, j, k
Rubio-Lago, L., Amaral, G. A., Arregui, A., González-Vázquez, J., and Bañares, L.: Imaging the molecular channel in acetaldehyde photodissociation: Roaming and transition state mechanisms, Phys. Chem. Chem. Phys., 14, 6067–6078, https://doi.org/10.1039/c2cp22231k, 2012. a
Ruscic, B. and Bross, D. H.: Active thermochemical tables (ATcT) values based on ver. 1.122p of the Thermochemical Network, available at: https://atct.anl.gov/ (last access: 8 April 2021), 2020. a, b, c
Ruscic, B., Pinzon, R. E., Von Laszewski, G., Kodeboyina, D., Burcat, A., Leahy, D., Montoy, D., and Wagner, A. F.: Active thermochemical tables: Thermochemistry for the 21st century, J. Phys. Conf. Ser., 16, 561–570, https://doi.org/10.1088/1742-6596/16/1/078, 2005. a, b, c
Saheb, V. and Zokaie, M.: Multichannel gas-phase unimolecular decomposition of acetone: Theoretical kinetic studies, J. Phys. Chem. A, 122, 5895–5904, https://doi.org/10.1021/acs.jpca.8b02423, 2018. a, b, c
Salter, R. J., Blitz, M. A., Heard, D. E., Pilling, M. J., and Seakins, P. W.: Pressure and temperature dependent photolysis of glyoxal in the 355-414 nm region: evidence for dissociation from multiple states., Phys. Chem. Chem. Phys., 15, 6516–6526, https://doi.org/10.1039/c3cp43596b, 2013. a
Schalk, O., Schuurman, M. S., Wu, G., Lang, P., Mucke, M., Feifel, R., and Stolow, A.: Internal conversion versus intersystem crossing: What drives the gas phase dynamics of cyclic α,β-enones?, J. Phys. Chem. A, 118, 2279–2287, https://doi.org/10.1021/jp4124937, 2014. a
Schuurman, M. S. and Stolow, A.: Dynamics at conical intersections, Annu. Rev. Phys. Chem., 69, 427–450, https://doi.org/10.1146/annurev-physchem-052516-050721, 2018. a
Seco, R., Peñuelas, J., and Filella, I.: Short-chain oxygenated VOCs: Emission and uptake by plants and atmospheric sources, sinks, and concentrations, Atmos. Environ., 41, 2477–2499, https://doi.org/10.1016/j.atmosenv.2006.11.029, 2007. a
Shaw, M. F., Sztáray, B., Whalley, L. K., Heard, D. E., Millet, D. B., Jordan, M. J., Osborn, D. L., and Kable, S. H.: Photo-tautomerization of acetaldehyde as a photochemical source of formic acid in the troposphere, Nat. Commun., 9, 1–7, https://doi.org/10.1038/s41467-018-04824-2, 2018. a, b, c, d, e, f, g, h, i, j
Simon, H., Beck, L., Bhave, P. V., Divita, F., Hsu, Y., Luecken, D., Mobley, J. D., Pouliot, G. A., Reff, A., Sarwar, G., and Strum, M.: The development and uses of EPA's SPECIATE database, Atmos. Pollut. Res., 1, 196–206, https://doi.org/10.5094/apr.2010.026, 2010. a
Skorobogatov, G. A., Meilakhs, A. G., Pogosyan, Y. I., and Khripun, V. K.: New mechanism of photodissociation of gaseous acetone, Russ. J. Gen. Chem., 72, 1271–1275, https://doi.org/10.1023/a:1020896200573, 2002. a
So, S., Wille, U., and da Silva, G.: Atmospheric chemistry of enols: A theoretical study of the vinyl alcohol + OH + O2 reaction mechanism, Environ. Sci. Technol., 48, 6694–6701, https://doi.org/10.1021/es500319q, 2014. a
So, S., Wille, U., and Da Silva, G.: A theoretical study of the photoisomerization of glycolaldehyde and subsequent OH radical-initiated oxidation of 1,2-ethenediol, J. Phys. Chem. A, 119, 9812–9820, https://doi.org/10.1021/acs.jpca.5b06854, 2015. a, b
So, S., Wille, U., and Da Silva, G.: Photoisomerization of methyl vinyl ketone and methacrolein in the troposphere: A theoretical investigation of ground-State reaction pathways, ACS Earth Space Chem., 2, 753–763, https://doi.org/10.1021/acsearthspacechem.8b00066, 2018. a, b, c, d, e
Tadić, J. M., Juranić, I. O., and Moortgat, G. K.: Photooxidation of n-heptanal in air: Norrish type I and II processes and quantum yield total pressure dependency, J. Chem. Soc. Perkin Trans., 2, 135–140, https://doi.org/10.1039/b106476m, 2002. a, b
Tadić, J. M., Moortgat, G. K., Bera, P. P., Loewenstein, M., Yates, E. L., and Lee, T. J.: Photochemistry and photophysics of n-butanal, 3-methylbutanal, and 3,3-dimethylbutanal: Experimental and theoretical study, J. Phys. Chem. A, 116, 5830–5839, https://doi.org/10.1021/jp208665v, 2012. a, b
Tanner, R. L., Zielinska, B., Uberna, E., Harshfield, G., and McNichol, A. P.: Concentrations of carbonyl compounds and the carbon isotopy of formaldehyde at a coastal site in Nova Scotia during the NARE summer intensive, J. Geophys. Res.-Atmos., 101, 28961–28970, https://doi.org/10.1029/95jd03574, 1996. a
Toulson, B. W., Kapnas, K. M., Fishman, D. A., and Murray, C.: Competing pathways in the near-UV photochemistry of acetaldehyde, Phys. Chem. Chem. Phys., 19, 14276–14288, https://doi.org/10.1039/c7cp02573d, 2017. a, b
Toulson, B. W., Fishman, D. A., and Murray, C.: Photodissociation dynamics of acetone studied by time-resolved ion imaging and photofragment excitation spectroscopy, Phys. Chem. Chem. Phys., 20, 2457–2469, https://doi.org/10.1039/C7CP07320H, 2018. a, b, c
Townsend, D., Lahankar, S. A., Lee, S. K., Chambreau, S. D., Suits, A. G., Zhang, X., Rheinecker, J., Harding, L. B., and Bowman, J. M.: The roaming atom: Straying from the reaction path in formaldehyde decomposition, Science, 306, 1158–1161, https://doi.org/10.1126/science.1104386, 2004. a, b, c
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016. a
Tsai, P.-Y., Li, H.-K., Kasai, T., and Lin, K.-C.: Roaming as the dominant mechanism for molecular products in the photodissociation of large aliphatic aldehydes, Phys. Chem. Chem. Phys., 17, 23112–23120, https://doi.org/10.1039/C5CP03408F, 2015. a
Valachovic, L. R., Tuchler, M. F., Dulligan, M., Droz-Georget, T., Zyrianov, M., Kolessov, A., Reisler, H., and Wittig, C.: Photoinitiated H2CO unimolecular decomposition: Accessing H + HCO products via S0 and T1 pathways, J. Chem. Phys., 112, 2752–2761, https://doi.org/10.1063/1.480849, 2000. a
Vigouroux, C., Hendrick, F., Stavrakou, T., Dils, B., De Smedt, I., Hermans, C., Merlaud, A., Scolas, F., Senten, C., Vanhaelewyn, G., Fally, S., Carleer, M., Metzger, J.-M., Müller, J.-F., Van Roozendael, M., and De Mazière, M.: Ground-based FTIR and MAX-DOAS observations of formaldehyde at Réunion Island and comparisons with satellite and model data, Atmos. Chem. Phys., 9, 9523–9544, https://doi.org/10.5194/acp-9-9523-2009, 2009. a
Wagner, P. J. and Klán, P.: Norrish type II photoelimination of ketones: Cleavage of 1,4-biradicals formed by γ-hydrogen abstraction, in: CRC Handbook of Organic Photochemistry and Photobiology, 2nd edn., edited by: Horspool, W. M. and Lenci, F., 52, 1–22, CRC Press, Boca Raton, 2004. a, b
Wagner, P. J. and Zepp, R. G.: Trapping by mercaptans of biradical intermediates in type II photoelemination, J. Am. Chem. Soc., 94, 287, https://doi.org/10.1021/ja00756a057, 1972. a, b
Wallington, T. J., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V., Mellouki, A., and Troe, J.: IUPAC task group on atmospheric chemical kinetic data evaluation, available at: http://iupac.pole-ether.fr/ (last access: 8 April 2021), 2018. a
Warneck, P. and Moortgat, G. K.: Quantum yields and photodissociation coefficients of acetaldehyde in the troposphere, Atmos. Environ., 62, 153–163, https://doi.org/10.1016/j.atmosenv.2012.08.024, 2012. a
Weigend, F. and Ahlrichs, R.: Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy., Phys. Chem. Chem. Phys., 7, 3297–305, https://doi.org/10.1039/b508541a, 2005. a
Whangbo, M. H. and Stewart, K. R.: On the thermodynamic stability and reactivity of alkyl-substituted alkenes, J. Org. Chem., 47, 736–738, https://doi.org/10.1021/jo00343a028, 1982. a, b
Wild, O., Zhu, X., and Prather, M. J.: Fast-J: Accurate simulation of in- and below-cloud photolysis in tropospheric chemical models, J. Atmos. Chem., 37, 245–282, https://doi.org/10.1023/A:1006415919030, 2000. a
Xiao, X., Prinn, R. G., Simmonds, P. G., Steele, L. P., Novelli, P. C., Huang, J., Langenfelds, R. L., O'Doherty, S., Krummel, P. B., Fraser, P. J., Porter, L. W., Weiss, R. F., Salameh, P., and Wang, R. H. J.: Optimal estimation of the soil uptake rate of molecular hydrogen from the advanced global atmospheric gases experiment and other measurements, J. Geophys. Res., 112, D07303, https://doi.org/10.1029/2006JD007241, 2007. a, b
Yang, C., Bhattacharyya, S., Liu, L., Fang, W.-H., and Liu, K.: Real-time tracking of the entangled pathways in the multichannel photodissociation of acetaldehyde, Phys. Chem. Chem. Phys., 11, 6423–6430, 2020. a
Yang, X., Xue, L., Wang, T., Wang, X., Gao, J., Lee, S., Blake, D. R., Chai, F., and Wang, W.: Observations and explicit modeling of summertime carbonyl formation in Beijing: Identification of key precursor species and their impact on atmospheric oxidation chemistry, J. Geophys. Res.-Atmos., 123, 1426–1440, https://doi.org/10.1002/2017jd027403, 2018. a
Yashiro, H., Sudo, K., Yonemura, S., and Takigawa, M.: The impact of soil uptake on the global distribution of molecular hydrogen: chemical transport model simulation, Atmos. Chem. Phys., 11, 6701–6719, https://doi.org/10.5194/acp-11-6701-2011, 2011. a
Yuan, B., Veres, P. R., Warneke, C., Roberts, J. M., Gilman, J. B., Koss, A., Edwards, P. M., Graus, M., Kuster, W. C., Li, S.-M., Wild, R. J., Brown, S. S., Dubé, W. P., Lerner, B. M., Williams, E. J., Johnson, J. E., Quinn, P. K., Bates, T. S., Lefer, B., Hayes, P. L., Jimenez, J. L., Weber, R. J., Zamora, R., Ervens, B., Millet, D. B., Rappenglück, B., and de Gouw, J. A.: Investigation of secondary formation of formic acid: urban environment vs. oil and gas producing region, Atmos. Chem. Phys., 15, 1975–1993, https://doi.org/10.5194/acp-15-1975-2015, 2015. a
Zahra, A. and Noyes, W. A.: The photochemistry of methyl isopropyl ketone, J. Phys. Chem., 69, 943–948, https://doi.org/10.1021/j100887a041, 1965. a, b, c
Zhang, R., Suh, I., Zhao, J., Zhang, D., Fortner, E. C., Tie, X., Molina, L. T., and Molina, M. J.: Atmospheric new particle formation enhanced by organic acids, Science, 304, 1487–1490, https://doi.org/10.1126/science.1095139, 2004. a
Zhu, C. and Zhu, L.: Photolysis of glycolaldehyde in the 280–340 nm region, J. Phys. Chem. A, 114, 8384–8390, https://doi.org/10.1021/jp104497d, 2010. a, b, c
Zhu, L., Tang, Y., Chen, Y., and Cronin, T.: Wavelength-dependent photolysis of C3-C7 aldehydes in the 280-330 nm region, Spectrosc. Lett., 42, 467–478, https://doi.org/10.1080/00387010903267195, 2009. a, b, c, d, e, f
- Abstract
- Introduction
- Computational methods
- Results and discussion
- Competition between carbonyl S0 reactions
- Tropospheric relevance of S0 reactions
- Conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Computational methods
- Results and discussion
- Competition between carbonyl S0 reactions
- Tropospheric relevance of S0 reactions
- Conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement