the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Jul 2022
| 04 Jul 2022
Investigation of the limonene photooxidation by OH at different NO concentrations in the atmospheric simulation chamber SAPHIR (Simulation of Atmospheric PHotochemistry In a large Reaction Chamber)
Jacky Yat Sing Pang
Anna Novelli
Martin Kaminski
Ismail-Hakki Acir
Birger Bohn
Philip T. M. Carlsson
Changmin Cho
Hans-Peter Dorn
Andreas Hofzumahaus
Anna Lutz
Sascha Nehr
David Reimer
Franz Rohrer
Ralf Tillmann
Robert Wegener
Astrid Kiendler-Scharr
Andreas Wahner
The oxidation of limonene by the hydroxyl (OH) radical and ozone (O3) was investigated in the atmospheric simulation chamber SAPHIR (Simulation of Atmospheric PHotochemistry In a large Reaction Chamber) in experiments performed at different nitric oxide (NO) mixing ratios from nearly 0 up to 10 ppbv. For the experiments dominated by OH oxidation, the formaldehyde (HCHO) yield was experimentally determined and found to be (12 ± 3), (13 ± 3), and (32 ± 5) % for experiments with low (∼ 0.1 ppbv), medium (∼ 0.3 ppbv), and high NO (5 to 10 ppbv), respectively. The yield in an ozonolysis-only experiment was (10 ± 1) %, which agrees with previous laboratory studies. The experimental yield of the first-generation organic nitrates from limonene–OH oxidation is calculated as (34 ± 5) %, about 11 % higher than the value in the Master Chemical Mechanism (MCM), which is derived from structure–activity relationships (SARs). Time series of measured radicals, trace-gas concentrations, and OH reactivity are compared to results from zero-dimensional chemical box model calculations applying MCM v3.3.1. Modeled OH reactivity is 5 to 10 s−1 (25 % to 33 % of the OH reactivity at the start of the experiment) higher than measured values at the end of the experiments under all chemical conditions investigated, suggesting either that there are unaccounted loss processes of limonene oxidation products or that products are less reactive toward OH. In addition, model calculations underestimate measured hydroperoxyl radical (HO2) concentrations by 20 % to 90 % and overestimate organic peroxyl radical (RO2) concentrations by 50 % to 300 %. The largest deviations are found in low-NO experiments and in the ozonolysis experiment. An OH radical budget analysis, which uses only measured quantities, shows that the budget is closed in most of the experiments. A similar budget analysis for RO2 radicals suggests that an additional RO2 loss rate constant of about (1–6) × 10−2 s−1 for first-generation RO2 is required to match the measured RO2 concentrations in all experiments. Sensitivity model runs indicate that additional reactions converting RO2 to HO2 at a rate constant of about (1.7–3.0) × 10−2 s−1 would improve the model–measurement agreement of NOx, HO2, and RO2 concentrations and OH reactivity. Reaction pathways that could lead to the production of additional OH and HO2 are discussed, which include isomerization reactions of RO2 from the oxidation of limonene, different branching ratios for the reaction of RO2 with HO2, and a faster rate constant for RO2 recombination reactions. As the exact chemical mechanisms of the additional HO2 and OH sources could not be identified, further work needs to focus on quantifying organic product species and organic peroxy radicals from limonene oxidation.
- Article
(7245 KB) - Full-text XML
-
Supplement
(2062 KB) - BibTeX
- EndNote





About 1000 Tg of biogenic volatile organic compounds (BVOCs) are emitted into the atmosphere every year. Approximately half of the emissions comprise isoprene, while 15 % consist of monoterpene species (Guenther et al., 2012). Among monoterpenes, limonene is the fourth-most abundant species, which comprises about 10 % of the total monoterpene emissions (Sindelarova et al., 2014). Apart from biogenic sources, limonene is also emitted from anthropogenic sources such as air fresheners and cleaning products (Liu et al., 2004; Nazaroff and Weschler, 2004; McDonald et al., 2018; Gkatzelis et al., 2021a, b). Limonene has two carbon–carbon double bonds, which make it reactive towards atmospheric oxidants such as ozone (O3), hydroxyl radicals (OH), and nitrate radicals (NO3) (Calogirou et al., 1999; Atkinson and Arey, 2003).
During daytime, the most important oxidant is OH. The reaction between the olefin group and the OH radical yields a β-hydroxyalkyl radical, which subsequently reacts with an oxygen molecule (O2) under atmospheric conditions to form an organic peroxy radical (RO2) (Fig. 1). In the presence of nitric oxide (NO), RO2 is converted to an alkoxy radical (RO), and thereby NO is oxidized to nitrogen dioxide (NO2). NO2 can be photolyzed back to NO together with an oxygen atom, which then reacts with an oxygen molecule producing ozone. This mechanism is the most relevant source of tropospheric ozone. Alkoxy radicals are very reactive, and they often quickly decompose, forming carbonyl products and a hydroperoxyl radical (HO2) or further organic radicals (Orlando et al., 2003). OH is then regenerated through the reaction between HO2 and NO, closing the radical reaction cycle and producing another ozone molecule from the photolysis of nitrogen dioxide. In polluted environments with high NOx (= NO + NO2) emissions such as in urban areas (NO concentration > 1000 pptv; e.g., Dusanter et al., 2009; Lu et al., 2017), the reaction between RO2 and NO is often the dominant pathway through which OH is regenerated. In remote regions with low NO mixing ratios (< 200 pptv; e.g., Ren et al., 2006; Whalley et al., 2011), RO2 predominantly reacts with HO2 and forms an organic peroxide molecule. This process terminates the radical chain. The organic peroxide produced during this process can potentially add to secondary organic aerosols (SOA) (Surratt et al., 2006). RO2 radicals can also react with other RO2 radicals. The reaction rate constant is usually low (k ranges from 10−15 to 10−11 cm3 s−1; Tyndall et al., 2001; Jenkin et al., 2019), and the RO2 loss rate is often small for atmospheric RO2 concentrations (RO2 ∼ 108 cm−3; Tan et al., 2018). Products of the RO2 recombination reactions are either alkoxy radicals or oxidized organic compounds containing alcohol or carbonyl functional groups, which terminate the radical reaction chain.
Figure 1Simplified mechanism of the limonene oxidation by OH and O3. If available, names of the species as they appear in MCM v3.3.1 are given. For simplicity the stereo-specificity of the intermediates, RO2 reactions with HO2 and other RO2, as well as the formation of organic nitrates are not shown here. Production of HCHO from the first oxidation step of limonene is labeled in red; HCHO production from the second oxidation step is labeled in blue; production of a HO2 radical is labeled in orange. The production of HCHO from the decomposition of LIMALBO is not classified, as it is not produced from the oxidation of the terminal C=C double bond. Most of the mechanism and branching ratios are taken from MCM v3.3.1, except for the branching ratios of the O3 addition at the external C=C bond of limonene (Wang and Wang, 2021) and the decomposition of LIMOOB to L5O2.
During the last decade, there has been increasing evidence from laboratory studies as well as theoretical studies that unimolecular reactions of RO2 can also be of importance in the atmosphere. Unimolecular H-shift reactions of RO2 can promote the formation of low-volatility organic compounds as the number of oxygen atoms in the molecule quickly increases during the process (Jokinen et al., 2014). In some of the unimolecular reactions, HOx (= OH + HO2) radicals can be regenerated, so that this reaction pathway becomes a radical propagation reaction that does not require the presence of NO. This has been, for example, shown for isoprene (Peeters and Müller, 2010; Crounse et al., 2011; Peeters et al., 2014; Berndt et al., 2019; Novelli et al., 2020) and methacrolein (Crounse et al., 2012; Fuchs et al., 2014). The reaction rate constant of H-shift unimolecular reactions depends on the position of the H atom relative to the peroxy group, the position of other functional groups to the H atom, and temperature (Crounse et al., 2013; Vereecken and Nozière, 2020). H-shift reactions are very slow (k < 10−3 s−1 at 298 K) in an aliphatic peroxy radical without an oxygenated function group (e.g., carbonyl, hydroxyl, alkoxy) attached to the carbon atom, from which the hydrogen is abstracted (Otkjær et al., 2018; Praske et al., 2019). Therefore, H-shift reactions typically cannot compete with bimolecular reactions under atmospheric conditions (kbi ∼ 10−2 s−1 for 50 pptv of NO and 5 × 108 cm−3 of HO2). Even if the RO2 radical contains a hydroxyl group, rate constants for H-shift reactions from the hydroxyl group are still low (k ∼ 10−2 to 10−1 s−1) (Vereecken and Nozière, 2020) and can only compete with bimolecular reactions in the atmosphere in remote areas or suburban regions with low to moderate NO concentrations (50–500 pptv). However, there are also fast H-shift reaction pathways that could compete with bimolecular reactions even in a moderately or heavily polluted scenario such as allylic H-shift (k ∼ 1 s−1) or aldehydic H-shift (k ∼ 1–100 s−1) reactions, in which the radical is stabilized by delocalized electrons or the carbonyl group, respectively (Otkjær et al., 2018; Zhang and Dibble, 2011; Vereecken and Nozière, 2020). Concerning limonene, which contains two carbon–carbon double bonds (Fig. 1), recent theoretical studies suggest that the RO2 radical formed after the reaction with OH or O3 undergoes rapid unimolecular reactions (reaction rate constants range from 1 to 102 s−1; Møller et al., 2020; Chen et al., 2021), which would be competitive with bimolecular reactions even in polluted environments. In this study, the atmospheric degradation of limonene was investigated in the atmospheric simulation chamber SAPHIR (Simulation of Atmospheric PHotochemistry In a large Reaction Chamber) at Forschungszentrum Jülich, Germany. Experiments were performed at three atmospherically relevant NO concentrations ranging from 0.1 to 10 ppbv. In addition, one ozonolysis experiment was conducted without the presence of NOx. Radiation and relative humidity during the experiments were also relevant to the conditions that are typically found in the atmosphere, which was an improvement compared to previous experiments that typically used artificial light sources or were conducted under very dry conditions (e.g., Larsen et al., 2001; Librando and Tringali, 2005).
The main objective of this work is to evaluate the performance of the current chemical model (Master Chemical Mechanism version 3.3.1, http://mcm.york.ac.uk/home.htt, last access: 27 June 2022) by comparing observations of trace gas and radical concentrations to model results. In particular, the question of whether the regeneration of OH from the radical cycle can be described by model calculations is investigated.
2.1 Atmospheric simulation chamber SAPHIR
SAPHIR is a large outdoor chamber located at Forschungszentrum Jülich, Germany. The cylindrical-shaped chamber (18 m length, 5 m diameter, volume 270 m3) is made of a double-wall Teflon (FEP) film, which minimizes wall loss and allows the transmission of the entire spectrum of solar radiation. A shutter system can be opened and closed to allow experiments in illuminated or dark conditions. All the experiments are performed in synthetic air produced by evaporating ultrapure liquid oxygen and nitrogen (Linde, purity > 99.9999 %). The air pressure inside the SAPHIR chamber is kept above the atmospheric pressure by 35 Pa to ensure that air outside the chamber cannot leak into the chamber, resulting in a typical dilution rate of trace gases of approximately 4 % h−1. The temperature in the chamber is similar to the ambient temperature. More details of the chamber can be found in previous publications: Rohrer et al. (2005) and Kaminski et al. (2017).
2.2 Instrumentation
Table 1 provides an overview of the quantities that were measured and the corresponding instruments. NO and NO2 concentrations were monitored with a chemiluminescence instrument (Eco Physics) with a blue-light photolytic converter that converts NO2 to NO. In some experiments, the zero value of the instrument was not accurately determined, as could be seen by a significant NO and NO2 mixing ratio (NO < 0.02, NO2 < 0.5 ppbv) that was measured in the dark, clean chamber. For these experiments, the instrumental zero was subtracted for the analysis of data in this work. Nitrous acid was measured in some experiments by a long-path absorption photometer (Li et al., 2014). Similar to the NOx instruments, non-zero HONO (< 100 pptv) was measured in the clean dark chamber in some experiments. This value was attributed to an unaccounted instrumental zero value and subtracted from the data.
Table 1Instrumentation for radical and trace-gas measurements in the chamber experiments.

a Differential optical absorption spectroscopy. b Laser-induced fluorescence. c Proton transfer reaction time-of-flight mass spectrometry. d Gas chromatography coupled with a flame ionization detector. e Differential optical absorption spectroscopy, available for the experiments in 2015. f Cavity ring-down spectroscopy, available for the experiments in 2019. g Not available in the experiments on 12 August 2013 and 5 June 2020. h Long-path absorption photometer.
O3 was measured with a UV absorption instrument (Ansyco). Photolysis frequencies were calculated from solar actinic flux densities measured by a spectroradiometer outside the chamber (Bohn et al., 2005; Bohn and Zilken, 2005).
OH was measured with a laser-induced fluorescence (LIF) instrument, in which OH is excited by laser light at a wavelength of 308 nm in a low-pressure cell (3.5 hPa) (Holland et al., 1995). HO2 was measured by the LIF instrument in a separate detection cell (HOx cell), in which HO2 is first converted to OH in the reaction with NO and the sum of OH and HO2 (= HOx) is detected by OH fluorescence. The HO2 concentration is then calculated by subtracting the OH concentration measured in the OH cell from the signal obtained in the HOx cell. Organic peroxy radicals (RO2) can potentially interfere in the measurement of HO2 due to the concurrent conversion of RO2 to HO2 after the reaction with NO. This interference is usually less than 5 % for simple alkoxy radicals (C1–C4) because of the slow conversion rate of the alkoxy radical produced in the reaction of RO2 with NO at the reduced O2 concentration in the low-pressure cell. However, β-hydroxy alkoxy radicals formed from the OH-initiated oxidation of larger alkene species (e.g., isoprene) could rapidly decompose and then react with O2 to form HO2, which adds to the sampled HO2 radical concentration (Fuchs et al., 2011; Lu et al., 2012). During the experiments in this work, the HO2 cell was operated with low NO concentrations to minimize the conversion of RO2 to HO2 radicals, but it cannot be excluded that a small fraction of limonene RO2 acted as an interference. The upper limit of such an interference would be around 15 %, as indicated by characterization experiments, which unfortunately did not allow us to determine an accurate number due to the limited precision of results.
RO2 was measured in the ROx-LIF system, which consists of a converter mounted on top of a fluorescence detection cell. In the converter, RO2 is converted to HO2 in the presence of added CO and NO at low total pressure (∼ 25 hPa). The HO2 is then passed into a low-pressure detection cell (3.5 hPa) where HO2 is converted into OH by reaction with a large excess of added NO. The RO2 concentration is calculated by subtracting the HOx concentration from the measured signal (Fuchs et al., 2008, 2011). The sensitivity of the LIF instrument to RO2 is regularly calibrated for methylperoxy radicals. Laboratory tests show that the instrument's sensitivity to RO2 radicals from limonene might be slightly reduced compared to the sensitivity for methylperoxy radicals (0.85 ± 0.05).
Apart from the detection by the LIF instrument, OH radical concentrations were measured by differential optical absorption spectroscopy (DOAS) (Dorn et al., 1995) in the experiments in 2015. The mean differences between the LIF-measured OH and DOAS-measured OH concentrations in the two experiments in 2015 were −13 % and +39 %. The difference between measurements in the latter experiment was higher than the combined accuracies of the measurements. Because DOAS is inherently a calibration-free method and it is regarded as a reference method (Schlosser et al., 2007, 2009), OH concentrations from this instrument are used for the evaluation of experiments in 2015.
Measurements of the OH reactivity (kOH), the inverse chemical lifetime of the OH radical, were achieved by an instrument making use of laser flash photolysis combined with the OH detection by LIF (Lou et al., 2010; Fuchs et al., 2017). In this instrument, air is sampled through a flow tube. A high concentration of OH is generated by ozone photolysis in the presence of water vapor using a short laser pulse at 266 nm, followed by the time-resolved measurement of the OH concentration, while the OH is consumed by OH reactants contained in the sampled air. The pseudo-first-order loss rate constant of the decay of the OH concentration gives the OH reactivity value.
Limonene concentrations were measured by gas chromatography coupled with a flame ionization detector (GC-FID, Kaminski, 2014) as well as by a proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS) (Lindinger et al., 1998; Jordan et al., 2009). Two GC-FIDs were used in the experiments in 2012. The differences between measurements by the two GC-derived measurements for limonene concentrations were about 12 %. Discrepancies between the limonene concentrations measured by GC-FID and PTR-MS were about 12 %, depending on which GC-FID measurement is used.
The amount of injected limonene can also be derived from the rapid increase in OH reactivity when limonene is injected. The injected limonene concentrations derived from the increase in OH reactivity agree with the PTR-MS measurements within 15 % in three experiments (8 and 10 August 2012 and 13 June 2015), but discrepancies were between 20 % and 55 % in the other experiments. For the analysis in this work, PTR-MS measurements were scaled in some experiments to match the limonene injections assuming that the measurement of OH reactivity is more accurate than PTR-MS measurements because no calibration of the instrument is required.
A time-of-flight chemical ionization mass spectrometer (ToF-CIMS) applying ionization with nitrate ions (15NO) detected some nitrated oxidized species in one limonene–OH oxidation experiment with high NO concentrations (12 August 2013) (Zhao et al., 2018). Although there was no calibration available to derive absolute concentrations, the time series of mass-to-charge ratios gave information about the relative abundance of these species.
Measurements of formaldehyde (HCHO) concentrations were available from either one of three instruments which included an instrument making use of the Hantzsch reaction (AL4021, Aero Laser GmbH), the DOAS instrument that is also used for the detection of OH, and a cavity ring-down spectroscopy (CRDS) analyzer (Picarro G2307) (Glowania et al., 2021). Measurements using the Hantzsch method were available in the experiments in 2012, measurements with DOAS were available in the experiments in 2015, and measurements with CRDS were available in the experiments in 2019. Measurements were corrected for an unaccounted instrumental zero value (< 0.5 ppbv) observed in the clean dark chamber in some experiments.
2.3 Limonene oxidation experiments
Before the start of an experiment, the chamber was cleaned by flushing dry, ultra-pure synthetic air through the chamber overnight to purge out trace gases that remained from previous experiments. To humidify the air at the start of the experiment, water vapor from boiling ultra-pure water (Milli-Q) was flushed into the chamber until the relative humidity reached about 70 %. The relative humidity gradually decreased to 30 %–50 % as a result of the increasing temperature in the chamber over the course of the experiment and the dilution by the dry replenishing air. Before the injection of limonene, the chamber roof was opened to allow sunlight to irradiate the clean chamber air (zero air phase). In the illuminated chamber, small amounts of nitrous acid (HONO), formaldehyde, and acetone were formed with a rate of a few hundred pptv h−1 (Rohrer et al., 2005), presumably from chamber wall reactions. Therefore, the primary source of OH and NO in most of the experiments was the photolysis of HONO, leading also to a continuous increase in nitrogen oxide concentrations (NOx).
In total, seven experiments investigating limonene oxidation were performed. Chemical conditions can be divided by the NO concentration levels (Table 2), for which the contributions of different RO2 loss reactions varied between radical propagation reactions (i.e., reaction with NO), radical termination channels (i.e., reactions with HO2 and RO2), and isomerization reactions.
Table 2Experimental conditions during the time period when the radical budget of limonene oxidation experiments is analyzed. kbi is the sum of the total loss rate constant of RO2 due to bimolecular reactions with NO, HO2, and other RO2 calculated from measured concentrations. Values are averages except for limonene concentrations, for which maximum concentrations after the injection are given. In experiments with multiple limonene injections, the range of the injections is noted.
In the experiments with low NO mixing ratios of 0.1 to 0.15 ppbv (1 September 2012, Fuchs et al., 2021a, 4 July 2019, Bohn et al., 2021b), about 3 to 4 ppbv of limonene was injected three times. Between each injection, limonene was oxidized for about 90 to 120 min, so that most of the limonene reacted away before the next injection. To suppress NO concentrations during the experiments, approximately 50 to 60 ppbv of O3 produced by a silent discharge ozonizer (O3Onia) was injected before opening the chamber roof.
In experiments with medium NO concentrations ranging from 0.25 to 0.4 ppbv (8 and 10 August 2012; Fuchs et al., 2021a, b), about 4 ppbv limonene was injected 2 h after opening the roof. No additional trace gases were added. Measured O3 mixing ratios increased from about 1 to 10 ppbv as a result of the photolysis of NO2, which was produced from the reaction between peroxy radicals and NO.
In the experiment with the high NO concentrations (3 August 2015, Bohn et al., 2021c), about 15 ppbv of NO was injected into the chamber before opening the chamber roof and 10 ppbv of limonene at later times. When most of the limonene was consumed within 2 h after the first injection, an additional injection of 10 ppbv of limonene was done. In this work, the HCHO yield is only analyzed based on the measurement before the second limonene injection (Sect. 3.1.1) because of the potential secondary production of HCHO from the oxidation of secondary products. The radical concentrations and OH reactivities are only analyzed after the second limonene injection (Sect. 3.2.3), because radical measurements failed during the first part of the experiment. A large fraction of NO was already titrated by ozone after the second limonene injection.
Lastly, a limonene ozonolysis experiment was conducted on 5 June 2020 (Bohn et al., 2021a), in which no NOx was present. This experiment intended to elucidate the ozonolysis chemistry. The chamber roof was closed at all times. In the first half of the experiment, about 4 ppbv of limonene was injected in addition to 45 ppbv O3. After 3 h about 4.5 ppbv of limonene was re-injected together with O3, so that O3 mixing ratios reached 70 ppbv; 100 ppmv of CO was added 30 min before the second injection to scavenge OH radicals that are produced from the limonene ozonolysis reaction.
2.4 Model calculations
The acquired measurements of trace gases and radicals are compared against a zero-dimensional box model applying MCM version 3.3.1 (Saunders et al., 2003; Jenkin et al., 2015). In addition to the chemistry from the MCM, chamber-specific processes including dilution and small productions of HONO, acetone, and formaldehyde in the presence of sunlight are included in the model. Dilution in the chamber is implemented as a first-order loss process. The rate constant is calculated based on the monitored replenishment flow rate. The parameterization for the production of chamber sources for nitrous acid, formaldehyde, and acetone follows the description in Rohrer et al. (2005) and Kaminski et al. (2017), in which production rates are parameterized as functions of temperature, relative humidity, and radiation. The source strengths for the production rates are scaled for each experiment from the observed increase in concentrations for the part of the experiment when the chamber roof was opened, but limonene was not present (zero-air phase).
In addition, a background OH reactivity in the range of 1 s−1 is present in the illuminated, clean chamber due to the presence of unmeasured OH reactants. In order to account for this background reactivity, an artificial OH reactant that behaves like CO is implemented in the model. Its concentration is adjusted in the model to match the observed OH reactivity during the zero-air phase and is assumed to be constant during the rest of the experiment.
The OH yield from limonene ozonolysis in the model calculations in this work is updated based on the IUPAC recommendations (Cox et al., 2020), decreasing the value from 87 % to 66 %. This is supported by multiple experimental studies (Aschmann et al., 2002; Herrmann et al., 2010; Forester and Wells, 2011) as well as theoretical studies (Wang and Wang, 2021). The decomposition product of the Criegee intermediate (MCM name: LIMOOB) is also updated, in which the terminal carbonyl group remains during the vinyl hydroperoxide mechanism (VHP). In addition, instead of forming a primary RO2 (MCM name: C923O2) by eliminating a carbonyl group, decomposition of LIMOOB leads to the production of a secondary β-oxo-substituted RO2 (L5O2, Fig. 1). This is supported by the theoretical investigation of the ozonolysis of cyclohexene that the departure of the carbonyl group is not competitive without any inducing functional group (e.g., β-hydroperoxyl) (Rissanen et al., 2014).
Three simulation model runs are performed for each experiment. In all model runs, physical parameters including temperature, pressure, photolysis frequencies, and the dilution rate of trace gases due to the replenishment flow are constrained to observations. If available, HONO concentrations are prescribed as measured to constrain the production from the chamber. In addition, ozone concentrations in the model are constrained to ensure that shortcomings of the model to predict ozone do not complicate the interpretation by an inappropriate fraction of limonene reacting with ozone. Time series of NO and NO2 concentrations are constrained to observations in the model runs except for the third set of simulations to avoid potential impacts of shortcomings of the model to describe these species.
In the first set of simulations (denoted as the “reference run”), model runs are performed without any further constraints or modifications. In the second set of simulations (denoted as the “constrained run”), HO2 concentrations are prescribed as measured to constrain the OH-production rate from the HO2+ NO reaction. In this model run, OH reactivity is also adjusted to the measurements to yield an OH loss rate as observed (Sect. 3.3.1 to 3.3.4). In the third set of simulations (Sect. 3.5), NO and NO2 concentrations are not constrained to investigate the fate of nitrogen oxides; radical concentrations and OH reactivity are also free parameters in the model.
OH reactivity from oxidation products tends to be overestimated by the model. Potential reasons include wall loss reactions of low-volatility compounds such as organic nitrates and peroxides, uncertainties from the reactivity of products, intermediates that are mostly derived using the structure–activity relationship (SAR) in the model (Jenkin et al., 1997; Saunders et al., 2003), and chemical loss due to reactions that are not included in the MCM (e.g., isomerization reactions). To reduce the OH reactivity in the model, it is assumed that organic nitrates and peroxides derived from limonene oxidation undergo a first-order loss process.
The additional loss rate for organic nitrates is estimated from the decaying signal at the corresponding mass charge ratio of the least oxidized C10 nitrate species (C10H17NO6) observed by the chemical ionization mass spectrometry (CIMS) instrument after the chamber roof was closed in the experiment with high NO concentrations. The lifetime of the least oxidized species C10 nitrate is used to estimate the loss rate, as the only C10 nitrate species from limonene oxidation that the MCM includes is C10H17NO4. The loss rate constant is equivalent to a lifetime of about 2 h (Supplement Fig. S1, Zhao et al., 2018), which is comparable to the chemical loss rate in the reaction with OH for conditions of these experiments (chemical lifetime of 0.5 to 4 h). The specific reason for the additional loss of nitrates could not be identified but might be due to wall loss or due to loss by hydrolysis reactions in the humidified air, similar to findings in an isoprene-rich forest (Romer et al., 2016).
Organic peroxides, however, could not be detected by the CIMS instrument, and hence the additional loss rate is adjusted such that the simulated OH reactivity agrees with measurements in the experiments with low and medium NO when there should have been significant production of organic peroxides. This requires a lifetime of 10 min, much shorter than the lifetime on the order of hours under typical atmospheric conditions or those in the current experiments.
3.1 Product yields from the oxidation of limonene
3.1.1 Formaldehyde and acetone yields from the oxidation of limonene by OH and O3
Formaldehyde (HCHO) is one of the oxidation products of the reaction of limonene with the OH radical as well as with ozone. It is mainly produced from the oxidation of the terminal double bond in limonene (Fig. 1). The product yield of HCHO from the oxidation of limonene by OH and O3 can be determined from the chamber experiments by comparing the measured concentrations of HCHO to the amount of oxidized limonene calculated from the measured limonene, OH, and O3 concentrations. As HCHO also has a small chamber source (Sect. 2.3) and is lost by its reaction with OH and by photolysis, its concentration needs to be corrected for these losses following the method presented in Kaminski et al. (2017) and Rolletter et al. (2019).
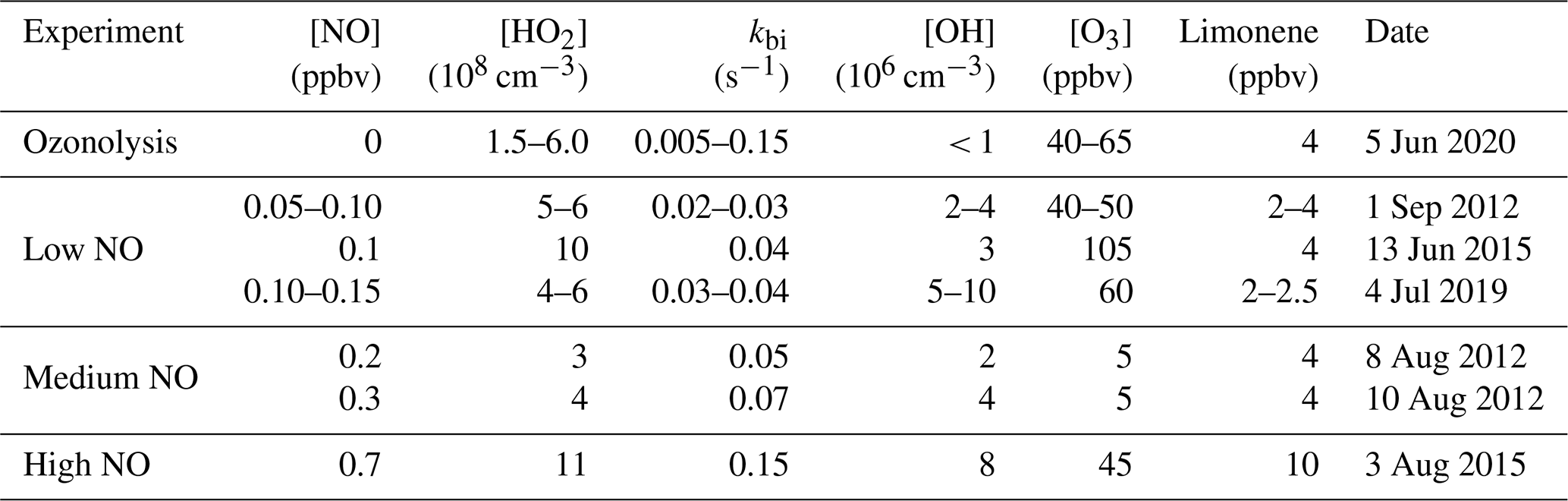
For the evaluation of the HCHO yields, only measurements from the first 2 h of the experiment after the first limonene injection are used, because further oxidation of accumulating organic products could contribute to the production of HCHO at later times of the experiment. This is demonstrated by a rapid increase in HCHO when 80 % of limonene had reacted away after a roughly linear relationship between HCHO and limonene concentrations (Figs. 1 and S2). To derive the HCHO yield, a linear regression is performed for data until 40 % of limonene is consumed and the slope of the linear regression gives the HCHO yield. This results in values of (12 ± 3) %, (13 ± 3) %, and (32 ± 5) % for the OH oxidation experiments with low, medium, and high NO mixing ratios, respectively (Fig. 2).
Figure 2HCHO concentrations divided by the injected limonene concentration plotted versus the fraction of reacted limonene for the first injection in the experiments with different NO levels. The regression includes data points up to 40 % of the fraction of reacted limonene.
Formaldehyde is only produced from one of the three major RO2 species formed in the initial reaction of limonene with OH. The SAR (Peeters et al., 2007) gives a yield of the formation of this RO2 radical (Fig. 1) that results from the OH addition to the terminal C=C double bond (MCM name: LIMCO2) of 37 % with an error of 15 %. When the LIMCO2 radical reacts with NO or RO2 radicals, an alkoxy radical is formed that subsequently forms HCHO from its decomposition together with a carbonyl compound (MCM name: LIMKET, Fig. 1). Therefore, the expected HCHO yield would be 37 % at maximum, from which the fraction of RO2 reacting with other reaction partners such as HO2 and the formation of organic nitrates in the reaction with NO (value determined in this work: (34 ± 5) %, Sect. 3.1.2) needed to be subtracted. The uncertainty in the yield of organic nitrates could be higher because the values are determined for all RO2 species from limonene oxidation by OH, but the nitrate yield for individual RO2 could vary. The fraction of RO2 reacting with NO is more than 85 % in the experiments with high and medium NO and 65 % in the experiment with low NO. The relative error of the fraction of RO2 reacting with NO could be up to 35 % due to the uncertainties of the reaction rate constants. Therefore, the HCHO yield expected from the SAR in the experiments with high and medium NO is (22 ± 9) %, and in the experiment with low NO it is (16 ± 6) %.
All observed HCHO yields agree with SAR-derived HCHO yields, which carry high relative uncertainties (40 %). This could explain the discrepancies between the observed yield for the experiment with high NO mixing ratios and the SAR-derived yield. The largest discrepancy could be observed for medium NO levels. A higher yield of LIMCO2 or a lower yield of organic nitrates for the reaction of LIMCO2 with NO could explain the discrepancies observed for the experiments with low NO. Another possible reason is that there are other reaction channels of the LIMCO2 peroxy radical that are competitive with the bimolecular reactions with NO and HO2 (kbi up to 0.07 s−1) for conditions of the experiments with low and medium NO, but they would not be relevant in the experiment with high NO mixing ratios (kbi up to 2 s−1). These reaction channels would not produce formaldehyde such as isomerization. Possible isomerization reactions of LIMCO2 are further discussed in Sect. 4.3.1.
The formation of HCHO at a later time in the experiments includes the production from further oxidation of first-generation products. Therefore, HCHO yields increase over the course of the experiment to values of 40 %–90 %, with the lowest yields in the experiments with low NO (Fig. S2). Part of the differences between the experiments could be due to the differences in the contributions of various product species, so that numbers are not necessarily comparable. The HCHO yield remains below 100 % in all the experiments. This is consistent with HCHO being only produced from the oxidation of the terminal C=C bond of the limonene structure on a short timescale of several hours in typical atmospheric conditions, so that total HCHO yield is limited to one in the limonene oxidation scheme.
Previous studies reported formaldehyde yields for the OH oxidation of limonene between 36 % and 43 % (Larsen et al., 2001; Lee et al., 2006) (Table 3). The experiments by Larsen et al. (2001) and Lee et al. (2006) were performed at high NO concentrations, but the yield of HCHO is higher than the HCHO yield determined in the experiment with high NO in this work. However, experiments by Larsen et al. (2001) and Lee et al. (2006) were performed with much higher concentrations of limonene and NO (Table 3), and it is not clear whether only HCHO from the first oxidation step is considered (Fig. 1), so that numbers may not be entirely comparable. In the study by Librando and Tringali (2005), formaldehyde yields were determined to be 27 % and 92 % in experiments with high limonene mixing ratios of 13 and 2 ppmv, respectively, but in the absence of NO. Yields in their experiments may not comparable to yields in this work, because the fate of organic peroxy radicals was likely dominated by atmospherically irrelevant RO2+ RO2 recombination reactions.
Table 3HCHO yields from the OH and O3 oxidation of limonene determined in the experiments in this work and reported in the literature. Maximum values for limonene, O3, and NO concentrations in the experiments are listed.
a Given as NOx concentration. b 200 ppmv cyclohexane. c 400 ppmv of 2-butanol or cyclohexane. d 60 ppmv CO.
The HCHO yield derived from the pure ozonolysis experiment in this work is (10 ± 1) % in the presence of the OH scavenger. The same value is obtained if the formaldehyde yield is determined from the part of the ozonolysis experiment without an OH scavenger, when approximately 40 % of limonene reacted with OH that is produced from the ozonolysis reaction. Similar to the OH reaction, formaldehyde production is expected from the subsequent chemistry after the ozone addition to the terminal C=C double bond. Because ozone preferably (87 %) adds to the endocyclic C=C double bond (Wang and Wang, 2021), a low yield can be expected. As there was no NO present during the experiment, production of formaldehyde from the reaction between limonene and OH could only be possible from the reaction of LIMCO2 with peroxy radicals or unimolecular reactions (e.g., isomerization). As the HCHO yield is the same independent of the addition of the OH scavenger, the yield from the limonene–OH reaction at zero NO must be similar to the HCHO yield from the limonene ozonolysis. The HCHO yield derived from the ozonolysis without the presence of the OH scavenger is similar to the HCHO yield in the experiments with low NO concentrations. This is expected because of the very low NO concentrations and the similar fraction of limonene that reacted with OH or O3 in both experiments.
The formaldehyde yield from the limonene ozonolysis derived in this work agrees well with the yield of 10 % determined in the work by Grosjean et al. (1993) performed with an OH scavenger. Yields were also determined in the work by Gong et al. (2018) from several experiments at various chemical conditions. Values of 5 %–11 % derived in their experiments with a limonene : O3 concentration ratio of 1:2 are consistent with results in this work. However, the HCHO yield was 11 %–27 % in the experiments, with a limonene : O3 concentration ratio of about 1:100. This higher yield might be explained by additional formaldehyde production from the ozonolysis of secondary products (Fig. 1). The effects of humidity and the presence of an OH scavenger on the HCHO yield were also investigated in Gong et al. (2018). In their experiments, the HCHO yield increases strongly with increasing humidity and in the absence of an OH scavenger when the limonene : O3 ratio was very low (1:100). On the other hand, the positive dependence of the HCHO yield on humidity and the absence of an OH scavenger is much less significant when the limonene : O3 concentration ratio was high (1:2). There is no significant impact of the OH scavenger on the HCHO yield found in this study, consistent with findings in the experiments in Gong et al. (2018).
In the experiments in this work, there was no significant acetone production from the oxidation of limonene, as the production rate of acetone from the chamber source (∼ 80 pptv h−1) could already explain the observed increase in the acetone concentration during the experiments. This suggests that, unlike other monoterpenes, acetone is not a significant product from limonene oxidation. This is consistent with findings by Lee et al. (2006) and Larsen et al. (2001). Also, the MCM does not predict acetone as a product in the limonene oxidation scheme.
3.1.2 Organic nitrate yield from the oxidation of limonene by OH
The organic nitrate yield of the RO2+ NO reaction for peroxy radicals produced from the limonene + OH reaction is determined for experiments in this work following the method described for the analysis of previous experiments in the SAPHIR chamber (Hantschke et al., 2021; Tan et al., 2021). The amount of organic nitrates (RONO2) in the chamber is estimated by the difference between the accumulated production of nitrogen oxides from the chamber source of HONO (QHONO), which is the only relevant source of nitrogen oxides in the experiments, and the total NOy (NOx+ HONO + HNO3) concentrations. This method assumes that the differences can be solely attributed to the production of peroxy acyl nitrate (PAN)-like species, which can act as a reservoir for nitrogen oxides and can be significant in the oxidation of specific organic compounds.
The source strength of HONO can be calculated from the photo-stationary state between HONO, NO, and OH using measured concentrations and measured photolysis frequencies:
The total number of organic nitrates can be calculated from the difference between the accumulated HONO production and the sum of other nitrogen oxide species, also considering their loss due to dilution in the chamber with rate constant kdil and the loss of NO2 in the reaction with OH (Hantschke et al., 2021):
Assuming that at least right after the injection of limonene, the production of organic nitrates is dominated by the reaction of NO with RO2 derived from limonene–OH, the organic nitrate yield Φlimonene+OH can be calculated:
The fraction of RO2 from limonene–OH oxidation to the measured total RO2 concentration is estimated based on the ratio of the OH reactivity from limonene to the OH reactivity from all OH reactants that are expected to produce RO2 in the reaction with OH (Tan et al., 2021). The latter can be calculated by subtracting the background OH reactivity (kOHbg) that includes the reactivities attributed to inorganic species and formaldehyde from the total measured OH reactivity (kOH):
Using Eqs. (3) and (4), the nitrate yield Φlimonene+OH is obtained as the slope of the linear regression between the calculated organic nitrate concentrations and the integrated turnover rate of the reaction between RO2 limonene and NO. Only experiments with medium NO mixing ratios are used for this analysis here, as HONO measurements were performed and more than 90 % of the limonene was oxidized by OH in these experiments. This results in a Φlimonene+OH of (34 ± 5) % (Fig. 3). The precision (∼ 15 %) of Φlimonene+OH is determined by the precision of the measurements with linear error propagation. The error of Φlimonene+OH is estimated to be about 30 %, which is mainly attributed to the accuracies of the reaction rate constants (∼ 30 %) and the measurements of HONO (10 %) and jHONO (18 %). The organic nitrate yield of the RO2 from limonene ozonolysis is not determined in this study as a considerable amount of PANs could be formed according to the MCM model. Therefore the organic nitrate yield analysis is not conducted for the experiments at low NO mixing ratios.
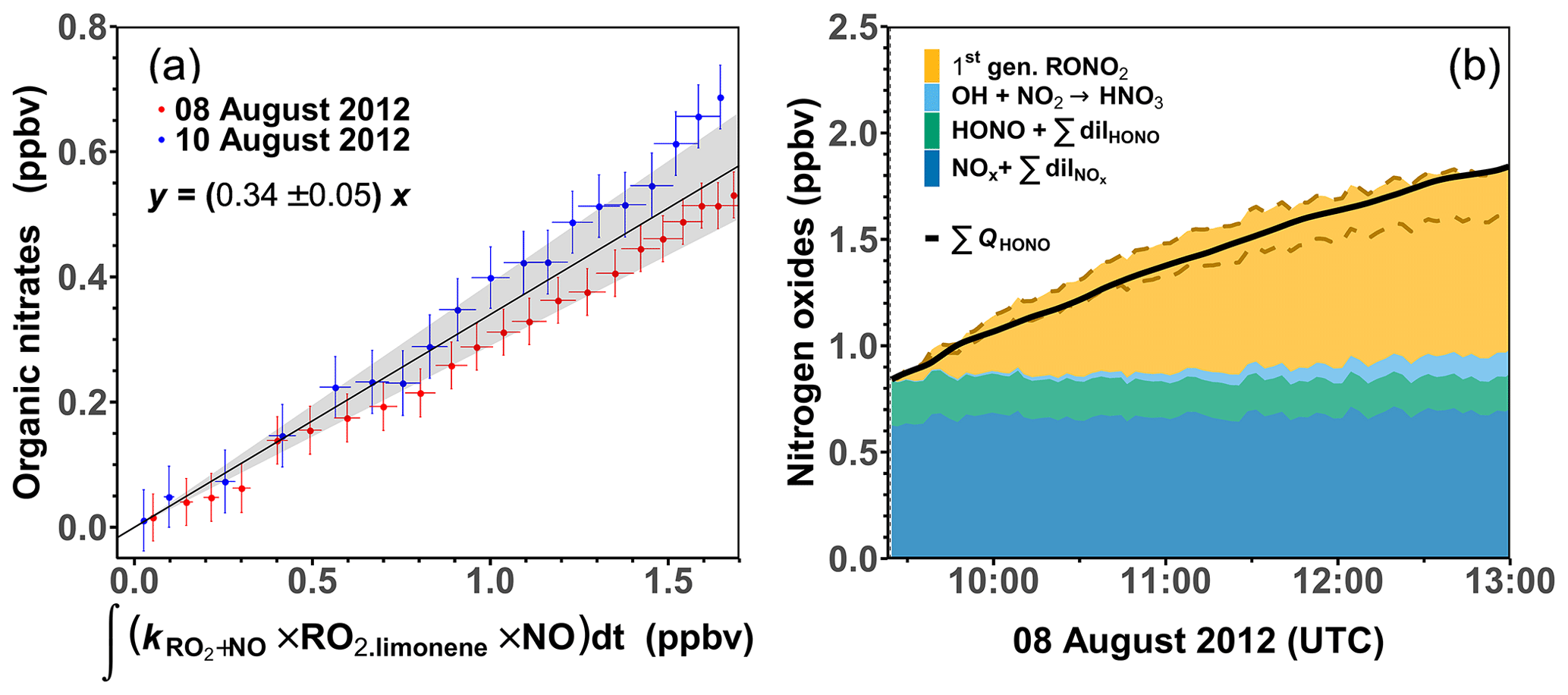
Figure 3(a) Regression between calculated organic nitrate mixing ratio and integrated turnover rate of the reaction of limonene-derived RO2 and NO. The regression only includes data when less than 60 % of the limonene from the first injection reacted away. (b) Concentrations of nitrogen oxide species after the first limonene injection in the experiment with medium NO on 8 August 2012 compared to the total production of HONO from the chamber source. The organic nitrate contribution is calculated using a nitrate yield of (34 ± 5) % for the reaction of limonene RO2 with NO; the uncertainty range is enclosed by the two dashed lines.
The organic nitrate yield in this study is higher than values estimated using different SAR approaches of 19 % (Jenkin et al., 2019) to 28 % (Arey et al., 2001; Leungsakul et al., 2005). However, the value agrees well with nitrate yield derived in experiments, in which the yield was derived from the analysis of the aerosol chemical composition produced from the oxidation of limonene (36 ± 6 %; Rollins et al., 2010).
3.2 Comparison of modeled and measured concentration time series
3.2.1 Model–measurement comparison for limonene photooxidation experiments at a low NO mixing ratio
Three experiments were performed at low NO concentrations on 1 September 2012 (Fig. 4), 13 August 2015, and 4 July 2019 (Supplement Figs. S3 and S4). Measured OH concentrations during the limonene oxidation were about (2–7) × 106 cm−3 in the experiments in 2012 and about (5–11) × 106 cm−3 in the experiments in 2019. OH concentrations were highest when most of the limonene was consumed. The overall higher OH concentrations in the experiments in 2019 were due to the higher NO mixing ratios (∼ 0.15 ppbv) compared to the experiment in 2012 (∼ 0.05 ppbv), which led to a faster OH production from the radical regeneration reaction of HO2 and NO. In these experiments, around 30 %–40 % of limonene reacted with O3 and 60 %–70 % of limonene reacted with OH. Measured HO2 concentrations were around (5–8) × 108 cm−3. Measured RO2 concentrations were around (2–5) × 108 cm−3 in the experiment in 2012 and (5–10) × 108 cm−3 in the experiments in 2019, respectively. The average chemical lifetime of RO2 to bimolecular reactions with NO, HO2, and RO2 can be calculated using measured NO, HO2, and RO2 concentrations, respectively, resulting in a chemical lifetime of 25 to 50 s; 60 %–70 % of the RO2 radicals reacted with NO, and 30 %–40 % reacted with HO2. Losses due to RO2+ RO2 recombination reactions are predicted not to play a major role if reaction rate constants are taken from the MCM that are much lower compared to the rate constants of the reactions with HO2.
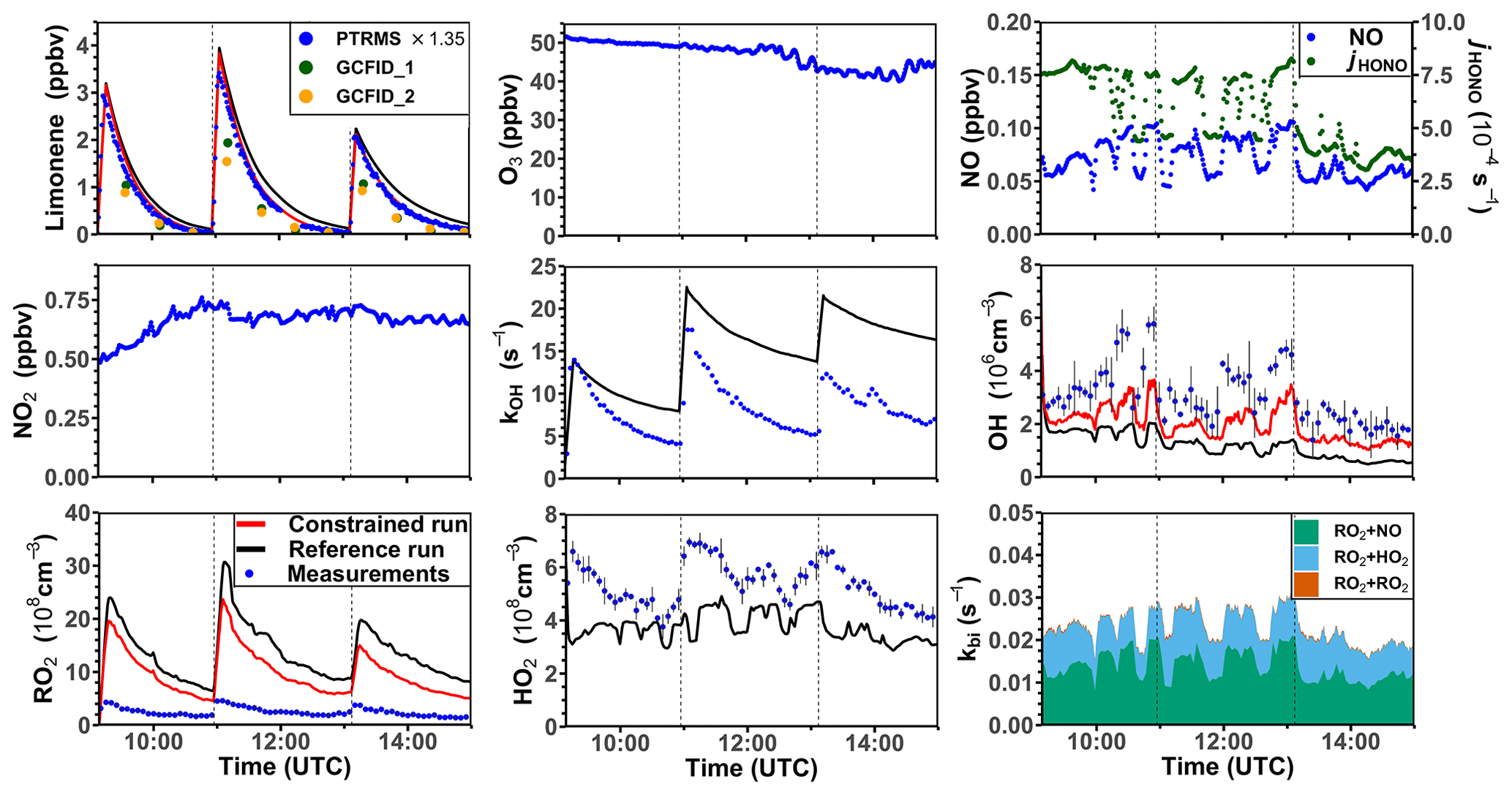
Figure 4Time series of radicals and inorganic and organic species in the limonene oxidation experiment with low NO mixing ratios on 1 September 2012. Limonene concentrations measured by the PTR-MS are scaled by a factor of 1.35 to match the increase in OH reactivity during the injections of limonene. The vertical dashed lines represent times when limonene was injected into the chamber. RO2 bimolecular reaction loss rate constants (kbi) are calculated based on the measured NO, HO2, and RO2 concentrations using the reaction rate constants as included in the MCM. In the constrained model run, HO2 concentrations are constrained to measurements, and the OH reactivity is adjusted by additional loss reactions to match measurements.
In the reference model run, which essentially uses the MCM chemistry without constraining radical production or destruction processes (Sect. 2.4), the OH reactivity is overestimated in the model by 3 to 5 s−1 after nearly all limonene has reacted away. Right after the limonene injections, HO2 concentrations are underestimated by 20 %–50 % and RO2 concentrations are overestimated by 500 %–700 %. OH is underestimated by at least 50 %, which could partly result from the overestimation of the OH reactivity and underestimated HO2 concentration (and thereby the reaction rate of HO2+ NO). The underestimated OH concentration also leads to a slightly slower decay of the modeled limonene, so that modeled limonene concentrations are about 0.1–0.2 ppbv higher than measured values at the end of the experiment.
In the model run with adjusted OH reactivity and constrained HO2 concentration (constrained model run), the discrepancy between modeled and measured OH concentrations is reduced. They are still underestimated by 33 % on average throughout the whole experiment and by about 50 % 1 h after the limonene injection, which could indicate a missing OH production process in the model. However, the temporal behavior of the modeled limonene concentration agrees with the behavior of the measurements, suggesting that measured OH concentrations are too high.
Maximum modeled RO2 concentrations are reduced by about 30 % compared to the reference run as a result of the increased HO2 concentration in the constrained run, leading to a higher RO2 loss by RO2+ HO2 recombination reactions. However, this improvement is insufficient to explain measured RO2 concentrations, which are still at least a factor of 4 lower than modeled values.
3.2.2 Model–measurement comparison for limonene photooxidation experiments at medium NO mixing ratios
Two oxidation experiments were performed at medium NO levels on 8 and 10 August 2012 (Figs. 5 and S5). Throughout both experiments, measured NO mixing ratios ranged from 0.25 to 0.4 ppbv. More than 90 % of the RO2 derived from the limonene–OH reaction reacted with NO. O3 mixing ratios increased gradually from about 1 to 10 ppbv as a result of the photolysis of NO2, which was produced from the reaction between organic peroxy radicals and HO2 with NO. At such low O3 concentrations, the reaction with OH was the dominant loss pathway for limonene (90 %). The measured OH concentration range was (2–5) × 106 cm−3, and the measured HO2 and RO2 concentrations were about (2–5) × 108 cm−3.
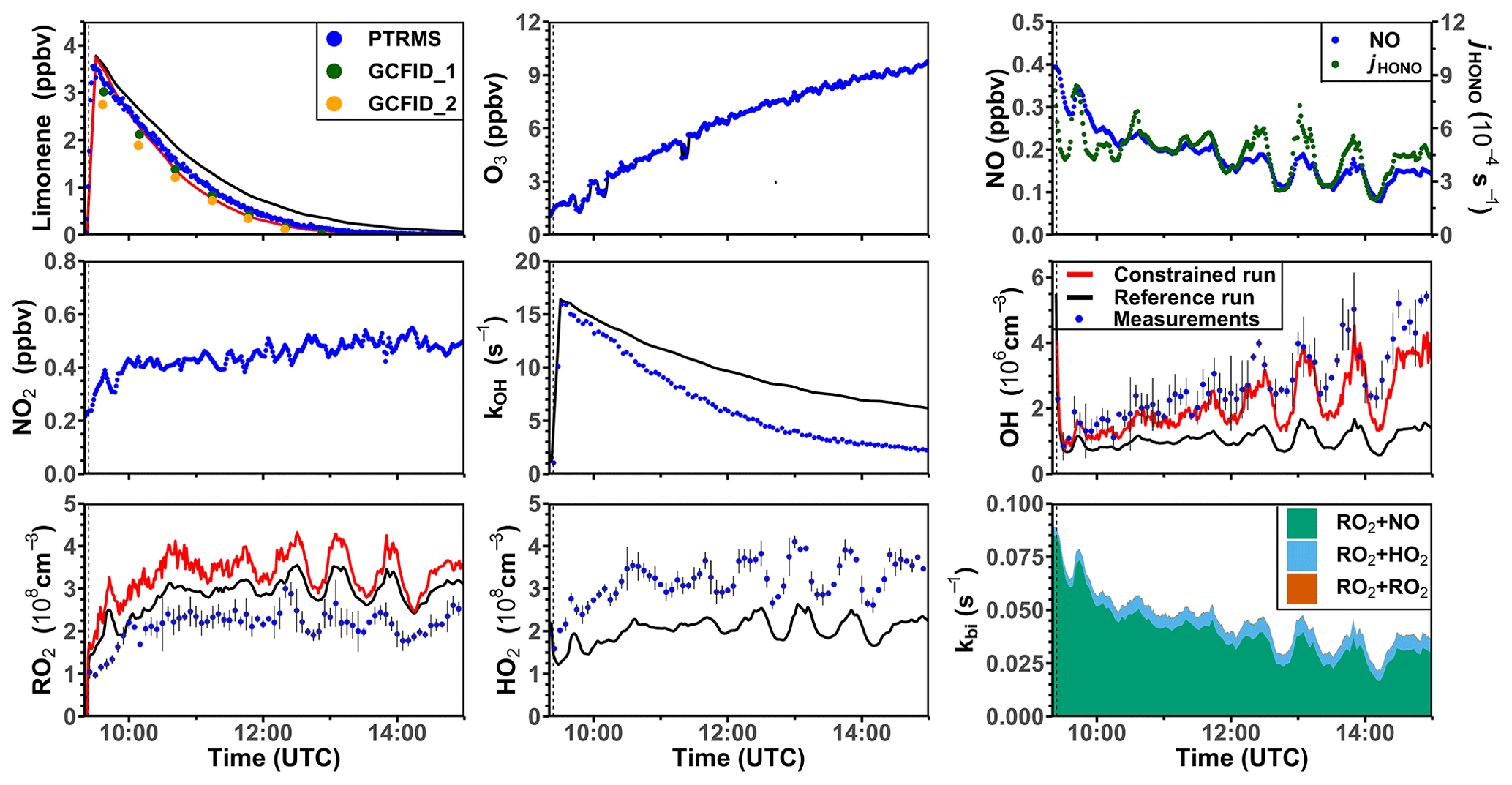
Figure 5Time series of radicals and inorganic and organic species in the limonene oxidation experiment at medium NO level on 8 August 2012. RO2 bimolecular reaction loss rate constants (kbi) are calculated based on the measured NO, HO2, and RO2 concentrations using the reaction rate constants from the MCM. In the constrained model run, HO2 concentrations are constrained to measurements, and the OH reactivity is adjusted by additional loss reactions to match measurements.
In the reference model run, the simulated OH reactivity increasingly deviates from measurements over the course of the experiment. The differences are about 2 to 4 s−1 after 2 to 3 h of oxidation, when all limonene has been consumed. Contributors to the OH reactivity in the model in addition to limonene are mostly oxidation products such as aldehydes and ketones (∼ 70 %), organic peroxides (∼ 15 %), and organic nitrates (∼ 15 %) (Supplement Fig. S6). Simulated HO2 concentrations are underestimated in the reference model run by around 10 %–30 %. Hence, the simulated OH concentration is underestimated by 30 %–80 % due to the slow regeneration rate from the reaction between HO2 and NO as well as the faster removal rate of OH. As a result, limonene concentrations are overestimated by the model by up to 0.3 ppbv throughout the experiment.
In the constrained model run, the simulated OH concentration is on average 20 % lower than measured values. This difference is within the uncertainties of OH measurements. The higher modeled OH concentrations compared to the reference model run lead to a faster consumption of limonene compared to the reference model. This temporal behavior better agrees with the temporal behavior observed by PTR-MS measurement, but the limonene consumption is slightly faster in the model than measurements suggest. Modeled RO2 concentrations are 50 %–100 % higher than measurements. Values are 10 %–20 % higher compared to the reference model run right after the injection of limonene, presumably due to the enhanced RO2 production in the constrained model run, in which OH concentrations are higher.
3.2.3 Model–measurement comparison for the limonene photooxidation experiment at high NO mixing ratios
One experiment was performed with high NO mixing ratios on 3 August 2015. In this experiment, limonene was injected twice. Unfortunately, measurements of radicals failed during the first part of the experiment, so that only the part of the experiment after the second limonene injection can be analyzed (Fig. 6). The ozone mixing ratio at the beginning of this part of the experiment was about 35 ppbv as a result of ozone production during the first part of the experiment. The OH concentration measured by DOAS was around (5–10) × 106 cm−3, so that around 70 % and 30 % of limonene reacted with OH and O3, respectively. The OH concentration measured by LIF was around (5–15) × 106 cm−3. The HO2 concentration was about 1 × 109 cm−3 throughout the experiment. The high NO and HO2 concentrations led to a short RO2 lifetime of 5 to 10 s, much shorter than in the other experiments with lower NO concentrations. The removal of RO2 by bimolecular reactions was mostly due to the reaction with NO (85 %) and HO2 (15 %).
In the reference model run, the model–measurement discrepancy of the OH reactivity is around 3 s−1 before the second limonene injection, and it further increases to 10 s−1 at the end of the experiment. The absolute discrepancy in OH reactivity is higher compared to other experiments because the total amount of limonene and therefore the production of oxygenated products were higher. Modeled OH and HO2 concentrations are both lower by 50 %–70 % than the measured values. The low OH concentration in the model again leads to a slower decay of modeled limonene concentrations compared to observed values. RO2 concentrations show a good agreement with the measurement, with a discrepancy of less than 20 %.
In the constrained model run, the simulated OH concentration is in good agreement with the measurement by DOAS in the first hour and only slightly underestimates the measurements by about 20 % at the end of the experiment. As a result, the agreement between modeled and measured time series of limonene concentrations improves compared to the reference model run. Differences between RO2 concentrations in the constrained and reference models are rather small except for the point in time when limonene is injected and RO2 concentrations in the constrained model run increase more rapidly than observations. This results in 30 % difference between modeled and measured values during the first hour of the experiment. This can be explained by the increased RO2 production from the limonene oxidation by the OH radical which is underestimated in the reference model run. Possible reasons for the overestimated RO2 concentration could include an underestimated loss rate of RO2 at the NO mixing ratio of about 1 ppbv (Sect. 3.4) or an overestimated production of RO2 from the further oxidation of products from the previous limonene injection.
3.2.4 Model–measurement comparison for the limonene ozonolysis experiment in the dark with and without an OH scavenger
The ozonolysis of limonene in the dark was investigated in a separate experiment on 5 June 2020 (Fig. 7). A significant OH concentration was observed with values up to 1.2 × 106 cm−3 after the first injection of limonene as a result of the OH production from the ozonolysis reaction. OH concentrations were below the limit of detection of both DOAS and LIF after 100 ppmv of CO had been injected as an OH scavenger right before the second limonene injection. The measured HO2 concentration was around 3 × 108 cm−3 after the first limonene injection and increased to maximum concentrations of 1 × 109 cm−3 after the second limonene injection due to the conversion of OH radicals formed from the ozonolysis in the reaction with excess CO. Because there was no limonene oxidation by OH, there was also less production of RO2 radicals than the first limonene injection. However, the observed maximum RO2 concentration after each limonene injection was about 1 × 109 cm−3.
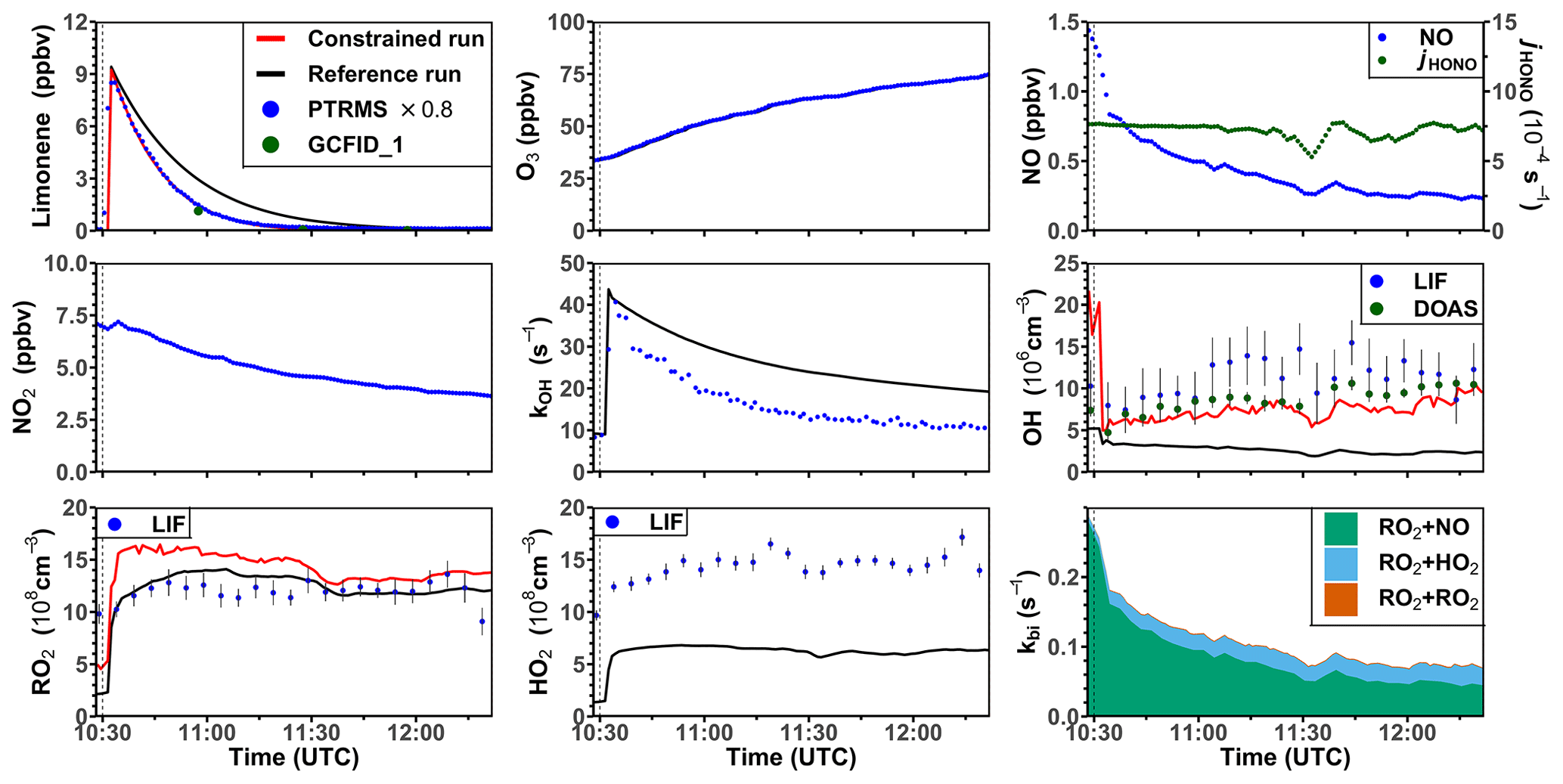
Figure 6Time series of radicals and inorganic and organic species during the limonene oxidation experiment at high NO mixing ratios on 3 August 2015. Limonene concentrations measured by the PTR-MS are scaled by a factor of 0.8 to match the increase in OH reactivity during the injection of limonene. RO2 bimolecular reaction loss rate constants (kbi) are calculated based on the measured NO, HO2, and RO2 concentrations using the reaction rate constants from the MCM. In the constrained model run, HO2 concentrations are constrained to measurements, and the OH reactivity is adjusted by additional loss reactions to match measurements.
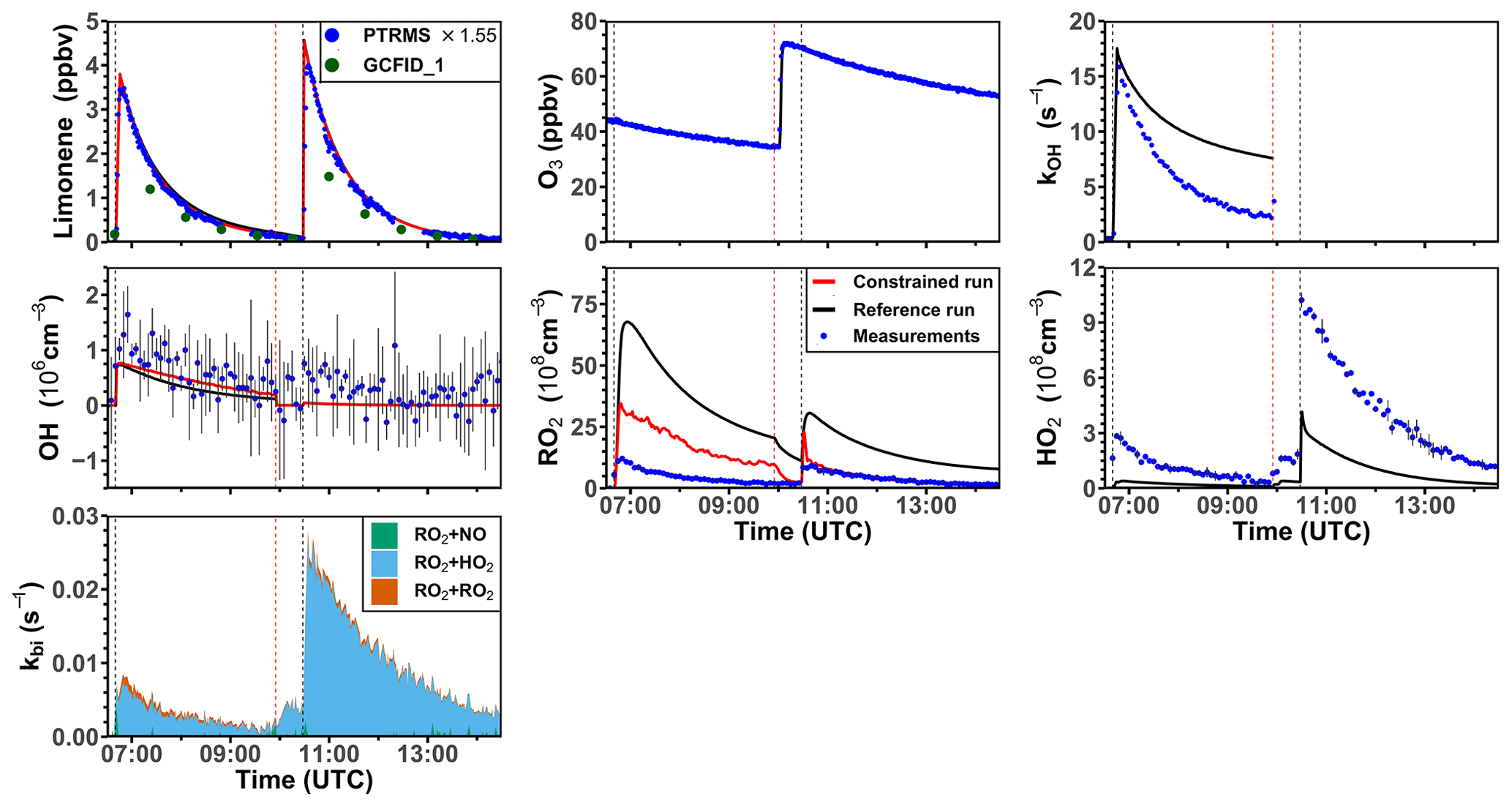
Figure 7Time series of radicals and inorganic and organic species during the limonene ozonolysis experiment in the dark on 5 June 2020. Limonene concentrations measured by the PTR-MS are scaled by a factor of 1.55 to match the increase in OH reactivity during the injection of limonene. The black vertical dashed lines represent when limonene was injected into the chamber; the red vertical dashed line represents the injection of 100 ppmv of CO. RO2 bimolecular reaction loss rate constants (kbi) are calculated based on the measured NO, HO2, and RO2 concentrations using the reaction rate constants in the MCM. In the constrained model run, HO2 concentrations are constrained to measurements, and the OH reactivity is adjusted by additional loss reactions to match measurements.
Similar to observations in the other experiments, modeled OH reactivity is higher than measured values by 5 s−1. The maximum discrepancies appear right before the injection of CO, after which OH reactivity could not be measured due to the high contribution of CO. In the reference model run, HO2 is underestimated by the model by around 90 % during the first part of the experiment, and modeled HO2 is also a factor of 3 lower than measurements during the pure ozonolysis part in the presence of CO as an OH scavenger. In contrast, measured RO2 concentrations are drastically overestimated by the model by a factor of 7 right after each limonene injection.
In the constrained model run, simulated OH concentrations are 25 % higher compared to the reference model as a result of the reduced OH reactivity in the model. The main difference between the results of the reference model run and the constrained model run is that RO2 concentrations are reduced by half in the constrained model. This is caused by an increased RO2 removal rate by the high HO2 concentration.
Using measured RO2 and HO2 concentrations, the maximum reaction rate constants of the reaction of RO2 with HO2 are about 0.005 and 0.02 s−1 for the first and second limonene injections, respectively. In contrast, the RO2 loss rate constant due to the recombination of organic peroxy radicals is calculated to be only 0.001 s−1, so that it is expected that RO2 reacted mainly with HO2 to form peroxides if only bimolecular reactions are considered. However, the loss due to RO2–RO2 reactions could be of similar importance to the loss due to RO2–HO2 reactions if low-HO2 and high-RO2 concentrations as predicted by the reference model run are used for the calculation. The large discrepancies between measured and modeled RO2 and HO2 radical concentrations and possible explanations are further discussed in Sect. 4.1 and 4.2.
3.3 Chemical OH radical budget using measured quantities
To evaluate whether unaccounted chemical processes are required to explain the total OH destruction rate in the experiments, the rates of OH-producing reactions are calculated using measured trace gas and radical concentrations. Turnover rates of single reactions are summed up and compared to the total OH destruction rate. Due to the short chemical lifetime of OH radicals (< 1 s), steady-state conditions are expected, so that the production and destruction rates must be balanced.
The total OH destruction rate is obtained by computing the product of the measured OH reactivity and OH concentration. In experiments in the year 2015, when OH was measured by both DOAS and LIF instruments, DOAS measurements are used for the calculation of the total OH destruction rate. The uncertainty of the OH destruction rate is 16 %, calculated by error propagation of the measurement uncertainties. The main OH production processes included in the chemical budget analysis are listed in Table 4. OH production by photolysis of HONO and O3, the reaction between HO2+ NO and between HO2+ O3, and the limonene ozonolysis reaction are considered. The uncertainties of these OH-producing processes range from 15 % (photolysis processes) to 20 % (HO2+ NO reaction and limonene ozonolysis reaction), which are derived from error propagation of the uncertainties of measurements (Table 1) and reaction rate constants (Atkinson et al., 2004; Cox et al., 2020).
Table 4Reactions that are included in the analysis of the OH production rate. Reaction rate constants are given for 298 K and 1 atm. The reaction rate constants that are used in the analysis are calculated for the measured temperature.

Figures 8 and S7 show the chemical budgets of OH radicals in the limonene oxidation experiments with different NO mixing ratios. The destruction and production rates of OH for experiments with medium (8 August 2012) and high NO (3 August 2015) are balanced within the uncertainties of measurements during all the experiments. This is consistent with the overall good agreement between modeled and measured OH radical concentrations obtained for the constrained model run (Figs. 5 and 6). In the experiment with high NO, the total OH turnover rate is higher (> 20 ppbv h−1) compared to the other experiments due to the high limonene concentration (10 ppbv) and the high NO concentration, both of which accelerate the turnover of radicals.
In experiments with medium and high NO concentrations, the main OH source after the limonene injection is the reaction between HO2 and NO (> 85 %), followed by the photolysis of HONO in the experiment with medium NO and ozone photolysis and limonene ozonolysis in the experiment with high NO, when 60 ppbv O3 was present. In total, about 85 % to 100 % of the OH radical production rate can be explained by the calculated processes that are listed in Table 4.
In the experiment with low NO on 1 September 2012 (Fig. 8b), the OH production rate is 6 ppbv h−1 after the injection of limonene, of which about 33 % can be attributed to OH production from limonene ozonolysis. The NO concentration varies over the course of the experiment, making OH regeneration from the HO2+ NO reaction an important OH source with a contribution of 15 %–50 % to the total OH production in addition to OH production from the photolysis of HONO and O3. About 1.0 ppbv h−1 (20 %–33 %) of the OH destruction rate is not explained by these OH production processes, which is consistent with the underestimation of OH concentrations in the constrained model run compared to measured values (Fig. 4). However, the gap between the OH destruction rate and destruction rate is about 1 ppbv h−1 throughout the whole experiment and does not vary with the amount of limonene present in the chamber. This suggests that either the missing OH source is not related to the oxidation of limonene or it is due to measurement artifacts, for example, in the OH measurements, which would lead to an overestimation of the OH destruction rate. An artifact in the OH measurement would also be consistent with what is observed in the constrained model run (Fig. 4), where the rapid decrease in the modeled limonene concentration suggests that the measured OH is too high in this experiment.
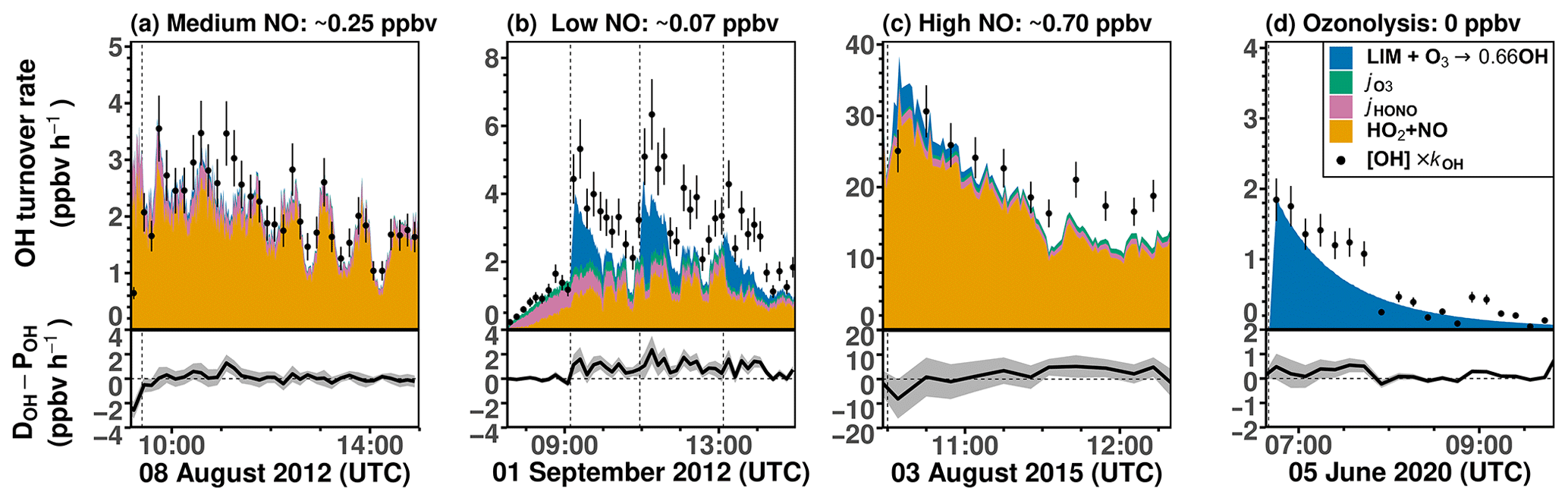
Figure 8Ten-minute average values of total OH destruction rates compared to the sum of OH production rates from the major OH sources that can be calculated from measurements in the (a) medium NO, (b) low NO, (c) high NO, and (d) ozonolysis experiments. Shaded areas in the difference plots give the uncertainties of the calculations. Production of OH from the reaction of O3 and HO2 is not included, because the contribution to the total OH production was negligible (< 0.01 ppbv h−1) for conditions of the experiments.
In the ozonolysis experiment, prior to the addition of CO as an OH scavenger (Fig. 8d), OH is only produced by the ozonolysis of limonene. The total OH production rate is about 2 to 3 ppbv h−1 at the beginning of the oxidation and gradually declines while limonene is being consumed. The total OH destruction rate is well-explained by the production from limonene ozonolysis, suggesting that OH production from further ozonolysis reactions of product species is not significant for conditions of this experiment.
In conclusion, the OH production rate from the four major OH sources that are included in the calculations (Table 4) is balanced by the OH destruction rate within the 25 % uncertainty of the calculation at the beginning of the experiment, when limonene oxidation is most important in the experiments with medium and high NO and in the ozonolysis experiment. In the experiments with low NO concentrations, imbalances of 20 %–33 % are observed, indicating that an additional OH production process with a rate of 1.0 ppbv h−1 would be required to explain the observed destruction rate, but there are indications that this could be due to a measurement artifact in the OH measurements.
In all the experiments, the OH production rates are lower than OH destruction rates at later times of the experiments, when secondary chemistry becomes important. However, differences are similar to the uncertainty of the calculations. These discrepancies may indicate that additional OH could be produced from unaccounted reactions of oxidation products, for example, from the photolysis of organic peroxides in the photooxidation experiments (Badali et al., 2015).
3.4 Chemical budget of first-generation peroxy radicals using measured quantities
The discrepancies between measured and simulated organic peroxy radical concentrations are much higher (up to a factor of 2) in the experiments with low NO concentrations and in the ozonolysis experiment compared to the experiments with medium or high NO concentrations. The analysis of the composition of the RO2 concentrations using model results from the constrained model run (Sect. 2.4) shows that the concentrations of RO2 produced in the initial reaction of limonene with OH and O3 already exceed the measured total RO2 concentrations (Figs. 9 and S8). Model–measurement percentage differences are at least a factor of 2 higher than the accuracy of the measured RO2 concentration (∼ 25 %). Therefore, the discrepancy suggests that additional loss pathways for RO2 have to be included in the model.
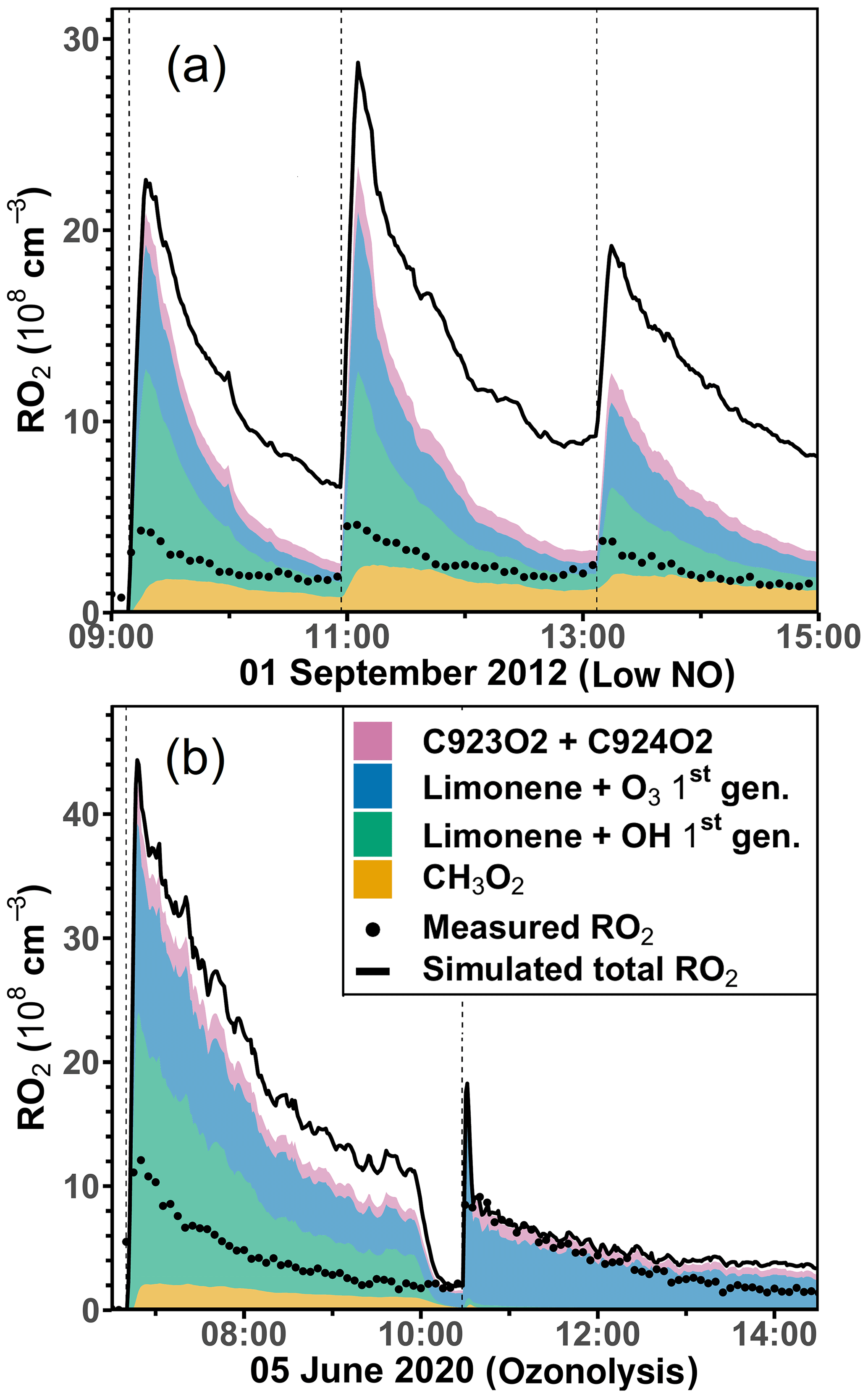
Figure 9Total RO2 radical concentrations and their speciation from model calculations (constrained model run) compared to the measured values (black dots) for the experiments with low NO (a) and the ozonolysis (b) experiment. Methylperoxy radicals (CH3O2) are mainly produced from the oxidation of HCHO. Radicals produced in the initial reactions of limonene with either OH or O3 are summed. C923O2 + C924O2 are RO2 radicals produced from the further oxidation of the first-generation oxidation products. Names are taken from the MCM model.
To examine the magnitude of the additional RO2 loss rate, a chemical budget analysis for RO2 radicals is performed similarly to the analysis for OH radicals (Sect. 3.3). As the chemical loss rate constants of peroxy radicals are within the range of 0.01 to 0.20 s−1, steady-state concentrations can be assumed. The production rate of the peroxy radicals produced right after the limonene injection is well-defined by the loss rate of limonene due to the reactions with OH and O3. Therefore, only measurements during the first 30 min after the first limonene injection are used for the analysis, so that calculations are not impacted much by additional RO2 production from the subsequent oxidation of organic products. The removal rate constant of peroxy radicals includes bimolecular reactions (kbi).
Values of the bimolecular reaction rate constants in the MCM for limonene-derived RO2 radicals are , ranging from 10−13 to 10−12 cm−3 s−1, × 10−11 cm3 s−1 at 298 K, and = 9.0 × 10−12 cm3 s−1 at 298 K. The additional loss rate constant (kadd) required to balance production () and destruction () rates can be calculated as
In this calculation, measured concentrations of radicals and trace gases are used. The RO2 yield of 0.66 from the limonene ozonolysis reaction assumes RO2 radicals are produced from the vinyl hydroperoxide (VHP) mechanism together with an OH radical (Wang and Wang, 2021).
Results from the five experiments are summarized in Table 5. The values of the additional loss rate constant, kadd, range from 0.01 to 0.06 s−1 in different experiments but are similar to the high relative uncertainty of at least 50 %. The large relative uncertainty is caused by the small differences between production and destruction rates, which also increase with increasing RO2 and NO concentrations (and thereby kbi). The relative uncertainties of the additional loss are less than 100 % in only three experiments, which include the ozonolysis experiment (5 June 2020) and the experiments with low (1 September 2012) and medium NO concentrations (8 August 2012). In the ozonolysis experiment, the additional RO2 loss is lower by a factor of 4 than in the other two experiments. The large difference in kadd could be attributed to the different RO2 species that are formed from the photooxidation reaction and the ozonolysis reaction. RO2 formed from the photooxidation reaction has retained its six-member ring moiety, whereas the majority of RO2 formed from the ozonolysis reaction is acyclic. In addition, the low temperature during the ozonolysis experiment could slow down the additional loss pathway.
Table 5Additional removal rate constants (kadd) that are required to balance the RO2 production and destruction rates in the different experiments together with conditions of the experiments such as the percentage of limonene that reacted with OH (LIM + OH) or O3 (LIM + O3). Only data from 30 min after the first limonene injection are analyzed.
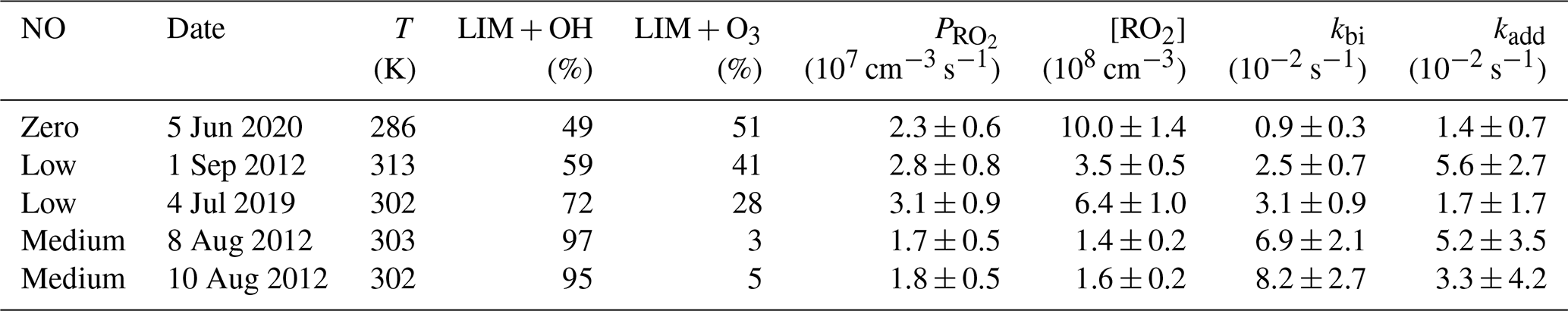
Potential additional loss pathways of RO2 radicals that are needed to balance the RO2 production rate in the experiments would be radical conversion reactions that can also lead to the production of OH radicals, so that these pathways are potentially connected to imbalances found in the chemical budget of OH radicals (Sect. 3.3). It is worth noting that discrepancies in the chemical budgets of RO2 and OH radicals are both smaller in the ozonolysis experiment compared to the photochemistry experiments at low and medium NO mixing ratios.
3.5 Impacts of RO2 model results on the modeled NOx concentrations
In the model runs described so far, NO and NO2 concentrations are constrained to measured values. If these are not constrained, NOx is underestimated at the beginning of the experiments, but values are overestimated at the end (Figs. 10 and S9). The discrepancy at the beginning can be mainly attributed to the overestimation of modeled RO2 concentration, which leads to an overestimation of the formation of organic nitrates that act as sinks for NOx on the timescale of the experiments. To illustrate the impact of RO2 concentrations on the modeled NOx concentrations, two model runs are compared: one with modeled RO2 concentrations (reference run) and the second with modeled RO2 concentrations adjusted to match the measurements.
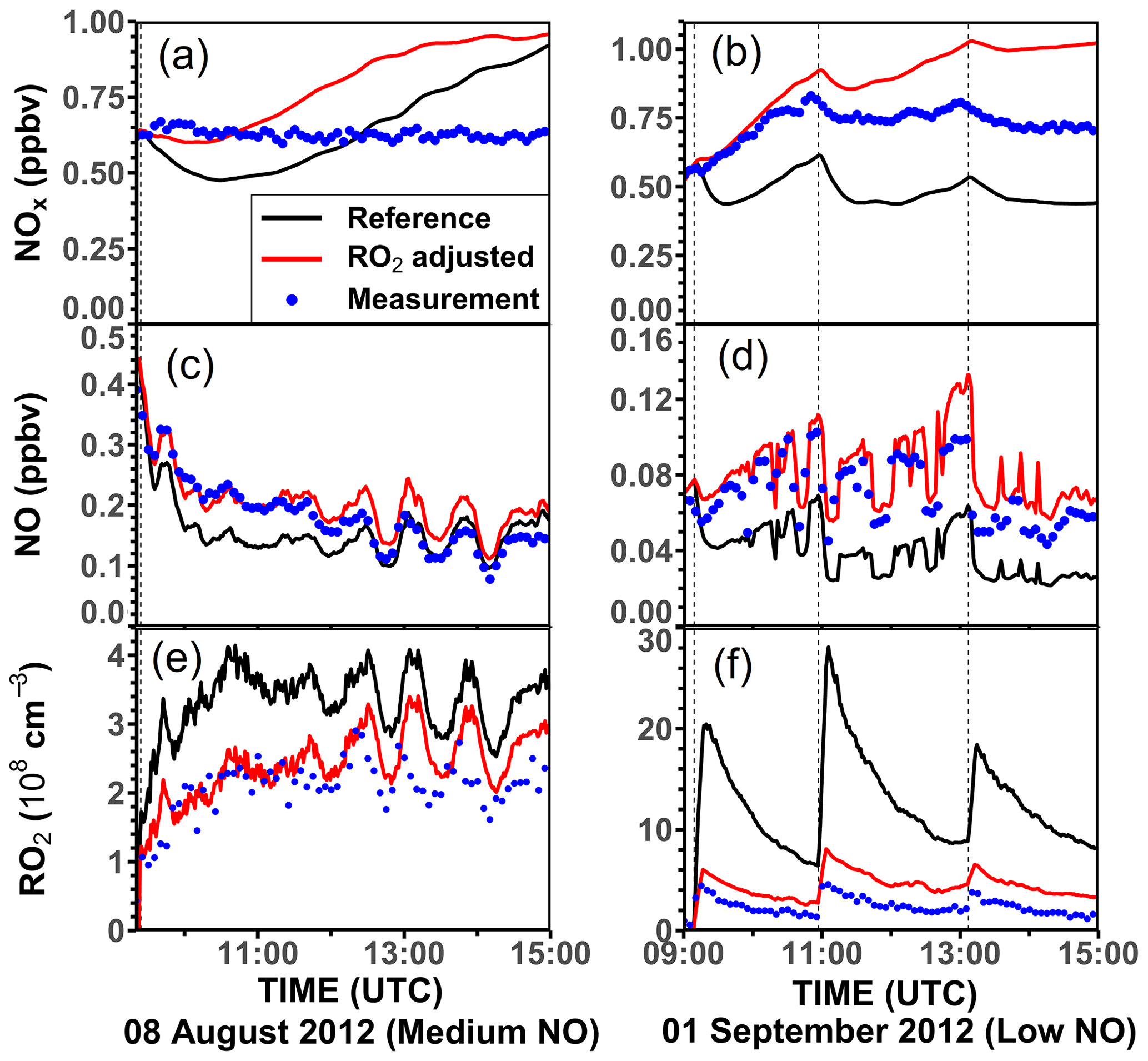
Figure 10Example of the impact of too high modeled RO2 (e, f) on the modeled NOx (a, b) and NO (c, d) concentrations in the experiments with medium-NO (8 August 2012) and low-NO (1 September 2012) concentrations. In both cases, the organic nitrate yield of 34 % from the reaction of NO with first-generation RO2 from the limonene + OH reaction is taken from the analysis in this work.
RO2 concentrations are adjusted by applying an additional loss with a fixed rate constant for all six first-generation RO2 derived from -OH and -O3 oxidation (Fig. 1). The additional loss rate constant for RO2 is around 0.01 to 0.06 s−1, similar to the loss rate constant derived in the analysis of the chemical budget for RO2 (kadd, Sect. 3.4). In both model runs, the organic nitrate yield of the RO2+ NO reaction for the first-generation RO2 radicals from limonene + OH is adjusted to the yield (34 %) that is found in the analysis of the experiments at a medium NO mixing ratio (Sect. 3.1.2). Measured , O3, and HO2 concentrations are used in both model runs to constrain the loss rate of NO2 and NO. It should be noted that only data within 1 h after the first limonene injection are evaluated as RO2 produced from the further oxidation of organic products is not considered.
Figure 10 shows the modeled NO and NOx concentrations for the two model runs. In the experiment with medium NO concentrations on 8 August 2012, RO2 radical concentrations are overestimated by about 50 %–100 % by the reference model and modeled NO, and NOx concentrations are 25 % lower than measurements. With an additional RO2 loss rate constant of 0.05 s−1 (Table 5), the fraction of RO2 that reacts with NO reduces from 80 %–90 % to 45 %–60 %. Therefore, the loss of NOx by the formation of organic nitrates is also reduced, so that the model–measurement agreement for NO and NOx improves for the first 2 h of the experiments.
Both model runs overestimate the NOx and NO concentrations when all limonene reacted away after 13:00 UTC (Fig. 5). The measured NOx concentration remains stable at around 0.6 ppbv throughout the whole experiment after the injection of limonene. However, NOx concentrations increase at a rate of about 0.15 ppbv h−1 in the reference model. The increase is reduced to less than 0.05 ppbv h−1 in the model run with the additional RO2 loss during the last 2 h of the experiment. The production of NOx in the model at later times of the experiment can be explained by the production of NO2 from the photolysis of the first-generation organic nitrates and their oxidation by OH. These effects are more important in the reference model run, when the modeled first-generation organic nitrates are high. To reconcile the difference in NOx concentrations between the model and measurements, a stronger nitrogen sink is required in the model. This may also suggest that the model underestimates the organic nitrate formation from the reaction of NO with RO2 from the oxidation of product species. Another explanation would be that the lifetime of limonene-derived organic nitrates from OH oxidation is too short in the model.
In the experiment with low NO concentrations on 1 September 2012, the fraction of RO2 that reacts with NO reduces from about 50 % to 16 % if an additional RO2 loss with a rate constant of 0.06 s−1 (Table 5) is applied. In this experiment, a large fraction of NOx in the model is lost due to the formation of PAN or PAN-like species from acyl peroxy radicals (e.g., CH3CO3 and C822CO3) that are formed in the radical chain reaction of the ozonolysis reaction of limonene (Fig. 1). The additional loss of the initially formed RO2 species competes with the reaction with NO2, and therefore the formation of PAN reduces if the additional loss is applied. This effect of reduced NOx loss in the ozonolysis reaction adds to the effect for a reduced organic nitrate formation discussed for the experiment on 8 September 2012 at medium NO.
Although the reduced NOx loss significantly improves the model–measurement agreement for the first part of the experiments, if an additional RO2 loss process is included in the model, NOx concentrations are overestimated by this model at later times, when the chemistry of product species gains in importance. This could be due to neglecting the impact of the subsequent chemistry of the additional RO2 loss reactions on nitrogen oxide concentrations. The chemistry of nitrogen oxide species in the experiment with low NO concentrations is more complex compared to the experiment with medium NO as a significant fraction of RO2 radicals is produced by the ozonolysis of limonene in addition to the reaction with OH. Further investigation will be required to specifically clarify the impact of the formation of PAN and PAN-like species from the ozonolysis of limonene (Fig. 1). To the best of our knowledge, there is no experimental study investigating PAN formation from the oxidation of limonene.
4.1 Sensitivity model runs including additional radical regeneration reactions for RO2 radicals
Additional radical regeneration reactions are further explored by sensitivity model runs. These model runs aim to reduce the discrepancies between measured and modeled OH, HO2, and RO2 concentrations in the reference model. This is achieved by implementing additional reaction pathways:
where RO2 is the peroxy radicals distinguished from their production in the reaction of limonene with either OH (limOH–RO2; i.e., LIMAO2, LIMBO2, and LIMCO2, Fig. 1) or O3 (limO3–RO2; i.e., LIMALAO2, LIMALBO2, and L5O2, Fig. 1), because these peroxy radicals are structurally similar to peroxy radicals from the OH reaction with a β-OH moiety of a six-carbon ring and limO3–RO2 being acyclic peroxy radicals with a β-oxo, an aldehyde, and an isopropenyl group. Therefore, it is assumed that they have similar reaction pathways. These reactions could involve a unknown reaction partner X, as used in Hofzumahaus et al. (2009), or could be unimolecular reactions.
Reaction rate constants for Reactions (R1a) and (R1b) (kR1a and kR1b) are implemented as pseudo-first-order reaction rate constants. RO2 within the same group (limOH–RO2 or limO3–RO2) is assumed to have the same rate constants. Reaction (R1a) would lead to HO2 production, and Reaction (R1b) would lead to OH production. In the sensitivity model runs, the reaction rate constants are optimized to minimize the model–measurement discrepancies for OH, HO2, RO2 concentrations and OH reactivity. The sum of kR1a and kR1b must be within the range of the additional RO2 loss rate constant kadd (Sect. 4.1, Table 5). A missing HO2 source is found in the reference model for all the experiments (Figs. 4–7). Assuming that the loss rate of the HO2 radical, which mainly reacts with NO, is correctly accounted for, an additional RO2 to HO2 conversion (Reaction R1a) is needed to bring measurement and model results into agreement. In contrast, missing OH is only found in the experiments with low NO, as evident from the analysis of the chemical budget of OH radicals (Sect. 3.3, Fig. 8). These observations indicate that additional RO2 to OH conversion (Reaction R1b) can only be competitive with other bimolecular reactions for NO mixing ratios of less than 0.05 ppbv, which is equivalent to a loss rate constant of k < 10−2 s−1.
The model–measurement agreement of radical concentrations is first optimized based on the second half of the ozonolysis experiment, when CO was added as an OH scavenger. In this case, only limO3–RO2 is present, but the conversion to either HO2 (Reaction R1a) or OH (Reaction R1b) cannot be distinguished, because OH rapidly converts to HO2. To achieve agreement between modeled and measured HO2 concentrations during this part of the ozonolysis experiment, the sum of the additional loss rate constants (kR1a+kR1b) would need to be (0.017 ± 0.008) s−1. The uncertainty is mainly due to the uncertainty in the measurement of HO2 concentrations (∼ 20 %). The upper limit for the rate constant kR1b for the loss of limO3–RO2 can be estimated from the first part of the ozonolysis experiment, when no OH scavenger was present. Since 80 %–100 % of the observed OH production can already be explained by OH production from the limonene ozonolysis reaction (Fig. 8d), the rate constant of kR1b for limO3–RO2 would need to be less than 0.004 s−1. This implies that the rate constant kR1b for limOH–RO2 is also less than ∼ 0.004 s−1, as about 40 % of limonene is oxidized by OH in the ozonolysis experiment without an OH scavenger.
The implementation of Reaction (1a) for limO3–RO2 cannot significantly improve the model–measurement discrepancies of HO2 concentrations in the experiments, when limonene is predominantly oxidized by OH. Also in the ozonolysis experiment, HO2 concentrations are still underestimated by about 40 % during the part of the experiment without an OH scavenger. Hence, the reaction rate constant kR1a for an additional loss of limOH–RO2 is also optimized to match the measured HO2 concentrations.
Optimization of the reaction rate constant kR1a for the additional loss of limOH–RO2 for individual experiments results in values that differ by 1 order of magnitude. For instance, the optimum rate constant is (0.006 ± 0.003) s−1 in the ozonolysis experiment without an OH scavenger, but it is (0.05 ± 0.03) s−1 in the experiment with medium NO concentrations on 8 August 2012. These optimized rate constants are consistent with the values of the loss rate constant kadd (Table 5), with the rate required in the ozonolysis experiment having a slower rate and the rate required in the experiment with medium NO having a faster rate. The exact reason for such large differences is not clear but could be related to the higher temperature (16–27 ∘C) in the photooxidation experiments, when the chamber air was exposed to sunlight. The average value of the rate constant for the conversion from RO2 to HO2 for the experiments in this work is 0.03 s−1. This value is applied to all sensitivity model runs in the following to illustrate its impacts on modeled RO2 and HO2 concentrations and OH reactivity (Table 6).
Table 6Modification of chemical reactions implemented in the sensitivity simulation runs.
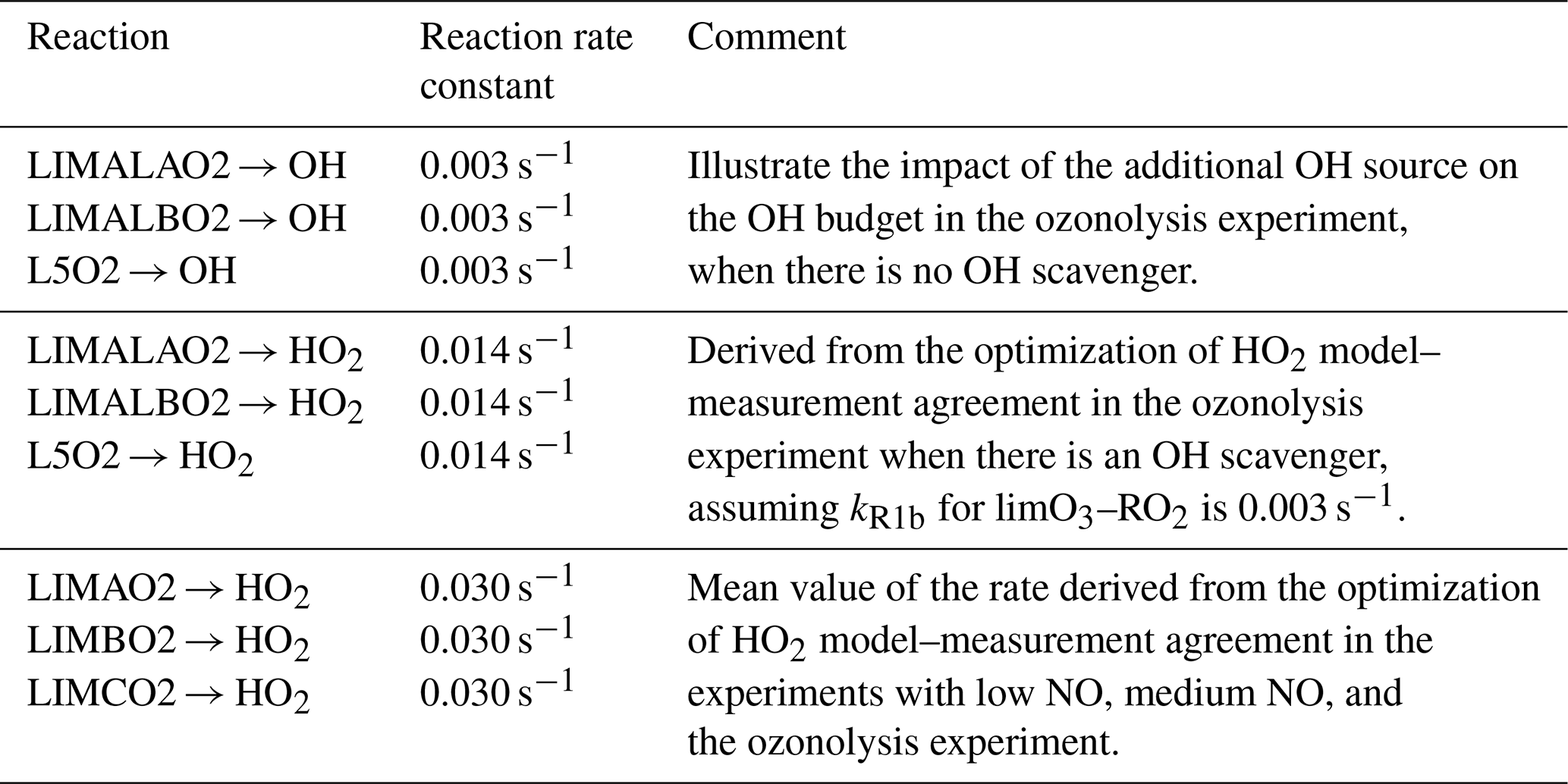
A summary of all reactions included for the sensitivity run is available in Table 6. Figure 11 shows the increase in the OH production rate in the sensitivity model runs that include the conversion of limO3–RO2 to OH at a rate constant of 0.004 s−1. The total OH production rate increases by about 0.2 ppbv h−1 in both experiments, corresponding to 5 % and 10 % increases, respectively. This reduces the imbalance between OH production and destruction rates in the experiment with low NO by about 20 % without significantly impacting the balance in the ozonolysis experiment. This demonstrates that the additional OH production from the conversion of first-generation RO2 from OH or O3 oxidation of limonene to OH is not sufficient to fully close the gap between OH production and destruction rates in the experiment with low NO, for which the discrepancy is largest among all experiments in this work.
Figures 12 to 14 and S10 to S12 show radical concentrations and OH reactivity obtained in the reference and sensitivity model runs. In the sensitivity model run, the model–measurement agreement for RO2 and HO2 concentrations improves compared to the reference model run, as can be expected from the adjustment of the reaction rate constant. In the experiment with low NO concentrations, however, an optimal agreement of both RO2 and HO2 concentrations cannot be simultaneously achieved. This suggests that some fraction of the additional RO2 loss pathway may not regenerate HO2 or OH radicals.
In the sensitivity model run the overestimation of the OH reactivity is reduced even without introducing additional loss pathways of oxidized products as implemented in the constrained model run (Sect. 2.4), because the production of organic peroxides is reduced due to the competition with the additional RO2 loss reaction. For example, the percentage of RO2 reacting with HO2 reduces from about 50 % to 25 % and from 90 % to 30 % in the experiment with low NO and in the ozonolysis experiment, respectively. In the sensitivity model, no closed-shell products are produced from the additional RO2 loss reaction. The good model–measurement agreement of the OH reactivity suggests that organic products from these reactions are not reactive or they are rapidly lost, for example, to the chamber wall. Therefore, no further conclusions about the type of products formed from Reaction (R1) can be drawn from these experiments.
4.2 Possible additional OH sources from RO2 reactions
Possible underlying mechanisms of the additional RO2 loss and the production of OH are discussed in the following section. It should be noted that the additional OH source discussed here is referring to the slow additional OH source (kR1b < 0.003 s−1) that could only slightly reduce the discrepancy between the OH production and destruction rates (Fig. 11). The larger discrepancy in the chemical OH budget that is observed in the experiment with low NO concentrations (Fig. 11a) compared to the other experiments cannot be explained by the additional conversion of RO2 to OH and, hence, it is not further discussed here. The rate constant of the additional RO2 to OH conversion, kR1b, is much slower than the total loss rate constant of RO2 due to bimolecular reactions (kbi > 0.01 s−1) or the additional RO2 to HO2 conversion (kR1a ∼ 0.017 s−1). Its contribution to the production rate of OH would also be small in all experiments (< 10 % of the total OH production rate). Therefore, there is no strong evidence for an additional conversion of RO2 to OH for experimental conditions in this study.
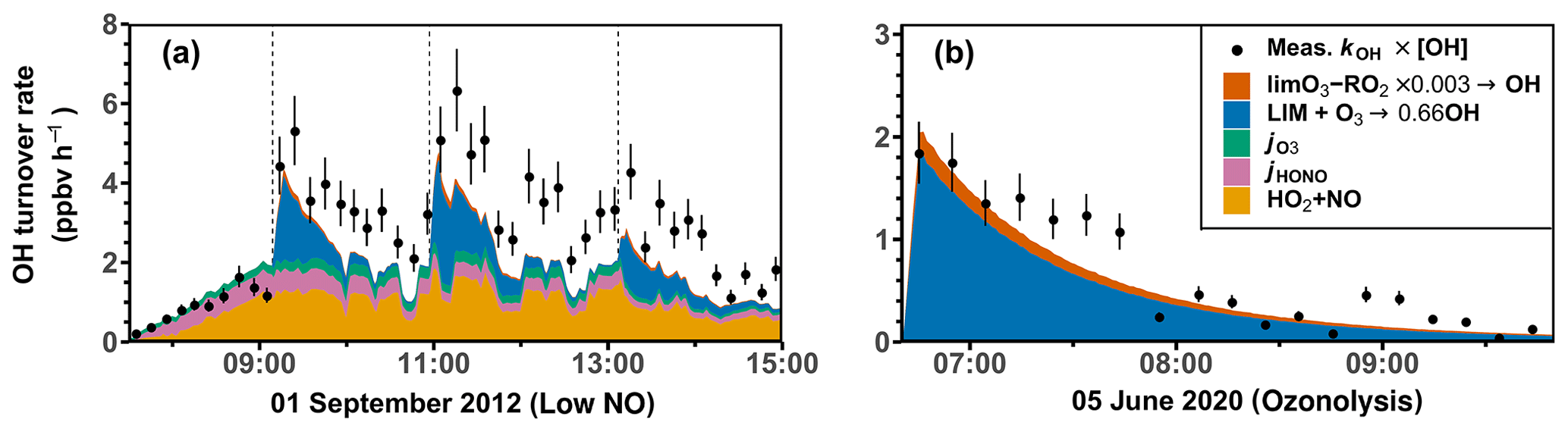
Figure 11Measured 10 min mean total OH production rate compared to the OH production rate from the main measured OH sources for (a) the experiment with low NO concentration on 1 September 2012 and (b) the ozonolysis experiment on 5 June 2020 for the sensitivity model run that includes additional OH production from the reaction of RO2 from limonene ozonolysis and HO2 (Reaction R2c).
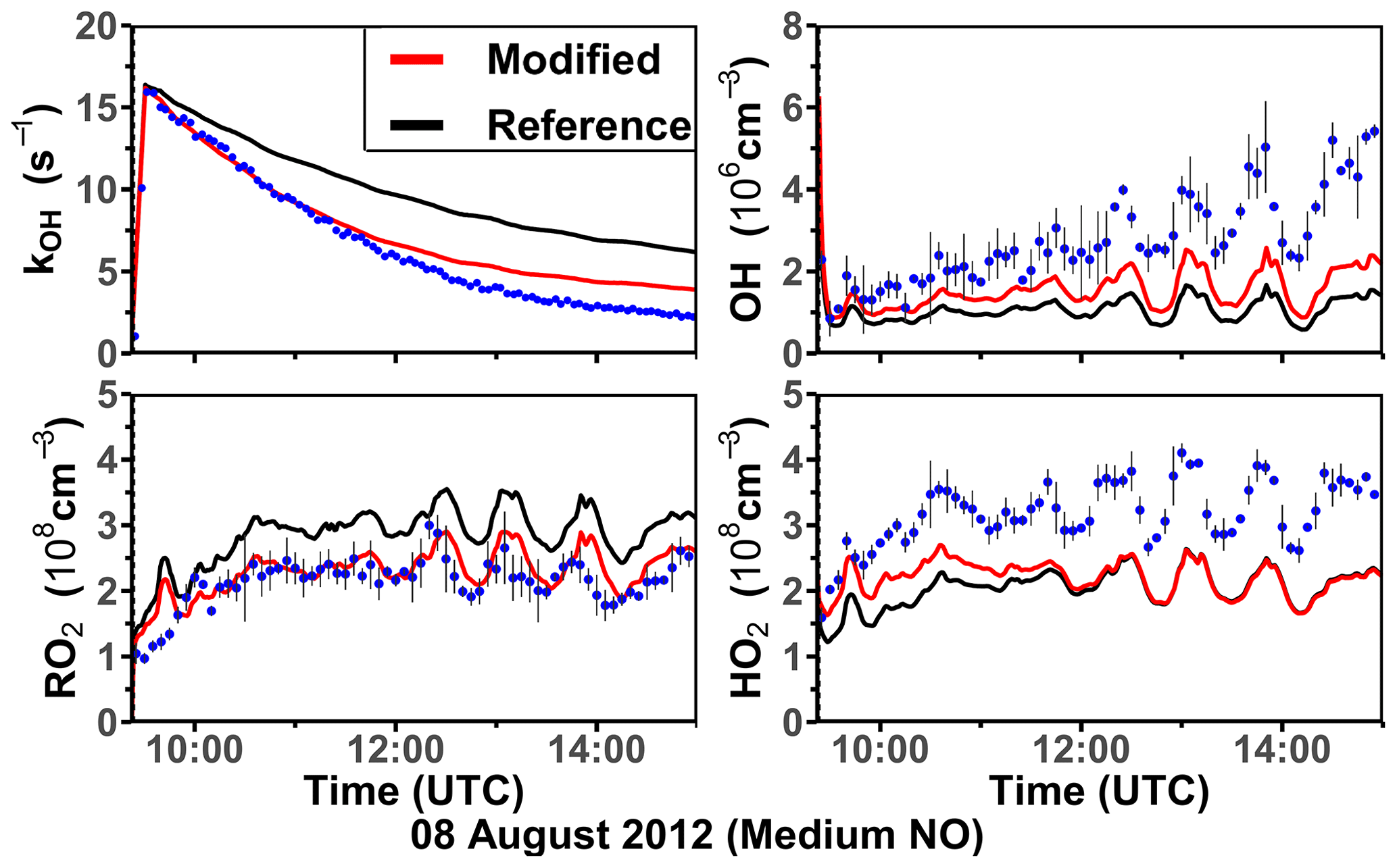
Figure 12Modeled and measured OH reactivity and HO2, RO2, and OH concentrations for the experiment with medium NO on 8 August 2012. Results from the reference model (reference) and the sensitivity model run (modified) that include additional RO2 loss processes producing OH and HO2 (Table 6) are compared.
4.2.1 RO2 isomerization reactions
Isomerization of RO2 could lead to the production of OH, which is shown in the OH oxidation of methacrolein (e.g., Crounse et al., 2012) and isoprene (e.g., Novelli et al., 2020). Isomerization reactions for RO2 from the limonene oxidation by OH and O3 are investigated by Møller et al. (2020) and Chen et al. (2021), respectively. Of the peroxy radical LIMAO2, LIMBO2, and LIMCO2 that are produced from the limonene oxidation by OH, it was calculated that LIMAO2 and LIMBO2 could undergo an -OH H shift with a rate constant of 10−3 to 10−4 s−1. LIMCO2 could undergo isomerization reactions that are competitive with bimolecular reactions for atmospheric conditions, which include a cyclization reaction with a rate constant of 0.2 to 0.8 s−1 and an allylic-H shift reaction with a rate constant of 0.1 to 1.7 s−1 (Fig. 15). On the other hand, peroxy radicals LIMALAO2, LIMALBO2, and L5O2 that are produced from the ozonolysis of limonene could all undergo much faster isomerization reactions (k > 0.5 s−1), such as aldehydic H-shift and cyclization reactions (Fig. 16) due to the loss of steric hinderance after the ring-opening ozonolysis reaction.
Subsequent reaction steps after the first isomerization reaction were not investigated in the work by Møller et al. (2020) and by Chen et al. (2021). Therefore, potential OH production from subsequent reactions is estimated with available SARs (e.g., Vereecken and Peeters, 2009; Vereecken and Nozière, 2020; Vereecken et al., 2021).

Figure 13Modeled and measured OH reactivity and HO2, RO2, and OH concentrations for the experiments with low NO concentrations on 1 September 2012. Results from the reference model (reference) and the sensitivity model run (modified) that includes additional RO2 loss processes producing OH and HO2 (Table 6) are compared.
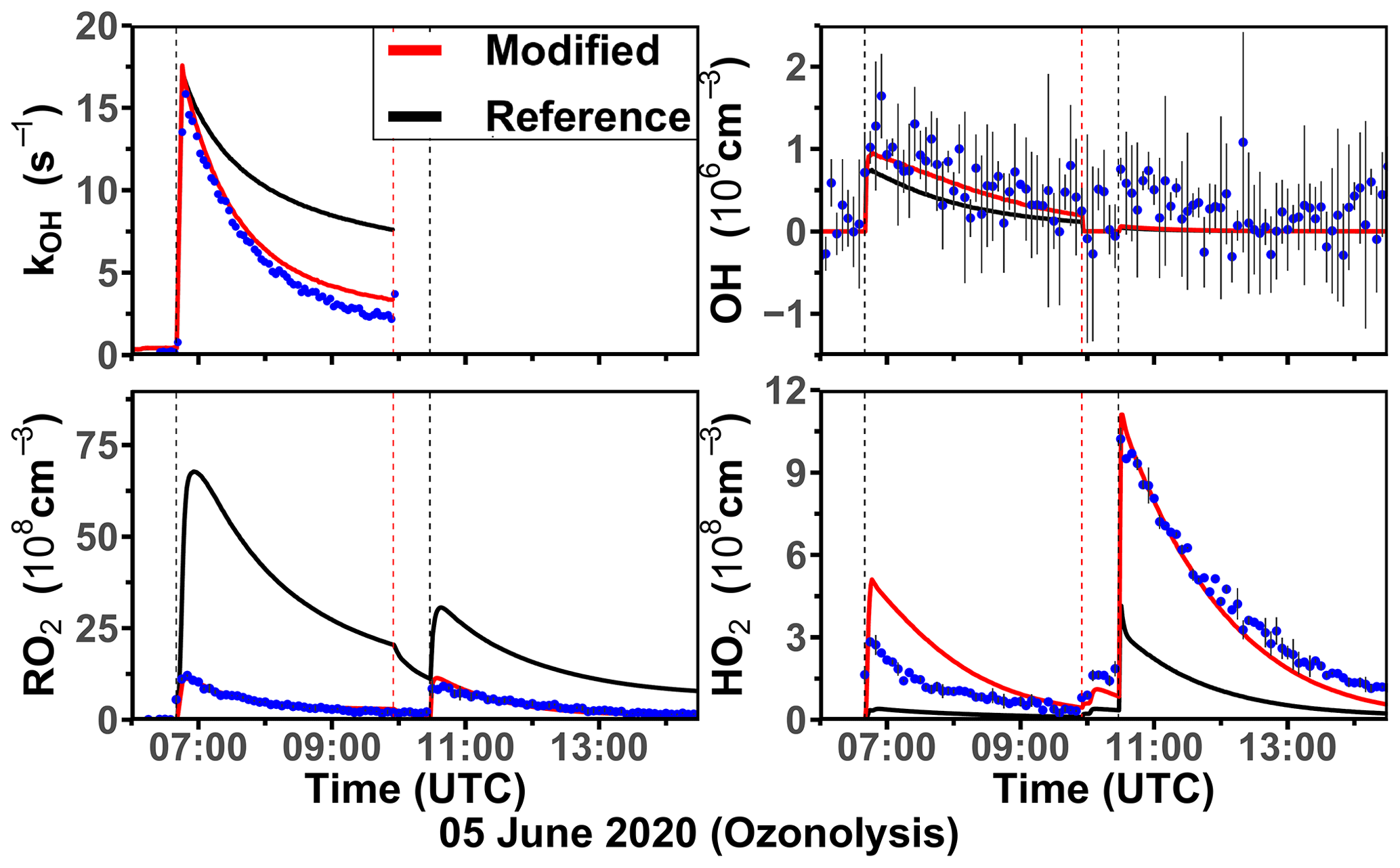
Figure 14Modeled and measured OH reactivity and HO2, RO2, and OH concentrations for the ozonolysis experiment on 5 June 2020. Results from the reference model (reference) and the sensitivity model run (modified) that includes additional RO2 loss processes producing OH and HO2 (Table 6) are compared.
The subsequent reaction of β-OOH-substituted alkoxy radicals, LIMA_15shift_O and LIMB_15shift_O, which are products of the 1,5-OH H-shift reactions of LIMAO2 and LIMBO2, respectively, is a ring-breaking decomposition reaction that produces an OH radical. For the cyclization product from the isomerization of LIMCO2 and LIMC_6cyc_O2, it may either undergo bimolecular reactions or isomerization by abstracting the α-H atom of the hydroxyl group (Fig. 15). Isomerization of LIMC_6cyc_O2 could result in an aldehyde together with an HO2 radical. However, it is uncertain whether the isomerization reaction of LIMC_6cyc_O2 can compete with other reaction channels, as the SAR does not apply to bicyclic compounds. The reaction between LIMC_6cyc_O2 and NO results in a bicyclic alkoxy radical (LIMC_6cyc_O), which then dissociates to a cyclic intermediate, analogous to the bicyclic alkoxy radical produced from the OH oxidation of β-pinene (Vereecken and Peeters, 2004). The dissociation of LIMC_6cyc_O may eventually produce an HO2 radical, which could not explain the production of OH at a zero NO condition.
For limO3–RO2, their subsequent reactions after the first isomerization reaction are also expected to be very fast (k > 0.1 s−1) because of the presence of either an aldehyde group or a C=C double bond. However, allylic H-shift, cyclization, and aldehydic H-shift reactions do not affect the RO2 and OH concentrations, as the resulting alkyl radicals do not decompose into a closed-shell product and an OH radical. Instead, an oxygen molecule rapidly adds to the alkyl radical site to form a highly oxidized RO2.
One of the possible additional OH sources from RO2 isomerization reactions could be from the H abstraction of the α-OOH group (Bianchi et al., 2019). In this case, the α-OOH-substituted alkyl radical could quickly decompose into a carbonyl product and an OH radical. Because of the fast first and second steps of the isomerization reaction, it is expected that limO3-RO2 could form RO2 with a hydroperoxide group that allows them to undergo an α-OOH H-shift reaction (e.g., LIMALA_17alde_O2, LIMALB_19alde_O2, L5_5cyc_17alde_O2; Fig. 16). The SAR-estimated α-OOH H-abstraction rate constant is about 10−3 to 10−2 s−1 (Vereecken and Nozière, 2020), which is slightly faster than the additional RO2 loss rate constant kR1b (< 0.003 s−1) applied in the sensitivity run. It should be noted that the SAR does not consider the impacts of neighboring functional groups on the α-OOH H-shift rate. All limO3–RO2 have a β-oxo group near the radical site, which could increase the H-abstraction rate near the β-oxo group by about 1 order of magnitude (e.g., Crounse et al., 2013), including the α-OOH H abstraction. However, RO2 derived from the isomerization of limO3–RO2 still has other possible isomerization reaction pathways that are even more competitive (k > 1 s−1) than the α-OOH H shift. One example is the H-shift reaction from the hydroperoxyl group to another peroxy group (Vereecken and Nozière, 2020). This complicates the chemistry of these highly oxidized peroxy radicals, and it is unclear whether they eventually undergo an α-OOH H abstraction.
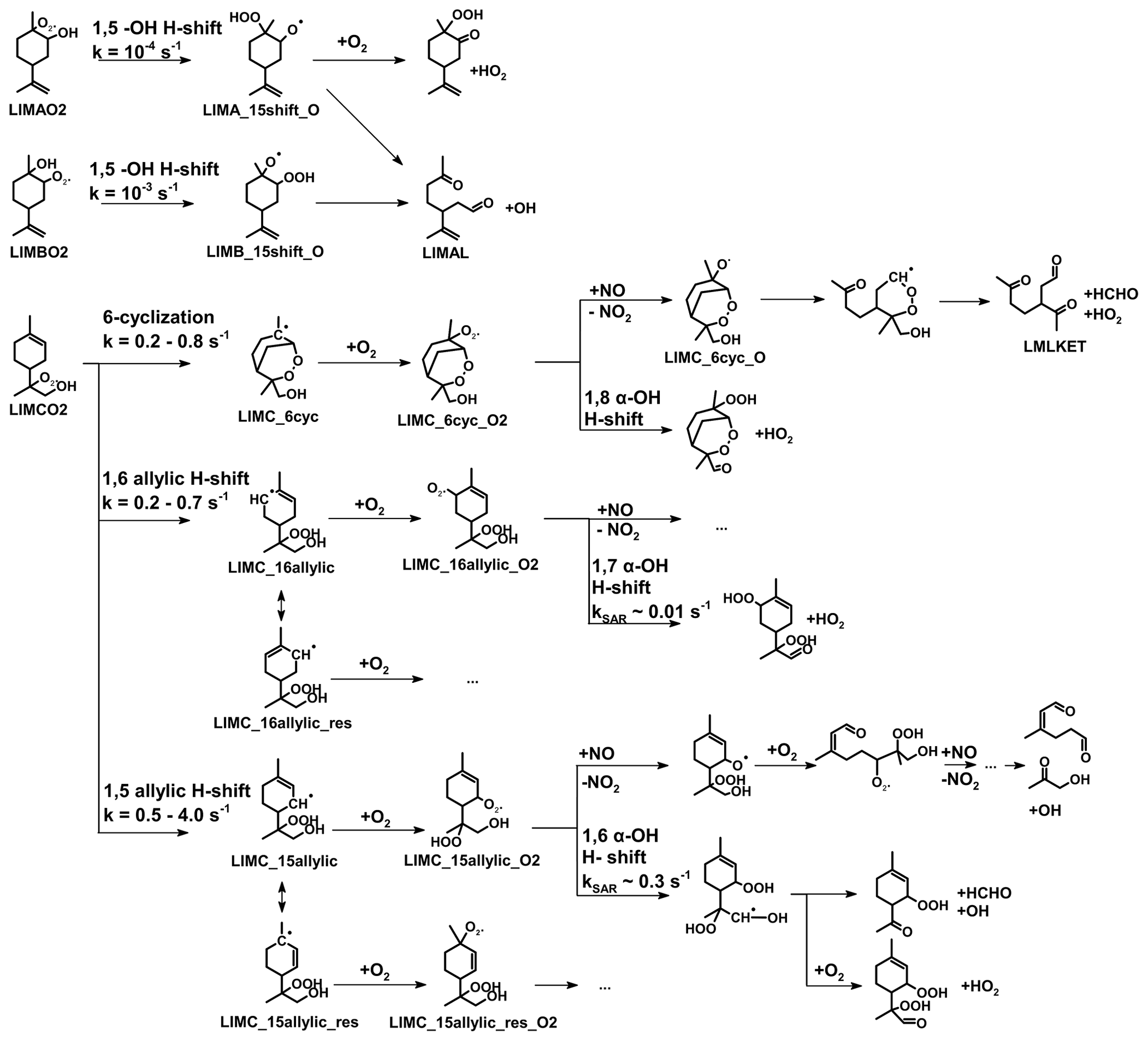
Figure 15Unimolecular reaction pathways of RO2 radicals from the reaction of limonene with OH reported in Møller et al. (2020) and subsequent anticipated reaction pathways. Reaction rate constants denoted with kSAR are directly taken from the SAR for peroxy radicals (Vereecken and Nozière, 2020). The unimolecular reaction rate constant for 1,8α-OH H shift is not available.
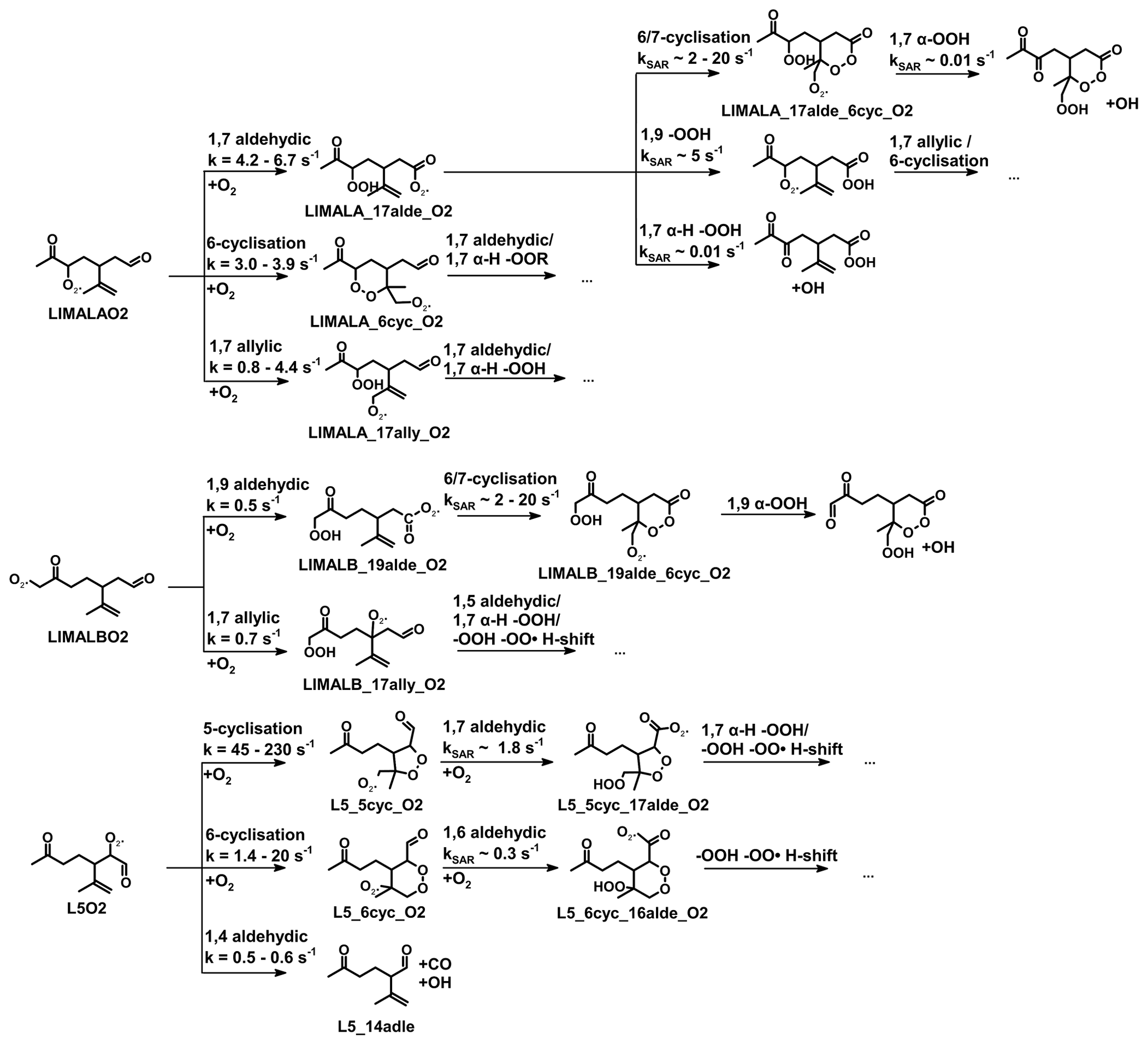
Figure 16Unimolecular reaction pathways for the major limonene-ozonolysis-derived RO2 radicals. Rate constants taken from calculations in Chen et al. (2021), in which stereoisomers are considered and therefore a range of values is given. Other rate constants (kSAR) are based on SARs in Vereecken and Nozière (2020) and Vereecken et al. (2021). It should be noted that their SAR does not address the effect of steric hindrance (in the case of a cyclic compound) or multiple functional groups near the abstracted H atom on the H-shift rate constant.
4.2.2 Reactions between RO2 and HO2
Another possible RO2 loss reaction that leads to OH production and that is not considered in the reference model is the reaction of β-oxo-substituted RO2 with HO2. Based on the work by Jenkin et al. (2019), RO2+ HO2 reactions can lead to different products depending on the functional groups nearby the peroxy group of the RO2 radical.
It has been shown in experimental studies investigating simple β-oxo-substituted RO2 that they can form an alkoxy radical together with an OH radical (Reaction R2c) instead of a hydroperoxide compound (Reaction 2a) (Jenkin et al., 2007; Hasson et al., 2012; Praske et al., 2015). With the branching ratios that are taken from the SAR, which is derived using simple β-oxo-substituted RO2, the OH yield of the RO2+ HO2 reaction for limO3–RO2 is about 30 % (Jenkin et al., 2019). This value is similar to the OH production rate in the sensitivity runs using an additional RO2 loss rate constant of kR1b=0.003 s−1. However, theoretical investigation by Iyer et al. (2018) suggests that the OH yield of the RO2+ HO2 reaction may not be high enough (< 1 %) based on the energy barriers that were calculated in that study for limO3–RO2. Currently, there is no laboratory study on the OH yield from the RO2+ HO2 reaction for large RO2, and therefore the OH yield of the RO2+ HO2 for large RO2 is highly uncertain.
4.3 Possible additional HO2 sources from RO2 reactions
4.3.1 RO2 isomerization reactions
Similar to the additional OH source, HO2 can also be produced from the isomerization reaction of RO2 radicals (e.g., Crounse et al., 2012; Peeters et al., 2014). Again, the possibility of additional conversion from RO2 to HO2 by isomerization is investigated using the isomerization pathways calculated for RO2 derived from limonene oxidation and SARs (Vereecken and Peeters, 2009; Møller et al., 2020; Vereecken and Nozière, 2020; Chen et al., 2021; Vereecken et al., 2021).
Peroxy radicals LIMAO2 and LIMBO2 that are produced from the OH oxidation of limonene can undergo a slow (k < 10−3 s−1) -OH H-shift reaction that is 1 order of magnitude slower than the rate of the RO2-to-HO2 conversion applied in the sensitivity run (Fig. 15). In addition, the production of HO2 through the H abstraction by O2 of the alkoxy radical LIMA_15shift_O is not as favorable as the ring-cleavage alkoxy dissociation that eventually produces an OH radical (Vereecken and Peeters, 2009). Therefore, even with the potential production of HO2 through the isomerization of RO2 derived from the isomerization of LIMCO2 (e.g., 1,8α-OH H shift of LIMC_6cyc_O2), the production of HO2 is limited by the 37 % yield of LIMCO2 from the oxidation of limonene by OH. In addition, the rate constants of the H-shift reaction for the intermediate radicals derived from LIMCO2 (e.g., LIMC_6cyc_O2, LIMC_ 16allylic_O2, LIMC_15allylic_O2) have a high uncertainty, as it is assumed that the SARs for acyclic compounds can be applied for cyclic RO2. For these reasons, isomerization reactions for limOH–RO2 are unlikely the reason for the additional HO2 production and RO2 loss required to match observed radical concentration measurements.
For limO3–RO2, reaction rate constants of the first two of the isomerization reaction are about 1 to 2 orders of magnitude faster than the RO2 to HO2 conversion rate used in the sensitivity run (Fig. 16). As discussed in Sect. 4.2.1, one of the possible RO2 loss mechanisms through the isomerization reaction is an α-OOH H-abstraction reaction. Although the value of the rate constant of the α-OOH H-abstraction reaction derived from SAR is of the same magnitude (∼ 10−2 s−1) as the RO2-to-HO2 conversion rate constant applied in the sensitivity run, abstraction of the hydrogen with an α-OOH group would lead to the production of an OH radical rather than a HO2 radical. Therefore, isomerization reactions of limO3–RO2 can also not explain the missing RO2 to HO2 conversion resulting from observations in the experiments in this work.
4.3.2 Reaction rate constant of the RO2 recombination reaction
Apart from isomerization, HO2 could also be produced from the dissociation of alkoxy radicals derived from RO2 from the reaction of limonene with OH. Alkoxy radicals could be produced from the recombination reaction of RO2 radicals in addition to the reaction of RO2 with NO.
The current knowledge about the branching ratio between Reactions (R3a) and (R3d) as well as the RO2 self- or cross-reaction rate constants is limited, especially for complex RO2 derived from monoterpenes. There are no specific investigations for RO2 from limonene. Reaction rate constants implemented in the MCM model are based on estimated cross-reaction rates between RO2 and methyl peroxy radicals (CH3O2) (Jenkin et al., 1997, 2019). The reaction rate constants of the RO2 recombination reactions, , for limonene-derived radicals are between 10−12 and 10−13 cm3 s−1 in the MCM, consistent with results for RO2 from methyl cyclohexene, which contain a trisubstituted endocyclic double bond like limonene (Boyd et al., 2003).
However, the reaction rate constant could be higher if the cross-reaction partners are other large limonene-derived radicals rather than CH3O2. For example, Berndt et al. (2018) investigated the self-reaction rate constants for RO2 derived from the reaction of α-pinene with OH after they undergo two steps of unimolecular reactions (i.e., C10H16OH(O2)2-O2). They found that values range between 1 and 4 × 10−11 cm3 s−1 in this case. However, it should be noted that these high rate constants are derived from the production rate of peroxide products (ROOR, Reaction R3d) rather than the loss rate of RO2.
Using the values of the reaction rate constants for RO2 from limonene oxidation from the MCM, the upper limit of the RO2 loss rate constant due to RO2–RO2 reactions is about 10−3 s−1 in the ozonolysis experiment and experiments with low NO and 2 × 10−4 s−1 in the experiments with medium NO. From the additional loss rate constant (∼ 10−2 s−1; Table 5) determined from the chemical budget analysis for RO2, the value of the reaction rate constant for the RO2–RO2 reaction that would be required to explain the observations () can be calculated. This results in values of that are about 3 × 10−10, 1 × 10−11, and 3 × 10−11 cm3 s−1 in the medium-NO, low-NO, and ozonolysis experiments, respectively. The uncertainties of the rate constants are about 50 %–60 %, which are derived from the error propagation of RO2 concentrations and optimal rate constants in the sensitivity model run (Table 6). It should be noted that these values are collective loss rate constants of all first-generation RO2 species from limonene oxidation before the formation of closed-shell products, including highly oxidized RO2 produced from potential auto-oxidation reactions.
The values of the reaction rate constant found in the low-NO experiment and ozonolysis experiment are on the same order of magnitude (10−11 to 10−10 cm3 s−1) as values reported by Berndt et al. (2018) for RO2 from α-pinene oxidation. Berndt et al. (2018) also showed that the reaction rate constant for the RO2–RO2 self-reaction increases when the RO2 becomes more oxidized. This hints that the importance of RO2 recombination reactions for RO2 derived from limonene oxidation could be higher than previously thought because of the rapid isomerization reaction of these radicals (Møller et al., 2020; Chen et al., 2021; Sect. 4.2.1).
In the experiment with low NO concentrations, the additional loss rate constant for RO2 radicals that is required to explain measured RO2 concentrations (k ∼ 0.06 s−1, Table 5) is higher than the rate constant of the additional RO2-to-HO2 conversion required to explain measured HO2 concentration (k ∼ 0.006–0.02 s−1). This would be consistent with a faster reaction rate constant for the RO2–RO2 reaction, because only a fraction of the RO2–RO2 reaction would lead to the formation of alkoxy and therefore HO2 radicals (Reaction 3).
However, it would be unclear why the reaction constant required in the experiment with a medium NO mixing ratio would be higher compared to the other experiments. It is also worth noting that the decomposition of alkoxy radicals produced from RO2 from the ozonolysis of limonene leads to the production of peroxy radicals, which does not lead to the production of HO2 in most of the RO2–RO2 reaction chain (Fig. 1). Therefore, the missing production of HO2 in the ozonolysis experiment cannot be explained by a higher than previously thought reaction rate constant of the RO2–RO2 reaction.
The photooxidation of limonene by OH and O3 was investigated in experiments for zero, low (∼ 0.1 ppbv), medium (∼ 0.4 ppbv), and high (∼ 1 ppbv) NO levels in the atmospheric simulation chamber SAPHIR. The experiments were conducted with limonene mixing ratios of 4–10 ppbv and O3 mixing ratios ranging from 0 to 50 ppbv.
The analysis of measured radical concentrations in the experiments revealed that current knowledge about the limonene oxidation as implemented in the MCM cannot explain observed values specifically concerning radical regeneration. Observed OH and HO2 concentrations were a factor of 2 to 3 higher than predicted by model calculations, whereas measured RO2 concentrations were at least 50 % lower than modeled values. The following processes in the limonene mechanism impacting radical concentrations could be identified that are not appropriately described.
-
The loss rate of OH radicals is too high in the model, as seen in higher-than-observed OH reactivity values. Although it cannot be excluded that chamber wall losses reduced the concentration of organic oxidation products, this hints at the reactivity of products species with respect to their reaction with OH being low or other products other than currently thought being produced, for example, by the competition of unaccounted radical reaction pathways.
-
The yield of organic nitrates from the reaction of RO2 radicals formed in the initial reaction of OH with limonene is found to be (34 ± 5) %, which agrees with the measurements by Rollins et al. (2010) but which is about 10 % higher than calculated from structure–activity relationships. The higher yield of organic nitrates reduces the efficiency of the radical regeneration in the limonene mechanism.
-
Formaldehyde is expected to be formed from the reaction chain after the addition of OH to the terminal C=C double bond if RO2 radicals react with NO, so that the formaldehyde yield would be similar to the yield of that RO2 species (37 %). The low formaldehyde yield of (13 ± 3) % in the experiments with medium NO concentrations suggests that there is an unaccounted RO2 loss reaction not producing formaldehyde that is competitive at 200 pptv NO.
-
OH production and destruction rates are balanced in most of the experiments if measured OH reactivity and measured HO2 concentrations are used for calculating reaction rates. This demonstrates that measured values are consistent and confirm the shortcomings of the limonene mechanism with regards to describing the HO2 production and OH reactivity.
-
An unaccounted RO2 loss process with a rate constant of 0.02 to 0.06 s−1 is required to balance the RO2 production rate from the reaction of OH with organic compounds. Formation of HO2 with a rate constant of 0.03 and 0.017 s−1 from an additional reaction of the RO2 from the reaction of limonene with OH and O3, respectively, can explain part of the model–measurement discrepancies for HO2.
-
An unaccounted RO2 loss process for RO2 from the ozonolysis of limonene that is competitive against the reaction with NO prevents the formation of NOx reservoir PAN and PAN-like species as suggested in the MCM model. The observed NOx concentrations do not exhibit a distinct temporal behavior that would be expected from the rapid loss of NOx species at the beginning of limonene oxidation when RO2 derived from limonene oxidation reacts with NOx and NOx reformation from the thermal decomposition of PAN species at the later times of the experiments when limonene has reacted away.
Overall, the results of the experiments clearly demonstrate that loss reactions of RO2 from the oxidation of limonene are not well understood. Unaccounted RO2 reactions lead to an enhanced radical regeneration. Organic products likely are less reactive than products that are currently thought to be formed. Time series of measured radical concentrations indicate that their further oxidation reactions need to be investigated to explain observed values at later times of the experiments when limonene had reacted away. The formation of NOx reservoir species (PAN/PAN-like species) is lower than expected. However, this is partly counteracted by a high yield of (34 ± 5) % of organic nitrates. Oxidation products are also likely to have a high organic nitrate yield, as indicated by the low measured NOx concentrations that would otherwise be expected to continuously increase over the course of the experiment due to the continuous emission of HONO by the chamber film.
Rate constants of RO2 isomerization reactions proposed by Møller et al. (2020) and Chen et al. (2021) are too low or too fast (k ∼ 10−4 to 10−3 and 1 to 102 s−1) to explain observed radical concentrations, and expected products are not consistent. A possible explanation could be that the reaction rate constant of RO2 recombination reactions for RO2 from limonene oxidation is higher than the reaction rate constants that are calculated by SARs implemented in the MCM. For example, experiments for RO2 from α-pinene oxidation by Berndt et al. (2018) show that the rate constant can be 1 to 2 orders of magnitude faster than implemented in the MCM model for α-pinene (10−13 to 10−12 cm3 s−1). These values are consistent with the additional loss rate required to explain radical concentrations in the experiments with limonene in this work. However, the importance of the alkoxy pathway of the RO2+ RO2 reaction for large monoterpene-derived RO2 is still unclear and needs further investigation.
Data from the experiments in the SAPHIR chamber used in this work are available on the EUROCHAMP data home page: https://doi.org/10.25326/JNMN-YC22 (Fuchs et al., 2021b); https://doi.org/10.25326/2PGS-FP66 (Fuchs et al., 2021c); https://doi.org/10.25326/77N2-ZK22 (Fuchs et al., 2021a); https://doi.org/10.25326/BP56-WP95 (Bohn et al., 2021c), https://doi.org/10.25326/C4SW-TP73 (Bohn et al., 2021b); https://doi.org/10.25326/7ZKX-347 (Bohn et al., 2021a).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-8497-2022-supplement.
JYSP, AN, and HF wrote the manuscript. MK, AN, and HF designed and led the experiments in the chamber. BB (radiation), RT, AL, IHA, and RW (organic compounds), XL (HONO), FR (ozone, nitrogen oxides), HPD, and PC (radicals), SN (OH reactivity), and CC and AN (radicals and OH reactivity) were responsible for measurements used in this work. All the co-authors commented on and discussed the manuscript and contributed to the writing of the manuscript.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors thank Luc Vereecken for the discussion on the chemical mechanism.
This research has been supported by Horizon 2020 (SARLEP, grant no. 681529) and Horizon 2020 (EUROCHAMP-2020, grant no. 730997).
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Ivan Kourtchev and reviewed by two anonymous referees.
Arey, J., Aschmann, S. M., Kwok, E. S. C., and Atkinson, R.: Alkyl Nitrate, Hydroxyalkyl Nitrate, and Hydroxycarbonyl Formation from the NOx-Air Photooxidations of C5−C8n-Alkanes, J. Phys. Chem. A, 105, 1020–1027, https://doi.org/10.1021/jp003292z, 2001.
Aschmann, S. M., Arey, J., and Atkinson, R.: OH radical formation from the gas-phase reactions of O3 with a series of terpenes, Atmos. Environ., 36, 4347–4355, https://doi.org/10.1016/S1352-2310(02)00355-2, 2002.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Badali, K. M., Zhou, S., Aljawhary, D., Antiñolo, M., Chen, W. J., Lok, A., Mungall, E., Wong, J. P. S., Zhao, R., and Abbatt, J. P. D.: Formation of hydroxyl radicals from photolysis of secondary organic aerosol material, Atmos. Chem. Phys., 15, 7831–7840, https://doi.org/10.5194/acp-15-7831-2015, 2015.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018.
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Chem. Commun., 2, 1–10, https://doi.org/10.1038/s42004-019-0120-9, 2019.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bohn, B. and Zilken, H.: Model-aided radiometric determination of photolysis frequencies in a sunlit atmosphere simulation chamber, Atmos. Chem. Phys., 5, 191–206, https://doi.org/10.5194/acp-5-191-2005, 2005.
Bohn, B., Rohrer, F., Brauers, T., and Wahner, A.: Actinometric measurements of NO2 photolysis frequencies in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 493–503, https://doi.org/10.5194/acp-5-493-2005, 2005.
Bohn, B., Dorn, H.-P., Rohrer, F., Tillmann, R., Fuchs, H., Novelli, A., and Cho, C.: Atmospheric simulation chamber study: limonene + O3 – Gas-phase oxidation – kinetic study, AERIS [data set], https://doi.org/10.25326/7ZKX-3472, 2021a.
Bohn, B., Dorn, H.-P., Rohrer, F., Tillmann, R., Fuchs, H., Novelli, A., and Cho, C.: Atmospheric simulation chamber study: limonene + OH – Gas-phase oxidation – kinetic study, AERIS [data set], https://doi.org/10.25326/C4SW-TP73, 2021b.
Bohn, B., Dorn, H.-P., Rohrer, F., Tillmann, R., Wegener, R., Li, X., and Fuchs, H.: Atmospheric simulation chamber study: limonene + OH – Gas-phase oxidation – kinetic study, AERIS [data set], https://doi.org/10.25326/BP56-WP95, 2021c.
Boyd, A. A., Villenave, E., and Lesclaux, R.: Self- and cross-reactions of β-hydroxyperoxy radicals of relevance to tropospheric monoterpene oxidation: structure–activity relationships for rate coefficients, Atmos. Environ., 37, 2751–2760, https://doi.org/10.1016/S1352-2310(03)00253-X, 2003.
Calogirou, A., Larsen, B. R., and Kotzias, D.: Gas-phase terpene oxidation products: a review, Atmos. Environ., 33, 1423–1439, https://doi.org/10.1016/S1352-2310(98)00277-5, 1999.
Chen, J., Møller, K. H., Wennberg, P. O., and Kjaergaard, H. G.: Unimolecular Reactions Following Indoor and Outdoor Limonene Ozonolysis, J. Phys. Chem. A, 125, 669–680, https://doi.org/10.1021/acs.jpca.0c09882, 2021.
Cox, R. A., Ammann, M., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 20, 13497–13519, https://doi.org/10.5194/acp-20-13497-2020, 2020.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/C1CP21330J, 2011.
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Dorn, H.-P., Neuroth, R., and Hofzumahaus, A.: Investigation of OH absorption cross sections of rotational transitions in the A2Σ+, , band under atmospheric conditions: Implications for tropospheric long-path absorption measurements, J. Geophys. Res.-Atmos., 100, 7397–7409, https://doi.org/10.1029/94JD03323, 1995.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., Molina, L. T., Baker, A., Meinardi, S., Blake, D., Sheehy, P., Merten, A., Zhang, R., Zheng, J., Fortner, E. C., Junkermann, W., Dubey, M., Rahn, T., Eichinger, B., Lewandowski, P., Prueger, J., and Holder, H.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 9, 6655–6675, https://doi.org/10.5194/acp-9-6655-2009, 2009.
Forester, C. and Wells, J.: Hydroxyl radical yields from reactions of terpene mixtures with ozone, Indoor air, 21, 400–9, https://doi.org/10.1111/j.1600-0668.2011.00718.x, 2011.
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 79, 084104, https://doi.org/10.1063/1.2968712, 2008.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P. Dorn, Häseler, R., Hofzumahaus, A., Holland, F., Kaminski, M., Li, X., Lu, K., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, and Wahner, A.: OH regeneration from methacrolein oxidation investigated in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 14, 7895–7908, https://doi.org/10.5194/acp-14-7895-2014, 2014.
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017.
Fuchs, H., Kaminski, M., Acir, I.-H., Bohn, B., Dorn, H.-P., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., and Li, X.: Atmospheric simulation chamber study: limonene + OH – Gas-phase oxidation – kinetic study, AERIS [data set], https://doi.org/10.25326/77N2-ZK22, 2021a.
Fuchs, H., Kaminski, M., Acir, I.-H., Bohn, B., Dorn, H.-P., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., and Li, X.: Atmospheric simulation chamber study: limonene + OH – Gas-phase oxidation – kinetic study, AERIS [data set], https://doi.org/10.25326/JNMN-YC22, 2021b.
Fuchs, H., Kaminski, M., Acir, I.-H., Bohn, B., Dorn, H.-P., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., and Li, X.: Atmospheric simulation chamber study: limonene + OH – Gas-phase oxidation – kinetic study, AERIS [data set], https://doi.org/10.25326/2PGS-FP66, 2021c.
Gkatzelis, G. I., Coggon, M. M., McDonald, B. C., Peischl, J., Aikin, K. C., Gilman, J. B., Trainer, M., and Warneke, C.: Identifying Volatile Chemical Product Tracer Compounds in U.S. Cities, Environ. Sci. Technol., 55, 188–199, https://doi.org/10.1021/acs.est.0c05467, 2021a.
Gkatzelis, G. I., Coggon, M. M., McDonald, B. C., Peischl, J., Gilman, J. B., Aikin, K. C., Robinson, M. A., Canonaco, F., Prevot, A. S. H., Trainer, M., and Warneke, C.: Observations Confirm that Volatile Chemical Products Are a Major Source of Petrochemical Emissions in U.S. Cities, Environ. Sci. Technol., 55, 4332–4343, https://doi.org/10.1021/acs.est.0c05471, 2021b.
Glowania, M., Rohrer, F., Dorn, H.-P., Hofzumahaus, A., Holland, F., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Comparison of formaldehyde measurements by Hantzsch, CRDS and DOAS in the SAPHIR chamber, Atmos. Meas. Tech., 14, 4239–4253, https://doi.org/10.5194/amt-14-4239-2021, 2021.
Gong, Y., Chen, Z., and Li, H.: The oxidation regime and SOA composition in limonene ozonolysis: roles of different double bonds, radicals, and water, Atmos. Chem. Phys., 18, 15105–15123, https://doi.org/10.5194/acp-18-15105-2018, 2018.
Grosjean, D., Williams, E. L., Grosjean, E., Andino, J. M., and Seinfeld, J. H.: Atmospheric oxidation of biogenic hydrocarbons: reaction of ozone with β-pinene, D-limonene and trans-caryophyllene, Environ. Sci. Technol., 27, 2754–2758, https://doi.org/10.1021/es00049a014, 1993.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hantschke, L., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., Glowania, M., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Atmospheric photooxidation and ozonolysis of Δ3-carene and 3-caronaldehyde: rate constants and product yields, Atmos. Chem. Phys., 21, 12665–12685, https://doi.org/10.5194/acp-21-12665-2021, 2021.
Hasson, A. S., Tyndall, G. S., Orlando, J. J., Singh, S., Hernandez, S. Q., Campbell, S., and Ibarra, Y.: Branching Ratios for the Reaction of Selected Carbonyl-Containing Peroxy Radicals with Hydroperoxy Radicals, J. Phys. Chem. A, 116, 6264–6281, https://doi.org/10.1021/jp211799c, 2012.
Herrmann, F., Winterhalter, R., Moortgat, G. K., and Williams, J.: Hydroxyl radical (OH) yields from the ozonolysis of both double bonds for five monoterpenes, Atmos. Environ., 44, 3458–3464, https://doi.org/10.1016/j.atmosenv.2010.05.011, 2010.
Holland, F., Hessling, M., and Hofzumahaus, A.: In Situ Measurement of Tropospheric OH Radicals by Laser-Induced Fluorescence – A Description of the KFA Instrument, J. Atmos. Sci., 52, 3393–3401, https://doi.org/10.1175/1520-0469(1995)052<3393:ISMOTO>2.0.CO;2, 1995.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified Trace Gas Removal in the Troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Iyer, S., Reiman, H., Møller, K. H., Rissanen, M. P., Kjaergaard, H. G., and Kurtén, T.: Computational Investigation of RO2+ HO2 and RO2+ RO2 Reactions of Monoterpene Derived First-Generation Peroxy Radicals Leading to Radical Recycling, J. Phys. Chem. A, 122, 9542–9552, https://doi.org/10.1021/acs.jpca.8b09241, 2018.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jenkin, M. E., Hurley, M. D., and Wallington, T. J.: Investigation of the radical product channel of the CH3COO2+ HO2 reaction in the gas phase, Phys. Chem. Chem. Phys., 9, 3149–3162, https://doi.org/10.1039/b702757e, 2007.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V.-M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, https://doi.org/10.1016/j.ijms.2009.07.005, 2009.
Kaminski, M.: Untersuchung des photochemischen Terpenoidabbaus in der Atmosphärensimulationskammer SAPHIR, Ph.D. thesis, Forschungszentrum Jülich GmbH Zentralbibliothek, Jülich, University of Cologne, Germany, 148 pp., ISBN 978-3-89336-967-6, 2014.
Kaminski, M., Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., and Wahner, A.: Investigation of the β-pinene photooxidation by OH in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 17, 6631–6650, https://doi.org/10.5194/acp-17-6631-2017, 2017.
Larsen, Bo. R., Di Bella, D., Glasius, M., Winterhalter, R., Jensen, N. R., and Hjorth, J.: Gas-Phase OH Oxidation of Monoterpenes: Gaseous and Particulate Products, J. Atmos. Chem., 38, 231–276, https://doi.org/10.1023/A:1006487530903, 2001.
Lee, A., Goldstein, A. H., Kroll, J. H., Ng, N. L., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes, J. Geophys. Res., 111, D17305, https://doi.org/10.1029/2006JD007050, 2006.
Leungsakul, S., Jaoui, M., and Kamens, R. M.: Kinetic Mechanism for Predicting Secondary Organic Aerosol Formation from the Reaction of d-Limonene with Ozone, Environ. Sci. Technol., 39, 9583–9594, https://doi.org/10.1021/es0492687, 2005.
Li, X., Rohrer, F., Hofzumahaus, A., Brauers, T., Häseler, R., Bohn, B., Broch, S., Fuchs, H., Gomm, S., Holland, F., Jäger, J., Kaiser, J., Keutsch, F. N., Lohse, I., Lu, K., Tillmann, R., Wegener, R., Wolfe, G. M., Mentel, T. F., Kiendler-Scharr, A., and Wahner, A.: Missing Gas-Phase Source of HONO Inferred from Zeppelin Measurements in the Troposphere, Science, 344, 292–296, https://doi.org/10.1126/science.1248999, 2014.
Librando, V. and Tringali, G.: Atmospheric fate of OH initiated oxidation of terpenes. Reaction mechanism of α-pinene degradation and secondary organic aerosol formation, J. Environ. Manage., 75, 275–282, https://doi.org/10.1016/j.jenvman.2005.01.001, 2005.
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research, Int. J. Mass Spectrom. Ion Process., 173, 191–241, https://doi.org/10.1016/S0168-1176(97)00281-4, 1998.
Liu, X., Mason, M., Krebs, K., and Sparks, L.: Full-scale chamber investigation and simulation of air freshener emissions in the presence of ozone, Environ. Sci. Technol., 38, 2802–2812, https://doi.org/10.1021/es030544b, 2004.
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Bohn, B., Brauers, T., Chang, C. C., Häseler, R., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S. R., Nehr, S., Shao, M., Zeng, L. M., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 12, 1541–1569, https://doi.org/10.5194/acp-12-1541-2012, 2012.
Lu, X., Chen, N., Wang, Y., Cao, W., Zhu, B., Yao, T., Fung, J. C. H., and Lau, A. K. H.: Radical budget and ozone chemistry during autumn in the atmosphere of an urban site in central China, J. Geophys. Res.-Atmos., 122, 3672–3685, https://doi.org/10.1002/2016JD025676, 2017.
McDonald, B. C., Gouw, J. A. de, Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L., Lee-Taylor, J., Hayes, P. L., McKeen, S. A., Cui, Y. Y., Kim, S.-W., Gentner, D. R., Isaacman-VanWertz, G., Goldstein, A. H., Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018.
Møller, K. H., Otkjær, R. V., Chen, J., and Kjaergaard, H. G.: Double Bonds Are Key to Fast Unimolecular Reactivity in First-Generation Monoterpene Hydroxy Peroxy Radicals, J. Phys. Chem. A, 124, 2885–2896, https://doi.org/10.1021/acs.jpca.0c01079, 2020.
Nazaroff, W. W. and Weschler, C. J.: Cleaning products and air fresheners: exposure to primary and secondary air pollutants, Atmos. Environ., 38, 2841–2865, https://doi.org/10.1016/j.atmosenv.2004.02.040, 2004.
Novelli, A., Vereecken, L., Bohn, B., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Reimer, D., Rohrer, F., Rosanka, S., Taraborrelli, D., Tillmann, R., Wegener, R., Yu, Z., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 3333–3355, https://doi.org/10.5194/acp-20-3333-2020, 2020.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 103, 4657–4690, https://doi.org/10.1021/cr020527p, 2003.
Otkjær, R. V., Jakobsen, H. H., Tram, C. M., and Kjaergaard, H. G.: Calculated Hydrogen Shift Rate Constants in Substituted Alkyl Peroxy Radicals, J. Phys. Chem. A, 122, 8665–8673, https://doi.org/10.1021/acs.jpca.8b06223, 2018.
Peeters, J. and Müller, J.-F.: HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: experimental evidence and global impact, Phys. Chem. Chem. Phys., 12, 14227–14235, https://doi.org/10.1039/C0CP00811G, 2010.
Peeters, J., Boullart, W., Pultau, V., Vandenberk, S., and Vereecken, L.: Structure–Activity Relationship for the Addition of OH to (Poly)alkenes: Site-Specific and Total Rate Constants, J. Phys. Chem. A, 111, 1618–1631, https://doi.org/10.1021/jp066973o, 2007.
Peeters, J., Müller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl Radical Recycling in Isoprene Oxidation Driven by Hydrogen Bonding and Hydrogen Tunneling: The Upgraded LIM1 Mechanism, J. Phys. Chem. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014.
Praske, E., Crounse, J. D., Bates, K. H., Kurtén, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Intramolecular Hydrogen Shift Chemistry of Hydroperoxy-Substituted Peroxy Radicals, J. Phys. Chem. A, 123, 590–600, https://doi.org/10.1021/acs.jpca.8b09745, 2019.
Ren, X., Brune, W. H., Oliger, A., Metcalf, A. R., Simpas, J. B., Shirley, T., Schwab, J. J., Bai, C., Roychowdhury, U., Li, Y., Cai, C., Demerjian, K. L., He, Y., Zhou, X., Gao, H., and Hou, J.: OH, HO2, and OH reactivity during the PMTACS–NY Whiteface Mountain 2002 campaign: Observations and model comparison, J. Geophys. Res.-Atmos., 111, D10S03, https://doi.org/10.1029/2005JD006126, 2006.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005.
Rolletter, M., Kaminski, M., Acir, I.-H., Bohn, B., Dorn, H.-P., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Investigation of the α-pinene photooxidation by OH in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 19, 11635–11649, https://doi.org/10.5194/acp-19-11635-2019, 2019.
Rollins, A. W., Smith, J. D., Wilson, K. R., and Cohen, R. C.: Real Time In Situ Detection of Organic Nitrates in Atmospheric Aerosols, Environ. Sci. Technol., 44, 5540–5545, https://doi.org/10.1021/es100926x, 2010.
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Allen, H. M., Ayres, B. R., Brown, S. S., Brune, W. H., Crounse, J. D., de Gouw, J., Draper, D. C., Feiner, P. A., Fry, J. L., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K., Teng, A. P., Wennberg, P. O., Wild, R. J., Zhang, L., and Cohen, R. C.: The lifetime of nitrogen oxides in an isoprene-dominated forest, Atmos. Chem. Phys., 16, 7623–7637, https://doi.org/10.5194/acp-16-7623-2016, 2016.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schlosser, E., Bohn, B., Brauers, T., Dorn, H.-P., Fuchs, H., Häseler, R., Hofzumahaus, A., Holland, F., Rohrer, F., Rupp, L. O., Siese, M., Tillmann, R., and Wahner, A.: Intercomparison of Two Hydroxyl Radical Measurement Techniques at the Atmosphere Simulation Chamber SAPHIR, J. Atmos. Chem., 56, 187–205, https://doi.org/10.1007/s10874-006-9049-3, 2007.
Schlosser, E., Brauers, T., Dorn, H.-P., Fuchs, H., Häseler, R., Hofzumahaus, A., Holland, F., Wahner, A., Kanaya, Y., Kajii, Y., Miyamoto, K., Nishida, S., Watanabe, K., Yoshino, A., Kubistin, D., Martinez, M., Rudolf, M., Harder, H., Berresheim, H., Elste, T., Plass-Dülmer, C., Stange, G., and Schurath, U.: Technical Note: Formal blind intercomparison of OH measurements: results from the international campaign HOxComp, Atmos. Chem. Phys., 9, 7923–7948, https://doi.org/10.5194/acp-9-7923-2009, 2009.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Tan, Z., Lu, K., Jiang, M., Su, R., Dong, H., Zeng, L., Xie, S., Tan, Q., and Zhang, Y.: Exploring ozone pollution in Chengdu, southwestern China: A case study from radical chemistry to O3-VOC-NOx sensitivity, Sci. Total Environ., 636, 775–786, https://doi.org/10.1016/j.scitotenv.2018.04.286, 2018.
Tan, Z., Hantschke, L., Kaminski, M., Acir, I.-H., Bohn, B., Cho, C., Dorn, H.-P., Li, X., Novelli, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Atmospheric photo-oxidation of myrcene: OH reaction rate constant, gas phase oxidation products and radical budgets, Atmos. Chem. Phys., 21, 16067–16091, https://doi.org/10.5194/acp-21-16067-2021, 2021.
Tyndall, G. S., Cox, R. A., Granier, C., Lesclaux, R., Moortgat, G. K., Pilling, M. J., Ravishankara, A. R., and Wallington, T. J.: Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res.-Atmos., 106, 12157–12182, https://doi.org/10.1029/2000JD900746, 2001.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Vereecken, L. and Peeters, J.: Nontraditional (Per)oxy Ring-Closure Paths in the Atmospheric Oxidation of Isoprene and Monoterpenes, J. Phys. Chem. A, 108, 5197–5204, https://doi.org/10.1021/jp049219g, 2004.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/B909712K, 2009.
Vereecken, L., Vu, G., Wahner, A., Kiendler-Scharr, A., and Nguyen, H. M. T.: A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 23, 16564–16576, https://doi.org/10.1039/D1CP02758A, 2021.
Wang, L. and Wang, L.: The oxidation mechanism of gas-phase ozonolysis of limonene in the atmosphere, Phys. Chem. Chem. Phys., 23, 9294–9303, https://doi.org/10.1039/D0CP05803C, 2021.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Zhang, F. and Dibble, T. S.: Effects of Olefin Group and Its Position on the Kinetics for Intramolecular H-Shift and HO2 Elimination of Alkenyl Peroxy Radicals, J. Phys. Chem. A, 115, 655–663, https://doi.org/10.1021/jp1111839, 2011.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.