the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Jun 2022
| 07 Jun 2022
Elucidating the critical oligomeric steps in secondary organic aerosol and brown carbon formation
Qiuju Shi
Xiaohui Ma
Lei Gao
Jiaxin Wang
Yixin Li
Yanpeng Gao
Guiying Li
Renyi Zhang
Taicheng An
Small α-dicarbonyls represent the major precursors of secondary organic aerosol (SOA) and brown carbon (BrC) in the atmosphere, but the chemical mechanisms leading to their formation remain unclear. Here we elucidate the fundamental kinetics and mechanisms for aqueous-phase oligomerization of glyoxal (GL) using quantum chemical and kinetic rate calculations. Our results identify several essential isomeric processes for GL, including protonation to yield diol tetrol and carbenium ions, nucleophilic addition of carbenium ions to diol tetrol as well as to free methylamine ammonia (MA AM), and deprotonation to propagate oligomers and N-heterocycles. Both protonation and nucleophilic addition occur without activation barriers and are dominantly driven by electrostatic attraction. Deprotonation proceeds readily via water molecules in the absence of MA AM but corresponds to the rate-limiting step for N-containing cationic intermediates to yield N-heterocycles. On the other hand, the latter occurs readily via a catalytic process by acidic anions (e.g., SO). A carbenium ion-mediated reaction rate of GL is 4.62 × 10−3 s−1 under atmospheric conditions, in good agreement with the experimental data. Our results provide essential mechanistic and kinetic data for accurate assessment of the role of small α-dicarbonyls in SOA and BrC formation.
- Article
(5765 KB) - Full-text XML
-
Supplement
(2259 KB) - BibTeX
- EndNote





Volatile organic compounds (VOCs) from biogenic and anthropogenic sources are of particular importance due to their chemical reactivity and high abundance in the atmosphere (Piccot, 1992; Acosta Navarro et al., 2014). Gas-phase reactions of VOCs associated with photochemical oxidant cycles generally produce secondary organic aerosol (SOA) particle mass and increase tropospheric ozone concentration (Lim et al., 2005; Ziemann and Atkinson, 2012; S. Wang et al., 2020; Y. Wang et al., 2020; Ge et al., 2021; Ma et al., 2021). Hence, the emission of VOCs is a key process in controlling the formation and growth of new particles over continental regions. Oxidation of biogenic terpenes from terrestrial vegetation and aromatics from human activities generates a large number of organic carbonyls, which are major precursors of SOA (Altieri et al., 2006; Volkamer et al., 2001; Gomez Alvarez et al., 2007; Sareen et al., 2016; Yang et al., 2020). These organic carbonyls engage in a variety of reactions, including new particle formation, condensation equilibrium partitioning, particle- and aqueous-phase reactions, leading to the formation of additional products in the particle phase (Loeffler et al., 2006; Kroll et al., 2005; Nozière et al., 2009; Marrero-Ortiz et al., 2019; Tuguldurova et al., 2019; Kua et al., 2011; Li et al., 2020; Shi et al., 2019; Xia et al., 2021).
Glyoxal (GL), an important and simple carbonyl compound, originates from the gas-phase oxidation of biogenic isoprene and anthropogenic aromatics (Volkamer et al., 2001; Gomez Alvarez et al., 2007; Lim et al., 2005). The global source of GL is estimated to be 45–56 Tg yr−1 (Fu et al., 2008; Myriokefalitakis et al., 2008), with a predicted contribution of 2.6 Tg C yr−1 to global SOA mass (Fu et al., 2008). GL and methylglyoxal contribute to 16 % of SOA mass during a severe haze episode in Hebei Province, China (J. Li et al., 2021). In Mexico City, GL contributes at least 15 % of SOA mass (Volkamer et al., 2007), and in the Pearl River Delta, China, 21 % of SOA formation originates from the heterogeneous reactions of GL and methylglyoxal (Ling et al., 2020). Hence, GL is a primary contributor to the rapid and efficient formation of SOA under urban environments. The aqueous-phase reaction of GL starts with hydration, subsequently forming several high-molecular-weight oligomers, such as dimers, trimers, tetramers, and pentamers (Hastings et al., 2005; Loeffler et al., 2006; Gomez et al., 2015; Kua et al., 2008; Avzianova and Brooks, 2013). Previous theoretical studies have suggested that the direct hydration of GL is thermodynamically and kinetically unfeasible (Shi et al., 2020; Ji et al., 2020). The rapid growth of SOA was observed in sulfuric acid nanoparticles (Jang et al., 2002; Huang et al., 2016; Surratt et al., 2007; Liggio et al., 2005b), while several other studies have revealed little effect of acids (such as sulfuric acid) on the formation of GL oligomers (Loeffler et al., 2006; Kroll et al., 2005). Hence, the aqueous-phase chemical reaction mechanism of GL and its role in SOA formation are still unclear.
On the other hand, brown carbon (BrC) is also generated by the aqueous-phase reaction of GL in the presence of amines or ammonium in the troposphere (Y. Li et al., 2021; Shapiro et al., 2009; Galloway et al., 2009; Lee et al., 2013; Maxut et al., 2015; Marrero-Ortiz et al., 2019; Tuguldurova et al., 2019; Kua et al., 2011). Zhang and co-workers have revealed a slight browning of glyoxal-amine mixtures (Marrero-Ortiz et al., 2019; Y. Li et al., 2021). They have also identified oligomers and N-heterocycles in NH4HSO4 and (NH4)2SO4, whereas only oligomers were detected in NaCl (Y. Li et al., 2021). Shapiro et al. have detected light-absorbing products, slowly formed from GL in a mildly acidic salt solution of (NH4)2SO4, using ultraviolet–visible (UV-vis) spectrophotometry, and matrix-assisted laser desorption ionization mass spectrometry (Shapiro et al., 2009). A chamber study of GL uptake to (NH4)2SO4 solution was performed using a high-resolution time-of-flight aerosol mass spectrometer and found that carbon–nitrogen (C-N) compounds are irreversibly produced in the solution (Galloway et al., 2009). However, De Haan et al. (2020) have described a rapid but reversible BrC formation in (NH4)2SO4 droplets in dry condition (RH <5 %) using cavity-attenuated phase shift single-scattering albedo spectrometry (De Haan et al., 2020). In addition, Powelson et al. (2014) found that the reaction of GL with methylamine is more effective than that with (NH4)2SO4 by using UV-vis and fluorescence spectroscopy (Powelson et al., 2014), while Lian et al. (2020) have proven that the synergistic effect of ammonium and amines contributes to the formation of imidazole in cloud processing (Lian et al., 2020). Most previous studies have shown that imidazole and high-molecular-weight light-absorbing C-N compounds are produced by different atmospheric chemical reactions of GL with amines ammonium, but the reaction mechanism is yet to be clarified.
In this work, the aqueous-phase chemistry of GL in the absence and presence of methylamine ammonia (MA AM) was systematically investigated using quantum chemical calculations. The fundamental chemical mechanisms of the formation of oligomers and N-heterocycles were investigated. The chemical composition and the product distribution were also estimated and characterized by conventional transition state theory considering solvent cage and diffusion-limited effects (Methods). The aerosol growth rate for the heterogeneous chemistry of GL was also evaluated under different atmospheric conditions. We paid special attention to the key factors in the formation of SOA and BrC from GL to provide insight into the important role of the aqueous-phase chemistry of GL.
All quantum chemical calculations were performed by means of the Gaussian 09 program (Frisch et al., 2009). The solvent effect of water in the aqueous phase was considered with a continuum solvation model (SMD) (Marenich et al., 2009). The solvation free energy includes two components: the bulk electrostatic contribution and the cavity-dispersion-solvent-structure contribution arising from short-range interactions between the solute and solvent molecules. Geometry optimization of all stationary points (SPs), such as reactants, transition states (TSs), intermediates, and products, was calculated using the M06-2X functional (Zhao and Truhlar, 2008) with the 6-311G(d,p) basis set (Ji et al., 2017, 2020), i.e., the M06-2X/6-311G(d,p) level, which has shown good performance in describing the geometrical optimization of the heterogeneous reactions of small α-dicarbonyls (Ji et al., 2020; Shi et al., 2020). Thermodynamic contributions and harmonic vibrational frequencies were calculated at the same level as that for geometry optimization to identify all SPs as either a TS (exactly with only one imaginary frequency) or the minima (zero imaginary frequency). Intrinsic reaction coordinate (IRC) calculations were implemented to construct the minimum energy pathway (MEP), verifying that each TS accurately connected the corresponding reactants and products. At the same level, TS was searched by examining the SP using the TS keyword in geometry optimization, while the absence of a TS was confirmed if no energy exceeded the bond dissociation energy along the reaction coordinate (Ji et al., 2020). The TSs for four deprotonation pathways in GL + MA AM reaction systems were identified at the M06-2X/6-311G(d) level because none of them was searched at the M06-2X/6-311G(d,p) level (detailed discussion in the Supplement). Pointwise potential curve (PPC) scanning was performed to further confirm a barrierless process at the M06-2X/6-311G(d,p) level (Hazra and Sinha, 2011). For this method, all other geometric parameters were fully optimized, except for fixing the internal breaking or forming bond length (detailed in the Supplement).
Based on the aforementioned optimized structures, single-point energy (SPE) calculation was performed to refine potential energy surface (PES) with a more flexible basis set 6-311 + G(3df,3pd), i.e., at the M06-2X/6-311 + G(3df,3pd) level. For simplicity, hereinafter they are denoted as the X//Y, i.e., M06-2X//M06-2X level, where Y is an SPE calculation at the M06-2X/6-311 + G(3df,3pd) level and X is the geometry optimized at the M06-2X/6-311G(d,p) level. To further evaluate the results at the M06-2X//M06-2X level, a higher-level calculation using the CCSD(T) method with the flexible 6-311 + G(2df,2p) basis set was performed to refine the PESs. The CCSD(T) method (Lutnaes et al., 2009; Raghavachari and Trucks, 1989), i.e., coupled cluster approach with single and double substitutions including a perturbative estimation of connected triple substitutions, corresponds to a higher electronic correlation method. As discussed in the Supplement, the M06-2X//M06-2X level is suitable for predicting energies and kinetics, and also represents a compromise between computational efficiency and accuracy.
The rate constants (k) of the pathways with TSs were calculated using the conventional transition state theory (TST) (Gao et al., 2014; Galano and Alvarez-Idaboy, 2009) based on the above PES information:
where h and kB are the Planck and Boltzmann constants, respectively, ΔG‡ represents the activation barrier energy with the thermodynamic contribution corrections and solvent cage effects, and σ is the reaction path degeneracy. To simulate realistic conditions in the solution, the rate constants are refined by using solvent cage effects (Okuno, 1997) and diffusion-limited effects (Collins and Kimball, 1949). For some of the reactions with low free-energy barriers, the rate constants are found to be close to the diffusion limit, which is calculated using the Collins–Kimball theory (Gao et al., 2014). The k values of the pathways without TSs are controlled by the diffusion-limit effect and thereby are equal to the diffusion-limited rate constants:
where R is the reaction distance, NA denotes the Avogadro number, and DAB represents the mutual diffusion coefficient of reactants. The branching ratio (Γ) was determined by the following equation:
where the k corresponds to the rate constant of each pathway, and the ktotal is the sum of the k value for each parallel pathway. A detailed description of the kinetics is provided in the Supplement.
3.1 Ion-mediated initial reaction of GL
The ion-mediated initial reactions of GL proceed via either proton-mediated (1) or hydroxyl ion (OH−)-mediated (1) hydration (Figs. 1a and S1a in the Supplement), yielding cationic (CIs) or anionic intermediates (AIs). As discussed in the Supplement, no TS is identified by PPC scanning for all pathways in ion-mediated reactions (Fig. S2a in the Supplement); that is, all ion-mediated pathways are barrierless processes unless otherwise stated. Figure 2 presents the natural population analysis of key species using the natural bond orbital (NBO) method (Glendening et al., 2011), and Fig. S3 in the Supplement lists the optimized geometries of all SPs. From a geometrical point of view, GL belongs to the C2h point group, and the positions of two carbonyl groups are equivalent. The two carbonyl O-atoms and C-atoms exhibit the most negative and positive natural charges of −0.532 and 0.383 e, respectively, indicating that the proton- and OH−-mediated reactions of GL start from carbonyl groups.
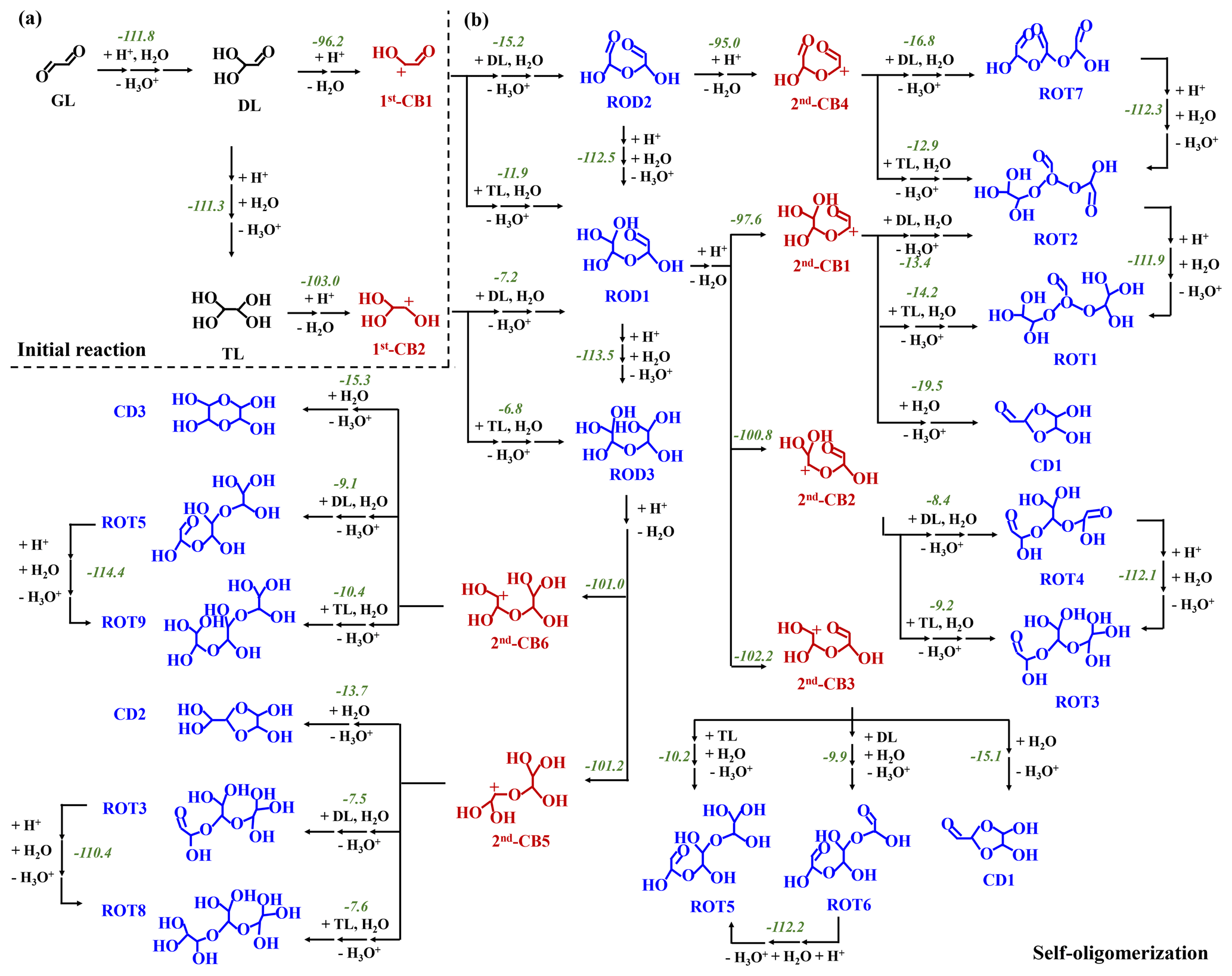
Figure 1PES of the GL oligomerization without MA AM (in kcal mol−1): (a) the initial reactions of GL and (b) the subsequent oligomerization to dimers and trimers. The number denotes the total ΔGr for each reaction.
The PESs of possible pathways in the ion-mediated reaction of GL are presented in Figs. 1a and S1. For proton-meditated pathways (1), protonation of carbonyl O-atom (11) is largely exothermic with a reaction energy (ΔGr) of −96.9 kcal mol−1, to form the CI11. Alternatively, protonation of GL can also be initiated by the hydronium ion (H3O+) with a barrierless process. As shown in Fig. S1 in the Supplement, water protonation can provide the corresponding ΔGr value of −111.0 kcal mol−1, and thus for the convenience of discussion, the following proton-mediated pathways refer specially to the hydrogen ion (H+)-initiated reactions. The nucleophilic attack of GL by OH− (11) is also exothermic with the ΔGr value of −13.4 kcal mol−1, to yield the AI11. The small exothermicity of 11 implies thermodynamically unfavorable formation of AI11. Table S1 in the Supplement lists the k values of 11 and 11 pathways as well as the ktotal of R1. Herein, the ktotal value of the ion-initiated reactions (the sum of the k values of 11 and 11 pathways) is 6.02 × 109 M−1 s−1, and the k of the 11 pathway contributes 30 % to the ktotal. Further, considering the moderately acidic condition inside fine particles (Liu et al., 2017; Guo et al., 2012), proton-mediated reaction of GL is of major significance in the troposphere, and, thus, the subsequent reactions of CIs are mainly considered in the following parts.
As shown in Fig. S3, the length of the C-O bond in CI11 is elongated to 1.24 Å, facilitating the subsequent hydration reaction (12). CI11 reacts via hydration and dehydration to yield diol (DL) with successive increasing in ΔGr values ranging from −8.0 to −1.0 kcal mol−1. Subsequently, DL protonation occurs at the carbonyl (21-1) or hydroxyl (22-1) O-atom, leading to the formation of tetrol (TL) and the first-generation carbenium ion (1st-CB1) (Figs. 1a and S1b). The 21-1 and 22-1 pathways to form CI21-1 and CI22-1 are also strongly exothermic with the ΔGr values of −103.0 and −104.4 kcal mol−1, respectively. The pathway for CI21-1 to TL proceeds via hydration and deprotonation with successive increasing in ΔGr values, and the CI22-1 to 1st-CB1 reaction via deprotonation corresponds to a slightly increasing ΔGr value, suggesting that both TL and 1st-CB1 are the dominant intermediates.
As shown in Table S1, the k values of 21-1 and 22-1 pathways are all 4.14 × 109 M−1 s−1 and their half-lives () are lower than s. The was calculated using the formula, (k × [H+]), where k is the rate constant of the 21-1 or 22-1 pathway and [H+] is the concentration of the hydrogen ion (10−6 M) in the weakly acidic solution. It implies the rapid conversion from DL to TL and 1st-CB1, in line with the experimental results showing that the abundance of GL monohydrate is lower than 2 % in acidic conditions (Malik and Joens, 2000). As discussed in the Supplement, it further confirms that the direct hydration (1 and 2) and OH−-mediated hydration (1 and 2) are kinetically and thermodynamically hindered. Hence, the cation-mediated initial reaction of GL, as the dominant route in the aqueous phase, is mainly focused on and explored in the following study.
3.2 Oligomerization mechanisms without methylamine ammonia
3.2.1 Dimerization
The dominant intermediates, TL and 1st-CB1, can subsequently conduct electrostatic attraction with each other. As shown in Fig. S3, the C-O(H) bond of 1st-CB1 after protonation is elongated by 0.05 Å relative to the C=O bond of GL, attributable to the presence of a carbenium ion center. The natural charge of the carbenium ion center in 1st-CB1 is 0.547 e (Fig. 2), implying that it is liable to nucleophilic addition with a negative natural charge center of TL via electrostatic attraction. All possible pathways involved in nucleophilic addition between 1st-CB1 and TL are constructed and depicted in Figs. 1b and S4a.
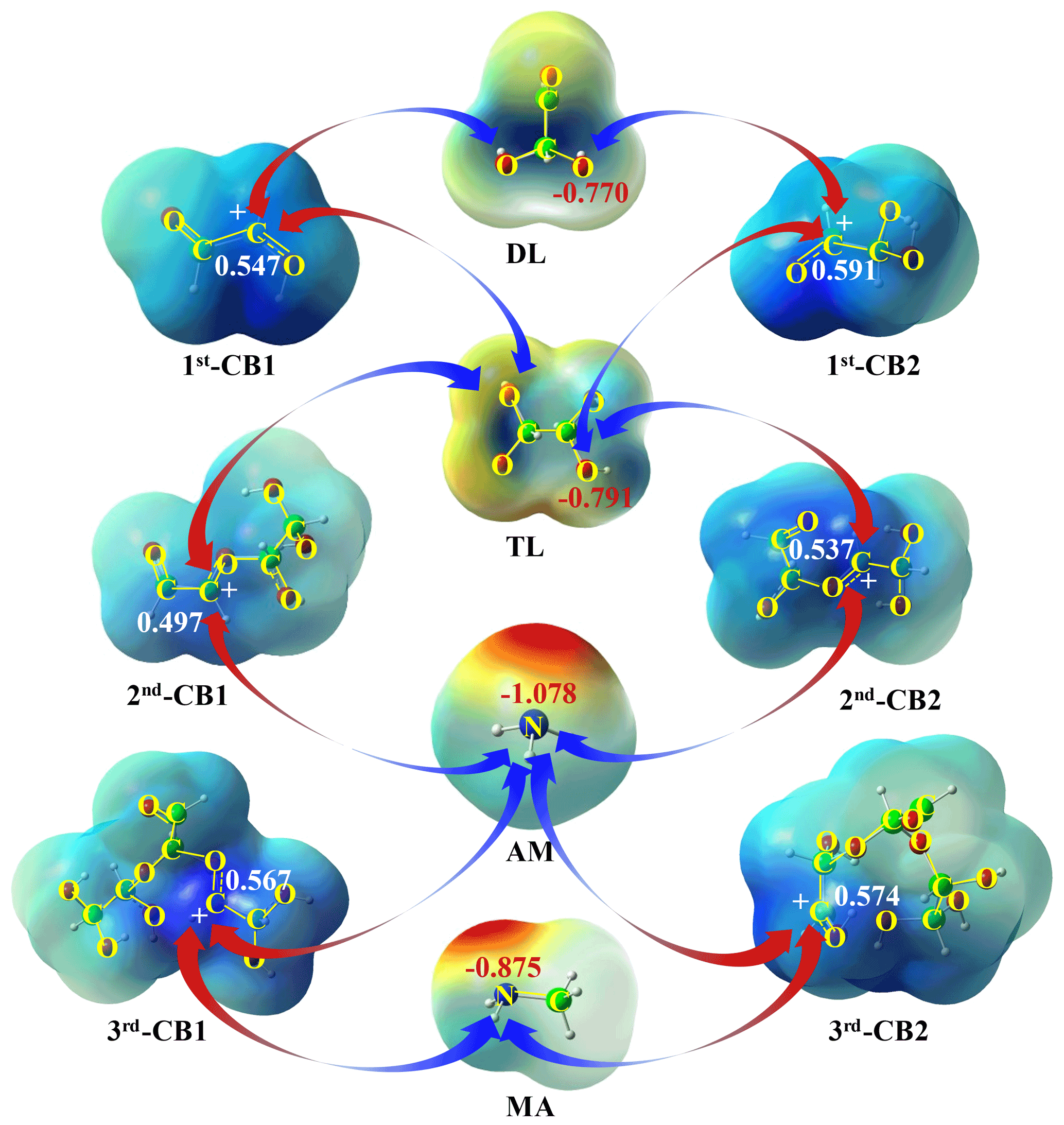
Figure 2The driving force for oligomerization without and with MA AM: the natural bond orbitals of DL, TL, MA, AM, and CBs (in e).
The nucleophilic addition of 1st-CB1 with TL (RTL41) is an exothermic process with the ΔGr value of −6.7 kcal mol−1. The subsequent hydration (RTL42) and deprotonation (RTL43) exhibit a small ΔGr value of −5.2 kcal mol−1, to yield a ring-opening dimer (ROD1) that has been identified under acidic condition using thermal desorption-ion drift-chemical ionization mass spectrometry (TD-ID-CIMS) (Y. Li et al., 2021). On the other hand, 1st-CB2 is produced via protonation (31) and dehydration (32) of TL, similar to the formation of 1st-CB1 (Fig. S1c). 1st-CB2 is also attacked by TL (RTL51) to form CITL51, which then proceeds via hydration (RTL52) and deprotonation (RTL53) to yield ROD3 (Fig. S4b in the Supplement). An intermolecular isomerization pathway exists from ROD1 to ROD3 (R6, Fig. S4c); that is, ROD3 can be formed from ROD1 via protonation (61), hydration (62 and 63), and deprotonation (64), with the total ΔGr value of −113.5 kcal mol−1. As shown in Fig. 2, the natural charge of C-atom in 1st-CB2 is more positive than that in 1st-CB1, implying a stronger electrostatic attraction between 1st-CB2 and TL. However, the ΔGr value of RTL51 is −3.1 kcal mol−1, which is higher than that of RTL41 (Fig. S4a–b). It indicates that the reactivity of the positive charge centers in carbenium ions is affected by both electrostatic attraction and steric effect.
The nucleophilic addition reaction of 1st-CBs with DL is also illuminated and presented in Figs. 1b and S4a–b, although DL is not the most dominant product in the aqueous-phase reaction of GL. Both ROD2 and ROD1 are generated from DL, respectively, through analogous pathways to RTL4 and RTL5. As shown in Fig. 2, the hydroxyl O-atom in TL has a larger negative natural charge than that in DL, implying that there is a stronger electrostatic attraction between 1st-CBs and TL. In addition, the ΔGr values of association reactions of 1st-CBs with DL are −5.3 (for 1st-CB1) and −1.8 (for 1st-CB2) kcal mol−1, respectively, which are less negative than those with TL. This indicates that the most abundant dimers correspond to the oligomeric pathways arising from 1st-CBs with TL in weakly acidic condition.
3.2.2 Trimerization and oligomerization
Similar to the formation of 1st-CBs, as shown in Fig. 1b, the ring-opening dimers then repeat protonation and dehydration to form six second-generation carbenium ions (2nd-CBs) (Fig. S5 in the Supplement), which further engage in the formation of cyclic dimers (CDs) or ring-opening trimers (ROTs). Figures S6–S7 in the Supplement present the schematic energy diagram for the formation of CDs and ROTs, and Fig. S8 in the Supplement depicts the optimized geometries of SPs involved in the aforementioned pathways. In total, three CDs and nine ROTs are produced and identified via R8–R20 routes, and the overall schematic diagram is shown in Fig. 1b. For example, protonation (81-1) and dehydration (81-2) of ROD1 yield 2nd-CB1, which undergoes intramolecular isomerization to produce CI111 (R111). CI111 is further hydrolyzed (R112) and deprotonated (R113) to yield CD1. Alternatively, the association of 2nd-CB1 with TL and DL yields CITL151 and CIDL151, respectively, and their subsequent pathways are similar to the pathways of the formation of RODs, resulting in the formation of ROT1 and ROT2 (Fig. S7a). Current results reveal that cyclic oligomers are difficult to be formed from the CBs with the positive charge center close to the O(H) atom, such as 2nd-CB2 and 2nd-CB4 in Fig. 1b. Moreover, the isomeric conversion can also occur among the nine ROTs via protonation, hydration, and deprotonation processes (R21–R27 in Fig. S9 in the Supplement).
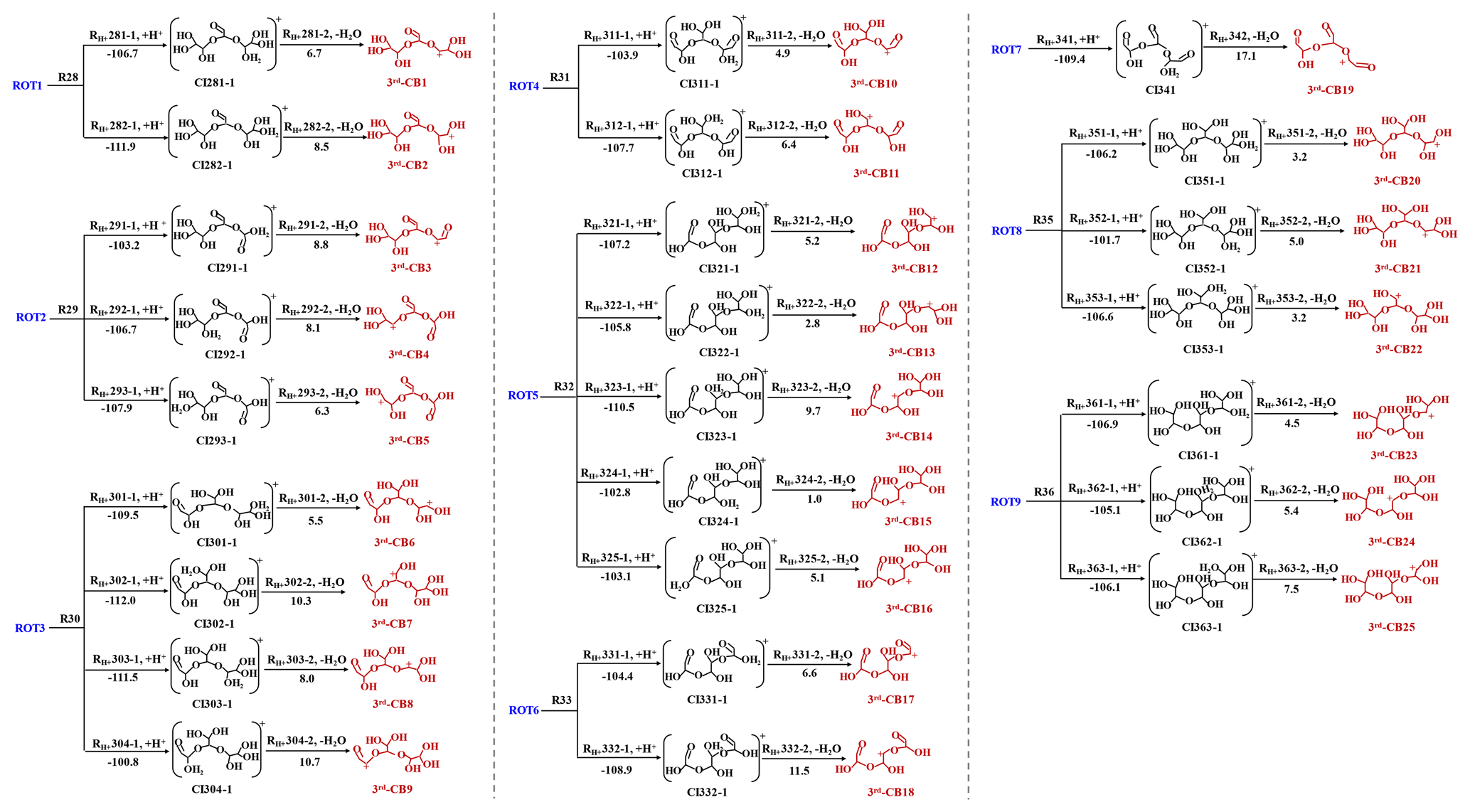
Figure 3The formation of 3rd-CBs from the ring-opening trimers (in kcal mol−1). The value represents the ΔGr of each step reaction.
Similar to dimerization pathways, the nine ROTs further engage in protonation and dehydration reactions to produce 25 third-generation carbenium ions (3rd-CBs) (R28–R36 in Fig. 3). In this study, the configurations of oligomers with the lowest energies are applied because many isomers of dimers and trimers can be yielded. Subsequently, 25 3rd-CBs undergo intramolecular isomerization, hydration, and deprotonation to further yield 12 cyclic trimers (CTs) (Fig. S10 in the Supplement and Table S2 in the Supplement). Based on kinetic rate calculations, the k values of dimer and trimer formation are ∼109 M−1 s−1 in the aqueous phase, limited by liquid-phase diffusion. It should be pointed out that the rate constants of dimer and trimer formation obtained from our theoretical calculations are distinct from those previously investigated by Ervens and Volkamer (Ervens and Volkamer, 2010). The rate constants obtained in this previous study are and ∼100 M−1 s−1 for dimer and trimer formation based on the direct nucleophilic addition between GL and GL or GL and GL hydrates. Our protonation-initiated cationic oligomerization involves nucleophilic addition of DL TL to CBs, which is fast and barrierless. Hence, the formation of various ring-opening or cyclic dimers and trimers is initiated by protonation and subsequently propagated via the electrostatic attraction, with the rate constants of ∼109 M−1 s−1, ultimately contributing to SOA formation.
3.3 Oligomerization mechanisms with methylamine ammonia
As shown in Fig. 2, a strong electrostatic attraction exists between 1st-CBs and AM or MA because N-atoms exhibit large negative natural charges (−0.875 e for MA and −1.078 e for AM). Hence, the carbenium ion-mediated oligomerization of GL with MA AM to form N-oligomers is simulated and presented in Fig. 4. Figures S11–S12 depict the optimized geometries of key SPs. Also, the involved cation-mediated pathways default to barrierless processes unless stated (Fig. S2b).

Figure 4The PES for formation of N-heterocycles starting from (a–b) 1st-CBs with MA AM to dimers, (c) dimers to N-1st-CBs, and (d–e) N-1st-CBs with MA AM to trimers (in kcal mol−1). The number denotes the ΔGr or ΔG‡ for each reaction step, and all energies are relative to the corresponding reactants.
3.3.1 The nucleophilic addition of 1st-CBs with MA
Attack of 1st-CBs by MA results in four N-containing ring-opening dimer (N-ROD) formation. For example, such multistep processes of 1st-CB1 with MA are shown as the following (Fig. 4a–b):
For the association pathway of 1st-CB1 with MA (RMA601), the ΔGr value is −43.2 kcal mol−1. In CIMA601, the length of the formed C-N bond is 1.50 Å (Fig. S12a in the Supplement). Unlike the oligomerization without MA, the subsequent deprotonation of CIMA601 is difficult to be initiated by H2O (Fig. S13 in the Supplement, as discussed in the Supplement). It implies that deprotonation from N-containing CIs needs to be initiated by species with larger electronegativity than H2O. Taking into account the real atmospheric conditions, in this study, deprotonation of the amino group in CIMA601 is initiated by SO (RMA602). As shown in Fig. S14 in the Supplement, the natural charge center of CIMA601 is located at the amino H-atom, which is readily abstracted by SO to form N-RODMA1. The RMA602 pathway proceeds via a TS with the small activation energy (ΔG‡) of −1.7 kcal mol−1. A pre-reactive complex (COMMA601) is identified prior to the TS and the corresponding ΔGr value is −1.0 kcal mol−1, which is lower than that of the corresponding reactants. As illustrated in the Supplement, the structure of COMMA601 is similar to that of the reactants, except for the broken bonds. As shown in Fig. 4b, protonation of N-RODMA1 at the hydroxyl group (RMA611) yields CIMA611 with the ΔGr value of −122.8 kcal mol−1. The subsequent dehydration of CIMA611 (RMA612) possesses small exothermicity with the Gr value of −5.7 kcal mol−1, to form CIMA612. Similar to the subsequent reaction of CIMA601, deprotonation of CIMA612 is also initiated by SO (RMA613), to form the other N-ROD (N-RODMA2), with the ΔG‡ value of −7.3 kcal mol−1. In addition, there also exists an intermolecular isomerization pathway from N-RODMA2 to N-RODMA3 via protonation, hydration, and deprotonation (Fig. 4c):
For the RMA62 pathway, the total ΔGr value for protonation, hydration, and deprotonation of N-RODMA2 is −108.1 kcal mol−1, yielding N-RODMA3. N-RODMA3 then repeats protonation and dehydration to yield N-containing CBs (N-1st-CBs), which subsequently engage in the nucleophilic addition with MA to form in sequence N-containing ring-opening trimers, N-ROTMA1 and N-ROTMA2, (Fig. 4c–e):
Subsequently, N-ROTMA2 undergoes the nucleophilic addition with 1st-CBs rather than protonation, attributing to geometric characteristics, i.e., no hydroxyl groups in N-ROTMA2 to protonation (Fig. S11 in the Supplement). For example, the reaction of N-ROTMA2 with 1st-CB1 to form N-heterocycles involves the following stepwise pathways (Fig. S15 in the Supplement):
The nucleophilic addition of N-ROTMA2 with 1st-CB1 (RMA66) is largely exothermic with the ΔGr value of −33.8 kcal mol−1 to overcome the barrier of subsequent intramolecular torsion and H-abstraction pathways. The intramolecular torsion from CIMA661 to CIMA662 proceeds via a TS, with the small ΔG‡ value of 5.1 kcal mol−1. The ΔG‡ value of the H-abstraction pathway (RMA663) is 18.4 kcal mol−1. Subsequently, protonation of N-IMMA1 occurs at the hydroxyl group to form CIMA671, which undergoes dehydration via a TS, yielding an N-containing cyclic tetramer (N-CTMA1, in Fig. S15). The ΔG‡ and ΔGr values of RMA672 are 17.3 and −89.4 kcal mol−1, respectively. The association reactions of 1st-CB2 with MA are also investigated and discussed in the Supplement, via the similar stepwise pathways of 1st-CB1 with MA (Figs. S16–S17), yielding an N-containing cyclic tetramer (N-CTMA2). The intermolecular isomerization reaction from N-CTMA1 to N-CTMA2 is also observed (Fig. S18a in the Supplement). All N-containing dimers, trimers, and tetramers subsequently contribute to N-heterocycles, which are the important precursors of BrC aerosols. Because all deprotonation reactions proceed via the corresponding TS in the presence of MA and their rate constants fall in the range of (1.17–1.30) × 109 M−1 s−1 (Table S3), deprotonation is the rate-limiting step to propagate N-heterocycles. It implies that N-heterocycle formation is more dependent on the content of inorganic compounds or inorganic salts in aerosol rather than on particle acidity.
3.3.2 The nucleophilic addition of 1st-CBs with AM
The carbenium ion-mediated reactions to N-heterocycles in the presence of AM involve the stepwise processes (Figs. 4 and S15–S17), similar to the nucleophilic addition of 1st-CBs with MA; that is, the formation of N-heterocycles from GL with AM involves three vital steps (Fig. S19 in the Supplement): (1) the nucleophilic reaction of AM with CBs to form N-RODs, (2) protonation and dehydration of N-RODs to yield N-containing CBs, and (3) the formation and propagation of N-heterocycles by the association reactions of N-containing CBs with AM. Alternatively, GL can be attacked by ammonium ion (NH) to produce CIAM601 due to the equilibrium reaction between AM and NH in solution, with the ΔGr value of −0.9 kcal mol−1 (Fig. 4a). CIAM601 engages in vital steps (2) and (3) to finally form N-heterocycles (N-CTAM1 and N-CTAM2). Different from the case in the presence of MA, N-CTAM1 and N-CTAM2 can subsequently proceed with deprotonation to form N-CTAM3 and N-CTAM4 (Fig. S18b), respectively, because of the presence of H-atoms in amino groups of N-CTAM1 and N-CTAM2 and the absence in N-CTMA1 and N-CTMA2. This explains that N-CTAM3 and N-CTAM4 are identified in the presence of ammonium salts (such as (NH4)2SO4) (Lee et al., 2013; Yu et al., 2011), while N-CTMA1 is observed in the presence of MA (De Haan et al., 2009). Hence, the carbenium ion-mediated mechanism also provides a key pathway for the formation of BrC from GL in the presence of ammonium salts.
Table 1Estimation of the rates for the oligomerization without and with MA AM and the growth rates to SOA and BrC under different atmospheric conditions.

a Numbers are the typical measured values of GL from Cerqueira et al. (2003), Lawson et al. (2015), Qian et al. (2019), and Munger
et al. (1995).
b The values are the rates of the oligomerization without and with
MA AM (in s−1).
c krate(total) is the sum of the krate values without and
with MA AM. The values in parentheses are from Liggio et al. (2005a).
d The values without and with MA AM are assumed as the growth rates
to SOA and to BrC, respectively. The values in parentheses are from Liggio et al. (2005a).
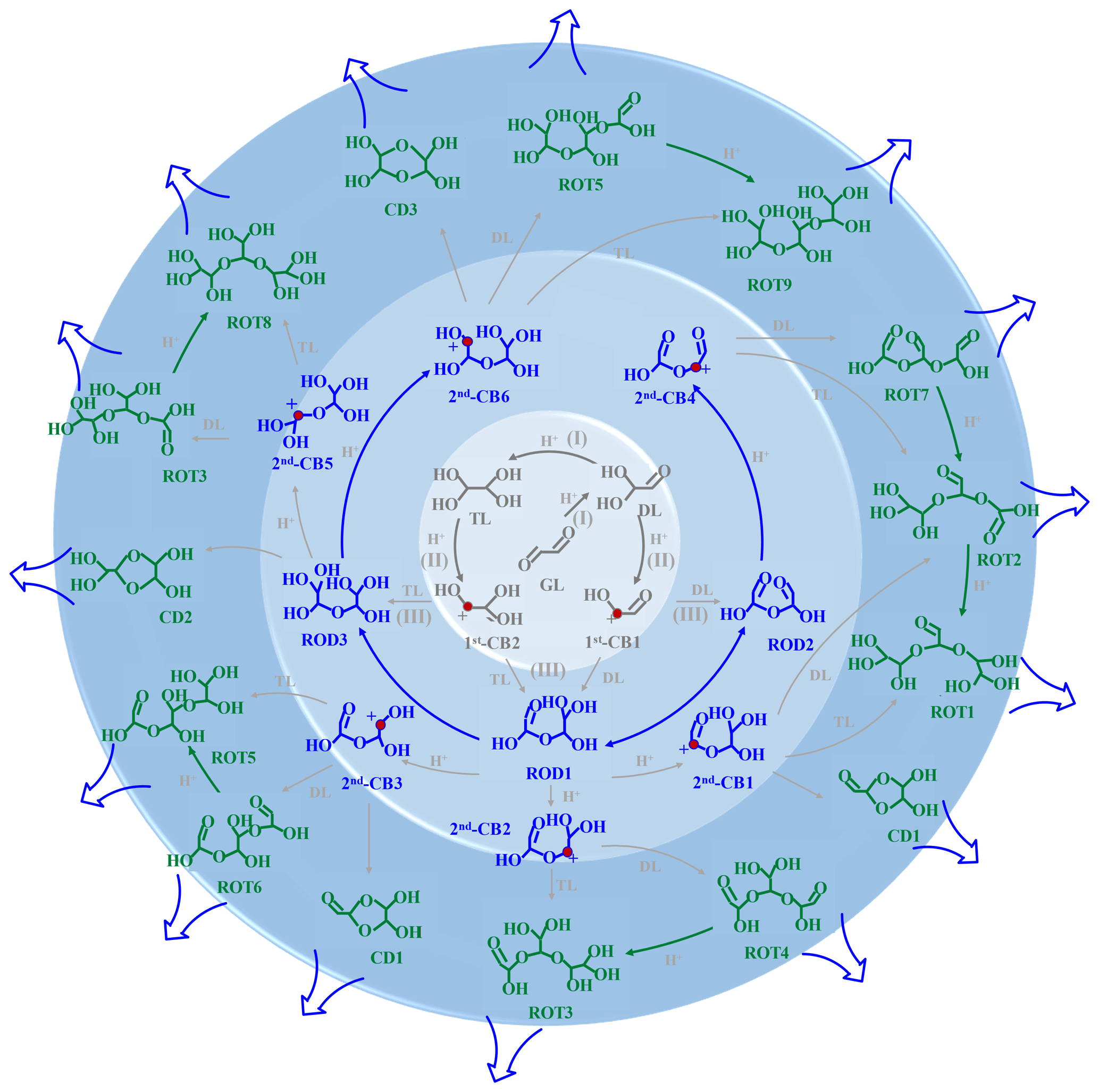
Figure 5Schematic diagram of formation and propagation of oligomers in the absence of MA AM. The inside circle ring represents the ion-mediated initial reaction of GL to yield DL, TL, and 1st-CBs; the middle circle ring corresponds to the formation of RODs and 2nd-CBs; the outer circle ring denotes the formation and propagation of ROTs from the association reactions of 2nd-CBs with DL TL.
3.4 Estimation of the heterogeneous GL reaction rates and growth rates of SOA and BrC formation
To evaluate the atmospheric regions where the heterogeneous reaction of GL will have significance, the heterogeneous GL reaction rates and the growth rates of SOA and BrC formation (GRSOA and GRBrC) are estimated under rural, remote, and urban conditions using the predicted carbenium ion-mediated reaction mechanism of GL mentioned earlier. First, the heterogeneous GL reaction rates without or with MA AM were driven by the expression as follows:
where Cg is the gas phase concentration under rural, remote, and urban conditions, k is the calculated rate constant of protonation reaction (4.20 × 109 M−1 s−1) in the absence of MA AM or deprotonation ( × 109 M−1 s−1) in the presence of MA AM, and γGL is the uptake coefficient of GL under the urban condition.
The heterogeneous GL reaction rate (krate(total)) is the sum of the rates without and with MA AM. Table 1 lists the krate without and with MA AM and krate(total) under rural, remote, and urban conditions as well as the available experimental data (Liggio et al., 2005a). The krate(total) values are 4.62 × 10−3, 9.25 × 10−4, and 1.85 × 10−3 s−1 under the three aforementioned conditions. The krate value under the urban condition almost agrees with that of the experimental data and is slightly larger than the values of the experimental data under other conditions (Liggio et al., 2005a). The lower values under remote and rural conditions are explained by the γGL used here, which is more suitable for the urban condition (Liggio et al., 2005a). Second, the growth rate (GRSOA) (Ji et al., 2020) is expressed as
where represents the rate of GL protonation given by krate × [H+] × HGL, where krate is the rate of heterogeneous GL reaction without MA AM (in Table 1), and [H+] is the concentration of the hydrogen ion (10−6 M) in the weakly acidic solution, and AWC is the aerosol water content (1.1 × 10−10) (Ji et al., 2020). The GRBrC is also calculated using Eq. (5), where is replaced by × [SO] × HGL, i.e.,
where krate is the rate of heterogeneous GL reaction with MA AM, and [SO] is the concentration of SO (5.0 × 10−7 M) in the weakly acidic solution. Considering that the deprotonation occurs readily by acidic anions (e.g., SO), the GRBrC values obtained in this study may be underestimated. The GRSOA and GRBrC values under the different atmospheric conditions are also presented in Table 1. The GRSOA is 1.41 µg m−3 h−1 under the urban condition, which is larger than the GRBrC (0.44 µg m−3 h−1 for MA and 0.45 µg m−3 h−1 for AM). The total growth rate (the sum of the GRSOA and GRBrC values) is also consistent with the experimental data (1.44 µg m−3 h−1) (Liggio et al., 2005a). Hence, it is reasonable to expect the occurrence of a carbenium ion-mediated mechanism in ambient aerosols as a means of SOA and BrC formation.
This study provides valuable insight into the aqueous chemistry of GL and also reveals the rate-limiting steps in the absence and presence of MA AM. In the absence of MA AM (Fig. 5), the cation-mediated oligomerization is characterized by barrierless pathways and strong electrostatic attraction as follows: (I) protonation, hydration, and deprotonation of GL to yield DL and TL, (II) protonation and dehydration to yield CBs, and (III) the formation of dimers from the association reactions of CBs with DL and TL. Each dimer repeats steps (II) and (III) to propagate the oligomerization. In the presence of MA AM, step (III) starts from nucleophilic addition of CBs with MA AM rather than with DL TL due to the stronger electrostatic attraction between CBs and MA AM (Fig. 2). However, the key mechanistic step in the propagation of N-heterocycles is deprotonation of N-containing cationic intermediates. Our results of two distinct mechanisms indicate that BrC formation is more dependent on the aerosol content of inorganic compounds or inorganic salts rather than particle acidity, compared with the formation of SOA.
On the other hand, there exist the competing pathways with the initial protonation pathway for the cationic oligomerization of GL without and with MA AM, (i.e., the association reactions of CIs with OH−). These competing pathways lead to CIs returning the corresponding reactants and affect the fate of GL. Figure S20 and Table S1 present the ΔGr and k values of the association reactions for some key CIs with OH−. Generally, the fate of CIs at each step is dominantly determined by the initial protonation rather than the reaction with OH−. For example, the ΔGr value of the association pathway of CI11 with OH− is −66.8 kcal mol−1, and its k value is 1.47 × 109 M−1 s−1. Compared with the protonation of GL to CI11, this reaction is thermodynamically and kinetically unfavorable. The branching ratios show that 70 % of GL proceeds via the protonation pathway to finally form DL. Because protonation is favorable in the acidic aerosol, the cation-mediated oligomerization of GL without and with MA AM can efficiently proceed to contribute to SOA and BrC under the atmospheric conditions.
Using our predicted heterogeneous GL reaction rates, the aqueous heterogeneous lifetime (τ) of GL is estimated to be 3.60 min under the urban conditions, somewhat shorter than that of experimental data (5.0 min) (Liggio et al., 2005a). However, the τ values are 89 and 61 min under rural and remote conditions due to low GL levels, respectively (Liggio et al., 2005a). It indicates a more important role of the aqueous heterogeneous reaction of GL in urban air quality relative to other conditions. On the other hand, the τ values determined here for rural, remote, and urban conditions are all lower than those of the photolysis (211 min) and photooxidation of GL (Liggio et al., 2005a). Especially, the τ value under the urban condition is significantly shorter than the total gas-phase loss (125 min) (Liggio et al., 2005a). The results indicate that even under relatively clean conditions, the heterogeneous GL loss rates are faster than the loss rates due to other gas-phase processes and are significantly rapid in polluted regions. Given that GL contributes to 6.9 % of the total radical production at midday (Aiello, 2003), the heterogeneous GL loss to particles implies the reduction of HOx and demands further study. Our work reveals the fundamental chemical mechanism of SOA and BrC formation from small α-dicarbonyls and also provides the kinetic and mechanistic data for atmospheric modeling to assess the budget of SOA and BrC formation on urban, regional, and global scales.
All raw data can be provided by the corresponding authors upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-7259-2022-supplement.
YJ and QS designed the research; YJ, QS, XM, LG, JW, and YL performed the research; YJ, QS, XM, LG, JW, YL, YG, GL, RZ, and TA analyzed the data; YJ and QS wrote the paper. YJ, XM, RZ, and TA reviewed and edited the paper.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work was financially supported by National Natural Science Foundation of China (grant nos. 42077189 and 4201001008), Natural Science Foundation of Guangdong Province, China (grant nos. 2019B151502064), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (grant nos. 2017BT01Z032), and Science and Technology Key Project of Guangdong Province, China (grant nos. 2019B110206002).
This paper was edited by Yafang Cheng and reviewed by Deming Xia and two anonymous referees.
Acosta Navarro, J. C., Smolander, S., Struthers, H., Zorita, E., Ekman, A. M., Kaplan, J. O., Guenther, A., Arneth, A., and Riipinen, I.: Global emissions of terpenoid VOCs from terrestrial vegetation in the last millennium, J. Geophys. Res.-Atmos., 119, 6867–6885, https://doi.org/10.1002/2013JD021238, 2014.
Aiello, M.: An automated instrument for the quantitation of atmospheric carbonyls: measurements and interpretation in southern ontario, Ph.D. thesis, York Univ., Toronto, Ont., Canada, 256 pp., 2003.
Altieri, K. E., Carlton, A. G., Lim, H. J., Turpin, B. J., and Seitzinger, S. P.: Evidence for oligomer formation in clouds: reactions of isoprene oxidation products, Environ. Sci. Technol., 40, 4956–4960, https://doi.org/10.1021/es052170n, 2006.
Avzianova, E. and Brooks, S. D.: Raman spectroscopy of glyoxal oligomers in aqueous solutions, Spectrochim. Acta., A, 101, 40–48, https://doi.org/10.1016/j.saa.2012.09.050, 2013.
Cerqueira, M. A., Pio, C. A., Gomes, P. A., Matos, J. S., and Nunes, T. V.: Volatile organic compounds in rural atmospheres of central Portugal, Sci. Total Environ., 313, 49–60, https://doi.org/10.1016/s0048-9697(03)00250-x, 2003.
Collins, F. C. and Kimball, G. E.: Diffusion-controlled reaction rates, J. Colloid Sci., 4, 425–437, https://doi.org/10.1016/0095-8522(49)90023-9, 1949.
De Haan, D. O., Tolbert, M. A., and Jimenez, J. L.: Atmospheric condensed-phase reactions of glyoxal with methylamine, Geophys. Res. Lett., 36, L11819, https://doi.org/10.1029/2009gl037441, 2009.
De Haan, D. O., Hawkins, L. N., Jansen, K., Welsh, H. G., Pednekar, R., de Loera, A., Jimenez, N. G., Tolbert, M. A., Cazaunau, M., Gratien, A., Bergé, A., Pangui, E., Formenti, P., and Doussin, J.-F.: Glyoxal's impact on dry ammonium salts: fast and reversible surface aerosol browning, Atmos. Chem. Phys., 20, 9581–9590, https://doi.org/10.5194/acp-20-9581-2020, 2020.
Ervens, B. and Volkamer, R.: Glyoxal processing by aerosol multiphase chemistry: towards a kinetic modeling framework of secondary organic aerosol formation in aqueous particles, Atmos. Chem. Phys., 10, 8219–8244, https://doi.org/10.5194/acp-10-8219-2010, 2010.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A., Peralta Jr., J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Keith, T., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M. Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J., and Fox, D. J.: Gaussian09, Revision D.01, Gaussian, Inc., Wallingford CT., USA, 2009.
Fu, T. M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res., 113, D15303, https://doi.org/10.1029/2007jd009505, 2008.
Galano, A. and Alvarez-Idaboy, J. R.: Guanosine + OH radical reaction in aqueous solution: A reinterpretation of the UV-vis data based on thermodynamic and kinetic calculations, Org. Lett., 11, 5114–5117, https://doi.org/10.1021/ol901862h, 2009.
Galloway, M. M., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Flagan, R. C., Seinfeld, J. H., and Keutsch, F. N.: Glyoxal uptake on ammonium sulphate seed aerosol: reaction products and reversibility of uptake under dark and irradiated conditions, Atmos. Chem. Phys., 9, 3331–3345, https://doi.org/10.5194/acp-9-3331-2009, 2009.
Gao, Y., Ji, Y., Li, G., and An, T.: Mechanism, kinetics and toxicity assessment of OH-initiated transformation of triclosan in aquatic environments, Water Res., 49, 360–370, https://doi.org/10.1016/j.watres.2013.10.027, 2014.
Ge, M., Tong, S., Wang, W., Zhang, W., Chen, M., Peng, C., Li, J., Zhou, L., Chen, Y., and Liu, M.: Important oxidants and their impact on the environmental effects of aerosols, J. Phys. Chem. A, 125, 3813–3825, https://doi.org/10.1021/acs.jpca.0c10236, 2021.
Glendening, E. D., Landis, C. R., and Weinhold, F.: Natural bond orbital methods, Wires Comput. Mol. Sci., 2, 1–42, https://doi.org/10.1002/wcms.51, 2011.
Gomez Alvarez, E., Viidanoja, J., Munoz, A., Wirtz, K., and Hjorth, J.: Experimental confirmation of the dicarbonyl route in the photo-oxidation of toluene and benzene, Environ. Sci. Technol., 41, 8362–8369, https://doi.org/10.1021/es0713274, 2007.
Gomez, M. E., Lin, Y., Guo, S., and Zhang, R.: Heterogeneous chemistry of glyoxal on acidic solutions. An oligomerization pathway for secondary organic aerosol formation, J. Phys. Chem. A, 119, 4457–4463, https://doi.org/10.1021/jp509916r, 2015.
Guo, J., Wang, Y., Shen, X., Wang, Z., Lee, T., Wang, X., Li, P., Sun, M., Collett, J. L., Wang, W., and Wang, T.: Characterization of cloud water chemistry at Mount Tai, China: Seasonal variation, anthropogenic impact, and cloud processing, Atmos. Environ., 60, 467–476, https://doi.org/10.1016/j.atmosenv.2012.07.016, 2012.
Hastings, W. P., Koehler, C. A., Bailey, E. L., and De Haan, D. O.: Secondary organic aerosol formation by glyoxal hydration and oligomer formation: humidity effects and equilibrium shifts during analysis, Environ. Sci. Technol., 39, 8728–8735, https://doi.org/10.1021/es050446l, 2005.
Hazra, M. K. and Sinha, A.: Formic acid catalyzed hydrolysis of SO3 in the gas phase: a barrierless mechanism for sulfuric acid production of potential atmospheric importance, J. Am. Chem. Soc., 133, 17444–17453, https://doi.org/10.1021/ja207393v, 2011.
Huang, M. Q, Cai, S. Y., Liao, Y. M., Zhao, W. X., Hu, C. J., Wang, Z. Y., and Zhang, W. J.: Theoretical studies on mechanism and rate constant of gas phase hydrolysis of glyoxal catalyzed by sulfuric acid, Chinese J. Chem. Phys., 29, 335–343, https://doi.org/10.1063/1674-0068/29/cjcp1509193, 2016.
Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, https://doi.org/10.1126/science.1075798, 2002.
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, P. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1715304114, 2017.
Ji, Y., Shi, Q., Li, Y., An, T., Zheng, J., Peng, J., Gao, Y., Chen, J., Li, G., Wang, Y., Zhang, F., Zhang, A. L., Zhao, J., Molina, M. J., and Zhang, R.: Carbenium ion-mediated oligomerization of methylglyoxal for secondary organic aerosol formation, P. Natl. Acad. Sci. USA, 117, 13294–13299, https://doi.org/10.1073/pnas.1912235117, 2020.
Kroll, J. H., Ng, N. L., Murphy, S. M., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Chamber studies of secondary organic aerosol growth by reactive uptake of simple carbonyl compounds, J. Geophys. Res., 110, D23207, https://doi.org/10.1029/2005jd006004, 2005.
Kua, J., Hanley, S. W., and de Haan, D. O.: Thermodynamics and kinetics of glyoxal dimer formation: A computational study, J. Phys. Chem. A, 112, 66–72, https://doi.org/10.1021/jp076573g, 2008.
Kua, J., Krizner, H. E., and De Haan, D. O.: Thermodynamics and kinetics of imidazole formation from glyoxal, methylamine, and formaldehyde: a computational study, J. Phys. Chem. A, 115, 1667–1675, https://doi.org/10.1021/jp111527x, 2011.
Lawson, S. J., Selleck, P. W., Galbally, I. E., Keywood, M. D., Harvey, M. J., Lerot, C., Helmig, D., and Ristovski, Z.: Seasonal in situ observations of glyoxal and methylglyoxal over the temperate oceans of the Southern Hemisphere, Atmos. Chem. Phys., 15, 223–240, https://doi.org/10.5194/acp-15-223-2015, 2015.
Lee, A. K., Zhao, R., Li, R., Liggio, J., Li, S. M., and Abbatt, J. P.: Formation of light absorbing organo-nitrogen species from evaporation of droplets containing glyoxal and ammonium sulfate, Environ. Sci. Technol., 47, 12819–12826, https://doi.org/10.1021/es402687w, 2013.
Lian, X., Zhang, G., Yang, Y., Lin, Q., Fu, Y., Jiang, F., Peng, L., Hu, X., Chen, D., Wang, X., Peng, P. A., Sheng, G., and Bi, X.: Evidence for the formation of imidazole from carbonyls and reduced nitrogen species at the individual particle level in the ambient atmosphere, Environ. Sci. Technol. Lett., 8, 9–15, https://doi.org/10.1021/acs.estlett.0c00722, 2020.
Liggio, J., Li, S. M., and McLaren, R.: Reactive uptake of glyoxal by particulate matter, J. Geophys. Res., 110, D10304, https://doi.org/10.1029/2004jd005113, 2005a.
Liggio, J., Li, S. M., and McLaren, R.: Heterogeneous reactions of glyoxal on particulate matter: identification of acetals and sulfate esters, Environ. Sci. Technol., 39, 1532–1541, https://doi.org/10.1021/es048375y, 2005b.
Li, J., Han, Z., Sun, Y., Li, J., and Liang, L.: Chemical formation pathways of secondary organic aerosols in the Beijing-Tianjin-Hebei region in wintertime, Atmos. Environ., 244, 117996, https://doi.org/10.1016/j.atmosenv.2020.117996, 2021.
Li, L., Zhong, J., Yan, Y., Zhang, J., Xu, J., Francisco, J. S., and Zeng, X. C.: Unraveling nucleation pathway in methane clathrate formation, P. Natl. Acad. Sci. USA, 117, 24701–24708, https://doi.org/10.1073/pnas.2011755117, 2020.
Lim, H. J., Carlton, A. G., and Turpin, B. J.: Isoprene forms secondary organic aerosol through cloud processing: model simulations, Environl. Sci. Technol., 39, 4441–4446, https://doi.org/10.1021/es048039h, 2005.
Ling, Z., Xie, Q., Shao, M., Wang, Z., Wang, T., Guo, H., and Wang, X.: Formation and sink of glyoxal and methylglyoxal in a polluted subtropical environment: observation-based photochemical analysis and impact evaluation, Atmos. Chem. Phys., 20, 11451–11467, https://doi.org/10.5194/acp-20-11451-2020, 2020.
Liu, M., Yu, S., Tian, Z., Xu, Z., and Tong, Z.: Fine particle pH during severe haze episodes in northern China, Geophys. Res. Lett., 44, 5213–5221, https://doi.org/10.1002/2017GL073210, 2017.
Li, Y., Ji, Y., Zhao, J., Wang, Y., Shi, Q., Peng, J., Wang, Y., Wang, C., Zhang, F., Wang, Y., Seinfeld, J. H., and Zhang, R.: Unexpected oligomerization of small alpha-dicarbonyls for secondary organic aerosol and brown carbon formation, Environ. Sci. Technol., 55, 4430–4439, https://doi.org/10.1021/acs.est.0c08066, 2021.
Loeffler, K. W., Koehler, C. A., Paul, N. M., and De Haan, D. O.: Oligomer formation in evaporating aqueous glyoxal and methyl glyoxal solutions, Environ. Sci. Technol., 40, 6318–6323, https://doi.org/10.1021/es060810w, 2006.
Lutnaes, O. B., Teale, A. M., Helgaker, T., Tozer, D. J., Ruud, K., and Gauss, J.: Benchmarking density-functional-theory calculations of rotational g tensors and magnetizabilities using accurate coupled-cluster calculations, J. Chem. Phys., 131, 144104, https://doi.org/10.1063/1.3242081, 2009.
Ma, F., Xie, H., Li, M., Wang, S., Zhang, R., and Chen, J.: Autoxidation mechanism for atmospheric oxidation of tertiary amines: Implications for secondary organic aerosol formation, Chemosphere, 273, 129207, https://doi.org/10.1016/j.chemosphere.2020.129207, 2021.
Malik, M. and Joens, J. A.: Temperature dependent near-UV molar absorptivities of glyoxal and gluteraldehyde in aqueous solution, Spectrochim. Acta. A, 56, 2653–2658, https://doi.org/10.1016/s1386-1425(00)00311-5, 2000.
Marenich, A. V., Cramer, C. J., and Truhlar, D. G.: Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 113, 6378–6396, https://doi.org/10.1021/jp810292n, 2009.
Marrero-Ortiz, W., Hu, M., Du, Z., Ji, Y., Wang, Y., Guo, S., Lin, Y., Gomez-Hermandez, M., Peng, J., Li, Y., Secrest, J., Zamora, M. L., Wang, Y., An, T., and Zhang, R.: Formation and optical properties of brown carbon from small alpha-dicarbonyls and amines, Environ. Sci. Technol., 53, 117–126, https://doi.org/10.1021/acs.est.8b03995, 2019.
Maxut, A., Nozière, B., Fenet, B., and Mechakra, H.: Formation mechanisms and yields of small imidazoles from reactions of glyoxal with NH in water at neutral pH, Phys. Chem. Chem. Phys., 17, 20416–20424, https://doi.org/10.1039/c5cp03113c, 2015.
Munger, J. W., Jacob, D. J., Daube, B. C., Horowitz, L. W., Keene, W. C., and Heikes, B. G.: Formaldehyde, glyoxal, and methylglyoxal in air and cloudwater at a rural mountain site in central Virginia, J. Geophys. Res.-Atmos., 100, 9325–9333, 1995.
Myriokefalitakis, S., Vrekoussis, M., Tsigaridis, K., Wittrock, F., Richter, A., Brühl, C., Volkamer, R., Burrows, J. P., and Kanakidou, M.: The influence of natural and anthropogenic secondary sources on the glyoxal global distribution, Atmos. Chem. Phys., 8, 4965–4981, https://doi.org/10.5194/acp-8-4965-2008, 2008.
Nozière, B., Dziedzic, P., and Córdova, A.: Products and kinetics of the liquid-phase reaction of glyoxal catalyzed by ammonium ions (NH), J. Phys. Chem. A, 113, 231–237, https://doi.org/10.1021/jp8078293, 2009.
Okuno, Y.: Theoretical investigation of the mechanism of the baeyer-villiger reaction in nonpolar solvents, Chem.-Eur. J., 3, 210–218, https://doi.org/10.1002/chem.19970030208, 1997.
Piccot, S. D.: A global inventory of volatile organic compound emissions from anthropogenic sources, J. Geophys. Res., 97, 9897–9991, https://doi.org/10.1029/92jd00682, 1992.
Powelson, M. H., Espelien, B. M., Hawkins, L. N., Galloway, M. M., and De Haan, D. O.: Brown carbon formation by aqueous-phase carbonyl compound reactions with amines and ammonium sulfate, Environ. Sci. Technol., 48, 985–993, https://doi.org/10.1021/es4038325, 2014.
Qian, X., Shen, H., and Chen, Z.: Characterizing summer and winter carbonyl compounds in Beijing atmosphere, Atmos. Environ., 214, 116845, https://doi.org/10.1016/j.atmosenv.2019.116845, 2019.
Raghavachari, K. and Trucks, G. W.: A fifth-order perturbation comparison of electron correlation theories, Chem. Phys. Lett., 157, 479–483, https://doi.org/10.1016/s0009-2614(89)87395-6, 1989.
Sareen, N., Carlton, A. G., Surratt, J. D., Gold, A., Lee, B., Lopez-Hilfiker, F. D., Mohr, C., Thornton, J. A., Zhang, Z., Lim, Y. B., and Turpin, B. J.: Identifying precursors and aqueous organic aerosol formation pathways during the SOAS campaign, Atmos. Chem. Phys., 16, 14409–14420, https://doi.org/10.5194/acp-16-14409-2016, 2016.
Shapiro, E. L., Szprengiel, J., Sareen, N., Jen, C. N., Giordano, M. R., and McNeill, V. F.: Light-absorbing secondary organic material formed by glyoxal in aqueous aerosol mimics, Atmos. Chem. Phys., 9, 2289–2300, https://doi.org/10.5194/acp-9-2289-2009, 2009.
Shi, G., Mahmood, A., Lu, G., Wang, X., Tong, S., Ge, M., Xie, X., and Sun, J.: Adsorption and photodegradation of acetaldehyde and ethylene on TiO2 (001) surface: experimental and first principle studies, Catal. Lett., 149, 2728–2738, https://doi.org/10.1007/s10562-019-02813-8, 2019.
Shi, Q., Zhang, W., Ji, Y., Wang, J., Qin, D., Chen, J., Gao, Y., Li, G., and An, T.: Enhanced uptake of glyoxal at the acidic nanoparticle interface: implications for secondary organic aerosol formation, Environ. Sci.-Nano, 7, 1126–1135, https://doi.org/10.1039/d0en00016g, 2020.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, https://doi.org/10.1021/es0704176, 2007.
Tuguldurova, V. P., Fateev, A. V., Poleshchuk, O. K., and Vodyankina, O. V.: Theoretical analysis of glyoxal condensation 495 with ammonia in aqueous solution, Phys. Chem. Chem. Phys., 21, 9326–9334, https://doi.org/10.1039/c8cp07270a, 2019.
Volkamer, R., Platt, U., and Wirtz, K.: Primary and secondary glyoxal formation from aromatics: Experimental evidence for the bicycloalkyl-radical pathway from Benzene, Toluene, and p-Xylene, J. Phys. Chem. A., 105, 7865–7874, https://doi.org/10.1021/jp010152w, 2001.
Volkamer, R., San Martini, F., Molina, L. T., Salcedo, D., Jimenez, J. L., and Molina, M. J.: A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol, Geophys. Res. Lett., 34, L19807, https://doi.org/10.1029/2007gl030752, 2007.
Wang, S., Du, L., Tsona, N. T., Jiang, X., You, B., Xu, L., Yang, Z., and Wang, W.: Effect of NOx and SO2 on the photooxidation of methylglyoxal: Implications in secondary aerosol formation, J. Environ. Sci. (China), 92, 151–162, https://doi.org/10.1016/j.jes.2020.02.011, 2020.
Wang, Y., Riva, M., Xie, H., Heikkinen, L., Schallhart, S., Zha, Q., Yan, C., He, X.-C., Peräkylä, O., and Ehn, M.: Formation of highly oxygenated organic molecules from chlorine-atom-initiated oxidation of alpha-pinene, Atmos. Chem. Phys., 20, 5145–5155, https://doi.org/10.5194/acp-20-5145-2020, 2020.
Xia, D., Chen, J., Wang, Y., Xu, T., Su, L., Xie, H., and Allen, D. T.: Organic acid-ammonia ion-induced nucleation pathways unveiled by quantum chemical calculation and kinetics modeling: a case study of 3-methyl-1,2,3-butanetricarboxylic acid, Chemosphere, 284, 131354, https://doi.org/10.1016/j.chemosphere.2021.131354, 2021.
Yang, Z., Tsona, N. T., Li, J., Wang, S., Xu, L., You, B., and Du, L.: Effects of NOx and SO2 on the secondary organic aerosol formation from the photooxidation of 1,3,5-trimethylbenzene: a new source of organosulfates, Environ. Pollut., 264, 114742, https://doi.org/10.1016/j.envpol.2020.114742, 2020.
Yu, G., Bayer, A. R., Galloway, M. M., Korshavn, K. J., Fry, C. G., and Keutsch, F. N.: Glyoxal in aqueous ammonium sulfate solutions: products, kinetics and hydration effects, Environ. Sci. Technol., 45, 6336–6342, https://doi.org/10.1021/es200989n, 2011.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Account., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.