the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 May 2022
| 11 May 2022
Estimation of mechanistic parameters in the gas-phase reactions of ozone with alkenes for use in automated mechanism construction
Mike J. Newland
Camille Mouchel-Vallon
Richard Valorso
Bernard Aumont
Luc Vereecken
Michael E. Jenkin
Andrew R. Rickard
Reaction with ozone is an important atmospheric removal process for alkenes. The ozonolysis reaction produces carbonyls and carbonyl oxides (Criegee intermediates, CI), which can rapidly decompose to yield a range of closed shell and radical products, including OH radicals. Consequently, it is essential to accurately represent the complex chemistry of Criegee intermediates in atmospheric models in order to fully understand the impact of alkene ozonolysis on atmospheric composition. A mechanism construction protocol is presented which is suitable for use in automatic mechanism generation. The protocol defines the critical parameters for describing the chemistry following the initial reaction, namely the primary carbonyl/CI yields from the primary ozonide fragmentation, the amount of stabilisation of the excited CI, the unimolecular decomposition pathways, rates and products of the CI, and the bimolecular rates and products of atmospherically important reactions of the stabilised CI (SCI). This analysis implicitly predicts the yield of OH from the alkene–ozone reaction. A comprehensive database of experimental OH, SCI and carbonyl yields has been collated using reported values in the literature and used to assess the reliability of the protocol. The protocol provides estimates of OH, SCI and carbonyl yields with root mean square errors of 0.13 and 0.12 and 0.14, respectively. Areas where new experimental and theoretical data would improve the protocol and its assessment are identified and discussed.
- Article
(3336 KB) - Full-text XML
-
Supplement
(1227 KB) - BibTeX
- EndNote





Reaction with ozone is an important atmospheric removal process for alkenes, competing with reaction with OH and NO3 radicals. The ozonolysis reaction produces carbonyls and carbonyl oxides, commonly denoted Criegee intermediates (CI), which can rapidly rearrange or decompose to yield a range of closed-shell and radical products (Johnson and Marston, 2008). Alkene ozonolysis has been shown to be an important non-photolytic source of OH radicals, with field measurements (Paulson and Orlando, 1996; Elshorbany et al., 2009) and modelling studies (e.g. Bey et al., 1997) suggesting it to be the dominant tropospheric OH source at night, in the winter (Heard et al., 2004; Emmerson et al., 2005), and in indoor environments (Carslaw, 2007). Unimolecular CI reactions (Ehn et al., 2014; Iyer et al., 2021) and bimolecular reactions of stabilised Criegee intermediates (SCI), with e.g. organic acids and peroxy radicals (e.g. Kristensen et al., 2014; Sakamoto et al., 2013; Zhao et al., 2015; Mackenzie-Rae et al., 2018), have been implicated in secondary organic aerosol formation. SCI can also act as an oxidant: this has been studied particularly for the reaction with SO2 (e.g. Welz et al., 2012; Mauldin et al., 2012; Caravan et al., 2020), which can lead to sulfate aerosol production and hence impact radiative forcing and climate (Pierce et al., 2013; Percival et al., 2013). However, both the SO2 and organic acid reactions, while important locally, are likely only of minor importance to global budgets of sulfate aerosol and organic acids (Welz et al., 2014; Newland et al., 2018). The dominant removal processes for most SCI in the troposphere are reaction with water vapour or unimolecular reaction (Vereecken et al., 2017). However, for certain structures, these reactions are sufficiently slow for bimolecular reactions with other trace gases to become important.
Understanding of the complex nature of the chemistry of Criegee intermediates has progressed rapidly in recent years, particularly with regard to the mechanisms and rates of decomposition of CI (i.e. SCI and chemically excited CI – CI∗) and the bimolecular reaction rates of SCI. This has been facilitated by direct experimental measurements of CI kinetics, generating CI through photolysis of di-iodo precursors (e.g. Welz et al., 2012; Chhantyal-Pun et al., 2020, and references therein), indirect measurements of CI kinetics during alkene ozonolysis experiments (e.g. Berndt et al., 2014a, b, 2015; Newland et al., 2015) and extensive theoretical studies (e.g. Vereecken et al., 2017, and references therein).
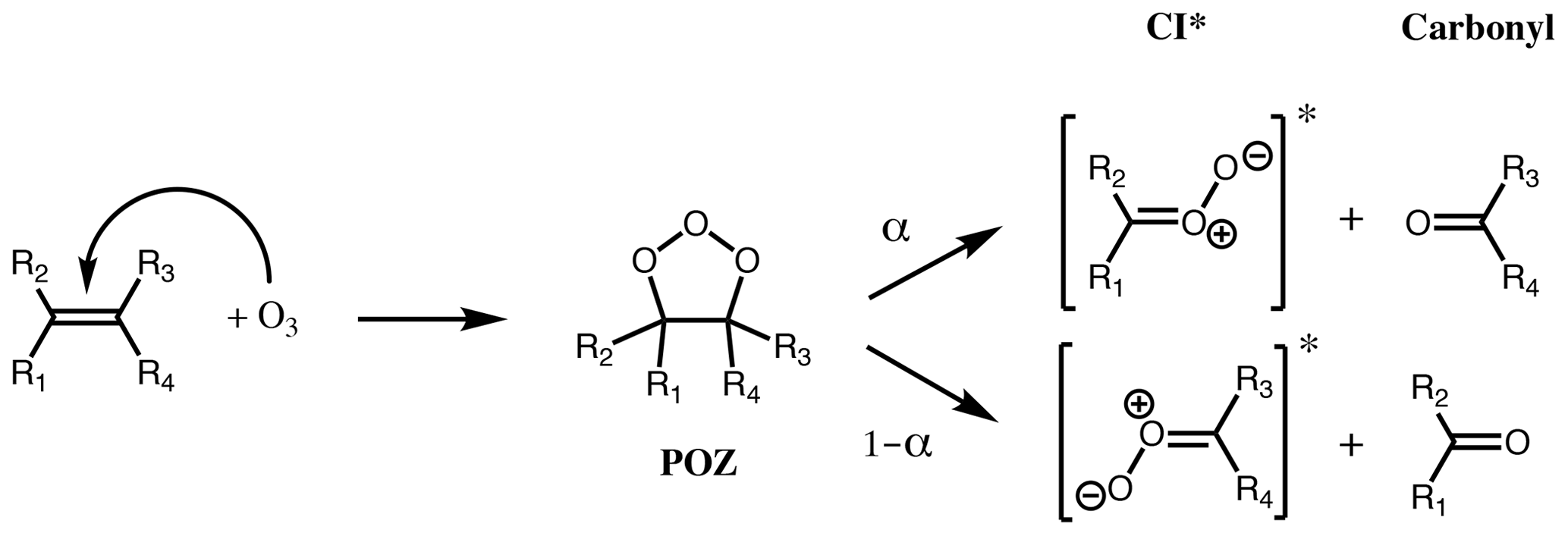
Figure 1First step of alkene ozonolysis. A primary ozonide (POZ) is formed which rapidly decomposes to yield a pair of chemically activated Criegee intermediates and carbonyl products.
The reaction of ozone with alkenes proceeds by a concerted addition to the C=C double bond, forming a short-lived primary ozonide (POZ). Typically, the POZ fragments into two pairs of carbonyls and Criegee intermediates (CI) (Fig. 1); for small- to medium-sized alkenes (C≤10) this POZ is vibrationally excited, decomposing promptly, while for large alkenes (e.g. C≥15, sesquiterpenes), theoretical studies suggest that the POZ can be collisionally stabilised prior to decomposition (Chuong et al., 2004; Nguyen et al., 2009a). Theoretical work also indicates that a small fraction of the POZ can rearrange to a carbonyl hydroperoxide when vinylic H atoms are present (Pfeifle et al., 2018); this mechanism is discussed separately below. It has also been suggested that different pathways may play a more significant role for a small number of systems, e.g. cyclohexadienes (Pinelo et al., 2013).
Criegee intermediates are generally zwitterionic in nature, as shown in Fig. 1, but the moiety is denoted simply as a > COO structure below (not to be confused with alkylperoxy radicals, ROO•). CI can be formed with the terminal oxygen of the carbonyl oxide moiety in either an E (anti) or Z (syn) configuration relative to a given substituent group. The two conformers are not in rapid equilibrium, with quantum calculations showing that the energy barrier to rotational interconversion for CH3CHOO is about 120 kJ mol−1 (Johnson and Marston, 2008, and references therein). This was confirmed by Vereecken et al. (2017), who calculated barriers exceeding 120 kJ mol−1 for saturated CI conformers. Isomeric CI conformers have been shown to have different unimolecular reaction rates (e.g. Vereecken et al., 2017), follow different unimolecular pathways (Herron and Huie, 1977; Niki et al., 1987; Martinez and Herron, 1987; Kidwell et al., 2016), and have very different reaction rates with water (e.g. Taatjes et al., 2013; Sheps et al., 2014; Huang et al., 2015). Therefore, these conformers must necessarily be considered separate species, irreversibly partitioned according to their nascent ratios, to accurately represent the effects of alkene ozonolysis on atmospheric composition.
Structure activity relationships (SARs) are commonly used to design the protocols needed to develop automated mechanism generation tools (Vereecken et al., 2018). This paper forms part of a series of articles devoted to the development of SARs for mechanism generation (Jenkin et al., 2018a, b, 2019, 2020). Updated SAR methods for the initial reactions of O3 with unsaturated organic compounds are presented in a companion paper (Jenkin et al., 2020), while in this work, a protocol is presented for the subsequent chemistry occurring following the initial O3 addition. This protocol details the yields of carbonyls and Criegee intermediates from the alkene + O3 reaction and the subsequent fate of the Criegee intermediates and accounts for the minor pathway by carbonyl-hydroperoxide radical formation. The protocol is based on available experimental data and theoretical data combined. For areas in which limited data exist, the protocol is set up to be easily updated as new experimental or theoretical results become available. These areas are highlighted in the paper and are recommended areas of further research. The protocol is currently being used to guide development of alkene ozonolysis chemistry in the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere, GECKO-A (Aumont et al., 2005), and the Master Chemical Mechanism, MCM (Jenkin et al., 1997, 2015; Saunders et al., 2003). It is noted that the protocol does not currently consider aromatic species that have been shown to react with ozone, such as catechols, for which the mechanism may be different to the Criegee mechanism described here.
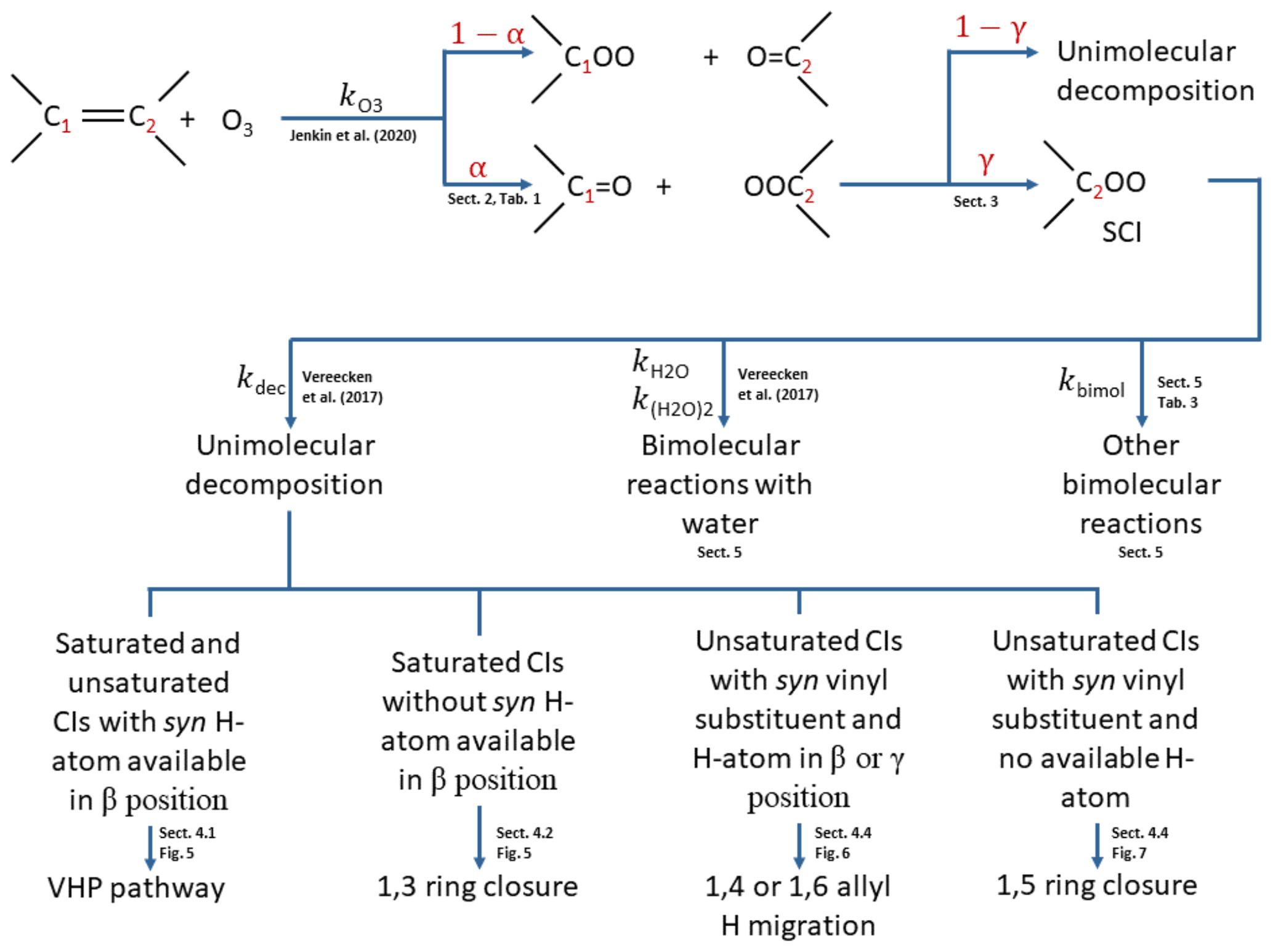
Figure 2Flow diagram for implementation of the protocol. α: branching ratios in POZ decomposition; γ: fraction of CI stabilised. > COO denotes the Criegee intermediate formed.
The methodology for applying the protocol described in this work is summarised in Fig. 2. The initial addition of ozone to the double bond follows the protocol described in the companion paper (Jenkin et al., 2020). The POZ formed from this protocol then decomposes according to the rules determined in Sect. 2 to give the primary carbonyl and the CI yields (α) and possibly a minor fraction of carbonyl hydroperoxide. A fraction (γ) of the CI is then stabilised (Sect. 3). Both the stabilised and chemically activated CI then follow the relevant set of rules from Vereecken et al. (2017) to ascribe them unimolecular decomposition mechanisms (and hence products) and rates (Sect. 4) and bimolecular reaction rates with water vapour (Sect. 5). Finally, bimolecular reaction rates with other atmospherically important species are assigned as a function of the SCI structure (Sect. 5).
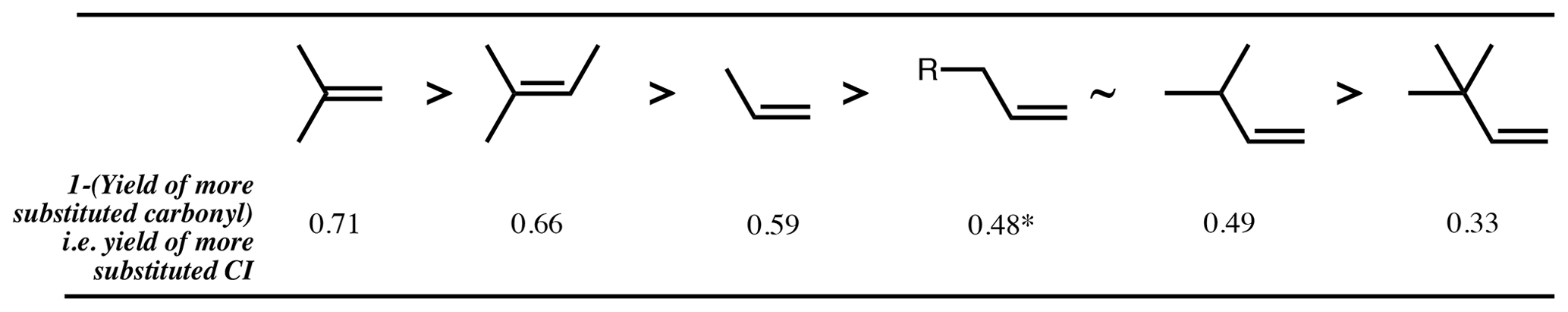
Figure 3Decreasing order of preference, from left to right, of more substituted CI formation from ozonolysis of example alkyl-substituted alkenes. Values are 1 (mean of measured yields of carbonyls) (Spreadsheet S1). ∗: mean measured yield of propanal (i.e. 1 – more substituted CI) formation from 1-butene is 0.35, but for all other 1-alkenes the yield of the larger primary carbonyl product ranges from 0.45 to 0.50.
2.1 Alkenes with aliphatic substituents
The fragmentation of the POZ has previously been parameterised based on the branching pattern around the double bond of the parent alkene (Jenkin et al., 1997; Rickard et al., 1999). Generally, it can be said that there is a preference for formation of the more substituted CI; e.g. the ozonolysis of 2-methyl propene yields ∼ 0.7 (CH3)2COO and ∼ 0.3 CH2OO (Rickard et al., 1999). However, consideration of just the immediate substituents of the double bond breaks down for more complex structures and for oxygenated substituents. There is clearly also an effect of substitution around the carbon adjacent to the double bond (the α-carbon atom). For instance, when there is a t-butyl group attached to the double bond, a strong preference is seen for formation of the opposing CI, as observed for yields of trimethylacetaldehyde from 3,3-dimethyl-1-butene (0.67) and trans-2,2-dimethyl-3-hexene (0.84) (Grosjean and Grosjean, 1997a). Using data from Grosjean and Grosjean (1997a), various homologous series of alkenes can be considered, such as the series with increasing methyl substitution on the α-carbon. For the 1-alkene series (Fig. 3), yields of the larger carbonyl of 0.35, 0.51 and 0.67 are determined for 1-butene, 3-methyl-1-butene and 3,3-dimethyl-1-butene, respectively.
Such relationships have been observed and discussed previously by Grosjean and Grosjean (1997a) in terms of (i) steric hindrance potentially weakening the O-O bond in the POZ on the side of the bulky substituent and (ii) the inductive effect of adjacent alkyl groups strengthening the O-O bonds in the POZ (Grosjean and Grosjean, 1997a). Earlier work considering POZ fragmentation in the aqueous phase (Fliszár and Renard, 1970; Fliszár and Granger, 1970; Fliszár et al., 1971) described similar relationships to those observed in the gas phase (i.e. that shown in Fig. 3), except in the case of terminal alkenes, for which the reverse trend was observed. In these studies, the observed trends are discussed in terms of stabilisation of the positive charge on the carbon in the POZ through (i) ”hyperconjugative stabilisation” in the transition state and (ii) the inductive effect during the POZ cleavage, with steric effects discounted as being unimportant in determining the POZ fragmentation pattern. Finally, Vereecken et al. (2017, Table S16 in their Supplement) analysed the stability of CI in terms of group additivity factors, showing that alkyl-substituted CI are more stable than H-substituted CI, but where the stability of the CI is inversely proportional to the branching on the β-carbon atom.
These works can be summarised by saying that it appears that a substituent with a partial negative charge, such as a methyl group, can stabilise the positive charge on the adjacent carbon in the POZ. This leads to a greater yield of the CI containing the more stabilising substituents. On the other hand, a substituent that leads to a partial positive charge on the α-carbon leads to a lower yield of that CI.
2.2 Oxygenated alkenes
Following the rationale discussed above, oxygenated substituents on the α-carbon might be expected to strongly influence the primary ozonide fragmentation pattern. The number of product yield studies on the ozonolysis of most classes of unsaturated oxygenates is rather limited. As discussed below, some oxygenated substituents appear to destabilise the positive charge on the carbon in the POZ (i.e. disadvantaging POZ fragmentation towards the production of the CI on the oxygenated side), particularly carbonyl groups, while others such as acrylate esters and carboxylic acids may stabilise the CI, favouring its formation. However, data are very limited and often ambiguous for most of the oxygenated classes. This is partly due to challenges in measuring products containing multiple oxygenated groups, partly because some of these classes are likely to be present in negligible amounts in the atmosphere and, for some, because ozonolysis will be a negligible atmospheric sink compared to e.g. reaction with OH or photolysis. The available data are provided in Spreadsheet S1 in the Supplement.
2.2.1 Enones/enals
Primary carbonyl yields have been reported for two α-β terminally unsaturated ketones (H2C=CHC(O)R). For methyl vinyl ketone (MVK), Grosjean et al. (1993a) and Ren et al. (2017) determined a strong preference for formation of the ketone-substituted product methyl glyoxal (0.87 and 0.71 ± 0.06 (with no OH scavenger), respectively). For ethyl vinyl ketone, primary carbonyl yields for formaldehyde (HCHO) and 2-oxobutanal have been determined to be 0.55 and 0.44 (Grosjean et al., 1996) and 0.37 and 0.49 (Kalalian et al., 2020), respectively, displaying no clear preference for either fragmentation pathway. For α-β unsaturated ketones (R1CH=CHC(O)R2), Grosjean and Grosjean (1999) measured the primary carbonyl yields from ozonolysis of 4-hexen-3-one to be acetaldehyde (CH3CHO) 0.51 ± 0.01 and 2-oxobutanal (CH3CH2C(O)CHO) 0.56 ± 0.02, while Wang et al. (2015) measured the primary carbonyl yields from ozonolysis of 3-methyl-3-buten-2-one (CH2=CR1C(O)R2) to be diacetyl (CH3COCOCH3) 0.30 ± 0.03 and HCHO 0.44 ± 0.05 and from 3-methyl-3-penten-2-one (R1CH=CR2C(O)R3) diacetyl 0.39 ± 0.04 and CH3CHO 0.61 ± 0.07. For ozonolysis of 2-enals, yields have been reported for crotonaldehyde (2-butenal) (CH3CHO 0.42, glyoxal 0.47) (Grosjean and Grosjean, 1997b) and trans-2-hexenal (butanal 0.53, glyoxal 0.56) (Grosjean et al., 1996). For the atmospherically important isoprene oxidation product methacrolein (2-methyl-prop-2-enal, MACR), Grosjean et al. (1993a) measured yields of methyl glyoxal of 0.58 ± 0.06 and HCHO of 0.12 ± 0.03. For 2-ethyl acrolein, the ethyl glyoxal yield has been measured to be 0.14 by Grosjean et al. (1994) and 0.49 ± 0.03 by O'Dwyer et al. (2010).
To summarise, the presence of a carbonyl group on a double bond appears to favour formation of the opposing CI. However, this effect is neutralised to an extent by the presence of an alkyl substituent on the same side of the double bond, e.g. in the case of 3-methyl-3-buten-2-one, methacrolein and 2-ethyl acrolein. There remain large uncertainties in the trends in these classes (it is noted that in some cases the sum of the measured primary carbonyl yields is well below one). They clearly warrant further study, owing to the significance of these classes of compounds in atmospheric chemistry (e.g. MACR and MVK from isoprene oxidation; Wennberg et al., 2018).
2.2.2 Enols/enol ethers
There has been very little experimental work on the atmospheric chemistry of enols due to difficulties in synthesis, storage, and measurement of these compounds. However, two recent theoretical studies examined the ozonolysis of enols. The first (Lei et al., 2020) on the simplest enol, vinyl alcohol (ethenol), suggested that formation of CH2OO + HCOOH is strongly favoured (∼ 78 %). The second (Wang et al., 2020), on the complex ketene–enol species 4-hydroxy-1,3-butadien-1-one, also suggests that formation of HCOOH and the corresponding CI is strongly favoured (84 %). By contrast, there have been several experimental studies on the product yields of the reactions of enol ethers (R1-O-CR2=CR3R4) with ozone. Most studies (Thiault et al., 2002; Klotz et al., 2004; Barnes et al., 2005; Zhou et al., 2006; Zhou, 2007; Al Mulla et al., 2010) have determined that the dominant POZ decomposition channel yields the formate (R1-O-C(O)R2) and the corresponding CI (R3R4COO), with measured yields of the formate ranging from 55 % to 89 % (see Spreadsheet S1). An exception to these studies is the work of Grosjean and Grosjean (1997b, 1999), which tended to find similar yields of the two primary carbonyl products.
2.2.3 Esters/acids
The primary carbonyl products of ozonolysis of the acrylate esters methyl acrylate, ethyl acrylate and methyl methacrylate were studied by Bernard et al. (2010). Grosjean and Grosjean (1997b) also studied methyl acrylate. There is no clear evidence of a preferential route for POZ fragmentation in these studies (see Spreadsheet S1). The primary carbonyl yields from vinyl acetate ozonolysis were measured to be 0.30 ± 0.04 and 0.70 ± 0.08 for HCHO and CH3C(O)OC(O)H, respectively, by Al Mulla et al. (2010) and 0.20 ± 0.06 and 0.97 ± 0.08 by Picquet-Varrault et al. (2010). These studies suggest a preference for formation of CH2OO and the anhydride. There are only two compounds reported for ozonolysis of α-β unsaturated acids: acrylic and methacrylic acid. For acrylic acid ozonolysis in the presence of formic acid as an SCI scavenger, Al Mulla et al. (2010) measured yields of 1.48 ± 0.2 and < 0.1 for HCHO and HC(O)C(O)OH, respectively, while in the absence of formic acid that group measured a yield of HCHO of 0.95 (Viero, 2008). For methacrylic acid, Al Mulla et al. (2010) measured yields of 0.77 ± 0.07 and 0.74 ± 0.10 for HCHO and CH3C(O)C(O)OH, respectively. It is difficult to rationalise these results: the acrylic acid experiments suggest a preference for formation of the CI with the acid moiety, but the methacrylic acid experiments suggest that the presence of a methyl group on the same side of the double bond as the acid reduces this preference, in contrast to most other systems where methyl substitution increases the yield of that CI. This is a recommended area for further study.
2.2.4 Alcohols
There are significant differences between measured primary carbonyl yields of α,β-unsaturated acyclic alcohols between studies by Grosjean and Grosjean (1997b), Le Person et al. (2009), O'Dwyer et al. (2010) and Kalalian et al. (2020). This is likely owing to different experimental set-ups between groups and the difficulty in quantitatively measuring compounds with multiple oxygenated substituents. Overall the data in Spreadsheet S1 suggest that the presence of a hydroxyl group in place of hydrogen on the α-carbon may lead to a slight preference for CI production on the other side of the double bond to the hydroxyl group.
2.3 Conjugated alkenes
The ozonolysis of conjugated alkenes leads to POZ with a vinyl substituent on the α-carbon. For non-symmetrical conjugated alkenes, the measurement of primary carbonyl yields can only be used to determine the POZ fragmentation if the relative contribution of reaction at each double bond to the overall reaction rate is known. For ozonolysis of the atmospherically important biogenic alkene isoprene, the primary carbonyl yields recommended by the IUPAC (Atkinson et al., 2006; iupac-aeris.ipsl.fr, last accessed 6 December 2021) are MVK 0.17, MACR 0.41 and HCHO 0.42. Based on reported product yields, the contribution of reaction to each double bond to the overall rate has been estimated to be 0.6 for the terminal double bond and 0.4 for the substituted double bond (Nguyen et al., 2016; Jenkin et al., 2020). However, to the authors' knowledge there has been no direct measurement of the reaction at each double bond, and this represents a significant uncertainty in one of the most important atmospheric ozonolysis systems. Based on this assumption and the recommended yields of MVK and MACR, the formation of MACR+CH2OO is favoured over methacrolein oxide (MACRO) + HCHO, and there is a slight preference for formation of methyl vinyl ketone oxide (MVKO) + HCHO compared to MVK+CH2OO. The MACR channel would suggest that the vinyl substituent is less favourable in the POZ decomposition compared to hydrogen. The methyl group present in MVKO stabilises the CI (see Sect. 2.1), leading to a preference for this channel. For symmetrical alkenes, the primary carbonyl yields should be directly representative of the POZ fragmentation. For 1,3-butadiene, an acrolein yield of 51 %–52 % has been measured (Niki et al., 1983; Kramp and Paulson, 2000), suggesting little preference for either POZ decomposition pathway, in contrast to the analogous MACR channel in isoprene. Lewin et al. (2001) reported complementary carbonyl yields from ozonolysis at the internal bond of (E)- and (Z)-penta-1,3-diene and 5-methylhexa-1,3-diene, which all showed a preference for formation of the unsaturated carbonyl (i.e. the saturated CI), suggesting that the vinyl group is less favourable than a methyl or isopropyl group, in agreement with the observations from isoprene. Note that, once the unsaturated CI is formed, the vinyl group can conjugate with the carbonyl oxide π system, leading to additional stabilisation such that vinyl CI are more stable than H-substituted CI (Vereecken et al., 2017); this is however a product-specific effect that is not available yet in the POZ decomposition.
2.4 Endocyclic alkenes
Decomposition of the POZ formed in the ozonolysis of endocyclic alkenes leads to a molecule containing both the carbonyl oxide and carbonyl moieties. Thus for non-substituted cycloalkenes (e.g. cyclopentene) there is only one possible CI species that can be formed (which can be in either the E or Z configuration). This means that there are no stable primary carbonyls formed, and so the relative contributions of the POZ decomposition pathways cannot be inferred from measured primary carbonyl yields as they can for aliphatic compounds. Even a simple endocyclic system such as cyclohexene gives a complex range of gas-phase (Aschmann et al., 2003; Hansel et al., 2018) and aerosol-phase (Kalberer et al., 2000; Ziemann, 2002) products, which can be attributed to decomposition of both the E and Z forms of hexanal carbonyl oxide. However, the measured OH yields can be used to give an estimate of the amount of CI decomposing via the vinylhydroperoxide (VHP) pathway (see Sect. 4.1). It is noted here that it has been proposed that alternative unimolecular pathways (that do not yield OH) are available to the CI formed from endocyclic alkenes (Chuong et al., 2004; Nguyen et al., 2009a; Long et al., 2019) but that these are only dominant for stabilised CI. Since the stabilised CI yield is low for endocyclic alkenes, at least up to C10 (monoterpenes) (Chuong et al., 2004), measured OH yields should give a fair representation of the relative amount of CI decomposing via the VHP pathway. For non-substituted cycloalkenes, OH yields have been compiled by Calvert et al. (2000) covering cyclo-pentene, -hexene, -heptene, -octene and -decene from a number of research groups (Spreadsheet S2). There is some spread in the data but no clear evidence favouring formation of (E) or (Z) CI; i.e. OH yields tend to centre around ∼ 0.5. For substituted cycloalkenes, Atkinson et al. (1995) measured an OH yield of 0.90 for 1-methyl-1-cyclohexene, suggesting either that the dominant CI formed is the di-substituted CI (which will then undergo decomposition via the VHP pathway to yield OH) or that the mono-substituted CI is formed predominantly as the syn conformer. The former must be considered more likely based on the observed trends in aliphatic alkenes for favouring formation of the more substituted CI and that there appears to be little preference for formation of syn-/anti-CI from non-substituted endocyclic alkenes. 1-methyl-1-cyclohexene is particularly important from the point of view of atmospheric chemistry as an analogue for the abundant biogenic monoterpenes α-pinene and limonene. OH yields from α-pinene and limonene ozonolysis have been measured by a number of groups and are also generally high (0.64–0.91) (Cox et al., 2020), similar to 1-methyl-1-cyclohexene.
2.5 Exocyclic alkenes
For exocyclic alkenes in which the double bond is attached to the ring, e.g. β-pinene, the data suggest that POZ fragmentation strongly favours formation of the ring-containing CI. For the monoterpene β-pinene, the mean measured yield of the C9 carbonyl, nopinone, is 0.21 (Grosjean et al., 1993b; Hakola et al., 1994; Rickard et al., 1999; Yu et al., 1999; Winterhalter et al., 2000; Hasson et al., 2001b; Lee et al., 2006; Ma and Marston, 2008), with theoretical work (Nguyen et al., 2009b) suggesting that some of this may be secondary and that the primary yield could be even lower. The other two compounds with a terminal double bond attached to the ring for which there are data are camphene (0.36 yield of C9 carbonyl; Hakola et al., 1994; Hasson et al., 2001b) and methylene cyclohexane (0.19 yield of C6 carbonyl; Hasson et al., 2001b). For the monoterpene sabinene, which has a terminal double bond attached to a C5 and C7 ring, the mean measured yield of the C9 carbonyl, sabinaketone, is 0.44. This is considerably higher than from those compounds where the double bond is on a C6 ring, probably demonstrating the impact of ring strain on the POZ fragmentation. The monoterpene terpinolene has a disubstituted double bond attached to a six-membered ring. Reported yields of the ring-containing carbonyl (0.40 ± 0.06, Hakola et al., 1994; 0.40 ± 0.08, Reissell et al., 1999; 0.45, Ma and Marston, 2009) suggest yields of the ring-containing CI of 0.60 and 0.55, respectively; this assumes 100 % reaction at the exocyclic double bond, with Hakola et al. (1994) measuring a yield of ≤ 2 % of the dicarbonyl expected as a product (though by no means the only one) from reaction at the endocyclic double bond. These CI yields are lower than for the exocyclic alkenes with terminal double bonds but are still considerably higher than most compounds which have a dimethyl substitution on the double bond, for which acetone yields tend to be ∼ 0.3. The presence of a ring clearly has a different effect than simply having two alkyl groups attached to the double bond, leading to much higher yields of the ring-containing CI.
For alkenes with a vinyl group attached to a ring, there are data only for vinyl cyclohexane and its aromatic analogue styrene. These have similar yields for the ring-containing carbonyl of 0.62 and 0.64, respectively (Grosjean and Grosjean, 1997a). There are no data for alkenes with double bonds more distant from a ring.
2.6 Yields of CI stereo-conformers
The formation of syn/anti conformers of CI in alkene ozonolysis was first discussed by Bauld et al. (1968) to explain the observed cis/trans yields of the secondary ozonide formed from ozonolysis in the aqueous phase. Their observations suggested that ozonolysis of cis-alkenes will predominantly form anti-CI, while for trans-alkenes the predominance was less clear and appeared to be dependent on alkene structure. In the gas phase, but-2-ene is the most studied system. Various experimental work has observed higher yields of OH from trans-but-2-ene compared to cis-but-2-ene (see Spreadsheet S3). Assuming that only (Z)-CI decomposition yields OH (see Sect. 4.1), this implies a higher nascent (Z):(E)-CH3CHOO ratio from decomposition of the POZ formed in trans-but-2-ene ozonolysis. Orzechowska and Paulson (2002) measured a ratio of 1.62 for the OH yields from trans-/cis-but-2-ene. They observed a similar relationship for trans-/cis-pent-2-ene and trans-/cis-hex-3-ene, with OH yield ratios determined as 1.80 and 1.51, respectively. Assuming that OH comes exclusively from (Z)-CH3CHOO implies a (Z):(E)-RCHOO ratio of 0.60:0.40–0.64:0.36 for these three systems. Kroll et al. (2002) determined a similar OH yield ratio for trans-/cis-hex-3-ene, but using isotopically labelled hydrogen atoms demonstrated that a fraction of this OH was not coming from the (Z)-CI. From their OH yield measurements, they inferred (Z):(E)-C2H5CHOO ratios of 50:50 for trans-3-hexene and 20:80 for cis-3-hexene. Campos-Pineda and Zhang (2018) reported direct measurements of the vinoxy radical formed in decomposition of syn-CH3CHOO from cis- and trans-but-2-ene ozonolysis, inferring a yield of syn-CH3CHOO of ∼ 0.5 from trans-but-2-ene and ∼ 0.3 from cis-but-2-ene, broadly in line with estimations from measured OH yields.
Early theoretical calculations considering the gas phase (Cremer, 1981a, b) suggested that (Z)-RCHOO is likely to be formed in greater yield for small alkenes but that (E)-RCHOO becomes more favoured in the ozonolysis of large alkenes. Calculations by Rathman et al. (1999) suggested that (Z)-CH3CHOO should be favoured in trans-but-2-ene ozonolysis but that conversely (E)-CH3CHOO would be favoured in cis-but-2-ene ozonolysis. Recent theoretical work (Watson, 2021) looking at POZ fragmentation for a series of disubstituted 2-alkenes (CH3CH=CHR) suggests formation of (E)-RCHOO will be strongly favoured in the ozonolysis of cis-alkenes (87 % for cis-but-2-ene, increasing to 93 % for cis-2-hexene), while there is a roughly equal split from ozonolysis of trans-alkenes. This is in qualitative agreement with the experimental work discussed above but suggests a stronger preference than observed in the direct measurements of the vinoxy radical by Campos-Pineda and Zhang (2018). For trisubstituted alkenes, Watson (2021) finds a strong preference for formation of (E)-RCHOO on the mono-substituted side of the double bond. For the C4-CI formed in isoprene ozonolysis, theoretical calculations have determined a relative split of 50:50 for the two conformers of MVKO (Kuwata et al., 2005) and 20:80 for syn-MACRO : anti-MACRO (Kuwata and Valin, 2008). This is in qualitative agreement with the observed low OH yield (0.08–0.13) from 1,3-butadiene (Atkinson and Aschmann, 1993; Kramp and Paulson, 2000) if it is assumed that decomposition of syn-MACRO will have a high OH yield, whereas anti-MACRO will not yield OH. To the authors' knowledge there is no other information on the relative yields of syn-/anti-R1R2COO (where R1 ≠R2).

2.7 POZ ring opening to a biradical
In addition to direct CI + carbonyl formation from the POZ, the possibility exists of ring opening of the POZ to a singlet alkoxy–peroxy biradical (> C(O•)-C(OO•) <) (O'Neal and Blumstein, 1973; Olzmann et al., 1997; Anglada et al., 1999; Fenske et al., 2000; Nguyen et al., 2015; Pfeifle et al., 2018) (Fig. 4). In addition to re-closing the ring to the POZ or decomposing to the CI + carbonyl, this alkoxy–peroxy biradical can migrate an H atom from the alkoxy-bearing carbon, forming a carbonyl hydroperoxide ( <); this pathway is only possible if the alkene has a vinylic H atom. The carbonyl hydroperoxide formed has a high energy content, over 400 kJ mol−1, and can eliminate an OH radical, forming a α-carbonyl–alkoxy radical that rapidly decomposes to an acyl radical and a carbonyl. This pathway has been invoked in theoretical studies as the main source of OH in the ozonolysis of ethene (in which OH cannot be formed via a VHP) (Nguyen et al., 2015; Pfeifle et al., 2018) and is expected to contribute somewhat to OH formation in other alkenes, though this has not yet been investigated experimentally or theoretically. Alternative proposed sources of OH in ethene ozonolysis all involve the CH2OO Criegee intermediate. However, theory has shown that direct OH formation from CH2OO by a 1,3-H migration involves too high a barrier (e.g. Nguyen et al., 2015; Pfeifle et al., 2018), while OH elimination from the hot formic acid formed in the 1,3 ring closure (see Sect. 4.2) is not competitive against formation of H2O + CO and H2 + CO2, as also borne out by HCOOH pyrolysis experiments (Chang et al., 2007; Vichietti et al., 2017). The carbonyl hydroperoxide route thus resolves an apparent discrepancy between ethene ozonolysis experiments, which observe significant OH yields, and experiments (Stone et al., 2018) and theoretical work (Nguyen et al., 2015; Pfeifle et al., 2018), which indicate very little OH formation from CH2OO. Pfeifle et al. (2018) calculated a yield of 12.3 % for the carbonyl hydroperoxide in ethene ozonolysis, while Nguyen et al. (2015) obtained 13 %, both at the low end of the current IUPAC-recommended OH yield (0.17 ± 0.05) for the reaction (Cox et al., 2020).
2.8 Protocol rules for POZ fragmentation
2.8.1 POZ fragmentation
A group contribution approach was designed to estimate POZ fragmentation yields. The approach assumes that the branching ratio for the two possible fragmentations of the POZ depends on the substituents of the R1a(R1b)C=C(R2b)R2a parent alkene. The general form of the relationship is given by
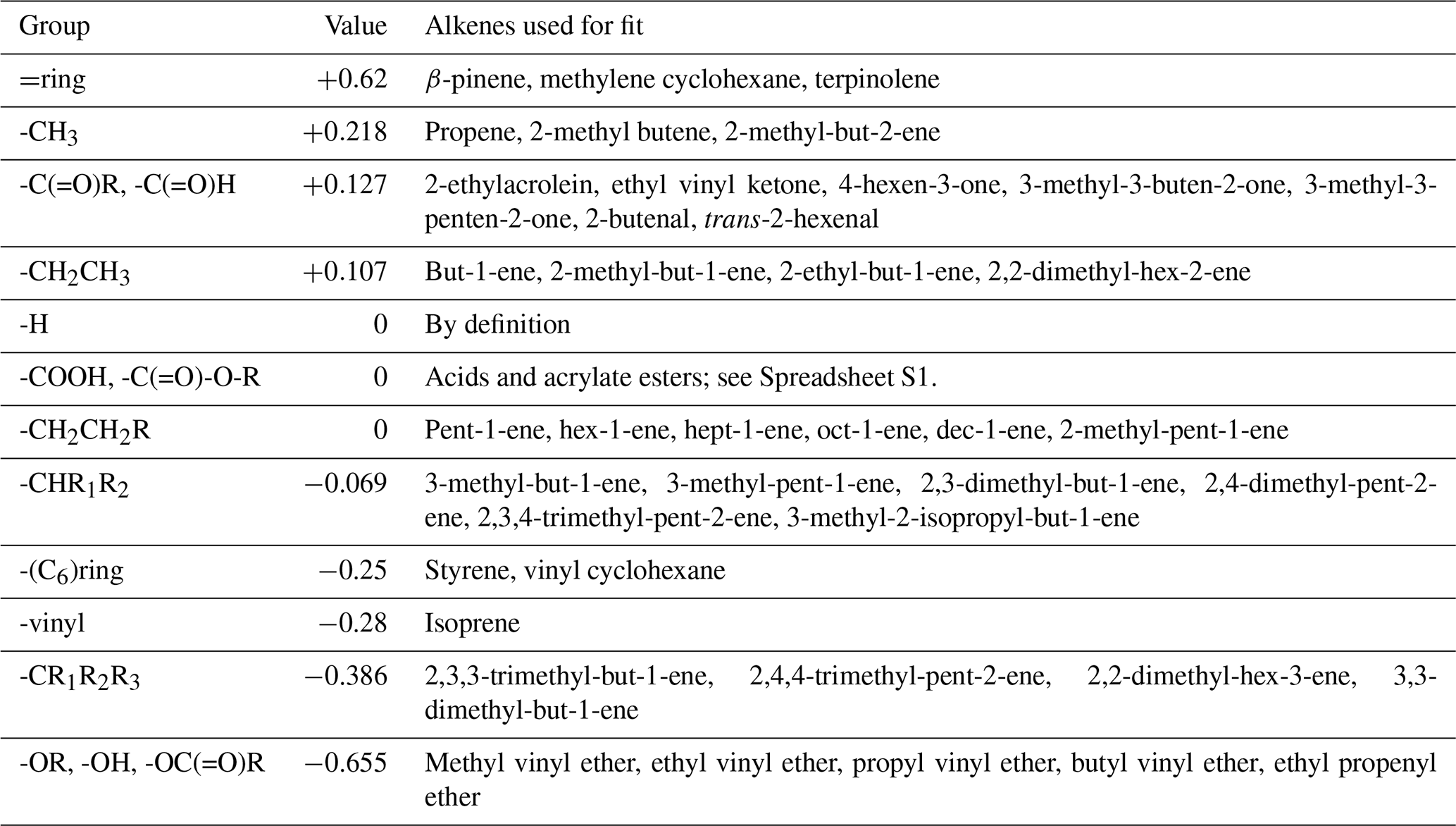
where YCIi is the CI production yield on the ith carbon and FR are the contributions for the four substituents on the C=C bond. The set of FR values is developed based on the observed primary carbonyl yields (Sect. S1 and Spreadsheet S1) and is based on a least squares fit to a relevant dataset of alkenes for each substituent (Figs. S1–S5 in the Supplement).
For a vinyl group, F is constrained to fit the IUPAC-recommended yields of MVK and MACR from isoprene ozonolysis, assuming that ozone reacts 60 % at the terminal double bond and 40 % at the substituted double bond (Nguyen et al., 2016; Jenkin et al., 2020). The presence of a carbonyl group adjacent to the double bond appears to strongly favour formation of the opposing CI in the case of MVK (i.e. -C(=O)CH3). However, this is not the case for other alkenes with the structure -C(=O)R in the database, for which there appears to be no clear preference for formation of either CI, with a fit to the data yielding a slightly positive F value of 0.127. The strongest negative effect (i.e. most strongly favouring formation of the carbonyl containing the functional group) observed in the database is for enol ethers (-OR), giving an F value of −0.655. This is assumed to also be the same case for enols (-OH) based on the theoretical calculations of Lei et al. (2020) and Wang et al. (2020) and for vinyl esters (-OC(=O)R), based on the observed values for vinyl acetate. By contrast, an acrylate ester (-C(=O)-OR) substituent adjacent to the double bond does not appear to have a strong effect on fragmentation, and F=0 is used. Similarly, the trend from the two unsaturated acids reported is unclear, and F=0 is also used here. An OH group on the alpha carbon appears to slightly decrease YCI compared to an H atom, but the data are currently too limited to recommend a group additivity value, so the OH group is treated as an H atom, i.e. . More distant oxygenated groups are not considered. The available data for exocyclic alkenes with the double bond attached to the ring are not able to take into account the effect of multiple rings, with F=ring being determined from only exocyclic alkenes with C6 rings (β-pinene, methylene cyclohexane and terpinolene). For rings with a vinyl group attached, is determined only from C6 rings, i.e. styrene and vinylcyclohexane. Endocyclic alkenes are assumed to follow the same fragmentation patterns as acyclic alkenes. For example, cyclohexene is considered to have the structure > CH2CH2CH=CHCH2CH2 <, 1-methyl cyclohexene > CH2CH2C(CH3)=CHCH2CH2 <, etc.
The group contribution value, F, is then used in Eq. (1) to determine the yield of CI1 (defined as having substituents 1a and 1b) from the general structure R1a(R1b)C=C(R2b)R2a. Generally, the measurement of the larger primary carbonyl was used to determine the primary carbonyl and CI yields. This is because, in some cases, the smaller carbonyl can be formed as a decomposition product of the larger CI and hence is not a true primary carbonyl yield.
2.8.2 conformer yields
In light of the current paucity of experimental and/or theoretical information on the relative yields, an equal 0.5:0.5 yield is assigned as a default value for isomers for all asymmetrical CI. The following two exceptions are nevertheless considered. For acyclic cis-RCH=CHR parent alkenes, a relative yield of 0.7:0.3 is set for (E):(Z) CI. For conjugated structures, formation of > C=C(R)-CHOO is assumed to be in a ratio of 0.8:0.2, based on the work of Kuwata et al. (2005) and Kuwata and Valin (2008).
2.8.3 Carbonyl-hydroperoxide route
While there is little information available on the stepwise carbonyl-hydroperoxide POZ decomposition mechanism (CHP, Fig. 4), it is needed to account for the radical yields observed in the ozonolysis of ethene as discussed above. There is no reason to assume it will not occur more generally for any alkenes with vinylic H atom(s), though perhaps with different fates of the intermediate biradical and/or carbonyl hydroperoxide (e.g. larger hydroperoxides could be more prone to collisional stabilisation and yield less prompt OH). Currently this channel is only included for the ethene–ozone reaction, for which it is assumed that 0.12 of the ethene–ozone reaction forms the biradical intermediate rather than the CI + carbonyl, using the contribution calculated for the carbonyl-hydroperoxide channel by Pfeifle et al. (2018). When more general data become available, assuming the channel is active for other systems, the protocol will be updated. The general structure of such a scheme might be that the POZ is assumed to break either of the O-O bonds with equal probability, forming one of two possible biradicals. If there is an available vinyl α-hydrogen, it is assumed that the H shift to the peroxy radical occurs, forming the carbonyl hydroperoxide (R1R2C(OOH)C(=O)R3), followed by loss of OH and scission of the C-C bond to yield the stable product R1R2C=O and the radical R3C•=O. If there is no available α-hydrogen, the biradical is assumed to yield the CI and carbonyl, either by C−C fragmentation or recyclisation to the POZ.
3.1 Excited vs. stabilised CI
Following decomposition of the primary ozonide, CI are formed with a broad range of internal energies (e.g. Drozd et al., 2011). Consequently, it is often useful to consider the mean energy of a population of CI. Those generated with a high internal energy, allowing prompt chemical reactions, are called excited or chemically activated CI (CI∗). Those without enough internal energy to undergo prompt decomposition are considered to be “stabilised” CI (SCI). Additionally, CI∗ can be collisionally stabilised. This has been demonstrated by experimental work showing that SCI yields are pressure-dependent (Drozd et al., 2011; Hakala and Donahue, 2016, 2018). Note that this pressure dependence is moderate and across the range of relevant atmospheric pressures not of primary concern; we base our analysis on the available data near 1 atm.
3.2 SCI yield
The total SCI yield for a given alkene is the sum of the fraction of the nascent CI population that is formed stabilised plus the fraction of CI∗ that is collisionally stabilised. The fate of the CI∗ is a competition between prompt unimolecular decay and collisional stabilisation, with the CI∗ having a lifetime of the order of nanoseconds against either of these processes (e.g. Drozd et al., 2017; Stephenson and Lester, 2020). Most alkenes will form a number of different CI∗, each with different lifetimes against unimolecular decay and collisional stabilisation. The rate of collisional stabilisation of a given CI∗ is dependent on the frequency of collisions (and hence pressure) and the efficiency of energy loss to the bath gas. The rate of unimolecular decay of a given CI∗ depends on (i) the energy of the CI∗ when formed, (ii) the activation energy for the most facile decay process/the energy required for tunnelling, and (iii) the relative density of states of the reactants and transition state, i.e. the entropy of the reaction. The dominant unimolecular decay mechanism is dependent on the structure of the CI; these mechanisms are discussed in Sect. 5.
Larger CI∗ will tend to be stabilised to a greater extent due to a greater density of states distributing the excess internal energy over a greater number of modes and so reducing the rate of unimolecular decay (Drozd and Donahue, 2011; Stephenson and Lester, 2020). Hence, as the size of the CI increases relative to the carbonyl co-product formed in POZ decomposition, the fraction of the energy taken by the CI from the POZ will increase somewhat (assuming the energy has time to become equally distributed throughout the POZ), but typically the mean excess energy per degree of freedom of the nascent CI population decreases, and hence the fraction of CI∗ with enough energy to undergo unimolecular decay also decreases (Fenske et al., 2000; Newland et al., 2020). This will lead to greater stabilisation, i.e. higher SCI yields. Similarly, for a given CI size, carbonyl co-products of increasing size will take a larger fraction of the excess energy, leaving the CI∗ moiety with less energy and thus also leading to higher SCI yields (Newland et al., 2020). Conversely, for endocyclic alkenes, decomposition of the POZ produces a single molecule containing both the carbonyl and carbonyl oxide moieties. Such CI have a high initial energy, with no energy lost from the POZ decomposition to the carbonyl or to relative motion of the fragments, and thus require many collisions to be quenched (Vereecken and Francisco, 2012). Consequently, endocyclic alkenes with ≤ C7 have little stabilisation (Hatakeyama et al., 1984; Campos-Pineda and Zhang, 2018; Drozd and Donahue, 2011). For the endocyclic C10 monoterpenes α-pinene and limonene, total SCI yields have been measured to be 0.13–0.22 (Hatakeyama et al., 1984; Taipale et al., 2014; Sipilä et al., 2014; Newland et al., 2018) and 0.23–0.27 (Sipilä et al., 2014; Newland et al., 2018), respectively. For the C15 sesquiterpene β-caryophyllene, a total SCI yield (including from decomposition of the stabilised POZ) of 0.74 was calculated by Nguyen et al. (2009a), with a value of > 0.6 determined experimentally (Winterhalter et al., 2009).
Total SCI yields have been measured experimentally for many alkene–ozone systems. These are generally determined indirectly by performing ozonolysis experiments in the presence of an SCI scavenging species (e.g. H2O, SO2, hexafluoroacetone). Measurements of scavenger removal, or formation of products from the SCI + scavenger reaction, are used to determine the SCI yield. Yields measured in such a way must be considered to be lower limits since, under most experimental conditions, a significant fraction of the SCI may undergo unimolecular decomposition based on recently reported fast SCI decomposition rates (e.g. Newland et al., 2015, 2018; Vereecken et al., 2017). The choice of scavenger species is also important. In some older experimental studies, water was used as an SCI scavenger, with H2O2 (e.g. Hasson et al., 2001a) or hydroxymethyl hydroperoxide (HMHP, e.g. Hasson et al., 2001a; Neeb et al., 1997) being the detected reaction products. For mono-substituted (E)-SCI or for CH2OO, this may be a reasonable assumption, with ∼ 102–103 at [H2O] = 5 × 1017 cm−3 (e.g. Vereecken et al., 2017). However, for (Z)-SCI, ∼ 10−2–10−1; i.e. the majority of the SCI will not be scavenged by H2O.
3.3 Protocol rules for CI stabilisation
The relationship between stabilisation of the CI∗ and size of the carbonyl co-product has been studied for CH2OO and (CH3)2COO by Newland et al. (2020) (Fig. 5). For CH2OO this relationship might be expected to represent a minimum for CI∗ that primarily decay via the 1,3 ring closure pathway (i.e. anti-CI∗; see Sect. 4.2), since larger CI∗ will have a slower decay rate due to a greater density of states. Similarly, the trend for (CH3)2COO can be assumed to be close to a minimum for CI∗ that primarily undergo the 1,4 VHP decomposition pathway (see Sect. 4.1), with only syn-CH3CHOO likely to have a lower density of states (and therefore faster decomposition) (Stephenson and Lester, 2020). With no further data available, the stabilisation trend of CH2OO is used for CI∗ that decompose via 1,3 ring closure, while that of (CH3)2COO is used for CI∗ that decay via the 1,4 VHP pathway. For other pathways, such as the 1,5 ring closure to a dioxole (see Sect. 4.4), important in isoprene ozonolysis, no information is available. CI∗ with a vinyl group syn to the terminal oxygen of the carbonyl oxide are considered to be syn-CI for the purposes of calculating stabilisation in the protocol.
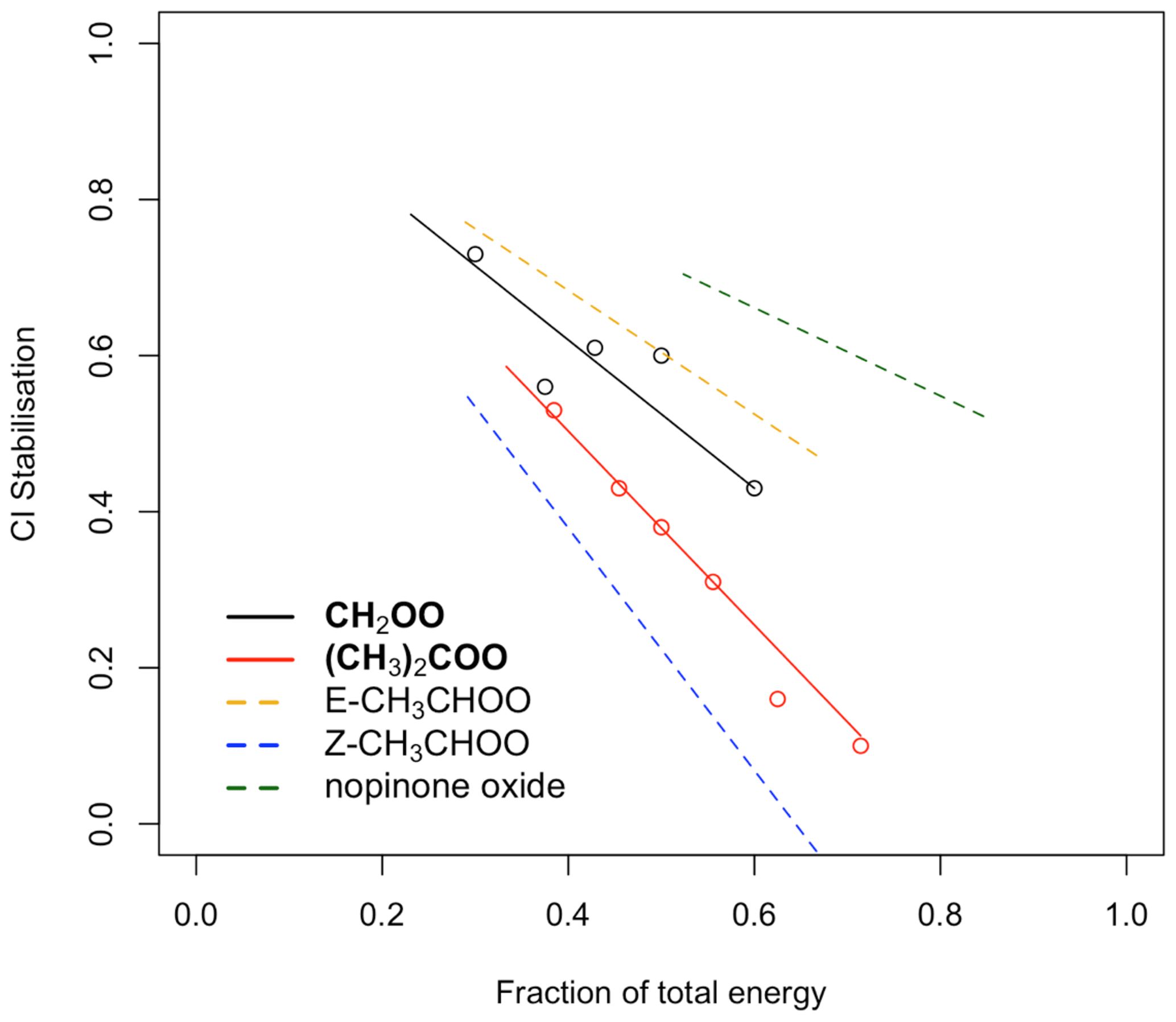
Figure 5Dependence of CI∗ stabilisation on the fraction of the total energy taken from the POZ. Black (CH2OO) and red ((CH3)2COO) points: measurements taken from Newland et al. (2020). Solid and dashed lines: fits calculated using Eq. (2).
An extension of Eq. (E7) in Newland et al. (2020) is used to estimate the CI stabilisation S:
where ACI is the total number of non-hydrogen atoms in the CI∗ and Atot is the total number of non-hydrogen atoms in the POZ. F13RC and FVHP are values determined for CH2OO and (CH3)2COO, based on the SCI yields for their symmetrical parent alkenes ethene and 2,3-dimethylbut-2-ene, respectively. For CH2OO this is 0.95 and for (CH3)2COO it is 1.24 (Newland et al., 2020). In this work, an additional term, zpath, is included to take into account the observed/predicted increased stabilisation of CI∗ with size. For CI∗ that decay via the 1,3 ring closure pathway, z13RC, is defined as , where is the total number of non-hydrogen atoms in CH2OO (i.e. 3), and x is an adjustable parameter. For CI∗ that decay via the 1,4 H shift, zVHP, is defined as , where = 5. In both terms, x=5, and it has been optimised to improve the fit between measured and calculated total SCI yields of larger alkenes (Newland et al., 2020).
Figure 5 shows the measured CI∗ stabilisation for CH2OO and (CH3)2COO as a function of the total energy taken from the POZ by the CI∗ from Newland et al. (2020). Fits to the measured data are calculated using Eq. (2). Also shown are the calculated stabilisation trends for (E)- and (Z)-CH3CHOO and nopinone oxide (the C9 CI∗ formed in β-pinene ozonolysis). Figure 5 shows that stabilisation of (E)-CI∗ is predicted to be considerably greater than for (Z)-CI∗ when formed with the same energy. For CH3CHOO it is noted that very little (0.11) stabilisation of (Z)-CH3CHOO∗ is predicted when produced from but-2-ene ozonolysis (fraction of total energy = = = 0.57), whereas a much greater stabilisation of (E)-CH3CHOO∗ is predicted. Using the -RCHOO yields given in Sect. 2.8.2 for cis- and trans-alkenes and the trends presented in Fig. 5, a total SCI yield of 0.33 for trans-but-2-ene and 0.42 for cis-but-2-ene is calculated, in good qualitative agreement with the relationship observed in Newland et al. (2015). The calculated values for nopinone oxide demonstrate the decreasing sensitivity of CI∗ stabilisation to the co-product size as the size of the CI∗ increases.
For endocyclic alkenes, an empirically derived sigmoid fit (Sect. S2, Eq. S1 and Fig. S6) is applied to the very limited dataset that shows YSCI≈0 for C ≤ 7, YSCI≈0.2 for monoterpenes and YSCI≈0.74 for sesquiterpenes.
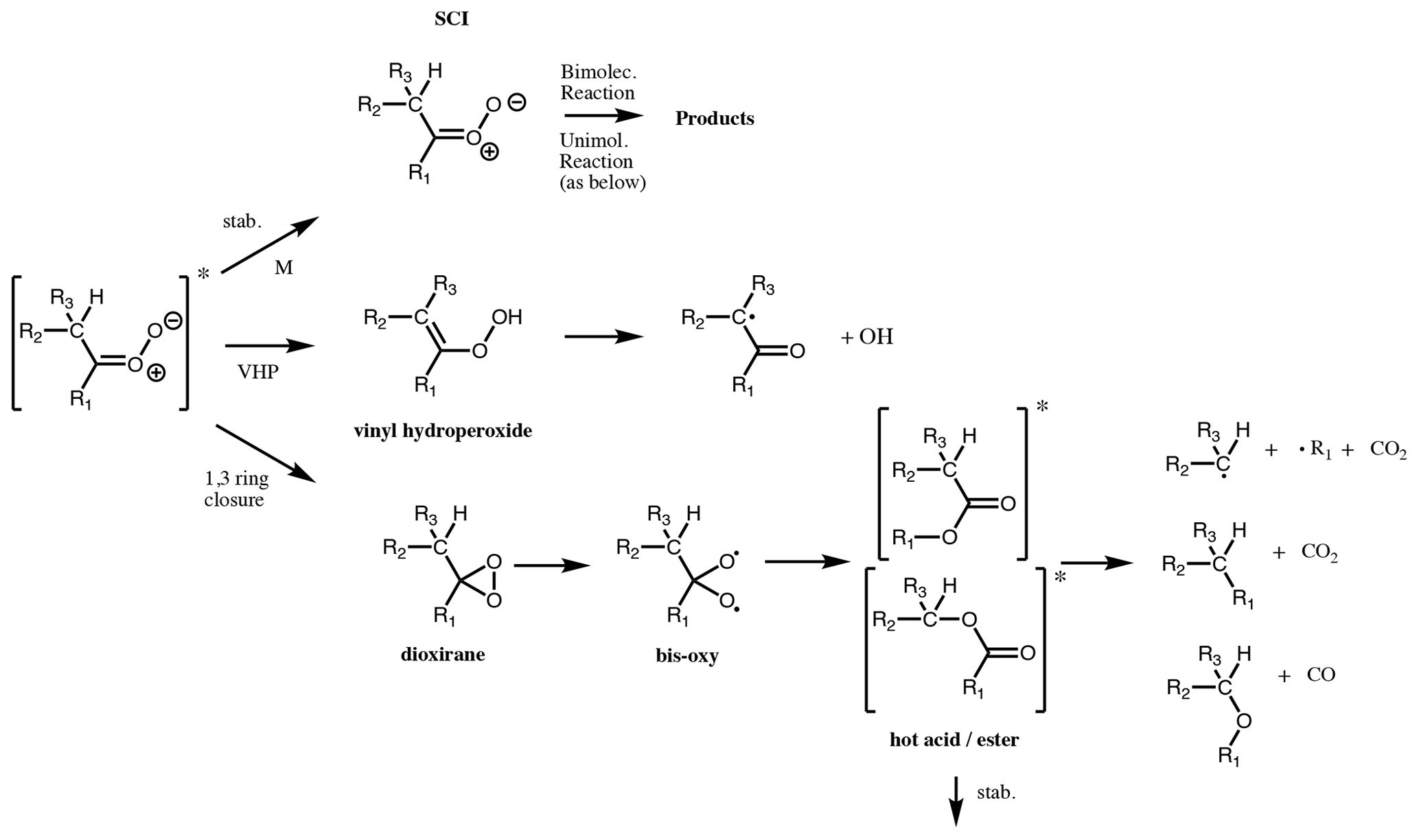
Figure 6Available pathways for a CI with a hydrogen atom available in the beta position to the carbonyl oxide. From top to bottom, the available pathways are the stabilisation (stab.) pathway, the vinylhydroperoxide (VHP) pathway and the 1,3 ring closure (hot acid/ester) pathway.
CI can undergo unimolecular isomerisation/decomposition. The unimolecular pathways available to SCI are assumed to be the same as those available to CI∗ (although it is noted that there is little evidence to back up this assumption). However, while for CI∗ these processes are prompt, occurring on a timescale of 10−9 s (Drozd et al., 2017), for SCI they occur at a range of rates such that their competition with atmospheric bimolecular reactions needs to be considered. A wide range of unimolecular isomerisation/decomposition pathways have been characterised for CI, but only two of these are believed to be important for saturated CI under atmospheric boundary layer conditions (Vereecken et al., 2017): a 1,4 H migration, i.e. the VHP pathway, and a 1,3 ring closure, i.e. the hot acid/ester pathway (Fig. 6). If the VHP pathway is available, then this will always be the dominant decomposition pathway as it is the energetically most facile, with only a slight entropic disadvantage compared to the 1,3 ring closure (Vereecken et al., 2017). Unsaturated CI have some additional pathways available (see Sect. 4.4).
Experimentally determined decomposition rates are available only for a limited number of SCI. Early estimates were considerably slower than more recent experimental evidence. Vereecken et al. (2017) recently published an extensive SAR providing temperature-dependent unimolecular rates and mechanisms for a wide range of SCI structures based on theoretical calculations tied to experimental work as well as group additivity relations.
4.1 VHP pathway
A CI with a β-hydrogen atom in a syn orientation to the terminal oxygen atom of the carbonyl oxide can isomerise to form a vinylhydroperoxide via a five-membered transition cycle (Fig. 6). This route is therefore available to monosubstituted (Z)-CI and disubstituted CI. The VHP formed has a short lifetime and promptly or thermally decomposes to form an OH radical and a β-acylalkyl (vinoxy) radical, in some cases with a small yield of β-acyl alcohols (Taatjes et al., 2017; Kuwata et al., 2018). The OH radicals are thus formed on a short timescale (e.g. Drozd et al., 2017) directly from the VHP decomposition. The β-acylalkyl radical reacts with O2 to form a β-acylperoxy radical. On a longer timescale, the subsequent chemistry of this peroxy radical can yield further HO2 and OH radicals (e.g. Nguyen et al., 2016).
The best-studied system that follows the 1,4 H-shift pathway is stabilised (CH3)2COO. Experimentally derived rates are fast (300–1000 s−1) (Berndt et al., 2014b; Newland et al., 2015; Chhantyal-Pun et al., 2017; Smith et al., 2016). The experimental evidence also shows a strong temperature dependence, with measured rates varying from 269 s−1 at 283 K to 916 s−1 at 323 K (Smith et al., 2016). This is in good agreement with the SAR of Vereecken et al. (2017), which shows that the rate of decomposition of saturated SCI is fastest (ca. 500 s−1) for those SCI with access to the VHP route. This SAR shows that the rate is slowed by more than an order of magnitude when only one H atom is available on the α-carbon and that the rates are also affected by the anti-substituent, with the presence of a vinyl group reducing rates by an order of magnitude and the presence of a carbonyl group reducing rates by 2 orders of magnitude.
This pathway may not be available to certain CI structures even though there is an available hydrogen on the α-carbon. This is the case for the bicyclic C9 CI formed in ozonolysis of the monoterpene β-pinene, with the terminal oxygen facing the four-membered ring. Calculations have shown that formation of the vinyl hydroperoxide is not possible for this CI due to the strain it would put on the ring, and so the dominant decomposition pathway is 1,3 ring closure (Nguyen et al., 2009b). This has also been shown to be the case for the cyclic C9 CI formed facing the three-membered ring in the ozonolysis of sabinene (Almatarneh et al., 2019).
4.2 1,3 ring closure
For monosubstituted (E)-CI and CH2OO (see Sect. 5.3), decomposition via a VHP is not available. Instead, unimolecular reaction proceeds predominantly via a 1,3 ring closure, with typical rates of ≤ 102 s−1 (Vereecken et al., 2017), to a chemically activated dioxirane species (Fig. 6). This breaks the weak O-O bond, giving a singlet bis-oxy radical (Wadt and Goddard, 1975; Herron and Huie, 1977, 1978). Various pathways have been proposed for the subsequent chemistry of this species based on observed product distributions (Chen et al., 2002). This pathway has been characterised best for CH2OO (Sect. 5.3). The dioxirane is thought to rearrange to a “hot” acid/ester, which can undergo decomposition to yield a range of products. As the size of the CI increases, the hot acid/ester is predicted to be more likely to be collisionally stabilised (Vereecken and Francisco, 2012).
There have been very few experimental studies to date on the products of isomerisation/decomposition of (E)-RCHOO. This is challenging experimentally as (E)-RCHOO will always be formed as a partner with (Z)-RCHOO. The most studied (E)-CI is (E)-CH3CHOO, with observed products from cis/trans-but-2-ene ozonolysis (which yields (E)- and (Z)-CH3CHOO as the CI products) of HCHO, CH3COOH, CH3OH, CH4, CHOCHO, ketene, CO and CO2 (e.g. Tuazon et al., 1997; Grosjean et al., 1994). With the exception of glyoxal, these can all be rationalised as decomposition products of “hot” (E)-CH3CHOO via various pathways (Reactions R1–R5). The relative proportion of each channel is based on the reported yields in Tuazon et al. (1997), except for CH3COOH, from Grosjean et al. (1994), although it is noted that CH3COOH may be a product of CH3CHOO + water vapour in their experimental set-up.
For R1R2COO decomposition via 1,3 ring closure, products are formed via a “hot” ester. There has been very little work on the relative contribution of decomposition channels and stabilisation for these species. For example, there is no experimental work to validate the predicted trend of increasing stabilisation of the hot acid/ester with size or at what size this becomes important. For the large terpenoid compounds β-pinene (Nguyen et al., 2009b) and β-caryophyllene (Nguyen et al., 2009a), the acids/lactones formed from isomerisation of the C9 dioxirane have been predicted to be fully stabilised.
4.3 CH2OO
CH2OO also follows the 1,3 ring closure pathway but is considered separately here as it has been the subject of a considerable body of work. Experimentally reported products from CH2OO decomposition include CO2, CO, H2, OH, HO2, H2O and HCOOH (e.g. Calvert et al., 2000). Recent theoretical (Nguyen et al., 2015; Stone et al., 2018; Peltola et al., 2020) works suggest that the only reaction pathway of the bis-oxy radical important under tropospheric conditions is isomerisation to “hot” formic acid, followed by decomposition to either H2 + CO2 or H2O + CO, in agreement with experimental and theoretical work on acid pyrolysis experiments (Chang et al., 2007; Vichietti et al., 2017). Due to the large excess energy and its small size, very little of the hot acid is stabilised, with measured HCOOH yields from ethene ozonolysis < 5 % (Calvert et al., 2000) (and the latter may be due to bimolecular reactions of SCI rather than stabilisation of the hot acid). Stone et al. (2018) and Peltola et al. (2020) considered the decomposition of stabilised CH2OO using master equation simulations, determining the major decomposition channel to be H2 + CO2 (64 % and 61 %, respectively), with the H2O + CO accounting for the remainder (36 %) in Stone et al. (2018), while Peltola et al. (2020) also found a small contribution (∼ 8 %) from the OH + HCO channel. It is noted that previous experimental work on ethene ozonolysis (Su et al., 1980; Horie et al., 1991; Neeb et al., 1998) has generally inferred a preference for the H2O + CO channel. This may be due to different pathways being followed by the dioxiranes formed from the excited CH2OO produced in the ozonolysis reaction compared to those formed from stabilised CH2OO, as suggested by work on larger systems (Nguyen et al., 2009a, b) and in the calculations of Nguyen et al. (2015) on excited CH2OO decomposition in ethene ozonolysis. A decomposition pathway to HCO + OH, proposed as the source of observed OH yields of 8 %–15 % in earlier experimental studies on the ozonolysis of ethene (Gutbrod et al., 1997; Rickard et al., 1999; Kroll et al., 2001; Alam et al., 2011) and larger alkenes (Kroll et al., 2002), has recently been determined experimentally to be negligible (Stone et al., 2018), accounting for less than 2 % of the overall decay. This is in agreement with earlier theoretical work (Olzmann et al., 1997; Nguyen et al., 2015) suggesting negligible OH yields from ethene ozonolysis. This apparent discrepancy between experiment and theory can be reconciled by invoking the possibility of OH formation via the carbonyl-hydroperoxide channel in the POZ decomposition, as discussed in Sect. 2.7.
The unimolecular decomposition rate of stabilised CH2OO has been experimentally determined to be very slow (< 12 s−1) (Berndt et al., 2015; Chhantyal-Pun et al., 2015; Newland et al., 2015; Stone et al., 2018; Peltola et al., 2020), with a current recommendation by IUPAC of ≤ 0.2 s−1 at 1 bar and 298 K (Cox et al., 2020). Even at the upper end of these estimates, decomposition is a negligible atmospheric fate for stabilised CH2OO compared to reaction with water vapour.
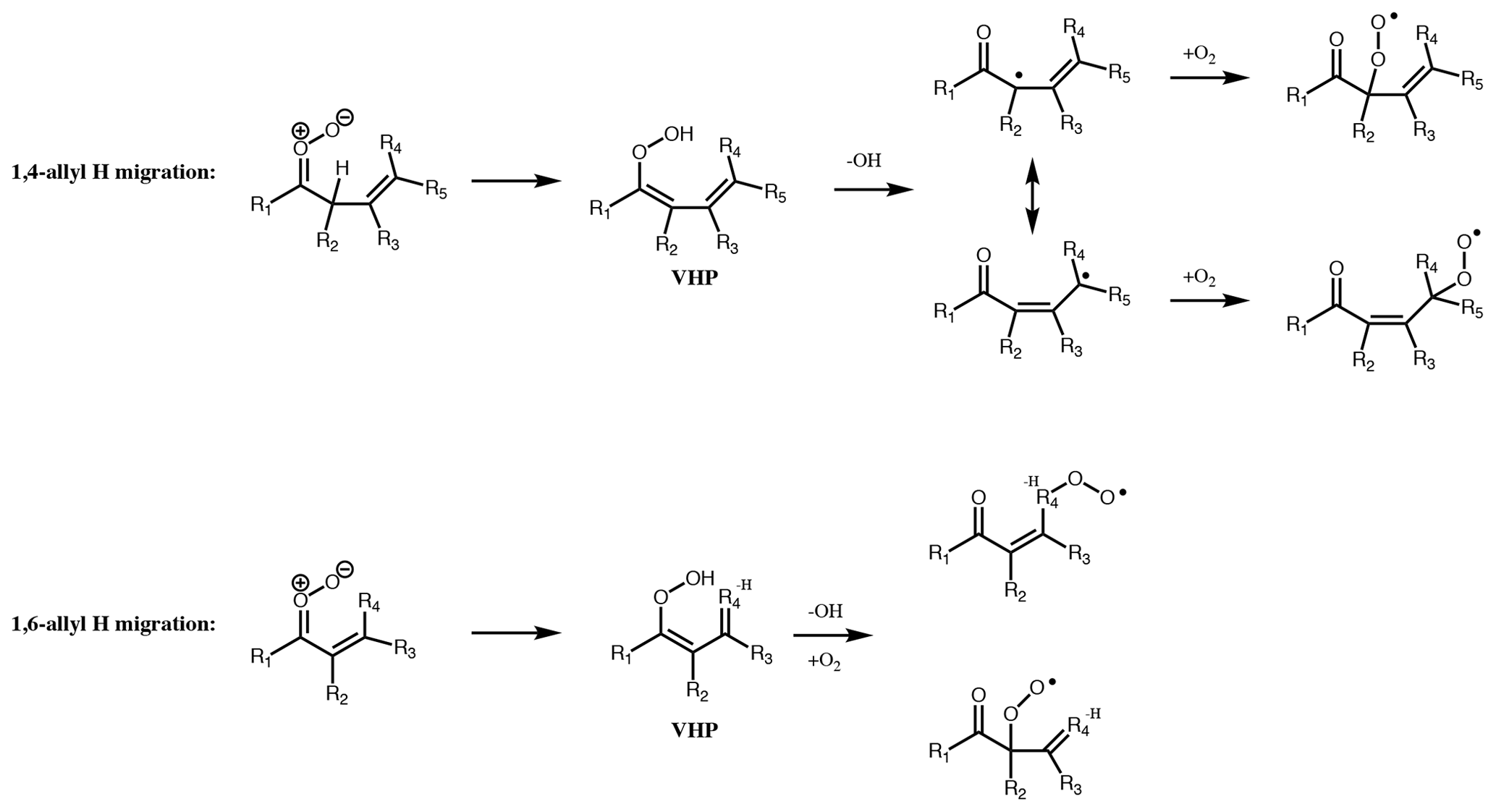
Figure 7Dominant unimolecular decomposition routes available to unsaturated CI with the terminal oxygen syn to an α or β vinyl group. Pathways available if terminal oxygen anti to a vinyl group is the same as for saturated CI. For 1,5 ring closure, see Fig. 8.
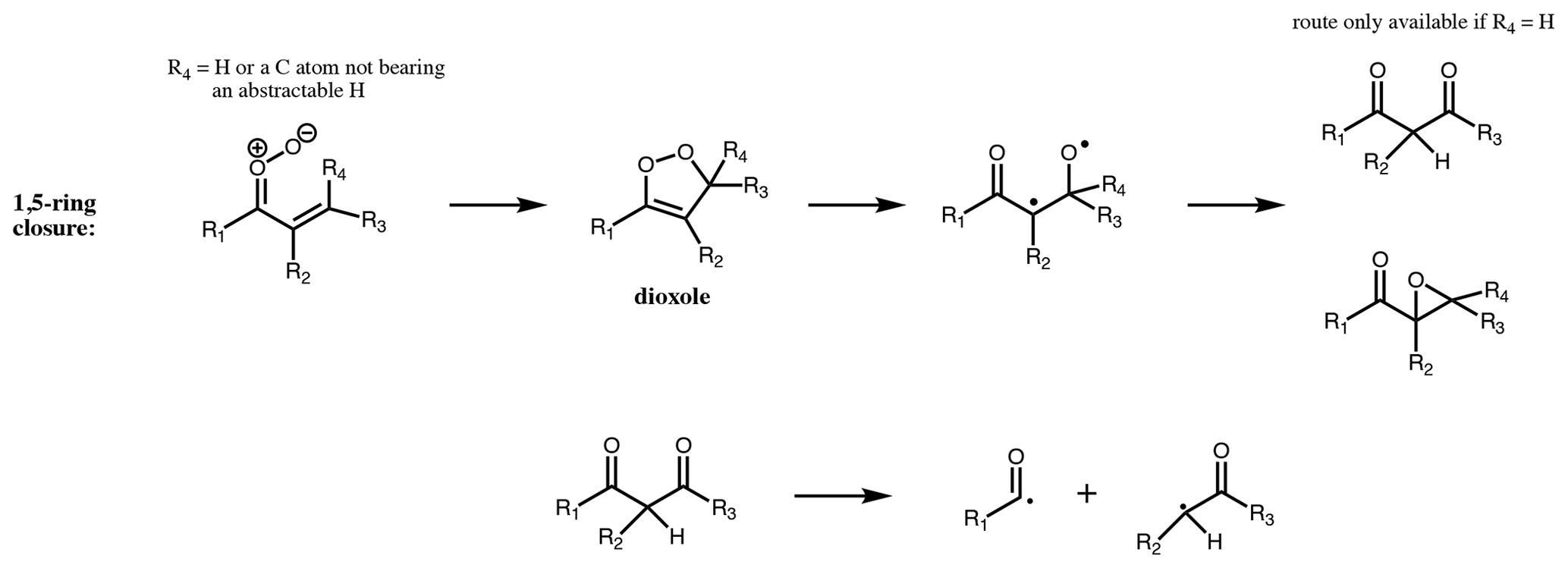
Figure 81,5 ring closure: dominant unimolecular pathway for unsaturated CI with the terminal oxygen syn to an α vinyl group, and R4 is not a carbon with an abstractable hydrogen.
4.4 Unimolecular reactions of unsaturated CI
The ozonolysis of conjugated alkenes proceeds via the same initial POZ mechanism as non-conjugated systems, but decomposition of the POZ leads to the formation of unsaturated CI and/or carbonyls. While many of the characteristics of the chemistry are expected to be similar, the theoretical work of Kuwata et al. (2005), Kuwata and Valin (2008) and Vereecken et al. (2017) has shown some important differences. Specifically, additional unimolecular decomposition channels (Figs. 7 and 8) become available, which in some cases are faster than the 1,4 H-shift channel.
If the vinyl group of an unsaturated CI is anti to the terminal oxygen of the carbonyl oxide, then the molecule will follow one of the two routes available to saturated CI but with a rate affected by the presence of the double bond. However, if the vinyl group is syn to the terminal oxygen, alternative mechanisms of decomposition are available. 1,4- and 1,6-allyl H migration (for the vinyl group in the β or α position, respectively) is available if an H atom is present on the α or γ carbon. These pathways lead to similar products to 1,4-alkyl H migration, with a vinylhydroperoxide intermediate decomposing to give OH and one of two possible unsaturated peroxy radicals. If no H atom is available for (Z)-β-unsaturated CI, then they follow the 1,3 ring closure channel with SCI decomposition rates ≤ 1 s−1. The rates of the 1,6-allyl H-migration channel for SCI are of the order of 106 s−1, while 1,4-allyl H migration of SCI has rates ranging from 101 to 104 s−1 depending on other substituents (Vereecken et al., 2017).
For CI with the carbonyl oxide syn to an α vinyl group and without an available hydrogen on the α carbon, the dominant decomposition mechanism is 1,5 ring closure, originally proposed by Kuwata et al. (2005) (Fig. 8). This forms an intermediate dioxole species with a five-membered ring. This is predicted to have high internal energy and to break the O-O bond, leading to an epoxy carbonyl, or, if R4 = H, to a dicarbonyl (Kuwata et al., 2005). The dicarbonyl has been predicted to undergo further prompt decomposition via various possible unimolecular channels, some of which appear to yield OH (Barber et al., 2018). Based on the stable product distribution from anti-MVKO decay, the decomposition of the dicarbonyl has been determined to be predominantly via C-C cleavage leading to two radicals (acetyl and vinoxy radicals in the case of anti-MVKO) (Vansco et al., 2020). These radicals will add O2, leading to RO2 radicals which may undergo further decomposition if formed chemically excited, ultimately to HCHO + OH + CO in both cases (Carr et al., 2011; Weidman et al., 2018; Vansco et al., 2020). For syn-MACRO, Vansco et al. (2020) determine a pathway via a dioxole analogous to that just described, leading to formyl and 2-methyl vinoxy radicals, the latter of which could ultimately yield CH3CHO + OH + CO. However, this accounts for only about half of the decomposition of the dicarbonyl, with the other half leading to acrolein via an unidentified unimolecular process. It is noted that Barber et al. (2018) and Vansco et al. (2020) did not consider the epoxide isomerisation pathway for the dioxole. The calculated unimolecular decay rates for the dioxole-forming pathways from syn-MACRO and anti-MVKO are fast; Vereecken et al. (2017, Table S25) reported rates of 2500 and 7700 s−1, respectively, with increasing substitution on the vinyl group accelerating the reaction further, while Barber et al. (2018) reported a somewhat slower rate for anti-MVKO of 2140 s−1. Decay of stabilised syn-MVKO is relatively slow at 33–50 s−1 (Vereecken et al., 2017; Barber et al., 2018), making it a potentially important bimolecular reaction partner in the atmosphere.
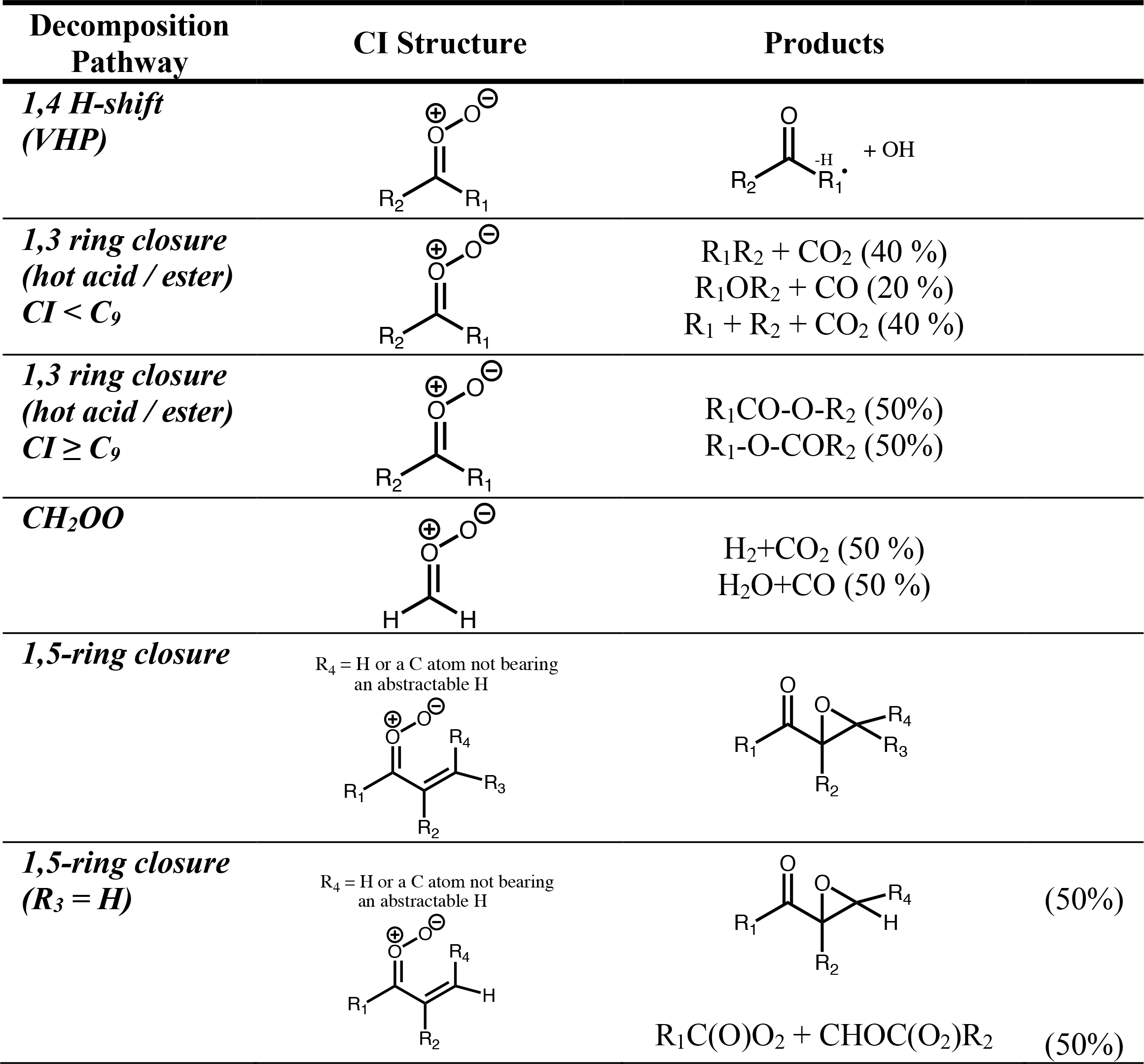
4.5 Protocol rules for CI decomposition
For unimolecular decomposition of CI, the SAR of Vereecken et al. (2017) is used to determine decomposition pathways and rates (for SCI). The products from each decomposition pathway are given in Table 2, where any secondary reactions such as recombination with O2 are already accounted for. The vinylhydroperoxide pathway is assumed to lead exclusively to a β-oxo alkyl radical and OH. For decomposition via 1,3 ring closure, the hot acid/ester formed is considered to decompose via one of the three major pathways determined for (E)-RCHOO; RH + CO2 (40 %), ROH + CO (20 %) and R + HO2 + CO2 (40 %), based on the observed product yields from cis and trans but-2-ene experiments by Tuazon et al. (1997). While it is noted that Grosjean et al. (1994) observed a CH3COOH yield of ∼ 20 %, this could also be a product of CH3CHOO + water vapour in their experimental set-up. For larger CI (≥ C9) the acid/ester is considered to be fully stabilised; if two esters can be formed, they are considered equally likely. This is recognised as an area where detailed experimental studies are required to establish the sensitivity of acid/ester stabilisation to CI size as well as identifying decomposition products for a range of CI sizes/structures and whether these are different for chemically activated/thermalised dioxiranes, as predicted (Anglada et al., 1998; Nguyen et al., 2009a, b). For CH2OO decomposition, the protocol assigns the products equally to two decomposition pathways: H2 + CO2 and H2O + CO; as discussed above, no OH is formed directly.
Table 3Bimolecular reaction rates with RCOOH, SO2, NO2 and inorganic acids applied to the four SCI structures. Rates are IUPAC recommendations (Cox et al., 2020) unless otherwise stated. Bimolecular reaction rates with water are taken from Vereecken et al. (2017); see main text.
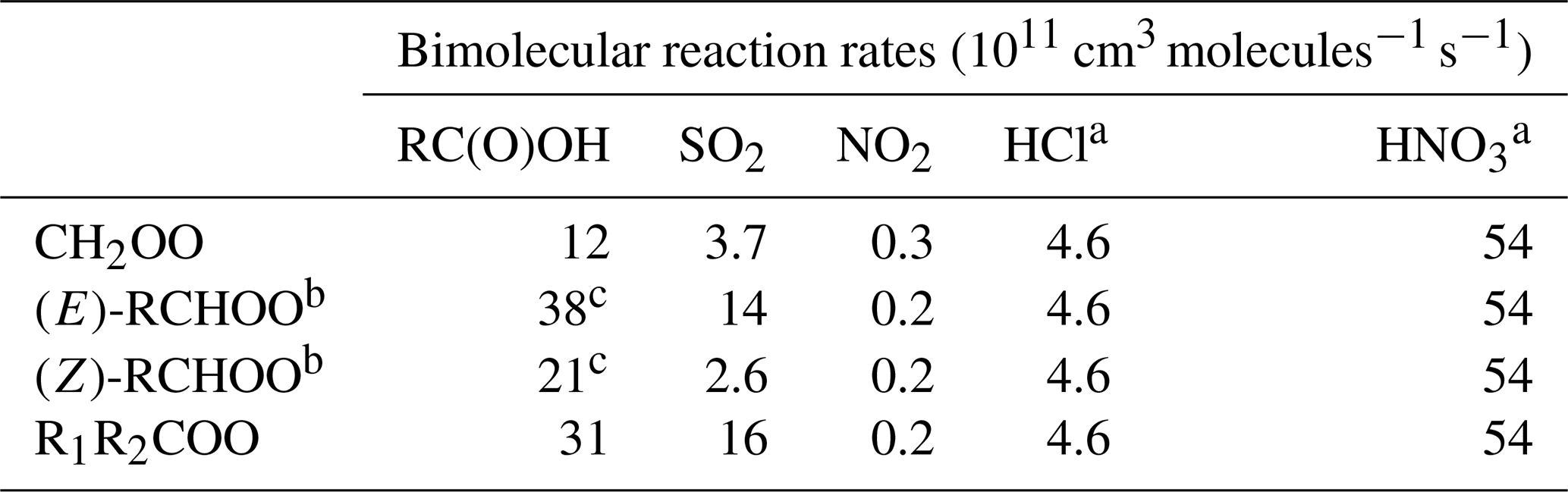
a All values for CH2OO reaction from Foreman et al. (2016). b IUPAC-recommended values for (E) and (Z)-CH3CHOO. c Mean of IUPAC-recommended values for reaction with HCOOH and CH3COOH.
For 1,4- and 1,6-allyl H migration in unsaturated CI (Fig. 7), formation of the alkyl radicals from each of the delocalised radical sites formed after OH elimination is assumed to be equally likely. The product yields given in Table 2 are for mechanisms that do not explicitly preserve stereo-specificity. For systems that track stereo-specific substitution on double bonds, H migration is only possible from the Z substituent, and the number of products is reduced accordingly, with a concomitant adjustment of the product yields.
For 1,5 ring closure (Fig. 8), formation of the epoxide or the dicarbonyl is considered equally likely. The dicarbonyl undergoes further decomposition to yield two RO2 following Barber et al. (2018). Unimolecular reaction rates for stabilised unsaturated CI are taken from the Vereecken et al. (2017) SAR. Clearly there remains much uncertainty in the proposed kinetics, and systematic experimental work on SCI yields and final product studies of ozonolysis of conjugated alkenes are required to improve the proposed protocol.
Based on the unimolecular pathways described in Sect. 5, many SCI have lifetimes against unimolecular reaction of the order of 10−3–10−1 s. These lifetimes are long enough to allow them to participate in bimolecular reactions with trace gases in the atmosphere under typical boundary layer conditions, where Vereecken et al. (2017) estimated that just under half of the CI in the atmosphere react with a co-reactant rather than unimolecularly. The co-reactants for which fast reactions, of potential tropospheric importance, have been demonstrated are H2O, (H2O)2, SO2, NO2 and organic and inorganic acids (Reactions R6–R11).
† Only available if R2 = H
Reactions with other trace gases have been investigated both experimentally and theoretically, but these are not included in the protocol at this time as they are not considered to be important under tropospheric conditions. Theoretical and experimental work has also shown that more complex bimolecular and unimolecular pathways may operate, forming heterocyclic molecules like cyclic peroxides and secondary ozonides (Chuong et al., 2004; Long et al., 2019). Again though, these reactions appear to be of negligible importance in the gas phase for SCI, with carbon numbers up to C10 (monoterpenes), and are not considered in this protocol. While only reactions relevant to the atmosphere are included in the protocol, reactions that are not expected to be relevant in the atmosphere are still maintained in the database since they may be useful for interpreting results of chamber simulations or other laboratory experiments (e.g. self-reaction or reaction with parent alkenes).
CH2OO and (E)-RCHOO react rapidly with H2O (Reaction R6a) (Welz et al., 2012; Taatjes et al., 2013; Stone et al., 2014) and with the water dimer, (H2O)2 (Reaction R6b) (Berndt et al., 2014a; Chao et al., 2015; Lewis et al., 2015; Lin et al., 2016), such that removal by water vapour is their predominant fate in the atmosphere. However, (Z)-RCHOO reacts slowly with H2O (Taatjes et al., 2013; Sheps et al., 2014; Huang et al., 2015), increasing the importance of bimolecular reactions with other atmospheric trace species such as acids and SO2 (Newland et al., 2018). The reaction of SCI with organic acids (Reaction R7) is also likely to be an important reaction in the atmosphere (Welz et al., 2014). The experimentally determined reaction rates for SCI + HCOOH and CH3COOH are 1–5 × 10−10 cm3 s−1 (Welz et al., 2014; Sipilä et al., 2014; Chung et al., 2019), close to the collisional limit. Other potentially important reactions in the atmosphere include those with SO2 (Reaction R8), NO2 (Reaction R9), and inorganic acids (Reactions R10 and R11). The rates of SCI + SO2 reaction have been the subject of several studies for the three smallest SCI, with good agreement between experiments. Larger SCI appear to have similar reaction rates with SO2 (Ahrens et al., 2014).
The products of many of the bimolecular reactions of SCI are still uncertain. This is the case for the most important bimolecular reactions in the atmosphere, those with H2O and (H2O)2. A recent experimental study (Sheps et al., 2017) of the reaction of CH2OO with (H2O)2, generating CH2OO from the photolysis of diiodomethane, determined yields of hydroxymethyl hydroperoxide (HMHP) (55 %), HCHO (40 %), and HCOOH (5 %). However, ozonolysis experiments (e.g. Nguyen et al., 2016) have generally found HMHP and HCOOH to be the main detected products, with negligible yields of HCHO. Based on results from isoprene ozonolysis chamber experiments, Nguyen et al. (2016) proposed yields from the CH2OO + H2O reaction of HMHP (73 %), HCOOH (21 %) and HCHO (6 %) and from the (H2O)2 reaction of HMHP (40 %), HCOOH (54 %) and HCHO (6 %). These low HCHO yields are in agreement with earlier work (Hasson et al., 2001b) that determined an HCHO yield of 6 %–9 %.
The products of SCI reaction with organic acids appear to be mainly hydroperoxide esters (Reaction R7). Hydroperoxy methyl formate (HPMF) has been detected as an intermediate in the CH2OO + HCOOH reaction (e.g. Neeb et al., 1995; Wolff et al., 1997; Hasson et al., 2001a; Chung et al., 2019), hydroperoxy methyl acetate in the CH2OO + CH3COOH reaction (Neeb et al., 1996) and hydroperoxy ethyl formate in the CH3CHOO + HCOOH reaction (Neeb et al., 1995, 1996; Cabezas and Endo, 2020). Theoretical calculations have predicted the formation of > 90 % HPMF for the reaction of CH2OO with HCOOH (Vereecken, 2017) and that the production of stabilised hydroperoxide esters will be even higher for larger SCI. The reaction with SO2 has been shown to form SO3 with close to unit yield (Reaction R8) (Kuwata et al., 2015). For NO2, while early experimental work (Ouyang et al., 2013) suggested SCI would oxidise NO2 to NO3, more recent experimental (Caravan et al., 2017) and theoretical (Vereecken and Nguyen, 2017) work has suggested the formation of a nitroalkylperoxy radical (R1R2C(O2)NO2). Subsequent reaction and formation of the alkoxy radical would be expected to yield a carbonyl and NO2. The main products of reaction of SCI with the inorganic acid HCl have been predicted to be chlorohydroperoxides (Reaction R10) (Foreman et al., 2016; Vereecken, 2017), with these products observed experimentally for CH2OO + HCl (Cabezas and Endo, 2017; Taatjes et al., 2021) and CH3CHOO + HCl (Cabezas and Endo, 2018). The main product of reaction with HNO3 has been predicted to be hydroperoxy nitrates (Reaction R11) (Foreman et al., 2016; Raghunath et al., 2017; Vereecken, 2017). Raghunath et al. (2017) further predicted decomposition of a fraction of the chemically activated hydroperoxy nitrates to CH2(O)NO3 + OH. This reaction has not yet been studied experimentally to the authors' knowledge.
Protocol rules for SCI bimolecular reactions
Bimolecular reaction rate coefficients for SCI are included for reaction with water vapour monomers and dimers, SO2, NO2, carboxylic acids and inorganic acids (HCl, HNO3) (Table 3). For the water vapour reactions, the rate coefficients are taken from the SAR of Vereecken et al. (2017), which provides values for 98 explicit structures. For bimolecular reactions of SCI with the other trace gases, four classes of SCI are considered: CH2OO, (Z)/(E)-RCHOO and R1R2COO (where R represents alkyl groups), based on the limited experimental data available. The rates are taken from IUPAC recommendations (Cox et al., 2020) where available and otherwise from sources as stated in Table 3. Where the structure does not fit into the defined classes, the CH2OO rate constant is attributed by default. Reaction products are as given in Reactions (R6)–(R11). In light of the current uncertainties of the product distribution of the reactions of SCI with water, here we assume the same products for the monomer and dimer reactions. We propose yields based on the direct study of Sheps et al. (2017) of α-hydroxy hydroperoxide (55 %), carbonyl (40 %) and acid (5 %), with the exception of R1R2COO, which cannot form the acid, for which we increase the α-hydroxy hydroperoxide to 60 %. These recommendations will be subject to change upon further experimental information becoming available.
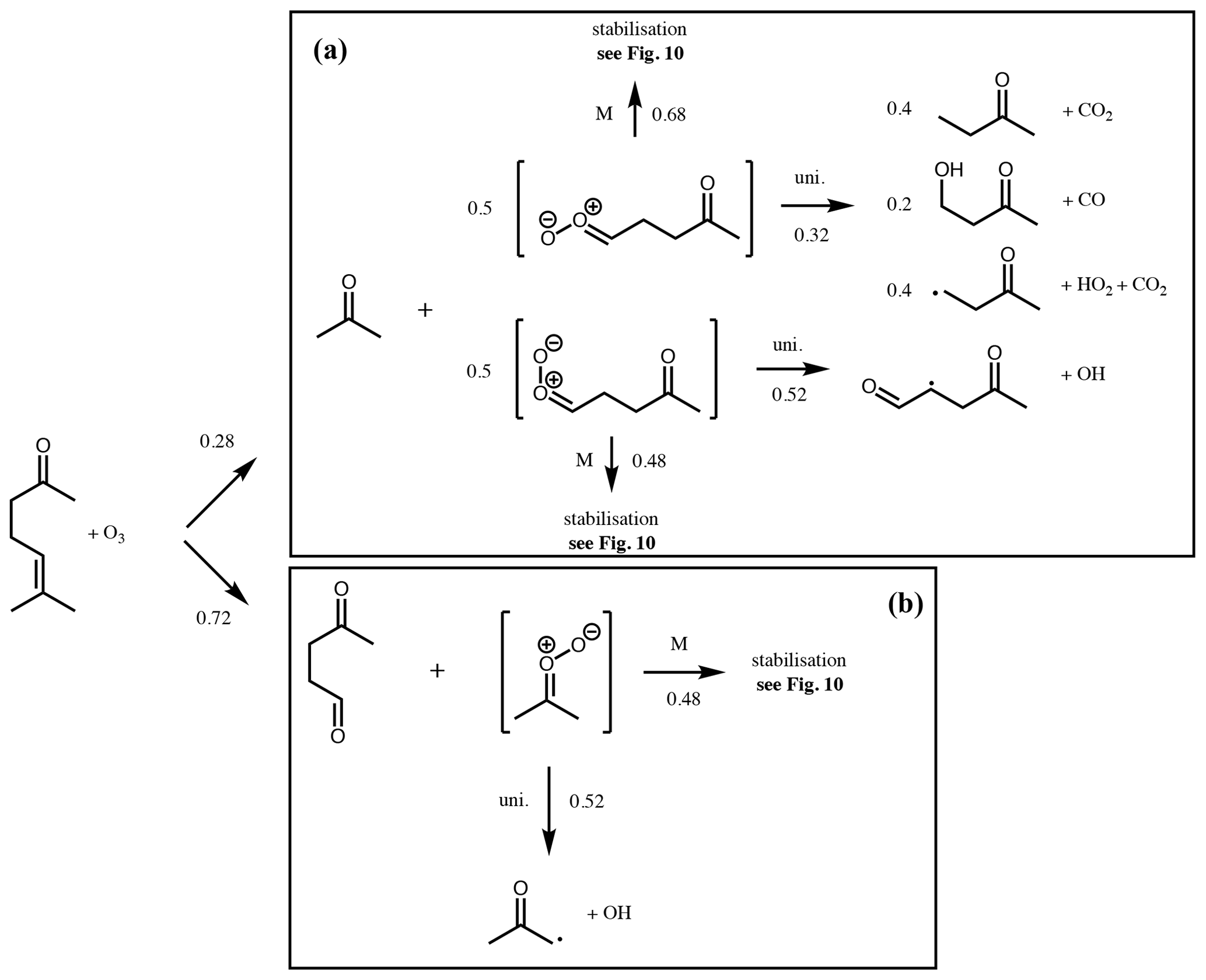
Figure 9Branching ratios and products of the CI decomposition produced following ozonolysis of 6-methyl-5-hepten-2-one.
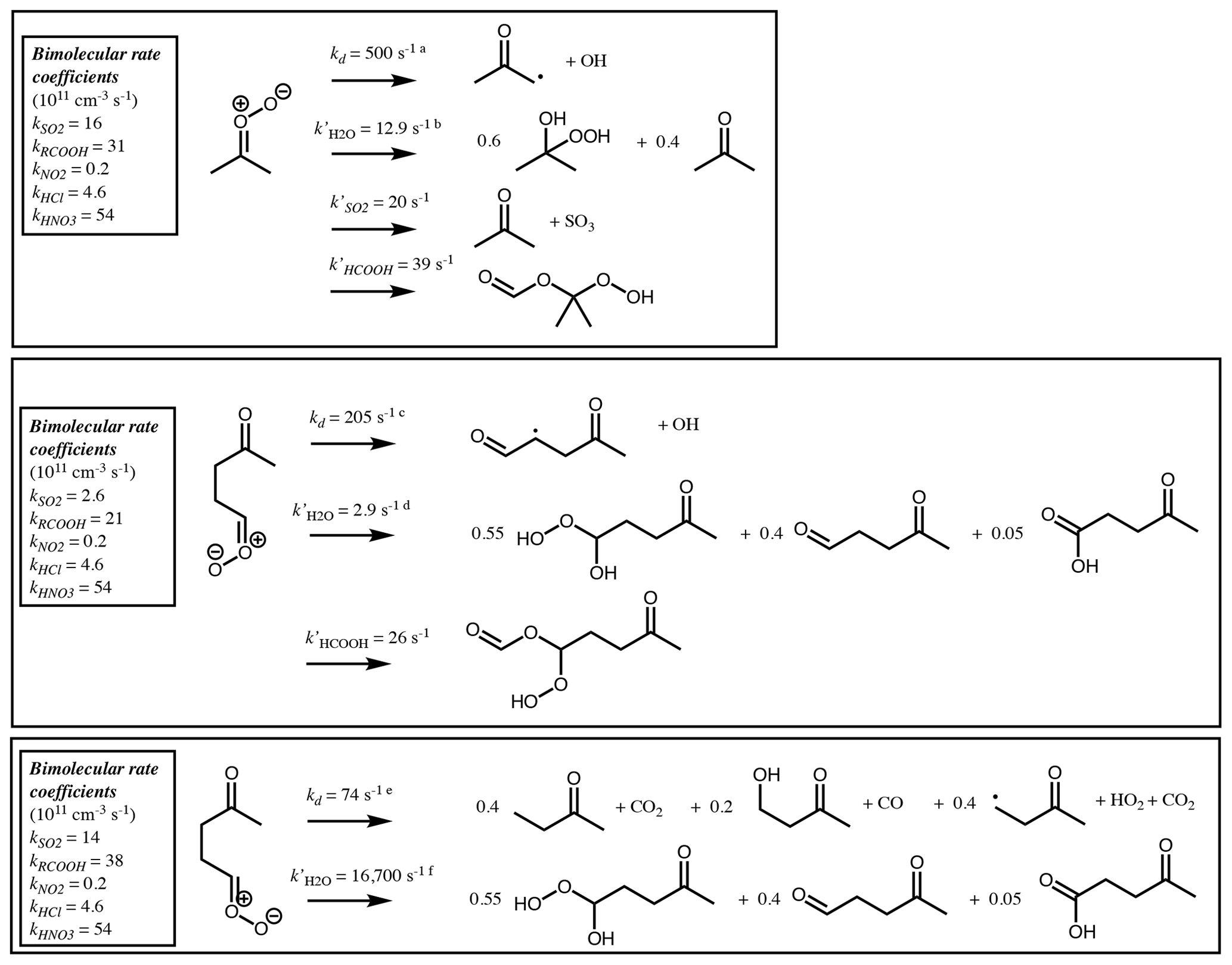
Figure 10Bimolecular rate coefficients (see Table 3) and products of the SCI produced following ozonolysis of 6-methyl-5-hepten-2-one at 298 K. Pseudo first-order loss rates (k′) and products are shown for decomposition and reaction with water vapour and for other pathways that contribute more than 1 % of the total loss assuming [SO2] = 5 ppbv, [RCOOH] = 5 ppbv, [H2O] = 5 × 1017 cm−3, [(H2O)2] = 5 × 1014 cm−3, [NO2] = 1 ppbv, [HCl] = 100 pptv and [HNO3] = 100 pptv. a 7.64 × 10−60 × . b Sum of first-order loss rates to water monomer (7.54 × 10−18 ⋅ [H2O]) and dimer (1.82 × 10−14 ⋅ [(H2O)2]). c 2.41 × 10−62 × . d Sum of first-order loss rates to water monomer (1.51 × 10−18 ⋅ [H2O]) and dimer (4.31 × 10−15 ⋅ [(H2O)2]). e 1.57 × 1010 × . f Sum of first-order loss rates to water monomer (1.58 × 10−14 ⋅ [H2O]) and dimer (1.75 × 10−11 ⋅ [(H2O)2]).
An example is described below for the unsaturated ketone, 6-methyl-5-hepten-2-one, and illustrated in Figs. 9 and 10. Further examples for α-pinene, cis-2-pentene, 2-methyl-1-pentene and 2-methyl-1,3-butadiene (isoprene) are given in the Supplement (Sect. S3). The initial rate of reaction with ozone is defined by the protocol in the companion paper (Jenkin et al., 2020). The branching ratio for formation of the disubstituted CI∗ is calculated to be 0.72 using the group additivity values in Table 2 and Eq. (1).
The syn- and anti-conformers of the two large CI∗ are formed with equal yield (0.14).
Stabilisation of each CI∗ is computed using Eq. (2).
The remaining (CH3)2COO∗ undergoes unimolecular decomposition via the VHP pathway to yield the acetonyl peroxy radical (CH3C(O)CH2OO) and OH. The remaining (Z)-CH3C(O)(CH2)2CHOO decomposes via the VHP pathway to yield CH3C(O)CH2CH(O2)CHO + OH, while (E)-CH3C(O)(CH2)2CHOO decomposes via 1,3 ring closure and yields CH3C(O)CH2CH3 + CO2 (40 %), CH3C(O)CH2CH2OH + CO (20 %) and CH3C(O)CH2CH2 + H + CO2 (40 %). Each stabilised CI can decompose via the same pathways as its respective CI∗, with temperature-dependent rates determined from Vereecken et al. (2017). At 298 K these are 478 s−1, 205 s−1 and 74 s−1 for (CH3)2COO, (Z)-CH3C(O)(CH2)2CHOO and (E)-CH3C(O)(CH2)2CHOO, respectively. Alternatively, they can undergo bimolecular reaction. Reaction rates with H2O and (H2O)2 are calculated using monomer and dimer reaction rates from Vereecken et al. (2017). Reaction rates with other trace gases are taken from Table 3 for the relevant CI structure. Figure 10 shows calculated pseudo first-order reaction rates for reaction with SO2 and RCOOH assuming atmospheric mixing ratios of [SO2] = 5 ppbv and [RCOOH] = 5 ppbv.
7.1 Experimental databases and assessment approach
A database of experimentally determined carbonyl yields, OH yields and SCI yields has been assembled to evaluate the new protocol (Spreadsheets S1–S3). Experimental conditions are also recorded in the database to enable some assessment of the validity of the assumptions inherent in the experimental set-up.
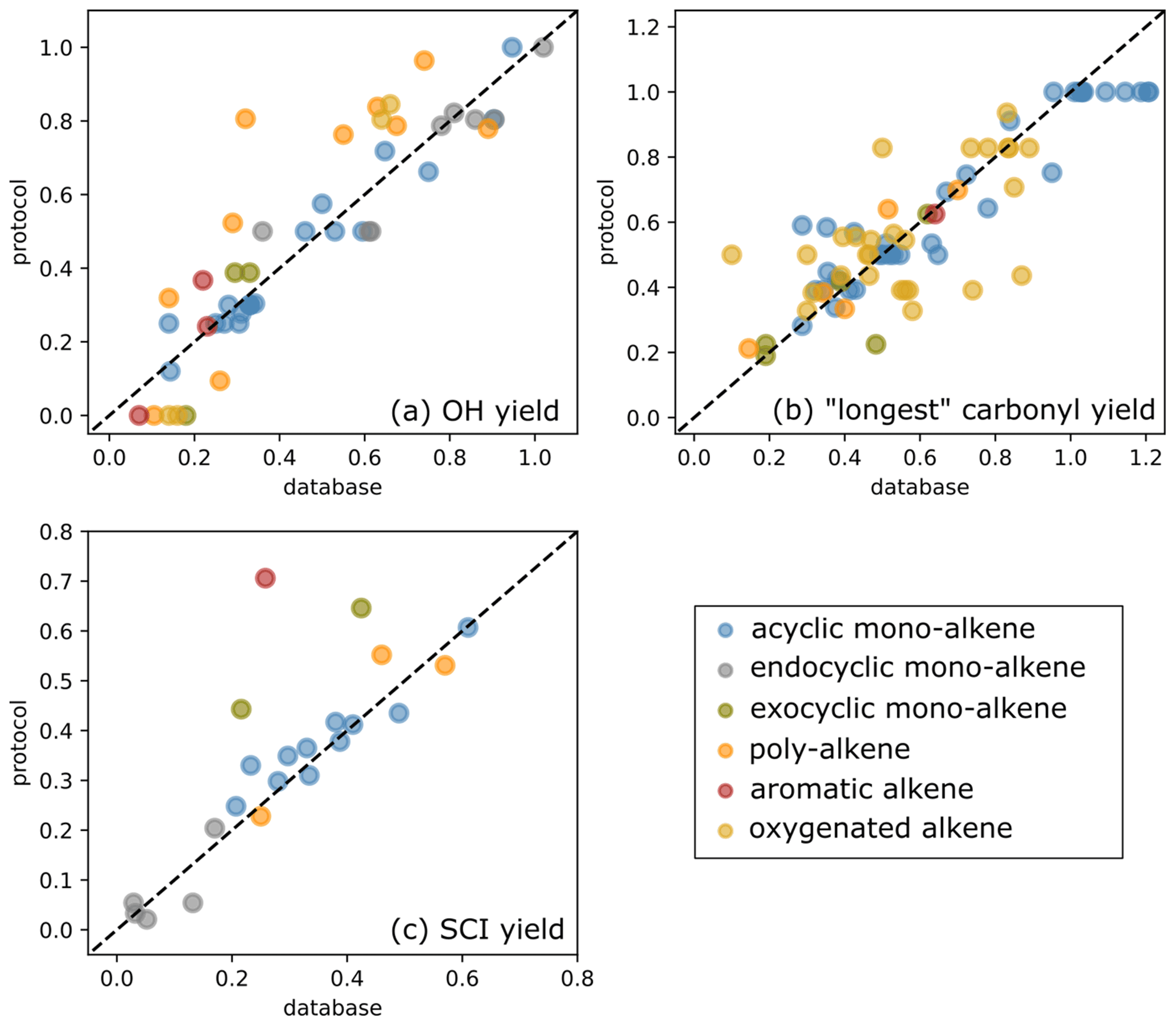
Figure 11Scatter plot of protocol yields vs. database yields: (a) OH yields, (b) yields for the “longest” carbonyl and (c) SCI yields.
The root mean squared error (RMSE) and the mean bias error (MBE) were examined to assess the reliability of the protocol. The RMSE and MBE are here defined as
where n is the number of species in the dataset. The databases were split into subsets to identify possible bias within a structural category of species (e.g. exocyclic vs. endocyclic monoalkenes). The various subsets examined and their corresponding number of species are summarised in Table 4. Three databases were used to perform the protocol assessment: carbonyl yield (Spreadsheet S1), SCI yield (Spreadsheet S2) and OH yield (Spreadsheet S3). The RMSE and MBE computed for the full databases and the various subsets are reported in Table 4. The scatter plots of protocol yields vs. database yields, by species category, are given in Fig. 11.
Table 4Number of species (n) in the database used to compute the mean bias error (MBE) and the root mean square error (RMSE) for the OH yields, SCI yields and “longest” carbonyl yield. not applicable – n/a.
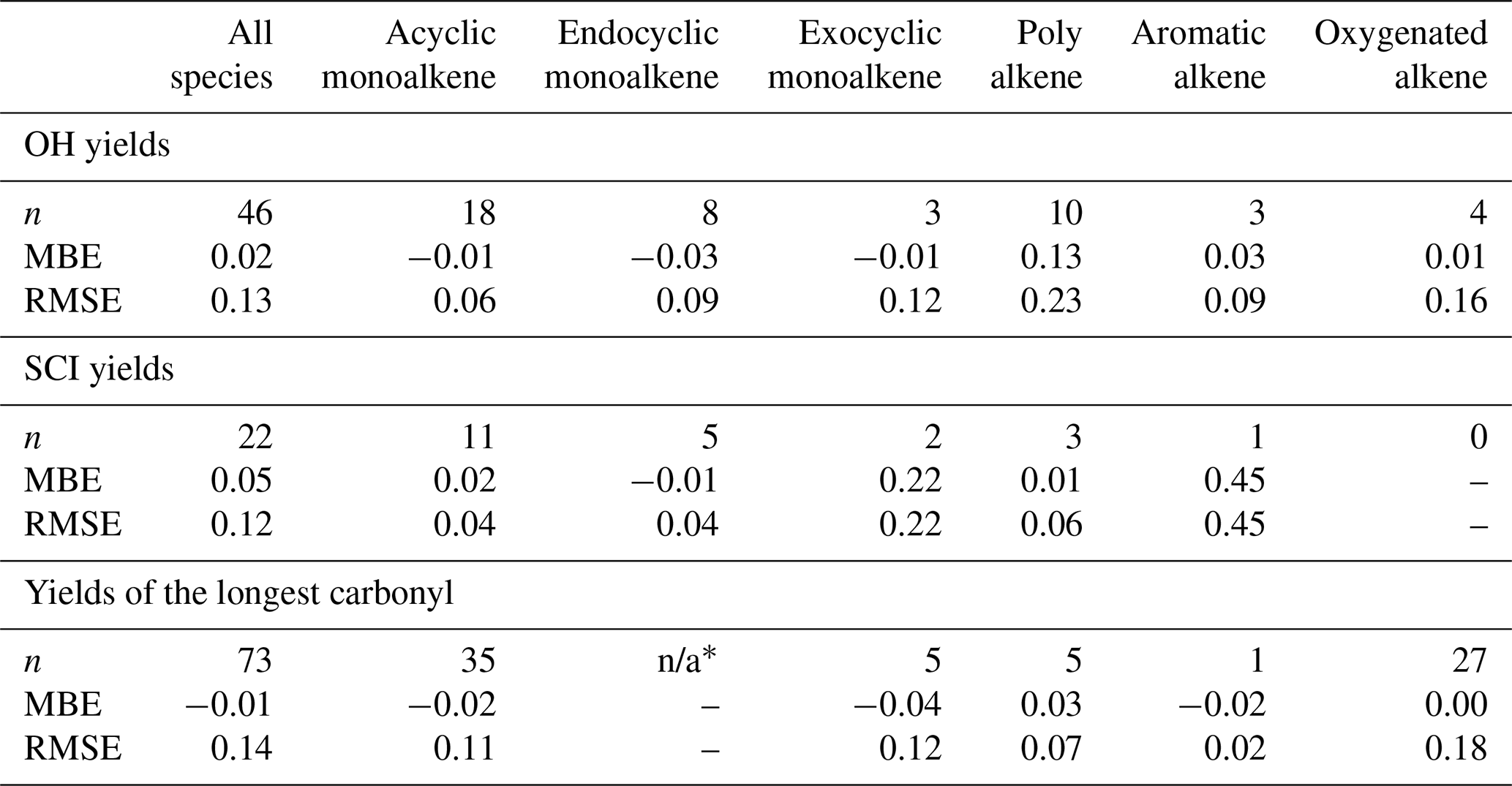
∗ Endocyclic alkenes do not produce a stable primary carbonyl as all possible molecules formed from the POZ fragmentation contain a carbonyl oxide moiety.
7.2 Primary carbonyl yields
The primary carbonyl yields from alkene ozonolysis are calculated in the protocol by assigning F values to different functional groups adjacent to the C=C bond that determine the relative fragmentation pattern of the POZ (Sect. 2). The calculated primary carbonyl yields can be compared to the measurements in the experimental database. For some functional groups, however, the number of data available are sparse and the carbonyl yields have been directly used to determine the F value. The carbonyl yields dataset should therefore rather be viewed as a training dataset than a validation dataset in this protocol assessment. Figure 11b shows the scatter plots for the calculated yields of the larger primary carbonyl (i.e. greater number of non-H atoms) formed in POZ decomposition compared to the experimentally reported values for each alkene in the database. No substantial bias is identified in the computed carbonyl yields (MBE = −0.01). For non-oxygenated alkenes, the fit is reasonably good, and the RMSE does not exceed 0.12 for the various hydrocarbon classes reported in Table 4. The major outlier is the yield of 4-ethyl-3-hexanone from 3,4-diethyl-2-hexene ozonolysis. This is based on one measurement (Grosjean and Grosjean, 1996a). It was noted in Jenkin et al. (2020) that the ozonolysis reaction rate reported by Grosjean and Grosjean (1996b) for this precursor compound is also a significant outlier from predicted trends. For symmetrical alkenes, the calculated primary carbonyl yield is unity, whereas measured yields tend to cluster slightly above one. This is likely due to a small amount of secondary formation of the carbonyls from bimolecular reactions of SCI. The poorest fitted class is oxygenated alkenes (RMSE = 0.18). This is likely due to a combination of factors. Firstly, the majority of these compounds have only one measurement. Secondly, measurements of multi-oxygenated VOCs are known to be more challenging than e.g. simple carbonyls. Thirdly, there are more likely to be additional chemical factors which are not yet understood in the ozonolysis of these more complex molecules influencing the POZ fragmentation. Two of the most significant outliers in the oxygenated alkenes are acrylic and methacrylic acid. As described in Sect. 2.2.3, it is difficult to reconcile the two available data points.
7.3 SCI yields
The yield of stabilised Criegee intermediates from an alkene–ozone reaction depends on the alkene structure (i.e. the POZ fragmentation pattern, Sect. 2), the dominant unimolecular decomposition route of the CI∗ (Sect. 3.2) and the size of the CI (Sect. 3.2). The yields calculated by the protocol are independent of the measurements in the database. SCI yields can therefore be considered a validation dataset to evaluate the reliability of the protocol. Total SCI yields have been measured for a number of alkenes, although the dataset is still relatively small. It should also be noted that many experimentally determined SCI yields have a large uncertainty associated with them, particularly earlier experiments where analysis techniques were less developed and the chemical models lacking. Figure 11c shows the scatter plot of the total SCI yields calculated by the protocol vs. experimental data. The data consist predominantly of acyclic monoalkenes, for which there is good agreement between the measurements and the calculated values (RMSE = 0.04). Figure 11c shows three major outliers for which the protocol overpredicts the measured SCI yield. These species are methylene cyclohexane and β-pinene (which constitute the subset of the exocyclic alkenes; RMSE = 0.22) and styrene, the only representative of the aromatic alkene class in this dataset (RMSE = 0.45). The methylene cyclohexane and styrene values are both based on one measurement (Hatakeyama et al., 1984), and the β-pinene value is based on two measurements (Hatakeyama et al., 1984; Newland et al., 2018) which are in poor agreement, giving values of 0.25 and 0.60, respectively. This clearly warrants revisiting experimentally, particularly with respect to the atmospherically important monoterpene β-pinene. Finally, the overall protocol SCI yields appear to be biased slightly high (+5 %), which is mainly explained by the overestimation described above for the exocyclic and aromatic alkenes.
7.4 OH yields
The reaction of alkenes with ozone yields OH through both primary (i.e. decomposition of CI via a vinylhydroperoxide) and secondary (i.e. peroxy radical chemistry) processes. The primary process can also be split: the decomposition of chemically activated CI∗, which under atmospheric conditions (and e.g. chamber laboratory experiment conditions) is assumed to happen at rates such that there is no competition with bimolecular reaction and the decomposition of stabilised CI, which occurs in competition with bimolecular reactions so that the OH yield depends on the unimolecular rate relative to the concentrations of possible co-reactants. The primary OH yield thus depends on the POZ fragmentation pattern (Sect. 2) and the decomposition pathways of the CI (Sect. 3.2).
Many studies have measured the OH yield for specific alkene–ozone reactions. As for the SCI yields above, the OH yield database can be viewed as a validation dataset to assess the reliability of the protocol since OH yields are not prescribed explicitly but are a product of the protocol rules for POZ fragmentation and CI decomposition pathways. For the comparison, protocol yields are computed assuming that all SCI produced undergo unimolecular decomposition (i.e. bimolecular reactions of SCI are ignored). Although many experiments will have been designed in such a way as to try to prevent bimolecular reactions, in reality a small fraction of the SCI will react bimolecularly, not producing OH, so the computed OH yield might be considered an upper limit. On the other hand, in many of the experiments there will likely be some contribution to the measured OH yield from peroxy radical chemistry (e.g. HO2 + O3), making the reported experimental yield an upper limit. No attempt is made here to determine the relative contribution from primary or secondary processes in the reported measurements, which is dependent on both experimental set-up and the particular alkene being studied, or to correct for possible bimolecular reactions. Therefore, a comparison between experimental and protocol OH yields clearly carries significant uncertainties.
With this in mind, the agreement between computed OH yields and the experimental values is very good (Fig. 11a). No substantial bias is observed on the complete dataset (MBE = 0.02). It is difficult to comment on some classes as they contain only one or two compounds (see Table 4). The protocol appears especially reliable for estimating the OH yields for monoalkenes (RMSE = 0.06) and endocyclic alkenes (RMSE = 0.09). The class for which the protocol does worst is polyalkenes (RMSE = 0.23), with a systematic overprediction at higher OH yields (MBE = 0.13). There are five compounds for which the protocol calculates an OH yield of zero (styrene, 1,3-butadiene, methyl vinyl ketone, methacrolein and camphene). The measured OH yields of these compounds are all below 0.2, and the measured OH could be a result of peroxy radical chemistry.
This paper provides a protocol by which the central features of alkene ozonolysis chemistry can be included in an explicit automatic chemical mechanism generator. It also serves to highlight the many gaps that remain in our knowledge of this complex, atmospherically important, process. This will hopefully help direct both experimental and theoretical research towards improving understanding in these areas. Some of the major areas of uncertainty identified in this work include the following:
- i.
The impact of oxygenated substituents on POZ fragmentation
- ii.
The impact of alkene structure on (E)/(Z)-CI conformer yields
- iii.
Products of the hot acid/ester channel and trends in the stabilisation of the hot acid/ester with size
- iv.
Further details of the mechanisms and products of non-Criegee ozonolysis chemistry, e.g. step-wise decomposition of the POZ via a carbonyl hydroperoxide
- v.
Product distributions of some of the major atmospheric SCI bimolecular reactions – e.g. the reaction of (Z)-RCHOO/CH2OO with H2O/(H2O)2
- vi.
Experimental evidence of the products of conjugated alkene ozonolysis
- vii.
Data on OH and SCI yields from alkenes with (multiple) functional groups
The reliability of the protocol designed in this work was assessed using experimental values for the OH, SCI and primary carbonyl yields, which are independent of the data used to derive the protocol. For these three datasets, the mean bias error (MBE) for the protocol-based yields is below 0.05, with no substantial bias identified. The protocol currently provides a fairly reliable estimate of the OH, SCI and primary carbonyl yields, with root mean squared errors (RMSEs) of 0.12, 0.13 and 0.15, respectively. The protocol thus appears robust in representing CI chemistry and its impact on atmospheric chemistry. However, the number of data available for some classes of compounds remain limited, such as oxygenated, exocyclic and poly-alkenes. The errors in the yields calculated for these species are also the most substantial, and additional experimental data for these categories of compound would be highly valuable to improve the protocol and its assessment.
All relevant data and supporting information have been provided in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-6167-2022-supplement.
All the authors defined the scope of the work. MJN and CM-V developed and applied the SAR methods with the help of LV, which were reviewed by all the co-authors. MJN drafted the manuscript with the help of ARR, which was reviewed by all the co-authors. RV and BA tested the SAR methods in GECKO-A and carried out the statistical analysis in Sect. 7.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
This work was performed as part of the MAGNIFY project (Mechanisms for Atmospheric chemistry: GeneratioN, Interpretation and FidelitY), with funding from the French National Research Agency (ANR) and the UKRI Natural Environment Research Council (NERC). It was also partially funded by the European Commission's EUROCHAMP-2020 project and the UK National Centre for Atmospheric Sciences (NCAS) Air Pollution Theme.
This research has been supported by the Natural Environment Research Council (grant no. NE/M013448/1), the Agence Nationale de la Recherche (grant no. ANR-14-CE01-0010) and the European Commission Horizon 2020 Framework Programme (grant no. EUROCHAMP-2020 – 730997).
This paper was edited by Kelley Barsanti and reviewed by two anonymous referees.
Ahrens, J., Carlsson, P. T. M., Hertl, N., Olzmann, M., Pfeifle, M., Wolf, J. L., and Zeuch, T.: Infrared Detection of Criegee Intermediates Formed during the Ozonolysis of β-pinene and Their Reactivity towards Sulfur Dioxide, Angew. Chem., 53, 715–719, 2014.
Alam, M. S., Camredon, M., Rickard, A. R., Carr, T., Wyche, K. P., Hornsby, K. E., Monks, P. S., and Bloss, W. J.: Total radical yields from tropospheric ethene ozonolysis, Phys. Chem. Chem. Phys., 13, 11002–11015, 2011.
Almatarneh, M. H., Elayan, I. A., Altarawneh, M., and Hollett, J. W.: A computational study of the ozonolysis of sabinene, Theor. Chem. Acc., 138, 1–14, 2019.
Al Mulla, I., Viera, L., Morris, R., Sidebottom, H., Treacy, J., and Mellouki, A.: Kinetics and Mechanisms for the Reactions of Ozone with Unsaturated Oxygenated Compounds, Chemphyschem, 11, 4069–4078, 2010.
Anglada, J. M., Bofill, J. M., Olivella, S., and Solé, A.: Theoretical Investigation of the Low-Lying Electronic States of Dioxirane: Ring Opening to Dioxymethane and Dissociation into CO2 and H2, J. Phys. Chem. A, 102, 3398–3406, 1998.
Anglada, J. M., Crehuet, R., and Bofill, J. M.: The Ozonolysis of Ethylene: A Theoretical Study of the Gas-Phase Reaction Mechanism, Chem.-Eur. J., 5, 1809–1822, 1999.
Aschmann, S. M., Tuazon, E. C., Arey, J., and Atkinson, R.: Products of the Gas-Phase Reaction of O3 with Cyclohexene, J. Phys. Chem. A, 107, 2247–2255, 2003.
Atkinson, R. and Aschmann, S. M.: OH radical production from the gas-phase reactions of O3 with a series of alkenes under atmospheric conditions, Environ. Sci. Technol., 27, 1357–1363, 1993.
Atkinson, R., Tuazon, E. C., and Aschmann, S. M.: Products of the gas-phase reactions of O3 with alkenes, Environ. Sci. Technol., 29, 1860–1866, 1995.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Barber, V. P., Shubhrangshu, P., Green, A. M., Trongsiriwat, N., Walsh, P. J., Klippenstein, S. J., and Lester, M. I.: Four-Carbon Criegee Intermediate from Isoprene Ozonoysis: Methyl Vinyl Ketone Oxide Synthesis, Infrared Spectrum, and OH Production, J. Am. Chem. Soc., 140, 10866–10880, 2018.
Barnes, I., Zhou, S. M., and Klotz, B.: Final Report of the EU project MOST, Contract EVK2-CT-2001-00114, European Union, Brussels, 2005.
Bauld, N. L., Thompson, J. A., Hudson, C. E., and Bailey, P. S.: Stereospecificity in Ozonide and Cross-Ozonide Formation, J. Am. Chem. Soc., 90, 1822–1830, 1968.
Bernard, F., Eyglunent, G., Daële, V., and Mellouki, A.: Kinetics and Products of Gas-Phase Reactions of Ozone with Methyl Methacrylate, Methyl Acrylate, and Ethyl Acrylate, J. Phys. Chem. A, 114, 8376–8383, 2010.
Berndt, T., Voigtländer, J., Stratmann, F., Junninen, H., Mauldin III, R. L., Sipilä, M., Kulmala, M., and Herrmann, H.: Competing atmospheric reactions of CH2OO with SO2 and water vapour, Phys. Chem. Chem. Phys., 16, 19130–19136, 2014a.
Berndt, T., Jokinen, T., Sipilä, M., Mauldin III, R. L., Herrmann, H., Stratmann, F., Junninen, H., and Kulmala, M.: H2SO4 formation from the gas-phase reaction of stabilized Criegee intermediates with SO2: Influence of water vapour content and temperature, Atmos. Environ., 89, 603–612, 2014b.
Berndt, T., Kaethner, R., Voigtländer, J., Stratmann, F., Pfeifle, M., Reichle, P., Sipilä, M., Kulmala, M., and Olzmann, M.: Kinetics of the unimolecular reaction of CH2OO and the bimolecular reactions with the water monomer, acetaldehyde and acetone under atmospheric conditions, Phys. Chem. Chem. Phys., 17, 19862–19873, 2015.
Bey, I., Aumont, B., and Toupance, G.: The nighttime production of OH radicals in the continental troposphere, Geophys. Res. Lett., 24, 1067–1070, 1997.
Cabezas, C. and Endo, Y.: Spectroscopic Characterization of the Reaction Products between the Criegee Intermediate CH2OO and HCl, Chemphyschem, 18, 1860–1863, 2017.
Cabezas, C. and Endo, Y.: The reaction between the methyl Criegee intermediate and hydrogen chloride: an FTMW spectroscopic study, Phys. Chem. Chem. Phys., 20, 22569–22575, 2018.
Cabezas, C. and Endo, Y.: Observation of hydroperoxyethyl formate from the reaction between the methyl Criegee intermediate and formic acid, Phys. Chem. Chem. Phys., 22, 446–454, 2020.
Calvert, J. G., Atkinson, R., Kerr, J. A., Madronich, S., Moortgat, G. K., Wallington, T. J., and Yarwood, G.: The Mechanism of Atmospheric Oxidation of the Alkenes, Oxford University Press, New York, USA, 552 pp., 2000.
Campos-Pineda, M. and Zhang, J.: Product yields of stabilized Criegee intermediates in the ozonolysis reactions of cis-2-butene, 2-methyl-2-butene, cyclopentene, and cyclohexene, Science China Chemistry, 61, 850–856, 2018.
Caravan, R. L., Khan, A. H. M., Rotavera, B., Papajak, E., Antonov, I. O., Chen, M.-W., Au, K., Chao, W., Osborn, D. L., Lin, J. J.-M., Percival, C. J., Shallcross, D. E., and Taatjes, C. E.: Products of Criegee intermediate reactions with NO2: experimental measurements and tropospheric implications, Faraday Discuss., 200, 313–330, 2017.
Caravan, R. L., Vansco, M. F., Au, K., Khan, M. A. H., Li, Y. L., Winiberg, F. A., Zuraski, K., Lin, Y. H., Chao, W., Trongsiriwat, N., and Walsh, P. J.: Direct kinetic measurements and theoretical predictions of an isoprene-derived Criegee intermediate, P. Natl. Acad. Sci. USA, 117, 9733–9740, 2020.
Carr, S. A., Glowacki, D. R., Liang, C.-H., Baeza-Romero, M. T., Blitz, M. A., Pilling, M. J., and Seakins, P. W.: Experimental and Modeling Studies of the Pressure and Temperature Dependences of the Kinetics and the OH Yields in the Acetyl + O2 Reaction, J. Phys. Chem. A., 115, 1069–1085, 2011.
Carslaw, N.: A new detailed chemical model for indoor air pollution, Atmos. Environ., 41, 1164–1179, 2007.
Chang, J.-G., Chen, H.-T., Xu, S., and Lin, M. C.: Computational study on the kinetics and mechanisms for the unimolecular decomposition of formic and oxalic acids, J. Phys. Chem. A., 111, 6789–6797, 2007.
Chhantyal-Pun, R., Welz, O., Savee, J. D., Eskola, A. J., Lee, E. P., Blacker, L., Hill, H. R., Ashcroft, M., Khan, M. A. H., Lloyd-Jones, G. C., and Evans, L.: Direct measurements of unimolecular and bimolecular reaction kinetics of the Criegee intermediate (CH3)2COO, J. Phys. Chem. A., 121, 4–15, 2017.
Chao, W., Hsieh, J.-T., Chang C.-H., and Lin J. J.-M.: Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapour, Science, 347, 751–754, https://doi.org/10.1126/science.1261549, 2015.
Chen, B.-Z., Anglada, J. M., Huang, M.-B., and Kong, F.: The Reaction of CH2 (X3B1) with O2 (): A Theoretical CASSCF/CASPT2 Investigation, J. Phys. Chem. A, 106, 1877–1884, 2002.
Chhantyal-Pun, R., Davey, A., Shallcross, D. E., Percival, C. J., and Orr-Ewing, A. J.: A kinetic study of the CH2OO Criegee intermediate self-reaction, reaction with SO2 and unimolecular reaction using cavity ring-down spectroscopy, Phys. Chem. Chem. Phys., 17, 3617–3626, 2015.
Chhantyal-Pun, R., Khan, M. A. H., Taatjes, C. A., Percival, C. J., Orr-Ewing, A. J., and Shallcross, D. E.: Criegee intermediates: production, detection and reactivity, Int. Rev. Phys. Chem., 39, 385–424, 2020.
Chung, C.-A., Su, J. W., and Lee, Y.-P.: Detailed mechanism and kinetics of the reaction of Criegee intermediate CH2OO with HCOOH investigated via infrared identification of conformers of hydroperoxymethyl formate and formic acid anhydride, Phys. Chem. Chem. Phys., 21, 21445–21455, 2019.
Chuong, B., Zhang, J., and Donahue, N. M.: Cycloalkene Ozonolysis: Collisionally Mediated Mechanistic Branching, J. Am. Chem. Soc., 126, 12363–12373, 2004.
Cox, R. A., Ammann, M., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 20, 13497–13519, https://doi.org/10.5194/acp-20-13497-2020, 2020.
Cremer, D.: Stereochemistry of the Ozonolysis of Alkenes: Ozonide versus Carbonyl Oxide Control, Angew. Chem., 20, 888–889, 1981a.
Cremer, D.: Theoretical Determination of Molecular Structure and Conformation. 8. Energetics of the Ozonolysis Reaction. Primary Ozonide vs. Carbonyl Oxide Control of Stereochemistry, J. Am. Chem. Soc., 103, 3627–3633, 1981b.
Drozd, G. T. and Donahue, N. M.: Pressure dependence of stabilized Criegee intermediate formation from a sequence of alkenes, J. Phys. Chem. A, 115, 4381–4387, 2011.
Drozd, G. T., Kroll, J., and Donahue, N. M.: 2,3-Dimethyl-2-butene (TME) ozonolysis: Pressure dependence of stabilized criegee intermediates and evidence of stabilized vinyl hydroperoxides, J. Phys. Chem. A, 115, 161–166, 2011.
Drozd, G. T., Kurtén, T., Donahue, N. M., and Lester, M. I.: Unimolecular decay of the dimethyl-substituted criegee intermediate in alkene ozonolysis: Decay time scales and the importance of tunnelling, J. Phys. Chem. A, 121, 6036–6045, 2017.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurteién, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Elshorbany, Y. F., Kurtenbach, R., Wiesen, P., Lissi, E., Rubio, M., Villena, G., Gramsch, E., Rickard, A. R., Pilling, M. J., and Kleffmann, J.: Oxidation capacity of the city air of Santiago, Chile, Atmos. Chem. Phys., 9, 2257–2273, https://doi.org/10.5194/acp-9-2257-2009, 2009.
Emmerson, K. M., Carslaw, N., and Pilling, M. J.: Urban Atmospheric Chemistry During the PUMA Campaign 2: Radical Budgets for OH, HO2 and RO2, J. Atmos. Chem., 52, 165–183, 2005.
Fenske, J. D., Hasson, A. S., Paulson, S. E., Kuwata, K. T., Ho, A., and Houk, K. N.: The Pressure Dependence of the OH Radical Yield from Ozone–Alkene Reactions, J. Phys. Chem. A, 104, 7821–7833, 2000.
Fliszár, S. and Granger, M.: A Quantitative Investigation of the Ozonolysis Reaction: XI. On the Effects of Substituents in Directing the Ozone Cleavage of trans-1,2-Disubstituted Ethylenes, J. Am. Chem. Soc., 92, 3361–3369, 1970.
Fliszár, S. and Renard, J.: Quantitative investigation of the ozonolysis reaction. XIV. A simple carbonium ion stabilization approach to the ozone cleavage of unsymmetrical olefins, J. Am. Chem. Soc., 93, 6953–6963, 1970.
Fliszár, S., Renard, J., and Simon, D. Z.: A Quantitative Investigation of the Ozonolysis Reaction. XV. Quantum Chemical Interpretation of the Substituent Effects on the Cleavage of l,2,3-Trioxolanes, J. Am. Chem. Soc., 93, 6953–6963, 1971.
Foreman, E. S., Kapnas, K. M., and Murray, C.: Reactions between Criegee Intermediates and the inorganic Acids HCl and HNO3: Kinetics and Atmospheric Implications, Angew. Chem. Int. Edit., 55, https://doi.org/10.1002/anie.201604662, 2016.
Grosjean, D., Williams, E. L., and Grosjean, E.: Atmospheric chemistry of isoprene and of its carbonyl products, Environ. Sci. Technol., 27, 830–840, 1993a.
Grosjean, D., Williams, E. L., Grosjean, E., Andino, J. M., and Seinfeld, J. H.: Atmospheric oxidation of biogenic hydrocarbons: reaction of ozone with b-pinene, d-limonene and trans-caryophyllene, Environ. Sci. Technol., 27, 2754–2758, 1993b.
Grosjean, D., Grosjean, E., and Williams, E. L.: Atmospheric Chemistry of Olefins: A Product Study of the Ozone-Alkene Reaction with Cyclohexane Added to Scavenge OH, Environ. Sci. Technol., 28, 188–196, 1994.
Grosjean, E. and Grosjean, D.: Carbonyl products of the gas phase reaction of ozone with 1,1-disubstituted alkenes. J. Atmos. Chem., 24, 141–156, https://doi.org/10.1007/BF00162408, 1996a.
Grosjean, E. and Grosjean, D.: Rate constants for the gas phase reaction of ozone with 1,1-disubstituted alkenes, Int. J. Chem. Kinet., 28, 911–918, 1996b.
Grosjean, E. and Grosjean, D.: Gas phase reaction of alkenes with ozone: Formation yields of primary carbonyls and biradicals, Environ. Sci. Technol., 31, 2421–2427, 1997a.
Grosjean, E. and Grosjean, D.: The Gas Phase Reaction of Unsaturated Oxygenates with Ozone: Carbonyl Products and Comparison with the Alkene-Ozone Reaction, J. Atmos. Chem., 27, 271–289, 1997b.
Grosjean, E. and Grosjean, D.: The Reaction of Unsaturated Aliphatic Oxygenates with Ozone, J. Atmos. Chem., 32, 205–232, 1999.
Grosjean, E., Grosjean, D., and Seinfeld, J. H.: Gas phase reaction of ozone with trans-2-hexenal, trans-2-hexenyl acetate, ethylvinyl ketone and 6-methyl-5-hepten-2-one, Int. J. Chem. Kinet., 28, 373–382, 1996.
Gutbrod, R., Meyer, S., Rahman, M. M., and Schindler, R. N.: On the use of CO as scavenger for OH radicals in the ozonolysis of simple alkenes and isoprene, Int. J. Chem. Kinet., 29, 717–723, 1997.
Hakala, J. P. and Donahue, N. M.: Pressure-Dependent Criegee Intermediate Stabilization from Alkene Ozonolysis, J. Phys. Chem. A, 120, 2173–2178, 2016.
Hakala, J. P. and Donahue, N. M.: Pressure Stabilization of Criegee Intermediates Formed from Symmetric trans-Alkene Ozonolysis, J. Phys. Chem. A, 122, 9426–9434, 2018.
Hakola, H., Arey, J., Aschmann, S. M., and Atkinson, R.: Product formation from the gas-phase reactions of OH radicals and O3 with a series of monoterpenes, J. Atmos. Chem., 18, 75–102, 1994.
Hansel, A., Scholz, W., Mentler, B., Fischer, L., and Berndt, T.: Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH4+ and acetate chemical ionization mass spectrometry, Atmos. Environ., 186, 248–255, https://doi.org/10.1016/j.atmosenv.2018.04.023, 2018.
Hasson, A. S., Orzechowska, G., and Paulson, S. E.: Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes 1. Ethene, trans-2-butene, and 2,3-dimethyl-2-butene, J. Geophys. Res., 106, 34131–34142, 2001a.
Hasson, A. S., Ho, A. W., Kuwata, K., and Paulson, S. E.: Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes 2. Asymmetric and biogenic alkenes, J. Geophys. Res., 106, 34143–34153, 2001b.
Hatakeyama, S., Kobayashi, H., and Akimoto, H.: Gas-phase oxidation of sulfur dioxide in the ozone-olefin reactions, J. Phys. Chem. 88, 4736–4739, 1984.
Heard, D. E., Carpenter, L. J., Creasey, D. J., Hopkins, J. R., Lee, J. R., Lewis, A. C., Pilling, M. J., Seakins, P. W., Carslaw, N., and Emmerson, K. M.: High levels of the hydroxyl radical in the winter urban troposphere, Geophys. Res. Lett., 31, L18112, https://doi.org/10.1029/2004GL020544, 2004.
Herron, J. T. and Huie, R. E.: Stopped-flow studies of the mechanisms of ozone-alkene reactions in the gas phase: propene and isobutene, J. Am. Chem. Soc., 99, 5430–5435, 1977.
Herron, J. T. and Huie, R. E.: Stopped-flow studies of the mechanisms of ozone-alkene reactions in the gas phase. I. Ethylene, J. Am. Chem. Soc., 99, 5430–5435, 1978.
Horie, O. and Moortgat, G. K.: Decomposition pathways of the excited Criegee intermediates in the ozonolysis of simple alkenes, Atmos. Environ. A-Gen., 25, 1881–1896, 1991.
Huang, H.-L., Chao, W., and Lin, J. J.-M.: Kinetics of a Criegee intermediate that would survive at high humidity and may oxidize atmospheric SO2, P. Natl. Acad. Sci. USA, 112, 10857–10862, 2015.
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in a-pinene ozonolysis, Nat. Commun., 12, 1–6, https://doi.org/10.1038/s41467-021-21172-w, 2021.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, 1997.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018a.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9329–9349, https://doi.org/10.5194/acp-18-9329-2018, 2018b.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jenkin, M. E., Valorso, R., Aumont, B., Newland, M. J., and Rickard, A. R.: Estimation of rate coefficients for the reactions of O3 with unsaturated organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 20, 12921–12937, https://doi.org/10.5194/acp-20-12921-2020, 2020.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, 2008.
Kalalian, C., Roth, E., El Dib, G., Singh, H. J., Rao, P. K., and Chakir, A.: Product investigation of the gas phase ozonolysis of 1-penten-3-ol, cis-2-penten-1-ol and trans-3-hexen-1-ol, Atmos. Environ., 238, 117732, 2020.
Kalberer, M., Yu, J., Cocker, D. R., Flagan, R. C., and Seinfeld, J. H.: Aerosol Formation in the Cyclohexene-Ozone System, Environ. Sci. Technol., 34, 4894–4901, 2000.
Kidwell, N. M., Li, H., Wang, X., Bowman, J. M., and Lester, M. I.: Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products, Nat. Chem., 8, 509–514, 2016.
Klotz, B., Barnes, I., and Imamura, T.: Product study of the gas phase reactions of O3, OH and NO3 reactions with methyl vinyl ether, Phys. Chem. Chem. Phys., 6, 1725–1734, 2004.
Kramp, F. and Paulson, S. E.: The gas phase reaction of ozone with 1, 3-butadiene: formation yields of some toxic products, Atmos. Environ., 34, 35–43, 2000.
Kristensen, K., Cui, T., Zhang, H., Gold, A., Glasius, M., and Surratt, J. D.: Dimers in α-pinene secondary organic aerosol: effect of hydroxyl radical, ozone, relative humidity and aerosol acidity, Atmos. Chem. Phys., 14, 4201–4218, https://doi.org/10.5194/acp-14-4201-2014, 2014.
Kroll, J. H., Hanisco, T. F., Donahue, N. M., Demerjian, K. L., and Anderson, J. G.: Accurate, direct measurements of OH yields from gas-phase ozone-alkene reactions using an in-situ LIF instrument, Geophys. Res. Lett., 28, 3863–3866, 2001.
Kroll, J., Donahue, N. M., Cee, V. J., Demerjian, K. L., and Anderson, J. G.: Gas-phase ozonolysis of alkenes: Formation of OH from anti carbonyl oxides, J. Am Chem. Soc., 124, 8518–8519, 2002.
Kuwata, K. T. and Valin, L. C.: Quantum chemical and RRKM/master equation studies of isoprene ozonolysis: Methacrolein and methacrolein oxide, Chem. Phys. Lett., 451, 186–191, 2008.
Kuwata, K. T., Valin, L. C., and Converse, A. D.: Quantum chemical and master equation studies of the methyl vinyl carbonyl oxides formed in isoprene ozonolysis, J. Phys. Chem. A, 109, 10710–10725, 2005.
Kuwata, K. T., Guinn, E. J., Hermes, M. R., Fernandez, J. A., Mathison, J. M., and Huang, K. A.: Computational Re-examination of the Criegee Intermediate-Sulfur Dioxide Reaction, J. Phys. Chem. A, 119, 10316–10335, 2015.
Kuwata, K. T., Luu, L., Weberg, A. B., Huang, K., Parsons, A. J., Peebles, L. A., Rackstraw, N. B., and Kim, M. J.: Quantum Chemical and Statistical Rate Theory Studies of the Vinyl Hydroperoxides Formed in trans-2-Butene and 2,3-Dimethyl-2-butene Ozonolysis, J. Phys. Chem. A, 122, 2485–2502, 2018.
Le Person, A., Solignac, G., Oussar, F., Daële, V., Mellouki A., Winterhalter, R., and Moortgat, G. K.: Gas phase reaction of allyl alcohol (2-propen-1-ol) with OH radicals and ozone, Phys. Chem. Chem. Phys., 11, 7619–7628, 2009.
Lee, A., Goldstein, A. H., Keywood, M. D., Gao, S., Varutbangkul, V., Bahreini, R., Ng, N. L., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary organic aerosol yields from the ozonolysis of ten different terpenes, J. Geophys. Res., 111, D07302, https://doi.org/10.1029/2005JD006437, 2006.
Lei, X., Wang, W., Gao, J., Wang, S. and Wang, W.: Atmospheric Chemistry of Enols: The Formation Mechanisms of Formic and Peroxyformic Acids in Ozonolysis of Vinyl Alcohol, J. Phys. Chem. A, 124, 4271–4279, 2020.
Lewin, A. G., Johnson, D., Price, D. W., and Marston, G.: Aspects of the kinetics and mechanism of the gas-phase reactions of ozone with conjugated dienes, Phys. Chem. Chem. Phys., 3, 1253–1261, 2001.
Lewis, T. R., Blitz, M. A., Heard, D. E., and Seakins, P. W.: Direct evidence for a substantive reaction between the Criegee intermediate, CH2OO, and the water vapour dimer, Phys. Chem. Chem. Phys., 17, 4859–4863, 2015.
Lin, L.-C., Chang, H., Chang, C., Chao, W., Smith, M. C., Chang, C., Lin, J. J., and Takahashi, K.: Competition between H2O and (H2O)2 reactions with CH2OO/CH3CHOO, Phys. Chem. Chem. Phys., 18, 4557–4568, 2016.
Long, B., Bao, J. L., and Truhlar, D. G.: Rapid unimolecular reaction of stabilized Criegee intermediates and implications for atmospheric chemistry, Nat. Commun., 10, 1–8, https://doi.org/10.1038/s41467-019-09948-7, 2019.
Ma, Y. and Marston G.: Multifunctional acid formation from the gas-phase ozonolysis of β-pinene, Phys. Chem. Chem. Phys., 10, 6115–6126, 2008.
Ma, Y. and Marston G.: Formation of organic acids from the gas-phase ozonolysis of terpinolene, Phys. Chem. Chem. Phys., 11, 4198–4209, 2009.
Mackenzie-Rae, F. A., Wallis, H. J., Rickard, A. R., Pereira, K. L., Saunders, S. M., Wang, X., and Hamilton, J. F.: Ozonolysis of α-phellandrene – Part 2: Compositional analysis of secondary organic aerosol highlights the role of stabilised Criegee intermediates, Atmos. Chem. Phys., 18, 4673–4693, https://doi.org/10.5194/acp-18-4673-2018, 2018.
Martinez, R. I. and Herron, J. T.: Stopped-flow studies of the mechanisms of alkene-ozone reactions in the gas-phase: tetramethylethylene, J. Phys. Chem. A, 91, 946–953, 1987.
Mauldin III, R. L., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V.-M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, https://doi.org/10.1038/nature11278, 2012.
Neeb, P., Horie, O., and Moortgat, G. K.: The nature of the transitory product in the gas-phase ozonolysis of ethene, Chem. Phys. Lett., 37, 150–156, 1995.
Neeb, P., Horie, O., and Moortgat, G. K.: Formation of secondary ozonides in the gas-phase ozonolysis of simple alkenes, Tetrahedron Lett., 246, 9297–9300, 1996.
Neeb, P., Sauer, F., Horie, O., and Moortgat, G. K.: Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour, Atmos. Environ., 31, 1417–1423, 1997.
Neeb, P., Horie, O., and Moortgat, G. K.: The ethene−ozone reaction in the gas phase, J. Phys. Chem. A., 102, 6778–6785, 1998.
Newland, M. J., Rickard, A. R., Alam, M. S., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: Kinetics of stabilised Criegee intermediates derived from alkene ozonolysis: reactions with SO2, H2O and decomposition under boundary layer conditions, Phys. Chem. Chem. Phys., 17, 4076–4088, 2015.
Newland, M. J., Rickard, A. R., Sherwen, T., Evans, M. J., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: The atmospheric impacts of monoterpene ozonolysis on global stabilised Criegee intermediate budgets and SO2 oxidation: experiment, theory and modelling, Atmos. Chem. Phys., 18, 6095–6120, https://doi.org/10.5194/acp-18-6095-2018, 2018.
Newland, M. J., Nelson B. S., Muñoz, A., Ródenas, M., Vera, T., Tarrega, J., and Rickard, A. R.: Trends in stabilisation of Criegee intermediates from alkene ozonolysis, Phys. Chem. Chem. Phys., 22, 13698–13706, 2020.
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., and Miller, D. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, 2016.
Nguyen, T. L., Winterhalter, R., Moortgat, G., Kanawati, B., Peeters, J., and Vereecken, L.: The gas-phase ozonolysis of β-caryophyllene (C15H24). Part II: A theoretical study, Phys. Chem. Chem. Phys., 11, 4173–4183, 2009a.
Nguyen, T. L., Peeters, J., and Vereecken, L.: Theoretical study of the gas-phase ozonolysis of β-pinene (C10H16), Phys. Chem. Chem. Phys., 11, 5643–5656, 2009b.
Nguyen, T. L., Lee, H., Matthews, D. A., McCarthy, M. C., and Stanton, J. F.: Stabilization of the Simplest Criegee Intermediate from the Reaction between Ozone and Ethylene: A High-Level Quantum Chemical and Kinetic Analysis of Ozonolysis, J. Phys. Chem. A, 119, 5524–5533, 2015.
Niki, H., Maker, P. D., Savage, C. M., and Breitenbach, L. P.: Atmospheric ozone-olefin reactions, Environ. Sci. Technol., 17, 312–322, 1983.
Niki, H., Maker, P. D., Savage, C. M., Breitenbach, L. P., and Hurley, M. D.: FTIR spectroscopic study of the mechanism for the gas-phase reaction between ozone and tetramethylethylene, J. Phys. Chem. A, 91, 941–946, 1987.
O'Dwyer, M. A., Carey, T. J., Healy, R. M., Wenger, J. C., Picquet-Varrault, B., and Doussin, J. F.: The Gas-phase Ozonolysis of 1-Penten-3-ol, (Z)-2-Penten-1-ol, and 1-Penten-3-one: Kinetics, Products and Secondary Organic Aerosol Formation, Z. Phys. Chem., 224, 1059–1080, 2010.
O'Neal, H. E. and Blumstein, C.: A new mechanism for gas phase ozone-olefin reactions, Int. J. Chem. Kinet., 5, 397–413, 1973.
Olzmann, M., Kraka, E., Cremer, D., Gutbrod, R., and Anderson, S.: Energetics, Kinetics, and Product Distributions of the Reactions of Ozone with Ethene and 2,3-Dimethyl-2-butene, J. Phys. Chem. A, 101, 9421–9429, 1997.
Orzechowska, G. E. and Paulson, S. E.: Production of OH radicals from the reactions of C4-C6 internal alkenes and styrenes with ozone in the gas phase, Atmos. Environ., 36, 571–581, 2002.
Ouyang, B., McLeod, M. W., Jones, R. L., and Bloss, W. J.: NO3 radical production from the reaction between the Criegee intermediate CH2OO and NO2, Phys. Chem. Chem. Phys., 15, 17070–17075, 2013.
Paulson, S. E. and Orlando, J. J.: The reactions of ozone with alkenes: An important source of HOx in the boundary layer, Geophys. Res. Letts., 23, 3727–3730, 1996.
Peltola, J., Seal, P., Inkilä, A., and Eskola, A.: Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: unimolecular decomposition of CH2OO and its reaction with formic acid, Phys. Chem. Chem. Phys., 22, 11797–11808, 2020.
Percival, C. J., Welz, O., Eskola, A. J., Savee, J. D., Osborn, D. L., Topping, D. O., Lowe, D., Utembe, S. R., Bacak, A., McFiggans, G., Cooke, M. C., Xiao, P., Archibald, A. T., Jenkin, M. E., Derwent, R. G., Riipinen, I., Mok, D. W. K., Lee, E. P. F., Dyke, J. M., Taatjes, C. A., and Shallcross, D. E.: Regional and global impacts of Criegee intermediates on atmospheric sulphuric acid concentrations and first steps of aerosol formation, Faraday Discuss., 165, 45–73, https://doi.org/10.1039/C3FD00048F, 2013.
Pfeifle, M., Ma, Y.-T., Jasper, A. W., Harding, L. B., Hase, W. L., and Klippenstein, S. J.: Nascent energy distribution of the Criegee intermediate CH2OO from direct dynamics calculations of primary ozonide dissociation, J. Chem. Phys., 148, 174306, https://doi.org/10.1063/1.5028117, 2018.
Picquet-Varrault, B., Scarfogliero, M, and Doussin, J.-F.: Atmospheric Reactivity of Vinyl Acetate: Kinetic and Mechanistic Study of Its Gas-Phase Oxidation by OH, O3, and NO3, Environ. Sci. Technol., 44, 4615–4621, 2010.
Pierce, J. R., Evans, M. J., Scott, C. E., D'Andrea, S. D., Farmer, D. K., Swietlicki, E., and Spracklen, D. V.: Weak global sensitivity of cloud condensation nuclei and the aerosol indirect effect to Criegee + SO2 chemistry, Atmos. Chem. Phys., 13, 3163–3176, https://doi.org/10.5194/acp-13-3163-2013, 2013.
Pinelo, L., Gudmundsdottir, A. D., and Ault, B. S.: Matrix Isolation Study of the Ozonolysis of 1,3- and 1,4-Cyclohexdiene: Identification of Novel Reaction Pathways, J. Phys. Chem. A, 117, 4174–4182, 2013.
Raghunath, P., Lee, Y.-P., and Lin, M. C.: Computational chemical kinetics for the reaction of Criegee intermediate CH2OO with HNO3 and its catalytic conversion to OH and HCO, J. Phys. Chem. A, 121, 3871–3878, 2017.
Rathman, W. C. D., Claxton, T. A., Rickard, A. R., and Marston, G.: A theoretical investigation of OH formation in the gas-phase ozonolysis of E-but-2-ene and Z-but-2-ene, Phys. Chem. Chem. Phys., 1, 3981–3985, 1999.
Ren, Y., Grosselin, B., Daële, V., and Mellouki, A.: Investigation of the reaction of ozone with isoprene, methacrolein and methyl vinyl ketone using the HELIOS chamber, Faraday Discussions, 22, 289–311, 2017.
Reissell, A., Harry, C., Aschmann, S. M., Atkinson, R., and Arey, J.: Formation of acetone from the OH radical- and O3-initiated reactions of a series of monoterpenes, J. Geophys. Res., 104, 13869–13879, 1999.
Rickard, A. R., Johnson, D., McGill, C. D., and Marston, G.: OH Yields in the Gas-Phase reactions of Ozone with Alkenes, J. Phys. Chem. A, 103, 7656–7664, 1999.
Sakamoto, Y., Inomata, S., and Hirokawa, J.: Oligomerization Reaction of the Criegee Intermediate Leads to Secondary Organic Aerosol Formation in Ethylene Ozonolysis, J. Phys. Chem. A, 117, 12912–12921, 2013.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Sheps, L., Scully, A. M., and Au, K.: UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO, Phys. Chem. Chem. Phys., 16, 26701–26706, 2014.
Sheps, L., Rotavera, B., Eskola, A. J., ., Osborn, D. L., Taatjes, C., Au, K., Shallcross, D. E., Khan, M. A. H., and Percival, C. J.: The reaction of Criegee intermediate CH2OO with water dimer: Primary products and atmospheric impact, Phys. Chem. Chem. Phys., 19, 21970–21979, 2017.
Sipilä, M., Jokinen, T., Berndt, T., Richters, S., Makkonen, R., Donahue, N. M., Mauldin III, R. L., Kurtén, T., Paasonen, P., Sarnela, N., Ehn, M., Junninen, H., Rissanen, M. P., Thornton, J., Stratmann, F., Herrmann, H., Worsnop, D. R., Kulmala, M., Kerminen, V.-M., and Petäjä, T.: Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids, Atmos. Chem. Phys., 14, 12143–12153, https://doi.org/10.5194/acp-14-12143-2014, 2014.
Smith, M. C., Chao, W., Takahashi, K., Boering, K. A., and Lin, J. J.-M.: Unimolecular Decomposition Rate of the Criegee Intermediate (CH3)2COO Measured Directly with UV Absorption Spectroscopy, J. Phys. Chem. A, 120, 4789–4798, https://doi.org/10.1021/acs.jpca.5b12124, 2016.
Stephenson, T. A. and Lester, M. I.: Unimolecular decay dynamics of Criegee intermediates: Energy-resolved rates, thermal rates, and their atmospheric impact, Int. Rev. Phys. Chem., 39, 1–33, 2020.
Stone, D., Blitz, M., Daubney, L., Howes, N. U. M., and Seakins, P.: Kinetics of CH2OO reactions with SO2, NO2, NO, H2O, and CH3CHO as a function of pressure, Phys. Chem. Chem. Phys., 16, 1139–1149, 2014.
Stone, D., Au, K., Sime, S., Medeiros, D. J., Blitz, M., Seakins, P., Decker. Z., and Sheps, L.: Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and CD2OO, Phys. Chem. Chem. Phys., 20, 24940–24954, 2018.
Su, F., Calvert, J. G., and Shaw, J. H.: A FT IR spectroscopic study of the ozone-ethene reaction mechanism in oxygen-rich mixtures, J. Phys. Chem., 84, 239–246, 1980.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Scheer, A. M., Shallcross, D. E., Rotavera, B., Lee, E. P. F., Dyke, J. M., Mok, D. K. W., Osborn, D. L., and Percival, C. J.: Direct Measurements of Conformer-Dependent Reactivity of the Criegee Intermediate CH3CHOO, Science, 340, 177–180, 2013.
Taatjes, C. A., Caravan, R. L., Winiberg, F. A. F., Zuraski, K., Au, K., Sheps, L., Osborn, D. L., Vereecken, L., and Percival, C. J.: Insertion products in the reaction of carbonyl oxide Criegee intermediates with acids: Chloro(hydroperoxy)methane formation from reaction of CH2OO with HCl and DCl, Mol. Phys., 119, e1975199, https://doi.org/10.1080/00268976.2021.1975199, 2021.
Taatjes, C. A., Liu, F., Rotavera, B., Kumar, M., Caravan, R., Osborn, D. L., Thompson, W. H., and Lester, M. I.: Hydroxyacetone production from C3 Criegee intermediates, J. Phys. Chem. A., 121, 6–23, 2017.
Taipale, R., Sarnela, N., Rissanen, M. P., Junninen, H., Rantala, P., Korhonen, F., Siivola, E., Berndt, T., Kulmala, M., Mauldin III, R., Petaja, T., and Sipilä, M. J.: New instrument for measuring atmospheric concentrations of non-OH oxidants of SO2, Boreal Environ. Res., 19, 55–70, 2014.
Thiault, G., Thévenet, R., Mellouki, A., and Le Bras, G.: OH and O3-initiated oxidation of ethyl vinyl ether, Phys. Chem. Chem. Phys., 4, 613–619, 2002.
Tuazon, E. C., Aschmann, S. M., Arey, J., and Atkinson, R.: Products of the Gas-Phase Reactions of O3 with a Series of Methyl-Substituted Ethenes, Environ. Sci. Technol., 31, 10, 3004–3009, 1997.
Vansco, M. F., Caravan, R. L., Zuraski, K., Winiberg, F. A. F., Au, K., Trongsiriwat, N., Walsh, P. J., Osborn, D. L., Percival, C. J., Khan, M. A. H., Shallcross, D. E., Taatjes, C. A., and Lester, M. I.: Experimental Evidence of Dioxole Unimolecular Decay Pathway for Isoprene-Derived Criegee Intermediates, J. Phys. Chem. A, 124, 3542–3554, 2020.
Vereecken, L.: The reaction of Criegee intermediates with acids and enols, Phys. Chem. Chem. Phys., 19, 28630–28640, 2017.
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 41, 6259–6293, 2012.
Vereecken, L. and Nguyen, H. M. T.: Theoretical Study of the Reaction of Carbonyl Oxide with Nitrogen Dioxide: CH2OO + NO2, Int. J. Chem. Kinet., 49, 752–760, 2017.
Vereecken, L., Novelli, A., and Taraborrelli, D.: Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 19, 31599–31612, 2017.
Vereecken, L., Aumont, B., Barnes, I., Bozzelli, J., Goldman, M., Green, W., Madronich, S., Mcgillen, M., Mellouki, A., Orlando, J., Picquet-Varrault, B., Rickard, A., Stockwell, W., Wallington, T., and Carter, W.: Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry. Int. J. Chem. Kinet., 50, 435–469, https://doi.org/10.1002/kin.21172, 2018.
Vichietti, R. M., Keidel Spada, R. F., Ferreira da Silva, A. B., Correto Machado, F. B., and Andrade Haiduke, R. L.: Accurate Calculations of Rate Constants for the Forward and Reverse H2O + CO ↔ HCOOH Reactions, ChemistrySelect, 2, 7267–7272, 2017.
Viero, L.: Kinetics and mechanisms for the oxidation of unsaturated organic acids and esters under atmospheric conditions, PhD thesis, University College Dublin, 2008.
Wadt, W. R. and Goddard, W. A.: The electronic structure of the Criegee intermediate. Ramifications for the mechanism of ozonolysis, J. Am. Chem. Soc., 97, 3004–3021, 1975.
Wang, J., Zhou, L., Wang, W., and Ge, M.: Gas-phase reaction of two unsaturated ketones with atomic Cl and O3: kinetics and products, Phys. Chem. Chem. Phys., 17, 12000–12012, 2015.
Wang, S., Newland, M. J., Deng, W., Rickard, A. R., Hamilton, J. F., Muñoz, A., Ródenas, M., Vázquez, M. M., Wang, L., and Wang, X.: Aromatic photo-oxidation, a new source of atmospheric acidity, Environ. Sci. Technol., 54, 7798–7806, 2020.
Watson, N. A. I.: An Analysis of the Sources and Sinks for Criegee Intermediates: An Extended Computational Study, PhD thesis, University of Cardiff, http://orca.cardiff.ac.uk/id/eprint/144742 (last access: 15 October 2021), 2021.
Weidman, J. D., Allen, R. T., Moore, K. B., and Schaefer, H. F.: High-level theoretical characterization of the vinoxy radical (•CH2CHO) + O2 reaction, J. Chem. Phys., 148, 184308, 2018.
Welz, O., Savee, J. D., Osborn, D. L., Vasu, S. S., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct Kinetic Measurements of Criegee Intermediate (CH2OO) Formed by Reaction of CH2I with O2, Science, 335, 204–207, 2012.
Welz, O., Eskola, A. J., Sheps, L., Rotavera, B., Savee, J. D., Scheer, A. M., Osborn, D. L., Lowe, D., Murray Booth, A., Xiao, P., Anwar H., Khan, M., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Rate coefficients of C1 and C2 Criegee intermediate reactions with formic and acetic acid near the collision limit: direct kinetics measurements and atmospheric implications, Angew. Chem. Int. Edit., 53, 4547–4750, 2014.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., and Smarte, M. D.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Winterhalter, R., Neeb, P., Grossmann, D., Kolloff, A., Horie, O., and Moortgat, G.: Products and mechanism of the gas phase reaction of ozone with β-pinene, J. Atmos. Chem., 35, 165–197, 2000.
Winterhalter, R., Herrmann, F., Kanawati, B., Nguyen, T. L., Peeters, J., Vereecken, L., and Moortgat, G.: The gas-phase ozonolysis of β-caryophyllene (C15H24). Part I: an experimental study, Phys. Chem. Chem. Phys., 11, 4152–4172, 2009.
Wolff, S., Boddenberg, A., Thamm, J., Turner, W. V., and Gräb, S.: Gas-phase ozonolysis of ethene in the presence of carbonyl-oxide scavengers, Atmos. Environ, 31, 2965–2969, 1997.
Yu, J. Z., Cocker, D. R., Griffin, R. J., Flagan, R. C., and Seinfeld, J. H.: Gas-phase ozone oxidation of monoterpenes: Gaseous and particulate products, J. Atmos. Chem., 34, 207–258, 1999.
Zhao, Y., Wingen, L. M., Perraud, V., Greaves, J., and Finlayson-Pitts, B. J.: Role of the reaction of stabilized Criegee intermediates with peroxy radicals in particle formation and growth in air, Phys. Chem. Chem. Phys., 17, 12500–12514, 2015.
Zhou, S.: Atmospheric Oxidation of Vinyl Ethers, PhD thesis, Bergische Universität Wuppertal, http://elpub.bib.uni-wuppertal.de (last access: 15 April 2021), 2007.
Zhou, S., Barnes, I., Zhu, T., Klotz, B., Albu, M., Bejan, I., and Benter, T.: Product Study of the OH, NO3, and O3 Initiated Atmospheric Photooxidation of Propyl Vinyl Ether, Environ. Sci. Technol., 40, 5415–5421, 2006.
Ziemann, P. J.: Evidence for Low-Volatility Diacyl Peroxides as a Nucleating Agent and Major Component of Aerosol Formed from Reactions of O3 with Cyclohexene and Homologous Compounds, J. Phys. Chem. A, 106, 4390–4402, 2002.
- Abstract
- Introduction
- Primary ozonide fragmentation
- Stabilisation of the Criegee intermediate
- Unimolecular reactions of CI∗ and SCI
- Bimolecular reactions of SCI
- Example of protocol application
- Protocol evaluation
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Primary ozonide fragmentation
- Stabilisation of the Criegee intermediate
- Unimolecular reactions of CI∗ and SCI
- Bimolecular reactions of SCI
- Example of protocol application
- Protocol evaluation
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement