the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Apr 2022
| 26 Apr 2022
Global simulations of monoterpene-derived peroxy radical fates and the distributions of highly oxygenated organic molecules (HOMs) and accretion products
Ruochong Xu
Joel A. Thornton
Ben H. Lee
Yanxu Zhang
Lyatt Jaeglé
Felipe D. Lopez-Hilfiker
Pekka Rantala
Tuukka Petäjä
We evaluate monoterpene-derived peroxy radical (MT-RO2) unimolecular autoxidation and self- and cross-reactions with other RO2 species in the GEOS-Chem global chemical transport model. The formation of associated highly oxygenated organic molecules (HOMs) and accretion products are tracked in competition with other bimolecular reactions. Autoxidation is the dominant fate up to 6–8 km for first-generation MT-RO2, which can undergo unimolecular H shifts. Reaction with NO can be a more common fate for H-shift rate constants < 0.1 s−1 or at altitudes higher than 8 km due to the imposed Arrhenius temperature dependence of unimolecular H shifts. For MT-derived HOM-RO2, generated by multistep autoxidation of first-generation MT-RO2, reaction with other RO2 species is predicted to be the major fate throughout most of the boreal and tropical forest regions, whereas reaction with NO dominates in the temperate and subtropical forests of the Northern Hemisphere. The newly added reactions result in an approximate 4 % global average decrease in HO2 and RO2, mainly due to faster self-/cross-reactions of MT-RO2, but the impact upon HO2, OH, and NOx abundances is only important in the planetary boundary layer (PBL) over portions of tropical forests. Predicted HOM concentrations in MT-rich regions and seasons can exceed total organic aerosol predicted by the standard version of the GEOS-Chem model depending on the parameters used. Comparisons to observations reveal that large uncertainties remain for key reaction parameters and processes, especially with respect to the photochemical lifetime and volatility of HOMs as well as the rates and branching of associated RO2 accretion products. Further observations and laboratory studies related to MT-RO2-derived HOMs and gas-phase RO2 accretion product formation kinetics – especially their atmospheric fate, such as gas–particle partitioning, multiphase chemistry, and net secondary organic aerosol formation – are needed.
- Article
(9442 KB) - Full-text XML
-
Supplement
(2510 KB) - BibTeX
- EndNote





Monoterpenes (MTs) are emitted by terrestrial vegetation at a rate of approximately 50–100 Tg yr−1 (Arneth et al., 2008; Guenther et al., 2012; Messina et al., 2016) and are a significant component of volatile consumer products (VCPs) (McDonald et al., 2018). Reaction of the more common monoterpenes, such as α- and β-pinene, Δ-3 carene, and limonene, with atmospheric oxidants is rapid, on the timescale of an hour, and produces a suite of semi-volatile (effective saturation concentration, C∗, is between 0.3 and 300 µg m−3), low-volatility ( µg m−3), and extremely low-volatility ( µg m−3) products that contribute to the nucleation and growth of aerosol particles through the formation of secondary organic aerosol (SOA) (Bianchi et al., 2019; Ehn et al., 2014; Hallquist et al., 2009; Kulmala et al., 2014; Palen et al., 1992; Pandis et al., 1992; Zhang et al., 1992). Recent work has shown that, even in some isoprene-dominated forest regions, monoterpene oxidation products can be the major component of fine particulate (PM2.5) SOA mass (Lee et al., 2020; Xu et al., 2018; Zhang et al., 2018).
Laboratory studies have shown that at least 30 %–50 % of the condensable mass produced during oxidation of α-pinene, by both the hydroxyl radical (OH) and ozone, is formed promptly in the first generation of oxidation (Berndt et al., 2016; Ehn et al., 2014; Jokinen et al., 2015; Mentel et al., 2015). This prompt formation of low-volatility mass stems from a fraction of the first-generation organic peroxy radicals (RO2) undergoing repeated unimolecular H-shift reactions followed by O2 addition, ultimately leading to highly oxygenated organic molecules (HOMs) that have low volatility or even extremely low volatility. The unimolecular H shifts are the rate-limiting steps to HOM formation and have been shown to exceed 1 s−1 at ∼ 296 K for certain RO2 species (Xu et al., 2019). At such timescales, bimolecular reactions of RO2 with the hydroperoxy radical (HO2), other RO2 species, and nitric oxide (NO), even if the latter is present at up to 1 ppb, are not competitive, and autoxidation to HOMs is expected to be a dominant fate for such RO2 in the atmosphere. Moreover, the rate constants of corresponding RO2 cross-reactions as well as the branching to accretion products, presumably organic peroxides (ROOR′), have been shown to be substantially larger than previous expectations (Berndt et al., 2018a, b) and important for new particle formation and growth (Bianchi et al., 2019). The ROOR′ products can be of low or extremely low volatility as well, even without substantial RO2 H-shift chemistry, but cross-reactions between isoprene-derived RO2 and MT-HOM RO2 specifically can be important in limiting ELVOC (extremely low-volatility organic compound) formation and, thus, nucleation (Öström et al., 2017; Roldin et al., 2019; McFiggans et al., 2019).
To date, relatively few studies have evaluated the global implications of such revisions to our understanding of the monoterpene-derived peroxy radical (MT-RO2) fate (Jokinen et al., 2015; Weber et al., 2020; Roldin et al., 2019; Zhu et al., 2019). Jokinen et al. (2015) showed the impact of MT-HOM formation at specified yields on SOA budgets and cloud condensation nuclei. Weber et al. (2020) used a condensed reaction mechanism to more explicitly treat the formation of HOMs through unimolecular MT-RO2 autoxidation and cross-reactions, but they did not conduct global online simulations. Roldin et al. (2019) used a similarly explicit mechanism in a 1-D column model to simulate HOMs over a boreal forest setting. Zhu et al. (2019) did not simulate autoxidation and used a less stringent definition of HOMs than recommended in Bianchi et al. (2019). Thus, global-scale simulations with online MT-RO2 chemistry and comparisons to observations, either using total organic aerosol mass as a constraint or more specific molecular composition measurements of gas- and aerosol-phase species, remain lacking. Moreover, the sensitivity of HOx, O3, and NOx abundances and lifetimes to such changes in RO2 chemistry have yet to be fully explored in global chemical transport models. The unimolecular MT-RO2 chemistry and faster RO2 cross-reactions have implications for HOx partitioning, OH recycling, and NOx lifetime in low-NOx forest regions. In addition, measurements of highly oxygenated organic nitrates can provide insights into the MT-RO2 reactivity governing the competition between autoxidation, RO2 cross-reactions, and RO2 reactions with nitric oxide (NO).
Herein, we use the GEOS-Chem global chemical transport model to evaluate the impact of MT-RO2 H shifts and cross-reactions on tropospheric hydrogen oxide radicals (HOx = OH + HO2) and total RO2 abundance, on ozone distributions, and to assess the potential contribution of MT-HOM and HOM nitrates to low-volatility and extremely low-volatility components and, by extension, the global budget of SOA. We update the GEOS-Chem mechanisms for MT oxidation using, where possible, laboratory-derived values of mechanistic parameters, such as MT-RO2 unimolecular H-shift rate constants, the fraction of MT-RO2 undergoing H shifts, and the rate constants for cross-reactions between MT-RO2 and other RO2 species, such as those derived from isoprene oxidation. We compare predicted HOMs and HOM nitrates to atmospheric observations in the gas and particle phases from two locations and conduct sensitivity studies to evaluate the impacts of uncertain kinetic parameters and mechanistic assumptions.
2.1 GEOS-Chem model
We use the GEOS-Chem chemical transport model (Bey et al., 2001) driven by assimilated MERRA-2 (Modern-Era Retrospective analysis for Research and Applications, Version 2) (Gelaro et al., 2017) meteorological fields. Simulations were conducted with a 2∘ × 2.5∘ (lat × long) horizontal resolution and 47 vertical levels for 28 months from March 2012 to June 2014. This time period provides the best overlap with available observations of monoterpene-derived oxidation products in the gas and particle phases made during the Southern Oxidant and Aerosol Study (SOAS) and Biogenic Aerosols – Effects on Clouds and Climate (BAECC) field campaigns, described in detail elsewhere (Carlton et al., 2018; Lee et al., 2016, 2018; Lopez-Hilfiker et al., 2016; Petäjä et al., 2016) and discussed further below. The first year of the simulation was for spin-up purposes, to allow for the accumulation of intermediate chemical reservoir species. For comparison to the observations, we sample the model in time and space corresponding to the lowest model grid box containing the location of the observations.
A reference simulation was conducted based on the public version 12.1.0 of GEOS-Chem (http://wiki.geos-chem.org/GEOS-Chem_12#12.1.0, last access: July 2019). The HOx–NOx–VOC–O3–BrOx tropospheric chemistry chemical mechanism in the reference simulation is described in Mao et al. (2010, 2013) with recent updates for biogenic volatile organic compound (VOC) chemistry (Fisher et al., 2016; Travis et al., 2016). Emissions of isoprene and monoterpenes are driven by the Model of Emissions of Gases and Aerosols from Nature v2.1 (MEGAN) (Guenther et al., 2012). Emissions in GEOS-Chem are based on the Harvard-NASA Emission Component (HEMCO) (Keller et al., 2014). Global anthropogenic emissions of NOx, SO2, CO, and various aerosol species are from the CEDS (Community Emission Data System) combined with MIX (Mosaic Asian anthropogenic emission inventory) in Asia, NEI (National Emission Inventory) in the USA, APEI (Air Pollutant Emissions Inventory) in Canada, BRAVO (United States–Mexico Emissions Inventory for the Big Bend Regional Aerosol and Visibility Observational Study) in Mexico, EMEP (European Monitoring and Evaluation Program) in Europe, and DICE (Diffuse and Inefficient Combustion Emissions in Africa). Open fire emissions are from the Global Fire Emissions Database (GFED4). Both gas and aerosol undergo dry deposition, with rates calculated online based on the resistance-in-series algorithm (Wesely, 1989; Zhang et al., 2001). Wet deposition is calculated for water-soluble aerosol and gas following Amos et al. (2012) and Liu et al. (2001).
2.2 Updates to the GEOS-Chem MT oxidation mechanism
Our goal for the mechanism updates was to preserve the current simplified framework for MT chemistry in GEOS-Chem as much as possible but also to include the essential features of the MT-RO2 unimolecular H shifts and bimolecular RO2 cross-reactions (see Fig. 1). Thus, RO2 and closed-shell products quickly became lumped into corresponding pools that loosely related to the dominant functional group character, such as carbonyl, alcohol, or nitrate. In this version, we restricted changes to chemistry stemming from OH reaction and ozonolysis only, and we did not consider nitrate radical (NO3) reactions of MTs. In what follows, to maintain a higher level of clarity, we mostly discuss the mechanism in general terms. Values of mechanistic parameters can be found in Tables S1–S5 and are discussed in more detail in the Supplement.
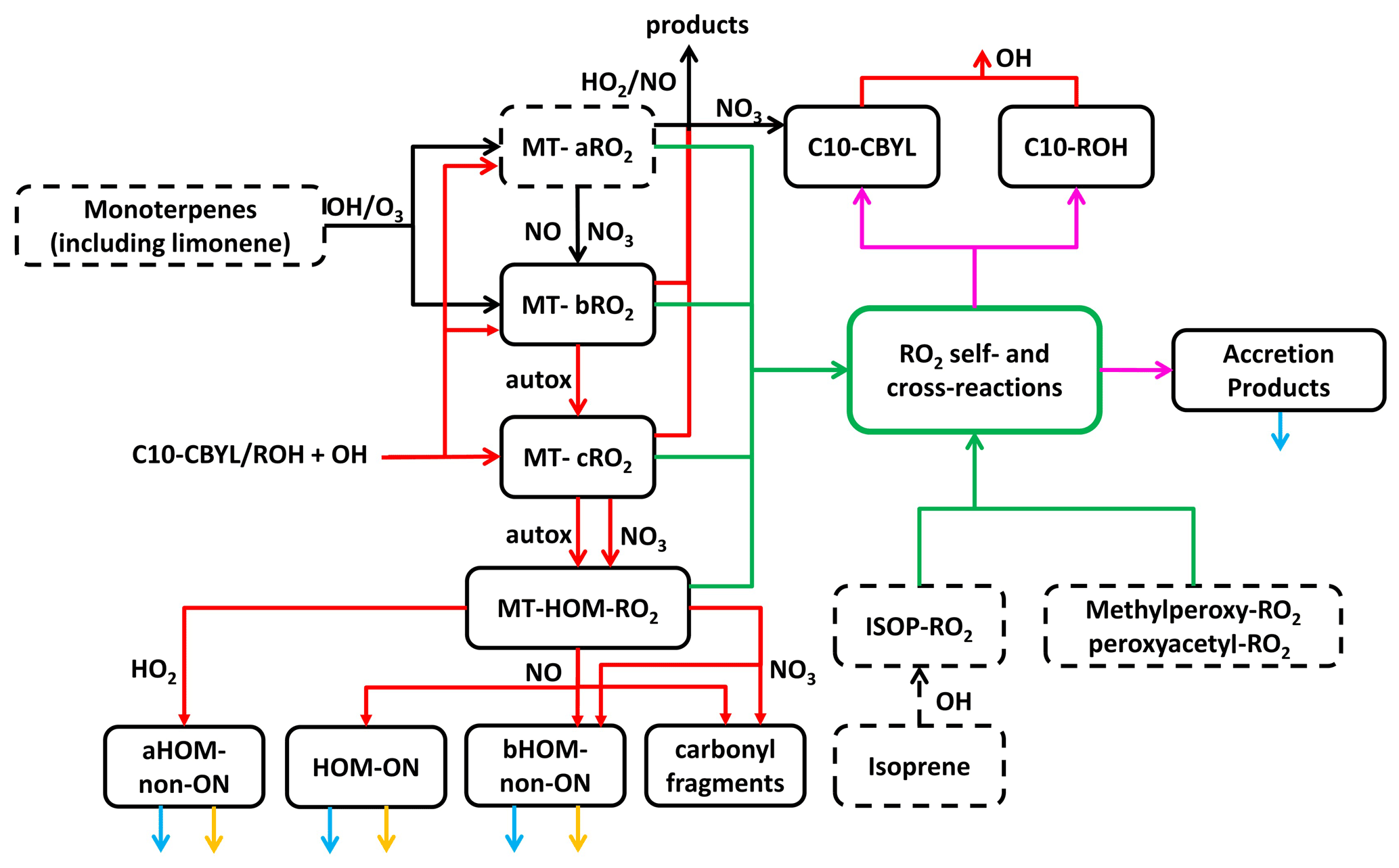
Figure 1The main reactions and processes included in the updated scheme are shown. Chemical species in solid boxes are newly added, whereas species in dashed boxes already exist in the GEOS-Chem mechanism. Dashed black arrows represent originally existing reactions without any modification, and solid black arrows represent those with certain modifications in the scheme. Red, green, and magenta arrows represent newly added RO2 formation and loss. Blue and yellow arrows represent wet/dry deposition and photolysis, respectively. More details are shown in the Supplement.
To account for MT-RO2 H-shift chemistry leading to HOMs (i.e., “autoxidation”), we split the first-generation MT-RO2, formed from the reaction of MTs with OH or ozone, into two pools, either MT-aRO2 or MT-bRO2. Both types of RO2 undergo the usual bimolecular reactions, but MT-aRO2 species do not undergo a unimolecular H shift whereas MT-bRO2 species do. The branching of MT-aRO2 or MT-bRO2 from MT + OH or MT + O3 reactions is based on laboratory-derived yields of MT-HOM and MT-nitrates; these laboratory studies typically find that the fraction of MT-RO2 undergoing autoxidation is <0.5 (Berndt et al., 2016; Kurten et al., 2015; Richters et al., 2016), although some researchers (Xu et al., 2019) have found it to be higher. The competitive yields of MT-HOM will be sensitive to the multiplicative product of H-shift rate constants and the fraction of first-generation RO2 able to undergo autoxidation. As a result, we vary this fraction for both OH and O3 reactions as part of sensitivity studies.
While MT-aRO2 species do not undergo unimolecular H shifts, we allow for a small fraction (5 %) of MT-aRO2 reactions involving NO or NO3 to produce MT-bRO2 in order to simulate the corresponding alkoxy radicals undergoing opening of the four-member ring that is often part of first-generation RO2 formed from α-pinene ozonolysis and OH reactions (Kurten et al., 2015; Roldin et al., 2019). This fractional fate of the corresponding alkoxy radical is much lower than assumed by Roldin et al. (2019); thus, our estimates in this regard might produce lower HOM concentrations. Iyer et al. (2021) show that prompt C4 ring opening from α-pinene ozonolysis is possible directly from the Criegee biradical; hence, reactions with NO or RO2 are not necessary for HOM formation. That said, opening of the four-member ring would lead to second-generation RO2 structures more amenable to unimolecular H-shift reactions (Iyer et al., 2021; Kurten et al., 2015), and it provides a means for enhancing or maintaining HOM formation in the presence of NO as suggested previously (Roldin et al., 2019; Pullinen et al., 2020), although the balance between alkoxy isomerization and decomposition remains uncertain.
The sequence of autoxidation and base H-shift rate constants, kauto, used in GEOS-Chem are similar to those described in Pye et al. (2019) for OH oxidation of MTs and in Jokinen et al. (2015) for MT + O3, which ultimately connect to experimental work in Berndt et al. (2016), Jokinen et al. (2015), and Ehn et al. (2014). The mole fraction of first-generation MT-RO2 able to undergo unimolecular H shifts and, thus, autoxidation (MT-bRO2) is 20 % from MT + OH and 3 % from MT + O3, in the base simulation (Table S3). The HOM mass yield can be 1.5–2 times larger than these molar values. We also note here that GEOS-Chem lumps MT, which can have very different HOM yields, especially from ozonolysis (Ehn et al., 2014). As such, we use the lower reported HOM yields from ozonolysis of α-pinene to reflect the fact that some of the lumped MTs will be β-pinene or other MTs lacking endocyclic double bonds. Our range of sensitivity studies likely captures the associated uncertainty range in these parameters.
A single temperature-dependent rate constant, kHshift, based upon recent laboratory studies and quantum chemical calculation of associated energy barriers is used to describe these H shifts. We use an activation energy of 17.7 kcal mol−1, based on the calculations in Berndt et al. (2016). Two values of kHshift, near 1.0 and 0.1 s−1 at 298 K, are tested in sensitivity studies. The H-shift reactions of MT-bRO2 are assumed to be followed by O2 addition to form a new peroxy radical, MT-cRO2, which, in turn, can also continue autoxidation to form a yet more oxidized MT-HOM-RO2. This sequence of RO2 autoxidation, occurring in competition with typical bimolecular reactions, simulates a first generation of MT-RO2, C10H15O4 or C10H17O3, undergoing two H-shift/O2-addition steps to form RO2 with compositions of C10H15O8 or C10H17O7, respectively, that are consistent with the current definition of HOMs (Bianchi et al., 2019). We do not track autoxidation of non-C10 RO2; thus, our definition of HOMs is specific to C10 MT products.
These MT-HOM-RO2 species undergo only bimolecular reactions with HO2, RO2, NO, and NO3. Except for RO2 cross-reactions, discussed further below, rate constants for such reactions are the default values used in GEOS-Chem for other RO2 species. The products of these reactions are split into four categories, three of which are HOMs. Reaction of MT-HOM-RO2 with HO2 is assumed to produce only HOM monomers (aHOM) without a nitrate group. Reaction with NO leads to HOM organic nitrates (HOM-ON), a second class of HOMs without a nitrate group (bHOM), and hydroxy carbonyl fragments assumed to be C5 species. The branching ratio for HOM-ON formation is assumed to be 0.2, determined using typical literature parameterizations based on the carbon number. We explicitly distinguish between non-nitrate HOMs that result from reaction of MT-HOM-RO2 with HO2 (aHOM) or NO (bHOM) to better account for the RO2 fate and the specific impact of NO on HOMs. The assumption that the alkoxy radical formation channel of MT-HOM-RO2 reactions with NO leads to HOMs is not well constrained, but it is typically a minor component of the HOM yield on a global average. We neglect further autoxidation reactions of RO2 and alkoxy radicals, and some of the fragmentation channels of the resulting alkoxy radicals may well produce C9 or C8 products that still meet the HOM definition. Reaction of MT-HOM-RO2 with the nitrate radical (NO3) is assumed to produce only an alkoxy radical product (and NO2), and the alkoxy radical either forms a bHOM (non-nitrate), similar to that from reaction of the RO2 with NO, or a C5 hydroxy carbonyl product to represent fragmentation into non-HOM products. We assume equal branching for these two pathways, which might lead to a slight overestimate of MT-HOM, but reaction with NO3 is a typically a minor fate for MT-HOM-RO2.
After the addition of RO2 H-shift chemistry, the next most significant change to monoterpene chemistry that we incorporated into GEOS-Chem involves the self- and cross-reactions of RO2. We specifically evaluated the impact of a higher rate constant and allowed for accretion product formation in competition with the more common alkoxy radical and disproportionation channels which led to lumped carbonyl (C10 CBYL) and alcohol (C10 OH) products following the typical lumping strategy in GEOS-Chem. Our basis for these changes includes the recent laboratory studies described in Berndt et al. (2018a, b) and Zhao et al. (2018), where cross-reaction rate constants were found to range from 10−12 to 10−10 cm3 molec.−1 s−1 and accretion product branching ranged from 4 % to >50 %. Given that there are only self-reactions for isoprene-derived RO2 in the current GEOS-Chem model without branching to accretion products, taking even the lower range from laboratory studies would represent a major shift in the RO2 fate, as we demonstrate in Sect. 3.
Important for regions with intense biogenic VOC emissions and relatively low NOx (such as regions of the Amazon), we specifically include cross-reactions between monoterpene- and isoprene-derived RO2. Our simulations include both low and high estimates of RO2 self- and cross-reaction rate constants to better demonstrate the range of possible impacts of these reactions, and we also apply different rate constants for highly oxidized RO2 (Table S4). For the rate constants considered, RO2 cross-reactions can become competitive or even dominant fates of RO2 and, thus, impact the abundance and recycling of HOx as well as the formation of low-volatility products that would contribute to organic aerosol. For accretion products, we use a conservative branching (4 %) from both self- and cross-reactions to produce C20 or C15 compounds, except for HOM-RO2 self- and cross-reactions, for which we also examine a larger 100 % branching to accretion products as suggested by some laboratory studies (Berndt et al., 2018a, b).
We assume that the dominant fate of gas-phase MT-HOM, as defined here, is to partition to existing aerosol particle mass and then become subject to (wet and dry) deposition or photolysis in the particle phase. While reaction with OH or other radical oxidants is possible, our assumption is that the vast majority of HOMs produced in this mechanism will be of low or extremely low volatility and, thus, be present predominantly in submicron aerosol particles. Our estimates of HOM mass concentrations are, therefore, possibly upper limits due to the uncertainty in HOM saturation vapor concentrations. As we do not explicitly consider gas–particle partitioning in this version, we use a single photolysis frequency equal to of to account for photochemical degradation of particle-phase HOMs. We do not treat heterogeneous oxidation explicitly, but we assume that our photolysis parameterization accounts for this process. The value of the photolysis frequency is based on how well the model reproduces HOM observations in the absence of further photochemical degradation as well as on laboratory chamber experiments showing loss of HOMs and associated MT SOA mass over time (Krapf et al., 2016; Pospisilova et al., 2020; Zawadowicz et al., 2020). The photochemical fate of HOMs remains one of the most uncertain aspects of the mechanism.
We parameterize HOM wet deposition following aerosol-phase organic nitrate in Fisher et al. (2016), and dry deposition is calculated online based on the resistance-in-series algorithm (Zhang et al., 2001) assuming HOMs behave similarly to SOA (particle dry deposition). Therefore, the global annually averaged dry deposition velocity of HOMs is about 0.06 cm s−1 on land. The parameters related to aerosol scavenging, rainout, and washout efficiency are listed in Table S5 following the parameterization of most SOA species in GEOS-Chem. We note that treating dry deposition of HOMs similar to submicron particles is possibly a small underestimate of the actual HOM dry deposition rate because HOMs likely condense to particles on timescales shorter than those of dry deposition for vapors in most cases. A typical condensation timescale to aerosol surface area is 15 min in the boundary layer, whereas a deposition velocity of 3 cm s−1 implies a boundary layer average timescale of several hours. Future updates to the mechanism could consider partitioning of HOMs to SOA based on more explicit tracking of composition–volatility relationships and, thus, better simulation of the net depositional scavenging.
2.3 Simulation design and configurations
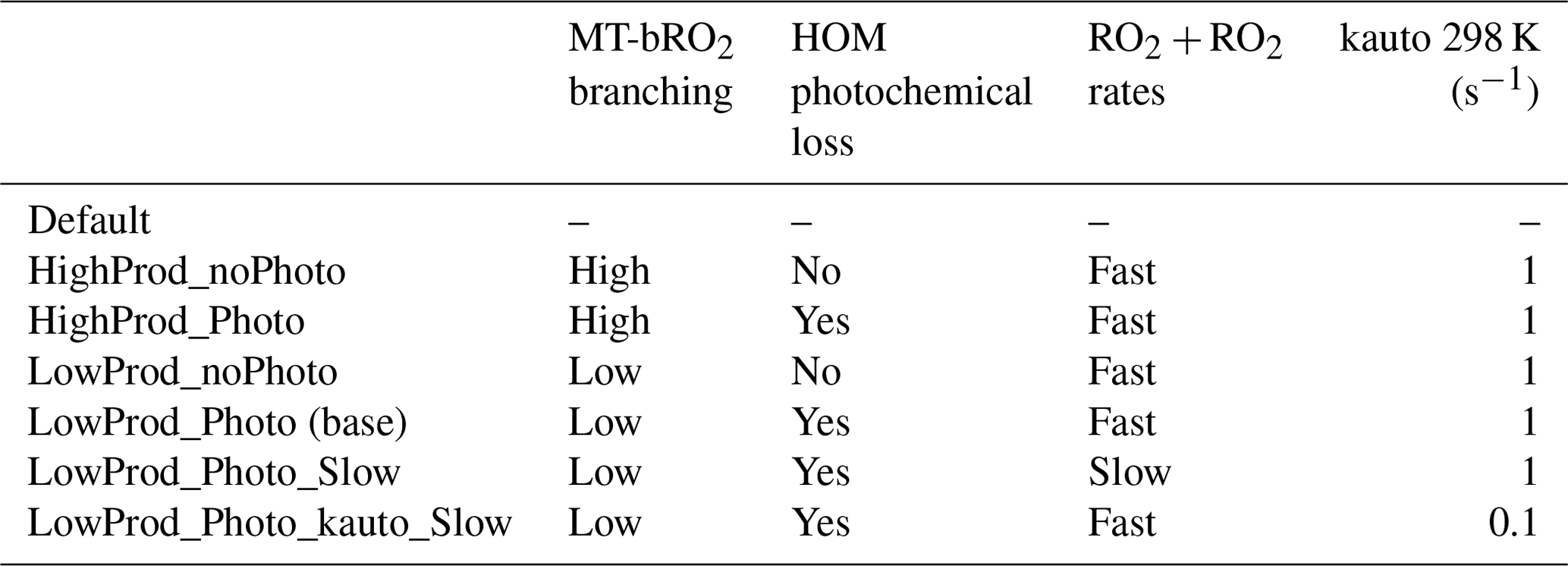
All simulation configurations are summarized in Table 1. A default simulation with no HOM formation nor any other newly added reactions was run for reference. The base simulation (LowProd_Photo) was run with relatively conservative MT-bRO2 branching and with HOM photolysis turned on, and another simulation with a larger branching ratio to MT-bRO2 was also run to better determine the HOM formation range (HighProd_Photo). Photolysis of HOMs was also turned off to test its impact in the LowProd_noPhoto and HighProd_noPhoto cases. Another two simulations configured with slow RO2 self- and cross-reaction rates (LowProd_Photo_Slow) and with a slow RO2 autoxidation rate (LowProd_Photo_kauto_Slow) were used to investigate the respective sensitivity of HOMs and accretion product formation to these rates. All simulations were conducted in the same way as described in Sect. 2.1 and 2.2. Results were output every month; however, when compared with observations, they are output with a 1 h resolution.
Table 1Simulations and the corresponding configurations. See the text for details.
2.4 Observations
Data from three campaigns, the Southern Oxidant and Aerosol Study (SOAS, 2013) in the southeastern USA; the Biogenic Aerosols – Effects on Clouds and Climate (BAECC, 2014) in Hyytiälä, Finland; and the Green Ocean Amazon Experiment (GoAmazon) in the Amazon, Brazil, were used for comparisons (Carlton et al., 2018; Martin et al., 2016; Petäjä et al., 2016). Measurements of organic aerosol mass concentrations from aerosol mass spectrometer (AMS) instruments (DeCarlo et al., 2006; Jayne et al., 2000) and gas- and particle-phase HOMs from high-resolution time-of-flight chemical ionization mass spectrometer (HRToF-CIMS) instruments were used when available (Lopez-Hilfiker et al., 2014). For HOM measurements, molecular formulas of compounds containing 10 carbon atoms and greater than or equal to 7 oxygen atoms were selected as HOMs for the comparisons. Those with one nitrate and without nitrate were compared to simulated HOM-ON and HOM-non-ON, respectively. We also compared predicted HOMs to total organic aerosol (OA) mass from aerosol mass spectrometer measurements, assuming that HOMs were present predominantly in submicron aerosol particles. Besides HOMs, closely related species in the scheme were also compared when available, including NO, O3, monoterpenes, and isoprene. Details on the measurements are presented in the Supplement, including the top contributing HOM species identified in data from the SENEX (Southeast Nexus) and BAECC campaigns (Tables S7 and S8).
3.1 MT and HOM RO2 fates
The largest change to the current mechanism was made to the fate of a fraction of MT-derived RO2, where we incorporated unimolecular autoxidation reactions for a subset of first-generation MT-derived RO2 (MT-bRO2 in the above scheme) as well as enhanced reaction rate constants for bimolecular RO2 self- and cross-reactions between MT and isoprene RO2. The fate of RO2 determines the volatility and reactivity of HOMs and, thus, of the potential for HOM contribution to aerosol formation and growth. In our simplified treatment, we assume that HOM-RO2 species only undergo bimolecular reactions. HOM-RO2 species that undergo unimolecular decomposition to a closed-shell product, such as by OH or HO2 elimination, may result in a non-HOM product. Thus, our flux of MTs to HOM-RO2 may be underestimated, but net HOM production may be more accurate.
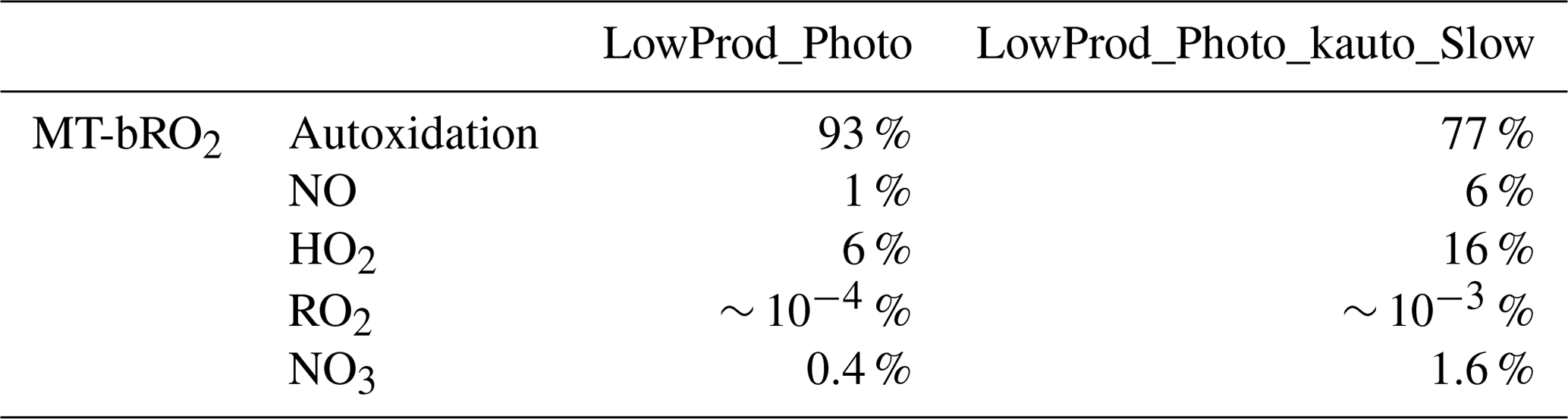
The spatial distribution of the annual average reaction fate of MT-bRO2 in the planetary boundary layer (PBL) is shown in Fig. 2 for two simulation cases: LowProd_Photo (panels a–b) and LowProd_Photo_kauto_Slow (panels c–d). The difference between these two simulations is the rate constant for the unimolecular RO2 H shift (∼ 1.0 s−1 vs. ∼ 0.1 s−1 at 298 K, respectively). For either case, unimolecular H shift and subsequent autoxidation is the dominant fate of the first-generation MT-bRO2 throughout the PBL on average. While likely dependent upon model resolution, when kauto is ∼ 0.1 s−1, the reaction with NO becomes a more common fate for MT-bRO2, although never more than 50 % of the total fate of this HOM-RO2 precursor, even in NOx-polluted regions such as the southeastern USA, eastern China, and western Europe. In Fig. 3, the annually averaged vertical profiles of the fate of MT-bRO2 are shown for two model grid points, one containing Centreville, AL, USA, and the other containing the Amazonian T3 site of the GoAmazon campaign. The dominance of unimolecular RO2 H shift and autoxidation as a fate for MT-bRO2 persists up to 6–8 km, even though its rate decreases exponentially with decreasing temperature. At both locations, reaction with NO at high altitudes becomes a major MT-bRO2 fate, especially over the southeastern USA; over the Amazon, reaction with HO2 and NO above 6 km are of similar importance, likely reflecting the combination of the activation energy required for the unimolecular H shift, decreases in temperature with altitude, and NO in the upper troposphere from lightning and convection.
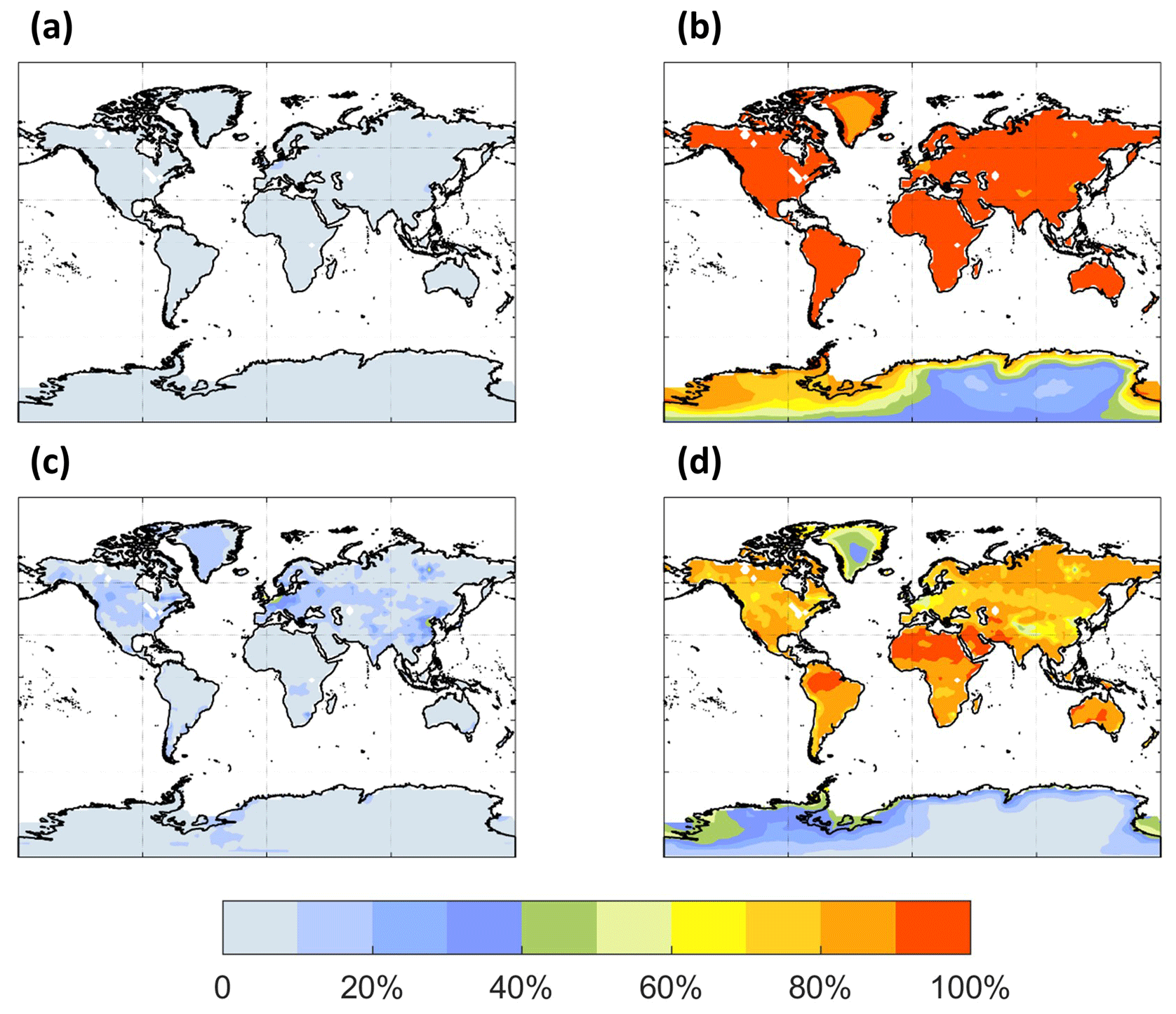
Figure 2The annual PBL-averaged MT-bRO2 consumption fractions by NO (a, c) and autoxidation (b, d) from the LowProd_Photo (a, b) and LowProd_Photo_ kauto_Slow (c, d) experiments. The autoxidation rate constant is ∼ 1.0 and ∼ 0.1 s−1 at 298 K for the respective experiments. The fractions by HO2, NO3, and RO2 are shown in Fig. S1.
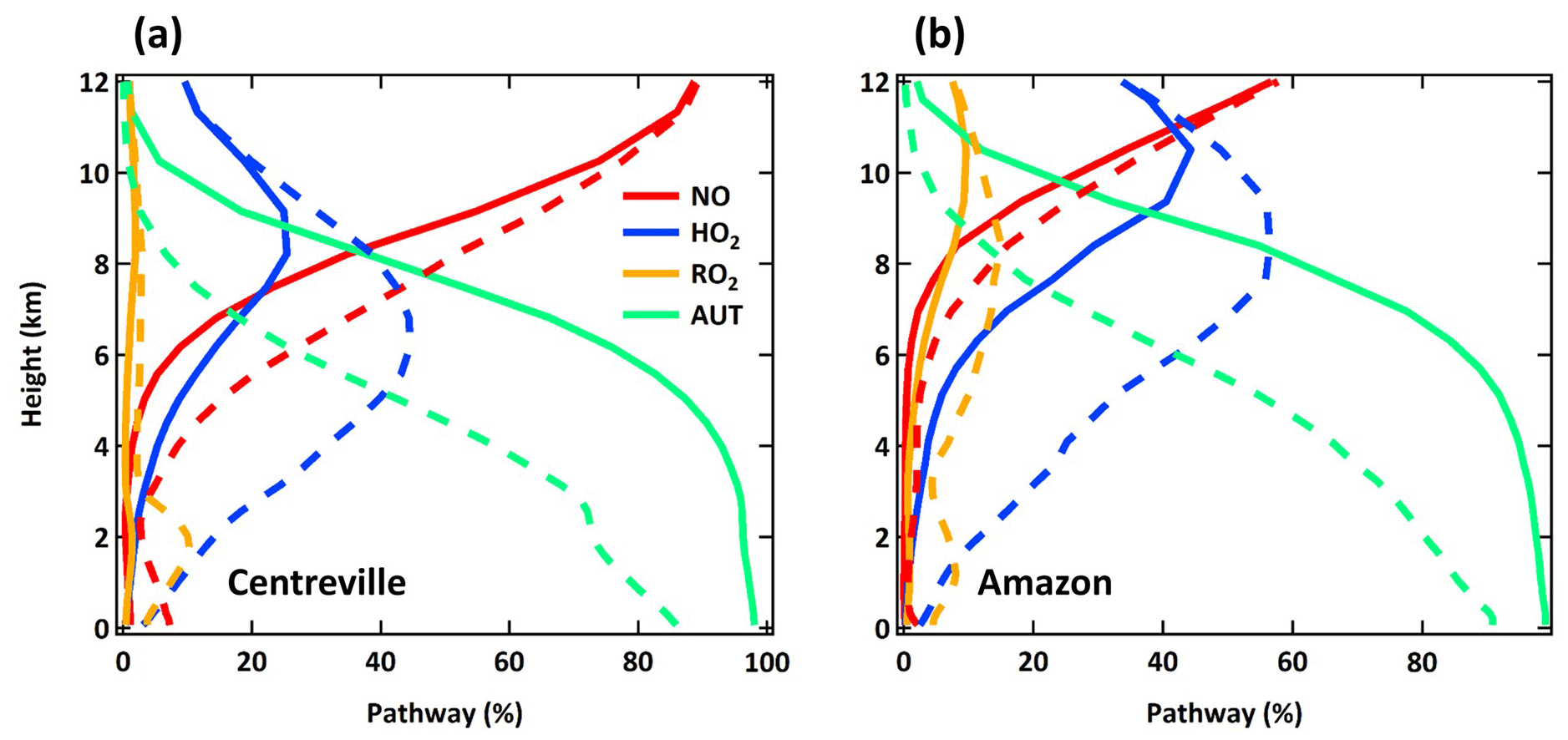
Figure 3Annual averaged vertical profiles of four dominant reaction pathways of MT-bRO2 at Centreville, AL, USA, and over the Amazon near Manaus from the LowProd_Photo (solid lines) and LowProd_Photo_kauto_Slow (dashed lines) simulations. Reaction with NO3 contributes less than 1 %; thus, it is not shown here.
Table 2Global PBL-averaged MT-bRO2 fates weighted by gridded MT-bRO2 concentrations on land.
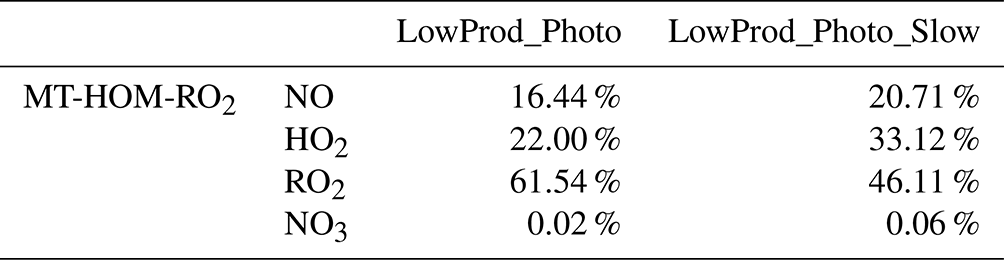
Figures 4 and 5 are similar to Figs. 3 and 4 but show the fates of HOM-RO2 instead of MT-bRO2. As HOM-RO2 species in the model do not undergo unimolecular reactions (see above), these fates are more similar to generic RO2 chemistry in the model with the important exception that the rate constants for self- and cross-reactions between HOM-RO2 and other RO2 species are generally much larger than those typical of other RO2 species in GEOS-Chem. In cases where HOM-RO2 rate constants for RO2 cross-reactions are relatively large (LowProd_Photo, Fig. 4a–c), e.g., as in Berndt et al. (2018a), reaction with RO2 is predicted to be the dominant HOM-RO2 fate throughout most of the boreal and tropical forest regions as well as portions of the southeastern USA. In temperate and subtropical forests of the Northern Hemisphere, reaction with NO is the major fate for HOM-RO2. The potential importance of reactions with RO2 being a dominant fate is two-fold. First, the branching of such reactions to accretion products is uncertain (see below) but likely also critical for the participation of biogenic VOCs in the nucleation of particles (Bianchi et al., 2019; Kulmala et al., 2014). However, the portion of such reactions that does not undergo accretion otherwise can result in less carbon mass moving to lower volatility due to C–C bond scission of alkoxy radical products (Orlando et al., 2003). In the simulation with slower RO2 cross-reactions (e.g., LowProd_Photo_Slow), the rate constants for which are near the lower limit of rate constant collections from several laboratory studies (Berndt et al., 2018a, b; Zhao et al., 2018), RO2 cross-reactions remain important (∼ 40 %) across boreal forests but are no longer dominant as a HOM-RO2 fate except in the tropical forest regions. Reactions with NO expand in importance in boreal forest regions in this simulation, at times being the dominant fate in regions of the North American boreal forest. While consistently significant, typically at 30 % to 40 % of HOM-RO2 fate, reaction of HOM-RO2 with HO2 is only rarely a majority fate in the PBL over forest regions.
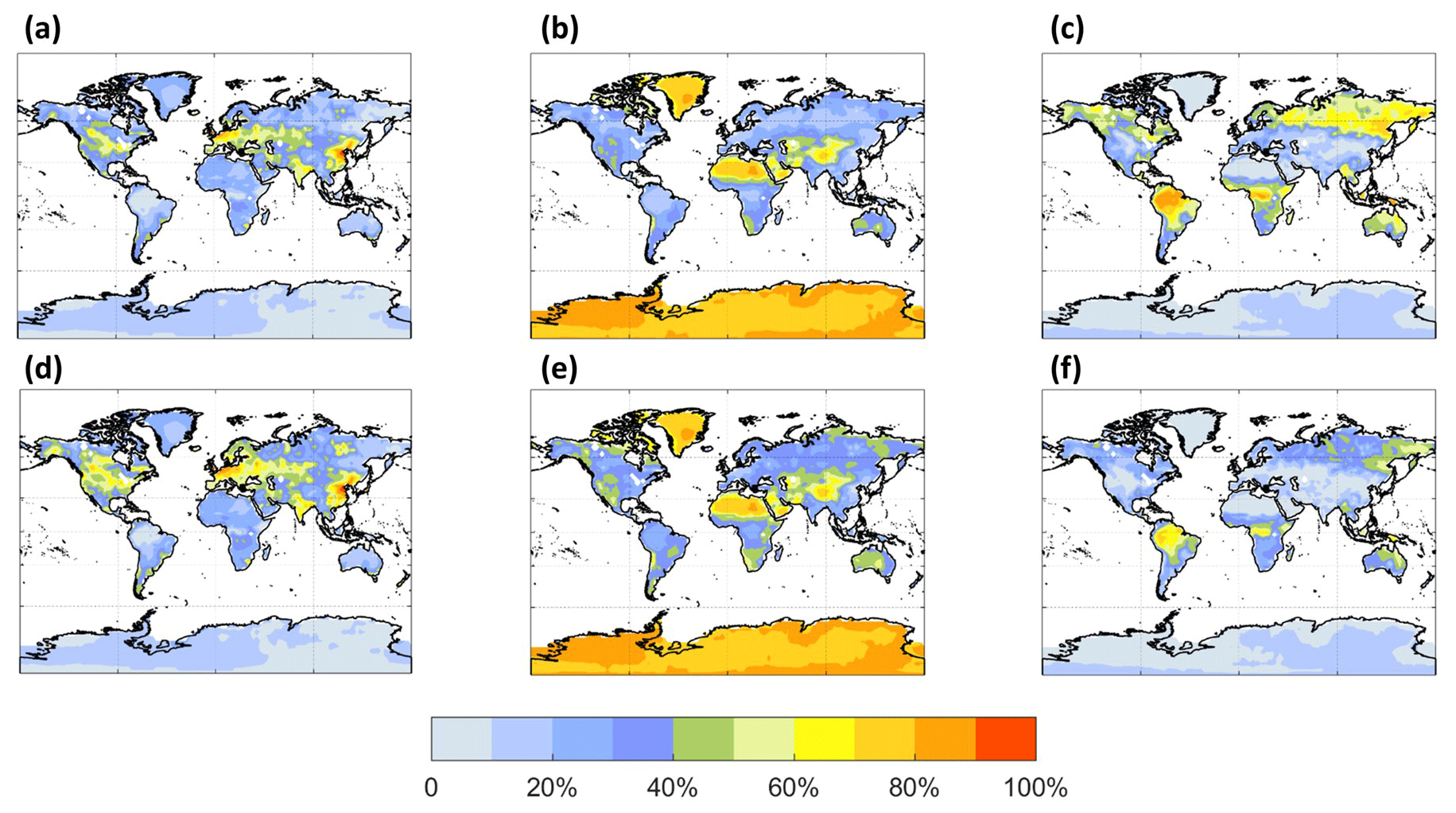
Figure 4The annual PBL-averaged MT-HOM-RO2 relative fates, including reaction with NO (a, d), HO2 (b, e), and RO2 (c, f), from the LowProd_Photo (a–c) and LowProd_Photo_Slow (d–f) simulations. Reaction with NO3 contributes <1 %; thus, it is not shown here.
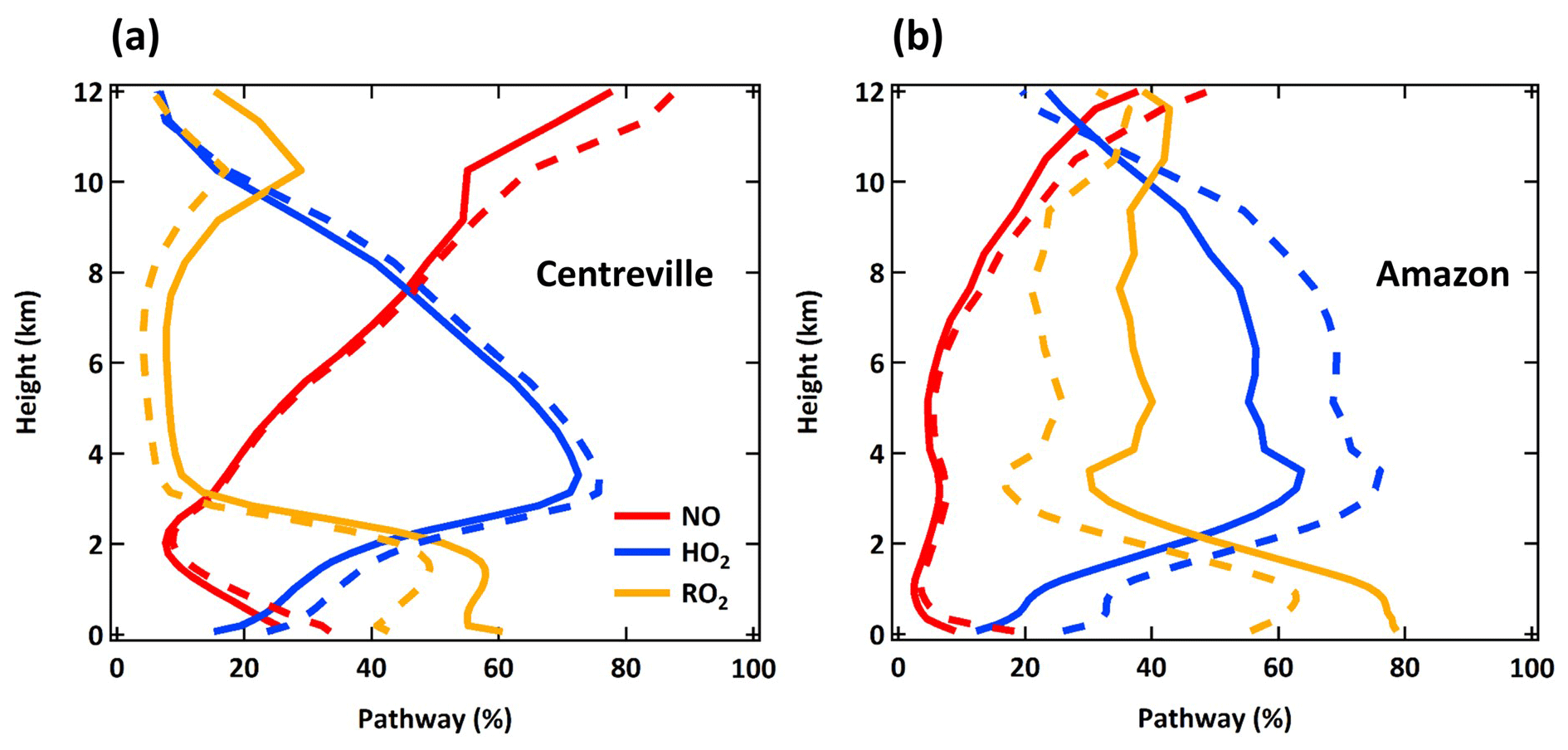
Figure 5Annual averaged vertical profiles of three dominant reaction pathways of MT-HOM-RO2 at Centreville, AL, USA, and over the Amazon near Manaus from the LowProd_Photo (solid lines) and LowProd_Photo_Slow (dashed lines) simulations. Reaction with NO3 contributes <1 %; thus, it is not shown here.
The annual averaged HOM-RO2 fate changes significantly between the boundary layer and free troposphere, as shown in Fig. 5 for the same two model locations as in Fig. 3. Throughout the low and middle troposphere at both locations, reaction with HO2 becomes the dominant HOM-RO2 fate, followed by RO2 over the Amazon, and NO over the southeastern USA. Reaction with NO becomes the dominant fate for HOM-RO2 in the upper troposphere over the southeastern USA, while NO, HO2, and RO2 reactions are predicted to be of similar importance over the Amazon.
Table 3Global PBL-averaged MT-HOM-RO2 fates weighted by gridded MT-HOM-RO2 concentrations on land.
3.2 Impact on HOx, NOx, and O3
By altering the fates of MT-derived RO2 chemistry and the interactions thereof with isoprene-derived RO2, we expect that the cycling and lifetime of HOx will be affected. Changes in HOx abundance and distribution will alter NOx cycling and fate, which will potentially impact tropospheric O3. MTs are not typically major components of OH reactivity, even in biogenically influenced regions, so these impacts are not expected to be large. As shown in Fig. 6, there are substantial decreases in the sum of HO2 and RO2 concentrations in certain regions averaged over the PBL, the height of which is taken from the MERRA-2 reanalysis data (Gelaro et al., 2017). HO2 and RO2 concentrations together decrease by as much as ∼ 20 % over boreal forests and up to 80 % over tropical forests. The global average decrease in the sum of HO2 and RO2 in this simulation compared with the default is 4 %. The updated description of RO2 self- and cross-reactions is the dominant driver of the shorter RO2 lifetime and, thus, of the calculated decreases. Given that most of these decreases in RO2 occur at locations with low NO, the impact upon HO2 and OH (Fig. 6b) is small globally, although not negligible in the PBL over the Amazon, reaching a ∼ 15 % decrease in OH. The lower OH predicted over the Amazon leads to longer NOx lifetimes there and, thus, a highly localized increase in NOx abundance. Otherwise, the effects on NOx and O3 are negligible globally.
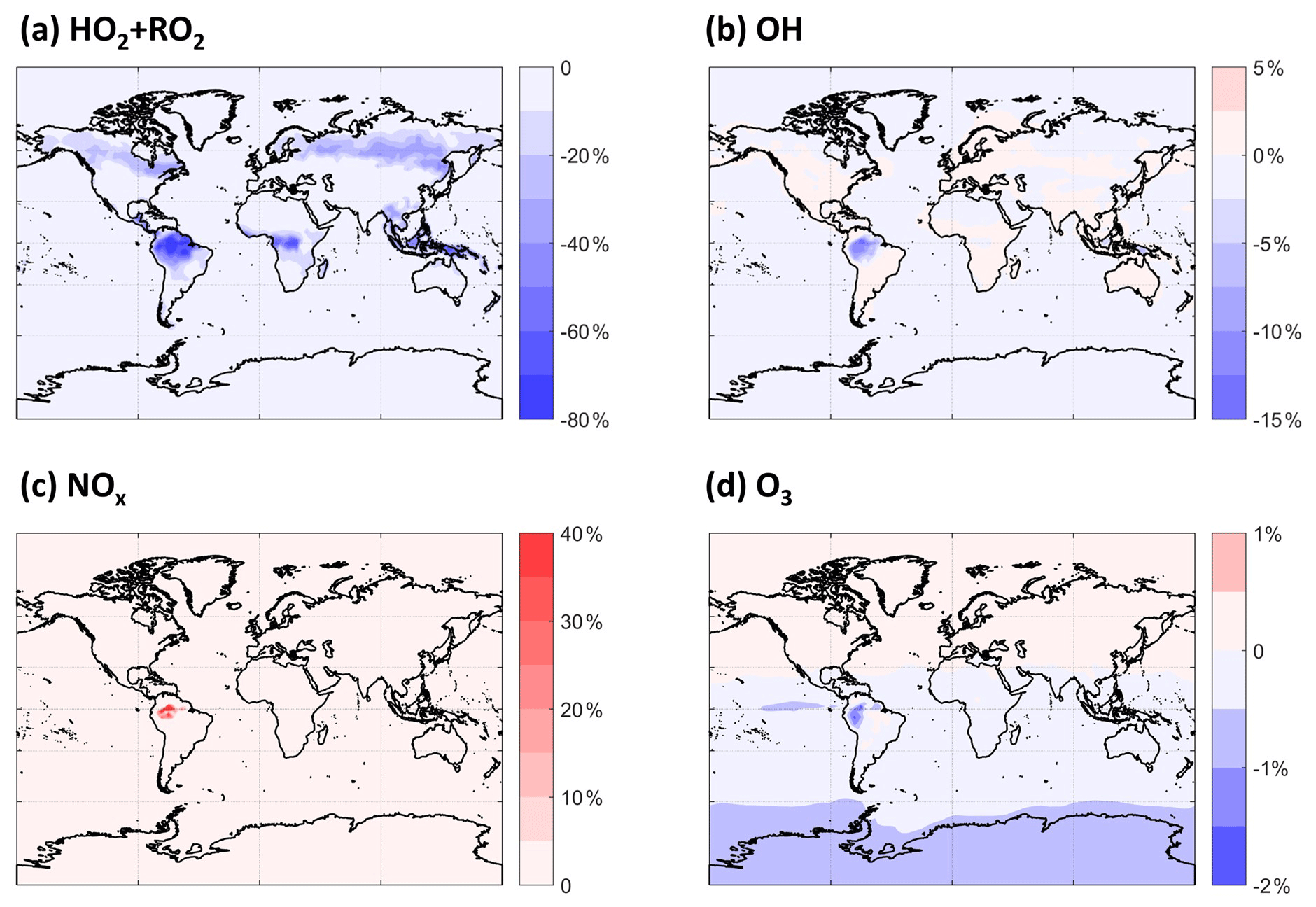
Figure 6The annual PBL-averaged relative differences in (a) HO2 + RO2, (b) OH, (c) NOx, and (d) O3 between the LowProd_Photo and Default simulations.
3.3 HOMs and associated accretion product distribution and concentrations
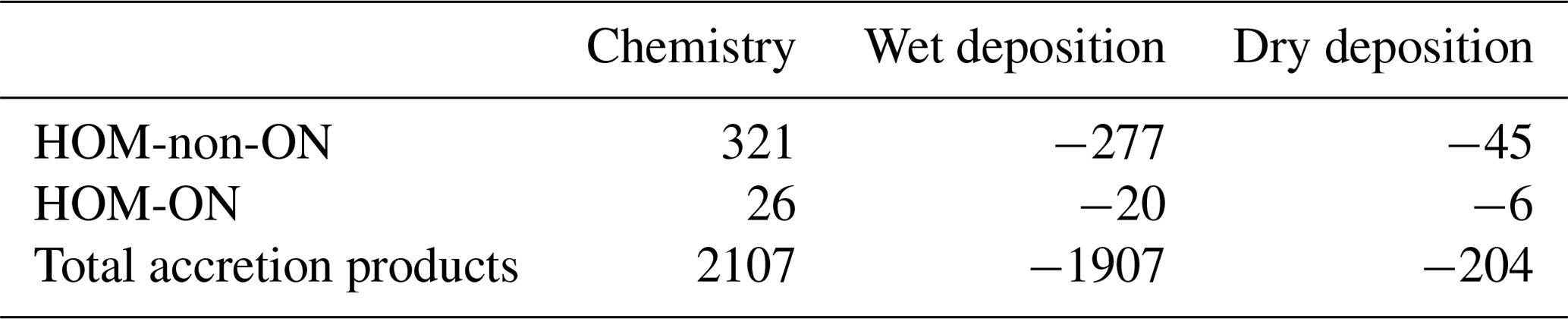
Global annual budgets for the chemistry (production + loss) and the wet and dry deposition of HOM-non-ON, HOM-ON, and MT-derived accretion products are summarized in Table 4 from the LowProd_Photo_kauto_Slow simulations. For even the slowest kauto used, the non-nitrate pathway for HOMs is more than factor of 10 that of HOM organic nitrates. Interestingly, even for a small branching to accretion products, MT-RO2-derived accretion products are a substantially more common fate than HOMs, suggesting either that the rates and branching are too high or that the chemical loss pathways of associated products are not well represented.
Table 4Global annual budgets of MT-HOM and MT-RO2 accretion products from March 2013 to February 2014 (in kt C).
The PBL-averaged mass concentrations (µg m−3) of HOMs predicted by the model are shown in Fig. 7a–d for the LowProd_noPhoto simulation, which produces middle- to upper-range estimates of HOM concentrations compared with the other scenarios tested. Maps from other sensitivity simulations are included in the Supplement (Figs. S2–S6). In this scenario, monoterpene-derived HOMs are predicted to average near 1 µg m−3 in the PBL over tropical forests with little seasonality, whereas HOMs reach 0.5–1 µg m−3 during summer months in the temperate and boreal forests of North America, Europe, and East Asia. In the LowProd_Photo scenarios, HOM concentrations are, on average, an order of magnitude lower than those shown in Fig. 7, although the spatial and seasonal patterns are similar. Given that HOMs with 10 carbons and 7 or more oxygens will have low or extremely low volatility, the majority of HOMs produced from monoterpene oxidation will likely contribute to SOA and, thus, to total OA. A background organic aerosol mass concentration in rural or remote forest regions of the order of 1 µg m−3 outside of biomass burning periods is not atypical (Jimenez et al., 2009).
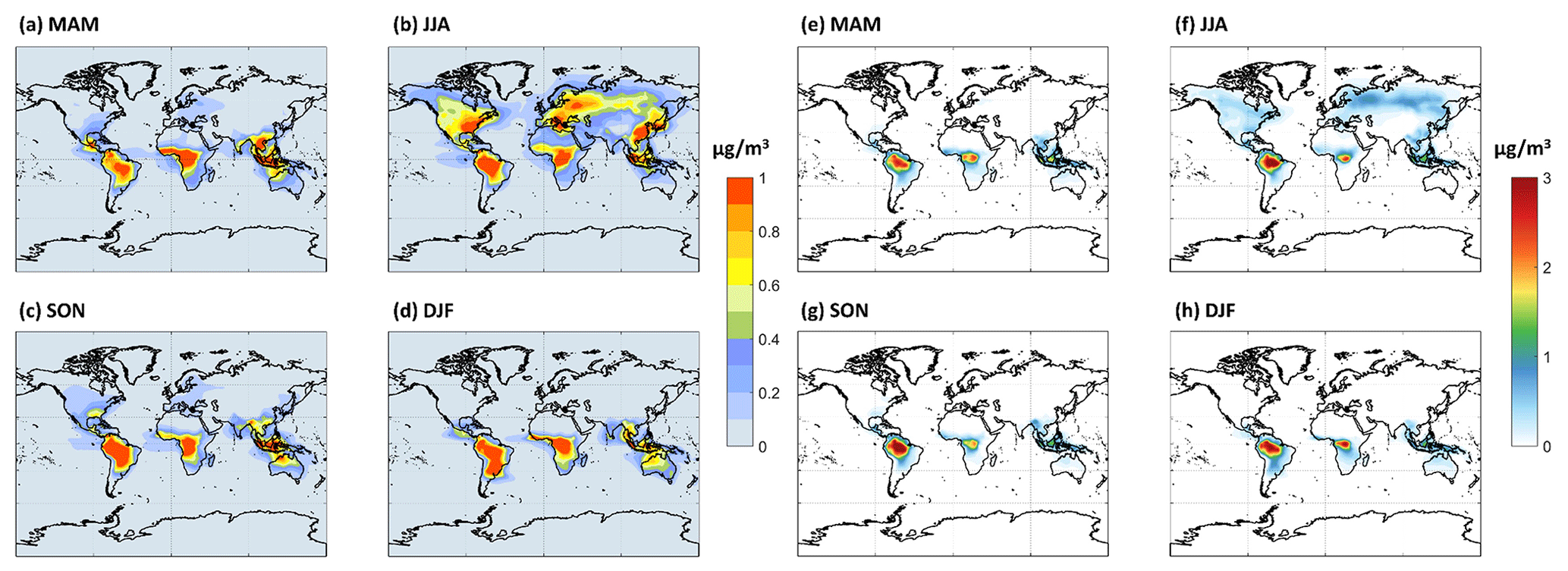
Figure 7The seasonal PBL-averaged total HOM mass concentrations for (a) March–April–May (MAM), (b) June–July–August (JJA), (c) September–October–November (SON), and (d) December–January–February (DJF) from the LowProd_noPhoto experiment. Seasonal PBL-averaged total C20 HOM accretion products are shown in panels (e)–(h), assuming that HOM-RO2 self- and cross-reactions produce accretion products at unit yield.
For comparison, we also show seasonal PBL distributions of HOM-RO2 self- or cross-reaction accretion products, assuming that C20 HOMs are formed at unit yield. This assumption provides an upper limit, although one that is supported by some laboratory studies (Berndt et al., 2018a, b). Throughout the tropical forests and boreal regions during summer, HOM-RO2 accretion products in this simulation reach 3 or 1 µg m−3, respectively. As total OA in some boreal and tropical forest measurements can be of this order (Jimenez et al., 2009; Lee et al., 2018; de Sá et al., 2018) outside of biomass burning periods, we conclude that C20 HOMs undergo particle-phase decomposition and/or that the HOM-RO2 self- and cross-reactions do not produce accretion products at unit yield or the model underestimates NO throughout boreal and tropical forest regions, which would suppress both HOMs and, more so, HOM accretion product concentrations. As shown in the Supplement (Fig. S7), assuming an accretion product branching of 4 % for all MT-RO2 self- or cross-reactions as in Zhao et al. (2018), including HOM-RO2, leads to significantly lower, although not unimportant, concentrations of accretion products. The total C15 + C20 accretion product concentrations in the PBL of tropical and boreal forest regions are typically less than 1 or 0.25 µg m−3 in this simulation, respectively.
Accretion products from HOM-RO2 reactions with other HOM-RO2 species are likely an important route to new particle formation, especially in the relatively warm PBL. Thus, to predict new particle formation in regions such as the remote temperate or boreal forests, such accretion products will need to be incorporated. As noted above, the self- and cross-reaction rates and accretion product branching in both cases are far larger than those commonly used in GEOS-Chem. Nucleation and growth of particles by MT-HOM and associated accretion products is beyond the scope of this work; however, in both treatments of accretion product formation, C20 HOM accretion products reach concentrations that are likely relevant for participation in new particle formation (e.g., 107–108 molec. cm−3) to the extent that it occurs in the PBL over forest regions (Bianchi et al., 2019). A remaining question is the extent to which MT-RO2-derived accretion products more generally form and contribute to OA mass.
Our results suggest that further refinement of HOM formation and loss kinetics is needed, as the range of our simulations suggests that HOMs either make relatively small contributions to regional OA or that they constitute the majority of OA outside of biomass burning periods over tropical forests year-round, as well as during summer months for temperate and boreal forests. Figure 8 illustrates that, for two of the sensitivity simulations which bound possible HOM formation and loss kinetics, MT-HOM concentrations alone are either 5 %–10 % of total OA predicted by the standard GEOS-Chem model or are more than a factor of 1.5 higher than the predicted total OA. Incorporating predicted MT-RO2-derived C15 and C20 accretion products as an OA source only increases the potential contribution of MTs to total OA. If the MT-RO2 accretion product branching is on average 4 %, accretion products can double the contribution of MTs to OA when HOMs are simulated in the LowProd_Photo case (see Fig. S7). If the accretion branching ratio is closer to unity, as expected for HOM-RO2, the contribution of HOM monomers and MT-HOM accretion products to OA is even larger, reaching or exceeding a mean ratio of 3 in tropical forests compared with GEOS-Chem-predicted OA. Thus, revising MT chemistry to incorporate gas-phase sources of low-volatility and extremely low-volatility pathways will likely increase, perhaps substantially, the total OA predicted by the GEOS-Chem model over forest regions.
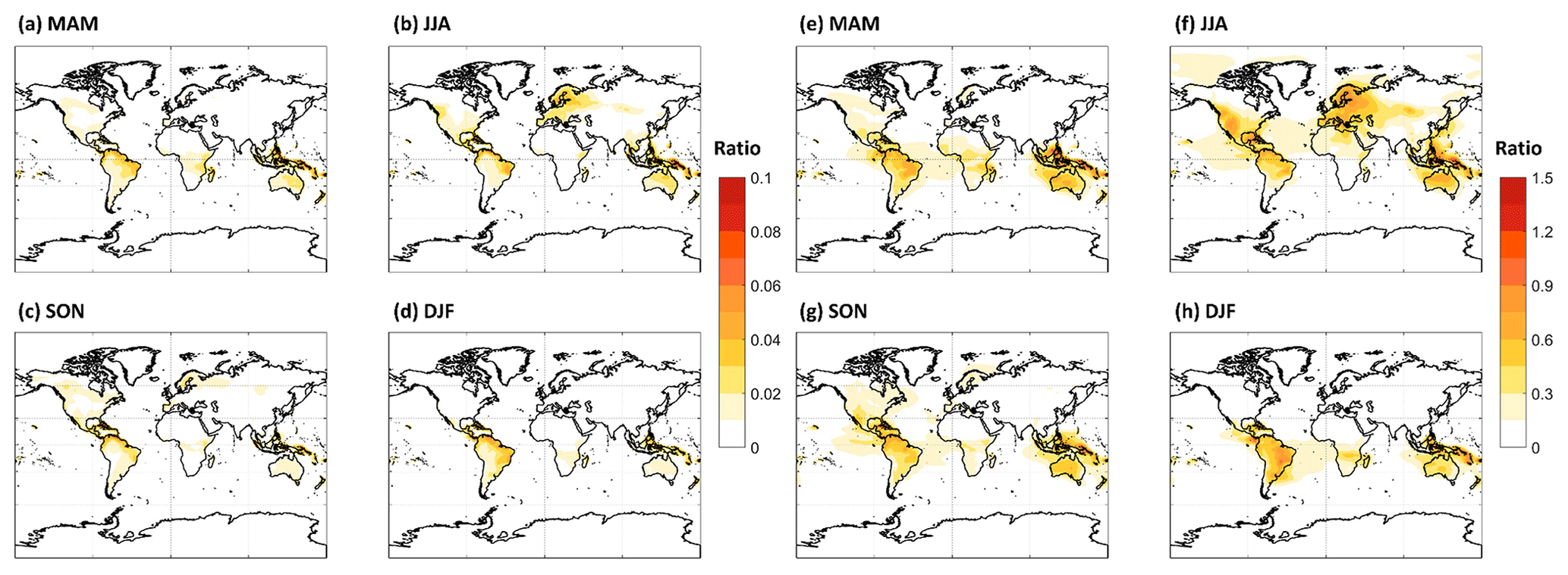
Figure 8The seasonal PBL-averaged total C10 HOM mass concentrations from the LowProd_Photo (a–d) or HighProd_noPhoto (e–h) simulations plotted relative to the total OA mass concentration predicted by GEOS-Chem for the same periods and locations. Note that the color scale for panels (a)–(d) (0 to 0.1) is about a factor of 10 lower than that for panels (e)–(h) (0 to 1.5).
There are limited observations of HOMs that can be used to investigate the validity of the different scenarios simulated here. First, the majority of HOMs will condense to form SOA, where they may further react to form products that might not be traceable to HOMs formed in the gas phase (Krapf et al., 2016; Lee et al., 2020; Pospisilova et al., 2020; Zawadowicz et al., 2020). Second, most HOMs have been observed only in the gas phase (Bianchi et al., 2016; Ehn et al., 2014; Massoli et al., 2018), which represents only a local steady state between the formation and condensation sink over small spatial scales compared with the current model resolution. The FIGAERO (Filter Inlet for Gases and Aerosols) HRToF-CIMS instrument measures some HOMs in both the gas and particle phases, while the aerosol mass spectrometer (AMS) provides an upper limit constraint on the total organic aerosol. In Fig. 8, we show observations from the FIGAERO HRToF-CIMS in a rural temperate and rural boreal forest, in Centreville, southeastern USA, and at the SMEAR II station in Hyytiälä, Finland, respectively, using only C10 compounds. In addition, we show AMS observations of total OA from these sites as well as from the T3 site of the GoAmazon campaign outside of Manaus, Brazil. The Centreville observations were obtained in June–July 2013, the SMEAR II observations were obtained from April–June 2014, and the GoAmazon observations were obtained from February–March 2014. More information can be found in the Supplement and related papers (Carlton et al., 2018; Martin et al., 2016; Petäjä et al., 2016). The FIGAERO HRToF-CIMS observations include both speciated HOM organic nitrates and non-nitrates.
We compare these observations to two simulations, HighProd_Photo and LowProd_Photo, where each includes photochemical losses of HOMs based on recent experimental work (Zawadowicz et al., 2020) but different yields of MT-bRO2 that can undergo unimolecular H shifts, as discussed above. The comparison is challenged for a number of reasons. First, monoterpene emissions are uncertain in a global sense but will also vary significantly at scales below the resolution of the model. Second, gas-phase HOMs will be sensitive to the local oxidant conditions, which will also depend on model-predicted NO concentrations and biogenic VOCs, while particulate HOMs potentially represent the integral of multiple days of formation, loss, and transport. Moreover, HOMs in the particle phase may react into non-HOM, be lost on instrument surfaces, or thermally decompose during the analysis, such that observations of total HOMs are possibly underestimated by the FIGAERO HRToF-CIMS instrument. To facilitate the comparison, we use the diurnal cycle in observations averaged over 4–6 weeks of observations to minimize the impact of meteorological variability. Addressing errors in the MEGAN emissions inventory is beyond the scope of this paper; therefore, we scale the predicted HOM concentrations in the lowest model level by the ratio of observed to predicted monoterpene concentrations in order to account for potential biases in the regional monoterpene emissions in the model (Fig. S8). For SOAS and GoAmazon, we use the hourly average measured monoterpene data for comparison with the hourly GEOS-Chem predictions, whereas we use the campaign average of measured monoterpene concentrations for the BAECC campaign at SMEAR II station. We separate HOM organic nitrates (ON) from HOM non-nitrates (non-ON) where possible but compare to the total measured gas + particle in each category.
As shown in Fig. 9, there is general order-of-magnitude agreement between the observed HOMs and those predicted by one of the model simulations when adjusted by the predicted and observed monoterpene concentrations, as described above. The HighProd_Photo simulation is better able to simulate the HOM-ON, but it overestimates the non-ON HOMs measured at the boreal forest location. In contrast, at the Centreville location, the HighProd_Photo simulation underestimates the measured HOM-ON but overestimates the measured non-ON HOMs. The general overestimation of observed non-ON HOMs could be due to the non-ON HOMs having reacted in the particle phase into components that are not detectable as HOMs due to the analytical limitations of the instrument used, which relies on thermal desorption and, thus, can be subject to the thermal decomposition of low-volatility components (Lopez-Hilfiker et al., 2014). We note that the HighProd_Photo simulation does not overestimate the observed fine-mode OA mass concentrations at any of the three locations, such that there is potential for a higher fraction of MT oxidation to result in HOMs and higher contributions of MT-HOM to OA than shown in Fig. 9. The reason for a low contribution of MT-HOM to OA predicted for the Amazon region remains unknown, but it is possibly related to errors in the modeled MT emission inventory, the limitations of comparing a relatively coarse model resolution to a single-location measurement, and/or the fact that the influences from isoprene, biomass burning, and other pathways are perhaps more important at this location.
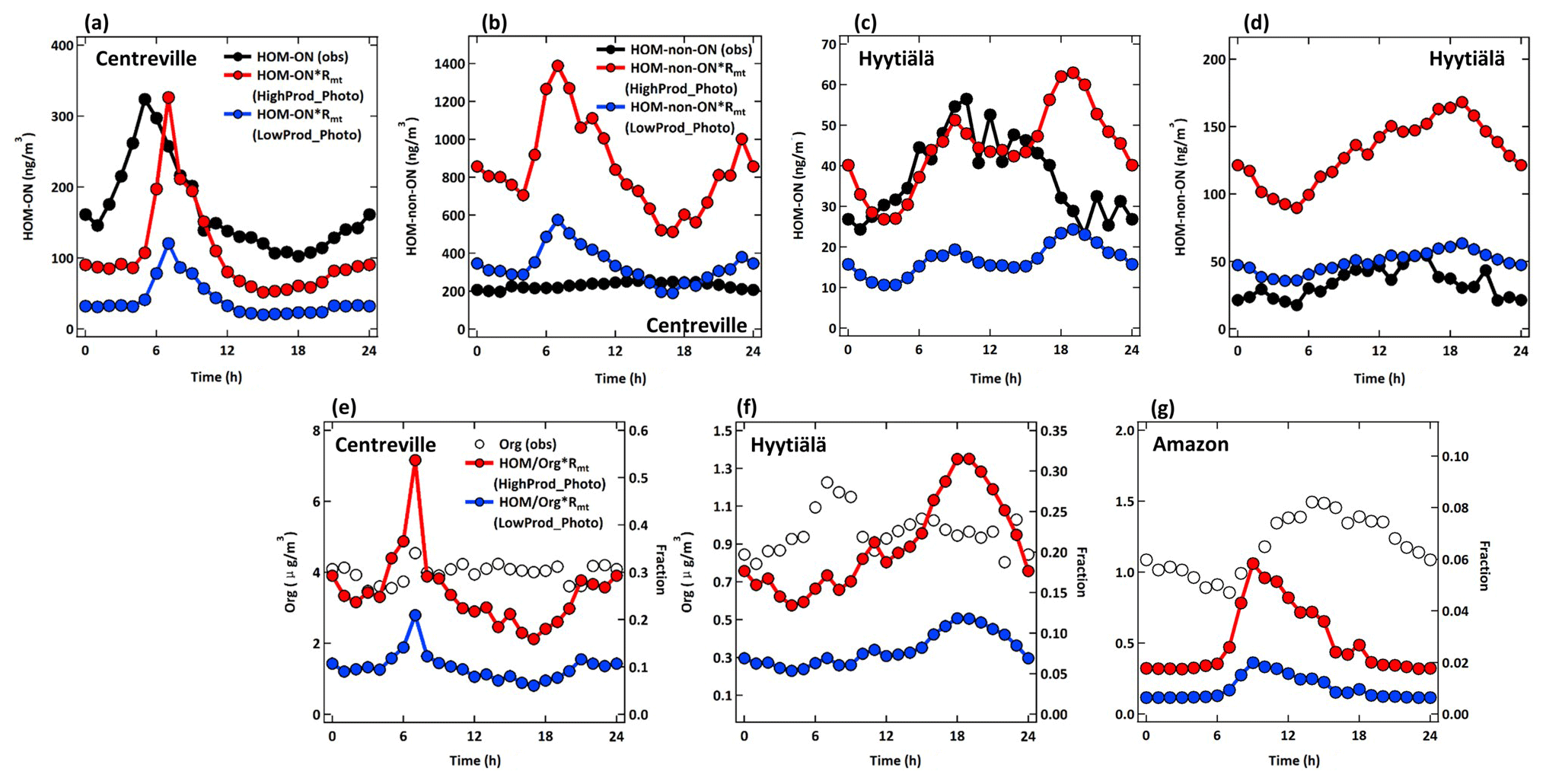
Figure 9Diurnal changes in observed (black line) and simulated (HighProd_Photo – red line; LowProd_Photo – blue line) (a) HOM-ON and (b) HOM-non-ON mass concentrations at the Centreville site. Panels (c) and (d) are the same as panels (a) and (b) but for the Hyytiälä site. (e–f) Diurnal changes in the observed organic aerosol mass concentrations (black hollow circle markers) and the fractions that simulated total HOMs account for by observed organic aerosols (HighProd_Photo – red; LowProd_Photo – blue) at the Centreville, Hyytiälä, and Amazon sites, respectively.
The general shape of the HOM diurnal cycle and HOMs relative to OA (Fig. 9) are typically well captured for each location, except for the late-evening and early-morning periods, which is possibly due to issues simulating the nocturnal layer relative to the emission height of monoterpenes. At the Amazon location, there is a clear late-afternoon peak in the measured OA that is not present in the predicted monoterpene-derived HOM concentrations. These comparisons suggest that, based on the current set of observations, we cannot conclude which set of HOM formation and loss kinetics is most appropriate for describing ambient HOMs. We can conclude that total HOM abundances, including both ON and non-ON HOMs, are potentially higher than those shown in Fig. 9, similar to those predicted by the HighProd_Photo case or the LowProd_noPhoto case, with PBL-averaged mass concentrations in MT-rich regions and seasons on the order of 0.5–1 µg m−3 (see, e.g., Figs. S9 and S10). Uncertainties in first-generation RO2 branching parameters, isomerization rate constants, and HOM chemical fate remain large, with limited observational constraints on total HOM concentrations (gas + particle).
3.4 Vertical profiles of HOMs
Figure 10 summarizes the vertical distribution of HOMs predicted by the LowProd_Photo simulation for two locations, one over the southeastern USA SOAS site and one over the Amazon region using the same grid that contains the GoAmazon T3 site (Martin et al., 2016). The expected predominance of MTs present within the PBL in all seasons and at all locations related to the surface vegetation source is evident. Furthermore, the different vertical profiles of HOM-ON compared with non-ON HOMs are also evident, with slower decays of non-ON HOMs with altitude up to 2–3 km above the surface during JJA in both the southeastern USA and Amazon regions, likely due to changes in the HOM- RO2 fate with altitude (see Sect. 3.1). Over the Amazon during JJA and SON, both non-ON HOM and HOM-ON concentrations are predicted to be relatively enhanced between 1 and 5 km compared with the lowest-altitude concentrations. The relative enhancement in this altitudinal region during JJA and SON compared with DJF and MAM likely reflects overall drier conditions but also significant vertical transport of HOM precursors during these seasons (e.g., through shallow convection). The relative enhancements specifically between 1 and 4 km compared with altitudes higher than 5 km could also reflect the temperature dependence of the unimolecular H-shift rate constant describing monoterpene-derived RO2 autoxidation and changing biomolecular reaction rates with altitude. This relative enhancement is not as obvious in the vertical profiles over the southeastern USA, which appear as smoother monotonic decays with altitude and are higher in abundance during summer months.
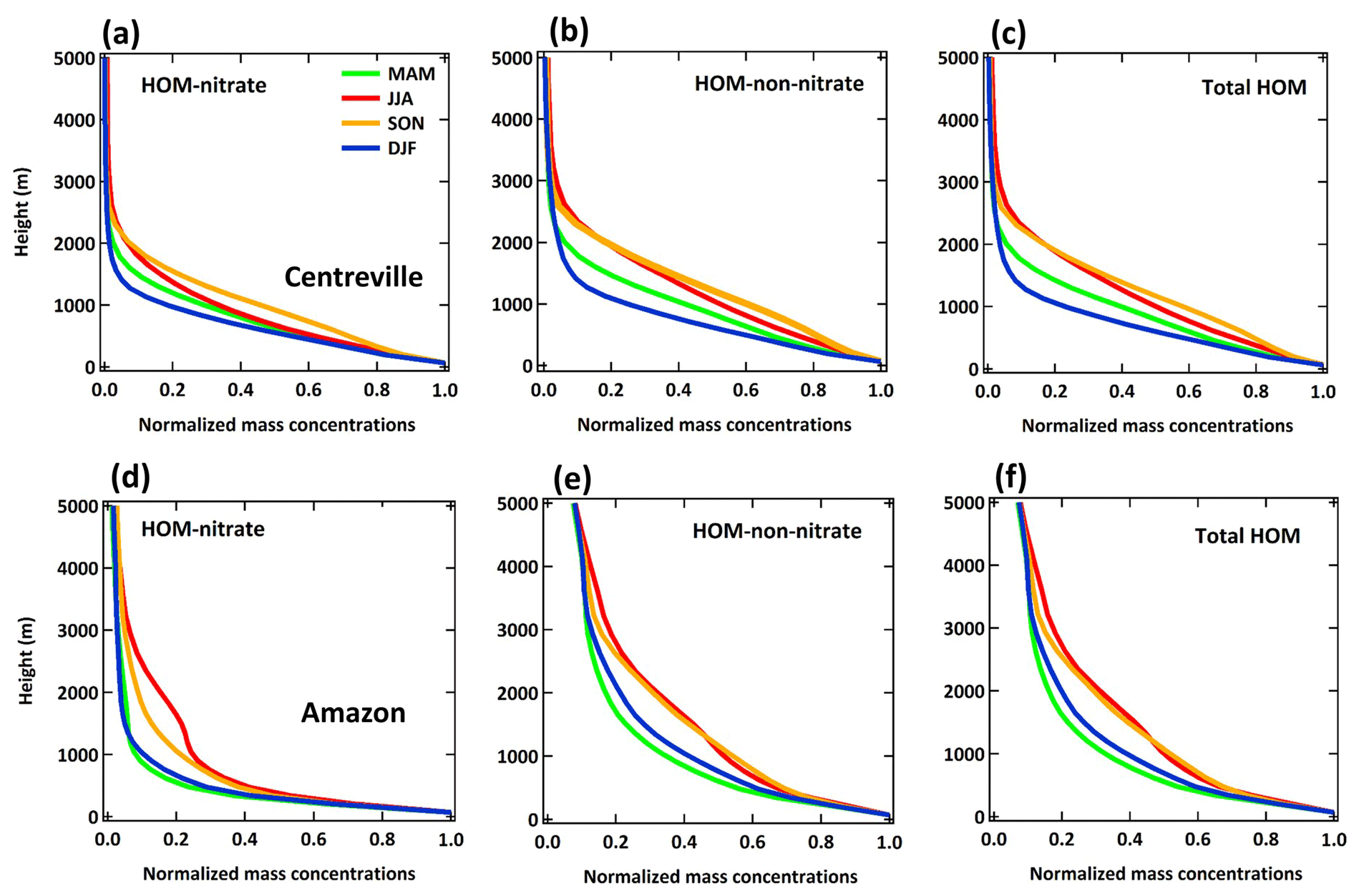
Figure 10The seasonal averaged vertical profiles of HOM-ON (a, d), HOM-non-ON (b, e), and total HOMs (c, f) at the Centreville (a–c) and Amazon (d–f) sites. All of the results are from the LowProd_Photo experiment. Values are normalized to the lowest-level values of each season. The profiles with absolute concentrations are shown in Fig. S9.
HOM-RO2 accretion products illustrate similar vertical profiles to the HOM monomers (see Figs. S12 and S13). If we use the rate constants reported by Berndt et al. (2018a, b) and a branching ratio of unity for HOM-RO2-derived accretion products, the seasonal mean abundance of predicted total C15 and C20 HOM accretion products reaches 1 and 5 µg m−3 in the PBL over the southeastern USA and tropical forest regions, respectively (see the Supplement), and decay reaches respective values of and µg m−3 (1×106 and 4×107 molec. cm−3) in the upper troposphere over these regions. Assuming a HOM-RO2 accretion product yield of 4 % instead, the predicted total of C15 and C20 HOM accretion products is 0.2 and 1 µg m−3 over the southeastern USA and tropical forests, respectively, decaying to respective values of and µg m−3 (3×105 and 1×107 molec. cm−3) in the upper troposphere. At such average concentrations in the upper troposphere over these regions, we conclude that either type of HOM accretion product will likely contribute significantly to new particle formation and growth, but uncertainty in the accretion product branching of HOM-RO2 reactions leads to a factor of 4 range in their estimated contribution.
We implemented a new mechanism to describe MT-derived RO2 chemistry in the GEOS-Chem global chemical transport model. The mechanism is relatively simple, adding 10 species and 37 reactions to the standard mechanism, without substantial addition of computation time. We focused on updating the representation of unimolecular H-shift reactions to form HOM-RO2 and the fate of HOM-RO2 as well as on the self- and cross-reactions of MT-derived RO2 and isoprene-derived RO2. Several sensitivity studies were conducted to evaluate the impact of various mechanism parameters and associated uncertainties; where possible, we also undertook a comparison with observations. The results from these sensitivity studies show that, for a model resolution of 2∘ × 2.5∘, uncertainty in the average H-shift rate constant is less important for predicted HOM concentrations than the fraction of MT reactions with OH or O3 to form RO2, which can undergo H shift and autoxidation, and the photochemical lifetime of HOMs. While a comprehensive comparison of HOM predictions with OA remains, the model predictions of HOMs did not exceed total measured OA mass concentrations at three locations, and this is currently the strongest constraint on HOMs. However, using HOM-ON measurements as a guide suggests that if the fraction of MT-RO2 that undergoes relatively rapid H shift (kauto>0.1 s−1) is greater than 0.25, significant photochemical losses of HOM mass from particles that are faster than the wet or dry deposition of particulate organics are required. Indeed, the current estimates of MT-derived HOM monomer and HOM accretion product formation rates from laboratory studies lead to mass concentrations on the same order as or even larger than OA mass concentrations predicted by the default GEOS-Chem model. However, uncertainties in emission inventories of biogenic VOCs and small absolute errors in NO or NO3 concentration fields in global-scale models contribute additional uncertainty with respect to the most appropriate set of parameters to use.
The majority of HOM production occurs in the continental boundary layer where MT emissions are significant, including boreal, temperate, and tropical regions. H shift and autoxidation are the major fate for the subset of MT-RO2 with that capability to undergo autoxidation, outcompeting reactions with NO, HO2, and RO2 up to 6 km altitude in relatively unpolluted regions. Autoxidation of first-generation MT-RO2 is significantly slower in the upper troposphere and likely uncompetitive with reactions with NO and HO2. As such, HOM formation in the outflow of deep convection is unlikely, although HOM formation from MTs detraining from shallow convection below 6 km is feasible.
Implementing faster self- and cross-reactions between RO2 in GEOS-Chem, as found by Zhao et al. (2020) and Berndt et al. (2018a), leads to significantly lower HO2 and RO2 concentrations in boreal and tropical forest regions (by 20 % or more compared with the standard mechanism), but average changes in OH, NOx, and O3 are negligible at the global scale. These reactions also alter the fate of MT-RO2, especially MT-derived HOM-RO2, for which reaction with MT-derived and other RO2 species (typically isoprene-derived species) is the dominant fate throughout the boundary layer, neglecting unimolecular HOM-RO2 reactions. While perhaps unexpected compared with previous RO2 fate assessments using slower RO2 self- and cross-reaction rate constants, such a situation can be supported in part by the molecular composition measurements of MT-HOM species, which show significant contributions of HOMs with H numbers less than 16 and odd numbers of O (e.g., C10H14O9). This evidence alone is not sufficient, as HOM-RO2 reactions with NO could also produce similar results. Future field campaigns that constrain the relevant NOx and oxidant fields along with HOMs in low-NOx regions could provide important constraints in this regard.
The branching to accretion products of RO2 self- and cross-reactions is a key parameter, with significantly different ranges produced by laboratory studies. The concentrations of C15 and C20 accretion products predicted using self- and cross-reaction rate constants of ∼ 10−11–10−10 cm3 molec.−1 s−1 with a conservative branching (4 %), from Zhao et al. (2018), are typically small compared with average OA mass concentrations, except in the tropical forest regions, where these accretion products alone are likely similar to background OA concentrations outside of biomass burning events. Using a larger branching to accretion products as supported by studies by Berndt et al. (2018a, b) leads to such accretion products likely dominating low-volatility products that could contribute to OA, with predicted mass concentrations well exceeding OA mass concentrations in remote tropical regions. Thus, further refinement of the rate constants and branching to gas-phase accretion products and their photochemical fates are needed, especially as these products from HOM-RO2 cross-reactions are likely essential in the contributions of MTs to new particle formation (Bianchi et al., 2019; McFiggans et al., 2019), especially over tropical forest regions (Andreae et al., 2018; Wang et al., 2016; Zhao et al., 2020).
The GEOS-Chem code used for the LowProd_Photo simulation is available publicly at https://doi.org/10.17605/OSF.IO/DNVZ4 (Thornton, 2022). The base GEOS-Chem code for version 12 is available from http://wiki.geos-chem.org/GEOS-Chem_12#12.1.0 (https://doi.org/10.5281/zenodo.1553349, International GEOS-Chem User Community, 2018).
Data used in this paper were obtained from the following repositories.
-
SOAS campaign: https://csl.noaa.gov/groups/csl7/measurements/2013senex/Ground/DataDownload/ (SOAS Science Team, 2020)
-
BAECC campaign: https://www.arm.gov/research/campaigns/amf2014baecc (U.S. Department of Energy Atmospheric Radiation Measurement Program, 2020a) and SMEAR II dataset https://smear.avaa.csc.fi/download (Keronen et al., 2020)
-
GoAmazon: https://www.arm.gov/research/campaigns/amf2014goamazon (U.S. Department of Energy Atmospheric Radiation Measurement Program, 2020b)
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-5477-2022-supplement.
JAT conceived of the project. RX and JAT developed the code. RX conducted the simulations and processed the output. BHL, FDLH, PR and TP provided data. YZ and LJ provided computing resources and support with GEOS-Chem. RX and JAT drafted the manuscript. All the authors read and commented on the manuscript.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Joel A. Thornton was supported by a grant from the National Science Foundation (grant no. CHE-1807204) and the U.S. Department of Energy Atmospheric Science Research program (grant no. DE-SC0021097). Ruochong Xu was supported by the Nanjing University with an undergraduate research fellowship.
This paper was edited by Andreas Hofzumahaus and reviewed by two anonymous referees.
Amos, H. M., Jacob, D. J., Holmes, C. D., Fisher, J. A., Wang, Q., Yantosca, R. M., Corbitt, E. S., Galarneau, E., Rutter, A. P., Gustin, M. S., Steffen, A., Schauer, J. J., Graydon, J. A., Louis, V. L. St., Talbot, R. W., Edgerton, E. S., Zhang, Y., and Sunderland, E. M.: Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition, Atmos. Chem. Phys., 12, 591–603, https://doi.org/10.5194/acp-12-591-2012, 2012.
Andreae, M. O., Afchine, A., Albrecht, R., Holanda, B. A., Artaxo, P., Barbosa, H. M. J., Borrmann, S., Cecchini, M. A., Costa, A., Dollner, M., Fütterer, D., Järvinen, E., Jurkat, T., Klimach, T., Konemann, T., Knote, C., Krämer, M., Krisna, T., Machado, L. A. T., Mertes, S., Minikin, A., Pöhlker, C., Pöhlker, M. L., Pöschl, U., Rosenfeld, D., Sauer, D., Schlager, H., Schnaiter, M., Schneider, J., Schulz, C., Spanu, A., Sperling, V. B., Voigt, C., Walser, A., Wang, J., Weinzierl, B., Wendisch, M., and Ziereis, H.: Aerosol characteristics and particle production in the upper troposphere over the Amazon Basin, Atmos. Chem. Phys., 18, 921–961, https://doi.org/10.5194/acp-18-921-2018, 2018.
Arneth, A., Monson, R. K., Schurgers, G., Niinemets, Ü., and Palmer, P. I.: Why are estimates of global terrestrial isoprene emissions so similar (and why is this not so for monoterpenes)?, Atmos. Chem. Phys., 8, 4605–4620, https://doi.org/10.5194/acp-8-4605-2008, 2008.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q.-B., Liu, H.-Y., Mickley, L. J., and Schultz, M. G.: Global Modeling of Tropospheric Chemistry with Assimilated Meteorology: Model Description and Evaluation, J. Geophys. Res., 106, 73–95, https://doi.org/10.1029/2001JD000807, 2001.
Bianchi, F., Trostl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kuerten, A., Manninen, H. E., Muench, S., Perakyla, O., Petaja, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, https://doi.org/10.1126/science.aad5456, 2016.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Carlton, A. G., de Gouw, J., Jimenez, J. L., Ambrose, J. L., Attwood, A. R., Brown, S., Baker, K. R., Brock, C., Cohen, R. C., Edgerton, S., Farkas, C. M., Farmer, D., Goldstein, A. H., Gratz, L., Guenther, A., Hunt, S., Jaeglé, L., Jaffe, D. A., Mak, J., McClure, C., Nenes, A., Nguyen, T. K., Pierce, J. R., de Sa, S., Selin, N. E., Shah, V., Shaw, S., Shepson, P. B., Song, S., Stutz, J., Surratt, J. D., Turpin, B. J., Warneke, C., Washenfelder, R. A., Wennberg, P. O., and Zhou, X.: Synthesis of the Southeast Atmosphere Studies: Investigating Fundamental Atmospheric Chemistry Questions, B. Am. Meteorol. Soc., 99, 547–567, https://doi.org/10.1175/BAMS-D-16-0048.1, 2018.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
de Sá, S. S., Palm, B. B., Campuzano-Jost, P., Day, D. A., Hu, W., Isaacman-VanWertz, G., Yee, L. D., Brito, J., Carbone, S., Ribeiro, I. O., Cirino, G. G., Liu, Y., Thalman, R., Sedlacek, A., Funk, A., Schumacher, C., Shilling, J. E., Schneider, J., Artaxo, P., Goldstein, A. H., Souza, R. A. F., Wang, J., McKinney, K. A., Barbosa, H., Alexander, M. L., Jimenez, J. L., and Martin, S. T.: Urban influence on the concentration and composition of submicron particulate matter in central Amazonia, Atmos. Chem. Phys., 18, 12185–12206, https://doi.org/10.5194/acp-18-12185-2018, 2018.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J. J., Mentel, T. F., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J. J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Gelaro, R., McCarty, W., Suárez, M. J., Todling, R., Molod, A., Takacs, L., Randles, C. A., Darmenov, A., Bosilovich, M. G., Reichle, R., Wargan, K., Coy, L., Cullather, R., Draper, C., Akella, S., Buchard, V., Conaty, A., da Silva, A. M., Gu, W., Kim, G., Koster, R., Lucchesi, R., Merkova, D., Nielsen, J. E., Partyka, G., Pawson, S., Putman, W., Rienecker, M., Schubert, S. D., Sienkiewicz, M., and Zhao, B.: The modern-era retrospective analysis for research and applications, version 2 (MERRA-2), J. Climate, 30, 5419–5454, 2017.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
International GEOS-Chem User Community: geoschem/geos-chem: GEOS-Chem 12.1.0 (12.1.0), Zenodo [code], https://doi.org/10.5281/zenodo.1553349, 2018.
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 12, 878, https://doi.org/10.1038/s41467-021-21172-w, 2021.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles, Aerosol Sci. Tech., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Keller, C. A., Long, M. S., Yantosca, R. M., Da Silva, A. M., Pawson, S., and Jacob, D. J.: HEMCO v1.0: a versatile, ESMF-compliant component for calculating emissions in atmospheric models, Geosci. Model Dev., 7, 1409–1417, https://doi.org/10.5194/gmd-7-1409-2014, 2014.
Keronen, P., Aalto, J., Rantala, P., Kolari, P., and Aalto, P.: SMEAR II Database, Fairdata.fi [data set], https://smear.avaa.csc.fi/download, last access: July 2020.
Krapf, M., El Haddad, I., Bruns, E. A., Molteni, U., Daellenbach, K. R., Prévôt, A. S. H., Baltensperger, U., and Dommen, J.: Labile Peroxides in Secondary Organic Aerosol, Chem, 1, 603–616, https://doi.org/10.1016/j.chempr.2016.09.007, 2016.
Kulmala, M., Petäjä, T., Ehn, M., Thornton, J., Sipilä, M., Worsnop, D. R., and Kerminen, V.-M.: Chemistry of Atmospheric Nucleation: On the Recent Advances on Precursor Characterization and Atmospheric Cluster Composition in Connection with Atmospheric New Particle Formation, Annu. Rev. Phys. Chem., 65, 21–37, https://doi.org/10.1146/annurev-physchem-040412-110014, 2014.
Kurten, T., Rissanen, M. P., Mackeprang, K., Thornton, J. A., Hyttinen, N., Jorgensen, S., Ehn, M., Kjaergaard, H. G., Kurtén, T., Rissanen, M. P., Mackeprang, K., Thornton, J. A., Hyttinen, N., Jørgensen, S., Ehn, M., and Kjaergaard, H. G.: Computational Study of Hydrogen Shifts and Ring-Opening Mechanisms in α-Pinene Ozonolysis Products, J. Phys. Chem. A, 119, 11366–11375, https://doi.org/10.1021/acs.jpca.5b08948, 2015.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., Thornton, J. A., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Lee, B. H., Lopez-Hilfiker, F. D., D'Ambro, E. L., Zhou, P., Boy, M., Petäjä, T., Hao, L., Virtanen, A., and Thornton, J. A.: Semi-volatile and highly oxygenated gaseous and particulate organic compounds observed above a boreal forest canopy, Atmos. Chem. Phys., 18, 11547–11562, https://doi.org/10.5194/acp-18-11547-2018, 2018.
Lee, B. H., D'Ambro, E. L., Lopez-Hilfiker, F. D., Schobesberger, S., Mohr, C., Zawadowicz, M. A., Liu, J., Shilling, J. E., Hu, W., Palm, B. B., Jimenez, J. L., Hao, L., Virtanen, A., Zhang, H., Goldstein, A. H., Pye, H. O. T., and Thornton, J. A.: Resolving Ambient Organic Aerosol Formation and Aging Pathways with Simultaneous Molecular Composition and Volatility Observations, ACS Earth Sp. Chem., 4, 391–402, https://doi.org/10.1021/acsearthspacechem.9b00302, 2020.
Liu, H., Jacob, D. J., Bey, I., and Yantosca, R. M.: Constraints from 210Pb and 7Be on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields, J. Geophys. Res.-Atmos., 106, 12109–12128, https://doi.org/10.1029/2000JD900839, 2001.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
Mao, J., Jacob, D. J., Evans, M. J., Olson, J. R., Ren, X., Brune, W. H., Clair, J. M. St., Crounse, J. D., Spencer, K. M., Beaver, M. R., Wennberg, P. O., Cubison, M. J., Jimenez, J. L., Fried, A., Weibring, P., Walega, J. G., Hall, S. R., Weinheimer, A. J., Cohen, R. C., Chen, G., Crawford, J. H., McNaughton, C., Clarke, A. D., Jaeglé, L., Fisher, J. A., Yantosca, R. M., Le Sager, P., and Carouge, C.: Chemistry of hydrogen oxide radicals (HOx) in the Arctic troposphere in spring, Atmos. Chem. Phys., 10, 5823–5838, https://doi.org/10.5194/acp-10-5823-2010, 2010.
Mao, J., Paulot, F., Jacob, D. J., Cohen, R. C., Crounse, J. D., Wennberg, P. O., Keller, C. A., Hudman, R. C., Barkley, M. P., and Horowitz, L. W.: Ozone and organic nitrates over the eastern United States: Sensitivity to isoprene chemistry, J. Geophys. Res.-Atmos., 118, 11256–11268, https://doi.org/10.1002/jgrd.50817, 2013.
Martin, S. T., Artaxo, P., Machado, L. A. T., Manzi, A. O., Souza, R. A. F., Schumacher, C., Wang, J., Andreae, M. O., Barbosa, H. M. J., Fan, J., Fisch, G., Goldstein, A. H., Guenther, A., Jimenez, J. L., Pöschl, U., Silva Dias, M. A., Smith, J. N., and Wendisch, M.: Introduction: Observations and Modeling of the Green Ocean Amazon (GoAmazon2014/5), Atmos. Chem. Phys., 16, 4785–4797, https://doi.org/10.5194/acp-16-4785-2016, 2016.
Massoli, P., Stark, H., Canagaratna, M. R., Krechmer, J. E., Xu, L., Ng, N. L., Mauldin, R. L., Yan, C., Kimmel, J., Misztal, P. K., Jimenez, J. L., Jayne, J. T., and Worsnop, D. R.: Ambient Measurements of Highly Oxidized Gas-Phase Molecules during the Southern Oxidant and Aerosol Study (SOAS) 2013, ACS Earth Sp. Chem., 2, 653–672, https://doi.org/10.1021/acsearthspacechem.8b00028, 2018.
McDonald, B. C., de Gouw, J. A., Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L., Lee-Taylor, J., Hayes, P. L., McKeen, S. A., Cui, Y. Y., Kim, S.-W., Gentner, D. R., Isaacman-VanWertz, G., Goldstein, A. H., Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, A. M., Simpson, D., Bergstroem, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Messina, P., Lathière, J., Sindelarova, K., Vuichard, N., Granier, C., Ghattas, J., Cozic, A., and Hauglustaine, D. A.: Global biogenic volatile organic compound emissions in the ORCHIDEE and MEGAN models and sensitivity to key parameters, Atmos. Chem. Phys., 16, 14169–14202, https://doi.org/10.5194/acp-16-14169-2016, 2016.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 103, 4657–4690, https://doi.org/10.1021/cr020527p, 2003.
Öström, E., Putian, Z., Schurgers, G., Mishurov, M., Kivekäs, N., Lihavainen, H., Ehn, M., Rissanen, M. P., Kurtén, T., Boy, M., Swietlicki, E., and Roldin, P.: Modeling the role of highly oxidized multifunctional organic molecules for the growth of new particles over the boreal forest region, Atmos. Chem. Phys., 17, 8887–8901, https://doi.org/10.5194/acp-17-8887-2017, 2017.
Palen, E. J., Allen, D. T., Pandis, S. N., Paulson, S. E., Seinfeld, J. H., and Flagan, R. C.: Fourier transform infrared analysis of aerosol formed in the photo-oxidation of isoprene and β-pinene, Atmos. Environ. A-Gen., 26, 1239–1251, https://doi.org/10.1016/0960-1686(92)90385-X, 1992.
Pandis, S. N., Harley, R. A., Cass, G. R., and Seinfeld, J. H.: Secondary organic aerosol formation and transport, Atmos. Environ. A-Gen., 26, 2269–2282, https://doi.org/10.1016/0960-1686(92)90358-R, 1992.
Petäjä, T., O'Connor, E. J., Moisseev, D., Sinclair, V. A. V. A., Manninen, A. J. A. J., Vaananen, R., von Lerber, A., Thorntoton, J. A., Nicocoll, K., Petersen, W., Chandrasekar, V., Smith, J. N., Winkler, P. M., Krueger, O., Hakola, H., Timonen, H., Brus, D., Laurila, T., Asmi, E., Riekkola, M.-L., Mona, L., Massoli, P., Engelmann, R., Komppppula, M., Wang, J., Kuang, C., Baeck, J., Virtanen, A., Levula, J., Ritsche, M., Hickmon, N., Petäjä, T., O'Connor, E. J., Moisseev, D., Sinclair, V. A. V. A., Manninen, A. J. A. J., Väänänen, R., von Lerber, A., Thornton, J. A., Nicoll, K., Petersen, W., Chandrasekar, V., Smith, J. N., Winkler, P. M., Krüger, O., Hakola, H., Timonen, H., Brus, D., Laurila, T., Asmi, E., Riekkola, M.-L., Mona, L., Massoli, P., Engelmann, R., Komppula, M., Wang, J., Kuang, C., Bäck, J., Virtanen, A., Levula, J., Ritsche, M., and Hickmon, N.: BAECC: A Field Campaign to Elucidate the Impact of Biogenic Aerosols on Clouds and Climate, B. Am. Meteorol. Soc., 97, 1909–1928, https://doi.org/10.1175/BAMS-D-14-00199.1, 2016.
Pospisilova, V., Lopez-Hilfiker, F. D., Bell, D. M., El Haddad, I., Mohr, C., Huang, W., Heikkinen, L., Xiao, M., Dommen, J., Prevot, A. S. H., Baltensperger, U., and Slowik, J. G.: On the fate of oxygenated organic molecules in atmospheric aerosol particles, Sci. Adv., 6, 11, https://doi.org/10.1126/sciadv.aax8922, 2020.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Richters, S., Herrmann, H., and Berndt, T.: Highly Oxidized RO2 Radicals and Consecutive Products from the Ozonolysis of Three Sesquiterpenes, Environ. Sci. Technol., 50, 2354–2362, https://doi.org/10.1021/acs.est.5b05321, 2016.
Roldin, P., Ehn, M., Kurtén, T., Olenius, T., Rissanen, M. P., Sarnela, N., Elm, J., Rantala, P., Hao, L., Hyttinen, N., Heikkinen, L., Worsnop, D. R., Pichelstorfer, L., Xavier, C., Clusius, P., Öström, E., Petäjä, T., Kulmala, M., Vehkamäki, H., Virtanen, A., Riipinen, I., and Boy, M.: The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system, Nat. Commun., 10, 4370, https://doi.org/10.1038/s41467-019-12338-8, 2019.
SOAS (Southern Oxidant and Aerosol Study) Science Team: SOAS 2013 Centreville Site Data Download, NOAA Chemical Sciences Laboratory [data set], https://csl.noaa.gov/groups/csl7/measurements/2013senex/Ground/DataDownload/, last access: July 2020.
Thornton, J. A.: GEOS-Chem Monoterpene Peroxy Radical Autoxidation Scheme, OSF Home [code], https://doi.org/10.17605/OSF.IO/DNVZ4, 2022.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
U.S. Department of Energy Atmospheric Radiation Measurement Program: Biogenic Aerosols – Effects on Clouds and Climate Campaign Data, ARM [data set], https://www.arm.gov/research/campaigns/amf2014baecc, last access: July 2020a.
U.S. Department of Energy Atmospheric Radiation Measurement Program: GoAmazon Data, ARM [data set], https://www.arm.gov/research/campaigns/amf2014goamazon, last access: July 2020b.
Wang, J., Krejci, R., Giangrandel, S., Kuang, C., Barbosa, H. M. J., Brito, J., Carbone, S., Chi, X., Comstock, J., Ditas, F., Lavric, J., Manninen, H. E., Mei, F., Moran-Zuloaga, D., Poehlker, C., Poehlker, M. L., Saturno, J., Schmid, B., Souza, R. A. F., Springston, S. R., Tomlinson, J. M., Toto, T., Walter, D., Wimmer, D., Smith, J. N., Kulmala, M., Machado, L. A. T., Artaxo, P., Andreae, M. O., Petaja, T., and Martin, S. T.: Amazon boundary layer aerosol concentration sustained by vertical transport during rainfall, Nature, 539, 416–419, https://doi.org/10.1038/nature19819, 2016.
Weber, J., Archer-Nicholls, S., Griffiths, P., Berndt, T., Jenkin, M., Gordon, H., Knote, C., and Archibald, A. T.: CRI-HOM: A novel chemical mechanism for simulating highly oxygenated organic molecules (HOMs) in global chemistry–aerosol–climate models, Atmos. Chem. Phys., 20, 10889–10910, https://doi.org/10.5194/acp-20-10889-2020, 2020.
Wesely, M. L.: Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ., 23, 1293–1304, https://doi.org/10.1016/0004-6981(89)90153-4, 1989.
Xu, L., Pye, H. O. T., He, J., Chen, Y., Murphy, B. N., and Ng, N. L.: Experimental and model estimates of the contributions from biogenic monoterpenes and sesquiterpenes to secondary organic aerosol in the southeastern United States, Atmos. Chem. Phys., 18, 12613–12637, https://doi.org/10.5194/acp-18-12613-2018, 2018.
Xu, L., Møller, K. H., Crounse, J. D., Otkjær, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular reactions of peroxy radicals formed in the oxidation of α-Pinene and β-Pinene by hydroxyl radicals, J. Phys. Chem. A, 123, 1661–1674, https://doi.org/10.1021/acs.jpca.8b11726, 2019.
Zawadowicz, M. A., Lee, B. H., Shrivastava, M., Zelenyuk, A., Zaveri, R. A., Flynn, C., Thornton, J. A., and Shilling, J. E.: Photolysis Controls Atmospheric Budgets of Biogenic Secondary Organic Aerosol, Environ. Sci. Technol., 54, 3861–3870, https://doi.org/10.1021/acs.est.9b07051, 2020.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V, Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhang, L., Gong, S., Padro, J., and Barrie, L.: A size-segregated particle dry deposition scheme for an atmospheric aerosol module, Atmos. Environ., 35, 549–560, https://doi.org/10.1016/S1352-2310(00)00326-5, 2001.
Zhang, S.-H., Shaw, M., Seinfeld, J. H., and Flagan, R. C.: Photochemical aerosol formation from α-pinene- and β-pinene, J. Geophys. Res.-Atmos., 97, 20717–20729, https://doi.org/10.1029/92JD02156, 1992.
Zhao, B., Shrivastava, M., Donahue, N. M., Gordon, H., Schervish, M., Shilling, J. E., Zaveri, R. A., Wang, J., Andreae, M. O., Zhao, C., Gaudet, B., Liu, Y., Fan, J., and Fast, J. D.: High concentration of ultrafine particles in the Amazon free troposphere produced by organic new particle formation, P. Natl. Acad. Sci. USA, 117, 25344–25351, https://doi.org/10.1073/pnas.2006716117, 2020.
Zhao, Y., Thornton, J. A., and Pye, H. O. T.: Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, P. Natl. Acad. Sci. USA, 115, 12142–12147, https://doi.org/10.1073/pnas.1812147115, 2018.
Zhu, J., Penner, J. E., Yu, F., Sillman, S., Andreae, M. O., and Coe, H.: Decrease in radiative forcing by organic aerosol nucleation, climate, and land use change, Nat. Commun., 10, 423, https://doi.org/10.1038/s41467-019-08407-7, 2019.