the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Mar 2022
| 08 Mar 2022
Analysis of reduced and oxidized nitrogen-containing organic compounds at a coastal site in summer and winter
Jenna C. Ditto
Jo Machesky
Drew R. Gentner
Nitrogen-containing organic compounds, which may be directly emitted into the atmosphere or which may form via reactions with prevalent reactive nitrogen species (e.g., NH3, NOx, NO3), have important but uncertain effects on climate and human health. Using gas and liquid chromatography with soft ionization and high-resolution mass spectrometry, we performed a molecular-level speciation of functionalized organic compounds at a coastal site on the Long Island Sound in summer (during the 2018 Long Island Sound Tropospheric Ozone Study – LISTOS – campaign) and winter. This region often experiences poor air quality due to the emissions of reactive anthropogenic, biogenic, and marine-derived compounds and their chemical transformation products. We observed a range of functionalized compounds containing oxygen, nitrogen, and/or sulfur atoms resulting from these direct emissions and chemical transformations, including photochemical and aqueous-phase processing that was more pronounced in summer and winter, respectively. In both summer and winter, nitrogen-containing organic aerosols dominated the observed distribution of functionalized particle-phase species ionized by our analytical techniques, with 85 % and 68 % of total measured ion abundance containing a nitrogen atom, respectively. Nitrogen-containing particles included reduced nitrogen functional groups (e.g., amines, imines, azoles) and common NOz contributors (e.g., organonitrates). Reduced nitrogen functional groups observed in the particle phase were frequently paired with oxygen-containing groups elsewhere on the molecule, and their prevalence often rivaled that of oxidized nitrogen groups detected by our methods. Supplemental gas-phase measurements, collected on adsorptive samplers and analyzed with a novel liquid chromatography-based method, suggest that gas-phase reduced nitrogen compounds are possible contributing precursors to the observed nitrogen-containing particles. Altogether, this work highlights the prevalence of reduced nitrogen-containing compounds in the less-studied northeastern US and potentially in other regions with similar anthropogenic, biogenic, and marine source signatures.
- Article
(3062 KB) - Full-text XML
-
Supplement
(5325 KB) - BibTeX
- EndNote





Coastal regions near the Long Island Sound often experience poor air quality due to a combination of biogenic and anthropogenic emissions from upwind metropolitan areas along the East Coast of the US. It is well established that these emissions undergo chemical transformations to form secondary pollutants during hours to days of over-water transport to downwind locations, including the states of Connecticut, Rhode Island, and Massachusetts (e.g., Cleveland et al., 1976). Emissions of gas-phase organic compounds (e.g., volatile, intermediate, and semi-volatile organic compounds – VOCs, IVOCs, and SVOCs) and primary organic aerosol (POA) are oxidized via numerous pathways in the atmosphere to yield ozone (O3) and secondary organic aerosol (SOA) (Hallquist et al., 2009). SOA constitutes a variable but significant fraction of particulate matter with a diameter of 2.5 µm or less (i.e., PM2.5). Both O3 and PM2.5 are of particular concern for human health and climate; O3 is known to cause an increase in respiratory-related illnesses (Di et al., 2017; Jerrett et al., 2009), while PM2.5 is known to cause adverse cardiovascular, respiratory, and cognitive effects and to impact climate forcings (Di et al., 2017; Hallquist et al., 2009; Kilian and Kitazawa, 2018; Pope and Dockery, 2006). Coupled with local emissions and chemistry, these incoming aged air parcels from coastal metropolitan areas contribute to the Long Island Sound region often entering non-attainment for O3 (United States Environmental Protection Agency, 2020), especially in the summer.
The chemistry and composition of organic compound emissions and secondary transformation products in the Long Island Sound area are historically understudied, though some past work has advanced our understanding of important sources and chemical pathways in the region. For example, VOC and sub-micron particulate matter composition were investigated during the 2002 New England Air Quality Study (de Gouw et al., 2005), with further VOC speciation in 2004 during the New England Air Quality Study – Intercontinental Transport and Chemical Transformation campaign (Warneke et al., 2007). More recently, a 2015 aircraft campaign in the northeastern US called WINTER characterized wintertime chemistry in the region and also investigated organic aerosol formation via aerosol mass spectrometry (Schroder et al., 2018). Finally, the Long Island Sound Tropospheric Ozone Study (LISTOS) campaign in 2018 focused on measuring and modeling O3 mixing ratios over the sound to investigate the dynamics of O3 formation linked to large metropolitan areas along the coast and the associated downwind impacts (Zhang et al., 2020).
However, little is known about the molecular-level chemical composition of the gas-phase IVOCs/SVOCs and functionalized organic aerosol formed in the northeastern US. This molecular-level speciation is key to understanding the physical/chemical properties of these compounds in the atmosphere and their chemical transformations, especially for classes of compounds containing reduced and oxidized nitrogen functional groups, whose emissions, lifetimes, and ultimate impacts are generally poorly understood. For example, nitrogen-containing compounds that serve as reservoir species for nitrogen oxides may increase the overall lifetimes of nitrogen oxides in the atmosphere via renoxification mechanisms (e.g., the photolysis of particulate nitrates, which has been studied in the marine boundary layer, Ye et al., 2016); some may act as light-absorbing chromophores (e.g., the brown carbon studied from a methylglyoxal and ammonium sulfate system, which yielded mostly N-containing chromophores, Lin et al., 2015), and some may have adverse but uncertain effects on human health (e.g., impacts on immune response to allergens, Ng et al., 2017).
There have been a wide range of measurements of organic nitrogen in the atmosphere, and many past studies have emphasized enhancements in the contribution of this organic nitrogen in various forms of water in the atmosphere, such as cloud water, fog water, rainwater, and aerosol liquid water. For example, in cloud water, observations of important contributions from nitrogen- and oxygen-containing organic compounds have been made using Fourier-transform ion cyclotron resonance mass spectrometry (FT ICR-MS) (Zhao et al., 2013). Across all the oxygenates (i.e., CHO), oxygen- and nitrogen-containing compounds (CHON), oxygen- and sulfur-containing compounds (CHOS), and oxygen-, nitrogen-, and sulfur-containing compounds (CHONS) that Zhao et al. (2013) observed in cloud water, roughly 65 % of ions (by number count) contained a nitrogen atom. Roughly half of all species observed were CHON compounds. Also, roughly half of the CHON species had low (<0.7) and were hypothesized to contain reduced nitrogen functional groups. Another example from a study in the southeastern US by Boone et al. (2015) showed that cloud water samples contained a large fraction of nitrogenated species relative to aerosol-phase samples. From a combination of direct-infusion electrospray ionization and nanospray desorption electrospray ionization measurements with high-resolution mass spectrometry, Boone et al. (2015) observed roughly 4 times more CHON molecular formulas in cloud water than in particle-phase samples, representing ∼20 % of all ions, by number count, in cloud water. They also suggested an important role for aqueous-phase reactions occurring between water-soluble oxygenated organic compounds and a diversity of nitrogen-containing species such as ammonium, nitrate, and small amines (Boone et al., 2015).
Similar observations have been made in fog water samples. For example, LeClair et al. (2012) discussed the importance of water-soluble organic nitrogen-containing compounds in fog water using FT ICR-MS. Roughly half of their observed compounds contained a nitrogen atom, and by tracking neutral losses, they identified that 50 %–83 % of their observed CHON species showed a neutral loss of HNO3 and thus likely contained a nitrate group. They noted that, in the absence of HNO3, CH3NO3, NO, or NO2 losses, the remaining nitrogen-containing ions observed likely contained reduced nitrogen groups such as amine, amino, or imine structures (LeClair et al., 2012). Another study of fog droplets with aerosol mass spectrometry by Kim et al. (2019) showed an enrichment of organic nitrogen in fog droplets, including observations of reduced nitrogen groups such as imidazoles and pyrazines (Kim et al., 2019). They observed fog water's ratio to be roughly 4 times greater than the ratio in oxygenated organic aerosol samples.
These trends extend to rainwater as well; FT ICR-MS measurements of rainwater in the northeastern US by Altieri et al. (2009) showed large contributions of nitrogen-containing organic compounds (Altieri et al., 2009). Approximately 70 % of their observed nitrogen-containing species were CHON species from positive mode ionization, which they suggested consisted largely of reduced nitrogen functional groups based on their detection in positive ionization mode and based on their elemental ratios. Similar enhancements in bulk organic nitrogen, made by measuring total nitrogen content and subtracting the contribution from inorganic nitrogen, were noted in both rainwater and aerosols collected on the Mediterranean coast (Mace et al., 2003a) and in both rainwater and cloud water in a Caribbean background marine environment (Gioda et al., 2011).
While Boone et al. (2015) showed enhanced nitrogen content in cloud water relative to aerosol particles, aerosol-phase samples have also been observed in other studies across the globe to contain high organic nitrogen content. For example, at another location in the southeastern US with strong marine and continental air influence, Lin et al. (2010) observed that organic nitrogen in PM2.5 contributed roughly 33 % of total PM2.5 nitrogen mass, which they computed by subtracting inorganic nitrogen contributions from total nitrogen content, as mentioned above (Lin et al., 2010). Similarly, 61 % of primary marine aerosols (magnitude-weighted) collected from a ship in the Atlantic Ocean and analyzed by FT ICR-MS were shown to contain nitrogen, and 54 % of these primary marine aerosol species were CHON compounds, with the remaining 7 % of nitrogen content distributed across CHONS and CHONP species (Wozniak et al., 2014). These primary marine aerosols typically had ratios less than 0.5 and were also likely reduced nitrogen-containing, consistent with Zhao et al. (2013). Other examples include bulk organic nitrogen measurements from aerosols collected inland during both the wet and dry seasons in the Amazon basin (Mace et al., 2003b), from aerosols sampled in Davis, California (Zhang et al., 2002), and from aerosols (and fog) in the Po Valley in Italy (Montero-Martínez et al., 2014).
Finally, a recent study of aerosols collected in a forest in Tokyo highlighted the role of aerosol liquid water as another important medium for the formation of water-soluble organic nitrogen-containing species and showed a positive correlation between the concentration of aerosol liquid water and water-soluble organic nitrogen (Xu et al., 2020).
Considering the coastal nature of our Long Island Sound site and general prevalence of water in the local/regional atmosphere (e.g., as cloud water, fog water, rainwater, and aerosol liquid water), the overall goal of this study was to examine the composition and contributions of nitrogen-containing organic compounds from mixed anthropogenic, biogenic, and marine sources as well as the possible roles of secondary product formation via aqueous-phase chemistry. We collected samples of organic gases and particles for detailed chemical speciation on the coast of the Long Island Sound in Guilford, Connecticut. We note that we used this site as a case study, but our observations of emissions and chemistry at this site are likely informative for other coastal urban and downwind regions due to the ubiquity of nitrogen-containing emissions from anthropogenic, biogenic, and marine sources.
Samples were collected during the summer and winter and analyzed via high-resolution mass spectrometry to speciate the complex mixture of emissions and chemical transformation products. These samples were taken alongside several targeted pollutant measurements including O3, nitrogen oxides (NOx), particulate matter with a diameter of ≤2.5 µm (PM2.5), and black carbon (BC), all to inform our chemically speciated analyses and to contribute to a longer-term characterization of this coastal area.
The specific objectives of this study were to (1) investigate compositional differences and possible chemical pathways contributing to measured summer and winter functionalized organic aerosols at this site, (2) examine the relative contributions of reduced and oxidized nitrogen groups to functionalized organic aerosol, and (3) use a novel sampling and liquid chromatography-based analytical approach to probe the molecular-level composition of functionalized gas-phase organic compounds and investigate possible nitrogen-containing gas-phase precursors to the observed reduced nitrogen-containing particles.
We collected measurements at the Yale Coastal Field Station (YCFS) in Guilford, Connecticut (41.26∘ N, 72.73∘ W) (Rogers et al., 2020). Inlets were positioned facing the Long Island Sound (i.e., south–southeast) to capture onshore flow. The YCFS often received aged urban incoming air from East Coast metropolitan areas, similar to known common air parcel trajectories in the region (Fig. S1 in the Supplement). However, due to extensive mixing in the northeastern corridor and over the Long Island Sound, along with extended collection times for offline gas- and particle-phase samples, we also observed considerable biogenic and anthropogenic influence from other areas of the northeastern US.
2.1 Offline samples of organic particles and gases analyzed via liquid and gas chromatography with mass spectrometry
We discuss three types of sampling and quadrupole time-of-flight mass spectrometry-based analyses here: particles collected on Teflon filters and analyzed using liquid chromatography with electrospray ionization, gases collected on packed adsorbent tubes and analyzed using gas chromatography with atmospheric pressure chemical ionization, and functionalized gases collected on cooled polyether ether ketone (PEEK) samplers and analyzed using liquid chromatography with electrospray ionization. Teflon filter and adsorbent tube measurements were collected at the YCFS during the summer as part of the 2018 LISTOS campaign from 9 July to 29 August 2018. Additional filter and adsorbent tube samples were collected during the following winter from 25 February to 5 March 2019. Supplemental wintertime gas-phase samples on PEEK tubing were collected briefly from 5 to 6 March 2020, prior to the COVID-19 shutdown. These sampling periods are discussed here as summer and winter case studies, but longer campaigns are warranted to assess full seasonal trends.
A custom filter and adsorbent tube housing was constructed to simultaneously collect particle- and gas-phase organic compounds, respectively (Sheu et al., 2018). The filter was positioned immediately upstream of the adsorbent tube to collect particles for analysis and to prevent particles from reaching the gas-phase adsorbent tube sample. The housing was designed to minimize spacing between the filter and adsorbent tube to reduce gas-phase losses to upstream surfaces and was built out of a modified passivated stainless steel filter holder (Pall) and an aluminum block with sealed 6.34 mm ( in.) holes for adsorbent tubes (Sheu et al., 2018).
For filter and adsorbent tube collection, we used a short inlet (0.9 m long, in. OD stainless steel tubing, positioned 2.5 m above the ground) upstream of the custom sampler to allow the sampling media to be housed in an air-conditioned trailer. A stainless steel mesh screen (84-mesh) was used at the opening of the inlet to limit particle size to ∼PM10 and to prevent large particles from entering the sampler (Ditto et al., 2018). Penetration efficiency through the mesh screen was computed for the 20 L min−1 flow rate using the screen thickness, mesh size (84-mesh), and wire diameter and accounting for the effects of diffusion, impaction, and interception. Based on this modeling, we expect roughly 50 % penetration efficiency at PM10 and 0 % at PM11 and larger.
2.1.1 Filter sampling, analysis, and data quality assurance and quality control (QA/QC)
Teflon filters (47 mm, 2.0 µm pores, Tisch Scientific) were used for particle-phase sampling. Filters were collected at 20 L min−1 for 8 h during the day (09:00–17:00 LT) and at night (21:00–05:00 LT). Samples were extracted in methanol and analyzed via liquid chromatography (LC) using an Agilent 1260 Infinity LC and an Agilent Poroshell 120 SB-Aq reverse-phase column (2.1×50 mm, 2.7 µm particle size). The LC was coupled to an electrospray ionization (ESI) source and a high-resolution mass spectrometer (Agilent 6550 Q-TOF) and operated following previously described methods (Ditto et al., 2018, 2020). The mass resolution () of the Q-TOF used in this work was ≥25 000–40 000, and the mass accuracy was 1–2 ppm. Our use of LC (or gas chromatography – GC) to separate compounds prior to their ionization and detection by the mass spectrometer reduced mass spectral interferences and thus enabled accurate molecular formula assignments beyond what would be possible by relying on the Q-TOF's mass resolution alone.
Filter extracts were run with MS (i.e., TOF-only, to identify molecular formulas) and MS/MS (i.e., tandem mass spectrometry, to identify functional groups) data acquisition, using both positive and negative mode electrospray ionization. These methods are hereafter referred to as “LC-ESI-MS” and “LC-ESI-MS/MS”, respectively. Acquisition and non-targeted analysis methods, including data quality assurance and quality control (QA/QC), are discussed in past work (Ditto et al., 2018, 2020). Briefly, for LC-ESI-MS analyses, any ion mass appearing in both a sample and its corresponding blank (matching ion mass with a tolerance of 5 ppm and matching ion retention time with a tolerance of 0.25 min – both tolerances were chosen to be quite conservative) was removed if its abundance in the sample was less than 5 times its abundance in the blank. Ions with greater sample : blank ratios were retained, and the abundance of the blank peaks was subtracted from the sample peaks. Positive and negative ionization mode data were combined, and any ions appearing in both modes were flagged; abundances were averaged, and the compound was only counted once. Ions from 50 to 600 were assigned formulas assuming hydrogen or sodium adducts in positive mode and acetate adducts or deprotonation in negative mode. We also allowed for the neutral loss of water. Only peaks that well surpassed instrument noise and that had strong peak quality scores (based on both liquid chromatography and mass spectrometry data) were selected for formula identification according to thresholds detailed in Ditto et al. (2018). Formulas were assigned with the following elemental constraints in Agilent's Mass Hunter software, , minimizing the parts-per-million mass difference between the observed and proposed ion mass and accounting for isotope distribution. Prior to non-targeted analysis, further QA/QC was performed on these formula identifications using custom R code. As discussed by Kind and Fiehn (2007), the number of elements was further constrained to 39 carbons and 72 hydrogens, and ratios were checked to ensure they fell within expected limits () (Kind and Fiehn, 2007). Formulas were then screened to ensure they agreed with the Nitrogen rule, to ensure that all double bond equivalent values were integers and to flag any large mass differences (>7 ppm) between the observed and proposed mass for a given molecular formula.
For MS/MS, any ions from the LC-ESI-MS analyses that passed these QA/QC steps were targeted for MS/MS fragmentation at 5, 10, 20, 30, and 40 V. We used SIRIUS with CSI:FingerID for functional group identification with a subset of compounds from MS analysis (Dührkop et al., 2015, 2019), as detailed in past work (Ditto et al., 2020). We assumed the same ionization behavior as discussed above, with the same elemental composition constraints and a conservative 7 ppm mass tolerance. Functional groups for the top-scoring candidate structure for each ion were tallied with the APRL Substructure Search Program (Ruggeri and Takahama, 2016). The exact position of each functional group was not considered, as the focus of our work was instead to assess the presence or absence of atmospherically relevant functional groups and their combinations across a large number of multifunctional compounds.
After stringent QA/QC for peak shape and accurate molecular formula determination, non-targeted compound identification from LC-ESI-MS identified an average of 200±56 and 167±47 compounds per sample analyzed in summer and winter, respectively, across 34 samples in summer and 15 in winter.
We note that filter ion abundance data are presented as combined positive and negative ionization mode data, which treat the compounds equally without corrections for ionization efficiency. As mentioned above, compounds were not double counted; any ion appearing in both positive and negative mode was flagged, its average abundance was computed, and it was tallied only once. While ionization efficiency differences between compound types exist, their exact effects for multifunctional compounds present in a complex mixture are uncertain. Thus, similarly to other studies and to our past work, we treat the intercomparison across compounds without adjusting for ionization efficiency differences (Ditto et al., 2018). We note that the figures in the main text are displayed as fractions of total observed ion abundance to consider variations in atmospheric abundance across the complex mixture of functionalized species. However, due to uncertainty in exact ionization efficiency, these are not intended to directly represent mass concentration. For comparison, identical figures represented by occurrence (i.e., unweighted by abundance) are presented in the Supplement (Figs. S4–S6, S8–S9, S11, and S13); general observations remain similar between abundance-weighted and occurrence results.
2.1.2 Adsorbent tube sampling, analysis, and data QA/QC
Gas-phase samples were collected on glass adsorbent tubes (6.35 mm OD, 88.9 mm long) packed with quartz wool, glass beads, Tenax TA, and Carbopack X (Sheu et al., 2018). Samples were collected at 200 mL min−1 for 2 h and sub-sampled off of the 20 L min−1 filter flow during the day (02:00–16:00 LT) and at night (02:00–04:00 LT). Adsorbent tubes were analyzed using a GERSTEL TD3.5+ thermal desorption unit and an Agilent 7890B gas chromatograph (GC) with a DB5-MS UI column (). The GC was coupled to an atmospheric pressure chemical ionization (APCI) source and the same Q-TOF as above, operated with MS (i.e., TOF-only) data acquisition and a positive ionization mode only. These methods are hereafter called “GC-APCI-MS”, and acquisition and analysis methods are discussed in past work (Ditto et al., 2021; Khare et al., 2019). After QA/QC (as detailed in Sect. 2.1.1), this non-targeted analysis yielded an average of 388±201 and 612±133 compounds per sample in summer and winter, respectively, across 34 samples in summer and 14 samples in winter.
2.1.3 PEEK collector sampling, analysis, and data QA/QC
Finally, as a supplemental analysis to probe the composition of functionalized gases that were not GC amenable and thus not measured using the adsorbent tube and thermal desorption–gas chromatography techniques mentioned above, we used PEEK-based sample collectors and liquid chromatography to trap and speciate oxygen-, nitrogen-, and/or sulfur-containing gases without thermal desorption. This method was designed to target functionalized gases, which represent important precursors, intermediates, and by-products in the atmospheric processing of emitted organic compounds but which are often challenging to speciate with traditional GC techniques due to their chemical functionality, reactivity, and/or thermal lability. Additionally, in many gas-phase measurement systems, primary emissions (i.e., hydrocarbons) can overwhelm the signal of more functionalized analytes, adding to the challenge of speciating these lower-abundance compounds.
Thus, to probe the chemical composition of these functionalized gases, we used a sampling approach, desorption method, separation method, and ionization technique that leveraged their relatively lower volatility and higher polarity. This included adsorptive sampling onto cooled PEEK tubing followed by direct inline desorption into the LC mobile phase for LC-ESI-MS analysis. ESI was specifically chosen here because it is sensitive to functionalized compounds. Testing was performed in positive and negative ionization mode, but field samples were run in positive mode only. Further details and discussion of this method, including method development and evaluation, can be found in Sect. S1 and Tables S1–S3 in the Supplement. Briefly, PEEK tubing was cooled to 2 ∘C and used as an adsorptive collector, with a Teflon filter positioned upstream of the PEEK tubing to remove particles. PEEK was selected due to its inert behavior, thus reducing the possibility of surface–analyte interactions that might inhibit effective inline solvent desorption and dissolution. PEEK is also compatible with the solvents used in the LC system and is frequently used in LC instruments. Field samples were collected on cooled PEEK tubing during the subsequent winter (5–6 March 2020) for 2 h each between 08:00 and 14:00 LT. For these 2 h (∼2.6 L) field samples, functionalized gases in a typical 100–250 g mol−1 molecular weight range were resolvable at ∼25–60 ppt in the atmosphere, based on instrument detection limits (Ditto et al., 2018). For analysis, each PEEK collector was installed in the LC system flow path, and analytes were directly desorbed using the LC mobile phase solvents and then trapped and focused on the LC column for 20 min before being analyzed using the same LC-ESI-MS system in positive ionization mode (Fig. 1). This inline mobile phase desorption step gently mobilized potentially fragile analytes from the PEEK collector and trapped and focused them on the LC column prior to chromatographic separation and mass spectral analysis. Additionally, this preconcentration step allows for the detection and characterization of lower-concentration species. Data were processed and QA/QC was performed as detailed in Sect. 2.1.1.
We note that there are other existing approaches for offline collection of highly functionalized organic gases and particles that are compatible with LC analysis, such as spray chambers, particle-into-liquid samplers, coated denuders, and polyurethane foam (PUF) sampling. This PEEK sampling method with inline desorption into the LC mobile phase was pursued to reduce sample preparation steps and thus possibilities of losses (e.g., during solvent extraction or evaporative preconcentration) as well as for its direct similarity to the filter-based particle-phase LC-ESI-MS analysis.
We also note that for all filter collection and LC analyses (filters and PEEK collectors), it is possible that some functional groups of interest may have undergone hydrolysis on the filter during 8 h filter collection periods or in the LC mobile phase, which was primarily water at the beginning of the LC solvent gradient. For example, organonitrates may be susceptible to hydrolysis depending on their structure; tertiary organonitrates can undergo hydrolysis on the timescale of minutes–hours depending on pH, while primary/secondary organonitrates are relatively stable. Hydrolysis occurs more quickly at low pH. The pH of the LC mobile phase (pH∼2) and the pH of the sampled aerosol (, Pye et al., 2020) are both acidic; alpha-pinene-derived organonitrates, for example, could have a lifetime of as low as roughly 8 min to 1.5 h across this pH range (Rindelaub et al., 2016). If hydrolysis occurred, some of the observed compounds could be by-products of other functionalized species. While we did not observe any of our nitrogen-containing test standards to hydrolyze over these timescales, standards were not available to reflect every functional group observed in these datasets.
2.2 Supporting measurements
O3, NOx, PM2.5, and BC concentrations were recorded concurrently during both the summer and winter sampling periods. O3 was measured with a 2B Tech Model 202 Ozone Monitor, NOx with a Thermo Scientific model 42i-TL analyzer, PM2.5 with a MetOne BAM-1020 instrument, and BC with a Magee Scientific AE33 aethalometer. O3 and NOx inlets were constructed from FEP tubing ( in. OD), with a Teflon filter housed in a PFA filter holder upstream to remove particles. The PM2.5 inlet was made of stainless steel tubing (1 in. OD), and the BC inlet was made of copper tubing ( in. OD). Both particle inlets were outfitted with a PM2.5 cyclone to limit particle size to 2.5 µm and below.
All inlets were mounted 3 m above the ground. Instrument flow rates were calibrated with an external mass flow controller. O3 and NOx monitors were zeroed with laboratory-generated zero air. The O3 monitor was calibrated against Connecticut Department of Energy and Environmental Protection instrumentation and further confirmed with an O3 generator in the lab. The NOx monitor was calibrated using a NO standard (AirGas, 2 ppm NO in nitrogen, ±5 %) diluted to 25 ppb with laboratory-generated nitrogen gas. The BC instrument was programmed to conduct an automatic performance check using particle-free air, and the PM2.5 instrument was zeroed following MetOne protocols with particle-free air. O3 and NOx data were collected at 1 s intervals, BC data were collected at 1 min intervals, and PM2.5 data were collected at 1 h intervals. BC data were saved directly from the instrument, while O3, NOx, and PM2.5 data were recorded with a LabJack T7 data logger and custom LabView code. In addition, hourly weather data (temperature, relative humidity, wind speed, wind direction) were collected with a WeatherHawk weather station on top of the 3 m tower.
During the summer, we also collected a small number of size-resolved particle samples on quartz filters using an eight-stage cascade impactor (Thermo Scientific Andersen Non-Viable Cascade Impactor). Sizes ranged from 0.43 to 10.0 µm (stage 0: 9.0–10.0 µm, stage 1: 5.8–9.0 µm, stage 2: 4.7–5.8 µm, stage 3: 3.3–4.7 µm, stage 4: 2.1–3.3 µm, stage 5: 1.1–2.2 µm, stage 6: 0.65–1.1 µm, stage 7: 0.43–0.65 µm). Quartz filters were extracted and analyzed following the same procedure as the Teflon filters discussed above, with the addition of a syringe filtration step to remove insoluble fibers. The cascade impactor was positioned on the roof of the trailer and pulled 28.3 L min−1 (GAST 1531-107B-G557X pump) through the inlet for periods of 8 h during the day and at night (same timing as above).
Finally, we computed 48 h backward trajectories for every hour during each offline sample collection period with the HYSPLIT Backward Trajectory Model (accessed online at https://www.ready.noaa.gov/HYSPLIT.php, last access: 1 June 2020), using GDAS1.0 meteorological data, the field site's coordinates as each trajectory's end point, and a final trajectory height of 50 m above the ground. We selected 48 h trajectories to focus on regional influence at the site, and we selected a final height of 50 m to be high enough to focus on the overall 48 h dynamics and reduce the possible influence of surface topography. Contributions from air parcels extending beyond 48 h likely exist but are outside of the regional scope of our study.
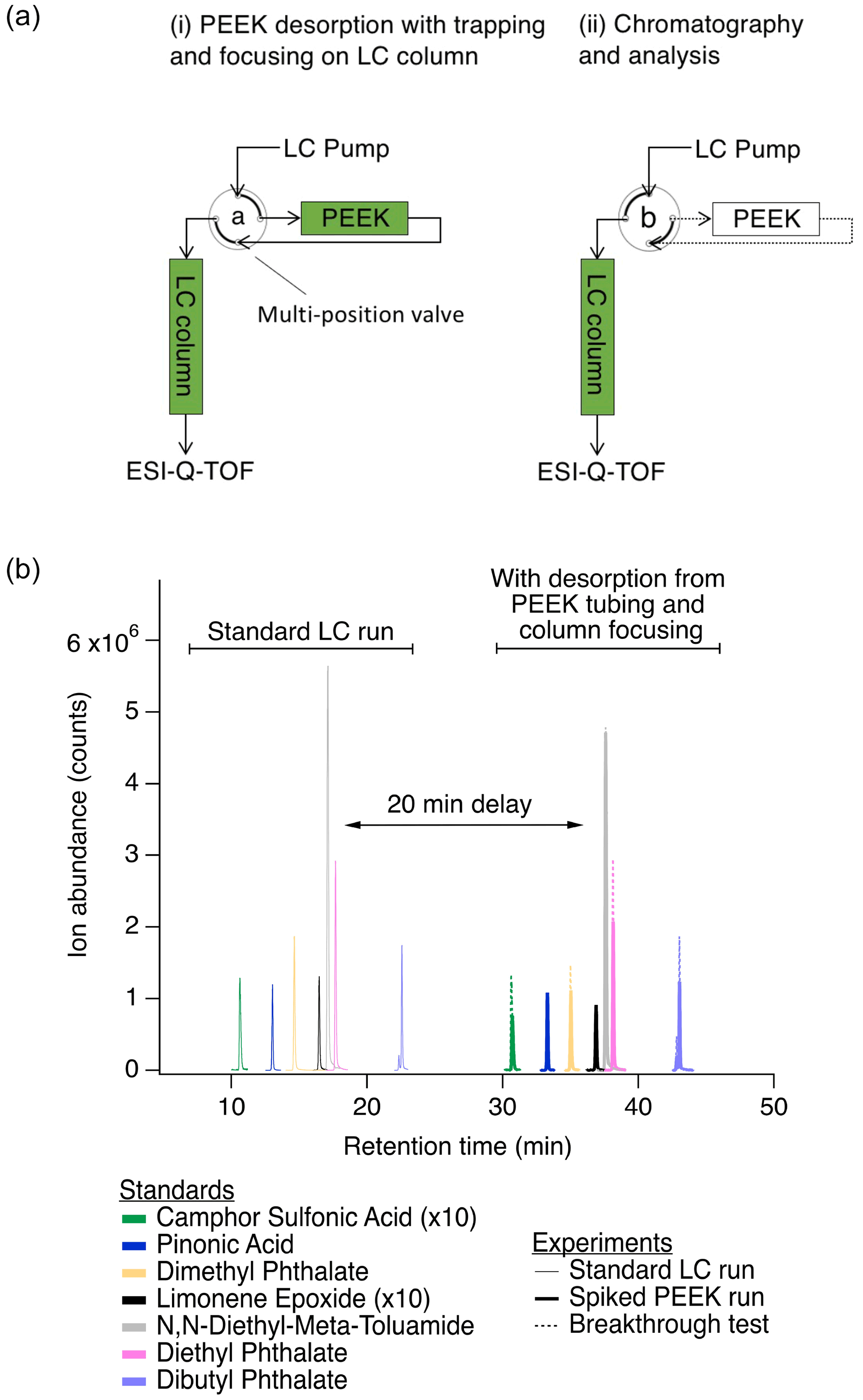
Figure 1(a) Simplified analytical system setup for functionalized gas-phase compounds, showing (i) desorption from the PEEK collector and trapping on the LC column in order to focus analytes prior to chromatographic separation and (ii) subsequent chromatographic separation and analysis (discussed in detail in Sect. S1). Green shading indicates active solvent flow through the PEEK collector and/or LC column. A multi-position valve was switched from position “a” (panel a.i) to position “b” (panel a.ii) to remove the PEEK collector from the flow path for chromatography and analysis. Table S1 describes the flow rates and solvents used in each of these steps. (b) Comparison of selected peaks from a typical LC run (solid traces from 10 to 23 min) to that from a PEEK collector spiked with a standard (bold traces from 30 to 43 min) demonstrates desorption, trapping/focusing, and similar chromatography. Comparable results from a 2 h breakthrough test at 2 ∘C with 22 mL min−1 air flow are also shown (overlayed dotted traces from 30 to 43 min). Spiked PEEK and breakthrough tests were performed to validate these sampling and analysis methods and are discussed further in Sect. S1. Test analytes were used across a range of functionalities, with examples shown here and the full list in Table S2.
3.1 Characteristics of the urban regional site
Backward trajectories for summertime and wintertime samples showed a strong urban influence. Summertime trajectories ranged from the northwest, west, and especially southwest (i.e., New York City and other coastal metropolitan areas, similar to well-established and expected air flow patterns near the Long Island Sound). In contrast, trajectories were almost exclusively from the northwest in the winter (Fig. S1). These air parcels brought a range of compounds from a mixture of anthropogenic, biogenic, and marine sources to the site, all with differences in gas- and particle-phase source profiles. However, due to the varied backward trajectories, dynamic variations in wind direction over the long-duration filter samples (Fig. S2 in the Supplement), and a high degree of mixing over the sound, our 8 h samples are representative of mixed regional conditions in summer and winter and are thus discussed in this context. Further detailed site characterization can be found in Sect. S2 and Figs. S1–S3 in the Supplement.
3.2 Summer and winter comparisons of functionalized organic aerosols
3.2.1 Summertime composition and the influence of photochemistry and NOx
During this period of active photochemistry, the observed distribution of particle-phase compounds in summertime samples spanned the intermediate-volatility (IVOC) to ultra-low-volatility organic compound (ULVOC) range, with a predominance of semivolatile (SVOC), low-volatility (LVOC), and extremely low-volatility organic compounds (ELVOC) as shown in Fig. 2a as a function of compound class. To assess differences in summer and winter volatility distributions, we used individual molecular formulas and the Li et al. (2016) parameterization to estimate the saturation mass concentration (log (C0)) of each observed compound (Li et al., 2016). Compounds were then classified into volatility bins following these definitions: ; ; ; ; ; (Donahue et al., 2011; Schervish and Donahue, 2020).
Due to elevated summertime O3 mixing ratios at the site (shown in Fig. S3, 8 h maximum mixing ratio in summer: 57±20 ppb vs. winter: 46±5 ppb, including day and night sampling periods), O3 may have influenced the photochemical processing of emitted volatile species, especially unsaturated biogenic VOCs which readily undergo ozonolysis due to their chemical structure. However, we did not observe a correlation between 8 h maximum (or 8 h average) O3 mixing ratios with average particle-phase volatility (as saturation mass concentration), carbon number, or (nor did we observe such relationships for gas-phase organic compounds). There were, however, weak relationships between NOx mixing ratios and each of these particle-phase characteristics in the summer. While average NOx mixing ratios were slightly lower during the summer (as shown in Fig. S3, 2.3±1.5 ppb in summer vs. 3.7±2.7 ppb in winter), NOx mixing ratios trended weakly with particle-phase (r∼0.45) and volatility (as saturation mass concentration, r∼0.49) and inversely with carbon number () in summer.
While our correlations and conclusions are somewhat limited by the 8 h filter sampling duration and the resulting highly regionally mixed samples, one possible hypothesis is that the presence of NOx could have promoted more fragmentation reactions in the gas phase (Loza et al., 2014) that decreased average carbon number and correspondingly increased volatility and . In fact, we observed highly oxidized C3–C6 compounds in the gas phase (from adsorbent tube measurements with GC-APCI, Sect. S2) that were possibly products of these fragmentation reactions of larger compounds. These trends of NOx mixing ratios with , volatility, and carbon number were not apparent for the observed complex mixture of gas-phase organic compounds. However, these highly oxidized gases may not have persisted in the gas phase and could have been taken up by the condensed/aqueous phase due to their water solubility, where they would have instead contributed to the observed trends of NOx with carbon number, volatility, and in the particle phase. We note that if there was significant uptake of gas-phase NOz to the particle phase, this may have in part contributed to the particle-phase correlations with NOx given that the chemiluminescence NOx analyzer used in this study is known to also respond to gas-phase NOz (Dunlea et al., 2007).
Additionally, NOx could have been involved in heterogeneous chemistry, promoting oxidation and/or nitrogen addition reactions, such as interaction with to yield organonitrates (Lim et al., 2016), formation and interaction with ambient nitrous acid (HONO) to yield nitrophenols (Vidović et al., 2018), or other pathways.
3.2.2 Comparison to wintertime composition and the role of aqueous-phase chemistry
In the winter, these same relationships between NOx and particle-phase characteristics were not observed. This is possibly due to the decreased role of photochemistry in the winter and the increased role of other competing physical and chemical processes, such as aqueous-phase chemistry. In the discussion of our results, we note that aqueous-phase chemistry is meant to be inclusive of aqueous processing in aerosols, in cloud water, and/or in fog water, all of which may have occurred upwind of the site during the 8 h sampling periods under variable local and regional weather conditions.
In the winter, we observed a generally higher average saturation mass concentration (summer: vs. winter: ). We note that this comparison of saturation mass concentrations was performed at a reference temperature of 300 K, and we discuss the expected wintertime volatility shift below. The wintertime was also slightly lower than summer (summer: vs. winter: ). In the winter, the observed chemical composition of the particle phase – in terms of both volatility and functional group distribution – suggests a relatively greater role for aqueous-phase processing. Our observations were similar to those made in past studies of higher-volatility products from fragmentation reactions in the aqueous phase, e.g., Brege et al. (2018), where they observed that aged fog water samples contained organic compounds with smaller carbon backbone structures than non-aqueous aged particles and linked this difference to aqueous-phase fragmentation reactions, the uptake of smaller water-soluble gases to the aqueous phase, and/or less oligomerization (Brege et al., 2018). Also, Yu et al. (2016), discussed the role of fragmentation in aging aqueous phenolic secondary organic aerosol (Yu et al., 2016), and Schurman et al. (2018) discussed the role of fragmentation and evaporation in cloud water (Schurman et al., 2018). Similarly, here we observed a shifted compound distribution that included smaller molecular weight and generally higher-volatility particle-phase species in winter compared to summer along with notably different functional group distributions, both of which could be attributed to aqueous chemistry.
We note that, for direct comparison, volatility bins in Fig. 2a and b were defined for the same reference temperature (i.e., 300 K, the average summertime sampling period temperature), though wintertime saturation mass concentrations for the observed compounds would shift approximately 2 orders of magnitude lower due to lower temperatures (i.e., 270 K). The dotted black line in Fig. 2b shows the shift in bins expected at 270 K. In the winter, compounds defining IVOCs or SVOCs at 300 K will expectedly exhibit a greater degree of partitioning to the particle phase, though the effect of this temperature shift on partitioning was likely more pronounced for the SVOCs than the IVOCs (Table S4 in the Supplement). Even when accounting for this shift, the mean saturation mass concentration of wintertime samples was , which is still higher than the mean summertime saturation mass concentration of and thus still demonstrates a volatility difference between summer and winter, with higher-volatility species in winter. This shift is also reflected in the carbon number distribution observed via the LC-ESI-MS/MS analysis of this sample set shown in Ditto et al. (2020), Fig. S5. In addition to this shift in molecular size and volatility, there was a distinct change in functional group composition from summer to winter discussed below.
To assess the potential contribution of aqueous-phase chemistry, we also estimated aerosol liquid water concentrations based on available data in Sect. S2.1 in the Supplement. We estimated a lower but still appreciable aerosol liquid water content in winter relative to summer, but with fewer photochemical processes in winter along with generally cloudier/foggier local weather (i.e., 44 % of summer sampling periods with partly cloudy or cloudy weather conditions vs. 67 % of winter sampling periods, from the Weather Underground archive), aqueous-phase processing likely remains an important pathway. We note that the compounds discussed here could have been formed locally or regionally, and thus the role of conditions at the site (aerosol liquid water, cloud cover, fog cover) is just as important as the conditions in the surrounding upwind region. As a result, it is challenging to pinpoint the exact contributions of aerosol liquid water, in-cloud, or in-fog processing, and we consider that all three may be occurring upwind or near the site.
Furthermore, from MS/MS analysis, we observed functional groups that were possible indicators of aqueous-phase processing, including the presence of nitrophenols during the winter, which may have formed via dark aqueous-phase reactions with HONO (Vidović et al., 2018), and relatively low contributions from carbonyls across seasons, possibly linked to carbonyl hydrolysis (Ditto et al., 2020). Based on laboratory studies, the presence of azole functional groups and other heterocyclic nitrogen species could also indicate aqueous-phase processing and may be formed from small carbonyl precursors such as glyoxal (DeHaan et al., 2009; Grace et al., 2019) and biacetyl (Grace et al., 2020) reacting with atmospheric ammonia or small amines. Many of the N-only-containing azoles observed here had similar substructures to those formed in the aqueous-phase reactions of small carbonyls with ammonia/amines (DeHaan et al., 2009; Grace et al., 2019). In addition, as discussed above, we observed many small gas-phase C3–C6 compounds at the site in the summer, which likely included multifunctional isoprene oxidation products (e.g., glycolaldehyde, hydroxyacetone, and isomers); these potential precursors could have reacted with atmospheric ammonia or species containing amino groups to form the observed azole-containing reaction products. We observed more azoles during the summer (Ditto et al., 2020), perhaps due to the increased prevalence of the C3–C6 precursors and overall prominence of atmospheric water (e.g., aerosol liquid water, cloud water, or fog water).
Lastly, the role of aqueous-phase chemistry in the region is further supported by prior summertime observations at Brookhaven National Laboratory (on the opposite side of the Long Island Sound), which examined a low-volatility oxygenated organic aerosol factor in the source apportionment of aerosol mass spectrometry measurements and showed a strong contribution from carboxylic acids and other ELVOCs that were attributed to aqueous-phase processing (Zhou et al., 2016).
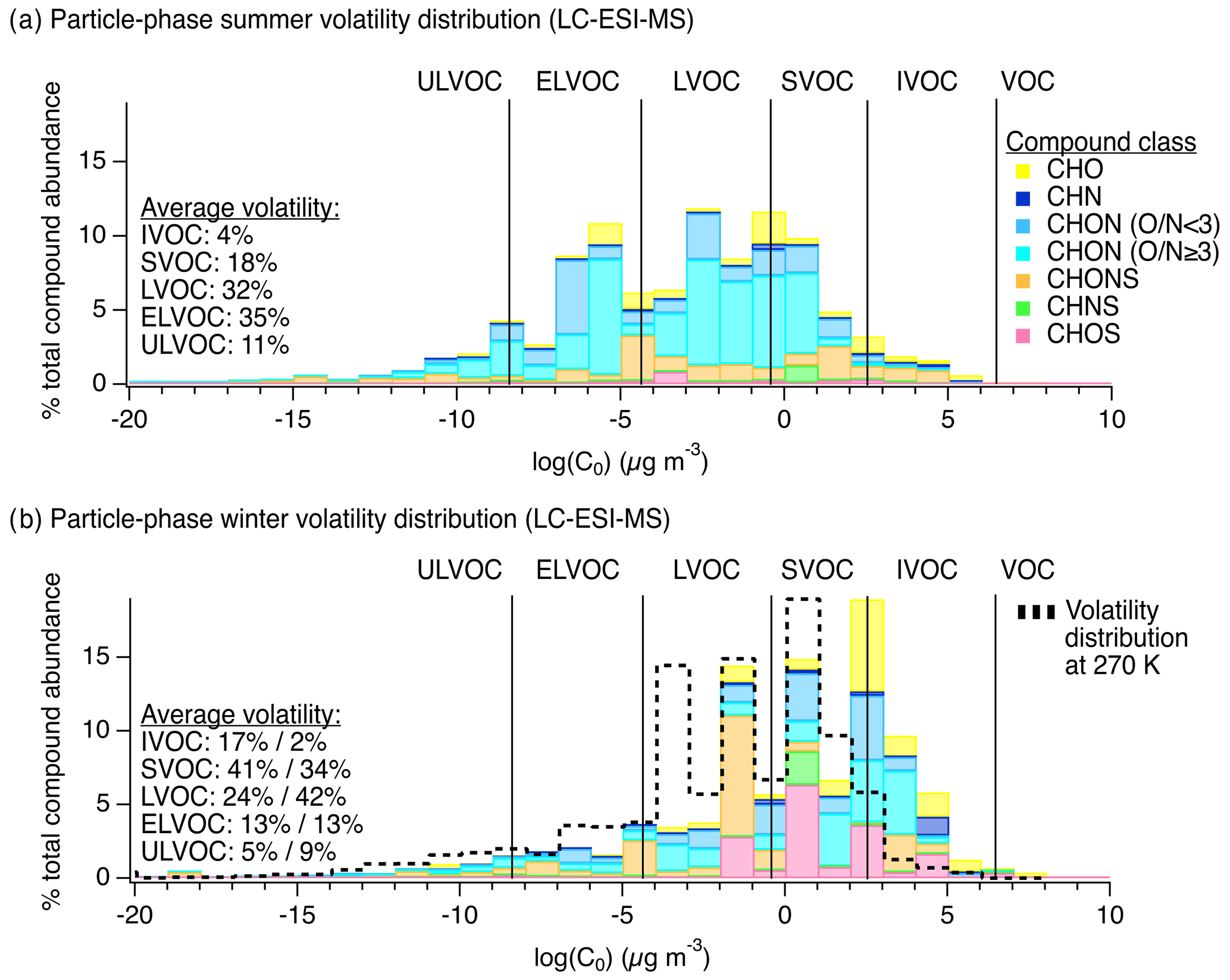
Figure 2Chemical composition of particle-phase organic compound mixtures at the YCFS from LC-ESI-MS measurements. Panels (a) and (b) show particle-phase volatility distributions by compound class in the summer (N=34) and winter (N=15), respectively, weighted by ion abundance. The same data tallied by occurrence are shown in Fig. S4 for comparison. For direct comparison, volatility bins were defined for the same reference temperature in (a) and (b) (i.e., 300 K, the average summertime sampling period temperature), though wintertime saturation mass concentrations for the observed compounds would shift approximately 2 orders of magnitude lower due to lower temperatures (i.e., 270 K). The dotted black line in (b) shows the shift in bins expected at 270 K, described further in Table S4. The average volatility distributions listed in (b) are shown at 300 K (%) followed by the estimate at 270 K (%).
3.2.3 Comparison to other sites using the same sampling and analytical methods
The distribution of compound classes observed at the YCFS was significantly different from observations at a range of field sites discussed in past studies (Fig. 3), including a remote forested site (i.e., the PROPHET site in northern Michigan), an urban inland site (i.e., near downtown Atlanta) across two seasons, and New York City (Ditto et al., 2018, 2019). We perform a direct comparison to our past studies here, because the same sampling and analytical methods were used, and thus we can compare the distribution of ions observed without any biases due to differences and uncertainties resulting from variations in sampling or ionization chemistry between instruments. While a more detailed site-to-site comparison is outside the scope of this work, the proximity of the YCFS to the ocean and thus the impact of marine emissions and over-water chemistry likely contributed to the differences between the YCFS and inland locations. In particular, at the YCFS, we observed notably smaller relative contributions from compounds containing carbon, hydrogen, and oxygen (i.e., CHO, 11 %–16 % of observed functionalized compounds here vs. 34 %–50 % at other sites), and the contributions from nitrogen-containing particle-phase compounds at the YCFS were in stark contrast to other sites. Here, 85 % of compounds (by ion abundance) in summer and 68 % of compounds in winter contained at least one nitrogen atom compared to 38 %–51 % at the other previously studied sites (Fig. 3). These nitrogen-containing species were comprised of compounds with various reduced and oxidized nitrogen-containing functional groups with varying oxygen-to-nitrogen ratios (), which are broadly classified and discussed below as compounds containing carbon, hydrogen, and nitrogen (i.e., CHN) and compounds containing carbon, hydrogen, oxygen, and nitrogen (i.e., CHON with ratio <3, and CHON, ratio ≥3). There were notably greater contributions at the site from nitrogen-containing compounds that also contained at least one oxygen atom, including CHON compounds with (19 %–20 % here vs. 10 %–15 % at other sites), CHON compounds with (24 %–44 % here vs. 14 %–19 % at other sites), as well as compounds containing oxygen, nitrogen, and sulfur (i.e., CHONS, 20 %–21 % here vs. 9 %–10 % at other sites) (Ditto et al., 2018).
We note that while these measurements were of PM10 aerosols, the observations of high nitrogen content were not biased by the inclusion of larger, primary (possibly biological) particles. Quartz filter samples collected with a cascade impactor at the site during the summer and analyzed with the same LC-ESI-MS methods did not show any significant differences between any of these nitrogen-containing compound classes as a function of particle size, across particles ranging from 0.4 to 10 µm (i.e., 69 %–71 % of ion abundance for PM≤2.2 µm and 69 %–73 % of ion abundance for PM ranging from 2.2 to 10 µm were nitrogen-containing species, summarized in Table S5 in the Supplement). This is consistent with past studies which have demonstrated that amines, as one example of a prominent nitrogen-containing functional group, are ubiquitous in size-resolved aerosol samples in urban and rural locations (VandenBoer et al., 2011).
The prevalence of nitrogen-containing species at the YCFS is consistent with the study at Brookhaven National Laboratory discussed above, where a dedicated nitrogen-enriched aerosol mass spectrometry factor was identified and contained prevalent signals from aliphatic amines and amides. However, in the Brookhaven study, the nitrogen-enriched factor was associated with industrial amine emissions that were enhanced during periods of southern/southwestern backward trajectory influence and that had correlations with tracers linked to industrial processes. In our study, there was no correlation between backward trajectory direction and the contribution of nitrogen-containing species. Also, wintertime air parcels arrived predominantly from directions other than south/southwest, suggesting that the nitrogen-containing species observed in our study were the result of mixed anthropogenic, biogenic, and marine precursors and their transformation products. This high nitrogen content at the YCFS, where aqueous-phase chemistry is expected to be important, is also consistent with the cloud water composition discussed in Zhao et al. (2013), which reported roughly 65 % of detected ions in their cloud water samples as containing a nitrogen atom, and the primary marine aerosol composition discussed in Wozniak et al. (2014), where 61 % of their observed compounds contained nitrogen and 54 % were CHON species.
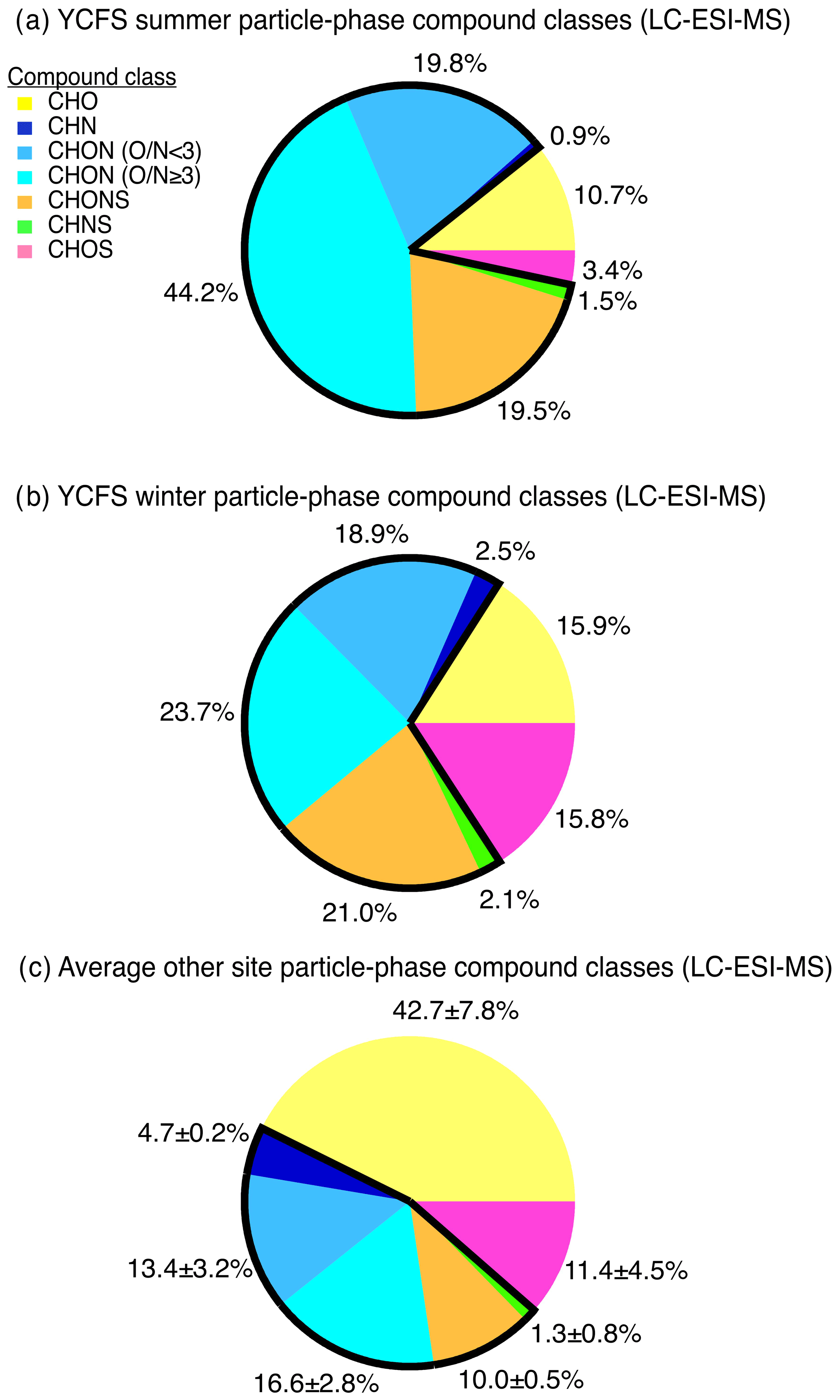
Figure 3Particle-phase compound class distributions shown as fractions of total detected ion signal in the (a) summer and (b) winter at the YCFS, weighted by compound abundance, in contrast with (c) the average compound class distribution from previously studied forested, urban inland, and urban coastal sites. The sites selected for comparison in (c) were chosen because the same sampling and analysis methods were used. Nitrogen-containing compound class contributions are outlined in black and are notably larger at the coastal site compared to other sites studied with these same filter collection and analysis methods. We note that, while a significant fraction of species contained nitrogen, individual compounds contained one to three nitrogen atoms, and the majority of the ion's molecular mass consisted of carbon and hydrogen atoms (mean in summer: 0.13±0.1, mean in winter: 0.22±0.19 for all N-containing ions). Note: CH and CHS species have poor ESI ionization efficiencies and are thus excluded here. Data tallied by occurrence are shown in Fig. S5 for comparison.
3.3 Speciating particle-phase multifunctional nitrogen-containing compounds
The observed particle-phase species were highly functionalized, were often multifunctional, and contained combinations of oxygen, nitrogen, and/or sulfur heteroatoms. Here, we discuss the functional groups present, broken up by the nitrogen-containing compound classes shown in Figs. 2 and 3, with additional discussion of other relevant compound classes in Sect. S3 in the Supplement.
3.3.1 CHN compounds
While nitrogen-containing compounds in general were very prominent at the site (Fig. 3a and b), CHN compounds were relatively less abundant in these samples of functionalized organic aerosol. Particle-phase CHN compounds represented just 1 % and 3 % of observed functionalized organic aerosol abundance in summer and winter, respectively, which was similar to observations at other ambient sites (∼5 % CHN) (Ditto et al., 2018).
In the summertime LC-ESI-MS/MS measurements, CHN particle-phase compounds were comprised primarily of amines (72 % of CHN species contained an amine group) and nitriles (28 % of CHN species contained a nitrile group), as shown in Fig. 4. In the winter, these compounds were nearly exclusively amines (present in 99 % of CHN species). Amines have many primary land-based sources (e.g., biogenic emissions, Kieloaho et al., 2013; agricultural activity, Ge et al., 2011; emissions from decomposing organic matter, Ge et al., 2011, Sintermann and Neftel, 2015; biomass burning, Ge et al., 2011; emissions from port activity, Gaston et al., 2013; chemical products, Khare and Gentner, 2018; vehicle exhaust, Sodeman et al., 2005), but their presence on the coast could also indicate marine contributions. Amines have been detected in bulk ocean water, the surface microlayer, and sea spray aerosol, and their emissions and chemical transformations in the marine environment have been the topic of many recent studies (e.g., Brean et al., 2021; Dall'Osto et al., 2019; Decesari et al., 2020; Di Lorenzo et al., 2018; van Pinxteren et al., 2012, 2019; Quinn et al., 2015; Wu et al., 2020). In the summer, biogenic and marine sources likely dominated the amine distribution, while in the winter, anthropogenic amine sources likely became more important.
Recent studies have also evaluated amine phase partitioning or formation in cloud/fog water (e.g., Chen et al., 2018; Youn et al., 2015) as well as condensed-phase or aqueous-phase pathways that may transform emitted amines (e.g., Ge et al., 2016; Lim et al., 2019; Tao et al., 2021). Interestingly, the observed amines at this site, as well as other reduced nitrogen groups like nitriles, imines, and enamines, were not present exclusively in CHN species and thus were a mix of both direct emissions and chemically processed compounds. Reduced nitrogen groups were often paired with hydroxyl groups, carboxylic acids, carbonyls, ethers, and esters as part of nitrogen- and oxygen-containing compounds with a range of ratios. This is consistent with other studies observing reduced nitrogen contributions to CHON compound classes, such as Zhao et al. (2013), LeClair et al. (2012), and Altieri et al. (2009) discussed above. As such, we discuss CHON species as a function of ratio to focus on differences between less-oxygenated () and more-oxygenated (, e.g., organonitrates) species, using a ratio of 3 to distinguish between the two as informed by the ratio of the organonitrate functional group.
3.3.2 CHON () compounds
CHON () compounds were notably more important at this site than other sites, representing 20 % and 19 % of observed functionalized organic aerosol abundance in summer and winter, respectively (Fig. 3a and b), compared to ∼13 % at other sites (from predominantly summer measurements). These CHON compounds included some functional groups that contained both oxygen and nitrogen, such as amide groups (12 % of this compound class's nitrogen content in summer vs. 1 % in winter, Fig. 4) and nitro groups (15 % of this nitrogen content in summer vs. 6 % in winter, Fig. 4). However, most CHON () compounds were comprised of a combination of nitrogen- or oxygen-containing groups rather than a functional group containing both nitrogen and oxygen. This included large contributions from hydroxyls and ethers across both seasons as well as important contributions from amines, isocyanates, and heterocyclic nitrogen, as shown in Ditto et al. (2020) (Fig. 1). The presence of these functional groups in the winter could be indicative of wood burning emissions in the region, which has been observed in the wintertime in past ambient sampling in the northeastern US (Sullivan et al., 2019). Isocyanates contributed notably to this compound class during the winter, which could similarly be linked to burning wood, other biomass, or building materials (Leslie et al., 2019; Priestley et al., 2018; Roberts et al., 2014) or could be photochemically produced via the oxidation of amines and amides (Borduas et al., 2015; Leslie et al., 2019). Importantly, levoglucosan, a common biomass burning tracer, was observed across nearly all daytime and nighttime winter particle-phase samples (verified with an authentic standard), supporting the influence of biomass burning compounds at the site. Together, the overall high prevalence of reduced nitrogen at this site could be influenced by the mixing of aged biomass burning plumes with marine air, which is consistent with past observations of very high alkylamine concentrations in biomass burning particles that mixed with marine air prior to sampling (Di Lorenzo et al., 2018).
3.3.3 CHON () compounds
CHON () compounds were the dominant compound class in the observed summertime distribution and played an important role in the wintertime distribution as well, comprising 44 % of observed functionalized organic aerosol abundance in summer vs. 24 % in winter (Fig. 3a and b). These contributions were far greater than the contributions of CHON () species at other sites, which typically ranged from 14 % to 19 % (predominantly from summertime measurement, Fig. 3c).
Similarly to CHON (), we observed some CHON () compounds with functional groups containing three oxygen atoms and one nitrogen atom, e.g., nitrophenols and organonitrates (Fig. 4), but also contributions from nitrogen-only functional groups paired with oxygen-containing groups. Notably, in the summer, there were important contributions from amines (47 % of this compound class's nitrogen content), imines (19 %), organonitrates (10 %), and azoles (16 %) (Fig. 4). In contrast, in the winter, nitrogen content in the CHON () compound class was dominated by IVOC/SVOC nitrophenols, comprising 64 % of the CHON () ion abundance.
NOx mixing ratios were typically low in both summer and winter (2.3±1.5 ppb in summer vs. 3.7±2.7 ppb in winter) but were slightly higher during winter. In the winter, CHON () compounds showed a weak positive relationship with NOx mixing ratios (r∼0.58) and a stronger correlation with NO mixing ratios (r∼0.81). This relationship between CHON () and NO (and NOx) suggests that many of these oxidized nitrogen species were products of NOx-related chemistry (i.e., NOz compounds). The enhancement in nitrophenols serves as one example of this, as NO mixing ratios also correlated with the contribution of nitrophenols in the winter (r∼0.69).
In past work, we discussed nitrophenol nighttime enhancements during winter and noted their reported aqueous formation pathways mentioned in prior laboratory studies (Ditto et al., 2020). Here, we demonstrate that nitrophenols were important contributors to the CHON () compound class and highlight their role as examples of NOz due to their possible formation via dark aqueous-phase nitration pathways of oxygenated aromatics with HONO (Vidović et al., 2018). While nitrophenols may have other sources (e.g., diesel exhaust), our observations of a clear nighttime enhancement during the winter suggest that these functional groups were most likely formed by secondary chemistry related to NOx oxidation, as this field site was removed from major roadways. Our wintertime observations suggest that HONO could have been derived from local wood burning and could have reacted away as the smoke plume aged to form stable products like nitrophenols, similarly to HONO transformation chemistry into other forms of oxidized nitrogen (e.g., particulate nitrates, PANs, organic nitrates) that has recently been observed in wildfire smoke (Juncosa Calahorrano et al., 2021).
Furthermore, the correlation between NO and CHON () could also be influenced by the daytime formation of organonitrates via reaction with OH• and NO (i.e., ) (Liebmann et al., 2019; Ng et al., 2017; Perring et al., 2013; Takeuchi and Ng, 2018), though organonitrates contributed a smaller fraction of CHON () species (i.e., 10 % of this compound class's nitrogen content across seasons).
3.3.4 Overall contributions of reduced and oxidized nitrogen groups
In the summer and winter, contributions from reduced nitrogen groups (e.g., groups shown in black/grey in Fig. 4) rivaled those of oxidized nitrogen groups in CHON compounds across a range of ratios. In the summer, reduced nitrogen groups contributed 50 % of all detected CHON () compounds by ion abundance, while in the winter they contributed 47 % (Fig. 4). For CHON compounds with , reduced nitrogen groups contributed 68 % of compound ion abundance in the summer (possibly related to marine influences, Wozniak et al., 2014), while in the winter they contributed just 13 %. Interestingly, 90 % of the dominant reduced nitrogen functional groups observed (amines and imines) were present in acyclic rather than cyclic structures, which may have been the result of either direct emissions or formation via reactions with ammonia or other small amines.
In contrast, possible NOz products (e.g., groups shown in blue in Fig. 4) were present in 18 % and 7 % of CHON () compounds in the summer and winter, respectively. For CHON () compounds, they were present in 18 % and 86 % in the summer and winter, respectively, with the latter wintertime increase in oxidized nitrogen groups largely driven by the presence of nitrophenols at night (Ditto et al., 2020). The remaining fraction of nitrogen-containing groups also contained oxygen but with a reduced nitrogen atom (e.g., amide, isocyanate, nitrogen-/oxygen-containing azole; shown in brown in Fig. 4). We note that CHONS compounds also represented a sizable fraction of observed organic nitrogen (Fig. 3) and contained a mix of reduced and oxidized functional groups (Sect. S3 and Figs. S7–S8 in the Supplement).
The importance of reduced nitrogen functional groups in CHON compounds highlights that not all oxygen- and nitrogen-containing species in the CHON () compound class were NOz, despite their apparent molecular formulas and the observed correlation observed between CHON () species with NO and NOx mixing ratios. For instance, many of the observed reduced nitrogen-containing functional groups co-occurred with several oxygen-containing groups like hydroxyls, carboxylic acids, esters, ethers, and carbonyls and thus had molecular formulas with , which could incorrectly be assumed to be an organonitrate or similar structure based on molecular formula alone.
We note that the relative distribution of reduced and oxidized nitrogen-containing groups shown here is subject to sampling and ionization conditions. While the electrospray ionization source used for the particle-phase analysis discussed here effectively ionized these nitrogen-containing groups, their relative sensitivity may differ because many of these functional groups were present in multifunctional compounds whose other features may also contribute to ionization behavior. Also, other aspects of the sample collection and extraction process could cause variability in the observed signal (e.g., PM size cut, organonitrate stability over long-duration samples). Thus, we emphasize that the observed relative abundances here are valuable because they suggest that fully reduced nitrogen-containing groups are important contributors to multifunctional CHON species, but their exact mass contributions remain uncertain.
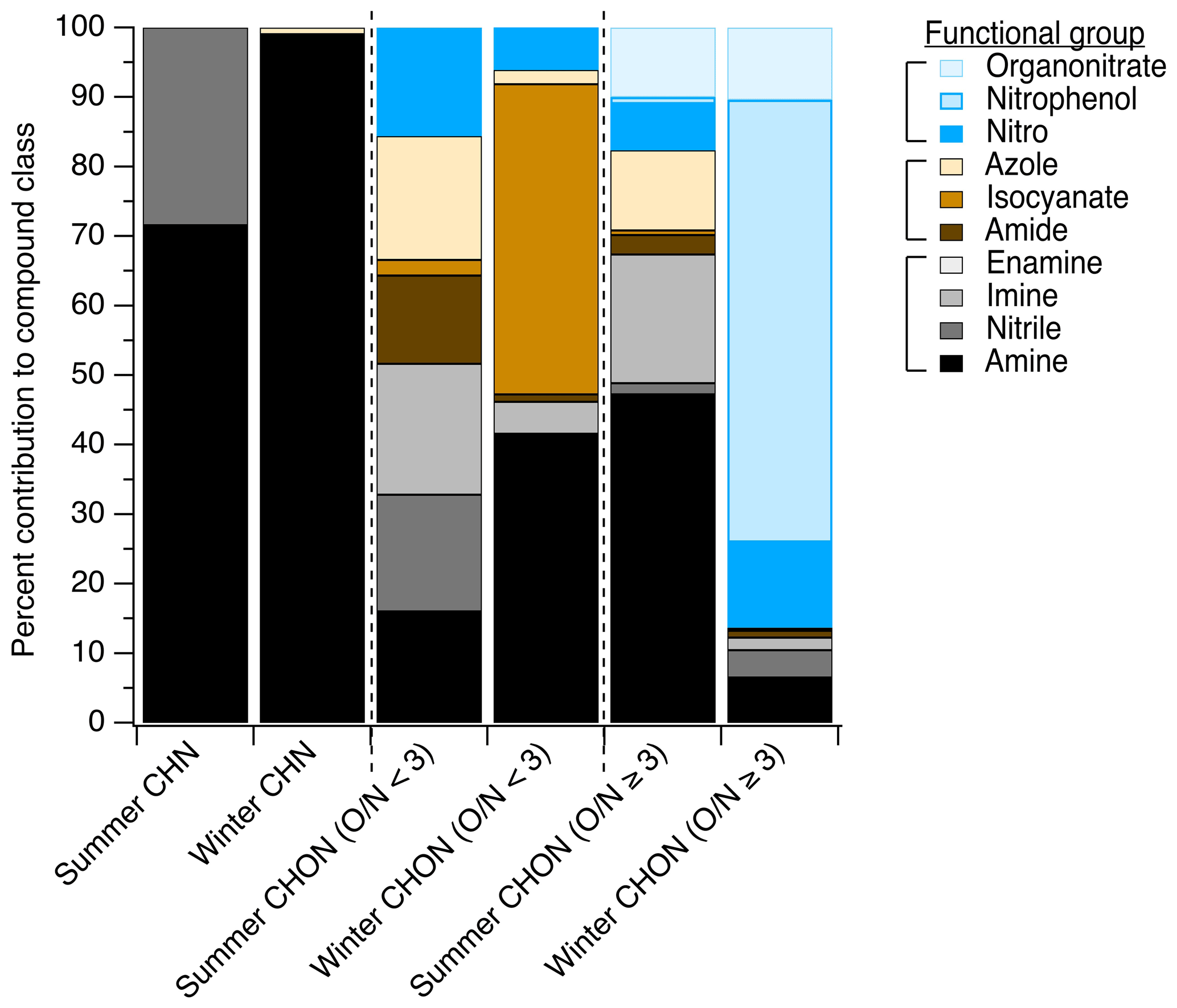
Figure 4The distribution of functional groups in particle-phase nitrogen-containing compounds measured via LC-ESI-MS/MS. The breakdown of CHN, CHON (), and CHON () compounds is shown as a function of contributions of each functional group to ion abundance, with possible NOz species shown in blue shades, fully reduced nitrogen-containing groups shown in black/grey shades, and groups containing both oxygen and nitrogen where the nitrogen atom itself is not oxidized shown in brown shades. The same data tallied by occurrence are shown in Fig. S6 for comparison. Figures S7 and S8 show the functional group distribution for CHNS and CHONS compound classes tallied by abundance and by occurrence, respectively.
3.4 Probing possible nitrogen-containing gas-phase precursors to observed nitrogen-containing particles with adsorptive sampling and LC-ESI-MS
The particle-phase volatility distribution in the winter ranged from IVOCs to ULVOCs. Of the observed compounds in winter, 68 % contained nitrogen; these likely included contributions from functionalized gas-phase precursors and likely were influenced by the active multiphase partitioning of these precursors and their gas- or particle-phase reaction products, with changes in organic aerosol loading, atmospheric liquid water concentrations, and temperature (Donahue et al., 2011; Ervens et al., 2011). This emphasizes the need to measure a broader range of these functionalized gas-phase compounds, which have known limitations with GC transmission but represent uncertain and important-to-measure SOA precursors.
However, despite evidence of higher-volatility particle-phase compounds with diverse nitrogen-containing functionalities that could dynamically partition between phases (Fig. 2a and b), the observed compound class distribution from gas-phase adsorbent tube measurements analyzed via GC-APCI-MS was dominated by hydrocarbons (i.e., CH, 24 % of detected ion abundance in summer vs. 18 % in winter) and oxygenates (i.e., CHO, 66 % in summer vs. 69 % in winter) (Figs. S10–S11 in the Supplement). These gas-phase species appeared to be lightly functionalized oxygenates (average : 0.12±0.13), showing minimal contributions from nitrogen (or sulfur) heteroatoms; only 9 % of detected ion abundance from gas-phase adsorbent tubes in summer and 11 % in winter contained a nitrogen heteroatom. This is likely due to measurement limitations: while GC-APCI techniques are extremely well suited for the analysis of less functionalized organic compounds from both instrument transmission and ionization efficiency perspectives, these techniques are not as effective for more polar, more functionalized, more thermally labile, or otherwise less-GC-amenable species. Thus, to examine a broader range of functionalized gas-phase compounds, we used an offline adsorptive sampling method on cooled PEEK tubing collectors and inline mobile phase desorption for LC-ESI-MS analysis (Fig. 1). CH and CHS compound classes were excluded from this gas-phase LC-ESI-MS analysis due to their poor ESI ionization efficiency.
Due to variations in trapping and desorption effectiveness (Sect. S1), this method was not intended to be used as a quantitative measurement of concentration but rather as a relative assessment of the distribution of nitrogen-containing gas-phase organic compounds. The variation between analytes in breakthrough testing does not influence our conclusions about the overall prevalence of observed gas-phase organic nitrogen. In laboratory tests, gas-phase sample collection, inline desorption to the mobile phase, trapping on the LC column, and chromatographic separation performed well. We observed limited breakthrough for most analytes during sampling, effective focusing prior to LC analysis, and similar separations for spiked collectors and breakthrough tests compared to standard LC runs (Fig. 1b).
Results from the application of this new method at the YCFS revealed a wide range of compounds with oxygen-, nitrogen-, and/or sulfur-containing functionality (Fig. 5) that existed at a lower average saturation mass concentration than the adsorbent tube methods during winter, with a log (C0) of for adsorbent tubes analyzed with GC-APCI-MS compared to for functionalized gases observed via LC-ESI-MS. This decrease in volatility corresponded to an increase in the average ratio of these functionalized gases to 0.24±0.24, which can partly be attributed to LC-ESI's poor ionization of CH compounds and to the collection system's design (targeting heteroatom-containing species and not higher-volatility hydrocarbons). This may be a lower limit of among functionalized compounds, as during testing with a mixture of standards, we often observed poor retention of high- sugars like xylitol and mannose on the LC analytical column (Table S2).
The gas-phase LC-ESI-MS data provide a valuable comparison to the wintertime particle-phase samples analyzed using the same instrument. These particle-phase samples had major contributions from CHO, CHON ( and ), CHONS, and CHOS compound classes (Fig. 3b). While not collected concurrently, the functionalized gas-phase samples in winter had similar contributions from CHO (20 %) and CHON () compounds (16 %), relatively more CHN (11 %) and CHON () (46 %) compounds, and fewer CHONS (2.7 %) and CHOS (4.4 %) compounds (Fig. 5a). The prevalence of gas-phase CHN, CHON (), and CHON () is of particular interest given the abundance of CHON compounds observed in the particle phase and the potential of these gases to partition to the particle phase and/or act as reactive precursors to other oxidized nitrogen-containing species.
The presence of these nitrogen-containing compounds in the gas phase also suggests that these compound classes observed in the particle phase at least partly originated in the gas phase and partitioned rather than formed exclusively as a result of particle-phase chemistry. These species could have also formed in the particle phase and partitioned to the gas phase with or without condensed-phase fragmentation (discussed above). In either scenario, these nitrogen-containing compounds likely actively partitioned between phases due to their volatility (e.g., IVOCs/SVOCs shown in Fig. 5a). Also, their polarity and high Henry's law coefficients (relative to non-functionalized hydrocarbons, Sander, 2015) suggest that these compounds could have been readily taken up by the aqueous phase. To check that these compounds were indeed gas-phase species under ambient conditions, we predicted the saturation mass concentration for individual compounds using individual ion formulas and estimated their gas–particle partitioning to a pre-existing condensed phase. While the range of compounds in Fig. 5a can be expected to dynamically partition, the results confirm that the overall suite of observed compounds would have predominantly existed as gases, with on average ∼80 % of observed ion abundance predicted to equilibrate to the gas phase across compound classes (Figs. S12–S13 in the Supplement).
Of all the gas-phase species observed with at least one nitrogen atom (i.e., CHN, CHON, CHONS, and CHNS) collected in winter, we note that 78 % of these compounds had an ratio of less than 3 (Fig. 5b), indicating that most of these gas-phase species were not organonitrates, nitrophenols, or other similar structures. This is similar to our particle-phase wintertime results, which showed important contributions from reduced nitrogen-containing groups paired with oxygen-containing groups in CHON () compounds. Notably, we observed an 11 % contribution of gas-phase CHN species with this gas-phase LC-ESI-MS method (Fig. 5a), in contrast to 2 % CHN in the wintertime particle-phase samples (Fig. 3). In the winter particle-phase samples, most CHN compounds contained amines (discussed above), and thus we postulate that these functionalized gas-phase CHN species were possibly also amines that acted as precursors to observed nitrogen-containing particle-phase compounds following oxidation and partitioning (or vice versa).
The substantial contribution from CHON with (46 %) to the functionalized gas-phase samples could be linked to less photochemical processing of CHON compounds relative to the particle phase and/or the emissions/oxidation of CHN or CHON compounds. Moreover, in the particle phase, we observed a weak negative relationship between CHN contribution and hydroxyl group prevalence in summertime measurements (), which may support the transformation of CHN to CHON compounds via the formation of hydroxyl-containing species. The elemental ratio distribution of these functionalized gases is summarized in Fig. S14 and Table S6 in the Supplement.
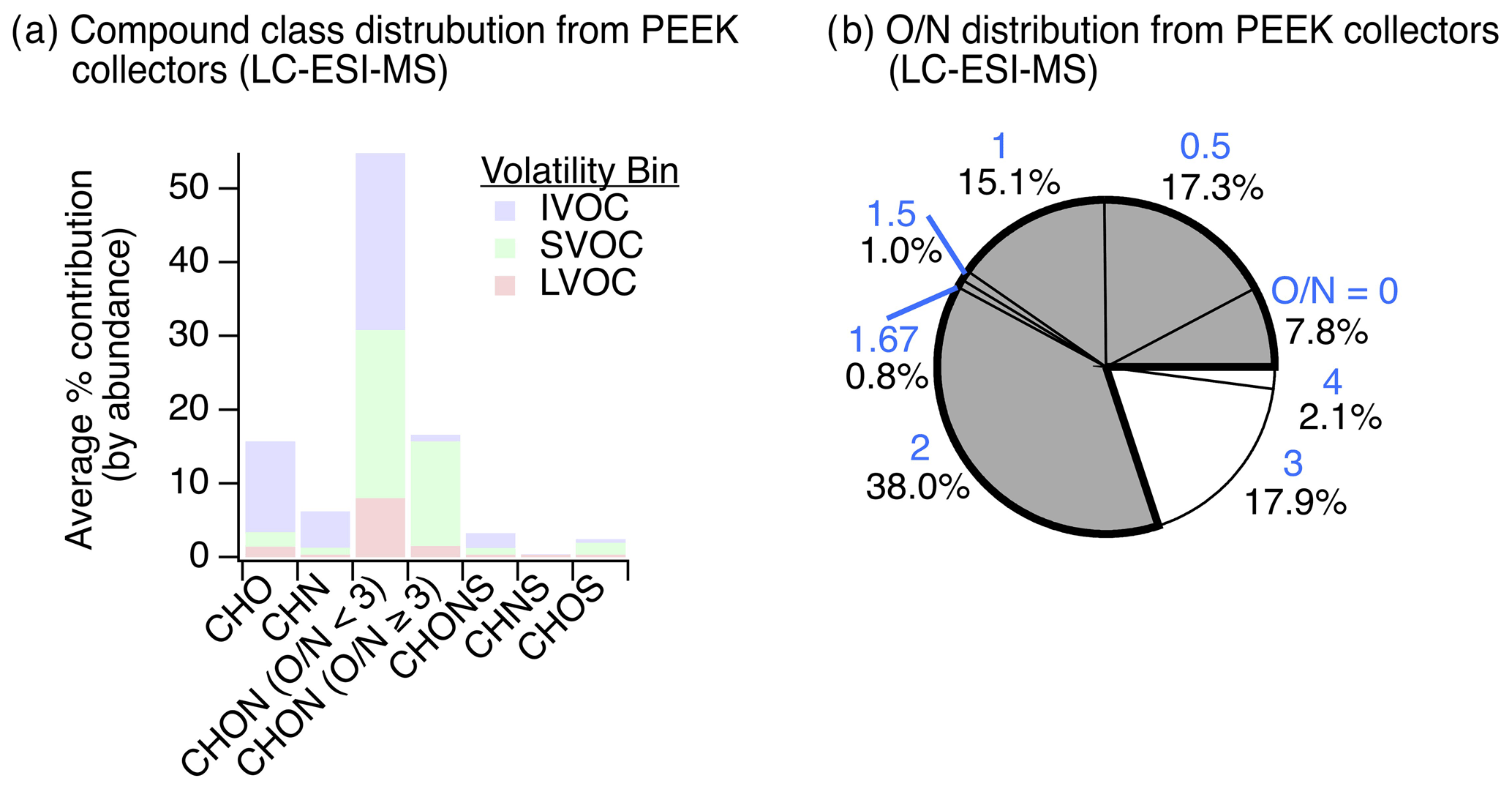
Figure 5Observations of gas-phase nitrogen-containing compounds. (a) The distribution of functionalized gases observed via sampling on PEEK collectors (N=6) and inline mobile phase desorption with non-targeted LC-ESI-MS analysis contained a diversity of oxygen-, nitrogen-, and/or sulfur-containing compounds in the IVOC–LVOC range (volatility assignment and grouping were the same as discussed in Fig. 2 at a reference temperature of 300 K for intercomparison). While we cannot rule out gas-phase LVOC contributions from evaporation off of the upstream particle filter, LVOC contributions were limited (∼12 %). (b) Oxygen-to-nitrogen () ratio distribution of observed gas-phase nitrogen-containing species where ratios <3 are colored grey and ratios ≥3 are colored white (blue text above each percentage signifies the ratio). The same data, tallied by occurrence, are shown in Fig. S9 for comparison.
Together, these results suggest that a mix of direct emissions and chemical processes during summer and winter in the Long Island Sound region resulted in a diverse mixture of multifunctional gases and particles, where more than two-thirds of observed particle-phase compounds contained at least one nitrogen atom.
The observed nitrogen-containing functional groups existed across a range of fully reduced (e.g., amines, imines) to oxidized (e.g., nitro, organonitrate) structures. These fully reduced nitrogen functional groups were prevalent across all nitrogen-containing compound classes, including CHON species, and we highlight their importance as contributors to these multifunctional compounds beyond typical NOz-type compounds that are commonly studied using online mass spectrometers and that share similar CHON molecular formulas. For instance, these gas- and particle-phase measurements of nitrogen-containing compounds are complementary to the measurements of these species made by chemical ionization mass spectrometers (CIMS) or by proton transfer reaction mass spectrometers (PTR-MS), whose ionization mechanisms can be tuned for sensitivity towards functionalized compounds of interest (Riva et al., 2019). While online mass spectrometers excel at high-time-resolution measurements that capture dynamic chemical processes in the atmosphere, their mass resolution is typically lower, and they normally do not utilize separations, so they largely depend on parent ion mass-to-charge ratios to assign molecular formulas without structural attribution. The offline methods used here cannot match the time resolution of online techniques. However, the use of chromatography to separate isomers, longer sampling times to increase sensitivity towards a greater range of compounds, and the use of higher-resolution mass spectrometers with MS/MS capabilities allow for improved compound identification and determination of functional group distribution at the molecular level. This enables us to distinguish between true NOz species and those that contain combinations of nitrogen and oxygen but that are not NOx oxidation products. Thus, both these online and offline methods should be employed together to differentiate a wider range of nitrogen-containing species and to achieve both temporal and chemical resolution.
As discussed throughout this work, the Long Island Sound region is affected by a mixture of anthropogenic, biogenic, and marine sources, all of which contain known emitters of organic nitrogen. Understanding the combined effect of these individual sources and their chemical transformations will be important in regions like the Long Island Sound, where a significant degree of mixing occurs over the sound before air parcels arrive inland. For example, past work has noted extremely high contributions from alkylamines in biomass-burning-influenced air mixed with marine air (Di Lorenzo et al., 2018). Similar enhancements could be expected when mixing other prominent sources of amines with marine air, such as in the aging urban outflow from the central Atlantic and northeastern US, which may be transported up the coast and impact states in the surrounding region.
As with any ambient site, these mixed emissions are chemically processed in the atmosphere via a multitude of pathways. Here, we observed evidence of photochemical and aqueous processes occurring in both seasons, but in the winter we observed various mixture-wide trends that suggested an enhanced role for aqueous-phase processing. These observations included higher overall particle-phase volatility and smaller carbon backbone sizes, which may indicate a more important role for aqueous-phase fragmentation reactions or aqueous uptake of water-soluble gases (Brege et al., 2018). We also observed key marker functional groups that may be formed via aqueous-phase chemistry (e.g., nitrophenols, azoles). The role of aqueous-phase chemistry and aqueous-phase uptake of gases is increasingly studied in laboratory and ambient contexts (Herrmann et al., 2015), and such chemistry should be further examined, especially in coastal and other humid regions.
For example, the aqueous-phase processing of atmospherically relevant nitrogen-containing species is particularly important to understand in ambient air due to the potential for brown carbon formation, which has significant impacts on climate forcing (Laskin et al., 2015). The role of ammonia and amines reacting with carbonyls is of interest for this type of chemistry (e.g., DeHaan et al., 2009; Grace et al., 2020; McNeill, 2015; Sareen et al., 2010) and should continue to be explored, particularly in coastal settings where concentrations of small gas-phase amines may be high due to their marine sources. As discussed above, our ambient observations of azoles could be indicative of such chemistry and should be explored in future comparisons of ambient and laboratory-generated species. Also, we observed a significant contribution from nitrophenols at our site, and while they are not formed by this same chemistry, they represent another important form of light-absorbing nitrogen-containing organic mass in the atmosphere (Hems and Abbatt, 2018). Finally, many of the nitrogen-containing functional groups observed in this work may be susceptible to hydrolysis, so the balance between hydrolysis and other aqueous pathways is important to consider and understand for appropriate representation of nitrogen-containing compounds in models for both aqueous aerosol and in-cloud/fog chemistry.
As another example, the greater prevalence overall of higher-volatility species observed in the winter particle-phase samples suggested possible dynamic partitioning or aqueous uptake of lighter gas-phase compounds; to explore the composition of these lighter gas-phase compounds that could exist as IVOCs/SVOCs and thus participate in phase partitioning, we supplemented our particle-phase analyses with a novel approach for investigating functionalized gases with LC-ESI-MS. Further investigation of these nitrogen-containing gases will facilitate new understanding of their gas–particle partitioning in the presence of atmospheric water and organic condensed species, and measurements across dynamic conditions will help elucidate the relative importance of both processes. For these types of measurements, further design iterations of the PEEK sampling system for functionalized gases and additional functionalized gas-phase samples for LC-ESI-MS analysis could be pursued. Concurrent high-volume filter samples could be collected for direct comparison to the particle phase, which was not possible in this study due to insufficient mass loading on the upstream filter during the short-duration functionalized gas sample (i.e., 2 h). Concurrent PEEK samples could also be collected for MS/MS analysis.
In summary, combinations of online and offline mass spectrometry to obtain temporal and chemical detail, further ambient observations of major organic nitrogen sources, a better understanding of the aqueous processing of nitrogen-containing compounds, and improved characterization of their gas–particle partitioning in the presence of atmospheric water will together allow for a more accurate representation of nitrogen-containing organic compounds in emission inventories and models and enhance our ability to predict their impacts on atmospheric composition, human health, and climate.
Data are available upon request to Drew R. Gentner (drew.gentner@yale.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-3045-2022-supplement.
JCD and DRG planned the field sampling and study. JCD collected and analyzed field samples and performed PEEK sampling and inline LC method development. JM performed inline LC method development. JCD and DRG wrote the manuscript with contributions from all the co-authors.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank GERSTEL for their collaboration with the thermal desorption unit used here. We also thank David Wheeler at the New York Department of Environmental Conservation, Pete Babich and Adam Augustine at the Connecticut Department of Energy and Environmental Protection, Luke Valin at the Environmental Protection Agency, and Jordan Peccia at Yale for the use of sampling equipment, as well as Paul Miller (NESCAUM) for organizing the LISTOS project. We thank Richard Boardman and the Yale Peabody Museum for enabling us to set up and collect samples at the YCFS site, and the help of Yale Peabody Museum EVOLUTIONS interns: Amir Bond, Ethan Weed, Paula Mock, and Aurea Orencia. We thank Trevor VandenBoer and Barbara Ervens for helpful feedback on the draft manuscript. Finally, we thank the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport and dispersion model.
This research has been supported by the National Science Foundation (grant no. AGS1764126) and Yale University (Natural Lands Program). Jo Machesky received support from the Goodyear Tire & Rubber Company and the Grumman Fellowship.
This paper was edited by Ryan Sullivan and reviewed by two anonymous referees.
Altieri, K. E., Turpin, B. J., and Seitzinger, S. P.: Composition of dissolved organic nitrogen in continental precipitation investigated by ultra-high resolution FT-ICR mass spectrometry, Environ. Sci. Technol., 43, 6950–6955, https://doi.org/10.1021/es9007849, 2009.
Boone, E. J., Laskin, A., Laskin, J., Wirth, C., Shepson, P. B., Stirm, B. H., and Pratt, K. A.: Aqueous Processing of Atmospheric Organic Particles in Cloud Water Collected via Aircraft Sampling, Environ. Sci. Technol., 49, 8523–8530, https://doi.org/10.1021/acs.est.5b01639, 2015.
Borduas, N., Da Silva, G., Murphy, J. G., and Abbatt, J. P. D.: Experimental and theoretical understanding of the gas phase oxidation of atmospheric amides with OH radicals: Kinetics, products, and mechanisms, J. Phys. Chem. A, 119, 4298–4308, https://doi.org/10.1021/jp503759f, 2015.
Brean, J., Dall'Osto, M., Simó, R., Shi, Z., Beddows, D. C. S., and Harrison, R. M.: Open ocean and coastal new particle formation from sulfuric acid and amines around the Antarctic Peninsula, Nat. Geosci., 14, 383–388, https://doi.org/10.1038/s41561-021-00751-y, 2021.
Brege, M., Paglione, M., Gilardoni, S., Decesari, S., Facchini, M. C., and Mazzoleni, L. R.: Molecular insights on aging and aqueous-phase processing from ambient biomass burning emissions-influenced Po Valley fog and aerosol, Atmos. Chem. Phys., 18, 13197–13214, https://doi.org/10.5194/acp-18-13197-2018, 2018.
Chen, C. L., Chen, S., Russell, L. M., Liu, J., Price, D. J., Betha, R., Sanchez, K. J., Lee, A. K. Y., Williams, L., Collier, S. C., Zhang, Q., Kumar, A., Kleeman, M. J., Zhang, X., and Cappa, C. D.: Organic Aerosol Particle Chemical Properties Associated With Residential Burning and Fog in Wintertime San Joaquin Valley (Fresno) and With Vehicle and Firework Emissions in Summertime South Coast Air Basin (Fontana), J. Geophys. Res.-Atmos., 123, 10707–10731, https://doi.org/10.1029/2018JD028374, 2018.
Cleveland, W. S., Kleiner, B., McRae, J. E., and Warner, J. L.: Photochemical air pollution: Transport from the New York City area into Connecticut and Massachusetts, Science, 191, 179–181, https://doi.org/10.1126/science.1246603, 1976.
Dall'Osto, M., Airs, R. L., Beale, R., Cree, C., Fitzsimons, M. F., Beddows, D., Harrison, R. M., Ceburnis, D., O'Dowd, C., Rinaldi, M., Paglione, M., Nenes, A., Decesari, S., and Simó, R.: Simultaneous Detection of Alkylamines in the Surface Ocean and Atmosphere of the Antarctic Sympagic Environment, ACS Earth Sp. Chem., 3, 854–862, https://doi.org/10.1021/acsearthspacechem.9b00028, 2019.
de Gouw, J. A., Middlebrook, A. M., Warneke, C., Goldan, P. D., Kuster, W. C., Roberts, J. M., Fehsenfeld, F. C., Worsnop, D. R., Canagaratna, M. R., Pszenny, A. A. P., Keene, W. C., Marchewka, M., Bertman, S. B., and Bates, T. S.: Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002, J. Geophys. Res.-Atmos., 110, 1–22, https://doi.org/10.1029/2004JD005623, 2005.
Decesari, S., Paglione, M., Rinaldi, M., Dall'Osto, M., Simó, R., Zanca, N., Volpi, F., Facchini, M. C., Hoffmann, T., Götz, S., Kampf, C. J., O'Dowd, C., Ceburnis, D., Ovadnevaite, J., and Tagliavini, E.: Shipborne measurements of Antarctic submicron organic aerosols: an NMR perspective linking multiple sources and bioregions, Atmos. Chem. Phys., 20, 4193–4207, https://doi.org/10.5194/acp-20-4193-2020, 2020.
DeHaan, D. O., Corrigan, A. L., Smith, K. W., Stroik, D. R., Turley, J. J., Lee, F. E., Tolbert, M. A., Jimenez, J. L., Cordova, K. E., Ferrell, G. R., De Haan, D. O., Corrigan, A. L., Smith, K. W., Stroik, D. R., Turley, J. J., Lee, F. E., Tolbert, M. A., Jimenez, J. L., Cordova, K. E., and Ferrell, G. R.: Secondary organic aerosol-forming reactions of glyoxal with amino acids, Environ. Sci. Technol., 43, 2818–2824, https://doi.org/10.1021/es803534f, 2009.
Di, Q., Wang, Y., Zanobetti, A., Wang, Y., Koutrakis, P., Choirat, C., Dominici, F., and Schwartz, J. D.: Air pollution and mortality in the medicare population, New Engl. J. Med., 376, 2513–2522, https://doi.org/10.1056/NEJMoa1702747, 2017.
Di Lorenzo, R. A., Place, B. K., VandenBoer, T. C., and Young, C. J.: Composition of Size-Resolved Aged Boreal Fire Aerosols: Brown Carbon, Biomass Burning Tracers, and Reduced Nitrogen, ACS Earth Sp. Chem., 2, 278–285, https://doi.org/10.1021/acsearthspacechem.7b00137, 2018.
Ditto, J. C., Barnes, E. B., Khare, P., Takeuchi, M., Joo, T., Bui, A. A. T., Lee-Taylor, J., Eris, G., Chen, Y., Aumont, B., Jimenez, J. L., Ng, N. L., Griffin, R. J., and Gentner, D. R.: An omnipresent diversity and variability in the chemical composition of atmospheric functionalized organic aerosol, Commun. Chem., 1, 75, https://doi.org/10.1038/s42004-018-0074-3, 2018.
Ditto, J. C., Joo, T., Khare, P., Sheu, R., Takeuchi, M., Chen, Y., Xu, W., Bui, A. A. T., Sun, Y., Ng, N. L., and Gentner, D. R.: Effects of Molecular-Level Compositional Variability in Organic Aerosol on Phase State and Thermodynamic Mixing Behavior, Environ. Sci. Technol., 53, 13009–13018, https://doi.org/10.1021/acs.est.9b02664, 2019.
Ditto, J. C., Joo, T., Slade, J. H., Shepson, P. B., Ng, N. L., and Gentner, D. R.: Nontargeted Tandem Mass Spectrometry Analysis Reveals Diversity and Variability in Aerosol Functional Groups across Multiple Sites, Seasons, and Times of Day, Environ. Sci. Tech. Let., 7, 60–69, https://doi.org/10.1021/acs.estlett.9b00702, 2020.
Ditto, J. C., He, M., Hass-Mitchell, T. N., Moussa, S. G., Hayden, K., Li, S.-M., Liggio, J., Leithead, A., Lee, P., Wheeler, M. J., Wentzell, J. J. B., and Gentner, D. R.: Atmospheric evolution of emissions from a boreal forest fire: the formation of highly functionalized oxygen-, nitrogen-, and sulfur-containing organic compounds, Atmos. Chem. Phys., 21, 255–267, https://doi.org/10.5194/acp-21-255-2021, 2021.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Dührkop, K., Shen, H., Meusel, M., Rousu, J., and Böcker, S.: Searching molecular structure databases with tandem mass spectra using CSI:FingerID, P. Natl. Acad. Sci. USA, 112, 12580–12585, https://doi.org/10.1073/pnas.1509788112, 2015.
Dührkop, K., Fleischauer, M., Ludwig, M., Aksenov, A. A., Melnik, A. V., Meusel, M., Dorrestein, P. C., Rousu, J., and Böcker, S.: SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information, Nat. Methods, 16, 299–302, https://doi.org/10.1038/s41592-019-0344-8, 2019.
Dunlea, E. J., Herndon, S. C., Nelson, D. D., Volkamer, R. M., San Martini, F., Sheehy, P. M., Zahniser, M. S., Shorter, J. H., Wormhoudt, J. C., Lamb, B. K., Allwine, E. J., Gaffney, J. S., Marley, N. A., Grutter, M., Marquez, C., Blanco, S., Cardenas, B., Retama, A., Ramos Villegas, C. R., Kolb, C. E., Molina, L. T., and Molina, M. J.: Evaluation of nitrogen dioxide chemiluminescence monitors in a polluted urban environment, Atmos. Chem. Phys., 7, 2691–2704, https://doi.org/10.5194/acp-7-2691-2007, 2007.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Gaston, C. J., Quinn, P. K., Bates, T. S., Gilman, J. B., Bon, D. M., Kuster, W. C., and Prather, K. A.: The impact of shipping, agricultural, and urban emissions on single particle chemistry observed aboard the R/V Atlantis during CalNex, J. Geophys. Res.-Atmos., 118, 5003–5017, https://doi.org/10.1002/jgrd.50427, 2013.
Ge, X., Wexler, A. S., and Clegg, S. L.: Atmospheric amines – Part I. A review, Atmos. Environ., 45, 524–546, https://doi.org/10.1016/j.atmosenv.2010.10.012, 2011.
Ge, Y., Liu, Y., Chu, B., He, H., Chen, T., Wang, S., Wei, W., and Cheng, S.: Ozonolysis of Trimethylamine Exchanged with Typical Ammonium Salts in the Particle Phase, Environ. Sci. Technol., 50, 11076–11084, https://doi.org/10.1021/acs.est.6b04375, 2016.
Gioda, A., Reyes-Rodríguez, G. J., Santos-Figueroa, G., Collett, J. L., Decesari, S., Ramos, M. D. C. K. V., Bezerra Netto, H. J. C., De Aquino Neto, F. R., and Mayol-Bracero, O. L.: Speciation of water-soluble inorganic, organic, and total nitrogen in a background marine environment: Cloud water, rainwater, and aerosol particles, J. Geophys. Res.-Atmos., 116, D05203, https://doi.org/10.1029/2010JD015010, 2011.
Grace, D. N., Sharp, J. R., Holappa, R. E., Lugos, E. N., Sebold, M. B., Griffith, D. R., Hendrickson, H. P., and Galloway, M. M.: Heterocyclic Product Formation in Aqueous Brown Carbon Systems, ACS Earth Sp. Chem., 3, 2472–2481, https://doi.org/10.1021/acsearthspacechem.9b00235, 2019.
Grace, D. N., Lugos, E. N., Ma, S., Griffith, D. R., Hendrickson, H. P., Woo, J. L., and Galloway, M. M.: Brown Carbon Formation Potential of the Biacetyl-Ammonium Sulfate Reaction System, ACS Earth Sp. Chem., 4, 1104–1113, https://doi.org/10.1021/acsearthspacechem.0c00096, 2020.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hems, R. F. and Abbatt, J. P. D.: Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Sp. Chem., 2, 225–234, https://doi.org/10.1021/acsearthspacechem.7b00123, 2018.
Herrmann, H., Schaefer, T., Tilgner, A., Styler, S. A., Weller, C., Teich, M., and Otto, T.: Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase, Chem. Rev., 115, 4259–4334, https://doi.org/10.1021/cr500447k, 2015.
Jerrett, M., Burnett, R. T., Pope, C. A., Ito, K., Thurston, G., Krewski, D., Shi, Y., Calle, E., and Thun, M.: Long-term ozone exposure and mortality, New Engl. J. Med., 360, 1085–1095, https://doi.org/10.1056/NEJMoa0803894, 2009.
Juncosa Calahorrano, J. F., Lindaas, J., O'Dell, K., Palm, B. B., Peng, Q., Flocke, F., Pollack, I. B., Garofalo, L. A., Farmer, D. K., Pierce, J. R., Collett, J. L., Weinheimer, A., Campos, T., Hornbrook, R. S., Hall, S. R., Ullmann, K., Pothier, M. A., Apel, E. C., Permar, W., Hu, L., Hills, A. J., Montzka, D., Tyndall, G., Thornton, J. A., and Fischer, E. V.: Daytime Oxidized Reactive Nitrogen Partitioning in Western U. S. Wildfire Smoke Plumes, J. Geophys. Res.-Atmos., 126, 1–22, https://doi.org/10.1029/2020JD033484, 2021.
Khare, P. and Gentner, D. R.: Considering the future of anthropogenic gas-phase organic compound emissions and the increasing influence of non-combustion sources on urban air quality, Atmos. Chem. Phys., 18, 5391–5413, https://doi.org/10.5194/acp-18-5391-2018, 2018.
Khare, P., Marcotte, A., Sheu, R., Walsh, A. N., Ditto, J. C., and Gentner, D. R.: Advances in offline approaches for trace measurements of complex organic compound mixtures via soft ionization and high-resolution tandem mass spectrometry, J. Chromatogr. A, 1598, 163–174, https://doi.org/10.1016/j.chroma.2019.03.037, 2019.
Kieloaho, A. J., Hellén, H., Hakola, H., Manninen, H. E., Nieminen, T., Kulmala, M., and Pihlatie, M.: Gas-phase alkylamines in a boreal Scots pine forest air, Atmos. Environ., 80, 369–377, https://doi.org/10.1016/j.atmosenv.2013.08.019, 2013.
Kilian, J. and Kitazawa, M.: The emerging risk of exposure to air pollution on cognitive decline and Alzheimer's disease – Evidence from epidemiological and animal studies, Biomed. J., 41, 141–162, https://doi.org/10.1016/j.bj.2018.06.001, 2018.
Kim, H., Collier, S., Ge, X., Xu, J., Sun, Y., Jiang, W., Wang, Y., Herckes, P., and Zhang, Q.: Chemical processing of water-soluble species and formation of secondary organic aerosol in fogs, Atmos. Environ., 200, 158–166, https://doi.org/10.1016/j.atmosenv.2018.11.062, 2019.
Kind, T. and Fiehn, O.: Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry, BMC Bioinformatics, 8, 105, https://doi.org/10.1186/1471-2105-8-105, 2007.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of Atmospheric Brown Carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
LeClair, J. P., Collett, J. L., and Mazzoleni, L. R.: Fragmentation analysis of water-soluble atmospheric organic matter using ultrahigh-resolution FT-ICR mass spectrometry, Environ. Sci. Technol., 46, 4312–4322, https://doi.org/10.1021/es203509b, 2012.
Leslie, M. D., Ridoli, M., Murphy, J. G., and Borduas-Dedekind, N.: Isocyanic acid (HNCO) and its fate in the atmosphere: A review, Environ. Sci. Process. Impacts, 21, 793–808, https://doi.org/10.1039/c9em00003h, 2019.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Liebmann, J., Sobanski, N., Schuladen, J., Karu, E., Hellén, H., Hakola, H., Zha, Q., Ehn, M., Riva, M., Heikkinen, L., Williams, J., Fischer, H., Lelieveld, J., and Crowley, J. N.: Alkyl nitrates in the boreal forest: formation via the NO3-, OH- and O3-induced oxidation of biogenic volatile organic compounds and ambient lifetimes, Atmos. Chem. Phys., 19, 10391–10403, https://doi.org/10.5194/acp-19-10391-2019, 2019.
Lim, S., McArdell, C. S., and von Gunten, U.: Reactions of aliphatic amines with ozone: Kinetics and mechanisms, Water Res., 157, 514–528, https://doi.org/10.1016/j.watres.2019.03.089, 2019.
Lim, Y. B., Kim, H., Kim, J. Y., and Turpin, B. J.: Photochemical organonitrate formation in wet aerosols, Atmos. Chem. Phys., 16, 12631–12647, https://doi.org/10.5194/acp-16-12631-2016, 2016.
Lin, M., Walker, J., Geron, C., and Khlystov, A.: Organic nitrogen in PM2.5 aerosol at a forest site in the Southeast US, Atmos. Chem. Phys., 10, 2145–2157, https://doi.org/10.5194/acp-10-2145-2010, 2010.
Lin, P., Laskin, J., Nizkorodov, S. A., and Laskin, A.: Revealing Brown Carbon Chromophores Produced in Reactions of Methylglyoxal with Ammonium Sulfate, Environ. Sci. Technol., 49, 14257–14266, https://doi.org/10.1021/acs.est.5b03608, 2015.
Loza, C. L., Craven, J. S., Yee, L. D., Coggon, M. M., Schwantes, R. H., Shiraiwa, M., Zhang, X., Schilling, K. A., Ng, N. L., Canagaratna, M. R., Ziemann, P. J., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol yields of 12-carbon alkanes, Atmos. Chem. Phys., 14, 1423–1439, https://doi.org/10.5194/acp-14-1423-2014, 2014.
Mace, K. A., Kubilay, N., and Duce, R. A.: Organic nitrogen in rain and aerosol in the eastern Mediterranean atmosphere: An association with atmospheric dust, J. Geophys. Res.-Atmos., 108, 4320, https://doi.org/10.1029/2002jd002997, 2003a.
Mace, K. A., Artaxo, P., and Duce, R. A.: Water-soluble organic nitrogen in Amazon Basin aerosols during the dry (biomass burning) and wet seasons, J. Geophys. Res.-Atmos., 108, 4512, https://doi.org/10.1029/2003jd003557, 2003b.
McNeill, V. F.: Aqueous organic chemistry in the atmosphere: Sources and chemical processing of organic aerosols, Environ. Sci. Technol., 49, 1237–1244, https://doi.org/10.1021/es5043707, 2015.
Montero-Martínez, G., Rinaldi, M., Gilardoni, S., Giulianelli, L., Paglione, M., Decesari, S., Fuzzi, S., and Facchini, M. C.: On the water-soluble organic nitrogen concentration and mass size distribution during the fog season in the Po Valley, Italy, Sci. Total Environ., 485–486, 103–109, https://doi.org/10.1016/j.scitotenv.2014.03.060, 2014.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Perring, A. E., Pusede, S. E., and Cohen, R. C.: An Observational Perspective on the Atmospheric Impacts of Alkyl and Multifunctional Nitrates on Ozone and Secondary Organic Aerosol, Chem. Rev., 113, 5848–5870, https://doi.org/10.1021/cr300520x, 2013.
Pope, C. A. and Dockery, D. D.: Health Effects of Fine Particulate Air Pollution: Lines that Connect, JAPCA J. Air Waste Ma., 56, 709–742, https://doi.org/10.1080/10473289.2006.10464485, 2006.
Priestley, M., Le Breton, M., Bannan, T. J., Leather, K. E., Bacak, A., Reyes-Villegas, E., De Vocht, F., Shallcross, B. M. A., Brazier, T., Khan, M. A., Allan, J., Shallcross, D. E., Coe, H., and Percival, C. J.: Observations of Isocyanate, Amide, Nitrate, and Nitro Compounds From an Anthropogenic Biomass Burning Event Using a ToF-CIMS, J. Geophys. Res.-Atmos., 123, 7687–7704, https://doi.org/10.1002/2017JD027316, 2018.
Pye, H. O. T., Nenes, A., Alexander, B., Ault, A. P., Barth, M. C., Clegg, S. L., Collett Jr., J. L., Fahey, K. M., Hennigan, C. J., Herrmann, H., Kanakidou, M., Kelly, J. T., Ku, I.-T., McNeill, V. F., Riemer, N., Schaefer, T., Shi, G., Tilgner, A., Walker, J. T., Wang, T., Weber, R., Xing, J., Zaveri, R. A., and Zuend, A.: The acidity of atmospheric particles and clouds, Atmos. Chem. Phys., 20, 4809–4888, https://doi.org/10.5194/acp-20-4809-2020, 2020.
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A., and Bates, T. S.: Chemistry and Related Properties of Freshly Emitted Sea Spray Aerosol, Chem. Rev., 115, 4383–4399, https://doi.org/10.1021/cr500713g, 2015.
Rindelaub, J. D., Borca, C. H., Hostetler, M. A., Slade, J. H., Lipton, M. A., Slipchenko, L. V., and Shepson, P. B.: The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact, Atmos. Chem. Phys., 16, 15425–15432, https://doi.org/10.5194/acp-16-15425-2016, 2016.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Roberts, J. M., Veres, P. R., VandenBoer, T. C., Warneke, C., Graus, M., Williams, E. J., Lefer, B., Brock, C. A., Bahreini, R., Ozturk, F., Middlebrook, A. M., Wagner, N. L., Dube, W. P., and DeGouw, J. A.: New insights into atmospheric sources and sinksof isocyanic acid, HNCO, from recent urbanand regional observations, J. Geophys. Res.-Atmos., 119, 1060–1072, https://doi.org/10.1002/2013JD019931, 2014.
Rogers, H. M., Ditto, J. C., and Gentner, D. R.: Evidence for impacts on surface-level air quality in the northeastern US from long-distance transport of smoke from North American fires during the Long Island Sound Tropospheric Ozone Study (LISTOS) 2018, Atmos. Chem. Phys., 20, 671–682, https://doi.org/10.5194/acp-20-671-2020, 2020.
Ruggeri, G. and Takahama, S.: Technical Note: Development of chemoinformatic tools to enumerate functional groups in molecules for organic aerosol characterization, Atmos. Chem. Phys., 16, 4401–4422, https://doi.org/10.5194/acp-16-4401-2016, 2016.
Sander, R.: Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 15, 4399–4981, https://doi.org/10.5194/acp-15-4399-2015, 2015.
Sareen, N., Schwier, A. N., Shapiro, E. L., Mitroo, D., and McNeill, V. F.: Secondary organic material formed by methylglyoxal in aqueous aerosol mimics, Atmos. Chem. Phys., 10, 997–1016, https://doi.org/10.5194/acp-10-997-2010, 2010.
Schervish, M. and Donahue, N. M.: Peroxy radical chemistry and the volatility basis set, Atmos. Chem. Phys., 20, 1183–1199, https://doi.org/10.5194/acp-20-1183-2020, 2020.
Schroder, J. C., Campuzano-Jost, P., Day, D. A., Shah, V., Larson, K., Sommers, J. M., Sullivan, A. P., Campos, T., Reeves, J. M., Hills, A., Hornbrook, R. S., Blake, N. J., Scheuer, E., Guo, H., Fibiger, D. L., McDuffie, E. E., Hayes, P. L., Weber, R. J., Dibb, J. E., Apel, E. C., Jaeglé, L., Brown, S. S., Thornton, J. A., and Jimenez, J. L.: Sources and Secondary Production of Organic Aerosols in the Northeastern United States during WINTER, J. Geophys. Res.-Atmos., 123, 7771–7796, https://doi.org/10.1029/2018JD028475, 2018.
Schurman, M. I., Boris, A., Desyaterik, Y., and Collett, J. L.: Aqueous secondary organic aerosol formation in ambient cloud water photo-oxidations, Aerosol Air Qual. Res., 18, 15–25, https://doi.org/10.4209/aaqr.2017.01.0029, 2018.
Sheu, R., Marcotte, A., Khare, P., Charan, S., Ditto, J. C., and Gentner, D. R.: Advances in offline approaches for chemically speciated measurements of trace gas-phase organic compounds via adsorbent tubes in an integrated sampling-to-analysis system, J. Chromatogr. A, 1575, 80–90, https://doi.org/10.1016/j.chroma.2018.09.014, 2018.
Sintermann, J. and Neftel, A.: Ideas and perspectives: on the emission of amines from terrestrial vegetation in the context of new atmospheric particle formation, Biogeosciences, 12, 3225–3240, https://doi.org/10.5194/bg-12-3225-2015, 2015.
Sodeman, D. A., Toner, S. M., and Prather, K. A.: Determination of Single Particle Mass Spectral Signatures from Light-Duty Vehicle Emissions, Environ. Sci. Technol., 39, 4569–4580, https://doi.org/10.1016/j.atmosenv.2007.01.025, 2005.
Sullivan, A. P., Guo, H., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., Campos, T., Shah, V., Jaeglé, L., Lee, B. H., Lopez-Hilfiker, F. D., Thornton, J. A., Brown, S. S., and Weber, R. J.: Biomass Burning Markers and Residential Burning in the WINTER Aircraft Campaign, J. Geophys. Res.-Atmos., 124, 1846–1861, https://doi.org/10.1029/2017JD028153, 2019.
Takeuchi, M. and Ng, N. L.: Organic nitrates and secondary organic aerosol (SOA) formation from oxidation of biogenic volatile organic compounds, ACS Symp. Ser. Multiph. Environ. Chem. Atmos., 1299, 105–125, https://doi.org/10.1021/bk-2018-1299.ch006, 2018.
Tao, Y., Liu, T., Yang, X., and Murphy, J. G.: Kinetics and Products of the Aqueous Phase Oxidation of Triethylamine by OH, ACS Earth Sp. Chem., 5, 1889–1895, https://doi.org/10.1021/acsearthspacechem.1c00162, 2021.
United States Environmental Protection Agency: Historical Exceedance Days in New England, Reg. 1 EPA New Engl., https://www3.epa.gov/region1/airquality/standard.html (last access: 14 August 2021), 2020.
van Pinxteren, M., Müller, C., Iinuma, Y., Stolle, C., and Herrmann, H.: Chemical Characterization of Dissolved Organic Compounds from Coastal Sea Surface Microlayers (Baltic Sea, Germany), Environ. Sci. Technol., 46, 10455–10462, https://doi.org/10.1021/es204492b, 2012.
van Pinxteren, M., Fomba, K. W., van Pinxteren, D., Triesch, N., Hoffmann, E. H., Cree, C. H. L., Fitzsimons, M. F., von Tümpling, W., and Herrmann, H.: Aliphatic amines at the Cape Verde Atmospheric Observatory: Abundance, origins and sea-air fluxes, Atmos. Environ., 203, 183–195, https://doi.org/10.1016/j.atmosenv.2019.02.011, 2019.
VandenBoer, T. C., Petroff, A., Markovic, M. Z., and Murphy, J. G.: Size distribution of alkyl amines in continental particulate matter and their online detection in the gas and particle phase, Atmos. Chem. Phys., 11, 4319–4332, https://doi.org/10.5194/acp-11-4319-2011, 2011.
Vidović, K., Lašič Jurković, D., Šala, M., Kroflič, A., and Grgić, I.: Nighttime Aqueous-Phase Formation of Nitrocatechols in the Atmospheric Condensed Phase, Environ. Sci. Technol., 52, 9722–9730, https://doi.org/10.1021/acs.est.8b01161, 2018.
Warneke, C., McKeen, S. A., de Gouw, J. A., Goldan, P. D., Kuster, W. C., Holloway, J. S., Williams, E. J., Lerner, B. M., Parrish, D. D., Trainer, M., Fehsenfeld, F. C., Kato, S., Atlas, E. L., Baker, A., and Blake, D. R.: Determination of urban volatile organic compound emission ratios and comparison with an emissions database, J. Geophys. Res.-Atmos., 112, 1–13, https://doi.org/10.1029/2006JD007930, 2007.
Wozniak, A. S., Willoughby, A. S., Gurganus, S. C., and Hatcher, P. G.: Distinguishing molecular characteristics of aerosol water soluble organic matter from the 2011 trans-North Atlantic US GEOTRACES cruise, Atmos. Chem. Phys., 14, 8419–8434, https://doi.org/10.5194/acp-14-8419-2014, 2014.
Wu, C., Wen, Y., Hua, L., Jiang, J., Xie, Y., Cao, Y., Chai, S., Hou, K., and Li, H.: Rapid and highly sensitive measurement of trimethylamine in seawater using dynamic purge-release and dopant-assisted atmospheric pressure photoionization mass spectrometry, Anal. Chim. Acta, 1137, 56–63, https://doi.org/10.1016/j.aca.2020.08.060, 2020.
Xu, Y., Miyazaki, Y., Tachibana, E., Sato, K., Ramasamy, S., Mochizuki, T., Sadanaga, Y., Nakashima, Y., Sakamoto, Y., Matsuda, K., and Kajii, Y.: Aerosol Liquid Water Promotes the Formation of Water-Soluble Organic Nitrogen in Submicrometer Aerosols in a Suburban Forest, Environ. Sci. Technol., 54, 1406–1414, https://doi.org/10.1021/acs.est.9b05849, 2020.
Ye, C., Zhou, X., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin, R. L., Campos, T., Weinheimer, A., Hornbrook, R. S., Apel, E. C., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J. N., Ortega, J., and Knote, C.: Rapid cycling of reactive nitrogen in the marine boundary layer, Nature, 532, 489–491, https://doi.org/10.1038/nature17195, 2016.
Youn, J. S., Crosbie, E., Maudlin, L. C., Wang, Z., and Sorooshian, A.: Dimethylamine as a major alkyl amine species in particles and cloud water: Observations in semi-arid and coastal regions, Atmos. Environ., 122, 250–258, https://doi.org/10.1016/j.atmosenv.2015.09.061, 2015.
Yu, L., Smith, J., Laskin, A., George, K. M., Anastasio, C., Laskin, J., Dillner, A. M., and Zhang, Q.: Molecular transformations of phenolic SOA during photochemical aging in the aqueous phase: competition among oligomerization, functionalization, and fragmentation, Atmos. Chem. Phys., 16, 4511–4527, https://doi.org/10.5194/acp-16-4511-2016, 2016.
Zhang, J., Ninneman, M., Joseph, E., Schwab, M. J., Shrestha, B., and Schwab, J. J.: Mobile Laboratory Measurements of High Surface Ozone Levels and Spatial Heterogeneity During LISTOS 2018: Evidence for Sea Breeze Influence, J. Geophys. Res.-Atmos., 125, 1–12, https://doi.org/10.1029/2019JD031961, 2020.
Zhang, Q., Anastasio, C., and Jimenez-Cruz, M.: Water-soluble organic nitrogen in atmospheric fine particles (PM2.5) from northern California, J. Geophys. Res.-Atmos., 107, 1–9, https://doi.org/10.1029/2001jd000870, 2002.
Zhao, Y., Hallar, A. G., and Mazzoleni, L. R.: Atmospheric organic matter in clouds: exact masses and molecular formula identification using ultrahigh-resolution FT-ICR mass spectrometry, Atmos. Chem. Phys., 13, 12343–12362, https://doi.org/10.5194/acp-13-12343-2013, 2013.
Zhou, S., Collier, S., Xu, J., Mei, F., Wang, J., Lee, Y.-N., Sedlacek, A. J., Springston, S. R., Sun, Y., and Zhang, Q.: Influences of upwind emission sources and atmospheric processing on aerosol chemistry and properties at a rural location in the Northeastern U. S., J. Geophys. Res.-Atmos., 121, 6049–6065, https://doi.org/10.1002/2015JD024568, 2016.