the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Feb 2022
| 11 Feb 2022
Pyruvic acid, an efficient catalyst in SO3 hydrolysis and effective clustering agent in sulfuric-acid-based new particle formation
Narcisse Tsona Tchinda
Xiuhui Zhang
The role of pyruvic acid (PA), one of the most abundant α-keto carboxylic acids in the atmosphere, was investigated both in the SO3 hydrolysis reaction to form sulfuric acid (SA) and in SA-based aerosol particle formation using quantum chemical calculations and a cluster dynamics model. We found that the PA-catalyzed SO3 hydrolysis is a thermodynamically driven transformation process, proceeding with a negative Gibbs free-energy barrier, ca. −1 kcal mol−1 at 298 K, ∼ 6.50 kcal mol−1 lower than that in the water-catalyzed SO3 hydrolysis. Results indicated that the PA-catalyzed reaction can potentially compete with the water-catalyzed SO3 reaction in SA production, especially in dry and polluted areas, where it is found to be ∼ 2 orders of magnitude more efficient that the water-catalyzed reaction. Given the effective stabilization of the PA-catalyzed SO3 hydrolysis product as SA•PA cluster, we proceeded to examine the PA clustering efficiency in a sulfuric-acid–pyruvic-acid–ammonia (SA-PA-NH3) system. Our thermodynamic data used in the Atmospheric Cluster Dynamics Code indicated that under relevant tropospheric temperatures and concentrations of SA (106 ), PA (1010 ) and NH3 (1011 and 5 × 1011 ), PA-enhanced particle formation involves clusters containing at most one PA molecule. Namely, under these monomer concentrations and 238 K, the (SA)2•PA•(NH3)2 cluster was found to contribute by ∼ 100 % to the net flux to aerosol particle formation. At higher temperatures (258 and 278 K), however, the net flux to the particle formation is dominated by pure SA-NH3 clusters, while PA would rather evaporate from the clusters at high temperatures and not contribute to the particle formation. The enhancing effect of PA was examined by evaluating the ratio of the ternary SA-PA-NH3 cluster formation rate to binary SA-NH3 cluster formation rate. Our results show that while the enhancement factor of PA to the particle formation rate is almost insensitive to investigated temperatures and concentrations, it can be as high as 4.7 × 102 at 238 K and [NH3] = 1.3 × 1011 . This indicates that PA may actively participate in aerosol formation, only in cold regions of the troposphere and highly NH3-polluted environments. The inclusion of this mechanism in aerosol models may reduce uncertainties that prevail in modeling the aerosol impact on climate.
- Article
(3183 KB) - Full-text XML
-
Supplement
(654 KB) - BibTeX
- EndNote





Understanding the detailed processes involved in secondary aerosol formation continues to retain the attention of many researchers around the world. This is due to the varied role aerosols play in degrading visibility and human health, as well in affecting climate by altering cloud properties and influencing the balance of solar radiation (Stocker et al., 2013). Sulfuric acid (H2SO4, SA), which is believed to be the key species driving aerosol formation in the atmosphere (Kulmala et al., 2000; Kulmala, 2003; Sipila et al., 2010; Sihto et al., 2006; Kuang et al., 2008), is primarily formed from the hydrolysis of sulfur trioxide (SO3). The main sources of atmospheric SO3 include electrically neutral SO2 oxidation by OH radicals and stabilized Criegee intermediates (Mauldin et al., 2012; Welz et al., 2012), whereas ion-induced oxidation constitutes a complementary source (Bork et al., 2013; Tsona et al., 2015a, 2016; Tsona and Du, 2019). The sinks of SO3 include its reaction with water to produce sulfuric acid and acid rain, as well as reactions with other organic and inorganic species including ammonia and methanol (Li et al., 2018; Liu et al., 2019).
The general mechanism for SO3 hydration to form sulfuric acid is a hydrogen atom transfer between H2O and SO3 within the SO3•(H2O)n≥2 cluster, assisted by a second water molecule (Hofmann-Sievert and Castleman, 1984; Holland and Castleman, 1978), according to the following reaction:
In the presence of a single water molecule, the above reaction was found to be prevented by a high energy barrier, around 30 kcal mol−1, whereas the barrier height gradually decreases to ca. ∼ 0 kcal mol−1 as the number of water molecules increases to 4 or more (Hofmann and Schleyer, 1994; Morokuma and Muguruma, 1994; Larson et al., 2000; Loerting and Liedl, 2000). In Reaction (R1), the second water molecule acting as a catalyst forms a bridge for the hydrogen atom to transfer from H2O to SO3. This reaction is considered as the major loss pathway for SO3 in the atmosphere. Beside a second water molecule, a number of studies have shown that the SO3 + H2O → H2SO4 reaction can be facilitated by organic and inorganic species including sulfuric acid, formic acid, nitric acid and oxalic acid (Torrent-Sucarrat et al., 2012; Hazra and Sinha, 2011; Daub et al., 2020; Long et al., 2013; Lv et al., 2019). In the presence of these species, the SO3 + H2O reaction can effectively proceed in a near barrierless mechanism, where they also act as bridge for hydrogen atom transfer. However, due to the low concentration of each of these catalysts, their overall effect on the rate of sulfuric acid formation from the SO3 + H2O reaction is not strong enough that they cannot effectively compete with the water-catalyzed reaction, given the relatively high concentration of water. An efficient catalyst would not only promote a fast hydrogen transfer between SO3 and H2O, but would also have a high enough concentration to induce a net higher reaction rate than the water.
Pyruvic acid (CH3C(O)COOH, PA), the simplest and one of the most abundant α-keto acids in the troposphere, is highly present in plants and in tropospheric air (Eisenreich et al., 2001; Jardine et al., 2010; Magel et al., 2006; Eger et al., 2020). Sources of PA in tropospheric air include photo-oxidation of isoprene, photolysis of methyglyoxal, reactions of peroxy radicals formed during the oxidation of propane and the photooxidation of aromatic compounds in the presence of NOx, as well as vegetation (Paulot et al., 2009; Jenkin et al., 1993; Warneck, 2005; Praplan et al., 2014; Talbot et al., 1990; Jardine et al., 2010). PA mixing ratios of up to 15, 25 and 96 ppt were reported in the free troposphere and forest canopy over central Amazonia, and in a Finnish boreal forest, respectively (Talbot et al., 1990; Eger et al., 2020). In a rural continental mountain top site over the eastern US, PA mixing ratios reaching 200 ppt were measured (Talbot et al., 1995), while levels as high as 800 ppt were found above the equatorial African rainforest (Helas et al., 1992). In regions highly affected by anthropogenic activities, PA levels of up to 500 ppt were observed, whereas significantly low levels were observed in the marine boundary layer over the Atlantic Ocean (63∘ N to 39∘ S) (Baboukas et al., 2000; Mattila et al., 2018). PA is expected to be removed from the atmosphere through photolysis and oxidation by OH radicals (Mellouki and Mu, 2003; Reed Harris et al., 2016, 2017a, b; Church et al., 2020).
The detection of PA in various media, including gas-phase, aerosol and aqueous-phase (Andreae et al., 1987; Talbot et al., 1990; Bardouki et al., 2003; Baboukas et al., 2000; Chebbi and Carlier, 1996; Kawamura et al., 1996, 2013; Kawamura and Bikkina, 2016), makes it a good candidate for atmospheric processes. Moreover, through its carboxyl and carbonyl functions, PA can partake not only in hydrogen atom transfer reactions but also in molecular clustering owing to its ability to form hydrogen bonds. In this study, we examine the catalytic effect of PA on the SO3 hydration to form SA and assess the subsequent clustering of the reaction products (PA•SA) with additional SA and ammonia molecules. The kinetics of the SO3 hydrolysis are determined, and the fate of the product cluster in atmospheric particle formation is evaluated from cluster dynamic simulations.
2.1 Quantum chemical calculations
The calculations in this study were divided into reaction mechanism and cluster formation parts, and all geometry optimizations were performed using the Gaussian 09 package (Frisch et al., 2013). In the reaction mechanism part, the configurations of different states of the scanned reaction pathways were initially optimized with the M06-2X density functional (Zhao and Truhlar, 2008) used in conjunction with the 6-31+G(d,p) basis set. Identified M06-2X/6-31+G(d,p) structures within 3 kcal mol−1 of the lowest energy structure were re-optimized, followed by vibrational frequency analysis at the M06-2X/6-311G(3df,3pd) level of theory, thereby yielding zero-point energies as well as thermal correction to Gibbs free energies. It should be noted that vibrational frequency calculations were performed under the harmonic oscillator and rigid rotor approximation at 298 K and 1 atm. Transition state configurations were determined using the synchronous transit quasi-Newton method (Peng et al., 1996) and confirmed by intrinsic reaction coordinate calculations (Fukui, 1981) to ensure they connected the reactants to desired products. Electronic energies of all M06-2X/6-311G(3df,3pd) optimized structures were corrected by the DLPNO-CCSD(T)/aug-cc-pVTZ method using Orca version 4.2.1 (Riplinger et al., 2013; Riplinger and Neese, 2013).
2.2 Kinetics
The kinetic analysis of the studied reactions was performed following the conventional transition state theory (Duchovic et al., 1996; Truhlar et al., 1996) with the Wigner tunneling correction and executed with the KiSThelP program (Canneaux et al., 2014). Starting from the initial reactants (SO3, H2O and PA), the different processes in these reactions include the collision of two separate reactants to form a binary complex that further interact with the third species to form the pre-reactive intermediate, the evaporation of the pre-reactive intermediate to initial reactants and its forward reaction to form the products, according to the following reaction:
Reactions (R2a) and (R2b) are representative examples of processes taking place in the SO3 hydrolysis with X as catalyst, where X is H2O or PA. It should be noted that the pre-reactive intermediate can also form from the formation of the binary complex between the catalyst and water, followed by its interaction with SO3. Recent studies showed that no matter the order in which the binary complex and the pre-reactive intermediate are formed and regardless of the specific reactants involved in the formation of the binary complex, the interactions between SO3, H2O and the catalyst lead to the formation of sulfuric acid plus catalyst (Weber et al., 2001; Jayne et al., 1997; Torrent-Sucarrat et al., 2012; Hazra and Sinha, 2011; Lv et al., 2019). Assuming equilibrium between the reactants and the complex, and steady-state approximation of the pre-reactive intermediate, the overall rate of Reaction (R2) with catalyst X is
where k1 is the collision frequency of SO3 and H2O to form the SO3⋯H2O binary complex, and k−1 is the evaporation rate constant of SO3⋯H2O back to initial reactants, k2 is the collision frequency of SO3⋯H2O and the catalyst to form the pre-reactive intermediate, k−2 is the evaporation rate constant of the pre-reactive intermediate back to its precursor reactants. Keq1 and Keq2 are equilibrium constants of formation of the binary complex and pre-reactive intermediate, respectively; kuni,x is the unimolecular rate constant of the reaction of the pre-reactive intermediate to the products; and kx is the overall rate constant of Reaction (R2) in the presence of catalyst X. [SO3], [H2O] and [X] are respective gas-phase concentrations of SO3, H2O and the catalyst (H2O or PA). The determination of equilibrium constants and unimolecular rate constants was executed using the KiSThelP program, and their numerical values are given in Tables S2 and S3 in the Supplement.
2.3 Cluster formation and dynamic simulations
As Reaction (R2) results in the formation of sulfuric acid complexed to the catalyst, we explored the thermodynamics of further clustering with more PA, sulfuric acid (SA) and ammonia (NH3) molecules. Although sulfuric acid as a monomer is known to cluster effectively with water at relevant atmospheric conditions, several studies have demonstrated that the binding strength of water significantly decreases as sulfuric acid is clustered to other sulfuric acid and base molecules (Temelso et al., 2012a, b; Henschel et al., 2014; Tsona et al., 2015b). Moreover, cluster dynamics simulations have shown that despite its clustering to sulfuric acid and sulfuric-acid-based clusters, water rapidly evaporates from the cluster owing to its high evaporation rate, particularly when base molecules are present in the cluster. As a result, most atmospheric sulfuric-acid-based clusters are detected in their dry state, exclusively (Almeida et al., 2013; Olenius et al., 2013b). It follows that though water would play a certain role in the cluster thermodynamics, its net effect on particle formation rate is negligible. Hence, water was not included in the studied clusters.
A number of initial configurations of SA–NH3 clusters were taken from previously published results (Ortega et al., 2012) and re-optimized with the M06-2X/6-31G(d,p), while those containing PA were built by the stepwise addition of monomers to the relevant cluster. On this basis, several starting configurations were generated manually by arranging the participating molecules or clusters in different directions. Depending on the cluster size, 10–30 initial configurations of each cluster were pre-optimized at the M062X/6-31+G(d) level of theory. All identified structures within 3 kcal mol−1 of the lowest energy structure were thereafter re-optimized with the M06-2X/6-31G(d,p) method, and the vibrational frequency analysis was subsequently performed at the same level of theory. It has been shown that the reduction from the 6-311G(3df,3pd) to 6-31G(d,p) basis set for sulfuric-acid-based cluster formation induces very little error in the thermal contribution to the Gibbs free energy, with no further substantial effect on the single-point energy, yet sufficiently reducing the computation cost (Elm and Mikkelsen, 2014). The electronic energies of M06-2X/6-31G(d,p) optimized structures were further corrected with the DLPNO-CCSD(T)/aug-cc-pVTZ method. To elucidate the role of PA in atmospheric particle formation, the influence of varying temperatures and monomer concentrations on SA-PA-NH3 clustering was examined using the Atmospheric Cluster Dynamics Code (ACDC; McGrath et al., 2012). The model cluster is (SA)s•(PA)p•(NH3)n (). The simulation box was set to 3 × 2, where “3” stands for the total number of acids (SA and PA) and “2” stands for the total number of NH3 molecules. The ACDC model, taking as input the thermodynamic data obtained from quantum chemical calculations, generates the time derivatives of all cluster concentrations and solves for the steady-state cluster distribution, using the MATLAB ode 15s routine for differential equations (Shampine and Reichelt, 1997). The time derivatives of cluster concentrations, also called birth–death equations, can be expressed as follows:
where Ci is the concentration of cluster i, βi,j is the collision coefficient of clusters i and j, and is the rate coefficient of cluster k evaporating into smaller clusters i and j. Qi and Si are the possible outside source term and sink term, respectively, for cluster i.
For two neutral clusters i and j, the collision coefficient under the assumption of hard-sphere and sticking collision was calculated according to the kinetic gas theory as
where mi and Vi are the respective mass and volume of cluster i, kB is the Boltzmann constant and T is the absolute temperature. The volume is calculated from atomic masses and densities of the compounds in the cluster.
The rate coefficient of the i+j cluster evaporating to i and j clusters was derived as
where Pref is the reference pressure at which Gibbs free energies are calculated, ΔGi is the Gibbs free energy of formation of cluster i from monomers. Further details on collision rate coefficients and evaporation rate coefficients evaluation as well as on ACDC simulations can be obtained from previous studies (McGrath et al., 2012; Ortega et al., 2012; Olenius et al., 2013a, b).
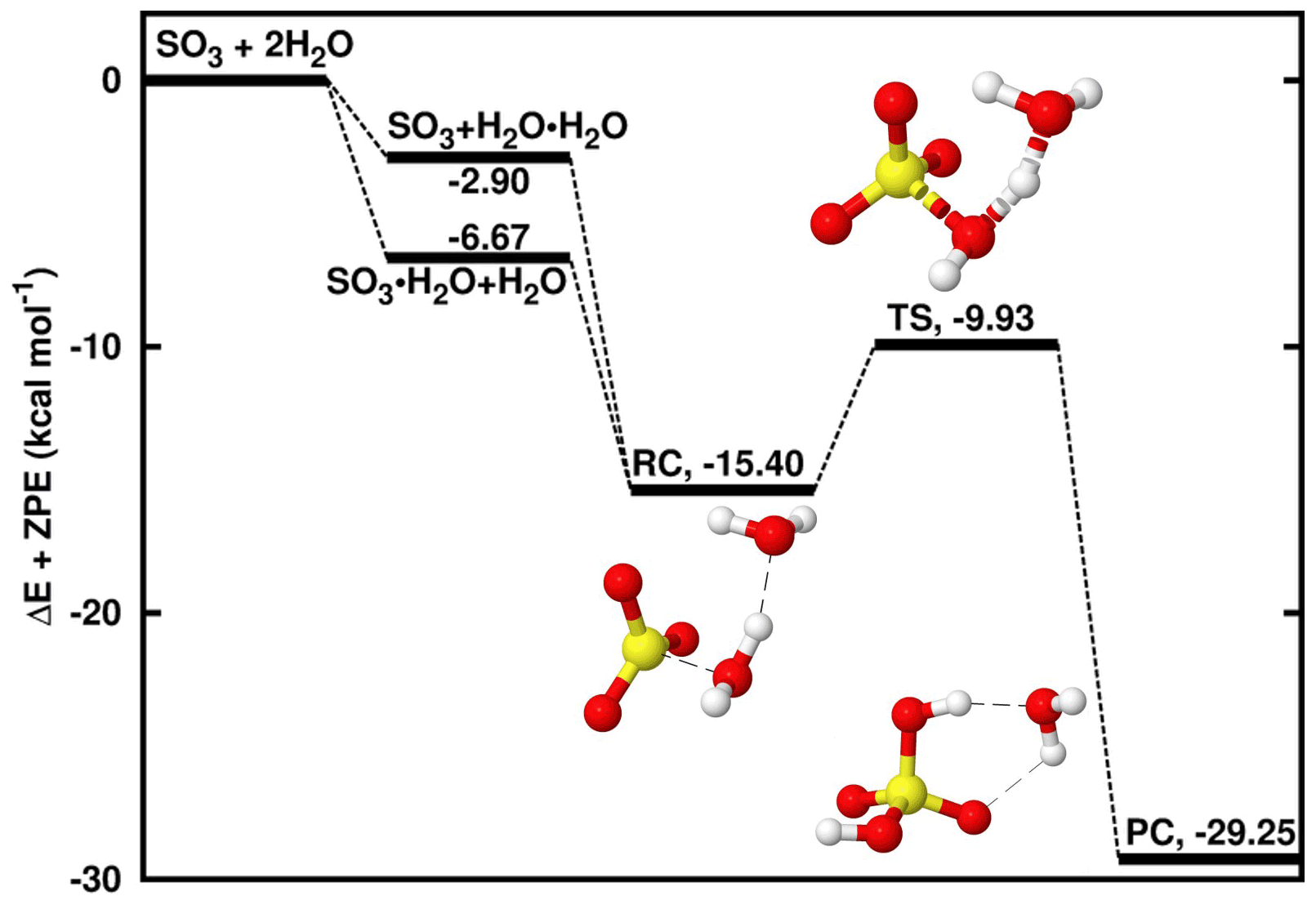
Figure 1Energy surface for the water-catalyzed SO3 hydrolysis. “RC” stands for pre-reactive intermediate; “TS” is transition state and “PC” is product complex. Color coding is yellow for sulfur atom, red for oxygen atom and white for hydrogen atom. Electronic energy values of all intermediates are indicated, and corresponding Gibbs free values are given in Table S4 in the Supplement.
3.1 Water-catalyzed SO3 hydrolysis
A number of studies have been dedicated to the SO3 + H2O → H2SO4 reaction, which was shown to be prevented by an electronic energy barrier as high as ∼ 30 kcal mol−1 under relevant atmospheric conditions. The presence of a second H2O molecule in this reaction lowers the energy barrier by favoring the formation of two kinds of binary hydrogen-bonded complexes, SO3⋯H2O and H2O⋯H2O, which can interact thereafter with the third species, H2O and SO3, respectively, to form a ternary pre-reactive intermediate, SO3⋯H2O⋯H2O. Indeed, due to the difficulty of termolecular interactions relative to biomolecular interactions (Buszek et al., 2012), the most likely situation is the interaction of two species to form a two-body complex followed by the interaction with the third species to form the pre-reactive intermediate. As shown on the energy surface in Fig. 1, though the formation of SO3⋯H2O is slightly more favorable than H2O⋯H2O formation, both complexes form SO3⋯H2O⋯H2O, henceforth denoted RC, that lies at 8.73 kcal mol−1 electronic energy below SO3⋯H2O and 12.50 kcal mol−1 below H2O⋯H2O. These energies are respectively within 0.17–1.81 and 0.47–1.06 kcal mol−1, similar to previously reported values for the same reaction (Hazra and Sinha, 2011; Torrent-Sucarrat et al., 2012; Long et al., 2013; Lv et al., 2019). The slight observed differences likely result from the differences in the computational approaches used in these studies. This intermediate overcomes an electronic energy barrier of 5.47 kcal mol−1 to form H2SO4⋯H2O. This energy barrier is ∼ 25 kcal mol−1 lower than the barrier in the reaction without the second water molecule and in good agreement with previous results (Torrent-Sucarrat et al., 2012; Hazra and Sinha, 2011; Long et al., 2013; Lv et al., 2019). The underlying mechanism is similar to those previously reported for the same reaction and is characterized by two hydrogen atom transfers to and from the second water molecule that acts as the catalyst.
The unimolecular decomposition of SO3⋯H2O⋯H2O to form H2SO4⋯H2O occurs at a rate constant of 1.35 × 108 s−1 at 298 K and, taking into account the collision and evaporation processes driving the formation of the binary complexes and SO3⋯H2O⋯H2O, the overall rate constant of the water-catalyzed SO3 hydrolysis at 298 K is determined to be 1.09 × 10−32 .
3.2 Pyruvic acid-catalyzed SO3 hydrolysis
Despite the demonstrated catalytic effect of water on SO3 hydrolysis, comparison of previous studies demonstrates that as many as four water molecules could be needed to achieve similar results to those obtained when a single molecule of other species is used as catalyst (Hazra and Sinha, 2011; Torrent-Sucarrat et al., 2012; Long et al., 2013; Lv et al., 2019; Daub et al., 2020; Larson et al., 2000). Some of these species include carboxylic acids, sulfuric acid and nitric acid. From these studies, while the electronic energy barrier could be reduced to ∼ 5.5 kcal mol−1 with water as the catalyst, it could be reduced to 3.7, 1.4, 0.6 and around 1 kcal mol−1, respectively, with HNO3, H2SO4, HCOOH and HOOC-COOH as catalysts. These results highlight the catalytic strength of carboxylic acids over other catalysts and further suggest that a second carboxyl function can have additional catalytic effects on the energy barrier.
In addition to the carboxyl group, PA possesses a ketone function at the α position. Church et al. have identified four stable conformational structures for PA that mainly differ by the orientation of the methyl group relative to the acidic OH group and that of the hydroxyl H atom relative to the ketone group, leading to trans-cis (Tc), trans-trans (Tt), cis-trans (Ct) and cis-cis (Cc) conformers, denoted as PATc, PATt, PACt and PACc conformers, respectively. These structures are shown in Fig. S1 in the Supplement, along with their energies given relative to the energy of the most stable conformer, PATc (Church et al., 2020). Only three of these PA conformations were able to form complexes with water: PATc, PATt and PACt. We also note that during geometry optimization, PATc could interact with H2O and be converted into PATt, forming the binary PATt⋯H2O complex. Henceforth, the PATt⋯H2O will represent the complex resulting from PATc + H2O and PATt + H2O interactions.
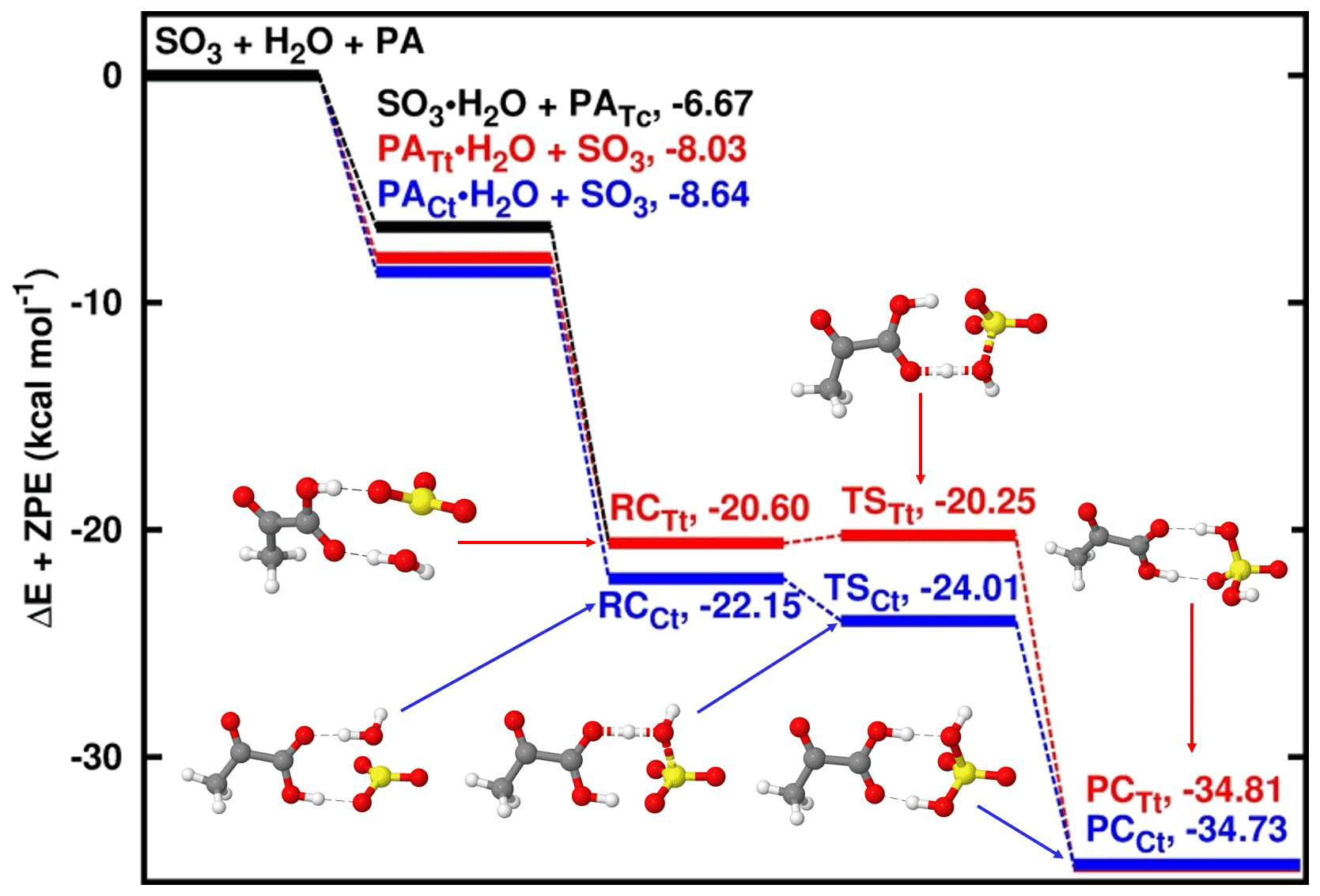
Figure 2Energy surfaces for the pyruvic-acid-catalyzed SO3 hydrolysis. “RC” stands for pre-reactive intermediate; “TS” is transition state and “PC” is product complex. Color coding is yellow for sulfur atom, red for oxygen atom, grey for carbon atom and white for hydrogen atom. Colored lines indicate the different paths involving the trans-trans and cis-trans conformers of pyruvic acid. Electronic energy values of all intermediates are indicated, and corresponding Gibbs free values are given in Table S4 in the Supplement.
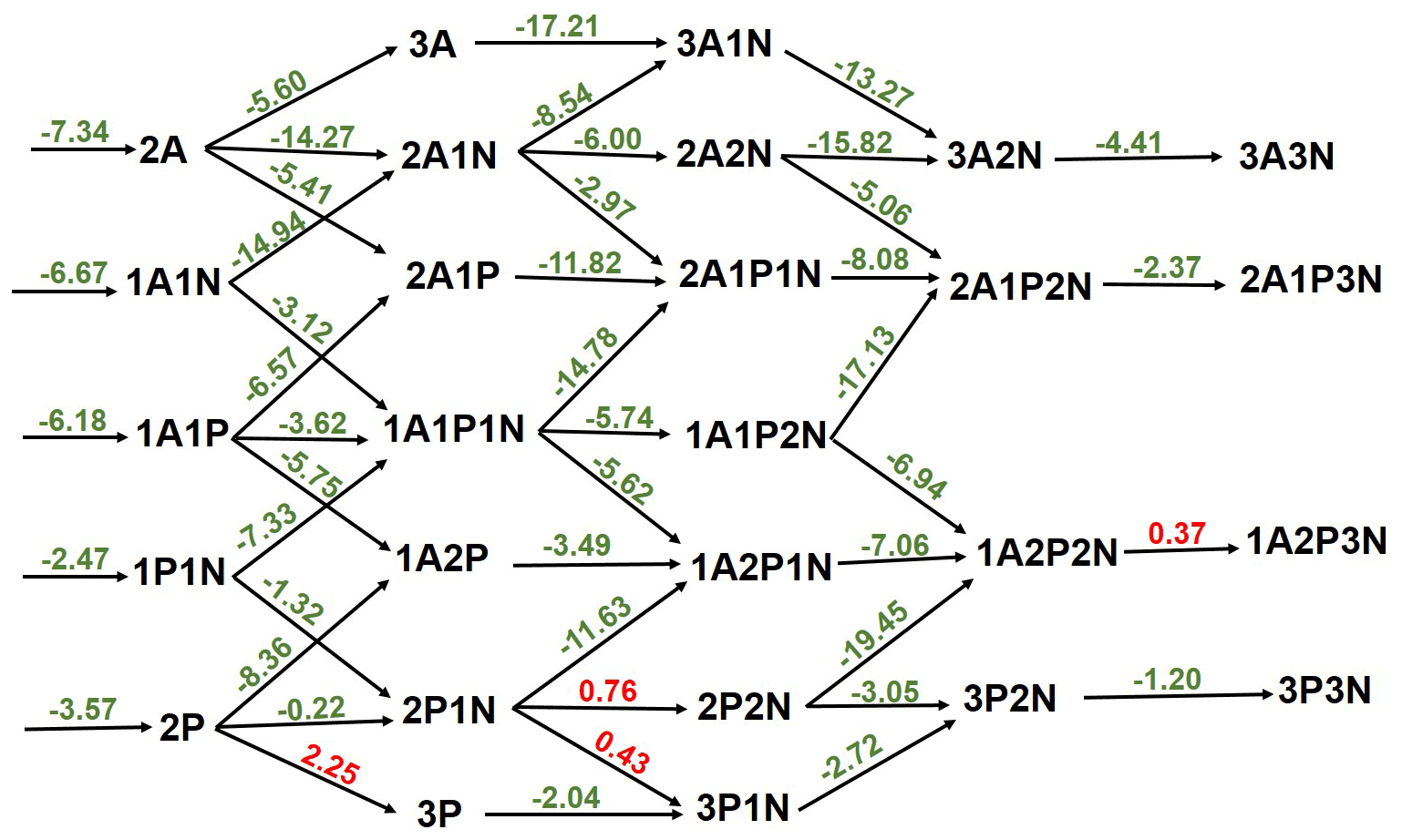
Figure 3Gibbs free-energy (in kcal mol−1) diagram of sulfuric-acid–pyruvic-acid–ammonia clusters at 298 K and 1 atm. “A” refers to sulfuric acid, “P” refers to pyruvic acid and “N” refers to ammonia. Green color indicates exergonic processes and red color indicates endergonic processes.
Similar to the SO3 hydrolysis where water acts as the catalyst, the reaction with PA acting as catalyst proceeds by formation of SO3⋯H2O and PA⋯H2O complexes prior to the formation of the PA⋯H2O⋯SO3 pre-reactive intermediate (see Fig. 2). While the pre-reactive intermediates formed with PATt and PACt conformers have almost equal electronic energy of formation, the PATt conformer is 0.23 kcal mol−1 more stable than the PACt conformer with respect to the Gibbs free energy at ambient conditions. Regardless of the PA conformation, the transformation of the pre-reactive intermediate to form PA⋯SA follows hydrogen transfer mechanisms where the hydrogen atom transfers from water to PA and then from PA to SO3, releasing the PA⋯SA complex, similar to the mechanisms where water, formic acid and oxalic acid act as catalysts. PATt and PACt conformers exhibit comparable binding strength with water at 1 atm and 298 K, with 2.75 and 2.29 kcal mol−1 Gibbs free-energy changes, respectively. This is a somewhat more favorable binding than the SO3 binding with water whose Gibbs free-energy change is 3.37 kcal mol−1 at similar conditions. It is obvious that the formation of PA⋯H2O⋯SO3 pre-reactive intermediate would preferably proceed through PA⋯H2O + SO3 rather than PA + H2O⋯SO3, although both interactions would lead to the same pre-reactive intermediate. Moreover, to evaluate the impact of each of these paths to the formation of the pre-reactive intermediate, we determined that among the two hydrates, PA⋯H2O will contribute by more than 99.99 % while SO3⋯H2O will contribute by less than 0.01 % to the formation of the pre-reactive intermediate regardless of the PA conformer (details are given in the Supplement).
The PATt⋯H2O⋯SO3 pre-reactive intermediate is formed with 0.84 and 0.24 kcal mol−1 Gibbs free-energy changes relative to PATt⋯H2O + SO3 and PATt + SO3⋯H2O interactions, respectively. While there is an electronic energy barrier as low as 0.45 kcal mol−1 separating PATt⋯H2O⋯SO3 from the product, this transformation is rather thermodynamically driven, with a negative Gibbs free-energy barrier of −0.92 kcal mol−1 and the product complex lying at 13.30 Gibbs free energy below PATt⋯H2O + SO3. Note that in the transformation of PATt⋯H2O⋯SO3 to the product complex (PCTt), the PATt conformer is isomerized to PACt. This phenomenon has also been observed in SO3 hydrolysis reactions catalyzed by oxalic acid (Lv et al., 2019).
The PACt⋯H2O⋯SO3 pre-reactive intermediate is formed with 0.20 and −0.86 kcal mol−1 Gibbs free-energy changes relative to PACt⋯H2O + SO3 and PACt + SO3⋯H2O interactions, respectively, and is separated from the product complex by a 0.09 kcal mol−1 electronic energy barrier. The negative Gibbs free-energy barrier of −1.15 kcal mol−1 in this path indicates the thermodynamically driven transformation process. This path is somewhat more favorable than the path catalyzed by PATt at atmospheric pressure and 298 K. Similar to the path catalyzed by PATt, PACt is isomerized to PATt during the PACt⋯H2O⋯SO3 → PCCt transformation process.
Based on the transition state energies, the unimolecular decompositions of PATt⋯H2O⋯SO3 and PACt⋯H2O⋯SO3 pre-reactive intermediates were determined to be 9.03 × 1011 and 1.80 × 1012 s−1, respectively, at 298 K. Corresponding overall rate constants for the PA-catalyzed SO3 + H2O reaction, calculated according to Eq. (1), are 2.95 × 10−27 and 3.52 × 10−26 , respectively. Considering the Gibbs free energies of PATc, PATt and PACt, and applying the law of mass action on the equilibria between PATc, PATt and PACt, the determined relative equilibrium distribution of PA conformers is 0.95 (PATc), 0.04 (PATt) and 0.01 (PACt) at 298 K (Table S1 in the Supplement). Considering this distribution, the weighted average rate constant of PA-catalyzed SO3 hydrolysis at 298 K is determined to be 3.10 × 10−27 , being 2.84 × 105 times higher than the rate constant of the water-catalyzed SO3 hydrolysis at 298 K. This ratio is much higher than observed with other catalysts, for example, 1.19 with nitric acid (Long et al., 2013), 102 with sulfuric acid (Torrent-Sucarrat et al., 2012), 103 with oxalic acid (Lv et al., 2019) and 104 with formic acid (Hazra and Sinha, 2011). However, given the low atmospheric concentrations of these species relative to water, the effective rate of the SO3 hydrolysis reactions where they act as catalysts are not high enough for them to effectively compete with the water-catalyzed reaction. For a reaction to compete with the water-catalyzed SO3 hydrolysis, not only should the catalyst be efficient in facilitating the hydrogen transfer between H2O and SO3, but its concentration must be high enough to cause a higher SA formation rate than the water-catalyzed reaction.

Figure 4Simulated steady-state clusters distribution in the sulfuric-acid–pyruvic-acid–ammonia system at 238 K, under different initial monomers concentrations. “A” refers to sulfuric acid, “P” refers to pyruvic acid and “N” refers to ammonia.
Comparing the rate constant of the PA-catalyzed SO3 hydrolysis reaction to the rate constants of the reaction catalyzed by other species mentioned above, it is obvious that PA is the most efficient catalyst. Consequently, PA may be an efficient partaker in SO3 hydrolysis in the atmosphere. To effectively compare the different catalytic effects of PA and water on the SO3 + H2O reaction, it is important to compare the overall rates of SA formation (given in Eq. 1) that take into account the concentrations of the catalysts. From Eq. (1), the ratio of the rates of SO3 hydrolysis reactions catalyzed by PA and water can be expressed as
where α is a measure of the relative efficiency of different catalysts and kx is the overall rate constant of the X-catalyzed SO3 hydrolysis (X = PA, H2O), given in Eq. (1). Assuming water concentrations within 1015–1017 that cover dry and humid conditions, and PA concentrations in the range 109–1011 that cover clean and polluted environments, our results show that the efficiency of PA as a catalyst lies in the range ∼ 10−2–102 relative to water. The H2O-catalyzed SO3 hydrolysis remains the main SO3 loss pathway under humid conditions, whereas the PA-catalyzed SO3 hydrolysis would be the dominant path under dry conditions and in polluted areas where PA concentrations can reach the ppb levels. It is estimated that under such conditions, the PA-catalyzed reaction can be around 2 orders of magnitude more efficient than the H2O-catalyzed SO3 hydrolysis to form sulfuric acid. This shows, in regard to the relatively high PA concentration and the high rate constant of the PA-catalyzed SO3 hydrolysis, that this reaction is more effective for SA production than reactions catalyzed by formic acid, sulfuric acid and oxalic acid. Given the renowned role of sulfuric acid in aerosol particle formation, the PA-catalyzed SO3 hydrolysis, stabilized as the PA⋯SA complex product, might provide an additional pathway for incorporating organic matter into aerosol particles.
3.3 Cluster thermodynamics and dynamic simulations
3.3.1 Cluster thermodynamics
The thermodynamics of further SA-PA clustering, with and without ammonia (NH3), was examined. In general, the cluster formation is thermodynamically favorable at various tropospheric temperatures as can be seen in Table S5 in the Supplement. The binding of PA to SA exhibits similar strength within 1 kcal mol−1 to the binding between two SA molecules, though this binding is weakened in the presence of NH3, likely as a result of the weaker acid nature of PA than SA. Figure 3 exhibits the free-energy diagram for cluster formation steps in the SA-PA-NH3 system at 298 K and 1 atm, and it shows that the stepwise formation of the studied clusters is in general exergonic, with only four moderate endergonic processes involving PA and NH3 additions. According to our thermodynamic data, PA additions to (PA)2 and (PA)2•NH3 are hindered by thermodynamic barriers of 2.25 and 0.43 kcal mol−1, respectively, while NH3 additions to (PA)2•NH3 and SA•(PA)2•(NH3)2 are hindered by 0.76 and 0.37 kcal mol−1 thermodynamic barriers, respectively. Similar additions to clusters containing SA instead of PA are much more exergonic, and this is as expected given the strong binding between SA and NH3, as compared with the binding between PA and NH3. The cluster formation depicted in the energy diagram of Fig. 3 is based on direct quantum chemical data at 298 K and 1 atm and does not take into account the actual concentrations of monomers. However, such processes in the real atmosphere depend on actual atmospheric conditions such as temperature and the concentrations of monomers participating in the process
3.3.2 Steady-state cluster concentrations and cluster formation rates
Time-dependent cluster concentrations were determined by solving the birth–death equations given in Eq. (2), using ACDC, under given concentrations of monomers. We found that under [SA] = 106–108 , [PA] = 107–1010 and [NH3] = 108–1011 conditions, the largest PA-containing cluster is (SA)2•PA•(NH3)2, with the highest concentration observed under 238 K, [SA] = 106 , [PA] = 1010 and [NH3] = 1011 conditions. This concentration is, however, still as low as ∼ 10 cm−3. Cluster equilibrium concentrations at selected representative conditions are shown in Fig. 4. Previous studies also found that in sulfuric-acid-based particle formation enhanced by methane sulfonic acid (MSA) and trifluoroacetic acid (TFA), the main cluster contributing to particle formation would bind a single molecule of MSA or TFA (Bork et al., 2014; Lu et al., 2020). This is due to the particular binding strength between the base molecule and SA, as compared to the binding with other acids.
The difficulty to form large clusters containing PA likely result from the low concentration of heterotrimers and tetramers containing PA. For example, it is seen from Fig. 4 that (SA)2•PA, SA•PA•NH3, SA•PA•(NH3)2 and (SA)2•PA•(NH3) all have steady-state concentrations below 10 cm−3, although the concentrations of heterodimers containing PA (SA•PA and PA•NH3) can reach 105 cm−3. These heterodimers likely undergo faster evaporation than, for example, SA•SA and SA•NH3, hence not effectively contributing to further growth. The highest concentrations of PA-containing clusters are found at low temperatures, exclusively, where the evaporation rates are reduced.
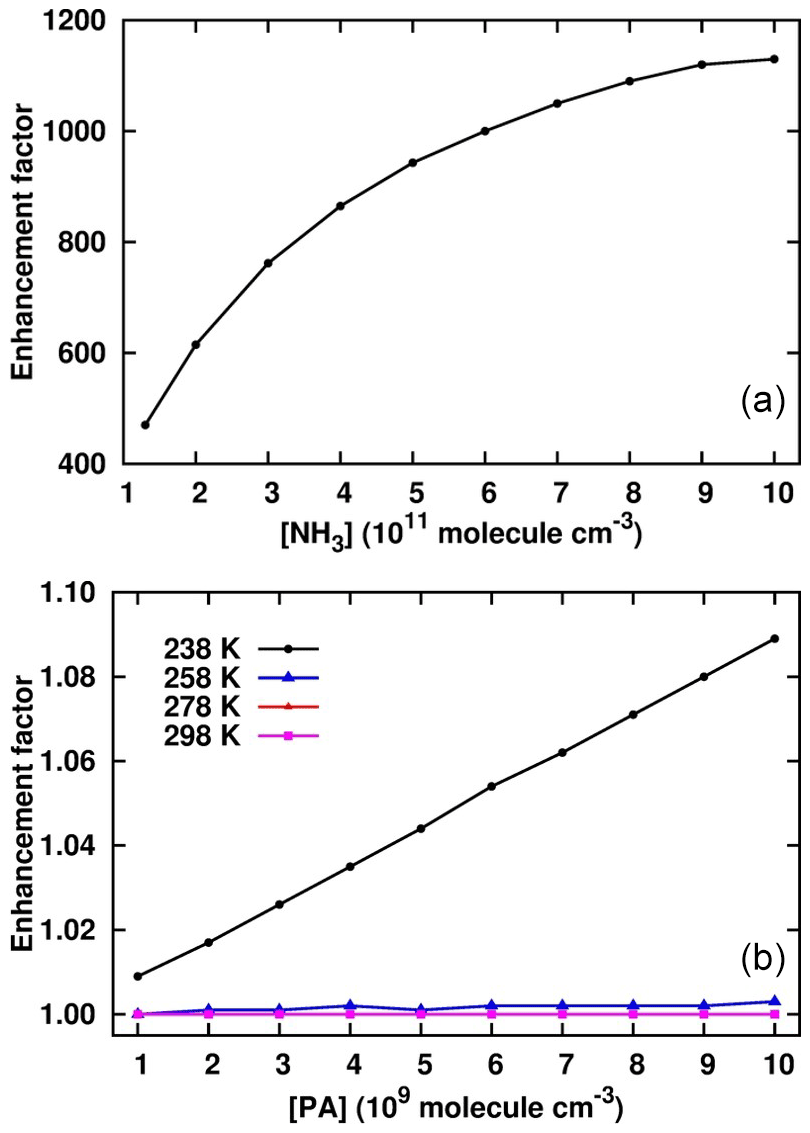
Figure 5Enhancement of PA in the cluster formation rate in the sulfuric-acid–pyruvic-acid–ammonia clusters at [SA] = 106 , [NH3] = 1010 , [PA] = 109–1010 and different temperatures (b), and T = 238 K, [SA] = 106 , [PA] = 1010 , [NH3] = 1011–1012 (a).
The contribution of PA to the particle formation was estimated by calculating the enhancement factor as
where J is the cluster formation rate calculated at fixed SA concentration (106 ), x = 107–1010 and y = 1010–1012 . Figure 5 shows the enhancement factor as a function of [PA] and [NH3]. At a fixed NH3 concentration of 1010 , the enhancement factor weakly increases with PA concentration within the temperature range considered. The trend was observed to be similar at lower and higher PA and NH3 concentrations, even when SA concentration increased to reach 108 . The only condition that could lead to significant PA enhancements was observed for [NH3] > 1011 and 238 K. Figure 5 shows that for 1011 < [NH3] < 1012 , the enhancement factor was much more pronounced, ranging between 4.7 × 102 and 1.15 × 103. Though this enhancement factor is much lower at warmer temperatures, it is obvious that PA would actively promote sulfuric-acid-based particle formation in NH3-polluted and cold environments. This is as expected since the promotion of new particle formation at cold temperatures has previously been evidenced (Lu et al., 2020; Liu et al., 2021). The implication of PA as participating agent in aerosol formation models would definitely reduce the errors existing in current aerosol models.
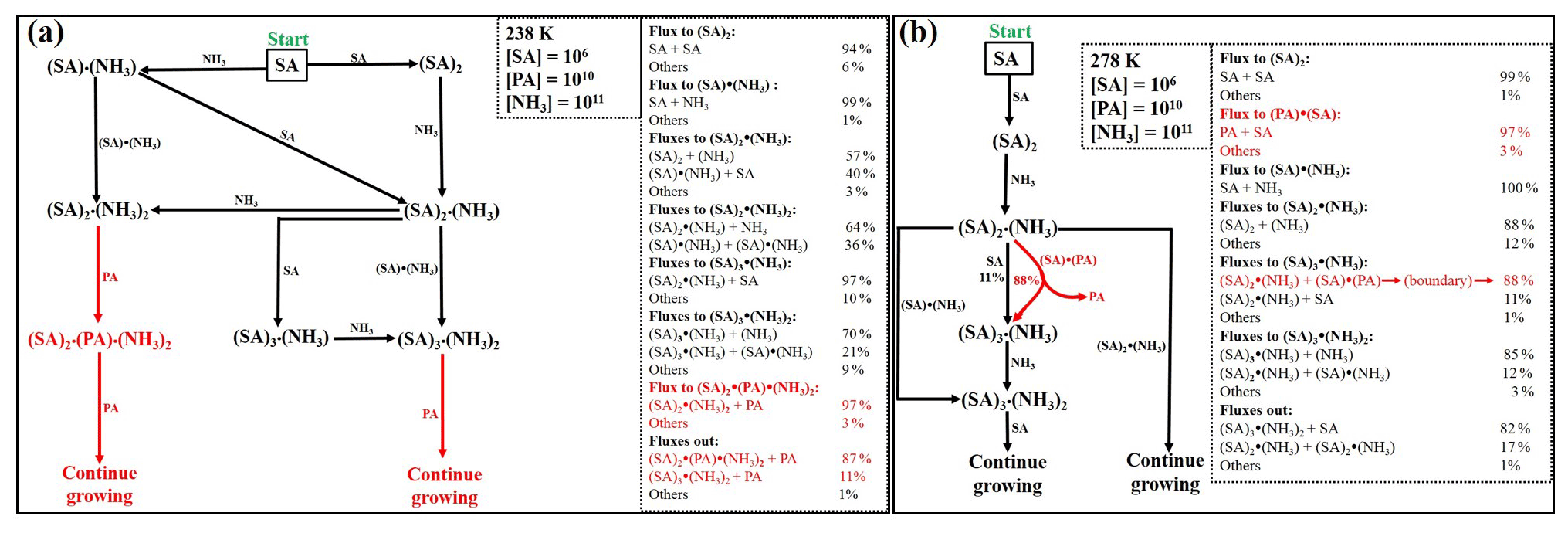
Figure 6Main cluster pathways at (a) 238 and (b) 278 K. Monomer concentrations are [SA] = 106 , [PA] = 1010 and [NH3] = 1011 . Red color indicates the path involving PA.
3.3.3 Cluster formation pathways
Gibbs free energies in Fig. 3 give information on whether the cluster formation is thermodynamically favorable at the reference pressure (1 atm); however, the concentrations of the clustering species participating in the process are not taken into account. The actual molecular clustering at given vapor concentrations and temperatures can be determined by performing ACDC simulations (McGrath et al., 2012; Olenius et al., 2013b). This is achieved by calculating the actual Gibbs free energies, that is, the vapor concentration-dependent Gibbs free energies of cluster formation, that are used to track the actual cluster formation pathways at given conditions. The temperatures considered in this study are 238, 258 and 278 K, which span most regions of the troposphere. Monomer concentrations were chosen to be [SA] = 106 , [PA] = 1010 , and [NH3] = 1011 and 5 × 1011 (Eger et al., 2020; Nair and Yu, 2020; Yao and Zhang, 2019; Zhang et al., 2021). As our simulation box size was set to 3 × 2, only clusters containing more than three acid molecules (SA and/or PA) with more than two NH3 molecules were allowed to grow out of the system and contribute to particle formation.
Clusters containing more than one PA molecule were found not to contribute to particle formation. While the cluster contribution to the growth is found to weakly depend on monomer concentrations, their temperature dependency is relatively stronger. Depending on the temperature, the clusters grew through the system via two main paths: one path involving pure SA-NH3 clusters and another one where PA also participates (see Figs. 6 and S2 in the Supplement). Cluster formation starts by SA collision with SA or NH3, forming (SA)2 or SA•NH3, followed by NH3 or SA addition to form (SA)2•(NH3). While PA can participate in the cluster formation as either a (PA)2 or SA•PA cluster, it can effectively contribute to the cluster growth at low temperature (238 K), exclusively. At higher temperatures (258 and 278 K), however, PA mainly evaporates from the clusters. At these temperatures, only (SA)3•(NH3)2 clusters will grow out of the system by clustering with SA and contribute to particle growth (See Figs. 6b and S2). At 238 K, the largest pure SA-NH3 clusters formed within the system are (SA)2•(NH3)2 and (SA)3•(NH3)2, and they grow out the system by consecutive uptake of PA monomers. The main interactions that contribute to particle formation with the participation of PA are (SA)3•(NH3)2 + PA and (SA)2•(PA)(NH3)2 + PA (see Fig. 6a).
We observed that with [SA] = 106 , [PA] = 1010 , and [NH3] =1011 , PA-containing clusters do not contribute to the particle formation at 258 and 278 K, whereas they predominantly contribute to the particle formation at 238 K, with ∼ 100 % of clusters growing out of the simulation box. It follows that under the investigated monomer concentration conditions, PA-containing clusters would completely dominate the particle formation at cold temperatures while pure SA-NH3 clusters will dominate at high temperatures.
The catalytic effect of pyruvic acid (PA) on SO3 hydrolysis to form sulfuric acid (SA) and its possible enhancement in atmospheric particle formation have been highlighted. Using quantum chemical calculations, we found that with PA as a catalyst, the SO3 hydrolysis occurs with a negative Gibbs free-energy barrier at 298 K and 1 atm, indicating a thermodynamically driven transformation process. Evaluation of the kinetics show that the rate constant of PA-catalyzed SO3 hydrolysis at 298 K is ∼ 3 × 105 times higher than that of water-catalyzed SO3 hydrolysis and 101–104 times higher than those of previously investigated SO3 hydrolysis processes with nitric acid, sulfuric acid, oxalic acid and formic acid acting as catalysts, hence highlighting the effective role of PA in the atmospheric chemistry of SO3. Overall, the determination of the reaction rates, taking into account the catalyst concentrations, indicates that water-catalyzed SO3 hydrolysis would be the main SO3 loss pathway to form sulfuric acid under humid conditions and clean areas, whereas PA-catalyzed SO3 hydrolysis would dominate the process under dry conditions and in polluted areas.
As the PA-catalyzed SO3 hydrolysis is highly stabilized by the formation of the SA•PA cluster, we further investigated the role of PA in the formation of SA-based molecular clusters. Using the quantum chemical data from our calculations and an Atmospheric Cluster Dynamics Code, we found that though PA is a weaker clustering agent to SA than SA itself, it effectively contributes to particle formation. Two main pathways were found to drive the cluster formation (one path forming pure SA-NH3 clusters and another one forming PA-containing clusters). Under [SA] = 106 , [PA] = 1010 and [NH3] = 1011 conditions, clusters containing more than one PA molecule were observed not to contribute to particle formation, and the main PA-containing cluster to contribute to particle formation is (SA)2•PA•(NH3)2 with the highest enhancement effect, 4.7 × 102, observed at 238 K and [NH3] = 1.3 × 1011 . This indicates that PA may actively participate in aerosol formation, especially in cold regions of the troposphere and highly NH3-polluted environments, and may readily be included in aerosol models.
All data from this research can be obtained upon request by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-1951-2022-supplement.
NTT designed the work and performed all quantum chemical calculations. LL performed the dynamic simulations. NTT, LD, LL and XZ analyzed the data. NTT wrote the paper. LD, LL and XZ reviewed and edited the paper.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We acknowledge the Wuxi Hengding Supercomputing Center Co., LTD for providing the computational resources.
This work was supported by the National Natural Science Foundation of China (grant no. 21876098), Shandong Society for Environmental Science (grant no. 202001), Youth Innovation Program of Universities in Shandong Province (grant no. 2019KJD007), and Fundamental Research Fund of Shandong University (grant no. 2020QNQT012).
This paper was edited by Timothy Bertram and reviewed by two anonymous referees.
Almeida, J., Schobesberger, S., Kuerten, A., Ortega, I. K., Kupiainen-Maatta, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurten, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppa, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petaja, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipila, M., Stozhkov, Y., Stratmann, F., Tome, A., Troestl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamaki, H., and Kirkby, J.: Molecular understanding of sulphuric acid-amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013.
Andreae, M. O., Talbot, R. W., and Li, S.-M.: Atmospheric measurements of pyruvic and formic acid, J. Geophys. Res.-Atmos., 92, 6635–6641, https://doi.org/10.1029/JD092iD06p06635, 1987.
Baboukas, E. D., Kanakidou, M., and Mihalopoulos, N.: Carboxylic acids in gas and particulate phase above the Atlantic Ocean, J. Geophys. Res.-Atmos., 105, 14459–14471, https://doi.org/10.1029/1999JD900977, 2000.
Bardouki, H., Liakakou, H., Economou, C., Sciare, J., Smolík, J., Ždímal, V., Eleftheriadis, K., Lazaridis, M., Dye, C., and Mihalopoulos, N.: Chemical composition of size-resolved atmospheric aerosols in the eastern Mediterranean during summer and winter, Atmos. Environ., 37, 195–208, https://doi.org/10.1016/S1352-2310(02)00859-2, 2003.
Bork, N., Kurtén, T., and Vehkamäki, H.: Exploring the atmospheric chemistry of and assessing the maximum turnover number of ion-catalysed H2SO4 formation, Atmos. Chem. Phys., 13, 3695–3703, https://doi.org/10.5194/acp-13-3695-2013, 2013.
Bork, N., Elm, J., Olenius, T., and Vehkamäki, H.: Methane sulfonic acid-enhanced formation of molecular clusters of sulfuric acid and dimethyl amine, Atmos. Chem. Phys., 14, 12023–12030, https://doi.org/10.5194/acp-14-12023-2014, 2014.
Buszek, R. J., Barker, J. R., and Francisco, J. S.: Water Effect on the OH plus HCl Reaction, J. Phys. Chem. A, 116, 4712–4719, https://doi.org/10.1021/jp3025107, 2012.
Canneaux, S., Bohr, F., and Henon, E.: KiSThelP: a program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 35, 82–93, https://doi.org/10.1002/jcc.23470, 2014.
Chebbi, A. and Carlier, P.: Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review, Atmos. Environ., 30, 4233–4249, https://doi.org/10.1016/1352-2310(96)00102-1, 1996.
Church, J. R., Vaida, V., and Skodje, R. T.: Gas-Phase Reaction Kinetics of Pyruvic Acid with OH Radicals: The Role of Tunneling, Complex Formation, and Conformational Structure, J. Phys. Chem. A, 124, 790–800, https://doi.org/10.1021/acs.jpca.9b09638, 2020.
Daub, C. D., Riccardi, E., Hänninen, V., and Halonen, L.: Path sampling for atmospheric reactions: formic acid catalysed conversion of SO3 + H2O to H2SO4, PeerJ Physical Chemistry, 2, e7, https://doi.org/10.7717/peerj-pchem.7, 2020.
Duchovic, R. J., Pettigrew, J. D., Welling, B., and Shipchandler, T.: Conventional transition state theory/Rice–Ramsperger–Kassel–Marcus theory calculations of thermal termolecular rate coefficients for H(D) + O2 + M, J. Chem. Phys., 105, 10367–10379, https://doi.org/10.1063/1.47299, 1996.
Eger, P. G., Schuladen, J., Sobanski, N., Fischer, H., Karu, E., Williams, J., Riva, M., Zha, Q., Ehn, M., Quéléver, L. L. J., Schallhart, S., Lelieveld, J., and Crowley, J. N.: Pyruvic acid in the boreal forest: gas-phase mixing ratios and impact on radical chemistry, Atmos. Chem. Phys., 20, 3697–3711, https://doi.org/10.5194/acp-20-3697-2020, 2020.
Eisenreich, W., Rohdich, F., and Bacher, A.: Deoxyxylulose phosphate pathway to terpenoids, Trends Plant Sci., 6, 78–84, https://doi.org/10.1016/S1360-1385(00)01812-4, 2001.
Elm, J. and Mikkelsen, K. V.: Computational approaches for efficiently modelling of small atmospheric clusters, Chem. Phys. Lett., 615, 26–29, https://doi.org/10.1016/j.cplett.2014.09.060, 2014.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams, Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, Ö., Foresman, J. B., and Fox, D. J.: Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2013.
Fukui, K.: The path of chemical reactions-the IRC approach, Accounts Chem. Res., 14, 363–368, 1981.
Hazra, M. K. and Sinha, A.: Formic acid catalyzed hydrolysis of SO3 in the gas phase: a barrierless mechanism for sulfuric acid production of potential atmospheric importance, J. Am. Chem. Soc., 133, 17444–17453, https://doi.org/10.1021/ja207393v, 2011.
Helas, G., Bingemer, H., and Andreae, M. O.: Organic acids over equatorial Africa: Results from DECAFE 88, J. Geophys. Res.-Atmos., 97, 6187–6193, https://doi.org/10.1029/91JD01438, 1992.
Henschel, H., Navarro, J. C. A., Yli-Juuti, T., Kupiainen-Määttä, O., Olenius, T., Ortega, I. K., Clegg, S. L., Kurtén, T., Riipinen, I., and Vehkamäki, H.: Hydration of Atmospherically Relevant Molecular Clusters: Computational Chemistry and Classical Thermodynamics, J. Phys. Chem. A, 118, 2599–2611, https://doi.org/10.1021/jp500712y, 2014.
Hofmann, M. and Schleyer, P. v. R.: Acid Rain: Ab Initio Investigation of the H2O.SO3 Complex and Its Conversion to H2SO4, J. Am. Chem. Soc., 116, 4947–4952, https://doi.org/10.1021/ja00090a045, 1994.
Hofmann-Sievert, R. and Castleman, A. W.: Reaction of sulfur trioxide with water clusters and the formation of sulfuric acid, J. Phys. Chem., 88, 3329–3333, https://doi.org/10.1021/j150659a038, 1984.
Holland, P. M. and Castleman, A. W.: Gas phase complexes: considerations of the stability of clusters in the sulfur trioxide-water system, Chem. Phys. Lett., 56, 511–514, https://doi.org/10.1016/0009-2614(78)89028-9, 1978.
Jardine, K. J., Sommer, E. D., Saleska, S. R., Huxman, T. E., Harley, P. C., and Abrell, L.: Gas Phase Measurements of Pyruvic Acid and Its Volatile Metabolites, Environ. Sci. Technol., 44, 2454–2460, https://doi.org/10.1021/es903544p, 2010.
Jayne, J. T., Pöschl, U., Chen, Y.-m., Dai, D., Molina, L. T., Worsnop, D. R., Kolb, C. E., and Molina, M. J.: Pressure and Temperature Dependence of the Gas-Phase Reaction of SO3 with H2O and the Heterogeneous Reaction of SO3 with H2O/H2SO4 Surfaces, J. Phys. Chem. A, 101, 10000–10011, https://doi.org/10.1021/jp972549z, 1997.
Jenkin, M. E., Cox, R. A., Emrich, M., and Moortgat, G. K.: Mechanisms of the Cl-atom-initiated oxidation of acetone and hydroxyacetone in air, J. Chem. Soc. Faraday T., 89, 2983–2991, https://doi.org/10.1039/FT9938902983, 1993.
Kawamura, K. and Bikkina, S.: A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation, Atmos. Res., 170, 140–160, https://doi.org/10.1016/j.atmosres.2015.11.018, 2016.
Kawamura, K., Kasukabe, H., and Barrie, L. A.: Source and reaction pathways of dicarboxylic acids, ketoacids and dicarbonyls in arctic aerosols: One year of observations, Atmos. Environ., 30, 1709–1722, https://doi.org/10.1016/1352-2310(95)00395-9, 1996.
Kawamura, K., Tachibana, E., Okuzawa, K., Aggarwal, S. G., Kanaya, Y., and Wang, Z. F.: High abundances of water-soluble dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in the mountaintop aerosols over the North China Plain during wheat burning season, Atmos. Chem. Phys., 13, 8285–8302, https://doi.org/10.5194/acp-13-8285-2013, 2013.
Kuang, C., McMurry, P., McCormick, A., and Eisele, F.: Dependence of nucleation rates on sulfuric acid vapor concentration in diverse atmospheric locations, J. Geophys. Res.-Atmos., 113, D10209, https://doi.org/10.1029/2007JD009253, 2008.
Kulmala, M.: How particles nucleate and grow, Science, 302, 1000–1001, 2003.
Kulmala, M., Pirjola, L., and Mäkelä, J. M.: Stable sulphate clusters as a source of new atmospheric particles, Nature, 404, 66–69, https://doi.org/10.1038/35003550, 2000.
Larson, L. J., Kuno, M., and Tao, F.-M.: Hydrolysis of sulfur trioxide to form sulfuric acid in small water clusters, J. Chem. Phys., 112, 8830–8838, 2000.
Li, H., Zhong, J., Vehkamäki, H., Kurtén, T., Wang, W., Ge, M., Zhang, S., Li, Z., Zhang, X., Francisco, J. S., and Zeng, X. C.: Self-Catalytic Reaction of SO3 and NH3 To Produce Sulfamic Acid and Its Implication to Atmospheric Particle Formation, J. Am. Chem. Soc., 140, 11020–11028, https://doi.org/10.1021/jacs.8b04928, 2018.
Liu, L., Zhong, J., Vehkamäki, H., Kurtén, T., Du, L., Zhang, X., Francisco, J. S., and Zeng, X. C.: Unexpected quenching effect on new particle formation from the atmospheric reaction of methanol with SO3, P. Natl. Acad. Sci. USA, 116, 24966, https://doi.org/10.1073/pnas.1915459116, 2019.
Liu, L., Yu, F., Tu, K., Yang, Z., and Zhang, X.: Influence of atmospheric conditions on the role of trifluoroacetic acid in atmospheric sulfuric acid–dimethylamine nucleation, Atmos. Chem. Phys., 21, 6221–6230, https://doi.org/10.5194/acp-21-6221-2021, 2021.
Loerting, T. and Liedl, K. R.: Toward elimination of discrepancies between theory and experiment: The rate constant of the atmospheric conversion of SO3 to H2SO4, P. Natl. Acad. Sci. USA, 97, 8874, https://doi.org/10.1073/pnas.97.16.8874, 2000.
Long, B., Chang, C.-R., Long, Z.-W., Wang, Y.-B., Tan, X.-F., and Zhang, W.-J.: Nitric acid catalyzed hydrolysis of SO3 in the formation of sulfuric acid: A theoretical study, Chem. Phys. Lett., 581, 26–29, https://doi.org/10.1016/j.cplett.2013.07.012, 2013.
Lu, Y., Liu, L., Ning, A., Yang, G., Liu, Y., Kurtén, T., Vehkamäki, H., Zhang, X., and Wang, L.: Atmospheric Sulfuric Acid-Dimethylamine Nucleation Enhanced by Trifluoroacetic Acid, Geophys. Res. Lett., 47, e2019GL085627, https://doi.org/10.1029/2019GL085627, 2020.
Lv, G., Sun, X., Zhang, C., and Li, M.: Understanding the catalytic role of oxalic acid in SO3 hydration to form H2SO4 in the atmosphere, Atmos. Chem. Phys., 19, 2833–2844, https://doi.org/10.5194/acp-19-2833-2019, 2019.
Magel, E., Mayrhofer, S., Müller, A., Zimmer, I., Hampp, R., and Schnitzler, J. P.: Photosynthesis and substrate supply for isoprene biosynthesis in poplar leaves, Atmos. Environ., 40, 138–151, https://doi.org/10.1016/j.atmosenv.2005.09.091, 2006.
Mattila, J. M., Brophy, P., Kirkland, J., Hall, S., Ullmann, K., Fischer, E. V., Brown, S., McDuffie, E., Tevlin, A., and Farmer, D. K.: Tropospheric sources and sinks of gas-phase acids in the Colorado Front Range, Atmos. Chem. Phys., 18, 12315–12327, https://doi.org/10.5194/acp-18-12315-2018, 2018.
Mauldin III, R., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V.-M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, https://doi.org/10.1038/nature11278, 2012.
McGrath, M. J., Olenius, T., Ortega, I. K., Loukonen, V., Paasonen, P., Kurtén, T., Kulmala, M., and Vehkamäki, H.: Atmospheric Cluster Dynamics Code: a flexible method for solution of the birth-death equations, Atmos. Chem. Phys., 12, 2345–2355, https://doi.org/10.5194/acp-12-2345-2012, 2012.
Mellouki, A. and Mu, Y.: On the atmospheric degradation of pyruvic acid in the gas phase, J. Photoch. Photobio. A, 157, 295–300, https://doi.org/10.1016/S1010-6030(03)00070-4, 2003.
Morokuma, K. and Muguruma, C.: Ab initio Molecular Orbital Study of the Mechanism of the Gas Phase Reaction SO3 + H2O: Importance of the Second Water Molecule, J. Am. Chem. Soc., 116, 10316–10317, https://doi.org/10.1021/ja00101a068, 1994.
Nair, A. A. and Yu, F.: Quantification of Atmospheric Ammonia Concentrations: A Review of Its Measurement and Modeling, Atmosphere, 11, 1092, https://doi.org/10.3390/atmos11101092, 2020.
Olenius, T., Kupiainen-Määttä, O., Ortega, I. K., Kurtén, T., and Vehkamäki, H.: Free energy barrier in the growth of sulfuric acid–ammonia and sulfuric acid–dimethylamine clusters, J. Chem. Phys., 139, 084312, https://doi.org/10.1063/1.4819024, 2013a.
Olenius, T., Schobesberger, S., Kupiainen-Määttä, O., Franchin, A., Junninen, H., Ortega, I. K., Kurtén, T., Loukonen, V., Worsnop, D. R., Kulmala, M., and Vehkamäki, H.: Comparing simulated and experimental molecular cluster distributions, Faraday Discuss., 165, 75–89, https://doi.org/10.1039/C3FD00031A, 2013b.
Ortega, I. K., Kupiainen, O., Kurtén, T., Olenius, T., Wilkman, O., McGrath, M. J., Loukonen, V., and Vehkamäki, H.: From quantum chemical formation free energies to evaporation rates, Atmos. Chem. Phys., 12, 225–235, https://doi.org/10.5194/acp-12-225-2012, 2012.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kroll, J. H., Seinfeld, J. H., and Wennberg, P. O.: Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 9, 1479–1501, https://doi.org/10.5194/acp-9-1479-2009, 2009.
Peng, C., Ayala, P. Y., Schlegel, H. B., and Frisch, M. J.: Using redundant internal coordinates to optimize equilibrium geometries and transition states, J. Comput. Chem., 17, 49–56, https://doi.org/10.1002/(SICI)1096-987X(19960115)17:1<49::AID-JCC5>3.0.CO;2-0, 1996.
Praplan, A. P., Hegyi-Gaeggeler, K., Barmet, P., Pfaffenberger, L., Dommen, J., and Baltensperger, U.: Online measurements of water-soluble organic acids in the gas and aerosol phase from the photooxidation of 1,3,5-trimethylbenzene, Atmos. Chem. Phys., 14, 8665–8677, https://doi.org/10.5194/acp-14-8665-2014, 2014.
Reed Harris, A. E., Doussin, J.-F., Carpenter, B. K., and Vaida, V.: Gas-Phase Photolysis of Pyruvic Acid: The Effect of Pressure on Reaction Rates and Products, J. Phys. Chem. A, 120, 10123–10133, https://doi.org/10.1021/acs.jpca.6b09058, 2016.
Reed Harris, A. E., Cazaunau, M., Gratien, A., Pangui, E., Doussin, J.-F., and Vaida, V.: Atmospheric Simulation Chamber Studies of the Gas-Phase Photolysis of Pyruvic Acid, J. Phys. Chem. A, 121, 8348–8358, https://doi.org/10.1021/acs.jpca.7b05139, 2017a.
Reed Harris, A. E., Pajunoja, A., Cazaunau, M., Gratien, A., Pangui, E., Monod, A., Griffith, E. C., Virtanen, A., Doussin, J.-F., and Vaida, V.: Multiphase Photochemistry of Pyruvic Acid under Atmospheric Conditions, J. Phys. Chem. A, 121, 3327–3339, https://doi.org/10.1021/acs.jpca.7b01107, 2017b.
Riplinger, C. and Neese, F.: An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 138, 034106, https://doi.org/10.1063/1.4773581, 2013.
Riplinger, C., Sandhoefer, B., Hansen, A., and Neese, F.: Natural triple excitations in local coupled cluster calculations with pair natural orbitals, J. Chem. Phys., 139, 134101, https://doi.org/10.1063/1.4821834, 2013.
Shampine, L. F. and Reichelt, M. W.: The MATLAB ODE Suite, SIAM J. Sci. Comput., 18, 1–22, https://doi.org/10.1137/S1064827594276424, 1997.
Sihto, S.-L., Kulmala, M., Kerminen, V.-M., Dal Maso, M., Petäjä, T., Riipinen, I., Korhonen, H., Arnold, F., Janson, R., Boy, M., Laaksonen, A., and Lehtinen, K. E. J.: Atmospheric sulphuric acid and aerosol formation: implications from atmospheric measurements for nucleation and early growth mechanisms, Atmos. Chem. Phys., 6, 4079–4091, https://doi.org/10.5194/acp-6-4079-2006, 2006.
Sipila, M., Berndt, T., Petaja, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin III, R. L., Hyvarinen, A.-P., Lihavainen, H., and Kulmala, M.: The Role of Sulfuric Acid in Atmospheric Nucleation, Science, 327, 1243–1246, https://doi.org/10.1126/science.1180315, 2010.
Stocker, T. F, Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P. M. (Eds.): IPCC, 2013: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 1585 pp., 2013.
Talbot, R. W., Andreae, M. O., Berresheim, H., Jacob, D. J., and Beecher, K. M.: Sources and sinks of formic, acetic, and pyruvic acids over central Amazonia: 2. Wet season, J. Geophys. Res.-Atmos., 95, 16799–16811, https://doi.org/10.1029/JD095iD10p16799, 1990.
Talbot, R. W., Mosher, B. W., Heikes, B. G., Jacob, D. J., Munger, J. W., Daube, B. C., Keene, W. C., Maben, J. R., and Artz, R. S.: Carboxylic acids in the rural continental atmosphere over the eastern United States during the Shenandoah Cloud and Photochemistry Experiment, J. Geophys. Res.-Atmos., 100, 9335–9343, https://doi.org/10.1029/95JD00507, 1995.
Temelso, B., Morrell, T. E., Shields, R. M., Allodi, M. A., Wood, E. K., Kirschner, K. N., Castonguay, T. C., Archer, K. A., and Shields, G. C.: Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications, J. Phys. Chem. A, 116, 2209–2224, https://doi.org/10.1021/jp2119026, 2012a.
Temelso, B., Phan, T. N., and Shields, G. C.: Computational Study of the Hydration of Sulfuric Acid Dimers: Implications for Acid Dissociation and Aerosol Formation, J. Phys. Chem. A, 116, 9745–9758, https://doi.org/10.1021/jp3054394, 2012b.
Torrent-Sucarrat, M., Francisco, J. S., and Anglada, J. M.: Sulfuric acid as autocatalyst in the formation of sulfuric acid, J. Am. Chem. Soc., 134, 20632–20644, https://doi.org/10.1021/ja307523b, 2012.
Truhlar, D. G., Garrett, B. C., and Klippenstein, S. J.: Current Status of Transition-State Theory, J. Phys. Chem., 100, 12771–12800, https://doi.org/10.1021/jp953748q, 1996.
Tsona, N. T. and Du, L.: A potential source of atmospheric sulfate from -induced SO2 oxidation by ozone, Atmos. Chem. Phys., 19, 649–661, https://doi.org/10.5194/acp-19-649-2019, 2019.
Tsona, N. T., Bork, N., and Vehkamäki, H.: Exploring the chemical fate of the sulfate radical anion by reaction with sulfur dioxide in the gas phase, Atmos. Chem. Phys., 15, 495–503, https://doi.org/10.5194/acp-15-495-2015, 2015a.
Tsona, N. T., Henschel, H., Bork, N., Loukonen, V., and Vehkamäki, H.: Structures, Hydration, and Electrical Mobilities of Bisulfate Ion–Sulfuric Acid–Ammonia/Dimethylamine Clusters: A Computational Study, J. Phys. Chem. A, 119, 9670–9679, 2015b.
Tsona, N. T., Bork, N., Loukonen, V., and Vehkamäki, H.: A Closure Study of the Reaction between Sulfur Dioxide and the Sulfate Radical Ion from First-Principles Molecular Dynamics Simulations, J. Phys. Chem. A, 120, 1046–1050, 2016.
Warneck, P.: Multi-Phase Chemistry of C2 and C3 Organic Compounds in the Marine Atmosphere, J. Atmos. Chem., 51, 119–159, https://doi.org/10.1007/s10874-005-5984-7, 2005.
Weber, R. J., Chen, G., Davis, D. D., Mauldin III, R. L., Tanner, D. J., Eisele, F. L., Clarke, A. D., Thornton, D. C., and Bandy, A. R.: Measurements of enhanced H2SO4 and 3–4 nm particles near a frontal cloud during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res.-Atmos., 106, 24107–24117, https://doi.org/10.1029/2000JD000109, 2001.
Welz, O., Savee, J. D., Osborn, D. L., Vasu, S. S., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct kinetic measurements of Criegee intermediate (CH2OO) formed by reaction of CH2I with O2, Science, 335, 204–207, 2012.
Yao, X. and Zhang, L.: Causes of Large Increases in Atmospheric Ammonia in the Last Decade across North America, ACS Omega, 4, 22133–22142, https://doi.org/10.1021/acsomega.9b03284, 2019.
Zhang, Y., Liu, X., Fang, Y., Liu, D., Tang, A., and Collett, J. L.: Atmospheric Ammonia in Beijing during the COVID-19 Outbreak: Concentrations, Sources, and Implications, Environ. Sci. Tech. Let., 8, 32–38, https://doi.org/10.1021/acs.estlett.0c00756, 2021.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.