the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Dec 2022
| 20 Dec 2022
Product distribution, kinetics, and aerosol formation from the OH oxidation of dimethyl sulfide under different RO2 regimes
Matthew B. Goss
Jordan E. Krechmer
Francesca Majluf
Alexander Zaytsev
Yaowei Li
Joseph R. Roscioli
Manjula Canagaratna
Frank N. Keutsch
Colette L. Heald
The atmospheric oxidation of dimethyl sulfide (DMS) represents a major natural source of atmospheric sulfate aerosols. However, there remain large uncertainties in our understanding of the underlying chemistry that governs the product distribution and sulfate yield from DMS oxidation. Here, chamber experiments were conducted to simulate gas-phase OH-initiated oxidation of DMS under a range of reaction conditions. Most importantly, the bimolecular lifetime (τbi) of the peroxy radical CH3SCH2OO was varied over several orders of magnitude, enabling the examination of the role of peroxy radical isomerization reactions on product formation. An array of analytical instruments was used to measure nearly all sulfur-containing species in the reaction mixture, and results were compared with a near-explicit chemical mechanism. When relative humidity was low, “sulfur closure” was achieved under both high-NO (τbi<0.1 s) and low-NO (τbi>10 s) conditions, though product distributions were substantially different in the two cases. Under high-NO conditions, approximately half the product sulfur was in the particle phase, as methane sulfonic acid (MSA) and sulfate, with most of the remainder as SO2 (which in the atmosphere would eventually oxidize to sulfate or be lost to deposition). Under low-NO conditions, hydroperoxymethyl thioformate (HPMTF, HOOCH2SCHO), formed from CH3SCH2OO isomerization, dominates the sulfur budget over the course of the experiment, suppressing or delaying the formation of SO2 and particulate matter. The isomerization rate constant of CH3SCH2OO at 295 K is found to be 0.13±0.03 s−1, in broad agreement with other recent laboratory measurements. The rate constants for the OH oxidation of key first-generation oxidation products (HPMTF and methyl thioformate, MTF) were also determined ( cm3 molec.−1 s−1, cm3 molec.−1 s−1). Product measurements agree reasonably well with mechanistic predictions in terms of total sulfur distribution and concentrations of most individual species, though the mechanism overpredicts sulfate and underpredicts MSA under high-NO conditions. Lastly, results from high-relative-humidity conditions suggest efficient heterogenous loss of at least some gas-phase products.
- Article
(2614 KB) - Full-text XML
-
Supplement
(3975 KB) - BibTeX
- EndNote




Dimethyl sulfide (DMS), emitted by marine phytoplankton, is an important natural source of sulfur to the atmosphere (Kloster et al., 2006; Lana et al., 2011). The atmospheric oxidation of DMS represents a dominant source of non-sea-salt sulfate aerosols and as such can play an important role in global aerosol climate effects (Charlson et al., 1987; Rap et al., 2013). The chemistry by which DMS oxidizes to form sulfate is highly complex: the mechanism includes multiple branch points and intermediate species, and many reaction rates and product yields are uncertain and/or highly dependent on reaction conditions (Barnes et al., 2006; Hoffmann et al., 2016). As a result, many large-scale models adopt a highly simplified DMS chemistry with fixed SO2 yields, usually without inclusion of other intermediates (Chin et al., 1996; Huijnen et al., 2010; Kloster et al., 2006; Lamarque et al., 2012). Such a simplified approach may lead to errors in predicted aerosol radiative effects, in the past, present, and future atmospheres (Fung et al., 2022).
The major daytime sink of DMS is its reaction with OH radicals. The detailed DMS + OH reaction scheme is shown in Fig. 1. A key branch point in DMS + OH is the methylthiomethylperoxy radical (CH3SCH2OO) formed from H-atom abstraction followed by O2 addition. The subsequent chemistry of this radical plays a determining role in the overall product distribution and thus likely influences the amount of sulfate aerosols that is ultimately formed. As with all large RO2 species, CH3SCH2OO radicals may undergo bimolecular reactions (with NO and HO2) or unimolecular reaction via a recently identified (Berndt et al., 2019; Veres et al., 2020; Wu et al., 2015; Ye et al., 2021; Jernigan et al., 2022a) isomerization channel:
The CH3SCH2O radical formed from the NO pathway (Reaction R1) forms SO2, sulfate, and methane sulfonic acid (MSA) (Barnes et al., 2006). The alkyl radical derived from Reaction (R3) will react with O2 to form OOCH2SCH2OOH, which will undergo a second isomerization reaction at a rate substantially faster than that of Reaction (R3) (Wu et al., 2015; Crounse et al., 2013), forming hydroperoxymethyl thioformate (HPMTF, HOOCH2SCHO), as shown in Fig. 1. In addition to Reactions (R1–R3), CH3SCH2OO may also react with other RO2 radicals (Barnes et al., 2006), though this process is likely to be minor under atmospheric conditions.

Figure 1Gas-phase DMS + OH oxidation mechanism. Measured closed-shell products are shown in bold. Reactions in black are taken from MCM (Master Chemical Mechanism); reactions in red, related to hydroperoxymethyl thioformate (HPMTF, HOOCH2SCHO) chemistry, are taken from Wu et al. (2015). Products that do not contain sulfur are not shown. The CH3SO2 radical (marked in blue) represents a link between addition and abstraction pathway products. Note that several products are shown multiple times.
The branching fraction of the CH3SCH2OO radical depends on the concentrations of NO and HO2 and the rate constants of Reactions (R1–R3). The rate constant for the isomerization reaction, kisom, is particularly uncertain, as values determined in previous studies span a very wide range, from ∼0.04 to ∼2 s−1 near room temperature (Berndt et al., 2019; Veres et al., 2020; Wu et al., 2015; Ye et al., 2021; Jernigan et al., 2022a). This highlights a major challenge in predicting CH3SCH2OO branching and the subsequent aerosol formation, both in the pristine atmosphere and in environments affected by anthropogenic emissions.
Most previous experimental studies investigating DMS oxidation have examined individual products and reaction steps in isolation (Barnes et al., 2006; Berndt et al., 2019; Jernigan et al., 2022a; Mihalopoulos et al., 1992; Patroescu et al., 1996); very few studies of the entire multiphase and multistep reaction system have been conducted, especially under conditions in which the recently discovered isomerization pathway (Reaction R3) may compete. Therefore, there have been relatively few experimental tests of our overall understanding of the reaction system, by comparison against predictions by state-of-the-art reaction mechanisms. Recently, we conducted laboratory measurements of a broad suite of organic sulfur products and sulfate aerosols from DMS + OH and estimated kisom to be 0.09 s−1 (0.03–0.3 s−1, 1σg) (Ye et al., 2021); however this was for a single reaction condition only (<5 % relative humidity, ∼1 ppb NO), and SO2 (a major inorganic sulfur-containing product) was not measured.
Here we extend our previous work by conducting a series of chamber experiments of DMS + OH under a wide range of values of the CH3SCH2OO bimolecular lifetime (τbi) and comprehensively characterizing sulfur-containing products (organic and inorganic, gas-phase and particulate), with the aim of accounting for all (or nearly all) reacted sulfur. Such “sulfur closure” measurements enable direct comparisons with predictions from a mechanistic model, in order to assess our current mechanistic understanding and identify possible gaps in this understanding. These measurements also enable the determination of key kinetic parameters in the reaction systems. In one experiment, we vary τbi over a wide range to estimate the kisom of the CH3SCH2OO radical, obtaining a kisom with a much smaller uncertainty range than in our previous study. The rate constants for the OH oxidation of key first-generation oxidation products (HPMTF and methyl thioformate, MTF) are also determined. Lastly, we investigate the effect of relative humidity (RH) on the DMS + OH product distributions.
Experiments were conducted in a 7.5 m3 temperature-controlled environmental chamber, held at 295 K (Hunter et al., 2014). The chamber is surrounded by 48 ultraviolet lights (Q-Lab) with a peak irradiance at 340 nm. Before each experiment, the chamber was flushed by zero air (AADCO, 737 series) for at least 12 h to ensure a clean gas and particle background. Throughout the course of each experiment, a constant flow of zero air was introduced into the chamber to replenish the flow drawn by the instruments. For high-RH experiments, the replenishment flow was first sent through a bubbler filled with Milli-Q water before entering the chamber. The rate of chamber dilution was derived by measuring the decay of acetonitrile, injected at low concentrations (5 ppb) in the beginning of each experiment. The overall dilution lifetime was approximately 10 h. Concentrations of all species reported below have been corrected for dilution.
The evolving chemical composition of the reaction mixture was monitored by a suite of real-time instruments located outside the chamber. The Supplement provides instrument details, as well as the sulfur species detected by each instrument (Table S2). Briefly, DMS and lightly oxygenated gaseous species were measured by a Vocus proton-transfer-reaction time-of-flight mass spectrometer (Vocus PTR-MS, Aerodyne Research Inc.) (Krechmer et al., 2018). More oxygenated gaseous species were measured by an iodide time-of-flight chemical ionization mass spectrometer (I− CIMS, Aerodyne Research Inc.) (Lee et al., 2014) and an ammonium time-of-flight chemical ionization mass spectrometer (NH CIMS, Ionicon Analytik) (Zaytsev et al., 2019). SO2 was detected by a compact tunable infrared laser direct absorption spectrometer (TILDAS, Aerodyne Research Inc.) (McManus et al., 2011, 1995). Particle-phase products, namely sulfate and MSA, were measured by an aerosol mass spectrometer (AMS, Aerodyne Research Inc.) (DeCarlo et al., 2006). The quantification of MSA was determined from the AMS tracer ion CH3SO (see the Supplement); this ion is believed to be unique to MSA (or methyl sulfonate), with negligible contributions from other sulfur-containing species (Hodshire et al., 2019; Huang et al., 2015). Our multi-instrument approach enables the measurement of essentially all closed-shelled sulfur products known in the DMS oxidation mechanism, except for carbonyl sulfide (OCS), which accounts for a very small (less than a couple percent) sulfur yield from DMS oxidation (Barnes et al., 1994; Jernigan et al., 2022a). Complementary instruments include an ozone monitor (2B Technologies), a NO–NO2–NOx analyzer (Thermo Scientific), a scanning mobility particle sizer (TSI), and a temperature and RH sensor (TE Connectivity). More details of the instruments, including their calibrations and measurement uncertainties, are provided in the Supplement.
Table 1Summary of experimental conditions.
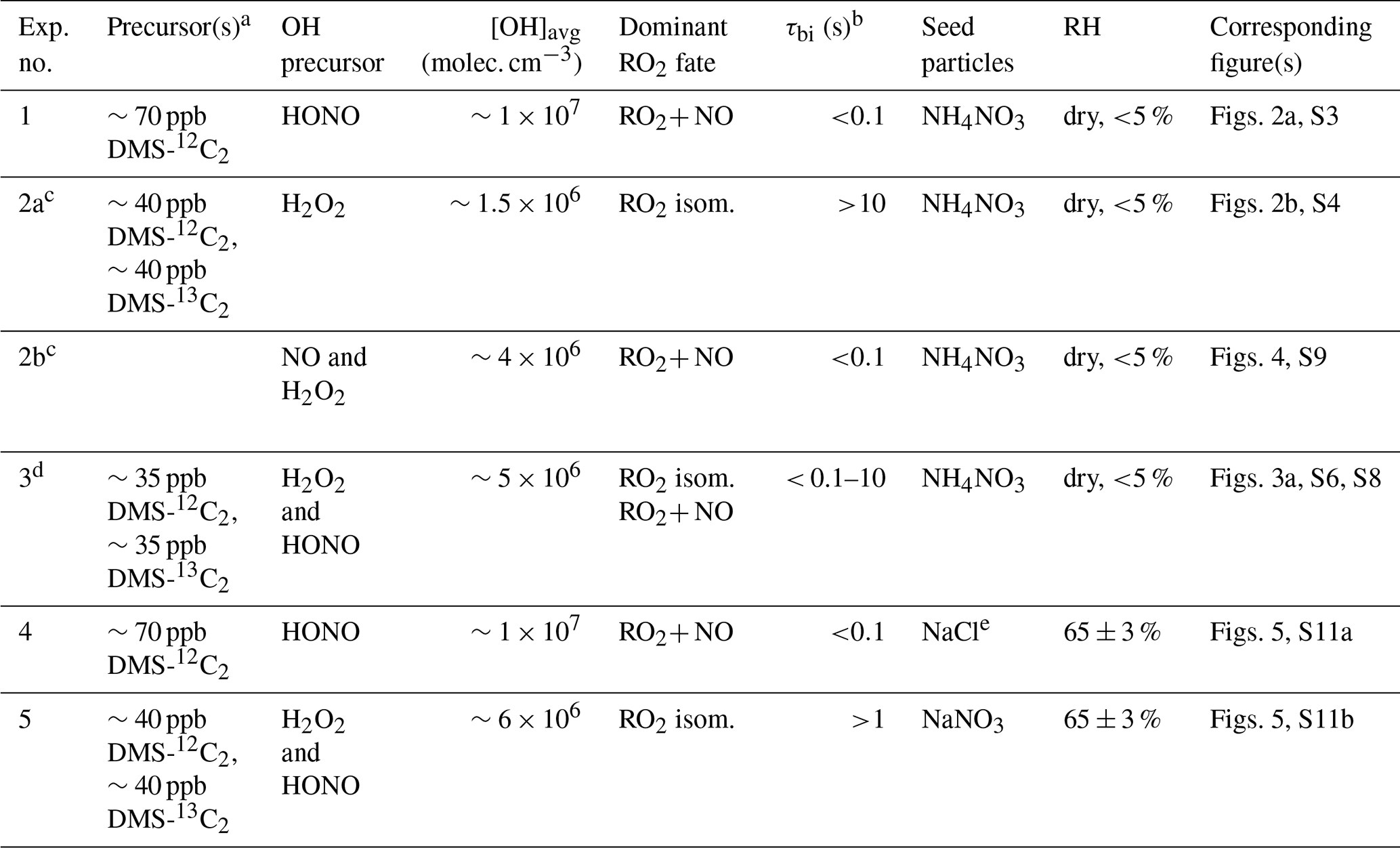
a To better separate HPMTF from N2O5, DMS-13C2 was used in low-NO experiments. b Bimolecular lifetime of the CH3SCH2OO radical, calculated as . c Experiments 2a and b were carried out as part of a single oxidation experiment; initially (Exp. 2a) OH was generated from H2O2 photolysis (low-NO conditions), and then (Exp. 2b) 70 ppb of NO was injected into the chamber. d 13C data in Experiment 3 were used to calculate kisom; HONO was added multiple times in the experiment. e The vaporizer in the AMS was operated at 800 ∘C. AMS calibration was done separately for 800 ∘C.
The experiments carried out in this study are listed in Table 1. At the beginning of each experiment, DMS, the acetonitrile dilution tracer, seed particles, and the OH precursor were added to the chamber and allowed to become well mixed. Total concentrations of DMS introduced to the chamber were similar among all experiments. In dry experiments, seed particles (ammonium nitrate) were added into the chamber via first atomization followed by drying, providing surface area for condensing vapors. In high-RH experiments, seed particles (sodium chloride and sodium nitrate) were introduced without drying, remaining as liquid particles under the chamber RH. Particle condensation timescales (seconds to tens of seconds) were much shorter than the condensation timescale of low-volatility species onto the chamber wall (∼2000 s, as determined previously for this chamber, Zaytsev et al., 2019). In these experiments, non-sulfate seeds were used to avoid interferences when quantifying secondary sulfate in the aerosols. For low-RH experiments (Exp. 1–3), ammonium nitrate seed particles were used, since dry ammonium nitrate particles are expected to be chemically inert. For the high-RH high-NO experiment (Exp. 4), NaCl particles were used. As discussed below, major products are similar to those in the high-NO dry experiment, suggesting that the NaCl seed particles in Exp. 4 have little to no effect on the product distribution in these experiments. More studies are needed to constrain the effects of different seed particles on the reactive uptake of DMS oxidation products (Jernigan et al., 2022b).
DMS was introduced by gently heating a known volume (1–2 µL) from a needle syringe, and the vapor was carried into the chamber by the dilution flow. For the long τbi experiments, in which HPMTF formation was expected (see Table 1), DMS-13C2 (99 atom % 13C, MilliporeSigma) was added as the precursor in addition to unlabeled DMS (>99 %, MilliporeSigma), in order to easily distinguish HPMTF (C2H4SO3 • I−, 234.893) from N2O5 (N2O5 • I−, 234.886) in the I− CIMS. The use of DMS-13C2 is expected to have little effect on the observed reaction kinetics in this study. For the high-NO (short τbi) experiments, HONO (tens of parts per billion) was added as the OH precursor, by passing air over a mixture of sodium nitrite and sulfuric acid into the chamber. For low-NO (long τbi) experiments, parts per million levels of H2O2 were added as the OH precursor, by vaporizing a known amount of 30 % H2O2 solution injected by a micro-syringe. The H2O2 concentration was derived based on the known photon flux in the chamber and the observed decay rate of NO. In some experiments (Exp. 2b, 3, and 5), aliquots of HONO or NO were added in the middle of the experiment to change reaction conditions. After all reagents were well mixed (>5 min), the UV lights were turned on to photolyze HONO and/or H2O2, generating OH radicals and initiating the reaction. The OH concentration was estimated from the decay of DMS (using cm3 molec.−1 s−1) (Jenkin et al., 1997; Saunders et al., 2003) and was used to determine the equivalent atmospheric OH exposure time, assuming [OH] molec. cm−3.
A 0-D model (the Framework for 0-D Atmospheric Modeling, F0AM) (Wolfe et al., 2016) coupled with the Master Chemical Mechanism (MCMv3.3.1) (Jenkin et al., 1997; Saunders et al., 2003) was used to simulate gas-phase DMS oxidation in each experiment. Here, the DMS scheme in the MCM was updated primarily based on Wu et al. (2015) with the isomerization rate constant of the CH3SCH2OO radical as 0.09 s−1, taken from our previous work (Ye et al., 2021). The complete reaction scheme is shown in Fig. 1. Newly added reactions with rate constants beyond the original MCM scheme are listed in Table S1. Model inputs, including concentrations of the precursor, oxidant, and chamber conditions including temperature, light intensity, and dilution rate were taken directly from the measurements. The uptake or heterogeneous reactions of water-soluble species (e.g., dimethyl sulfoxide (DMSO), dimethyl sulfone (DMSO2), methane sulfinic acid (MSIA), and HPMTF) are not considered in this modeling, though as described below such processes may occur. In the high-NO experiments, model NO concentrations were constrained to values measured by the NO–NO2–NOx analyzer. In the low-NO experiment (Exp. 2a) in which the sub-ppb-level NO concentration was near or below the detection limit (0.4 ppb) of the NOx analyzer, the model was used to constrain background NO concentration by matching the modeled DMS decay to the measured decay (Ye et al., 2021). The estimated [NO] in Exp. 2a was ∼10 ppt.
3.1 Comprehensive measurements of S-containing products
Figure 2a and b shows the measured product evolution from Experiments 1 and 2a under dry conditions. A range of sulfur-containing products were measured in both the gas and aerosol phases, shown as stacked colored traces. Changes in concentrations are given in parts per billion of sulfur (Δ ppb S) and are presented as a function of atmosphere-equivalent OH exposure time. Shown in grey is the amount of DMS oxidized over the course of the experiment. By the end of the experiment, only a fraction of the DMS had been consumed, since OH exposures were not high enough to fully deplete the DMS. In Exp. 1 (high-NO conditions, Fig. 2a), HONO was used as the OH precursor, and the NO was kept at ∼50 ppb by continuous addition, ensuring that the dominant fate of the RO2 radicals was reaction with NO (τbi<0.1 s). After ∼12 h of atmosphere-equivalent OH exposure, 104 % (100 %–124 %, 1σ) of the reacted sulfur was measured as products, indicating excellent sulfur closure. The uncertainty in sulfur closure includes uncertainty in both gas-phase and particle-phase measurements (see Supplement for more details). The initial dip in the first 2 h may be due to loss of products to surfaces such as the chamber wall or sampling lines. It is likely that there is an equilibrium between the sampling line and the gas phase. This drop, of 1–2 ppb S, represents a relatively small portion of the total sulfur reacted by the end of the experiment.

Figure 2Stacked plots showing the total sulfur measured (a and b) and modeled (c and d) under high-NO (a and c) and low-NO (b and d) conditions. Panels (a) and (c) are for Exp. 1, and panels (b) and (d) are for Exp. 2a. Data shown in panel (b) are from DMS-12C2 and DMS-13C2 combined. Products with a formula of C2H6SO2 may be DMSO2 and/or CH3SCH2OOH; under high-NO conditions, they are likely to be predominantly DMSO2. Minor products detected but not listed in the legend due to their very low concentrations include CH2SO2 (a sulfene or thioacid) and CH3SO6N (likely methanesulfonyl peroxynitrate). Note that y axes denote the changes in concentrations of the precursor and products.
Major sulfur-containing products in Exp. 1 were SO2, particulate MSA, and particulate sulfate, with 48 % of the product sulfur found in the particle phase. The measured MSA : sulfate ratio () is in broad agreement with those reported in Chen et al. (2012). Minor species observed included dimethyl sulfoxide (DMSO), C2H6SO2 (likely dimethyl sulfone, DMSO2), and methane sulfinic acid (MSIA), known products from the addition channel, as well as CH2SO2 (likely a thioacid, which may be formed as an OH oxidation product of HPMTF, Jernigan et al., 2022a) and CH3SO6N (likely methanesulfonyl peroxynitrate, formed from CH3S(O)2OO + NO2). No HPMTF was observed in these experiments, which is expected given the short bimolecular RO2 lifetime.
In Exp. 2a (low-NO conditions, Fig. 2b), H2O2 was the OH precursor, and NO and HO2 levels were sufficiently low (∼10 and 100 ppt, respectively) enough for RO2 isomerization to dominate (τbi>10 s). HO2 generated from H2O2+ OH is expected to promote the formation of CH3SCH2OOH from Reaction (R2); however, we cannot distinguish CH3SCH2OOH from its isomer, DMSO2. Product distributions are dramatically different than those under high-NO conditions. The total sulfur products measured accounted for nearly all (90 % (64 %–118 %)) of the reacted DMS sulfur; this sulfur closure is good but slightly worse than in Exp. 1. The larger uncertainty range is due to the uncertainty of the HPMTF calibration in the I− CIMS. However, the near-sulfur closure, derived from multiple independently calibrated instruments, combined with the HPMTF yields (discussed in Sect. 3.3) suggest that our estimated sensitivity is reasonably accurate, and thus our overall uncertainty of total sulfur may be an overestimate.
Due to the long RO2 bimolecular lifetime (τbi>10 s), the dominant product is HPMTF from CH3SCH2OO isomerization; this accounts for about half of the reacted sulfur (60 % of the measured product sulfur). It is expected that a negligible amount (1 % or less) of HPMTF was lost to the chamber walls under the experimental condition here based on its estimated vapor pressure (see the Supplement). The time series of C2H4SO3-12C2 in the I− CIMS (C2H4SO3 • I−) and in the NH CIMS (C2H4SO3 • NH), shown in Fig. S2, match very well. This indicates that there was negligible N2O5 formation from the residual NOx in the chamber, since N2O5 is not measurable by the NH CIMS, and therefore our quantification of HPMTF-12C2 in Exp. 2a with I− CIMS is free of N2O5 interferences. Only 3.3 % (3.1 %–5.4 %) of the reacted sulfur was found in the aerosol by the end of the experiment.
3.2 Model–measurement comparison
The (near-)sulfur closure of the experiments, in which virtually all the reacted sulfur was measured as products, enables a comparison with the mechanistic model. MCM predictions for the two experiments described above (Exp. 1 and 2a) are shown in Fig. 2c and d; individual species are also compared in Figs. S3 and S4. Under high-NO conditions, measurements and model predictions (Figs. 2a and c, S3) agree well for gas-phase species and for total particulate sulfur. However, the two differ greatly in terms of particle-phase composition: AMS measurements indicate ∼70 % of the particle-phase sulfur is MSA, with the remainder as sulfate; by contrast, the model predicts that sulfate dominates, with a negligible (∼0.1 %) contribution from MSA. This suggests the mechanism may underestimate the rate of MSA formation (a result consistent with recent studies; Wolleson de Jonge et al., 2021; Shen et al., 2022) and/or overestimate the rate of sulfuric acid formation.
In the MCM, both MSA and sulfuric acid are formed from reactions of the CH3S(O)2O radical:
Reaction (R5) generates sulfur trioxide (SO3), which will quickly hydrolyze to form sulfuric acid. SO3 can also be formed by the OH oxidation of SO2, but this reaction would occur over 50 h of OH exposure, much longer than the oxidation timescale in Exp. 1. Since the measured and modeled total particulate sulfur (MSA + sulfate) agree well, the model–measurement differences in the ratio of MSA to sulfuric acid (or sulfate) may relate to the relative rates of these CH3S(O)2O reactions. It is possible that the rate constant of Reaction (R4) is underestimated in the mechanisms, but even if it is increased it to a gas-kinetic rate ( cm3 molec.−1 s−1), MSA is still not predicted to dominate over sulfuric acid. Instead, the decomposition of CH3S(O)2O (Reaction R5), which has received little study, might be slower than the value used in the mechanism (∼0.09 s−1), leading to slower sulfuric acid formation. Alternatively, MSA might be formed by the reaction of CH3S(O)2O with species other than HO2, such as DMS or HCHO (Barnes et al., 2006; Yin et al., 1990). While such reactions are unlikely to be important in the atmosphere, they might occur in laboratory experiments, which have relatively high concentrations of organic species. However, the kinetics of such reactions are not well known and warrant future research.
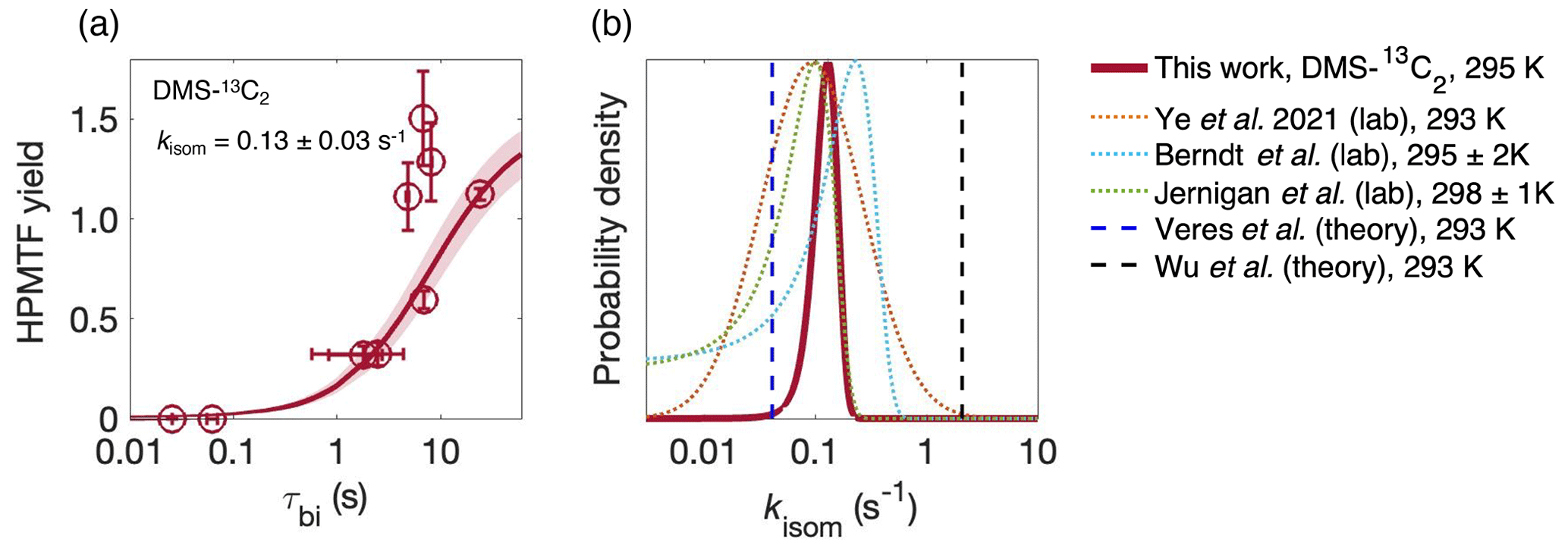
Figure 3(a) The yield of HPMTF in the abstraction channel as a function of the bimolecular lifetime τbi of CH3SCH2OO from the DMS-13C2 data. The shaded area is 1σ of the fit, which takes into account uncertainty in both τbi (arising from errors in [NO] and [HO2]) on the x axis, and instrument noise on the y axis. Uncertainty in the CIMS sensitivity to HPMTF affects the absolute measurements but not the inflection point of the curve or the derived value of kisom. (b) Comparison of kisom from this work with previous determinations of kisom at 293–298 K (Berndt et al., 2019; Jernigan et al., 2022a; Veres et al., 2020; Wu et al., 2015; Ye et al., 2021).
Another potential source of MSA is the oxidation of MSIA by OH (Yin et al., 1990; Lucas and Prinn, 2002; von Glasow and Crutzen, 2004; Wollesen de Jonge et al., 2021; Shen et al., 2022). This pathway is currently not included in the MCM, which has MSIA reacting with OH to form SO2 and CH3 (Fig. 1). It has been suggested (Yin et al., 1990) that the reaction may occur via abstraction of the acidic hydrogen:
As shown in Fig. 1, the resulting CH3S(O)O radical may react with ozone to form CH3S(O)2O, which can react further to form MSA or SO3 (Reactions R4–R5). However, inclusion of this reaction in the model increases MSA formation only slightly, and the model–measurement discrepancy remains large (Fig. S5). Alternatively, OH might add to MSIA (Lucas and Prinn, 2002; Arsene et al., 2002; Shen et al., 2022), forming the intermediate CH3SO(OH)2 that can react with O2 to produce MSA:
Including these reactions into the mechanism, using the rate constant for MSIA + OH suggested by the MCM ( cm3 molec.−1 s−1) substantially increases the predicted MSA but at the same time decreases the predicted SO2 concentration, worsening the model–measurement agreement for SO2, and does not change predicted sulfate formation, leading to an overestimate in total aerosol production (Fig. S5). Taken together, while the OH oxidation of MSIA (Reactions R6–R8) may contribute to MSA formation, it appears not to be the only (or major) source for the MSA model–measurement discrepancy in the present experiments.
In the low-NO case (Figs. 2b and d, S4), measured and modeled concentrations also broadly agree. The predicted concentration of HPMTF is lower (by ∼30 %) than what was measured. This could be due to the uncertainty in the sensitivity of HPMTF in the I− CIMS and/or in the kisom value used in the model. The kisom value used, 0.09 s−1, is derived from our previous study (Ye et al., 2021); as discussed below, this value agrees with that determined in this work. Compared to measurements, the model also predicts somewhat higher concentrations of minor sulfur-containing products, such as DMSO, C2H6SO2 (DMSO2 + CH3SCH2OOH), MSIA, and MTF. This could be caused by overestimates of instruments' sensitivities, uncertainties in the rate constants in the model, or some losses to surfaces. Nevertheless, overall the model and measurements agree quite well, with product formation dominated by HPMTF and little aerosol formation since low-volatility species (MSA and sulfuric acid) are formed only as later-generation products.
3.3 Determination of kisom
The fate of the CH3SCH2OO radical, and hence the product distribution of DMS oxidation, relies critically on the isomerization rate constant of the CH3SCH2OO radical (kisom). In our previous work we determined kisom from a single reaction condition (at one value of τbi), and the kisom value had a large uncertainty due to the poorly constrained sensitivity of HPMTF in the CIMS. Here, we determine kisom by examining product formation at multiple values of τbi, similar to previous measurements of isomerization rates of terpene-derived RO2 radicals (Xu et al., 2019). MCM modeling suggests that RO2 + RO2 reactions represent ∼1 % of the RO2 sink in the experiments, and therefore the only bimolecular reactions considered are RO2 + NO and RO2 + HO2. HONO or NO was added to the chamber several times during the experiment (Fig. S6), perturbing the branching of the CH3SCH2OO radical (isomerization vs. bimolecular reactions). The total S measurements are shown in Fig. S8. The yield of HPMTF in the abstraction channel (Δ[HPMTF] Δ[DMS]abs) was calculated for each perturbation as a function of τbi after taking into account the of loss via OH oxidation ( cm3 molec.−1 s−1; see Sect. 3.4). The detailed calculation is described in the Supplement (Eqs. S1–S4). Figure 3a shows the HPMTF yield as a function of τbi. As expected, the yield increases dramatically with τbi, and fitting these data to Eq. S4 (given in the Supplement) enables the determination of kisom. The best-fit value for kisom is 0.13±0.03 s−1. The uncertainty is much smaller than in our previous determination (Ye et al., 2021) since the fit depends only on the shape (the inflection point) of the curve and not the absolute yield values and thus is insensitive to the uncertain HPMTF calibration factor. Nonetheless, since the asymptotic (high τbi) value is close to 1 (1.5), our estimated calibration factor appears to be reasonably accurate. The three data points with higher HPMTF yields (top of Fig. 3a) were collected in the latter half of the experiment, after HPMTF had built up in the chamber, and therefore correcting for OH loss resulted in an increased HPMTF yield. Because of their larger measurement uncertainties, these data points have smaller effects on the overall fit to Eq. (S4). If the OH loss is not included, s−1 (Fig. S7).
Figure 3b compares our value of kisom with previous measurements and theoretical determinations (T= 293–298 K) (Berndt et al., 2019; Jernigan et al., 2022a; Veres et al., 2020; Wu et al., 2015; Ye et al., 2021). Our measured value of kisom is consistent with our previous (single τbi) measurement (Ye et al., 2021) though with a much reduced uncertainty and is also in broad agreement with measured values from Berndt et al. (2019) (0.23±0.12 s−1) and Jernigan et al. (2022) (0.1±0.05 s−1).
3.4 Reaction rates of OH with HPMTF and MTF
Here we examine the oxidation of HPMTF and MTF, two species whose chemical fates are not well known. Both were formed only under low-NO conditions (Exp. 2a); because of the relatively low OH concentrations of that experiment, their concentrations increased throughout the entire experiment, with no subsequent decay. Thus, to estimate kOH+HPMTF and kOH+MTF, high concentrations of NO (∼70 ppb) were introduced at the end of Experiment 2 (denoted as Exp. 2b, shown in Fig. 4a). The large amount of NO essentially terminated the production of HPMTF and MTF and at the same time increased the OH concentration in the chamber. The total sulfur product distribution for Exp. 2 is shown in Fig. S9. The loss of HPMTF during this period, shown in Fig. 4b, is expected to be dominated by OH reaction because the high level of NO precluded substantial oxidation by O3 and NO3. Photolysis of HPMTF is also unlikely to contribute to the observed decay: by assuming that its photolytic cross sections are equal to the summed cross section of aldehydes and organic peroxides taken from MCM (Khan et al., 2021), we estimate that photolysis accounted for only 4 % of the HPMTF loss in our chamber. Using the cross section for MTF measured by Patroescu et al. (1996), we obtain an even lower photolysis rate, with photolysis accounting for less than 2 % of HPMTF loss in the chamber.

Figure 4(a) NO concentration measured by the NO–NO2–NOx analyzer in Exp. 2. At OH exposure ∼5.8 h, 70 ppb of NO was injected into the chamber. (b) Time series of HPMTF-12C2 and MTF-12C2 in Exp. 2. The decay of HPMTF and MTF was used to estimate their reaction rate coefficients with OH.
By calculating [OH] using the decay of DMS after the addition of NO, we fit the decay of HPMTF (Figs. 4b and S10) to derive kOH+HPMTF of 2.1 (2.0–2.2) cm3 molec.−1 s−1. This is in agreement with recent measurements of Jernigan et al. (2022) (1.4 (0.27–2.4) cm3 molec.−1 s−1); both experimental values are an order of magnitude higher than an earlier theoretical estimate of the rate ( cm3 molec.−1 s−1) (Wu et al., 2015). Using this lower value, Khan et al. (2021) estimated that photolysis loss dominates HPMTF sink in the global marine sulfur budget, with OH oxidation only accounting for 10 % of HPMTF loss. This higher OH rate constant suggests that OH oxidation is in fact likely to be an important loss process for HPMTF, at least when liquid water is not present (Fung et al., 2022; Vermeuel et al., 2020; Novak et al., 2021).
MTF is formed predominantly as a second-generation DMS oxidation product from CH3SCH2OOH + OH in low-NO conditions in the mechanism. Using a similar method as kOH+HPMTF (Figs. 4b and S10), kOH+MTF is estimated to be 1.35 (1.3–1.4) cm3 molec.−1 s−1, which agrees with the only other measurement of kOH+MTF, cm3 molec.−1 s−1, by Patroescu et al. (1996).
3.5 Role of relative humidity
The experiments described above were carried out under dry conditions and thus focus only on homogenous gas-phase chemistry; in the atmosphere, heterogeneous and aqueous-phase processes may also be important contributors to DMS oxidation chemistry (Hoffmann et al., 2016). Thus, Experiments 4 and 5 were carried out at 65 % RH, under high- and low-NO levels, respectively. These experiments were carried out over longer timescales (higher OH exposures) than the corresponding dry experiments to better probe multi-generational product formation.
Results from Exp. 4 (in which 50–100 ppb NO was maintained in the chamber) are shown in Fig. 5a. The overall product distribution is similar to that under dry conditions (Fig. 2a), with SO2, MSA, and sulfate being the major reaction products. The modeled product distribution shown in Fig. S11a is largely the same as that in the dry experiment (Fig. 2c), as water does not play a role in the gas-phase oxidation mechanism shown in Fig. 1. Even though this experiment was carried out over longer timescales, the measured sulfur closure is quite good, 107 % (99 %–171 %) of the reacted DMS at the end of the experiment.

Figure 5Results from the high-humidity (65 % RH) DMS oxidation experiments. (a) Product formation under high-NO conditions (Exp. 4). (b) Product formation under low-NO conditions (Exp. 5). Because of instrument downtime, no data were collected for the first 4 h of equivalent OH exposure. (c) Comparison of major species between the low-RH (Exp. 1) and high-RH experiment (Exp. 4) under high-NO conditions. (d) Comparison of major species between the low-RH (Exp. 2) and high-RH experiment (Exp. 5) under low-NO conditions. Changes in product concentrations are plotted against changes in DMS concentration over the initial 6 h of OH exposure, when data from both the dry and high-RH experiments were available.
Figure 5c compares the evolving concentrations of major product species under high- and low-RH conditions, presented as change in product concentration relative to change in DMS concentration, over the initial OH exposure (corresponding to that of Exp. 1). Over these timescales, species such as DMSO, SO2, and MSA showed a relatively small effect of RH. By contrast, almost no C2H6SO2 (likely DMSO2) was measured in the gas phase under high-RH conditions. Within the timescale of the experiments, our measurements do not suggest conversion of MSA to sulfate in the aerosol phase, as predicted in some modeling studies (Fung et al., 2022; Chen et al., 2018). This difference may arise from low particle-phase OH concentrations in our experiments.
Figure 5b shows products from Exp. 5 (65% RH, low-NO conditions, τbi>1 s). As in the low-RH, long-τbi case (Exp. 2a, Fig. 2b), HPMTF and SO2 are the dominant measured products, and little aerosol formation is observed. One minor new product, with formula SO6, was detected in the I− CIMS in this experiment; it is likely an adduct (i.e., O3 • SO3 • I−) or a fragment formed in the instrument, but the parent species is unknown. In contrast to the high-NO experiment (Exp. 4), sulfur closure was markedly worse than under dry conditions. In the first 6 h of equivalent OH exposure (the timescale of the dry experiment), only 74 % (53 %–97%) of the reacted sulfur was detected as products. This sulfur closure degraded still further as the experiment proceeded and was only 23 % (18 %–31 %) at the end of the experiment. Here, I− CIMS sensitivities derived from the dry calibration were used for species quantification and therefore may underestimate the concentration under high-RH conditions (Lee et al., 2014; Veres et al., 2020). However, these differences would have to be dramatic (by factor of 5 or more) to account for all the reacted sulfur, and therefore such calibration errors are unlikely to explain the decreased sulfur closure.
Figure 5d shows differences for key product species formed in the long-τbi experiments under the high- and low-RH conditions, again over the timescales of the dry experiment (the first 6 h of equivalent OH exposure). Over these timescales, the initial yields of DMSO, C2H6SO2, and HPMTF are not substantially different in the humid and dry cases. SO2 concentrations were lower under humid conditions but with an absolute difference of only ∼2 ppb. Thus the production rates of these species are not affected dramatically by RH level. Instead the poor sulfur closure at high-RH conditions suggests that extra losses over longer timescales may be most likely by uptake to surfaces. The low aerosol concentration towards the end of the experiment (due to particle wall loss over the long experimental time, ∼17 h) could lead to substantial chamber wall loss of low-volatility products, which would contribute to this gap in measured sulfur. Such surface losses are likely exacerbated at high-RH conditions, due to uptake into the aqueous phase. The initial aerosol liquid water content (LWC) in the high-RH experiment was 10–100 µg m3, orders of magnitude lower than LWC in maritime clouds (Wallace and Hobbs, 2006). Therefore, such losses may play an even more important role in the real atmosphere. Indeed, studies have suggested that uptake to cloud water may be an important sink of gas-phase HPMTF. Using in situ measurements, Vermeuel et al. (2020) and Novak et al. (2021) have shown that HPMTF is lost to clouds and aerosols effectively in the marine boundary layer. Similarly, using a global model, Fung et al. (2022) found that including cloud uptake into a global model substantially decreases the global burden of HPMTF, by up to 86 %. This uptake of water-soluble intermediate species (e.g., MSIA, DMSO2, and HPMTF) into cloud droplets may then contribute to the condensed-phase production of MSA and sulfate (Hoffmann et al., 2021; Novak et al., 2021), but such processes are not accessed in the present chamber experiment.
In this study, we conducted a series of chamber experiments to investigate the total product distribution from DMS oxidation at different RO2 fates and relative humidities. Under dry conditions, good sulfur closure was obtained, suggesting most of the sulfur-containing product species were accounted for. Under high-NO conditions (τbi<0.1 s), major products are SO2, MSA, and sulfate, whereas under low-NO conditions (τbi>10 s), HPMTF formed from RO2 isomerization makes up about half of the product sulfur, with very little MSA or sulfate formation. Comparisons between measurements and MCM predictions show relatively good agreement for most species and total aerosol formation. However, under high-NO conditions, the model predicts much more sulfate and less MSA than was measured; this might indicate errors in the kinetics of the reactions that lead to rapid (first-generation) MSA or sulfate formation. This work also provides new measurements of the rate constants (at 295 K) of key reactions in the DMS oxidation mechanism, including kisom (0.13±0.03 s−1), kHPMTF+OH ( cm3 molec.−1 s−1), and kMTF+OH ( cm3 molec.−1 s−1). Our measured value of kHPMTF+OH, which is consistent with that of Jernigan et al. (2022a), suggests that OH is a more important gas-phase sink of HPMTF than photolysis. Lastly, results from high-RH conditions suggest heterogeneous losses of at least some of the products, indicating that uptake into the atmospheric aqueous phase (e.g., cloud droplets) may be an important sink as well.
Taken together, our results show that RO2 fate has a controlling influence on the distribution of sulfur-containing products from DMS oxidation. In particular, the formation of HPMTF from RO2 isomerization suppresses (or at least delays) the gas-phase formation of SO2, sulfate, and MSA. Additional studies are needed to constrain the temperature dependence of kisom to predict the formation of HPMTF (and other products) in warmer or colder environments, as well as to characterize the full multiphase product distribution under higher-RH conditions. In addition, experiments carried out over longer oxidation timescales and with different oxidants are needed to better understand the amount and rate of aerosol formation over days of oxidation. A related need is improved constraints on the atmospheric fate of HPMTF and other key reaction intermediates (e.g., DMSO, MSIA), including rates and products of gas-phase oxidation, aqueous-phase oxidation, and photolysis, as well as rates of physical loss (deposition and uptake).
Chamber data and species concentrations for all experiments and model outputs have been archived and are available via the Kroll Group publication website at http://krollgroup.mit.edu/publications.html (Kroll Group, 2022). The F0AM model used in this work is publicly available at https://github.com/AirChem/F0AM/releases/tag/v3.2 (Wolfe, 2019; Wolfe et al., 2016), and the latest release is available at https://zenodo.org/record/6984581 (Wolfe et al., 2022).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-16003-2022-supplement.
QY, MBG, JEK, FM, AZ, YL, and JRR collected the data. QY and MBG analyzed the data. MBG performed box model simulations. QY and JHK wrote the manuscript. MC, FNK, CLH, and JHK provided project guidance. All authors were involved in helpful discussion and contributed to the manuscript.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors thank Timothy Bertram, Gordon Novak, and Chris Jernigan at the University of Wisconsin–Madison for insightful discussions.
This work was supported by the U.S. Department of Energy Biological and Environmental Research program (grant no. DE-SC0018934) and the Harvard Global Institute.
This paper was edited by Sergey A. Nizkorodov and reviewed by three anonymous referees.
Arsene, C., Barnes, I., Becker, K. H., Schneider, W. F., Wallington, T. T., Mihalopoulos, N., and Patroescu-Klotz, I. V.: Formation of methane sulfinic acid in the gas-phase OH-radical initiated oxidation of dimethyl sulfoxide, Environ. Sci. Technol, 36, 5155–5163, 2002.
Barnes, I., Becker, K. H., and Patroescu, I.: The tropospheric oxidation of dimethy sulfide: a new source of carbonyl sulfide, Geophys. Res. Lett., 21, 2389–2392, 1994.
Barnes, I., Hjorth, J., and Mihalapoulos, N.: Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529+, 2006.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019.
Charlson, R. J., Lovelock, J. E., Andreae, M. O., and Warren, S. G.: Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate, Nature, 326, 665–661, 1987.
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018.
Chen, T. and Jang, M.: Chamber simulation of photooxidation of dimethyl sulfide and isoprene in the presence of NOx, Atmos. Chem. Phys., 12, 10257–10269, https://doi.org/10.5194/acp-12-10257-2012, 2012.
Chin, M., Jacob, D. J., Gardner, G. M., Foreman-fowler, M. S., Spiro, P. A., and Savoie, D. L.: A global three-dimensional model of tropospheric sulfate, J. Geophys. Res., 101, 18667–18690, 1996.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, 2013.
Decarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Fung, K. M., Heald, C. L., Kroll, J. H., Wang, S., Jo, D. S., Gettelman, A., Lu, Z., Liu, X., Zaveri, R. A., Apel, E. C., Blake, D. R., Jimenez, J.-L., Campuzano-Jost, P., Veres, P. R., Bates, T. S., Shilling, J. E., and Zawadowicz, M.: Exploring dimethyl sulfide (DMS) oxidation and implications for global aerosol radiative forcing, Atmos. Chem. Phys., 22, 1549–1573, https://doi.org/10.5194/acp-22-1549-2022, 2022.
Hodshire, A. L., Campuzano-Jost, P., Kodros, J. K., Croft, B., Nault, B. A., Schroder, J. C., Jimenez, J. L., and Pierce, J. R.: The potential role of methanesulfonic acid (MSA) in aerosol formation and growth and the associated radiative forcings, Atmos. Chem. Phys., 19, 3137–3160, https://doi.org/10.5194/acp-19-3137-2019, 2019.
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016.
Hoffmann, E. H., Heinold, B., Kubin, A., Tegen, I., and Herrmann, H.: The Importance of the Representation of DMS Oxidation in Global Chemistry-Climate Simulations, Geophys. Res. Lett., 48, e2021GL094068, https://doi.org/10.1029/2021GL094068, 2021.
Huang, D. D., Li, Y. J., Lee, B. P., and Chan, C. K.: Analysis of Organic Sulfur Compounds in Atmospheric Aerosols at the HKUST Supersite in Hong Kong Using HR-ToF-AMS, Environ. Sci. Technol., 49, 3672–3679, https://doi.org/10.1021/es5056269, 2015.
Huijnen, V., Williams, J., van Weele, M., van Noije, T., Krol, M., Dentener, F., Segers, A., Houweling, S., Peters, W., de Laat, J., Boersma, F., Bergamaschi, P., van Velthoven, P., Le Sager, P., Eskes, H., Alkemade, F., Scheele, R., Nédélec, P., and Pätz, H.-W.: The global chemistry transport model TM5: description and evaluation of the tropospheric chemistry version 3.0, Geosci. Model Dev., 3, 445–473, https://doi.org/10.5194/gmd-3-445-2010, 2010.
Hunter, J. F., Carrasquillo, A. J., Daumit, K. E., and Kroll, J. H.: Secondary Organic Aerosol Formation from Acyclic, Monocyclic, and Polycyclic Alkanes, Environ. Sci. Technol., 48, 10227–10234, 2014.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, 1997.
Jernigan, C. M., Fite, C. H., Vereecken, L., Berkelhammer, M. B., Rollins, A. W., Rickly, P. S., Novelli, A., Taraborrelli, D., Holmes, C. D., and Bertram, T. H.: Efficient production of carbonyl sulfide in the low-NOx oxidation of dimethyl sulfide, Geophys. Res. Lett., 49, https://doi.org/10.1029/2021GL096838, 2022a.
Jernigan, C. M., Christopher, D. C., and Timothy H. B.: Reactive Uptake of Hydroperoxymethyl Thioformate to Sodium Chloride and Sodium Iodide Aerosol Particles, J. Phys. Chem. A, 126, 4476–4481, 2022b.
Khan, M. A. H., Bannan, T. J., Holland, R., Shallcross, D. E., Archibald, A. T., Matthews, E., Back, A., Allan, J., Coe, H., Artaxo, P., and Percival, C. J.: Impacts of Hydroperoxymethyl Thioformate on the Global Marine Sulfur Budget, ACS Earth Sp. Chem., 5, 2577–2586, 2021.
Kloster, S., Feichter, J., Maier-Reimer, E., Six, K. D., Stier, P., and Wetzel, P.: DMS cycle in the marine ocean-atmosphere system – a global model study, Biogeosciences, 3, 29–51, https://doi.org/10.5194/bg-3-29-2006, 2006.
Krechmer, E. J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and Gouw, J. De: Evaluation of a New Reagent-Ion Source and Focusing Ion- Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Kroll Group: Chamber data and species concentrations, http://krollgroup.mit.edu/publications.html, last access: 2022.
Lamarque, J.-F., Emmons, L. K., Hess, P. G., Kinnison, D. E., Tilmes, S., Vitt, F., Heald, C. L., Holland, E. A., Lauritzen, P. H., Neu, J., Orlando, J. J., Rasch, P. J., and Tyndall, G. K.: CAM-chem: description and evaluation of interactive atmospheric chemistry in the Community Earth System Model, Geosci. Model Dev., 5, 369–411, https://doi.org/10.5194/gmd-5-369-2012, 2012.
Lucas, D. D. and Prinn, R. G.: Mechanistic studies of dimethylsulfide oxidation products using an observationally constrained model, J. Geophys. Res.-Atmos., 107, ACH-12, https://doi.org/10.1029/2001JD000843, 2002.
Lana, A., Bell, T. G., Simo, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrationsand emission fluxes in the global ocean, Global Biogeochem. Cycles, 25, GB1004, https://doi.org/10.1029/2010GB003850, 2011.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
McManus, J. B., Kebabian, P. L., and Zahniser, M.S.: Astigmatic mirror multipass absorption cells for long-path-length spectroscopy, Appl. Optics, 34, 3336–3348, 1995.
McManus, J. B., Zahniser, M. S., Nelson, D. D., McGovern, R. M., Agnese, M., and Brown, W. F.: Compact Quantum Cascade Laser Instrument for High Precision Trace Gas Measurements, in: Optical Instrumentation for Energy and Environmental Applications (p. EThC2), Optica Publishing Group, https://doi.org/10.1364/E2.2011.EThC2, 2011.
Mihalopoulos, N., Barnes, I., and Becker, K. H.: Infrared absorption spectra and integrated band intensities for gaseous methanesulphonic acid (MSA), Atmos. Environ., 25, 807–812, 1992.
Novak, G. A., Fite, C. H., Holmes, C. D., Veres, P. R., Neuman, J. A., Faloona, I., Thornton, J. A., Wolfe, G. M., Vermeuel, M. P., Jernigan, C. M., Peischl, J., Ryerson, T. B., Thompson, C. R., Bourgeois, I., Warneke, C., Gkatzelis, G. I., Coggon, M. M., Sekimoto, K., Bui, T. P., Dean-Day, J., Diskin, G. S., DiGangi, J. P., Nowak, J. B., Moore, R. H., Wiggins, E. B., Winstead, E. L., Robinson, C., Thornhill, K. L., Sanchez, K. J., Hall, S. R., Ullmann, K., Dollner, M., Weinzierl, B., Blake, D. R., and Bertram, T. H.: Rapid cloud removal of dimethyl sulfide oxidation products limits SO2 and cloud condensation nuclei production in the marine atmosphere, P. Natl. Acad. Sci., 118, e2110472118, https://doi.org/10.1073/PNAS.2110472118, 2021.
Patroescu, I. V., Barnes, I., and Becker, K. H.: FTIR kinetic and mechanistic study of the atmospheric chemistry of methyl thiolformate, J. Phys. Chem., 100, 17207–17217, https://doi.org/10.1021/jp961452u, 1996.
Rap, A., Scott, C. E., Spracklen, D. V., Bellouin, N., Forster, P. M., Carslaw, K. S., Schmidt, A., and Mann, G.: Natural aerosol direct and indirect radiative effects, Geophys. Res. Lett., 40, 3297–3301, https://doi.org/10.1002/grl.50441, 2013.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Shen, J., Scholz, W., He, X. C., Zhou, P., Marie, G., Wang, M., Marten, R., Surdu, M., Rörup, B., Baalbaki, R. and Amorim, A, Ataei, F., Bell, D. M., Bertozzi, B., Brasseur, Z., Caudillo, L., Chen, D., Chu, B., Dada, L., Duplissy, J., Finkenzeller, H., Granzin, M., Guida, R., Heinritzi, M., Hofbauer, V., Iyer, S., Kemppainen, D., Kong, W., Krechmer, J. E., Kürten, A., Lamkaddam, H., Lee. C. P., Lopez, B., Mahfouz, N. G., Manninen, H. E., Massabò, D., Mauldin, R. L., Mentler, B., Müller, T., Pfeifer, J., Phillppov, M., Piedehierro, A. A., Roldin, P., Schobesberger, S., Simon, M., Stolzenburg, D., Tham, Y. J., Tomé, A., Umo, N. S., Wang, D., Wang, Y., Weber, S. K., Welti, A., Wollesen de Jonge, R., Wu, Y., Zauner-Wieczorek, M., Zust, F., Baltensperger, U., Curtius, J., Flagan, R. C., Hansel, A., Möhler, O., Petäjä, T., Volkamer, R., Kulmala, M., Lehtipalo, K., Rissanen, M., Kirkby, J., El-Haddad, I., Bianchi, F., Silipä, M., Donahue, N. M., and Worsnop, D. R.: High Gas-Phase Methanesulfonic Acid Production in the OH-Initiated Oxidation of Dimethyl Sulfide at Low Temperatures, Environ. Sci. Technol., 56, 13931–13944, 2022.
Veres, P. R., Andrew Neuman, J., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J. F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J., Paul Bui, T., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, P. Natl. Acad. Sci. USA, 117, 4505–4510, https://doi.org/10.1073/pnas.1919344117, 2020.
Vermeuel, M. P., Novak, G. A., Jernigan, C. M., and Bertram, T. H.: Diel profile of hydroperoxymethyl thioformate: Evidence for surface deposition and multiphase chemistry, Environ. Sci. Technol, 54, 12521–12529, 2020.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
Wallace, J. M. and Hobbs, P. V.: Atmospheric science: an introductory survey, U. K. Elsevier Inc., ISBN 9780127329512, 2006.
Wolfe, G.: F0AM model, https://github.com/AirChem/F0AM/releases/tag/v3.2, last access: 2019.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wolfe, G., Haskins, J., and srobe: AirChem/F0AM: v4.2.2 (v4.2.2), Zenodo [code], https://doi.org/10.5281/zenodo.6984581, 2022.
Wollesen de Jonge, R., Elm, J., Rosati, B., Christiansen, S., Hyttinen, N., Lüdemann, D., Bilde, M., and Roldin, P.: Secondary aerosol formation from dimethyl sulfide – improved mechanistic understanding based on smog chamber experiments and modelling, Atmos. Chem. Phys., 21, 9955–9976, https://doi.org/10.5194/acp-21-9955-2021, 2021.
Wu, R., Wang, S., and Wang, L.: New mechanism for the atmospheric oxidation of dimethyl sulfide. The importance of intramolecular hydrogen shift in a CH3SCH2OO radical, J. Phys. Chem. A, 119, 112–117, https://doi.org/10.1021/jp511616j, 2015.
Xu, L., Møller, K. H., Crounse, J. D., Otkjær, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular reactions of peroxy radicals formed in the oxidation of α-Pinene and β-Pinene by hydroxyl radicals, J. Phys. Chem. A, 123, 1661–1674, https://doi.org/10.1021/acs.jpca.8b11726, 2019.
Ye, Q., Goss, M. B., Isaacman-Vanwertz, G., Zaytsev, A., Massoli, P., Lim, C., Croteau, P., Canagaratna, M., Knopf, D. A., Keutsch, F. N., Heald, C. L., and Kroll, J. H.: Organic Sulfur Products and Peroxy Radical Isomerization in the OH Oxidation of Dimethyl Sulfide, ACS Earth Sp. Chem., 5, 2013–2020, https://doi.org/10.1021/acsearthspacechem.1c00108, 2021.
Yin, F., Grosjean, D., and Seinfeld, J. H.: Photooxidation of Dimethyl Sulfide and Dimethyl Disulfide. I: Mechanism Development, J. Atmos. Chem., 11, 309–365, 1990.
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019.