the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Nov 2022
| 04 Nov 2022
Chamber investigation of the formation and transformation of secondary organic aerosol in mixtures of biogenic and anthropogenic volatile organic compounds
Aristeidis Voliotis
Yunqi Shao
M. Rami Alfarra
Thomas J. Bannan
Dawei Hu
Kelly L. Pereira
Jaqueline F. Hamilton
Mattias Hallquist
Thomas F. Mentel
Gordon McFiggans
A comprehensive chamber investigation of photochemical secondary organic aerosol (SOA) formation and transformation in mixtures of anthropogenic (o-cresol) and biogenic (α-pinene and isoprene) volatile organic compound (VOC) precursors in the presence of NOx and inorganic seed particles was conducted. To enable direct comparison across systems, the initial concentration (hence reactivity) of the systems towards the dominant OH oxidant was adjusted. Comparing experiments conducted in single-precursor systems at various initial reactivity levels (referenced to a nominal base case VOC concentration, e.g. halving the initial concentration for a initial reactivity experiment) as well as their binary and ternary mixtures, we show that the molecular interactions from the mixing of the precursors can be investigated and discuss challenges in their interpretation. The observed average SOA particle mass yields (the organic particle mass produced for a mass of VOC consumed) in descending order were found for the following systems: α-pinene (32 ± 7 %), α-pinene–o-cresol (28 ± 9 %), α-pinene at initial reactivity (21 ± 5 %), α-pinene–isoprene (16 ± 1 %), α-pinene at initial reactivity (15 ± 4 %), o-cresol (13 ± 3 %), α-pinene–o-cresol–isoprene (11 ± 4 %), o-cresol at initial reactivity (11 ± 3 %), o-cresol–isoprene (6 ± 2 %), and isoprene (0 ± 0 %). We find a clear suppression of the SOA mass yield from α-pinene when it is mixed with isoprene, whilst no suppression or enhancement of SOA particle yield from o-cresol was found when it was similarly mixed with isoprene. The α-pinene–o-cresol system yield appeared to be increased compared to that calculated based on the additivity, whilst in the α-pinene–o-cresol–isoprene system the measured and predicted yields were comparable. However, in mixtures in which more than one precursor contributes to the SOA particle mass it is unclear whether changes in the SOA formation potential are attributable to physical or chemical interactions, since the reference basis for the comparison is complex. Online and offline chemical composition as well as SOA particle volatility, water uptake, and “phase” behaviour measurements that were used to interpret the SOA formation and behaviour are introduced and detailed elsewhere.
- Article
(3010 KB) - Full-text XML
- Companion paper 1
- Companion paper 2
- Companion paper 3
-
Supplement
(1207 KB) - BibTeX
- EndNote





The fine fraction of particulate matter (PM) plays the dominant role in the impact of air pollution on human health and of aerosol on climate through direct radiative effects and cloud adjustments. Ambient PM2.5 was the fifth-ranking global mortality risk factor in 2015, with exposure to it causing 4.2 million deaths and 103.1 million disability-adjusted life years (DALYs), which is 7.6 % of total global deaths and 4.2 % of global DALYs (Cohen et al., 2017). Moreover, fine PM is responsible for the aerosol effects that make the single greatest contributory uncertainty to radiative forcing (IPCC, 2021).
Organic material makes a major contribution to the mass of fine PM in the atmosphere (Jimenez et al., 2009), and secondary organic aerosol (SOA) is the major contributor (Hallquist et al., 2009). Nonetheless, our ability to predict the atmospheric burden and hence impacts of fine secondary aerosol particles (Kanakidou et al., 2005; Tsigaridis and Kanakidou, 2018) has been limited by basic understanding of the formation of this organic component (Hallquist et al., 2009). Photochemistry dictates the levels of NO2 and O3, and it influences the SOA fraction of PM. It is relatively straightforward to understand and control primary pollutants. However, these secondary pollutants make a substantial contribution to air quality degradation and are set to become increasingly important as primary pollutants are cleaned up. However, understanding the rate and extent of SOA formation in the real atmosphere presents several challenges. There are tens of thousands of organic compounds in the atmosphere, ranging across more than 12 orders of magnitude in volatility (Goldstein and Galbally, 2007) with possible oxidation products running to millions (Aumont et al., 2005). They have extensive biogenic and anthropogenic sources and are spatially heterogeneous. Their reactivity ranges over many orders of magnitude, and their lifetimes at ambient oxidant levels range from less than a second to several years. An unknown but substantial proportion of the organic compounds have the potential to act as SOA precursors, and the degree to which this is influenced by the complex atmospheric mixture is unclear.
It has long been established that prediction of the formation of secondary gaseous pollutants in the troposphere requires knowledge of the nature of the mixture of volatile organic compounds (VOCs), their abundance, chemical regime, and air mass history (see e.g. Jenkin et al., 2017). In contrast, the formation of SOA in the real atmosphere is conventionally considered less mechanistically (see Shrivastava et al., 2017, for a discussion of neglected complexities and non-linearities) and frequently more empirically. Against the backdrop of this massive complexity of the atmospheric mixture of SOA precursors, the basis for our understanding of SOA formation has been primarily derived from experimental investigations of single-component systems (Thornton et al., 2020; Donahue et al., 2012; Jenkin et al., 2012). A wealth of literature derived from chamber experiments on biogenic (Thornton et al., 2020; Carlton et al., 2009) and anthropogenic (Schwantes et al., 2017; Nakao et al., 2012) precursors under a range of chemical environments combined with fundamental kinetic studies (Ziemann and Atkinson, 2012; Cash et al., 2016) has enabled numerous ways of representing the formation and transformation of atmospheric SOA at varying levels of detail (Shrivastava et al., 2017; Charan et al., 2019). There have additionally been studies of SOA formation in source-oriented mixtures from diesel (Weitkamp et al., 2007; Nakao et al., 2011a) and gasoline (Nordin et al., 2013; Platt et al., 2013) exhaust, woodburning (Tiitta et al., 2016), cooking (Reyes-Villegas et al., 2018; Kaltsonoudis et al., 2017), and macroalgal (McFiggans et al., 2004) and plant (Joutsensaari et al., 2005; VanReken et al., 2006; Pinto et al., 2007; Mentel et al., 2009; Hao et al., 2009; Wyche et al., 2014) emissions. Building on a well-established framework first suggested by Pankow (1994) to account for absorptive partitioning of mixtures of organic components in the atmosphere, attempts have been made to provide empirically constrained and mechanistically augmented conceptual frameworks of organic aerosol (Donahue et al., 2006, 2011; Schervish and Donahue, 2020), but there is currently no universally accepted mechanistic basis for SOA understanding in the complex atmosphere.
It was established that the scavenging of the OH radical by addition of isoprene significantly inhibits new particle formation in plant chamber studies (Kiendler-Scharr et al., 2009), and subsequently Berndt et al. (2018) showed that the presence of isoprene could inhibit the production of dimers that can be involved in nucleation or early particle formation. More recent studies have indicated that the mass and yield of SOA formed from individual precursors may similarly be influenced by the presence of other VOCs (McFiggans et al., 2019), suggesting that it is necessary to take a more mechanistic approach in order to capture observed behaviour of SOA formation in mixtures. In addition to the observed suppression of particle mass and yield from α-pinene being attributed to the established scavenging of oxidant by the lower SOA yield isoprene, it was found that C5 peroxy radical isoprene oxidation products scavenged highly oxygenated C10 peroxy radicals that would otherwise form extremely low-volatility condensable compounds. This had also been reported by Berndt et al. (2018), who demonstrated a similar scavenging of C10 radical products in the presence of ethylene and isoprene. McFiggans et al. (2019) further reported such product scavenging and consequent reduction in α-pinene SOA yield by CO and CH4. Such interactions should be no surprise, given our understanding of peroxy radical cross-reactions and termination fates in controlling production of secondary gaseous pollutants. Moreover, the existence of such interactions in mixtures is somewhat obvious in light of the understanding that has emerged over recent years (e.g. Bianchi et al., 2019) of the roles of atmospheric autoxidation of VOCs producing highly oxygenated organic molecules (HOMs). Following the early postulations of the role of autoxidation in atmospheric VOC degradation (Crounse et al., 2013), the importance of HOMs in SOA formation has been widely established and quantified in monoterpene oxidation (Ehn et al., 2012, 2014; Jokinen et al., 2015; Berndt et al., 2016; Berndt et al., 2018; Bianchi et al., 2019). The termination of the RO2 formed via autoxidation (“HOM–RO2”) will depend on the chemical conditions (the abundance of NOx, HO2, and the numerous other RO2 species present in the mixture) as well as the formation rate of HOM–RO2. Schervish and Donahue (2020) provide a discussion of the determinants of the fate of HOM–RO2 and potential consequences for the distribution of low-, extremely low-, and ultra-low-volatility organic compound (LVOC, ELVOC, and ULVOC, respectively) products and hence for SOA formation in α-pinene oxidation. Autoxidation is widespread and common, and, in atmospheric systems, it is unlikely to be restricted to monoterpenes (though unsaturated compounds may be expected to be more susceptible; see Bianchi et al., 2019). Indeed, the application of recently developed mass spectrometric techniques has revealed the presence of HOMs in the oxidation of aromatic compounds (Wang et al., 2017, 2020; Molteni et al., 2018; Tsiligiannis et al., 2019; Garmash et al., 2020; Mehra et al., 2020; Priestley et al., 2021). The degree to which the distribution of highly oxygenated compounds results from direct autoxidation or from multigenerational pathways is unclear, though the prevalence of large highly oxygenated RO2 during aromatic oxidation is clear. The attention on aromatic compounds has been driven by their relevance as anthropogenic VOC emissions, making significant manmade contributions to the VOC burden in the polluted troposphere. Given the prevalence and diversity of autoxidation mechanisms, it is probable that HOMs would be detected through the application of modern mass spectrometry to the investigation of many classes of atmospheric VOCs. The recent focus on the products of autoxidation in mixtures highlights the need to incorporate them alongside the more well-established products of the oxidation of VOCs when understanding gas-phase photochemistry. The implications for production of aerosol particle precursors are less well studied. Since the fractional contribution of HOMs to particle mass is unclear, the role of autoxidation and influences of interactions for HOM contributions to SOA mass formation in mixtures is unquantified. It is therefore important to additionally maintain a focus on influences of the non-HOM components on SOA formation in oxidation of VOC mixtures.
Given the potential diversity in VOC sources contributing to the pollutant mixture in the ambient atmosphere, it is important to establish the experimental basis for an understanding of SOA formation beyond the recently studied “simple” biogenic mixed systems (Berndt et al., 2018; McFiggans et al., 2019; Shilling et al., 2019). The importance of establishing a framework to understand interactions in systems of mixed anthropogenic and biogenic VOCs stretches well beyond speculative curiosity. Such a framework may be a key to explaining observed non-linearities when natural and manmade pollution mixes (Spracklen et al., 2011; Emanuelsson et al., 2013). Using such considerations as motivation, a series of experiments was conceived to explore SOA formation from typical biogenic and anthropogenic VOC precursors as well as their binary and ternary mixtures. The binary α-pinene–isoprene system reported elsewhere was used as a biogenic mixture with established mechanistic interactions in its photo-oxidation. o-Cresol was chosen as a representative anthropogenic VOC, being both emitted as a primary pollutant and formed through oxidation of other aromatic compounds. The construction of the study is detailed in the Methodology section below. The experiments were conducted in the 18 m3 Manchester Aerosol Chamber (MAC), a Teflon photochemical chamber operating in batch mode. The design of experiments on mixed VOCs is complex and requires consideration of aspects not encountered in single-VOC experiments. This paper explores many of these elements and associated challenges with a view to inform best practice in their planning and executions. The objectives were broad and diverse, and they aimed to do the following:
- i.
establish the suitability of the experimental design for investigating SOA formation in VOC mixtures;
- ii.
establish the suitability of conventionally reported metrics such as SOA mass yield for quantifying SOA formation in VOC mixtures;
- iii.
use these metrics to quantify any interactions in VOC mixtures leading to changes in SOA formation that may be expected based on single-precursor experiments; and
- iv.
use a suite of online and offline measurements of chemical and physical properties to probe the chemistry and physics leading to these interactions and the resulting particle properties of potential atmospheric significance.
This paper will address the first three of these and provide an introduction to the findings related to the fourth, which have been detailed in a number of published companion papers (Du et al., 2022a; Shao et al., 2022a, b; Voliotis et al., 2021; Wang et al., 2021, 2022; Voliotis et al., 2022) and (Du et al., 2022b; Shao et al., 2022d). It is envisaged that the experimental programme will act as a springboard to investigate the detailed mechanisms of interactions in mixtures of VOCs involved in ambient SOA formation.
Importantly, this paper aims to explore whether SOA experiments using multiple VOC precursors can reveal aspects of the multiphase atmospheric systems inaccessible to experiments using a single precursor species.
The mixture of atmospheric VOCs and variability in the prevailing oxidising environment are complex, and the choice of precursors, concentrations, and experimental conditions cannot be comprehensive. In addition to representativeness of atmospheric complexity and concentrations in the multiphase system, numerous infrastructural and measurement considerations must be accommodated. There are additional compromises in fully addressing specific objectives of the study. In this section, these criteria and the bounds of the study are addressed, and the experimental design in response to these requirements is discussed along with the choices made. Finally, the capabilities, consequences, and compromises inherent in these choices are listed before outlining the methodology employed.
2.1 VOC mixture and oxidant choices
Of the tens of thousands (or perhaps millions) of atmospheric organic compounds present in the vapour phase, a handful have been studied in previous chamber SOA experiments and fewer in mixtures. The selection of compounds for the study of mixed SOA precursors is simultaneously simple and complex in that study of a random mixture might be expected to yield novel results. However, care is required to interpret their importance and atmospheric relevance. Disregarding chlorine atoms and the Criegee radicals as “exotic”, the dominant oxidants at night are the nitrate radical (NO3) and ozone, and during the day, they are the hydroxyl radical (OH) and ozone. Whilst arguments (such as possible dominance of daytime oxidising capacity and pathways to SOA over night-time) have sometimes been used for oxidant selection, this is fairly arbitrary, and we do not invoke such justification for the choice of daytime oxidation conditions for the present study. Similarly arbitrary is the selection of NOx regime. Whilst molar VOC : NOx ratio dependence of SOA formation from some precursors has been well studied (e.g. Pullinen et al., 2020), this is not the case for mixtures of VOCs. It has been contended for certain systems that SOA formation pathways in low- and high-NOx regimes change dramatically and consequently influence SOA particle mass yield (e.g. Sarrafzadeh et al., 2016). Differences in experimental configurations and conditions lead to apparently conflicting findings, and controversies remain surrounding the influence of NOx on SOA formation, requiring unambiguous mechanistic resolution. Truly low-NOx regimes occur in limited locations in the ambient atmosphere, and not in the perturbed Northern Hemisphere. With inclusion of an anthropogenic SOA precursor in the mixture, it would be unreasonable to expect there to be a complete absence of NOx. This study uses moderate VOC : NOx ratios (molar ratios between roughly 3 and 8) broadly corresponding to a perturbed background chemical regime.
In this study we initiate photochemistry through photolysis of NO2 under the simulated solar irradiation in the MAC to form O3 such that it rapidly moves towards photostationary state (PSS) with NO, according to Reactions (R1), (R2), and (R3).
The O3 is photolysed at wavelengths below 310 nm to yield O(3P) and O(1D), with the latter Reaction (R4) reacting with available water vapour to form the hydroxyl radical, OH (Reaction R5).
The O3 will attack the double bond in unsaturated compounds such as α-pinene and isoprene, initiating oxidation and yielding secondary OH. The OH will attack all VOCs in the system, either by hydrogen abstraction or OH addition. A currently unquantified contribution to the OH concentration will likely be contributed by the release from the walls of nitrous acid (HONO), as has been seen in previous studies in Teflon reactors under illumination (Rohrer et al., 2005; Ródenas et al., 2013). This is under investigation and will influence the photochemical environment in the MAC, but it does not directly change the results reported here.
The reactivity of gaseous VOCs towards ambient oxidants spans several orders of magnitude. The ratio of reactivity towards each oxidant varies substantially. Reactivity towards ozone will be negligible for saturated VOCs but can lead to lifetimes of a few minutes for reactive sesquiterpenes, for example. In order for each VOC in a mixture to contribute significantly to the distribution of oxidation products in a chamber experiment and thereby influence the pathways to and potential for SOA formation, it is desirable for them to have comparable reactivity towards the available oxidants. In such a mixture, the oxidation of similar amounts of each VOC might be reasonably expected to provide a comparable rate of change of oxidation products into the mix. These would then have the opportunity to contribute to SOA production pathways, serving as a useful basis for the current experiments.
Choice of the VOCs determines the level and mixture of atmospheric components that are employed to approximately represent a given atmospheric situation. McFiggans et al. (2019), in their choice of α-pinene and isoprene, looked at binary mixtures of abundant biogenically emitted VOCs (as did Jaoui and Kamens, 2003, in looking at mixtures of α- and β-pinene). Much of the global atmosphere is perturbed by anthropogenic pollutants, and contributions from natural and manmade sources will depend upon the mixture and strength of, as well as distance from, upwind sources (amongst many factors). This study builds on the previous binary mixture of low- and high-yield biogenic SOA precursors (isoprene and α-pinene), adding a moderate-yield anthropogenic VOC. Studies could equally be focused on a range of high-, low-, or moderate-SOA-yield anthropogenic VOCs or any other combinations. Extension and broadening of the current approach is of substantial interest, but the experiments in this study were designed solely to investigate one anthropogenic VOC and two biogenic VOCs as well as their mixtures. The biogenic VOCs that were chosen were α-pinene and isoprene to enable comparison of batch reactor experiments with previous flow tube (Berndt et al., 2018), well-mixed, continuously stirred tank reactor (McFiggans et al., 2019) or flow-through chamber studies (Shilling et al., 2019). The anthropogenic aromatic OVOC o-cresol was chosen for its fairly rapid rate constant with OH ( cm3 molec.−1 s−1) such that it would exhibit comparable reactivity towards OH as a similar concentration of α-pinene or isoprene. A concept of “initial isoreactivity” towards OH was employed to select ratios of the initial concentrations of the VOCs. A lower concentration of a more reactive VOC was injected than of a less reactive one in a mixture, whilst the overall initial mixture reactivity was preserved. The intention behind this approach was to ensure that the production rates of first-generation oxidation products were broadly comparable and available to participate in SOA particle mass formation: the initial destruction rate of each VOC and their first-generation oxidation product production rate will be the same, enabling the opportunity to explore interactions in the mixed systems. This means that the amounts are added in an α-pinene : isoprene : o-cresol ratio of 309 : 164 : 400 based on the ratios of the inverse of their IUPAC-recommended rate constants at 298 K (Atkinson et al., 2006; Mellouki et al., 2021). It should be noted that, owing to the means of initiating photochemistry and indirect production of OH, it was not guaranteed at the outset that most of the VOC consumption would be attributable to OH reaction owing to the reactivity towards ozone of the unsaturated biogenic VOCs. Further, the turnover of products from each VOC will depend on this loss rate and the reactivity of their oxidation products to the prevailing oxidants, so isoreactivity will not be preserved throughout the experiments. In order to construct a systematic investigation of the single-precursor, binary, and ternary mixtures, individual VOC experiments were attempted at full reactivity, half-reactivity, and one-third reactivity for comparison of SOA particle mass, composition, and properties. This was to enable comparison of the gas or particle components present in the half-reactivity individual VOC experiment with those from this same VOC in a binary experiment or those in the one-third-reactivity individual VOC experiment with those from this same VOC in a ternary experiment.
As with reactivity, the SOA particle mass yield of VOCs can vary widely, from unmeasurably low to several tens of percent. Intuitively, the presence of a higher-yield VOC might be expected to contribute more mass than a lower-yield VOC and consequently increase the yield of the mixture. What is less clear is the degree to which a lower-yield component might reduce the yields of a higher-yield VOC and vice versa – i.e. how strongly is the chemistry coupled, and how do such interactions influence SOA particle formation? More fundamentally, the reference for and calculation of yields in VOC mixtures are challenging depending on the often arbitrarily selected definition; does consumption of all VOCs count, or just of those expected to be SOA precursors (see the results and discussion in Sects. 4 and 5)? Using previously reported values for the SOA particle mass yields, it was expected that an initial full-reactivity mixing ratio in the hundreds of parts per billion by volume range would provide the 10 µg m−3 (o-cresol, Henry et al. 2008; Nakao et al., 2011b) to a few hundred micrograms per cubic metre (α-pinene, Eddingsaas et al., 2012) required to provide enough particle mass on the filter at the end of an experiment (at a final chamber volume of around 10 m3) for molecular characterisation.
2.2 Infrastructural and instrumentational considerations
The SOA precursors in this study were mixtures of VOCs – i.e. compounds almost entirely in the vapour phase under normal temperate surface atmospheric conditions. Use of VOCs straightforwardly enables use of the heated glass-bulb arrangement in the MAC for the injection and vaporisation of small quantities of liquid organic components. This widespread approach enables comparability with other studies using conventional SOA precursors. Modification of the injection system could enable use of solid stock for studies of mixtures of “unconventional” SOA precursors that have had much recent attention, such as those falling into the intermediate-volatility (IVOC) and semi-volatile (SVOC) classes.
All experiments employed ammonium sulfate seed particles. Ammonium sulfate is one of the most abundant electrolytes in the atmosphere and a component readily and reliably determined by instrumentation reliant on its volatilisation (to contrast with, for example, sodium chloride). Use of seed particles was primarily to enable greater reproducibility by providing sufficient particulate mass to act as a condensation sink for partitioning compounds, suppressing nucleation and its attendant high sensitivities to small perturbations in experimental conditions. In chamber experiments, there is a competition for condensable vapours between the walls and any existing particles such that seeded experiments enable earlier formation of, and hence greater, SOA particle mass. Moreover, detection of this particle mass and determination of its composition and properties are more straightforward with instruments that require particles to be greater than a certain size. Whilst we previously pioneered the technique of generation of SOA particle seeds for use in chamber experiments (Hamilton et al., 2011), use of ammonium sulfate avoids the additional complication of the chemistry by residual VOC oxidation products. Ammonium sulfate solution is nebulised into a stainless-steel retaining drum prior to mixing into the final fill cycle for the experiment (see Sect. 2.4 below). It is recognised that inorganic seeds may not present the most effective absorptive medium for condensational uptake of organic vapours. Our seed generation process is highly reproducible, and an implicit assumption of the likelihood of comparable efficiency as an absorptive medium across the systems of choice is made. All seeds were nebulised from stock solutions, likely existing as metastable aqueous particles at the nominal relative humidity (RH) of 50 % in the chamber. At the varying SOA : seed ratio across the systems, the partitioning through uptake to the strong aqueous solutions as well as subsequent dissolution and absorption may well differ. Moreover, acidic seeds are not accessible in the MAC because uptake of background ambient NH3 leads to neutralisation of H+ at any reasonable seed mass loading. Acid-catalysed reactions may be of greater or lesser importance across systems. Because of these complications, the assumption of similar efficiency of inorganic seeds as an absorptive medium may not hold and should be a focus of further studies.
Instrumental detection limits and sampling requirements dictate the accessible range of concentrations for the experiments, though many of the techniques employed are capable of high-precision and high-time-resolution measurement at low concentrations. Most employ online or semi-continuous operation, and low concentrations could be readily accommodated by increasing instrument integration times. Filters are collected for molecular determination of SOA particle composition by the evacuation of the entire chamber through pre-fired quartz filters (Sect. 3). Collection of sufficient mass on the filter provides a lower limit to the mass concentration loadings at the end of the experiment and dictates moderately high injected VOC concentration (dependent on the particle mass yield of the mixture). This is to maximise the molecular identification and quantification since the detection limits for the components are enhanced by maximising the total particle mass collected. The consequences of higher-than-ambient VOC and PM mass loadings are discussed in Sect. 5.
Very low-NOx studies are not accessible in the MAC owing to variable and sometimes elevated NOx concentrations in our chamber laboratory in the centre of Manchester. Such conditions limit the ability of our Purafil scrubber to completely remove NOx at our high inlet flow and may potentially lead to the increase in NOx by diffusion through the Teflon film. Such effects would be non-negligible when attempting sub-ppbv NOx experiments but present at worst modest challenges at higher NOx conditions and are unnoticeable under the chosen VOC concentrations and moderate VOC : NOx ratios of the current programme.
The chemical conditions in all experiments were controlled by photo-oxidation under a simulated solar spectrum (Shao et al., 2022a) at moderate VOC : NOx ratio, with NOx injected as NO2. The seed particles were injected to provide a condensation sink sufficiently large to compete effectively with the wall for condensable vapours. The interaction of Teflon chamber walls with vapours is the subject of considerable ongoing investigation (e.g. Zhang et al., 2014). Vapour–wall interactions have not been fully quantified in the MAC. They will vary across systems with the vapour pressure of individual condensing oxidation products. However, injected seed was sufficient to suppress observable particle formation. Without seeds, nucleation and growth were found to occur in all α-pinene-containing systems and the o-cresol system at our concentrations. Suppression by the presence of seeds is an experimental demonstration that the injected concentrations compete effectively with the walls for the vapours. Nonetheless, the walls will potentially influence the measured particle mass yields in the lower-yield systems more than in higher-yield systems owing to the lower condensation sink provided by the available particles. This is discussed in Sect. 4. Table 1 shows the initial conditions of all experiments.
Table 1List of initial conditions for all the experiments in the campaign.
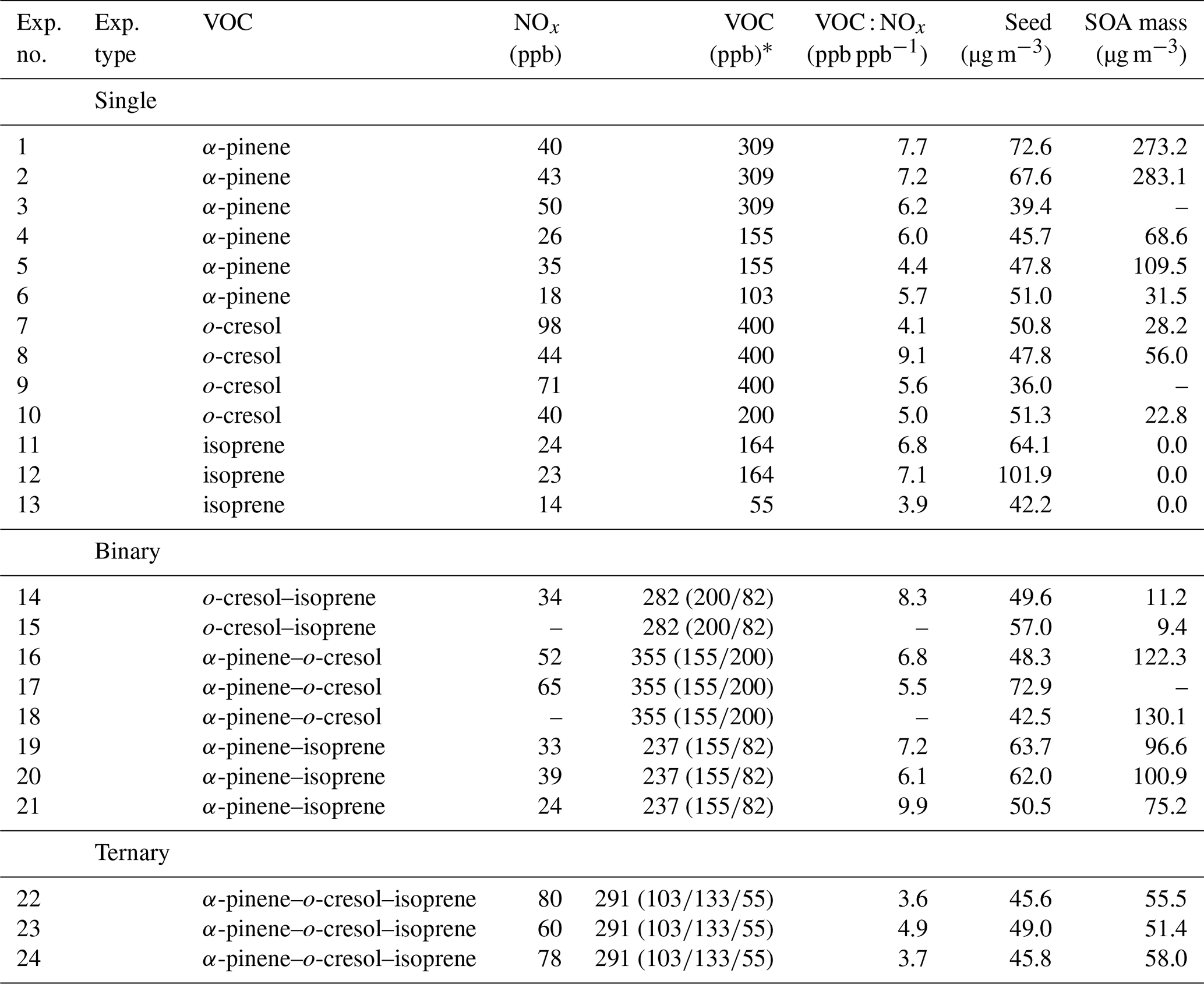
* All nominal reported initial VOC concentrations have a ±15 % injection uncertainty; this was verified using several calibrated GC-MS runs for components injected at the same concentrations in individual experiments and mixtures. The individual VOC concentration in the binary and ternary mixtures shown in brackets corresponds to the precursor VOC listing. A dash indicates missing data owing to instrument problems.
It is not the intention to quantitatively establish the extent and nature of interactions between VOC precursors in the photochemical processes leading to SOA particle formation in ambient mixtures and the consequent impacts on SOA composition and properties in the real atmosphere. The VOC mixtures and set of experimental conditions represent a small sample across a large chemical space and range of temperatures. The current programme aims to reveal examples of the behaviours in mixed systems and provide indicative quantifications of potential interactions and consequences. Focus is placed on the formation of particle mass as well as the physical properties and chemical composition of the evolving particle distribution as it transforms through the experiment. Less emphasis is placed on the quantification of the radical chemistry and oxidative environment, although some characterisation of products in the gas phase and their partitioning to the particle phase is attempted. The experimental design and instrumentational suite are used so far as possible to contribute to addressing the objectives outlined in Sect. 1.
2.3 Methodology
All experiments were performed in the MAC, which consists of an 18 m3 FEP Teflon bag mounted on three pairs of rectangular extruded aluminium frames housed in an air-conditioned enclosure, and details of the experimental procedure can be found in Shao et al. (2022a). In the MAC, ground-level solar illumination is simulated using two 6 kW Xenon arc lamps and a bank of halogen lamps mounted on the inner aluminium wall of the enclosure, which is lined with reflective Mylar film to maximise and homogenise the light intensity throughout the chamber. Removal of unwanted heat from the lamps is provided by the temperature and RH conditioned air introduced between the bag and the enclosure at 3 m3 s−1 as well as active water cooling of the mounting bars of the halogen lamps and of the filter in front of the arc lamps. In addition to removing the heat from the arc lamp, the water in the filters removes unwanted IR radiation, and the 4 mm quartz plates forming the filter windows remove all UV radiation below 290 nm and increasingly transmit light up to 100 % above 305 nm. The chamber inlet system comprises a high-flow-rate blower feeding dried laboratory air through 50 mm diameter stainless-steel pipework to a series of high-capacity filters and variously via a series of two- and three-way electropneumatic valves to a humidifier, ozoniser, and aerosol generation drum before delivering it to the chamber through a Teflon manifold. This is mounted on the top frame of the central rigidly fixed pair of frames. The upper and lower pairs of frames are counterweighted and free to track vertically, allowing the bag to expand and collapse as it is filled and flushed by switching the valve positioning.
All controls are automated, and a series of pre- and post-experimental procedures have been programmed, comprising repeatable, characterised sequences of filling and flushing. The pre-experiment sequence is conducted prior to each experiment to ensure an adequately low background of indicative particulate and gaseous contaminants, monitored by CPC, NOx, and O3 analysers. A 1 h long chamber background characterisation procedure, following the pre-experiment sequence, is conducted to ensure that a baseline contamination level has been established. This is followed by injection of the VOCs, NOx, and seed particles as well as a 1 h collection of data from the experimental background in the dark, during which the chamber conditions and all instrumentation are stabilised. A post-experiment sequence is conducted after each experiment to flush the chamber of all residual contaminants and leave a clean bag for the next experiment. The final fill of the post-experiment sequence contains parts per million levels of ozone, which is used to soak the chamber overnight between subsequent experiments. A weekly vigorous clean is conducted using full illumination with no UV filter on the arc lamps as well as parts per million levels of ozone at high RH for maximum OH production.
Ammonium sulfate seed particles are atomised into a 50 L stainless-steel drum for pre-concentration prior to injection of the polydisperse population into the chamber. The seed concentration in the chamber is controlled by altering the injection time into the drum and the concentration of the solution. After the final pre-experiment flush cycle, the fill flow is diverted through the drum. Liquid α-pinene, isoprene, and o-cresol are injected as required through the septum of a heated glass bulb and evaporated into an N2 carrier flow into this final fill along with NOx as NO2 from a cylinder, also carried by N2. Photochemistry is initiated by irradiating the chamber mixture with the selected VOC and NOx concentrations using the lamps as described above. Online instrumentation is used to continuously monitor the concentration of NOx, O3, particle number, and mass throughout each experiment. It should be noted that o-cresol was found to interfere with O3 measurement as a result of its UV absorption. The decay of o-cresol, measured by a chemical ionisation mass spectrometer (CIMS), was used to correct the O3 data in all o-cresol-containing experiments. The dark o-cresol measured before introduction of O3 into the MAC was used to calibrate the O3 analyser signal for absorption by o-cresol. It should be noted that any UV absorption from the oxidation products of o-cresol cannot be captured, and O3 is thus reported as an upper limit in o-cresol-containing systems. It should also be noted that there will be an uncorrected influence of NOy in the NO2 channel of the 42i NOx analyser owing to the molybdenum converter employed to produce NO prior to detection by chemiluminescence (see e.g. Villena et al., 2012). This should be recognised in the interpretation of our experiments with the photochemical production of HNO3 and organic nitrates.
The instrumentation includes a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; Aerodyne Inc.) to measure particle composition by mass, a Filter Inlet for Gases and AEROsols coupled to an iodide chemical ionisation mass spectrometer (FIGAERO-I−-CIMS; Aerodyne Inc.) for gas- and particle-phase oxygenated component measurement, a scanning mobility particle sizer (SMPS; TSI Inc.) for particle size distribution retrieval, a home-built hygroscopicity tandem differential mobility analyser (HTDMA) for hygroscopic growth factor determination, a cloud condensation nucleus counter (CCNc; DMT Inc.) for cloud droplet potential evaluation, and a home-built three-arm bounce impactor for particle rebound determination. Some of the online instrumentation was changed after several hours of reaction to cycle between sampling after a home-built thermodenuder (TD) and directly from the chamber. Finally, a semi-continuous two-trap gas chromatograph with mass spectrometric detection (GC-MS; Agilent) was used to monitor VOC concentrations. Table 2 provides the list of all instrumentation employed throughout the programme. At the end of each 6 h experiment, the entire remaining contents of the bag are flushed through a Whatman quartz microfibre filter (pre-fired at 550 ∘C for 5.5 h) to collect the particles. The filters were then wrapped in foil and stored at −18 ∘C prior to analysis by LC-electrospray Orbitrap MS.
Table 2List of instrumentation employed over the course of the study. n/a: not applicable.
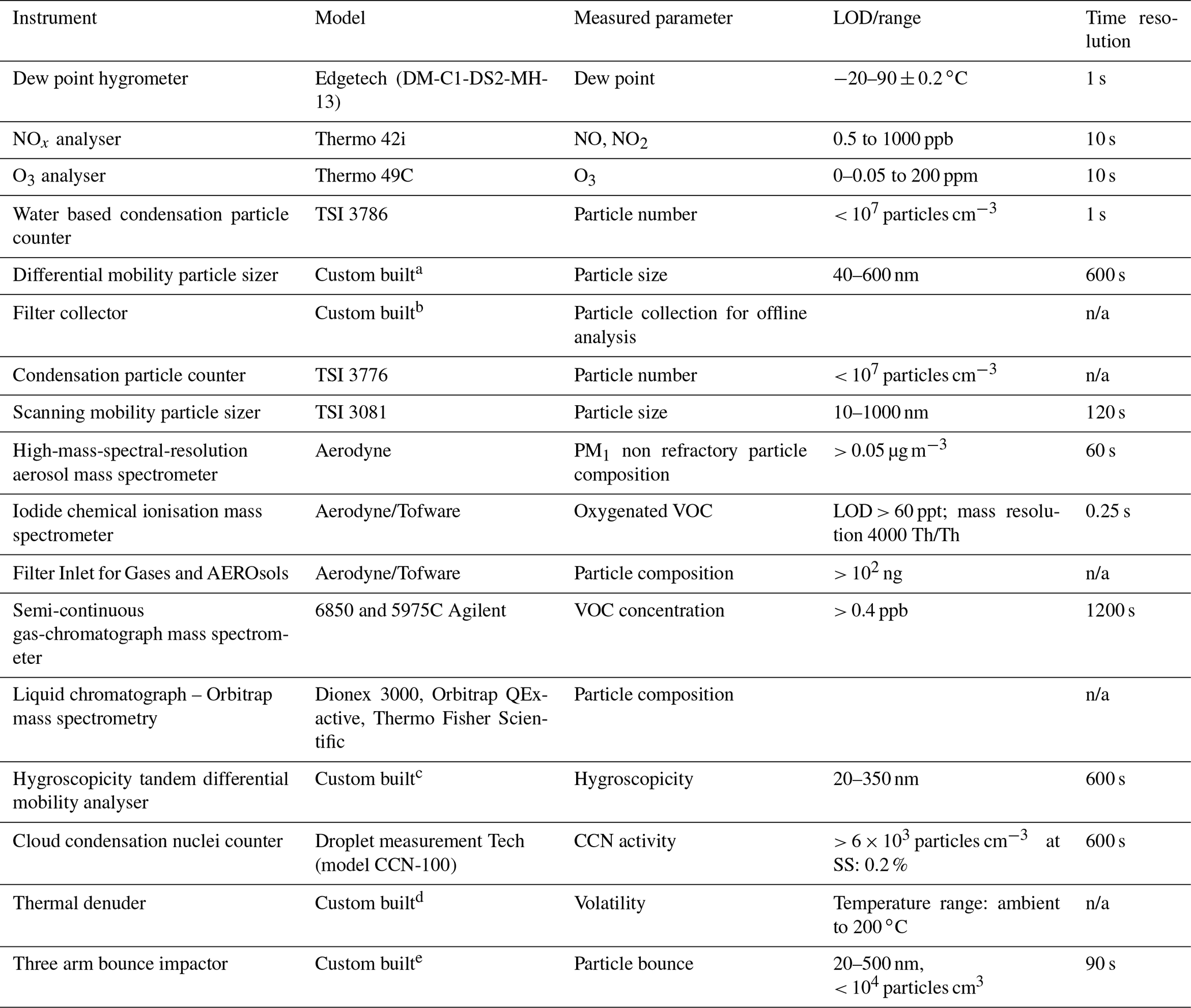
a Alfarra et al. (2012). b Hamilton et al. (2011). c Good et al. (2010). d Voliotis et al. (2021). e Liu et al. (2017).
As described in Shao et al. (2022a), actinometry and off-gassing experiments were conducted regularly through the programme in order to establish the consistency of the chamber's performance, evaluate the effectiveness of the cleaning procedure, and confirm cleanliness of the chamber. Background filters were collected from the actinometry and off-gassing experiments.
2.4 Compromises in the experimental plan and consequences
OH and O3 are the dominant oxidants under the chosen photo-oxidation conditions. The steady-state concentration of the nitrate radical under our illumination was calculated to be too low for significant VOC consumption. However, the unsaturated biogenic compounds will each react with ozone with appreciable reactivity, whilst o-cresol exhibits negligible reactivity towards ozone. Ozone is the first oxidant to be formed through photolysis of NO2, and there will be appreciable formation of biogenic oxidation products prior to formation of those from o-cresol. Consumption of VOCs by different oxidants is presented in Sect. 3.
In addition to the differential reactivity towards the two dominant oxidants, there will be differences in the product formation rates in the single-VOC experiments with varying concentrations owing to the use of a single VOC : NOx ratio. This will result from the reduced NOx at reduced VOC concentrations, which will lead to reduced OH concentration owing to reduction in the NO + HO2 flux. Though there will also be a reduced rate of production of ozone in these experiments, non-linearity leads to changes in the O3 : OH ratio and hence changes in the contributions of the oxidation pathways for the biogenic VOCs. There may be less expected influence on the pathways for o-cresol oxidation, though the rate of oxidation will nevertheless be slowed.
Atmospheric OH reactivity can be estimated from the quantification of VOC abundance or from direct measurements of OH lifetime. Using the latter, Whalley et al. (2016) reported central London diurnal average morning peaks of around 27 s−1 and a campaign maximum of around 116 s−1. Our full-reactivity experiments have an OH reactivity around 465 s−1, which is some 4 times higher with only one VOC, two VOCs, or three VOCs for single-precursor, binary, or ternary experiments, respectively. It would be desirable to work with lower concentrations, but a comparable turnover of oxidant can be achieved at atmospherically reasonable oxidant concentrations at our chosen VOC reactivity. In addition to challenges associated with maintaining oxidant levels at such high VOC concentrations, there is the likelihood that the chemical regime, particularly with respect to radical concentrations, may introduce biases in observed system behaviour. In our systems, OH reactivity is provided by C5, C7, and C10 VOC compounds in addition to that provided by NO2 and VOC oxidation products formed by the oxidation (e.g. Yang et al., 2016). This will lead to a high abundance of relatively large peroxy radical, RO2, species. OH reactivity in the ambient atmosphere is likely to be distributed amongst a high diversity of small (including CO and CH4) and larger components (including carbonyls and other oxygenates). The distribution of RO2 and the ratio of RO2 : HO2 may be substantially different. Whilst this has not been explored systematically in chamber experiments to our knowledge, Peng and Jimenez (2020) have discussed such chemical regime dependence in oxidation flow reactors. Nonetheless, it is not the intention or objective of the current study to mimic the atmospheric chemical regime, but more to explore the potential for mixed VOC systems to reveal interactions in SOA formation processes.
A further consideration is the selection of non-acidic seed experiments. The systems studied will more reflect direct partitioning of gaseous oxidation products rather than accounting for products of condensed phase reactions, particularly those that are acid-catalysed. Again, the intention is not to comprehensively mimic the atmosphere, and this is recognised in the interpretation and discussed in further detail in Sect. 4.
3.1 Photochemistry, VOC consumption, and SOA particle mass formation
To provide the photochemical context for example systems, Fig. 1 shows time series of NO2, NO, and O3 from experiments in the following four systems: α-pinene, isoprene, α-pinene–isoprene, and o-cresol. Note that, in the presence of o-cresol, O3 measurement by UV absorption was influenced by UV absorption by o-cresol, and Fig. 1d is corrected as explained in the Methodology section. Figure S1 shows NO2, NO, and O3 for all systems, similarly corrected for o-cresol-containing systems.
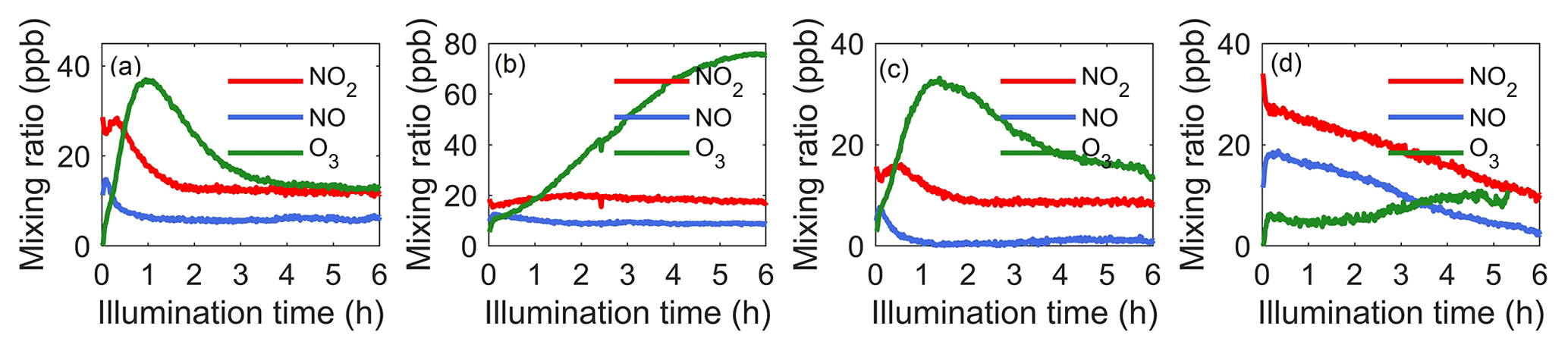
Figure 1NO2, NO, and O3 time series in example single-VOC and mixed-VOC experiments with (a) α-pinene, (b) isoprene, (c) α-pinene–isoprene, and (d) o-cresol. Note that, in the presence of o-cresol, O3 measurement by UV absorption was influenced by UV absorption by o-cresol, and O3 data in (d) were corrected as explained in the Methodology section.
Some insight into the trajectory towards PSS across the systems can be gained by inspection of Supplement Fig. S2, which provides a comparative summary of the Leighton ratio (Leighton, 1961) for all systems and the O3 calculated assuming PSS, i.e. Leighton ratio of 1, shown for the o-cresol-containing systems. The Leighton ratio is given by
where is the photolysis rate of NO2 (i.e. rate of Reaction R1), is the rate of reaction of O3 with NO (Reaction R3), and [NO2], [O3], and [NO] are the molecular concentrations of NO2, O3, and NO, respectively.
In the polluted high-NOx atmosphere, PSS universally applies and φ is unity. In moderately polluted conditions, radical reactions are increasingly important in conversion of NO to NO2 and typically φ > 1. This is because HO2 and RO2 will compete with the O3 reaction for NO (Reaction R3) according to Reactions (R6) and (R7):
where RO2 arises from biogenic VOCs or o-cresol. Note that, in the ambient atmosphere, RO2 can stem from a very wide pool of VOCs. In principle, it is possible to calculate the combined reactivity of HO2 and RO2 towards NO that would be required to explain the positive deviation from unity. Further deviation from PSS and consequent increase in φ will occur when O3 loss processes other than Reaction (R3) become important. These can include reactions of O3 with NO2, alkenes, and radicals. In our chamber systems, the O3 reactions with α-pinene and isoprene as well as radical reactions with NO are competitive and provide substantial deviation from PSS. The NOx and O3 trajectories in the various systems can be seen to vary substantially. Nevertheless, in all systems, illumination of the chamber leads to the expected onset of significant photochemistry to initiate the VOC photo-oxidation.
From Fig. 2, it can be seen that the rate of decay is not the same in each experiment, despite the attempt at initial isoreactivity, as explained in Sect. 2.5. Oxidation products from each VOC will nevertheless be present in appreciable quantities in each experiment, satisfying the objectives of the design to enable exploration of interactions of oxidation products in SOA particle formation.
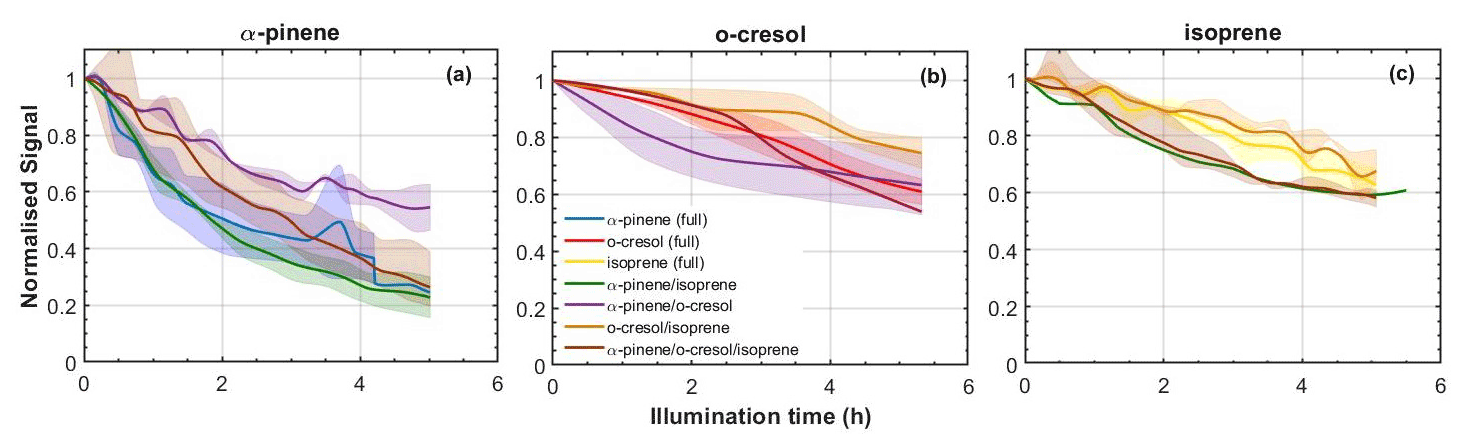
Figure 2Decay rates of each VOC across all mixtures: (a) α-pinene, (b) o-cresol, and (c) isoprene. All lines (and shaded areas) containing blue correspond to experiments including α-pinene, containing red to those including o-cresol, and yellow to isoprene. For example, purple lines correspond to an α-pinene–o-cresol binary mixture and brown to the ternary mixture. The shaded areas represent the envelope of measurements from all repeat experiments around the solid line mean.
Figure 3 shows the time series of SOA particle mass in all experiments (with shading representing the full range of the measurement across all repeat experiments) in all precursor systems studied. It can be seen that the greatest SOA mass was generated in α-pinene-containing systems, which is unsurprising given the known efficiency with which it forms particle mass. It was found that o-cresol and o-cresol-containing mixtures were the next most efficient at producing particle mass. This might be expected with its reported moderate SOA-particle-producing potential (Henry et al., 2008). Negligible SOA particles were formed in all single-VOC isoprene experiments. Only when isoprene was in a mixture was any mass formed, and in all cases, this was lower than the mass formed from the other VOC alone. Again, this is not too surprising given the low or negligible particle mass yield reported for isoprene on neutral particle seeds. It can be seen in Fig. 2 that VOC consumption was incomplete in all experiments, and Fig. 3 shows that the particle mass may not have fully peaked in all o-cresol and o-cresol–isoprene experiments. Nevertheless, the mass peak was observed in most experiments before they were completed, and the chamber contents were flushed through the filter for compositional analysis. Figure 3b shows the SOA particle mass corrected for the losses of particles to the chamber walls. This was conducted by calculating the exponential decay of particles of each size from a targeted ammonium sulfate wall loss correction experiment performed close to the experiment of interest. Details of the wall loss correction can be found in Shao et al. (2021a).
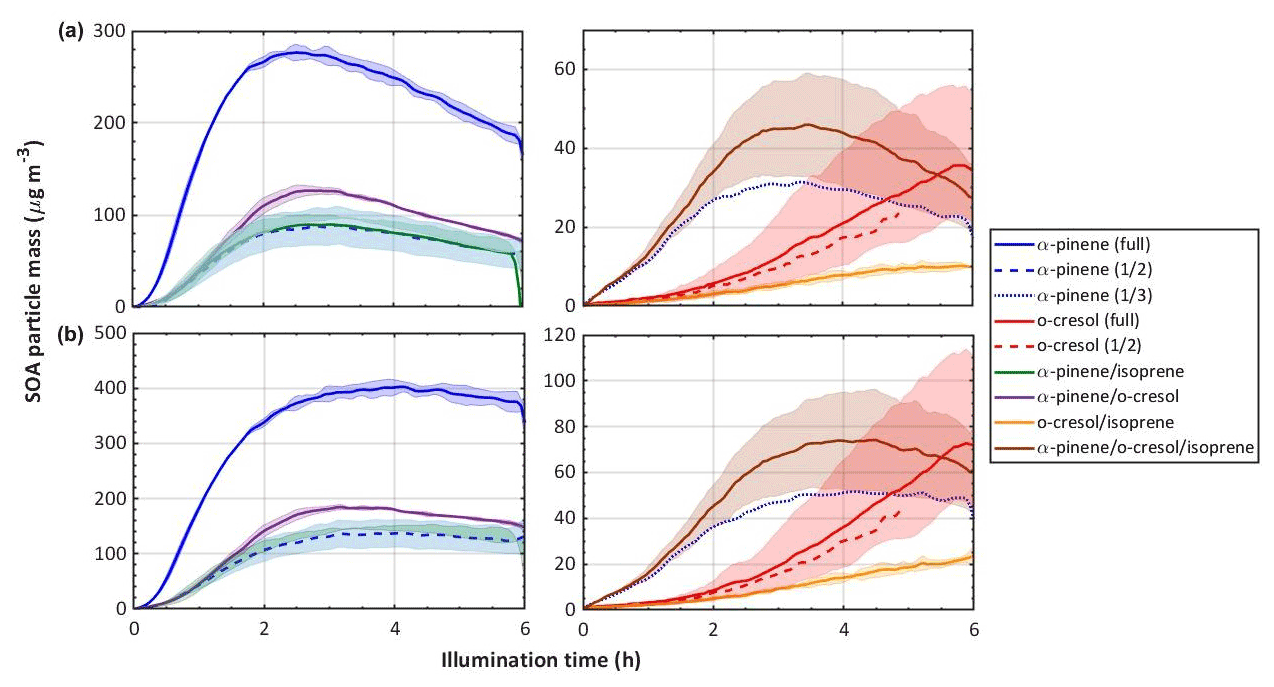
Figure 3SOA particle mass in each system, (a) raw measurements, and (b) particle wall-loss-corrected mass. Note that this is organic mass determined from AMS measurements and therefore does not include the ammonium sulfate seed particles. The same colour scheme as in Fig. 2 is used; the solid line is the mean, and the shaded area represents the envelope of the measurements from all repeat experiments.
3.2 SOA particle mass yield in individual VOC systems and mixtures
SOA particle mass yield is a widely used metric that aims to represent the effectiveness of a VOC in forming organic particle mass. In single-precursor systems it is straightforwardly the SOA particle mass formed per unit of VOC consumed:
Frequently, the yield is reported as a single number and this may be taken from measurements at (a) the maximum SOA particle mass formed in the experiment, (b) the maximum VOC consumed, or, perhaps more arbitrarily, (c) the end of an experiment. Such approaches may be reasonably applied for comparisons between precursors in identical oxidation conditions in the same chamber. Owing to significant particle losses to chamber walls, it can be seen from Fig. 3 that it is important to correct the SOA particle mass formed in the yield calculations for such losses. It is noted that the yields also need to be corrected for VOC or OVOC product losses to the walls, as discussed in Sect. 4. Table 3 shows the calculated yields for all individual VOC and mixture experiments. The first nine rows show the yields for single-VOC experiments. Following the SOA particle mass plots shown in Fig. 3, it is clear that the efficiency in forming mass is in the order α-pinene > o-cresol > isoprene, with no SOA particle mass ever measured in isoprene oxidation, though appreciable isoprene consumption was observed as shown in Fig. 2. For α-pinene and o-cresol it can be seen that the yield increased with increasing initial concentration. The o-cresol and o-cresol–isoprene experiments were not continued to the point of maximum mass formation (as shown in Fig. 3), so the maximum VOC consumption and mass formation both correspond to the end of the experiments in these systems. Figure 4 shows the yield plotted against SOA particle mass for α-pinene and o-cresol for all individual VOC experiments, indicating that the yield increases monotonically with SOA particle mass within propagated experimental uncertainties. Such behaviour is expected and is consistent with absorptive partitioning considerations (Bowman et al., 1997). Note that organic mass formation in the “full-reactivity” (i.e. 309 ppb α-pinene) experiment is so rapid that VOC data are only available for yield calculation beyond 100 µg m−3. Supplement Fig. S4 alternatively shows the SOA particle mass yield plotted against the total particle mass in the chamber, including the ammonium sulfate seed particles that provide a condensation sink for the condensable vapours produced by VOC oxidation.

Figure 4SOA particle mass yield as a function of mass formed in the single-precursor α-pinene and o-cresol experiments at all initial concentrations. Each curve is comprised of multiple data points measured over the duration of each experiment. Error bars represent the propagated uncertainties in all measurements and in the particle wall loss corrections applied.
Since there is no SOA particle mass formed from isoprene, Table 3 additionally presents yield data omitting the consumption of isoprene in the denominator of Eq. (2), accordingly increasing the yield. Figure 5 shows the yield curves for typical experiments in all systems. The yield curves allow comparison between the systems, both including and excluding isoprene in the denominator of Eq. (2). The two-product model (Odum et al., 1996) was used to fit the yield curves for α-pinene and o-cresol. These are included to guide the eye. Here, the two-product model parameterised the relation of overall SOA yield and the adsorptive mass assuming only two products to exist in the system. The equation of the two-product model is shown in Eq. (3). α, Kp, and COA represent the stoichiometric factor, the partitioning coefficient of product, and the total absorbing organic mass, respectively. The α1, α2, Kp,1, and Kp,2 can be fitted from the yield curves.
Table 3Measured and particle wall-loss-corrected SOA particle mass yields for all systems calculated at maximum particle mass and maximum VOC consumed. For mixtures containing isoprene, which had zero yield on the neutral seeds injected, the yields were calculated excluding the consumption of isoprene in the system, allowing them to be compared to the mixtures without isoprene. Yield was calculated with the density of organic matter of 1.4 µg m−3. The maximum mass used in the yield at maximum mass calculation is given in Table 1. The uncertainties in SOA particle mass yield were calculated by propagating the ±1σ uncertainties of measured ΔSOA and ΔVOC. A dash indicates missing data owing to experiment problems.
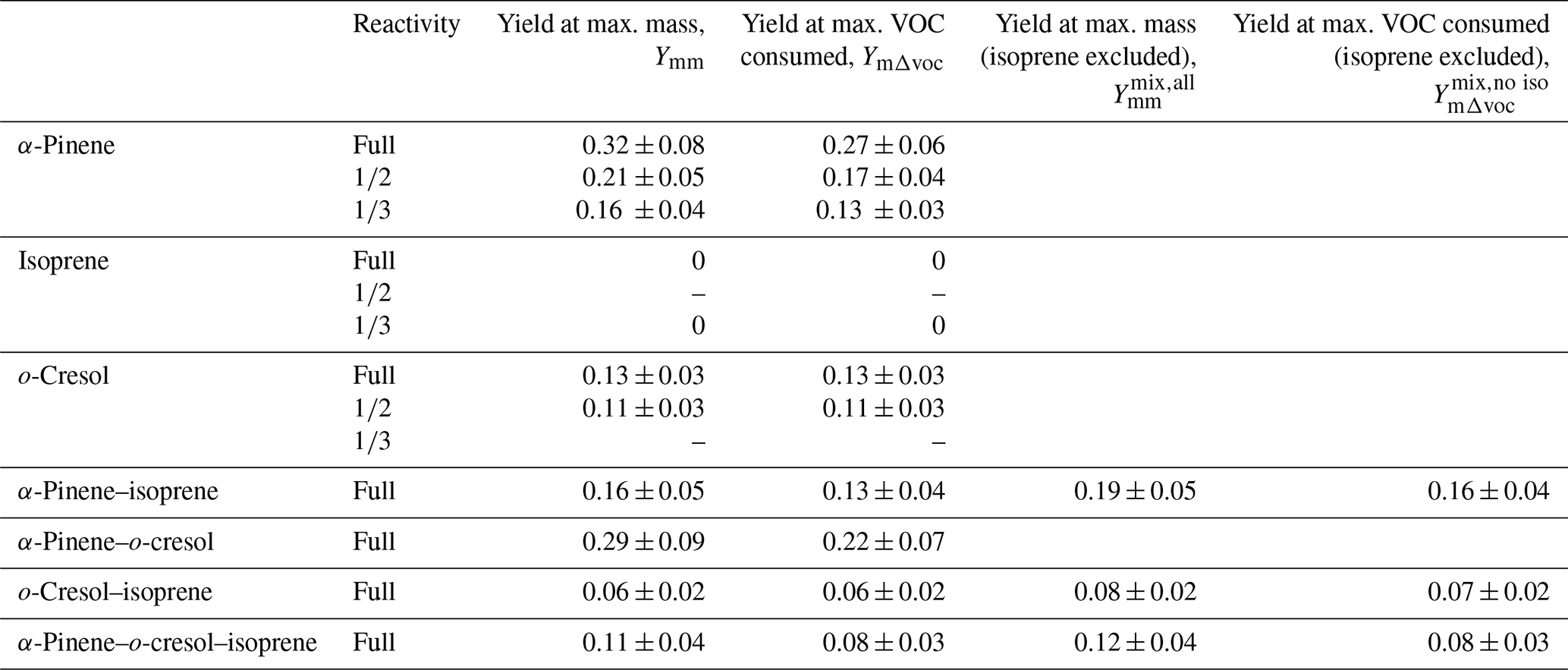
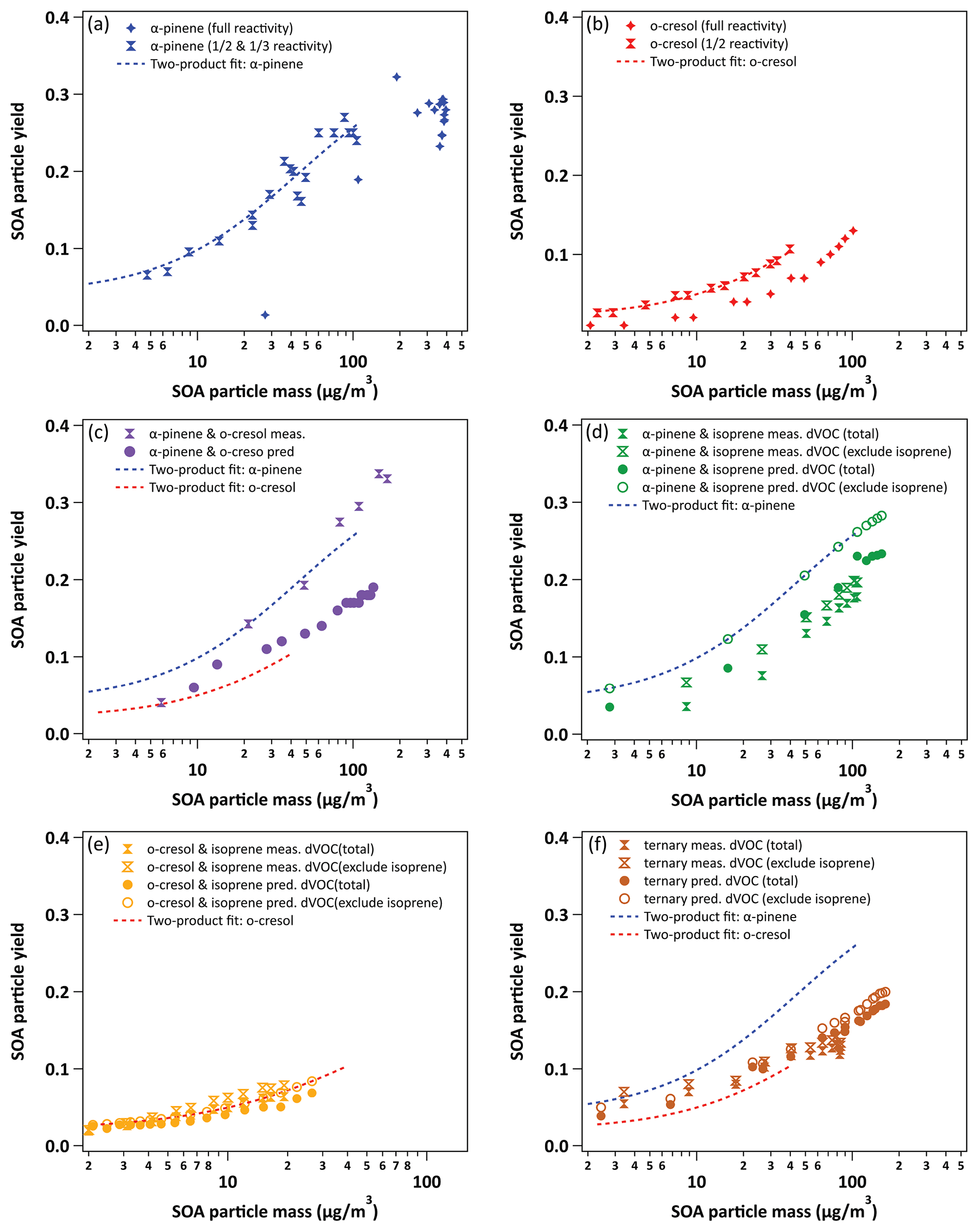
Figure 5Yield data for selected representative experiments in all systems (with two-product yield curves for α-pinene and o-cresol single-VOC experiments). Panel (a) shows the binary α-pinene–o-cresol mixture and its constituents, (b) the binary α-pinene–isoprene mixture, (c) the o-cresol–isoprene mixture (expanded y-axis plot shown in Fig. S5), and (d) the ternary α-pinene–o-cresol–isoprene mixture. Yields “predicted” from the linear combination of yields from the individual VOC experiments using Eq. (4) are shown for each mixture.
In addition to the measured points, Fig. 5 shows “predicted” yields for the mixtures assuming additivity based on the SOA particle mass at the same VOC consumption measured in the single-VOC experiments using Eqs. (3) and (4).
where ΔVOCn is the consumption of VOCs and is the SOA particle mass at this consumption.
The parameters (α1, α2, Kp,1, Kp,2) from the two-product fit by Eq. (3) from the single-VOC half-reactivity and one-third-reactivity experiments were used to generate a yield–mass–ΔVOC look-up table. This was then used to calculate the hypothetical SOA particle mass formed from each precursor at the consumption of the VOCs at each point in the binary and ternary systems as shown in Fig. 5 (and expanded for the o-cresol–isoprene system in Fig. S5). The predicted yield in the binary α-pinene–o-cresol mixture is below that measured and slightly higher than that measured for o-cresol at 50 % reactivity. The yield measured in the mixture is comparable to that of α-pinene measured at one-half and one-third reference reactivity. For the isoprene-containing binary mixtures, clearly the predicted yield excluding isoprene in the denominator, , is identical to that of the two-product curve fitted to the single-VOC experiment derived yield of other components in the mixtures. In the α-pinene–isoprene system, the predicted yield accounting for the consumption of both α-pinene and isoprene, , is higher than in that measured but, as shown in Tables 3 and 4, comparable to the measured yield calculated from the consumption of α-pinene only, . In the o-cresol–isoprene system, the predicted yield accounting for the consumption of both o-cresol and isoprene, , is lower than that measured, (at higher mass loadings); there is little difference within uncertainty at a lower mass. The predicted yield accounting for the consumption of o-cresol alone, , is lower than that measured when calculated using the same total VOC consumption, , but comparable to the measured yield using the consumption of both o-cresol and isoprene, . Finally, in the ternary system, the predicted yield excluding isoprene consumption, , is similarly (and obviously) higher than that including its consumption, , but both predictions are between the yields of single α-pinene and o-cresol experiment measurements. The measured yields accounting for only α-pinene and o-cresol consumption in the ternary mixture, , are also similarly (and obviously) higher than that including isoprene consumption, . There is insufficient difference between the measurements or the predictions to state whether inclusion or exclusion of isoprene consumption gives much better agreement, though there is an indication that the predicted yields show a steeper gradient with SOA particle mass than the measured slope, which more closely follows that of o-cresol than that of α-pinene. The measured single-precursor yields shown in Table 3, including the ones at maximum VOC consumed used in the predictions, are plotted for clarity in Fig. S6. Table 4 distils the predictions based on the yield from single-VOC experimental data at the same consumption as in the mixtures into single values predicted at maximum SOA particle mass and maximum VOC consumption, both with and without isoprene decay in the calculation (plotted for clarity in Fig. 6).
Table 4SOA predicted yield for the four mixtures at maximum SOA particle mass produced and maximum VOC consumed. The uncertainties in SOA particle mass yield were calculated by propagating the ±1σ uncertainties of predicted SOA and measured VOC.

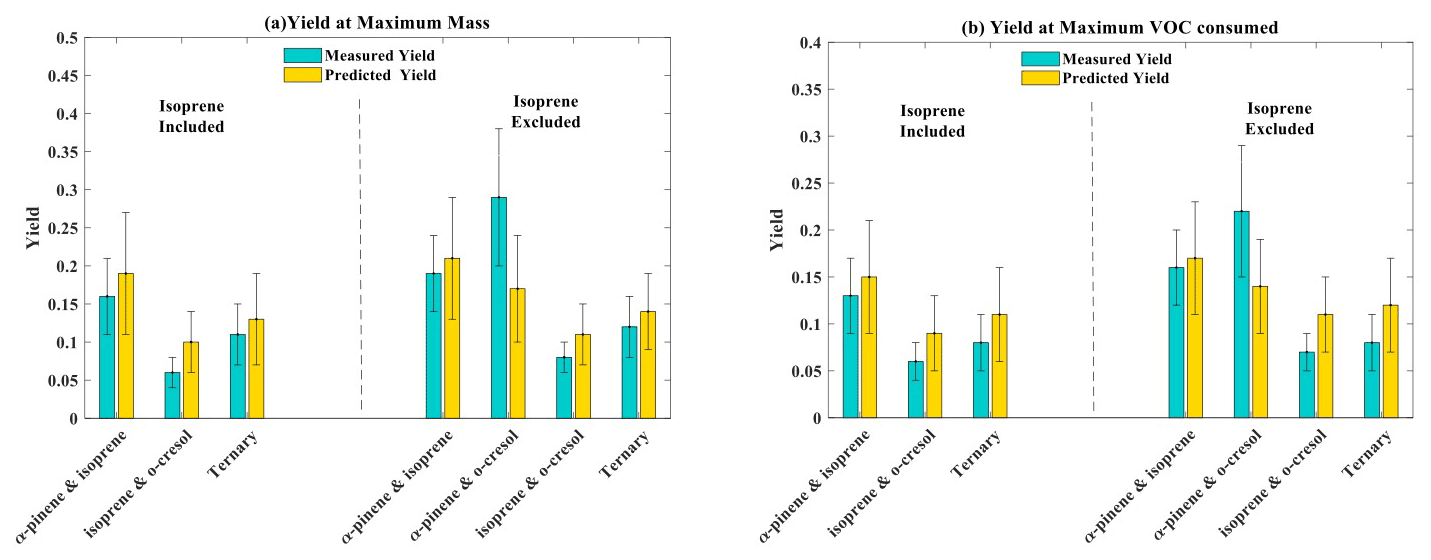
Figure 6(a) Measured and predicted yield for all mixture precursor systems calculated at maximum particle mass. (b) Measured and predicted yield for all mixture precursor systems calculated at maximum VOC consumed. For mixtures containing isoprene, which had zero yield on the neutral seeds injected, the yields were calculated excluding the consumption of isoprene in the system, allowing them to be referenced to the mixtures without isoprene. Yield was calculated with the density of organic matter of 1.4 µg m−3. The maximum mass used in the yield at maximum mass calculation is given in Table 1. The uncertainties in SOA particle mass yield were calculated by propagating ±1σ uncertainties of predicted SOA and measured VOC.
3.3 Oxidant contributions to VOC decay
To explore the likely fate of the parent VOCs in the MAC, we used the measured O3 concentrations and VOC decay rates, and we exploited the differential reactivities of the VOCs to investigate the decay attributable to each oxidant and also overcome measurement difficulties encountered in the experiments. For example, knowing that o-cresol has a negligible rate of reaction with O3, we are able to calculate the OH concentration from the o-cresol decay curve. This can then be used to attribute the fraction of the decay of isoprene and α-pinene to OH and O3 in their binary mixtures with o-cresol. This is important, since the measurement of O3 by UV absorption in the presence of o-cresol is challenging and requires correction for the additional absorption by o-cresol. In systems without o-cresol, the stepwise decrease attributable to O3 can be constructed for α-pinene and isoprene using the O3 measurements (reaction rates of 9.6 and cm3 molec.−1 s−1, respectively; Cox et al., 2020). By comparing with the actual α-pinene and isoprene, the residual decay can be attributed to OH. An example of this oxidant reconciliation is shown in Fig. 7 for the binary α-pinene–isoprene system.
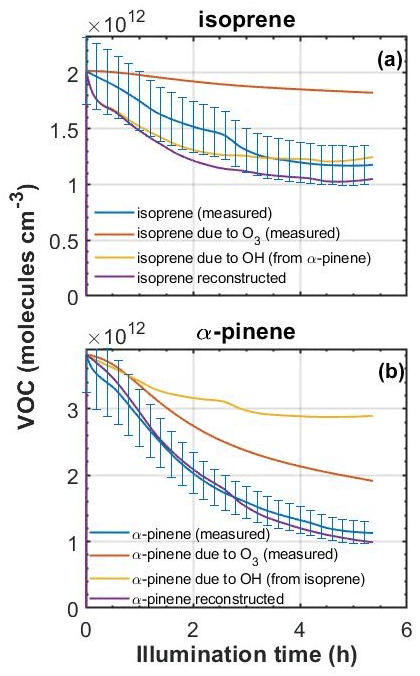
Figure 7Measured (±15 % error) and reconstructed decays of (a) isoprene and (b) α-pinene in an α-pinene–isoprene binary experiment. First, in each case the decay of the VOC due to ozone was predicted based on the O3 concentration, the reaction rate of each VOC toward O3, and the initial VOC concentration. Second, the OH concentration from each VOC was estimated from the difference in the VOC decay attributed to O3 and the measured VOC decay. Finally, the OH concentration from each precursor was used to reconstruct the decay of the other (i.e. OH from isoprene was used to reconstruct the decay of α-pinene and vice versa).
From Fig. 7, it can be seen that roughly twice as much loss of α-pinene can be attributed to O3 as to OH and roughly 4 times less isoprene loss attributed to O3 than to OH. The inability to control isoreactivity towards all oxidants through controlling the initial VOC concentrations will influence prediction of SOA particle mass yields owing to the differences in the tendencies of oxidation products from different oxidants to condense. These aspects are discussed in Sect. 4.
3.4 Supporting measurements of SOA particle composition and properties
The measurements presented so far are central to the first three objectives outlined in the Introduction. This section provides descriptions of additional measurements presented in the Supplement and detailed in companion papers aiming to address the fourth objective – to probe the chemistry and physics leading to interactions in the mixed VOC systems and the properties of particles resulting from them of potential atmospheric significance.
The differences in the SOA particle oxygenation trajectory between the systems are illustrated in Figs. S7 and S8 by the percentages of the AMS total signal at (f44) and 43 (f43) to represent more and less oxygenated contributions, respectively, to the SOA particle mass. A comprehensive analysis of the carbon oxidation state of the SOA particle components using data from high-resolution AMS, online FIGAERO-I−-CIMS, and offline analysis of filter samples by LC-Orbitrap MS is presented in Shao et al., 2022d).
Further insight into the changes in the chemical composition can be provided by the time series of gaseous and particulate components from the FIGAERO-I−-CIMS measurements. Figure S9 shows the changes in particulate mass spectra of single α-pinene and o-cresol experiments as well as their mixture, showing a clear increase in signal through the experiment with the increasing SOA particle mass in all systems and the appearance of peaks uniquely found in the mass spectra of the mixture. In conjunction with offline LC-Orbitrap MS analysis of filter samples, peak assignment of these mass spectra has been used to attribute signal to the molecular formulae and hence to broad chemical groupings in all single-VOC and mixed systems; full analysis and discussion can be found in Du et al. (2022a) and Du et al. (2022b).
Figure S10 summarises the attribution of the components of SOA particles sampled on filters collected at the end of a ternary o-cresol–α-pinene–isoprene mixture experiment, taking advantage of the separation and accurate mass resolution afforded by the LC-Orbitrap MS technique. Signal is attributed to the individual parent VOC (by matching the attributed formulae to those identified in the individual precursor experiments) and to molecules found only in the mixture and plotted according to their fractional signal contribution. Clearly a substantial fraction of the unique-to-mixture signal is found at carbon number greater than any precursor VOC (in negative ionisation mode 57 % of the signal in 48 individual peaks with nC > 10 and in positive mode 60 % in 115). Whether such components result from gaseous or condensed phase reactions cannot be determined from these measurements, but they clearly result from interactions in the mixture. A detailed discussion of the untargeted ultra-performance liquid chromatography–Orbitrap–mass spectrometer (UPLC-Orbitrap-MS) data analysis can be found in Pereira et al. (2021), Shao et al. (2022a), and Du et al. (2022a), and an analysis and discussion of the composition from all individual precursor systems and their mixtures is presented in Shao et al. (2022a).
Combinations of online and offline analyses can help guide interpretation of the differences in composition between the systems. Figure S11 examines the presence of C7H7NO4 (methyl-nitrocatechol and its isomers) in o-cresol-containing systems. Panel (a) expands on the molecular attribution by LC-Orbitrap MS to demonstrate the ability to separate isomeric contributions to the signal normalised to the total detected signal by retention time in single-precursor and mixed systems. The isomer at an LC retention time of 9 min dominates the signal in all systems, whilst the isomer at a retention time of 6.1 min displays negligible signal fractions in all systems. In contrast, the I−-CIMS is incapable of separating structural isomers. Panel (b) shows the time series of the total signal at 296 (normalised to the total attributed signal) corresponding to the combined signal from all C7H7NO4 isomers in each system. Du et al. (2022a) compare and explore the advantages of the combination of online and offline techniques and demonstrate how clustering analysis can be used to interpret the composition evolution in precursor mixtures. Du et al. (2022b) apply this combination of techniques to all individual systems and their mixtures.
The attribution of elemental composition in the high-resolution FIGAERO-I−-CIMS analysis enables comprehensive mapping of the particulate oxygen to carbon ratio (O : C) as a function of carbon number, as shown in Fig. S12 for each system. The majority of signal in the o-cresol single-VOC system comprises compounds at the same carbon number, C7, as the parent. On average, the O : C ratios of these C7 compounds in the o-cresol single-VOC system are slightly higher than C10 products dominating the α-pinene single-VOC system. A significant fraction of the signal in all α-pinene-containing systems is found at carbon numbers corresponding to the “dimer” range, which are all found predominantly in the particle phase (i.e. implying a low volatility). These products are absent in the mixtures containing α-pinene and o-cresol, suggesting an increase in the overall volatility. A range of new products is present in mixtures (e.g. α-pinene–o-cresol, o-cresol–isoprene) with higher carbon numbers (nC=11-16), lower O : C (O : C < 0.5), and varying volatility (log10C 1–3), indicating that the molecular interactions influence the overall SOA particle volatility. A more complete discussion of the chemical composition and volatility variation across the mixtures in the gas and particle phases can be found in Du et al. (2022b) and Voliotis et al. (2021, 2022).
The volatility distribution of oxidation products can be assembled using either C* of all identified formulae from the gas-to-particle ratio of their ions in partitioning calculations or the AMS mass fraction remaining after thermal denuding (Voliotis et al., 2021, 2022). The distributions using the former technique are shown for the single VOCs as well as their binary and ternary mixtures in Fig. S13. The volatility distributions of particles in the mixture experiments can be similar (α-pinene–isoprene) or quite different (o-cresol–isoprene) to those in the experiments using the single precursor, suggesting that mixing precursors can have a varying effect on the resultant particle volatility. A full discussion of the chemical composition and its influence on volatility in all systems can be found in Voliotis et al. (2022).
Figure S14 shows the behaviour of particle water uptake and the rebound fraction of particles in an impactor in three example systems. These have been chosen for their differences in the rate of formation of SOA particle mass and, in our seeded experiments, the associated organic : inorganic particle mass ratio. The rate of decrease of particle hygroscopicity and of increase of rebound fraction at higher RH clearly follows the rate of SOA particle mass increase in the system, indicating the influence of the mixing of precursors on the change in particle properties through the change in the rate of formation of condensable material (as explored in Wang et al., 2021, 2022).
4.1 Experimental conditions and photochemistry
The control of photochemical conditions in VOC mixtures is challenging. The chemical space is multidimensional and non-linear, and it is unclear how best to define the regime for direct comparison across systems. The approach taken in the current study, to start with concentrations of each VOC isoreactive towards a single oxidant, cannot account for the differential reactivity towards the two oxidants, O3 and OH, present in the system. This is particularly true when the production of oxidants is tightly coupled to the VOC oxidation itself and secondary oxidant production can be at a comparable level to primary production. The combination of oxidants leads to significantly different VOC decay and hence turnover of oxidants and products. This is an unavoidable feature of VOC mixtures that will occur in the real atmosphere, and such differences need to be carefully considered in the interpretation of the results.
It can be seen from Figs. 1 and S1 that the time profiles of NOx and O3 vary substantially across the single-precursor experiments and the mixtures. This is a characteristic of the method of initiating photochemistry in our experiments through photolysis of a VOC : NOx mixture in the initial absence of O3. Within the same single-VOC systems, the O3 concentration temporal profile can vary with VOC concentration at similar VOC : NOx values. It is particularly notable in the α-pinene experiments owing to its reactivity towards O3. As expected, there is a higher ozone concentration in some systems (notably those containing isoprene). The Leighton ratios shown in Fig. S2 indicate wide variation in the deviation from photostationary state across the systems, with the high-concentration single-VOC o-cresol experiments and the α-pinene–o-cresol and α-pinene–isoprene binary mixtures showing the greatest positive deviation (i.e. greatest ability for RO2 and HO2 to compete with O3 for NO, whereas the single-VOC isoprene and α-pinene, binary isoprene–o-cresol, and ternary systems all move towards low φ values. Values below 1 could arise from a wall source of NO. Alternatively, errors propagated from the determination of from actinometry experiments and measurements of NO, NO2, and O3, as well as the heterogeneity of the owing to variation in light intensity across the chamber (not reflected in the point measurements of the gases) could lead to errors in the calculated Leighton ratio, but this must be the subject of further work. The implications would be that the absolute values of the Leighton ratio may carry unknown errors, but their trends and relative values between experiments should be reliable.
It is shown in Fig. 2 that the decay rate of each VOC was different, likely resulting from the differential reactivity of each VOC towards the different oxidants present in the systems at any one time. However, it can be seen that the decay of each individual VOC was comparable across each system (with the notable exceptions of decreased α-pinene decay in its binary mixture with o-cresol and slightly increased o-cresol decay in the ternary mixture). This may be (i) coincidental, (ii) indicative that the oxidant regimes remain comparable between experiments, or (iii) indicative that the decay is relatively insensitive to the change in oxidant regimes between the mixtures in our experiments. Figures 1 and S1 show the differences in O3 across systems, so the differences in oxidants are non-negligible (option ii). In relation to the decay with respect to each oxidant in a mixed oxidant system, it should be borne in mind when comparing the results from different studies that results may diverge if the dominant oxidant changes. For example, Fig. 7 shows that in the MAC α-pinene–isoprene experiments, roughly twice as much α-pinene consumption was due to O3 reaction as to OH consumption. By contrast, in the experiments in the Jülich Plant Atmosphere Chamber (JPAC) reported in McFiggans et al. (2019) around 90 % of the consumption was estimated to result from OH reaction. This variation in the oxidants controlling VOC decay between experiments in our study and between studies demonstrates a need to explore across the full oxidant regime from OH-only to O3-only to fully unravel the mechanisms of formation of SOA.
Control of the oxidant regime in chamber experiments is strongly dictated by the goals of the study. It is perhaps most straightforward in a dark system in the absence of NOx, wherein only O3 is introduced with a single unsaturated VOC. Even then, the chemical regime can change as the oxidant is consumed and moves from excess to limited. Moreover, the yield of OH from ozonolysis can be very significant such that the VOC consumption from the OH oxidation must be considered. A dark OH source (such as ozonolysis of tetramethylethylene, TME) can be used for oxidation of a saturated VOC to enable the OH to decay relatively “cleanly”, though the possibility for interaction between the TME- and SOA-precursor-derived RO2 must be remembered. Dark oxidation by continuously injected NO3 (from the headspace above frozen N2O5) in the absence of O3 can also provide a “clean” system with relatively few interfering pathways. Controlled NO3 generated from O3 reaction with NO2 starts to become quite complex. The competition between oxidants in establishing the dark consumption of unsaturated VOC will be challenging, though the NO3 reaction can dominate in systems containing phenolic compounds such as o-cresol. In illuminated systems, the oxidant regime is also complex. Photo-oxidation in the absence of NOx can enable isolation of OH consumption of saturated compounds more straightforwardly. For unsaturated VOCs, ozonolysis (and its associated secondary OH source) must be considered. As is evident from the presented experiments, photo-oxidation in the presence of NOx introduces about as much complexity as can be envisaged. Photo-oxidation of multiple VOCs in the presence of NOx multiplies this complexity. The study of McFiggans et al. (2019) avoided many of the challenges faced in the current study by considering the mixture of biogenic VOCs in the absence of NOx and in a regime dominated by OH oxidation. This can be considered reasonable, since mixtures of these VOCs might exist in the clean troposphere, remote from anthropogenic sources. In studying mixtures of anthropogenic and biogenic VOCs, it might be considered less reasonable since anthropogenic VOCs seldom exist in the absence of appreciable NOx. Nonetheless, mechanistic insight from such experiments would be useful. Related to this, it should also be noted that, whilst maintaining the VOC : NOx ratio relatively constant across the systems is desirable, with reduced VOC concentrations this implies reduced NO concentrations. This can lead to reduced OH concentrations and hence VOC turnover. This is of importance when comparing the single-VOC experiments at different concentrations and also when considering the initially isoreactive mixtures. Whilst the VOCs have been chosen for broadly comparable reactivity, their concentrations vary by a factor of 2.44 at comparable mixture reactivity. Consideration of the approach of using scavengers, such as n-butanol to scavenge OH, is also worthwhile. Supplemented with experiments using a non-O3 OH source such as injection of H2O2 could enable valuable isolation of the VOC consumption by individual oxidants. However, as noted in our study, the assumed reference state for yield calculation would be clouded by the scavenger addition – should its concentration be included in the denominator of the yield in mixtures? The oxidation products of the scavenging will potentially interact through termination of radicals, forming partitioning products, and cannot be ignored a priori.
One feature of chamber experiments using mixtures of VOCs is the ability to exploit their differential reactivity towards the oxidants present in the system to attribute their decay to each of them. This reconciliation of the VOC decay, as shown in Fig. 7, is potentially a powerful interpretive tool for diagnosis of the concentration of oxidants in the system and understanding of the turnover of VOCs in the system. In our systems when o-cresol is present, there are limitations owing to the interference of the direct O3 measurements by absorption of UV by o-cresol. However, the reconciliation in these cases can be used to aid validation of the correction based on CIMS measurement of o-cresol (see methods in Sect. 2.3). It should be noted any absorption by o-cresol oxidation products can still lead to bias in the reconciliation since O3 will be underestimated. Alternative O3 measurement by chemiluminescence suffered from instrument failure in our experiments but would be useful in future programmes. Clearly, direct measurement of OH could provide additional confidence in the oxidant field, but some constraint can be provided by the indirect method described here.
A final consideration relates to the exploratory nature of the current study, which aims to establish the suitability of the approach to reveal aspects of SOA formation in mixed VOC atmospheric systems inaccessible to experiments using a single precursor species. The broad suite of analytical techniques employed to investigate these aspects have varying requirements, and some compromises have been necessary. Most importantly, the offline filter analyses require a minimum particle mass loading with the corresponding consumption of VOCs with varying yield. This has necessitated the use of initial VOC concentrations above ambient levels. Whilst such concentrations have been commonplace in chamber experiments, they do introduce limitations. Further developments and increases in mass resolution of online mass spectrometric techniques will help this aspect of future study design. Figure S2 indicates that the deviation from PSS is likely to be significantly affected by the reactions of RO2 (and/or HO2) with NO in some of the binary mixtures. One further important limitation arises from the non-scalability involved with processes that are higher than first order. Peroxy radical terminations often involve their cross-reaction, which will be in competition with other bimolecular pathways; so, reactions that are second order in peroxy radical concentration will be in competition with those that are first order. This will be important in the termination of peroxy radicals formed during autoxidation leading to HOM formation. In particular, the reactions of large HOM–RO2 with other large peroxy radicals leading to gas-phase formation of accretion products in the “dimer” range may be overrepresented at higher concentrations. Under clean ambient conditions, their reaction partners would likely be dominated by HO2 from CO oxidation or smaller organic peroxy radicals such as CH3O2 from CH4 oxidation. Under more polluted conditions, the alkoxy pathways from NO reaction will be competitive or even dominant. Each of these aspects should also be considered when interpreting the interactions in mixed VOC systems.
4.2 Yield calculation and reporting
The representation of effectiveness of a VOC to produce SOA particle mass in a given system by its incremental yield is conceptually simpler when there is only one VOC being oxidised than in mixtures, but even then, it has complications. Equation (2) can be applied, but with some need to consider the oxidant regime. Assuming there are no interactions between the oxidation products solely formed from reaction with each oxidant, the yield will be the sum of the total SOA particle mass formed in the system divided by the total consumption of VOCs by all oxidants. If there is a greater yield through reaction with one oxidant than with another, the yield in mixed oxidants will be enhanced in experiments with a greater proportion of the decay from the first oxidant, so the total yield of the VOC will not be the same in experiments with different oxidant ratios. There are a number of further considerations in the calculation and presentation of yield that should be discussed for single-VOC experiments and several more that are of relevance for mixtures.
In Fig. 3 we present a summary of the SOA particle mass in the various single-VOC systems and their mixtures. It can be seen that the corrections accounting for the losses of particles to the chamber walls are substantial, with peak mass concentrations typically 30 % to 40 % higher than those measured, and no substantial decay after the peak mass has been formed (as expected). Whilst not without uncertainty, the loss of particles to the walls can be relatively straightforwardly estimated. This is not the case for the interactions of the walls with vapours, which has been the subject of extensive debate (Zhang et al., 2014; Loza et al., 2010; Ye et al., 2016; Krechmer et al., 2020). A full discussion of this subject and characterisation of vapour interaction with the Teflon chamber walls is not provided here (fluxes to and from the chamber walls). We acknowledge that these effects will likely be substantial and that these are unquantified in our reported SOA particle mass corrections and yields. An extensive study of the losses of vapours to walls across multiple chambers within the EUROCHAMP2020 network was conducted, and data from our MAC facility have been included. A full analysis of these data is ongoing, and the results will enable reinterpretation of yield data from across the consortium. It would be premature to pre-empt such an analysis. In the comparisons between different systems in our chamber, it must therefore be noted that we are implicitly assuming that the vapour–wall interactions are comparable. This will introduce unquantifiable errors, since there are likely significant vapour pressure differences for the products in different experiments, and there are also likely vapour pressure dependencies of wall interactions. Experiments producing different amounts of SOA, either because of the presence of higher-yield VOCs or increased concentrations, would allow partitioning of higher-volatility vapours to the particles. These would be affected to a different degree by the walls. Most importantly, it means that, whilst the comparisons may (or may not) be valid across systems in our chamber, our reported yields are very likely not comparable with those in other chambers without further considerations. More broadly, any comparison of yields between chambers should be approached with extreme caution without full confidence that the vapour interactions with the walls of the specific chamber for the specific system have been well quantified.
Yields presented in Table 3 calculated as single values at either maximum SOA particle mass (Ymm) or maximum VOC consumed (YmΔvoc) can provide useful classifications for comparisons of systems wherein temporal and mass dependence of yields are comparable. The mass-dependent curves shown in Figs. 4 and 5 capture further details of the behaviour throughout the experiments, and fitting the yield vs. absorptive mass relationship after Odum et al. (1996) to the single-VOC experiments enables predictions of the hypothetical yields presented in the figures and as single values in Table 4 (as described in Sect. 3). There are a number of considerations when assessing whether it should be expected that such predictions are valid. The first of these is the inclusion of the decay of all VOCs in the yield calculation. Whether the consumption of all VOCs in a mixture should be considered in calculating the mixed yield depends on the question being addressed. It may be inappropriate to consider the consumption of a VOC that would not contribute to formation of SOA particles when investigating the change in yield of a VOC mixture. In the present study and consistent with previous work, isoprene was consistently found to generate no measurable SOA particles above the background chamber concentration (< 1 µg m−3) at any initial isoprene concentration studied and hence exhibited negligible yield. Therefore, under the conditions of our experiments, when replacing half the reactivity of a higher-yield VOC with isoprene in a binary mixture, it may be considered obvious that the SOA particle mass will be reduced. This is the case in the α-pinene–isoprene and o-cresol–isoprene mixtures. Instead, it may be more insightful to consider whether the yield of the α-pinene and o-cresol in the binary mixtures is reduced in the presence of isoprene by comparing them to their single-VOC system yields. This follows the approach for neutral seeded experiments in McFiggans et al. (2019). As reported in Table 4 and shown in Fig. 6, the measured binary yield is indeed below that of α-pinene alone (either including or excluding isoprene in the calculation). For the o-cresol–isoprene mixture, the effect is more ambiguous, mainly because the SOA mass had not finished increasing at the conclusion of the experiments, but also because the yields are lower and deviations between the yields more difficult to measure. It should be mentioned that the particle mass yield of isoprene has been measured to be substantial in the presence of acidic seeds (which were not used in our experiments) and cannot be ignored. This raises an interesting question: should there be a threshold below which the reference for mixture yield should exclude a component? Clearly a zero-yield or extremely low-SOA-yield component such as CO or CH4 (Bianchi et al., 2016) would not be expected to contribute any particle mass in a mixture and would be expected to reduce the overall mixture yield when replacing reactivity of a non-negligible yield SOA precursor. In such cases, and in the case of isoprene with a neutral seed, it appears to make sense to inspect the yield at the consumption of a known SOA precursor with reference to that of the precursor for any enhancement or suppression. In mixtures of multiple VOCs with non-negligible individual particle mass yields, it makes sense to compare the particle mass yield of the mixture to the linear combination of the individual precursor's yields at the same VOC consumption. The measurement in the α-pinene–o-cresol binary mixture is higher than the “prediction” based on the individual precursor experimental yields, possibly indicating enhancement of the mass in the mixed experiment above that which would result from the yields of the individual VOCs. The “predictions” with and without isoprene consumption are comparable to the measurements in the ternary mixture. Without additional composition measurements, this could be interpreted as no interactions taking place or possibly as the suppression of yield by the presence of isoprene offsetting the enhancement of the mass resulting from the combined oxidation of α-pinene and o-cresol. However, such predictions may be overly simplistic. Clearly the oxidation conditions vary significantly across the mixtures (as seen from Figs. 1 and S1), and the differences in the chemical trajectories and the time series of the ratios of oxidants between the systems give reason to question the validity of the yield predictions. This is likely to be important in α-pinene-containing systems. Here the particle mass yields from ozonolysis and OH consumption have been reported to differ substantially, and Fig. 7 shows that about twice as much α-pinene results from consumption by O3 as that by OH in the α-pinene–isoprene system. In the single-VOC α-pinene system, the consumption by each oxidant was found to comprise roughly 50 % of the total based on the assumption that the consumption not attributable to the measured O3 was attributable to OH (not shown). Such differences in yields from VOC consumption by different oxidants necessitate the consideration of the changing ozone production potential and the secondary formation of OH from ozonolysis or nitrate radical attack of the components in a mixture. Irrespective of whether the presence of a VOC in a mixture has a substantial direct impact on the SOA yield, its influence on the relative concentrations of oxidants may indirectly impact the mixture's SOA particle mass yield.
A final consideration when reporting yields relates to the use of seeds as absorptive mass. Conventionally, SOA particle yield curves are plotted against absorptive mass, which, in nucleation experiments, is equivalent to the SOA particle mass. In the seeded experiments in the current study, the seeds are used to provide a condensation sink to suppress nucleation, to compete effectively with the walls for condensable vapour, and to facilitate measurement by ensuring particles are of a detectable size for composition measurement from early in the experiments. The seeds are nebulised from solution and introduced (and presumably maintained) as metastable aqueous electrolyte solution aerosol at the RH of the experiment. Nevertheless, it is clear that such strong solution particles act as absorptive mass, since organic mass is observed early in the experiments by AMS (Figs. S3, S8 and S14) and earlier than the onset of nucleation in experiments at comparable VOC concentrations (not shown). Moreover, they are internally mixed throughout the experiments, as evidenced by HTDMA measurements (shown in Fig. S14 and detailed in Wang et al., 2022). Whether the total inorganic mass should be included when interpreting yield data is not clear. Figure S4 depicts the yield behaviour with the inclusion of inorganic seed in the total particle mass, assuming it acts as an effective mass for absorptive partitioning. The added insight provided by such a plot is unclear, though there is a strong argument for dependence on total absorptive mass in the representation of absorptive partitioning even if this is reduced by a factor (analogous to a mass-based activity coefficient to account for non-ideal mixing effects). Note here that inorganic nitrate formation from OH oxidation of NO2 and subsequent neutralisation of the HNO3 by available NH3 lead to the changes in inorganic components throughout the experiments, as shown in the top panels of Fig. S14. The effects of changing inorganic seed composition represent an important topic for future studies.
4.3 Chemical composition
The addition of measurements of particle chemical composition provide important insights into the development of the photo-oxidation in mixtures and the resulting particle yields.
Online measurements by AMS in Figs. S7 and S8 reveal interesting differences in the degree of oxygenation of the SOA in different systems. The full-reactivity single-VOC α-pinene experiment has the lowest f44 and highest f43 of all systems. Systems that do not contain α-pinene can be seen to comprise a persistently higher f44 and lower f43 than all α-pinene-containing systems. This is attributable to a higher contribution of the mass from products of α-pinene with a lower degree of oxygenation when it is present in the mixture. However, there is a substantial difference in the fractional contributions of the fragments with concentration in the single-VOC α-pinene experiments, with higher f44 in the lower-concentration experiments forming lower mass. This may be partly explained in terms of absorptive partitioning, with lower absorptive mass enabling condensation of only the lower-volatility (and more oxidised and higher-f44) products. However, such effects would likely be small since the mass reduction is not large. Moreover, the difference between f44 at full reactivity and half-reactivity might be expected to be smaller than that between half-reactivity and one-third reactivity since the mass difference is smaller between the former pair of experiments. However, the reverse is true and the reason for this is not obvious. Single-VOC o-cresol and its binary mixture with isoprene rapidly exhibit, and then maintain through to significant particle mass production, a higher degree of oxygenation than the other systems as represented by the f44. The high f44 generated in the o-cresol systems and persisting through to high masses later in the experiments is notable and suggests that a high degree of oxygenation is required to enable partitioning of these compounds to the particles (Emanuelsson et al., 2013). This contrasts sharply with the lower f44 of all α-pinene-containing systems. This may indicate the requirement for molecules derived from the smaller C7 precursor to be more oxygenated than those from its C10 counterpart. Since α-pinene frequently dominates the mass owing to its high yield, this may offset any o-cresol-derived high f44 in systems containing both α-pinene and o-cresol. Clearly a less coarse diagnostic of composition is required to determine the reasons for the observed differences. Shao et al. (2022d) examine the oxidation state of carbon in all systems using combined analyses of high-resolution AMS, FIGAERO-I−-CIMS, and LC-Orbitrap MS data. The attribution of the 44 fragment to di- and polycarboxylic acids is interesting in this context given the high fractional signal contribution of C7H7NO4 isomers from FIGAERO-I−-CIMS and LC-Orbitrap MS; see text below and Fig. S11.
The FIGAERO-I−-CIMS data shown in Fig. S9 provide an indication of the sorts of analytical products that can aid this interpretation. Whilst the additional analyses presented in Du et al. (2022a) and Du et al. (2022b) can more fully explore the impacts of the mixing of SOA precursors, several features can be directly observed from the mass spectra. First, there is an evident increase in the contribution of higher-mass ions with time in the α-pinene single-VOC system that is not observed in either the o-cresol single-VOC system or their binary mixture. Second, there is clear evidence for signal contributions from components in the mixture that are not present in the individual systems. This is expanded upon in Fig. S10, which shows that much of the signal in analyses of the filters taken at the end in the ternary mixture is not found at masses corresponding to those found in individual-component VOC oxidation. Such high-resolution analyses are required to unambiguously identify the specific products resulting from interactions in the system and to postulate mechanisms leading to their formation. It is not yet possible to state whether the molecules uniquely found in the mixtures are formed by gas-phase cross-reaction or condensed phase accretion in the particles or on the filter, and it has been known for some time that reactions on filters can lead to potential artefacts (Turpin et al., 1994; Liu et al., 2014). Neither is it currently possible to unambiguously state that this signal corresponds to the same fraction of the mass owing to potential differential sensitivity. These aspects are discussed in detail in Shao et al. (2022b). Nonetheless, it can be stated that these compounds are only found in the mixed systems. An important challenge with such identification and attributions is the requirement for substantial sampled mass. This may provide a lower limit to the yield and/or initial concentration of precursors that can be studied in this way. Nevertheless, the power of such analyses is further exploited in separating the isomeric contributions to particle components as shown in Fig. S11a. Combining the power of offline and online analyses, this separation of C7H7NO4 isomers in all o-cresol-containing systems may be used to interpret the significant differences in the time trends of C7H7NO4 shown in the FIGAERO-I−-CIMS data in Fig. S11b. Whilst this may be indicative of mechanistic differences, there are confounding differences in the rate of SOA particle mass formation between the systems (and hence abundance of absorptive mass) in addition to potential differences in the sensitivity in the different systems and total signal used for the normalisation because of the different FIGAERO filter loadings. These considerations are discussed in detail in Du et al. (2022b) for all systems. Nonetheless, the isolation of the fractional isomeric contributions and the differences in the time series reveal changes in the behaviour of the C7H7NO4 contribution between the systems, even if this behaviour has yet to be fully reconciled.
4.4 Properties: volatility, water uptake, and phase
Figure S12 exploits the high-resolution analysis of the FIGAERO-CIMS data to show the O : C ratio of detected particle compounds as a function of their carbon number in all systems at the end of the experiments. Combining this with the ratio of signal in the particle and gas samples is used to indicate likely influences of the mixing of precursors on particle volatility. Whilst there are uncertainties associated with the partitioning and hence volatility derivations, it is evident that changes in the components present in mixtures can lead to change in the volatility and potentially yield, as reported in detail in Voliotis et al. (2021, 2022). This is extended in Fig. S13, which expresses the FIGAERO-I−-CIMS data as volatility distributions, further highlighting the differences between the particle volatility in the mixture and in the single-VOC systems. This is clearly complex but is broadly consistent with the observed yield behaviour for most systems. For example, the SOA particle volatility in the single α-pinene experiment at half-reactivity is comparable to that measured in the binary system with isoprene, consistent with their comparable yields. Similarly, the measured volatility in the α-pinene and o-cresol binary systems and in the ternary systems appear to be roughly between those obtained in the single-precursor experiments, in line with the measured yields that were found to be higher than those in single o-cresol but equal to or lower than those obtained in the single α-pinene experiments. As has been described in detail elsewhere (see Stark et al., 2017, Voliotis et al., 2021, and Du et al., 2022a), the quantification of the effective saturation concentration from the FIGAERO-CIMS is challenging for compounds with high or low C* due to signal-to-noise limitations of the technique. This results in a narrowing of the retrieved volatility distributions. Nonetheless, Voliotis et al. (2021) showed that the technique can reasonably capture the volatility changes between the different systems. Substantial apparent inconsistencies were found in the o-cresol and isoprene binary system, wherein the measured volatility appeared to be substantially higher than that in the single o-cresol system, consistent with the lower SOA particle yields measured (Table 3).
The differences in the rates of formation of SOA in the particles and the total mass formed directly influence the rates of change of particle properties as expected. Since the particles are grown on inorganic seeds, the physical properties in all systems are initially dominated by the inorganic components. For clarity, three example systems (o-cresol–isoprene, o-cresol, and α-pinene) are shown in Fig. S14. This shows the relationship between the multicomponent particle composition, the hygroscopicity parameter, κ (under subsaturated and supersaturated conditions from HTDMA and CCNc, respectively), and the rebound fraction (BF) of particles (indicative of their phase state) as the SOA to inorganic mass fraction of the particles develops in the example systems. Here, aerosol particles tend to be liquid at BF below 0.2, non-liquid above 0.8, and in a transition phase between 0.2 and 0.8. The hygroscopicity much more rapidly decreases in systems wherein organic material is more rapidly formed, and the transition to a higher fraction of particles rebounding on a filter at high RH is similarly more rapid. It is clear that the organic-to-inorganic ratio plays a controlling role in the physical behaviour of the particles. The evolution of both these properties across all systems is discussed in detail in Wang et al. (2021, 2022).
4.5 Further work
Whilst it is important to explore mixtures including multiple SOA particle precursors under controlled conditions to understand behaviour in the real atmosphere, and measurements in such mixtures can reveal features inaccessible to single-VOC experiment, the experimental design will determine their usefulness. Alternative and supplementary methodologies may allow more direct resolution of outstanding questions related to precursor mixing. The effects of mixed and variable oxidant concentrations throughout chamber experiments are both problematic and insightful. Clearly secondary production of oxidants (both OH and ozone) may differentially influence the decay of VOCs in the mixture and it may not be possible to scavenge secondary oxidants without influencing the oxidation product distribution. In any case, secondary oxidant formation occurs in the real atmosphere and cannot be overlooked. More control over the primary oxidants may be achieved through use of “cleaner” sources, such as photolysis of H2O2 since avoidance of O3 from injection or NO2 photolysis may be advantageous. Nevertheless, secondary O3 will react with any unsaturated compounds present. Selection of VOCs with low OH yields from ozonolysis may allow isolation of its pathways but will limit the choice of mixtures. Augmentation of broadband illumination with discrete intense light sources such as in the JPAC in the McFiggans et al. (2019) study may increase the OH : O3 ratio such that the OH channels dominate, though care is necessary to avoid excessively high OH concentrations. Unless conducted at very low NOx concentrations, significant VOC consumption in batch-mode chamber experiments will lead to variable VOC : NOx ratios. This will lead to variable production rates of ozone as well as changes in the relative importance of the NO-mediated alkoxy radical pathways. Mechanistic interpretation of such experiments must account for these changes. In any case, isolation of individual mechanistic pathways and probing details of kinetics and product yields may be more suited to more targeted flow tube studies than to chamber experiments. If it is not possible to control the oxidising environment across complex mixtures, then systematic exploration of mixtures with direct and more comprehensive oxidant and peroxy radical measurements would be of benefit. Such well-instrumented studies will be able to take full advantage of the trajectories through chemical space afforded by relatively long batch-mode chamber experiments. Movement towards more atmospherically representative chemical regimes would benefit from lower-VOC-concentration experiments. However, moving towards a more realistic radical termination regime would present detection limit challenges to some techniques for composition measurements. More comprehensive coverage of speciated VOC and OVOC measurements using modern online mass spectrometry methods and improved mass spectral resolution of such temporally resolved instruments may avoid the need to combine online and offline techniques. Whilst they reveal important impacts of the temporal changes in VOC mixtures, the batch reactor experiments in the present study are complex. Use of well-mixed flow reactor experiments to study multiple steady states as in McFiggans et al. (2019) can enable focus of batch reactor experiments on sensitive areas of the chemical regime. The range of potential mixtures is vast, so there is a need to focus on targeted areas of importance. It is not immediately obvious how this should be done, though it should be recognised that oxidation and SOA formation will occur during both day and night, and night-time oxidation in mixtures by O3 and NO3 should be considered.
A comprehensive suite of instrumentation was deployed to investigate the formation of SOA particulate mass on inorganic seeds in chamber photo-oxidation of anthropogenic (o-cresol) and biogenic (α-pinene and isoprene) VOCs as well as their binary and ternary mixtures in the presence of NOx. Whilst compromises were necessary in the experimental design and the complexity of the systems introduced substantial challenges to their interpretation, several important observations were possible.
First, the photochemical trajectory as defined by the changing deviation from photostationary state and of the changing ratio of oxidants is understandably system-dependent, and the rates of consumption of each VOC by each oxidant are consequently variable.
Second, the yield concept typically used for an individual VOC requires careful consideration when adapting it for use with complex mixtures. In order to account for the dependence of condensed mass on available adsorptive mass, it will be necessary to use the individual component yield at the same mass as in the mixtures. However, in order to compare the mass formed at the same VOC consumption, it is necessary to reference this to the corresponding single-precursor experiment at that same VOC consumption and therefore not that calculated at the same adsorptive mass.
In mixtures in which the majority of the mass is contributed by only one component precursor, it is possible to identify and quantify any interactions in terms of SOA mass or yield. Thus, as shown in Fig. 5, there is clear indication of suppression of the yield of α-pinene in its mixture with isoprene, but as with the o-cresol–isoprene mixture there is a possible indication of enhancement, though this is too small to be unambiguous.
It is as straightforward to make a comparison between the predicted and measured yield in mixtures of precursors with appreciable yield as it is to compare the measured yield of a single component with its yield in a mixture with a precursor of zero yield. When two components with appreciable yield are mixed with another VOC with no yield the reference point for the comparison can be complex, and our approach to “prediction” of the yield would depend on the assumed baseline for the individual components.
The mixed α-pinene–o-cresol system is measured to have an enhanced yield above that expected from additivity of the individual VOC yield at the same consumption. In such mixtures, in which there is a significant contribution to the SOA mass from more than one component precursor, we are unable to unambiguously attribute any discrepancies in predicted and measured mass to physical or chemical interactions. This is because yield and partitioned mass will depend on available adsorptive mass and on the rate of consumption of individual VOCs as well as the production of condensable oxidation products. In the ternary system, the measured yield is comparable to that calculated from additivity of the component yields. It is unclear whether this is attributable to cancelling of suppression and enhancement effects, but chemical interactions are evident from the components uniquely found in the mixtures. Importantly, more work is required to understand the influence of Teflon wall interactions with vapours on comparison of yields in systems wherein different SOA particle masses are formed.
The physical properties such as water uptake and phase behaviour of the particles will depend on the SOA particle mass formed, so changes in the change in particle composition in mixtures will control changes in particle physical properties.
Mixing experiments are crucial and highly beneficial for our understanding of atmospheric chemical interactions. However, the interpretation quickly becomes complex, and both the experimental design and evaluation need to be scrutinised carefully. Here, advanced online and offline compositional measurements can reveal substantial additional information to aid in the interpretation of yield data, including components uniquely found in mixtures and physicochemical property changes in the SOA formed from mixtures of VOCs.
All the data used in this study for each of the experiments shown in Table 1 are openly available at the EUROCHAMP database (https://data.eurochamp.org/data-access/chamber-experiments/#/, EUROCHAMP 2020, 2022). Their DOIs are listed in Table S1. The remaining data discussed here and shown in Figs. S7 to S14 are available upon request from the corresponding author(s).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-14147-2022-supplement.
GM, MRA, MH, TFM AV, YW, YS, and MD conceived the study. AV, YW, YS, and MD conducted the experiments and data analysis. TJB, DH, and KP provided on-site support with the instrument deployment and data analysis procedures. JFH provided scientific advice. GM, AV, YW, YS, and MD wrote the paper with inputs from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The Manchester Aerosol Chamber received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 730997, which supports the EUROCHAMP2020 research programme. Aristeidis Voliotis and Mao Du acknowledge the Presidents Doctoral Scholarship from the University of Manchester, and Aristeidis Voliotis acknowledges support from the Natural Environment Research Council (NERC) EAO Doctoral Training Partnership (grant no. NE/L002469/1). Yu Wang acknowledges CSC scholarship support. M. Rami Alfarra acknowledges funding support from the Natural Environment Research Council (NERC) through the UK National Centre for Atmospheric Science (NCAS). Instrumentational support was funded through the NERC Atmospheric Measurement and Observational Facility (AMOF). We acknowledge Zhijun Wu from Peking University, China, for providing the bounce behaviour instrument through Trans-National Access (TNA) of EUROCHAMP-2020. Mattias Hallquist and Thomas F. Mentel acknowledge the strategic research area MERGE, the Swedish Research Council (grant no. 2018-04430), and Formas (grant no. 2019-586). Thomas F. Mentel acknowledges support from the EU project FORCeS (grant agreement no. 821205).
This research has been supported by the Natural Environment Research Council (grant no. NE/L002469/1).
This paper was edited by Ivan Kourtchev and reviewed by three anonymous referees.
Alfarra, M. R., Hamilton, J. F., Wyche, K. P., Good, N., Ward, M. W., Carr, T., Barley, M. H., Monks, P. S., Jenkin, M. E., Lewis, A. C., and McFiggans, G. B.: The effect of photochemical ageing and initial precursor concentration on the composition and hygroscopic properties of β-caryophyllene secondary organic aerosol, Atmos. Chem. Phys., 12, 6417–6436, https://doi.org/10.5194/acp-12-6417-2012, 2012.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018.
Bianchi, F., Barmet, P., Stirnweis, L., El Haddad, I., Platt, S. M., Saurer, M., Lötscher, C., Siegwolf, R., Bigi, A., Hoyle, C. R., DeCarlo, P. F., Slowik, J. G., Prévôt, A. S. H., Baltensperger, U., and Dommen, J.: Contribution of methane to aerosol carbon mass, Atmos. Environ., 141, 41–47, https://doi.org/10.1016/j.atmosenv.2016.06.036, 2016.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bowman, F. M., Odum, J. R., Seinfeld, J. H., and Pandis, S. N.: Mathematical model for gas-particle partitioning of secondary organic aerosols, Atmos. Environ., 31, 3921–3931, https://doi.org/10.1016/S1352-2310(97)00245-8, 1997.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Cash, J. M., Heal, M. R., Langford, B., and Drewer, J.: A review of stereochemical implications in the generation of secondary organic aerosol from isoprene oxidation, Environ. Sci.-Proc. Imp., 18, 1369–1380, https://doi.org/10.1039/C6EM00354K, 2016.
Charan, S. M., Huang, Y., and Seinfeld, J. H.: Computational Simulation of Secondary Organic Aerosol Formation in Laboratory Chambers, Chem. Rev., 119, 11912–11944, https://doi.org/10.1021/acs.chemrev.9b00358, 2019.
Cohen, A. J., Brauer, M., Burnett, R., Anderson, H. R., Frostad, J., Estep, K., Balakrishnan, K., Brunekreef, B., Dandona, L., Dandona, R., Feigin, V., Freedman, G., Hubbell, B., Jobling, A., Kan, H., Knibbs, L., Liu, Y., Martin, R., Morawska, L., Pope, C. A., III, Shin, H., Straif, K., Shaddick, G., Thomas, M., van Dingenen, R., van Donkelaar, A., Vos, T., Murray, C. J. L., and Forouzanfar, M. H.: Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015, The Lancet, 389, 1907–1918, https://doi.org/10.1016/S0140-6736(17)30505-6, 2017.
Cox, R. A., Ammann, M., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 20, 13497–13519, https://doi.org/10.5194/acp-20-13497-2020, 2020.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, The J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, 2006.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K.-H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012.
Du, M., Voliotis, A., Shao, Y., Wang, Y., Bannan, T. J., Pereira, K. L., Hamilton, J. F., Percival, C. J., Alfarra, M. R., and McFiggans, G.: Combined application of online FIGAERO-CIMS and offline LC-Orbitrap mass spectrometry (MS) to characterize the chemical composition of secondary organic aerosol (SOA) in smog chamber studies, Atmos. Meas. Tech., 15, 4385–4406, https://doi.org/10.5194/amt-15-4385-2022, 2022a.
Du, M., Voliotis, A., Shao, Y., Wang, Y., Bannan, T. J., Pereira, K. L., Hamilton, J. F., Percival, C. J., Rami Alfarra, M., and McFiggans, G.: Chemical characterization and clustering behaviour of oxidation products generated from the mixed anthropogenic and biogenic VOCs in chamber studies, Atmos. Chem. Phys., in preparation, 2022b.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012.
Ehn, M., Kleist, E., Junninen, H., Petäjä, T., Lönn, G., Schobesberger, S., Dal Maso, M., Trimborn, A., Kulmala, M., Worsnop, D. R., Wahner, A., Wildt, J., and Mentel, Th. F.: Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 12, 5113–5127, https://doi.org/10.5194/acp-12-5113-2012, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Emanuelsson, E. U., Hallquist, M., Kristensen, K., Glasius, M., Bohn, B., Fuchs, H., Kammer, B., Kiendler-Scharr, A., Nehr, S., Rubach, F., Tillmann, R., Wahner, A., Wu, H.-C., and Mentel, Th. F.: Formation of anthropogenic secondary organic aerosol (SOA) and its influence on biogenic SOA properties, Atmos. Chem. Phys., 13, 2837–2855, https://doi.org/10.5194/acp-13-2837-2013, 2013.
EUROCHAMP 2020: The Database of Atmospheric Simulation Chamber Studies (DASCS), https://data.eurochamp.org/data-access/chamber-experiments/#/, last access: 24 October 2022.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, 2007.
Good, N., Coe, H., and McFiggans, G.: Instrumentational operation and analytical methodology for the reconciliation of aerosol water uptake under sub- and supersaturated conditions, Atmos. Meas. Tech., 3, 1241–1254, https://doi.org/10.5194/amt-3-1241-2010, 2010.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hamilton, J. F., Rami Alfarra, M., Wyche, K. P., Ward, M. W., Lewis, A. C., McFiggans, G. B., Good, N., Monks, P. S., Carr, T., White, I. R., and Purvis, R. M.: Investigating the use of secondary organic aerosol as seed particles in simulation chamber experiments, Atmos. Chem. Phys., 11, 5917–5929, https://doi.org/10.5194/acp-11-5917-2011, 2011.
Hao, L. Q., Yli-Pirilä, P., Tiitta, P., Romakkaniemi, S., Vaattovaara, P., Kajos, M. K., Rinne, J., Heijari, J., Kortelainen, A., Miettinen, P., Kroll, J. H., Holopainen, J. K., Smith, J. N., Joutsensaari, J., Kulmala, M., Worsnop, D. R., and Laaksonen, A.: New particle formation from the oxidation of direct emissions of pine seedlings, Atmos. Chem. Phys., 9, 8121–8137, https://doi.org/10.5194/acp-9-8121-2009, 2009.
Henry, F., Coeur-Tourneur, C., Ledoux, F., Tomas, A., and Menu, D.: Secondary organic aerosol formation from the gas phase reaction of hydroxyl radicals with m-, o- and p-cresol, Atmos. Environ., 42, 3035–3045, https://doi.org/10.1016/j.atmosenv.2007.12.043, 2008.
IPCC: Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M. I., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R., and Zhou, B. (Eds.): Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, in press, 2021.
Jaoui, M. and Kamens, R. M.: Gaseous and Particulate Oxidation Products Analysis of a Mixture of α-pinene +β-pinene/O3/Air in the Absence of Light and α-pinene +β-pinene/NOx/Air in the Presence of Natural Sunlight, J. Atmos. Chem., 44, 259–297, https://doi.org/10.1023/A:1022977427523, 2003.
Jenkin, M. E., Wyche, K. P., Evans, C. J., Carr, T., Monks, P. S., Alfarra, M. R., Barley, M. H., McFiggans, G. B., Young, J. C., and Rickard, A. R.: Development and chamber evaluation of the MCM v3.2 degradation scheme for β-caryophyllene, Atmos. Chem. Phys., 12, 5275–5308, https://doi.org/10.5194/acp-12-5275-2012, 2012.
Jenkin, M. E., Derwent, R. G., and Wallington, T. J.: Photochemical ozone creation potentials for volatile organic compounds: rationalization and estimation, Atmos. Environ. 163, 128–137, https://doi.org/10.1016/j.atmosenv.2017.05.024, 2017.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525, https://doi.org/10.1126/science.1180353, 2009.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Joutsensaari, J., Loivamäki, M., Vuorinen, T., Miettinen, P., Nerg, A.-M., Holopainen, J. K., and Laaksonen, A.: Nanoparticle formation by ozonolysis of inducible plant volatiles, Atmos. Chem. Phys., 5, 1489–1495, https://doi.org/10.5194/acp-5-1489-2005, 2005.
Kaltsonoudis, C., Kostenidou, E., Louvaris, E., Psichoudaki, M., Tsiligiannis, E., Florou, K., Liangou, A., and Pandis, S. N.: Characterization of fresh and aged organic aerosol emissions from meat charbroiling, Atmos. Chem. Phys., 17, 7143–7155, https://doi.org/10.5194/acp-17-7143-2017, 2017.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kiendler-Scharr, A., Wildt, J., Maso, M. D., Hohaus, T., Kleist, E., Mentel, T. F., Tillmann, R., Uerlings, R., Schurr, U., and Wahner, A.: New particle formation in forests inhibited by isoprene emissions, Nature, 461, 381–384, https://doi.org/10.1038/nature08292, 2009.
Krechmer, J. E., Day, D. A., and Jimenez, J. L.: Always Lost but Never Forgotten: Gas-Phase Wall Losses Are Important in All Teflon Environmental Chambers, Environ. Sci. Technol., 54, 12890–12897, https://doi.org/10.1021/acs.est.0c03381, 2020.
Leighton, P. A.: Photochemistry of air pollution, Phys. Chem., 9, 152–183, 1961.
Liu, C.-N., Lin, S.-F., Awasthi, A., Tsai, C.-J., Wu, Y.-C., and Chen, C.-F.: Sampling and conditioning artifacts of PM2.5 in filter-based samplers, Atmos. Environ., 85, 48–53, s10.1016/j.atmosenv.2013.11.075, 2014.
Liu, Y., Wu, Z., Wang, Y., Xiao, Y., Gu, F., Zheng, J., Tan, T., Shang, D., Wu, Y., Zeng, L., Hu, M., Bateman, A. P., and Martin, S. T.: Submicrometer Particles Are in the Liquid State during Heavy Haze Episodes in the Urban Atmosphere of Beijing, China, Environ. Sci. Tech. Lett., 4, 427–432, https://doi.org/10.1021/acs.estlett.7b00352, 2017.
Loza, C. L., Chan, A. W. H., Galloway, M. M., Keutsch, F. N., Flagan, R. C., and Seinfeld, J. H.: Characterization of Vapor Wall Loss in Laboratory Chambers, Environ. Sci. Technol., 44, 5074–5078, https://doi.org/10.1021/es100727v, 2010.
McFiggans, G., Coe, H., Burgess, R., Allan, J., Cubison, M., Alfarra, M. R., Saunders, R., Saiz-Lopez, A., Plane, J. M. C., Wevill, D., Carpenter, L., Rickard, A. R., and Monks, P. S.: Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine, Atmos. Chem. Phys., 4, 701–713, https://doi.org/10.5194/acp-4-701-2004, 2004.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, A. M., Simpson, D., Bergstrom, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mehra, A., Wang, Y., Krechmer, J. E., Lambe, A., Majluf, F., Morris, M. A., Priestley, M., Bannan, T. J., Bryant, D. J., Pereira, K. L., Hamilton, J. F., Rickard, A. R., Newland, M. J., Stark, H., Croteau, P., Jayne, J. T., Worsnop, D. R., Canagaratna, M. R., Wang, L., and Coe, H.: Evaluation of the chemical composition of gas- and particle-phase products of aromatic oxidation, Atmos. Chem. Phys., 20, 9783–9803, https://doi.org/10.5194/acp-20-9783-2020, 2020.
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥ C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021.
Mentel, Th. F., Wildt, J., Kiendler-Scharr, A., Kleist, E., Tillmann, R., Dal Maso, M., Fisseha, R., Hohaus, Th., Spahn, H., Uerlings, R., Wegener, R., Griffiths, P. T., Dinar, E., Rudich, Y., and Wahner, A.: Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 9, 4387–4406, https://doi.org/10.5194/acp-9-4387-2009, 2009.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Nakao, S., Shrivastava, M., Nguyen, A., Jung, H., and Cocker III, D. R.: Interpretation of Secondary Organic Aerosol Formation from Diesel Exhaust Photooxidation in an Environmental Chamber, Aerosol Sci. Tech., 45, 964–972, https://doi.org/10.1080/02786826.2011.573510, 2011a.
Nakao, S., Clark, C., Tang, P., Sato, K., and Cocker III, D.: Secondary organic aerosol formation from phenolic compounds in the absence of NOx, Atmos. Chem. Phys., 11, 10649–10660, https://doi.org/10.5194/acp-11-10649-2011, 2011b.
Nakao, S., Liu, Y., Tang, P., Chen, C.-L., Zhang, J., and Cocker III, D. R.: Chamber studies of SOA formation from aromatic hydrocarbons: observation of limited glyoxal uptake, Atmos. Chem. Phys., 12, 3927–3937, https://doi.org/10.5194/acp-12-3927-2012, 2012.
Nordin, E. Z., Eriksson, A. C., Roldin, P., Nilsson, P. T., Carlsson, J. E., Kajos, M. K., Hellén, H., Wittbom, C., Rissler, J., Löndahl, J., Swietlicki, E., Svenningsson, B., Bohgard, M., Kulmala, M., Hallquist, M., and Pagels, J. H.: Secondary organic aerosol formation from idling gasoline passenger vehicle emissions investigated in a smog chamber, Atmos. Chem. Phys., 13, 6101–6116, https://doi.org/10.5194/acp-13-6101-2013, 2013.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
Pankow, J. F.: An absorption-model of gas-particle partitioning of organic-compounds in the atmosphere, Atmos. Environ., 28, 185–188, https://doi.org/10.1016/1352-2310(94)90093-0, 1994.
Peng, Z. and Jimenez, J. L.: Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 49, 2570–2616, https://doi.org/10.1039/C9CS00766K, 2020.
Pereira K.L., M. W. Ward, J. L. Wilkinson, J. B. Sallach, D. J. Bryant, W. J. Dixon, J. F. Hamilton and A. C. Lewis, An Automated Methodology for Non-targeted Compositional Analysis of Small Molecules in High Complexity Environmental Matrices Using Coupled Ultra Performance Liquid Chromatography Orbitrap Mass Spectrometry. Environ. Sci. Technol, 18, 7365–7375, 10.1021/acs.est.0c08208, 2021.
Pinto, D. M., Tiiva, P., Miettinen, P., Joutsensaari, J., Kokkola, H., Nerg, A.-M., Laaksonen, A., and Holopainen, J. K.: The effects of increasing atmospheric ozone on biogenic monoterpene profiles and the formation of secondary aerosols, Atmos. Environ., 41, 4877–4887, https://doi.org/10.1016/j.atmosenv.2007.02.006, 2007.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Močnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Priestley, M., Bannan, T. J., Le Breton, M., Worrall, S. D., Kang, S., Pullinen, I., Schmitt, S., Tillmann, R., Kleist, E., Zhao, D., Wildt, J., Garmash, O., Mehra, A., Bacak, A., Shallcross, D. E., Kiendler-Scharr, A., Hallquist, Å. M., Ehn, M., Coe, H., Percival, C. J., Hallquist, M., Mentel, T. F., and McFiggans, G.: Chemical characterisation of benzene oxidation products under high- and low-NOx conditions using chemical ionisation mass spectrometry, Atmos. Chem. Phys., 21, 3473–3490, https://doi.org/10.5194/acp-21-3473-2021, 2021.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Reyes-Villegas, E., Bannan, T., Le Breton, M., Mehra, A., Priestley, M., Percival, C., Coe, H., and Allan, J. D.: Online Chemical Characterization of Food-Cooking Organic Aerosols: Implications for Source Apportionment, Environ. Sci. Technol., 52, 5308–5318, https://doi.org/10.1021/acs.est.7b06278, 2018.
Ródenas, M., Munoz, A., Alacreu, F., Brauers, T., Dorn, H. P., Kleffmann, J., and Bloss, W.: Assessment of HONO Measurements: The FIONA Campaign at EUPHORE, in: Disposal of Dangerous Chemicals in Urban Areas and Mega Cities: Role of Oxides and Acids of Nitrogen in Atmospheric Chemistry, edited by: Barnes, I. and Rudzinski, K. J., Springer, Dordrecht, 45–58, https://doi.org/10.1007/978-94-007-5034-0_4, 2013.
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Schervish, M. and Donahue, N. M.: Peroxy radical chemistry and the volatility basis set, Atmos. Chem. Phys., 20, 1183–1199, https://doi.org/10.5194/acp-20-1183-2020, 2020.
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017.
Shao, Y., Wang, Y., Du, M., Voliotis, A., Alfarra, M. R., O'Meara, S. P., Turner, S. F., and McFiggans, G.: Characterisation of the Manchester Aerosol Chamber facility, Atmos. Meas. Tech., 15, 539–559, https://doi.org/10.5194/amt-15-539-2022, 2022a.
Shao, Y., Voliotis, A., Du, M., Wang, Y., Pereira, K., Hamilton, J., Alfarra, M. R., and McFiggans, G.: Chemical composition of secondary organic aerosol particles formed from mixtures of anthropogenic and biogenic precursors, Atmos. Chem. Phys., 22, 9799–9826, https://doi.org/10.5194/acp-22-9799-2022, 2022b.
Shao, Y., Voliotis, A., Du, M., Wang, Y., Pereira, K., Hamilton, J., Alfarra, M. R., and McFiggans, G.: Characterisation of the oxidation state of secondary organic aerosols in mixed precursor systems, Atmos. Chem. Phys., in preparation, 2022c.
Shao, Y., Voliotis, A., Du, M., Wang, Y., Pereira, K., Hamilton, J., Alfarra, M. R., and McFiggans, G.: The evolution of carbon oxidation state during secondary organic aerosol formation from individual and mixed organic precursors, Atmos. Chem. Phys., in preparation, 2022d.
Shilling, J. E., Zawadowicz, M. A., Liu, J. M., Zaveri, R. A., and Zelenyuk, A.: Photochemical Aging Alters Secondary Organic Aerosol Partitioning Behavior, ACS Earth Space Chem., 3, 2704–2716, https://doi.org/10.1021/acsearthspacechem.9b00248, 2019.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T. Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Thornton, J. A., Shilling, J. E., Shrivastava, M., D'Ambro, E. L., Zawadowicz, M. A., and Liu, J.: A Near-Explicit Mechanistic Evaluation of Isoprene Photochemical Secondary Organic Aerosol Formation and Evolution: Simulations of Multiple Chamber Experiments with and without Added NOx, ACS Earth Space Chem., 4, 1161–1181, https://doi.org/10.1021/acsearthspacechem.0c00118, 2020.
Tiitta, P., Leskinen, A., Hao, L., Yli-Pirilä, P., Kortelainen, M., Grigonyte, J., Tissari, J., Lamberg, H., Hartikainen, A., Kuuspalo, K., Kortelainen, A.-M., Virtanen, A., Lehtinen, K. E. J., Komppula, M., Pieber, S., Prévôt, A. S. H., Onasch, T. B., Worsnop, D. R., Czech, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Transformation of logwood combustion emissions in a smog chamber: formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging, Atmos. Chem. Phys., 16, 13251–13269, https://doi.org/10.5194/acp-16-13251-2016, 2016.
Tsigaridis, K. and Kanakidou, M.: The Present and Future of Secondary Organic Aerosol Direct Forcing on Climate, Current Climate Change Reports, 4, 84–98, https://doi.org/10.1007/s40641-018-0092-3, 2018.
Tsiligiannis, E., Hammes, J., Salvador, C. M., Mentel, T. F., and Hallquist, M.: Effect of NOx on 1,3,5-trimethylbenzene (TMB) oxidation product distribution and particle formation, Atmos. Chem. Phys., 19, 15073–15086, https://doi.org/10.5194/acp-19-15073-2019, 2019.
Turpin, B. J., Huntzicker, J. J., and Hering, S. V.: Investigation of organic aerosol sampling artifacts in the los angeles basin, Atmos. Environ., 28, 3061–3071, https://doi.org/10.1016/1352-2310(94)00133-6, 1994.
VanReken, T. M., Greenberg, J. P., Harley, P. C., Guenther, A. B., and Smith, J. N.: Direct measurement of particle formation and growth from the oxidation of biogenic emissions, Atmos. Chem. Phys., 6, 4403–4413, https://doi.org/10.5194/acp-6-4403-2006, 2006.
Villena, G., Bejan, I., Kurtenbach, R., Wiesen, P., and Kleffmann, J.: Interferences of commercial NO2 instruments in the urban atmosphere and in a smog chamber, Atmos. Meas. Tech., 5, 149–159, https://doi.org/10.5194/amt-5-149-2012, 2012.
Voliotis, A., Wang, Y., Shao, Y., Du, M., Bannan, T. J., Percival, C. J., Pandis, S. N., Alfarra, M. R., and McFiggans, G.: Exploring the composition and volatility of secondary organic aerosols in mixed anthropogenic and biogenic precursor systems, Atmos. Chem. Phys., 21, 14251–14273, https://doi.org/10.5194/acp-21-14251-2021, 2021.
Voliotis, A., Du, M., Wang, Y., Shao, Y., Bannan, T. J., Flynn, M., Pandis, S. N., Percival, C. J., Alfarra, M. R., and McFiggans, G.: The influence of the addition of isoprene on the volatility of particles formed from the photo-oxidation of anthropogenic–biogenic mixtures, Atmos. Chem. Phys., 22, 13677–13693, https://doi.org/10.5194/acp-22-13677-2022, 2022.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: autoxidation and accretion, Atmos. Chem. Phys., 20, 9563–9579, https://doi.org/10.5194/acp-20-9563-2020, 2020.
Wang, Y., Voliotis, A., Shao, Y., Zong, T., Meng, X., Du, M., Hu, D., Chen, Y., Wu, Z., Alfarra, M. R., and McFiggans, G.: Phase state of secondary organic aerosol in chamber photo-oxidation of mixed precursors, Atmos. Chem. Phys., 21, 11303–11316, https://doi.org/10.5194/acp-21-11303-2021, 2021.
Wang, Y., Voliotis, A., Hu, D., Shao, Y., Du, M., Chen, Y., Kleinheins, J., Marcolli, C., Alfarra, M. R., and McFiggans, G.: On the evolution of sub- and super-saturated water uptake of secondary organic aerosol in chamber experiments from mixed precursors, Atmos. Chem. Phys., 22, 4149–4166, https://doi.org/10.5194/acp-22-4149-2022, 2022.
Weitkamp, E. A., Sage, A. M., Pierce, J. R., Donahue, N. M., and Robinson, A. L.: Organic Aerosol Formation from Photochemical Oxidation of Diesel Exhaust in a Smog Chamber, Environ. Sci. Technol., 41, 6969–6975, https://doi.org/10.1021/es070193r, 2007.
Whalley, L. K., Stone, D., Bandy, B., Dunmore, R., Hamilton, J. F., Hopkins, J., Lee, J. D., Lewis, A. C., and Heard, D. E.: Atmospheric OH reactivity in central London: observations, model predictions and estimates of in situ ozone production, Atmos. Chem. Phys., 16, 2109–2122, https://doi.org/10.5194/acp-16-2109-2016, 2016.
Wyche, K. P., Ryan, A. C., Hewitt, C. N., Alfarra, M. R., McFiggans, G., Carr, T., Monks, P. S., Smallbone, K. L., Capes, G., Hamilton, J. F., Pugh, T. A. M., and MacKenzie, A. R.: Emissions of biogenic volatile organic compounds and subsequent photochemical production of secondary organic aerosol in mesocosm studies of temperate and tropical plant species, Atmos. Chem. Phys., 14, 12781–12801, https://doi.org/10.5194/acp-14-12781-2014, 2014.
Yang, Y., Shao, M., Wang, X., Nölscher, A. C., Kessel, S., Guenther, A., and Williams, J.: Towards a quantitative understanding of total OH reactivity: A review, Atmos. Environ., 134, 147–161, https://doi.org/10.1016/j.atmosenv.2016.03.010, 2016.
Ye, P., Ding, X., Hakala, J., Hofbauer, V., Robinson, E. S., and Donahue, N. M.: Vapor wall loss of semi-volatile organic compounds in a Teflon chamber, Aerosol Sci. Tech., 50, 822–834, https://doi.org/10.1080/02786826.2016.1195905, 2016.
Zhang, X., Cappa, C. D., Jathar, S. H., Mcvay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/C2CS35122F, 2012.
- Article
(3010 KB) - Full-text XML