the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Jun 2021
| 14 Jun 2021
Towards a chemical mechanism of the oxidation of aqueous sulfur dioxide via isoprene hydroxyl hydroperoxides (ISOPOOH)
Kelvin H. Bates
Jean C. Rivera-Rios
Joshua L. Cox
Joshua D. Shutter
Frank N. Keutsch
In-cloud chemistry has important ramifications for atmospheric particulate matter formation and gas-phase chemistry. Recent work has shown that, like hydrogen peroxide (H2O2), the two main isomers of isoprene hydroxyl hydroperoxide (ISOPOOH) oxidize sulfur dioxide dissolved in cloud droplets (SO2,aq) to sulfate. The work revealed that the pathway of SO2,aq oxidation with ISOPOOH differs from that of H2O2. We investigate the chemical mechanisms of oxidation of SO2,aq with ISOPOOH in the cloud-relevant pH range of 3–6 and compare them with the previously reported mechanisms of oxidation of SO2,aq with H2O2, methyl hydroperoxide and peroxyacetic acid. The organic products of the reaction are identified, and two pathways are proposed. For 1,2-ISOPOOH, a higher yield pathway via proposed radical intermediates yields methyl vinyl ketone (MVK) and formaldehyde, which can react to hydroxymethanesulfonate (HMS) when SO2,aq is present. A lower yield non-fragmentation oxygen addition pathway is proposed that results in the formation of isoprene-derived diols (ISOPOH). Based on global simulations, this mechanism is not a significant pathway for formation of MVK and formaldehyde relative to their gas-phase formation but, as previously reported, it can be regionally important for sulfate production. The study adds to previous work that highlights similarities and differences between gas-phase and cloud-droplet processing of reactive organic carbon.
- Article
(513 KB) - Full-text XML
-
Supplement
(1298 KB) - BibTeX
- EndNote





Isoprene (C5H8) is the main non-methane biogenic volatile organic compound emitted to the atmosphere with global emission estimates of ∼ 470 Tg C (Guenther et al., 2006, 2012; St. Clair et al., 2016). In the atmosphere, C5H8 primarily reacts with hydroxyl radicals (OH) forming peroxy radicals (RO2) after oxygen addition (Wennberg et al., 2018). Under pristine, HO2-dominated (low-NO) conditions, isoprene RO2 reacts with hydroperoxyl radicals (HO2) to form multifunctional organic hydroperoxides, isoprene hydroxyl hydroperoxides (ISOPOOH, C5H10O3), of which 1-hydroxyl-2-hydroperoxyl- and 4-hydroxyl-3-hydroperoxyl-ISOPOOH (1,2-ISOPOOH and 4,3-ISOPOOH, respectively) are the most abundant isomers (Rivera-Rios et al., 2014; Krechmer et al., 2015; St. Clair et al., 2016). ISOPOOH mixing ratios of up to 1 ppb and up to ∼ 2 ppb have been reported in the Amazon rainforest and the Blodgett Forest Research Station in California, respectively (Worton et al., 2013; Liu et al., 2016).
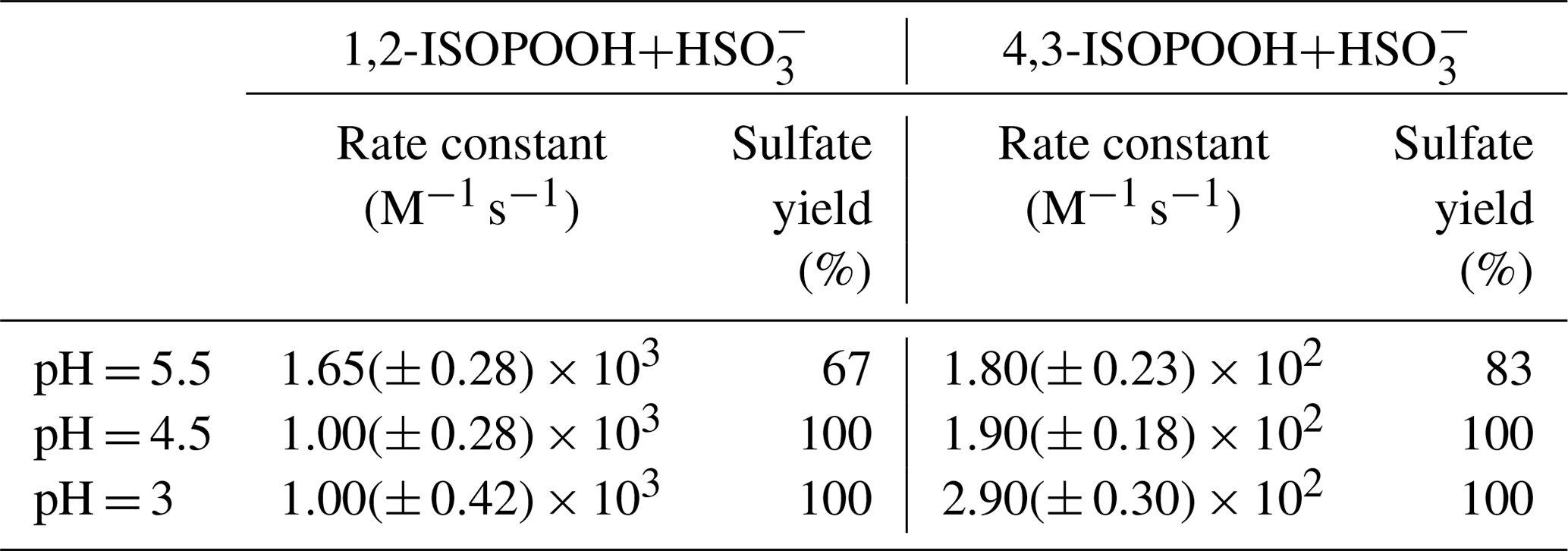
Table 1Sulfate production rate constants and yields in the aqueous phase due to oxidation of SO2,aq via 1,2-ISOPOOH and 4,3-ISOPOOH (Dovrou et al., 2019b).
In the gas phase, ISOPOOH is oxidized by OH to primarily produce isoprene epoxydiols (IEPOX), which contribute to secondary organic aerosol (SOA) mass (Paulot et al., 2009a; Surratt et al., 2010; Lin et al., 2012; Nguyen et al., 2014; McNeill, 2015; Zhang et al., 2018). St Clair et al. (2016) investigated the gas-phase oxidation mechanism of ISOPOOH and elucidated some non-IEPOX oxidation pathways, and Krechmer et al. (2015) observed the formation of non-IEPOX low volatile organic compounds that contribute to SOA. These pathways are summarized and compiled into a complete gas phase mechanism in the work of Wennberg et al. (2018).
Having Henry's law constants on the order of 105 M atm−1, ISOPOOH isomers can partition into cloud and fog water and participate in condensed-phase reactions (Rivera-Rios, 2018). Dovrou et al. (2019b) showed that ISOPOOH oxidizes sulfur dioxide (SO2,aq) in cloud and fog water, producing sulfate (). SO2,aq reacts with water to form sulfurous acid (SO2⋅H2O), which dissociates to bisulfite () when pH > 2. At higher pH (pH > 6), subsequently dissociates to form sulfite (). In cloud pH range of 3–6 the dominant form of SO2,aq is , and our study investigated pH values of 3, 4.5 and 5.5 (Fig. S1 in the Supplement). Aqueous hydrogen peroxide (H2O2) oxidizes SO2,aq to sulfate with a 100 % yield with a rate constant that increases with decreasing pH (Lind et al., 1987). Dovrou et al. (2019b) reported that at pH = 5.5 the rate constant for oxidation of SO2,aq by 1,2-ISOPOOH is equal, within uncertainty, to that of oxidation of SO2,aq by H2O2 at pH = 5.5, while the rate constant of the oxidation for SO2,aq by 4,3-ISOPOOH is an order of magnitude lower. The rate constants for the reactions of both ISOPOOH isomers with SO2,aq have a much smaller pH dependence than that of H2O2 + SO2,aq. Similar to H2O2, the sulfate yield at pH = 4.5 and 3 is 100 % for both ISOPOOH isomers. However, Dovrou et al. (2019b) showed that for pH = 5.5 the sulfate yield is only 67 % and 83 % of the reacted SO2,aq for 1,2-ISOPOOH and 4,3-ISOPOOH, respectively (Table 1). Due to its higher abundance, solubility and rate of reaction with SO2,aq, 1,2-ISOPOOH contributes much more to atmospheric sulfate production than the 4,3 isomer (Dovrou et al., 2019b).
The differences between both the sulfate yield and the pH-dependence of rate constants suggest that the chemical mechanisms of SO2,aq oxidation via H2O2 and ISOPOOH are different. Hoffmann and Edwards (1975) proposed a mechanism for the oxidation of by H2O2:
with Reaction (R2), rearrangement of peroxysulfite, being the rate-limiting step. To compare by H2O2 and understand the mechanism of SO2,aq oxidation with ISOPOOH, time-dependent proton nuclear resonance (1H-NMR) spectra of the reaction were obtained.
2.1 Chemical and sample preparation
Sodium metabisulfite was purchased from Sigma Aldrich (purity ≥ 99 %) and used as the source of bisulfite () in the solutions (Dovrou et al., 2019b). Formaldehyde solution (37 wt. % in H2O, 10 %–15 % methanol as stabilizer), methyl vinyl ketone (MVK, purity 99 %), formaldehyde-sodium bisulfite adduct (hydroxymethanesulfonate, HMS, purity 95 %), acetic acid (purity ≥ 99.7 %), dimethyl sulfoxide (DMSO, purity ≥ 99.7 %), 2-methyl-2-vinyloxirane (purity 95 %) and 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (purity 97 %) were purchased from Sigma Aldrich. The two main ISOPOOH isomers, 1,2-ISOPOOH and 4,3-ISOPOOH, were synthesized in the lab according to the procedures described by Rivera-Rios (2018). Hydrogen peroxide (H2O2) was purchased from Sigma Aldrich (30 wt. % in H2O), and filtered Milli-Q water and deuterium oxide (D2O) were used as solvents.
2-Methyl-3-butene-1,2-diol (1,2-ISOPOH) was synthesized, following the procedure in Zhang et al. (2012) and Bates et al. (2014), by hydrolysing 2-methyl-2-vinyloxirane. Approximately 5400 mg of 2-methyl-2-vinyloxirane was dissolved in 54 mL of 0.1 M HCl at 50 ∘C for 30 min. The procedure was modified by saturating the reaction mixture with sodium chloride (NaCl), instead of lyophilizing it. Subsequently, the diol was isolated via extraction with diethyl ether. The diethyl ether solution was then evaporated under reduced pressure to isolate the diol. The purity of the diol was determined to be 70 % using NMR analysis, and the identified impurities were diethyl ether, water, and the 1,2-ISOPOH dimer.
Samples for kinetics experiments were prepared with a ratio of and , as discussed in Dovrou et al. (2019b). The concentrations of the ion chromatography (IC) and NMR spectrometry experiments were 10 s of µM and a few mM, respectively, based on the sensitivity of the methods. For the NMR analysis of standards, 1 mM concentrations were used. After completion of reactions, 0.1–2 mM standards were added to identify proposed products by observing increases in the intensity of the corresponding peaks in the spectra.
Table 2Carbon budget of the 1,2-ISOPOOH+SO2,aq reaction and molar yields. Carbon concentrations are in mM. Hydrated formaldehyde could not be quantified as it is under the large D2O signal. At lower pH, acetic acid may be lost to the gas phase.

2.2 Ion chromatography and NMR analysis
A Dionex ICS-5000+ IC system and an Agilent I500B NMR were used to analyse the samples and quantify sulfate produced. The AG12A guard column and the AS12A analytical column (Dionex Ionpac) were selected in order to separate sulfur-containing species (Dovrou et al., 2019a). The mobile phase was 4.5 mM:1.4 mM sodium carbonate/sodium bicarbonate with a flow rate of 1.5 mL min−1 (Dovrou et al., 2019b). 1H-NMR was used to identify the organic reaction products. For the conducted experiments, 16 and 32 scan averages were chosen, with a relaxation delay of 45 s and at proton operating frequency of 400 MHz.
All experiments were repeated at least 4 times, each sample was prepared separately prior to analysis, and temperature and pH were monitored. DMSO, methanol and 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt ((CH3)3Si(CH2)3SO3Na) were used as internal standards for quantitative yields.
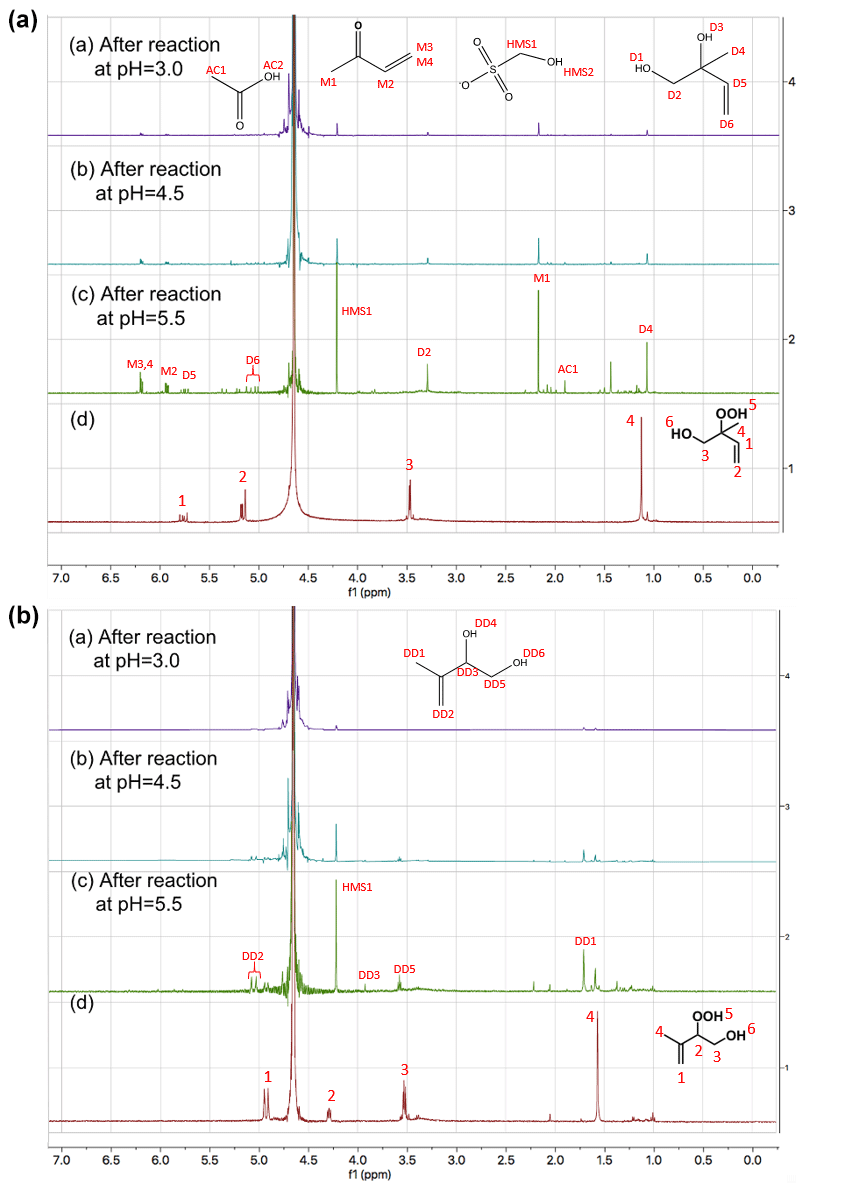
Figure 1Proton NMR spectra (1H-NMR, 400 MHz, D2O) of (A) 1,2-ISOPOOH (d) and the products of the reaction of SO2,aq + 1,2-ISOPOOH at (a) pH = 3, (b) pH = 4.5 and (c) pH = 5.5 and (B) 4,3-ISOPOOH (d) and the products of the reactions of SO2,aq + 4,3-ISOPOOH at (a) pH = 3, (b) pH = 4.5 and (c) pH = 5.5. The concentration of the ISOPOOH isomers was 1 mM, and the concentration of SO2,aq was 2 mM. The spectra are focused on the areas of change. Chemical structures of (a,d) 1,2-ISOPOOH and (b,d) 4,3-ISOPOOH are presented. The labels at each peak represent the hydrogens of the compounds and the spectra are shifted for D2O at 4.67 ppm.
3.1 Product identification
Figure 1 shows the NMR spectra of 1,2- and 4,3-ISOPOOH standards in D2O (broad peak at 4.7 ppm) together with the NMR spectra after reaction of ISOPOOH with a two-fold excess of SO2,aq at three different pH values. After completion of the SO2,aq + ISOPOOH reaction at pH = 5.5 the ISOPOOH precursors are not observable and a number of high-intensity 1H-NMR signals are observed. Signals with the same shifts are observed under more acidic conditions but with slightly lower intensity, while some of the smaller peaks from the pH = 5.5 spectra of both isomers are not observed in the pH = 4.5 and 3 spectra. This is consistent with the hypothesis that the reaction and product distribution have a pH dependence and suggests competition between different reaction paths. These different paths may also explain the pH dependence of the observed sulfate yields.
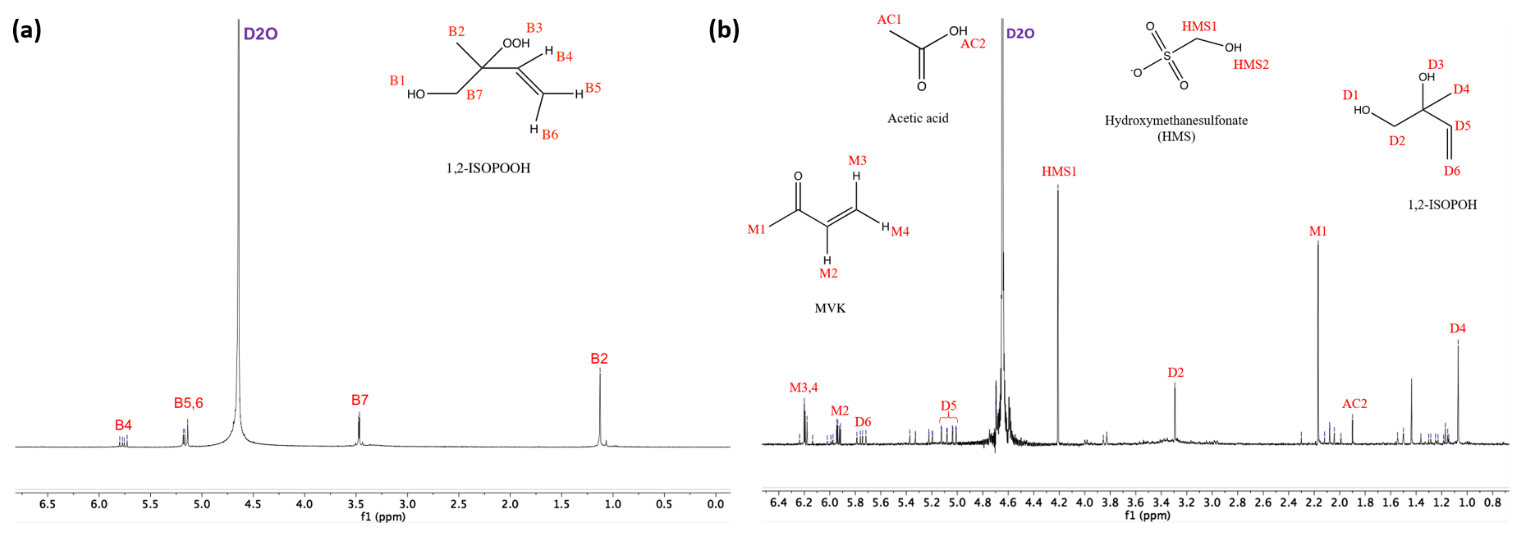
Figure 2Proton NMR spectra (1H-NMR, 400 MHz, D2O) for 1,2-ISOPOOH (a), and products of the reaction of SO2,aq + 1,2-ISOPOOH (b). The marked peaks represent the position of the hydrogens for each molecule. The pH of the samples was 5.5 and the concentrations used were [1,2-ISOPOOH] = 1 mM and [SO2,aq] = 2 mM. D2O shift at 4.67 ppm.
Products were identified via comparison of 1H-NMR spectra of standards as well as spiking SO2,aq + ISOPOOH mixtures after completion of reaction with standards of the proposed products. Methyl vinyl ketone (MVK), methacrolein (MACR) and (hydrated) formaldehyde (HCHO), all major products of gas-phase oxidation, were identified by their 1H-NMR chemical shifts (St. Clair et al., 2016). Due to its higher importance, the 1,2-ISOPOOH reaction and products were examined in more detail. For this reaction at pH = 5.5 MVK (30 % molar yield), hydrated formaldehyde (CH2(OH)2), HMS (30 % molar yield), 1,2-ISOPOH (22 % molar yield) and acetic acid (5 % molar yield) were the main products (Fig. 2a and Table 2). The products of the SO2,aq + 4,3-ISOPOOH reaction included CH2(OH)2, 4,3-ISOPOH and HMS (Fig. 2b). In both reactions, CH2(OH)2 is under the large D2O peak, δ ≈ 4.74 ppm, which prevents quantification (Sect. 3.2). 2-Methyl-2-vinyloxirane was also used as a standard, but it was not observed as a product (Fig. S2 in the Supplement). The standard spectrum showed that 2-methyl-2-vinyloxirane hydrolyzed to 1,2-ISOPOH under the experimental conditions, although not very rapidly. Thus, we cannot exclude that 2-methyl-2-vinyloxirane is formed and rapidly hydrolyzed to 1,2-ISOPOH, although the timescale of the experiments should have allowed its observation. Some minor product signals could not be identified; for example, a signal in the SO2,aq + 1,2-ISOPOOH pH = 5.5 spectrum at δ = 1.43 ppm (Fig. 1) is likely a methyl group not adjacent to a carbonyl group based on its chemical shift; however, we were not able to identify the corresponding compound.
3.2 Proposed reaction pathways
It is instructive to consider the previously studied oxidation of SO2,aq with H2O2 (Reactions R1–R2) and methyl hydroperoxide (CH3OOH; Reaction R3), and to lesser degree peroxyacetic acid (CH3C(O)OOH; Reaction R4) (Lind et al., 1987).
Reaction (3) has two pathways oxidizing S(IV) to S(VI): a higher yield pathway (Reaction R3a), forming sulfate and methanol and a lower yield pathway (Reaction R3b), forming methyl bisulfite.
Lind et al.'s (1987) results for CH3OOH and CH3(O)OOH are consistent with a fast initial equilibrium, like Reaction (R1), followed by rearrangement to the products. In addition, Lind et al. (1987) stated that at pH values above 4.8 the CH3(O)OOH reaction had another mechanism, but the authors did not elaborate, only stated that the mechanism was first-order in both reactants. Reactions (R3a) and (R4) are effectively oxygen atom transfer reactions, analogous to the net (R1) and (R2) Reactions of H2O2. Reaction (R3b) forms an organosulfate as the stable reaction product (Lind et al., 1987). In our discussion of ISOPOOH reaction pathways we focus on the atmospherically more important 1,2-ISOPOOH. Based on the product distribution we propose a non-fragmentation pathway for 1,2-ISOPOOH forming 1,2-ISOPOH (22 % molar yield at pH = 5.5) and two fragmentation pathways producing MVK (30 % molar yield at pH = 5.5) together with formaldehyde/HMS (> 30 % molar yield at pH = 5.5) and acetic acid (5 % molar yield at pH = 5.5) with other small species such as CO. These pathways, accounting for 57 % of 1,2-ISOPOOH reaction at pH = 5.5, will be discussed below.
3.2.1 Non-fragmentation pathway
Analogous to Reaction (R1), we also consider an initial fast equilibrium of ISOPOOH with SO2,aq. However, because of the lower symmetry than H2O2, there are two possibilities, depending on which peroxy O is involved in the nucleophilic attack on sulfur.
Peroxysulfite from Reaction (R5a) will rearrange to sulfate following Reaction (R2). The organo-peroxysulfite from Reaction (R5b) could potentially rearrange to form an organosulfate (Reaction R6a) which is known to rapidly hydrolyze, forming sulfate and ISOPOH (Darer et al., 2011; Hu et al., 2011). This rapid hydrolysis of tertiary sulfate likely also applies to the organo-peroxysulfite. The hydrolysis product (Reaction R6b) is again ISOPOH and peroxysulfite, which will rearrange to sulfate following Reaction (R2).
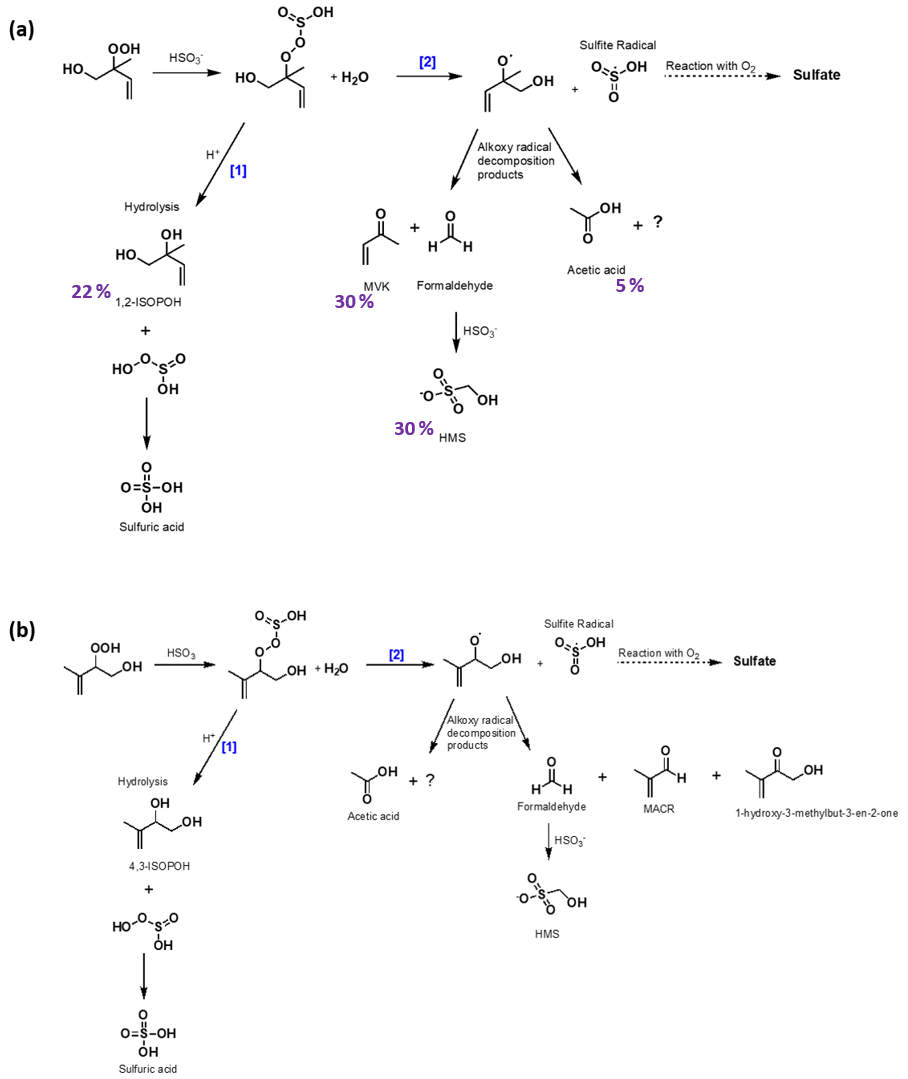
Figure 3Proposed chemical mechanisms of the oxidation of SO2,aq by (a) 1,2-ISOPOOH with carbon yields of identified products at pH = 5.5 (Table 2) presented in purple and (b) 4,3-ISOPOOH. There are two competing mechanisms: after ISOPOOH reacts with SO2,aq, displacing water, a hydrolysis reaction [1] or an O−O bond breakage [2] is taking place. In mechanism [1], the product hydrolysis results in the same intermediate that the reaction of SO2,aq with H2O2 is forming, and either a formation of a diol or an epoxide is being generated (Fig. S2). In mechanism [2], an alkoxy radical and sulfite radical are formed leading to the production of MVK, MACR, HCHO and other products. Considering the reaction of SO2,aq with 1,2-ISOPOOH (a), the sum of the products' carbon yields is 57 % and the remaining 43 % is attributed to hydrated formaldehyde, repartition of MVK to the gas phase, and possible formation of CO or other small molecules that have repartitioned to the gas phase, as discussed in Sect. 3.2.2.
The net outcome of all pathways is effectively an oxygen atom transfer from 1,2-ISOPOOH to sulfite to form sulfate and 1,2-ISOPOH. This net mechanism is consistent with the NMR spectra and IC analysis, which show clear evidence for ISOPOH and sulfate production. This effective reaction path is shown in the left-hand side of Fig. 3a[1]. This pathway accounts for 22 % of 1,2-ISOPOOH reaction at pH = 5.5.
3.2.2 Fragmentation pathways
1,2-ISOPOH yield is only about two-thirds of the yield of MVK at all pH values, which is strong evidence that an additional pathway, likely not present for CH3OOH, exists. MVK and formaldehyde, either as its hydrate or as the adduct with the excess SO2,aq, are in fact the highest yield products. These products have to involve breaking of the carbon backbone, which strongly suggests a radical mechanism. The products are identical to those resulting from oxidation of isoprene by OH under high-NO conditions, which involves an alkoxy radical (1,2-ISOPO, Fig. 3). In addition, the products are identical to those observed for gas-phase ISOPOOH reacting on metal surfaces (Rivera-Rios et al., 2014). For the latter process, Rivera-Rios et al. (2014) proposed cleavage of the 1,2-ISOPOOH peroxy bond producing 1,2-ISOPO, which rapidly forms MVK and formaldehyde. The high sulfate yield at all pH values implies that both the ISOPOH and the fragmentation channel have to produce sulfate. In combination with the previous results mentioned above, we propose that the second pathway involves cleavage of the peroxy bond of the organo-peroxysulfite resulting in 1,2-ISOPO, which rapidly forms MVK/formaldehyde, and a sulfite radical. The sulfite radical rapidly reacts with O2 producing a peroxysulfate radical, which can undergo further reaction, e.g., with SO2,aq or organic molecules to form sulfate and oxidized organics (Neta and Huie, 1985; Yang et al., 2015). The subsequent (secondary) reaction of HCHO with SO2,aq explains the formation of HMS, and thus the pathway forms MVK, HCHO and the sulfite radical as first-generation products with the sulfite radical likely forming sulfate subsequently (Fig. 3) (Dovrou et al., 2019a; Munger et al., 1984, 1986). This pathway accounts for 30 % of 1,2-ISOPOOH reaction at pH = 5.5.
The observation of acetic acid also requires a fragmentation of the backbone. This pathway likely involves formation of additional formaldehyde and CO, which are both not quantifiable in our experiments. For this reason, this is the most uncertain pathway, although it is clear that it accounts for 5 % of 1,2-ISOPOOH reaction at pH = 5.5, which makes the sum of the non-fragmentation and the two fragmentation pathways 57 % at pH = 5.5 (Rivera-Rios et al., 2014; Paulot et al., 2009b).
There likely are three contributors to the carbon yield of less than 100 %. Hydrated formaldehyde could not be quantified due to interference with D2O although this likely is already accounted for in the MVK and acetic acid pathways. However, MVK could also repartition to the gas phase, as discussed in Sect. 4; thus, its aqueous contribution might be underestimated. It is likely that formation of CO or other small molecules that potentially partition to the gas phase are not detected by NMR. Lastly, it is also possible that numerous small-yield products are formed that are below the NMR detection limit.
3.3 pH dependence
The sulfate yield increases while the observed carbon decreases with decreasing pH, and the ratio of ISOPOH to MVK is pH independent. Moreover, at lower pH, ISOPOH and MVK molar yields are reduced by no more than 10 %, and the acetic acid path is greatly reduced. We propose two explanations for the fact that the observed carbon balance decreases with decreasing pH. Kok et al. (1986) showed that the formation of HMS is a factor of 10 slower at pH = 4 than pH = 5, which could explain that at pH≤4.5 the sulfate yield observed in the oxidation of SO2,aq with ISOPOOH is 100 %, indicating that SO2,aq is oxidized faster to sulfate than when reacting with formaldehyde to form HMS, while at pH = 5.5 the sulfate yield is lower. Thus, the decreased HMS yield is attributed to slow HMS formation so that more formaldehyde stays in the hydrated form which could not be quantified. Second, at lower pH acetic acid may partition to the gas phase, and thus it cannot be observed. The results do not provide a clear explanation for the lower sulfate yield at pH = 5.5. The non-fragmentation (ISOPOH) pathway, which is more important at pH = 5.5, cannot explain this as it should have a 100 % sulfate yield. Thus, the cause has to result from one of the fragmentation pathways. It is possible that at higher pH the sulfite radical has a lower yield of forming sulfate; however, studying this goes beyond the scope of this work.
In addition, S(IV) speciation might affect the sulfate yield at high pH. While the dominant form of SO2,aq in our experiments is bisulfite, at the highest pH value of our experiments, pH = 5.5, sulfite is likely formed and could potentially react with ISOPOOH (Fig. S1). The ratio of bisulfite to sulfite at pH = 5.5 is approximately 60. However, if the rate constant of ISOPOOH with sulfite is orders of magnitude faster than the reaction of ISOPOOH with bisulfite, and the reaction mechanism is different for sulfite than bisulfite, this could contribute to changes in product distributions. Our experiments do not allow unambiguous and quantitative analysis of this aspect. Performing experiments at higher pH, exceeding the cloud-relevant pH, to shift the equilibrium to sulfite has the significant complication that formaldehyde will react rapidly with it to form HMS.
In summary, we observe clear evidence for a non-fragmentation pathway that likely is similar to the mechanism of H2O2 and CH3OOH. A difference to CH3OOH is that in contrast to methyl bisulfate the organo-sulfates from ISOPOOH hydrolyze easily, especially the tertiary one from 1,2-ISOPOOH. The non-fragmentation pathway effectively corresponds to an oxygen atom transfer, and in contrast to H2O2 and CH3OOH this is not the major pathway. We observe fragmentation pathways that we hypothesize involve cleavage of the peroxy bond based on previous work on ISOPOOH. Such a pathway was not observed for CH3OOH as the product of this channel would be neither methanol nor methyl bisulfate. There are at least two fragmentation pathways, with the major observed one producing MVK and HCHO, which subsequently forms HMS, and a minor one forming acetic acid and probably small molecules including CO. It is likely that additional smaller molecules that partition to the gas phase are formed as well as other molecules at low concentration that cannot be detected via NMR, which could include organo-sulfur compounds from the reaction of the sulfite radical with organics.
In the atmosphere, MVK and MACR will partition to the gas phase as they have high vapour pressures of 11 and 16 kPa and low Henry's law constants of 41 and 6.5 M atm−1, respectively (Iraci et al., 1999). The diffusion time of MVK in a cloud droplet is less than 25 s. The rate of the reaction of MVK with SO2,aq was estimated using NMR analysis, from which we estimate a lower limit for the rate constant of k ≥ 104 . However, considering the diffusion time, SO2,aq and expected MVK concentrations, the formation of the organosulfur compound by reaction of MVK with SO2,aq is unlikely in the atmosphere.
The annual production of the products formed via the reaction of ISOPOOH with SO2,aq in the atmosphere was calculated using the estimated ISOPOOH amount participating in this reaction from GEOS-Chem (Bey et al., 2001; Park et al., 2004; Alexander et al., 2012; Dovrou et al., 2019b). The results from the simulations performed in the work of Dovrou et al. (2019b) were re-analysed with respect to the amount of ISOPOOH reacting in each pathway. Dovrou et al. (2019b) did not consider the organic products of the ISOPOOH+SO2,aq reaction as they focused on an estimate of the globally and regionally produced sulfate (see Dovrou et al., 2019b, for all details on model configurations). This work focused on the organic reaction products, primarily to provide the first insight into this for multifunctional organic hydroperoxides, and to evaluate whether in addition to the clear regional importance for sulfate, the reactions are important for the organic carbon budget. To be specific, the amount of ISOPOOH reacting in each pathway was calculated from the simulations and was adjusted for each organic product using the product yields. The results were scaled by the relative molecular weights in order to calculate the total production of the compounds of interest.
Globally, it is estimated that, under current conditions, 4.2 Tg yr−1 of gas-phase 1,2-ISOPOOH reacts with SO2,aq, representing 3.2 % of the global loss of the ISOPOOH isomer and 1.7 % of the global production of sulfate (Dovrou et al., 2019b). The 1,2-ISOPOOH pathway contributes to the production of 0.40–0.53, 0.02–0.06, 0.28–0.36, 0.95–1.31 and 1.40–1.52 Tg yr−1 of MVK, acetic acid, HCHO, HMS and 1,2-ISOPOH, respectively (ranges are for the differences in yields across the experimental pH range). The production of MVK from these ISOPOOH+SO2,aq oxidation pathways is equivalent to 0.5 % of the total global production under present conditions. Under pre-industrial conditions, the total atmospheric burden of SO2 decreases; thus, the amount of ISOPOOH reacting with SO2,aq decreases somewhat: 2.4 Tg yr−1 of gas-phase 1,2-ISOPOOH reacts with SO2,aq, representing 1.6 % of the global respective losses of the isomer and 1.0 % of the global production of sulfate. The reaction of 1,2-ISOPOOH+SO2,aq results in 0.23–0.30, 0.01–0.04, 0.16–0.21, 0.55–0.76 and 0.80–0.88 Tg yr−1 of MVK, acetic acid, HCHO, HMS and 1,2-ISOPOH, respectively. The production of MVK from these ISOPOOH+ SO2,aq oxidation pathways is equivalent to 3.7 % of the total global production under pre-industrial conditions. These results show that the importance of these pathways lies in the production of sulfate on regional scales, especially for isoprene-dominated regions, rather than the formation of the organic reaction products. The conversion of bisulfite to sulfate via the 1,2-ISOPOOH pathway can contribute up to ∼ 50 % of total sulfate production in pristine regions such as the Amazon, and ∼ 3 % in more polluted regions such as the southeastern United States under current atmospheric conditions (Dovrou et al., 2019b). The southeastern United States is an area of interest as NOx levels are decreasing, creating an atmosphere where hydroperoxide formation can be favoured, which may increase their contribution to sulfate formation; in a simulation without anthropogenic emissions, the contribution of ISOPOOH to sulfate formation increases from ∼ 3 % to ∼ 20 % (Dovrou et al., 2019b).
This work is primarily useful as a foundation for the mechanistic understanding of the reactivity of multifunctional organic hydroperoxides with SO2,aq, for which there is no previous data in the literature. The results show that multifunctional organic hydroperoxides have a significantly more complex behaviour with channels analogous to H2O2 and CH3OOH but also fragmentation channels. Dovrou et al. (2019b) demonstrated the importance of these pathways in the production of sulfate under isoprene-dominated regions compared to the formation of the organic reaction products. Although the reactions do not play an important role for the organic carbon budget, our work advances understanding of the atmospheric chemistry of multifunctional organic hydroperoxides and adds new pathways to the isoprene oxidation mechanism. Furthermore, the results can help clarify the composition of cloud droplet residual aerosols.
The data used in this work are available upon request. Please email Eleni Dovrou (dovrouel@gmail.com).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-8999-2021-supplement.
ED conducted the ion chromatography experiments and ED and JCRR conducted the NMR experiments. ED, KHB and FNK analysed the data. JCRR synthesized the two ISOPOOH isomers. JLC and JDS synthesized the 1,2-ISOPOH to provide the 1H-NMR standard, and JLC provided the 2-methyl-2-vinyloxirane and acetic acid 1H-NMR standards. ED prepared the paper with contributions from KHB, JCRR, JLC, JDS and FNK.
The authors declare that they have no conflict of interest.
This work was supported by the Harvard Global Institute. Eleni Dovrou is grateful for the Onassis Foundation scholarship for Hellenes.
This research has been supported by the Harvard Global Institute.
The article processing charges for this open-access
publication were covered by the Max Planck Society.
This paper was edited by Ryan Sullivan and reviewed by Becky Alexander and one anonymous referee.
Alexander, B., Allman, D. J., Amos, H. M., Fairlie, T. D., Dachs, J., Hegg, D. A., and Sletten, R. S.: Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean, J. Geophys. Res., 117, 1–17, https://doi.org/10.1029/2011JD016773, 2012.
Bates, K. H., Crounse, J. D., St. Clair, J. M., Bennett, N. B., Nguyen, T. B., Seinfeld, J. H., Stoltz, B. M., and Wennberg, P. O.: Gas phase production and loss of isoprene epoxydiols, J. Phys. Chem. A, 118, 1237–1246, https://doi.org/10.1021/jp4107958, 2014.
Bey, I., Jacob, J., Yantosca, R. M., Logan, A., Field, B. D., Fiore, A. M., Li, Q., Liu, H. Y., Mickley, J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res.-Atmos., 106, 73–95, https://doi.org/10.1029/2001JD000807, 2001.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and stability of atmospherically relevant isoprene-derived organosulfates and organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
Dovrou, E., Lim, C. Y., Canagaratna, M. R., Kroll, J. H., Worsnop, D. R., and Keutsch, F. N.: Measurement techniques for identifying and quantifying hydroxymethanesulfonate (HMS) in an aqueous matrix and particulate matter using aerosol mass spectrometry and ion chromatography, Atmos. Meas. Tech., 12, 5303–5315, https://doi.org/10.5194/amt-12-5303-2019, 2019a.
Dovrou, E., Rivera-Rios, J. C., Bates, K. H., and Keutsch, F. N.: Sulfate Formation via Cloud Processing from Isoprene Hydroxyl Hydroperoxides (ISOPOOH), Environ. Sci. Technol., 53, 12476–12484, https://doi.org/10.1021/acs.est.9b04645, 2019b.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hoffmann, M. R. and Edwards, J. O.: Kinetics of the oxidation of sulfite by hydrogen peroxide in acidic solution, J. Phys. Chem., 79, 2096–2098, https://doi.org/10.1021/j100587a005, 1975.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Iraci, L. T., Baker, B. M., Tyndall, G. S., and Orlando, J. J.: Measurements of the Henry's law coefficients of 2-methyl-3-buten-2-ol, methacrolein, and methylvinyl ketone, J. Atmos. Chem., 33, 321–330, https://doi.org/10.1023/A:1006169029230, 1999.
Kok, G. L., Gitlin, S. N., and Lazrus, A. L.: Kinetics of the Formation and Decomposition of Hydroxymethanesulfonate, J. Geophys. Res., 91, 2801–2804, https://doi.org/10.1029/JD091iD02p02801, 1986.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., St. Clair, J. M., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015.
Lin, Y. H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene epoxydiols as precursors to secondary organic aerosol formation: Acid-catalyzed reactive uptake studies with authentic compounds, Environ. Sci. Technol., 46, 250–258, https://doi.org/10.1021/es202554c, 2012.
Lind, J. A., Lazrus, A. L., and Kok, G. L.: Aqueous Phase Oxidation of Sulfur(Iv) By Hydrogen-Peroxide, Methylhydroperoxide, and Peroxyacetic Acid, J. Geophys. Res., 92, 4171–4177, https://doi.org/10.1029/JD092iD04p04171, 1987.
Liu, Y., Brito, J., Dorris, M. R., Rivera-Rios, J. C., Seco, R., Bates, K. H., Artaxo, P., Duvoisin Jr., S., Keutsch, F. N., Kim, S., Goldstein, A. H., Guenther, A. B., Manzi, A. O., Souza, R. A. F., Springston, S. R., Watson, T. B., McKinney, K. A., and Martin, A. T.: Isoprene photochemistry over the Amazon rainforest, P. Natl. Acad. Sci. USA, 113, 6125-6130, https://doi.org/10.1073/pnas.1524136113, 2016.
McNeill, V. F.: Aqueous organic chemistry in the atmosphere: Sources and chemical processing of organic aerosols, Environ. Sci. Technol., 49, 1237–1244, https://doi.org/10.1021/es5043707, 2015.
Munger, J. W., Jacob, D. J., and Hoffmann, M. R.: The occurrence of bisulfite-aldehyde addition products in fog- and cloudwater, J. Atmos. Chem., 1, 335–350, https://doi.org/10.1007/BF00053799, 1984.
Munger, J. W., Tiller, C., and Hoffmann, M. R.: Identification of hydroxymethanesulfonate in fog water, Science, 231, 247–249, https://doi.org/10.1126/science.231.4735.247, 1986.
Neta, P. and Huie, R. E.: Free-radical chemistry of sulfite, Environ. Health Persp., 64, 209–217, https://doi.org/10.2307/3430011, 1985.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014.
Park, R. J., Jacob, D. J., Field, B. D., and Yantosca, R. M.: Natural and transboundary pollution influences on sulfate-nitrate- ammonium aerosols in the United States: Implications for policy, J. Geophys. Res., 109, 1–17, https://doi.org/10.1029/2003JD004473, 2004.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., Clair, J. M. S., Seinfeld, J. H., and Wennberg, P. O.: Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 325, 730–734, 2009a.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kroll, J. H., Seinfeld, J. H., and Wennberg, P. O.: Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 9, 1479–1501, https://doi.org/10.5194/acp-9-1479-2009, 2009b.
Rivera-Rios, J. C.: Atmospheric Chemistry of Isoprene Hydroxyhydroperoxides, PhD dissertation, Harvard University, Cambridge MA, May 2018.
Rivera-Rios, J. C., Nguyen, T. B., Crounse, J. D., Jud, W., St. Clair, J. M., Mikoviny, T., Gilman, J. B., Lerner, B. M., Kaiser, J. B., De Gouw, J., Wisthaler, A., Hansel, A., Wennberg, P. O., Seinfeld, J. H., and Keutsch, F. N.: Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 41, 8645–8651, https://doi.org/10.1002/2014GL061919, 2014.
St. Clair, J. M., Rivera-Rios, J. C., Crounse, J. D., Praske, E., Kim, M. J., Wolfe, G. M., Keutsch, F. N., Wennberg, P. O., and Hanisco, T. F.: Investigation of a potential HCHO measurement artifact from ISOPOOH, Atmos. Meas. Tech., 9, 4561–4568, https://doi.org/10.5194/amt-9-4561-2016, 2016.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M. N., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., Mcvay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., Clair, J. M. S., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Worton, D. R., Surratt, J. D., Lafranchi, B. W., Chan, A. W. H., Zhao, Y., Weber, R. J., Park, J. H., Gilman, J. B., De Gouw, J., Park, C., Schade, G., Beaver, M., Clair, J. M. S., Crounse, J., Wennberg, P., Wolfe, G. M., Harrold, S., Thornton, J. A., Farmer, D. K., Docherty, K. S., Cubison, M. J., Jimenez, J. L., Frossard, A. A., Russell, L. M., Kristensen, K., Glasius, M., Mao, J., Ren, X., Brune, W., Browne, E. C., Pusede, S. E., Cohen, R. C., Seinfeld, J. H., and Goldstein, A. H.: Observational insights into aerosol formation from isoprene, Environ. Sci. Technol., 47, 11403–11413, https://doi.org/10.1021/es4011064, 2013.
Yang, Y., Jiang, J., Lu, X., Ma, J., and Liu, Y.: Production of Sulfate Radical and Hydroxyl Radical by Reaction of Ozone with Peroxymonosulfate: A Novel Advanced Oxidation Process, Environ. Sci. Technol., 49, 73307339, https://doi.org/10.1021/es506362e, 2015.
Zhang, Y., Chen, Y., Lambe, A. T., Olson, N. E., Lei, Z., Craig, R. L., Zhang, Z., Gold, A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., Gaston, C. J., Thornton, J. A., Vizuete, W., Ault, A. P., and Surratt, J. D.: Effect of the Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX), Environ. Sci. Tech. Let., 5, 167–174, https://doi.org/10.1021/acs.estlett.8b00044, 2018.
Zhang, Z., Lin, Y. H., Zhang, H., Surratt, J. D., Ball, L. M., and Gold, A.: Technical Note: Synthesis of isoprene atmospheric oxidation products: Isomeric epoxydiols and the rearrangement products cis- and trans-3-methyl-3,4-dihydroxytetrahydrofuran, Atmos. Chem. Phys., 12, 8529–8535, https://doi.org/10.5194/acp-12-8529-2012, 2012.