the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Apr 2021
| 21 Apr 2021
Temperature and acidity dependence of secondary organic aerosol formation from α-pinene ozonolysis with a compact chamber system
Yange Deng
Sathiyamurthi Ramasamy
Yu Morino
Shinichi Enami
Hiroshi Tanimoto
Secondary organic aerosols (SOAs) affect human health and climate change prediction; however, the factors (e.g., temperature, acidity of pre-existing particles, and oxidants) influencing their formation are not sufficiently resolved. Using a compact chamber, the temperature and acidity dependence of SOA yields and chemical components in SOA from α-pinene ozonolysis were systematically investigated under 278, 288, and 298 K temperatures using neutral ((NH4)2SO4) and acidic (H2SO4+((NH4)2SO4)) seed aerosols. SOA components with less than 400 were analyzed using negative electrospray ionization liquid-chromatography time-of-flight mass spectrometry. Based on the slightly negative temperature dependence of the SOA yields, the enthalpies of vaporization under neutral and acidic seed conditions were estimated to be 25 and 44 kJ mol−1, respectively. In addition, SOA yields increased with an increase in the acidity of seed particles (solid/near-solid state) at low SOA mass loadings, when compared with the seed particle amounts. Acidity dependence analysis of the chemical formula, molecular mass, and O:C ratio of the detected compounds indicated the enhanced formation of multiple oligomers in the wide molecular mass range with a wide range of O:C ratios under acidic seed conditions. The peak abundances of some chemical compounds increased with an increase in the acidity of seed particles (e.g., 197, 311, 313, 339, 355, and 383), while decreases in the peak abundances of some chemical compounds were observed (e.g., 171, 185, 215, 343, and 357). The acidity dependence could be explained by acid-catalyzed heterogeneous reactions or acid-catalyzed decomposition of hydroperoxides. In addition, organosulfate (OS) formation was observed under acidic seed conditions. Six out of the 11 detected OSs were potentially formed via the aldehyde + HSO pathway.
- Article
(1362 KB) - Full-text XML
-
Supplement
(1808 KB) - BibTeX
- EndNote





Secondary organic aerosol (SOA) in the atmosphere is a complex set of organic compounds which are formed through oxidation of precursor volatile organic compounds (VOCs) of either biogenic (e.g., monoterpene and isoprene) or anthropogenic (e.g., alkanes and aromatics) origins, or both (Hallquist et al., 2009). SOA plays important roles in the aerosol effect on climate (Tilmes et al., 2019), air quality (Parrish et al., 2011), and human health (Shiraiwa et al., 2017). Nevertheless, it is noted in the IPCC Fifth Assessment Report (AR5) that the formation of SOA has not been included in the estimation of the radiative forcing from aerosols because the formation is influenced by a variety of factors not yet sufficiently quantified (Stocker et al., 2014). However, in association with the advance of research technologies, processes that influence the growth of SOA particles to sizes relevant for clouds and radiative forcing have been intensively investigated (Shrivastava et al., 2017).
The importance of the formation of low-volatility (and/or extremely low-volatility) organic compounds (LVOCs) with saturation concentrations less than 10−0.5 µg m−3 through heterogeneous/multiphase accretion processes has been highlighted in SOA formation mechanisms in recent studies (Ziemann and Atkinson, 2012; Shrivastava et al., 2017). Semi-volatile organic compounds (SVOCs) are generated from oxidation reactions of VOCs in the gas phase. Heterogenous/multiphase reactions of SVOCs on particles are thought to contribute to the formation of LVOCs in SOA. Earlier acid-catalyzed heterogeneous reaction studies (Jang et al., 2002; Hallquist et al., 2009; Ziemann and Atkinson, 2012) proposed the formation of hemiacetals, aldol products, and organosulfates, among others, in the presence of acidic seed particles, which prompted the notion that the acidity of pre-existing particles is one of the key factors that influence SOA formation.
The influence of the acidity of pre-existing particles on SOA formation has been investigated in both chamber experiments and field measurements. From chamber experiments, Eddingsaas et al. (2012) observed the clear uptake of several SVOCs (e.g., α-pinene oxide and α-pinene hydroxy hydroperoxides) after the injection of acidic particles into the α-pinene OH oxidation system under low-NOx conditions (photooxidation for 4 h, lights off, and contents in the dark for 2 h followed by the injection). However, no apparent uptake was observed after the injection of neutral particles. Shiraiwa et al. (2013a) observed the evident formation of peroxyhemiacetals after gaseous tridecanal was injected into the dodecane photooxidation system under dry, ammonium sulfate seed particles and low-NOx conditions (photooxidation for 4 h, lights off, and contents in the dark for 2 h followed by the injection), which was believed to have been catalyzed by the presence of acids generated in the low-NOx dodecane mechanism. However, the influence of the acidity of pre-existing particles on SOA yields from chamber experiments is poorly understood. Previous studies have reported complex results. For example, Eddingsaas et al. (2012) reported greater SOA yields under acidic than under neutral seed conditions from photooxidation of α-pinene under high-NOx conditions and no influence of seed particle acidity on SOA yields under low-NOx conditions. Field studies also reported inconsistent results on the influence of acidity on SOA formation. Some researchers reported that SOA formation was enhanced under more acidic conditions (Chu et al., 2004; Lewandowski et al., 2007; Zhang et al., 2007; Hinkley et al., 2008; Rengarajan et al., 2011; Zhou et al., 2012), whereas others reported little or no enhancement under acidic conditions (Takahama et al., 2006; Peltier et al., 2007; Tanner et al., 2009). Other factors, such as temperature, humidity, NOx concentration level, and oxidation agents, might have also affected the results of the aforementioned studies in addition to the acidity of pre-existing particles (Jang et al., 2008), but they are also not well understood. For example, the relative importance of the temperature dependencies of the volatilities of oxidation products and of the gas-phase and multiphase chemical reactions, which could result in different temperature dependence of SOA yields (Pathak et al., 2007b; von Hessberg et al., 2009), is still not well constrained. This motivated the current study to develop a new compact chamber system, in which SOA formation reactions under controlled temperature, humidity, oxidation agents, and seed particle acidity can be easily performed.
Monoterpenes are known to be a large source of SOA in the global atmosphere (Kelly et al., 2018). α-Pinene is the dominant monoterpene and the second most emitted VOC following isoprene (Guenther et al., 2012; Messina et al., 2016). It can react rapidly with atmospheric oxidants including O3, OH, and NO3 radicals, and ozonolysis is the major atmospheric oxidation pathway, which is estimated to account for 46 % of reacted α-pinene (Capouet et al., 2008). Previous studies regarding α-pinene ozonolysis indicate that both SOA yields and chemical compositions in SOA are influenced by the air temperature and aerosol acidity (Czoschke et al., 2003; Gao et al., 2004; Iinuma et al., 2004, 2005; Czoschke and Jang, 2006; Jang et al., 2006; Northcross and Jang, 2007; Surratt et al., 2007, 2008; Hallquist et al., 2009; Saathoff et al., 2009; Kristensen et al., 2014, 2017). Pathak et al. (2007b) found that the SOA yields show a weak temperature dependence in the range of 15 to 40 ∘C and a stronger temperature dependence between 0 and 15 ∘C. Saathoff et al. (2009) parameterized the temperature dependence of the two-product SOA yield parameters using a dataset including several previous studies as well as their own comprehensive measurements from 243 to 313 K. In general, a negative temperature dependence of the α-pinene ozonolysis SOA yields could be confirmed. While enhancements of SOA yields from α-pinene ozonolysis reactions under acidic seed aerosol conditions compared with neutral seed conditions have been generally observed in previous studies (Czoschke et al., 2003; Gao et al., 2004; Iinuma et al., 2004, 2005; Czoschke and Jang, 2006; Jang et al., 2006; Northcross and Jang, 2007), the degree of enhancement varied probably because of the different experimental settings among the studies. For example, the study of Czoschke et al. (2003), Iinuma et al. (2004), Czoschke and Jang (2006), Jang et al. (2006), and Northcross and Jang (2007) reported enhancement ranges of 21 %–87 % from weak acid seed conditions to high acid seed conditions. Interestingly, studies of Gao et al. (2004) reported smaller enhancement (8 %–15 %) of SOA yields when the initial α-pinene concentrations were high, and the study of Iinuma et al. (2005) reported both increases and decreases in SOA yields under acidic seed conditions. It turned out that OH radical scavengers, which are known to play an important role in influencing SOA yields (Iinuma et al., 2005; Na et al., 2007), were applied in the studies of Gao et al. (2004) and Iinuma et al. (2005), but not applied in the studies of others.
With respect to chemical compositions, Kristensen et al. (2017) compared the chemical compositions of α-pinene ozonolysis SOA formed at temperatures of 293 and 258 K and found that the mass fraction of carboxylic acids increased at 258 K compared to 293 K, while the formation of dimer esters was suppressed at the sub-zero reaction temperature. Compared with neutral seed conditions, enhanced formation of large molecules under acidic seed conditions has been reported by Gao et al. (2004) and Iinuma et al. (2004). Gao et al. (2004) additionally reported less abundant small oligomers (e.g., a compound with molecular mass of 358) under acidic seed conditions. This research indicates that systematic studies of α-pinene ozonolysis SOA formation under specific experimental settings are warranted to clarify temperature and acidity dependence.
Furthermore, enhanced formation of organosulfates (OSs) from α-pinene oxidation under acidic seed conditions has been suggested by previous studies (Surratt et al., 2007, 2008; Iinuma et al., 2009; Duporte et al., 2020). OS has been regarded as an important aerosol component, accounting for up to 30 % of organic mass in PM10 and also as an anthropogenic pollution marker in the past 2 decades (Iinuma et al., 2007, 2009; Surratt et al., 2007, 2008; Riva et al., 2015, 2016; Duporte et al., 2016, 2020; Brüggemann et al., 2020). The formation mechanisms of OS from α-pinene oxidations have been studied under different experimental settings. For example, Surratt et al. (2007) proposed OS formation through esterification of hydroxyl or carbonyl groups in photooxidation experiments. Surratt et al. (2008) proposed nitrooxy organosulfate formation through esterification of hydroxyl groups in nighttime oxidation (i.e., NO3-initiated oxidation under dark conditions). Iinuma et al. (2009) proposed the acid-catalyzed ring opening of an epoxide mechanism through the α-pinene oxide–acidic sulfate particle experiment. Nozière et al. (2010) proposed a sulfate-radical-initiated OS formation process of α-pinene in irradiated sulfate solutions. For α-pinene ozonolysis experiments, the formation of OS through reactions between SO2 and stabilized Criegee intermediates under dry conditions or organic peroxides in the aqueous phase has been recently suggested (Ye et al., 2018; Stangl et al., 2019; Wang et al., 2019). However, based on our knowledge, no study concerning OS formation from α-pinene ozonolysis in the presence of sulfate particles exists.
In this study, α-pinene ozonolysis experiments have been conducted under dark conditions in the presence of seed particles and an OH scavenger utilizing a self-made compact-type chamber system. The study aimed to characterize the newly developed chamber as well as to study the temperature and acidity dependence of the yield and chemical composition of α-pinene ozonolysis SOA. Moreover, OS compounds and their possible formation mechanisms have been targeted during the analysis.
2.1 Chamber description and operation
A temperature-controllable chamber system has been developed for the simulation of SOA formation (Fig. S1 in the Supplement). The chamber is a cuboid shape Teflon bag (FEP; 0.7 m3 volume, 900 mm × 600 mm × 1300 mm; 50 µm thickness; Takesue, Japan) contained in a constant temperature cabinet (HCLP-1240; W1200 mm × D703 mm × H1466 mm; NK System, Japan). The temperature inside the cabinet was measured using a thermocouple attached to the inside of the cabinet (T3 in Fig. S1). The achievable operating temperature range of the chamber was 5–40∘. A detailed evaluation of the thermostat capacity of the chamber under dark conditions is presented in Text S1 in the Supplement, which indicates that the temperature inside the chamber was well controlled (varied within ±1 ∘C). The chamber is collapsible and is operated at atmosphere pressure.
For a typical experimental run, a total volume of 0.6 standard cubic meters (sm3) of G3 pure air (CO2 < 1 ppmv, CO < 1 ppmv, THC < 1 ppmv, and dew temperature < −70 ∘C) was introduced into the Teflon chamber at a flow rate of 20 slpm for 30 min. The relative humidity (RH) of the chamber air was adjusted by passing the G3 pure air through Milli-Q water (resistivity of 18.2 MΩ cm, total organic carbon content ≤ 5 ppb) before it entered the Teflon bag. The RH of the chamber air was measured after the experimental run by pumping the remaining chamber air into a separate small Teflon bag, to which a Vaisala RH&T probe (model, HMP76) equipped with a measurement indicator (model, M170) was attached. Particle number concentration and VOC mixing ratio measurements indicate that the chamber background concentrations of particles and VOCs were negligible, and no extra contamination was observed by the humidification process of the G3 air (Text S2). α-Pinene liquid (Wako Chemicals, Japan) was injected into the G3 pure air line through a septum equipped in a Swagelok PFA Tee connector using a micro-syringe (ITO Corporation, Japan) in the middle of the injection of dry pure G3 air. Diethyl ether in nitrogen gas (mass fraction of 0.4 %; Takachiho, Japan) used as an OH radical scavenger was introduced to the chamber in excess amounts (approximately 53 ppmv; 164–1963 times the initial concentration of α-pinene) after the introduction of pure air. Seed aerosol particles generated by a commercial atomizer (ATM220S, Topas GmbH, Germany) were subsequently introduced into the Teflon chamber after being dried by a diffusion dryer containing silica gel. Neutral seed aerosols were generated from a mol L−1 (NH4)2SO4 solution, and acidic seed aerosols were generated using a solution mixture of 0.25 mol L−1 (NH4)2SO4 and 0.25 mol L−1 H2SO4.
After 30 min of stabilization, the initial concentrations of α-pinene and seed aerosol particles were measured with a quadrupole-type proton transfer reaction mass spectrometry instrument (PTR-QMS500, Ionicon Analytik GmbH, Innsbruck, Austria) and a scanning mobility particle sizer (SMPS, TSI classifier model 3082; differential mobility analyzer model 3081; condensation particle counter model 3772; USA), respectively. The PTR-MS was operated at a flow rate of approximately 250 cm3 s−1 under a field strength (, where E is the electric field strength (V cm−1) and N is the buffer gas number density (molecule cm−3) of the drift tube) of 106 Td. The length of the drift tube was 9.2 cm. The drift voltage was set to 400 V. The temperatures of the inlet and drift tubes were set to 105 ∘C, and the pressure at the drift tube was set to 2.1 mbar. The signal intensities of ions with values of 21, 30, 32, 37, 45, 46, 75, 81, and 137 were recorded approximately every 4.5 s. The detection sensitivity of α-pinene was 3.3 ± 0.6 ncps ppbv−1 (ncps means normalized counts per second to 106 cps of H3O+). The SMPS was contained in a smaller constant temperature cabinet (LP-280-E, NK System, Japan), whose temperature was adjusted to be the same as the cabinet containing the Teflon chamber. The sheath and sample flow rates of the SMPS were 3.0 and 0.3 L min−1, respectively. The measured diameter range of the SMPS was 13.8–697.8 nm, and the data were collected every 5 min.
After obtaining the initial concentration of α-pinene and seed particles, excess ozone produced by irradiation of pure O2 with vacuum ultraviolet light from a low-pressure mercury lamp ozone generator (model 600, Jelight Compony Inc., USA) was introduced into the chamber at a flow rate of 200 slpm for 1.5 min, to initiate the ozonolysis reactions. After the ozone generator was turned off, the introduction of pure O2 continued for another minute to purge all generated ozone into the Teflon bag. Subsequently, the G3 pure air was introduced for 1 min to facilitate the mixing of the chamber air. The ozone concentration in the chamber was measured with an ozone monitor (model 1200, Dylec, Japan) immediately after its introduction. For some experiments, the order of the introduction of α-pinene and O3 was inverted (see Tables S1 and S2 in the Supplement). In the experiments in which O3 was first introduced, the introduction of G3 pure air was sustained for one more minute after the injection of α-pinene to purge all α-pinene into the Teflon bag and to facilitate the mixing of the chamber air. In the latter case, the maximum α-pinene concentrations appeared within 55 s of its introduction, which indicated that the mixing by introducing air with a flow rate of 20 slpm was probably completed within 55 s. The concentrations of both the α-pinene and aerosol particles were continually measured until the end of the experiment, which is defined in this study as 90 min after the start of the α-pinene ozonolysis reaction. The concentration of ozone after 90 min of ozonolysis reactions was also measured.
In total, 40 experimental runs were executed under neutral or acidic seed aerosol conditions at temperatures of 278, 288, or 298 K (Tables S1 and S2). Notably, when the chamber temperature was set to 278 K, the temperature in the small cabinet was set to 280 K, which is the lowest work temperature of the SMPS. In the present study, we did not investigate the influence of humidity on SOA yield. Since very dry conditions are not realistic in ambient air, we carried out the experiments at medium humidity (26 %–55 % RH, Tables S1 and S2). The differences in RH among experiments in the present study would not influence SOA formation significantly, as explained in the following section. First, nucleation would be negligible in all experiments because of the high concentrations of seed particles applied. Consequently, the influence of RH on SOA formation would be reflected in the particle phase (Kristensen et al., 2014). In addition, because the seed particles were dried into effloresced states before being introduced into the chamber, all particles would be in solid (neutral seed conditions) or near-solid states (acidic seed conditions) (Tang and Munkelwitz, 1977). Therefore, the influence of water on the particle phase through physical partitioning or chemical reactions would be minor (Faust et al., 2017). Before each experimental run, the Teflon bag was cleaned by filling it with pure G3 air and then evacuating all the air from the bag at least three times, which took approximately 40 min. The very low chamber background particle concentrations indicate that the bag was sufficiently cleaned (Text S2).
One Teflon filter (PF020, 47 mm diameter, Advantec MFS) aerosol sample was collected for each different combination of seed and temperature conditions. The sample volume was 0.5 m3 for each sample. In total, six aerosol samples were collected, and they were subjected to negative electrospray ionization liquid-chromatography time-of-flight mass spectrometry analyses (Sect. 2.2). A blank filter was also analyzed using a procedure similar to that of the sample filters. The results confirmed no substantial contamination in the filter and the filter analysis procedure (Text S2). The acidity of the seed particles was measured in a separate experiment where the seed particles were sampled on a Teflon filter which was then extracted into 10 mL of Milli-Q water. The pH of the water solution was measured with a pH meter (FPH70, AS ONE, Japan). The H+ concentration was ∼ 220 nmol m−3 under the acidic seed conditions.
2.2 (−) ESI LC-TOF-MS analysis
Chemical composition analysis of the Teflon filter samples was conducted using electrospray ionization liquid-chromatography time-of-flight mass spectrometry (ESI LC-TOF-MS) (Agilent Technologies, UK) similarly to in previous studies (Sato et al., 2018, 2019) except that negative mode was used in this study whereas positive mode was used in those previous studies. The key configuration parameter settings were as follows: nebulizer pressure was 0.21 MPa, the voltage in the spray chamber was −3500 V, the drying nitrogen gas temperature was 325 ∘C and flow rate was 5 L min−1, and the fragmentor voltage was 175 V. The mass calibration and lock-mass correction were conducted using G1969-85000 and G1969-85001 tuning mixtures (Agilent Technologies, UK), respectively. The mass resolution of the mass spectrometer (full width at half maximum) was > 20 000. For the analysis, the Teflon filter sample was sonicated in 5 mL methanol for 30 min after the addition of internal standard (i.e., sodium ethyl-d5 sulfate methanol solution, Sect. 3.2). The filter extract was concentrated to near dryness under a stream of nitrogen (∼ 1 L min−1). A 1 mL formic-acid–methanol–water solution () was added to the concentrated extract to obtain the analytical sample. A 10 µL aliquot of the analytical sample was injected into the LC-TOF-MS instrument and separated with an octadecyl silica gel column (Inertsil ODS-3; GL Science, Japan; 0.5 µm × 3.0 mm × 150 mm). A formic-acid–water solution (0.05 % and methanol were used as mobile phases. The total flow of the mobile phases was 0.4 mL min−1. The methanol fraction during each analysis was set at 10 % (0 min), 90 % (30 min), 90 % (40 min), 10 % (45 min), and 10 % (60 min). As reported previously (Sato et al., 2007), the recovery of malic acid, whose saturation concentration was estimated to be 157 µg m−3, was determined to be > 90 %, suggesting that evaporation loss during pre-treatment is negligible for molecules with saturation concentrations of ∼ 102 µg m−3 or less. We tentatively determined the molecular formulae and signal intensities of 362 products (including 11 organosulfates) with different (Table S3) based on retention times and interpretation of mass spectra. In addition, the tentatively determined molecular structures and compound names of some major products based on literature data and the results of this study (Sect. 4.2) are presented in Table S4.
2.3 Evaluation of wall loss
The wall-loss rate of particles in the chamber was evaluated by measuring the time evolution of the volume–size distributions of seed-only particles using the SMPS. The measurements were carried out whenever a new Teflon bag was used or the experimental conditions (i.e., temperature or seed particle acidity) were changed under humid air conditions. The latest measured bulk wall-loss rate (Sect. 3.1) was applied for each SOA formation experiment.
In Fig. S4, size-resolved particle wall-loss rates, which were determined assuming first-order wall-loss constants (Wang et al., 2018a), were shown for seed particles of different size distributions. Large wall loss was observed for particles with mobility diameters less than 100 nm and larger than 200 nm. The size distributions of the measured particle wall-loss rates presented shapes similar to that of a 0.83 m3 Teflon chamber (Hu et al., 2014), whereas in the latter, the lowest wall-loss rates appeared in the smaller size end (∼ 70–110 nm) and were greater (∼ 0.2 h−1) than those in the present study. The large apparent wall-loss rates of sub-100 nm particles were also similar to those of a 1.5 m3 Teflon reactor (Wang et al., 2018a). Model simulation (Text S3) and literature survey results revealed that the high wall-loss rates of sub-100 nm particles were mainly caused by particle coagulation (Nah et al., 2017; Wang et al., 2018a), and those of super-200 nm particles were likely the result of turbulent deposition (Lai and Nazaroff, 2000). Figure S4 also indicates that the wall-loss rates of super-200 nm particles were relatively high when the mean diameter of the seed particles was relatively small. Therefore, we waited for 30 min after the introduction of seed particles to start the ozonolysis reaction so that the size distribution of the seed particles could shift to the larger size end due to coagulation and loss of small particles. In addition, we used high-concentration solutions for the generation of seed particles to produce larger particles in this study (Sect. 2.1).
Wall loss of gas-phase organic compounds in the Teflon chamber could also cause the underestimation of SOA yields (Zhang et al., 2014; Krechmer et al., 2016). Although not experimentally determined in the present study, the influence of gas-phase wall loss on SOA yields will be discussed based on the studies of Zhang et al. (2014) and Krechmer et al. (2016) in Sect. 4.1.
3.1 Derivation of SOA yield
SOA yield (Y) is defined as the ratio of the mass concentration of SOA (mSOA, µg cm−3) to that of the reacted α-pinene (ΔVOC, µg cm−3) in each experimental run.
where mSOA was calculated as the product of the increased volume of aerosol particles from the volume of seed particles and a SOA density of 1.34 g cm−3 (Sato et al., 2018), and the arithmetic mean of the last three data of each experimental run was applied here. It was corrected for particle-phase wall loss using the bulk-volume wall-loss rate, assuming a first-order wall-loss constant which is independent of particle size and reaction time (Pathak et al., 2007b). The size-resolved wall-loss rates were not applied because the bulk wall-loss rates were very close to the size-resolved rates at approximately 300 nm (Fig. S4). The mode diameters of the volume–size distributions of the seed particles (Fig. S5) and of aerosol particles at the end of the ozonolysis reactions were also approximately 300 nm (Fig. S5). A detailed explanation of the derivation of mSOA is presented in Text S4. Note that when the mass loadings of SOA are low, the obtained mSOA and related yields retain greater uncertainties because the subtracted volume concentrations of seed particles from the measured volume concentrations are large (Mei et al., 2013). The influence of gas-phase wall loss on SOA yield is discussed qualitatively in Sect. 4.1.
In addition, a four-product volatility basis-set (VBS) gas–particle partitioning absorption model (Eq. 2, Donahue et al., 2006; Lane et al., 2008) was applied to assist the interpretation of the observed responses of Y to the chamber temperature and the acidity of the seed aerosol.
where αi is the mass-based stoichiometric yield for product i, and is the effective saturation concentration of i in µg m−3. In this study, αi is assumed to be temperature-independent (Pathak et al., 2007a) whereas the temperature dependence of is accounted for using the Clausius–Clapeyron equation as follows:
where T0 is the reference temperature, which is 298 K in this study; is the effective saturation concentration of i at T0; R is the ideal gas constant; and ΔHvap,i is the effective enthalpy of vaporization. The temperature dependence of Y is then represented by the substitution of Eq. (3) for in Eq. (2). We further assume a constant effective ΔHvap for all condensable organic compounds. Thus, the four-product basis set has five free parameters: α1, α2, α3, α4, and ΔHvap. Here, µg m−3, which was set based on the measured range of mSOA (Sect. 4.1) in this study. Microsoft Excel Solver GRG Nonlinear engine was used for the derivation of the five parameters under neutral or acidic seed particle conditions.
3.2 Derivation of the ethyl-d5-sulfate equivalent (EDSeq.) yield of OS
Before the extraction of filter samples, 20 µL of sodium ethyl-d5 sulfate (EDS) methanol solution (50 µg mL−1) was added to each sample filter as an internal standard for the quantification of OS. The ethyl-d5-sulfate equivalent (EDSeq.) masses of OSs were determined by comparing the total chromatographic peak areas of OSs to that of the EDS standard with known mass. The EDSeq. masses of OSs were divided by the corresponding air volumes collected to obtain the EDSeq. concentrations of OSs. The EDSeq. molecular yield of OS is defined as the ratio between the estimated EDSeq. concentration of OS (mOS, µg cm−3) and the reacted mass concentration of α-pinene (ΔVOC, µg cm−3). Note that the sensitivity of ESI mass spectrometry is compound-specific; therefore, the calculated EDSeq. yield includes the uncertainties that result from compound-specific sensitivities.
3.3 Volatility distribution analysis
SOA compounds identified from the six filter samples through LC-TOF-MS analysis were subjected to volatility distribution analysis. The saturation concentration (C∗) of each chemical compound was calculated and then ascribed to the volatility basis set (Donahue et al., 2006). Again, 298 K was used as the reference temperature (T0). For compounds whose chemical structures have been suggested by previous researchers, the SPARC online calculator (Hilal et al., 2003; Sato et al., 2018) was used for the derivation of their . For other compounds, including organosulfates, the following equation from Li et al. (2016) was applied:
where is the reference carbon number; nC, nO, and nS are the numbers of carbon, oxygen, and sulfur atoms in the molecule, respectively; bC, bO, and bS are the respective contribution of each atom to ; and bCO is the carbon–oxygen nonideality. For compounds containing only C, H, and O atoms, the values for , bC, bO, and bCO are 22.66, 0.4481, 1.656, and −0.7790, respectively. For OS compounds that contain C, H, O, and S atoms, the values for , bC, bO, bCO, and bS are 24.06, 0.3637, 1.327, −0.3988, and 0.7579, respectively. The of the 362 compounds determined by LC-TOF-MS analysis are presented in Table S3. As has been noted previously, the sensitivity of ESI mass spectrometry is compound-specific; thus the calculated distribution includes the uncertainties that result from compound-specific sensitivities. The estimated volatility distributions were further used to estimate the influence of gas-phase wall loss on SOA yields following the method suggested by Krechmer et al. (2016) (Text S5).
4.1 Performance of the Teflon chamber
An example experimental run of SOA formation from α-pinene ozonolysis is presented in Fig. 1 (exp. no. 27). The initial concentrations of α-pinene and ozone were 145 and > 824 ppbv, respectively. The number concentration of seed aerosol particles was 9.6 × 103 cm−3, and they were concentrated in the diameter range of 80–200 nm. SOA was formed while α-pinene was consumed immediately at the introduction of excess O3. The mass concentration of SOA reached its maximum while α-pinene was almost totally consumed approximately 50 min after the introduction of ozone. The time variation in α-pinene conforms to an α-pinene-limited first-order chemical reaction (5τ=47 min). With the particle-phase wall-loss correction, the SOA loading at the end of this experiment was calculated to be 200 ± 34 µg m−3, which resulted in a final SOA yield of 26 ± 7 % (Table S2). As the corrected SOA particle concentration was constant after 50 min, we consider the wall-loss correction applied here to be reasonable (Ng et al., 2006). Without particle-phase wall-loss correction, the concentration of SOA particles at 90 min would be underestimated by approximately 40 %. The observed particle number–size distribution shifted to a much greater but narrower size range of 200–300 nm at the end of the experiment. Evolution of the particle number–size distribution is presented in Fig. S6.
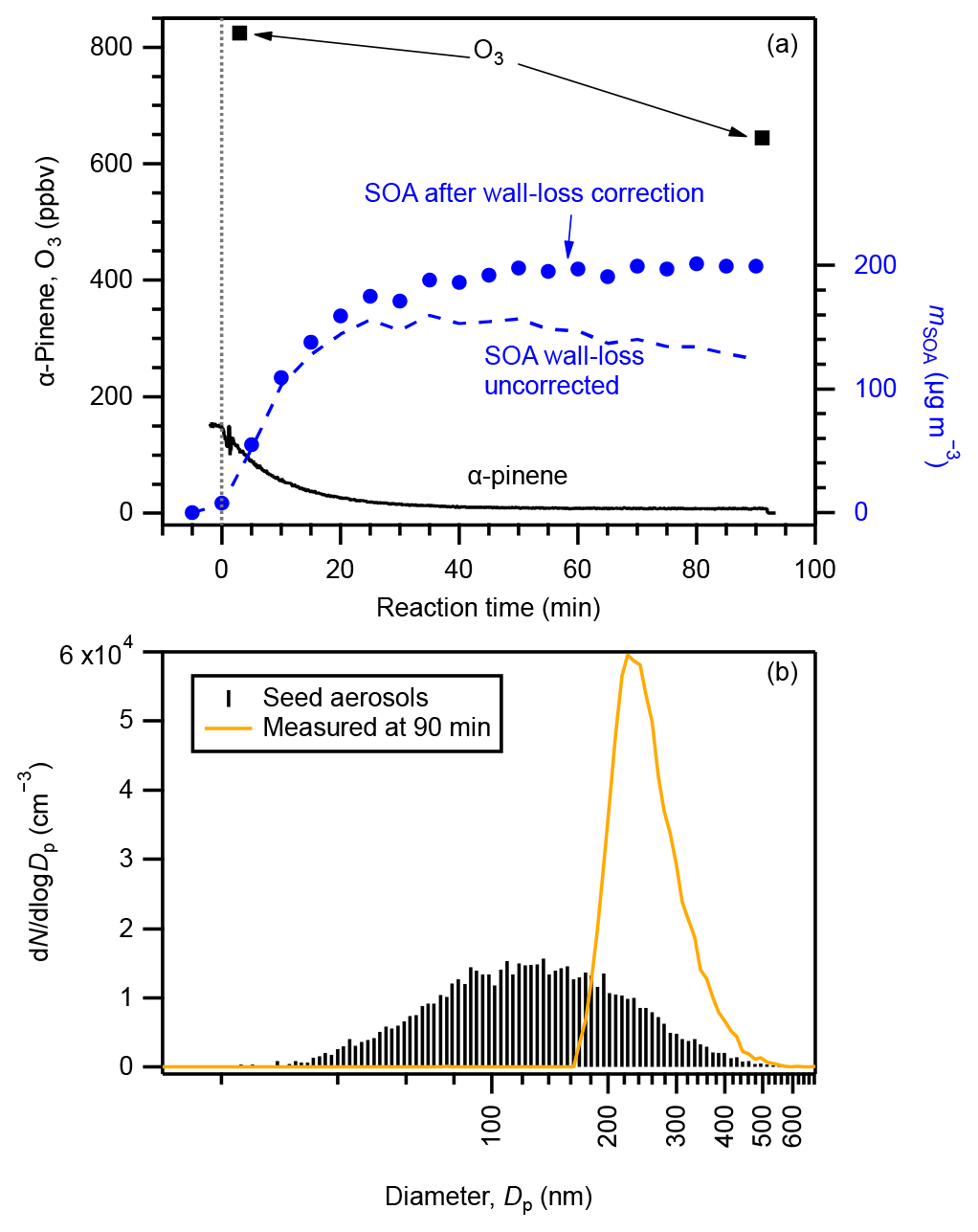
Figure 1Example experimental run: (a) concentrations of α-pinene (black solid curve), O3 (square markers), and SOA before (blue dashed line) and after (circle markers) particle-phase wall-loss correction compared with reaction time and (b) number–size distributions of seed aerosol particles (black sticks) and aerosol particles at the end of the experimental run (brown curve). The vertical dotted line in panel (a) indicates the starting time that O3 was injected. Data shown in this figure are from exp. no. 27 (Table S2).
The measured SOA mass yields at 298 K under neutral seed conditions were compared with those of Pathak et al. (2007a) and other experimental studies (Fig. 2). In this study, seven experiments with different initial α-pinene concentrations (54–323 ppbv) at 298 K and neutral seeds were conducted under a RH condition of approximately 26 %–27 % (Table S1). In Pathak et al. (2007a), the SOA mass yields from pre-existing studies under low-NOx dark ozonolysis conditions were summarized and categorized into two groups according to experimental RH conditions: RH < 10 % and RH = 50 %–73 %. They further fitted the data in each group using the multiple product basis-set approach (Pathak et al., 2007a). The four-product basis-set fitting was adopted to compare with the experiment results of this study (Fig. 2). The SOA yields in this study were 25 %–60 % lower than that of Pathak et al. (2007a). Possible reasons may include lack of consideration of the wall loss of oxidized organic vapors because the surface-to-volume ratio of the chamber used in this study (7.1 m−1) was much larger than those of previous studies (< 3 m−1; Pathak et al., 2007a, and references therein). According to Zhang et al. (2014), the vapor wall-loss bias factor, Rwall (defined as the ratio of the SOA mass when the vapor wall loss was assumed to be zero to the SOA mass when the optimal vapor wall loss rate was applied), was reported to be ∼ 4 at the initial seed surface area of ∼ 2 × 103 µm2 cm−3 (seed-to-chamber surface area ratio ∼ 1 × 10−3) and ∼ 2 at the initial seed surface area of > 6 × 103 µm2 cm−3 (seed-to-chamber surface area ratio > 3 × 10−3) during the photooxidation of toluene. As the initial seed surface area in the present study was in the range of (1–3) × 103 µm2 cm−3 (seed-to-chamber surface area ratio (1–4) × 10−4), Rwall in the Teflon bag might be at least twice that of the large chambers. This leads to the underestimation of the SOA yield of 50 % compared with the values obtained from the large chambers. A low limit correction of the gas-phase wall-loss influence for exp. no. 2 and other experimental runs in which chemical composition analysis had been conducted based on the method of Krechmer et al. (2016) (Text S5, Fig. 2), which confirmed that gas-phase wall loss is one reason for the lower SOA yields in the present study compared with Pathak et al. (2007a) and other previous studies presented in Fig. 2. We note that this is a shortcoming of the compact chamber with a 0.7 m3 volume used in this study. We also note that the chamber is aimed at exploratory research, where multiple experiments under different temperature, seed particle, relative humidity, oxidant, and radiation conditions can be executed within relatively short periods. Furthermore, the vapor wall-loss correction factors of the SOA mass presented no obvious temperature dependence (Text S5). Therefore, the temperature dependence of SOA yields will be discussed, assuming that the underestimation of the SOA yield due to the wall loss of oxidized organic vapors does not affect the temperature dependence.
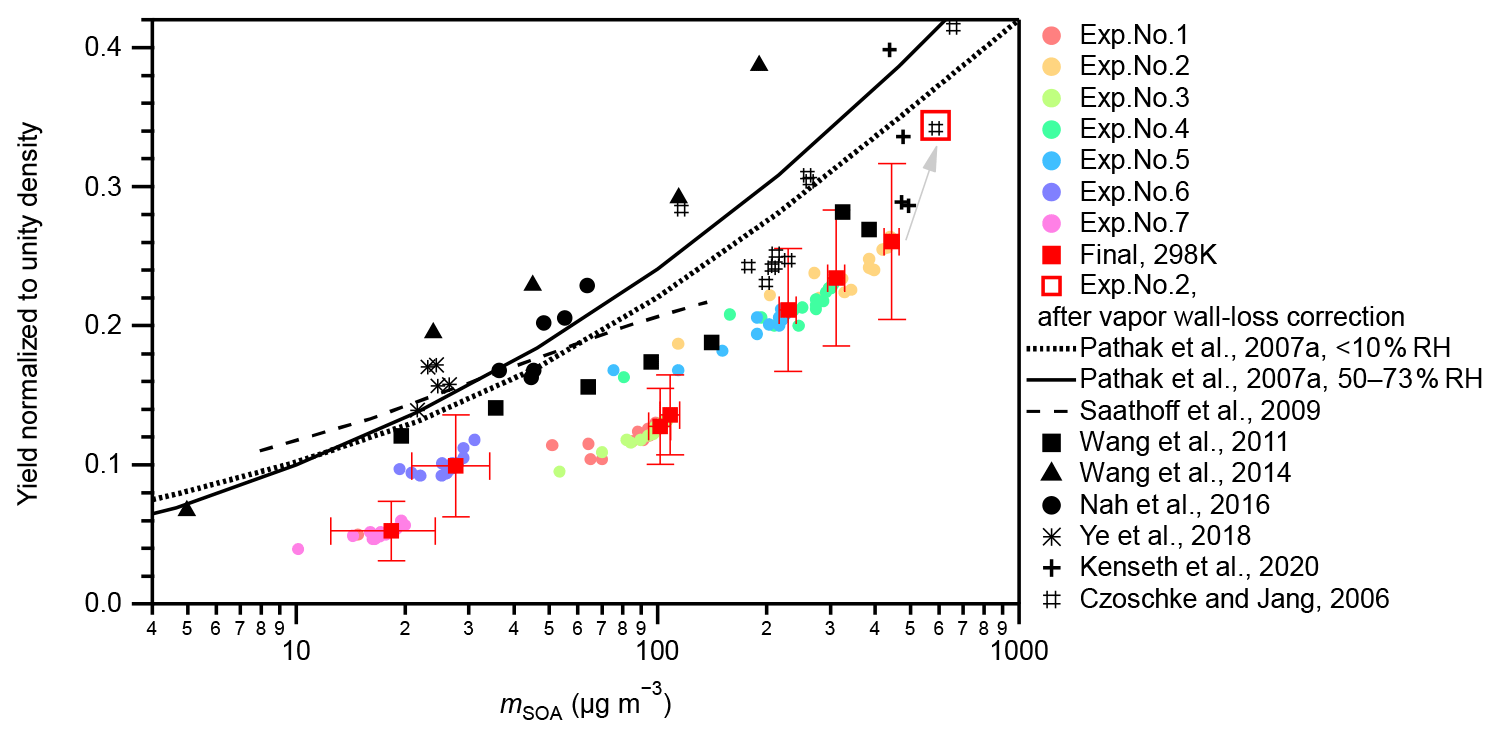
Figure 2Yield comparison. SOA mass yields measured at 298 K under neutral seed conditions in the present study were compared to those of previous studies. Colored markers represent the results of this study. Colored circular markers represent the real-time SOA yields, i.e., the SOA yields along with the α-pinene ozonolysis reactions from 0 to 90 min. Different experimental runs are differentiated by colors. Red solid square markers represent the final SOA yields of the seven experiments. Horizontal error bars indicate the uncertainties of the final SOA concentrations; vertical error bars indicate the uncertainties of the final SOA yields. However, the systematic errors from vapor wall loss are not included. The open square indicates the result of exp. no. 2 after gas-phase wall-loss correction (Text S5). Black markers and curves represent results of previous studies. Black markers represent experimental results. The solid and dotted black curves represent the parameterized results from the four-product volatility basis-set fittings of previous α-pinene ozonolysis experiments under low-NOx and dark conditions summarized in Pathak et al. (2007a). The solid curve represents results under a 50 %–73 % RH range, and the dotted curve represents results under RH < 10 %. The dashed curve represents the results calculated using Eqs. (1), (6), (7), (10), and (11) at 303 K in Saathoff et al., 2009. The experiments of Saathoff et al. (2009) were carried out at 303 K and 48 %–37 % and 0.02 % RH, without or with OH scavenger (cyclohexane or 2-butanol); experiments of Wang et al. (2011) were carried out at 295 K and < 1 % RH, without OH scavenger; experiments of Wang et al. (2014) were carried out at 295 K and < 5 % RH, without OH scavenger; experiments of Nah et al. (2016) were carried out at 298 K and < 5 % RH, with cyclohexane as OH scavenger; experiments of Ye et al. (2018) were carried out at 296 K and 12 %–14 % and 48 %–49 % RH, with cyclohexane as OH scavenger; experiments of Kenseth et al. (2020) were carried out at 295 K and < 5 % RH, without OH scavenger; and experiments of Czoschke and Jang (2006) were carried out at 294–300 K and 14 %–67 % RH, without OH scavenger. Note that all data presented in this figure are normalized to unity density (1 g cm−3).
4.2 Temperature and acidity dependence of SOA yield
The yields of SOA from α-pinene ozonolysis under different experimental conditions in this study are summarized in Fig. 3. Results under neutral and acidic seed conditions are presented separately in panels (a) and (b). In each panel, the measured SOA yields as a function of SOA mass loadings are indicated by markers, and the four-product VBS model fitting results are shown by curves. The fitted parameters with the four-product VBS model under neutral and acidic seed conditions are summarized in Table 1. Under neutral seed conditions, the fitted temperature-independent stoichiometric yields and ΔHvap is 25 kJ mol−1. Under acidic seed conditions, and ΔHvap is 44 kJ mol−1. The fitting results pointed out that most of the detected SOA compounds are of relatively high saturation concentration (i.e., c∗ at 298 K of 1000 µg m−3) under both neutral and acidic seed conditions. Weak increases in SOA yields with decreases in the chamber temperature can be observed under both seed conditions. This is consistent with the result in Pahtak et al. (2007b), which found a weak dependence of SOA yields on temperature in the range of 288–303 K. As temperature decreases, SVOCs tend to partition into the particle phase because of the lowering of their volatilities.
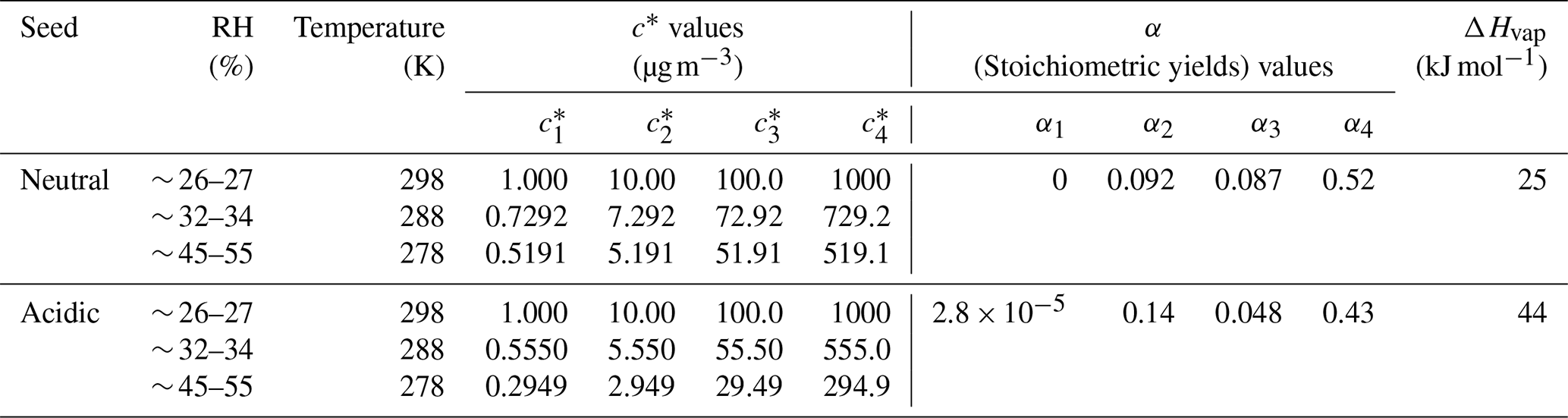
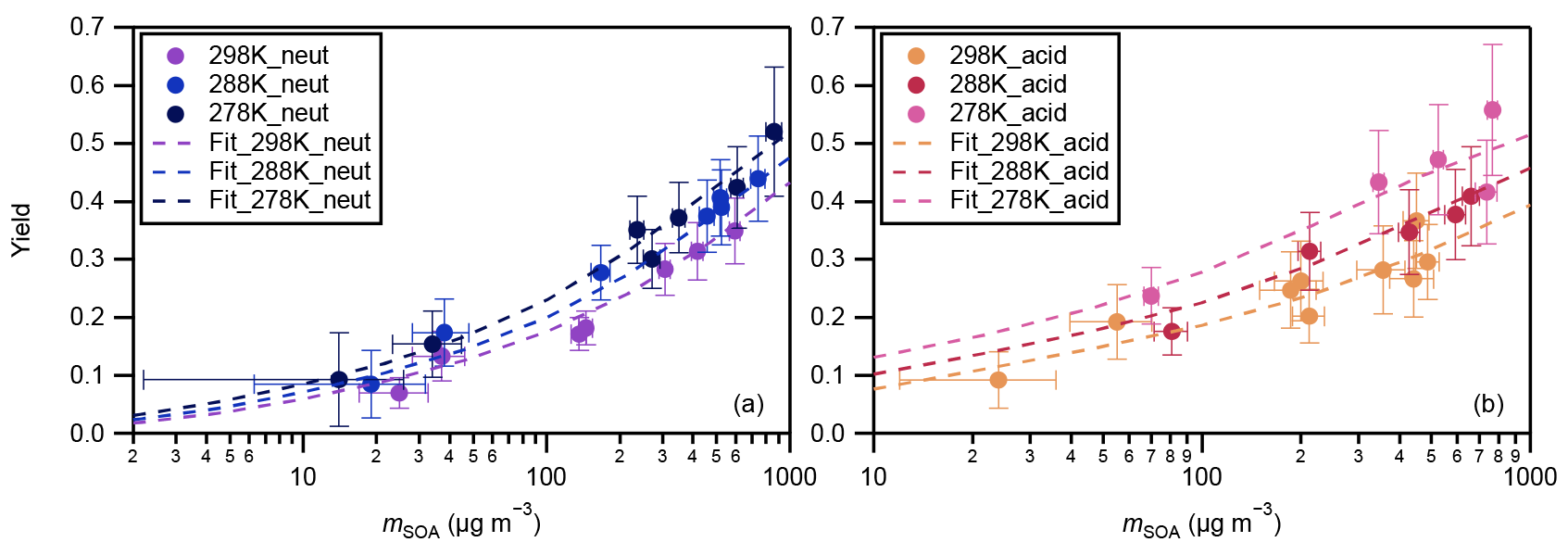
Figure 3Mass yields of SOA for increasing SOA mass loadings. Markers and whiskers are measured data and their uncertainties (Text S4; the uncertainties of the data during the reaction time of 85–90 min are presented); dashed curves are fitting of measured data using a four-product VBS model (Donahue et al., 2006; Lane et al., 2008). Panel (a) presents results under neutral seed conditions, and panel (b) presents results under acidic seed conditions.
The effective ΔHvap of α-pinene ozonolysis SOA derived from the four-product VBS fitting in this study was compared with previous studies (Table 2, Fig. S7). The ΔHvap values derived in this study were comparable to those in Saathoff et al. (2009) and Pathak et al. (2007a) where the experiments were executed in large chambers of 10–200 m3. It may support our assumption that the temperature dependence of SOA yields was not influenced by vapor wall loss. It is also in agreement with the ΔHvap of 40 kJ mol−1 applied in the CMAQv4.7 model (Carlton et al., 2010). However, they are lower than those of Saha and Grieshop (2016) and much lower than those of Epstein et al. (2010). While the ΔHvap in Saha and Grieshop. (2016) was derived based on thermodenuder measurements which attributed 20 %–40 % of SOA mass to low-volatility material (C∗ < 0.3 µg m−3), most of the measured SOA in this study is of relatively high volatility as aforementioned. The differences in the volatility of SOA and the derived ΔHvap between Saha and Grieshop (2016) and this study are consistent with the general phenomenon that ΔHvap is conversely related with volatility (Epstein et al., 2010). The ΔHvap in Epstein et al. (2010) was derived from published experimental vapor pressure data of organic compounds. The reason for the difference in ΔHvap between this study and Epstein et al. (2010) cannot be currently explained. Notably, sensitivity analyses achieved by fixing the stoichiometric yields αi while changing ΔHvap and comparing the resulting VBS curves with measured data (Fig. S8) indicated that the effective ΔHvap could be in the 0 to 70 and 0 to 80 kJ mol−1 ranges for neutral and acidic seed conditions, respectively.
Figure S9 presents the comparisons of the SOA yields under neutral and acidic seed conditions at different temperatures. This indicates that the SOA yields were enhanced under acidic seed conditions when the SOA loadings were low. When the SOA loadings were high, the enhancement disappeared. This is consistent with the results of Gao et al. (2004), which reported obvious initial α-pinene concentration dependence of the enhancement of SOA yields under acidic conditions when compared with neutral seed conditions. For the initial α-pinene concentrations of 12, 25, 48, 52, 96, 120, and 135 ppbv, the relative enhancements of SOA yields were 37 %, 34 %, 26 %, 24 %, 15 %, 10 %, and 8 %, respectively (Gao et al., 2004). This is probably because the SOA components can be of high viscosity under conditions where RH is smaller than around 50 %, and if high SOA mass loadings coated the seed particles, the acid-catalyzed heterogeneous SOA formation reactions could be impeded (Shiraiwa et al., 2013b; Zhou et al., 2013). In this study, the initial concentrations of α-pinene were 54–323 ppbv at 298 K, suggesting that the enhancement could be less than 24 %. When the SOA volume loading was 50 µm3 cm−3, the fitted SOA yields under acidic conditions were enhanced by 11 %, 17 %, and 25 % when compared to the neutral seed conditions under 298, 288, and 278 K, respectively. This is consistent with the findings of Gao et al. (2004) and is also comparable to the results of Iinuma et al. (2005). In Iinuma et al. (2005), the experiment with 2-butanol as an OH radical scavenger under room temperature (294–298 K) reported an enhancement of 19 %, with a final SOA volume concentration of approximately 50 µm3 cm−3. Furthermore, the degree of acidity of the seed aerosols could have also influenced the enhancement (Gao et al., 2004; Czoschke and Jang, 2006). Further comprehensive studies are warranted (including the consideration of the particle viscosity and phase separation) on the influence of seed aerosol acidity on α-pinene ozonolysis SOA formation.
Table 2Comparison of ΔHvap in this study with previous studies.

4.3 Temperature and acidity dependence of SOA composition
Among the 362 compounds identified through LC-TOF-MS analysis in this study (Table S3), 331 compounds were ascribed to VBS bin ranges of −8 to 3. The other 31 compounds were ascribed to higher VBS bin ranges of 4–6. Only the former 331 compounds are targeted in the following discussions for two reasons. First, less than a half of the compounds that belong to VBS bins 4 or greater could exist in the particle phase (Donahue et al., 2006), which would introduce large uncertainties for the estimation of the mass concentrations of the compounds in the gas phase from the particle phase. In addition, LC-TOF-MS analysis of pure compounds indicated that fragmentation of high molecular compounds during the ionization could occur; e.g., pinic acid (C9H14O4) could be fragmented into C8H14O2, and the latter was assigned to VBS bin 6 (Fig. S10). Note that due to the potentially high viscosity of the newly formed SOA, high-volatility compounds formed inside the aerosol particle could have been wrapped into the particle phase and detected (Shiraiwa et al., 2013b).
Figure 4 presents the volatility distributions of the identified compounds. The measured intensities of particle-phase compounds were normalized by their total intensity for each experiment and are presented in Fig. 4a. Compounds that were attributed to VBS bins between 0 and 3 are known as SVOCs (Li et al., 2016). Those in bins 2 and 3 generally presented a decreasing tendency with the increase in experimental temperatures, and there was no obvious temperature dependence for those in bins 0 and 1 (Fig. 4a). The corresponding gas-phase concentrations of each compound were derived assuming gas–particle partitioning equilibrium (Odum et al., 1996). The ΔHvap values derived in this study (Table 1) were used to calculate the saturation concentration under 278 and 288 K following Eq. (3). The intensities of both the particle and gas phases were then normalized by their total amount and are presented in Fig. 4b. With the inclusion of the corresponding gaseous phase compounds, the temperature dependence of compounds in VBS bin 3 changed to positive (i.e., increased with temperature) whereas the tendency in bin 2 remained negative (Fig. 4b). As the α-pinene ozonolysis rate constant at the temperature range of 278–298 K does not vary considerably (within 15 %; IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation, http://iupac.pole-ether.fr, last access: 1 February 2021), α-pinene was completely consumed at the reaction time of 90 min, and the temperature dependence of gas-phase wall loss was considered insignificant (Text S5), the total amounts of SVOCs formed should be similar at the three temperatures. Hence, the total amounts of gas- and particle-phase compounds in each VBS bin should be independent of experimental temperatures. If a larger ΔHvap were applied for the derivation of the intensity of gas-phase compounds (e.g., the semiempirical equation in Epstein et al., 2010), the total amount in VBS bin 3 at 298 K would be much higher than at other temperatures, which is unreasonable. This suggested the appropriateness of the ΔHvap values derived from the four-product VBS fitting of the SOA yields in Sect. 4.1. In addition, we note that the volatility distribution pattern derived in this study is similar to that of the experimental runs 1 and 6 (derived using the same method) of Sato et al. (2018), although positive electrospray ionization analysis was used and the α-pinene ozonolysis experiments were carried out under dry conditions in the latter. According to Morino et al. (2020), both the root-mean-square errors between the observed and simulated SOA concentrations for the formation experiments and between the observed and simulated volume fraction remaining for the heating experiments were minimized in the case of the C∗ distribution reported by Sato et al. (2018). In Fig. 4b, the volatility distributions determined by normalizing the stoichiometric SOA yields (Table 2) based on the total mass fractions of compounds in VBS bins 0 to 3 are also presented. The mass fractions determined from the LC-TOF-MS data at VBS bins 1 and 3 and 0 and 2 were smaller and larger, respectively, than those determined from the SOA stoichiometric yields. These differences are probably caused by uncertainties in saturation concentration and sensitivity parameterization as well as the existence of undetected molecules in the LC-TOF-MS analysis (Sato et al., 2018).
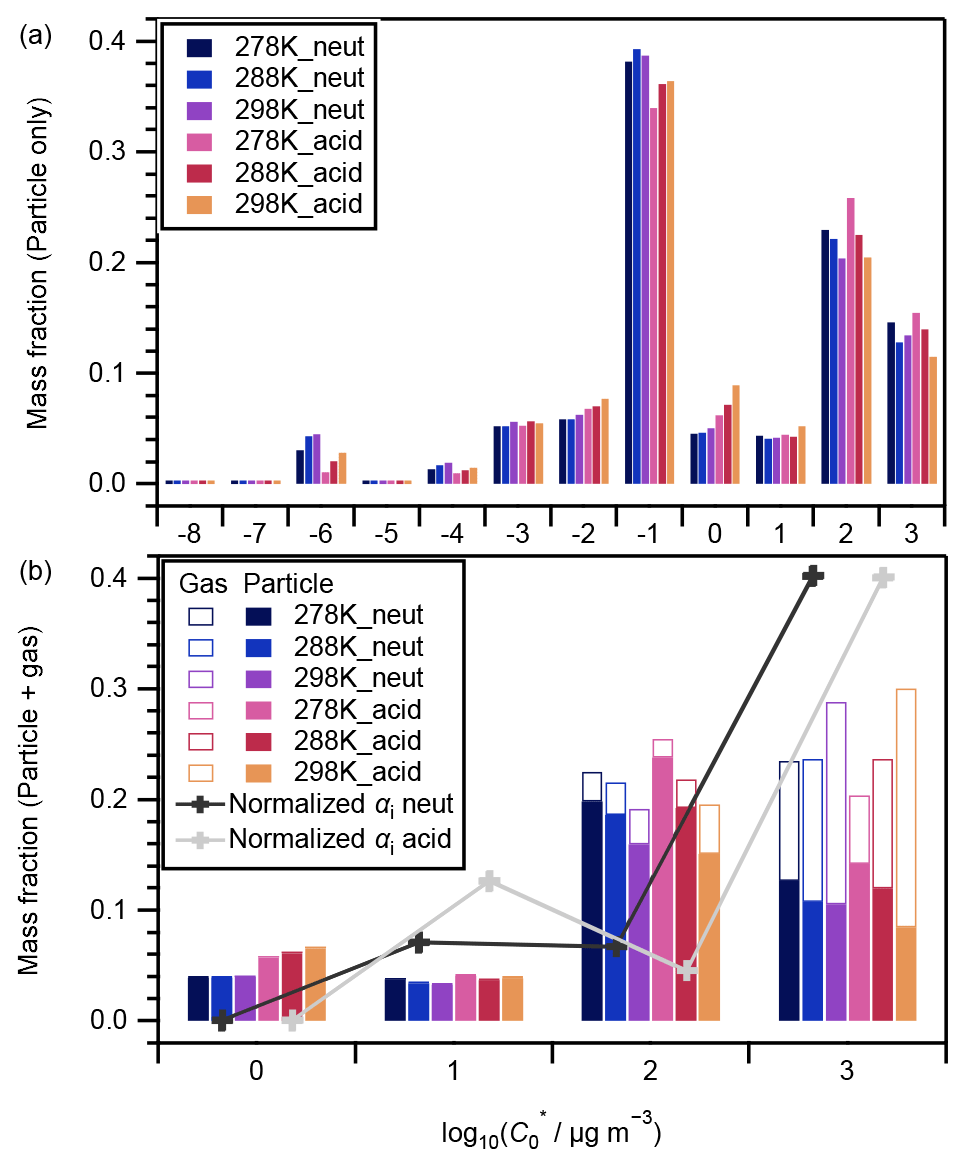
Figure 4Volatility distributions of chemical components in (a) particle-only phase and (b) particle + gas phases. In panel (a), data were normalized by the total signal intensity of the particle phase of each experiment; in panel (b), data were normalized by the total intensity of the particle phase and its equilibrium gas phase in each experiment. Curves and markers imposed in panel (b) are the volatility distributions under neutral (black) and acidic (grey) seed conditions determined by normalizing the stoichiometric SOA yields αi by the total mass fraction of compounds in VBS bins 0 to 3 (Sato et al., 2018).
To gain more insights into the acidity dependence of the α-pinene ozonolysis SOA, the relative intensities of the compounds identified by LC-TOF-MS under acidic and neutral seed conditions were compared. For compounds whose intensity under acidic seed conditions was more than 1.1 times that of neutral seed conditions, monomers with a chemical formula of C10H9−18O4−14 accounted for 32 %, and oligomers with a chemical formula of C11−22H7−35O2−12 accounted for 68 % of the total intensity. Conversely, for compounds whose intensity under acidic conditions was less than 0.9 times that of neutral seed conditions, monomers with a chemical formula of C6−10H7−15O4−6 accounted for 87 %, and oligomers with a chemical formula of C11−21H9−27O5−10 accounted for 13 % of the total intensity.
Figure 5 presents the particle-phase mass distributions of compounds whose mass fractions under acidic seed conditions were more than 1.1 (or less than 0.9) times those of neutral seed conditions. The molecular mass of those compounds with greater mass fractions under acidic seed conditions was generally distributed in the higher mass end compared with those with greater mass fractions under neutral seed conditions. The mass-fraction-weighted mean molecular mass of those compounds presenting greater intensities under acidic seed conditions was 284 ± 14 (mean ± standard deviation) g mol−1, whereas that of those presenting greater intensities under neutral seed conditions was 204 ± 4 g mol−1.
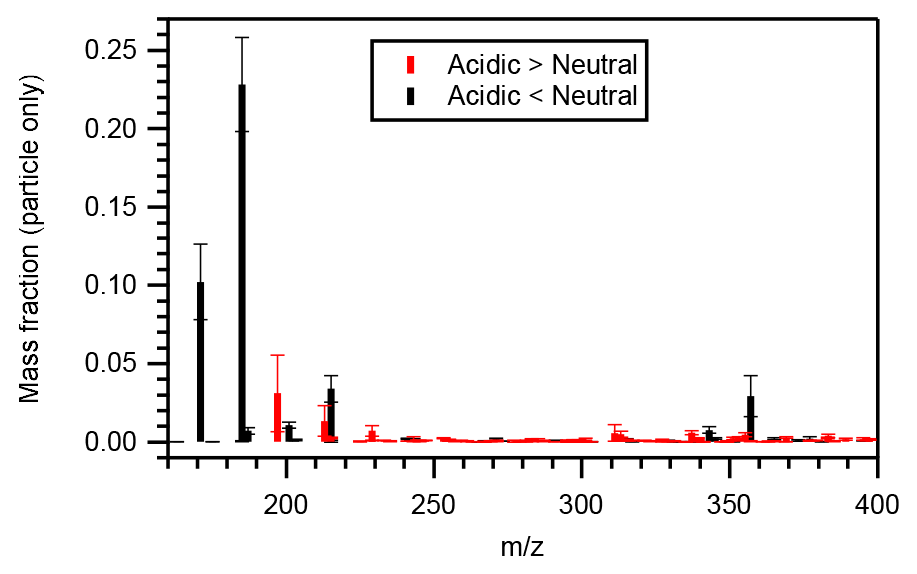
Figure 5Mean particle-phase mass fraction distributions of compounds whose particle-phase mass fractions under acidic seed conditions were more than 1.1 (red symbols) or less than 0.9 (black symbols) times those of neutral seed conditions. Whiskers represent the standard deviations of the three experiments at different temperatures.
Figure 6 presents the identified compounds whose intensity under acidic seed conditions was more than 1.1 (or less than 0.9) times that of neutral seed conditions in the atomic H-to-C ratio (H:C) versus O-to-C ratio (O:C) space. The color scale indicates the mean mass fraction of identified compounds across all six experiments. It is suggested that compounds with a lower intensity under acidic seed conditions were concentrated in the O:C ratio range of 0.4–0.75 and H:C ratio range of 1.1–1.7, whereas those with higher intensity under acidic seed conditions were more broadly distributed in the H:C versus O:C space. Compounds with O:C ratios less than 0.4 were oligomers with a chemical formula of C11−22H19−35O2−7 and were distributed in the VBS bins of −3 to 3. Compounds with O:C ratios greater than 0.75 were likely to be highly oxidized molecules with a chemical formula of C10−14H9−21O9−14 and were attributed to VBS bins −8 to −2. Furthermore, oligomers with O:C ratios of less than 0.4 accounted for 61 % of those oligomers with high relative intensity under acidic conditions, whereas those with O:C ratios of greater than 0.75 accounted for only 1 %. This, together with the aforementioned chemical formula and molecular mass distributions, indicated that the formation of many oligomers, especially with small O:C ratios, was enhanced under acidic seed conditions.
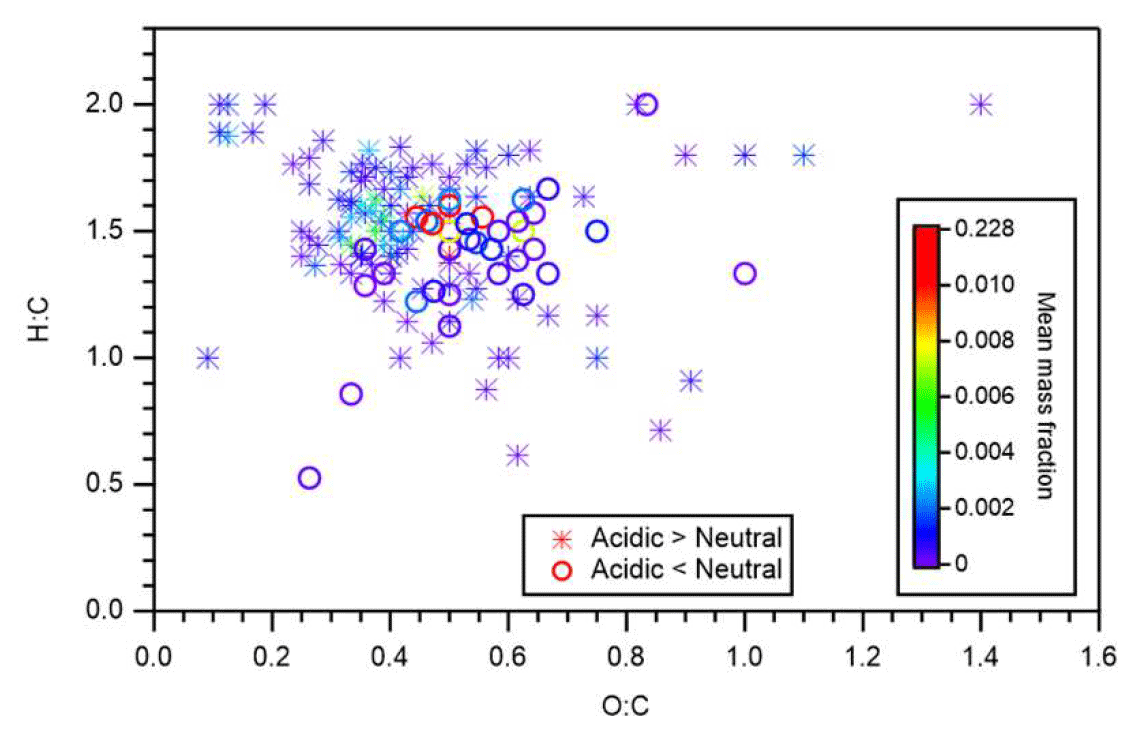
Figure 6Atomic H-to-C ratio (H:C) versus O-to-C ratio (O:C). Stars and circles respectively indicate compounds whose particle-phase mass fractions under acidic seed conditions were more than 1.1 and less than 0.9 times those of neutral seed conditions. The color scale indicates the mean mass fraction of identified compounds across all six experiments. Notably, organosulfates were not presented.
Figure 7 presents the compounds whose normalized intensity under acidic seed conditions was less than 0.9 (panel a) or more than 1.1 (panel b) times that of neutral seed conditions in the VBS space. The acidity dependence of the major compounds in VBS bins was tentatively explained from the viewpoints of acid-catalyzed decomposition reactions or acid-catalyzed heterogenous reactions. Compounds that presented lower intensities under acidic than neutral seed conditions were mainly distributed in VBS bins 3, 2, −1, −4, and −6 (Fig. 7a). The respective compounds that presented the highest intensity in VBS bins 3, 2, −1, −4, and −6 were 215.091 (C10H15O5), 171.065 (C8H11O4), 185.081 (C9H13O4), 343.139 (C16H23O8), and 357.154 (C17H25O8), and they accounted for on average 25, 46, 62, 54, and 99 % of the total intensity of the compounds determined in the respective bins. The structures of 215 have been proposed by Zhang et al. (2017) as hydroperoxides from α-pinene ozonolysis reactions. They can decompose under acidic seed conditions to 197.081 (C10H13O4, VBS bin 2) (Scheme 1a) as well as 155.010 (C8H11O3, VBS bin 5) (Scheme 1b) (acid-catalyzed decomposition of hydroperoxides; Seubold and Vaughan, 1953). Next, previous studies indicate that 171 and 185 were monomer precursors of dimers 343 and 357, and all four products exist in both laboratory α-pinene ozonolysis SOA samples and field samples (Gao et al., 2004; Yasmeen et al., 2010; Kristensen et al., 2014; Zhang et al., 2015). The chemical structure of 185 has been proposed as pinic acid (Gao et al., 2004; Yasmeen et al., 2010; Kristensen et al., 2014) and 171 as terpenylic acid (Yasmeen et al., 2010; Kristensen et al., 2014) and norpinic acid (Gao et al., 2004). In this study, excess OH scavengers could have minimized the formation of an OH functional group in the α-pinene ozonolysis products, whereas the aldehydes should dominate the products (Gaona-Colmán et al., 2017). Thus, it is possible that the acid-catalyzed heterogenous esterification between acid products such as 171 and 185 and aldehyde products such as pinonaldehyde (Hallquist et al., 2009) led to the decreased intensity of 171 and 185 under acidic seed conditions, which further led to the decreased intensity of the related dimers of 343 and 357. Scheme 2 presents a possible esterification reaction between terpenylic acid (a possible isomer of 171) and pinonaldehyde. The product, 339.180 (C18H27O6), ascribed to VBS bin −1, presented higher relative intensity under acidic seed conditions.
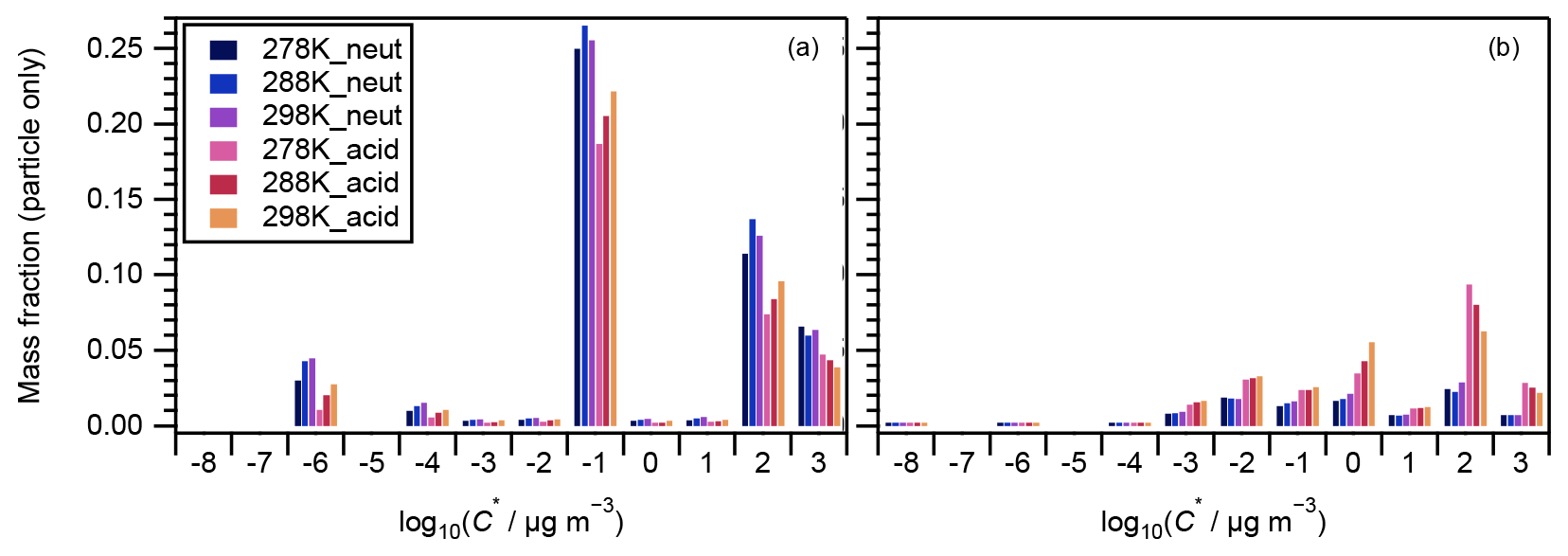
Figure 7Volatility distribution of SOA compounds whose particle-phase mass fractions under acidic seed conditions were (a) less than 0.9 and (b) more than 1.1 times those of neutral seed conditions. Note the mass fractions presented here are the same as those in Fig. 4a.
The compounds with higher contributions under acidic than neutral seed conditions were mainly distributed in VBS bins −3, −2, 0, and 2 (Fig. 7b). The most abundant compounds of those whose intensity under acidic seed conditions was more than 1.1 times that of neutral conditions in bins 2, −2, and −3 were 197 (C10H13O4), 355.175 (C18H27O7), and 383.170 (C19H27O8), respectively. Their contributions to the total signal of their respective bins were 14 %, 7 %, and 5 %, and the ratios of the intensities under acidic seed conditions to those under neutral seed conditions were 5.4, 1.8, and 1.3, respectively. The higher intensity of 197 under acidic than neutral conditions might be explained partly by the acid-catalyzed decomposition of 215 (Scheme 1a). Zhang et al. (2015) proposed 355 as a dimer ester with a hydroperoxide function. Another possible assignment of 355 is an ester from the reaction of a Criegee intermediate (C10H16O3) with terpenylic acid and/or norpinic acid (C8H12O4) (Kristensen et al., 2017). The latter assignment of the product also has a hydroperoxide group. The formation of hydroperoxides involving Criegee intermediates has been observed in the gas phase (Sekimoto et al., 2020) and on the surface of aqueous droplets (Enami and Colussi, 2017). The relatively high intensity of 355 under acidic seed conditions might be explained by the fact that the hydrolysis of ester hydroperoxides becomes slower under acidic conditions (Zhao et al., 2018). Kristensen et al. (2017) proposed 383 as a dimer ester, whereas the molecular structure is to be identified. In bin 0, the intensities of 311.149 (C16H23O6) and 313.165 (C16H25O6) were the highest (contributing on average 10 % and 9 % of the total intensity of the bin, respectively), and under acidic seed conditions were 4.8 and 1.5 times those of neutral seed conditions, respectively. Although the structure of 311 is yet to be identified, 313 was proposed as a dimer ester by Zhang et al. (2015). These dimer esters at 383 and 313 might be formed through acid-catalyzed heterogeneous reactions.
In addition, the 11 identified organosulfate compounds were ascribed to the VBS bins of −3 to 0 (Table 3). All of them presented greater intensity on average under acidic conditions than under neutral seed conditions (Fig. S11). Four of the OSs were in VBS bin 0, which together contributed 9.2 % of the intensity of that bin. The contributions of the two, three, and two OS compounds (which respectively belong to VBS bins of −1, −2, and −3) to their respective bins were 0.4 %, 4.7 %, and 2.6 %. The formation mechanisms of those OS compounds will be discussed in the next section. It should be noted that the intensities of OS compounds in this study might be underestimated because methanol, which could react with carbonyl and carboxylic compounds in the SOA (Bateman et al., 2008), was used as the extraction solution. Acetonitrile should be used as the extraction solution in future studies to confirm the influence (Bateman et al., 2008).
Table 3Organosulfates identified in this study, their respective and formula of [M−H]−, assigned VBS bin number, proposed OS structure, and proposed precursor oxidation product structure.
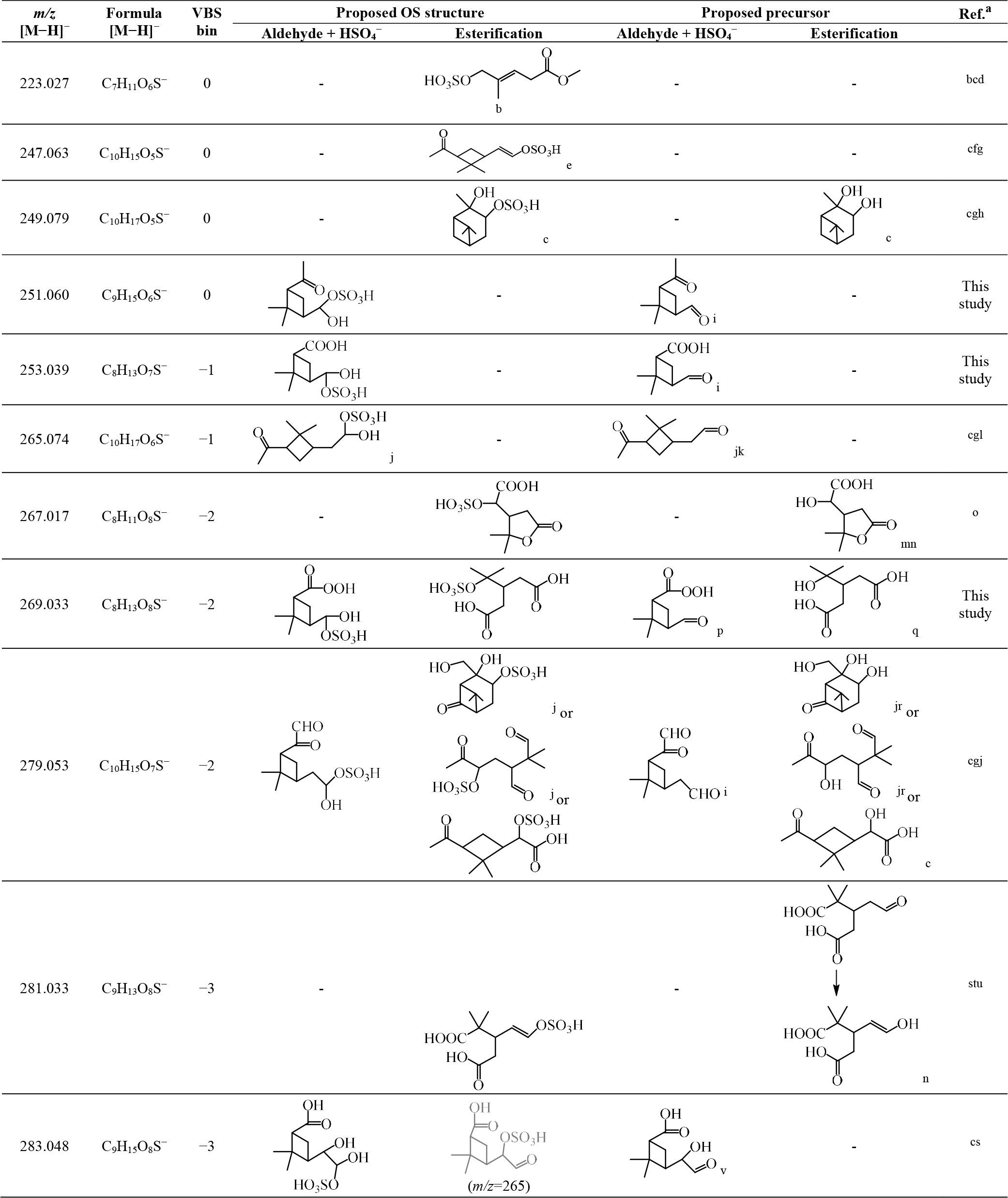
a Selected laboratory and/or field studies where the corresponding OS has been identified. b Hettiyadura et al. (2019). c Surratt et al. (2008). d Yassine et al. (2012). e Zhang et al. (2015). f Tao et al. (2014). g Ma et al. (2014). h Wang et al. (2018b). i Yu et al. (1999). j Surratt et al. (2007). k Jackson et al. (2017). l Liggio and Li (2006). m Kahnt et al. (2014a). n Kahnt et al. (2014b). o Meade et al. (2016). p Reinnig et al. (2009). q Kristensen et al. (2014). r Aschmann et al. (1998, 2002). s Brüggemann et al. (2019). t Wang et al. (2016). u Brüggemann et al. (2017). v Ma et al. (2008).
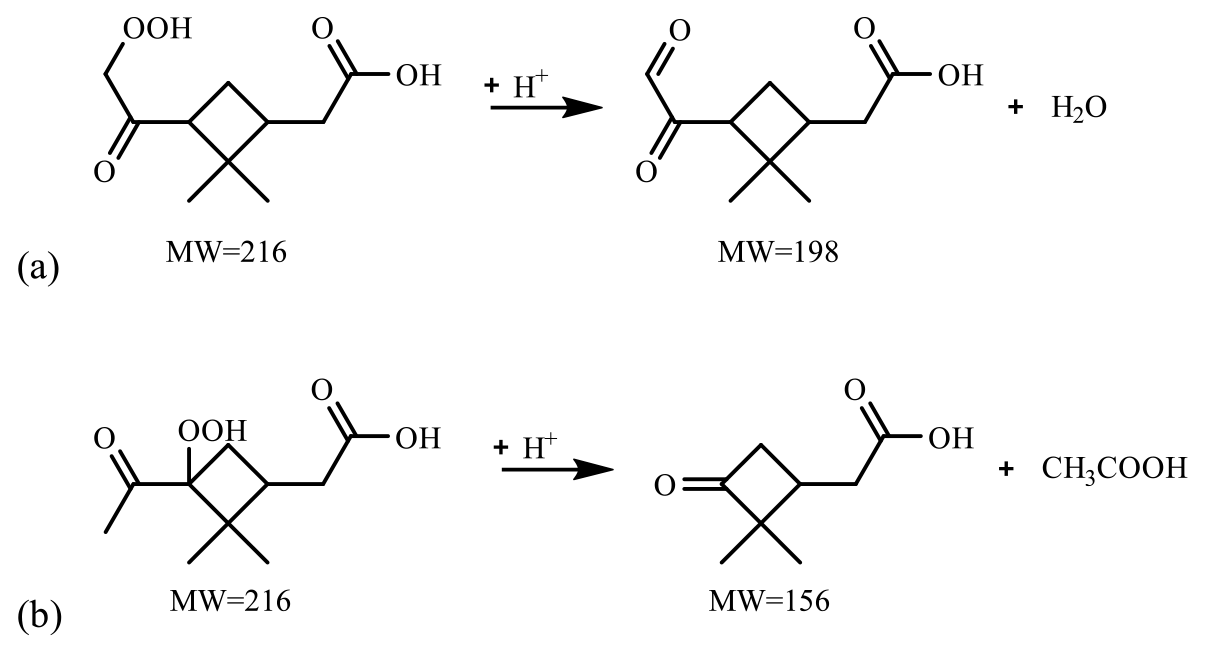
Scheme 1Proposed acid-catalyzed decomposition reactions of hydroperoxides at 215 in this study.
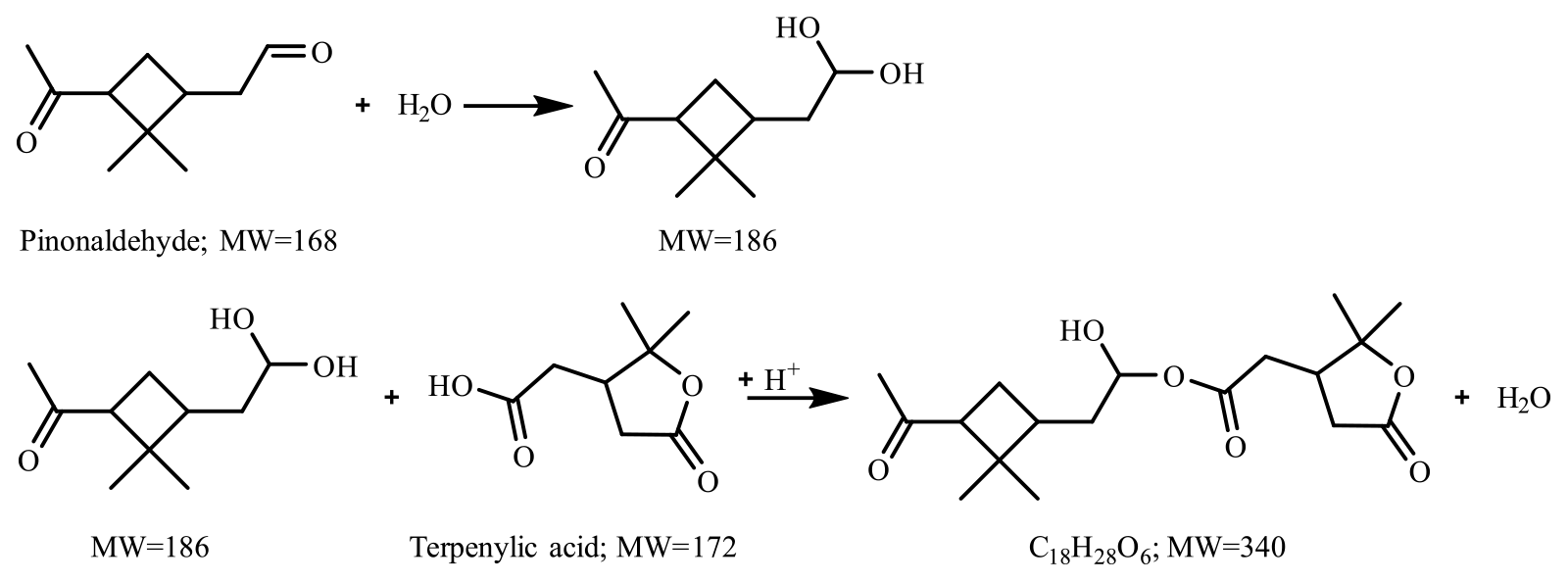
Scheme 2Possible acid-catalyzed esterification reaction of terpenylic acid ( 171) with pinonaldehyde.
4.4 OS formation under acidic conditions
In this study, significant signals were identified for 11 organosulfates under acidic seed conditions (Fig. S11). Figure 8 presents the ethyl-d5-sulfate equivalent (EDSeq.) molecular yields of those OS compounds estimated from the LC-TOF-MS analysis. 223 (C7H12O6S) was the most abundant OS identified at 298 k, followed by 279 (C10H16O7S), 281 (C9H14O8S), 269 (C8H14O8S), 283 (C9H16O8S), 265 (C10H18O6S), 253 (C8H14O7S), 267 (C8H12O8S), 251 (C9H16O6S), 249 (C10H18O5S), and 247 (C10H16O5S).
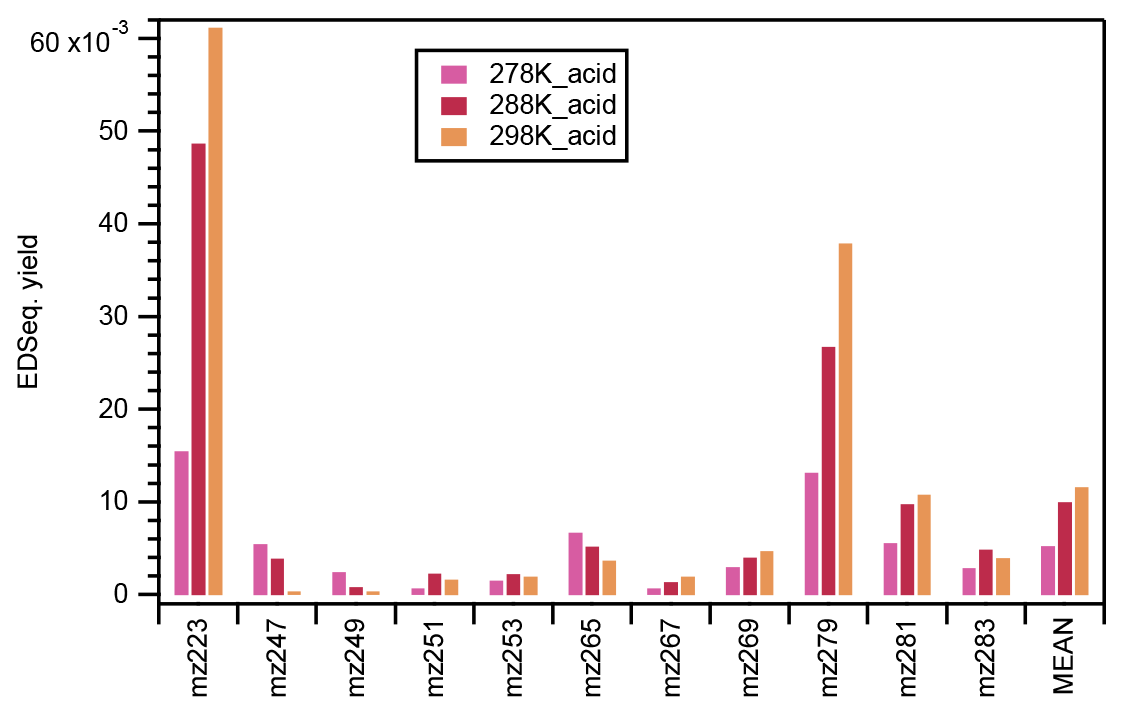
Figure 8The ethyl-d5-sulfate equivalent (EDSeq.) yield of the 11 OS compounds and their arithmetic mean under acidic seed conditions at different temperatures. The retention time range used for the calculation of the intensity of each OS compound is indicated in Fig. S11.
The potential formation mechanisms of the OS compounds were proposed based on literature data in combination with the results of the experiments of this study. To our knowledge, while the formation of OS through α-pinene ozonolysis in the presence of an OH radical scavenger has been reported very recently (Ye et al., 2018; Stangl et al., 2019; Wang et al., 2019), these studies focused on the reactions between α-pinene ozonolysis products and SO2 rather the identification of OS compounds. Earlier studies on the identification and formation mechanisms of OS with α-pinene as the precursor VOC were related to photooxidation (i.e., OH-initiated oxidation) or nighttime oxidation (i.e., NO3-initiated oxidation under dark conditions) (Surratt et al., 2007, 2008). In this study, the formation of OS from α-pinene ozonolysis in the presence of an OH radical scavenger and acidic seed particles was investigated.
Table 3 summarizes the possible chemical structures of the 11 detected OS compounds and their precursor α-pinene oxidation products. Among the 11 OS compounds detected in this study, eight of them have been detected in previous laboratory and/or field studies of α-pinene oxidations, and the other three ( 251, 253, and 269) were detected for the first time in this study (Table 3). While all 11 OS compounds detected in this study are formed from α-pinene ozonolysis due to the presence of excess OH radical scavenger, five of the previously identified OS compounds ( 223, 247, 249, 265, and 279) were detected in α-pinene photooxidation and/or nighttime oxidation experiments (Surratt et al., 2008), and the other three ( 267, 281, and 283) were only identified in ambient aerosols.
The chemical structures of five of the OS compounds (i.e., 223, 247, 249, 265, and 279) have been proposed in previous studies. Four of them ( 223, 247, 249, and 279) were likely to be sulfate esters formed from alcohols and sulfate (Surratt et al., 2007, 2008; Zhang et al., 2015; Hettiyadura et al., 2019), and one ( 265) was said to be from the sulfation of pinonaldehyde (Liggio and Li, 2006; Surratt et al., 2007). The former is referred to hereafter as the alcohol pathway and the latter the aldehyde pathway (aldehyde + HSO; Surratt et al., 2007) (Scheme S1). Because aldehydes should dominate the products from α-pinene ozonolysis in the presence of an OH scavenger (Gaona-Colmán et al., 2017) and pinonaldehyde has been detected in previous α-pinene ozonolysis studies with an OH scavenger (Jackson et al., 2017), we propose that the aldehyde pathway could be one of the dominant OS formation pathways in this study. In addition to 265, we propose possible aldehyde precursors formed in α-pinene ozonolysis experiments for five other OS compounds identified in this study (i.e., 251, 253, 269, 279, and 283). Except for the study where the aldehyde precursor of 269 was found (Reinnig et al., 2009), an OH scavenger was used in all other studies where the aldehyde precursors were found (Yu et al., 1999; Ma et al., 2008; Jackson et al., 2017). Presently, we suppose that the other five OS compounds were possibly formed through the alcohol pathway although no relevant precursor information has been found for 223 and 247. Notably, another possible precursor with a hydroxyl function, which is formed in α-pinene ozonolysis experiments with an OH scavenger, has also been found for the OS at 269 (Kristensen et al., 2014). Additionally, the proposed precursor for 283 (Table 3) also contains a hydroxyl function, which can form another OS of 265.038 (C9H13O7S) through the alcohol pathway. A weak signal of this OS was observed (not presented in Fig. S11).
As shown in Fig. 8, the ethyl-d5-sulfate equivalent (EDSeq.) yields of 247, 249, and 265 decreased with the increase in temperature. In fact, the OS at 247 and 249 could only be observed at the lower temperatures. Conversely, the EDSeq. yields of other OS compounds increased with the temperature, or at least the yields at 278 K were the lowest among the three temperature conditions. The temperature dependence of OS EDSeq. yields seems not to be directly related to the formation mechanisms of either the alcohol pathway or the aldehyde pathway. Nevertheless, the mean EDSeq. yields of the 11 OS compounds, which were dominated by 223 and 279, increased with the increase in reaction temperature. As 11 OSs (including three unreported) were observed in the α-pinene ozonolysis reactions with an OH scavenger and acidic seed particles, the identification of the structures of the OSs using high-resolution ion mobility mass spectrometry is planned as the next step. When the structures of the OSs can be determined and the precursor compounds can be assumed, the formation mechanisms of OSs in α-pinene ozonolysis with acidic seeds may be confirmed.
Using the compact chamber system, SOA formation from α-pinene ozonolysis was studied with diethyl ether as an OH radical scavenger at temperatures of 278, 288, and 298 K, with acidic and neutral seed aerosol particles. The SOA yields and compounds with a molecular mass of less than 400 Da determined using a LC-TOF-MS were analyzed from the perspectives of temperature and seed particle acidity dependence.
The SOA yield increased slightly with the decrease in chamber temperature. The enthalpies of vaporization under neutral and acidic seed conditions were estimated to be 25 and 44 kJ mol−1, respectively. The acidity dependence of the SOA yields at low SOA loadings were comparable to those reported by Gao et al. (2004) and Iinuma et al. (2005). However, the enhancement of the SOA yields under acidic conditions would be limited if the SOA mass loadings are much greater than the amounts of pre-existing particles.
Among the 362 compounds identified, the volatility of 331 was distributed in the VBS bins between −8 and 3. The temperature dependence of the volatility distribution of those identified compounds (particle phase + gas phase) could be consistently explained by the enthalpies of vaporization derived in this study.
The compounds whose intensities under acidic seed conditions were less than 0.9 times those under neutral seed conditions were dominated by monomers, whereas the compounds whose intensities under acidic seed conditions were more than 1.1 times those under neutral seed conditions were dominated by oligomers. The O:C ratios of the former were concentrated in the range of 0.4–0.75. The O:C ratios of the latter were broadly distributed. The compounds with O:C ratios less than 0.4 were all oligomers, which accounted for 61 % of the oligomers with high relative intensity under acidic conditions, whereas those with O:C ratios of greater than 0.75 were highly oxidized molecules and only contributed to 1 % of the oligomers. In addition, the mean molecular mass of the former compounds (204 ± 4 g mol−1) was evidently lower than that of the latter (284 ± 14 g mol−1). The differences indicated that the formation of many oligomers, especially with low O:C ratios, was enhanced under acidic seed conditions. The acidity dependence of certain major compounds could be explained by acid-catalyzed heterogenous reactions (e.g., 171, 185, 343, and 357) or acid-catalyzed decomposition reactions (e.g., 215 and 197), which suggests that little or no enhancement of SOA under acidic conditions in field observations could occur when acid-catalyzed decomposition is dominant.
For the first time, organosulfate compounds were studied for α-pinene ozonolysis reaction in the presence of an OH scavenger and acidic seed particles. Eleven OS compounds were determined from LC-TOF-MS analysis. All of them on average presented higher yields under acidic than under neutral seed conditions. Six of the OS compounds were potentially formed via the aldehyde + HSO pathway, which should be confirmed in future studies through high-resolution mass spectrometry analyses.
Finally, the organosulfates and the oligomers that increased with an increase in acidity of the seed particles could be indicators of the acidity of pre-existing particles in the field, and the new findings obtained from this study should be confirmed using more complex and larger chambers.
All the final data supporting the findings of this study are available in the manuscript or in the Supplement. Raw data used to derive the final data are available on request to the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-5983-2021-supplement.
SI, YD, SE, and HT performed chamber experiments. KS and SR carried out LC-TOF-MS analyses. SI, YD, SE, and MY performed data analyses. YD, SI, and HT wrote the manuscript, and all authors contributed to the revisions of the manuscript.
The authors declare that they have no conflict of interest.
We thank Sumiko Komori, Yoshikatsu Takazawa, and Tomoharu Sano for the technical support. This work was supported by NIES research funding (Type A) and JSPS KAKENHI grant no. 19H01154.
This research has been supported by the National Institute for Environmental Studies (NIES research funding (Type A)) and the Japan Society for the Promotion of Science (grant no. 19H01154).
This paper was edited by Sergey A. Nizkorodov and reviewed by two anonymous referees.
Aschmann, S. M., Reissell, A., Atkinson, R., and Arey, J.: Products of the gas phase reactions of the OH radical with α- and β-pinene in the presence of NO, J. Geophys. Res.-Atmos., 103, 25553–25561, https://doi.org/10.1029/98jd01676, 1998.
Aschmann, S. M., Atkinson, R., and Arey, J.: Products of reaction of OH radicals with α-pinene, J. Geophys. Res.-Atmos., 107, 4191, https://doi.org/10.1029/2001jd001098, 2002.
Bateman, A. P., Walser, M. L., Desyaterik, Y., Laskin, J., Laskin, A., and Nizkorodov, S. A.: The effect of solvent on the analysis of secondary organic aerosol using electrospray ionization mass spectrometry, Environ. Sci. Technol., 42, 7341–7346, https://doi.org/10.1021/es801226w, 2008.
Brüggemann, M., Poulain, L., Held, A., Stelzer, T., Zuth, C., Richters, S., Mutzel, A., van Pinxteren, D., Iinuma, Y., Katkevica, S., Rabe, R., Herrmann, H., and Hoffmann, T.: Real-time detection of highly oxidized organosulfates and BSOA marker compounds during the F-BEACh 2014 field study, Atmos. Chem. Phys., 17, 1453–1469, https://doi.org/10.5194/acp-17-1453-2017, 2017.
Brüggemann, M., van Pinxteren, D., Wang, Y. C., Yu, J. Z., and Herrmann, H.: Quantification of known and unknown terpenoid organosulfates in PM10 using untargeted LC-HRMS/MS: contrasting summertime rural Germany and the North China Plain, Environ. Chem., 16, 333–346, https://doi.org/10.1071/en19089, 2019.
Brüggemann, M., Xu, R. S., Tilgner, A., Kwong, K. C., Mutzel, A., Poon, H. Y., Otto, T., Schaefer, T., Poulain, L., Chan, M. N., and Herrmann, H.: Organosulfates in Ambient Aerosol: State of Knowledge and Future Research Directions on Formation, Abundance, Fate, and Importance, Environ. Sci. Technol., 54, 3767–3782, https://doi.org/10.1021/acs.est.9b06751, 2020.
Capouet, M., Mueller, J. F., Ceulemans, K., Compernolle, S., Vereecken, L., and Peeters, J.: Modeling aerosol formation in α-pinene photo-oxidation experiments, J. Geophys. Res.-Atmos., 113, D02308, https://doi.org/10.1029/2007jd008995, 2008.
Carlton, A. G., Bhave, P. V., Napelenok, S. L., Edney, E. D., Sarwar, G., Pinder, R. W., Pouliot, G. A., and Houyoux, M.: Model Representation of Secondary Organic Aerosol in CMAQv4.7, Environ. Sci. Technol., 44, 8553–8560, https://doi.org/10.1021/es100636q, 2010.
Chu, S. H., Paisie, J. W., and Jang, B. W. L.: PM data analysis – a comparison of two urban areas: Fresno and Atlanta, Atmos. Environ., 38, 3155–3164, https://doi.org/10.1016/j.atmosenv.2004.03.018, 2004.
Czoschke, N. M. and Jang, M.: Acidity effects on the formation of α-pinene ozone SOA in the presence of inorganic seed, Atmos. Environ., 40, 4370–4380, https://doi.org/10.1016/j.atmosenv.2006.03.030, 2006.
Czoschke, N. M., Jang, M., and Kamens, R. M.: Effect of acidic seed on biogenic secondary organic aerosol growth, Atmos. Environ., 37, 4287–4299, https://doi.org/10.1016/s1352-2310(03)00511-9, 2003.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Duporte, G., Flaud, P. M., Geneste, E., Augagneur, S., Pangui, E., Lamkaddam, H., Gratien, A., Doussin, J. F., Budzinski, H., Villenave, E., and Perraudin, E.: Experimental Study of the Formation of Organosulfates from α-Pinene Oxidation, Part I: Product Identification, Formation Mechanisms and Effect of Relative Humidity, J. Phys. Chem. A, 120, 7909–7923, https://doi.org/10.1021/acs.jpca.6b08504, 2016.
Duporte, G., Flaud, P. M., Kammer, J., Geneste, E., Augagneur, S., Pangui, E., Lamkaddam, H., Gratien, A., Doussin, J. F., Budzinski, H., Villenave, E., and Perraudin, E.: Experimental Study of the Formation of Organosulfates from α-Pinene Oxidation, 2. Time Evolution and Effect of Particle Acidity, J. Phys. Chem. A, 124, 409–421, https://doi.org/10.1021/acs.jpca.9b07156, 2020.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012.
Enami, S. and Colussi, A. J.: Criegee Chemistry on Aqueous Organic Surfaces, J. Phys. Chem. Lett., 8, 1615–1623, https://doi.org/10.1021/acs.jpclett.7b00434, 2017.
Epstein, S. A., Riipinen, I., and Donahue, N. M.: A Semiempirical Correlation between Enthalpy of Vaporization and Saturation Concentration for Organic Aerosol, Environ. Sci. Technol., 44, 743–748, https://doi.org/10.1021/es902497z, 2010.
Faust, J. A., Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Role of Aerosol Liquid Water in Secondary Organic Aerosol Formation from Volatile Organic Compounds, Environ. Sci. Technol., 51, 1405–1413, https://doi.org/10.1021/acs.est.6b04700, 2017.
Gao, S., Ng, N. L., Keywood, M., Varutbangkul, V., Bahreini, R., Nenes, A., He, J. W., Yoo, K. Y., Beauchamp, J. L., Hodyss, R. P., Flagan, R. C., and Seinfeld, J. H.: Particle phase acidity and oligomer formation in secondary organic aerosol, Environ. Sci. Technol., 38, 6582–6589, https://doi.org/10.1021/es049125k, 2004.
Gaona-Colmán, E., Blanco, M. B., Barnes, I., Wiesen, P., and Teruel, M. A.: OH- and O3-initiated atmospheric degradation of camphene: temperature dependent rate coefficients, product yields and mechanisms, RSC Adv., 7, 2733–2744, https://doi.org/10.1039/c6ra26656h, 2017.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hettiyadura, A. P. S., Al-Naiema, I. M., Hughes, D. D., Fang, T., and Stone, E. A.: Organosulfates in Atlanta, Georgia: anthropogenic influences on biogenic secondary organic aerosol formation, Atmos. Chem. Phys., 19, 3191–3206, https://doi.org/10.5194/acp-19-3191-2019, 2019.
Hilal, S. H., Karickhoff, S. W., and Carreira, L. A.: Prediction of the vapor pressure boiling point, heat of vaporization and diffusion coefficient of organic compounds, QSAR Comb. Sci., 22, 565–574, https://doi.org/10.1002/qsar.200330812, 2003.
Hinkley, J. T., Bridgman, H. A., Buhre, B. J. P., Gupta, R. P., Nelson, P. F., and Wall, T. F.: Semi-quantitative characterisation of ambient ultrafine aerosols resulting from emissions of coal fired power stations, Sci. Total Environ., 391, 104–113, https://doi.org/10.1016/j.scitotenv.2007.10.017, 2008.
Hu, C. J., Cheng, Y., Pan, G., Gai, Y. B., Gu, X. J., Zhao, W. X., Wang, Z. Y., Zhang, W. J., Chen, J., Liu, F. Y., Shan, X. B., and Sheng, L. S.: A Smog Chamber Facility for Qualitative and Quantitative Study on Atmospheric Chemistry and Secondary Organic Aerosol, Chinese J. Chem. Phys., 27, 631–639, https://doi.org/10.1063/1674-0068/27/06/631-639, 2014.
Iinuma, Y., Boge, O., Gnauk, T., and Herrmann, H.: Aerosol-chamber study of the α-pinene/O3 reaction: influence of particle acidity on aerosol yields and products, Atmos. Environ., 38, 761–773, https://doi.org/10.1016/j.atmosenv.2003.10.015, 2004.
Iinuma, Y., Boge, O., Miao, Y., Sierau, B., Gnauk, T., and Herrmann, H.: Laboratory studies on secondary organic aerosol formation from terpenes, Faraday Discuss., 130, 279–294, https://doi.org/10.1039/b502160j, 2005.
Iinuma, Y., Müller, C., Boge, O., Gnauk, T., and Herrmann, H.: The formation of organic sulfate esters in the limonene ozonolysis secondary organic aerosol (SOA) under acidic conditions, Atmos. Environ., 41, 5571–5583, https://doi.org/10.1016/j.atmosenv.2007.03.007, 2007.
Iinuma, Y., Boge, O., Kahnt, A., and Herrmann, H.: Laboratory chamber studies on the formation of organosulfates from reactive uptake of monoterpene oxides, Phys. Chem. Chem. Phys., 11, 7985–7997, https://doi.org/10.1039/b904025k, 2009.
Jackson, S. R., Ham, J. E., Harrison, J. C., and Wells, J. R.: Identification and quantification of carbonyl-containing α-pinene ozonolysis products using O-tert-butylhydroxylamine hydrochloride, J. Atmos. Chem., 74, 325–338, https://doi.org/10.1007/s10874-016-9344-6, 2017.
Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, https://doi.org/10.1126/science.1075798, 2002.
Jang, M., Czoschke, N. M., Northcross, A. L., Cao, G., and Shaof, D.: SOA formation from partitioning and heterogeneous reactions: Model study in the presence of inorganic species, Environ. Sci. Technol., 40, 3013–3022, https://doi.org/10.1021/es0511220, 2006.
Jang, M., Cao, G., and Paul, J.: Colorimetric particle acidity analysis of secondary organic aerosol coating on submicron acidic aerosols, Aerosol Sci. Tech., 42, 409–420, https://doi.org/10.1080/02786820802154861, 2008.
Kahnt, A., Iinuma, Y., Blockhuys, F., Mutzel, A., Vermeylen, R., Kleindienst, T. E., Jaoui, M., Offenberg, J. H., Lewandowski, M., Boge, O., Herrmann, H., Maenhaut, W., and Claeys, M.: 2-Hydroxyterpenylic Acid: An Oxygenated Marker Compound for α-Pinene Secondary Organic Aerosol in Ambient Fine Aerosol, Environ. Sci. Technol., 48, 4901–4908, https://doi.org/10.1021/es500377d, 2014a.
Kahnt, A., Iinuma, Y., Mutzel, A., Böge, O., Claeys, M., and Herrmann, H.: Campholenic aldehyde ozonolysis: a mechanism leading to specific biogenic secondary organic aerosol constituents, Atmos. Chem. Phys., 14, 719–736, https://doi.org/10.5194/acp-14-719-2014, 2014b.
Kelly, J. M., Doherty, R. M., O'Connor, F. M., and Mann, G. W.: The impact of biogenic, anthropogenic, and biomass burning volatile organic compound emissions on regional and seasonal variations in secondary organic aerosol, Atmos. Chem. Phys., 18, 7393–7422, https://doi.org/10.5194/acp-18-7393-2018, 2018.
Kenseth, C. M., Hafeman, N. J., Huang, Y. L., Dalleska, N. F., Stoltz, B. M., and Seinfeld, J. H.: Synthesis of Carboxylic Acid and Dimer Ester Surrogates to Constrain the Abundance and Distribution of Molecular Products in α-Pinene and β-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 54, 12829–12839, https://doi.org/10.1021/acs.est.0c01566, 2020.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kristensen, K., Cui, T., Zhang, H., Gold, A., Glasius, M., and Surratt, J. D.: Dimers in α-pinene secondary organic aerosol: effect of hydroxyl radical, ozone, relative humidity and aerosol acidity, Atmos. Chem. Phys., 14, 4201–4218, https://doi.org/10.5194/acp-14-4201-2014, 2014.
Kristensen, K., Jensen, L. N., Glasius, M., and Bilde, M.: The effect of sub-zero temperature on the formation and composition of secondary organic aerosol from ozonolysis of α-pinene, Environ. Sci.-Proc. Imp., 19, 1220–1234, https://doi.org/10.1039/c7em00231a, 2017.
Lai, A. C. K. and Nazaroff, W. W.: Modeling indoor particle deposition from turbulent flow onto smooth surfaces, J. Aerosol Sci., 31, 463–476, 2000.
Lane, T. E., Donahue, N. M., and Pandis, S. N.: Simulating secondary organic aerosol formation using the volatility basis-set approach in a chemical transport model, Atmos. Environ., 42, 7439–7451, https://doi.org/10.1016/j.atmosenv.2008.06.026, 2008.
Lewandowski, M., Jaoui, M., Kleindienst, T. E., Offenberg, J. H., and Edney, E. O.: Composition of PM2.5 during the summer of 2003 in Research Triangle Park, North Carolina, Atmos. Environ., 41, 4073–4083, https://doi.org/10.1016/j.atmosenv.2007.01.012, 2007.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Liggio, J. and Li, S. M.: Organosulfate formation during the uptake of pinonaldehyde on acidic sulfate aerosols, Geophys. Res. Lett., 33, L13808, https://doi.org/10.1029/2006gl026079, 2006.
Ma, Y., Russell, A. T., and Marston, G.: Mechanisms for the formation of secondary organic aerosol components from the gas-phase ozonolysis of α-pinene, Phys. Chem. Chem. Phys., 10, 4294–4312, https://doi.org/10.1039/b803283a, 2008.
Ma, Y., Xu, X. K., Song, W. H., Geng, F. H., and Wang, L.: Seasonal and diurnal variations of particulate organosulfates in urban Shanghai, China, Atmos. Environ., 85, 152–160, https://doi.org/10.1016/j.atmosenv.2013.12.017, 2014.
Meade, L. E., Riva, M., Blomberg, M. Z., Brock, A. K., Qualters, E. M., Siejack, R. A., Ramakrishnan, K., Surratt, J. D., and Kautzman, K. E.: Seasonal variations of fine particulate organosulfates derived from biogenic and anthropogenic hydrocarbons in the mid-Atlantic United States, Atmos. Environ., 145, 405–414, https://doi.org/10.1016/j.atmosenv.2016.09.028, 2016.
Mei, F., Hayes, P. L., Ortega, A., Taylor, J. W., Allan, J. D., Gilman, J., Kuster, W., de Gouw, J., Jimenez, J. L., and Wang, J.: Droplet activation properties of organic aerosols observed at an urban site during CalNex-LA, J. Geophys. Res.-Atmos., 118, 2903–2917, https://doi.org/10.1002/jgrd.50285, 2013.
Messina, P., Lathière, J., Sindelarova, K., Vuichard, N., Granier, C., Ghattas, J., Cozic, A., and Hauglustaine, D. A.: Global biogenic volatile organic compound emissions in the ORCHIDEE and MEGAN models and sensitivity to key parameters, Atmos. Chem. Phys., 16, 14169–14202, https://doi.org/10.5194/acp-16-14169-2016, 2016.
Morino, Y., Sato, K., Jathar, S. H., Tanabe, K., Inomata, S., Fujitani, Y., Ramasamy, S., and Cappa, C. D.: Modeling the Effects of Dimerization and Bulk Diffusion on the Evaporative Behavior of Secondary Organic Aerosol Formed from α-Pinene and 1,3,5-Trimethylbenzene, ACS Earth Space Chem., 4, 1931–1946, https://doi.org/10.1021/acsearthspacechem.0c00106, 2020.
Na, K., Song, C., Switzer, C., and Cocker, D. R.: Effect of ammonia on secondary organic aerosol formation from α-Pinene ozonolysis in dry and humid conditions, Environ. Sci. Technol., 41, 6096–6102, https://doi.org/10.1021/es061956y, 2007.
Nah, T., McVay, R. C., Zhang, X., Boyd, C. M., Seinfeld, J. H., and Ng, N. L.: Influence of seed aerosol surface area and oxidation rate on vapor wall deposition and SOA mass yields: a case study with α-pinene ozonolysis, Atmos. Chem. Phys., 16, 9361–9379, https://doi.org/10.5194/acp-16-9361-2016, 2016.
Nah, T., McVay, R. C., Pierce, J. R., Seinfeld, J. H., and Ng, N. L.: Constraining uncertainties in particle-wall deposition correction during SOA formation in chamber experiments, Atmos. Chem. Phys., 17, 2297–2310, https://doi.org/10.5194/acp-17-2297-2017, 2017.
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Contribution of first- versus second-generation products to secondary organic aerosols formed in the oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, https://doi.org/10.1021/es052269u, 2006.
Northcross, A. L. and Jang, M.: Heterogeneous SOA yield from ozonolysis of monoterpenes in the presence of inorganic acid, Atmos. Environ., 41, 1483–1493, https://doi.org/10.1016/j.atmosenv.2006.10.009, 2007.
Nozière, B., Ekstrom, S., Alsberg, T., and Holmstrom, S.: Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols, Geophys. Res. Lett., 37, L05806, https://doi.org/10.1029/2009gl041683, 2010.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
Parrish, D. D., Singh, H. B., Molina, L., and Madronich, S.: Air quality progress in North American megacities: A review, Atmos. Environ., 45, 7015–7025, https://doi.org/10.1016/j.atmosenv.2011.09.039, 2011.
Pathak, R. K., Presto, A. A., Lane, T. E., Stanier, C. O., Donahue, N. M., and Pandis, S. N.: Ozonolysis of α-pinene: parameterization of secondary organic aerosol mass fraction, Atmos. Chem. Phys., 7, 3811–3821, https://doi.org/10.5194/acp-7-3811-2007, 2007a.
Pathak, R. K., Stanier, C. O., Donahue, N. M., and Pandis, S. N.: Ozonolysis of α-pinene at atmospherically relevant concentrations: Temperature dependence of aerosol mass fractions (yields), J. Geophys. Res.-Atmos., 112, D03201, https://doi.org/10.1029/2006jd007436, 2007b.
Peltier, R. E., Sullivan, A. P., Weber, R. J., Brock, C. A., Wollny, A. G., Holloway, J. S., de Gouw, J. A., and Warneke, C.: Fine aerosol bulk composition measured on WP-3D research aircraft in vicinity of the Northeastern United States – results from NEAQS, Atmos. Chem. Phys., 7, 3231–3247, https://doi.org/10.5194/acp-7-3231-2007, 2007.
Reinnig, M. C., Warnke, J., and Hoffmann, T.: Identification of organic hydroperoxides and hydroperoxy acids in secondary organic aerosol formed during the ozonolysis of different monoterpenes and sesquiterpenes by on-line analysis using atmospheric pressure chemical ionization ion trap mass spectrometry, Rapid Commun. Mass Sp., 23, 1735–1741, https://doi.org/10.1002/rcm.4065, 2009.
Rengarajan, R., Sudheer, A. K., and Sarin, M. M.: Aerosol acidity and secondary organic aerosol formation during wintertime over urban environment in western India, Atmos. Environ., 45, 1940–1945, https://doi.org/10.1016/j.atmosenv.2011.01.026, 2011.
Riva, M., Tomaz, S., Cui, T. Q., Lin, Y. H., Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an Unrecognized Secondary Anthropogenic Source of Organosulfates and Sulfonates: Gas-Phase Oxidation of Polycyclic Aromatic Hydrocarbons in the Presence of Sulfate Aerosol, Environ. Sci. Technol., 49, 6654–6664, https://doi.org/10.1021/acs.est.5b00836, 2015.
Riva, M., Da Silva Barbosa, T., Lin, Y.-H., Stone, E. A., Gold, A., and Surratt, J. D.: Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes, Atmos. Chem. Phys., 16, 11001–11018, https://doi.org/10.5194/acp-16-11001-2016, 2016.
Saathoff, H., Naumann, K.-H., Möhler, O., Jonsson, Å. M., Hallquist, M., Kiendler-Scharr, A., Mentel, Th. F., Tillmann, R., and Schurath, U.: Temperature dependence of yields of secondary organic aerosols from the ozonolysis of α-pinene and limonene, Atmos. Chem. Phys., 9, 1551–1577, https://doi.org/10.5194/acp-9-1551-2009, 2009.
Saha, P. K. and Grieshop, A. P.: Exploring Divergent Volatility Properties from Yield and Thermodenuder Measurements of Secondary Organic Aerosol from α-Pinene Ozonolysis, Environ. Sci. Technol., 50, 5740–5749, https://doi.org/10.1021/acs.est.6b00303, 2016.
Sato, K., Hatakeyama, S., and Imamura, T.: Secondary organic aerosol formation during the photooxidation of toluene: NOx dependence of chemical composition, J. Phys. Chem. A, 111, 9796–9808, https://doi.org/10.1021/jp071419f, 2007.
Sato, K., Fujitani, Y., Inomata, S., Morino, Y., Tanabe, K., Ramasamy, S., Hikida, T., Shimono, A., Takami, A., Fushimi, A., Kondo, Y., Imamura, T., Tanimoto, H., and Sugata, S.: Studying volatility from composition, dilution, and heating measurements of secondary organic aerosols formed during α-pinene ozonolysis, Atmos. Chem. Phys., 18, 5455–5466, https://doi.org/10.5194/acp-18-5455-2018, 2018.
Sato, K., Fujitani, Y., Inomata, S., Morino, Y., Tanabe, K., Hikida, T., Shimono, A., Takami, A., Fushimi, A., Kondo, Y., Imamura, T., Tanimoto, H., and Sugata, S.: A study of volatility by composition, heating, and dilution measurements of secondary organic aerosol from 1,3,5-trimethylbenzene, Atmos. Chem. Phys., 19, 14901–14915, https://doi.org/10.5194/acp-19-14901-2019, 2019.
Sekimoto, K., Fukuyama, D., and Inomata, S.: Accurate identification of dimers from α-pinene oxidation using high-resolution collision-induced dissociation mass spectrometry, J. Mass Spectrom., 55, e4508, https://doi.org/10.1002/jms.4508, 2020.
Seubold, F. H. and Vaughan, W. E.: Acid-catalyzed decomposition of cumene hydroproxide, J. Am. Chem. Soc., 75, 3790–3792, https://doi.org/10.1021/ja01111a055, 1953.
Shiraiwa, M., Yee, L. D., Schilling, K. A., Loza, C. L., Craven, J. S., Zuend, A., Ziemann, P. J., and Seinfeld, J. H.: Size distribution dynamics reveal particle-phase chemistry in organic aerosol formation, P. Natl. Acad. Sci. USA, 110, 11746–11750, https://doi.org/10.1073/pnas.1307501110, 2013a.
Shiraiwa, M., Zuend, A., Bertram, A. K., and Seinfeld, J. H.: Gas-particle partitioning of atmospheric aerosols: interplay of physical state, non-ideal mixing and morphology, Phys. Chem. Chem. Phys., 15, 11441–11453, https://doi.org/10.1039/c3cp51595h, 2013b.
Shiraiwa, M., Ueda, K., Pozzer, A., Lammel, G., Kampf, C. J., Fushimi, A., Enami, S., Arangio, A. M., Frohlich-Nowoisky, J., Fujitani, Y., Furuyama, A., Lakey, P. S. J., Lelieveld, J., Lucas, K., Morino, Y., Poschl, U., Takaharna, S., Takami, A., Tong, H. J., Weber, B., Yoshino, A., and Sato, K.: Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 51, 13545–13567, https://doi.org/10.1021/acs.est.7b04417, 2017.
Shrivastava, M., Cappa, C. D., Fan, J. W., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Stangl, C. M., Krasnomowitz, J. M., Apsokardu, M. J., Tiszenkel, L., Ouyang, Q., Lee, S., and Johnston, M. V.: Sulfur Dioxide Modifies Aerosol Particle Formation and Growth by Ozonolysis of Monoterpenes and Isoprene, J. Geophys. Res.-Atmos., 124, 4800–4811, https://doi.org/10.1029/2018jd030064, 2019.
Stocker, T. F., Qin, D. H., Plattner, G. K., Alexander, L. V., Allen, S. K., Bindoff, N. L., Breon, F. M., Church, J. A., Cubasch, U., Emori, S., Forster, P., Friedlingstein, P., Gillett, N., Gregory, J. M., Hartmann, D. L., Jansen, E., Kirtman, B., Knutti, R., Kanikicharla, K. K., Lemke, P., Marotzke, J., Masson-Delmotte, V., Meehl, G. A., Mokhov, I., Piao, S. L., Ramaswamy, V., Randall, D., Rhein, M., Rojas, M., Sabine, C., Shindell, D., Talley, L. D., Vaughan, D. G., and Xie, S. P.: Climate Change 2013 – The Physical Science Basis, Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, USA, 1552 pp., 2014.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 41, 517–527, https://doi.org/10.1021/es062081q, 2007.
Surratt, J. D., Gomez-Gonzalez, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate formation in biogenic secondary organic aerosol, J. Phys. Chem. A, 112, 8345–8378, https://doi.org/10.1021/jp802310p, 2008.
Takahama, S., Davidson, C. I., and Pandis, S. N.: Semicontinuous measurements of organic carbon and acidity during the Pittsburgh air quality study: Implications for acid-catalyzed organic aerosol formation, Environ. Sci. Technol., 40, 2191–2199, https://doi.org/10.1021/es050856+, 2006.
Tang, I. N. and Munkelwitz, H. R.: Aerosol growth studies – III ammonium bisulfate aerosols in a moist atmosphere, J. Aerosol Sci., 8, 321–330, https://doi.org/10.1016/0021-8502(77)90019-2, 1977.
Tanner, R. L., Olszyna, K. J., Edgerton, E. S., Knipping, E., and Shaw, S. L.: Searching for evidence of acid-catalyzed enhancement of secondary organic aerosol formation using ambient aerosol data, Atmos. Environ., 43, 3440–3444, https://doi.org/10.1016/j.atmosenv.2009.03.045, 2009.
Tao, S., Lu, X., Levac, N., Bateman, A. P., Nguyen, T. B., Bones, D. L., Nizkorodov, S. A., Laskin, J., Laskin, A., and Yang, X.: Molecular Characterization of Organosulfates in Organic Aerosols from Shanghai and Los Angeles Urban Areas by Nanospray-Desorption Electrospray Ionization High-Resolution Mass Spectrometry, Environ. Sci. Technol., 48, 10993–11001, https://doi.org/10.1021/es5024674, 2014.
Tilmes, S., Hodzic, A., Emmons, L. K., Mills, M. J., Gettelman, A., Kinnison, D. E., Park, M., Lamarque, J. F., Vitt, F., Shrivastava, M., Campuzano-Jost, P., Jimenez, J. L., and Liu, X.: Climate Forcing and Trends of Organic Aerosols in the Community Earth System Model (CESM2), J. Adv. Model. Earth Sy., 11, 4323–4351, https://doi.org/10.1029/2019ms001827, 2019.
von Hessberg, C., von Hessberg, P., Pöschl, U., Bilde, M., Nielsen, O. J., and Moortgat, G. K.: Temperature and humidity dependence of secondary organic aerosol yield from the ozonolysis of β-pinene, Atmos. Chem. Phys., 9, 3583–3599, https://doi.org/10.5194/acp-9-3583-2009, 2009.
Wang, J., Doussin, J. F., Perrier, S., Perraudin, E., Katrib, Y., Pangui, E., and Picquet-Varrault, B.: Design of a new multi-phase experimental simulation chamber for atmospheric photosmog, aerosol and cloud chemistry research, Atmos. Meas. Tech., 4, 2465–2494, https://doi.org/10.5194/amt-4-2465-2011, 2011.
Wang, N., Jorga, S. D., Pierce, J. R., Donahue, N. M., and Pandis, S. N.: Particle wall-loss correction methods in smog chamber experiments, Atmos. Meas. Tech., 11, 6577–6588, https://doi.org/10.5194/amt-11-6577-2018, 2018a.
Wang, S. Y., Zhou, S. M., Tao, Y., Tsui, W. G., Ye, J. H., Yu, J. Z., Murphy, J. G., McNeill, V. F., Abbatt, J. P. D., and Chan, A. W. H.: Organic Peroxides and Sulfur Dioxide in Aerosol: Source of Particulate Sulfate, Environ. Sci. Technol., 53, 10695–10704, https://doi.org/10.1021/acs.est.9b02591, 2019.
Wang, X., Liu, T., Bernard, F., Ding, X., Wen, S., Zhang, Y., Zhang, Z., He, Q., Lü, S., Chen, J., Saunders, S., and Yu, J.: Design and characterization of a smog chamber for studying gas-phase chemical mechanisms and aerosol formation, Atmos. Meas. Tech., 7, 301–313, https://doi.org/10.5194/amt-7-301-2014, 2014.
Wang, X. K., Rossignol, S., Ma, Y., Yao, L., Wang, M. Y., Chen, J. M., George, C., and Wang, L.: Molecular characterization of atmospheric particulate organosulfates in three megacities at the middle and lower reaches of the Yangtze River, Atmos. Chem. Phys., 16, 2285–2298, https://doi.org/10.5194/acp-16-2285-2016, 2016.
Wang, Y., Hu, M., Guo, S., Wang, Y., Zheng, J., Yang, Y., Zhu, W., Tang, R., Li, X., Liu, Y., Le Breton, M., Du, Z., Shang, D., Wu, Y., Wu, Z., Song, Y., Lou, S., Hallquist, M., and Yu, J.: The secondary formation of organosulfates under interactions between biogenic emissions and anthropogenic pollutants in summer in Beijing, Atmos. Chem. Phys., 18, 10693–10713, https://doi.org/10.5194/acp-18-10693-2018, 2018b.
Yasmeen, F., Vermeylen, R., Szmigielski, R., Iinuma, Y., Böge, O., Herrmann, H., Maenhaut, W., and Claeys, M.: Terpenylic acid and related compounds: precursors for dimers in secondary organic aerosol from the ozonolysis of α- and β-pinene, Atmos. Chem. Phys., 10, 9383–9392, https://doi.org/10.5194/acp-10-9383-2010, 2010.
Yassine, M. M., Dabek-Zlotorzynska, E., Harir, M., and Schmitt-Kopplin, P.: Identification of Weak and Strong Organic Acids in Atmospheric Aerosols by Capillary Electrophoresis/Mass Spectrometry and Ultra-High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, Anal. Chem., 84, 6586–6594, https://doi.org/10.1021/ac300798g, 2012.
Ye, J., Abbatt, J. P. D., and Chan, A. W. H.: Novel pathway of SO2 oxidation in the atmosphere: reactions with monoterpene ozonolysis intermediates and secondary organic aerosol, Atmos. Chem. Phys., 18, 5549–5565, https://doi.org/10.5194/acp-18-5549-2018, 2018.
Yu, J. Z., Cocker, D. R., Griffin, R. J., Flagan, R. C., and Seinfeld, J. H.: Gas-phase ozone oxidation of monoterpenes: Gaseous and particulate products, J. Atmos. Chem., 34, 207–258, https://doi.org/10.1023/a:1006254930583, 1999.
Zhang, Q., Jimenez, J. L., Worsnop, D. R., and Canagaratna, M.: A case study of urban particle acidity and its influence on secondary organic aerosol, Environ. Sci. Technol., 41, 3213–3219, https://doi.org/10.1021/es061812j, 2007.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhang, X., McVay, R. C., Huang, D. D., Dalleska, N. F., Aumont, B., Flagan, R. C., and Seinfeld, J. H.: Formation and evolution of molecular products in α-pinene secondary organic aerosol, P. Natl. Acad. Sci. USA, 112, 14168–14173, https://doi.org/10.1073/pnas.1517742112, 2015.
Zhang, X., Lambe, A. T., Upshur, M. A., Brooks, W. A., Be, A. G., Thomson, R. J., Geiger, F. M., Surratt, J. D., Zhang, Z. F., Gold, A., Graf, S., Cubison, M. J., Groessl, M., Jayne, J. T., Worsnop, D. R., and Canagaratna, M. R.: Highly Oxygenated Multifunctional Compounds in α-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 51, 5932–5940, https://doi.org/10.1021/acs.est.6b06588, 2017.
Zhao, R., Kenseth, C. M., Huang, Y. L., Dalleska, N. F., Kuang, X. B. M., Chen, J. R., Paulson, S. E., and Seinfeld, J. H.: Rapid Aqueous-Phase Hydrolysis of Ester Hydroperoxides Arising from Criegee Intermediates and Organic Acids, J. Phys. Chem. A, 122, 5190–5201, https://doi.org/10.1021/acs.jpca.8b02195, 2018.
Zhou, S. M., Shiraiwa, M., McWhinney, R. D., Poschl, U., and Abbatt, J. P. D.: Kinetic limitations in gas-particle reactions arising from slow diffusion in secondary organic aerosol, Faraday Discuss., 165, 391–406, https://doi.org/10.1039/c3fd00030c, 2013.
Zhou, S. Z., Wang, Z., Gao, R., Xue, L. K., Yuan, C., Wang, T., Gao, X. M., Wang, X. F., Nie, W., Xu, Z., Zhang, Q. Z., and Wang, W. X.: Formation of secondary organic carbon and long-range transport of carbonaceous aerosols at Mount Heng in South China, Atmos. Environ., 63, 203–212, https://doi.org/10.1016/j.atmosenv.2012.09.021, 2012.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.