the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Mar 2021
| 05 Mar 2021
Chemical characterisation of benzene oxidation products under high- and low-NOx conditions using chemical ionisation mass spectrometry
Michael Priestley
Thomas J. Bannan
Michael Le Breton
Stephen D. Worrall
Sungah Kang
Iida Pullinen
Sebastian Schmitt
Ralf Tillmann
Einhard Kleist
Defeng Zhao
Jürgen Wildt
Olga Garmash
Archit Mehra
Asan Bacak
Dudley E. Shallcross
Astrid Kiendler-Scharr
Åsa M. Hallquist
Mikael Ehn
Carl J. Percival
Mattias Hallquist
Thomas F. Mentel
Gordon McFiggans
Aromatic hydrocarbons are a class of volatile organic compounds associated with anthropogenic activity and make up a significant fraction of urban volatile organic compound (VOC) emissions that contribute to the formation of secondary organic aerosol (SOA). Benzene is one of the most abundant species emitted from vehicles, biomass burning and industry. An iodide time-of-flight chemical ionisation mass spectrometer (ToF-CIMS) and nitrate ToF-CIMS were deployed at the Jülich Plant Atmosphere Chamber as part of a series of experiments examining benzene oxidation by OH under high- and low-NOx conditions, where a range of organic oxidation products were detected. The nitrate scheme detects many oxidation products with high masses, ranging from intermediate volatile organic compounds (IVOCs) to extremely low volatile organic compounds (ELVOCs), including C12 dimers. In comparison, very few species with C≥6 and O≥8 were detected with the iodide scheme, which detected many more IVOCs and semi-volatile organic compounds (SVOCs) but very few ELVOCs and low volatile organic compounds (LVOCs). A total of 132 and 195 CHO and CHON oxidation products are detected by the iodide ToF-CIMS in the low- and high-NOx experiments respectively. Ring-breaking products make up the dominant fraction of detected signal and 21 and 26 of the products listed in the Master Chemical Mechanism (MCM) were detected. The time series of highly oxidised (O≥6) and ring-retaining oxidation products (C6 and double-bond equivalent = 4) equilibrate quickly, characterised by a square form profile, compared to MCM and ring-breaking products which increase throughout oxidation, exhibiting sawtooth profiles. Under low-NOx conditions, all CHO formulae attributed to radical termination reactions of first-generation benzene products, and first-generation auto-oxidation products are observed. Several N-containing species that are either first-generation benzene products or first-generation auto-oxidation products are also observed under high-NOx conditions. Hierarchical cluster analysis finds four clusters, of which two describe photo-oxidation. Cluster 2 shows a negative dependency on the ratio, indicating it is sensitive to NO concentration and thus likely to contain NO addition products and alkoxy-derived termination products. This cluster has the highest average carbon oxidation state ( and the lowest average carbon number. Where nitrogen is present in a cluster member of cluster 2, the oxygen number is even, as expected for alkoxy-derived products. In contrast, cluster 1 shows no dependency on the ratio and so is likely to contain more NO2 addition and peroxy-derived termination products. This cluster contains fewer fragmented species, as the average carbon number is higher and lower than cluster 2, and more species with an odd number of oxygen atoms. This suggests that clustering of time series which have features pertaining to distinct chemical regimes, for example, perturbations, coupled with a priori knowledge, can provide insight into identification of potential functionality.
- Article
(1040 KB) - Full-text XML
-
Supplement
(746 KB) - BibTeX
- EndNote





Benzene is an aromatic volatile organic compound (VOC) commonly used as a vehicular fuel additive (Verma and Des Tombe, 2002) and as a chemical intermediate in the manufacture of a range of products, e.g. detergents (Oyoroko and Ogamba, 2017), lubricants (Rodriguez et al., 2018), dyes (Guo et al., 2018), and pesticides (Wang et al., 2014). Whilst current global estimates of benzenoid (molecules containing a benzene ring) emission to the atmosphere by vegetation are of a comparable order to those of anthropogenic activity (∼ 5 times lower; Misztal et al., 2015), and background concentrations of benzene are enhanced by increased biomass burning (Archibald et al., 2015), emission of benzene to the urban atmosphere is dominated by vehicle exhausts (Gentner et al., 2012) and solvent evaporation (Hoyt and Raun, 2015). As well as a toxin (Snyder et al., 1975) and carcinogen (Bird et al., 2010; Ruchirawat et al., 2005, 2007), benzene is photochemically active and contributes to the formation of ozone (O3) and secondary organic aerosol (SOA), both of which act to modify the climate and contribute to poor air quality (Henze et al., 2008; Ng et al., 2007). SOA formation from benzene has previously been quantified with a focus on the contribution from smaller mass, ring-breaking reaction products such as epoxides (Glowacki et al., 2009) and dicarbonyl aldehydes (e.g. O'Brien et al., 1983; Johnson et al., 2005). For example, 37 % of the glyoxal formed in the LA basin is estimated to derive from aromatic sources (Knote et al., 2014). These smaller ring-breaking products typically make up a larger fraction of SOA mass than the ring-retaining products (Borrás and Tortajada-Genaro, 2012) and so have traditionally been the main focus for SOA quantification. Other major, toxic benzene oxidation products such as catechol, nitrophenol and maleic anhydride are also known components of SOA (Borrás and Tortajada-Genaro, 2012).
More recently the auto-oxidation mechanism has been demonstrated to occur in aromatic systems (Wang et al., 2017, 2020; Molteni et al., 2018; Tsiligiannis et al., 2019; Garmash et al., 2020; Mehra et al., 2020), producing highly oxygenated organic molecules (HOMs, defined as containing 6 or more oxygen atoms which is a product of the auto-oxidation mechanism) and incorporating up to 11 oxygen atoms (Bianchi et al., 2019). The auto-oxidation mechanism of intra-molecular H shifts from the carbon backbone to the peroxy radical centre forming peroxide groups is consistent with other VOC systems (Rissanen et al., 2014) initiated by a variety of different oxidants (e.g. Mentel et al., 2015; Berndt et al., 2016). The inclusion of an alkyl group to a benzene ring facilitates HOM formation (Wang et al., 2017) as a consequence of the greater degrees of freedom afforded to the molecule. Whilst benzene is not the most reactive of aromatic VOCs, τOH≈9.5 d for benzene vs. τOH≈6–10 h for xylenes ([OH] = 2.0 × 106 molecules cm−3; Atkinson and Arey, 2007), it is ubiquitous in the urban environment and is the simplest C6 aromatic ring system to study.
Oxidation of benzene occurs nearly exclusively via hydroxyl radical (OH) addition to form the cyclohexadienyl radical (benzene-OH adduct), which subsequently adds O2 to form the hydroxycyclohexadiene peroxy radical (C6H6–OH–O2) (Volkamer et al., 2002) (Fig. 1). Two subsequent reaction pathways are postulated for this peroxy radical: either elimination of HO2 yields phenol, and secondary OH attack must occur again, or an endocyclic di-oxygen-bridged carbon-centred radical intermediate is formed by the addition of the peroxy group to (typically) a β-carbon (Glowacki et al., 2009). This di-oxygen-bridged carbon-centred radical may add another O2 and form a peroxy radical (named as BZBIPERO2 in the Master Chemical Mechanism, MCM; Saunders et al., 2003). However it is not known if auto-oxidation may continue to form a second oxygen-bridged radical, described as type II auto-oxidation (Molteni et al., 2018; Fig. 1, BZBIPERO2-diB), or form a hydroperoxide carbon-centred radical, formed through intra-molecular hydrogen abstraction (type I auto-oxidation). At each step, termination of the peroxy radical to hydroxyl, hydroperoxyl, nitrate, or peroxy nitrate or reduction to an alkoxy radical is possible, from which further termination via formation of a nitrite, nitro, or acyl group can occur. The dominant fate of this alkoxy radical is ring-breaking, which can further form a variety of SOA-relevant compounds such as HOMs, glyoxal and various C4 and C5 compounds (Xu et al., 2020).
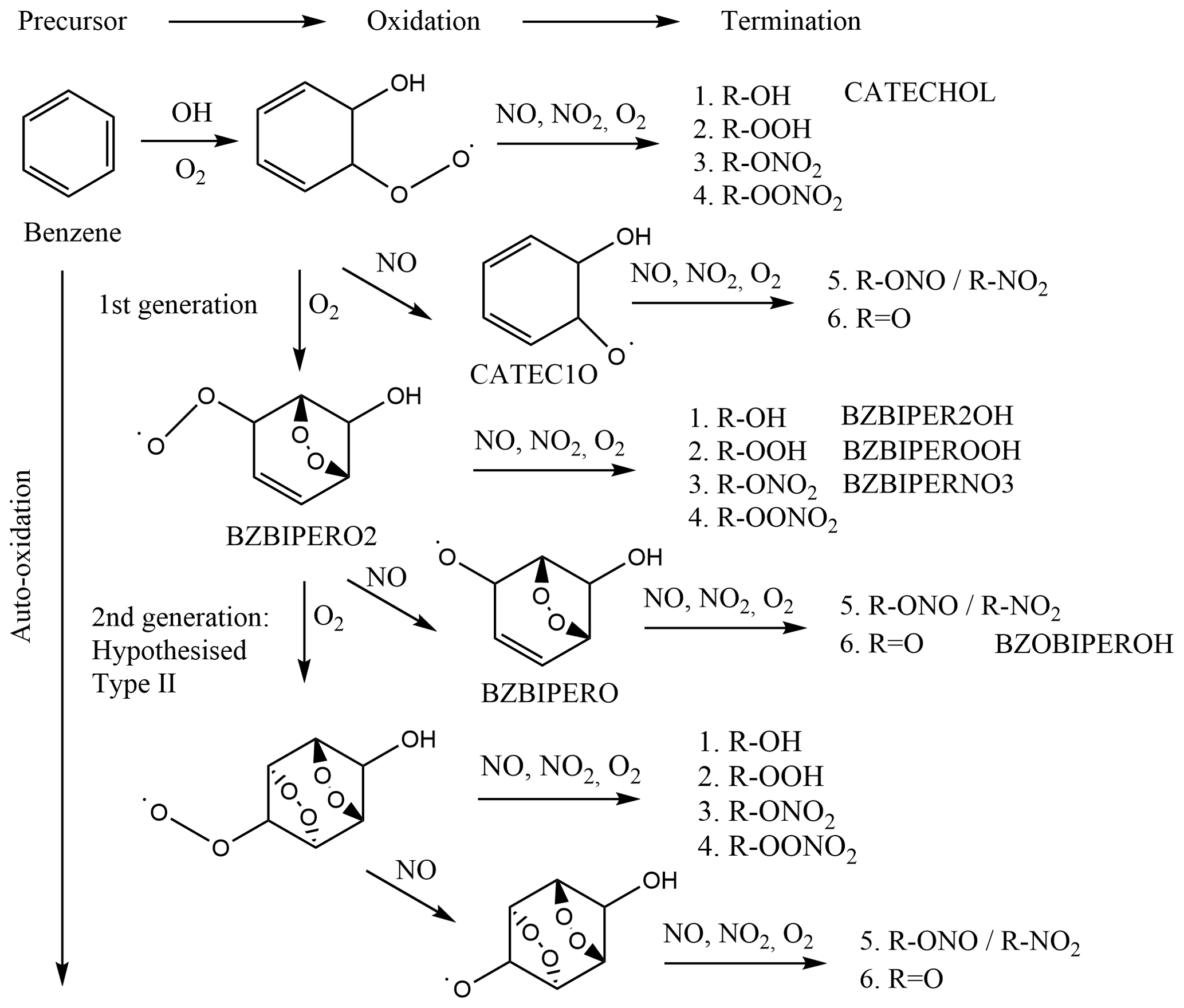
Figure 1Reaction scheme of benzene oxidation by OH with proposed di-bridged species, adapted from Molteni et al. (2018). Species present in the MCM are labelled.
Typically the presence of NOx alters the product distribution of VOC oxidation and reduces SOA formation (e.g. Stirnweis et al., 2017). With other atmospherically relevant VOC precursors of SOA, e.g. isoprene or monoterpenes, high-NO conditions can suppress SOA formation (Sarrafzadeh et al., 2016; Wildt et al., 2014) as reduction of RO2 to RO ultimately leads to fragmentation of the RO species (Nguyen et al., 2015; Surratt et al., 2006), but it can also form epoxides, aldehydes, and hydroperoxides, which readily partition to the aerosol phase and contribute to SOA growth (Surratt et al., 2010).
The further reaction of NOx with oxygenated VOCs produces species including nitro organics, nitrates, peroxy nitrates, and peroxyacyl nitrates (Atkinson, 2000) that can also condense and contribute to SOA formation. Under conditions where NOx is present in moderate amounts, the HO2 : OH ratio is low as HO2 is reduced by NO to form OH. This allows more OH oxidation to occur and less termination of RO2 by HO2 to occur, whereas under low-NOx conditions VOC consumption is lower, as OH recycling from HO2 relies on the slower HO2+ O3 reaction (Atkinson, 2000). Here low and high NOx are relative terms and are defined by the available VOCs. This reduction of the HO2 : OH ratio as a function of NO has been observed at Mace Head, Ireland, where clean low-NOx marine-influenced air can be contrasted with polluted NOx-containing continental air (Creasey et al., 2002). In that instance, an increase in NO concentration from 0.01 to 1 ppb reduces the HO2 : OH ratio from 200:1 to 10:1.
The auto-oxidation mechanism is known to form biogenic HOMs and, as a consequence, SOA in ambient rural environments (e.g. Ehn et al., 2014). The mechanism is increasingly a competitive process in urban and suburban environments where NOx concentrations have seen significant reductions in recent years (Praske et al., 2018). These low concentrations of NOx (0.1–1 ppb) have been shown to enhance local oxidant production and drive an increase in biogenic HOM formation (Pye et al., 2019). Aromatic HOM formation by OH at relevant urban NOx concentrations has been demonstrated (Garmash et al., 2020; Mehra et al., 2020; Tsiligiannis et al., 2019; Zaytsev et al., 2019).
ToF-CIMS is frequently used to probe VOC oxidation due to the ability to detect many low-concentration compounds simultaneously in real time (e.g. Chhabra et al., 2015). As a result of the sensitivity of the nitrate ionisation scheme towards HOMs, this reagent ion is typically used to study the auto-oxidation mechanism and HOM formation. However, to observe carbon in a diverse range of forms, e.g. different oxidation states, and of different functionalities, multiple ionisation schemes are required as they are sensitive towards different oxygenated volatile organic compounds (OVOCs) (e.g. Isaacman-Vanwertz et al., 2017; Riva et al., 2019). These methods, combined with other measurement techniques, have been demonstrated to enable the complete observation of all the reacted carbon within a system (carbon closure) (Isaacman-Vanwertz et al., 2018).
In this study, the oxidation of benzene and its oxidation products by OH under high- and low-NOx conditions are investigated in the Jülich Plant Atmosphere Chamber (JPAC) (Mentel et al., 2009, 2015) with two time-of-flight chemical ionisation mass spectrometers (ToF-CIMS) using the iodide and nitrate ionisation schemes. The ionisation schemes are compared to assess the similarities in detected oxidation products in terms of molecular identification and bulk properties. Mechanistic investigations to assess the ability of the iodide CIMS to detect species that currently are not accounted for; i.e. HOMs and those described in the MCM are investigated. Additionally, high- and low-NOx conditions are probed. As sensitivities for the iodide CIMS are lacking, an exploration of product descriptions is made using bulk analysis, as well as broad groupings defined by bulk properties. Finally, the application of hierarchical clustering analysis (HCA) is used to provide the basis of a methodology to assess the time series behaviour of oxidation products that can further aid molecular identification, e.g. by inferring potential functionality.
The experiments were performed in a 1450 L borosilicate chamber of the Jülich Plant Atmospheric Chamber (JPAC) described elsewhere (Mentel et al., 2009). The chamber is operated as a continuously stirred tank reactor, with modifications as described in Mentel et al. (2015). A fan made of Teflon provides typical mixing time of less than 2 min. The total flow in and out the chamber was 30 L min−1, resulting in an average residence time in the chamber of ∼ 2900 s or 48 min. Benzene (Merck, ≥99.7 %) was introduced into the chamber by flowing purified ambient air over a diffusion source to maintain a constant concentration and was monitored by recording the inlet and outlet concentrations by a quadrupole proton transfer mass spectrometer (PTR-MS). The PTR-MS (IONICON) was calibrated using a benzene diffusion source (Garmash et al., 2020). The difference between the outlet and inlet concentrations, measured at a 2 min time resolution and switched every 25 min, describes the reacted benzene, from which the OH concentration is calculated. The loss of benzene by photolysis was found to be negligible as measured by Q-PTR during experiments where only benzene was introduced into the chamber, in accordance with observations in the literature (Fally et al., 2009; Kamps et al., 1993).
OH is generated by photolysis of O3 by a UV lamp (λ<254 nm, Philips, TUV 40W, herein termed TUV) and the subsequent O(1D) reaction with H2O. O3 is introduced to the chamber by a separate line to the benzene and was monitored by UV absorption (Model 49, Thermo Environmental instruments). OH concentrations are calculated when steady-state conditions (ss) are reached according to the equation below and are described in detail elsewhere (Garmash et al., 2020). The chamber volume (V=1450 L) divided by the flow rate (F = 30 slm) is the residence time in the chamber (t=2900 s). The rate coefficient for the reaction of benzene with OH, cm3 s−1, was taken from IUPAC (2008). Verification experiments were done by introducing 1,8-cineole into the chamber, which confirmed the calculated OH concentrations to be within 6 %–12 % (Garmash et al., 2020). Integrating the OH concentration over the residence time gives an equivalent OH dosage that may be compared to atmospheric levels. Here OH equivalent doses are equivalent to approximately 5 d of oxidation at a relevant atmospheric OH concentration (106 molecules cm−3).
For the high-NOx experiment, NO (Linde, 120 ± 5 ppm NO in N2) was injected into the chamber via a separate perfluoroalkoxy alkane (PFA) line. The NO input of 20 ppb was measured by chemiluminescence (CLD 770 and PLC 760, Eco Physics). A total of 12 discharge lamps termed UVA (HQI, 400 W/D; Osram, Munich, Germany) were activated to photolyse NO2 ( s−1) to increase the ratio, although NO2 remains the dominant NOx compound. More than half of the NOx is consumed by reaction of NO2 with OH and organic nitrate formation. About 10 % of the remaining NOx exists as NO for the given O3 and . Considering reactions of NO with RO2, this fraction is even less. At the end of the experiment, the TUV lamp was kept on and the UVA lamps switched off. Temperature and humidity were maintained at 288 ± 1 K and 67 ± 2 % through the experiments. The experimental conditions are summarised in Table 1. It is expected that as the NOx concentration decreases, peroxy radical lifetime is much longer than the required timescale for auto-oxidation hydrogen shifts (Praske et al., 2018), and so auto-oxidation products can be expected in this low-NOx case here (≤300 pptv). Where the UVA lamps are active, the NO NOx ratio is increased, thus increasing the ratio of RO ROx (RO + RO2). The high-NOx condition for the iodide ToF-CIMS comparison (Sect. 3.3) is performed when s−1.
Table 1Summary of selected experiments performed at JPAC used in this study. Two photolysis states are reported for the NOx-containing studies. The first describes steady-state concentrations with (UVA) and ultraviolet (TUV) light on, the second with only the (TUV) light on. OH equivalent dose is based on 106 OH molecules cm−3. * NO2, background from VOC catalyser. ** not measured, estimated from experiment with similar NOx addition, O3 and OH.

2.1 ToF-CIMS instrumentation
2.1.1 Iodide
The University of Manchester ToF-CIMS has been described by Priestley et al. (2018). The iodide ToF-CIMS ion molecule reaction region (IMR) was held at a constant pressure of 100 mbar by a scroll pump (Agilent SH-112) controlled with a servo control valve placed between the scroll pump and the IMR. The short segmented quadrupole (SSQ) region was held at 1.30 mbar by a second scroll pump (Triscroll 600). Chamber air was drawn into the IMR via a critical orifice at 2.2 slm, where it mixed with I− ions. The reagent I− ions were created by flowing 10 standard cubic centimetres per minute (sccm) of nitrogen (N2) over a methyl iodide (CH3I) permeation tube made of PFA and held at 40 ∘C. This flow met 2 slm N2 and was flowed through a 210Po 10 mCi alpha-emitting reactive ion source (NRD Inc.). The mass calibration was performed for seven known masses: NO, I−, I−⋅H2O, I−⋅HCOOH, I−⋅HNO3, I, and I, covering a mass range of 62 to 381 . The mass calibration was fitted to a third-order polynomial and was accurate to within 2 ppm, ensuring peak identification was accurate below 20 ppm. The resolution was 3231 at 127 and 3720 at 381 The inlet to the I− ToF-CIMS sampled from the middle of the chamber and was comprised of 50 cm O.D. PFA tubing, giving a residence time of <1.5 s.
Formic acid calibrations and chamber and instrumental backgrounds were taken before every experiment to assess instrument sensitivity changes. Backgrounds were performed by overflowing the CIMS inlet with N2, and calibrations were performed by flowing 50 sccm N2 over a formic acid permeation tube held at 40 ∘C. The formic acid sensitivity was 3.15±0.26 Hz ppt−1 (2σ), measured as a 5 min average normalised to 106 Hz iodide. The background was 2.72 ± 0.66 ppt (2σ), measured as a 5 min average normalised to 106 Hz iodide. The limit of detection (3σ) was 100 ppt. Calibration of all detected masses was not possible, so data are presented either as signal (Hz) from the instrument or as a number of species.
It is not possible to identify a compound from its formula alone. Oxidation is initiated with OH addition to the benzene ring, but fragmentation occurs, and so the OH moiety may be lost; additionally, multiple OH attacks make the counting of oxygen more difficult (Garmash et al., 2020). It is for these reasons that RNOx species are described more generally, and any discussion of compound classes is speculative. Data analysis was performed using Tofware (v3.1.0; Stark et al., 2015).
2.1.2 Nitrate
The Jülich ToF-CIMS with nitrate ionisation is described elsewhere (Garmash et al., 2020). Chamber air was drawn into an Eisele-type inlet at 9 slm, where it merges with a 20 slm sheath flow of nitrogen that contains nitrate ions (NO3) for approximately 200 ms. These ions are generated by flowing 10 sccm dry nitrogen through a wash bottle containing nitric acid (HNO3, Aldrich, 70 %) that passes through a 241Am alpha-emitting source. The inlet flow enters the instrument through a 300 µm diameter aperture, after which the ions are collimated and energetically homogenised through a series of differentially pumped chambers.
Mass calibration is performed on nitrate, its dimer and trimer ions, NO3, NO3⋅HNO3, and NO3⋅(HNO3)2, and hexafluoropentanoic acid (NO3⋅HFPA adduct), providing an accurate mass range up to 326 . The resolution was ∼ 4000 at 125 . The inlet to the instrument sampled from the middle of the chamber by a 1 m O.D stainless steel tube.
2.2 Calculation of average carbon oxidation state ()
Average carbon oxidation state (Kroll et al., 2011) is commonly used to describe the degree of oxidation within a complex oxidation reaction. It is calculated using the oxygen to carbon ratio (O:C), hydrogen to carbon ratio (H:C) and the nitrogen to carbon ratio (N:C) and assumes nitrogen is in the form of nitrate where the oxidation state of N (OSN) is +5:
It is more difficult to justify this assumption for gas-phase data, and as this work explores different N-containing functional groups, this nitrate assumption cannot be used. Instead of assuming all N is in the form of nitrate, a range of OSN (5+, 3+ and 0) are considered, and is calculated as a range from a theoretical minimum, where all N is considered nitrate (5+), to a theoretical maximum, where all N is considered cyanate (0). Nitro and nitroso compounds are also considered (3+), but amines and amides are not as they are not expected to be present in the system. As neither the maximum or minimum OSN is a likely or an accurate description of the system, they merely represent theoretical limits to calculate ; the true value will lie somewhere in the middle, in all likelihood between the mean and lower limit as most species are expected not to be cyanates. The reported here is the mean average of three scenarios, where all N is considered to exist in 0, 3+, or 5+ oxidation states. This methodology reduces the accuracy of the reported , but it clearly and accurately presents the range.
In this document is reported as a function of carbon number. The variation in between all CHON of the same carbon number is much greater than the variability in of a single CHON compound as a consequence of varying OSN for that compound.
Where a species has the form RNO1,2, the species is considered to be either a cyanate or hydroxy cyanate, and the OSN is set to 0. For RNO3 species the nitrogen is considered to be either a dihydroxy cyanate or a hydroxy RONO or RNO2 compound, and so OSN is set to either 0 or 3+. RNO>3 have OSN set to 0, 3+, or 5+. This leads to the modification of the parameterisation:
2.2.1 Hierarchical cluster analysis (HCA)
Hierarchical cluster analysis (HCA) is an analytical technique used to simplify datasets containing large numbers of individual observations into groups or clusters defined by their similarity, with the aim of increasing interpretability of the dataset. HCA is an agglomerative technique which allows for the successive merging of individuals or clusters to form larger clusters. Due to the complex nature of atmospheric oxidation and the detection of many oxidation products by mass spectrometry, it is common practice to reduce the dimensionality of mass spectrometry datasets in order to better describe bulk properties of the process being studied (Buchholz et al., 2020; Isokääntä et al., 2020; Koss et al., 2020; Sekimoto et al., 2018), with HCA being one such method. HCA is independent of calibration as it relies on relative differences between time series rather than exact concentrations, making it a suitable technique to apply to mass spectrometry data, where authentic standards do not exist, and individual calibrations are next to impossible. The final number of clusters is selected based on the distance between them and is determined by the user. Here we use HCA to group the time series of benzene oxidation products to investigate its utility as a tool for investigating oxidation processes or product properties where the reaction occurs in a continually stirred flow reactor.
In order to use HCA, the linkage criterion and distance metric must be selected. The root of the sum of the squares of a pair of observations (time series), A and B (i.e. the Euclidean distance), is chosen as the distance metric to define the similarity of the time series at each time step, t.
The Ward linkage criterion was chosen to determine the distance between sets of observations (U, V, which can be single or multi-member clusters) as it gave similar or more interpretable results compared with other linkage criteria. Here, the sum of the squares of the cluster members (xi, where i iterates over the members U, V, or their agglomeration) from their cluster means (mU, mV, or mU∪V) is calculated. This is performed for the two candidate clusters for agglomerating (U, V) and for the new cluster they would form through agglomeration (U∪V). The difference in the sum of the squares between this potential merged cluster and the initial two candidate clusters is the distance used to assess whether the agglomeration of the candidate clusters is acceptable:
When the increase in the sum of the squares (i.e. distances) is minimal after agglomerating the two candidate clusters, the new cluster is formed. Here, the time series for all CHO and CHON compounds from the iodide ToF-CIMS measurements were selected for clustering. Time series were scaled between 0–1 to remove the effect of their differing magnitudes and focus on the changes in trends. The scaled time series were then smoothed to a 10 min average to remove noise that may affect the results of the HCA. This treatment is similar to other studies using HCA with mass spectrometric data (e.g. Koss et al., 2020). The analysis was implemented using the cluster.hierarchy module from SciPy (1.2.0) scientific Python library using Python 3.6.
We detect a range of benzene oxidation products, including many with the same formula as those listed in the MCM, as well as highly oxidised species with both ToF-CIMS instruments. Well-resolved peaks at a given unit mass were frequently observed in the I− spectrum. The separation between the I− ion cluster with its large negative mass defect and the positive defect (excess) of the high-H-containing deprotonated species allows for good peak separation at the resolution of the ToF-CIMS (Fig. 2). Beyond I− and H2O, the largest adduct signals we identified with the I− CIMS were low-mass species: formic acid (CH2O2), nitric acid (HNO3), CH2O3, C3H6O3, HONO, C4H4O3, and C3H8O3. The prevalence of these ions is likely due to instrument sensitivity rather than chemistry.
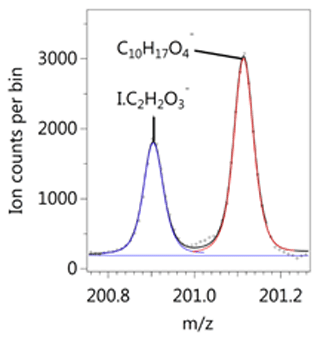
Figure 2Example of peak separation between an iodide adduct I.C2H2O and deprotonated signal C10H17O detected during background measurements before the low-NOx experiment.
3.1 Organic product distributions: comparison of nitrate and iodide ionisation
A total of 42 formulae are found to be common in the low-NOx experiment between the two instruments, representing the overlap of oxidation product signals, of which 20 are C6 compounds and the rest have lower carbon numbers. The time series behaviour of the signals at the start of oxidation is highly variable and is specific to each instrument and ion measured by that instrument as oxidant levels and wall partitioning equilibrates. Typically, the level of highly oxidised species increases significantly from the outset but relaxes to lower levels rapidly before it grows again to reach steady state. This early transient maximum spiking behaviour has been observed in other studies investigating VOC oxidation using the nitrate ToF-CIMS (e.g. Ehn et al., 2014) and is thought to be a consequence of a mixture of two phenomena that could include a latent aerosol sink shifting the equilibrium species from the gas to the aerosol phase as well as the dynamic nature of oxidant concentrations (mainly adaptation of O3) before the steady state is reached and mixing is totally homogenous. The iodide CIMS measures a delayed response from wall equilibration processes (Krechmer et al., 2016; Pagonis et al., 2017), making the detection of these rapid changes more challenging. For the range of species measured here, the variability between the two measurements for the same species varies (Fig. S1). In general the nitrate observes square waveforms, whereas the iodide observes sawtooth waveforms. Where the agreement is particularly bad, it is most likely that different isomers contribute to the different signals measured on the two instruments.
The nitrate ionisation scheme detects a greater number of higher mass species with a higher oxygen content (Fig. 3c, e) and higher (Fig. 3h, 0.84 vs. 0.42), whereas the iodide ionisation scheme detects lower mass compounds with a lower oxygen number and lower , as has been reported elsewhere in different VOC oxidation systems (e.g. Isaacman-VanWertz et al., 2017). The average mass of a nitrate adduct (not including the reagent ion) is ∼ 240 g mol−1, with ∼ 30 % of that attributable to oxygen, whereas the average mass of an iodide adduct is 140 g mol−1, with ∼ 20 % attributable to oxygen (Fig. 3g).

Figure 3Summary of CHO and CHON statistics of detected oxidation products during the high-NOx (20 ppb) benzene oxidation for iodide (purple) and nitrate (green) ionisation schemes. (a, b, c, d) Frequency distributions of the atoms C, H, O, and N for the detected oxidation products. (e) Average number of atoms per detected oxidation product split into C, H, O, and N. (f) Van Krevelen diagram (O:C vs. H:C). (g) The average mass of a detected oxidation product detected by iodide and nitrate ionisation. (h) The average carbon oxidation state of oxidation products detected by iodide and nitrate ionisation. Limits represent minimum and maximum as a function of minimum and maximum OSN; see Sect. 2.3.
The nitrate CIMS is able to detect more high-mass species greater than C6, including a large number of dimers (C12), whereas the iodide scheme did not detect many species greater than C6 in this instance but did measure a greater number of low C molecules compared to the nitrate (Fig. 3a). Few species greater than C12 are detected with either reagent ion, suggesting that in this system their formation was not likely, as both the nitrate and iodide ionisation schemes are capable of detecting > C20 compounds (e.g. Mohr et al., 2019). The iodide scheme detects a maximum number of molecules containing O3 and O4 and very few with more than O7 (Fig. 3c). The nitrate scheme observes a broad range of oxygen numbers, peaking between O9 and O11 but up to O18. Iodide detects an average of ∼ 1.5, whereas nitrate detects of approximately 1.1 due to higher O:C ratios (Fig. 3f). Detection of molecules with greater than C6 that are not C12 dimers are likely formed through peroxy radical cross-reactions.
With both nitrate and iodide ionisation, the maximum variation in oxygen number per molecule is detected for C6 compounds (Fig. 4a), although for nitrate this is closely followed by C12, i.e. C6 dimers as well as C5, C9 and C10 compounds. C5 compounds are shown to form from the ring opening of the di-oxygen-bridged alkoxy radical (Xu et al., 2020) and could subsequently be involved in dimer formation of C9 and C10 compounds.

Figure 4Comparison of organic oxidation products from the iodide (purple) and nitrate (green) ionisation schemes. (a) Carbon number vs. oxygen number. (b) Average carbon oxidation state ( vs. carbon number. (c) Saturation concentration (C*) based on Mohr et al. (2019).
The nitrate CIMS detects approximately half the number of low-oxygen-content (O2−4) (and N-containing) oxidation products compared to the iodide CIMS. Subsequently, as a function of carbon number differs between the ionisation schemes at higher carbon numbers (Fig. 4b). Below C8 the broadly agrees; however above C8 the iodide observes negative , whereas nitrate observes positive values. This is likely due to the detection of different isomers, which become more prevalent at these higher masses.
The saturation concentrations (C*) of the detected species were calculated using the relation of Mohr et al. (2019). The C* indicates that there is significant overlap of semi-volatile organic compound (SVOC) and intermediate volatile organic compound (IVOC) detection between the two instruments (Fig. 4c). Whilst the iodide ionisation scheme detected HOMs, it did not detect any ELVOCs (extremely low volatile compounds) and very few LVOCs (low volatile compounds), detecting instead mainly IVOCs, SVOCs, and some VOCs (Fig. 4c). This is in contrast to the nitrate scheme which detected mainly ELVOCs, SVOCs, and some IVOCs, although fewer than the iodide. This highlights that sampling with both nitrate and iodide schemes captures a large range of gas-phase oxidation products, as has been shown in other systems (e.g. Riva et al., 2019).
3.2 Mechanistic investigation
Nitrate CIMS is routinely used to detect HOMs, but iodide CIMS is also able to detect high-mass compounds (e.g. Mohr et al., 2019). To test the extent to which the iodide CIMS is able to detect HOMs that could potentially originate from auto-oxidative processes, a number of theoretically suggested formulae based on the auto-oxidation mechanism with propagation and termination steps were devised and then searched for within the spectra. The applied mechanism includes the benzene oxidation scheme from the MCM, with two additional auto-oxidation steps added in. It consists of auto-oxidative intra-molecular hydrogen shifts from the carbon backbone to a peroxy radical group forming a hydroperoxide group, followed by O2 addition to form a new peroxy radical (Fig. 1). The peroxy radical groups were terminated to hydroxyl, peroxyl, nitrate, or peroxy nitrate groups or reduced to form the alkoxy equivalent, which then terminates to a carbonyl group or a nitro group (isomeric with nitrite). This was repeated for phenol and catechol precursors as products of subsequent OH attack (Garmash et al., 2020). To reduce complexity, this subsequent OH attack is only considered at the beginning of the mechanism. This provides a total of 53 individual oxidation products with 21 unique formula. All the suggestions of potential mechanistic routes to formation are speculative and set against the mechanistic paradigm laid out here.
We observe 14 and 21 individual deprotonated and adduct signals in the low- and high-NOx spectra that correspond to 9 and 11 unique formula respectively. These 9 and 11 unique formula correspond to a maximum of 27 and 33 individual oxidation products (Table S1). At least one signal (adduct or deprotonated) for all theoretic CHO products is observed in the low-NOx case apart from C6H8O9 and C6H8O10, although these are observed in the nitrate spectrum. No derived CHON products are observed in the low-NOx case, indicating that the incorporation of nitrogen into compounds observed under these conditions occurs at a later point. All three are found in the low-NOx case, and C6H8O7 and C6H8O8 are found in the high-NOx case. C6H8O8 is an exclusively second-generation auto-oxidation product that has been observed previously in benzene oxidation studies (Molteni et al., 2018). C6H8O9 and C6H8O10 are not observed in the high-NOx case. C6H7NO4, C6H7NO6, and C6H7NO7 are the only NOx-containing species observed in the high-NOx case. The formation of C6H7NO4 has two mechanistic routes, the nitration of the initial hydroxy benzene peroxy radical or the addition of a peroxy nitrate group to the phenol, whereas C6H7NO6 and C6H7NO7 have numerous routes to formation, with the only non-auto-oxidation route being the nitration of phenol and catechol respectively. It should be noted that these species are identified in Xu et al. (2020) as products of an epoxidation channel of the alkoxy phenol radical. Exclusively second-generation high-oxygen-content CHON species C6H7NO8, C6H7NO9, C6H7NO10, and C6H7NO11 are not observed with iodide but are observed with nitrate. Figure 5 summarises these ions in the mass spectra.

Figure 5Unit mass spectra of (a) low-NOx and (b) high-NOx experiments from the iodide CIMS. Detected ions from the mechanism are highlighted as CHO (blue) or CHON (red).
HOMs and auto-oxidation are currently not a common inclusion in chemical box models, although some mechanisms are available (e.g. Weber et al., 2020). Due to its usage and optimisation for HOM detection, the nitrate CIMS does not typically provide insight into traditional oxidation chemistry, as its sensitivity to low-molecular-weight species is poor. In order to assess the traditional oxidation chemistry of this system, we describe which oxidation products from the MCM (Bloss et al., 2005; Jenkin et al., 2003) are detectable by the iodide CIMS.
MCM products
The MCM lists 85 multigenerational oxidation products of benzene, including important intermediate and radical species (Bloss et al., 2005; Jenkin et al., 2003). The iodide ToF-CIMS detects 19 (∼ 25 %) and 26 (∼ 30 %) of these 85 oxidation products under low- and high-NOx conditions (Fig. 6). These signals are a mixture of adducts and non-adduct peaks that match the exact masses of the MCM oxidation products to within 20 ppm error. Whilst only 19 and 26 of the species with a formula cited in the MCM are detected, signals increase for 132 and 195 peaks in the mass spectrum under low- and high-NOx conditions respectively, indicating that many more products are formed than are explicitly accounted for. The distributions of the number of detected C, H, O, and N broadly follow the available MCM C, H, O, and N; for example, the number of C3 and C5 compounds in this reaction scheme are much fewer than C, which is reflected proportionally in the number of detected species. This suggests that distributed over the entire mechanism, there is no preference for detection of any specific CHON configurations. A list of all detected species can be found in Table S2.
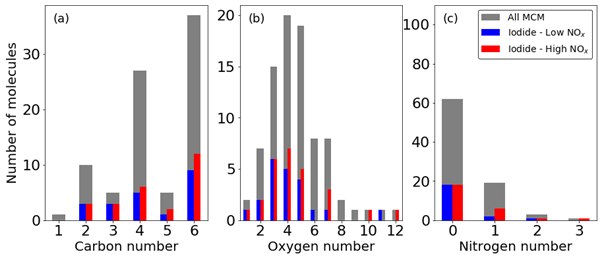
Figure 6Fraction of ions observed from the MCM in the high- and low-NOx experiments expressed as the number of molecules containing atom numbers for (a) carbon, (b) oxygen, and (c) nitrogen.
Without calibration, much of the oxidation product descriptions must be either tentative, when making direct identifications, or broad, and describe only bulk properties. In the absence of direct calibration, we describe the bulk properties of iodide-CIMS-detected CHO and CHON compounds and group them according to chemical composition criteria in order to present an overall depiction of all detectable oxidation products.
3.3 Iodide-CIMS-detected CHO and CHON under low- and high-NOx conditions
A total of 132 and 195 adduct and deprotonated peaks were identified as CHO or CHON in the low- and high-NOx experiments respectively using the iodide CIMS, of which 126 are common between the experiments (Fig. 7). The number of detected compounds is much higher in the high-NOx case compared with the low-NOx case. This is expected as the complexity of the system increases, and additional reaction pathways are viable. This is especially true regarding the detection of N-containing compounds. As the chamber had a NOx background of ∼ 300 ppt, formation of N-containing compounds still occurred during the low-NOx experiment. A total of 24 N-containing products were detected during the low-NOx experiment compared with 70 detected during the high-NOx experiment (20 ppb NOx), of which 23 were common.
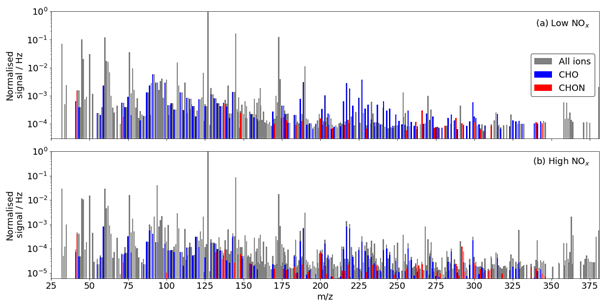
Figure 7(a) Unit mass spectrum during benzene oxidation by OH under (a) low-NOx conditions and (b) under high-NOx conditions from the iodide CIMS. Identified CHO and CHON compounds are highlighted.
The majority of species detected are C6 and C4 compounds (Fig. 8a) in both cases, with O3−4 also most abundant. The atom distributions (Fig. 8a–d) appear very similar, with the largest difference being the number of detected N-containing compounds (Fig. 8d), where more than twice as many are detected in the high-NOx case. The average carbon and hydrogen atom number for the detected species are marginally higher for the low-NOx case than for the high-NOx case (Fig. 8e). This observation is further enhanced when weighting the average composition by signal rather than merely counting detected species. This is consistent with greater fragmentation occurring under higher NOx conditions and is also reflected in the average masses of the reaction products when signal-weighted (Fig. 8g). The O:C and H:C ratios are very similar between the two experiments (H:C and O:C compared with H:C and O: C; Fig. 8h), and this is reflected in , which is marginally higher in the high-NOx case (0.36 vs. 0.42), although the range of possible for both experiments has significant overlap (0.31–0.42 for the low-NOx case and 0.28–0.60 for the high-NOx case; Fig. 8h). The small differences in oxidation product characteristics between the two NOx conditions indicate bulk analysis of all oxidation products is not sensitive enough. In order to better understand the differences between oxidation products from the different NOx cases, the ions are grouped by preference to formation under high- or low-NOx conditions (defined as the regime in which signal enhancement is greatest) and re-analysed (Fig. S2). Despite having a very similar average mass, those products that preferentially formed under high-NOx conditions have a lower compared to the low-NOx conditions, where .
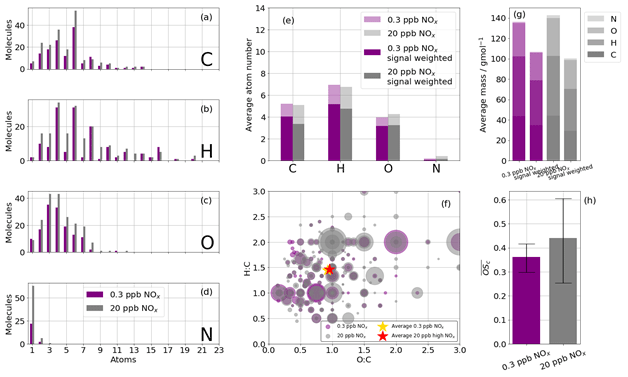
Figure 8Summary of CHO and CHON statistics of detected oxidation products during high-NOx (20 ppb, grey) and low-NOx (0.3 ppb, purple) benzene oxidation with the iodide ionisation scheme. (a, b, c, d) Frequency distributions of the atoms C, H, O, and N for the detected oxidation products. (e) The average number of atoms per detected oxidation product split into C, H, O, and N atoms; also shown is the signal-weighted average atom number. (f) Van Krevelen diagram (O:C vs. H:C) sized by signal. (g) The average mass of a detected oxidation product for high- and low-NOx conditions; also shown are the signal-weighted average masses. (f) The mean average carbon oxidation state ( of the detected oxidation products for high- and low-NOx conditions. Limits represent minimum and maximum OSC as a function of minimum and maximum OSN; see Sect. 2.3.
In order to better describe these oxidation products, five different groupings of compounds are used to categorise detected oxidation products. Background species refer to those present in the chamber before oxidation and comprise <0.05 % of total signal. Compounds of formulae that match species described in the benzene oxidation mechanism of the MCM are denoted “MCM” and comprise <∼10 % of signal, whilst highly oxidised products are identified as species containing 6 or more oxygen atoms and comprise <∼0.3 % of signal. These highly oxidised products can include ring-breaking and ring-retaining products. The rest of the products are termed “remainder” and are either ring-retaining or ring-breaking based on the number of carbon atoms present. Where 6 carbon atoms are present and the double-bond equivalent (DBE) equals 4, then the species is ring-retaining, and where these criteria are not met, it is defined as a ring-breaking product (Mehra et al., 2020). Remaining ring-breaking products make up the most of the remaining signal (∼ 90 %), leaving ring-retaining products contributing <∼ 4 %.
MCM and ring-breaking products are observed to grow continually over the time period of oxidation, whereas highly oxidised and ring-retaining products equilibrate and remain flat for the duration of the experiment (Fig. 9). This is also true of the high-NOx experiment, although when NO2 photolysis stops halfway through the oxidation, the growth of signal is diminished and the sawtooth profile less apparent. The MCM and ring-breaking groups contain small, oxidised molecules that are near the end of the oxidative chain, representing large sink of the carbon in the system, and so continually grow. The ring-retaining and highly oxidised species reach an equilibrium quickly as they tend to be earlier generation products, and so their profiles remain flat.
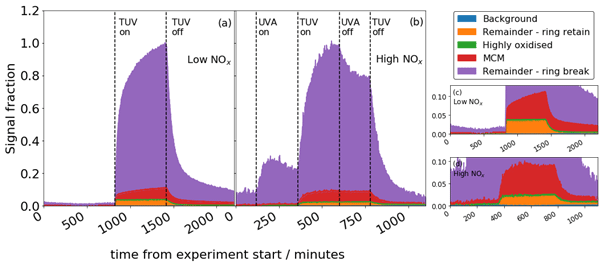
Figure 9Total signal from CHON + CHO compounds for the low-NOx (a) and high-NOx (b) experiments. Panels (c) and (d) show the same data in (a) and (b) with a reduced y scale; y and x scale have the same units.
Ring retention and ring-breaking products
In terms of signal, most of the ring-retaining products are O, of which O1,2 is dominated by the formation of phenol and catechol. Conversely, O3 is not dominated by any one product but instead comprises both CHO and CHON products in both NOx conditions (Fig. 10). Although O1−7 species are observed in both cases, higher O numbers of 10, 11, and 12 are only visible in the high-NOx case due to the presence of nitrogen groups that can incorporate more oxygen.
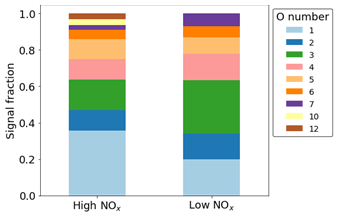
Figure 10Normalised signal fraction of ring-retaining products under high- and low-NOx conditions. Signal for phenol and catechol dominate for both conditions and make up the O1 and O2 fractions.
For the ring-breaking products, the majority of signal can be attributed to the presence of species containing 3 and 4 oxygen atoms, although at lower carbon numbers of 1 and 2, lower oxygen numbers of 1 and 2 become important (Fig. 11). The majority of signal for both cases is due to C compounds. C3 compounds show the greatest signal contribution for the largest range of oxygen numbers. This is especially true in the high- NOx case where signal C3 compounds have a greater contribution from O>4. Similarly at higher carbon numbers, e.g. 5 and 6, higher oxygen numbers of between 6 and 12 are more readily observed; again where N is incorporated in the high-NOx case, a greater inclusion of oxygen is also observed. The form of this organic nitrogen is perturbed throughout the experiment by altering the NO NOx fraction by photolysing NO2. It is stressed that as these values are reported as uncalibrated instrument responses, they cannot be used to directly assess chemical pathways. However they allow for the comparison of product distributions between high- and low-NOx conditions in terms of sensitivity to iodide CIMS measurements (Figs. 10 and 11).
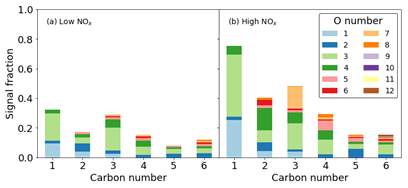
Figure 11Signal fraction as a function of carbon number for ring-breaking products for the (a) high-NOx and (b) low-NOx conditions.
Descriptions of bulk properties and groupings into large families are a useful method to assess broad trends within the system. However it may be possible to produce less granularity in identified species by looking at specific atomic composition and attempting to infer potential functionality.
3.4 Hierarchical cluster analysis to infer CHON functionality
A four-cluster solution was chosen from analysis of the dendrogram (Fig. S3) and is sufficient to provide enough detail that can be explained but not so much that interpretation becomes unclear. Further refinement of clusters greater than four offers no more insight into time series behaviour due to oxidation behaviour but rather describes signal delay, likely due to gas wall partitioning (Krechmer et al., 2016; Pagonis et al., 2017). This behaviour is still observed in the four-cluster solution; however, other affects pertaining to chemistry rather than partitioning are more visible at this low cluster number. Therefore, the four-cluster solution is chosen (Fig. 12). See Table S2 for a list of MCM-defined products and their assigned clusters.
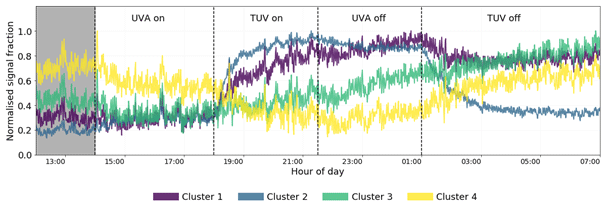
Figure 12Time series of the four cluster means for the high-NOx experiment. Clusters 3 and 4 represent slow-formation and background clusters and give no information regarding the effect of NO2 photolysis. Clusters 1 and 2 display similar behaviours when the TUV light is switched on but different behaviours when the UVA light is switched off, indicating their members are sensitive to the change of the NO NOx ratio determined by the UVA light state. The shaded area is the dark state before any photolysis occurs.
Cluster 3 and cluster 4 do not display much effect of NO2 photolysis. Cluster 3 is a slow-formation cluster that grows as photo-oxidation begins. This is likely to represent semi-volatile material that partitions to walls and equilibrates with the instrument lines and IMR. Cluster 4 is a background cluster that decays when photo-oxidation begins (TUV on) and recovers when photo-oxidation stops. The background cluster 4 has the lowest number of RNOx-containing members, and the slow-growth, semi-volatile cluster 3 has the greatest number (Fig. 13c). Cluster 4 contains the greatest number of species with high carbon numbers with a negative . These characteristics are reflected in cluster 3 but to a lesser extent as carbon numbers are smaller and less negative. It is also notable that cluster 1 products remain at elevated levels in the chamber after all photochemistry has stopped. This again may be indicative of more semi-volatile material formed. This is anecdotally corroborated by the higher oxygen and carbon numbers of cluster 1 compared to cluster 2 (Fig. 13a). Cluster 2 shares time series features that are similar to clusters 1 and 3. It has the same long-range response as cluster 3 but the same short-range increase from photo-oxidation as cluster 1. This is reflected in Fig. 13b, where cluster 2 overlaps with clusters 1 and 3, which themselves are well separated. Both these clusters have lower average carbon numbers and higher, positive compared with clusters 3 and 4 (Fig. 13b).
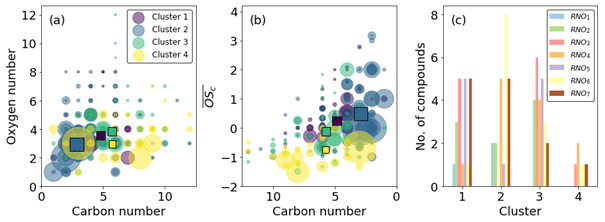
Figure 13Cluster characteristics. (a) Carbon vs. oxygen number. (b) Carbon number vs. average carbon oxidation state. Squares indicate cluster averages. (c) Number of RNOx species. Clustering based on time series similarity provides good separation in vs. C space (b) and splits RNOodd and RNOeven in clusters 1 and 2 (c).
Clusters 1 and 2 represent the formation of oxidation products. They increase similarly when photo-oxidation is initiated; however when the ratio is increased, cluster 1 continues to increase, but cluster 2 decreases. This suggests cluster 1 products are independent of the ratio at these high-NO2 fractions; the growth curve is independent of the decrease in NO concentration, suggesting this cluster is characterised by more reactions of NO2 and peroxy radicals. Conversely to cluster 1, cluster 2 decreases when the increases, suggesting NO is an important route to formation, either from NO addition products or the increased alkoxy radical fraction , which is supported by cluster 2 products having higher and lower carbon numbers than cluster 1 products.
Both clusters 1 and 2 contain a similar number of RNO and RNO2 species, but cluster 1 has a greater, odd number of oxygen as RNO3 and RNO5, whereas cluster 2 has a greater even number of oxygen in RNO4 and RNO6. The inclusion of these groupings in one cluster leads to a lack of them in the other; no RNO4 or RNO6 is found in cluster 1, nor is there a large amount of RNO5 or any RNO3 in cluster 2.
Ideally, NO and NO2 addition to peroxy precursors (cluster 1) would produce nitrates and peroxy acyl nitrates (PANs) that have an odd number of oxygens (RNO3 and RNO5), and addition to alkoxy precursors (cluster 2) would produce nitro- and nitrite compounds that have an even number of oxygens (RNO2). This is indeed observed; however in reality, this distinction is not so clear-cut, as OH addition vs. abstraction would reverse this. Additionally, the presence of a pathway to form N-containing ring-retaining epoxides from the alkoxy phenyl radical recently described by Xu et al. (2020) further demonstrates the difficulty of structural assignments and accounting for the position of oxygen atoms. C6H7NO4 and C6H7NO6 are two formulae that could represent ring-retaining nitrogen-containing epoxides; however as these two formulae are found in cluster 3, they do not impact the discussion on the effect of x ratios on clusters 1 and 2 here.
So although these observations are broadly followed, they are not exact resulting in a blurring of RNOx groupings, especially at higher O numbers (Fig. 13c). Both clusters 1 and 2 have the same number of RNO7 species, despite their time series profiles showing a different dependency on the ratio. It is likely that these RNO7 are structurally different, and the oxygen is incorporated into their structures in different ways. For example, cluster 1 RNO7, which are all C5 and C6 compounds, may contain more PAN-like compounds as these are formed from peroxy acyl radicals and NO2 addition, e.g. to BZEMUCCO3 (MCM), which is the peroxy radical formed from BZEPOXMUC, a first-generation ring-opening product of benzene, to form BZEMUCPAN (C6H5NO7). In contrast, cluster 2 RNO7 are smaller C3 and C4 compounds, which may require the fragmentation of larger organic precursors enhanced by the presence of NO, e.g. the unimolecular decomposition of the alkoxy radical MALANHYO to form HCOCOHCO3 and then HCOCOPAN (C3H3NO7).
Two ToF-CIMS instruments using iodide and nitrate ionisation schemes were deployed at the Jülich Plant Atmosphere Chamber as part of a series of experiments examining benzene oxidation by OH under high-NOx (20 ppb) and low-NOx (0.3 ppb) conditions.
Both ionisation schemes detect benzene oxidation products including highly oxidised organic molecules. Nitrate CIMS detects many C12 dimers and a greater number of species with high oxygen number (O9-O11). This translates to higher , especially at higher carbon numbers (C≥8), and lower C*, indicating the detection of ELVOCs, SVOCs, and IVOCs. In contrast, the iodide CIMS detects no dimers but does detect a wider range of monomers and ring-breaking products (C≤6) than the nitrate scheme, with most common oxygen numbers of (O3-O4). The of high carbon species is lower, although at lower carbon numbers (C<6) between the two ionisation schemes broadly agrees due to the increased likelihood of measuring the same species, rather than isomers. The corresponding C* measured by the iodide CIMS suggests no ELVOC was detected, but more SVOCs and IVOCs and some VOCs were measured when compared to the nitrate CIMS. Thus the two ionisation schemes cover a large range of volatilities.
CHO species (132) and CHON species (195) are detected in the low- and high-NOx experiments, respectively. Of these, 126 are common. In both cases these are mostly C4 and C6 compounds. A greater number of oxidation products are measured in the high-NOx case, including 70 N-containing compounds compared to 24 in the low-NOx case, of which all but one are common. Splitting oxidation products into five categories, the contribution to signal increases sequentially from background, highly oxidised products, ring-retaining, MCM and ring-breaking. The signal assigned to MCM products represents between and of the 85 listed multigenerational oxidation products detected under low- and high-NOx conditions and is proportionally distributed across all carbon numbers. Highly oxidised and ring-retaining products equilibrate quickly within the system and remain throughout the experiments, whereas MCM and ring-breaking products continue to grow throughout the experiments. Highly oxidised and ring-retaining products are earlier generation products which form quickly and equilibrate as loss and formation processes equalise. MCM and ring-breaking products contain smaller, more oxidised molecules that are further down the oxidation chain and represent the largest destination of carbon within the system. Signal from ring-retaining products is dominated by phenol (O1) and catechol (O2), whereas O3 compounds comprise a number of different species. Ring-breaking products are dominated by C1, C2, and C3 compounds with similar oxygen numbers. C3 compounds show the greatest variability of oxygen atom number. For both retention and breaking products, high O numbers of up to 7 are identified; however in the high-NOx case where incorporation of RNOx is greater, higher O numbers of 10, 11, and 12 are observed. Within the context of the theoretical mechanistic investigation (Sect. 3.3), iodide ionisation is able to detect 27 and 33 species belonging to potential auto-oxidation reaction pathways. These detected species include one previously observed, exclusively second-generation auto-oxidation product, C6H8O8, and all its derived first-generation auto-oxidation products. However, it is noted that only two of these first-generation product steps (C6H8O6, C6H8O7) are formed exclusively through auto-oxidation, whilst the rest have other routes to formation. Higher oxygen-content products C6H8O9 and C6H8O10 are not observed in either NOx case by iodide but are observed with the nitrate CIMS.
Clustering the time series in the high-NOx experiment into four clusters distinguishes two clusters that contain products formed from photo-oxidation. One of these clusters (cluster 1) is independent of the ratio, whereas a second cluster (cluster 2) decreases as a result of NO-dependent formation, either through addition and/or an increased fraction. Cluster 2 has higher and lower carbon numbers than cluster 1, suggesting it consists of more oxidised and fragmented compounds consistent with an increased . Cluster 2 contains RNO4 and RNO6 but no RNO3 and little RNO5, whereas the opposite is true for cluster 1. This somewhat agrees with theoretical RNOx product distributions as NOx addition to alkoxy radicals (cluster 2) is more likely to produce even oxygen-content RNOx (through the formation of nitrites and nitro compounds) and odd oxygen RNOx through addition to peroxy radicals (cluster 1, such as nitrates and PANs). For species with larger oxygen content, e.g. RNO7, which is detected in both clusters, the carbon number is lower for cluster 2 (C3,4) compared to cluster 1 (C5,6), indicating more fragmentation has occurred, again implying a greater contribution from the alkoxy channel. It is noted that the effect of OH addition, rather than H abstraction as the initiation step to the reaction, will reverse this pattern and, along with other unimolecular rearrangements, may explain some of the RNOx cluster variability observed. This methodology used in conjunction with an internal calibration procedure such as voltage scanning (Lopez-Hilfiker et al., 2016) could then be used to quantitatively describe shifts in CHON composition as a function of NO NOx and further investigate the relative importance of isomers and their formation pathways to a mass spectrometric signal.
Data are available on request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-3473-2021-supplement.
MP analysed nitrate and iodide ToF-CIMS data and wrote the manuscript. OG analysed nitrate ToF-CIMS data and provided peak list data. TJB and MLB operated the iodide CIMS. CJP, GM, HC, TFM, DES, AM, SDW, and AB contributed to iodide CIMS analysis and manuscript development. CJP and GM led the development of the experiments presented here. TFM, AKS, CJP, MH, GM, and ÅMH designed the experiments. SK, IP, SS, RT, EK, DZ, and JW operated the nitrate CIMS and performed the experiments.
The authors declare that they have no conflict of interest.
This work was conducted during a PhD study supported by the Natural Environment Research Council (NERC) EAO Doctoral Training Partnership and is fully funded by NERC, whose support is gratefully acknowledged (grant ref. no. NE/L002469/1). The study was funded by the European Research Council (grant no. 638703) and supported by the Doctoral School in Atmospheric Sciences at the University of Helsinki (ATM-DP). This work was supported by the Academy of Finland (grant nos. 317380 and 320094) and the Swedish Research Council (grant no. 2014-5332). Åsa M. Hallquist acknowledges Formas (grant no. 214-2013-1430) and VINNOVA, Sweden's Innovation Agency (grant no. 2013-03058), including support for her research stay at Forschungszentrum Jülich. Part of the research in this study was performed at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration.
This research has been supported by the Natural Environment Research Council (grant no. NE/L002469/1), the European Research Council (grant no. 638703), the Vetenskapsrådet (grant no. 2014-5332), the Svenska Forskningsrådet Formas (grant no. 214-2013-1430), VINNOVA (grant no. 2013-03058), and the Academy of Finland (grant nos. 317380 and 320094).
The article processing charges for this open-access
publication were covered by the Gothenburg University Library.
This paper was edited by Lea Hildebrandt Ruiz and reviewed by two anonymous referees.
Archibald, A. T., Witham, C. S., Ashfold, M. J., Manning, A. J., O'Doherty, S., Greally, B. R., Young, D., and Shallcross, D. E.: Long-term high frequency measurements of ethane, benzene and methyl chloride at Ragged Point, Barbados: Identification of long-range transport events, Elem. Sci. Anthr., 3, 000068, https://doi.org/10.12952/journal.elementa.000068, 2015.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/S1352-2310(99)00460-4, 2000.
Atkinson, R. and Arey, J.: Mechanisms of the gas-phase reactions of aromatic hydrocarbons and PAHs with OH and NO3 Radicals, Polycycl. Aromat. Compd., 27, 15–40, https://doi.org/10.1080/10406630601134243, 2007.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bird, M. G., Greim, H., Kaden, D. A., Rice, J. M., and Snyder, R.: BENZENE 2009-Health effects and mechanisms of bone marrow toxicity: Implications for t-AML and the mode of action framework, Chem. Biol. Interact., 184, 3–6, https://doi.org/10.1016/j.cbi.2009.12.001, 2010.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005.
Borrás, E. and Tortajada-Genaro, L. A.: Secondary organic aerosol formation from the photo-oxidation of benzene, Atmos. Environ., 47, 154–163, https://doi.org/10.1016/j.atmosenv.2011.11.020, 2012.
Buchholz, A., Ylisirniö, A., Huang, W., Mohr, C., Canagaratna, M., Worsnop, D. R., Schobesberger, S., and Virtanen, A.: Deconvolution of FIGAERO–CIMS thermal desorption profiles using positive matrix factorisation to identify chemical and physical processes during particle evaporation, Atmos. Chem. Phys., 20, 7693–7716, https://doi.org/10.5194/acp-20-7693-2020, 2020.
Chhabra, P. S., Lambe, A. T., Canagaratna, M. R., Stark, H., Jayne, J. T., Onasch, T. B., Davidovits, P., Kimmel, J. R., and Worsnop, D. R.: Application of high-resolution time-of-flight chemical ionization mass spectrometry measurements to estimate volatility distributions of α-pinene and naphthalene oxidation products, Atmos. Meas. Tech., 8, 1–18, https://doi.org/10.5194/amt-8-1-2015, 2015.
Creasey, D. J., Heard, D. E., and Lee, J. D.: Eastern Atlantic Spring Experiment 1997 (EASE97) 1. Measurements of OH and HO2 concentrations at Mace Head, Ireland, J. Geophys. Res.-Atmos., 107, ACH 3-1–ACH 3-15, https://doi.org/10.1029/2001JD000892, 2002.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Fally, S., Carleer, M. and Vandaele, A. C.: UV Fourier transform absorption cross sections of benzene, toluene, meta-, ortho-, and para-xylene, J. Quant. Spectrosc. Radiat. Transf., 110, 766–782, https://doi.org/10.1016/j.jqsrt.2008.11.014, 2009.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Gentner, D. R., Isaacman, G., Worton, D. R., Chan, A. W. H., Dallmann, T. R., Davis, L., Liu, S., Day, D. A., Russell, L. M., Wilson, K. R., Weber, R., Guha, A., Harley, R. A., and Goldstein, A. H.: Elucidating secondary organic aerosol from diesel and gasoline vehicles through detailed characterization of organic carbon emissions, P. Natl. Acad. Sci., 109, 18318–18323, https://doi.org/10.1073/pnas.1212272109, 2012.
Glowacki, D. R., Wang, L., and Pilling, M. J.: Evidence of formation of bicyclic species in the early stages of atmospheric benzene oxidation, J. Phys. Chem. A, 113, 5385–5396, https://doi.org/10.1021/jp9001466, 2009.
Guo, Y., Xue, Q., Zhang, H., Wang, N., Chang, S., Wang, H., Pang, H., and Chen, H.: Treatment of real benzene dye intermediates wastewater by the Fenton method: characteristics and multi-response optimization, RSC Adv., 8, 80–90, https://doi.org/10.1039/C7RA09404C, 2018.
Henze, D. K., Seinfeld, J. H., Ng, N. L., Kroll, J. H., Fu, T.-M., Jacob, D. J., and Heald, C. L.: Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high- vs. low-yield pathways, Atmos. Chem. Phys., 8, 2405–2420, https://doi.org/10.5194/acp-8-2405-2008, 2008.
Hoyt, D. and Raun, L. H.: Measured and estimated benzene and volatile organic carbon (VOC) emissions at a major U.S. refinery/chemical plant: Comparison and prioritization, J. Air Waste Manag. Assoc., 65, 1020–1031, https://doi.org/10.1080/10962247.2015.1058304, 2015.
Isaacman-VanWertz, G., Massoli, P., O'Brien, R. E., Nowak, J. B., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., Su, L., Knopf, D. A., Misztal, P. K., Arata, C., Goldstein, A. H., and Kroll, J. H.: Using advanced mass spectrometry techniques to fully characterize atmospheric organic carbon: current capabilities and remaining gaps, Faraday Discuss., 200, 579–598, https://doi.org/10.1039/C7FD00021A, 2017.
Isaacman-Vanwertz, G., Massoli, P., O'Brien, R., Lim, C., Franklin, J. P., Moss, J. A., Hunter, J. F., Nowak, J. B., Canagaratna, M. R., Misztal, P. K., Arata, C., Roscioli, J. R., Herndon, S. T., Onasch, T. B., Lambe, A. T., Jayne, J. T., Su, L., Knopf, D. A., Goldstein, A. H., Worsnop, D. R., and Kroll, J. H.: Chemical evolution of atmospheric organic carbon over multiple generations of oxidation, Nat. Chem., 10, 462–468, https://doi.org/10.1038/s41557-018-0002-2, 2018.
Isokääntä, S., Kari, E., Buchholz, A., Hao, L., Schobesberger, S., Virtanen, A., and Mikkonen, S.: Comparison of dimension reduction techniques in the analysis of mass spectrometry data, Atmos. Meas. Tech., 13, 2995–3022, https://doi.org/10.5194/amt-13-2995-2020, 2020.
IUPAC: IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation – Data Sheet HOx_AROM1, IUPAC, available at: http://iupac.pole-ether.fr/htdocs/datasheets/pdf/HOx_AROM1_HO_benzene.pdf (last access: 12 October 2020), 2008.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Johnson, D., Jenkin, M. E., Wirtz, K., and Martin-Reviejo, M.: Simulating the Formation of Secondary Organic Aerosol from the Photooxidation of Aromatic Hydrocarbons, Environ. Chem., 2, 35, https://doi.org/10.1071/EN04079, 2005.
Kamps, R., Müller, H., Schmitt, M., Sommer, S., Wang, Z., and Kleinermanns, K.: Photooxidation of exhaust pollutants. I. Degradation efficiencies, quantum yields and products of benzene photooxidation, Chemosphere, 27, 2127–2142, https://doi.org/10.1016/0045-6535(93)90125-O, 1993.
Knote, C., Hodzic, A., Jimenez, J. L., Volkamer, R., Orlando, J. J., Baidar, S., Brioude, J., Fast, J., Gentner, D. R., Goldstein, A. H., Hayes, P. L., Knighton, W. B., Oetjen, H., Setyan, A., Stark, H., Thalman, R., Tyndall, G., Washenfelder, R., Waxman, E., and Zhang, Q.: Simulation of semi-explicit mechanisms of SOA formation from glyoxal in aerosol in a 3-D model, Atmos. Chem. Phys., 14, 6213–6239, https://doi.org/10.5194/acp-14-6213-2014, 2014.
Koss, A. R., Canagaratna, M. R., Zaytsev, A., Krechmer, J. E., Breitenlechner, M., Nihill, K. J., Lim, C. Y., Rowe, J. C., Roscioli, J. R., Keutsch, F. N., and Kroll, J. H.: Dimensionality-reduction techniques for complex mass spectrometric datasets: application to laboratory atmospheric organic oxidation experiments, Atmos. Chem. Phys., 20, 1021–1041, https://doi.org/10.5194/acp-20-1021-2020, 2020.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016.
Mehra, A., Wang, Y., Krechmer, J. E., Lambe, A., Majluf, F., Morris, M. A., Priestley, M., Bannan, T. J., Bryant, D. J., Pereira, K. L., Hamilton, J. F., Rickard, A. R., Newland, M. J., Stark, H., Croteau, P., Jayne, J. T., Worsnop, D. R., Canagaratna, M. R., Wang, L., and Coe, H.: Evaluation of the chemical composition of gas- and particle-phase products of aromatic oxidation, Atmos. Chem. Phys., 20, 9783–9803, https://doi.org/10.5194/acp-20-9783-2020, 2020.
Mentel, Th. F., Wildt, J., Kiendler-Scharr, A., Kleist, E., Tillmann, R., Dal Maso, M., Fisseha, R., Hohaus, Th., Spahn, H., Uerlings, R., Wegener, R., Griffiths, P. T., Dinar, E., Rudich, Y., and Wahner, A.: Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 9, 4387–4406, https://doi.org/10.5194/acp-9-4387-2009, 2009.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Misztal, P. K., Hewitt, C. N., Wildt, J., Blande, J. D., Eller, A. S. D., Fares, S., Gentner, D. R., Gilman, J. B., Graus, M., Greenberg, J., Guenther, A. B., Hansel, A., Harley, P., Huang, M., Jardine, K., Karl, T., Kaser, L., Keutsch, F. N., Kiendler-Scharr, A., Kleist, E., Lerner, B. M., Li, T., Mak, J., Nölscher, A. C., Schnitzhofer, R., Sinha, V., Thornton, B., Warneke, C., Wegener, F., Werner, C., Williams, J., Worton, D. R., Yassaa, N., and Goldstein, A. H.: Atmospheric benzenoid emissions from plants rival those from fossil fuels, Sci. Rep., 5, 1–10, https://doi.org/10.1038/srep12064, 2015.
Mohr, C., Thornton, J. A., Heitto, A., Lopez-Hilfiker, F. D., Lutz, A., Riipinen, I., Hong, J., Donahue, N. M., Hallquist, M., Petäjä, T., Kulmala, M., and Yli-Juuti, T.: Molecular identification of organic vapors driving atmospheric nanoparticle growth, Nat. Commun., 10, 1–7, https://doi.org/10.1038/s41467-019-12473-2, 2019.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci., 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
O'Brien, R. J., Green, P. J., Nguyen, N. L., Doty, R. A., and Dumdel, B. E.: Carbon Balances in Simulated Atmospheric Reactions: Aromatic Hydrocarbons, Environ. Sci. Technol., 17, 183–186, https://doi.org/10.1021/es00109a013, 1983.
Oyoroko, E. and Ogamba, E. N.: Effects of detergent containing linear alkyl benzene sulphonate on behavioural response of Heterobranchus bidorsalis, Clarias gariepinus and Heteroclarias, Biotechnol. Res., 3, 59–64, 2017.
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci., 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Priestley, M., Le Breton, M., Bannan, T. J., Leather, K. E., Bacak, A., Reyes-Villegas, E., De Vocht, F., Shallcross, B. M. A., Brazier, T., Anwar Khan, M., Allan, J., Shallcross, D. E., Coe, H., and Percival, C. J.: Observations of Isocyanate, Amide, Nitrate, and Nitro Compounds From an Anthropogenic Biomass Burning Event Using a ToF-CIMS, J. Geophys. Res.-Atmos., 2, 1–26, https://doi.org/10.1002/2017JD027316, 2018.
Pye, H. O. T., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Rodriguez, P. G., Dral, A. P., van den Nieuwenhuijzen, K. J. H., Lette, W., Schipper, D. J., and ten Elshof, J. E.: Thermochemical stability and friction properties of soft organosilica networks for solid lubrication, Materials (Basel)., 11, 1–12, https://doi.org/10.3390/ma11020180, 2018.
Ruchirawat, M., Navasumrit, P., Settachan, D., Tuntaviroon, J., Buthbumrung, N., and Sharma, S.: Measurement of genotoxic air pollutant exposures in street vendors and school children in and near Bangkok, Toxicol. Appl. Pharmacol., 206, 207–214, https://doi.org/10.1016/j.taap.2004.11.025, 2005.
Ruchirawat, M., Settachan, D., Navasumrit, P., Tuntawiroon, J., and Autrup, H.: Assessment of potential cancer risk in children exposed to urban air pollution in Bangkok, Thailand, Toxicol. Lett., 168, 200–209, https://doi.org/10.1016/j.toxlet.2006.09.013, 2007.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Sekimoto, K., Koss, A. R., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Warneke, C., Yokelson, R. J., Roberts, J. M., and de Gouw, J.: High- and low-temperature pyrolysis profiles describe volatile organic compound emissions from western US wildfire fuels, Atmos. Chem. Phys., 18, 9263–9281, https://doi.org/10.5194/acp-18-9263-2018, 2018.
Snyder, R., Kocsis, J. J., and Drew, R.: Current concepts of chronic benzene toxicity, Crit. Rev. Toxicol., 3, 265–288, https://doi.org/10.3109/10408447509079860, 1975.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kimmel, J. R., Cubison, M. J., Chhabra, P. S., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Methods to extract molecular and bulk chemical information from series of complex mass spectra with limited mass resolution, Int. J. Mass Spectrom., 389, 26–38, https://doi.org/10.1016/j.ijms.2015.08.011, 2015.
Stirnweis, L., Marcolli, C., Dommen, J., Barmet, P., Frege, C., Platt, S. M., Bruns, E. A., Krapf, M., Slowik, J. G., Wolf, R., Prévôt, A. S. H., Baltensperger, U., and El-Haddad, I.: Assessing the influence of NOx concentrations and relative humidity on secondary organic aerosol yields from α-pinene photo-oxidation through smog chamber experiments and modelling calculations, Atmos. Chem. Phys., 17, 5035–5061, https://doi.org/10.5194/acp-17-5035-2017, 2017.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci., 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Tsiligiannis, E., Hammes, J., Salvador, C. M., Mentel, T. F., and Hallquist, M.: Effect of NOx on 1,3,5-trimethylbenzene (TMB) oxidation product distribution and particle formation, Atmos. Chem. Phys., 19, 15073–15086, https://doi.org/10.5194/acp-19-15073-2019, 2019.
Verma, D. K. and Des Tombe, K.: Benzene in gasoline and crude oil: Occupational and environmental implications, Am. Ind. Hyg. Assoc. J., 63, 225–230, https://doi.org/10.1080/15428110208984708, 2002.
Volkamer, R., Klotz, B., Barnes, I., Imamura, T., Wirtz, K., Washida, N., Becker, K. H., and Platt, U.: OH-initiated oxidation of benzene, Phys. Chem. Chem. Phys., 4, 1598–1610, https://doi.org/10.1039/b108747a, 2002.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, T., Bo, P., Bing, T., Zhaoyun, Z., Liyu, D., and Yonglong, L.: Benzene homologues in environmental matrixes from a pesticide chemical region in China: Occurrence, health risk and management, Ecotoxicol. Environ. Saf., 104, 357–364, https://doi.org/10.1016/j.ecoenv.2014.01.035, 2014.
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: autoxidation and accretion, Atmos. Chem. Phys., 20, 9563–9579, https://doi.org/10.5194/acp-20-9563-2020, 2020.
Weber, J., Archer-Nicholls, S., Griffiths, P., Berndt, T., Jenkin, M., Gordon, H., Knote, C., and Archibald, A. T.: CRI-HOM: A novel chemical mechanism for simulating highly oxygenated organic molecules (HOMs) in global chemistry–aerosol–climate models, Atmos. Chem. Phys., 20, 10889–10910, https://doi.org/10.5194/acp-20-10889-2020, 2020.
Wildt, J., Mentel, T. F., Kiendler-Scharr, A., Hoffmann, T., Andres, S., Ehn, M., Kleist, E., Müsgen, P., Rohrer, F., Rudich, Y., Springer, M., Tillmann, R., and Wahner, A.: Suppression of new particle formation from monoterpene oxidation by NOx, Atmos. Chem. Phys., 14, 2789–2804, https://doi.org/10.5194/acp-14-2789-2014, 2014.
Xu, L., Møller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New insights into the radical chemistry and product distribution in the OH-initiated oxidation of benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020.
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019.