the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Mar 2021
| 04 Mar 2021
Halogen activation in the plume of Masaya volcano: field observations and box model investigations
Julian Rüdiger
Alexandra Gutmann
Nicole Bobrowski
Marcello Liotta
J. Maarten de Moor
Rolf Sander
Florian Dinger
Jan-Lukas Tirpitz
Martha Ibarra
Armando Saballos
María Martínez
Elvis Mendoza
Arnoldo Ferrufino
John Stix
Juan Valdés
Jonathan M. Castro
Thorsten Hoffmann
Volcanic emissions are a source of halogens in the atmosphere. Rapid reactions convert the initially emitted hydrogen halides (HCl, HBr, and HI) into reactive species such as BrO, Br2, BrCl, ClO, OClO, and IO. The activation reaction mechanisms in the plume consume ozone (O3), which is entrained by ambient air that is mixed into the plume. In this study, we present observations of the oxidation of bromine, chlorine, and iodine during the first 11 min following emission, examining the plume from Santiago crater of the Masaya volcano in Nicaragua. Two field campaigns were conducted: one in July 2016 and one in September 2016. The sum of the reactive species of each halogen was determined by gas diffusion denuder sampling followed by gas chromatography–mass spectrometry (GC-MS) analysis, whereas the total halogens and sulfur concentrations were obtained by alkaline trap sampling with subsequent ion chromatography (IC) and inductively coupled plasma mass spectrometry (ICP-MS) measurements. Both ground and airborne sampling with an unoccupied aerial vehicle (carrying a denuder sampler in combination with an electrochemical SO2 sensor) were conducted at varying distances from the crater rim. The in situ measurements were accompanied by remote sensing observations (differential optical absorption spectroscopy; DOAS). The reactive fraction of bromine increased from 0.20 ± 0.13 at the crater rim to 0.76 ± 0.26 at 2.8 km downwind, whereas chlorine showed an increase in the reactive fraction from (2.7 ± 0.7) × 10−4 to (11 ± 3) × 10−4 in the first 750 m. Additionally, a reactive iodine fraction of 0.3 at the crater rim and 0.9 at 2.8 km downwind was measured. No significant change in BrO SO2 molar ratios was observed with the estimated age of the observed plume ranging from 1.4 to 11.1 min. This study presents a large complementary data set of different halogen compounds at Masaya volcano that allowed for the quantification of reactive bromine in the plume of Masaya volcano at different plume ages. With the observed field data, a chemistry box model (Chemistry As A Boxmodel Application Module Efficiently Calculating the Chemistry of the Atmosphere; CAABA/MECCA) allowed us to reproduce the observed trend in the ratio of the reactive bromine to total bromine ratio. An observed contribution of BrO to the reactive bromine fraction of about 10 % was reproduced in the first few minutes of the model run.
- Article
(3836 KB) - Full-text XML
-
Supplement
(18003 KB) - BibTeX
- EndNote





Volcanoes are known to be important emitters of atmospheric trace gases and aerosols, both through explosive eruptions and persistent quiescent degassing (von Glasow et al., 2009). The most abundant gases in volcanic emissions are water, carbon dioxide, sulfur compounds, and hydrogen halides (Symonds et al., 1994). Typically, halogen emissions are largely dominated by chlorine (HCl) and fluorine (HF), whereas bromine (HBr) and iodine (HI) are 3 and 5 orders of magnitude less abundant than chlorine and fluorine, respectively (e.g., Aiuppa et al., 2005; Pyle and Mather, 2009). Despite their low abundance, the heavy halogens (bromine and iodine) can have a significant impact on the chemistry of the atmosphere (e.g., von Glasow, 2010; Saiz-Lopez and von Glasow, 2012; Platt and Bobrowski, 2015). The chemical composition of volcanic plumes is the subject of a large number of studies, which are often aimed at gaining insights into subsurface processes, such as the degassing of magma in connection with changes in volcanic activity (e.g., Aiuppa et al., 2007). In addition, the effects of volcanic gases on the atmosphere and biosphere at local, regional, and global scales are also of interest, including acid deposition (wet and dry), nutrient input (e.g., Delmelle, 2003), aerosol formation, and the effects on the solar radiation balance (e.g., Mather et al., 2013; Malavelle et al., 2017).
Volcanic halogen emissions have been studied for years (e.g., Noguchi and Kamiya, 1963; Giggenbach, 1975), and the determination of chlorine and sulfur is a common procedure in such gas geochemical investigations. Bromine only attracted more attention in later years, when the reactive bromine species BrO was observed in volcanic plumes (e.g., Bobrowski et al., 2003; Oppenheimer et al., 2006). This also proved that not only sulfur species (H2S, SO2) undergo oxidation by ambient reactants (such as OH and O3), and it laid the foundation for various studies on oxidized halogen species (such as BrO, ClO, OClO, and IO) (e.g., Lee et al., 2005; Theys et al., 2014; General et al., 2015; Gliß et al., 2015; Schönhardt et al., 2017). Despite the low abundance of bromine in volcanic gas emissions, the relatively simple detection of BrO by differential optical absorption spectroscopy (DOAS) promoted research on the origin and fate of BrO in volcanic plumes. Based on thermodynamic modeling, Gerlach (2004) hypothesized that BrO is not primarily emitted by volcanoes and is instead formed only after the initial emissions are mixed with entrained ambient air. As SO2 can also be easily measured by DOAS, and the oxidation of SO2 plays a minor role over a period of minutes to hours (McGonigle et al., 2004), the ratio of BrO to SO2 is used as a dilution-compensated observation parameter.
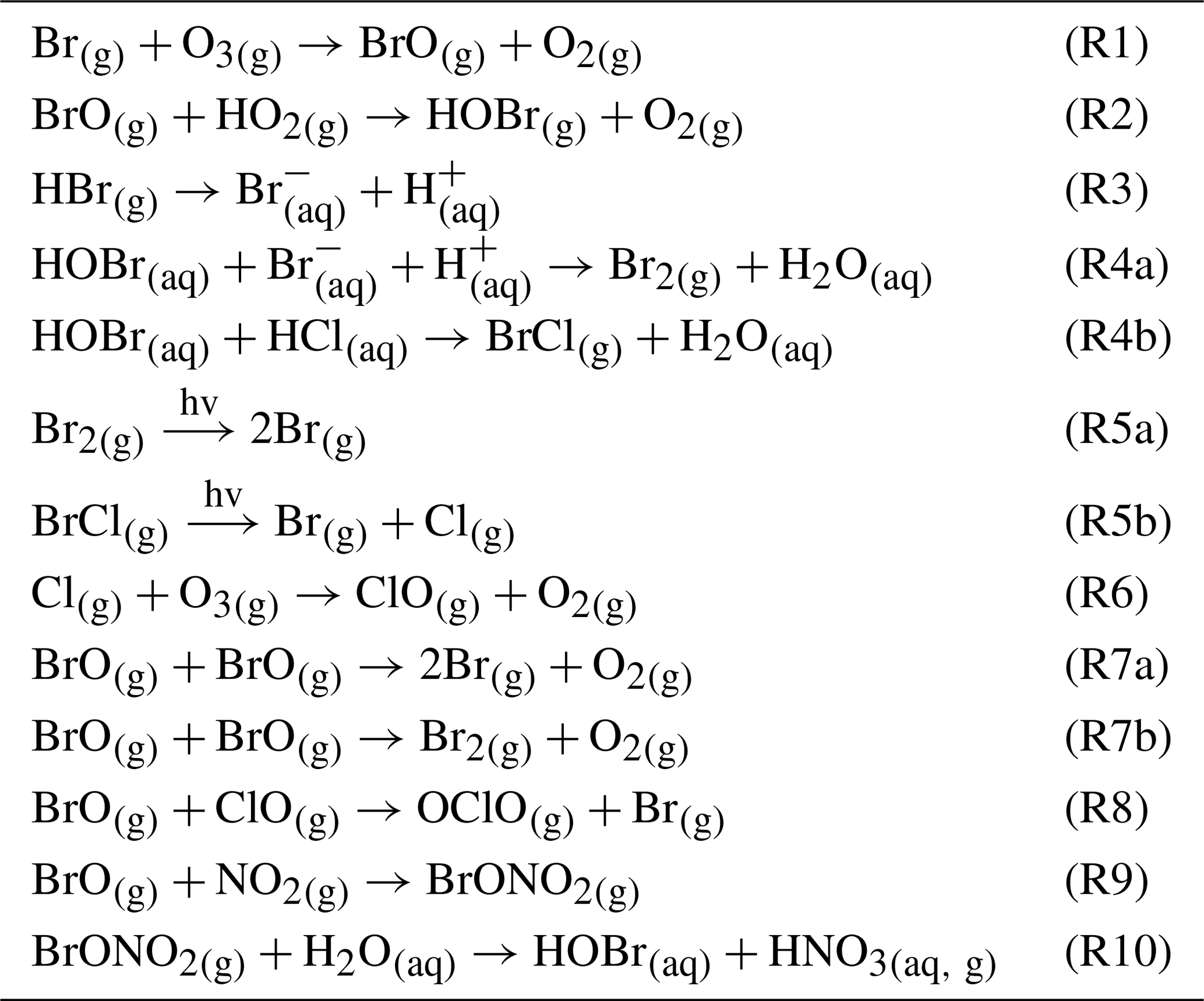
An increase in the BrO SO2 ratio with increasing distance from the emitting vent has been observed at various volcanoes (e.g., Bobrowski et al., 2007; Vogel, 2012; Gliß et al., 2015) as have variations in BrO SO2 in a lateral plume dimension with higher ratios at the edges of the plume (e.g., Bobrowski et al., 2007; Louban et al., 2009; General et al., 2015; Kern and Lyons, 2018). This has been explained by a limited transfer of atmospheric O3 to the center of the plume, which is thought to promote the formation of BrO in a chain reaction mechanism involving heterogeneous chemistry. Shortly after the discovery of the reactive bromine species BrO, reactive chlorine species, ClO and OClO, were also observed using the same DOAS techniques (e.g., Lee et al., 2005; Bobrowski et al., 2007; Donovan et al., 2014; Theys et al., 2014; General et al., 2015; Gliß et al., 2015; Kern and Lyons, 2018). It was found that the abundance of ClO and OClO is of the same order of magnitude as BrO, in contrast to total chlorine, which is typically 3 orders of magnitude more abundant than bromine. The formation of reactive chlorine species is considered to be a secondary product of the activation cycle of bromine (see Table 1). Recently, reactive iodine species have also been detected by satellite observations in the plume of Kasatochi (Schönhardt et al., 2017), but they could not be confirmed by ground-based measurements to date.
Table 1Overview of halogen reactions in volcanic plumes (X = Cl, Br).
Both the transformation of halogen species in the plume and their fate in the atmosphere are of interest. In particular, clarification regarding the amounts emitted into the atmosphere and the distribution of the halogens emitted by quiescent (i.e., passive, non-eruptive) and eruptive degassing is of interest. The global volcanic SO2 flux has been estimated as 23 Tg yr−1 for the period from 2004 to 2016 (Carn et al., 2017), resulting in estimated halogen fluxes of the same order for chlorine and 3 orders of magnitude lower for bromine, taking global mean sulfur / halogen ratios into account (Aiuppa et al., 2009).
Bromine from various sources (e.g., polar regions, salt lakes, and volcanoes) is involved in tropospheric and stratospheric ozone depletion (e.g., Wennberg, 1999; Rose et al., 2006; Simpson et al., 2007). Tropospheric ozone depletion has also been observed in volcanic plumes (e.g., Hobbs et al., 1982; Kelly et al., 2013; Surl et al., 2015; Roberts, 2018), which supports the proposed reaction mechanisms for BrO formation via autocatalytic chain reactions. Recent observations of halogen oxides by satellites (e.g., Theys et al., 2009; Carn et al., 2016) and aircraft missions (Heue et al., 2011) confirm that some large volcanic eruptions may inject a proportion of their volcanic halogens into the free troposphere or even to the stratosphere, thereby confirming their potential impact on stratospheric ozone. In addition to the effects of volcanic degassing on atmospheric chemistry, measurements of volcanic emission have become an important and well-established tool in the assessment of volcanic hazard, and gas monitoring is used at many volcanoes around the world (e.g., Carroll and Holloway, 1994; Aiuppa et al., 2007; de Moor et al., 2016).
It has been also observed that the BrO SO2 gas ratio changes with the activity of volcanoes. Bobrowski and Giuffrida (2012) observed lower BrO SO2 ratios in Etna's plume during eruptive phases. Moveover, long-term DOAS observations by Lübcke et al. (2014), who used stationary spectrometers within the Network for Observation of Volcanic and Atmospheric Change (NOVAC; Galle et al., 2010), showed a decrease in the BrO SO2 ratio before and during explosive activity at Nevado del Ruiz volcano. More recently, a study by Dinger et al. (2018) at the Cotopaxi volcano (Ecuador) showed low BrO SO2 ratios at the beginning of eruptive activity compared with higher ratios present during declining volcanic activity. Finally, Warnach et al. (2019) found a low BrO SO2 ratio during high explosive periods and an increased BrO SO2 ratio during less explosive periods at Tungurahua volcano.
However, it is not yet clear whether the BrO SO2 ratio can be used as a robust diagnostic tool for forecasting volcanic activity. As BrO is a reactive secondary gas species, its concentration in a volcanic plume potentially depends on atmospheric variables such as humidity, oxidant abundance, solar radiation, and aerosol surface. The BrO SO2 ratio might not always or may only partially be controlled by the total bromine emission at a particular volcano under study (Roberts et al., 2018). Therefore, further knowledge of the chemistry that drives halogen activation is required.
Following the Introduction, an overview of the volcanic plume halogen chemistry and related model studies is given in Sect. 2. The comprehensive data set obtained during two field campaigns at Masaya volcano using several measurement techniques, including DOAS (e.g, Bobrowski et al., 2003), Multi-GAS (Shinohara, 2005; Aiuppa et al., 2006), alkaline traps (e.g., Wittmer et al., 2014), and gas diffusion denuder sampling (e.g., Rüdiger et al., 2017) is then presented in Sect. 3. In Sect. 4, we outline the use of an unoccupied aerial vehicle (UAV; e.g., Rüdiger et al., 2018; Stix et al., 2018) that enabled the sampling of a downwind plume for the investigation of halogen-induced plume-aging processes; this process is then reproduced by the atmospheric modeling of plume halogen chemistry. In Sect. 5, the outcome of the modeling is compared with the field measurement results before Sect. 6 draws the conclusions and provides an outlook for future studies.
Besides numerous field surveys at various volcanoes, several atmospheric modeling studies have been conducted that have improved our understanding of the complex chemical reactions in volcanic plumes marking the interface between volcanic trace gases (and aerosols) and ambient air. Two different models have been developed by researchers to simulate the in-plume chemistry, the microphysical marine boundary layer model (MISTRA; Bobrowski et al., 2007; von Glasow, 2010) and PlumeChem (Roberts et al., 2009; Roberts et al., 2014). While MISTRA is a 1-D box model including multiphase chemistry, PlumeChem additionally includes plume dispersion and 3-D simulation by employing a multiple grid box mode, and the multiphase chemistry is parameterized using uptake coefficients rather than being modeled explicitly in the aqueous phase. The models used in this study (MISTRA; PlumeChem; and Chemistry As A Boxmodel Application Module Efficiently Calculating the Chemistry of the Atmosphere, CAABA/MECCA) are initialized with the gas composition of a so-called “effective source region”. This gas composition is typically derived from a thermodynamic equilibrium model (HSC, Outotec, Finland; e.g., Gerlach, 2004; Martin et al., 2006). Different mixtures of magmatic gas and ambient air yield the hot gas mixture of the effective source region, which is quenched to ambient temperature and then mixed with ambient air including O3, OH, and NOx. The suitability of the HSC model to represent high-temperature chemistry in the plume has been debated, and it has recently been shown that the chemistry should be better represented by a kinetics model (Martin et al., 2012; Roberts et al., 2019). The limitations of HSC with respect to representing the high-temperature chemistry include the assumption of thermodynamic equilibria in the hot plume region, which is quite improbable. The choices for temperature, a mixing ratio of volcanic and magmatic gas, and a “quenching factor” are rather arbitrary and do not necessarily reflect reality. The kinetics model studies show that the timescale for substantial NOx to be formed thermally appears longer than a reasonable lifetime of a hot plume (Martin et al., 2012). Moreover, Roberts et al. (2019) showed differences in the predicted formation of HxOy in a kinetics model compared with those assumed by thermodynamics, in terms of magnitudes and speciation. Therefore, conclusions drawn from atmospheric modeling that depend on the HSC initializations are inherently limited by the uncertainties and limitation of HSC. Nevertheless, we follow the former studies using HSC, despite its limitations, because there is currently no kinetics model with a more comprehensive chemistry available as an alternative.
The initially emitted HBr is converted into reactive species via an autocatalytic mechanism, involving multiphase reactions, which constitute a so-called “bromine explosion” (von Glasow et al., 2009). Under ozone consumption, Br radicals – formed by high-temperature dissociation in the effective source region – react to BrO (Table 1, Reaction R1), which in turn reacts with HO2 or NO2 to form HOBr (Reaction R2) or BrNO3, respectively. A subsequent uptake into aerosol enables the conversion of HBr into Br2, which partitions into the gas phase and is photolyzed to give two Br radicals and start the cycle again (Reaction R5a). The self-reaction of two BrO to give two Br (with Br2 as a secondary product) and O2 is suggested to be the major ozone-depleting channel at high bromine concentrations, as in a young plume (von Glasow, 2009). Once HBr becomes depleted, the uptake of HOBr BrNO3 may promote the formation of BrCl, which also consumes O3 and forms reactive chlorine species such as ClO and OClO (Reactions R6, R8). The major reaction pathways that involve the formation and degradation of BrO in volcanic plumes are shown in Table 1.
An extensive review of the advances of bromine speciation in volcanic plumes including a comparison of different model approaches has recently been presented by Gutmann et al. (2018). In this study, we present in situ measurements along with remote sensing data on the activation of Br, Cl, and I in the volcanic plume of Masaya and further investigate the involved halogen species by atmospheric model simulations using the CAABA/MECCA box model. Although fluorine has been measured as well, it is not discussed in detail in this study due to the high water solubility and the non-reactivity of fluoride towards oxidation.
3.1 Site description and flight/sample strategy
Masaya volcano in Nicaragua is a shield volcano with a caldera size of 6 km × 11 km. The caldera hosts a set of vents, of which the Santiago pit crater, which formed in 1858–1859, is currently active (McBirney, 1956). Since mid-November 2015, the Santiago crater has contained a persistent superficial lava lake (∼ 40 m × 40 m). The lava lake has been associated with large volcanic gas emissions, making it one of the largest contributors of SO2 emissions in the Central American volcanic arc (Martin et al., 2010; de Moor et al., 2017; Aiuppa et al., 2018). Due to the high emission rates and the low-altitude ground-hugging plume, Masaya volcano has a severe environmental impact on the downwind areas, affecting human and animal health and vegetation (Delmelle et al., 2002; van Manen, 2014). With its easy accessibility by car and low altitude, the emissions of Masaya volcano have been studied extensively throughout the last decades. Of particular note is the establishment of molar halogen-to-sulfur ratios, determined to be of the order of 0.3–0.7 for chlorine and 3 × 10−4 for bromine (e.g., Witt et al., 2008; Martin et al., 2010; de Moor et al., 2013). These halogen values are considered to be on the high end observed in magmas and plumes, yet they are rather typical for arc volcanism (Aiuppa, 2009; Gutmann et al., 2018). Measurements of reactive bromine species (BrO) have been reported in the past (Bobrowski and Platt, 2007; Kern et al., 2009). Continuous composition monitoring (by Multi-GAS) has been realized since 2014, and gas data for the onset of the superficial lava lake were presented by Aiuppa et al. (2018).
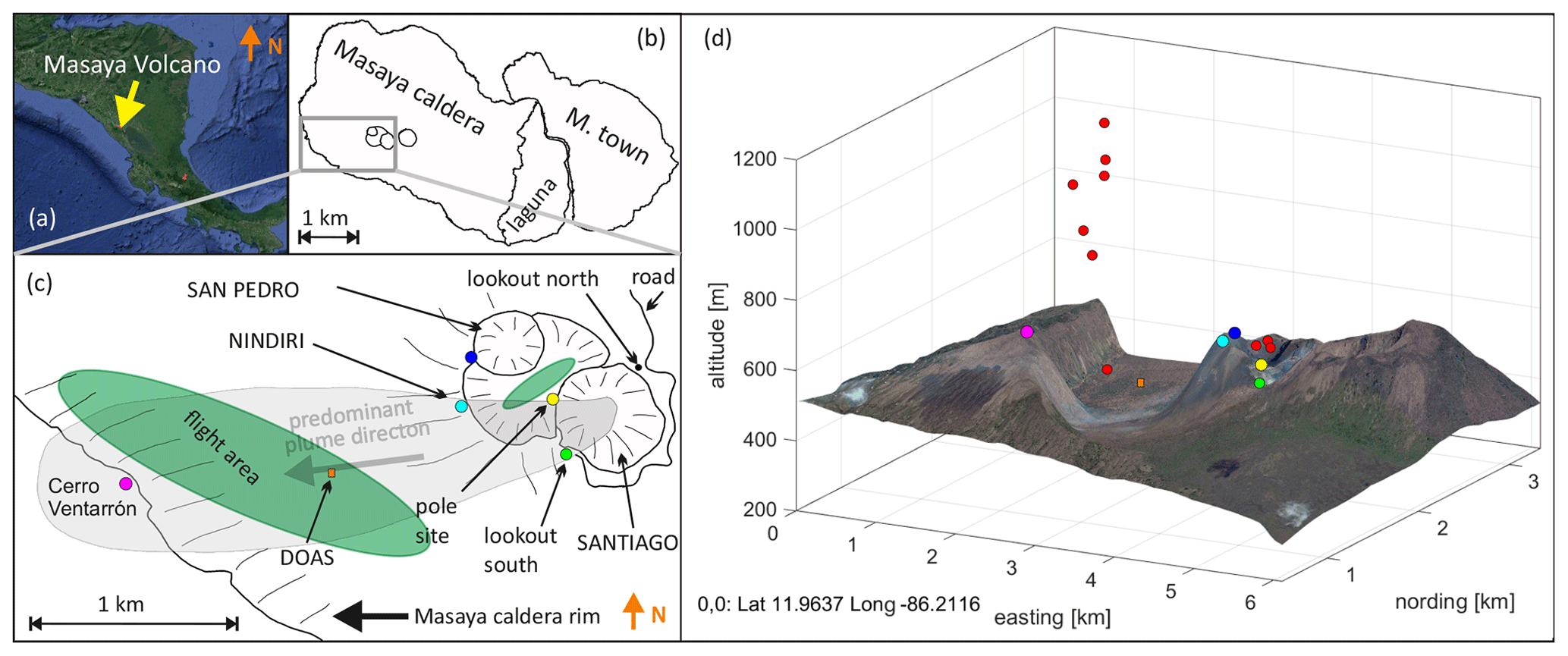
Figure 1(a) Location of the Masaya volcano in Central America (© Google Maps). (b) The Masaya pit crater system in the Masaya caldera. The flight area (green patch and red points) and sampling locations marked by colored circles in a sketched map (c) and 3-D plot (d).
In our field campaigns in 2016, UAV-based and ground-based sampling approaches were undertaken to study the plume of Masaya volcano with a focus on halogen emissions and atmospheric reactions of the emitted halogens. Samples were taken on the ground level at the edge of Santiago crater (Fig. 1) at two locations (lookout south and pole site), at the top of the rim of Nindirí crater (Nindirí rim) and at Cerro Ventarrón. UAV-based sampling was conducted in the plume hovering over the Nindirí crater and above the caldera bottom and caldera rim (red points in Fig. 1d). Using ground-based and UAV-based methods, the plume was sampled over a distance of about 2.8 km, covering an estimated age of 10 min, depending on the wind velocity.
3.2 Alkaline traps
Total halogen amounts were obtained by ground-based sampling using alkaline traps (Raschig tubes, RTs, and Drechsel bottles, DBs) (Liotta et al., 2012; Wittmer et al., 2014) at the locations marked in Fig. 1. The alkaline solution quantitatively captures acidic gas species, due to an acid–base reaction, and enables the determination of total halogens (F, Cl, Br, and I) and sulfur (S) concentrations. The sampled solutions were measured by ion chromatography (IC) and inductively coupled plasma mass spectrometry (ICP-MS) at the Geochemistry Laboratories of the Istituto Nazionale di Geofisica e Vulcanologia, Palermo (Italy). A 1 M NaOH solution was used in the RTs and a 4 M NaOH solution was used in the DBs. Both solutions were made from 99 % purity NaOH (Merck, Germany) in 18.2 MΩ cm−1 water. The plume samples were pumped through the RTs using a GilAir Plus pump (Sensidyne, USA) for about 1 h at 2.8–4 L min−1. Total volume data logging enabled mixing ratio calculation of the RT samples. A custom-built pump (without data logging) was used to pump approx. 1 L min−1 of gas through the DBs for between 18 and 30 h each. These samples were used for gas ratio comparisons over a longer time period.
3.3 Gas diffusion denuder sampling
Reactive halogen species (RHS) were sampled by gas diffusion denuder samplers using 1,3,5-trimethoxybenzene as a reactive coating (Rüdiger et al., 2017) on borosilicate brown-glass tubes with a diameter of 0.9 cm. An electrophilic substitution reaction occurs within this coating, effectively trapping halogen species with an oxidation state (OS) of +1 or 0 (HalX, e.g., Br2 (OS 0) or BrCl, BrNO3, or BrO (OS + 1)), which are considered as reactive species in contrast to the -1 OS species Br or HBr(g). However, it is not yet experimentally evidenced that the denuder technique is completely naïve to halogen radicals, although following the chemical reaction mechanism, the reaction of the denuder coating with halogen radicals is unlikely. Ground-based denuder measurements employed a serial setup of two denuders (2 × 50 cm) at a flow rate of 250 mL min−1 using a GilAir Plus pump and were conducted simultaneously to the RT sampling for 60 min to give the ratios of reactive species to total halogens (e.g., HalX Br) or total sulfur (e.g., HalX S). For the UAV-based sampling, a remotely controlled sampler (called Black Box) was used and is described in detail in Rüdiger et al. (2018). The typical sampling flow rate was about 180 mL min−1 for 5 to 15 min. The Black Box enabled logging of the sampling duration and SO2 mixing ratios via the built-in SO2 electrochemical sensor (CiTiceL 3MST/F, City Technology, Portsmouth, United Kingdom). Furthermore, the SO2 sensor signal was transmitted to the remote control, which helped to identify regions of high SO2 concentrations in real time. The SO2 signal of the sensor was time integrated over the sampling period of the denuders to derive the HalX S ratios at the location where the UAV hovered during sampling.
3.4 Unoccupied aerial vehicle sampler
The UAV used for this study is a small four-rotor multicopter with foldable arms (Black Snapper, Globe Flight, Germany) called RAVEN (Rüdiger et al., 2018). We achieved flight times of up to 15 min with a payload of approximately 1 kg, depending on the sampling setup. Global Positioning System (GPS) data of the flights were recorded on board using the micro-SD data logger (Core 2, Flytrex, Aviation, Tel Aviv, Israel) with a 2 Hz time resolution. The four batteries of the UAV were charged in the field with a car battery, enabling up to eight flights per day.
3.5 DOAS
DOAS measurements of SO2 and BrO were performed by a scanning-DOAS station from NOVAC (Galle et al., 2010), which is located approximately 1.5 km west-southwest of Santiago crater at an altitude of 387 m a.s.l. (above sea level, Aiuppa et al., 2018). This UV spectrometer records the intensity spectra of the diffuse solar radiation over a wavelength range from 280 to 450 nm for different viewing angles by scanning the sky from horizon to horizon at steps of 3.6∘. For most of the time, the volcanic plume transects the scan plane nearly orthogonally. The slant column densities (SCDs) are retrieved from these spectra via the DOAS method (Platt and Stutz, 2008). Due to the rather high BrO detection limit, spectral and arithmetical averaging is required for a reliable retrieval of the BrO SCDs and, ultimately, the calculated BrO SO2 molar ratios. As a drawback, the temporal resolution of the BrO and BrO SO2 data is reduced to a data point roughly every 30 min. For a detailed methodological description, see Lübcke et al. (2014) and Dinger (2019). Due to a data gap caused by an instrument outage, DOAS data for July 2016 were not available; therefore, the times series only covers the later part of the field study. The obtained BrO SO2 ratios were investigated for a period between 6 August and 30 September 2016. The plume age was estimated by employing wind speed data obtained at the airport of Managua (Iowa State University, 2018), 18 km north of Masaya volcano. With an estimated plume height of 600 to 1000 m a.s.l. above the DOAS instrument (observed by UAV measurements), an estimation for the plume transition path length from the vent to the zenith position above the DOAS was calculated to be about 1.4 km. This distance was used to estimate plume ages for BrO SO2 ratios, by dividing 1.4 km by the wind speed obtained for the respective BrO SO2 ratios. These calculations resulted in plume ages between 1.4 and 11.1 min (see Fig. S1 in the Supplement), which is reasonable compared to the estimated plume age for the UAV- and ground-based data. An average plume age for the 2.8 km distance (Cerro Ventarrón) to the vent was estimated to be 9.3 min using a speed of 5 m s−1. This wind speed was based on ground-based measurements with a handheld anemometer that were taken during the field campaigns at the rims of Santiago and Nindirí craters. It has to be stated that the wind speed was often unstable during the sampling procedures, and the applied data are based on our spot-check measurements are a source of uncertainty.
In order to compare the results of the field measurements of RHS with theoretical predictions, the CAABA/MECCA (Sander et al., 2011) box model was used. In its base configuration, CAABA/MECCA simulates the chemistry of an atmospheric air parcel. In this study, however, it was adapted to the conditions of a volcanic plume with a focus on the chemistry of the bromine, chlorine, and iodine. The atmospheric box model was initialized with the gas composition of the effective source region that was calculated by the thermodynamic equilibrium model HSC (HSC 6.1, Outotec, Finland) and then quenched with ambient air to start the atmospheric model, similar to earlier works (e.g., Gerlach, 2004; Bobrowski et al., 2007; Roberts et al., 2009, 2014).
4.1 Thermodynamic equilibrium model (HSC)
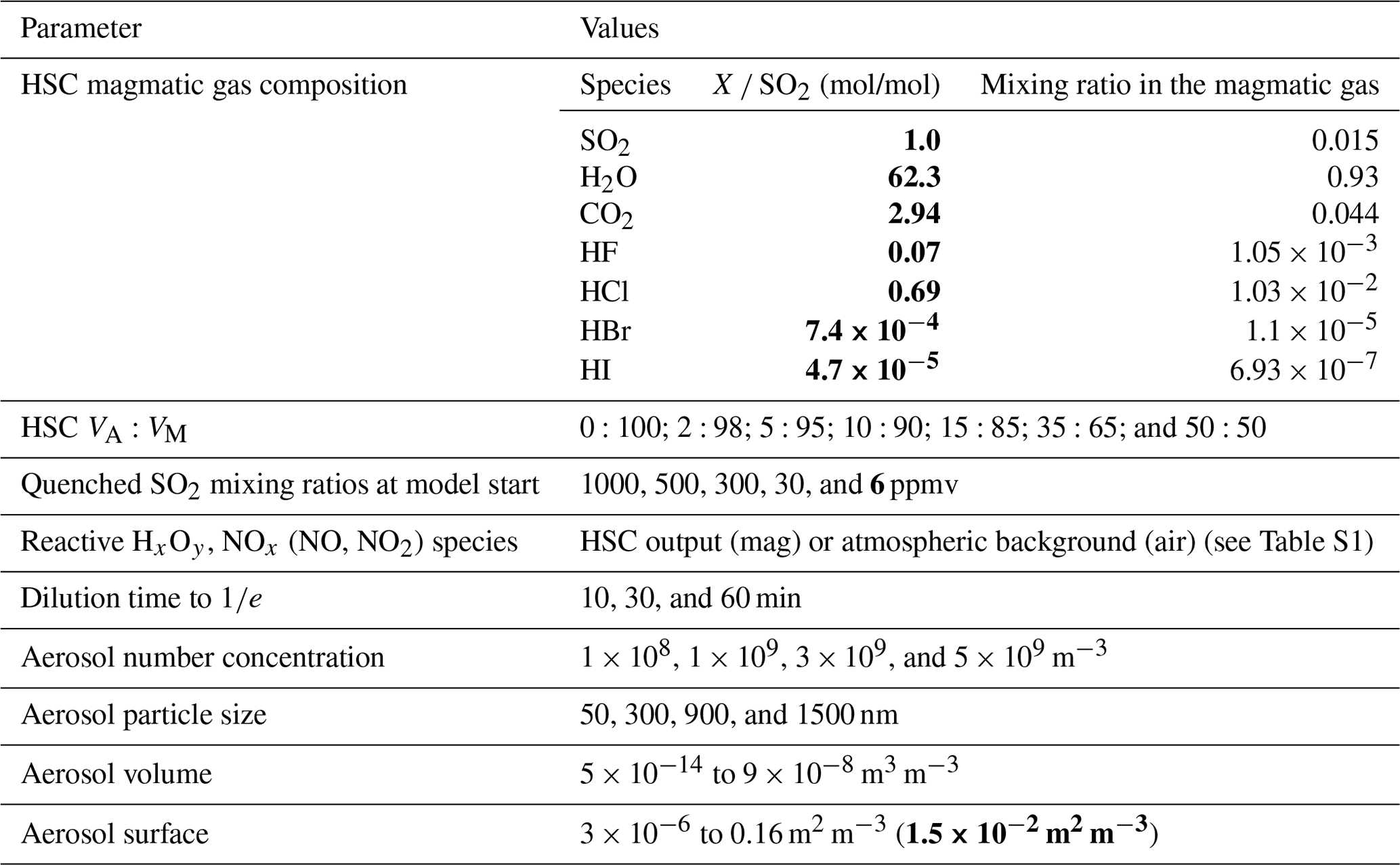
Data from field measurements in 2016 determined the initial conditions for the model runs. SO2, CO2, and H2O mixing ratios were derived from Multi-GAS measurements (de Moor et al., 2017; Rüdiger et al., 2018; Stix et al., 2018), and halogen amounts were derived from the alkaline trap sampling. The sums of all gas mixing ratios were set to 100 % to estimate the magmatic gas composition. H2S and H2 were not detectable (H2S SO2 ratio < 0.01) by the Multi-GAS measurements and were, therefore, neglected in the magmatic gas contribution. The high-temperature magmatic gas composition was mixed with different percentages of ambient atmospheric background air resulting in different atmospheric–magmatic gas ratios (VA:VM), according to the calculations of Martin et al. (2006). The atmospheric background gas composition was taken from Roberts et al. (2014) (who used atmospheric background data for the Etna volcano, Italy), as no detailed data on the atmospheric background composition at Masaya volcano were available, regarding trace gas species. The HSC model was reduced to produce gas species containing the C, S, O, N, H, F, Cl, Br, and I elements, and the temperature in the HSC model was arbitrarily set to 1000 ∘C. Similar to Roberts et al. (2009), this magmatic–ambient gas mixture of the effective source region was quenched with ambient air in order to obtain mixing ratios of all gas species (SO2 mixing ratios are used as a proxy) for the initialization of the CAABA/MECCA model (Fig. 2).
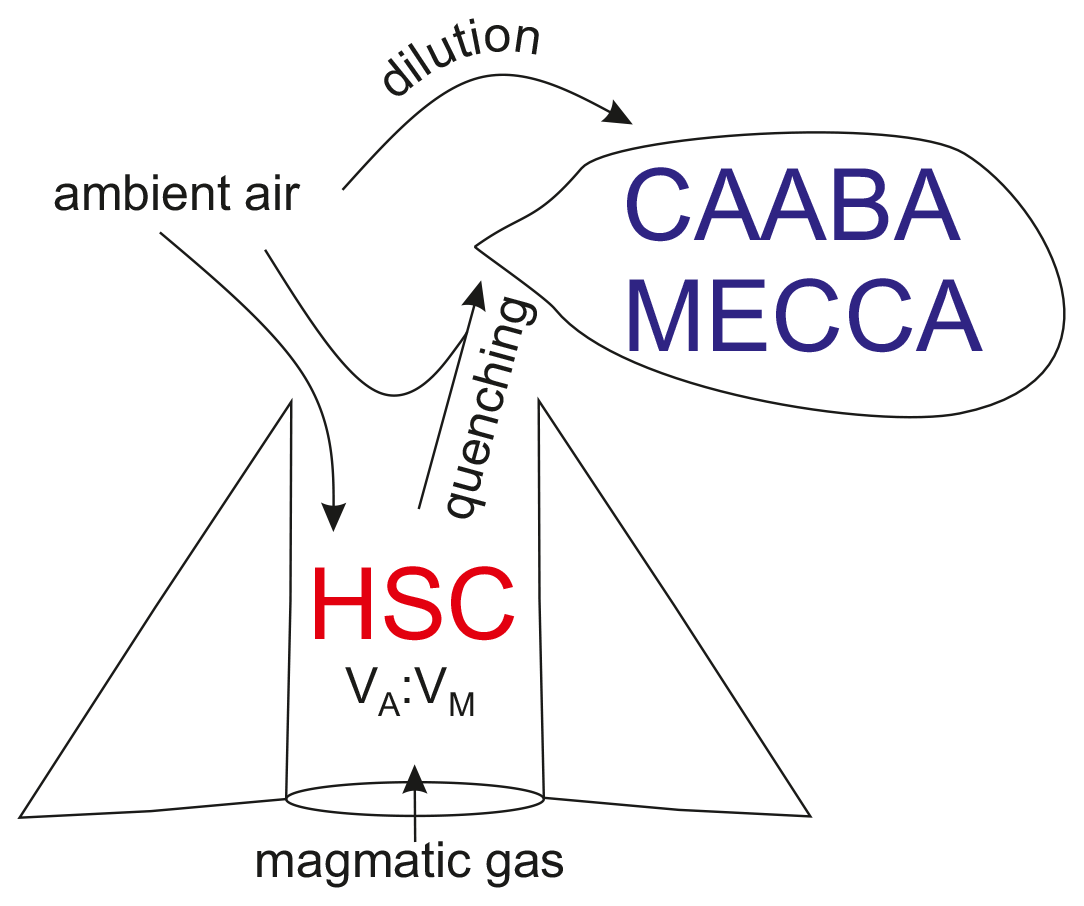
A list of the input and output species for the thermodynamic modeling of the high-temperature effective source region using the HSC model can be found in the Supplement along with the atmospheric background composition (Table S1).
4.2 Atmospheric box model (CAABA/MECCA)
The start point of the atmospheric chemistry box model was set to be within Santiago crater, so the plume reaching the crater rim has already experienced chemical reactions. Thus, we were able to compare our field measurement results with the model output.
Throughout the box model run, further dilution with the same ambient air mixture was employed to entrain oxidants into the plume. In the box model, the dilution was achieved by adding an amount of ambient air, mixing it, and then removing the same amount of mixed plume at a rate that achieves dilution to (0.37) over the dilution times listed in Table 3. For the dilution, the composition of the ambient air was the same as in Sect. 4.1 – taken from Roberts et al. (2014). Aerosol data were taken from optical particle counter measurements at Masaya (Stix et al., 2018), which showed a bimodal distribution of particles with diameters of 1.1 and 2.4 µm and PM2.5 values reaching up to 5.4 × 103 µg m−3. However, only a unimodal aerosol distribution was employed in CAABA/MECCA. The aerosol particle number concentrations and particle radii were varied in the model runs (see Table 2) to cover various particles' masses and surfaces, including those observed in the field measurements. The aerosol chemical composition was set to be a 1 : 1 sulfuric acid / sulfate aerosol composition with ion concentrations according to the Köhler equation (Laaksonen et al., 1998) and given radii, temperature, and relative humidity. Other parameters used in the CAABA/MECCA box model were a temperature of 298 K, a relative humidity of 80 %, and a pressure of 960 hPa. The actinic flux was determined using the JVAL photolysis module (Sander et al., 2014) initialized for solar noon on 1 August at Masaya's latitude (11.98∘ N). The runtime of the model was 25 min with a time step of 2 s. With the initialization of the model, particulate sulfur accounts for less than 1 % of the total sulfur content. Comparison of the measurement data with the box model data was conducted by identifying model scenarios that produce the formation and progression of the bromine species and their ratios to total bromine and sulfur amounts that are comparable to what was measured. In order to simplify the selection of model scenarios that fit best with the observations, a computational procedure was chosen that compares the fit of model results for the progression of selected ratios (e.g., BrX Br or BrO SO2) with the fit of the respective field measurements' data (the reader is referred to the Supplement for more details).

Table 3Thermodynamic (HSC) and box (CAABA/MECCA) model parameters. Bold text is used to represent parameters measured at the Santiago crater rim.
Two approaches were investigated: (1) molecular reactive bromine species BrX, including Br2, BrCl, BrNO2, BrNO3, HOBr, and BrO; and (2) all reactive bromine species (r-Br), which also includes Br radicals (r-Br = BrX + Br radicals; see Table 2). The progressions of the respective reactive bromine species (BrX and r-Br) were fitted over the estimated plume age, and the fit coefficients were compared with the coefficients from the field data to find the best agreement by minimizing the deviation of the respective fit parameters.
For samples taken on the ground, for example at the Santiago or Nindirí crater rims, the data include denuder (reactive halogens such as Br2, BrCl, or, in general, BrX and ClX) and RT samples (total halogens such as total bromine, Br, chlorine, Cl), whereas for aerial samples (e.g., caldera valley), RT data are not available. Ratios of the reactive halogens to sulfur or total halogen amounts were derived by employing the RT data and, in the case of aerial samples, data from the SO2 electrochemical sensor. A comparison of an RT sample simultaneously taken with Multi-GAS measurements resulted in 4.18 ± 0.22 ppm of SO2 in the RT sample and an average mixing ratio of 3.95 ± 0.20 ppm SO2 for the Multi-GAS data. Based on Multi-GAS measurements conducted during the field campaign, which showed no presence of H2S, it is assumed that the sulfur content of the alkaline trap samples originates mostly from SO2. Particulate S could also be entrained by the alkaline trap, but it is thought to contribute only a minor fraction of total S, as shown for Masaya by Martin et al. (2010) (SO2 SO = 190). Therefore, we regard the alkaline trap sulfur and the electrochemically sensed SO2 as equivalent and use measured SO2 mixing ratios as a plume dilution marker. The uncertainties of the obtained ratios are derived by the propagation of the errors of the analytical procedure and the sampling parameters. This includes the errors of gas chromatography–mass spectrometry (GC-MS), IC, and ICP-MS measurements as well as uncertainties in the sampling flow rate and the solution volume.
5.1 Total halogens
Sampling activities for a period of 9 d in July 2016 and 5 d in September 2016 gave 36 sample sets consisting of different combinations of alkaline traps, denuder, and SO2 sensor data (see Table S2). The alkaline trap samples were analyzed by IC and ICP-MS: sulfur, fluorine, and chlorine amounts were obtained by IC, and bromine and iodine amounts were derived from ICP-MS analysis. The average molar (mol/mol) halogen to sulfur ratios (Hal S) from samples taken at the Santiago crater rim are 0.07 ± 0.03 for fluorine, 0.69 ± 0.08 for chlorine, 7.4 × 10−4 ± 1.7 × 10−4 for bromine, and 4.6 × 10−5 ± 1.0 × 10−5 for iodine (see Table 4).
Table 4Median halogen / sulfur ratios measured at Santiago crater and downwind at Nindirí rim compared with downwind samples from a denser plume S > 1 ppmv; the uncertainty is given as the median average deviation.
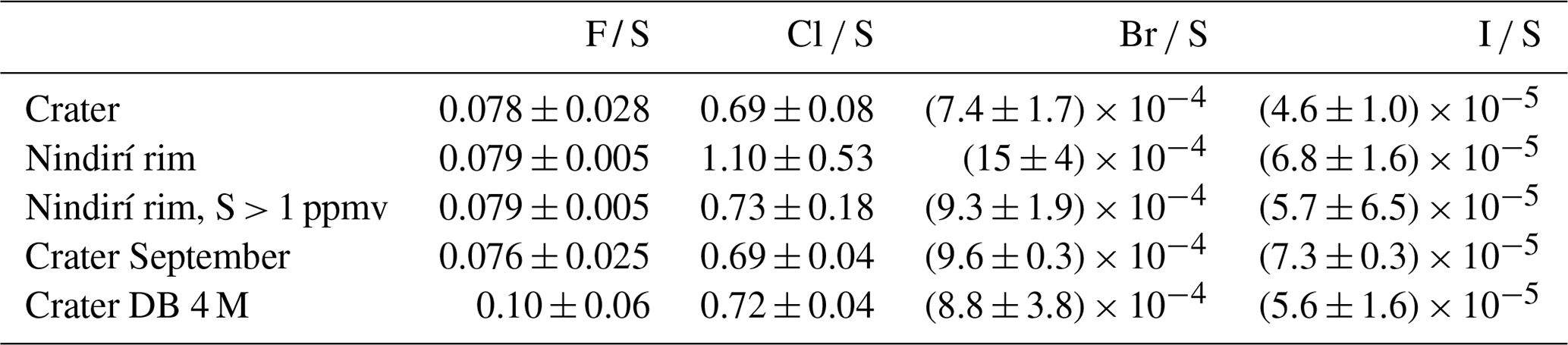
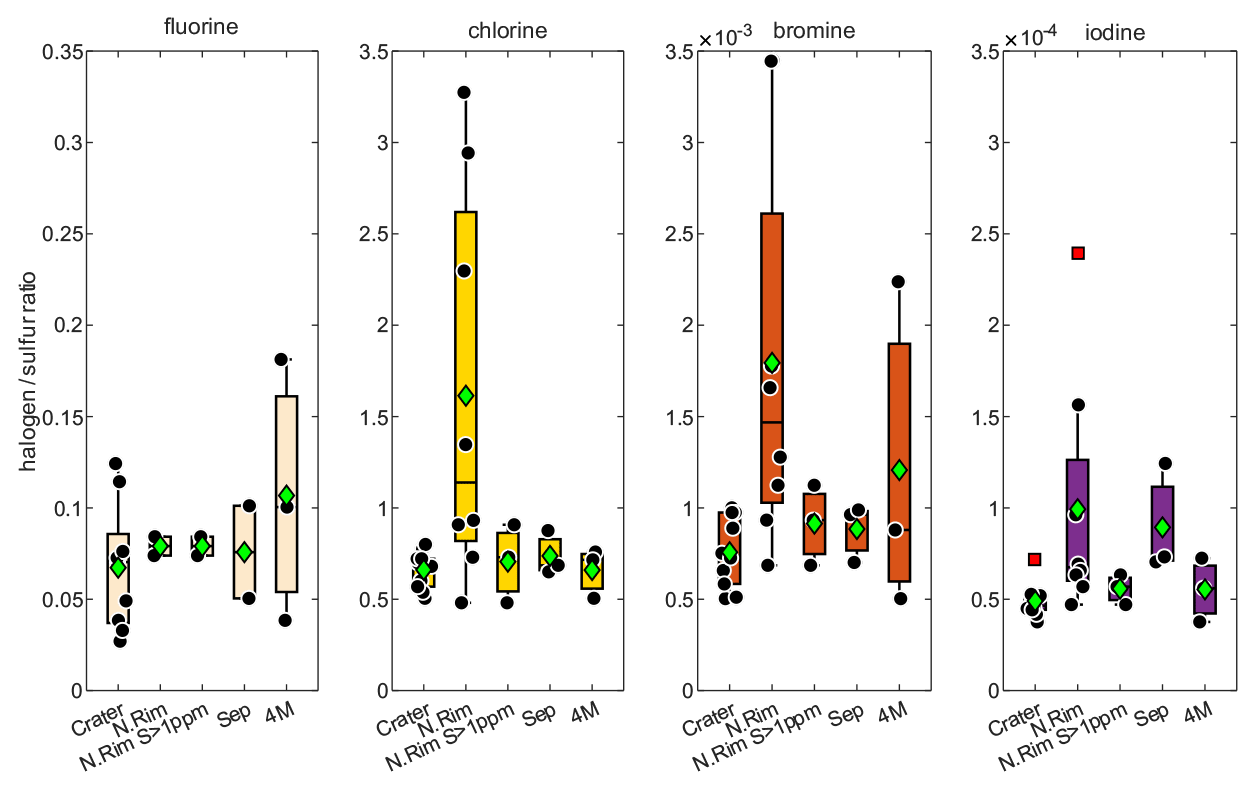
Figure 3Evaluation of the measured halogen to sulfur ratios for the Crater : Raschig tube in July at Santiago crater, the N.Rim : RT in July at Nindirí rim, the N.Rim S > 1 ppmv : RT in July at Nindirí rim without samples with less than 1 ppmv sulfur, the Sep : RT in September at Santiago crater, and the 4 M : Drechsel bottle samples in July at Santiago crater with 4 M NaOH solution. For each box, the black dots are the individual data points, the central mark is the median, the green diamond is the mean, the box extends vertically between the 25th and 75th percentiles, the whiskers extend to the most extreme data that are not considered outliers, and the outliers are plotted individually marked by red squares.
The halogen to sulfur ratios (Hal S) obtained during the field campaign were categorized into groups with respect to their different sampling location, date, and method (see Fig. 3). For fluorine, bromine, and iodine, the deviation within the 4 M NaOH solution samples (Drechsel samples) is larger than with chlorine. Whereas chlorine shows only a 6 % deviation, deviations of 29 % for I, 43 % for Br, and 62 % for F are determined for the samples taken with DBs. The 4 M solutions were typically left at the site for overnight sampling (18–24 h). During this time, precipitation events might have affected the incorporated plume Hal S ratios due to the different scavenging efficiencies for each halogen compound and the water solubilities of the respective gases. Duffel et al. (2003) also reported a high variability in HF data (obtained by open-path Fourier transform infrared spectroscopy; OP-FTIR) and associated it with scavenging of the soluble HF by a condensed plume during overnight measurements. Different deposition rates of particulate and gas phases could cause a loss of particles prior to sampling and, therefore, a larger variability in the Hal S ratios in overnight samples. Regarding the sampling location, at a first glance, a difference in the Hal S ratio can be noted between the Santiago crater rim and Nindirí rim. However, excluding samples that were taken under rather diluted plume conditions with a mean sulfur mixing ratio of < 1 ppmv, the difference in the Hal S ratio between the Santiago crater rim and Nindirí rim becomes negligible. While halogen and sulfur amounts in the considered samples were above the detection limit, one potential reason for discrepancies in the ratios may derive from contamination by entrained ash in the RTs, although attention had been paid to pointing the RT entrance away from the source. With lower overall plume enrichment in certain samples halogen-laden particle uptake could be a reason for an increased Hal S ratio in the Nindirí rim samples. A significant change in the I S ratio is observable between July and September (higher values in September), whereas the other Hal S ratios do not change largely over this period (see Fig. S6). Using an average flux of 3029 ± 1396 t d−1 (1σ) of SO2, obtained by car DOAS traverses during the field work in July 2016 (de Moor et al., 2017), the following halogen fluxes were calculated for July 2016: 66 ± 40 t d−1 of HF, 1190 ± 130 t d−1 of HCl, 2.8 ± 0.7 t d−1 of HBr, and 0.28 ± 0.06 t d−1 of HI. The cumulative error of the halogen fluxes is derived from the propagation of the SO2 flux and the halogen / sulfur ratio uncertainties. The data set for halogens obtained in this study complements the measurements (filter packs) by Witt et al. (2008) in 2006 and Martin et al. (2010) in 2009 and is the first detailed data set on halogens for Masaya since the appearance of the lava lake (see Table 5). The Cl S ratio is within the uncertainty of that reported by Martin et al. (2010) and twice as high as Witt et al. (2008). While the 2016 F S ratio was about half of what was measured in 2009, the abundances of the heavier halogens have increased by a factor of ∼ 2 for Br and ∼ 3 for I compared with 2009; therefore, they are significantly higher than the ratios in 2006 and 2009. Based on the CO2 SO2 ratio, Aiuppa et al. (2018) reported increased CO2 emissions associated with the increased level and extension of the lava lake. For the period prior to the appearance of the lava lake, the CO2 SO2 ratios were already higher than the ratios reported for 2006 and 2009 and those determined for the end of July 2016 until March 2017. Aiuppa et al. (2018) presented an average CO2 SO2 ratio of 5.5 ± 1.9 during the period of our measurements compared with 3.4 ± 0.5 in 2006 (Witt et al., 2008) and 2.7 ± 0.3 in 2009 (Martin et al., 2010) or 2.3–2.5 in 1998 (Burton et al., 2000). Alongside the halogen measurements in this study, CO2 SO2 ratios were obtained in July 2016 (de Moor et al., 2017; Rüdiger et al., 2018) that represent rather short snapshot periods of few hours and resulted in values of 4.0 ± 0.6 and 3.6 ± 0.6, respectively – so lower than the average of Aiuppa et al. (2018). Compared with 2006 and 2009, the increased Br S and I S ratios in 2016 go along with observations of increased CO2 SO2 ratios (Table 5; Aiuppa et al., 2018), whereas Cl S ratios in 2016 are similar to 2009. Aiuppa et al. (2018) pointed out that the formation of the lava lake was associated with upward magma migration and deep rising CO2-rich gas bubbles. Observations by Bobrowski et al. (2017) hinted at the potential increase in the ratio of heavier halogens to sulfur with higher CO2 SO2 ratios; conversely, Cadoux et al. (2018) presented laboratory experiments with melts that indicated a contradicting behavior.
Table 5Halogen / sulfur and inter-halogen ratios of Masaya's gas emissions between 2006 and 2016. “n.d.” denotes not detected.
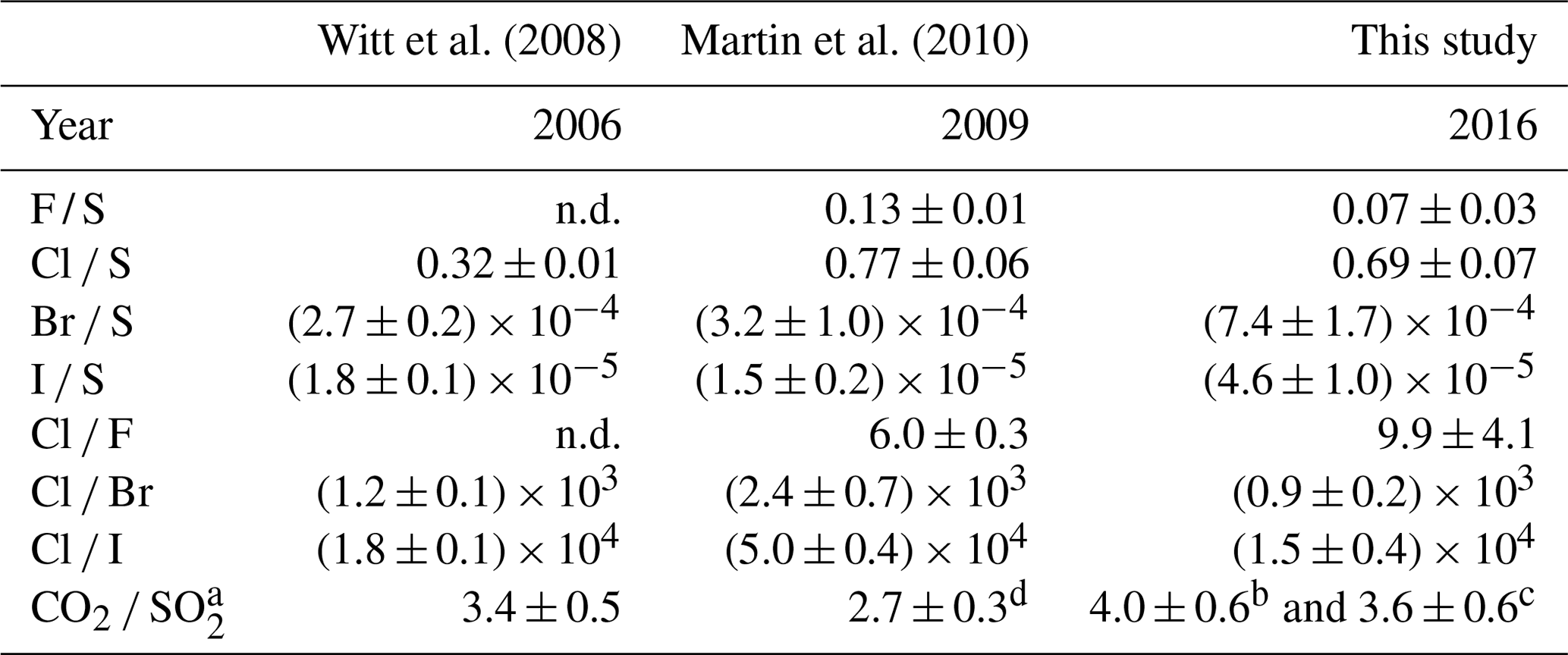
Halogen data from Witt et al. (2008) and Martin et al. (2010) were derived from filter pack measurements. a Multi-GAS (MG) data. b MG data from de Moor et al. (2017). c MG data from Rüdiger et al. (2018). d OP-FTIR data.
5.2 Reactive halogens
Reactive halogens were measured by gas diffusion denuder sampling. The reactive halogen data are categorized by their sampling location, and the median of the species ratios for each location was calculated along with their propagated uncertainties (Table 6). For each location, a distance to the vent was estimated based on the path of the downwind-drifting plume and GPS data of the sampling locations. For aerial samples, the GPS coordinates of the highest measured SO2 mixing ratios were chosen as a representative location. The uncertainties in the distance were estimated for each location based on the spatial distribution of the respective samples. Figure 4 shows the reactive halogen / sulfur (HalX S) and reactive halogen / total halogen (HalX Hal) ratios as a function of the distance to the vent. For bromine, whose activation in volcanic plumes has been studied extensively in the past (e.g., Oppenheimer et al., 2006; Bobrowski et al., 2007; Bobrowski and Platt, 2007; Bobrowski and Giuffrida, 2012), an increase in the BrX S and BrX Br over distance and, therefore, the plume age is clearly observable. The ratio of BrX S increases from (1.3 ± 0.6) × 10−4 at the Santiago crater rim up to (20 ± 14) × 10−4 at a downwind location above the caldera valley (red dots in Fig. 1). Furthermore, the BrX Br ratio increases from 0.20 ± 0.13 at the Santiago crater rim to 0.76 ± 0.26 at Cerro Ventarrón (purple dot). The ratios obtained for the Santiago crater rim are in the range of what was recently measured at the Etna (Rüdiger et al., 2017), Nyamulagira (Bobrowski et al., 2017), and Stromboli (Rüdiger et al., 2018) volcanoes using the same method and at similar distances from the vents. Although the Br S ratio increased from 2009 to 2016, the average BrO SO2 ratio from August to September 2016, 3.4 × 10−5, is similar to observations by Kern et al. (2009) (average of 3.0 × 10−5), who conducted measurements at the crater rim of Santiago crater in 2007. However, measurements of BrO SO2 ratios in 2003 were significantly lower – about 1.1 × 10−5 (Platt and Bobrowski, 2015).
Table 6RHS / sulfur and total halogen ratios at five different locations and estimated plume ages at 5 m s−1 wind speed (median values). “bdl” denotes that the concentration was below the detection limit.
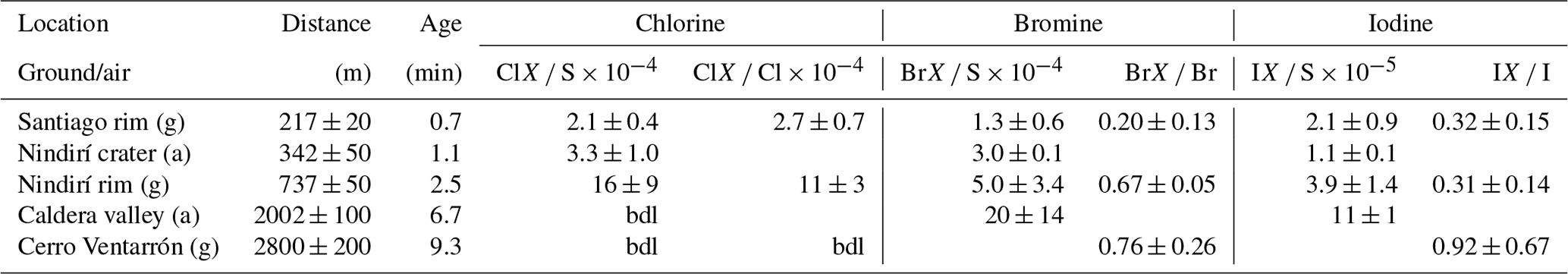
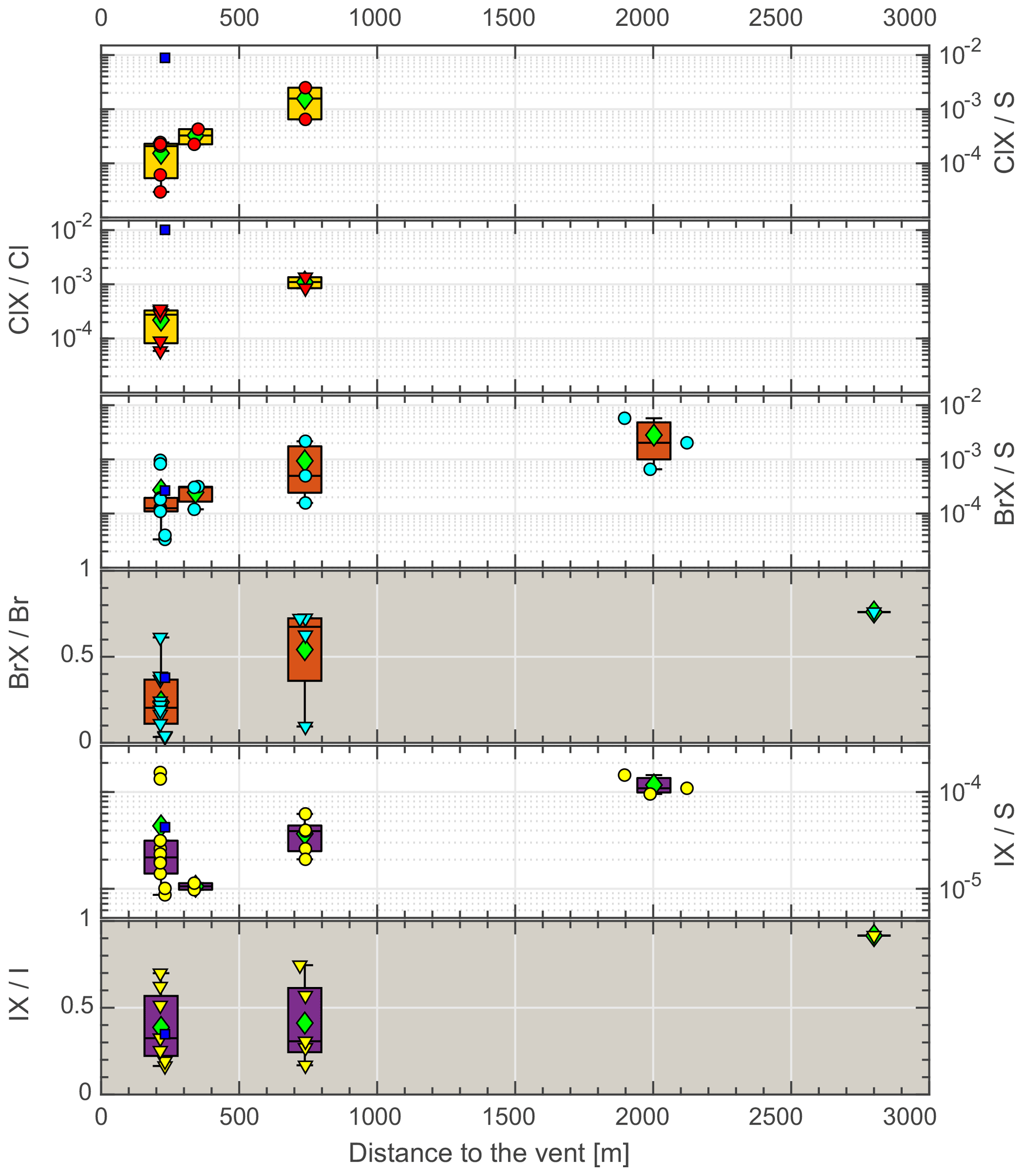
Figure 4Reactive halogen species (RHS) progression during plume aging. Note the non-logarithmic scale (gray background). Circles denote the Hal S ratio, triangles denote the HalX Hal ratio, diamonds denote the mean, the boxes show the 25th and 75th percentiles and the median, blue squares denote night samples (not included in mean and median calculation).
Due to technical problems causing an instrument outage, DOAS data were only available for the period shortly after the field campaign in July 2016, but we assume that no significant changes in the degassing behavior occurred between July and September 2016, which is plausible in light of the stable CO2 SO2 gas ratios presented by Aiuppa et al. (2018) for that period. Therefore, a comparison of the BrO SO2 and BrX S ratios is feasible. For the plume measured by the DOAS instrument in the zenith orientation, we estimated a distance to the vent of 1.4 km and employed wind speed data to derive an estimated plume age, which is presented in Table 7. By using those data, BrO accounts for approximately 10 % of the reactive bromine species for plume ages (< 5 min.) (see Fig. 5). The apparent increase in the BrX S ratio after 6 min needs to be regarded with caution, as the downwind BrX S ratio was obtained in a diluted plume, which is a source of uncertainty due to low SO2 mixing ratios. The denuder sampling enriches reactive species on the coating and, therefore, achieves a better detection limit with time. The electrochemical sensor signal, on the other hand, is limited by the instrumental detection limit, and time-integrated SO2 mixing ratios may not include phases in which SO2 was present but was below the limit of detection (0.1 ppmv). Therefore, SO2 might be underestimated, and the BrX S ratio might be overestimated in the diluted plume. These uncertainties are included in the error estimation given by the error bars of this respective sample. The farthest downwind BrX S ratio was obtained by a calculation that employs the BrX Br ratio and the total Br S ratio, due to sulfur being below the detection limit with the IC method. The propagation of the respective uncertainty is included in the error bars as well. However, as the uncertainties are known and estimated, the resulting BrX S progression still provides insights into the chemistry of the plume. The comparison with the BrO SO2 ratios underlines the postulated reaction mechanism, in which BrO is an intermediate product that further reacts to form other reactive species, as shown by the reaction paths in Table 1. Therefore, BrO might reach a rather steady state of being formed and reacting with other compounds (e.g., Bobrowski and Giuffrida, 2012; Gutmann et al., 2018).
Table 7BrO SO2 ratios from a stationary NOVAC DOAS instrument at the estimated plume ages obtained from wind speed (see Sect. 3.5 and Fig. S2). Values marked with “*” are single data points with the uncertainty of the single measurement. The other uncertainties are derived from multiple measurements and the respective standard deviation.

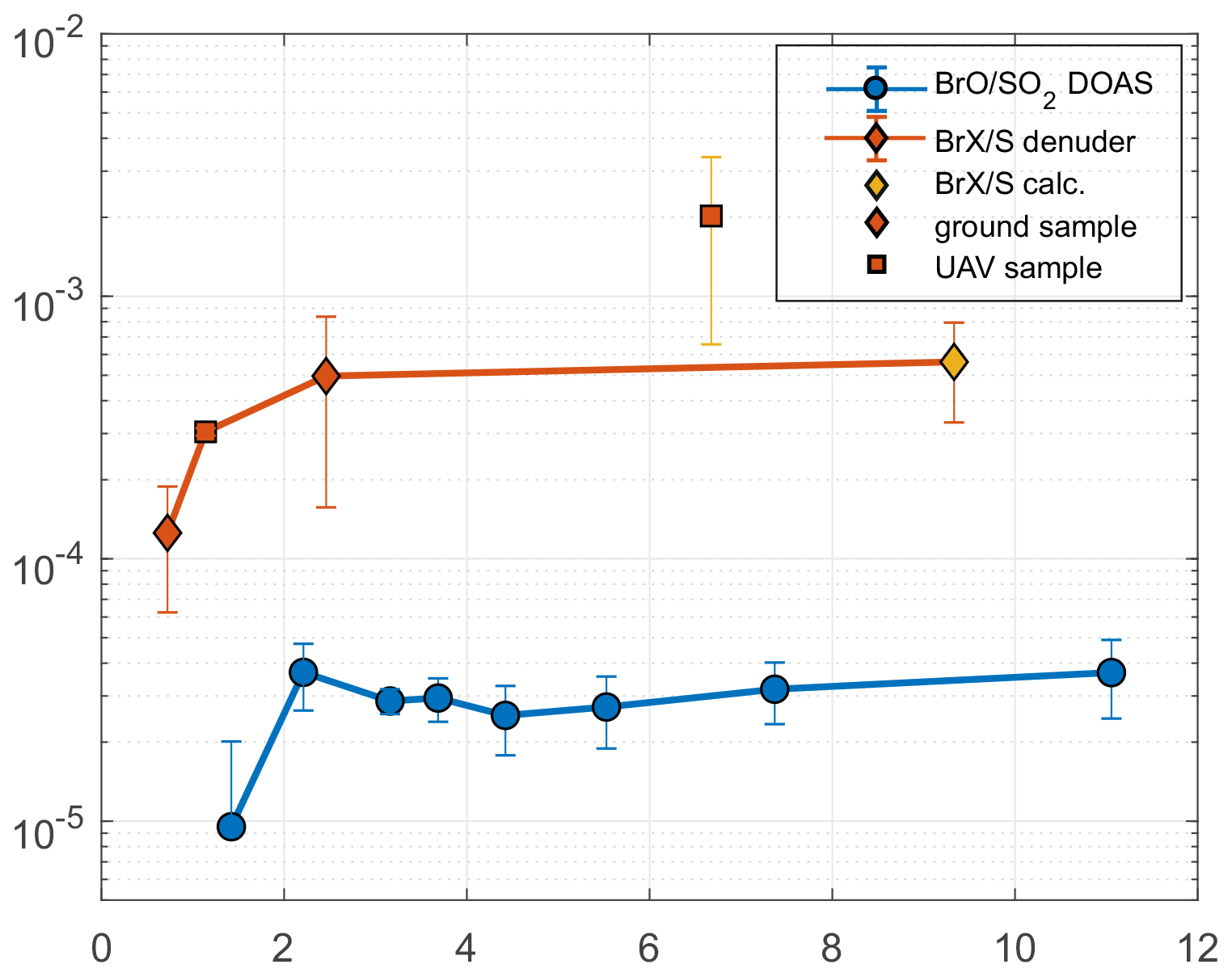
Figure 5Average RHS / S ratios with plume age: BrO SO2 ratios measured by a stationary DOAS instrument and BrX S ratios obtained by ground- and aerial-based denuder sampling; the calculated BrX S value was derived from the BrX Br ratio at this plume age and Santiago crater rim Br S value. The UAV sample could be affected by the underestimation of SO2 (see text).
Furthermore, an activation of chlorine was observed, which has also been detected in the past by remote sensing techniques at other volcanoes but not at Masaya (e.g., Lee et al., 2005 – Sakurajima; Bobrowski et al., 2007 – Etna; Donovan et al., 2014 – Soufrière Hills; Gliß et al., 2015 – Etna). Regarding reactive chlorine ClX (Cl2, ClNO2, ClNO3, HOCl, ClO, and OClO), both the ClX S and ClX Cl ratios increase with distance from the vent. While bromine activation is widespread, chlorine activation is only observed to be of the order of 10−4 of total chlorine. Due to the higher total chlorine abundance ClX, mixing ratios still show similar values to BrX. Roberts (2018) pointed out that significant Cl activation would only occur if HBr had already been transformed to BrX; in this case, the reaction channels (R4b), (R5b), (R6), and (R8), which lead to the activation of chlorine via BrCl and Cl radicals to form ClO with ozone and OClO from ClO and BrO, would play a more important role. Recently, Kern and Lyons (2018) observed a lack of OClO in the center of a volcanic plume by DOAS measurements, while it was increased (relative to SO2) at the edges. They attributed this observation to the incomplete activation of Br in the plume center and the dominance of Br2 formation (Reaction R4a) over BrCl formation (Reaction R4b) with an undepleted reservoir of particulate Br. However, Kern et al. (2009) did not detect ClO nor OClO in the plume of Masaya close to the vent, but they presented a detection limit for ClO SO2 and OClO SO2 of 5 × 10−3 and 7 × 10−6, respectively. As the ClX S ratios potentially include ClO and OClO as reactive chlorine species, we applied a calculation shown in Kern et al. (2009, Eq. 2) to compare our results with their detection limit (for long-path DOAS). Under the assumption that ClX is made up of only ClO and no OClO, and BrO is 10 % of the measured BrX at the Santiago crater rim, a potential OClO SO2 ratio of 3.5 × 10−5 was calculated by employing the rate constant and photolysis frequency for the formation and depletion of OClO at an average SO2 mixing ratio of 6 ppmv at the Santiago crater rim measurement site. The calculated OClO SO2 ratio is above the estimated detection limit for OClO by Kern et al. (2009) by a factor of 5. With ClO accounting for 20 % of ClX, which is a high estimate according to the model run presented in Sect. 5.4.3 and Fig. 9, the OClO SO2 ratio (7 × 10−6) would be in agreement with the DOAS measurements conducted in 2007 by Kern et al. (2009).
As iodine is the least abundant halogen in volcanic gases, it has not been observed in many previous studies. The detection of reactive iodine in volcanic plumes was limited to one satellite-based study of IO (Schönhardt et al., 2017) in the plume of Mt. Kasatochi in 2008. Due to its low abundance, it is challenging to determine iodine in alkaline trap samples or denuder samples. The ratios of activated iodine to activated bromine (0.16 at Santiago crater and 0.12 further downwind) are of the same order as those presented by Schönhardt et al. (2017) for IO BrO (0.09), and iodine activation shows a similar trend to bromine (Fig. 4). The IX I ratio increases from 0.3 close to the emission source to 0.9 about 10 min downwind, whereas the IX S ratio increases by a factor of 5 to 10 over the same distance (Table 6).
5.3 Nighttime sample anomaly
One simultaneous denuder and RT sample was taken during a nighttime visit to the Santiago crater rim in 2016 (sampling duration approx. 1 h). This sample shows an anomalous value for reactive chlorine (see Fig. 4). The values for reactive bromine and iodine are similar to those measured at the same location during the day. One possibility is that this value is an artifact caused during sampling or analysis. However, the absolute signal of 1-chloro-2,4,6-trimethoxybenzene (Cl-TMB) on the respective denuder was above the average signal produced by the highest concentrated calibration standard. Therefore, contamination by a calibration standard during the analysis can be ruled out. As the sample was measured in triplicate, random instrument error is also unlikely. Potential contamination by a different chemical compound during sampling in the field or in the laboratory would have needed to produce the same retention time and ratio as Cl-TMB, which we assume to be unlikely.
A potential explanation for this high reactive chlorine value is the enhancement of chlorine species at night that are otherwise photolyzed at daytime. A fraction of the HalX species is already formed by high-temperature reactions on the surface of the lava lake (e.g., Br2 and Cl2) (Martin et al., 2006) and can be measured at the Santiago crater rim without involving photochemistry. For example, the HSC equilibrium model (see Sect. 4.1) for bromine speciation at 1000 ∘C gives a Br2 HBr ratio of 10−4 for an air entrainment into plume gas of 2 %–5 %, which is in the range of the nighttime sample. Regarding reactive chlorine, the HSC model predicts a substantial fraction of Cl atoms (0.1 % of HCl). The Cl atoms can react with each other to form Cl2 by recombination (Hippler and Troe, 1976), which is more than 2 orders of magnitudes faster than the reaction of Cl with methane (Bryukov et al., 2002). Therefore, formation of Cl2 in the cooled plume gas mixture, after the emission at the lava lake surface and prior to sampling at the Santiago crater rim, would induce a larger nighttime signal compared with daytime, with photolysis occurring under the assumption that elemental chlorine is not trapped by the currently used denuder coating (Rüdiger et al., 2017).
5.4 Comparison with box model results
A two-stage chemistry modeling approach (see Sect. 1) was applied to analyze the field observations. The two major objectives were (1) investigating the field data for plausibility and (2) applying the CAABA/MECCA box model in the field of volcanic plume chemistry. The output of the HSC model gives 110 gas species; from these, 42 were used as input for the CAABA/MECCA box model and are listed in Table S1 (also including the halogen radicals). By iterating the various parameters for the model start conditions that are shown in Table 3, 3816 different model runs of were performed. Each model run simulated the progression of the set of model species during the first 25 min. As reactive bromine species are the most thoroughly studied reactive halogens in volcanic plumes, this section is mostly focused on Br. The measurement data (denuder) give the sum of reactive bromine species, and the model data provide detailed speciation information. The comparison was conducted by a script-based routine, which was required to evaluate the large number of model runs. The routine compares the fit parameters of the progression of the measurement data with the respective model speciation output for bromine (see Sect. 4.2 and Fig. S2). Several ensembles of start parameters match the measurement data with good agreement. Although the progressions of BrX Br and r-Br Br deviate in certain model runs quite substantially, only the BrX Br progressions are discussed in more detail for the sake of clarity. Please see the Supplement for more details on r-Br Br progressions.
5.4.1 Bromine chemistry
For the BrX Br progression, the best matching scenarios are presented in Fig. 6 (dashed lines). The BrO SO2 progressions were also fitted separately, and the best matching model runs with regards to BrO SO2 are presented as well (dotted lines). In Fig. 6, the solid lines represent the model runs that show the smallest deviation between the measured data and modeled data for both the BrX Br and BrO SO2 ratios.

Figure 6The temporal evolution of different modeled bromine species' ratios (molar) over the model time of 25 min with the respective measurement data (if available). Each subfigure shows the model runs that are closest to the measurement data, derived by the fit comparison.
BrX Br progressions
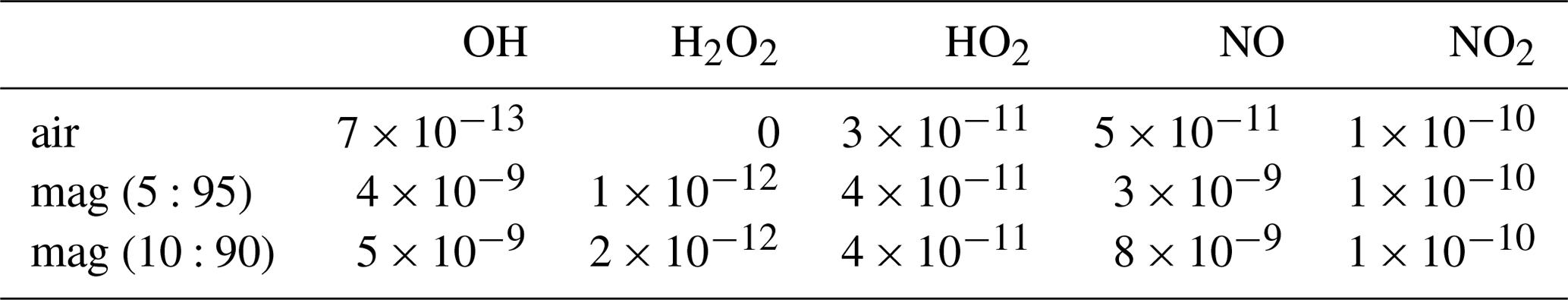
Figure 6 shows the progression of the bromine species over the model time of 25 min and the corresponding field measurements. The best matching model scenarios imply atmospheric gas to magmatic gas ratios (VA:VM) of 15:85 or less. Roberts et al. (2014) considered a VA:VM of 10:90 or less as likely for a system like the Etna volcano. For Masaya volcano with its active lava lake, we consider a VA:VM of up to 15:85 as realistic with respect to the potentially stronger mixing of atmospheric and magmatic gases at the lava lake–atmosphere interface. Besides the initial halogen speciation, changes in VA:VM also affect the initial HxOy NOx mixing ratios (see Table 8). The model runs were either conducted with the HxOy NOx mixing ratios produced by the HSC model (magmatic scenario: more HxOy NOx) or the respective atmospheric background mixing ratios (atmospheric scenario: less HxOy NOx) at the model initialization. Therefore, the best matching model runs are shown for both initial magmatic (red lines) and atmospheric (blue lines) HxOy NOx scenarios in Fig. 6. Roberts et al. (2014) has already discussed the discrepancy between a kinetically limited formation of NOx from background N2 and the contradicting observations of HxOy NOx compounds at the crater rim (e.g., Oppenheimer et al., 2010; Carn et al., 2011; Martin et al., 2012). The HSC model might overpredict NOx, as its formation is kinetically limited as a result of the high bond strength of N2 that is entrained in the plume. Roberts et al. (2014) alluded to the need for an alternative explanation for NOx at volcanoes, where it has been observed. In a recent study Roberts et al. (2019) presented a time-resolved chemical kinetics model for the high-temperature near-source chemistry of volcanic emissions that is an improvement on the HSC model. In contrast to HSC, Roberts et al. (2019) reproduced reduced gas species and high-temperature formation of HO2, OH, and H2O2 but has not yet included NOx chemistry. Therefore, two scenarios with magmatic and atmospheric HxOy NOx composition are investigated as extremes, representing the HSC output and the atmospheric background composition, respectively. It has to be noted that the chemical compositions of these two scenarios represent actual extremes rather than reality, which should be somewhere in between. As the HSC output is biased by the limitations of the model (see Sect. 2) and the choices of the initialization parameters used, the atmospheric background composition at Masaya volcano (boundary layer) might be different from the one used in this study taken from Roberts et al. (2014) for Etna volcano (free troposphere).
Table 8HxOy and NOx species used for the initialization of the CAABA/MECCA model runs.
BrX S progressions
For the progressions of the BrX Br and BrO SO2 ratios, model scenarios with a good agreement with the measurement data have been identified. However, for the BrX S ratios (Fig. 6b), the model underpredicts the field observations. In the modeled BrX S ratios, S represents the total sulfur content (all S species), which is constant, except for dilution. The prediction of the BrX Br progressions are in better agreement with the measured data. As mentioned above (Sect. 5.2), we think that the measured BrX S ratios could be overestimated due to the underestimation of SO2, especially in the samples obtained by UAV measurements, which can be explained as follows: an underestimation of sulfur could occur when the plume is diluted and SO2 is below the detection limit of electrochemical sensor. Under these circumstances, the sensor does not detect SO2 while the denuder is still trapping small amounts of reactive Br, resulting in an overestimated BrX S ratio. Other hypotheses addressing the discrepancy in observed and measured BrX S progressions could be related to the heterogeneity of the actual plume caused by “puffs” with high and low concentrations and, therefore, different chemistry compared with the modeled “bulk plume” or different background ozone (NOx, HOx) levels than that assumed for this study.
BrO SO2 progressions
The measured BrO SO2 progression (Fig. 6c) could be reproduced by various model runs with different VA:VM ratios, HxOy NOx mixing ratios, and initial start concentrations. The BrO SO2 ratio is only slightly overpredicted in the model runs that fitted best for the BrX Br progression. This overprediction is smaller for the model run with the magmatic HxOy NOxscenario (see Table 8). Potentially, a higher abundance of HxOy and NOx species could promote the loss of BrO to form HOBr and BrNO3. The model runs that show good agreement with the BrO SO2 progression (Fig. 6c) are able to reproduce the fraction of BrO of the total reactive bromine (5 %–15 %) (Fig. 6d) that is comparable to what was observed in the field data (∼ 10 %). Employing a conversion of total Br to reactive Br of 67 % for a plume age of 2.5 min (Table 6) and a BrO fraction on the BrX of 10 %, BrO accounts for roughly 7 % of the total bromine in our case. Recently, Gutmann et al. (2018) compiled data on the extent of the BrO Br fraction at Masaya and presented values of 5 %–15 % increasing with the distance to the crater.
Detailed bromine speciation
Figure 7 gives an overview of the detailed bromine speciation during the first 25 min of the model runs that fitted best with the observations of the reactive bromine progressions and the BrO SO2 data. For both approaches (with and without including Br radicals to the species measured by the denuders) one atmospheric and one magmatic scenario are presented in detail. It is noticeable that the BrCl fraction is more elevated in the scenarios with a magmatic HxOy NOx composition compared with the atmospheric scenarios, whereas the Br is less prominent. BrCl is photolyzed slower than Br2 (Maric et al., 1994); therefore, a larger fraction of BrCl, compared with Br2, could cause a slower formation of Br. The BrCl fractions might also be enhanced due to larger abundances of ClX, including ClO, OClO, and Cl2.
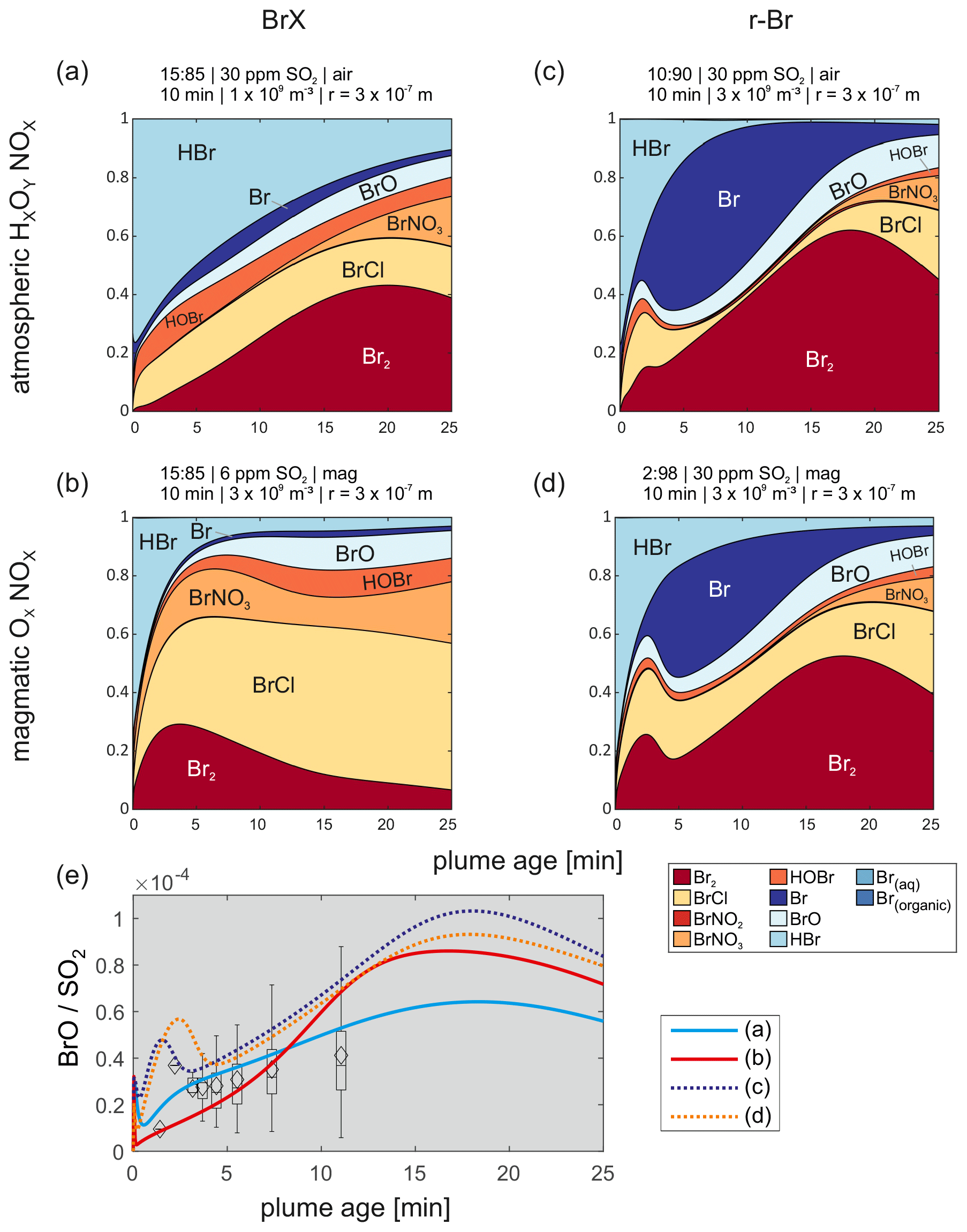
Figure 7(a–d) Detailed bromine speciation for the first 25 min of the four selected model runs closest to the measured BrX Br, r-Br Br, and BrO SO2 progressions. Four different model scenarios are shown, which fit best to the measurement data, using the progressions of BrX Br and r-Br Br with magmatic and ambient air oxidant amounts, respectively. Panel (e) shows the corresponding BrO SO2 ratios for the model runs in panels (a)–(d).
In all four runs in Fig. 7, the HBr mixing ratios decrease rapidly after initialization as it is transformed to reactive species or taken up by the aerosol. Less aerosol (particle number concentration and diameter) leads to a slower loss of HBr (Fig. 7a). Only a small amount of bromine is present as aqueous Br. Regarding the heterogeneous reaction mechanism of the bromine explosion, aqueous Br and HOBr(aq) are needed to form Br2, which is then emitted from the particle. As the modeled outgassing of Br2 is faster than the uptake of HBr and HOBr, bromide is depleted from the particles. At low levels of aerosol bromide, Reaction (R4b) produces BrCl and represents another sink for HOBr. Formation of BrCl by the mentioned Reaction (R4b) is also known for other systems, such as simulated ice surfaces (e.g., Fickert et al., 1999; Huff and Abbatt, 2000).
Comparison with other volcanic plume modeling
We compared our model study to earlier studies that applied the MISTRA (von Glasow, 2010) and PlumeChem (Roberts et al., 2014) models, which were based on volcanic plume measurements at Mt. Etna. All three studies succeed in simulating a bromine activation of the magnitude measured by our denuder sampling technique. However, the differences in the specific species making up reactive bromine are substantial. While similar to our results, PlumeChem reproduces a large fraction of Br2 as well as a local maximum in the young plume (∼ 5 min). MISTRA, on the other hand, shows a constant fraction of approximately 5 %–10 %. The modeled HOBr in PlumeChem is also in agreement with our CAABA/MECCA output for the first 10 min. However, regarding BrO, our finding is comparable to the results of MISTRA which produces less than 10 % BrO of the total Br in the young plume. PlumeChem, in comparison, models a varying BrO fraction of between 10 % and 50 % of the total Br, depending on the initial total Br S ratio and BrO reaching a fraction of ∼ 50 % of the reactive bromine in all its scenarios. Furthermore, the substantial contribution of BrCl that we simulated was not presented by MISTRA or PlumeChem. BrNO3 (formed by BrO and NO2), on the other hand, is present in PlumeChem and CAABA/MECCA, whereas MISTRA produces BrNO2 as a reactive bromine–nitrous species. In our model run in Fig. 7b, the fraction of BrNO3 is larger compared with the other scenarios, although NO2 mixing ratios are similar to the atmospheric background in the magmatic scenario. However, in Fig. 7b, NO is more abundant and can form NO2 under O3 consumption to react with BrO to BrNO3. Nevertheless, the formation of BrNO3 is limited by the photolabile NO2 and by a competitive reaction of BrO with HO2, ClO, and itself (Reactions R2, R7a, R7b, and R8 in Table 1). In order to explore the reason for those model differences in more detail, the reaction mechanisms used by the three models need to be compared, and similarities and differences in reaction rates require evaluation. This, however, lies beyond the scope of this work.
5.4.2 Input parameter sensitivity analysis
The so-called base run of the CAABA/MECCA box model, which encompasses the set of parameters from Table 3 that produced the most proximal model recreation of the field observations of the BrX Br was chosen to study the sensitivity of the model with respect to changes in the initial start conditions. These start conditions are
-
the initial volume ratio of atmospheric gas to magmatic gas (VA:VM),
-
the quenching factor (QF) for the initial quenching of the mixture of high-temperature gases to low-temperature conditions with ambient air (e.g., quenching to 6 ppmv SO2),
-
the dilution time (τ) of the plume, within the initial mixing ratio of an inert species gets diluted by a factor of (0.37),
-
the quantity of reactive oxidants HxOy and NOx,
-
the number concentration (NC) of particles per cubic meter,
-
the radii of the particles.
The model run shown in Fig. 7c was chosen as the model base run. This is based on two considerations. First, an atmospheric HxOy NOx scenario is more likely than a magmatic scenario due to the kinetically limited formation of NOx species by high-temperature chemistry in the effective source region. Second, even though the potential measurement of Br radicals by the denuder technique cannot be ruled out, the discrepancy between the progressions with and without Br radicals in Fig. 7c is still within the deviation of the measurement data. Therefore, the base run (the best fit in Fig. 7 with the r-Br Br progression; 10:90; 30 ppmv SO2; air; 10 min; 3 × 109 particles m−3; r = 3 × 10−7) was used with permuted start conditions to evaluate the influence of these conditions on the specific model outcome. The results of these tests are shown in Fig. 8.
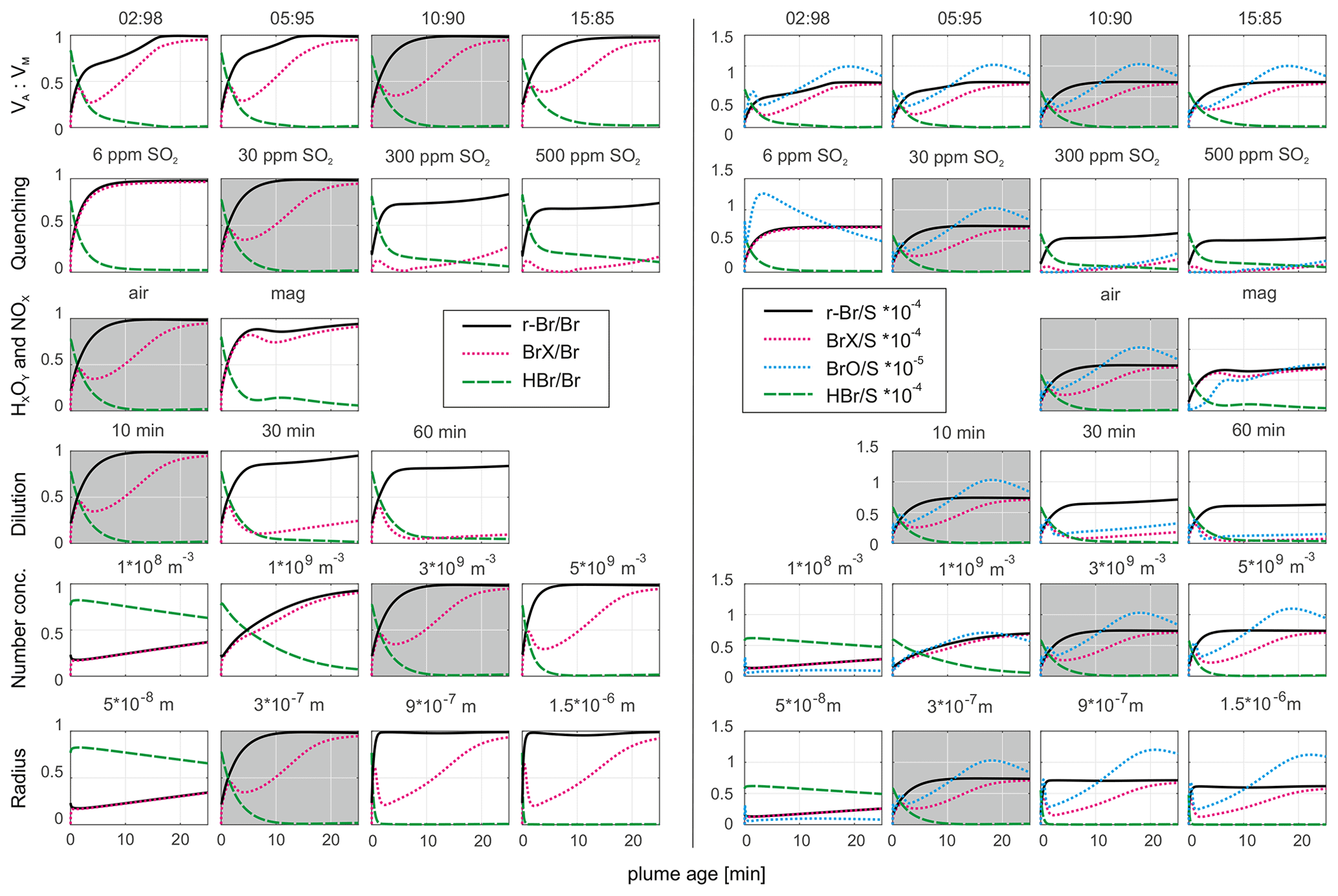
Figure 8The progression of selected bromine species over a model runtime of 25 min related to total bromine (left half) and sulfur (right half) abundances. The best fit to the measurement data from Fig. 7c (10:90; 30 ppmv SO2; air; 10 min; 3 × 109 particles m−3; r = 3 × 10−7 m) is shown using a gray background, and within the specific rows, the one model parameter is varied over different values in order to explore the model output answer to the model input change.
Changes in the initial VA:VM ratio (in the case of VM ≥ 85 %) has little effect on the BrX Br or BrX S ratios, whereas r-Br Br is slightly smaller for VM ≥ 95 %. In the VM ≥ 95 % scenario, potentially less Br radical is formed with less atmospheric gas in the HSC model. The QF shows a significant impact: in the base run a quenching to 30 ppmv SO2 reduces the presence of Br radicals and almost equals r-Br and BrX. Less quenching, indicated by higher SO2 mixing ratios, leads to less r-Br formation (relative to total Br) and increases the fraction of Br radicals. The relative BrO abundance is also reduced with less quenching due to the larger abundance of bromine in the modeled scenario, while the O3 entrainment remains fixed. Therefore, more Br is available to consume the same amount of O3, resulting in less relative formation of BrX. Likewise, a slower dilution results in slower in-mixing of O3 into the plume, causing the slower formation of BrX and BrO. The shape of the BrX species' progression in the scenarios with less quenching (30 to 500 ppmv SO2) and slower dilution time (30 and 60 min) is related to a substantial consumption of ozone in the modeled plume (see Fig. S5). In the 30 ppmv SO2 scenario, ozone is already decimated almost completely at around 2 min after plume release, causing a temporarily decrease in BrX followed by an increase, whereas the ozone concentration recovers. With a quenching to 6 ppm SO2, which actually corresponds to the measured value, sufficient O3 is provided which results in the almost complete reaction of Br radicals to other BrX species. The scenarios with 300 and 500 ppmv SO2 show a rather complete depletion of ozone during the simulation time but are not very realistic. A dilution time of 10 min (to a factor of (0.37) of the initial SO2 mixing ratio) fits best with the measured average SO2 mixing ratios in the aged plume.
With an initial magmatic HxOy NOx abundance, r-Br consists of less Br radicals and more other BrX species. The presence of NOx (Fig. S5) promotes the formation of reactive bromine reservoir species and causes less ozone consumption compared with the atmospheric HxOy NOx case. The aerosol number concentration (NC) at a given aerosol size affects the Br activation. After reaching a certain threshold, enough aerosol particles are present such that the Br activation is not limited by this parameter. This is also true for the radius of the aerosol particles at the number concentration of the base run. Regarding the base run scenario, a surface area of 3.4 × 10−3 m2 m−3 (3 × 109 particles m−3 at 300 nm radius) appears to be appropriate for the observed Br activation. Otherwise, fewer particles with the same radius lead to a slower activation, similar to the same number of smaller particles. However, a larger surface area per volume leads to an increased activation pace. Similar to our observations, Roberts et al. (2014) showed the impact of higher and lower aerosol surface on the Br activation, leading to larger and smaller activation, respectively, in their PlumeChem model. However, the necessary surface area is easily provided by the volcanic aerosol at Masaya, considering the optical particle counter data on the particle sizes and abundances obtained, which reached up to approximately 1.5 × 10−2 m2 m−3 calculated with data from Stix et al. (2018) – so about 5 times as much as used in our base run scenario.
5.4.3 Chlorine and iodine chemistry
The HSC model produces reactive chlorine and iodine species. A typical output for chlorine is a Cl2 Cl ratio of 8 × 10−5 and a ClO / Cl ratio of 3 × 10−6. ClONO2 and OClO are formed over the model runtime, and the measured reactive chlorine species are of the order of the model predictions (∼ 0.3 × 10−3 at 2–3 min), although the model base run shows a decrease in reactive species during the first 7–8 min (see Fig. 9).
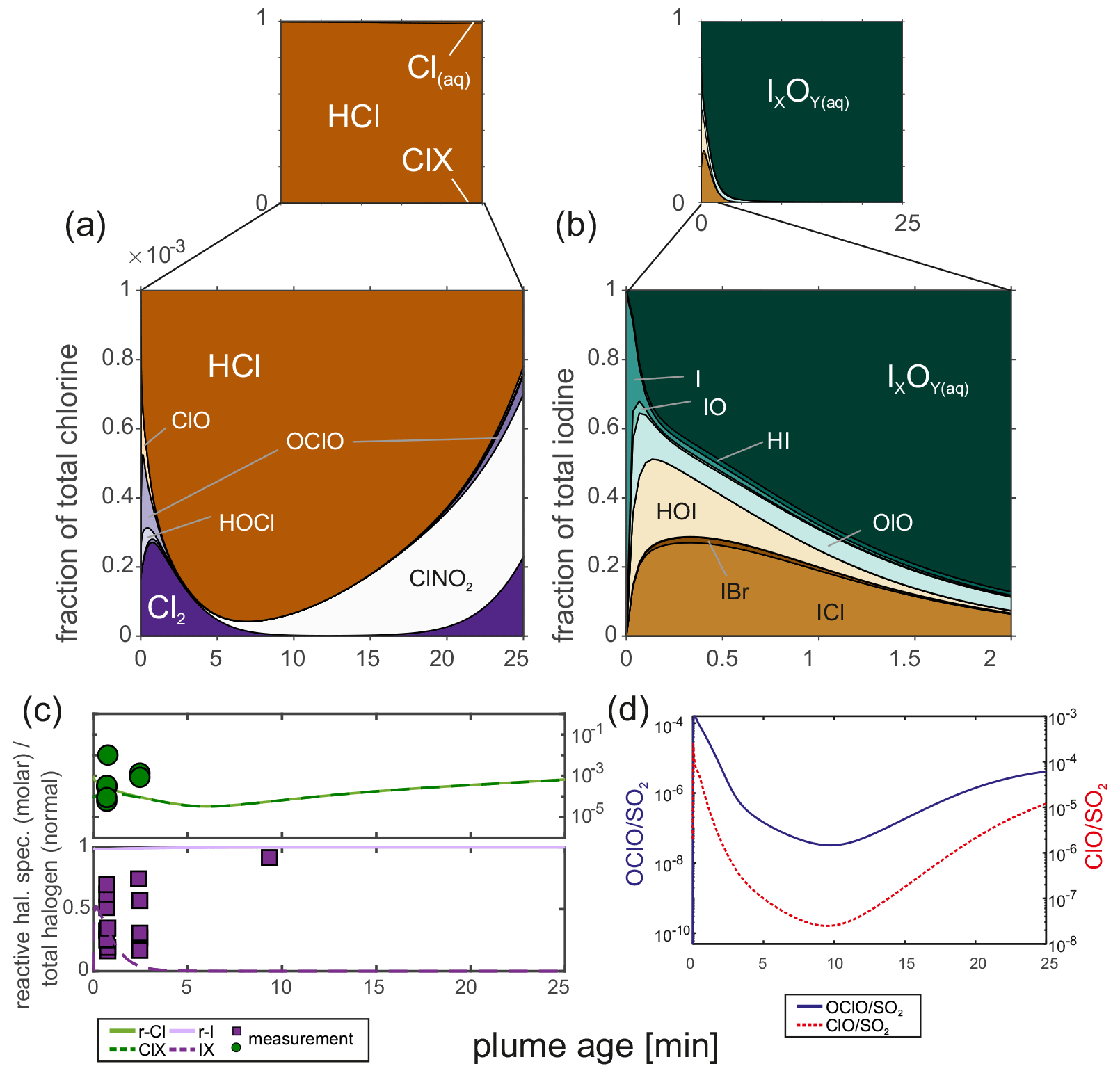
Figure 9(a) Chlorine and (b) iodine speciation of the base model run (10:90; 30 ppmv SO2; air; 10 min; 3 × 109 particles m−3; r = 3 × 10−7 m); (c) comparison of the measured and modeled reactive halogen to total halogens; (d) molar OClO SO2 and ClO SO2 ratios.
Regarding iodine, the modeled speciation reflects the field observations, albeit only partially. Similar to Martin et al. (2006), 99 % of the iodine is present as atomic I in the HSC model. Diatomic iodine species are formed during the first minute of the box model simulation alongside HOI by reactions analogous to Reactions (R1) and (R2). Eventually IO is further oxidized, similar to Reaction (R8), to form OIO, which is known to condense on preexisting particles and undergo new particle formation (e.g., Hoffmann et al., 2001; O'Dowd et al., 2002; Saiz-Lopez et al., 2012). CAABA/MECCA assumes a loss rate of OIO for new particle formation and uptake on existing aerosol that is combined to IxOy(aq). The kinetics of this process and the fate of the respective iodine species are not constrained very well. However, our measurements suggest a reactive iodine fraction (IX: I2, IO, INO2, IONO2, HOI, OIO, HIO3, ICl, IBr; r-I: IX + I) of 0.32 ± 0.15 (0.16 to 0.70) at the Santiago crater rim, which is in agreement with the model result, and despite the high conversion of HI emission into reactive iodine, there is a low proportion (∼ 4 %) of reactive iodine as IO. For a more distant position, the measurements and the model diverge (Fig. 9c), although a smaller loss rate of OIO and further aqueous chemistry of IxOy(aq) could potentially explain the measured reactive iodine species downwind. Furthermore, ultrafine and newly formed particles (< 10 nm) consisting of IxOy could also diffuse to the denuder walls and react with the coating, thereby inducing a false reactive iodine signal.
In this study, we present an innovative approach using a combination of ground-based and UAV-based measurements of halogen speciation in the plume of Masaya volcano over an estimated plume age of 1–11 min. By using an UAV, we were able to sample the plume at an age that is typically not accessible. Additionally, the application of different techniques allowed for the most detailed observation of changes in the halogen speciation during the first 11 min following the gas release. This led us to new observations: (1) the quantification of reactive bromine in the plume of Masaya at different plume ages and (2) the first quantification of reactive iodine and chlorine in the volcanic plume of Masaya. In addition, we provide the largest complementary data set to specify different halogen compounds at a volcano. These include alkaline traps (total amount of halogens), denuders (quantification of reactive halogens), and DOAS data (BrO), with the latter two being applied to different plume ages. With these data, we finally succeeded in demonstrating, through field studies, that a large part of the emitted bromine is converted into reactive species (76 % is already converted after 11.1 min), which was previously only assumed by model studies. The proportion of BrO (about 10 %) to total bromine and the behavior of BrO SO2 (rapidly reaching a plateau) is in line with earlier studies (e.g., Gutmann et al., 2018). We have also shown that a large part of iodine (92 %) is converted into reactive iodine compounds and that only a very small fraction (about 0.1 %) of chlorine is converted into reactive chlorine.
In addition to our field studies, we applied the CAABA/MECCA box model to test if our current understanding of bromine chemistry in volcanic plumes fits our experimental results, assuming reasonable input data for the species not measured. The BrO contribution to r-Br, determined by measurement data, could be reproduced by our box model simulations. The overall progression of the BrX Br and r-Br Br ratios were reflected by various model runs encompassing different plausible starting parameters. Although the bromine activation was reproduced by CAABA/MECCA, differences in reactive bromine to sulfur ratios occurred between observation and model values, which might stem from an underestimation of SO2 in the electrochemical sensor data for the diluted plume. Alongside more detailed observational studies, extension of knowledge on the chemical mechanisms of reactions occurring in volcanic plume environments is still needed. Further data on O3, HxOy, and NOx in the plume would help to pinpoint a more detailed set of start parameters. The measured and modeled chlorine speciation are comparable. For iodine, the implementations of iodine chemistry, such as the knowledge on iodine oxide particle formation, into the model are necessary to enable a qualified comparison with the observed iodine data. Within the range of model parameters that we studied, the dilution time and the quenching factor were shown to have a large effect on the plume chemistry, whereas the initial volume ratio of atmospheric and magmatic gases (in the HSC model) seemed to play a less important role. The presence of initial oxidants was shown to influence the relative abundance of the single Br species (e.g., Br2, BrCl, BrNO3, BrO, and Br) while not largely affecting the overall reactive bromine abundance.
Our method was able to determine the sum of reactive species for the respective halogens, which presents an important step forward with respect to our knowledge on volcanic plume halogen chemistry. However, more detailed speciation within the reactive fraction is a desirable topic for future research. For this purpose, other selective denuder coatings will be developed and applied to further distinguish between species such as Br2, BrCl, or BrNO3 and Br radicals. Although we estimated that BrO contributes up to 10 % of r-Br and r-Br accounts for up to 76 % of total Br after 10 min, the BrO formation seems to be more sensitive to changing model parameters than the overall r-Br formation. As BrO detection is possible with DOAS spectrometers and has already been conducted at numerous volcanoes, the influencing factors on the extent of its formation need to be studied further, in particular with respect to its potential as a volcanic forecasting parameter and its use to estimate total bromine emissions. Detailed measurements in the field and further studies in controlled environments, like atmospheric simulation chambers, will help to further assess bromine activation in volcanic plumes.
CAABA/MECCA is a community model published under the GNU General Public License, for further information see http://www.mecca.messy-interface.org (last access: 1 March 2021). A frozen version of the code used in this study (including the adaptation to the Masaya volcano) is included in the Supplement.
The HSC Chemistry model is commercially available from Metso Outotec Corporation, Finland.
The data sets used in this study are included in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-3371-2021-supplement.
JR, NB, and TH designed the research. JR, AG, NB, JMdM, MI, MM, AF, and JS performed field sample and data collection. JR, AG, and ML conducted laboratory sample analyses. FD provided the BrO SO2 NOVAC results. JR, JLT, AS, EM, JV, and JMC provided instrumental and logistical support. RS provided the CAABA/MECCA program and helped to perform model runs. All authors contributed to the paper.
The authors declare that they have no conflict of interest.
Julian Rüdiger, Nicole Bobrowski, Alexandra Gutmann, and Thorsten Hoffmann acknowledge support from the “Volcanoes and Atmosphere in Magmatic, Open Systems” (VAMOS) research center at the University of Mainz, Germany. Julian Rüdiger is thankful for funding from the German Academic Exchange Service (DAAD) and support from INETER (Nicaragua) and OVSICORI Universidad Nacional (Costa Rica). Julian Rüdiger and Nicole Bobrowski are grateful to the DFG “HALVIRE” project for financial support during data evaluation, modeling, and writing. The authors also wish to thank Feiyue Wang from the Centre for Earth Observation Science (CEOS) and the Department of Environment and Geography of the University of Manitoba, Canada, for supporting field work at the Masaya complex. We would like to express our gratitude to the referees Christoph Kern, Tjarda Roberts, and the anonymous referee for their comprehensive reviews that helped to substantially improve the paper.
This research has been supported by the Deutsche Forschungsgemeinschaft (grant nos. BO 3611/2-1, HE 5214/8-1, and PL 193/18-1).
This open-access publication was funded
by Johannes Gutenberg University Mainz.
This paper was edited by Rolf Müller and reviewed by Christoph Kern, Tjarda Roberts, and one anonymous referee.
Aiuppa, A.: Degassing of halogens from basaltic volcanism: Insights from volcanic gas observations, Chem. Geol., 263, 99–109, https://doi.org/10.1016/j.chemgeo.2008.08.022, 2009.
Aiuppa, A., Federico, C., Franco, A., Giudice, G., Gurrieri, S., Inguaggiato, S., Liuzzo, M., McGonigle, A. J. S., and Valenza, M.: Emission of bromine and iodine from Mount Etna volcano, Geochem. Geophy. Geosy., 6, Q08008, https://doi.org/10.1029/2005GC000965, 2005.
Aiuppa, A., Federico, C., Giudice, G., Gurrieri, S., Liuzzo, M., Shinohara, H., Favara, R., and Valenza, M.: Rates of carbon dioxide plume degassing from Mount Etna volcano, J. Geophys. Res., 111, B09207, https://doi.org/10.1029/2006JB004307, 2006.
Aiuppa, A., Moretti, R., Federico, C., Giudice, G., Gurrieri, S., Liuzzo, M., Papale, P., Shinohara, H., and Valenza, M.: Forecasting Etna eruptions by real-time observation of volcanic gas composition, Geology, 35, 1115–1118, https://doi.org/10.1130/G24149A.1, 2007.
Aiuppa, A., Baker, D. R., and Webster, J. D.: Halogens in volcanic systems, Chem. Geol., 263, 1–18, https://doi.org/10.1016/j.chemgeo.2008.10.005, 2009.
Aiuppa, A., de Moor, J. M., Arellano, S., Coppola, D., Francofonte, V., Galle, B., Giudice, G., Liuzzo, M., Mendoza, E., Saballos, A., Tamburello, G., Battaglia, A., Bitetto, M., Gurrieri, S., Laiolo, M., Mastrolia, A., and Moretti, R.: Tracking Formation of a Lava Lake From Ground and Space: Masaya Volcano (Nicaragua), 2014–2017, Geochem. Geophy. Geosy., 19, 496–515, https://doi.org/10.1002/2017GC007227, 2018.
Bobrowski, N. and Giuffrida, G.: Bromine monoxide / sulphur dioxide ratios in relation to volcanological observations at Mt. Etna 2006–2009, Solid Earth, 3, 433–445, https://doi.org/10.5194/se-3-433-2012, 2012.
Bobrowski, N. and Platt, U.: SO2/BrO ratios studied in five volcanic plumes, J. Volcanol. Geoth. Res., 166, 147–160, 2007.
Bobrowski, N., Hönninger, G., Galle, B., and Platt, U.: Detection of bromine monoxide in a volcanic plume, Nature, 423, 273–276, 2003.
Bobrowski, N., von Glasow, R., Aiuppa, A., Inguaggiato, S., Louban, I., Ibrahim, O. W., and Platt, U.: Reactive halogen chemistry in volcanic plumes, J. Geophys. Res., 112, https://doi.org/10.1029/2006JD007206, 2007.
Bobrowski, N., Giuffrida, G. B., Arellano, S., Yalire, M., Liotta, M., Brusca, L., Calabrese, S., Scaglione, S., Rüdiger, J., Castro, J. M., Galle, B., and Tedesco, D.: Plume composition and volatile flux of Nyamulagira volcano, Democratic Republic of Congo, during birth and evolution of the lava lake, 2014–2015, B. Volcanol., 79, B09207, https://doi.org/10.1007/s00445-017-1174-0, 2017.
Bryukov, M. G., Slagle, I. R., and Knyazev, V. D.: Kinetics of Reactions of Cl Atoms with Methane and Chlorinated Methanes, J. Phys. Chem., 106, 10532–10542, https://doi.org/10.1021/jp0257909, 2002.
Burton, M. R., Oppenheimer, C., Horrocks, L. A., and Francis, P. W.: Remote sensing of CO2 and H2O emission rates from Masaya volcano, Nicaragua, Geology, 28, 915–918, https://doi.org/10.1130/0091-7613(2000)28<915:RSOCAH>2.0.CO;2, 2000.
Cadoux, A., Iacono-Marziano, G., Scaillet, B., Aiuppa, A., Mather, T. A., Pyle, D. M., Deloule, E., Gennaro, E., and Paonita, A.: The role of melt composition on aqueous fluid vs. silicate melt partitioning of bromine in magmas, Earth Planet. Sci. Lett., 498, 450–463, https://doi.org/10.1016/j.epsl.2018.06.038, 2018.
Carn, S. A., Froyd, K. D., Anderson, B. E., Wennberg, P., Crounse, J., Spencer, K., Dibb, J. E., Krotkov, N. A., Browell, E. V., Hair, J. W., Diskin, G., Sachse, G., and Vay, S. A.: In situ measurements of tropospheric volcanic plumes in Ecuador and Colombia during TC 4, J. Geophys. Res., 116, 86, https://doi.org/10.1029/2010JD014718, 2011.
Carn, S. A., Clarisse, L., and Prata, A. J.: Multi-decadal satellite measurements of global volcanic degassing, J. Volcanol. Geoth. Res., 311, 99–134, https://doi.org/10.1016/j.jvolgeores.2016.01.002, 2016.
Carn, S. A., Fioletov, V. E., McLinden, C. A., Li, C., and Krotkov, N. A.: A decade of global volcanic SO2 emissions measured from space, Sci. Rep., 7, 44095, https://doi.org/10.1038/srep44095, 2017.
Carroll, R. and Holloway, J. R. (Eds.): Volatiles in magmas, in: Reviews in Mineralogy & Geochemistry, 30, De Gruyter, https://doi.org/10.1515/9781501509674, 1994.
Delmelle, P.: Environmental impacts of tropospheric volcanic gas plumes, Geol. Soc. London Sp. Pub., 213, 381–399, https://doi.org/10.1144/GSL.SP.2003.213.01.23, 2003.
Delmelle, P., Stix, J., Baxter, P., Garcia-Alvarez, J., and Barquero, J.: Atmospheric dispersion, environmental effects and potential health hazard associated with the low-altitude gas plume of Masaya volcano, Nicaragua, B. Volcanol., 64, 423–434, https://doi.org/10.1007/s00445-002-0221-6, 2002.
de Moor, J. M., Fischer, T. P., Sharp, Z. D., King, P. L., Wilke, M., Botcharnikov, R. E., Cottrell, E., Zelenski, M., Marty, B., Klimm, K., Rivard, C., Ayalew, D., Ramirez, C., and Kelley, K. A.: Sulfur degassing at Erta Ale (Ethiopia) and Masaya (Nicaragua) volcanoes: Implications for degassing processes and oxygen fugacities of basaltic systems, Geochem. Geophy. Geosy., 14, 4076–4108, https://doi.org/10.1002/ggge.20255, 2013.
de Moor, J. M., Aiuppa, A., Avard, G., Wehrmann, H., Dunbar, N., Muller, C., Tamburello, G., Giudice, G., Liuzzo, M., Moretti, R., Conde, V., and Galle, B.: Turmoil at Turrialba Volcano (Costa Rica): Degassing and eruptive processes inferred from high-frequency gas monitoring, J. Geophys. Res.-Sol. Ea., 121, 5761–5775, https://doi.org/10.1002/2016JB013150, 2016.
de Moor, J. M., Kern, C., Avard, G., Muller, C., Aiuppa, A., Saballos, A., Ibarra, M., LaFemina, P., Protti, M., and Fischer, T. P.: A New Sulfur and Carbon Degassing Inventory for the Southern Central American Volcanic Arc: The Importance of Accurate Time-Series Datasets and Possible Tectonic Processes Responsible for Temporal Variations in Arc-Scale Volatile Emissions, Geochem. Geophy. Geosy., 18, 4437–4468, https://doi.org/10.1002/2017GC007141, 2017.
Dinger, F.: On long-term variations in the BrO/SO2 molar ratios in volcanic gas plumes, PhD thesis, Johannes Gutenberg-University Mainz, Mainz, Germany, 251 pp., 2019.
Dinger, F., Bobrowski, N., Warnach, S., Bredemeyer, S., Hidalgo, S., Arellano, S., Galle, B., Platt, U., and Wagner, T.: Periodicity in the BrO SO2 molar ratios in the volcanic gas plume of Cotopaxi and its correlation with the Earth tides during the eruption in 2015, Solid Earth, 9, 247–266, https://doi.org/10.5194/se-9-247-2018, 2018.
Donovan, A., Tsanev, V., Oppenheimer, C., and Edmonds, M.: Reactive halogens (BrO and OClO) detected in the plume of Soufrière Hills Volcano during an eruption hiatus, Geochem. Geophy. Geosy., 15, 3346–3363, https://doi.org/10.1002/2014GC005419, 2014.
Duffell, H. J., Oppenheimer, C., Pyle, D. M., Galle, B., McGonigle, A. J. S., and Burton, M. R.: Changes in gas composition prior to a minor explosive eruption at Masaya volcano, Nicaragua, J. Volcanol. Geoth. Res., 126, 327–339, https://doi.org/10.1016/S0377-0273(03)00156-2, 2003.
Fickert, S., Adams, J. W., and Crowley, J. N.: Activation of Br2 and BrCl via uptake of HOBr onto aqueous salt solutions, J. Geophys. Res.-Atmos., 104, 23719–23727, https://doi.org/10.1029/1999JD900359, 1999.
Galle, B., Johansson, M., Rivera, C., Zhang, Y., Kihlman, M., Kern, C., Lehmann, T., Platt, U., Arellano, S., and Hidalgo, S.: Network for Observation of Volcanic and Atmospheric Change (NOVAC) – A global network for volcanic gas monitoring: Network layout and instrument description, J. Geophys. Res., 115, D05304, https://doi.org/10.1029/2009JD011823, 2010.
General, S., Bobrowski, N., Pöhler, D., Weber, K., Fischer, C., and Platt, U.: Airborne I-DOAS measurements at Mt. Etna: BrO and OClO evolution in the plume, J. Volcanol. Geoth. Res., 300, 175–186, https://doi.org/10.1016/j.jvolgeores.2014.05.012, 2015.
Gerlach, T. M.: Volcanic sources of tropospheric ozone-depleting trace gases, Geochem. Geophy. Geosy., 5, Q09007, https://doi.org/10.1029/2004GC000747, 2004.
Giggenbach, W. F.: Variations in the carbon, sulfur and chlorine contents of volcanic gas discharges from White Island, New Zealand, B. Volcanol., 39, 15–27, https://doi.org/10.1007/BF02596943, 1975.
Gliß, J., Bobrowski, N., Vogel, L., Pöhler, D., and Platt, U.: OClO and BrO observations in the volcanic plume of Mt. Etna – implications on the chemistry of chlorine and bromine species in volcanic plumes, Atmos. Chem. Phys., 15, 5659–5681, https://doi.org/10.5194/acp-15-5659-2015, 2015.
Gutmann, A., Bobrowski, N., Roberts, T., Rüdiger, J., and Hoffmann, T.: Advances in bromine speciation in volcanic plumes, Front. Earth Sci., 6, 213, https://doi.org/10.3389/feart.2018.00213, 2018.
Heue, K.-P., Brenninkmeijer, C. A. M., Baker, A. K., Rauthe-Schöch, A., Walter, D., Wagner, T., Hörmann, C., Sihler, H., Dix, B., Frieß, U., Platt, U., Martinsson, B. G., van Velthoven, P. F. J., Zahn, A., and Ebinghaus, R.: SO2 and BrO observation in the plume of the Eyjafjallajökull volcano 2010: CARIBIC and GOME-2 retrievals, Atmos. Chem. Phys., 11, 2973–2989, https://doi.org/10.5194/acp-11-2973-2011, 2011.
Hippler, H. and Troe, J.: Flash photolysis study of the recombination of chlorine atoms in the presence of various inert gases and NO, Int. J. Chem. Kinet., 8, 501–510, https://doi.org/10.1002/kin.550080404, 1976.
Hobbs, P. V., Tuell, J. P., Hegg, D. A., Radke, L. F., and Eltgroth, M. W.: Particles and gases in the emissions from the 1980–1981 volcanic eruptions of Mt. St. Helens, J. Geophys. Res., 87, 11062, https://doi.org/10.1029/JC087iC13p11062, 1982.
Hoffmann, T., O'Dowd, C. D., and Seinfeld, J. H.: Iodine oxide homogeneous nucleation: An explanation for coastal new particle production, Geophys. Res. Lett., 28, 1949–1952, https://doi.org/10.1029/2000GL012399, 2001.
Huff, A. K. and Abbatt, J. P. D.: Gas-Phase Br2 Production in Heterogeneous Reactions of Cl2 HOCl, and BrCl with Halide-Ice Surfaces, J. Phys. Chem., 104, 7284–7293, https://doi.org/10.1021/jp001155w, 2000.
Iowa State University: Iowa Environmental Mesonet: ASOS-AWOS-METAR Data, availabne at: https://mesonet.agron.iastate.edu/request/download.phtml?network=NI_ASOS, last access: 20 October 2018.
Kelly, P. J., Kern, C., Roberts, T. J., Lopez, T., Werner, C., and Aiuppa, A.: Rapid chemical evolution of tropospheric volcanic emissions from Redoubt Volcano, Alaska, based on observations of ozone and halogen-containing gases, J. Volcanol. Geoth. Res., 259, 317–333, https://doi.org/10.1016/j.jvolgeores.2012.04.023, 2013.
Kern, C. and Lyons, J. J.: Spatial distribution of halogen oxides in the plume of Mount Pagan volcano, Mariana Islands, Geophys. Res. Lett., 45, 9588–9596, https://doi.org/10.1029/2018GL079245, 2018.
Kern, C., Sihler, H., Vogel, L., Rivera, C., Herrera, M., and Platt, U.: Halogen oxide measurements at Masaya Volcano, Nicaragua using active long path differential optical absorption spectroscopy, B. Volcanol., 71, 659–670, https://doi.org/10.1007/s00445-008-0252-8, 2009.
Laaksonen, A., Korhonen, P., Kulmala, M., and Charlson, R. J.: Modification of the Köhler Equation to Include Soluble Trace Gases and Slightly Soluble Substances, J. Atmos. Sci., 55, 853–862, https://doi.org/10.1175/1520-0469(1998)055<0853:MOTKHE>2.0.CO;2, 1998.
Lee, C., Kim, Y. J., Tanimoto, H., Bobrowski, N., Platt, U., Mori, T., Yamamoto, K., and Hong, C. S.: High ClO and ozone depletion observed in the plume of Sakurajima volcano, Japan, Geophys. Res. Lett., 32, L21809, https://doi.org/10.1029/2005GL023785, 2005.
Liotta, M., Rizzo, A., Paonita, A., Caracausi, A., and Martelli, M.: Sulfur isotopic compositions of fumarolic and plume gases at Mount Etna (Italy) and inferences on their magmatic source, Geochem. Geophy. Geosy., 13, Q05015, https://doi.org/10.1029/2012GC004118, 2012.
Louban, I., Bobrowski, N., Rouwet, D., Inguaggiato, S., and Platt, U.: Imaging DOAS for volcanological applications, B. Volcanol., 71, 753–765, https://doi.org/10.1007/s00445-008-0262-6, 2009.
Lübcke, P., Bobrowski, N., Arellano, S., Galle, B., Garzón, G., Vogel, L., and Platt, U.: BrO/SO2 molar ratios from scanning DOAS measurements in the NOVAC network, Solid Earth, 5, 409–424, https://doi.org/10.5194/se-5-409-2014, 2014.
Malavelle, F. F., Haywood, J. M., Jones, A., Gettelman, A., Clarisse, L., Bauduin, S., Allan, R. P., Karset, I. H. H., Kristjánsson, J. E., Oreopoulos, L., Cho, N., Lee, D., Bellouin, N., Boucher, O., Grosvenor, D. P., Carslaw, K. S., Dhomse, S., Mann, G. W., Schmidt, A., Coe, H., Hartley, M. E., Dalvi, M., Hill, A. A., Johnson, B. T., Johnson, C. E., Knight, J. R., O'Connor, F. M., Partridge, D. G., Stier, P., Myhre, G., Platnick, S., Stephens, G. L., Takahashi, H., and Thordarson, T.: Strong constraints on aerosol-cloud interactions from volcanic eruptions, Nature, 546, 485, https://doi.org/10.1038/nature22974, 2017.
Maric, D., Burrows, J. P., and Moortgat, G. K.: A study of the UV-visible absorption spectra of Br2 and BrCl, J. Photoch. Photobio. A, 83, 179–192, https://doi.org/10.1016/1010-6030(94)03823-6, 1994.
Martin, R. S., Mather, T. A., and Pyle, D. M.: High-temperature mixtures of magmatic and atmospheric gases, Geochem. Geophy. Geosy., 7, Q04006, https://doi.org/10.1029/2005GC001186, 2006.
Martin, R. S., Sawyer, G. M., Spampinato, L., Salerno, G. G., Ramirez, C., Ilyinskaya, E., Witt, M. L. I., Mather, T. A., Watson, I. M., Phillips, J. C., and Oppenheimer, C.: A total volatile inventory for Masaya Volcano, Nicaragua, J. Geophys. Res., 115, B09215, https://doi.org/10.1029/2010JB007480, 2010.
Martin, R. S., Ilyinskaya, E., and Oppenheimer, C.: The enigma of reactive nitrogen in volcanic emissions, Geochim. Cosmochim. Ac., 95, 93–105, https://doi.org/10.1016/j.gca.2012.07.027, 2012.
Mather, T. A., Pyle, D. M., and Oppenheimer, C.: Tropospheric Volcanic Aerosol, in: Volcanism and the Earth's Atmosphere, edited by: Robock, A and Oppenheimer, C., Wiley, Washington, D.C., USA, 189–212, https://doi.org/10.1029/139GM12, 2013.
McBirney, A. R.: The Nicaraguan volcano Masaya and its caldera, Eos Trans. AGU, 37, 83–96, https://doi.org/10.1029/TR037i001p00083, 1956.
McGonigle, A. J. S., Delmelle, P., Oppenheimer, C., Tsanev, V. I., Delfosse, T., Williams-Jones, G., Horton, K., and Mather, T. A.: SO2 depletion in tropospheric volcanic plumes, Geophys. Res. Lett., 31, L13201, https://doi.org/10.1029/2004GL019990, 2004.
Noguchi, K. and Kamiya, H.: Prediction of volcanic eruption by measuring the chemical composition and amounts of gases, B. Volcanol., 26, 367–378, https://doi.org/10.1007/BF02597298, 1963.
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hämeri, K., Pirjola, L., Kulmala, M., Jennings, S. G., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions, Nature, 417, 632, https://doi.org/10.1038/nature00775, 2002.
Oppenheimer, C., Tsanev, V. I., Braban, C. F., Cox, R. A., Adams, J. W., Aiuppa, A., Bobrowski, N., Delmelle, P., Barclay, J., and McGonigle, A. J. S.: BrO formation in volcanic plumes, Geochim. Cosmochim. Ac., 70, 2935–2941, https://doi.org/10.1016/j.gca.2006.04.001, 2006.
Oppenheimer, C., Kyle, P., Eisele, F., Crawford, J., Huey, G., Tanner, D., Kim, S., Mauldin, L., Blake, D., Beyersdorf, A., Buhr, M., and Davis, D.: Atmospheric chemistry of an Antarctic volcanic plume, J. Geophys. Res., 115, 5473, https://doi.org/10.1029/2009JD011910, 2010.
Platt, U. and Bobrowski, N.: Quantification of volcanic reactive halogen emissions, in: Volcanism and global environmental change, edited by: Schmidt, A., Fristad, K., and Elkins-Tanton, L. T., Cambridge University Press, Cambridge, UK, 115–132, 2015.
Platt, U. and Stutz, J.: Differential absorption spectroscopy, in: Differential Optical Absorption Spectroscopy, Springer, Berlin and Heidelberg, Germany, 135–174, 2008.
Pyle, D. M. and Mather, T. A.: Halogens in igneous processes and their fluxes to the atmosphere and oceans from volcanic activity: A review, Chem. Geol., 263, 110–121, https://doi.org/10.1016/j.chemgeo.2008.11.013, 2009.
Roberts, T.: Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions, Geosciences, 8, 68, https://doi.org/10.3390/geosciences8020068, 2018.
Roberts, T. J., Braban, C. F., Martin, R. S., Oppenheimer, C., Adams, J. W., Cox, R. A., Jones, R. L., and Griffiths, P. T.: Modelling reactive halogen formation and ozone depletion in volcanic plumes, Chem. Geol., 263, 151–163, https://doi.org/10.1016/j.chemgeo.2008.11.012, 2009.
Roberts, T. J., Martin, R. S., and Jourdain, L.: Reactive bromine chemistry in Mount Etna's volcanic plume: the influence of total Br, high-temperature processing, aerosol loading and plume–air mixing, Atmos. Chem. Phys., 14, 11201–11219, https://doi.org/10.5194/acp-14-11201-2014, 2014.
Roberts, T. J., Vignelles, D., Liuzzo, M., Giudice, G., Aiuppa, A., Coltelli, M., Salerno, G., Chartier, M., Couté, B., Berthet, G., Lurton, T., Dulac, F., and Renard, J.-B.: The primary volcanic aerosol emission from Mt Etna: Size-resolved particles with SO2 and role in plume reactive halogen chemistry, Geochim. Cosmochim. Ac., 222, 74–93, https://doi.org/10.1016/j.gca.2017.09.040, 2018.
Roberts, T. J., Dayma, G., and Oppenheimer, C.: Reaction Rates Control High-Temperature Chemistry of Volcanic Gases in Air, Front. Earth Sci., 7, 1441, https://doi.org/10.3389/feart.2019.00154, 2019.
Rose, W. I., Millard, G. A., Mather, T. A., Hunton, D. E., Anderson, B., Oppenheimer, C., Thornton, B. F., Gerlach, T. M., Viggiano, A. A., Kondo, Y., Miller, T. M., and Ballenthin, J. O.: Atmospheric chemistry of a 33–34 hour old volcanic cloud from Hekla Volcano (Iceland): Insights from direct sampling and the application of chemical box modeling, J. Geophys. Res., 111, Q08008, https://doi.org/10.1029/2005JD006872, 2006.
Rüdiger, J., Bobrowski, N., Liotta, M., and Hoffmann, T.: Development and application of a sampling method for the determination of reactive halogen species in volcanic gas emissions, Anal. Bioanal. Chem., 52, 325, https://doi.org/10.1007/s00216-017-0525-1, 2017.
Rüdiger, J., Tirpitz, J.-L., de Moor, J. M., Bobrowski, N., Gutmann, A., Liuzzo, M., Ibarra, M., and Hoffmann, T.: Implementation of electrochemical, optical and denuder-based sensors and sampling techniques on UAV for volcanic gas measurements: examples from Masaya, Turrialba and Stromboli volcanoes, Atmos. Meas. Tech., 11, 2441–2457, https://doi.org/10.5194/amt-11-2441-2018, 2018.
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/c2cs35208g, 2012.
Saiz-Lopez, A., Plane, J. M. C., Baker, A. R., Carpenter, L. J., Glasow, R. V., Martín, J. C. G., McFiggans, G., and Saunders, R. W.: Atmospheric chemistry of iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012.
Sander, R., Baumgaertner, A., Gromov, S., Harder, H., Jöckel, P., Kerkweg, A., Kubistin, D., Regelin, E., Riede, H., Sandu, A., Taraborrelli, D., Tost, H., and Xie, Z.-Q.: The atmospheric chemistry box model CAABA/MECCA-3.0, Geosci. Model Dev., 4, 373–380, https://doi.org/10.5194/gmd-4-373-2011, 2011.
Sander, R., Jöckel, P., Kirner, O., Kunert, A. T., Landgraf, J., and Pozzer, A.: The photolysis module JVAL-14, compatible with the MESSy standard, and the JVal PreProcessor (JVPP), Geosci. Model Dev., 7, 2653–2662, https://doi.org/10.5194/gmd-7-2653-2014, 2014.
Schönhardt, A., Richter, A., Theys, N., and Burrows, J. P.: Space-based observation of volcanic iodine monoxide, Atmos. Chem. Phys., 17, 4857–4870, https://doi.org/10.5194/acp-17-4857-2017, 2017.
Shinohara, H.: A new technique to estimate volcanic gas composition: Plume measurements with a portable multi-sensor system, J. Volcanol. Geoth. Res., 143, 319–333, https://doi.org/10.1016/j.jvolgeores.2004.12.004, 2005.
Simpson, W. R., von Glasow, R., Riedel, K., Anderson, P., Ariya, P., Bottenheim, J., Burrows, J., Carpenter, L. J., Frieß, U., Goodsite, M. E., Heard, D., Hutterli, M., Jacobi, H.-W., Kaleschke, L., Neff, B., Plane, J., Platt, U., Richter, A., Roscoe, H., Sander, R., Shepson, P., Sodeau, J., Steffen, A., Wagner, T., and Wolff, E.: Halogens and their role in polar boundary-layer ozone depletion, Atmos. Chem. Phys., 7, 4375–4418, https://doi.org/10.5194/acp-7-4375-2007, 2007.
Stix, J., de Moor, J. M., Rüdiger, J., Alan, A., Corrales, E., D'Arcy, F., Diaz, J. A., and Liotta, M.: Using Drones and Miniaturized Instrumentation to Study Degassing at Turrialba and Masaya Volcanoes, Central America, J. Geophys. Res.-Sol. Ea., 123, 6501–6520, https://doi.org/10.1029/2018JB015655, 2018.
Surl, L., Donohoue, D., Aiuppa, A., Bobrowski, N., and von Glasow, R.: Quantification of the depletion of ozone in the plume of Mount Etna, Atmos. Chem. Phys., 15, 2613–2628, https://doi.org/10.5194/acp-15-2613-2015, 2015.
Symonds, R. B., Rose, W. I., Bluth, G. J. S., and Gerlach, T. M.: Volcanic-gas studies; methods, results, and applications, Rev. Mineral. Geochem., 30, 1–66, 1994.
Theys, N., van Roozendael, M., Dils, B., Hendrick, F., Hao, N., and de Mazière, M.: First satellite detection of volcanic bromine monoxide emission after the Kasatochi eruption, Geophys. Res. Lett., 36, L03809, https://doi.org/10.1029/2008GL036552, 2009.
Theys, N., de Smedt, I., van Roozendael, M., Froidevaux, L., Clarisse, L., and Hendrick, F.: First satellite detection of volcanic OClO after the eruption of Puyehue-Cordón Caulle, Geophys. Res. Lett., 41, 667–672, https://doi.org/10.1002/2013GL058416, 2014.
van Manen, S. M.: Perception of a chronic volcanic hazard: Persistent degassing at Masaya volcano, Nicaragua, J. Appl. Volcanol., 3, 155, https://doi.org/10.1186/s13617-014-0009-3, 2014.
Vogel, L.: Volcanic plumes: Evaluation of spectroscopic measurements, early detection, and bromine chemistry, Heidelberg University Library, Heidelberg, Germany, 2012.
von Glasow, R.: Atmospheric chemistry in volcanic plumes, P. Natl. Acad. Sci. USA, 107, 6594–6599, https://doi.org/10.1073/pnas.0913164107, 2010.
von Glasow, R., Bobrowski, N., and Kern, C.: The effects of volcanic eruptions on atmospheric chemistry, Chem. Geol., 263, 131–142, https://doi.org/10.1016/j.chemgeo.2008.08.020, 2009.
Warnach, S., Bobrowski, N., Hidalgo, S., Arellano, S., Sihler, H., Dinger, F., Lübcke, P., Battaglia, J., Steele, A., Galle, B., Platt, U., and Wagner, T.: Variation of the BrO SO2 Molar Ratio in the Plume of Tungurahua Volcano Between 2007 and 2017 and Its Relationship to Volcanic Activity, Front. Earth Sci., 7, 99, https://doi.org/10.3389/feart.2019.00132, 2019.
Wennberg, P.: Atmospheric chemistry: Bromine explosion, Nature, 397, 299–301, 1999.
Witt, M. L. I., Mather, T. A., Pyle, D. M., Aiuppa, A., Bagnato, E., and Tsanev, V. I.: Mercury and halogen emissions from Masaya and Telica volcanoes, Nicaragua, J. Geophys. Res., 113, B06203, https://doi.org/10.1029/2007JB005401, 2008.
Wittmer, J., Bobrowski, N., Liotta, M., Giuffrida, G., Calabrese, S., and Platt, U.: Active alkaline traps to determine acidic-gas ratios in volcanic plumes: Sampling techniques and analytical methods, Geochem. Geophy. Geosy., 15, 2797–2820, https://doi.org/10.1002/2013GC005133, 2014.