the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Dec 2021
| 21 Dec 2021
Biomass burning plume chemistry: OH-radical-initiated oxidation of 3-penten-2-one and its main oxidation product 2-hydroxypropanal
Iulia Patroescu-Klotz
Peter Wiesen
In order to enlarge our understanding of biomass burning plume chemistry, the OH-radical-initiated oxidation of 3-penten-2-one (3P2), identified in biomass burning emissions, and 2-hydroxypropanal (2HPr) was investigated at 298 ± 3 K and 990 ± 15 mbar in two atmospheric simulation chambers using long-path FTIR spectroscopy. The rate coefficient of 3P2 + OH was determined to be (6.2 ± 1.0) × 10−11 cm3 molec.−1 s−1 and the molar first-generation yields for acetaldehyde, methyl glyoxal, 2HPr, and the sum of peroxyacetyl nitrate (PAN) and CO2, used to determine the CH3C(O) radical yield, were 0.39 ± 0.07, 0.32 ± 0.08, 0.68 ± 0.27, and 0.56 ± 0.14, respectively, under conditions where the 3P2-derived peroxy radicals react solely with NO. The 2HPr + OH reaction was investigated using 3P2 + OH as a source of the α-hydroxyaldehyde adjusting the experimental conditions to shift the reaction system towards secondary oxidation processes. The rate coefficient was estimated to be (2.2 ± 0.6) × 10−11 cm3 molec.−1 s−1. Employing a simple chemical mechanism to analyse the temporal behaviour of the experiments, the further oxidation of 2HPr was shown to form methyl glyoxal, acetaldehyde, and CO2 with estimated yields of 0.27 ± 0.08, 0.73 ± 0.08, and 0.73 ± 0.08, respectively.
- Article
(2664 KB) - Full-text XML
-
Supplement
(519 KB) - BibTeX
- EndNote





Unsaturated ketones are of increasing interest as more sources for their atmospheric burden are uncovered. They are potentially formed in the atmospheric oxidation of terpenes involving OH radicals or O3 molecules. 3-Buten-2-one (methyl vinyl ketone) is the most famous representative formed through the gas-phase oxidation of the most abundantly emitted non-methane hydrocarbons (NMHC), namely isoprene (Calvert et al., 2000). Other α,β-unsaturated ketones, however, are used in the food and fragrances industry (Bickers et al., 2003). Ciccioli et al. (2001) investigated controlled biomass burning emissions representative for the Mediterranean vegetation and identified 3-penten-2-one (3P2) in both the flaming and smoldering of pine wood. Hatch et al. (2017) identified 3P2 among various oxygenated species in the gas-phase biomass burning emissions of Chinese rice straw, Indonesian peat, and boughs of ponderosa pine and black spruce during the FLAME-4 campaign. Besides, they also identified 2-methylpenta-1,3-diene (Hatch et al., 2017), which potentially yields 3P2 in the further gas-phase oxidation. All these findings pinpoint 3P2 as a likely constituent of most biomass burning (BB) plumes.
Understanding the BB plume chemistry is of global interest since their photochemical ageing is believed to be a potentially significant contributor to ozone and organic aerosol formation (Jaffe et al., 2008; Yokelson et al., 2009; Alvarado et al., 2015). For instance, Jaffe et al. (2008) found a correlation between the interannual variation in O3 and burned area in the western USA. During the next decades a drier climate, expected due to global warming, will likely result in an increase in both the number and intensity of fire events worldwide, which could consequently enlarge the influence of biomass burning on air quality. Some field measurements of BB plumes have shown that wildfires may significantly increase the ozone enhancement ratios (ΔOCO) (Mauzerall et al., 1998; Honrath et al., 2004), while in others a correlation between O3 and CO could not be observed (Alvarado et al., 2010). Given typical emission ratios from biomass burning, ozone formation is mainly limited by the availability of NOx, which, in turn, depends on the fuel nitrogen content and the combustion efficiency (Jaffe and Wigder, 2012 and references therein). Alvarado et al. (2010) were the first to observe a fast peroxyacetyl nitrate (PAN) production in a young boreal smoke plume within the first hours after emission. Simulations of a young BB plume from a prescribed fire showed the evolution of secondary organic aerosol and O3 to be sensitive to unidentified volatile organic compounds (VOCs) whose chemistry is likely characterized, amongst others, through (a) OH rate coefficients in the order of 10−11 cm3 molec.−1 s−1, (b) RO2 + NO reactions resulting mainly in fragmentation, and (c) an efficient HO2 regeneration (Alvarado et al., 2015). However, this resulted still in a significant overestimation of downwind PAN formation (Alvarado et al., 2015). Accordingly, given the complexity of biomass burning smoke and the various conditions possible within both a young and an aged plume, the chemistry is still not well characterized. In particular, the oxidation of very reactive organic species seems to be crucial for a comprehensive picture.
The present work therefore contributes to expanding our understanding of BB plume chemistry by studying the oxidation processes of single species, identified in gas-phase emissions, in simulation chamber experiments. In this respect we investigated the OH-radical-initiated oxidation of 3P2 and the fate of its main oxidation product. Up to now, to the best of our knowledge, only kinetic data were reported for the reactions of 3P2 with OH radicals and Cl atoms (Blanco et al., 2012), NO3 radicals (Canosa-Mas et al., 2005), and O3 (Greene and Atkinson, 1994; Sato et al., 2004; Illmann et al., 2021a).
Kinetic and product study experiments were conducted in two indoor simulation chambers at 298 ± 3 K and in 990 ± 15 mbar of synthetic air. In both chambers OH radicals were generated by the photolysis of methyl nitrite in the presence of sufficient amounts of NO to suppress any ozone formation and consequently the generation of NO3 radicals. Methyl nitrite has been synthesized by the dropwise addition of sulfuric acid to a saturated aqueous solution of sodium nitrite in methanol according to a method previously outlined by Taylor et al. (1980). The product was collected and stored in a cooling trap at 195 K. Its purity was verified via Fourier-transform infrared (FTIR) spectroscopy.
2.1 480 L chamber
The cylindrical borosilicate glass tube with a length of 3 m and a 0.45 m inner diameter is surrounded by 32 superactinic fluorescent lamps (Philips TL A 40W: 300–460 nm, Imax at 360 nm) and closed at both ends by aluminium flanges. These contain various ports for the introduction of reactants and bath gas and the coupling with analytical devices. The pumping system consists of a rotary vane pump and a roots pump yielding an end vacuum up to 10−3 mbar. A White-type mirror system is installed inside the chamber (optical path length: 50.4 ± 0.2 m) and coupled to a Nicolet 6700 FTIR spectrometer to monitor reactants and products. Spectra are recorded in the spectral range 4000–700 cm−1 with a resolution of 1 cm−1. The present set-up of the chamber is described with further details in the recent literature (Illmann et al., 2021b).
The initial mixing ratios in the 480 L chamber experiments in ppmV (1 ppmV = 2.46 × 1013 molec. cm−3 at 298 K) were 5.8–9.4 for 3P2, 5.0 for isoprene, 8.3 for E2-butene, 10–16 for methyl nitrite, and 20–27 for NO.
2.2 1080 L chamber
The 1080 L chamber consists of two joint quartz-glass tubes with a total length of 6.2 m and a 0.47 m inner diameter. It is surrounded by 32 superactinic fluorescent lamps (Philips TL05 40W: 300–460 nm, Imax at 360 nm) and 32 low-pressure mercury vapour lamps (Philips TUV 40W, Imax at 254 nm) which can be switched individually. The pumping system consists of a turbo-molecular pump backed by a double-stage rotary fore pump to yield an end vacuum of 10−4 mbar. The White-type mirror system installed inside the chamber is operated at a total optical path length of 484.7 ± 0.8 m and coupled to a Nicolet iS50 FTIR spectrometer recording FTIR spectra in the range 4000–700 cm−1 with a resolution of 1 cm−1. A more detailed description of the chamber can be found in the recent literature (Illmann et al., 2021b).
The initial mixing ratios in the 1080 L chamber experiments, in ppmV (1 ppmV = 2.46 × 1013 molec. cm−3 at 298 K), were 1.1–1.3 for 3P2, 0.9–1.1 for isoprene, 1.3–1.5 for E2-butene, 0.9–1.9 for methyl nitrite, and 2.0–3.7 for NO, 1.5–1.7 for 3-buten-2-ol, 1.3–1.4 for 3-penten-2-ol, and 13 000–17 000 for CO.
2.3 Methods
The rate coefficient of the 3P2 + OH reaction has been determined using the relative-rate technique, thus by relating the consumption of 3-penten-2-one to the consumption of a reference compound. If reactions other than presented below (Reactions R1–R3) are negligible in the experimental set-up,
the following equation can be used to determine the rate coefficient k3P2:
where [X]t is the concentration of the species X at time t. The rate coefficient ratio used to calculate k3P2 were thus obtained from regression analysis after plotting against .
Mixing ratios of identified species in the product study experiments were obtained by subtracting calibrated reference FTIR spectra of the target species. The cross sections we used for calibration were taken either from the literature, in the case of methyl glyoxal (Profeta et al., 2011; Talukdar et al., 2011) and peroxyacetyl nitrate (Allen et al., 2005), the internal Wuppertal laboratory database (acetaldehyde), or determined within this work (3-penten-2-one, 2-hydroxypropanal). CO2 was quantified by the integration of the absorption features in the spectral range 2400–2349 cm−1 and a polynomial calibration function derived from the injection of various volumes of CO2 using a calibrated gas-tight syringe. Uncorrected molar formation yields of the reaction products were calculated by plotting the mixing ratio of formed product against the mixing ratio of the consumed 3-penten-2-one. In order to obtain first-generation yields (yields without impact of secondary reactions in the experimental set-up), the temporal behaviour of all quantified species was simulated following the approach previously outlined in the recent literature (Illmann et al., 2021b). Here, the differential equations are constructed based on the simplified reaction sequence of each species and solved by the Euler method using calculation software like Microsoft Excel. Input parameters are the rate coefficients of each of the sequence's reaction, the initial concentration of 3P2, and the time-dependent OH concentration calculated based on the 3P2 consumption. The molar formation yields for products of the target reaction are included as variable parameters to be modified until the simulated temporal behaviour of each species matches the experimental data. The details are provided within Sect. 3.4.
Cross sections and reference FTIR spectra of 2HPr were obtained by the in situ generation of the aldehyde through the ozonolysis of 3-buten-2-ol and 3-penten-2-ol, respectively, in the presence of sufficient CO to scavenge any OH radical formed during the O3 reaction, in the 1080 L chamber. In order to investigate the OH oxidation of 2HPr, the 3P2 + OH reaction was used as a source of the α-hydroxyaldehyde. An estimation of the rate coefficient was obtained following the procedure outlined previously by Baker et al. (2004). Branching ratios for the product formation were derived from modelling the temporal behaviour of the relevant species using our recently presented approach (Illmann et al., 2021b).
Typically, 15 spectra were recorded per experiment, and the first five spectra were collected in the dark to determine potential wall losses in each experiment. In the product study experiments five spectra were additionally recorded after the OH reaction was terminated (the lamps were switched off) in order to check for the wall loss rates of each formed reaction product. For the 2HPr + OH investigations, a second methyl nitrite and NO injection occurred after the first irradiation period, in the dark, followed by further irradiation. About 50–70 scans were co-added per spectrum which leads to averaging periods of about 80–115 s. Time intervals for both irradiation and dark periods of the experiments were in the order of 15 min.
For economic reasons, the housing which enfolds the transfer optics between FTIR spectrometer and chamber is flushed with purified dry air. Therefore, quantification of CO2 is, due to a slight variability in the dry air supply, unreliable under normal laboratory conditions. To be able to quantify CO2, in selected product study experiments, the transfer optics housing was flushed with ultrapure N2 evaporated from a liquid nitrogen tank.
2.4 Materials
The following chemicals have been used without further purification and purities as stated by the suppliers: 3-penten-2-one (Alfa Aesar, technical grade 85 %), trans-2-butene (Messer, 99 %), isoprene (Aldrich, 99 %), 3-buten-2-ol (Alfa Aesar, 97 %), 3-penten-2-ol (Sigma Aldrich, 96 %), NO (Air Liquide, 99.5 %), CO (Air Liquide, 99.97 %), and synthetic air (Messer, 99.9999 %). While the supplier states the predominance of the trans isomer for 3-penten-2-ol, the cis trans isomer ratio is not specified for 3-penten-2-one. The latter compound is not commercially available with purities higher than 85 %. Another sample of 70 % purity (technical grade) contains, as stated by the supplier, 30 % 4-methyl-3-penten-2-one as impurity. However, the gas-phase FTIR spectra of the 70 % sample after subtraction of the 4-methyl-3-penten-2-one content are identical to the spectra recorded for the 85 % sample. It is therefore reasonable to assume that no absorptions other than those belonging to 3-penten-2-one are present in the spectra of the sample used.
The first-order wall loss of 3P2 was < 1 × 10−5 s−1 in all 480 L chamber experiments and in the range of (5–10) × 10−5 s−1 in the 1080 L chamber experiments, respectively. Typically, the consumption through OH radicals was about 1 order of magnitude faster than the wall loss. The reference compounds did not show any wall loss. Photolysis and dark reactions between 3P2 and the radical source were found to be negligible under all experimental conditions.

Figure 1Relative-rate plots of all experiments using isoprene (green) and E2-butene (blue) as references. Different experimental runs for each reference are denoted with different symbols. The error bars consist of a systematic uncertainty and an additional 10 % relative error to cover uncertainties derived from the experimental and evaluation procedure, respectively.
3.1 3-Penten-2-one + OH kinetics
Kinetic experiments were performed in both chambers under varying light intensity using isoprene and E2-butene as references. The relative-rate plots according to Eq. (1) are presented in Fig. 1 for all performed experiments. The relative ratios , determined for each individual experiment following regression analysis, agree within < 13 % using isoprene and < 9 % using E2-butene as a reference compound, respectively. Intercepts of the regression lines were found to be zero within a 2σ statistical error and the correlation coefficients were R2 > 0.99. Given the latest IUPAC recommendations (Mellouki et al., 2021) for the rate coefficients of the OH radical reactions with E2-butene (k= (7.1 ± 1.1) × 10−11 cm3 molec.−1 s−1) and isoprene (k= (1.0 ± 0.2) × 10−10 cm3 molec.−1 s−1), respectively, the calculated rate constants for 3P2 derived from both references are in excellent agreement. All this suggests that secondary processes other than wall loss can be neglected in the present experimental set-up. Table 1 summarizes the results obtained from all conducted kinetic experiments. The weighted average rate coefficient is (6.2 ± 1.0) × 10−11 cm3 molec.−1 s−1 where the quoted error results from the 2σ statistical error of the weighted mean and an additional 10 % relative error to cover uncertainties derived from the experimental and evaluation procedure.
Table 1Results of the 3P2 + OH kinetic experiments.
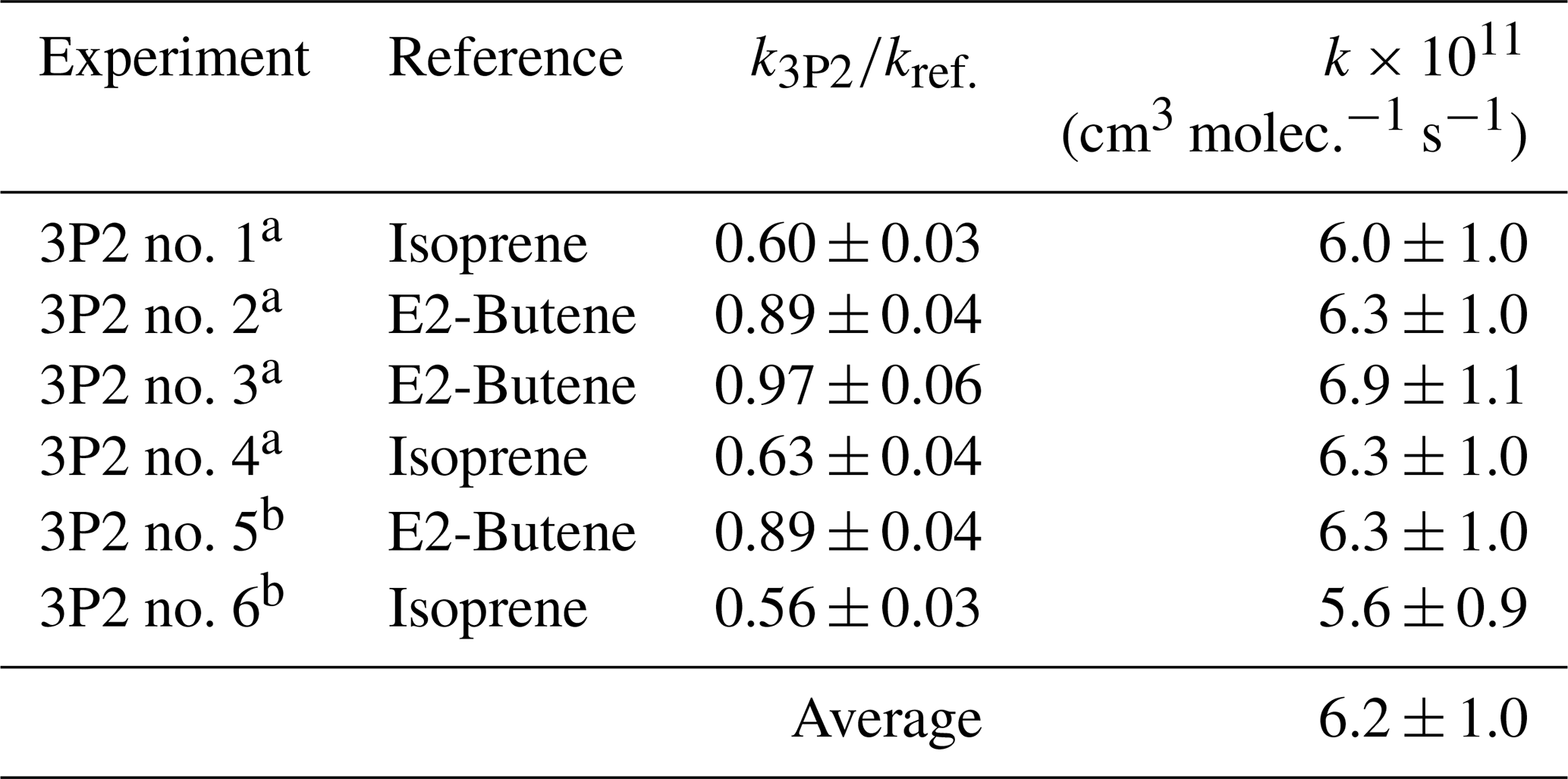
a Performed in the 1080 L chamber. b Performed in the 480 L chamber.
The rate coefficient of 3P2 has been determined previously in our laboratory (k3P2= (7.22 ± 1.74) × 10−11 cm3 molec.−1 s−1) based on experiments employing a 3P2 sample, which contained about 30 % mesityl oxide as major impurity (Blanco et al., 2012). Nevertheless, both determinations agree within 20 % and the value obtained here is within the uncertainties of the former study.
3.2 In situ generation of 2-hydroxypropanal
2-Hydroxypropanal (2HPr) is one of the expected main products in the OH-radical-initiated oxidation of 3P2. This α-hydroxyaldehyde, commonly known as lactaldehyde, is available commercially only as 1 M aqueous solutions, where various dimer species coexist (Takahashi et al., 1983). Therefore, in order to obtain gas-phase FTIR spectra and cross sections of 2HPr, the ozonolysis reactions of 3-buten-2-ol (3B2OL) and 3-penten-2-ol (3P2OL) were used to generate the α-hydroxyaldehyde in situ in the 1080 L chamber. Given the wall loss of 2HPr, observed in most of the 3P2 product study experiments in the 1080 L chamber (see Sect. 3.3), the 3B2OL ozonolysis experiments were optimized to ensure large product formation ratios with negligible losses.
It is well established that ozonolysis reactions proceed through a 1,3-dipolar cycloaddition forming initially a five-membered primary ozonide (POZ), which readily decomposes through bond scission between the carbon atoms of the former C=C bond and one of the O−O bonds. Hence, assuming this to be the only reaction pathway taking place, each of the two possible decomposition channels results in the formation of a stable carbonyl species and a Criegee intermediate (CI) as shown in Fig. 2. 3-Buten-2-ol ozonolysis leads to formaldehyde as one of the primary carbonyls. When comparing the product spectra obtained here to infrared spectra of 1,2-epoxybutane, there is no indication for epoxide formation in the 3B2OL ozonolysis spectra thus giving confidence in the correctness of the initial assumption. Since the sum of the yields of both HCHO and 2HPr, expected to be the other primary carbonyl (Fig. 2), should accordingly be equal to unity, then the 2HPr yield (y2HPr) is defined as 1−yHCHO. This allows us to derive a correlation between the integrated absorption of 2HPr and its concentration. Beside HCHO and 2HPr acetaldehyde formation can be observed unambiguously in the 3B2OL ozonolysis system. The acetaldehyde yield was reproducible throughout all 3B2OL experiments. The formation can only be explained by decomposition of the CI formed according to channel (a) (Fig. 2). Our results show that acetaldehyde accounts for 36 ± 10 % of the CI decomposition in the 3B2OL ozonolysis. However, the fate of CIs is sensitive to the degree of excitation, which is not necessarily the same for different ozonolysis systems. Given that the same CI is formed in the 3P2OL ozonolysis (Fig. 2) acting as a potential secondary source of acetaldehyde, this experimental system turned out to be improper for a cross section determination of 2HPr. The 3P2OL ozonolysis has therefore only been used for confirmation of the 2HPr spectral features by comparison with the 3B2OL experimental system (see Sect. 3.3).
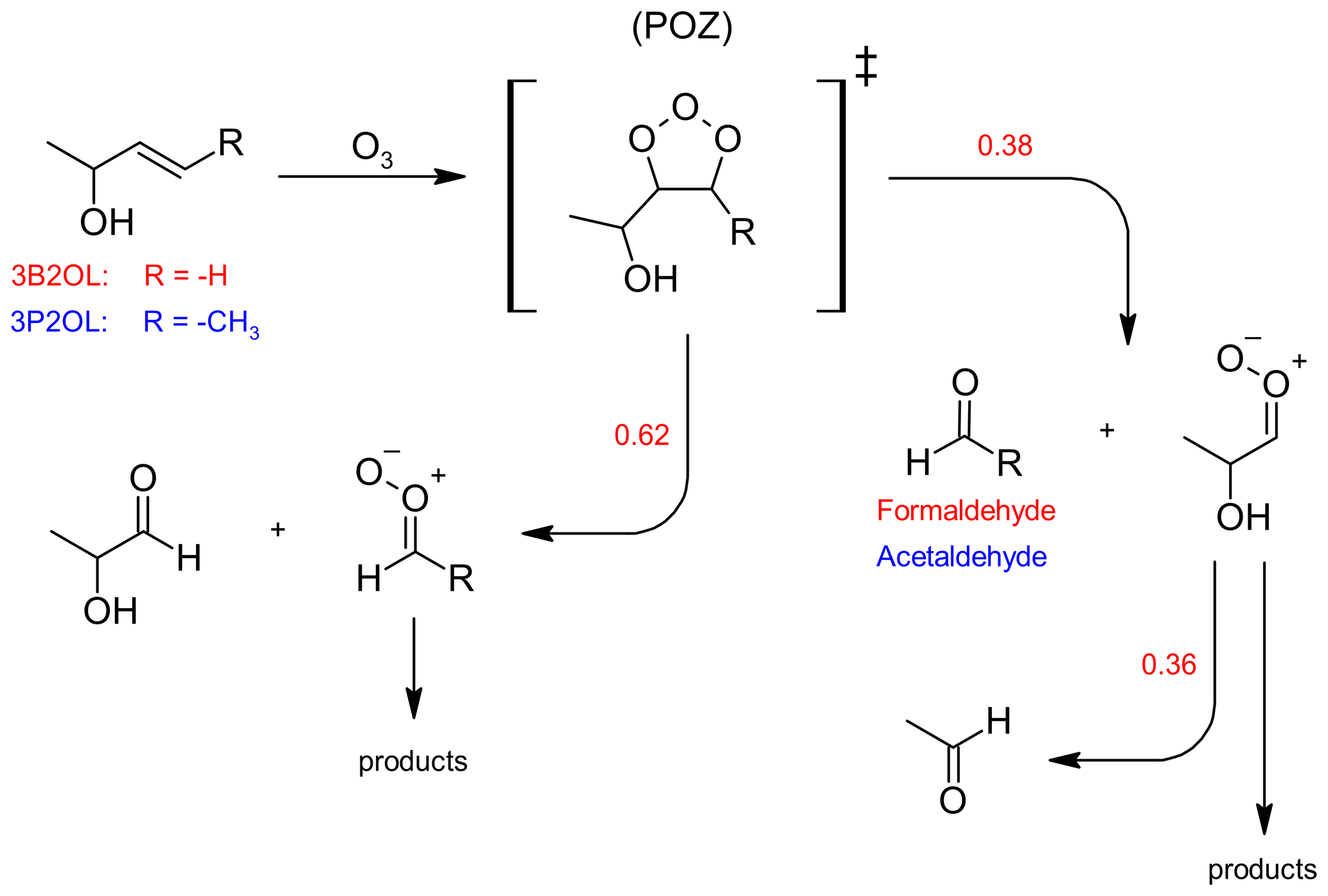
Figure 2Formation of 2-hydroxypropanal through the ozonolysis of 3-buten-2-ol (3B2OL, red) and 3-penten-2-ol (3P2OL, blue) and respective average branching ratios. For readability reasons only one stereo-isomer is drawn for each Criegee intermediate.
The observed yield for the primary aldehyde HCHO (0.38 ± 0.06) is well below 0.5, thus indicating a preference of the POZ decomposition towards 2HPr formation. Since the OH absorption band centred around 3550 cm−1 (integration range: 3580–3500 cm−1) is expected to be free from interferences of other unidentified species, this band was chosen to determine a cross section for 2HPr based on the comparison of FTIR spectra of both ozonolysis systems and residuals of the 3P2 + OH system. A plot of the integrated OH absorption versus the concentration of 2HPr, calculated based on the yields as discussed above, is shown in Fig. S1 in the Supplement. The cross section for the integrated OH absorption band was determined as (5.0 ± 1.5) × 10−18 cm molec.−1 (base e) by averaging the results of all individual experiments. The experimental results agree within 9 % but given the error of the HCHO yield and the wall loss of 2HPr, we prefer, however, to assign an expanded uncertainty of 30 %.
3.3 3-Penten-2-one + OH mechanism
Product study experiments were carried out in both simulation chambers. After subtraction of 3P2 and the species related to the methyl nitrite photolysis itself (methyl nitrite, methyl nitrate, NO, NO2, HONO, HNO3, and HCHO) the FTIR spectra contain absorption features that can be attributed unambiguously to acetaldehyde, methyl glyoxal, and peroxyacetyl nitrate (PAN). CO2 formation is observed by the absorption features in the range 2400–2250 cm−1. Plots of the identified products (in ppmV without corrections) versus the consumed 3P2 corrected for the wall loss (in ppmV) are presented in Fig. 3 for all conducted experiments. In the case of methyl glyoxal and acetaldehyde, these correlations exhibit a very high linearity. This indicates clearly their formation as primary products in the OH-initiated oxidation of 3P2. Molar formation yields based on averaging the results of the regression analysis of each experiment are 0.40 ± 0.07 and 0.29 ± 0.09 for acetaldehyde and methyl glyoxal, respectively, without corrections other than the 3P2 wall loss. The errors represent the corresponding precision errors. The larger relative error associated with methyl glyoxal results from a larger scattering in the experimental data.
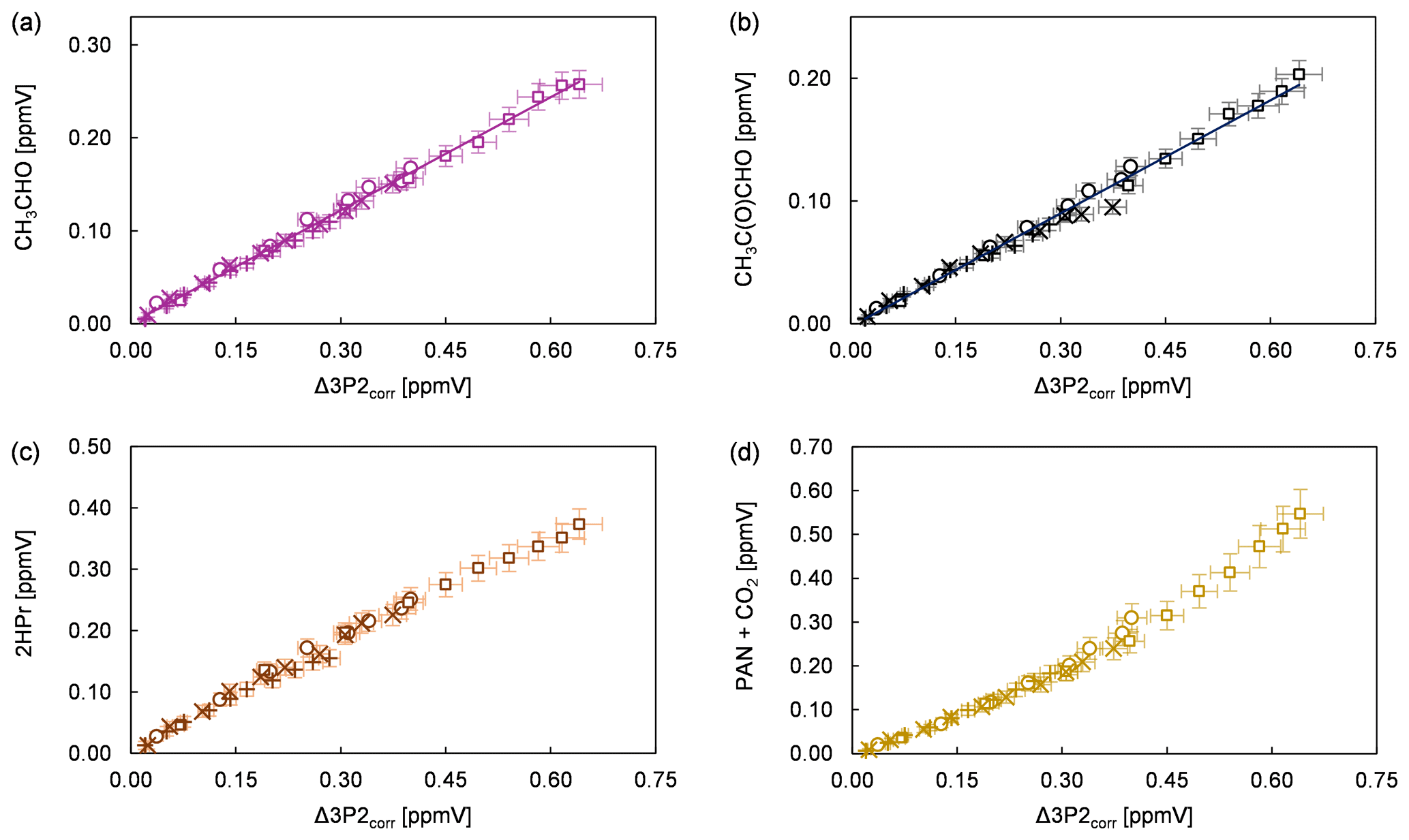
Figure 3Yield plots for (a) acetaldehyde, (b) methyl glyoxal, (c) 2-hydroxypropanal, and (d) the sum of PAN and CO2 for all conducted experiments corrected for the wall loss of 3P2. The error bars consist of the corresponding precision error. The data of the 480 L chamber experiments are multiplied with a factor of 0.1 to fit within the scale of 1080 L chamber experiments. Different experimental runs are denoted with different symbols.
Based on the structure–activity relationship (SAR) approach provided by Kwok and Atkinson (1995) the group rate constant is in the order of 10−13 cm3 molec.−1 s−1 for H-atom abstraction from the terminal methyl groups, whereas addition to the C=C double bond accounts for about 10−11 cm3 molec.−1 s−1. Hence, it is very likely that the OH radical will add almost exclusively to either the α- or β-carbon atom to form the corresponding α- or β-hydroxyalkyl radical. Under atmospheric conditions these radicals will react immediately with molecular oxygen yielding the corresponding hydroxyperoxy radicals. By employing an excess of NO virtually all RO2 radicals will react with NO and form mainly hydroxyalkoxy radicals (Fig. 4). A fraction of the RO2 + NO reaction might also produce organic nitrates (RONO2). The β-RO radical could react either with oxygen to yield a 1,3-dicarbonyl species (Fig. 4, pathway β2) or dissociate to form acetaldehyde. The co-built hydroxyalkyl radical will react subsequently with oxygen yielding methyl glyoxal, in agreement with our experimental observations.

Figure 4Proposed mechanism for the OH-radical-initiated oxidation of 3P2. The reaction pathways are named according to the position where the oxygen adds to form the peroxy radical.
Formation of PAN originates from CH3C(O) radicals, generated in the reaction system through the subsequent reactions with oxygen and NO2. However, we recently pointed out that under the typical experimental conditions when the methyl nitrite photolysis is used as source for OH radicals, PAN formation accounts only for up to one-third of the fate of acetyl radicals (Illmann et al., 2021b). The main fate of the readily formed acetylperoxy radical will be the reaction with NO which eventually yields CO2 and HCHO. Since HCHO is formed in the methyl nitrite photolysis itself, the sum of PAN and CO2 has been used to estimate the formation yield of CH3C(O) radicals. The correlation between (PAN + CO2) and Δ3P2 is strongly linear up to a consumption of about 30 % (Fig. 3) and becomes precisely non-linear with higher levels of the 3P2 consumption. This indicates strongly, on the one hand, the formation of CH3C(O) radicals due to the OH reaction of 3P2. On the other hand, secondary processes like further oxidation of the first-generation products increase the formation rate of acetyl radicals at longer reaction times. The primary generation of CH3C(O) radicals results from the bond scission between Cα and the carbonyl carbon atom of the α-RO radical (pathway α1, Fig. 4). The molar formation yield of the sum PAN + CO2, without corrections, is 0.63 ± 0.14 based on averaging the results of the regression analysis of each experiment over the linear range.
In contrast to daytime conditions within the troposphere, the photolysis of the generated acetaldehyde and methyl glyoxal under our experimental conditions was negligible compared to the further oxidation through OH radicals. The reaction of both aldehydes with OH was shown to proceed nearly exclusively via the abstraction of the aldehydic H atom, therefore being a secondary source of CH3C(O) radicals with strength of 95 %–100 % (Calvert et al., 2011).
Similarly to the β-RO, the α-RO radical could also react with oxygen according to pathway α3 to form a hydroxydicarbonyl species (Fig. 4). However, based on the observed spectral features there is no evidence to support further transformation pathways of both RO radicals other than the decomposition channels. This is in agreement with the product studies of structurally similar α,β-unsaturated ketones conducted under conditions where RO radicals are formed solely through the reaction of RO2 + NO (Tuazon and Atkinson, 1989; Galloway et al., 2011; Praske et al., 2015; Illmann et al., 2021b). The exothermicity of RO2 + NO results in RO radicals that are chemically activated and prone to decomposition (Orlando et al., 2003). For 1,2-hydroxyalkoxy radicals it was also shown that the reaction with O2 cannot compete with the dissociation channel (Atkinson, 2007).
2-Hydroxypropanal is a co-product of CH3C(O) radicals in the pathway α1 (Fig. 4). Residual spectra of the 3P2 + OH system after subtraction of acetaldehyde, methyl glyoxal, and PAN are shown in Fig. 5 together with spectra recorded during the ozonolysis of 3B2OL and 3P2OL (see Sect. 3.2). Absorption bands centred on 3640 cm−1 and in the range 1120–1090 cm−1 and centred on 3640 and 980 cm−1, and in the range 1120–1090 cm−1 are present only in the residual spectra of the 3P2OL and 3B2OL ozonolysis experiments, respectively, indicating additional unidentified reaction products, which likely result from CI decomposition processes. However, other spectral ranges centred on 3550 cm−1 (O−H stretching vibration), 1750 cm−1 (C=O stretching vibration), 1370 cm−1 ( bending vibration), 830 cm−1 (C−H bending vibration), and the main parts of the characteristic absorption pattern in the range 3040–2640 cm−1 (C−H stretching vibration) agree within the spectra in both position and relative intensity, thus giving confidence in the identification of 2HPr.
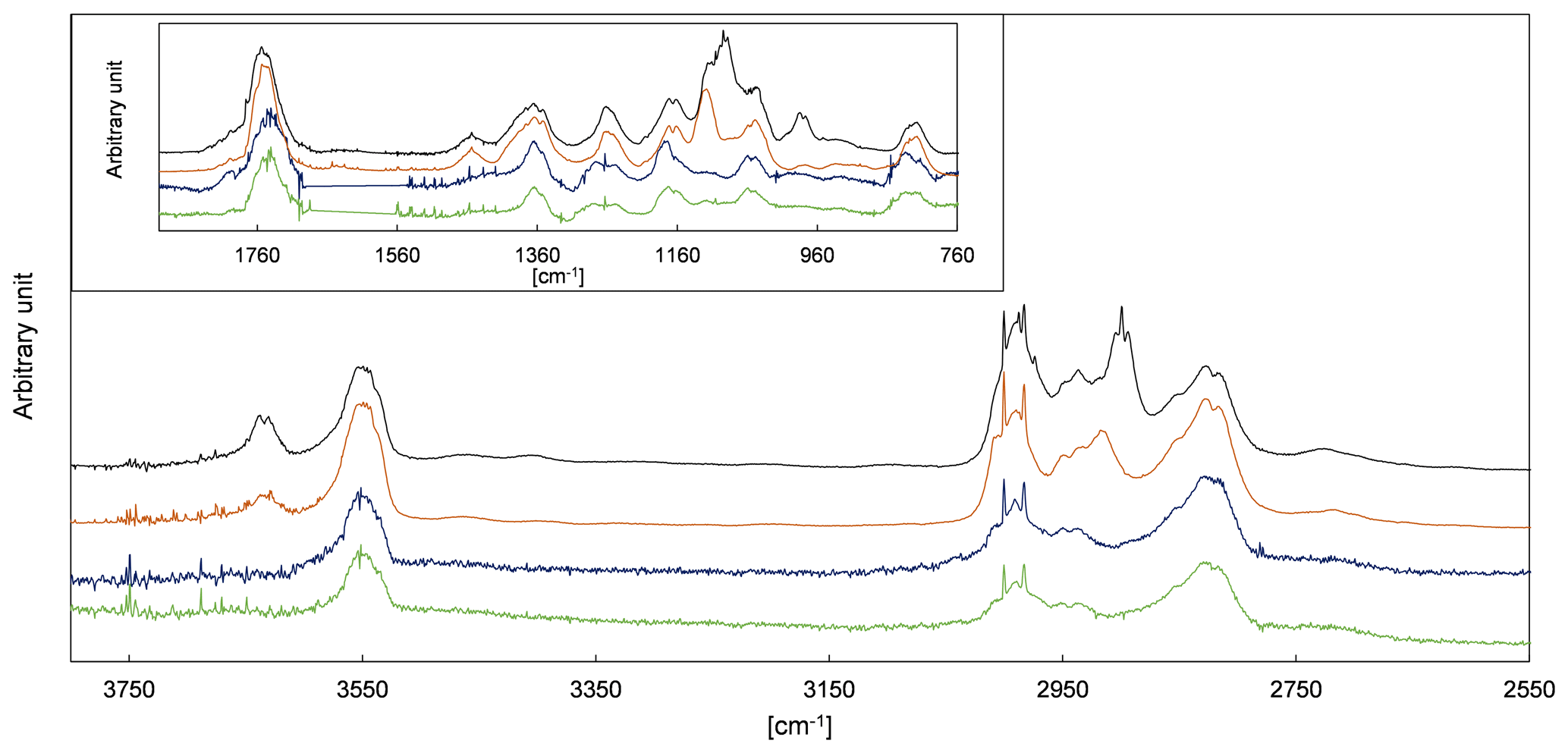
Figure 5FTIR spectra of the residuals assigned to 2HPr obtained in the 1080 L chamber (green) and 480 L chamber (blue) 3P2 + OH experiments and residuals obtained from the ozonolysis of 3-penten-2-ol (brown) and 3-buten-2-ol (black).
Based on the integrated absorption cross section determined within this work, an averaged molar yield of 0.59 ± 0.25 is derived for 2HPr in the 3P2 + OH reaction, from the regression analysis of all experiments without further corrections. One should note that the 2HPr yields of all experiments, performed in both chambers, agree within 12 %, and the major uncertainty is derived from the accuracy of the absorption cross section. Nevertheless, the 2HPr yield is similar to the molar yield of PAN + CO2, which is reflected by the proposed mechanism since these species are formed accordingly in the same reaction pathway (Fig. 4). On the other hand, the yield plots of 2HPr show a small but precise curvature in each single experiment (Fig. 3). The wall loss of the α-hydroxyaldehyde was found to be < 10 × 10−5 s−1 in the 1080 L chamber and < 4 × 10−5 s−1 in the 480 L chamber, respectively. This all indicates that further oxidation of the α-hydroxyaldehyde is significant under the experimental conditions.
3.4 2-Hydroxypropanal + OH and modelling
Among the class of α-hydroxyaldehydes mechanistic information on the OH reaction and photolysis at atmospheric pressure were reported merely for glycolaldehyde (Niki et al., 1987; Bacher et al., 2001; Magneron et al., 2005). Baker et al. (2004) reported rate coefficients for the OH reaction of a series of hydroxyaldehydes synthesized in situ via the reaction of OH radicals with precursor alcohols. The authors obtained k= (1.7 ± 0.2) × 10−11 cm3 molec.−1 s−1 for 2HPr + OH through a non-linear least squares analysis of the data of the 2-methyl-2,4-pentanediol oxidation (Baker et al., 2004). Under tropospheric daytime conditions both photolysis and OH-initiated oxidation are important removal processes for glycolaldehyde (Bacher et al., 2001). However, assuming a similar behaviour for 2HPr, photolysis is expected to be negligible under the experimental conditions of the present study, since the OH radical level is much higher while photolysis frequencies are lower than within the troposphere. Including the above rate constant of 2HPr + OH into the model described previously (Illmann et al., 2021b) results in a corrected average yield of 0.68 ± 0.27, which is about 15 % higher than determined from the yield plot, without proper corrections (Fig. 3). Hence, a significant fraction of the α-hydroxyaldehyde is subject to OH-radical-initiated oxidation.
Based on the SAR approach by Kwok and Atkinson (1995) and the mechanistic information reported for the glycolaldehyde oxidation (Niki et al., 1987; Bacher et al., 2001; Magneron et al., 2005), one would expect abstraction of the aldehydic H atom to dominate compared to abstraction of the carbon-bonded H atom of the entity for the OH reaction of 2HPr, as presented in Fig. 6. The abstraction from the terminal −CH3 group and the −OH group is expected to be negligible due to the much lower group rate constants. The hydroxypropionyl radical formed according to channel (a) will either eliminate carbon monoxide and react with O2 to form acetaldehyde or react with oxygen to form a hydroxypropionylperoxy radical (Fig. 6). The latter radical, resulting from channel (2a), may either yield peroxyhydroxypropionyl nitrate or will be converted to the corresponding RO radical (Fig. 6). This species will readily eliminate CO2 and finally form acetaldehyde as well. A theoretical investigation on the C5-acylperoxy radical indicates that H migration reactions (1,5-H, 1,6-H or 1,7-H shift) of larger acylperoxy radicals might be fast enough to compete with the bimolecular reactions at low parts-per-billion levels of NO (Knap and Jørgensen, 2017). However, based on the predicted effects of the substitution pattern on the reactivity towards H migration reactions (Vereecken and Nozière, 2020), one would not expect the unimolecular reaction of the smaller hydroxypropionylperoxy radical to be competitive, at least not under our experimental conditions. By analogy to the OH-initiated oxidation of 3-hydroxy-2-butanone (Aschmann et al., 2000) one would expect reaction with oxygen to predominate over decomposition for the hydroxyalkyl radical formed following channel (b), thus leading to methyl glyoxal (Fig. 6). Hence, the 2HPr + OH reaction appears to be a secondary source of acetaldehyde and methyl glyoxal in the experimental system.
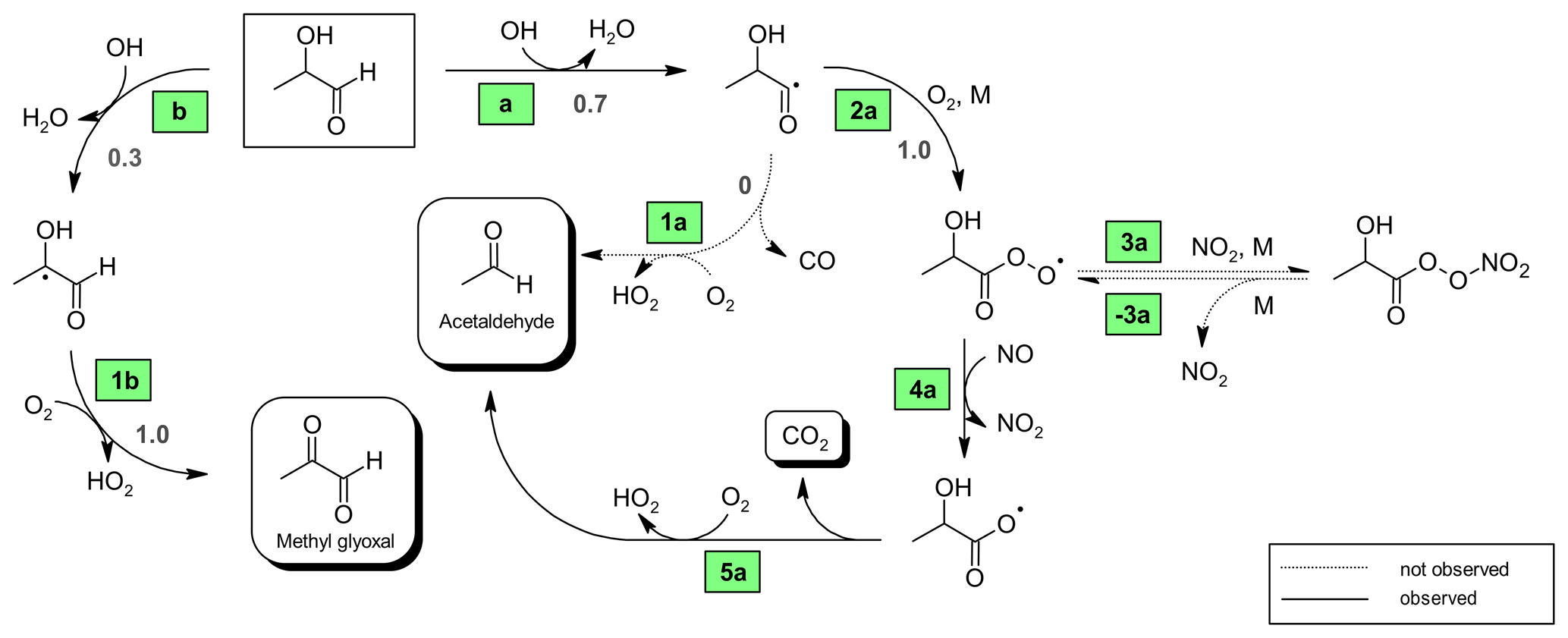
Figure 6Proposed mechanism for the OH-radical-initiated oxidation of 2-hydroxypropanal (2HPr). Relevant branching ratios estimated within this work are denoted in grey next to the reaction channel.
In order to investigate the 2HPr + OH reaction, methyl nitrite and NO were added for a second time, after a 3P2 consumption of about 70 %, to shift the reaction system towards secondary oxidation processes. Applying the approach presented by Baker et al. (2004) to these experiments, as shown in Fig. S2 in the Supplement, allows us to estimate a value of (2.2 ± 0.6) × 10−11 cm3 molec.−1 s−1 for the rate coefficient of 2HPr + OH. Our estimation is about 30 % larger than previously reported (Baker et al., 2004). Taking into account that both determinations are based on the in situ generation of the α-hydroxyaldehyde, this is still an excellent agreement. In panel (c) of Fig. 7 it can be observed that the mixing ratio of acetaldehyde (purple circles) increases continuously over the second irradiation period, while that of methyl glyoxal (black circles) is reaching a plateau relatively fast. This is in qualitative agreement with the proposed mechanism. Peroxy nitrates other than PAN, formed through 3P2 + OH, could not be detected. Traces of the analogue peroxyhydroxyacyl nitrate resulting from glycolaldehyde oxidation have only been previously observed when the corresponding RO2 radical was generated through the reaction of glycolaldehyde with Cl atoms in the presence of NO2 (Niki et al., 1987). Magneron et al. (2005) did not detect any PAN-type species in the glycolaldehyde +OH system and therefore concluded that this species is probably unstable and readily dissociates. Hence, abstraction of the aldehydic H atom following channel (a) will likely result exclusively in the formation of acetaldehyde irrespective of the branching ratio between the (1a) and the (2a) channel (Fig. 6).
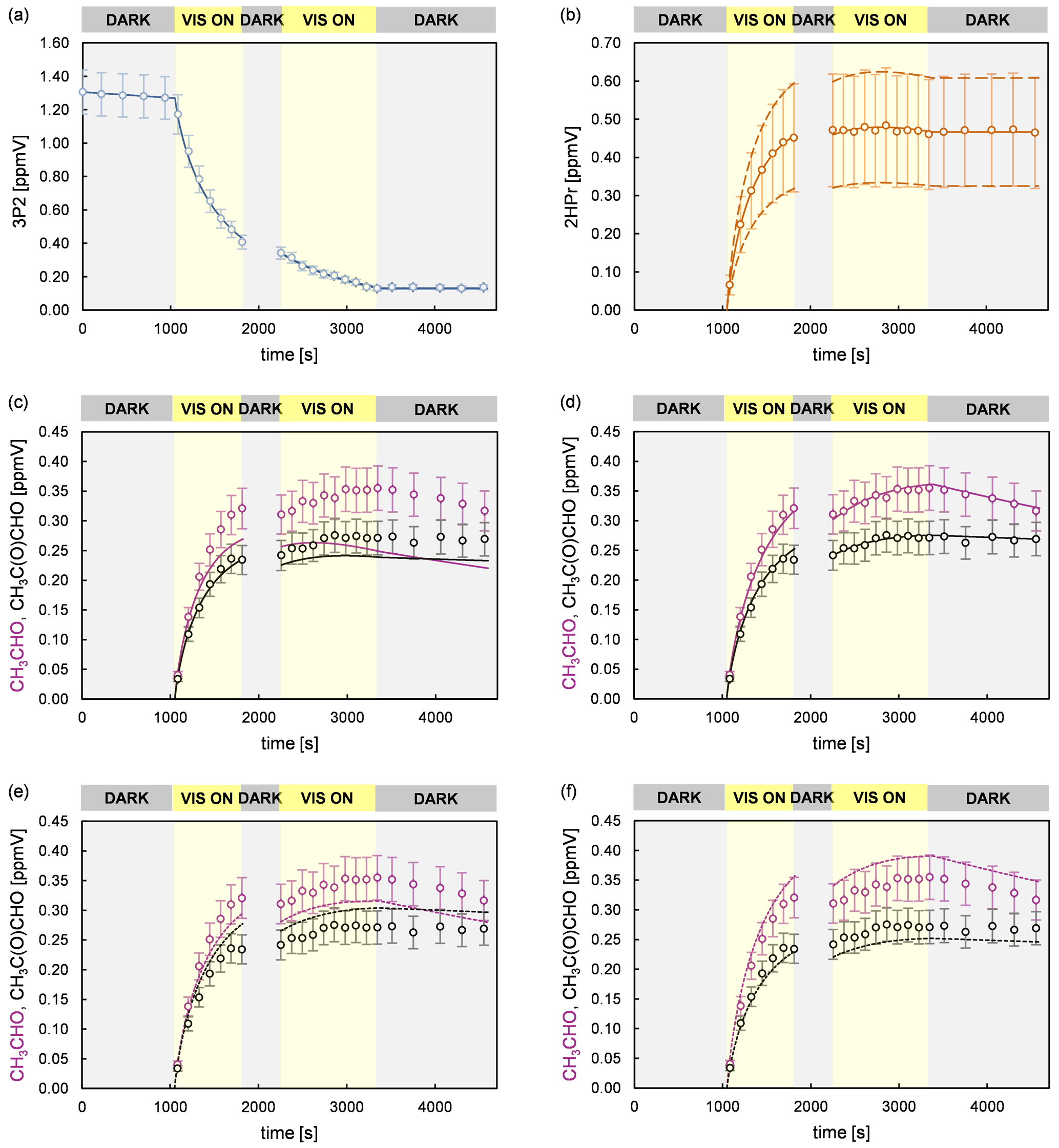
Figure 7Experimental and simulated time profiles for a 3P2 + OH experiment, performed in the 1080 L chamber, with a supplementary addition of methyl nitrite and NO during the second dark phase of the experiment. The experimental set-up did not allow the quantification of CO2. The circles represent the experimental data. The error bars represent the accuracy error for each species. The lines show the simulated profiles of (a) 3P2 and (b) 2HPr assuming the average yield (solid line) and the upper and lower limit (dashed lines). The simulated profiles of acetaldehyde and methyl glyoxal are shown considering the average 2HPr yield (c) without their secondary formation, (d) with the optimum parameters used to reproduce the experimental data, (e) for the lower and upper limit of acetaldehyde and methyl glyoxal (scenario 1), respectively, and (f) for the upper and lower limit of acetaldehyde and methyl glyoxal (scenario 2), respectively.
The molar formation yields of acetaldehyde and methyl glyoxal, derived from 3P2 + OH as well as from 2HPr + OH were included as parameters in a simplified model (Illmann et al., 2021b) and varied until the experimental time profiles are reproduced by the simulation. Since the OH-initiated oxidation of 2HPr is expected to proceed solely through the channels (a) and (b), the product yields of acetaldehyde and methyl glyoxal, from 2HPr + OH, should correspond to the branching ratios ka and kb, respectively (Fig. 6). Their sum should, in turn, equal unity. Table 2 shows the simplified reaction sequences and the rate coefficients needed to describe the reaction system. These sequences do not follow the nomenclature used in the proposed mechanisms (Figs. 4 and 6) since the simplified model does not differentiate whether a product is formed directly from a parent compound through more than one pathway.
Table 2Simplified reaction sequence used for the modelling of the temporal behaviour of experimentally quantified species in order to obtain first-generation yields for the respective reactions (3P2 + OH, 2HPr + OH).

a Input parameter. b Rate coefficient determined within this work. c Average branching ratio obtained through modelling. d Average branching ratio (R4d1 + R4d2) R4 = 0.58 obtained through modelling. e Determined in each individual experiment. f Rate coefficient from Atkinson et al. (2006). g Branching ratio (R6a + R6b) R6 = 0.95 from Atkinson et al. (2006). h Rate coefficient from Atkinson et al. (2006). i Branching ratio (R8a + R8b) R8 = 1.0 from Atkinson et al. (2006). j Rate coefficient from Baker et al. (2004).
Figure 7 summarizes an analysis of the model sensitivity, observing also the accuracy of all quantified species. For all species but 2HPr accuracy was defined as a 10 % relative error plus the corresponding detection limit. The accuracy of 2HPr is given as a 30 % relative error plus the detection limit due to the uncertainty of the cross section determination. Panels (c)–(f) show different model runs for acetaldehyde and methyl glyoxal in which the 2HPr yield was set to 0.66, represented by the solid line in panel (b). As can be seen in panel (c), without considering the 2HPr + OH reaction the simulated profile represents roughly the experimental methyl glyoxal data during the first irradiation period. By contrast, the acetaldehyde profile matches the experimental data only in the beginning of the first irradiation corresponding to a 3P2 consumption of < 30 %. The temporal profiles of both species completely fail in reproducing the measured data during the second irradiation, where more than 70 % of the 3P2 is already consumed. This demonstrates unambiguously that a secondary source for both acetaldehyde and methyl glyoxal is needed to describe the experimental system, namely the α-hydroxyaldehyde oxidation. However, the match between the simulated and experimental time profiles in the beginning of the first irradiation allows us to set values for the first-generation yields of acetaldehyde and methyl glyoxal from 3P2 + OH in these experiments. Panel (d) shows the optimum model run that allows a simultaneous fit of the experimental time profiles for both acetaldehyde and methyl glyoxal. In order to assess the errors for the branching ratios ka and kb, two scenarios were defined which represent the limiting cases and thus enable us to determine the maximum variation in ka and kb. Accordingly, panel (e) shows a model run in which acetaldehyde is simulated for its lower limit of the accuracy error and methyl glyoxal for the upper limit, respectively, (scenario 1), while panel (f) represents the inverse case (scenario 2). These scenarios were modelled for different strengths of the secondary source of acetaldehyde and methyl glyoxal, meaning that the 2HPr yield from 3P2 + OH was varied within the limits imposed by the accuracy of the 2HPr measurement, as shown in panel (b). For both scenarios, the obtained first-generation yields of acetaldehyde and methyl glyoxal from 3P2 + OH were found to be independent from the 2HPr yield. Since the formation of acetaldehyde and methyl glyoxal from the 3P2 + OH reaction does not necessarily depend on 2HPr, this observation is rather self-consistent and serves merely as a validation of our model. Based on the proposed 3P2 + OH mechanism one would expect their yields to be the same; thus their ratio should equal unity. This does correspond to scenario 1, while an acetaldehyde methyl glyoxal ratio > 1 is observed for scenario 2 (Fig. S3 in the Supplement). Although within the accuracy errors this indicates a small bias between the acetaldehyde and methyl glyoxal quantification. The sum of the acetaldehyde and methyl glyoxal yield from 2HPr + OH correlates with the 2HPr yield from the 3P2 + OH reaction, where larger values are observed when the input 2HPr yield is lowered (Fig. S4 in the Supplement). In order to reproduce the entire time profiles of acetaldehyde and methyl glyoxal, an overestimation of the 2HPr mixing ratio and hence the strength of the secondary acetaldehyde and methyl glyoxal source is compensated for by an underestimation of the acetaldehyde and methyl glyoxal yield in the model. Hence, this behaviour can be rationalized in terms of an antagonistic effect. The sum of the acetaldehyde and methyl glyoxal yields becomes unity when a 2HPr yield of about 0.54 and 0.61 is used in the model for scenarios 1 and 2, respectively. Considering the yield of 0.66, used to match the experimental data in panel (b), this might indicate an overestimation of the 2HPr mixing ratios. However, the differences are within the accuracy due to the rather uncertain 2HPr cross section. The branching ratios ka and kb were obtained by scaling of the acetaldehyde and methyl glyoxal yield. These were found to be independent from the 2HPr yield within the 2HPr accuracy limits and almost indistinguishable in between scenarios 1 and 2 (Fig. S5 in the Supplement). Accordingly, the average branching ratios ka and kb are 0.73 ± 0.08 and 0.27 ± 0.08, respectively. Within the uncertainties, this is in agreement with SAR predictions (Kwok and Atkinson, 1995) which estimate 0.8 and 0.2 for the branching ratios, respectively, as well as previous results on the OH reaction of glycolaldehyde at atmospheric pressure (Niki et al., 1987; Bacher et al., 2001; Magneron et al., 2005).
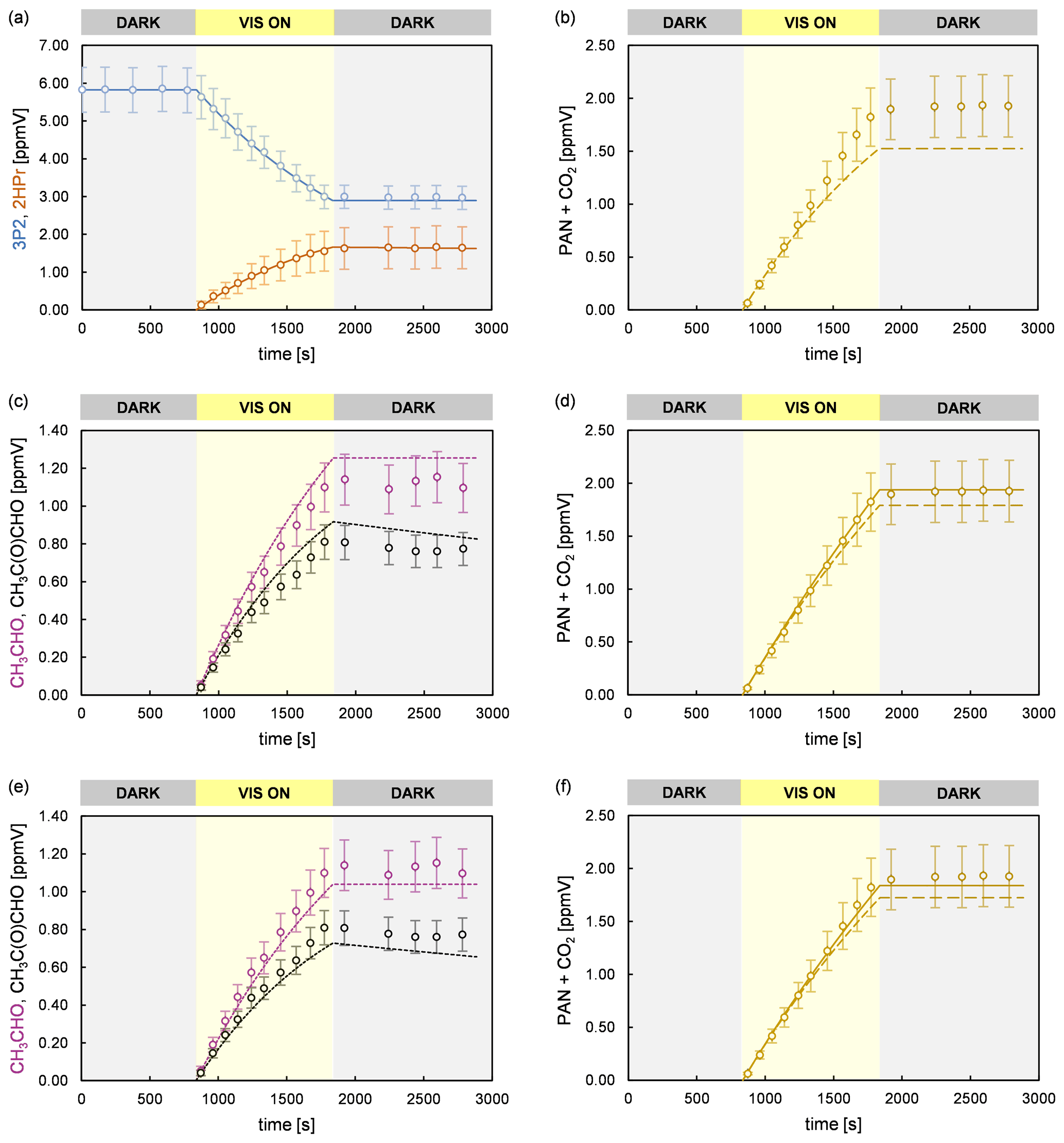
Figure 8Experimental (circles) and simulated (lines) temporal behaviour of 3P2, 2HPr, acetaldehyde, methyl glyoxal, and PAN + CO2 in an experiment performed in the 480 L chamber. The error bars of the experimental data represent the accuracy error. Model runs of the sum parameter PAN + CO2 are shown considering only the formation of PAN and CO2 due to CH3C(O) radicals formed in the reaction system (dashed line) and including additionally the CO2 source from 2HPr + OH (solid line). The model run in panel (b) considers only primary PAN and CO2 formation from 3P2 + OH. Panel (c) and (d) represent model runs according to scenario 3 (acetaldehyde and methyl glyoxal at the upper limit of the accuracy error), and panel (e) and (f) represent those according to scenario 4 (acetaldehyde and methyl glyoxal at the lower limit of the accuracy error), respectively.
Based on these results, the temporal profiles of acetaldehyde and methyl glyoxal are well-reproduced for all conducted experiments. Their corrected yields in the 3P2 + OH reaction are 0.39 ± 0.07 and 0.32 ± 0.08, respectively. Hence, while larger molar yields were observed for acetaldehyde than for methyl glyoxal without proper corrections the model predicts both first-generation yields to be the same within the accuracy errors, which indicate their formation according to the same reaction channel. The branching ratios of the simplified reaction scheme, obtained through modelling, are given in Table 2.
Figure 8 shows time profiles obtained from an experiment performed in the 480 L chamber, in which PAN and CO2 were quantified, as well as simulated profiles from different model runs. As presented in panel (b), the experimental data are reproduced solely for less than the first half of the irradiation period if only PAN and CO2 formation from 3P2 + OH are considered in the model. This corresponds to a 3P2 consumption of < 30 %, which is consistent with the non-linearity of the yield plot observed for higher 3P2 consumption levels (Fig. 3). As discussed before, PAN and CO2 formation are affected from the further oxidation of acetaldehyde and methyl glyoxal. However, CO2 elimination from the 2HPr-derived RO radical (Fig. 6) is an additional source of CO2 in the experimental system according to pathway (5a). Given that abstraction of the aldehydic H atom of 2HPr is expectedly leading solely to acetaldehyde, the yield of CO2 from the 2HPr oxidation depends only on the ratio between decomposition of the hydroxypropionyl radical and its reaction with oxygen (Fig. 6). In order to assess the uncertainty on the sum parameter PAN + CO2 due to secondary chemistry, the temporal profile of PAN + CO2 was simulated assuming both acetaldehyde and methyl glyoxal at the upper (scenario 3) and lower limit (scenario 4) of the measurement accuracy (Fig. 8). Hence, the strength of the secondary sources of CH3C(O) radicals in the experimental system was either maximized or minimized in the model. Moreover, the temporal behaviour of PAN + CO2 was simulated without considering CO2 formation from 2HPr + OH (dashed lines) and assuming the CO2 yield to equal the acetaldehyde yield (solid lines). In both scenarios (panels d and f of Fig. 8) the temporal profiles are nearly indistinguishable during the first half of the irradiation time, and one obtains the same first-generation yield for the sum parameter PAN + CO2, used to determine the CH3C(O) radical yield. The entire profile is reproduced solely when the CO2 yield from 2HPr + OH is equalized to the acetaldehyde yield in scenario 3 (panel d). In scenario 4, where the secondary formation of CH3C(O) radicals was set to the lower limit, the model slightly underestimates the sum of PAN and CO2 at the end of the experiment (panel f). However, in both scenarios the model predicts the sum of PAN + CO2 to be significantly lower than experimentally observed at the end of the irradiation period, when the CO2 formation from 2HPr is set to 0 (dashed lines). When introducing larger PAN + CO2 yields for 3P2 + OH, it is possible to match the observed profile for the second half of the experiment. However, in this case the model overestimates PAN + CO2 formation in the first half of the experiment, in which secondary formation is expected to be almost negligible.
Table 3Uncorrected yields and first-generation yields (yields corrected for secondary processes) of species identified in the 3P2 + OH reaction.

For the hydroxyacetyl radical Méreau et al. (2001) concluded, based on ab initio calculations, that decomposition cannot compete with the O2 reaction. Niki et al. (1987) observed CO2 instead of CO formation in the glycolaldehyde oxidation when secondary oxidation processes were minimized in the experimental system. These findings together with the significant discrepancy of the simulated and experimental time profile for PAN + CO2 at long irradiation times, when a CO2 formation from 2HPr oxidation is not included in the model, suggest that decomposition of the hydroxypropionyl radical is negligible and (see Fig. 6). Including the additional CO2 source in the model significantly improves the consistency between the simulated and experimental PAN + CO2 profile at long irradiation times, although slight discrepancies remain in some experiments. One should note that in this regard the time profile no longer represents merely the formation of CH3C(O) radicals. However, given that both the simulations with and without the additional CO2 source are indistinguishable in the first part of the irradiation period (Fig. 8), it is still possible to derive the corrected average yield for PAN + CO2 (0.56 ± 0.14) representing the yield of CH3C(O) radicals.
As for acetaldehyde and methyl glyoxal, the yields for 2HPr and PAN + CO2 are the same within the assigned accuracy, thus indicating their formation in the same reaction channel. Since carbon dioxide formation might be easily affected from processes on the chamber walls, the corrected yield for PAN + CO2 should, therefore, be still regarded as an upper limit. A build-up of CO2 from the walls might become relevant at longer irradiation times, and this supposedly explains the remaining small discrepancies at irradiation times > 10 min in some experiments. However, the reproducibility of the yields is essentially the same as for 2HPr for experiments performed in both chambers. Besides, separate control experiments, in which synthetic air was irradiated with the same set of lamps, did not show significant CO2 production. Therefore, the influence of off-gasing processes on its temporal behaviour is probably negligible in the beginning of the experiments, when the formation of the products in the target reaction dominates over secondary chemistry. An overestimation of the CH3C(O) radical yield is thus unlikely. Uncorrected and corrected molar yields, namely first-generation yields, of all quantified products are summarized in Table 3.
Combining the yields of the 3P2 oxidation products leads to a carbon balance close to unity (0.98 ± 0.18). The branching ratios for the pathways αON and βON (Fig. 4) forming RONO2 species are expectedly very minor channels. This is in agreement with previous findings in our laboratory, where the production of RONO2 species in the OH oxidation of α,β-unsaturated ketones was observed only in conjunction with the formation of tertiary RO2 radicals (Illmann et al., 2021b). Further, Praske et al. (2015) reported a low overall RONO2 yield of 0.040 ± 0.006 for methyl vinyl ketone oxidation.
The atmospheric lifetime of 3P2 with respect to OH radicals, defined as (k3P2 × [OH]), is about 4.5 h when assuming a global average OH radical concentration of 1.0 × 106 cm−3 within the troposphere (Bloss et al., 2005). Calvert et al. (2011), however, recommended an OH radical level of 2.5 × 106 cm−3 for the calculation of atmospheric lifetimes for short-lived species, which consequently leads to an even shorter lifetime. Since our experiments indicate no measurable photolysis, the OH reaction is the dominant degradation process during the daytime. 3P2 is thus oxidized close to the emission/formation source. A larger influence on atmospheric processes is indicated by the primary formation of CH3C(O) radicals, which account for 22 ± 6 % of the 3P2 oxidation. As acetyl radicals ultimately yield PAN, depending on the ratio, 3P2 exhibits a huge potential of forming NOx reservoir species. The gas-phase oxidation of the first-generation product 2HPr by OH radicals would generate mainly acetaldehyde (∼ 80 %), hence increasing the potential of forming NOx reservoirs. However, by comparison with glycolaldehyde, photolysis of 2HPr may also be competitive. On the other hand, since 2HPr is highly soluble in water, uptake into the aqueous phase (aerosols) may also be an important loss process. Hydration would significantly increase the lifetime towards photolysis and potentially lead to the formation of organic acids, as discussed previously for glycolaldehyde (Calvert et al., 2011).
Given both the short lifetime and the mechanism of the OH-initiated oxidation, 3P2 is an example of species whose chemistry explains the rapid PAN formation in young biomass burning plumes, as found previously in field observations (Alvarado et al., 2010). Acetaldehyde and methyl glyoxal also form acetyl radicals in their further gas-phase oxidation (Calvert et al., 2011). Besides, methyl glyoxal is also known as a source of secondary organic aerosol (Fu et al., 2008). Box models yielded high average OH radical concentrations of about 7.5 × 106 cm−3 within young biomass burning plumes (Müller et al., 2016). Based on that, once emitted from biomass burning, more than 50 % of the 3P2 carbon is converted into CH3C(O) radicals in less than 3 h. Assuming gas-phase products only in the further oxidation pathways, possibly up to two PAN molecules are formed per 3P2 molecule consumed. The α,β-unsaturated ketone also fits well into the characteristics of unknown VOCs as deployed by Alvarado et al. (2015) to elucidate the evolution of O3 and secondary organic aerosol in a plume of a prescribed fire in California since (a) the OH rate coefficient of 3P2 is in the order of 10−11 cm3 molec.−1 s−1, (b) the RO2+ NO reactions of 3P2-derived peroxy radicals result exclusively in fragmentation of the molecules, and (c) the mechanism proposed in this study predicts a high HO2 regeneration level. Therefore, single-component studies as the present one contribute to a better understanding of the complex biomass burning plume chemistry.
Data can be provided upon request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-18557-2021-supplement.
NI conducted the experiments and processed the data. All authors contributed to the preparation of the paper.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
The authors gratefully acknowledge support from the EU Horizon 2020 research and innovation programme through the EUROCHAMP-2020 Infrastructure Activity (grant agreement no. 730997) and the Deutsche Forschungsgemeinschaft (DFG) through the grant agreement WI 958/18-1.
This research has been supported by the Deutsche Forschungsgemeinschaft (DFG) (grant no. WI 958/18-1) and the European Commission Horizon 2020 Framework Programme (EUROCHAMP-2020 (grant no. 730997)).
This paper was edited by Jason Surratt and reviewed by Aparajeo Chattopadhyay and two anonymous referees.
Allen, G., Remedios, J. J., Newnham, D. A., Smith, K. M., and Monks, P. S.: Improved mid-infrared cross-sections for peroxyacetyl nitrate (PAN) vapour, Atmos. Chem. Phys., 5, 47–56, https://doi.org/10.5194/acp-5-47-2005, 2005.
Alvarado, M. J., Logan, J. A., Mao, J., Apel, E., Riemer, D., Blake, D., Cohen, R. C., Min, K.-E., Perring, A. E., Browne, E. C., Wooldridge, P. J., Diskin, G. S., Sachse, G. W., Fuelberg, H., Sessions, W. R., Harrigan, D. L., Huey, G., Liao, J., Case-Hanks, A., Jimenez, J. L., Cubison, M. J., Vay, S. A., Weinheimer, A. J., Knapp, D. J., Montzka, D. D., Flocke, F. M., Pollack, I. B., Wennberg, P. O., Kurten, A., Crounse, J., Clair, J. M. St., Wisthaler, A., Mikoviny, T., Yantosca, R. M., Carouge, C. C., and Le Sager, P.: Nitrogen oxides and PAN in plumes from boreal fires during ARCTAS-B and their impact on ozone: an integrated analysis of aircraft and satellite observations, Atmos. Chem. Phys., 10, 9739–9760, https://doi.org/10.5194/acp-10-9739-2010, 2010.
Alvarado, M. J., Lonsdale, C. R., Yokelson, R. J., Akagi, S. K., Coe, H., Craven, J. S., Fischer, E. V., McMeeking, G. R., Seinfeld, J. H., Soni, T., Taylor, J. W., Weise, D. R., and Wold, C. E.: Investigating the links between ozone and organic aerosol chemistry in a biomass burning plume from a prescribed fire in California chaparral, Atmos. Chem. Phys., 15, 6667–6688, https://doi.org/10.5194/acp-15-6667-2015, 2015.
Aschmann, S. M., Arey, J., and Atkinson, R.: Atmospheric Chemistry of Selected Hydroxycarbonyls, J. Phys. Chem. A, 104, 3998–4003, https://doi.org/10.1021/jp9939874, 2000.
Atkinson, R.: Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 41, 8468–8485, https://doi.org/10.1016/j.atmosenv.2007.07.002, 2007.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Bacher, C., Tyndall, G. S., and Orlando, J. J.: The Atmospheric Chemistry of Glycolaldehyde, J. Atmos. Chem., 39, 171–189, https://doi.org/10.1023/A:1010689706869, 2001.
Baker, J., Arey, J., and Atkinson, R.: Rate Constants for the Gas-Phase Reactions of OH Radicals with a Series of Hydroxyaldeydes at 296 ± 2 K, J. Phys. Chem. A, 108, 7032–7037, https://doi.org/10.1021/jp048979o, 2004.
Bickers, D. R., Calow, P., Greim, H. A., Hanifin, J. M., Rogers, A. E., Saurat, J.-H., Sipes, I. G., Smith, R. L., and Tagami, H.: The safety assessment of fragrance materials, Regul. Toxicol. Pharmacol., 37, 218–273, https://doi.org/10.1016/S0273-2300(03)00003-5, 2003.
Blanco, M. B., Barnes, I., and Wiesen, P.: Kinetic Investigation of the OH Radical and Cl Atom Initiated Degradation of Unsaturated Ketones at Atmospheric Pressure and 298 K, J. Phys. Chem. A, 116, 6033–6040, https://doi.org/10.1021/jp2109972, 2012.
Bloss, W. J., Evans, M. J., Lee, J. D., Sommariva, R., Heard, D. E., and Piling, M. J.: The oxidative capacity of the troposphere: Coupling of field measurements of OH and a global chemistry transport model, Faraday Discuss., 130, 425–436, https://doi.org/10.1039/B419090D, 2005.
Calvert, J. G., Mellouki, A., Orlando, J. J., Pilling, M. J., and Wallington, T. J.: The mechanisms of atmospheric oxidation of the oxygenates, Oxford University Press, New York, ISBN-13 978-0199767076, 2011.
Calvert, J. G., Atkinson, R., Kerr, J. A., Madronich, S., Moortgat, G. K., Wallington, T. J., and Yarwood, G.: The mechanisms of atmospheric oxidation of the alkenes, Oxford University Press, New York, ISBN-13 978-0195131772, 2000.
Canosa-Mas, C. E., Flugge, M. L., King, M. D., and Wayne, R. P.: An experimental study of the gas-phase reaction of the NO3 radical with α,β-unsaturated carbonyl compounds, Phys. Chem. Chem. Phys., 7, 643–650, https://doi.org/10.1039/B416574H, 2005.
Ciccioli, P., Brancaleoni, E., Frattoni, M., Cecinato, A., and Pinciarelli, L.: Determinations of volatile organic compounds (VOC) emitted from biomass burning of mediterranean vegetation species by GC-MS, Anal. Lett., 34, 937–955, https://doi.org/10.1081/AL-100103604, 2001.
Fu, T.-M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res.-Atmos., 113, 1–17, https://doi.org/10.1029/2007JD009505, 2008.
Galloway, M. M., Huisman, A. J., Yee, L. D., Chan, A. W. H., Loza, C. L., Seinfeld, J. H., and Keutsch, F. N.: Yields of oxidized volatile organic compounds during the OH radical initiated oxidation of isoprene, methyl vinyl ketone, and methacrolein under high-NOx conditions, Atmos. Chem. Phys., 11, 10779–10790, https://doi.org/10.5194/acp-11-10779-2011, 2011.
Greene, C. R. and Atkinson, R.: Rate constants for the gas-phase reactions of O3 with a series of cycloalkenes and α,β-unsaturated ketones at 296 ± 2 K, Int. J. Chem. Kinet., 26, 37–44, https://doi.org/10.1002/kin.550260106, 1994.
Hatch, L. E., Yokelson, R. J., Stockwell, C. E., Veres, P. R., Simpson, I. J., Blake, D. R., Orlando, J. J., and Barsanti, K. C.: Multi-instrument comparison and compilation of non-methane organic gas emissions from biomass burning and implications for smoke-derived secondary organic aerosol precursors, Atmos. Chem. Phys., 17, 1471–1489, https://doi.org/10.5194/acp-17-1471-2017, 2017.
Honrath, R. E., Owen, R. C., Val Martín, M., Reid, J. S., Lapina, K., Fialho, P., Dziobak, M. P., Kleissl, J., and Westphal, D. L.: Regional and hemispheric impacts of anthropogenic and biomass burning emissions on summertime CO and O3 in the North Atlantic lower free troposphere, J. Geophys. Res.-Atmos., 109, D24310, https://doi.org/10.1029/2004JD005147, 2004.
Illmann, J. N., Patroescu-Klotz, I., and Wiesen, P.: Gas-phase reactivity of acyclic α,β-unsaturated carbonyls towards ozone, Phys. Chem. Chem. Phys., 23, 3455–3466, https://doi.org/10.1039/D0CP05881E, 2021a.
Illmann, J. N., Gibilisco, R. G., Bejan, I. G., Patroescu-Klotz, I., and Wiesen, P.: Atmospheric oxidation of α,β-unsaturated ketones: kinetics and mechanism of the OH radical reaction, Atmos. Chem. Phys., 21, 13667–13686, https://doi.org/10.5194/acp-21-13667-2021, 2021b.
Jaffe, D., Chand., D., Hafner, W., Westerling, A., and Spracklen, D.: Influence of Fires on O3 Concentrations in the Western U.S., Environ. Sci. Technol., 42, 5885–5891, https://doi.org/10.1021/es800084k, 2008.
Jaffe, D. A. and Wigder, N. L.: Ozone production from wildfires: A critical review, Atmos. Environ., 51, 1–10, https://doi.org/10.1016/j.atmosenv.2011.11.063, 2012.
Knap, H. C. and Jørgensen, S.: Rapid Hydrogen Shift Reactions in Acyl Peroxy Radicals, J. Phys. Chem. A, 121, 1470–1479, https://doi.org/10.1021/acs.jpca.6b12787, 2017.
Kwok, E. S. C. and Atkinson, R.: Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 29, 1685–1695, https://doi.org/10.1016/1352-2310(95)00069-B, 1995.
Magneron, I., Mellouki, A., Le Bras, G., Moortgat, G. K., Horowitz, A., and Wirtz, K.: Photolysis and OH-Initiated Oxidation of Glycolaldehyde under Atmospheric Conditions, J. Phys. Chem. A, 109, 4552–4561, https://doi.org/10.1021/jp044346y, 2005.
Mauzerall, D. L., Logan, J. A., Jacob, D. J., Anderson, B. E., Blake, D. R., Bradshaw, J. D., Heikes, B., Sachse, G. W., Singh, H., and Talbot, B.: Photochemistry in biomass burning plumes and implications for tropospheric ozone over the tropical South Atlantic, J. Geophys. Res.-Atmos., 103, 8401–8423, https://doi.org/10.1029/97JD02612, 1998.
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥ C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021.
Méreau, R., Rayez, M.-T., Rayez, J.-C., Caralp, F., and Lesclaux, R.: Theoretical study on the atmospheric fate of carbonyl radicals: kinetics of decomposition reactions, Phys. Chem. Chem. Phys., 3, 4712–4717, https://doi.org/10.1039/B105824J, 2001.
Müller, M., Anderson, B. E., Beyersdorf, A. J., Crawford, J. H., Diskin, G. S., Eichler, P., Fried, A., Keutsch, F. N., Mikoviny, T., Thornhill, K. L., Walega, J. G., Weinheimer, A. J., Yang, M., Yokelson, R. J., and Wisthaler, A.: In situ measurements and modeling of reactive trace gases in a small biomass burning plume, Atmos. Chem. Phys., 16, 3813–3824, https://doi.org/10.5194/acp-16-3813-2016, 2016.
Niki, H., Maker, P. D., Savage, C. M., and Hurley, M. D.: Fourier Transform Infrared Study of the Kinetics and Mechanisms for the Cl-Atom and HO-Radical-Initiated Oxidation of Glycolaldehyde, J. Phys. Chem., 91, 2174–2178, https://doi.org/10.1021/j100292a038 , 1987.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 103, 4657–4689, https://doi.org/10.1021/cr020527p, 2003.
Praske, E., Crounse, J. D., Bates, K. H., Kurtén, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015.
Profeta, L. T. M., Sams, R. L., and Johnson, T. J.: Quantitative Infrared Intensity Studies of Vapor-Phase Glyoxal, Methylglyoxal, and 2,3-Butanedione (Diacetyl), with Vibrational Assignments, J. Phys. Chem. A, 115, 9886–9900, https://doi.org/10.1021/jp204532x, 2011.
Sato, K., Klotz, B., Taketsuga, T., and Takaynagi, T.: Kinetic measurments for the reactions of ozone with crotonaldehyde and its methyl derivatives and calculations of transition-state theory, Phys. Chem. Chem. Phys., 6, 3696–3976, https://doi.org/10.1039/B402496F, 2004.
Takahashi, H., Kobayashi, Y., and Kaneko, N.: Conformational studies of DL-lactaldehyde by 1H-NMR, Raman and i.r. spectroscopy, Spectrochim. Acta A, 39, 569–572, https://doi.org/10.1016/0584-8539(83)80108-1, 1983.
Talukdar, R. K., Zhu, L., Feierabend, K. J., and Burkholder, J. B.: Rate coefficients for the reaction of methylglyoxal (CH3COCHO) with OH and NO3 and glyoxal (HCO)2 with NO3, Atmos. Chem. Phys., 11, 10837–10851, https://doi.org/10.5194/acp-11-10837-2011, 2011.
Taylor, W. D., Allston, T. D., Moscato, M. J., Fazekas, G. B., Kozlowski, R., and Takacs, G. A.: Atmospheric photodissociation lifetimes for nitromethane, methyl nitrite, and methyl nitrate, Int. J. Chem. Kinet., 12, 231–240, https://doi.org/10.1002/kin.550120404, 1980.
Tuazon, E. C. and Atkinson, R.: A Product Study of the Gas-Phase Reaction of Methyl Vinyl Ketone with the OH Radical in the Presence of NOX, Int. J. Chem. Kinet., 21, 1141–1152, https://doi.org/10.1002/kin.550211207, 1989.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Yokelson, R. J., Crounse, J. D., DeCarlo, P. F., Karl, T., Urbanski, S., Atlas, E., Campos, T., Shinozuka, Y., Kapustin, V., Clarke, A. D., Weinheimer, A., Knapp, D. J., Montzka, D. D., Holloway, J., Weibring, P., Flocke, F., Zheng, W., Toohey, D., Wennberg, P. O., Wiedinmyer, C., Mauldin, L., Fried, A., Richter, D., Walega, J., Jimenez, J. L., Adachi, K., Buseck, P. R., Hall, S. R., and Shetter, R.: Emissions from biomass burning in the Yucatan, Atmos. Chem. Phys., 9, 5785–5812, https://doi.org/10.5194/acp-9-5785-2009, 2009.