the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Oct 2021
| 04 Oct 2021
Evolution of OH reactivity in NO-free volatile organic compound photooxidation investigated by the fully explicit GECKO-A model
Julia Lee-Taylor
Harald Stark
John J. Orlando
Bernard Aumont
Jose L. Jimenez
OH reactivity (OHR) is an important control on the oxidative capacity in the atmosphere but remains poorly constrained in many environments, such as remote, rural, and urban atmospheres, as well as laboratory experiment setups under low-NO conditions. For an improved understanding of OHR, its evolution during oxidation of volatile organic compounds (VOCs) is a major aspect requiring better quantification. We use the fully explicit Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) model to study the OHR evolution in the NO-free photooxidation of several VOCs, including decane (an alkane), m-xylene (an aromatic), and isoprene (an alkene). Oxidation progressively produces more saturated and functionalized species. Total organic OHR (including precursor and products, OHRVOC) first increases for decane (as functionalization increases OH rate coefficients) and m-xylene (as much more reactive oxygenated alkenes are formed). For isoprene, C=C bond consumption leads to a rapid drop in OHRVOC before significant production of the first main saturated multifunctional product, i.e., isoprene epoxydiol. The saturated multifunctional species in the oxidation of different precursors have similar average OHRVOC per C atom. The latter oxidation follows a similar course for different precursors, involving fragmentation of multifunctional species to eventual oxidation of C1 and C2 fragments to CO2, leading to a similar evolution of OHRVOC per C atom. An upper limit of the total OH consumption during complete oxidation to CO2 is roughly three per C atom. We also explore the trends in radical recycling ratios. We show that differences in the evolution of OHRVOC between the atmosphere and an environmental chamber, and between the atmosphere and an oxidation flow reactor (OFR), can be substantial, with the former being even larger, but these differences are often smaller than between precursors. The Teflon wall losses of oxygenated VOCs in chambers result in large deviations of OHRVOC from atmospheric conditions, especially for the oxidation of larger precursors, where multifunctional species may suffer substantial wall losses, resulting in significant underestimation of OHRVOC. For OFR, the deviations of OHRVOC evolution from the atmospheric case are mainly due to significant OHR contribution from RO2 and lack of efficient organic photolysis. The former can be avoided by lowering the UV lamp setting in OFR, while the latter is shown to be very difficult to avoid. However, the former may significantly offset the slowdown in fragmentation of multifunctional species due to lack of efficient organic photolysis.
- Article
(3008 KB) - Full-text XML
-
Supplement
(1490 KB) - BibTeX
- EndNote





Photooxidation is a key process altering the concentrations of trace gases in the atmosphere (Levy II, 1971; Atkinson and Arey, 2003). It is also the main contributor to the formation of O3 and secondary aerosols (Haagen-Smit, 1952; Chameides et al., 1988; Hallquist et al., 2009). Both products are major tropospheric pollutants (Nel, 2005; Cohen et al., 2017), and the latter have large climate impacts (Stocker et al., 2014).
Hydroxyl radical (OH) is the primary oxidizing agent in atmospheric photooxidation (Levy II, 1971). Its atmospheric fate is governed by the species that it reacts with, i.e., OH reactants. The first-order rate constant of OH consumption by an OH reactant is often called its OH reactivity (OHR), equal to the product of the reactant concentration and second-order rate constant with OH. Total OHR (OHRtot), i.e., the sum of OHR across all OH reactants (OHRtot = , where ki and ci are the second-order rate constant with OH and concentration of the ith OH reactant), is the real first-order loss rate constant of OH.
OHR has been measured for over 20 years (Kovacs and Brune, 2001) in various settings, e.g., urban areas (Lu et al., 2013; Whalley et al., 2016), forested areas (Nölscher et al., 2016; Zannoni et al., 2016), and environmental chambers (Fuchs et al., 2013, 2017; Nölscher et al., 2014; Novelli et al., 2018). Despite numerous measurements and remarkable technical developments (Yang et al., 2016; Fuchs et al., 2017), a sizable fraction of total OHR in most measurements has not been chemically speciated, leading to so-called “missing reactivity” (Williams and Brune, 2015; Yang et al., 2016). Multiple studies (Nölscher et al., 2016; Whalley et al., 2016; Sato et al., 2017) have attributed missing reactivity to the highly complex mixture of intermediates and products of volatile organic compound (VOC) oxidation, most of which are oxygenated VOCs (OVOCs). Primary VOCs themselves have been found to be the largest contributor of the speciated OHR in many studies (Yang et al., 2016). In order to well understand ambient OHR, the evolution of OHR (including that from OVOCs) during primary VOC photooxidation thus needs to be investigated.
Experimentally, this can be done in environmental chambers. However, only a few such experiments have been published (Nakashima et al., 2012; Nehr et al., 2014; Nölscher et al., 2014; Sato et al., 2017), all under high-NO conditions, where the key organic radical intermediate in VOC oxidation, i.e., organic peroxy radical (RO2), mainly reacts with NO. To our knowledge, no experiment of this type at low NO, where RO2 can substantially react with hydroperoxy radical (HO2), has been published so far, potentially partially due to the difficulty in experimentally ensuring that low-NO conditions are achieved in chambers (Nguyen et al., 2014). Also, many OVOCs, which may account for missing reactivity, have sufficiently low volatility to significantly partition to chamber walls (Matsunaga and Ziemann, 2010; Krechmer et al., 2016), further complicating these experiments. The OVOC wall losses also often limit operation times of chamber experiments to a few hours, after which the wall losses are so large that meaningful interpretation of experimental results would be difficult. Therefore, the highest equivalent photochemical age that can be reached in chamber experiments is also typically hours and far shorter than would be needed to explore the OHR evolution in later stages of VOC oxidation.
Oxidation flow reactors (OFR) are an alternative to chambers with much smaller volume, shorter residence time (and thus smaller wall losses of trace gases), and stronger oxidative capacity (Kang et al., 2007; Brune, 2019; Peng and Jimenez, 2020). The most common version of OFR is equipped with low-pressure Hg lamps emitting UV at 185 and 254 nm, which photolyzes water vapor, O2, and O3 and generates a large amount of OH both directly and through subsequent radical reactions. High OH concentration in OFR often leads to equivalent photochemical age of days to weeks (Li et al., 2015; Peng et al., 2015). In principle, OFR can also be employed to explore OHR evolution in VOC oxidation. However, OHR from VOC (OHRVOC, from both precursor and oxidation intermediates/products, in which we include CO as an “organic” product of VOC oxidation) can have strong impacts on oxidative capacity (particularly OH concentration) and hence radical chemistry in OFR at both low (Li et al., 2015; Peng et al., 2015) and high NO (Peng and Jimenez, 2017; Peng et al., 2018). Peng and Jimenez (2020) have called for highly chemically detailed modeling of gas-phase organic chemistry in OFR to assess the impacts of organic OH reactants on OH in a more quantitative manner.
In this study, we explore for the first time the OHR evolution in entire NO-free VOC photooxidation processes by modeling. Since chemical mechanism incompleteness causes other models to unsatisfactorily simulate measured OHRtot (Williams and Brune, 2015), we use the fully chemically explicit model GECKO-A (Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere) (Aumont et al., 2005). We simulate the photooxidation of different types of VOCs in the atmosphere, in chamber, and in OFR to find out general trends of OHR evolution in VOC oxidation and whether VOC oxidation chemistries in chamber and OFR are representative of that in the atmosphere in terms of OHR evolution.
Here we first discuss the VOC precursor types and conditions selected for the model cases in this study. Then we describe the GECKO-A model and present our additional mechanism, model, and software development required for this study.
Table 1Conditions and integration time steps of the simulations in the present work.
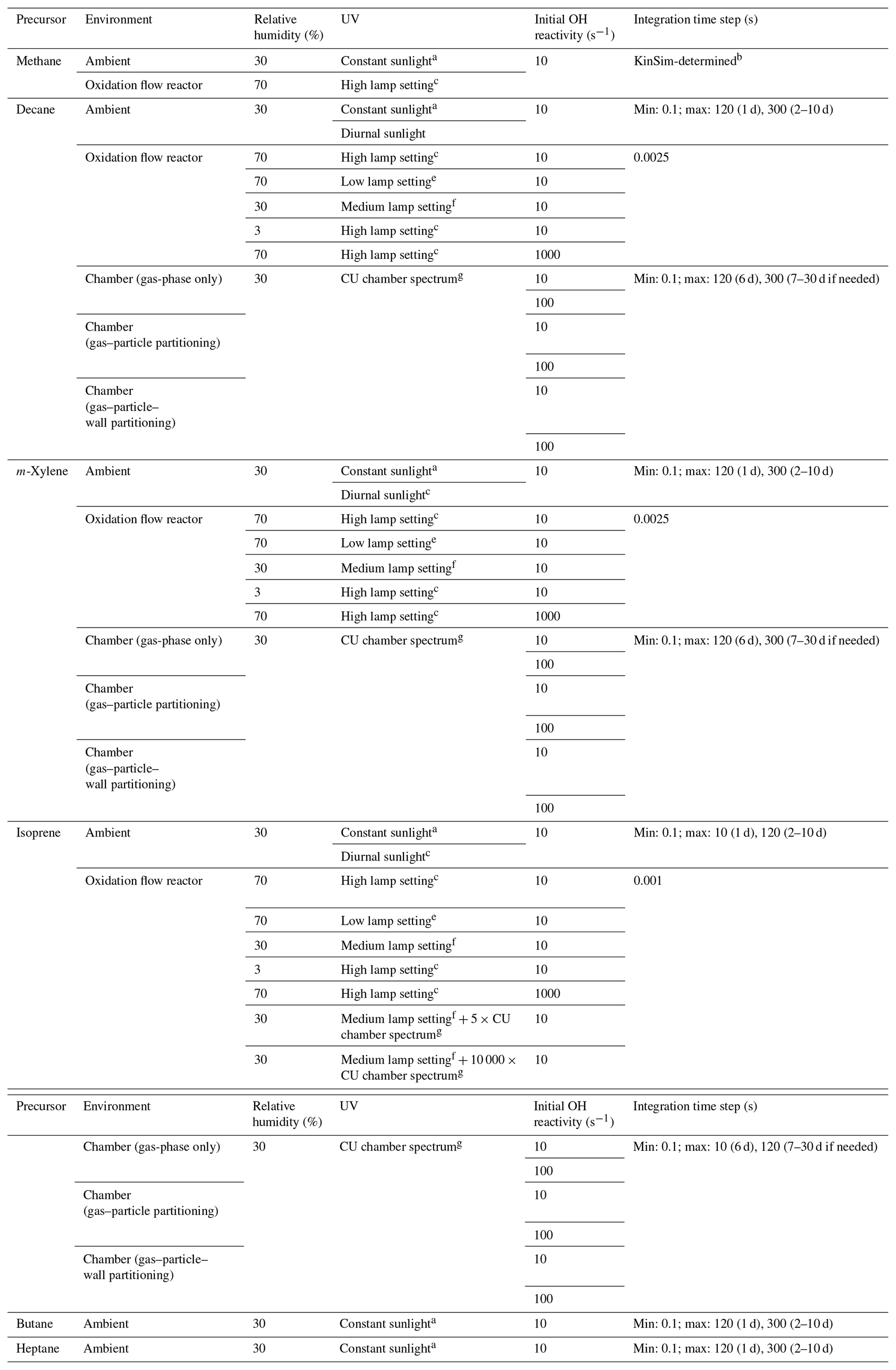
a At solar zenith angle of 45∘. b Simulation performed in the solver KinSim, which fully controls its integration time step selection. c Diurnal variation between solar zenith angles of 0 and 90∘. d UV at 185 nm = 1 × 1014 ; UV at 254 nm = 8.5 × 1015 . e UV at 185 nm = 1 × 1011 ; UV at 254 nm = 4.2 × 1013 . f UV at 185 nm = 1 × 1013 ; UV at 254 nm = 1.4 × 1015 . g UV source spectrum of the University of Colorado Environmental Chamber Facility (Krechmer et al., 2017).
2.1 Model cases
The photooxidation of an alkane (decane), an alkene (isoprene), and an aromatic (m-xylene) is investigated under a variety of conditions without any NO. In pristine regions such as open oceans, NO has typical concentrations on the order of 1 ppt (Wofsy et al., 2021) and hence contributes only a few percent to RO2 loss (Peng et al., 2019). For simplicity, we choose not to maintain such a low NO level in the simulations but to model zero-NO cases instead. The model cases are listed in Table 1: (i) two cases under ambient conditions, one with constant sunlight at solar zenith angle of 45∘ and the other with diurnally varying solar radiation and a noontime solar zenith angle of 0∘; (ii) six cases under typical chamber conditions, i.e., low (10 s−1)/high (100 s−1) precursor OHR without gas–particle–wall partitioning, with gas–particle partitioning (no wall) and with gas–particle–wall partitioning; and (iii) five cases under OFR conditions, of which two conditions resulting in significant non-tropospheric organic photolysis (Peng et al., 2016) and one leading to remarkable deviations of RO2 fate from that in the troposphere are not recommended in practice but are still included for completeness since they are similar to conditions in some literature studies (Table 1). For the UV source in chamber cases, we adopt the spectrum of the blacklight and fluorescence light array in the University of Colorado Environmental Chamber Facility (CU chamber; Krechmer et al., 2017). The CU chamber has a volume of ∼ 20 m3, a surface area of ∼ 65 m2, and an estimated wall condensation timescale of ∼ 1000 s (Krechmer et al., 2016). The parameterization for the reversible gas–wall partitioning is taken from Krechmer et al. (2016) with updates of Liu et al. (2019). Wall partitioning in chambers at equilibrium is a function of the surface-to-volume ratio (Krechmer et al., 2016). The timescale to approach equilibrium is expected to be larger in larger chambers but still far shorter than the long experiments needed to investigate high photochemical ages. Therefore differences in wall partitioning timescale are not important for this study. Figure S9 of Krechmer et al. (2016) compared the CU chamber and a few other well-known chambers (including very large ones such as EUPHORE, Siese et al., 2001, and SAPHIR, Rohrer et al., 2005), showing relatively small differences (within a factor of ∼ 2 in terms of surface-to-volume ratio). Therefore the conclusions about wall partitioning in this study should be approximately applicable to most chambers. The cases under ambient conditions and chamber conditions with low/high precursor OHR are simulated for 10 and 6/30 d, respectively, to encompass an equivalent photochemical age of > 10 d in each case (given a typical average ambient OH concentration of 1.5 × 106 molecules cm−3 in the real atmosphere, Mao et al., 2009; see Fig. 1 for the correspondence between equivalent photochemical age and OH exposure; OHexp, i.e., the integral of OH concentration over time). The simulated OFR in the present work employs the light source parametrization obtained by Li et al. (2015) and Peng et al. (2015). UV at both 185 and 254 nm is used to generate OH, i.e., the OFR185 mode of operation. The residence time in the OFR is always 3 min. Wall losses in the OFR should be smaller than in the chamber, due to reduced wall contact (Brune 2019), and are not simulated here. As several key parameters of the chamber and OFR cases were obtained experimentally at room temperature and atmospheric pressure in Boulder, Colorado, USA (typically 295 K and 835 mbar), for an easier comparison, we use these values for the temperature and atmospheric pressure of all model cases.
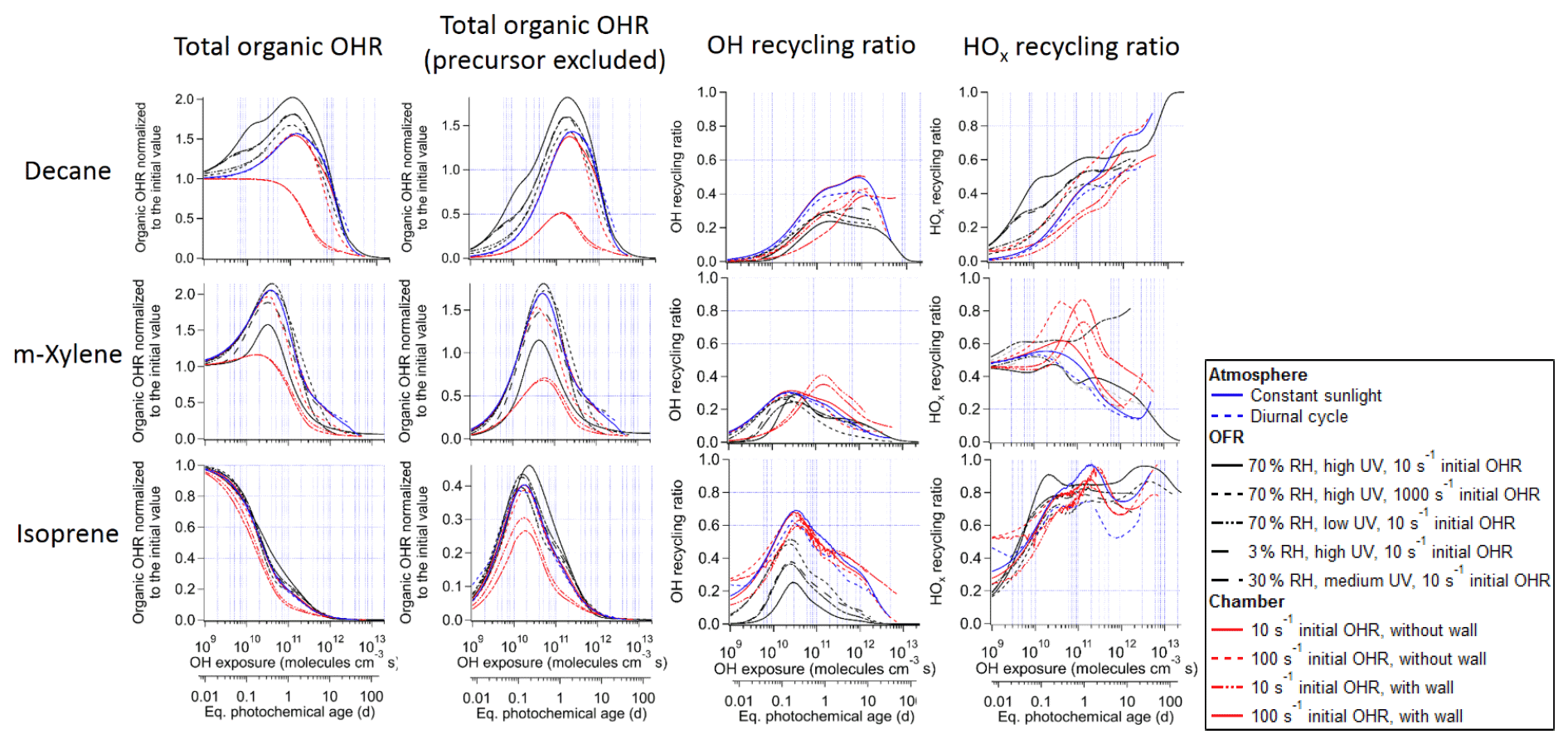
Figure 1Total organic OH reactivity (OHR) with and without the contribution of the precursor, OH recycling ratio (β1), and HOx recycling ratio (β2) as a function of OH exposure (or equivalent photochemical age; second x axis) in the photooxidations of decane, isoprene, and m-xylene under different conditions in the atmosphere, oxidation flow reactor (OFR), and chamber.
In addition, we simulate illustrative cases of methane oxidation, under ambient and OFR conditions (Table 1 and Sect. 3.1). Note that these two simulations are performed using the GECKO-A-generated mechanism (see Sect. 2.2) in KinSim (Peng and Jimenez, 2019), a chemical-kinetics solver that is not GECKO-A's default, to avoid possible numerical issues in the GECKO-A internal solver, as methane oxidation by OH is very slow (Atkinson and Arey, 2003) and very long runs are needed. To characterize trends of OHR evolution (see Sect. 3.5), the ambient cases with constant sunlight are simulated for two more alkanes, i.e., butane and heptane (Table 1). To explore the effects of UV sources in OFR (see Sect. 3.4), two simulations under a typical OFR condition with an additional broad-spectrum UV source (5 and 10 000 times the chamber UV source in this study, respectively) are performed for isoprene (Table 1).
2.2 The GECKO-A model
GECKO-A (Aumont et al., 2005; with updates as described by Camredon et al., 2007; Valorso et al., 2011; Lee-Taylor et al., 2015) is an explicit chemical model which uses known mechanisms and rates supplemented with experimentally based structure–activity relationships (SARs) to generate comprehensive atmospheric oxidation mechanisms for organic species. The mechanisms are implemented within a box model with a two-step solver (Verwer, 1994; Verwer et al., 1996). In mechanism generation, isomer lumping for mechanism reduction purposes is applied to certain products with branching ratios < 1 % (here typically N-containing products, which are not relevant for our simulations). It has a negligible impact on the results.
The core isoprene scheme in GECKO-A is adopted from the Master Chemical Mechanism (MCM) v3.3.1 (Jenkin et al., 2015), while the meta-xylene oxidation mechanism follows MCM v3.2 (Jenkin et al., 2003, Bloss et al., 2005), typically until ring-breaking occurs, whereupon the GECKO-A mechanism generator implements the standard SAR protocols as described by Aumont et al. (2005), Camredon et al. (2007), and Lee-Taylor et al. (2015). Under the zero-NO conditions employed in this study, we find that, in two of the four m-xylene reaction channels (xylenol, 17 %; and MXYLO2, 4 %), some product species persist anomalously owing to a lack of alternative reaction pathways in the MCM. We therefore allow GECKO-A to apply the standard SARs to two cyclic non-aromatic products of xylenol (MXYOLO2 and MXYOLOOH in the 51 % xylenol OH-oxidation channel; see Scheme S1 in the Supplement). We also introduce OH oxidation of MXYCATECH and MXY1OOH (in the 42 % and 7 % xylenol OH-oxidation channels) and of MXYLOOH and MXYLAL (in the MXYLO2 channel), assuming similarity to the MCM OH oxidation of xylenol to MXYOLO2, and with net OH rate constants estimated using the EPA EPI Suite software package (US EPA, 2012). MXYLOOH, MXCATECH, and MXYLAL each yield between two and six bicyclic non-aromatic substituted peroxy radicals, with net OH rate constants of 1.77 × 10−11, 1.56 × 10−10, and 8.6 × 10−13 cm3 molec−1 s−1, respectively. (The MXYLOOH OH rate also includes MXYLAL production). MXY1OOH is assigned a substituted single-ring hydroxy-ketone product, with OH rate constant 3.26 × 10−11 cm3 molec−1 s−1. The early part of the meta-xylene reaction scheme used in this work is shown in Scheme S1.
We tested the effect of solver integration time step length on output precision. The output species concentrations in all simulations but for isoprene OFR (Table 1) converge well as integration time step decreases (Fig. S1 in the Supplement). In the isoprene OFR test cases, the output values oscillate over a small range (≲ 5 %) for integration time steps < 0.01 s (Fig. S1). Since this numerical error is smaller than typical rate constant measurement uncertainties (from ∼ 10 % to a factor of 2–3; Burkholder et al., 2015), as well as the uncertainties related to the SARs used in GECKO-A, it is deemed acceptable for the relevant simulations in this study. The integration time step for each simulation in the present work is reported in Table 1.
We allow mechanism generation to proceed through to CO2 production in most cases in this study. The only exception is for extremely-low-volatility species (saturation vapor pressure < 10−13 atm) which are considered to be completely and irreversibly partitioned to the particle phase. Particle- and wall-phase species are no longer considered in the OHR budget, since heterogeneous oxidation is much slower than gas-phase oxidation (e.g., George and Abbatt, 2010). Gas–particle–wall partitioning is activated only for the chamber cases where wall effects are considered. For the ambient cases and the chamber cases without gas–wall partitioning, gas–particle partitioning is also disabled to avoid artificial condensation of gases into the particle phase. In environments with very low NO (e.g., remote atmosphere), organic aerosol concentration is typically 0.2 µg m−3 (Hodzic et al., 2020), while most major intermediates/products have higher saturation concentrations (C*) and hence largely stay in the gas phase. C* is calculated using the parameterization of Nannoolal et al. (2008) (default option of GECKO-A). Although SIMPOL (Pankow and Asher, 2008) was recommended by Krechmer et al. (2016) to estimate C* for the chamber wall partitioning treatment using their parameterization, the C* estimates by the Nannoolal and SIMPOL parameterizations are close (generally within a factor of 2) for the species that can reversibly partition between the gas and wall phases (C* ∼ 0.1–1000 µg m−3) in this study. This difference is smaller than the uncertainties of the Krechmer et al. (2016) parameterization. Therefore, the use of the parameterization of Nannoolal et al. (2008) for C* estimation is acceptable.
Concerns have previously been expressed about non-conservation of carbon in GECKO-A (Mouchel-Vallon et al., 2020). This has proven in the current simulations to be almost entirely due to lack of accounting for product CO2 in some handwritten reactions. We edited the handwritten isoprene and m-xylene schemes (see above and Sect. 2.2.3) for carbon balance, which reduced simulation-end carbon losses in the m-xylene and isoprene ambient cases with constant UV from 4 % and 9 %, respectively, to negligible levels (< 0.4 %; Fig. S2).
For the current study, we have made several updates to GECKO-A, i.e., (i) inclusion of key OFR-specific radical reactions, (ii) extension of the UV range considered to cover 185 and 254 nm, and (iii) updates to the NO-free m-xylene oxidation mechanism, so that GECKO-A is able to simulate OFR chemistry and the entire process of NO-free m-xylene photooxidation (until CO/CO2). We will describe these three updates below.
2.2.1 Key radical reactions in oxidation flow reactor
We have added several reactions that are unimportant in the troposphere but that are required to fully represent the radical chemistry within the OFR (Li et al., 2015). The most important inorganic reactions are H2O + hν (185 nm) → H + OH, O2 + hν (185 nm) → 2O(3P), and O3 + hν (254 nm) → O(1D) + O2. These three reactions, together with O(3P) + O2 + M → O3 + M and O(1D) + H2O → 2OH, which are already in the GECKO-A inorganic radical chemistry scheme, are responsible for the OH generation in OFR. The OFR radical chemistry has previously been modeled in detail using KinSim (Peng and Jimenez, 2019), which was validated against experimental observations (Li et al., 2015; Peng et al., 2015). A comparison between KinSim and GECKO-A for a range of OFR conditions shows typical agreement between the two models within 2 % for key outputs.
Due to high OH in OFR, reaction of RO2 with OH is also included in mechanism generation, with an assumed rate constant of 1 × 1010 cm3 molec−1 s−1 (Peng et al., 2019). The products of this type of reaction are assumed to be RO (alkoxy radical) + HO2 for alkyl RO2 and R (alkyl radical) + CO2 + HO2 for acyl RO2. Although these reactions for certain RO2 may have reaction intermediates, the reactions of the intermediates (with OH) are believed to be very fast under OFR conditions where OH is much higher than in the atmosphere (Peng and Jimenez, 2020), and hence only the probable final products (no intermediates) of these reactions are included in mechanism generation. The reaction of RO2 with OH is not included in the mechanisms for the ambient and chamber simulations due to low contribution of this pathway to the RO2 fate in those cases.
2.2.2 Organic photolysis at 185 and 254 nm
Organic photolysis is assessed in GECKO-A via a lookup table of j values for reference chromophores pre-calculated at different solar zenith angles with the Tropospheric Ultraviolet and Visible (TUV) 1-D radiative transfer model (Madronich and Flocke, 1999). The reference cross sections used in the model generally do not cover the UV wavelengths at which OFR operates (with narrow peaks at 185 and 254 nm) since they are not tropospherically relevant. Thus it was necessary to extend to 185 nm the relevant reference absorption cross sections. We have done this using literature values via the Mainz UV–Vis spectral atlas (Keller-Rudek et al., 2020) or by extrapolating the available cross-section data, using other similar chromophores as references. Details of all cross-section extensions are given in Table S1 in the Supplement. Where quantum yield information was not available, we assume values of unity since photons at 185 and 254 nm are usually sufficiently energetic to make photolysis occur (Ausloos and Lias, 1971). In case of multiple product channels for a photolyzed molecule, the branching ratios of those channels at 185 and 254 nm are estimated through extrapolation of branching ratio data from available ranges followed by a renormalization. Finally, we apply the OFR UV spectrum within TUV to calculate OFR-relevant j-value lookup tables.
2.2.3 Mechanism of NO-free m-xylene oxidation
The meta-xylene oxidation mechanism in GECKO-A follows MCM v3.2 until all aromatic, epoxy, or bridged-peroxy rings are broken (See Scheme S1). Since the MCM was designed for typical urban environments with abundant NOx, it omits some reaction pathways for other oxidants, assuming them to be of negligible importance. The relevant photolysis loss pathways are slow under ambient conditions and inactive in the OFR case. This leads to persistence and accumulation of certain hydroperoxides and their interconverting peroxy radicals under NO-free conditions. We added two NO-free oxidation reactions to the xylenol branch of the meta-xylene oxidation scheme, Scheme S1. In the 51 % branch, we allow the unsaturated bicyclic peroxide MXYLOOH to react with, sequentially, OH (estimating kVOC+OH cm3 molec−1 s−1) and HO2 (estimating cm3 per molecule per second), to produce a saturated bicyclic peroxide (denoted “TT8001” in Scheme S1). In the 42 % branch, we add a competing O3 reaction with the alkoxy radical MXCATEC1O, producing an unsaturated carbonyl alkoxy radical 1T8000, which eliminates CH3 to form the unsaturated cyclic hydroxy dicarbonyl TU7000. Both products are then further oxidized via the standard GECKO SARs.
2.3 GECKO Loader and Plotter
To allow GECKO-A outputs, which are usually highly complex and voluminous, to be explored and visualized in detail on standard (non-UNIX) personal computers, we have developed the GECKO Loader and Plotter based in the data-analyzing and graphic-making package Igor Pro 8.0 (WaveMetrics, Lake Oswego, Oregon, USA). This tool assists in the rapid and detailed analysis of model-chamber/OFR comparison studies.
Specifically, the GECKO Loader and Plotter facilitates: (i) filtering the (sometimes extremely large and finely resolved) model results time series to examine specific characteristics, (ii) identifying the most abundant and/or influential species in each phase (gas, particle, and wall), (iii) selecting species by specific chemical identity (molecular formula, specific formula, and/or functional group identity), (iv) plotting time series of individual species and their formation/destruction rates, (v) assessing and displaying aggregated properties (volatility distribution, mass spectrum, Henry's law constant distribution) of the product mixture and subsets thereof, and (vi) calculating bulk characteristics of the simulation (OHexp, OHRVOC, light intensity, elemental ratios, etc.) and relating species abundances to them.
In this section, we will show the evolution of OHRVOC in the photooxidation of different precursors under various conditions. To aid the presentation of this evolution for larger precursors, whose oxidation is more complex, the oxidation of the simplest VOC, i.e., methane, will be first discussed. After presenting the results of individual precursors, we will compare the results between conditions and between precursors to illustrate the general trends. Along with the OHR evolution, the OH recycling ratio (β1, defined as number of OH molecules generated from organic reactions per OH consumed by organics) and HOx (= OH + HO2) recycling ratio (β2, defined as number of OH and HO2 molecules generated from organic reactions per OH consumed by organics) will also be discussed, as they are important parameters that may considerably affect the budget of atmospheric oxidizing agents (Stone et al., 2012) and the HO2-to-OH ratio and RO2 chemistry in OFR (Peng et al., 2015, 2019).
3.1 Methane
To explain one of the main features in the OHR evolution in VOC photooxidation, i.e., OHRVOC peaking at a certain OHexp, the oxidation of CH4 is employed as an example because of its simpler mechanism (Scheme S2). The results of this oxidation under the ambient condition show that OHRVOC peaks at an OHexp of about 1 × 1013 molecules cm−3 s (Fig. S3). As the OHR of the precursor always decreases during its oxidation, the appearance of such a peak of OHRVOC before all VOCs are finally oxidized to CO2 indicates that the OHR increase from intermediates and products is faster than the OHR decrease of the precursor. This is obviously the case for CH4 oxidation, as there is no significant CH4 loss before OHexp ∼ 1013 molecules cm−3 s by its very slow reaction with OH (rate constant on the order of 10−15 cm3 molec−1 s−1; Atkinson and Arey, 2003) and all the non-CO2 intermediates/products of the oxidation (CH3OOH, CH3OH, HCHO, and CO) are orders of magnitude more reactive toward OH than is CH4 (Atkinson and Arey, 2003). This large difference in precursor and intermediate/product oxidation timescales allows the oxidations of intermediates/products (including CO, whose reaction rate constant with OH is ∼ 2 × 10−13 cm3 molec−1 s−1; Burkholder et al., 2015) to establish a steady state, whereby the OHR of the intermediates/products is proportional to the concentration/OHR of CH4. After OHexp ∼ 1013 molecules cm−3 s, CH4 concentration decay and consequently that of all intermediates/products become significant, giving the OHRVOC peak around 1 × 1013 molecules cm−3 s.
We also performed a simulation under a typical OFR condition. The OHRVOC peak also appears around 1 × 1013 molecules cm−3 s in this case for the same reasons discussed above, but its height is almost twice that of the ambient case (Fig. S3). The OHR of CO in both cases is similar, while that of CH3OH is higher in the ambient case but those of CH3OOH and HCHO are significantly higher in the OFR case. This is because the relative importance of the various reactions involved in CH4 oxidation (Scheme S2) depends on the conditions in each reactor.
In the OFR case, OH and HO2 concentrations are ∼ 4 and ∼ 3 orders of magnitude higher than typical ambient values, respectively (Peng et al., 2015). The reactions of two intermediates, CH3OOH and HCHO, with OH and the reaction of the only major RO2 involved, CH3OO, with HO2 are much faster than their photolysis or the self-reaction of CH3OO (Scheme S2). Neglecting organic photolysis and CH3OO self-reaction (and thus CH3OH as a product of the latter), the mechanism of CH4 oxidation can be simplified to an OH-driven chain (CH4 → CH3OOH → HCHO → CO → CO2) with a fast steady-state branch on CH3OOH (CH3OOH ↔ CH3OO). For a simple chain, as we show in Appendix A that the OHR of precursor and that of each intermediate are equal. At the OHRVOC peak, the OHR of HCHO and CO are very close to that of CH4, while that of CH3OOH is larger, because the branch reaction CH3OOH + OH → CH3OO + H2O also contributes to OHR but does not affect the chain (and hence the OHR of the downstream species). With such an idealized chain mechanism, the OHRVOC peak height equals the precursor OHR multiplied by the number of steps needed to produce CO2.
In contrast, the OHRVOC peak height (and its composition) in the ambient case cannot be explained by the simple chain. Both HCHO photolysis and CH3OO self-reaction play a major role in the oxidation in this case and are significantly faster than the reactions in the simple chain (HCHO + OH and CH3OO + HO2). In terms of the relationship of these two reactions with the chain, HCHO photolysis bypasses HCHO + OH in converting HCHO to CO, while CH3OO self-reaction bypasses CH3OOH, in effect short-circuiting the involvement of OH in the oxidation and hence lowering OHRVOC (Appendix A). Since the only reaction of CO is CO + OH, its OHR at the OHRVOC peak is essentially unaltered.
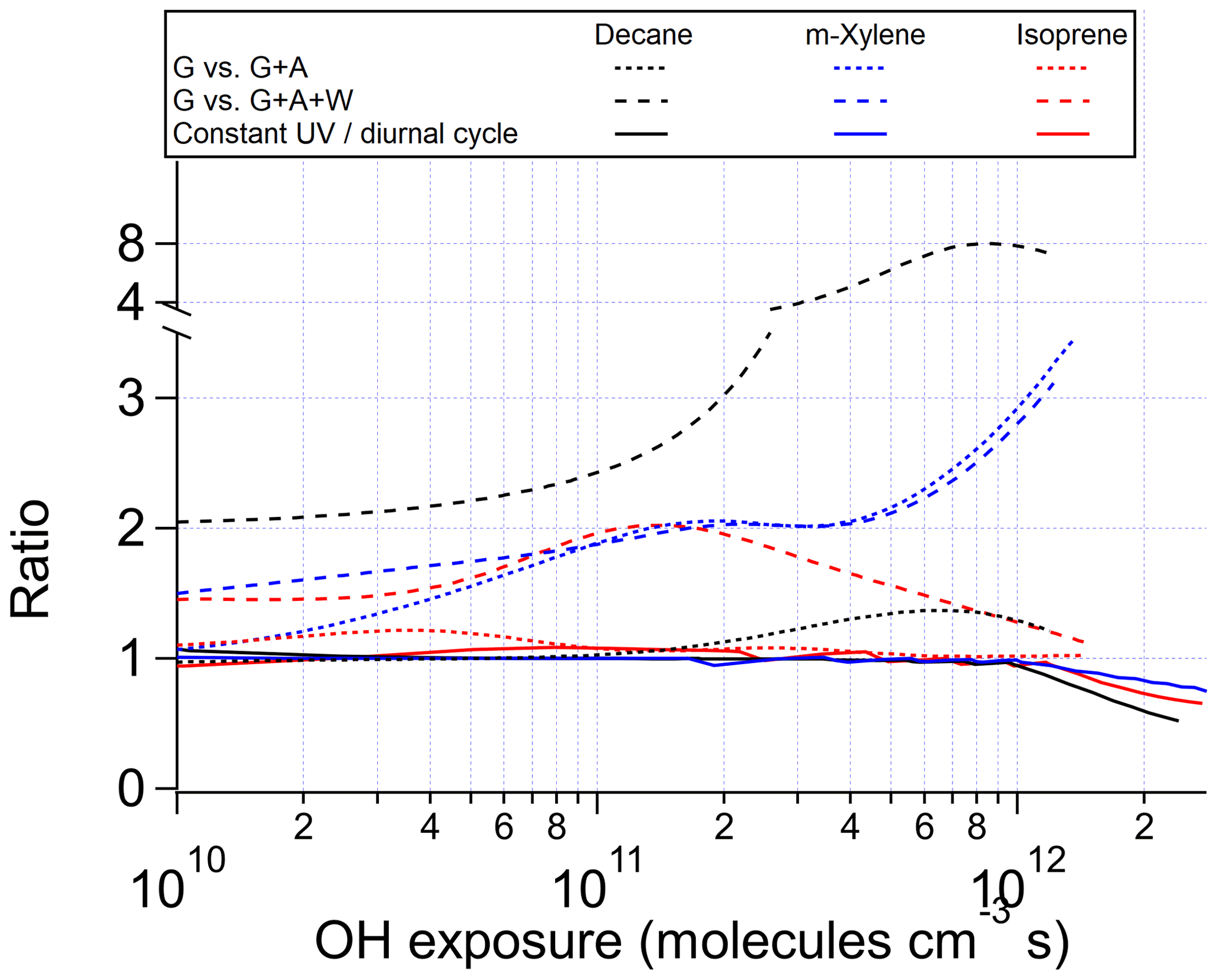
Figure 2Ratios of OHR of the products present in the gas phase between the chamber cases without gas–particle–wall partitioning and (i) with gas–particle (G vs. G + A) or (ii) gas–particle–wall partitioning (G vs. G + A+W) at initial OHR of 10 s−1, as well as between the ambient cases with constant and diurnal sunlight for the photooxidations of decane, m-xylene, and isoprene as a function of OH exposure.
3.2 Decane
3.2.1 Ambient and OFR cases
The evolution of OHRVOC during the oxidation of another alkane, decane, and under ambient and OFR conditions exhibits a smaller difference and smaller peak enhancements than those in the methane cases (Fig. 1), although the chain lengths of the decane oxidation to CO2 should be much longer than that of methane. The ambient cases with constant and diurnal solar radiation have almost the same OHRVOC evolution as a function of OHexp (Figs. 1 and 2).
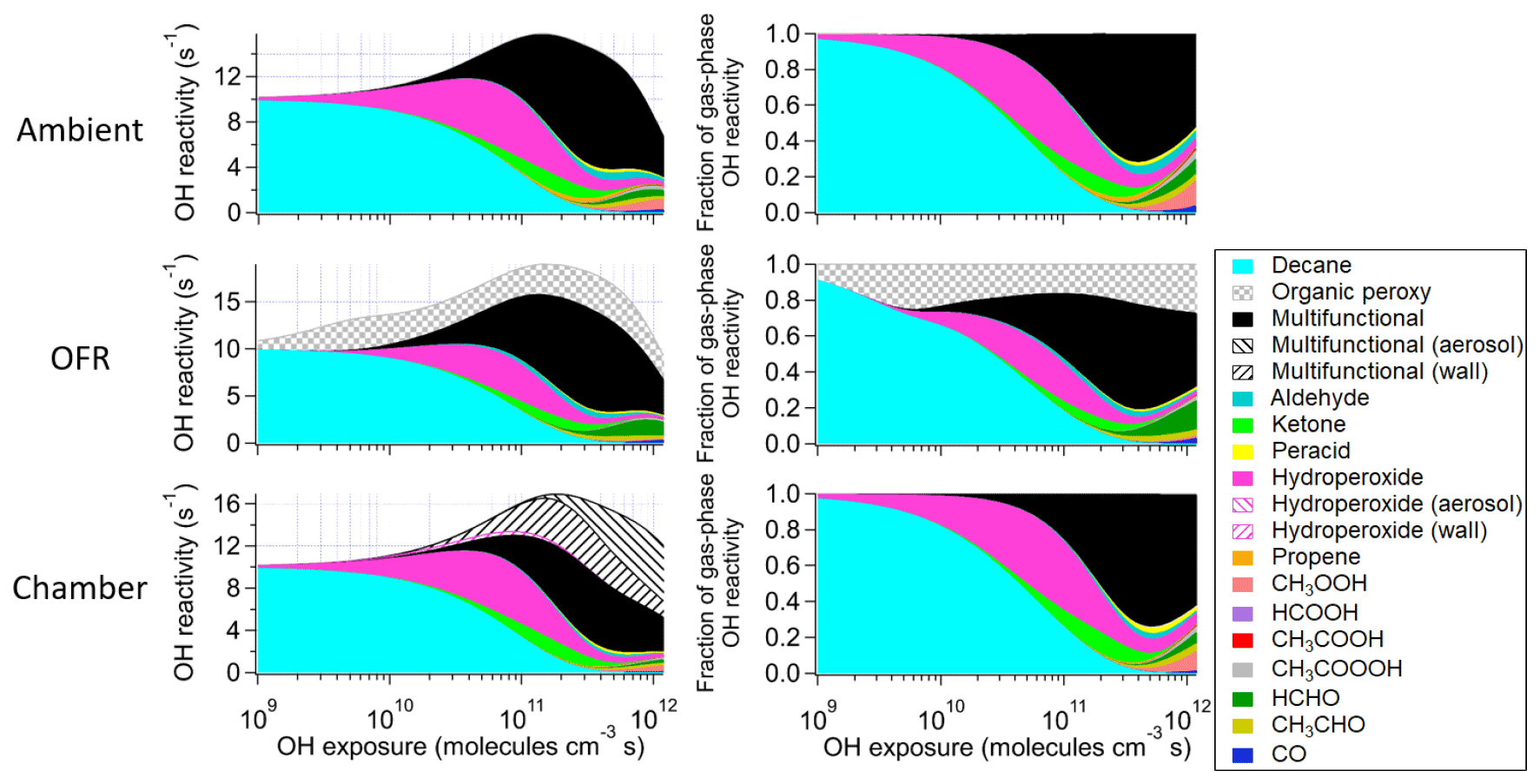
Figure 3Absolute and fractional contributions to the organic OHR during decane photooxidation of the main species and types of species as a function of OH exposure in the ambient case with constant sunlight; the OFR case with relative humidity of 30 %, medium UV lamp setting, and initial OHR of 10 s−1; and the chamber case with initial OHR of 10 s−1 and gas–wall partitioning. The types of species shown in this figure exclude the C1 and C2 species listed separately. The OHR of the particle- and wall-phase species are the values as if those species were gas-phase OHR contributors, although they actually do not react with OH in the simulations.
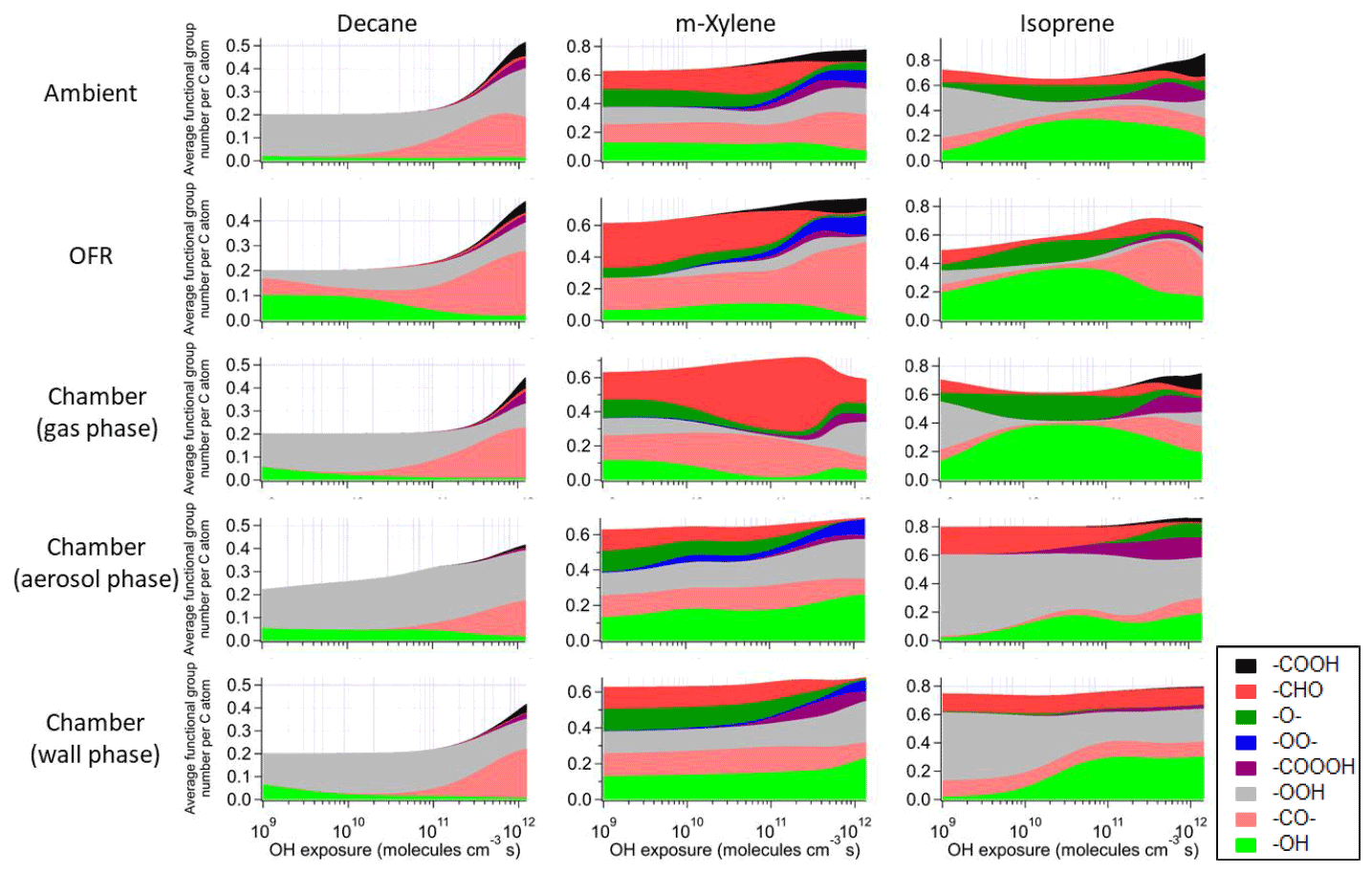
Figure 4Average number of functional group per C atom as a function of OH exposure in the saturated multifunctional species in the ambient case with constant sunlight, the OFR case with relative humidity of 70 %, high UV lamp setting, and initial OHR of 10 s−1, as well as the gas, aerosol, and wall phases in the chamber case with initial OHR of 10 s−1 and gas–particle–wall partitioning of the photooxidations of decane, m-xylene, and isoprene. Note that the functional group “–O–” represents ether, ester, and epoxy groups in the GECKO-A model.
These differences from the methane cases arise because a key assumption of the simple reaction chain model, i.e., slow precursor decay allowing intermediates/products to build up and reach a steady state, no longer holds in decane oxidation. The main first-generation products, i.e., secondary decyl hydroperoxides, react with OH only less-than-3-times more rapidly (in terms of the rate constant of the whole molecule) than does decane, as the significant activation effect of the –OOH group only applies to the α-H, and all other H atoms in this long chain alkyl, though less reactive, can be abstracted by OH (Kwok and Atkinson, 1995; Aumont et al., 2005). When decyl hydroperoxides are present in significant amounts (OHexp ∼ 5 × 1010 molecules cm−3 s), decane loss is also significant (Fig. 3). Also, oxidation of monohydroperoxides to ketones, their most likely fate (due to the activated α-H), lowers OHRVOC, as the oxidation removes the most activated H (Kwok and Atkinson, 1995). The multifunctional products of further oxidation in the middle OHexp range (before ∼ 2 × 1011 molecules cm−3 s) mainly have –OOH and –CO– (Fig. 4), which do not further increase OHR substantially with respect to monohydroperoxides, for similar reasons as the comparison of monohydroperoxides with decane. After the OHRVOC peak, the precursor is largely consumed and –CH(OOH)– groups become increasingly oxidized to –CO– in both monohydroperoxides and multifunctional hydroperoxides (Figs. 3 and 4), which rapidly decreases OHRVOC. Since the decane oxidation chain does not reach a steady state, it results in only limited OHRVOC enhancement at peak.
In the absence of steady state for the nodes (stable species) in the decane oxidation chains (nodes far downstream insufficiently populated), organic photolysis and RO2 self- and cross-reactions only help move OHR contributors to downstream nodes but do not significantly change their total concentrations. This is shown by the relatively small differences in the composition of stable OHR contributors between the ambient and OFR cases (Fig. 3). The remarkable difference between these cases is the contribution of RO2 to OHR, which is as high as ∼ 3 s−1 in the OFR case shown in Fig. 3, while estimated to be only up to ∼ 0.1 s−1 in the ambient case, given the RO2 concentration in the simulation.
It is known that RO2 + OH can be a significant RO2 loss pathway in OFR, especially when OH and HO2 production is relatively strong (higher relative humidity (RH) and UV). We have previously advised to avoid such conditions in low-NO chemistry based on simplified modeling, because of its high-NO-like organic product (Peng et al., 2019). Here our chemically explicit modeling results show that the limitation for OFR chemistry caused by RO2 + OH may not be as serious as suggested by Peng et al. (2019), at least in terms of OHRVOC and, to some extent, of organic composition (Fig. 3). The condition of the OFR case shown in Fig. 3 (30 % RH, medium UV, and 10 s−1 initial OHR) is a compromise between the goals of reaching an equivalent photochemical age of ∼ 10 d, avoiding significant non-tropospheric organic photolysis, and keeping a more atmospherically relevant RO2 chemistry (Peng et al., 2016; Peng et al., 2019; Peng and Jimenez, 2020). In this case, the fractional contribution of RO2 + OH to RO2 fate is still sizable (> 30 %). However, the evolution of the composition of monofunctional species in this OFR case before the OHRVOC peak equivalent age is similar to that in the ambient case (Fig. 3), as hydroperoxide production through RO2 + HO2 is still the main loss pathway of the first-generation RO2 and RO produced from RO2 + OH can also form ketones, i.e., the main second-generation products. The other main fate of RO, i.e., isomerization, leads to slightly faster production of multifunctional species, since the product of the recombination of the immediate product of this isomerization, i.e., an alkyl radical, with O2 is already a bifunctional RO2. This isomerization also creates a hydroxyl group on the C backbone, resulting in a relatively high share of hydroxyl in the functional groups of the multifunctional species (Fig. 4).
Before the OHRVOC peak, as OHexp increases, carbonyls accumulate. They are prone to Norrish-type photochemistry (Turro et al., 2009), which plays a major role in the OHR evolution after the OHRVOC peak in the ambient cases by breaking C10 species into smaller molecules (Fig. 3). Alkenes, which can only be produced via Norrish type II reaction in this case (Turro et al., 2009), are non-negligible OHR contributors around 3 × 1011 molecules cm−3 s. A set of oxygenated C1 and C2 species are also largely produced through organic photolysis followed by reactions with (O2 and) HO2. Organic photolysis, along with OH reaction pathways can also produce RO2. Self- and cross-reactions of acylperoxy radicals, which are formed in significant amounts in this OHexp range, can rapidly generate alkoxy radicals (Orlando and Tyndall, 2012), which may decompose subsequently (Ziemann and Atkinson, 2012). At ∼ 1 × 1012 molecules cm−3 s, the small species produced account for about half of OHRVOC in the ambient cases (Fig. 3). In the OFR cases, organic photolysis is usually much weaker than in the atmosphere (Peng et al., 2016; Peng and Jimenez, 2020), as the negligible OHR of alkenes in the OFR case in Fig. 3 also indicates. However, fragmentation of multifunctional species does not appear to be significantly weaker in the OFR case than in the ambient case shown in Fig. 3. This is largely due to fast RO2 + OH. The reactions of acylperoxys with OH lead to direct fragmentation (Orlando and Tyndall, 2012). Highly functionalized RO can also form from the reactions of multifunctional RO2 with OH and then often rapidly decompose. RO2 + OH also results in a major difference of the OFR case from the ambient case at high equivalent ages (Fig. 3), i.e., lower (higher) OHR contribution from CH3OOH (HCHO) in OFR than in the atmosphere. Most CH3OO reacts with OH to produce CH3O and then HCHO in the OFR case, leaving a minor fraction of CH3OO reacting with HO2 to form CH3OOH.
3.2.2 Chamber cases
Three types of chamber simulations, without gas–particle–wall partitioning, with gas–particle partitioning only (no wall), and with gas–particle–wall partitioning, are considered in this study. The first type has almost the same results as the ambient cases in terms of the evolution of OHRVOC and its composition as a function of OHexp (Fig. 1). Despite different spectra, sunlight and chamber lights cover the same wavelength range and usually generate oxidizing agent radicals (e.g., OH and HO2) in similar amounts. Therefore, all key parameters are very similar between the ambient case with constant solar radiation and the chamber case (with 10 s−1 initial OHR) and without gas–particle–wall partitioning. This explains the high similarities between the results of the ambient cases and the chamber cases without wall partitioning. Unfortunately, the lack of wall partitioning is not realistic for current chambers.
The chamber cases with gas–particle–wall partitioning at lower initial OHR, which are realistic, show very large deviations from the ambient cases that cannot be explained by gas–particle partitioning only (Figs. 1, 3 and S4). These deviations are mostly due to wall partitioning of OVOCs around the OHexp of the OHR peak and at lower OHexp. In this OHexp range, the wall removes about half of the OHR of decane oxidation intermediates/products (Figs. 1 and 2) and hence also almost removes the OHRVOC peak in the relevant chamber cases (Fig. 1). In detail, some decyl hydroperoxides partition to the wall in the chamber case shown in Fig. 3, as decyl is a relatively large alkyl and leads to hydroperoxides of sufficiently low volatility to promote wall partitioning. The C10 ketones, usually of higher volatility than the corresponding hydroperoxides, do not show significant wall partitioning (Fig. 3), while about half of the multifunctional C10 species, of even lower volatility than the corresponding hydroperoxides, are partitioned to the wall.
At higher OHexp (> 2 × 1011 molecules cm−3 s), more multifunctional species stay in the gas phase or partition to the particle phase (Figs. 3 and S4). Those in the gas phase are formed via C10 fragmentation and are thus of higher volatility (Fig. 3). Those partitioned to the particle phase instead of the wall are due to a higher organic aerosol concentration resulting from accumulation during a long oxidation. The substantial partitioning of multifunctional species to aerosol and the wall also slows down their oxidative evolution in the gas phase relative to the ambient cases (Fig. 4). The degree of oxidation of products partitioned to the particle phase increases since low OHexp. This is due to a volatility fractionation caused by the lower ability of the particle phase to absorb condensable organic gases than that of the wall phase at this OHexp. At low organic aerosol loading, the relative potential of particle partitioning to wall partitioning for organic gases of higher functionalization is higher than for those of lower functionalization.
At higher initial OHR (100 s−1), and hence higher organic aerosol loading, condensable gases have a significantly higher tendency of partitioning to the particle phase. The reduction of OHR of the higher initial OHR chamber case with aerosol partitioning only (no wall partitioning) relative to the purely gas-phase case is comparable to the lower initial OHR case with gas–particle–wall partitioning at low OHexp (before the OHR peak) (Fig. S4). At high OHexp, this OHR reduction is even stronger than in the lower initial OHR case with gas–particle–wall partitioning, as partitioning of OVOCs to the particle phase dominates over that to the walls.
As OHexp increases and large multifunctional species are formed in increasing amounts from oxidation, their substantial partitioning to aerosol and the wall decreases the OHR of decane oxidation intermediates/products by a factor up to 8 around 1 × 1012 molecules cm−3 s compared to the chamber cases without gas–particle–wall partitioning (Figs. 2 and S4). At higher OHexp (long oxidation times) gas-phase concentrations of partitioning species decline, allowing reverse partitioning back from the particle phase and the wall which then serves as a source rather than a sink. As a result, the ratio of the OHR of oxidation intermediates/products in the chamber case with gas–particle–wall partitioning to that without this partitioning decreases (Fig. 2).
3.2.3 OH and HOx recycling ratios
As discussed above, we also compute OH (β1) and HOx (β2) recycling ratios in decane oxidation. Note that these quantities also include OH and HO2 generated as a result of organic photolysis. The differences in these recycling ratios between the simulated cases are relatively small. β1 is close to 0 at OHexp ≲ 1 × 1010 molecules cm−3 s (Fig. 1), as the initial reaction of decane with OH only produces an RO2 and subsequently C10 hydroperoxides and no HOx. Then β1 undergoes a fast increase between ∼ 1 × 1010 and 1 × 1011 molecules cm−3 s (Fig. 1), as the further oxidation of C10 hydroperoxides to ketones fully recycles OH (R1-CH(OOH)-R2 + OH → R1-CO-R2 + H2O + OH) in the ambient and chamber cases. Nevertheless, β1 only increases up to ∼ 0.4 at this stage in the ambient and chamber cases, as oxidation of C10 hydroperoxides to dihydroperoxy species and precursor oxidation also account for a substantial fraction of OH loss but do not recycle it. In the OFR cases, β1 only increases up to ∼ 0.2–0.3 at this stage, since RO2 + OH starts to be active but does not recycle OH. Then, β1 roughly plateaus up to ∼ 1 × 1012 molecules cm−3 s, as the overall effect of the decrease in hydroperoxy concentration, reducing OH recycling, and the increase in the concentration of acylperoxy, enhancing OH recycling through its reaction with HO2 (Orlando and Tyndall, 2012), is relatively small. Finally, β1 gradually decreases to 0 (Fig. 1), as all OVOCs degrade to highly oxidized C1 species, i.e., HCHO, HCOOH, and CO, which only have HO2 recycling but no OH recycling, and the unreactive CO2.
The HOx recycling ratio (β2) in decane oxidation is similar to β1 before ∼ 1 × 1011 molecules cm−3 s for the ambient and chamber cases, as only OH (but not HO2) is recycled at this stage. β2 is a little higher in the OFR cases than in the other cases at this stage because of the HO2 recycling by RO2 + OH. However, at higher OHexp, β2 continues to increase with OHexp to a final value of 1 (Fig. 1). This difference between β1 and β2 is by definition due to HO2 recycling. Its significance rises in parallel with that of organic photolysis, which can often produce HCO radicals and acylperoxy radicals. The former extremely rapidly undergoes HCO + O2 → CO + HO2; the latter can rapidly convert peroxy radicals to alkoxy radicals (Orlando and Tyndall, 2012), which may then react with O2 to generate HO2 (Ziemann and Atkinson, 2012). At very high OHexp (1012–1013 molecules cm−3 s), reactive highly oxidized small VOCs are the dominant OHR contributors, and many of them recycle HO2 during their oxidation by OH (Fig. 3). Finally, once CO becomes the only remaining OHR contributor, β2 is 1.
3.3 m-Xylene
Most features in m-xylene oxidation can be explained based on similar discussions as for decane oxidation in Sect. 3.2. OHRVOC also has a maximum during the oxidation (Figs. 1 and S4), as most of the direct products of m-xylene oxidation by OH, i.e., the unsaturated carbonyl (MXYEPOXMUC in MCM v 3.2 notation; see Scheme S1), the unsaturated endo-cyclic peroxide (MXYBPEROOH), and xylenols, are more reactive toward OH than m-xylene. The OHR of these initial products is enhanced much more during the oxidation of m-xylene than of decane, owing to the creation of C=C bonds in many post-aromatic (ring-opening) products; hence the OHRVOC peak enhancement in m-xylene oxidation is larger than in decane oxidation. Because the reaction rate constant of m-xylene with OH slightly exceeds that of decane, the OHRVOC peak in m-xylene oxidation occurs at slightly lower OHexp than in decane oxidation (Fig. 1). In the OFR case under the same condition as the decane case shown in Fig. 3, the evolution of OHR of the stable organic species is again similar to that in the ambient case. And OHRVOC is higher in the OFR case again mainly due to OHR from RO2 (Figs. 1 and S4). Several main first- and second-generation products are already highly functionalized through fast O2 addition (Scheme S1), and they are also often unsaturated and prone to further functionalization. Therefore, the degree of functionalization in saturated aliphatic multifunctional species is much higher in m-xylene than in decane oxidation (Fig. 4). Also, as several aromatic-scheme-specific reaction types occur in the early stages of m-xylene oxidation, e.g., endo-O2 addition (creating –OO–) and ring-opening (creating –CO–, –CHO, etc.), multifunctional species functionality is more diverse than in decane oxidation (Fig. 4). Photolysis again plays a role in species fragmentation and the production of highly oxidized C1 and C2 species after the OHRVOC peak (Fig. S4).
At low OHexp and that of the OHRVOC peak, particle and wall partitioning also substantially reduces the OHRVOC in the relevant chamber cases of m-xylene oxidation while the OHRVOC reduction due to partitioning to the particle phase is smaller than that due to the wall (Figs. 1, 2, S4 and S5). The precursor (m-xylene) is a C8 species, and even many first-generation products of its oxidation are highly oxygenated (Scheme S1) lower-volatility species. The relative reduction of OHR of the intermediates/products also increases with OHexp before the OHexp of the OHR peak, as volatile species are oxidized and become more prone to wall partitioning (Figs. 2 and S4). At higher OHexp, more condensed organics are partitioned to the particle phase because of high organic aerosol concentration (Fig. S4), and the wall and aerosol again serve as OVOC source (Fig. 2).
The evolution of β1 and β2 in m-xylene oxidation is somewhat different than in decane oxidation (Fig. 1). In the ambient cases, they are non-negligible even at OHexp as low as 1 × 109 molecules cm−3 s (∼ 0.05 and ∼ 0.45, respectively). OH is mainly recycled from one of the endo-cyclic peroxide routes (m-xylene + OH + 2O2 → MXYBIPERO2; MXYBIPERO2 + HO2 → MXYBPEROOH + O2; MXYBPEROOH + OH → MXYOBPEROH + H2O + OH, Scheme S1), which involve various functional groups and open the possibility of radical recycling. The third step of this route is very fast (with a rate constant on the order of 10−10 cm3 molec−1 s−1). Once the second step takes place, the third step contributes to OH recycling. However, in the OFR cases with strong water vapor photolysis (not in the other OFR cases), the third step does not play a significant role, and β1 is ∼ 0 at very low OHexp (Fig. 1) due to the relatively slow second step (RO2 + HO2). In the former cases, this is due to the relatively slow second step (RO2 + HO2), while in the latter cases, the highly oxygenated compounds partition to the wall even more rapidly (in hundreds of seconds; Krechmer et al., 2016) than their reactions with HOx. Strong HO2 recycling occurs in all simulated cases from the beginning of the oxidation (Fig. 1), since two of the three major channels of m-xylene + OH (i.e., those forming MXYEPOXMUC and xylenol, respectively) produce HO2 as well.
As more multifunctional species are formed (particularly through ring-opening) near the OHexp of the peak OHRVOC, HOx recycling is also active, with β1 increasing and β2 remaining high (Fig. 1). There is a high peak in β2 for the chamber case with high initial OHR (100 s−1) and no aerosol or wall partitioning. It results from RO2 cross-reactions, many of which produce alkoxy radicals that subsequently yield carbonyls and HO2 through reactions with O2 (Orlando and Tyndall, 2012). RO2 cross-reactions are significant in that OHexp range also because (i) high precursor concentration translates into higher RO2 concentration and (ii) acylperoxy radicals, whose reactions with other RO2 are fast (Orlando and Tyndall, 2012), are rapidly formed from the oxidation of –CHO groups in the ring-opening products (Scheme S1). The chamber case with high initial OHR and gas–particle–wall partitioning does not have such a high β2 peak, because of fast partitioning of the oxidation products containing –CHO groups to the aerosol and wall phases, which significantly reduces acylperoxy radical concentration around the OHexp of the peak OHRVOC. At higher OHexp, the calculated β1 and β2 become less reliable, since remaining apparent OHR contributors may in fact be persistent artifacts of the incompleteness of the (handwritten) m-xylene oxidation mechanism which may substantially bias β1 and β2 when the concentrations of remaining OHR contributors should be generally low. Therefore, we do not try to interpret the features in β1 and β2 at high OHexp for m-xylene oxidation.
3.4 Isoprene
The most salient difference of the OHRVOC evolution in the photooxidation of isoprene from that of the other precursors in this study is the lack of OHRVOC peak in the isoprene cases (Figs. 1 and S5). The decrease in OHRVOC all along this photooxidation is expected since the reaction of isoprene with OH is very fast (at 1 × 10−10 cm3 molec−1 s−1; Atkinson and Arey, 2003), and all intermediates/products of this photooxidation react with OH more slowly than isoprene. The OHRVOC of the intermediates/products peaks slightly after an OHexp of 1 × 1010 molecules cm−3 s (Fig. 1). At this OHexp, the main type of the first-generation products, oxygenated unsaturated species (e.g., isoprene-derived unsaturated hydroxyl hydroperoxides, ISOPOOH), are largely produced from isoprene + OH and their loss rates (with rate constant with OH slightly lower than that of isoprene) reach the maxima (Fig. S5). Further oxidation leads to the loss of all C=C bonds in the isoprene C backbone and thus a substantial drop of the OHR of the molecule.
Before OHexp∼ 5 × 1010 molecules cm−3 s in isoprene photooxidation, the main deviations from the ambient cases shown by the chamber cases with wall partitioning are again caused by wall partitioning of multifunctional species, but their relative magnitudes are different than in the photooxidations of decane and m-xylene, with the impacts of wall partitioning being smaller (Fig. S5). Oxygenated species derived from isoprene, a C5 species, should be generally more volatile and less prone to wall partitioning than those derived from decane and m-xylene. On the other hand, isoprene reacts with OH much more rapidly than do decane or m-xylene, creating a larger deviation from the steady state for RO2 directly derived from isoprene and a more remarkable decrease in the OHR of the first-generation products (Fig. S5). In the OFR case shown in Fig. S5, RO2 contributes negligibly to OHRVOC, since many first-generation isoprene-derived RO2 have other very fast loss pathways and the very fast decay of isoprene cannot sustain RO2 production at OHexp ≳ 1 × 1010 molecules cm−3 s.
After OHexp∼ 5 × 1010 molecules cm−3 s, the deviation caused by chamber wall partitioning becomes more significant as highly oxidized and lower-volatility multifunctional species (Fig. 4) are formed in significant amounts (Figs. 2 and S6). However, aerosol partitioning does not become more significant as, in the chamber cases of isoprene oxidation, aerosol formation is always so small that partitioning to the particle phase never competes with that to the walls. At very high OHexp, the wall again acts as a source of OVOCs in isoprene oxidation, as in those of the other precursors (Fig. 2). The deviations of OFR cases from the ambient cases are mainly caused by RO2 + OH and lack of organic photolysis. These two effects lead to too much HCHO produced and inefficient production of other C1 and C2 species (Fig. S5).
To test whether one of the issues, i.e., lack of organic photolysis in OFR, can be mitigated by adding tropospherically relevant UV sources, we perform two additional simulations. Adding the emissions corresponding to the high Hg lamp setting with 5 times the UV of the CU chamber (a rough upper limit for experimental implementation) has a negligible effect (Fig. S6). To reach a ratio between tropospherically relevant UV (UVA+UVB) intensity and OH concentration similar to that in the ambient case with constant sunlight requires addition of a chamber light ∼ 10 000 times stronger than the CU chamber light. Such a strong UV source is obviously not realistic, and, while it does increase both early organic photolysis and the relative contribution of C1 and C2 photoproducts to OHRVOC around 2 × 1011 molecules cm−3 s (Fig. S6), it increases the deviation of this OFR case from the ambient cases at very high OHexp, where oxidation of C1 and C2 species to CO proceeds much more rapidly than in the atmosphere.
Product functionality in isoprene oxidation is more diverse than in decane oxidation (Fig. 4). This is due to both the active isomerization and the propensity of the isoprene C = C bonds prone to addition of various groups (Wennberg et al., 2018). Notably, epoxy groups in species such as isoprene-derived epoxydiol (IEPOX) account for a large fraction of saturated product functionality (Fig. 4), particularly at OHexp on the order of 1010 molecules cm−3 s. In the gas phase of the chamber cases with wall partitioning, the overwhelming majority of saturated multifunctional organic molecules are IEPOX up to 1 × 1011 molecules cm−3 s (Fig. 4), as more highly oxidized species mostly partition to the wall.
IEPOX formation from isoprene-derived hydroperoxide (ISOPOOH) oxidation by OH (ISOPOOH + OH → IEPOX + OH) leads to the peak of OH recycling around 3 × 1010 molecules cm−3 s (Fig. 1). OH recycling is active even at very low OHexp (1 × 109 molecules cm−3 s) because a significant amount of ISOPOOH forms early and can recycle OH through its oxidation, except in the OFR cases with strong water vapor photolysis, where ISOPOOH cannot be efficiently formed from first-generation RO2. HO2 recycling is also active in the entire course of the photooxidation (Fig. 1), because of a number of isomerization and photolysis pathways that form alkoxy radicals and highly oxidized C1 species such as HCOOH, HCHO, and CO at very high OHexp (Fig. S5).
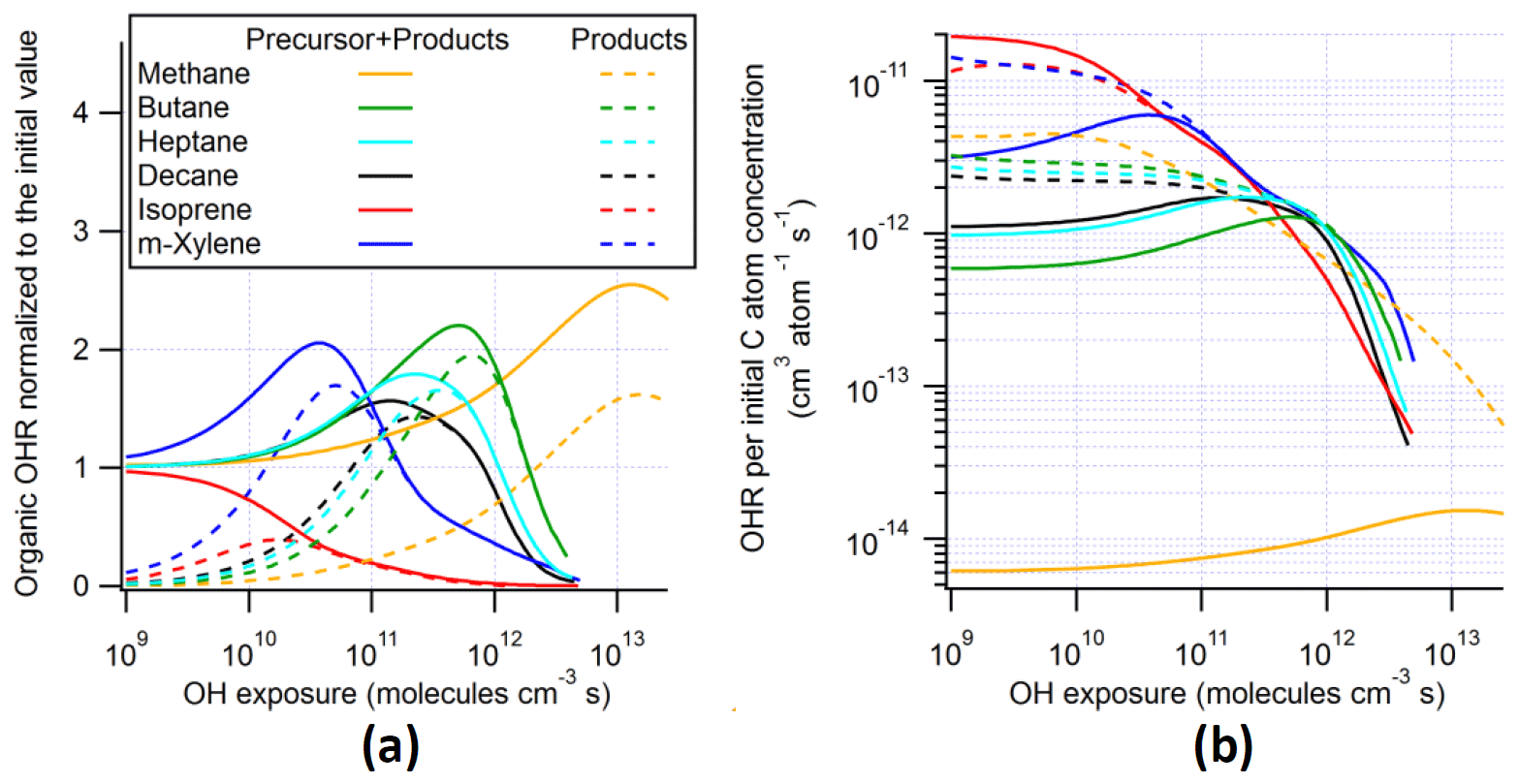
Figure 5(a) OHR and (b) OHR per initial C atom concentration of the organics (including and excluding the precursor) as a function of OH exposure in the ambient cases with constant sunlight of the photooxidation of methane, butane, heptane, decane, isoprene, and m-xylene.
3.5 Trends in OHR per C atom
To explore some general trends of OHR evolution in VOC photooxidation, simulations are performed for the ambient cases with constant UV for two additional alkanes between methane and decane, i.e., butane and heptane. The results of these simulations are compared to the existing analogous cases in Fig. 5. For all cases, the OHRVOC peak height decreases and the OHexp of the OHRVOC peak shifts towards lower OHexp, as the C number of the precursor alkane increases. This can be explained by the fact that the OH rate constants of these alkanes increase with C number and suggests a possible general trend between OHR peak location and C number.
To explore these trends further, we calculate the OHR per unit starting concentration of C atom (in the precursor) in all ambient cases with constant UV in this study (Fig. 5b). In this study, CO2 is not included initially but produced during the oxidation. Therefore, C atoms in the produced CO2 are taken into account in the calculation of OHR per C atom. For real atmospheric cases, initial CO2 is present but should not be considered in this calculation. Note that OHR per C atom has a unit of cm3 per atom per second and represents the average contribution to the rate constant with OH of all considered C atoms. Despite large differences among the reactivities of these precursors, the OHR per C atom in the simulations of all precursors but methane converges near an OHexp of 3 × 1011 molecules cm−3 s and then follows a very similar downward trend (Fig. 5b). This OHexp value is roughly where saturated multifunctional species have their maximal relative contribution to the OHRVOC (Figs. 3 and S5). Even in the ambient cases of m-xylene oxidation, saturated multifunctional species also account for about half of OHRVOC when the contribution of aromatics, some of which may artificially persist due to mechanism incompleteness, is excluded (Fig. S4). Also, at OHexp >∼ 3 × 1011 molecules cm−3 s, a C atom in saturated multifunctional species on average has at least 0.3 functional groups in the ambient cases (Fig. 4), and the functional group composition is relatively diverse at this OHexp. Therefore, the convergence value of OHR per C atom of ∼ 2 × 10−12 cm3 per atom per second at ∼ 3 × 1011 molecules cm−3 s can be largely regarded as a relatively invariant average of those of secondary H and α-H of various O-containing functional groups. Note that this average is for NO-free conditions and can be lower at high NO due to deactivating effects of N-containing groups formed during oxidation (Isaacman-VanWertz and Aumont, 2021).
Before the convergence, isoprene has the highest OHR per C atom (on the order of 10−11 cm3 per atom per second) among the precursors and intermediates/products (Fig. 5b), because of its conjugated C=C bonds. The OHR per C atom of its first-generation oxidation products is slightly lower and close to that of the oxidation intermediates/products of m-xylene, as the main contributors in both cases are oxygenated monoalkenes. The average OHR per C atom of the studied alkanes increases with C number (Fig. 5b), with the upper limit around 1 × 10−12 cm3 per atom per second consistent with Kwok and Atkinson (1995), since the less-reactive -CH3 groups (with OHR per C atom of ∼ 1 × 10−13 cm3 per atom per second) contribute proportionally less to molecular OHR as C number increases. Conversely, the early-stage products of alkane oxidation (mainly alkyl monohydroperoxides) show higher average OHR per C atom for shorter molecules (Fig. 5b), owing to the activating (increasing OHR) contribution of the –OOH group.
Following the convergence of OHR per C atom, this quantity in all non-methane ambient cases in this study sees a similar decay (Fig. 5b). This coincides with multifunctional species broken into small highly oxidized C1 and C2 compounds. Although among them there are species with OHR per C atom > 5 × 10−12 cm3 per atom per second (e.g., CH3OOH, CH3CHO, and HCHO), the average OHR per C atom of these C1 and C2 species is mainly governed by those reacting more slowly (e.g., HCOOH and particularly CO) and hence reaching higher concentrations amid the fast decay of multifunctional species. The similar fast drop of OHR per C atom after OHexp∼ 1 × 1012 molecules cm−3 s for various precursors implies a transition from OHR from saturated multifunctional molecules to OHR from CO before the final oxidation to CO2 (which has zero OHR).
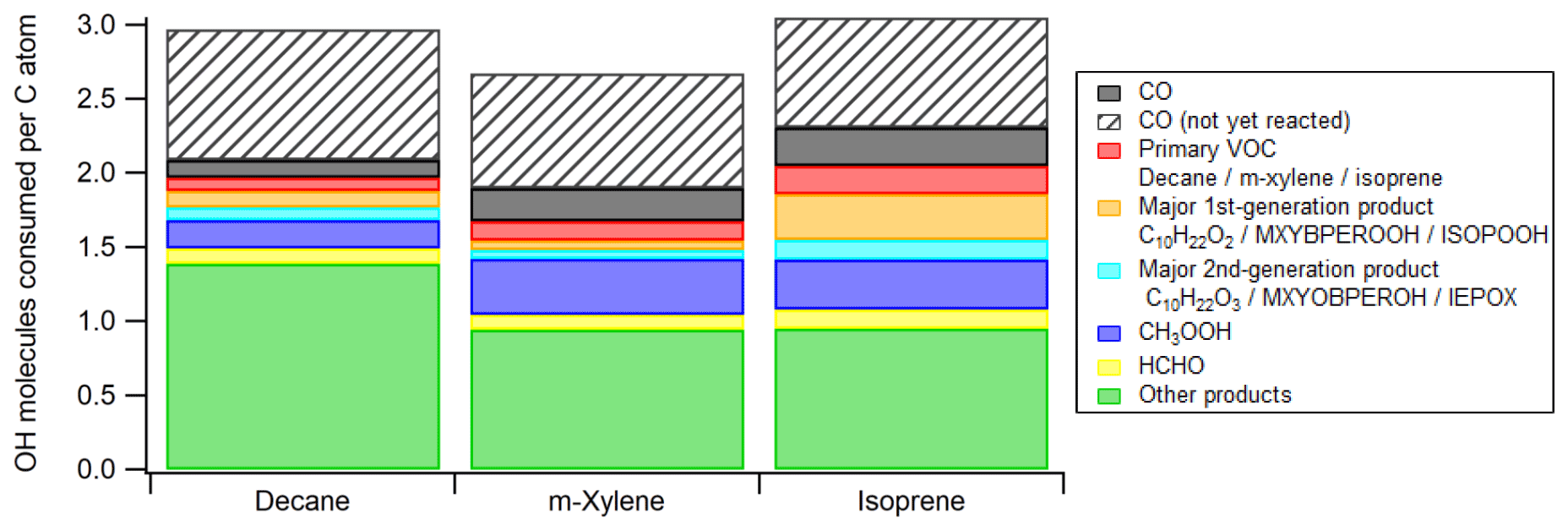
Figure 6Average numbers of OH molecules consumed per C atom in the ambient cases with constant sunlight during photooxidation of isoprene, decane, and m-xylene. The contribution from CO that is not yet oxidized by OH at the end of simulation is also added to ensure that each CO molecule consumed one OH radical. ISOPOOH, IEPOX, C10H22O2, and C10H22O3 are isoprene hydroxyl hydroperoxides, isoprene epoxydiols, decyl hydroperoxides, and hydroxydecyl hydroperoxides, respectively. See Scheme S1 for the structures of MXYBPEROOH and MXYOBPEROH.
3.6 Total OH consumption for each precursor
Integrating OHR per C atom over OHexp allows us to assess the average number of OH molecules consumed by each C atom during the entire course of oxidation. This quantity can also be apportioned to the contributions of different OH reactants (Fig. 6). Due to incomplete oxidation of several species, especially CO, the value of this quantity for an oxidation with all C atoms ending up with CO2 should be higher than those at simulation end (OHexp∼ 4 × 1012 molecules cm−3 s). We correct this in Fig. 6 by including additional contribution of CO to make its total contribution 1, since CO, the typical penultimate product, consumes one OH molecule in its final oxidation but is still present in significant quantities at the end of our simulations. Thus, each C atom reacts with OH around three times in the course of the oxidation of isoprene and decane to CO2 (Fig. 6). A simplistic and chemically intuitive explanation for this number is that the average oxidation state () of both isoprene and decane C atoms is ∼ −2 and needs to increase to the value of +4 in CO2 at the end of the oxidation. A C1 unit reacting once with OH likely increases its by ∼ 2. This increase is usually realized by an abstraction of H atom by OH or an addition of OH (), followed by an abstraction of H atom by O2 or an addition of O2 (). Note that ∼ 3 OH consumed per C atom oxidized to CO2 is likely an upper limit, since the mechanisms in this study do not include RO2 autoxidation (Crounse et al., 2013; Ehn et al., 2014), which reduces the number of OH needed for complete VOC oxidation. Also, in a real low-NO environment, NO is still present in low concentrations and converts RO2 to RO. RO may undergo H abstraction through isomerization or reaction with O2, which also lowers the number of OH needed, although the effect is usually small. The number of OH consumed per C atom in m-xylene oxidation is slightly lower than three (Fig. 6) because of the multiple addition of O2 following a single OH addition in the initiation reaction, i.e., m-xylene + OH.
Using the fully explicit GECKO-A model, we simulated OHR evolution in the photooxidation of several types of VOCs (i.e., alkane, alkene, and aromatic) without NO until very high equivalent photochemical ages (> 10 d) under a variety of conditions (in the atmosphere, chamber, and OFR). We analyzed the simulations in detail and found a number of common features as well as some differences resulting from certain precursors. These features are summarized below.
-
All simulated non-methane cases very roughly follow this general oxidation chain pattern: precursor → first-generation products → (second-generation products →) multifunctional species → highly oxidized C1 and C2 species → CO (or HCOOH) → CO2. These species are generally not at steady state and gain significance/predominance one after another in the entire course of oxidation. Simulation results suggest that fragmentation products are not formed in significant amounts until the late stage of the oxidation, which would be a key difference from studies of OHR evolution in high-NO VOC oxidation (Nakashima et al., 2012; Sato et al., 2017).
-
In methane oxidation, the intermediates do not gain dominance in sequence. Instead, they simultaneously increase as the oxidation proceeds and then simultaneously decrease when the methane decay becomes significant. The OHR evolution in methane oxidation is close to the idealized steady-state chain model, as the reaction of methane with OH is orders of magnitude slower than those of its oxidation intermediates, which allows the intermediates to reach their steady state.
The following discussion refers to the non-methane cases.
-
Where different types of species dominate OHRVOC in sequence, OHRVOC increases after the current dominant type converts to one with a higher average OHR per C atom, and vice versa. Photooxidations of alkanes and aromatics follow the increasing trend from precursor to saturated multifunctional species (via alkyl monohydroperoxides) and from precursor to unsaturated oxygenated species, respectively. The increase in aromatic oxidation is likely to be more significant, since unsaturated oxygenated species are more reactive than saturated multifunctional species. The conversions from multifunctional species to CO2 lead to a decay of OHRVOC in both alkane and aromatic photooxidations. OHRVOC in alkene photooxidation is likely to always drop rapidly during C=C bond oxidation and more slowly afterwards.
-
A relatively weak enhancement of OHR per C atom of a C atom with –OOH substitution can explain the large range spanned by the precursors and their intermediates/products in this study at low OHexp. Around an OHexp of 3 × 1011 molecules cm−3 s, precursors are largely converted to saturated multifunctional species (e.g., by addition to C=C bonds in unsaturated precursors and abstraction of H atoms in saturated precursors), and the reactive mixtures of those precursors thus have similar OHR per C atom. They then all follow the course: multifunctional species → highly oxidized C1 and C2 species → CO (or HCOOH) → CO2 and show similar decays of OHR per C atom.
-
In decane and isoprene oxidation, our simulations show that each C atom consumes at most around three OH molecules in the course of its oxidation to CO2. This can be simplistically explained as three occurrences of oxidation by OH that increase, by two each time, the of decane and isoprene (∼ −2) to that of CO2 (+4). The total number of OH consumed by each C atom is likely to be lower when RO2 autoxidation can be included in the mechanism generation.
In general, the OHR evolution differences resulting from different precursors are larger than those due to different conditions. The difference in OHRVOC between the ambient cases with constant and diurnal sunlight is small. Nevertheless, physical conditions may still lead to significant differences, which are summarized below.
-
In current chambers, gas–wall partitioning can be a prominent issue that causes substantial wall partitioning of certain OVOCs of lower volatility, depending on the chemical system under study. The clearest example in this study is the substantial wall losses of C10 multifunctional species from the gas phase in decane oxidation and hence the remarkably lowered OHRVOC peak height in the chamber simulation. The wall also preferentially absorbs more oxidized (and thus lower-volatility) species, which alters the functional composition of gas-phase multifunctional species. The wall can even serve as a source of multifunctional species at very high OHexp, when the gas-phase concentrations of those species are very low. The magnitude of the effects of wall partitioning also depends on the size of the precursor, with the oxidation of larger precursors in chambers suffering larger impacts of wall partitioning.
-
The strong wall losses have important implications, as they can change our modeling results substantially. Systematic OVOC gas–particle–wall partitioning corrections must be made for low-NO oxidation chamber experiments that study OHRVOC. In case of large precursors, highly chemically explicit modeling will likely be necessary to infer the OHR of multifunctional species, which may account for a large fraction of missing reactivity but suffer substantial wall losses. Although the few existing chamber studies on OHRVOC evolution were all under high-NO conditions, which may result in more fragmentation and higher-volatility products, the magnitude of wall partitioning of large multifunctional species in this study is so substantial that we believe this effect would also be important at high NO. Schwantes et al. (2017) considered wall partitioning in their modeling of o-cresol oxidation based on MCM v3.3.1 but still could not achieve good agreement with the measurements for a number of products. Considering this, one should not assume that it is appropriate to neglect gas–particle–wall partitioning in high-NO chamber experiments, just based on agreement between the high-NO chamber experiments and modeling with MCM-based schemes and without gas–particle–wall partitioning corrections. Even for OHR studies with less surface loss issues, e.g., ambient studies, a combination of gas-phase-only OHR measurement and modeling may still not be adequate as reduction of OHR due to OVOC condensation on aerosols can also be significant in some situations (Fig. S4). Therefore, condensed phases (particle and wall) need to be included in future OHR studies to better assess the deviation of the actual OHR from a purely gas-phase picture.
-
OFR has two issues under certain conditions that can cause deviations from the ambient cases in terms of OHRVOC. Strong RO2 + OH may significantly contribute to OHRVOC. Interestingly, this type of reaction does not seem to be able to substantially alter the composition of OHRVOC before the OHRVOC peak. Besides, the conditions resulting in strong water vapor photolysis have already been identified as those leading to atmospherically irrelevant RO2 chemistry in low-NO OFR in previous studies (Peng et al., 2019; Peng and Jimenez, 2020). As long as OFR users follow the guidelines for experimental planning provided in those studies (and use a much lower UV lamp setting), strong RO2 + OH can be avoided, as shown in Fig. 1.
-
The other main issue of OFR is the lack of efficient organic photolysis, particularly at high OHexp, when multifunctional species break into highly oxidized C1 and C2 compounds. This problem has been highlighted in previous studies (Peng et al., 2016; Peng and Jimenez, 2020) and been shown again in the present work to be extremely difficult to avoid if a high OHexp is desired. However, the conversion of multifunctional species into highly oxidized C1 and C2 species may not be much slower in OFR than in the atmosphere, since RO2 + OH, leading to RO formation and subsequently its decomposition, may also play a major role in this conversion. This also results in significantly higher (lower) production of HCHO (CH3OOH) in OFR than in the atmosphere at high equivalent ages.
With all the key findings in this study presented above, we believe that we have, to some extent, addressed the issues of missing reactivity, of model limitations, and of OHR in remote areas for OHR studies raised by Williams and Brune (2015). With the fully explicit GECKO-A model, we speciated the likely source of the missing reactivity, i.e., multifunctional OVOCs. A contrast between the technical issues in some isoprene and m-xylene simulations and the high consistency in the other cases highlights the importance of the completeness of the mechanism (even beyond the MCM level) in OHR-related modeling studies. Substantial wall partitioning of OVOCs in some chamber experiments highlights the importance of better constraining gas-to-surface deposition terms. More studies, both modeling (with highly chemically explicit mechanisms) and experimental (particularly low-NO), are needed to achieve better model–experiment closure. Finally, this study may have opened up the possibility of parameterizing the OHR evolution in (at least low-NO) VOC photooxidation as a function of OHexp only with the often-available knowledge on the first- and second-generation products and the relevant SARs such as Kwok and Atkinson (1995), as the OHR evolution beyond multifunctional species has been shown to be similar for most VOC oxidations. This parametrization may be utilized in regional and global models to better constrain OHR at high equivalent photochemical ages, e.g., in remote regions.
-
Consider the following reaction chain, where OH is the only oxidant:
A1 + OH → A2, k1,
A2 + OH → A3, k2,
A3 + OH → A4, k3,
etc.
At steady state, k1[A1][OH] = k2[A2][OH] = k3[A3] [OH] = … = C (C is a constant).
Then OHR due to individual species, OHRi, is equal to and is identical for all species.
-
Consider a parallel conversion of A1 to A2 by a means other than reaction with OH:
A1 + OH → A2, k1,
A1 + B → A2, , B ≠ OH.
At steady state, k1[A1][OH] + [A1][B] = C.
Therefore, OHRA1 = k1[A1] < C/[OH].
-
Now consider a reaction converting A1 directly to A3 occurring in parallel to reaction chain 1:
A1 + D → A3, , D ≠ OH,
A1 + OH → A2, k1,
A2 + OH → A3, k2.
At steady state, [A1][D] + k2[A2][OH] = C.
And OHRA1 < C/[OH], since k1[A1][OH] = k2[A2] [OH].
The chemical mechanisms generated and the outputs of the GECKO-A simulations in this study are available upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-14649-2021-supplement.
ZP and JLJ conceived the study. ZP designed the study. JLT and ZP performed the simulations. HS, JLT, ZP, and JLJ developed the GECKO Loader and Plotter. ZP, JLT, JJO, and BA made updates and developments for GECKO-A. ZP, JLT, JJO, BA, and JLJ analyzed the results. ZP led the manuscript writing with inputs from all authors.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank Sasha Madronich and Alma Hodzic for useful discussions. We would like to acknowledge high-performance computing support from Cheyenne (https://doi.org/10.5065/D6RX99HX) provided by NCAR's Computational and Information Systems Laboratory, sponsored by the National Science Foundation.
This research has been supported by the National Science Foundation (grant nos. AGS-1822664 and AGS-1740610).
This paper was edited by Andreas Hofzumahaus and reviewed by two anonymous referees.
Atkinson, R. and Arey, J.: Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Ausloos, P. J. and Lias, S. G.: Photochemistry in the Far Ultraviolet, Annu. Rev. Phys. Chem., 22, 85–107, https://doi.org/10.1146/annurev.pc.22.100171.000505, 1971.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005.
Brune, W. H.: The Chamber Wall Index for Gas–Wall Interactions in Atmospheric Environmental Enclosures, Environ. Sci. Technol., 53, 3645–3652, https://doi.org/10.1021/acs.est.8b06260, 2019.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies: Evaluation Number 18, JPL Publication 15-10, Pasadena, CA, USA, available at: http://jpldataeval.jpl.nasa.gov/ (last access: 1 July 2021), 2015.
Camredon, M., Aumont, B., Lee-Taylor, J., and Madronich, S.: The system: an explicit model of secondary organic aerosol formation, Atmos. Chem. Phys., 7, 5599–5610, https://doi.org/10.5194/acp-7-5599-2007, 2007.
Chameides, W., Lindsay, R., Richardson, J., and Kiang, C.: The role of biogenic hydrocarbons in urban photochemical smog: Atlanta as a case study, Science, 241, 1473–1475, https://doi.org/10.1126/science.3420404, 1988.
Cohen, A. J., Brauer, M., Burnett, R., Anderson, H. R., Frostad, J., Estep, K., Balakrishnan, K., Brunekreef, B., Dandona, L., Dandona, R., Feigin, V., Freedman, G., Hubbell, B., Jobling, A., Kan, H., Knibbs, L., Liu, Y., Martin, R., Morawska, L., Pope, C. A., Shin, H., Straif, K., Shaddick, G., Thomas, M., van Dingenen, R., van Donkelaar, A., Vos, T., Murray, C. J. L., and Forouzanfar, M. H.: Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015, Lancet, 389, 1907–1918, https://doi.org/10.1016/S0140-6736(17)30505-6, 2017.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Haseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/Ngeo1964, 2013.
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017.
George, I. J. and Abbatt, J. P. D.: Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nat. Chem., 2, 713–22, https://doi.org/10.1038/nchem.806, 2010.
Haagen-Smit, A. J.: Chemistry and Physiology of Los Angeles Smog, Ind. Eng. Chem., 44, 1342–1346, https://doi.org/10.1021/ie50510a045, 1952.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hodzic, A., Campuzano-Jost, P., Bian, H., Chin, M., Colarco, P. R., Day, D. A., Froyd, K. D., Heinold, B., Jo, D. S., Katich, J. M., Kodros, J. K., Nault, B. A., Pierce, J. R., Ray, E., Schacht, J., Schill, G. P., Schroder, J. C., Schwarz, J. P., Sueper, D. T., Tegen, I., Tilmes, S., Tsigaridis, K., Yu, P., and Jimenez, J. L.: Characterization of organic aerosol across the global remote troposphere: a comparison of ATom measurements and global chemistry models, Atmos. Chem. Phys., 20, 4607–4635, https://doi.org/10.5194/acp-20-4607-2020, 2020.
Isaacman-VanWertz, G. and Aumont, B.: Impact of organic molecular structure on the estimation of atmospherically relevant physicochemical parameters, Atmos. Chem. Phys., 21, 6541–6563, https://doi.org/10.5194/acp-21-6541-2021, 2021.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Keller-Rudek, H., Moortgat, G. K., Sander, R., and Sörensen, R.: The MPI-Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest, available at: http://www.uv-vis-spectral-atlas-mainz.org (last access: 26 March 2019), 2020.
Kovacs, T. A. and Brune, W. H.: Total OH loss rate measurement, J. Atmos. Chem., 39, 105–122, https://doi.org/10.1023/A:1010614113786, 2001.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Krechmer, J. E., Day, D. A., Ziemann, P. J., and Jimenez, J. L.: Direct Measurements of Gas/Particle Partitioning and Mass Accommodation Coefficients in Environmental Chambers, Environ. Sci. Technol., 51, 11867–11875, https://doi.org/10.1021/acs.est.7b02144, 2017.
Kwok, E. and Atkinson, R.: Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 29, 1685–1695, https://doi.org/10.1016/1352-2310(95)00069-B, 1995.
Lee-Taylor, J., Hodzic, A., Madronich, S., Aumont, B., Camredon, M., and Valorso, R.: Multiday production of condensing organic aerosol mass in urban and forest outflow, Atmos. Chem. Phys., 15, 595–615, https://doi.org/10.5194/acp-15-595-2015, 2015.
Levy II, H.: Normal atmosphere: large radical and formaldehyde concentrations predicted, Science, 173, 141–143, https://doi.org/10.1126/science.173.3992.141, 1971.
Li, R., Palm, B. B., Ortega, A. M., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., de Gouw, J., and Jimenez, J. L.: Modeling the radical chemistry in an Oxidation Flow Reactor (OFR): radical formation and recycling, sensitivities, and OH exposure estimation equation, J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Liu, X., Day, D. A., Krechmer, J. E., Brown, W., Peng, Z., Ziemann, P. J., and Jimenez, J. L.: Direct measurements of semi-volatile organic compound dynamics show near-unity mass accommodation coefficients for diverse aerosols, Communications Chemistry, 2, 98, https://doi.org/10.1038/s42004-019-0200-x, 2019.
Lu, K. D., Hofzumahaus, A., Holland, F., Bohn, B., Brauers, T., Fuchs, H., Hu, M., Häseler, R., Kita, K., Kondo, Y., Li, X., Lou, S. R., Oebel, A., Shao, M., Zeng, L. M., Wahner, A., Zhu, T., Zhang, Y. H., and Rohrer, F.: Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006, Atmos. Chem. Phys., 13, 1057–1080, https://doi.org/10.5194/acp-13-1057-2013, 2013.
Madronich, S. and Flocke, S.: The Role of Solar Radiation in Atmospheric Chemistry, in: Environmental Photochemistry. The Handbook of Environmental Chemistry (Reactions and Processes), vol 2/2L, edited by: Boule, P., Springer, Berlin, Heidelberg, pp. 1–26, 1999.
Mao, J., Ren, X., Brune, W. H., Olson, J. R., Crawford, J. H., Fried, A., Huey, L. G., Cohen, R. C., Heikes, B., Singh, H. B., Blake, D. R., Sachse, G. W., Diskin, G. S., Hall, S. R., and Shetter, R. E.: Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 9, 163–173, https://doi.org/10.5194/acp-9-163-2009, 2009.
Matsunaga, A. and Ziemann, P. J.: Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements, Aerosol Sci. Tech., 44, 881–892, https://doi.org/10.1080/02786826.2010.501044, 2010.
Mouchel-Vallon, C., Lee-Taylor, J., Hodzic, A., Artaxo, P., Aumont, B., Camredon, M., Gurarie, D., Jimenez, J.-L., Lenschow, D. H., Martin, S. T., Nascimento, J., Orlando, J. J., Palm, B. B., Shilling, J. E., Shrivastava, M., and Madronich, S.: Exploration of oxidative chemistry and secondary organic aerosol formation in the Amazon during the wet season: explicit modeling of the Manaus urban plume with GECKO-A, Atmos. Chem. Phys., 20, 5995–6014, https://doi.org/10.5194/acp-20-5995-2020, 2020.
Nakashima, Y., Tsurumaru, H., Imamura, T., Bejan, I., Wenger, J. C., and Kajii, Y.: Total OH reactivity measurements in laboratory studies of the photooxidation of isoprene, Atmos. Environ., 62, 243–247, https://doi.org/10.1016/j.atmosenv.2012.08.033, 2012.
Nannoolal, Y., Rarey, J., and Ramjugernath, D.: Estimation of pure component properties part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contribution and group interactions, Fluid Phase Equilibr., 269, 117–133, https://doi.org/10.1016/j.fluid.2008.04.020, 2008.
Nehr, S., Bohn, B., Dorn, H.-P., Fuchs, H., Häseler, R., Hofzumahaus, A., Li, X., Rohrer, F., Tillmann, R., and Wahner, A.: Atmospheric photochemistry of aromatic hydrocarbons: OH budgets during SAPHIR chamber experiments, Atmos. Chem. Phys., 14, 6941–6952, https://doi.org/10.5194/acp-14-6941-2014, 2014.
Nel, A.: Air Pollution-Related Illness: Effects of Particles, Science, 308, 804–806, https://doi.org/10.1126/science.1108752, 2005.
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, 2014.
Nölscher, A. C., Butler, T., Auld, J., Veres, P., Muñoz, A., Taraborrelli, D., Vereecken, L., Lelieveld, J., and Williams, J.: Using total OH reactivity to assess isoprene photooxidation via measurement and model, Atmos. Environ., 89, 453–463, https://doi.org/10.1016/j.atmosenv.2014.02.024, 2014.
Nölscher, A. C., Yañez-Serrano, A. M., Wolff, S., de Araujo, A. C., Lavrič, J. V., Kesselmeier, J., and Williams, J.: Unexpected seasonality in quantity and composition of Amazon rainforest air reactivity, Nat. Commun., 7, 10383, https://doi.org/10.1038/ncomms10383, 2016.
Novelli, A., Kaminski, M., Rolletter, M., Acir, I.-H., Bohn, B., Dorn, H.-P., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Holland, F., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Evaluation of OH and HO2 concentrations and their budgets during photooxidation of 2-methyl-3-butene-2-ol (MBO) in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 18, 11409–11422, https://doi.org/10.5194/acp-18-11409-2018, 2018.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Peng, Z. and Jimenez, J. L.: Modeling of the chemistry in oxidation flow reactors with high initial NO, Atmos. Chem. Phys., 17, 11991–12010, https://doi.org/10.5194/acp-17-11991-2017, 2017.
Peng, Z. and Jimenez, J. L.: KinSim: A Research-Grade, User-Friendly, Visual Kinetics Simulator for Chemical-Kinetics and Environmental-Chemistry Teaching, J. Chem. Educ., 96, 806–811, https://doi.org/10.1021/acs.jchemed.9b00033, 2019.
Peng, Z. and Jimenez, J. L.: Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 49, 2570–2616, https://doi.org/10.1039/C9CS00766K, 2020.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Peng, Z., Palm, B. B., Day, D. A., Talukdar, R. K., Hu, W., Lambe, A. T., Brune, W. H., and Jimenez, J. L.: Model Evaluation of New Techniques for Maintaining High-NO Conditions in Oxidation Flow Reactors for the Study of OH-Initiated Atmospheric Chemistry, ACS Earth Space Chem., 2, 72–86, https://doi.org/10.1021/acsearthspacechem.7b00070, 2018.
Peng, Z., Lee-Taylor, J., Orlando, J. J., Tyndall, G. S., and Jimenez, J. L.: Organic peroxy radical chemistry in oxidation flow reactors and environmental chambers and their atmospheric relevance, Atmos. Chem. Phys., 19, 813–834, https://doi.org/10.5194/acp-19-813-2019, 2019.
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005.
Sato, K., Nakashima, Y., Morino, Y., Imamura, T., Kurokawa, J., and Kajii, Y.: Total OH reactivity measurements for the OH-initiated oxidation of aromatic hydrocarbons in the presence of NOx, Atmos. Environ., 171, 272–278, https://doi.org/10.1016/j.atmosenv.2017.10.036, 2017.
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017.
Siese, M., Becker, K. H., Brockmann, K. J., Geiger, H., Hofzumahaus, A., Holland, F., Mihelcic, D., and Wirtz, K.: Direct measurement of OH radicals from ozonolysis of selected alkenes: a EUPHORE simulation chamber study, Environ. Sci. Technol., 35, 4660–4667, 2001.
Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M.: Climate Change 2013 – The Physical Science Basis, edited by: Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2014.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348, https://doi.org/10.1039/c2cs35140d, 2012.
Turro, N. J., Ramamurthy, V., and Scaiano, J. C.: Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, CA, USA, available at: https://uscibooks.aip.org/books/principles-of-molecular-photochemistry-an-introduction/ (last access: 25 January 2021), 2009.
US EPA: Estimation Programs Interface SuiteTM, available at: https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface (last access: 1 July 2021), 2012.
Valorso, R., Aumont, B., Camredon, M., Raventos-Duran, T., Mouchel-Vallon, C., Ng, N. L., Seinfeld, J. H., Lee-Taylor, J., and Madronich, S.: Explicit modelling of SOA formation from α-pinene photooxidation: sensitivity to vapour pressure estimation, Atmos. Chem. Phys., 11, 6895–6910, https://doi.org/10.5194/acp-11-6895-2011, 2011.
Verwer, J. G.: Gauss–Seidel Iteration for Stiff ODES from Chemical Kinetics, SIAM J. Sci. Comput., 15, 1243–1250, https://doi.org/10.1137/0915076, 1994.
Verwer, J. G., Blom, J. G., van Loon, M., and Spee, E. J.: A comparison of stiff ODE solvers for atmospheric chemistry problems, Atmos. Environ., 30, 49–58, https://doi.org/10.1016/1352-2310(95)00283-5, 1996.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Whalley, L. K., Stone, D., Bandy, B., Dunmore, R., Hamilton, J. F., Hopkins, J., Lee, J. D., Lewis, A. C., and Heard, D. E.: Atmospheric OH reactivity in central London: observations, model predictions and estimates of in situ ozone production, Atmos. Chem. Phys., 16, 2109–2122, https://doi.org/10.5194/acp-16-2109-2016, 2016.
Williams, J. and Brune, W.: A roadmap for OH reactivity research, Atmos. Environ., 106, 371–372, https://doi.org/10.1016/j.atmosenv.2015.02.017, 2015.
Wofsy, S. C., Apel, E., Blake, D. R., Brock, C. A., Brune, W. H., Bui, T. P., Daube, B. C., Dibb, J. E., Diskin, G. S., Elkiins, J. W., Froyd, K., Hall, S. R., Hanisco, T. F., Huey, L. G., Jimenez, J. L., McKain, K., Montzka, S. A., Ryerson, T. B., Schwarz, J. P., Stephens, B. B., Weinzierl, B., and Wennberg, P.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, Oak Ridge National Laboratory Distributed Active Archive Center, Oak Ridge, Tennessee, USA, 2021.
Yang, Y., Shao, M., Wang, X., Nölscher, A. C., Kessel, S., Guenther, A., and Williams, J.: Towards a quantitative understanding of total OH reactivity: A review, Atmos. Environ., 134, 147–161, https://doi.org/10.1016/j.atmosenv.2016.03.010, 2016.
Zannoni, N., Gros, V., Lanza, M., Sarda, R., Bonsang, B., Kalogridis, C., Preunkert, S., Legrand, M., Jambert, C., Boissard, C., and Lathiere, J.: OH reactivity and concentrations of biogenic volatile organic compounds in a Mediterranean forest of downy oak trees, Atmos. Chem. Phys., 16, 1619–1636, https://doi.org/10.5194/acp-16-1619-2016, 2016.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.
- Abstract
- Introduction
- Methods
- Results and discussions
- Summary and conclusions
- Appendix A: The effect on OH reactivity of non-OH reactions in an OH-driven reaction chain
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Methods
- Results and discussions
- Summary and conclusions
- Appendix A: The effect on OH reactivity of non-OH reactions in an OH-driven reaction chain
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement