the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Feb 2021
| 03 Feb 2021
Seasonal variation and origins of volatile organic compounds observed during 2 years at a western Mediterranean remote background site (Ersa, Cape Corsica)
Cécile Debevec
Stéphane Sauvage
Valérie Gros
Thérèse Salameh
Jean Sciare
François Dulac
Nadine Locoge
An original time series of about 300 atmospheric measurements of a wide range of volatile organic compounds (VOCs) was obtained at a remote Mediterranean station on the northern tip of Corsica (Ersa, France) over 25 months from June 2012 to June 2014. This study presents the seasonal variabilities of 35 selected VOCs and their various associated sources. The VOC abundance was largely dominated by oxygenated VOCs (OVOCs) along with primary anthropogenic VOCs with a long lifetime in the atmosphere. VOC temporal variations were then examined. Primarily of local origin, biogenic VOCs exhibited notable seasonal and interannual variations, related to temperature and solar radiation. Anthropogenic compounds showed increased concentrations in winter (JFM months) followed by a decrease in spring/summer (AMJ/JAS months) and higher winter concentration levels in 2013 than in 2014 by up to 0.3 µg m−3 in the cases of propane, acetylene and benzene. OVOC concentrations were generally high in summertime, mainly due to secondary anthropogenic/biogenic and primary biogenic sources, whereas their lower concentrations during autumn and winter were potentially more influenced by primary/secondary anthropogenic sources. Moreover, an apportionment factorial analysis was applied to a database comprising a selection of 14 individual or grouped VOCs by means of the positive matrix factorization (PMF) technique. A PMF five-factor solution was taken on. It includes an anthropogenic factor (which contributed 39 % to the total concentration of the VOCs selected in the PMF analysis) connected to the regional background pollution, three other anthropogenic factors (namely short-lived anthropogenic sources, evaporative sources, and long-lived combustion sources, which together accounted for 57 %) originating from either nearby or more distant emission areas (such as Italy and south of France), and a local biogenic source (4 %). Variations in these main sources impacting VOC concentrations observed at the Ersa station were also investigated at seasonal and interannual scales. In spring and summer, VOC concentrations observed at Ersa were the lowest in the 2-year period, despite higher biogenic source contributions. During these seasons, anthropogenic sources advected to Ersa were largely influenced by chemical transformations and vertical dispersion phenomena and were mainly of regional origins. During autumn and winter, anthropogenic sources showed higher contributions when European air masses were advected to Ersa and could be associated with potential emission areas located in Italy and possibly more distant ones in central Europe. Higher VOC winter concentrations in 2013 than in 2014 could be related to contribution variations in anthropogenic sources probably governed by their emission strength with external parameters, i.e. weaker dispersion phenomena and the pollutant depletion. High-frequency observations collected during several intensive field campaigns conducted at Ersa during the three summers 2012–2014 confirmed findings drawn from bi-weekly samples of the 2-year period in terms of summer concentration levels and source apportionment. However, they also suggested that higher sampling frequency and temporal resolution, in particular to observe VOC concentration variations during the daily cycle, would have been necessary to confirm the deconvolution of the different anthropogenic sources identified following the PMF approach. Finally, comparisons of the 25 months of Ersa observations with VOC measurements conducted at 17 other European monitoring stations highlighted the representativeness of the Ersa station for monitoring seasonal variations in VOC regional pollution impacting continental Europe. Nevertheless, VOC winter concentration levels can significantly vary between sites, pointing out spatial variations in anthropogenic source contributions. As a result, Ersa concentration variations in winter were more representative of VOC regional pollution impacting central Europe. Moreover, interannual and spatial variations in VOC winter concentration levels were significantly impacted by synoptic phenomena influencing meteorological conditions observed in continental Europe, suggesting that short observation periods may reflect the variability of the identified parameters under the specific meteorological conditions of the study period.
- Article
(7886 KB) - Full-text XML
-
Supplement
(5675 KB) - BibTeX
- EndNote





The main trace pollutants in the atmosphere encompass a multitude of volatile organic compounds (VOCs), with lifetimes varying from minutes to months (e.g. Atkinson, 2000). Their distribution is principally due to (i) multiple natural and anthropogenic sources, which release VOCs directly into the atmosphere. At a global scale, natural emissions are quantitatively larger than anthropogenic ones (Guenther et al., 2000), and the largest natural source is considered to be the vegetation (Finlayson-Pitts and Pitts, 2000; Guenther et al., 2000, 2006). In urban areas, numerous anthropogenic sources can abundantly emit various VOCs (Friedrich and Obermeier, 1999). Once in the atmosphere, VOC temporal and spatial variabilities are notably influenced by (ii) mixing processes along with (iii) removal processes or chemical transformations (Atkinson, 2000; Atkinson and Arey, 2003). Accordingly, with a view to thoroughly characterizing VOC sources, it is meaningful to examine their chemical composition and identify the factors controlling their variations at different timescales.
VOC regional distributions change considerably as a result of various confounding factors, namely the emission strength of numerous potential sources, diverse atmospheric lifetimes and removal mechanisms, transport processes and fluctuations in meteorological conditions. Therefore, these elements underline the necessity of carrying out long-term VOC measurements. Growing efforts are currently being made to conduct European background measurements over several seasons (e.g. Seco et al., 2011), 1 year (such as Helmig et al., 2008; Legreid et al., 2008) and even several years (Solberg et al., 1996, 2001, and Tørseth et al., 2012, at several European sites; Hakola et al., 2006, and Hellén et al., 2015, in Scandinavia; Dollard et al., 2007; Grant et al., 2011, and Malley et al., 2015, in the United Kingdom; Borbon et al., 2004; Sauvage et al., 2009, and Waked et al., 2016, in France; Plass-Dülmer et al., 2002, in Germany; Navazo et al., 2008, in the Iberian Peninsula; Lo Vullo et al., 2016 in Italy). These research studies principally explored the effectiveness of emission regulations and links between tropospheric ozone production and VOC concentration levels. They also assessed seasonal variations and regional distributions of VOC concentrations. Nonetheless, investigations of principal factors governing temporal and spatial variations in VOC concentration levels in the European background atmosphere remain scarce. However, the consideration of the influence of (i) source emission strength variations (built upon a factorial analysis – e.g. Lanz et al., 2009; Lo Vullo et al., 2016), (ii) long-range transport of pollution (e.g. by the examination of air-mass trajectories combined with concentrations measured at a study site; Sauvage et al., 2009) and (iii) fluctuations in meteorological conditions (which are prone to dispersing the pollutants over long distances by convective and advective transport) can supply relevant information to deal more in depth with the evaluation of seasonal variations and regional distribution of VOC concentrations in the European background atmosphere.
Particulate and gaseous pollutants detrimentally affect the Mediterranean atmosphere. Accordingly, they are prone to increasing aerosol and/or ozone concentration levels in the Mediterranean, regularly higher compared to most regions of continental Europe, and primarily during summer (Doche et al., 2014; Nabat et al., 2013; Safieddine et al., 2014). The Mediterranean region is known to be a noteworthy climate change “hotspot” which is expected to go through severe warming and drying in the 21st century (Giorgi, 2006; Kopf, 2010; Lelieveld et al., 2014). As a consequence, this can have serious consequences for the release of VOCs from biogenic and anthropogenic sources along with their fate in the atmosphere, with uncertain predicted impacts (Colette et al., 2012, 2013; Jaidan et al., 2018). Actually, the examination of air composition, concentration levels and trends in the Mediterranean region continues to be challenging, primarily due to the lack of extensive in situ observations. Given this context, as part of the multidisciplinary regional research programme MISTRALS (Mediterranean Integrated Studies at Regional and Local Scales; http://mistrals-home.org/, last access: 11 October 2020), the project ChArMEx (the Chemistry-Aerosol Mediterranean Experiment; Dulac, 2014) aims at assessing the current and future state of the atmospheric environment in the Mediterranean along with examining its repercussions for the regional climate, air quality and marine biogeochemistry. Within the framework of ChArMEx, several observation periods were conducted at the Ersa station, a remote site considered to be representative of the north-western Mediterranean basin, in order to explain variations in VOC concentrations affecting the western Mediterranean atmosphere. Michoud et al. (2017) characterized the variations in VOC concentrations observed at Ersa in summer 2013 (from 15 July to 5 August 2013) by identifying and examining their sources.
In this article, we have presented and discussed the factors controlling seasonal and interannual variations of a selection of VOCs observed at the Ersa station over more than 2 years (from early June 2012 to late June 2014). To this end, this study describes (i) the concentration levels of the targeted VOCs, (ii) their temporal variations at seasonal and interannual scales, (iii) the identification and characteristics of their main sources by statistical modelling, (iv) the evaluation of their source contributions on seasonal bases, together with (v) the representativeness of the Ersa station in terms of seasonal variations in VOC concentrations impacting continental Europe.
2.1 Study site
Located in the north-western part of the Mediterranean Sea, Corsica is a French territory situated 11 km north of the Sardinian coasts, 90 km east of Tuscany (Italy) and 170 km south of the French Riviera (France). The fourth largest Mediterranean island, its land corresponds to an area of 8681 km2 encompassed by around 1000 km of coastline (Encyclopædia Britannica, 2018). Corsica contrasts to other Mediterranean islands due to the importance of its forest cover (about a fifth of the island).
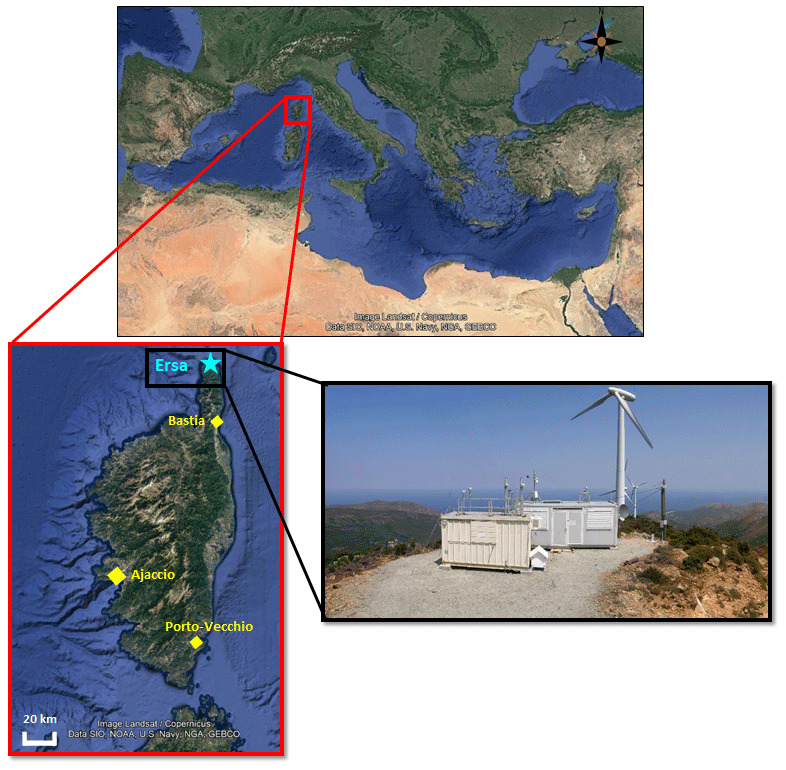
Figure 1Maps of the Mediterranean region and Corsica (source Google Earth) and view of the sampling station. (a) Position of Corsica in the Mediterranean region. (b) The sampling site and major Corsican agglomerations are displayed as a blue star and yellow diamonds, respectively. (c) Picture of the sampling site, during the 2-year observation period. Maps provided by Google Earth Pro software (v.7.3.3; image Landsat/Copernicus; data SIO, NOAA, U.S. Navy, NGA, GEBCO; © Google Earth).
Within the framework of the ChArMEx project, an enhanced observation period was set up at a ground-based station in the north of Corsica (Ersa; 42.969∘ N, 9.380∘ E) over 25 months, from early June 2012 to late June 2014. The aim was to provide a high-quality controlled climatically relevant gas/aerosol database following the recommendations and criteria of international atmospheric chemistry networks, i.e. the Aerosol, Clouds and Trace gases Research Infrastructure (ACTRIS – https://www.actris.eu/, last access: 11 October 2020), the European Monitoring and Evaluation Program (EMEP – http://www.emep.int/, last access: 11 October 2020; Tørseth et al., 2012), and the Global Atmosphere Watch of the World Meteorological Organization (WMO-GAW – http://www.wmo.int/pages/prog/arep/gaw/gaw_home_en.html, last access: 11 October 2020). The Ersa remote site is part of the Corsican Observatory for Research and Studies on Climate and Atmosphere-ocean environment (CORSiCA – https://corsica.obs-mip.fr/, last access: 11 October 2020; Lambert et al., 2011) and is located on the highest point of a ridge equipped with windmills (see the orographic description of the surroundings in Cholakian et al., 2018), at an altitude of 533 m above sea level (a.s.l.). Given its position in the north of the 40 km long Cape Corsica peninsula (Fig. 1), the Mediterranean Sea is clearly visible from the sampling site on the western, northern, and eastern sides (2.5–6 km from the sea; see also the figure presented in Michoud et al., 2017). The station was initially set up in order to monitor and examine pollution advected to Ersa by air masses transported over the Mediterranean and originating from the Marseille–Fos–Berre region (France; Cachier et al., 2005), the Rhone Valley (France), and the Po Valley (Italy; Royer et al., 2010), namely largely industrialized regions. The Ersa station is about 30 km north of Bastia (Fig. 1), the second largest Corsican city (44121 inhabitants; census 2012) and the main harbour. An international airport (Bastia-Poretta) is located 16 km further south of Bastia city centre. More than 2 million passengers transited in Corsica via Bastia during the tourist season (May–September) in 2013 (ORT Corse, 2013). However, as the Cape Corsican peninsula benefits in the south from a mountain range (peaking between 1000 and 1500 m a.s.l.) acting as a natural barrier, the sampling site is therefore not affected by transported pollution originating from Bastia. Only small rural villages and a small local fishing harbour (Centuri) are found within 5 km of the measurement site. Additionally, the Ersa station is accessible by a dead-end road serving only the windmill site, surrounded by vegetation made up of Mediterranean maquis, a shrubland biome characteristically consisting of densely growing evergreen shrubs, and also roamed by a herd of goats from a nearby farm. Some forests (78 % of holm oaks, with some cork oaks and chestnuts) are also located nearby, thus ensuring that local anthropogenic pollution does not contaminate in situ observations. As a result, the Ersa station can be characterized as a remote background Mediterranean site.
2.2 Experimental set-up
2.2.1 VOC measurements
During a study period of 2 years, non-methane hydrocarbons (NMHCs) and OVOCs (carbonyl compounds) were measured, routinely employing complementary offline methods. Four-hour-integrated (09:00–13:00 or 12:00–16:00 UTC) ambient air samples were collected bi-weekly (every Monday and Thursday) into steel canisters and on sorbent cartridges. The inlets were roughly 1.5 m above the roof of a container mainly housing trace gas analysers. Table 1 describes VOC measurements set up throughout the observation period and Fig. S1 specifies their collection periods.
Table 1Technical details of the set-up for VOC measurements during the field campaign from June 2012 to June 2014. Air samples were collected bi-weekly (every Monday and Thursday) at Ersa from 09:00 to 13:00 UTC (from early November 2012 to late December 2012 and from early November 2013 to late June 2014) or from 12:00 to 16:00 UTC (from early June 2012 to late October 2012 and from early January 2013 to late October 2013). VOCs are explicitly listed in Sect. S1 of the Supplement.

As generally realized in the EMEP network, 24 C2–C9 NMHCs were collected into Silcosteel canisters of a volume of 6 L, conforming to the TO-14 technique, which is considered adequate for the measurement of many non-polar VOCs (US-EPA, 1997); 152 air samples were realized with a homemade device (PRECOV) for sampling air at a steady flow rate regulated to 24 mL min−1 by canisters previously placed under vacuum. NMHC analysis was performed by a gas chromatograph coupled with a flame ionization detector (GC-FID) within 3 weeks following the sampling. Separation was performed by a system of dual-capillary columns supplied with a switching device: the first column was a CP Sil5CB (50 m × 0.25 mm × 1 µm), suitable for the elution of VOCs from six to nine carbon atoms, and the other one was a plot Al2O3 ∕ Na2SO4 (50 m × 0.32 mm × 5 µm), in order to effectively elute VOCs from two to five carbon atoms. Four main steps constituted the quality assurance/quality control programme: (i) the implementation of standard operating procedures, (ii) canister cleaning and certification (blank levels < 0.02 ppb), (iii) regular intercomparison exercises and (iv) sampling tests carried out in field conditions and concomitant with in situ measurements (Sauvage et al., 2009).
About 150 air samples were gathered using sorbent cartridges (63 air samples on multi-sorbent cartridges and 89 additional ones on 2,4-dinitrophenylhydrazine – DNPH – cartridges), by means of an automatic clean room sampling system (ACROSS, TERA Environment, Crolles, France). C1–C16 VOCs were collected via a 0.635 cm diameter 3 m long PFA line. They were then trapped into one of the two cartridge types: a multi-sorbent one consisting of carbopack C (200 mg) and carbopack B (200 mg; marketed under the name of carbotrap 202 by Perkin-Elmer, Wellesley, Massachusetts, USA) or a Sep-Pak DNPH-Silica one (proposed by Waters Corporation, Milford, Massachusetts, USA). These offline techniques are further characterized in Detournay et al. (2011), and their satisfying use in situ has already been discussed by Detournay et al. (2013) and Ait-Helal et al. (2014). Succinctly here, the sampling of 44 C5–C16 NMHCs, comprising alkanes, alkenes, aromatic compounds and six monoterpenes, as well as six C6–C11 n-aldehydes, was conducted at a flow rate fixed at 200 mL min−1 and using the multi-sorbent cartridges. These latter ones were preliminary prepared by means of a RTA oven (French abbreviation for “régénérateur d'adsorbant thermique” – manufactured by TERA Environment, Crolles, France) in order to condition them during 24 h with purified air heated to 250 ∘C and at a flow rate regulated at 10 mL min−1. Concomitantly, 15 additional C1–C8 carbonyl compounds were collected at a flow rate fixed at 1.5 L min−1 using the DNPH cartridges. During the field campaign, several ozone scrubbers were successively inserted into the sampling lines in order to limit any eventual ozonolysis of the measured VOCs: a MnO2 ozone scrubber was retained for the multi-sorbent cartridges, while KI ozone scrubber was placed upstream of the DNPH cartridges. Moreover, stainless-steel particle filters of 2 µm diameter porosity (Swagelok) were installed in order to prevent particle sampling. Then, VOC samples were transferred to the laboratory to be analysed within 6 weeks of their collection using a GC-FID (for the multi-adsorbent cartridges) or by a high-performance liquid chromatograph connected to an ultraviolet detector (HPLC-UV; for the DNPH cartridges).
The reproducibility of each analytical instrument was frequently checked by analysing a standard and examining results by plotting them on a control chart realized for each compound. The VOC detection limit was determined as 3 times the standard deviation of the blank variation. Detection limits in this study were all below 0.05 µg m−3 for the steel canisters and the DNPH cartridges and 0.01 µg m−3 for the multi-sorbent cartridges. The uncertainties for each species were evaluated respecting the ACTRIS-2 guidelines for the uncertainty evaluation (Reimann et al., 2018), considering precision, detection limit and systematic errors in the measurements. Relative uncertainties assessed in this study ranged from 7 % to 43 % for the steel canisters, from 7 % to 73 % for the multi-sorbent cartridges and from 6 % to 41 % for the DNPH cartridges. Finally, the VOC dataset was validated following the ACTRIS protocol (Reimann et al., 2018).
Among the 71 different VOCs monitored at Ersa during the observation period, 35 VOCs were finally selected in this study following the methodology described in Sect. S1 of the Supplement.
2.2.2 Ancillary measurements
Other trace gases (CO and O3) and meteorological parameters were ancillary monitored at the Ersa site during the observation period. CO was measured from 22 November 2012 to 16 December 2013 by a cavity ring-down spectroscopy analyser (G2401; Picarro, Santa Clara, California, USA) at a time resolution of 5 min. O3 was measured from 31 May 2012 to 26 December 2013 using an ultraviolet absorption analyser (TEI 49i manufactured by Thermo Environmental Instruments Inc., Waltham, Massachusetts, USA) at a time resolution of 5 min. Meteorological parameters (temperature, pressure, relative humidity, wind speed, wind direction and total – direct and diffuse – solar radiation) were measured every minute from 8 June to 14 August 2012 and every 5 min from 15 August 2012 to 11 July 2014, with a weather station (CR1000 manufactured by Campbell Scientific Europe, Antony, France) placed at approximately 1.5 m above an adjacent container roof to the one which housed trace gas instruments. Note that ancillary trace gas and meteorological parameter results presented in this study are 4 h averages concurrent with VOC sampling periods (see Fig. S1).
2.3 Identification and contribution of major sources of VOCs
In order to characterize NMHC concentrations measured at Ersa with steel canisters (the reasons for this VOC selection are presented in Sect. S2 of the Supplement), we apportioned them within their sources in this study using the positive matrix factorization approach (PMF; Paatero, 1997; Paatero and Tapper, 1994). The PMF mathematical theory has already been presented in Debevec et al. (2017) and is therefore mentioned in Sect. S2. We used the PMF version 5.0, an enhanced tool developed by the Environmental Protection Agency (EPA) and including the multilinear engine program version 2 (ME-2; Paatero, 1999), and followed the guidance on its use (Norris et al., 2014). Using NMHC inputs composed of 152 atmospheric data points of 14 variables (13 single primary NMHCs and another 1 resulting from the grouping of C8 aromatic compounds) and following the methodology presented in Sect. S2, a five-factor PMF solution has been selected in this study.
2.4 Geographical origins of VOC sources
2.4.1 Classification of air-mass origins
In order to identify and classify air-mass origins, we analysed back-trajectories calculated by the online version of the HYSPLIT Lagrangian model (the Hybrid Single Particle Lagrangian Integrated Trajectory Model developed by the National Oceanic and Atmospheric Administration – NOAA – Air Resources Laboratory; Draxler and Hess, 1998; Stein et al., 2015) using Ersa as the receptor site (arrival altitude: 600 m a.s.l.). For each 4 h-atmospheric data point of the field campaign used for the factorial analysis, five back-trajectories of 48 h were computed using GDAS one-degree resolution meteorological data, in order to follow the same methodology as Michoud et al. (2017). The first back-trajectory of a set corresponds to the hour when the air sampling was initiated (i.e. 09:00 or 12:00 UTC – see Table 1) and the four other ones were calculated every following hour. The time step between each point along the back-trajectories was fixed at 1 h.
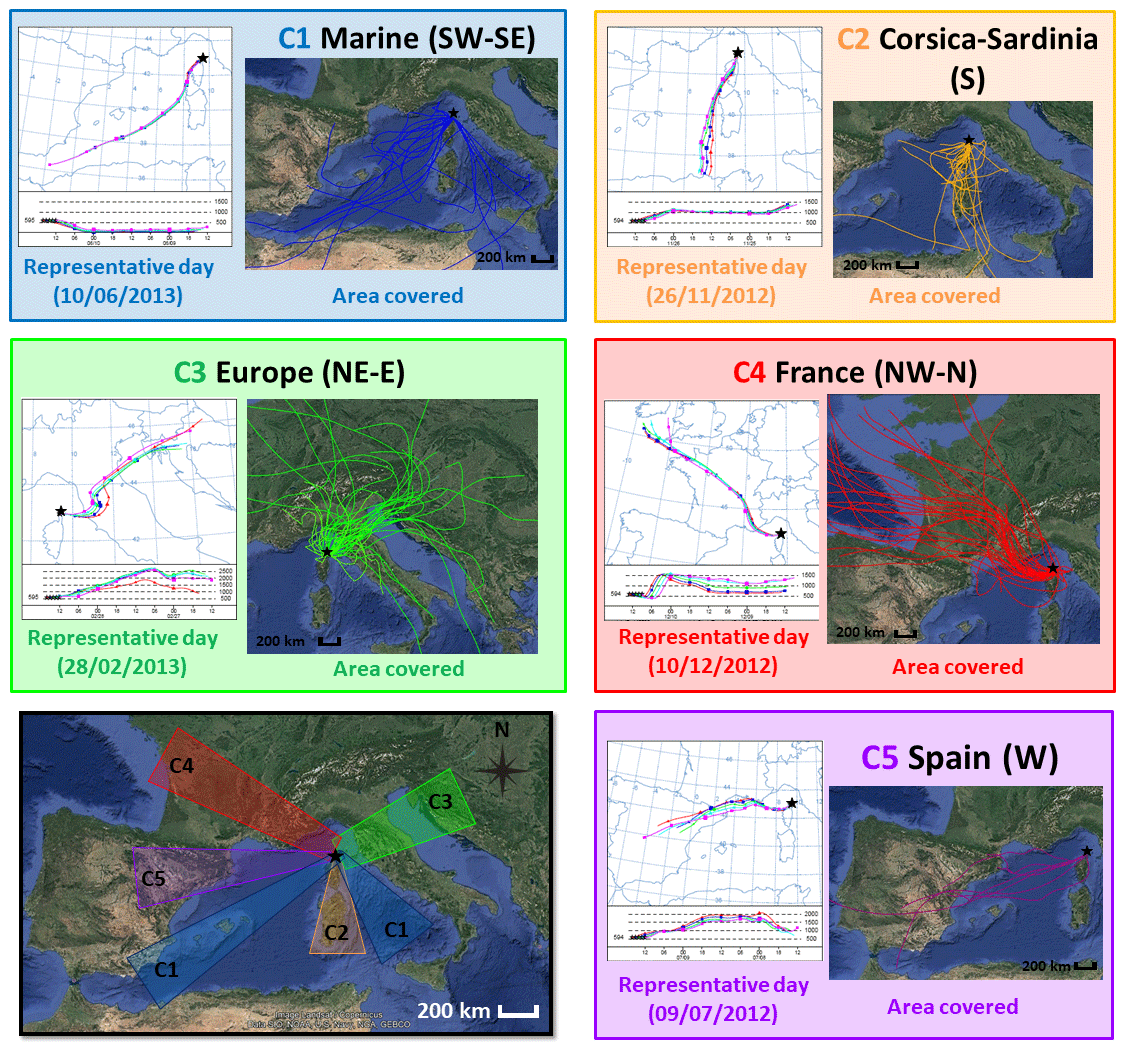
Figure 2Classification of air masses which impacted the Ersa site during the 2-year observation period as a function of their trajectory. Back-trajectories simulated with the HYSPLIT Lagrangian model were classified into five clusters: Marine (cluster 1 – wind sectors SW and SE), Corsica–Sardinia (cluster 2 – S), Europe (cluster 3 – NE–E), France (cluster 4 – NW–N) and Spain (cluster 5 – W). These five clusters were illustrated by example maps with five trajectories (interval of 1 h between each, time of arrival indicated by different colours of trajectory, the Ersa station represented by a black star) for 5 d that are representative of an isolated cluster. Finally, areas covered by back-trajectories of each cluster are also indicated. Maps provided by Google Earth Pro software (v.7.3.3; image Landsat/Copernicus; data SIO, NOAA, U.S. Navy, NGA, GEBCO; © Google Earth).
Then, back-trajectories were visually classified. Having several back-trajectories per sample allowed us to check whether air masses transported to the station over 4 h were of the same origin. Samples associated with air masses showing contrasted trajectories (e.g. due to a transitory state between two different origins) were classified as being of mixed origins (9 % of the air masses) and discarded from this study. Remaining air masses were then manually classified into five trajectory clusters (marine, Corsica–Sardinia, Europe, France and Spain – Fig. 2 and Table 2) depending on their pathway when they reached the Ersa station, their residence time over each potential source region and the length of their trajectories. Additionally, air masses of each cluster were subdivided depending on their distance travelled during their 48 h course in order to highlight potential more distant sources from local ones. This subdivision is also given in Table 2 in order to pinpoint differences in transport times.
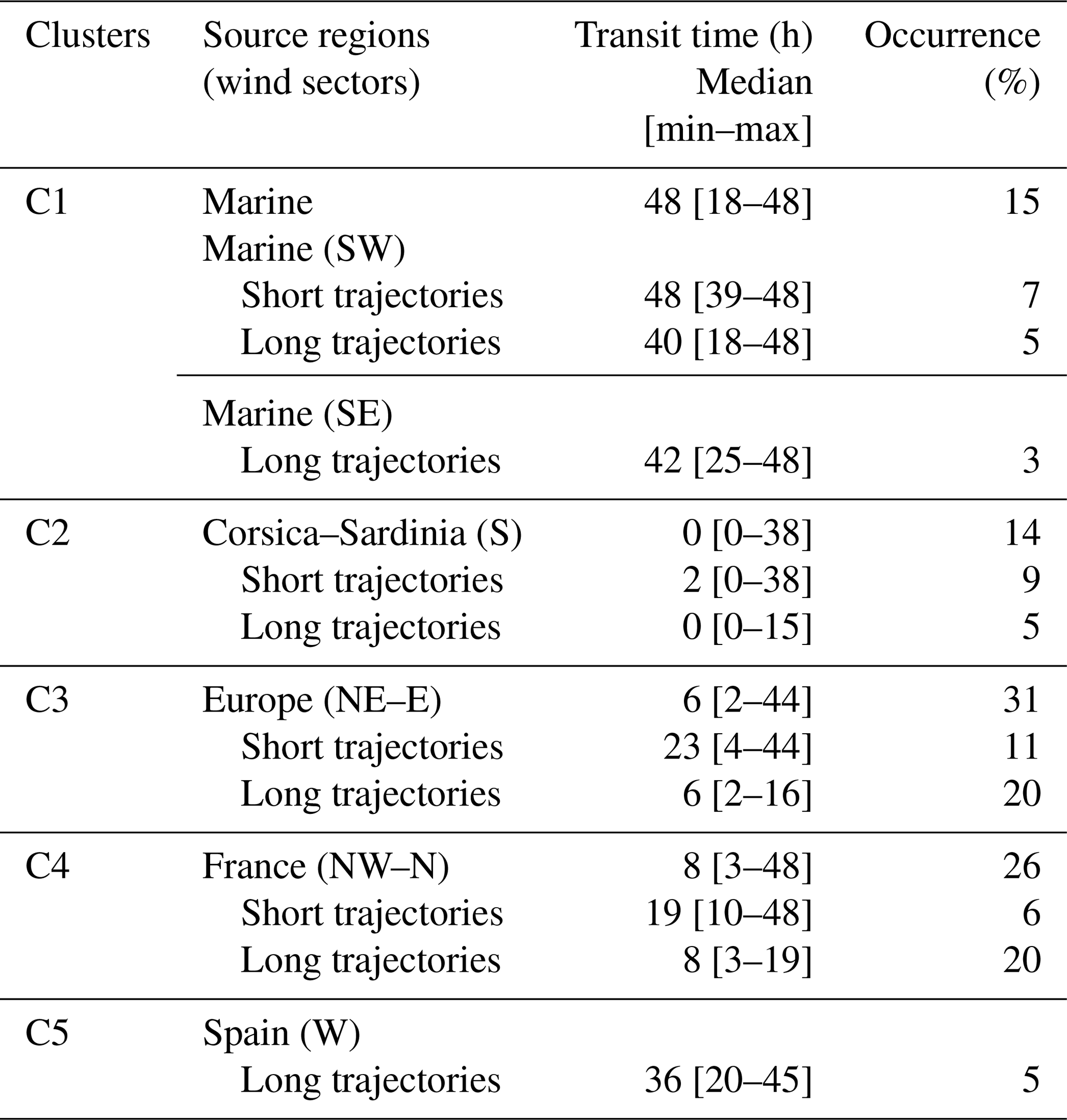
Table 2Back-trajectory clusters for air masses observed at Ersa from June 2012 to June 2014. The transit time (expressed in hours) corresponds to the time spent since the last anthropogenic contamination, i.e. since air masses left continental coasts.
2.4.2 Identification of potential emission areas
PMF source contributions were coupled with back-trajectories in order to investigate potential emission regions contributing to long-distance pollution transport to the Ersa site. To achieve this, the concentration field (CF) statistical method established by Seibert et al. (1994) was chosen in the present study. The CF principle has already been presented in Debevec et al. (2017) and is therefore mentioned in Sect. S3 of the Supplement.
Seventy-two-hour back-trajectories together with meteorological parameters of interest (i.e. precipitation) were retrieved from the GDAS meteorological fields with a PC-based version of the HYSPLIT Lagrangian model (version 4 revised in April 2018), following the same methodology as that used for the 48 h back-trajectories previously presented. The arrival time of trajectories at the Ersa station corresponds to the hour when half of the sampling was carried out (i.e. 11:00 or 14:00 UTC – see Table 1). Note that longer back-trajectories were considered for CF analyses than those for air-mass origin classification (Sect. 2.4.1), in order to be in the same conditions as Michoud et al. (2017) and hence to have comparative results between the two Ersa VOC studies.
CF analyses applied to VOC source contributions were carried out by means of the ZeFir tool (version 3.70; Petit et al., 2017). Back-trajectories were shortened (i.e. the ZeFir tool considered shorter back-trajectories than 72 h) when precipitation higher than 0.1 mm was encountered along the trajectory (Bressi et al., 2014). As done by Michoud et al. (2017), back-trajectories have also been shortened when air-mass altitudes exceeded 1500 m a.s.l. in order to discard biases related to the significant dilution impacting air masses reaching the free troposphere. A better statistical significance of the CF results is commonly considered for grid cells with a high number of crossing trajectory points. As a result, some studies applied an empirical weighing function so as to limit the possible influence of elevated concentrations, which may be observed during occasional episodes (e.g. Bressi et al., 2014; Waked et al., 2014, 2018), on cells with a low number of trajectory points. We initially tried to apply this weighing function in this study. Exploratory tests revealed that CF results with the empirical weighing function only highlighted local contributions, given the number of air masses of this study. The farther a cell was from the Ersa station, the lower was its corresponding nij value (number of points of the total number of back-trajectories contained in the ijth grid cell, Sect. S3 of the Supplement) and the more the weighing function tended toward downweighting the results related to this cell. Therefore, CF results discussed in this study were realized without weighing, and these limitations should be taken into account when examining CF analyses, which are hence considered to be indicative information.
Finally, the spatial coverage of grid cells was set from (9∘ W, 32∘ N) to (27∘ E, 54∘ N), with a grid resolution of 0.3∘ × 0.3∘. Allocated contributions were smoothed following a factor (corresponding to the strength of a Gaussian filter) set to 5 to take into account the uncertainties in the back-trajectory path (Charron et al., 2000).
3.1 Meteorological conditions
Monthly variations in meteorological parameters are depicted in Fig. 3. As the field measurement period covered 2 years (i.e. from June 2012 to June 2014), their interannual variations are also shown in Fig. 3b.
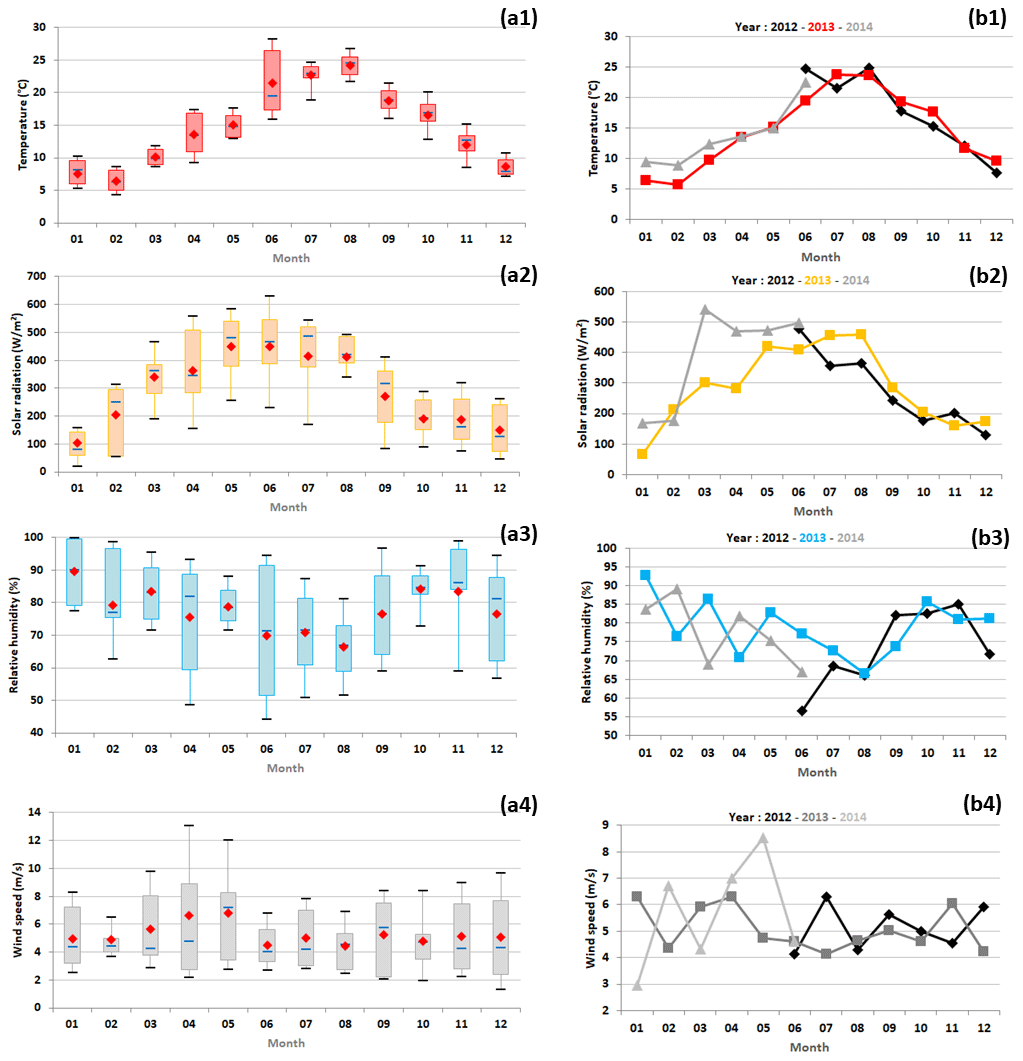
Figure 3(a) Monthly variations in meteorological parameters (temperature expressed in ∘C, global solar radiation in W m−2, relative humidity in % and wind speed in m s−1) represented by box plots; the blue solid line, the red marker, and the box represent the median, the mean, and the interquartile range of the values, respectively. The bottom and top of the box depict the first and third quartiles and the ends of the whiskers correspond to the first and ninth deciles. (b) Their monthly average concentrations as a function of the year. Note that meteorological parameter data used in this study were restricted to periods when VOC measurements were realized.
Air temperatures showed typical seasonal variations, i.e. the highest recorded in summer (i.e. the months of July to September) and the lowest in winter (i.e. the months of January to March). They were globally in the range of normal values over the period 1981–2010 determined by Météo-France (the French national meteorological service; normal values correspond to minimal and maximal mean values determined for Bastia and are available for consultation at http://www.meteofrance.fr/climat-passe-et-futur/climathd, last access: 11 October 2020). Moreover, June temperatures were colder in 2013 than in 2012 and 2014 (mean temperatures of 24.7 ± 5.8, 19.4 ± 4.1 and 22.5 ± 5.4 ∘C in 2012, 2013 and 2014, respectively), which could have influenced biogenic emissions. Winter temperatures were colder in 2013 than in 2014 (mean temperatures of 7.0 ± 4.1 and 9.7 ± 1.5 ∘C in 2013 and 2014, respectively). This finding could be explained by different winter climatic events which affected a large part of continental Europe in 2013 and 2014. On the one hand, the European winter was particularly harsh in 2013, caused by changes in air-flux orientation originally due to the sudden stratospheric warming of the stratospheric polar vortex (Coy and Pawson, 2015). On the other hand, most of the western European countries experienced a mild winter in 2014 characterized by its lack of cold outbreaks and nights and caused by an anomalous atmospheric circulation (Rasmijn et al., 2016; Van Oldenborgh et al., 2015; Watson et al., 2016).
Solar radiation followed typical seasonal variations, with the highest recorded from May to August and the lowest in December and January. Spring (i.e. the months of April to June) solar radiations were higher in 2014 than in 2013 (average values of 371 ± 157 and 478 ± 153 W m−2 in 2013 and 2014, respectively). Summer solar radiations were higher in 2013 than in 2012 (average values of 332 ± 164 and 395 ± 128 W m−2 in 2012 and 2013, respectively). These solar radiation variations may have affected biogenic VOC (BVOC) emissions and photochemical reactions.
Relative humidity followed opposite seasonal variations in temperature and solar radiation. Air in June was drier in 2012 than in 2013 and 2014 (mean relative humidities of 57 ± 15 %, 77 ± 16 % and 67 ± 33 % in 2012, 2013 and 2014, respectively). The wind speed did not show a clear seasonal variation over the 2 years studied. Wind speeds were slightly higher in April and May, which could induce higher dispersion of air pollutants and favour their advection to the Ersa station by the most distant sources.
3.2 Air-mass origins
Occurrences of air-mass origins which influenced Ersa throughout the observation period are indicated in Table 2. The Ersa station was predominantly influenced by continental air masses coming from Europe (corresponding to cluster 3, 31 %), France (cluster 4, 26 %), Corsica–Sardinia (cluster 2, 14 %) and Spain (cluster 5, 5 %) and to a lesser extent by air masses predominantly of marine origin (cluster 1, 15 %). Each of these five clusters is mostly associated with a particular trajectory sector (e.g. south for air masses originating from Corsica and/or Sardinia; see Fig. 2) and is defined by a different transit time from continental coasts. According to Michoud et al. (2017), transit time can be viewed as an indicator of the last time when an air mass could have been enriched in anthropogenic sources (Table 2). Continental air masses spent less time over the sea than marine ones. Transit times of continental air masses over the sea differed depending on how they were categorized. Air masses originating from Corsica–Sardinia, France and Europe spent 0–8 h (median values – Table 2) above the sea before reaching the Ersa station, while the air masses originating from Spain spent about 36 h. These contrasting transit times may denote both distinctive atmospheric processing times for air masses and different oceanic source influences on VOC concentrations observed at the Ersa station.
European and French air masses showed lower transit times over the sea (median values of 6 and 8 h, respectively; Table 2) when their trajectories were categorized as long, compared to short ones (23 and 19 h, respectively). These findings are based on the fact that an air-mass trajectory classified as short has a closer distance between two of its successive trajectory points compared to another one classified as long. Due to the Ersa location in the Mediterranean Sea, air masses with trajectories categorized as long spent longer periods above the sea before reaching the Ersa site. Note that European and French air masses were more frequently characterized by long trajectories (accounting for 20 % of the air masses observed at Ersa during the studied period, for each) than short ones (11 % and 6 %, respectively). Moreover, marine air masses with short and long trajectories both showed long transit times (median values of 40–48 h – Table 2). Corsican–Sardinian air masses were only characterized by long trajectories.
3.3 VOC mixing ratios
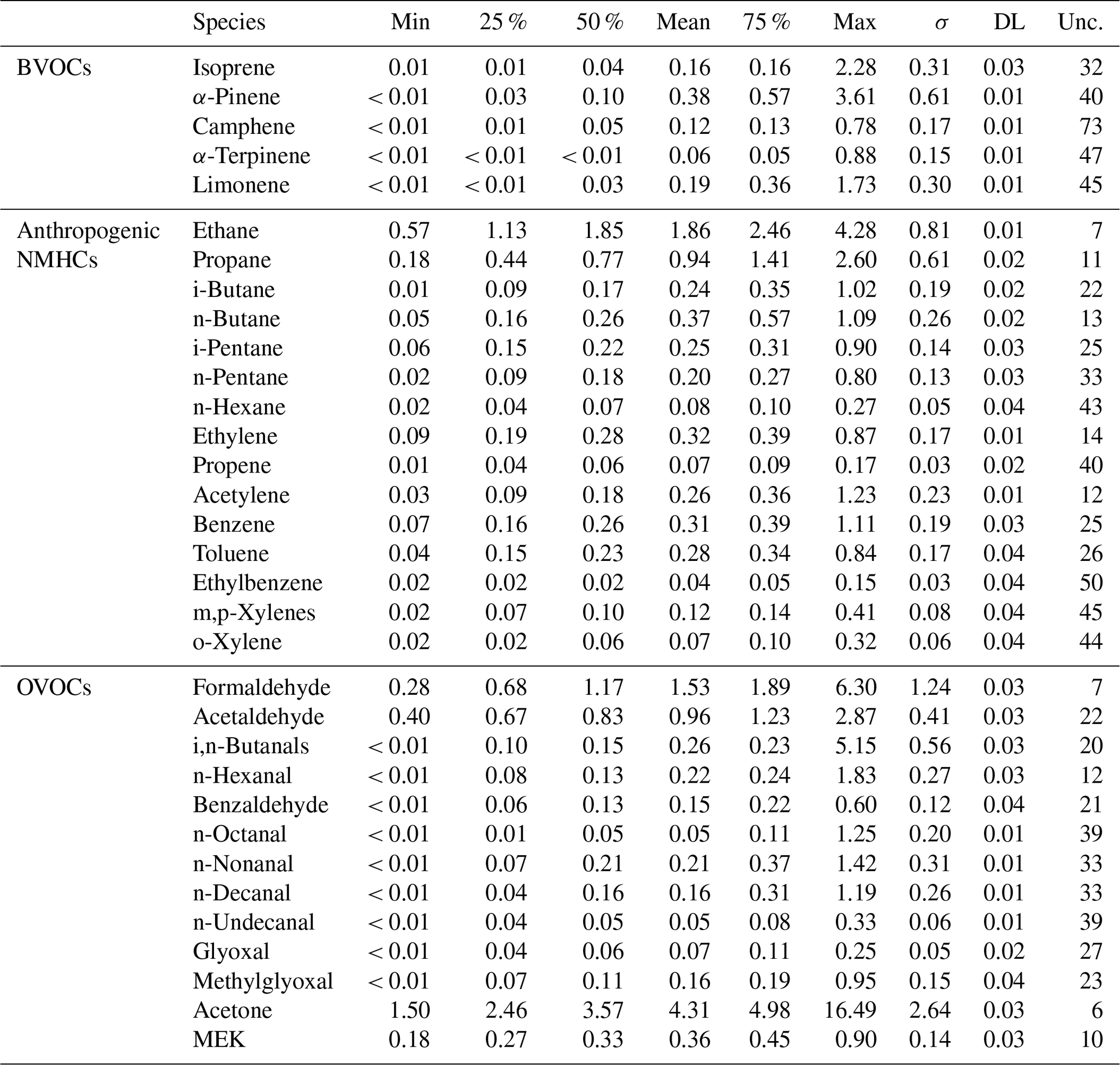
Statistical results of concentrations of the 35 VOCs selected in this study (see Sect. S1 in the Supplement) are summarized in Table 3. Their average concentration levels as a function of the measurement sampling time (09:00–13:00 or 12:00–16:00 UTC) are also indicated in Table S1. These VOCs were organized into three principal categories: biogenic, anthropogenic, and oxygenated VOCs (5, 16 and 14 targeted species, respectively; Table 3). Isoprene and four monoterpenes were classified into BVOCs, while the other primary hydrocarbons (alkanes, alkenes, alkynes and aromatic compounds) were included in anthropogenic NMHCs, since their emissions are especially in connection with human activities. OVOCs have been presented separately, as these compounds come from both biogenic and anthropogenic (primary and secondary) sources. OVOCs were the most abundant, accounting for 65 % of the total concentration of the 35 VOCs selected in this study. They were mainly composed of acetone (contribution of 51 % to the OVOC cumulative concentration). Anthropogenic NMHCs also contributed significantly (26 %) to the total VOC concentration and principally consisted of ethane and propane (which represented 34 % and 17 % of the anthropogenic NMHC cumulative concentration, respectively) as well as n-butane (7 %). The high contribution of species which generally have the longest lifetimes in the atmosphere (see Sect. 3.5) is consistent with the remote location of the Ersa site and is in agreement with Michoud et al. (2017). BVOCs contributed little to the total VOC concentration (9 % on annual average, 13 % in summer). They were mainly composed of isoprene and α-pinene (contributions of 44 % and 32 % to the BVOC mass, respectively). These compounds are among the major BVOCs in terms of emission intensity for the Mediterranean vegetation (Owen et al., 2001) and accounted for half of isoprenoid concentrations recorded during the intensive field campaign conducted at Ersa in summer 2013 (Debevec et al., 2018; Kalogridis, 2014). By contrast, a larger α-terpinene contribution was noticed during the summer 2013 intensive field campaign than the 2-year observation period. Note that speciated monoterpenes were measured differently during the summer 2013 field campaign by means of an automatic analyser (see Sect. S4 in the Supplement).
Table 3Statistics (µg m−3), standard deviations (σ – µg m−3), detection limits (DL – µg m−3) and relative uncertainties (Unc. – %) of selected VOC concentrations measured at the site from June 2012 to June 2014.
3.4 VOC variability
Monthly and interannual variations of primary (anthropogenic and biogenic) NMHCs along with OVOCs selected in this study (Sect. S1) are discussed in this section. Seasonal VOC concentration levels are indicated in Table 4. Note that the results of comparison between the VOC monitoring measurements investigated in this study and concurrent field campaign measurements performed during the summers 2012–2014 (presented in Sect. S4 of the Supplement) supported the representativeness of the 2-year observation period with regard to its summer concentration levels.

3.4.1 Biogenic VOCs
Concentration variations of three selected BVOCs, isoprene, α-pinene and camphene, were analysed at different timescales (monthly/interannual variations; Fig. 4). These BVOCs exhibited high concentrations from June to August consistently with temperature and solar radiation variations (see Sect. 3.1). Indeed, throughout the summer 2013 observation period, Michoud et al. (2017) and Kalogridis (2014) observed that emissions of isoprene and the sum of monoterpenes were mainly governed by temperature and solar radiation, supported both by the diurnal variations in their concentrations (Geron et al., 2000a, b; Guenther et al., 2000) and their correlations with environmental parameters. Additionally, significant concentrations of α-pinene were noticed from September to November (Fig. 4), while isoprene concentrations were close to the detection limit and temperature and solar radiation were decreasing. However, solar radiation decreased much faster than temperature during these months (Fig. 3), which could suggest that additional emissions (Laothawornkitkul et al., 2009), dependent only on temperature contrary to those prevailing in summer, have influenced α-pinene concentrations during these months. Ozone concentrations, lower in autumn (i.e. the months of October to December) than in summer (O3 concentration variations are depicted in Fig. S2 of the Supplement), also pointed out a weaker degradation of α-pinene in autumn.
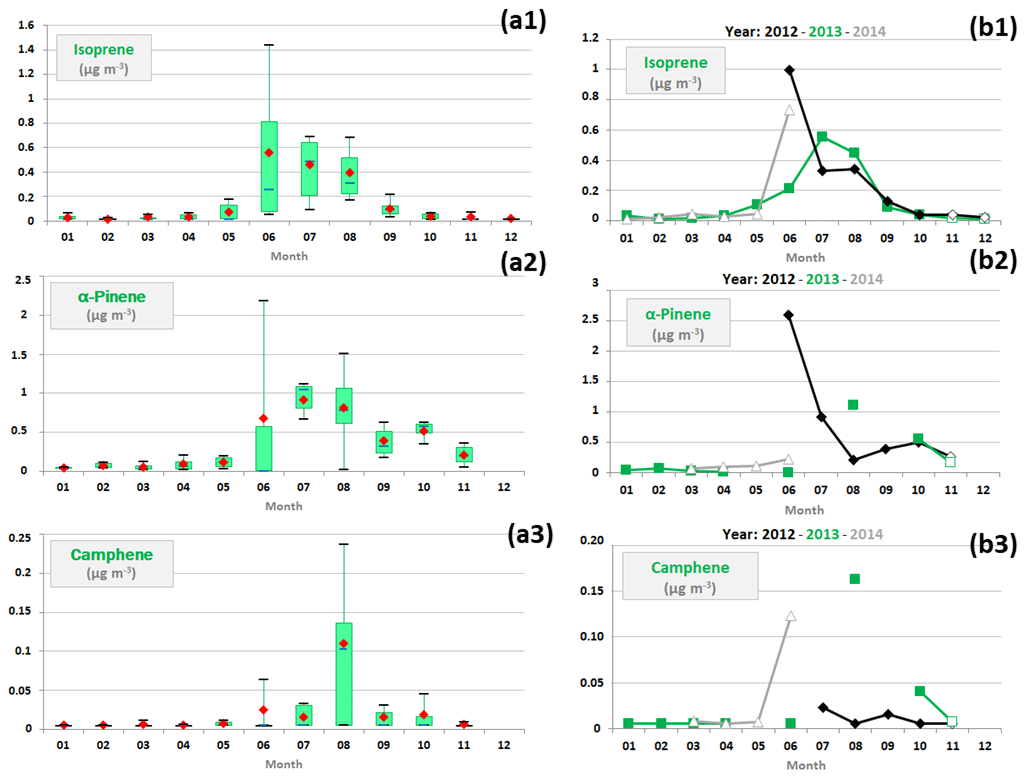
Figure 4(a) Monthly variations in a selection of biogenic VOC concentrations (expressed in µg m−3) represented by box plots; the blue solid line, the red marker, and the box represent the median, the mean, and the interquartile range of the values, respectively. The bottom and top of the box depict the first and third quartiles and the ends of the whiskers correspond to the first and ninth deciles. (b) Their monthly average concentrations as a function of the year; full markers indicate months when VOC samples were collected from 12:00 to 16:00 UTC and empty markers those when VOC samples were collected from 09:00 to 13:00 UTC.
Biogenic compounds showed significant interannual variations over the study period, linked to temperature and solar radiation variations. Higher June concentrations of isoprene and α-pinene were noticed in 2012 (average concentrations of 1.0 ± 1.1 and 2.6 ± 1.4 µg m−3 for isoprene and α-pinene, respectively) and 2014 (0.7 ± 0.5 and 0.2 µg m−3) than in 2013 (0.2 ± 0.2 and < 0.1 µg m−3). Higher June concentrations of camphene (and α-terpinene; not shown) were also noticed in 2014 than in 2013 (Fig. 4). These concentration levels may be related to the fact that temperature and solar radiation were more favourable to enhancing June biogenic emissions in 2012 and 2014 than in 2013 (Sect 3.1). Due to air relative humidity values observed in June (Sect. 3.1), we cannot rule out that an increase in BVOC concentrations may be linked to a transient modification of BVOC emissions induced by drought stress (Ferracci et al., 2020; Loreto and Schnitzler, 2010; Niinemets et al., 2004). Moreover, isoprene and α-pinene concentrations in July and August were higher in 2013 (average concentrations of 0.5 ± 0.3 and 1.1 ± 0.4 µg m−3 for isoprene and α-pinene, respectively) than in 2012 (0.3 ± 0.2 and 0.6 ± 0.3 µg m−3). High concentrations of camphene and α-terpinene were also noticed in August 2013 (0.2 ± 0.1 and 0.3 ± 0.3 µg m−3, respectively; Fig. 4). Solar radiations were lower in July and August 2012, temperatures were slightly lower in July 2012 and mean wind speed was slightly higher in July 2012 (Fig. 3). These meteorological conditions may have affected biogenic emissions and have favoured their dispersion and their dilution by marine air masses, owing to the position of the Ersa station (Sect. 2.1).
Note that the interpretation of interannual variations in BVOC measurements is based on a limited number of sampling days during the study period and different collection times (Table 1 and Sect. 2.2.1). It should therefore be considered cautiously, given variable day-to-day and strong diurnal BVOC concentration variations which were observed during the summer 2013 observation period (Kalogridis, 2014).
3.4.2 Anthropogenic VOCs
Variations of a selection of NMHCs are depicted in Fig. 5. These compounds illustrate contrasted reactivity, based on their atmospheric lifetimes (estimated according to their photochemical reaction rates with OH radicals defined in Atkinson, 1990, and Atkinson and Arey, 2003). Despite lifetimes in the atmosphere ranging from a few hours to some days, all selected NMHCs were characterized by similar seasonal variations, with an increasing winter trend followed by a decrease in spring/summer (Fig. 5 and Table 4), with the exception of n-hexane, propene and C8 aromatics (which were the most reactive species of the NMHCs selected in this study and had the lowest concentrations; Tables 3 and 4). NMHC concentrations were higher in winter than in summer, up to 5 times higher in the case of acetylene (Table 4). Note that ethane concentration levels were relatively important during summer (mean concentration of 1.0 ± 0.2 µg m−3), while other NMHCs showed concentrations below 0.4 µg m−3.
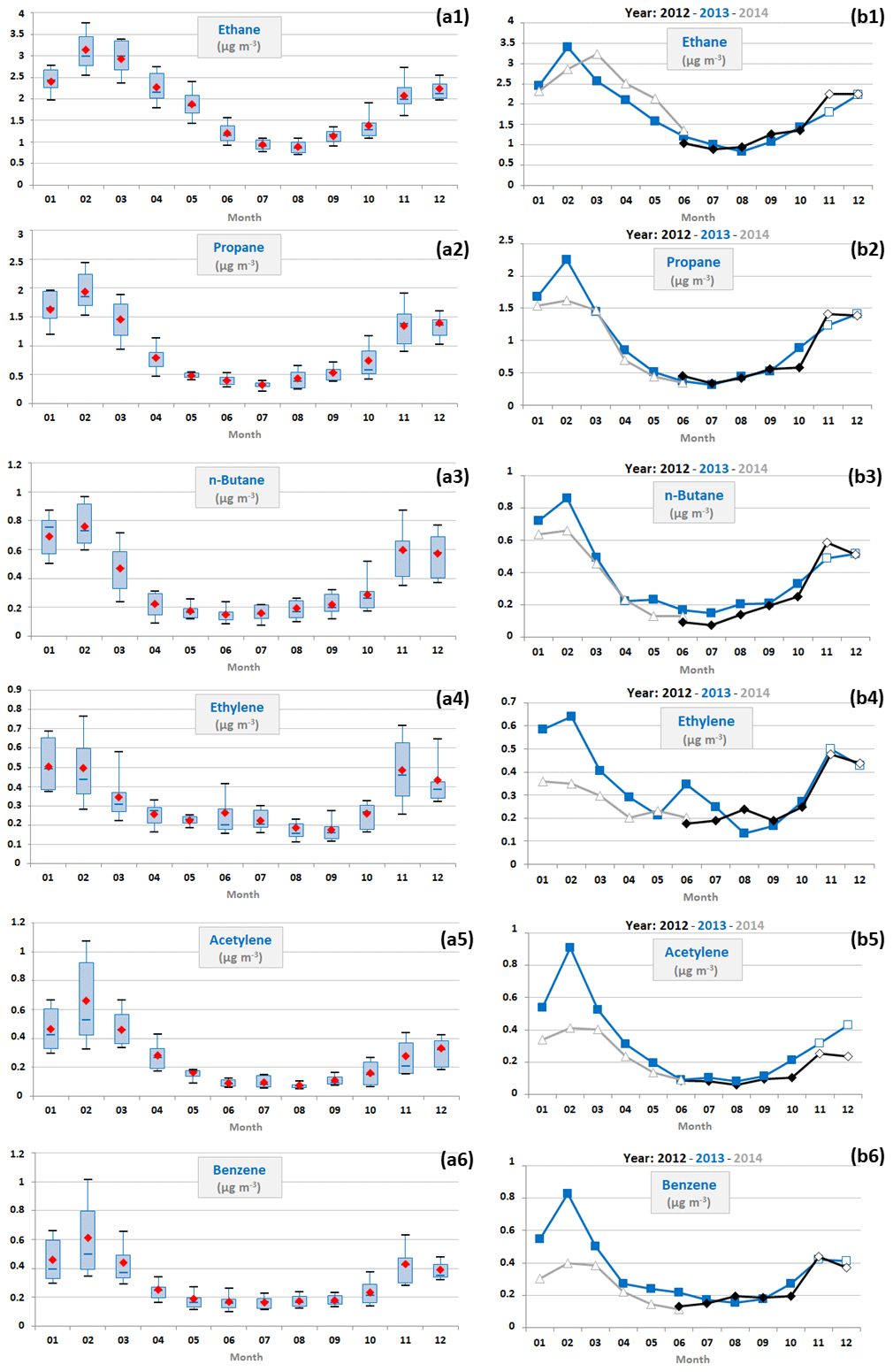
Figure 5(a) Monthly variations in a selection of anthropogenic VOC concentrations (expressed in µg m−3) represented by box plots; the blue solid line, the red marker, and the box represent the median, the mean, and the interquartile range of the values, respectively. The bottom and top of the box depict the first and third quartiles and the ends of the whiskers correspond to the first and ninth deciles. (b) Their monthly average concentrations as a function of the year; full markers indicate months when VOC samples were collected from 12:00 to 16:00 UTC and empty markers those when VOC samples were collected from 09:00 to 13:00 UTC.
NMHCs exhibited different concentration levels during the two studied winter periods (Fig. 5). Mean winter NMHC concentrations were higher in 2013 than in 2014 by up to 0.3 µg m−3 in the cases of propane, acetylene and benzene (relative differences of 15 %, 42 % and 42 %, respectively). These compounds and ethane had the longest lifetimes among those selected in this study. However, ethane concentrations recorded at Ersa did not show any interannual variation over the study period (Fig. 5).
3.4.3 Oxygenated VOCs
Variations of selected OVOCs, illustrating contrasted reactivity (following the same methodology as that applied to NMHCs; Sect. 3.4.2), are presented in Fig. 6. Formaldehyde, acetaldehyde, glyoxal, methylglyoxal and C6–C11 aldehydes have relatively short lifetimes into the atmosphere, and hence they can result from relatively close sources. On the other hand, acetone and methyl ethyl ketone (MEK) have the longest atmospheric lifetime among the OVOCs selected in this study, and hence they can also result from distant sources and/or be formed within emission-enriched air masses advected to the Ersa station.
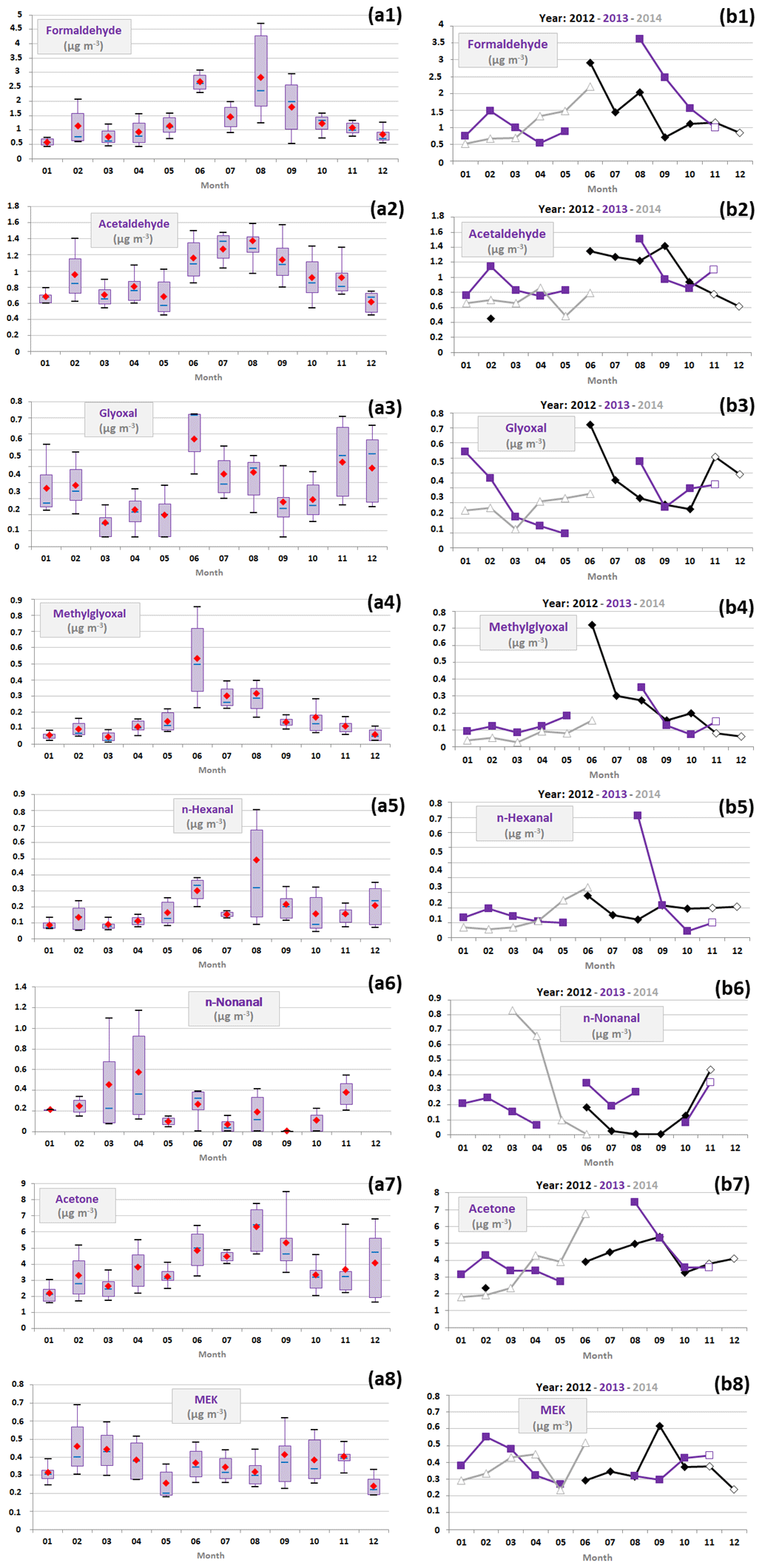
Figure 6(a) Monthly variations in a selection of oxygenated VOC concentrations (expressed in µg m−3) represented by box plots; the blue solid line, the red marker, and the box represent the median, the mean, and the interquartile range of the values, respectively. The bottom and top of the box depict the first and third quartiles and the ends of the whiskers correspond to the first and ninth deciles. (b) Their monthly average concentrations as a function of the year; full markers indicate months when VOC samples were collected from 12:00 to 16:00 UTC and empty markers those when VOC samples were collected from 09:00 to 13:00 UTC.
Firstly, formaldehyde, methylglyoxal and n-hexanal have shown similar seasonal variations (Fig. 6), with high summer and spring concentrations (Table 4), suggesting an important contribution of primary/secondary biogenic sources to their concentrations. Fu et al. (2008) found that the largest global sources for methylglyoxal were isoprene and to a lesser extent acetone. Besides photochemical production, n-hexanal and formaldehyde can be notably emitted by many plant species (Guenther et al., 2000; Kesselmeier and Staudt, 1999; Wildt et al., 2003). Interannual variations in formaldehyde, methylglyoxal and n-hexanal summer concentrations confirmed their links with biogenic sources. For instance, the highest concentrations of methylglyoxal were observed in June 2012 (average concentration of 0.7 µg m−3), similarly to isoprene (Sect. 3.4.1). Monthly concentrations of n-hexanal peaked at 0.7 µg m−3 in August 2013, in agreement with monoterpenes, especially camphene and α-terpinene (Sect. 3.4.1). Formaldehyde showed high concentrations in both June 2012 and August 2013 (2.9 and 3.6 µg m−3, respectively).
Acetaldehyde and acetone have shown an increase in their concentrations more marked in summer than in winter (Fig. 6), suggesting they were probably mainly of both secondary (anthropogenic/biogenic) and primary biogenic origins. Acetaldehyde is known to be mainly produced through the chemical transformation of anthropogenic and biogenic VOCs (Rottenberger et al., 2004; Schade and Goldstein, 2001; Seco et al., 2007; Wolfe et al., 2016), particularly in clean and remote areas. Acetaldehyde can also be released by plants (Jardine et al., 2008; Rottenberger et al., 2008; Winters et al., 2009). Acetone emissions are thought to be globally of biogenic rather than anthropogenic origin (Goldstein and Schade, 2000; Schade and Goldstein, 2006). Acetone can also result from the oxidation of various VOCs (Goldstein and Schade, 2000; Jacob et al., 2002; Singh et al., 2004), and roughly half of its concentrations measured at diverse urban or rural sites have been assigned to regional background pollution by several studies (e.g. Debevec et al., 2017; de Gouw et al., 2005; Legreid et al., 2007; with regional contributions at a scale of hundreds of kilometres). Additionally, acetaldehyde and acetone concentration variations in winter (e.g. mean February concentration in 2013 was 0.5 and 2.4 µg m−3 higher than in 2014, respectively) also pinpointed primary/secondary anthropogenic origins (Sect. 3.4.2).
Glyoxal and MEK showed an increase in their concentrations in both summer and winter (Fig. 6 and Table 4), suggesting they were probably produced by several biogenic and anthropogenic sources. Glyoxal increases were in similar proportions (Fig. 6 and Table 4), while the MEK increase in winter was more marked than in summer, which may indicate that primary/secondary anthropogenic sources primarily contributed to MEK concentrations. Fu et al. (2008) found that the largest global sources for glyoxal were isoprene and to a lesser extent acetylene and direct emissions. MEK can be emitted from both vegetation and numerous anthropogenic sources and can result from the oxidation of various VOCs (Yáñez-Serrano et al., 2016, and references therein). Glyoxal and MEK exhibited different concentration levels during the two studied winter periods since their mean February concentration in 2013 was 65 %–75 % higher than in 2014, confirming their links with anthropogenic sources. These two OVOCs also showed different interannual variations in late spring and summer (Fig. 6). The highest concentrations of glyoxal were measured in June 2012 (average concentration of 0.2 µg m−3), similarly to isoprene and α-pinene (Sect. 3.4.1), when MEK concentrations were among their lowest (0.3 µg m−3).
Finally, n-nonanal did not show clear seasonal variations. Its March and April concentrations were higher in 2014 than in 2013. An increase in MEK concentrations was also noticed in March and April 2014 in lower proportion. These findings suggest contributions from different sources in winter/early spring, compared to the rest of the year. Moreover, a slight increase in n-nonanal concentrations was noted in August 2013, which is consistent with a biogenic source contribution (Matsunaga et al., 2003; Wildt et al., 2003).
3.5 Major NMHC sources
In the coming section, major sources which have impacted primary NMHC concentrations measured at Ersa were identified using a PMF five-factor solution (from simulations presented in Sect. S2). Figure 7 depicts factor contributions to the species chosen as inputs for the PMF tool along with NMHC contributions to the five factors defined by the factorial analysis. Figure 8 and Table 5 show factor contribution time series and their relative contributions to the total concentration of the NMHCs selected in this factorial analysis, respectively. Average factor relative contributions as a function of the measurement sampling time are also indicated in Table 5. In the present section, lifetimes were assessed from kinetic rate constants of the reactions of the selected NMHCs with OH (Atkinson and Arey, 2003) given an average OH concentration of 0.5 × 106 and 2.5 × 106 molecules cm−3 in winter and summer, respectively (Spivakovsky et al., 2000).
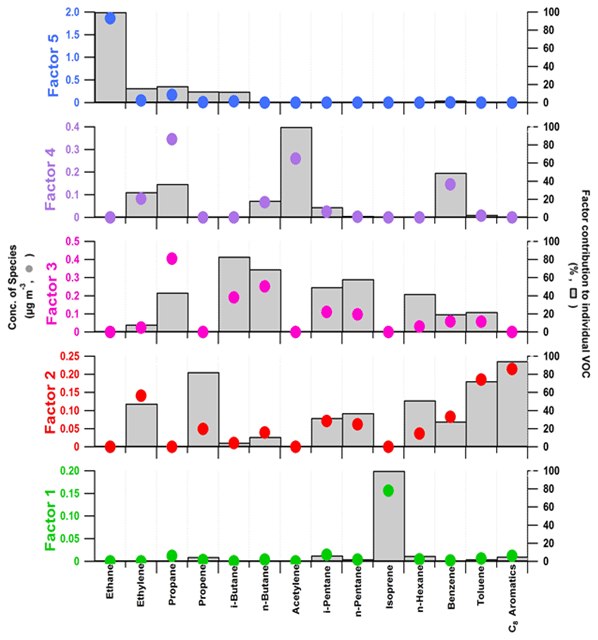
Figure 7Chemical profiles of the five-factor PMF solution (14 variables). Factor contributions to each species (µg m−3) and the percent of each species apportioned to the factor are displayed as a grey bar and a coloured circle, respectively. Factor 1 – local biogenic source; factor 2 – short-lived anthropogenic sources; factor 3 – evaporative sources; factor 4 – long-lived combustion sources; factor 5 – regional background.
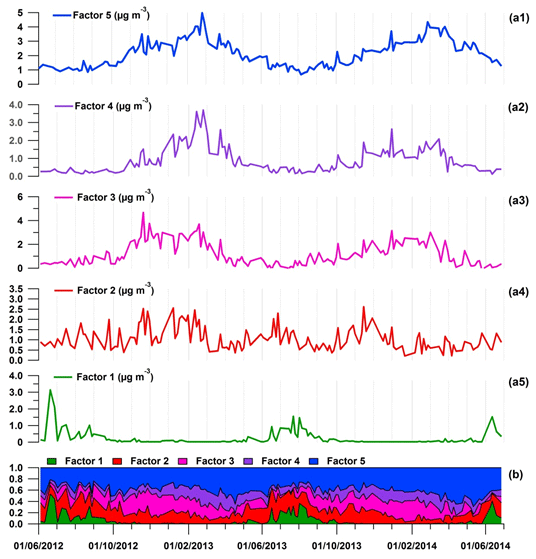
Figure 8(a) Time series of factor contributions (µg m−3) and (b) accumulated relative NMHC contributions. Factor 1 – local biogenic source; factor 2 – short-lived anthropogenic sources; factor 3 – evaporative sources; factor 4 – long-lived combustion sources; factor 5 – regional background. Note that the NMHC dataset used for the PMF analysis included different sampling time hours (09:00–13:00 or 12:00–16:00 UTC) following shifts during the 2-year period (see Table 1).
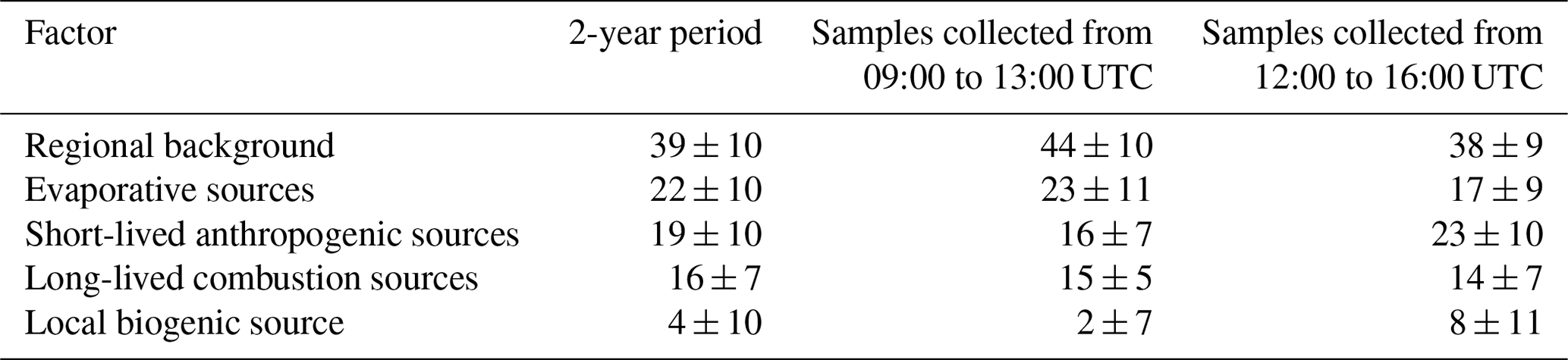
Table 5Average relative factor contributions ± standard deviations (%) for the whole period and as a function of the measurement sampling time (see Table 1).
As VOC concentrations resulted from direct emissions, chemistry, transport and mixing, each individual computed factor cannot be attributed solely to one source category, especially for a remote receptor site like Ersa. A part of them may not be specifically associated with emission profiles but should rather be considered aged profiles originating from several source regions comprising several source categories (Sauvage et al., 2009). The PMF analysis was hence performed in this study to define co-variation factors of NMHCs that were characteristic of aged or local primary emissions along with secondary photochemical transformations taking place during the transport of air masses observed at this remote site (Michoud et al., 2017).
Since the low number of NMHCs considered in the factorial analysis of this study, PMF result relevance was checked, benefiting from a previous PMF analysis performed with the Ersa VOC summer 2013 dataset (Michoud et al., 2017; see Sect. 3.5.6). Experimental strategies to characterize VOC concentration variations at a remote background such as Ersa are also discussed in Sect. 3.5.6.
3.5.1 Local biogenic source (factor 1)
Factor 1 contributions to measured concentrations of the NMHCs selected in the factorial analysis were 0.2 ± 0.4 µg m−3 on average (corresponding to 4 % of the sum of these NMHCs – Table 5) and peaked at 3.1 µg m−3 on 20 June 2012. In late spring/summer, it was one of the main factors observed (16 % on average and up to 53 %; Fig. 8). The chemical profile of factor 1 depicts an elevated contribution of isoprene, known as a chemical marker for biogenic emissions, with its variability fully related to this factor. The isoprene relative contribution to factor 1 is 70 %. The estimated tropospheric lifetime of isoprene was quite short (winter: 6 h and summer: ∼ 1 h), indicating that this compound was emitted mostly by local vegetation. Consequently, factor 1 is labelled “local biogenic source”. Note that the factor 1 contribution to selected NMHC concentrations may be slightly influenced by the two different sampling times used during the 2-year observation period (Table 5) as well as the number of VOCs and data points considered in the PMF analysis (see Sects. 3.4.1 and S2). However, Michoud et al. (2017) provided additional information on this local primary biogenic source in summer, like the contributions of additional primary BVOCs (the sum of monoterpenes) and some OVOCs (carboxylic acids, methanol and acetone), and presented its clear diurnal variations.
3.5.2 Short-lived anthropogenic sources (factor 2)
Factor 2 contributed 19 % to the sum of the selected NMHCs (Table 5; average contribution of 1.0 ± 0.5 µg m−3). This factor mainly consists of toluene (73 % of its variability attributed to this factor; Fig. 7), C8 aromatic compounds (93 %), ethylene (48 %) and propene (83 %), typically emitted by combustion processes and with short-to-medium lifetimes (winter: 1-4 d; summer: 5–20 h). The cumulative relative contribution of these VOCs to factor 2 is 66 %. Besides road traffic, toluene is also a good marker for solvents generated by industrial sources (Buzcu and Fraser, 2006; Leuchner et al., 2015; Zhang et al., 2014), suggesting that this profile could also be imputed to industrial sources. Additionally, a significant proportion of C5–C6 alkanes, i.e. i-pentane (32 % explained), n-pentane (37 %) and n-hexane (51 %), typically emitted by gasoline evaporation and with medium lifetimes (winter: 4–6 d; summer: 21–31 h), also contributes 19 % to this factor. Factor 2 did not show a good correlation with CO (Pearson correlation coefficient only of 0.2). As a conclusion, factor 2 is hence attributed to the grouping of several short-lived anthropogenic sources, partly related to gasoline combustion and/or evaporation and solvent use. Note that the PMF model did not reconstruct well 5 of the species comprising this factor (propene, n-pentane, n-hexane, toluene, and C8 aromatics; Sect. S2 in the Supplement), especially their concentration variations over short periods. As a result, factor 2 contributions over short periods may be underestimated. However, given the objectives of this study, the examination of factor 2 contribution variations will be limited to seasonal and interannual ones (Sect. 4). Factor 2 contribution variations over a short period were previously investigated in Michoud et al. (2017).
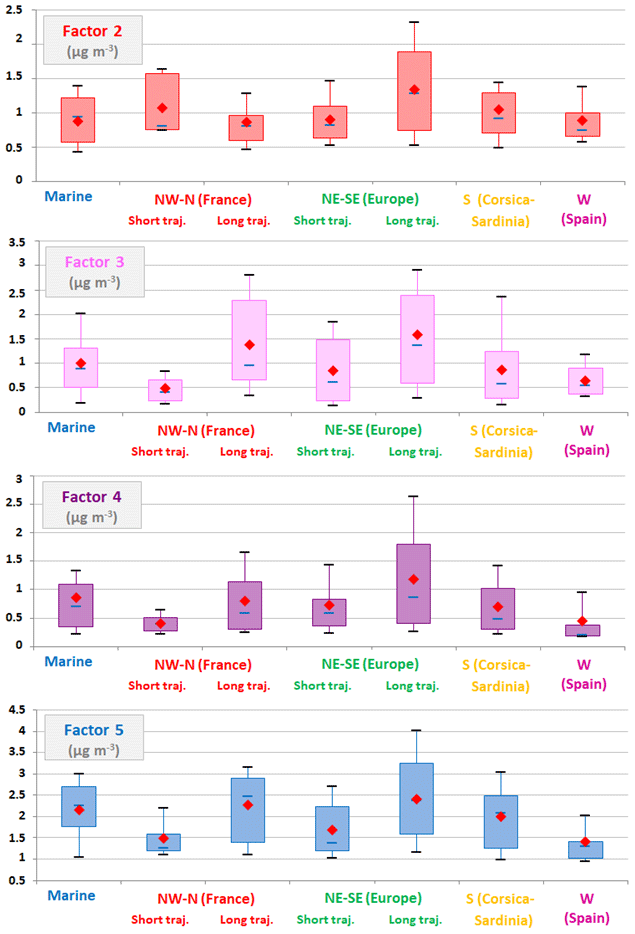
Figure 9NMHC factor contributions (µg m−3) as a function of air-mass origin represented by box plots; the blue solid line, the red marker, and the box represent the median, the mean, and the interquartile range of the values, respectively. The bottom and top of the box depict the first and third quartiles and the ends of the whiskers correspond to the first and ninth deciles. Factor 2 – short-lived anthropogenic sources; factor 3 – evaporative sources; factor 4 – long-lived combustion sources; factor 5 – regional background. Air masses originating from France and Europe are subdivided into short and long trajectories to highlight local and more distant contributions (see Sect. 3.2). Note that the NMHC dataset used for the PMF analysis included different sampling time hours (09:00–13:00 or 12:00–16:00 UTC) following shifts during the 2-year period (see Table 1).
Regarding factor 2 contributions coupled with air-mass origins (Fig. 9), more elevated contributions were noticed at Ersa under the influence of continental air masses coming from France and Europe. The distinction of short trajectories from long ones (see Sect. 3.2) highlighted that factor 2 was potentially influenced by relatively close sources when Ersa received air masses from continental France, whereas other continental European sources were probably more distant (Fig. 9). Furthermore, CF analysis applied to factor 2 contributions (see Fig. S3 of the Supplement) confirmed that this factor was influenced by various potential emission areas, either located in Italy (the Po Valley and central Italy), France (south-eastern region) or possibly in central Europe (western Hungary, Croatia and Slovenia). Given the CF results, ship emission contribution cannot be discarded as well, as already suggested in Michoud et al. (2017). Indeed, ship emissions are predominantly composed of light alkenes, aromatic compounds and heavy alkanes (> C6 compounds; Eyring et al., 2005).
3.5.3 Evaporative sources (factor 3)
Factor 3 contributions to the selected NMHC concentrations were 1.2 ± 1.0 µg m−3 on average (22 % of their sum; Table 5). The profile of this anthropogenic factor displays an important contribution from alkanes, principally i-/n-butanes (with lifetimes of 10–11 d in winter and ∼ 2 d in summer), with more than 69 % of their variabilities explained by factor 3 (Fig. 7), along with i-/n-pentanes, n-hexane and propane (42 %–59 %; lifetimes of 4–21 d in winter and 21 h–4 d in summer). The cumulative relative contribution of these alkanes to factor 3 is up to 88 %. C3–C6 alkanes are identified in the gasoline composition and evaporation sources (storage, extraction and distribution of gasoline or liquid petroleum gas; Sauvage et al., 2009, and references therein). Additionally, propane can be viewed as a relevant profile signature of natural gas transport, storage and use (Leuchner et al., 2015). Consequently, this factor can be viewed as “evaporative sources”.
Regarding factor contributions as a function of air-mass origins (Fig. 9), more elevated contributions were noticed at the Ersa station when aged air masses originated from France and Europe and were probably transported toward Ersa by relatively distant sources. According to the CF analysis applied to factor 3 contributions (Fig. S3 of the Supplement), the Po Valley (especially Emilia-Romagna, an Italian region centre for food and automobile production), central Italy, the south-east of France and the Sardinian region seemed to be identified as the main potential emission areas for factor 3, as well as possibly more distant emission areas in central Europe such as western Hungary (i.e. the western Transdanubian region specializing in the automotive and machinery industries).
3.5.4 Long-lived combustion sources (factor 4)
The average contribution of factor 4 to the selected NMHC concentrations is evaluated at 0.9 ± 0.7 µg m−3 (corresponding to 16 % of their sum; Table 5). Its profile displays an important contribution from acetylene (100 % explained; Fig. 7), benzene (49 %) and propane (37 %), with lifetimes of 19–26 d in winter and 4–5 d in summer. The average cumulative relative contribution of these VOCs to factor 4 is 80 %. Aromatic compounds and acetylene are generally associated with combustion sources, such as vehicle exhaust (e.g. Badol et al., 2008; Pang et al., 2014). However, factor 4 is characterized by a loading of benzene far superior to toluene's (49 % and 3 %, respectively), suggesting it is more related to a residential heating source than a traffic one (Elbir et al., 2007; Leuchner et al., 2015; Sauvage et al., 2009). The factor 4 profile, mainly composed of long-lived species together with a low contribution of shorter-lived species, may indicate that it is related to partly aged air masses which were advected towards the Ersa station. These suggestions are consistent with the fact that this factor correlated particularly well with CO (Pearson correlation coefficient of 0.8). As a result, this factor can be viewed as “long-lived combustion sources”, including residential heating.
Factor 4 showed higher contributions when the Ersa station received European air masses (see Fig. 9), particularly those with a long trajectory, which is consistent with factor 4 profile examination. The CF analysis depicted in Fig. S3 of the Supplement only pointed out western Hungary and to a lesser extent the Po Valley as the main potential emission areas for factor 4.
3.5.5 Regional background (factor 5)
Contributing 39 % to the selected NMHC concentrations (average contribution of 2.2 ± 1.0 µg m−3), factor 5 corresponds to the dominant source detected at Ersa during the study period. Its profile is principally composed of ethane (with its variability fully explained by factor 5; Fig. 7) and propane (18 % explained; lifetimes of 21–93 d in winter and 4–19 d in summer). Their relative contribution to factor 5 is up to 96 % and they typically result from the transport, storage and use of natural gas. Additional anthropogenic NMHCs with shorter lifetimes are attributed to this factor, including ethylene and propene (12 %–16 % explained; lifetimes of 21 h–3 d in winter and 4–13 h in summer), despite a low contribution to factor 5 mass (∼ 3 %). Given the high abundance of long-lived species in its profile, factor 5 can be viewed as a regional contribution of diverse remote sources of the Mediterranean region, thus indicating the continental regional background (Hellén et al., 2003; Leuchner et al., 2015; Sauvage et al., 2009; regional contributions at a scale of hundreds of kilometres). As a conclusion, we associated factor 5 with the “regional background”.
Factor 5 showed slightly higher contributions when the Ersa station was under the influence of European air masses, particularly those with a long trajectory (potentially connected to distant emission areas; Fig. 9). As expected, the CF analysis applied to factor 5 contributions did not clearly pinpoint a specific potential emission area (Fig. S3 of the Supplement), apart from maybe western Hungary and to a lesser extent the Po Valley.
3.5.6 Towards the best experimental strategy to characterize variations in VOC concentrations observed at a remote background site
In this study, we decided to restrain the number of VOCs in the factorial analysis in order to have a better temporal representativeness of factor contributions (see Sect. S2). This choice has undoubtedly had some consequences for the PMF solution results of this study (in terms of factor number, chemical profiles and variations). Consequently, to support our results, we compared our five-factor PMF solution modelled with the 2-year NMHC dataset (14 variables, 152 4 h resolution observations), with the six-factor PMF solution modelled with the summer 2013 VOC dataset (42 variables, 329 1.5 h resolution observations; Michoud et al., 2017). The comparative study (see Sect. S5 in the Supplement) pointed out a good representativeness of the primary sources identified in this study. Overall, sources identified in both factorial analyses as influencing NMHC concentrations at Ersa had similar chemical compositions, regarding NMHCs in common. This finding supports the relevance of the 17 NMHCs selected in the factorial analysis of this study to identify major sources which have influenced Ersa NMHC concentrations over 2 years and suggests that they include key source tracers.
In order to support these statements, we computed a summer 2013 PMF solution only with the NMHCs selected in the factorial analysis of this study, and we compared it with the summer 2013 PMF solution modelled with a higher VOC number by Michoud et al. (2017). Results (see Sect. S6 in the Supplement) revealed a global consistency of the two summer 2013 PMF solutions in terms of factor chemical composition, regarding NMHCs in common between the two factorial analyses, and contribution variations, with the exception of those of short-lived anthropogenic sources (which may also depend on additional VOCs; Sect. S6). PMF factorial analyses computed with a limited VOC number therefore show limitations in the investigation of short-lived anthropogenic sources influencing Ersa VOC concentrations given the remote location of the Ersa station.
The two studies on Ersa VOC measurements also helped to discuss here the best experimental strategy to characterize variations in VOC concentrations measured at a remote background site surrounded by vegetation such as Ersa (Sect. 2.1). Firstly, the consideration of monoterpenes in the 2-year PMF analysis would significantly increase the contribution of primary biogenic sources (see Sect. 4.1). A better temporal resolution and coverage of the VOC measurements would have been beneficial in this study to further assess the contribution of local biogenic sources, considering BVOC day-to-day variations (as assessed with the summer 2013 dataset; Sect. 3.4.1). Additional measurements of speciated monoterpenes, sesquiterpenes and BVOC oxidation products would even help in identifying different local sources associated with various emitting Mediterranean plants (Owen et al., 2002). This statement is supported by the fact that different interannual variations in BVOC concentrations were noticed between isoprene and selected speciated monoterpenes in this study (Sect. 3.4.1). Contrarily, concentrations of non-speciated monoterpenes and isoprene measured in summer 2013 were mainly attributed to the same primary biogenic source in Michoud et al. (2017). However, concentration variations of the sum of monoterpenes may have mirrored those related to abundant biogenic species such as α-pinene and thus masked those characterizing more reactive species such as α-terpinene (Kalogridis, 2014; Debevec et al., 2018).
The limited number of VOCs included in our PMF study had the advantage of a better deconvolution by the PMF model of primary sources and a better reconstruction of measured VOCs with low concentrations (Sects. S2 and S6), without the need to minimize the weight of species with elevated background levels (unlike ethane, methanol and acetone in Michoud et al., 2017). These low concentrations thus represented a higher proportion of the total VOC concentrations that the model was supposed to explain. The longer timescale of VOC measurements (i.e. the 2-year period) even helped to deconvolve long-lived combustion sources from regional background (Sect. S5). However, PMF comparison results also raised the importance of a finer time resolution and a higher temporal coverage of VOC measurements conducted over a long period at a remote background site such as Ersa to support source apportionment results, in terms of decorrelation of anthropogenic sources. Indeed, anthropogenic sources identified in this study with bi-weekly observations have shown some significant correlations between them (Sect. S5) as a result of similar seasonal variations in concentrations of NMHCs composing them (Sects. 3.4.2 and 4). The consideration of diurnal variations in the 2-year factorial analysis would help in limiting this potential statistical bias.
The incorporation of 19 OVOCs in the source apportionment of Michoud et al. (2017) had little impact on the deconvolution of the main sources influencing NMHC concentrations observed at the Ersa station (Sects. S5 and S6). However, it can modify their relative contributions to the total VOC concentration, emphasizing those of local biogenic/anthropogenic sources and decreasing those of regional anthropogenic sources (Michoud et al., 2017). VOCs were largely dominated at Ersa by OVOCs (Sect. 3.3), especially in summer (Michoud et al., 2017). OVOC abundance observed at Ersa was further increased by the measurement of alcohols and carboxylic acids, carried out only in summer 2013 with automatic analysers (Sect. S4). The incorporation of OVOCs in the 2-year PMF analysis would have helped to better identify co-variations between these species (Sect. 3.4.3) and their proportion resulting from primary sources, but the limited number of samples realized with DNPH cartridges did not favour it (Fig. S1 and Sect. S2). Formaldehyde has shown in this study high concentration levels (Table 3) and clear seasonal variations (Sect. 3.4.3) and would have been of high interest in the summer 2013 PMF analysis.
In this section, seasonal accumulated concentrations of the 35 VOCs selected in this study (21 NMHCs and 14 OVOCs; Sect. S1) are examined (Fig. 10) so as to identify the prevailing drivers of their variations. Measured concentrations of the 17 NMHCs selected in the factorial analysis (Sect. 3.5) were distributed among their major sources in Fig. 10. Figure S4 presents separately variations in NMHC factor contributions. Based on the work of Michoud et al. (2017) and Kalogridis (2014), it has been assumed in this study that the concentrations of the four monoterpenes (Table 1), not included in the PMF analysis, were fully emitted by the local vegetation. Biogenic sources refer here to the grouping of the PMF biogenic source and speciated monoterpenes. The coming discussion has been structured in two steps: on the one hand the factors explaining the VOC concentration levels and their variations in spring and summer and on the other hand the factors controlling those in autumn and winter.
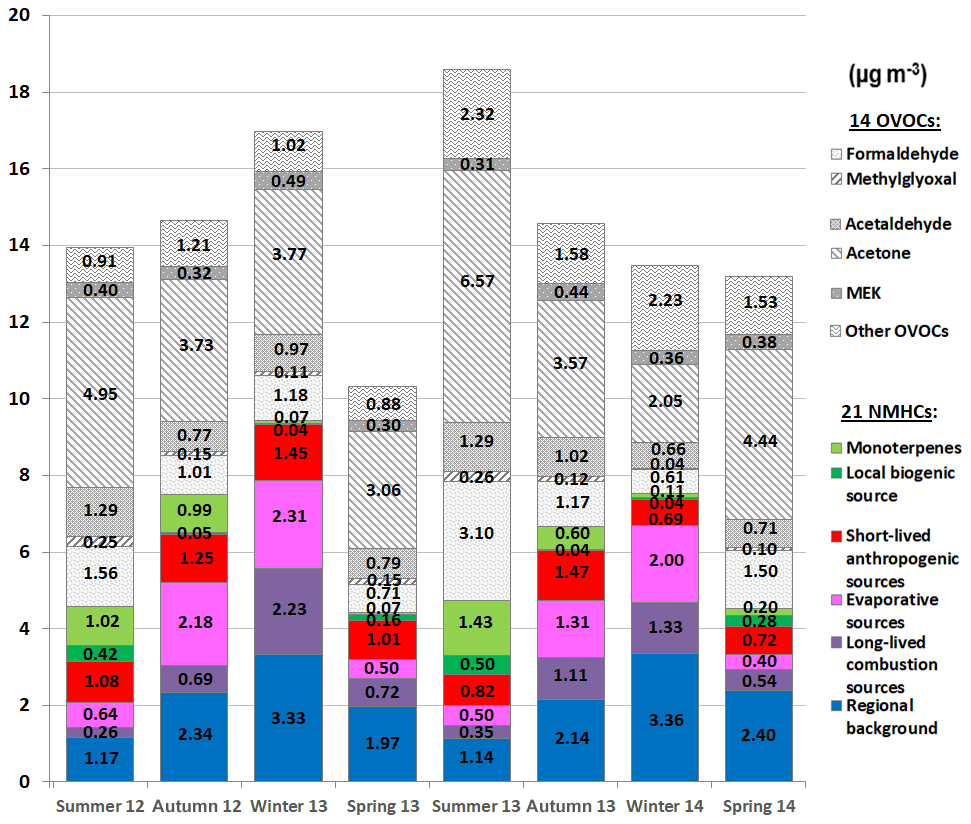
Figure 10Variations in seasonal averaged accumulated concentrations (expressed in µg m−3) of the 35 VOCs selected in this study. The 17 NHMCs selected in the factorial analysis were apportioned to the five modelled NMHC sources. NMHC seasonal measured concentrations which were not modelled by the PMF tool were lower than 0.09 µg m−3 and are not reported here. Winter: 1 January–31 March periods – spring: 1 April–30 June periods – summer: 1 July–30 September periods – autumn: 1 October–31 December periods. Note that the VOC dataset included different sampling time hours (09:00–13:00 or 12:00–16:00 UTC) following shifts during the 2-year period (see Table 1).
4.1 VOC concentration variations in spring and summer
Firstly, the 21 NMHCs selected in this study have shown low concentrations during spring and summer periods (average seasonal accumulated concentration of 4.6 ± 0.1 µg m−3; Fig. 10), while the 14 OVOCs exhibited high concentrations (8.0 ± 1.8 µg m−3). PBL height can be high in these seasons (von Engeln and Teixeira, 2013), favouring vertical dispersion. OVOC concentration levels in spring and summer are in agreement with a high photochemical decay of VOCs, which were rapidly depleted. Consequently, the most reactive VOCs composing emission profiles of the anthropogenic sources can have reacted and/or been dispersed before reaching the Ersa station. The clear seasonal cycles of these sources, with a decline in their contributions in spring and summer (Fig. S4), confirmed this finding.
Regional background contributed in spring and summer 24 % to 53 % to total NMHC concentrations. As natural gas sources were attributed to the regional background (Sect. 3.5.5), a decrease in their emissions can presumably occur in the hot season, enhancing the decline in regional background contributions (Fig. S4). These regional background contributions also suggest that aged emissions advected by air masses to the Ersa station significantly influenced VOC concentrations observed during these seasons. This statement is also in agreement with the high acetone concentration levels in spring and summer (4.8 ± 1.5 µg m−3; Fig. 10). As a reminder, this carbonyl compound can result from distant sources and/or be formed within polluted air masses before reaching the Ersa station (Sect. 3.4.3). As shown in Fig. 10, biogenic sources contributed to total NMHC concentrations especially in summer (up to 41 %), in connection with temperature and solar radiation variations (Sect. 3.4.1). This finding is also supported by the high summer concentration levels of formaldehyde, methylglyoxal, acetaldehyde and acetone (Fig. 10; Sect. 3.4.3). Short-lived anthropogenic sources explained in spring and summer 16 % to 24 % of the NMHC concentrations. Despite the high reactivity of the VOCs comprising these anthropogenic sources (Sect. 3.5.2), their contributions were not reduced as sharply as those of other anthropogenic sources (Fig. S4), which may be related to high influences of ship transport and relatively close potential emission areas (Sect. 3.5.2). Evaporative and long-lived combustion sources only contributed 6 % to 16 % in spring and summer, as they likely originated from emission areas relatively distant from the Ersa station (Sects. 3.5.3 and 3.5.4). Low contributions of long-lived combustion sources can also be partly explained by a lower source strength (especially for a residential heating source; Sect. 3.5.4).
Looking now at the interannual variations during the spring and summer periods, Fig. 10 highlights that total NMHC concentrations were in the same range during the two summer periods as well as the two spring ones (absolute difference was 0.2 µg m−3 between summers 2012 and 2013 and below 0.1 µg m−3 between springs 2013 and 2014). As depicted in Fig. S4, spring and summer contributions of evaporative sources, long-lived combustion sources, and regional background were in the same range over the study period, as well as MEK concentrations (0.3–0.4 µg m−3; Fig. 10), which have been attributed mostly to anthropogenic (primary/secondary) sources (Sect. 3.4.3). These findings can suggest that these anthropogenic sources, originating from distant emission areas, were largely influenced during these seasons by pollutant depletion and vertical/horizontal dispersion. Spring and summer contributions of short-lived anthropogenic sources appear to be more variable over the study period (0.7–1.1 µg m−3; Figs. 10 and S4). This finding suggests that these sources were largely influenced by origins of air masses, which hence advected to Ersa numerous emissions, potentially of variable strength and from various locations relatively close to Ersa. Furthermore, biogenic sources showed slightly higher summer contributions in 2013 than in 2012 (1.9 and 1.4 µg m−3, respectively), especially in connection with concentrations of monoterpenes (Fig. 10) and consistent with temperature and solar radiation variations (Sect. 3.4.1). Summer concentrations of formaldehyde and acetone were also higher in 2013 than in 2012 (differences of 1.5–1.6 µg m−3), while acetaldehyde and methylglyoxal ones were in the same range (Fig. 10), in agreement with isoprene. Considering regional background contributions and MEK concentrations in summer periods, interannual variations in acetone summer concentrations seem to be related to a change in its biogenic primary/secondary contributions rather than a change in its background levels. These findings suggest enhanced emissions from the local vegetation in summer 2013, partly related to the release of monoterpenes, and/or higher biogenic secondary contributions. For similar reasons, spring contributions of biogenic sources were slightly higher in 2014 than in 2013 (Fig. 10). Transient drought stress of the vegetation could also have influenced BVOC emissions in spring 2014 (Sect. 3.4.1).
4.2 VOC concentration variations in autumn and winter
During autumn and winter periods, total NMHC concentrations increased (7.8 ± 1.2 µg m−3), while OVOC concentrations declined (7.1 ± 0.8 µg m−3). The decline in OVOC concentration levels mainly results from the decrease in acetone and formaldehyde concentrations (3.3 ± 0.8 µg m−3 and 1.0 ± 0.3 µg m−3, contributing 34 %–52 % and 10 %–16 % to total OVOC concentrations, respectively), partly related to a weaker biogenic activity. Chemical reaction rates also dropped in autumn and winter as a consequence of the decrease in OH concentrations related to the decline in available UV light and temperatures (Fig. 3). As a result, VOCs were not removed from the atmosphere as quickly as in spring and summer, in agreement with OVOC concentration levels. PBL height also decreased during these seasons, impacting pollutant vertical dispersion. These conditions, favouring pollutant advection and accumulation, explained the higher contributions of anthropogenic sources perceived at Ersa in autumn and winter, especially those from sources mainly of regional origins (Fig. S4).
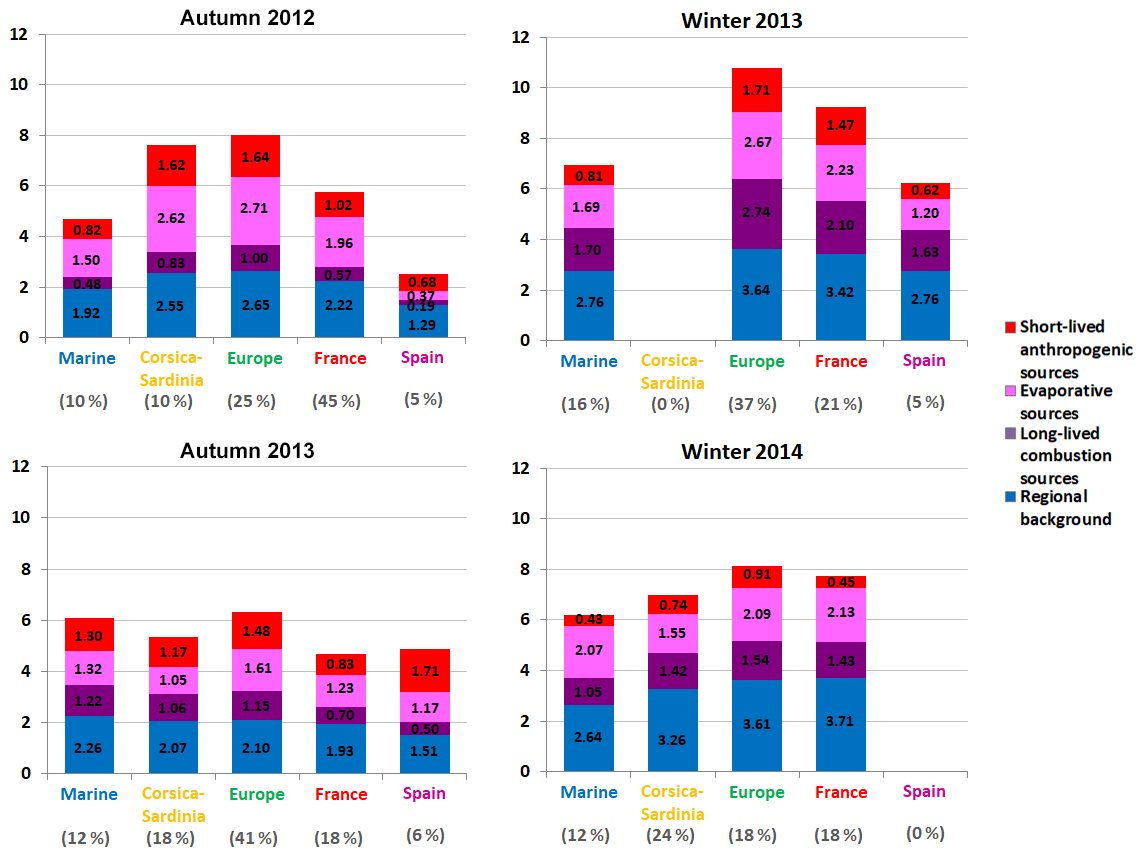
Figure 11Average accumulated contributions (expressed in µg m−3) of the NMHC anthropogenic sources (Sect. 3.5) in autumn and winter as a function of air-mass origin (Sect. 3.2). Winter: 1 January–31 March periods – autumn: 1 October–31 December periods.
The regional background and evaporative sources were identified as the dominant contributors to total NMHC concentrations in autumn and winter (contributions of 31 %–45 % and 22 %–29 %, respectively). Long-lived combustion sources also contributed, particularly in winter (explaining 18 %–24 % of total NMHC concentrations), since the low ambient temperatures typical of winter (Fig. 3) may lead to an increased use of residential heating (Sect. 3.5.4). In order to better identify regional influences, average contributions of the anthropogenic sources in autumn and winter were investigated as a function of air-mass origin in Fig. 11. During these seasons, the Ersa station was mostly influenced by continental air masses coming from Europe and France (their cumulative occurrences were 36 % to 70 %). NMHC anthropogenic sources showed higher accumulated contributions when European air masses were advected to Ersa (Fig. 11), and they could be attributed to potential emission areas located in Italy and possibly more distant ones in central Europe (Sect. 3.5). To a lesser extent, high accumulated contributions of anthropogenic sources were also noticed when Ersa received air masses originating from continental France (potential emission areas located in the south-east of France) and Corsica–Sardinia.
Figure 10 highlights notable interannual variations in VOC concentrations in winter (absolute difference of 3.5 µg m−3), while they were in the same range in autumn (difference below 0.1 µg m−3). Indeed, NMHC winter concentrations were higher in 2013 (9.4 µg m−3) than in 2014 by up to 1.9 µg m−3, similarly to OVOC winter concentrations (7.5 and 6.0 µg m−3 in 2013 and 2014, respectively). Even though winter contributions of long-lived combustion sources, short-lived anthropogenic sources and evaporative sources were significantly reduced in 2014 compared to in 2013 (absolute differences from 0.3 to 0.9 µg m−3; Fig. 10), the seasonal patterns of their variations were similar in 2013 and 2014, as depicted in Fig. S4. These findings could be evidence of homogenous regional background pollution distribution at a synoptic scale. Regional background winter contributions observed in 2013 and 2014 (absolute difference below 0.1 µg m−3; Fig. 10) are also in agreement with this suggestion. Acetone and MEK winter concentrations were lower in 2014 than in 2013 (Fig. 10), which could be related to variations in their anthropogenic primary/secondary sources rather than in their background levels.
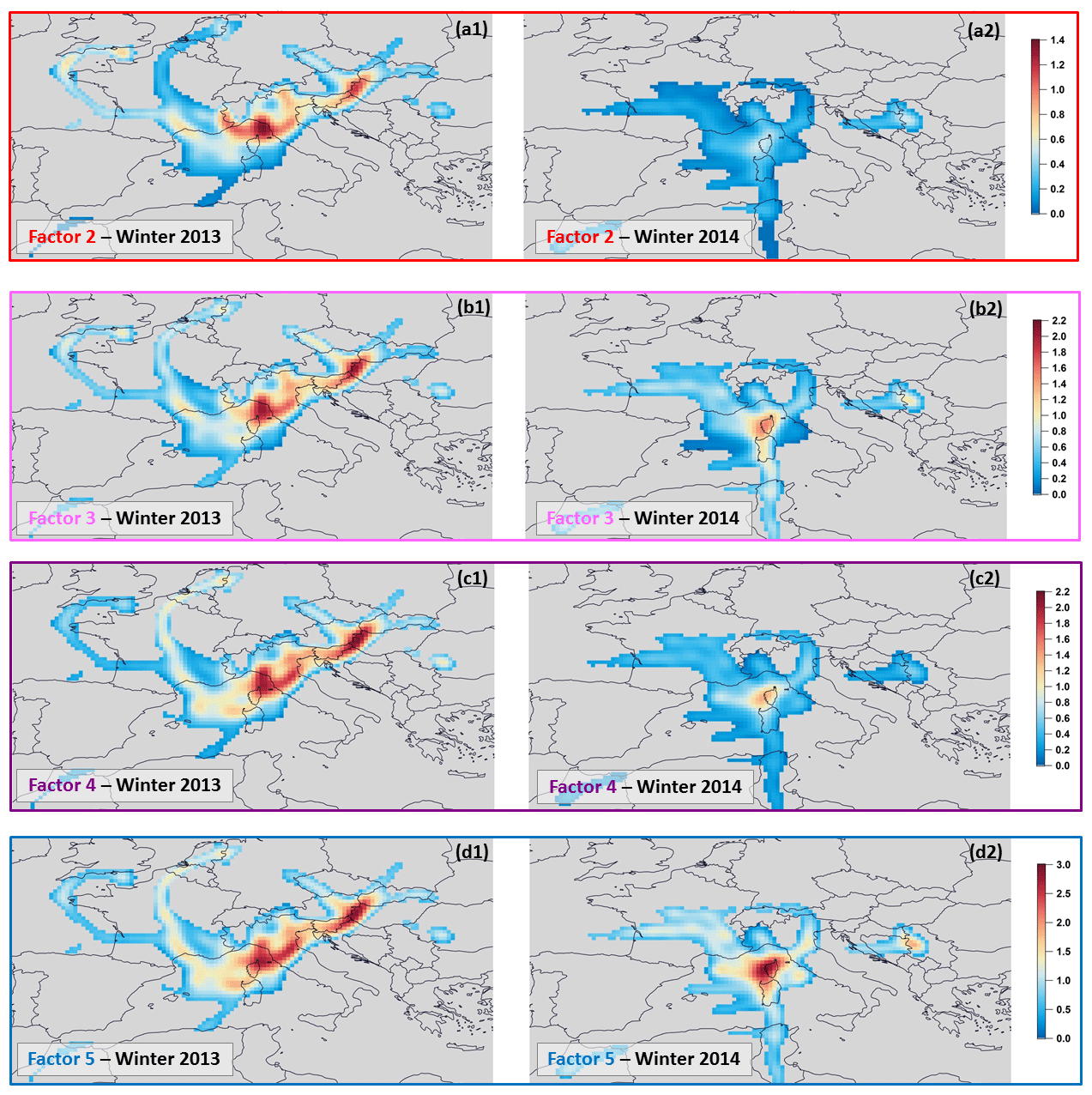
Figure 12Potential source area contributions to NMHC anthropogenic sources during winters 2013 and 2014. Contributions are expressed in µg m−3. Factor 2 – short-lived anthropogenic sources; factor 3 – evaporative sources; factor 4 – long-lived combustion sources; factor 5 – regional background. Winter: 1 January–31 March periods.
The interannual variations in VOC winter concentrations could be partly explained by occurrences and origins of air masses advected to the Ersa station, their enrichment in different anthropogenic sources (based on potential emission areas observed in winters 2013 and 2014; Fig. 12) and their respective contributions (Fig. 11). On the one hand, Ersa was more under the influence of European air masses in winter 2013 than in winter 2014 (occurrences of 37 % and 18 %, respectively). When continental European and French air masses were advected to Ersa, anthropogenic sources showed higher accumulated winter contributions in 2013 (10.8 and 9.6 µg m−3, respectively; Fig. 11) than in 2014 (8.2 and 7.8 µg m−3). In winter 2013, the main potential emission areas for the anthropogenic NMHC sources were located in Italy (Tuscan coasts and the Po Valley), central Europe (Slovenia and western Hungary) and to a lesser extent the south of France. Contrariwise, in winter 2014, Ersa did not seem to be influenced at all by air masses originating from central Europe (especially from Slovenia and Hungary; Fig. 12), and potential emission areas located in the Po Valley and the south-east of France did not contribute significantly to VOC concentrations. On the other hand, the Ersa station received air masses originating from Corsica–Sardinia, i.e. relatively close potential emission areas, more frequently in winter 2014 than in winter 2013 (occurrences of 24 % and 0 %, respectively). The cumulative contribution of anthropogenic sources in winter 2014 under Corsican–Sardinian air-mass influence was only 7.0 µg m−3 (Fig. 12). These findings can suggest less aged (but rather low) emissions have influenced Ersa concentrations in winter 2014, partly explaining why OVOC winter concentrations observed at Ersa were lower in 2014 than in 2013.
Moreover, most countries of western Europe experienced different winters in 2013 and 2014 (see Sect. 3.1 and Fig. S5 of the Supplement). As a reminder, winter 2013 was considered rather rigorous, since e.g. French temperatures were lower by up to 1–1.5 ∘C than the average value for 1981–2010 according to Météo France (http://www.meteofrance.fr/climat-passe-et-futur/bilans-climatiques/bilan-2013/bilan-de-lhiver-2012-2013, last access: 11 October 2020). On the other hand, winter 2014 was rather mild and temperatures were the hottest of the 1951–2014 period (the mean temperature for Europe was ∼ 2.6 ∘C higher than the pre-industrial period; EEA, 2015). As a consequence, the interannual variations in VOC winter concentrations may also result from different influences of meteorological conditions. Indeed, meteorological winter conditions were probably more favourable to inducing higher OH concentrations in 2014 than in 2013, leading to higher photochemical decay and thus lower NMHC concentrations observed at the Ersa station. Nevertheless, OVOC winter concentrations were higher in 2013 than in 2014, which can therefore highlight that they were mainly due to increased contributions from anthropogenic sources of regional origins, thus offsetting the lower degradation of VOCs in winter 2013. Moreover, winter 2014 temperatures along with the lack of cold nights (Sect. 3.1) may have affected the source strength, especially of long-lived combustion sources. Rain event intensities and occurrences in winters 2013 and 2014 could also have impacted enrichment in anthropogenic (primary/secondary) sources of air masses advected to Ersa and hence have influenced Ersa VOC concentrations. Note that a very high monthly rainfall was recorded in northern Italy in winter 2014 (300 % higher than the seasonal average values for the 1981–2010 period; see Fig. S5 of the Supplement). Abundant rainfalls were also noticed in the south-east of France during winter 2014 (the highest ones recorded over the 1959–2014 period, according to Météo France; http://www.meteofrance.fr/climat-passe-et-futur/bilans-climatiques/bilan-2014/bilan-climatique-de-l-hiver-2013-2014, last access: 11 October 2020). As a consequence, these meteorological conditions may have reduced anthropogenic source contributions resulting from potential emission areas located in the Po Valley and the south-east of France in winter 2014.
From June 2012 to June 2014, NMHC measurements were concurrently conducted at 17 other European background monitoring stations (described in Sect. S7), allowing us (i) to examine the representativeness of the Ersa station in terms of seasonal variations in NMHC concentrations impacting continental Europe and (ii) to provide some insights into dominant drivers for VOC concentration variations in Europe built on what we have learned from Ersa's VOC observations.
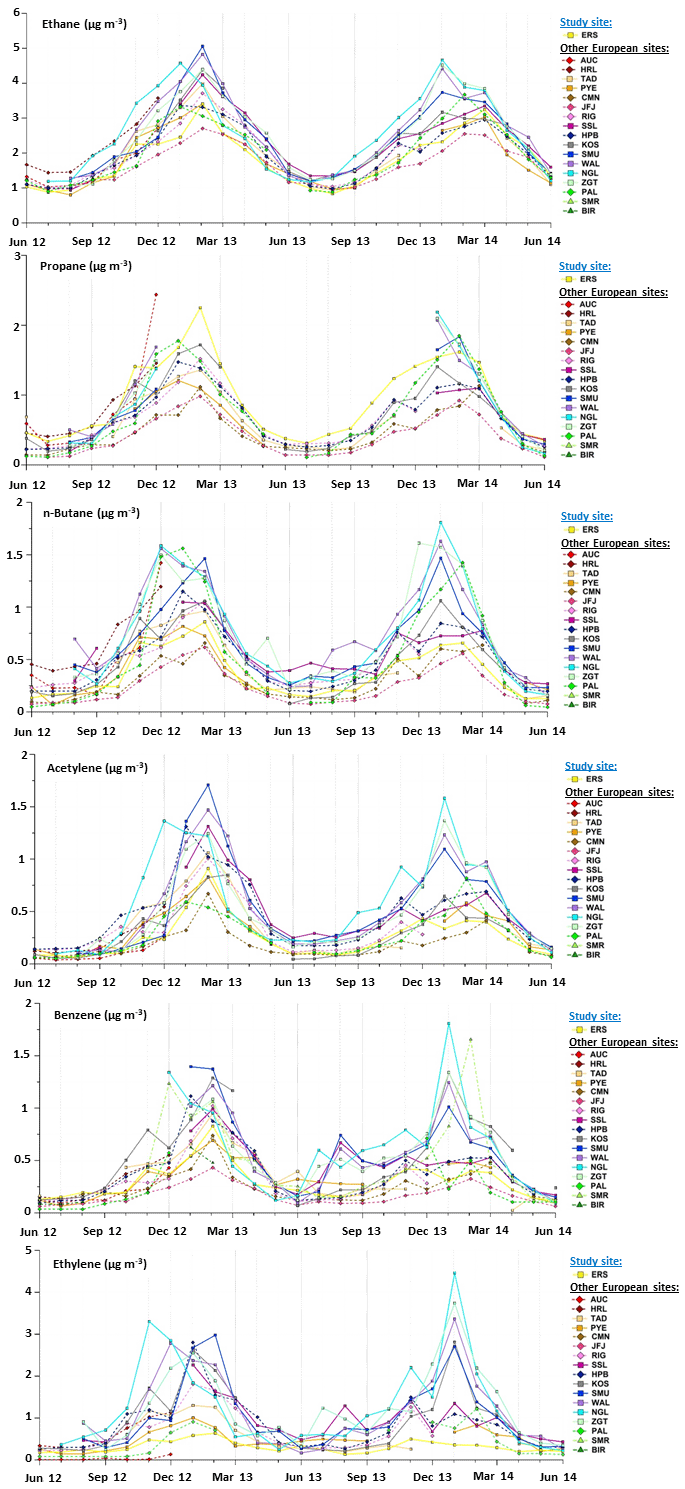
Figure 13Monthly concentration time series of a selection of NMHCs (expressed in µg m−3) measured at Ersa and 17 other European monitoring stations. Stations are indicated according to their GAW identification (see Sect. S7). “ERS” refers to the study site.
Figure 13 depicts monthly concentration time series of a selection of NMHCs measured at the 18 European monitoring stations (including Ersa). Concentrations of NMHCs with typically medium-to-long lifetimes in the atmosphere, i.e. ethane, propane, n-butane, acetylene and benzene, were examined here since they can be significantly influenced by regional contributions, as observed in this study (Sect. 3.5) and elsewhere (Debevec et al., 2017; Michoud et al., 2017; Sauvage et al., 2009). As a reminder, concentrations observed at Ersa were mainly explained in this study by regional background for ethane, long-lived combustion sources for propane, acetylene and benzene, and evaporative sources for n-butane (see Sect. 3.5 and Fig. 7). The study of concentration variations of these source tracers can thus help to highlight temporal and spatial variations in source contributions to NMHC concentrations observed in most of continental Europe. In addition, despite its shorter lifetime compared to other NMHCs of the selection, Fig. 13 also presents ethylene concentration variations in order to investigate short-lived anthropogenic source importance and variability in continental Europe.
Globally, concentrations of these selected NMHCs measured at the 17 European stations showed the same seasonal variations as observed at Ersa (Fig. 13 and Sect 3.4.2), hence ensuring the representativeness of the Ersa station for monitoring regional pollution in continental Europe. On the one hand, NMHC monthly concentration levels in summer were low and relatively homogeneous Europe-wide whatever the location and the typology of the station. The highest absolute differences between NMHC summer concentrations measured at two stations was 0.4–0.7 µg m−3 for ethane, 0.1–0.2 µg m−3 for acetylene, 0.1–0.7 µg m−3 for propane and benzene, 0.2–0.6 µg m−3 for n-butane and 0.3–1.2 µg m−3 for ethylene. They suggest that the temperature was the main driver in regulating summer concentration levels, linked to photochemistry processes and the vertical dispersion (Sect. 4.1). Ethane concentration levels were still relatively important during summer (above 1.0 µg m−3), suggesting that long-range transport (up to intercontinental pollution transport) was among the main parameters governing VOC summer concentrations in continental Europe. On the other hand, anthropogenic NMHC monthly concentration levels in winter appear to be more spatially variable in continental Europe. The highest absolute differences between NMHC winter concentrations measured at two stations were 1.3–2.6 µg m−3 for ethane, 0.6–1.6 µg m−3 for propane, acetylene and benzene, 0.6–1.4 µg m−3 for n-butane, and 1.2–4.1 µg m−3 for ethylene. They may probably highlight spatial variations in anthropogenic source contributions to NMHC winter concentrations observed in continental Europe. The lowest NMHC winter concentrations were observed at stations located in southern and western Europe, including Ersa, other French sites and high-altitude ones (see Fig. 13 and Sect. S7). Note that high-altitude sites may have the particularity of being frequently in free-tropospheric conditions compared to the other European sites. Additionally, south-western France and the Po Valley experienced a wet winter in both 2013 and 2014 (see Sect. 4.2 and Fig. S5 of the Supplement) that may have had a significant impact on the enrichment in NMHC anthropogenic sources of air masses advected to these regions and hence can have participated in the decrease in NMHC winter concentrations monitored at nearby stations. NMHC winter concentrations tended to be more elevated at stations located in central Europe (i.e. those located in Switzerland, Germany and the Czech Republic – see Sect. S7) than those in southern and western Europe. This was especially noticed for NMHCs mainly explained by long-lived combustion sources, evaporative sources and short-lived anthropogenic sources, which could suggest that these central European stations were under different influences. These findings are consistent with Ersa NMHC source contribution variations as a function of air-mass origin and CF analyses examined in this study (Sect. 4.2 and Figs. 9 and S3 of the Supplement). Furthermore, precipitation in central Europe was less frequent and/or less intense in winter than average values for the 1981–2010 period (see Fig. S5 of the Supplement) in both 2013 and 2014, which may have favoured VOC source contribution accumulation and transport and hence can have induced higher VOC winter concentrations measured at nearby monitoring stations. Additionally, high NMHC winter concentrations were also observed in northern Europe, especially for NMHCs mainly explained by evaporative sources and long-lived combustion sources.
Figure 14Average accumulated concentrations of a selection of 15 VOCs (expressed in µg m−3) measured at Ersa and 13 other European monitoring stations in winters 2013 and 2014. Stations are indicated according to their GAW identification (see Sect. S7). “ERS” refers to the study site and its location and results are highlighted. Selected VOCs are those taken into account in the factorial analysis of this study. Note that NMHC measurements did not cover the winter 2014 period at the TAG and RIG stations. For the AUC, HRL, BIR and SMR stations, represented by grey markers, NMHC measurements were not realized both during winters 2013 and 2014 or were conducted with a PTR-MS, and hence we considered accumulated concentrations only of aromatic compounds to be not sufficiently representative of interannual variations in NMHC winter concentrations. Map provided by Google Earth Pro software (v.7.3.3 image Landsat/Copernicus – IBCAO; data SIO, NOAA, U.S. Navy, NGA, GEBCO; © Google Earth).
To go further, Fig. 14 depicts accumulated winter concentrations of 15 selected NMHCs measured in 2013 and 2014 at 14 European monitoring stations (including Ersa) in order to investigate dominant drivers for VOC winter concentration variations in Europe built on what we have learned from Ersa's VOC observations discussed in Sect. 4.2. These selected NMHCs are those taken into account in the PMF analysis of this study (with the exception of ethylbenzene and o-xylene; Sect. S2). Firstly, stations located in south-western France and the Po Valley showed similar NMHC winter concentrations in 2013 and 2014 (total differences ranged from −0.1 to 0.4 µg m−3). At these sites, winter concentrations of NMHCs potentially explained by long-lived combustion sources slightly decreased in 2014 compared to in 2013 (reduction of 0.1–0.4 µg m−3, i.e. of 7 %–24 %), which is consistent with synoptic phenomena (Sect. 3.1) inducing warmer temperatures in winter 2014 than average values for the 1981–2010 period (Fig. S5 of the Supplement). However, NMHC winter concentration levels observed at the Ersa and north-western French stations, along with their interannual variations, were more similar to those in central Europe than those in south-western France and southern Europe (Fig. 14). Indeed, NMHC winter concentrations measured in central Europe were generally significantly higher in 2013 than in 2014 (total differences of 2.6–3.2 µg m−3), with the exception of stations located in north-eastern Germany (i.e. WAL, NGL and ZGT stations – Sect. S7; total differences of −2.8–0.7 µg m−3). These interannual variations seem to be particularly linked to contribution variations of long-lived combustion sources (reduction of 0.7–1.1 µg m−3, i.e. 21 %–44 %) and short-lived anthropogenic sources (reduction of 0.6–1.3 µg m−3, i.e. of 21 %–43 %). NMHC winter concentrations related to evaporative sources also decreased in 2014, but to a lesser extent (reduction of 0.3–0.5 µg m−3, i.e. of 13 %–24 %). These findings are consistent with winter variations in anthropogenic source contributions impacting VOC concentrations at Ersa (Sect. 4.2), especially when the stations received air masses originating from central Europe (Fig. 11). They also highlight that interannual variations in NMHC winter concentrations observed in central Europe were influenced by both regional and local contributions. Furthermore, due to synoptic phenomena that occurred in winters 2013 and 2014 (Sect. 3.1), temperatures in central Europe were, respectively, colder and warmer compared to average values for the 1981–2010 period (Fig. S5 of the Supplement). Interannual variations in winter temperatures were more marked in central Europe than in southern France and southern Europe (Fig. S5 of the Supplement), which could partly explain their respective interannual variations. Moreover, NMHC winter concentrations monitored in Scandinavia (represented by PAL station results in Fig. 14) were higher in 2014 than in 2013 (total difference of 0.7 µg m−3) as well as those measured at stations located in north-eastern Germany (especially NGL and ZGT stations; differences of 1.7–2.8 µg m−3). These increases in winter 2014 in these regions concerned in particular NMHCs related to long-lived combustion (0.2–0.5 µg m−3, i.e. 9 %–25 %) and evaporative sources (0.3–0.4 µg m−3, i.e. 11 %–18 %). Even though these regions experienced a cold winter in 2013 (Fig. S5 of the Supplement), early winter 2014 in northern Europe was also colder than average values for the 1981–2010 period, due to an intense cold wave resulting from a strong anticyclone centred on western Russia and extending from Finland to Crimea. Additionally, winter precipitation in these regions was less frequent and/or intense than average values for the 1981–2010 period (Fig. S5 of the Supplement) in 2014 than in 2013, which may have favoured accumulation and transport and hence induced higher NMHC concentrations measured at nearby monitoring stations.
Within the framework of the ChArMEx project, a monitoring station has been set up and operated from June 2012 to June 2014 at a remote background site of Corsica (Ersa) in the north-western part of the Mediterranean in order to examine seasonal variabilities of 35 selected VOCs (21 NMHCs and 14 carbonyl compounds).
Biogenic VOCs were principally imputed to the local vegetation and exhibited high concentrations and interannual variations in late spring and summer, related to temperature and solar radiation variations. Anthropogenic NMHCs, with atmospheric lifetimes ranging from a few hours to some days, have shown low concentrations in spring and summer since the Ersa station was mostly influenced by aged advected air masses. Anthropogenic sources identified in this study were hence largely influenced by the pollutant depletion and vertical dispersion before reaching the Ersa station, especially those originating from distant emission areas. As a result, the regional background and short-lived anthropogenic sources (from various locations relatively close to Ersa) mainly contributed to NMHC concentrations in spring and summer. Anthropogenic NMHCs were also characterized by high concentration levels in autumn and winter and winter concentrations higher in 2013 than in 2014. The weaker pollutant depletion and vertical dispersion in autumn and winter have favoured the advection to Ersa of air masses enriched in primary anthropogenic sources originating from more distant emission areas (located in France, Italy and to a lesser extent central Europe) than in spring and summer. As a result, the regional background, evaporative sources and long-lived combustion sources were identified as the dominant contributors to NMHC autumn and winter concentrations. Interannual variations in their winter contributions were mainly governed by occurrences and origins of air masses advected to the Ersa station as well as by meteorological conditions (temperature and precipitation) occurring before they reached the Ersa station.
OVOCs largely prevailed in the VOC abundance during the 2-year monitoring period. They have also shown different seasonal and interannual variations, suggesting several source contributions. OVOC concentrations measured at Ersa were generally higher in summer, which could be the result of a high contribution of secondary anthropogenic and primary/secondary biogenic sources, whereas their concentrations during autumn and winter declined and were potentially more influenced by primary/secondary anthropogenic sources, more specifically for MEK and glyoxal. OVOCs showed higher winter concentrations in 2013 than in 2014 as a result of the enrichment of air masses advected to Ersa in primary anthropogenic sources of regional origins.
Selected NMHC variabilities in continental Europe supported Ersa being considered a good regional representative station. However, NMHC winter concentration levels can significantly vary temporally and spatially, pointing out spatial variations in anthropogenic source contributions to NMHC concentrations observed in continental Europe in relation to meteorological conditions. Ersa winter concentration variations were found to be more representative of central Europe than southern/western or northern Europe. These findings point out the interest in conducting multi-site and multi-year measurements that are sufficiently representative of interannual and spatial variations in regional pollution impacting continental Europe in winter. They also pointed out that the PMF-derived factors controlling VOC concentration variations at remote sites may be mainly controlled by the meteorological conditions that occurred during the study period when issued from short observation periods (i.e. up to 2 months).
Concurrent datasets of VOC concentrations from three summer campaigns performed at Ersa helped to support the representativeness of the 2-year monitoring period in terms of summer concentration levels, variations, and source apportionment. They also highlight limitations of this study in the investigation of seasonal variations in Ersa VOC concentrations, in connection with the VOC range and the temporal resolution of their measurements considered in the PMF analysis. After this work, some questions remain regarding the contribution of local BVOC and OVOC sources at seasonal and interannual scales. It would be interesting to conduct additional long-term VOC measurements at Ersa, including OVOCs and tracers of various primary sources, at a finer time resolution and a higher temporal coverage. This would help to complete the understanding of determinants governing VOC concentration variations initiated by both Michoud et al. (2017) and this study.
Access to the 2-year (data from steel canisters: https://mistrals.sedoo.fr/?editDatsId=723&datsId=723&project_name=ChArMEx, Sauvage, 2021a; data from DNPH cartridges: https://mistrals.sedoo.fr/?editDatsId=813&datsId=813&project_name=ChArMEx, Sauvage, 2021b; data from multi-sorbent cartridges: https://mistrals.sedoo.fr/?editDatsId=1272&datsId=1272&project_name=ChArMEx, Sauvage, 2021c) and summer 2013 (data from PTR-TOF-MS: https://mistrals.sedoo.fr/?editDatsId=1261&datsId=1261&project_name=ChArMEx, Dusanter, 2021; data from GC-FID-FID: https://mistrals.sedoo.fr/?editDatsId=1262&datsId=1262&project_name=ChArMEx, Sauvage, 2021d; data from GC-FID-MS: https://mistrals.sedoo.fr/?editDatsId=1263&datsId=1263&project_name=ChArMEx, Sauvage, 2021e; data from DNPH cartridges: https://mistrals.sedoo.fr/?editDatsId=1265&datsId=1265&project_name=ChArMEx, Sauvage, 2021f) VOC datasets used for this publication is open to registered users following the data and publication policy of the ChArMEx programme (http://mistrals.sedoo.fr/ChArMEx/Data-Policy/ChArMEx_DataPolicy.pdf, last access: 11 October 2020). VOC datasets from the Ersa summer field campaigns of 2012 and 2014 are available upon request. Please contact Stéphane Sauvage (stephane.sauvage@imt-lille-douai.fr) for further information.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-1449-2021-supplement.
SS, NL, JS and FD designed the research and were involved in the logistics and the collection of VOC samples in the field. TS calculated uncertainties related to VOC measurements conducted with DNPH cartridges and steel canisters and validated them following the ACTRIS protocol. CD, SS, VG and NL analysed VOC data, conducted and interpreted the VOC PMF analysis and examined dominant factors controlling VOC concentrations. CD wrote the manuscript. All the co-authors were involved in data discussion and edited the paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “CHemistry and AeRosols Mediterranean EXperiments (ChArMEx) (ACP/AMT inter-journal SI)”. It is not associated with a conference.
The authors are thankful for the O3 and CO datasets made available, respectively, by François Gheusi from Laboratoire d'Aérologie and by the ICOS France monitoring network. The authors also want to thank Thierry Leonardis, Emmanuel Tison, Vincent Gaudion, Laurence Depelchin and Isabelle Fronval for their contributions to obtaining VOC datasets, from the organization and the setting of instruments at Ersa to the analysis of VOC samples at the laboratory, but also Thierry Bourianne and the Qualitair Corse team for their involvement on site concerning VOC sample routines as well as the associated logistic procedure. The authors finally thank the ChArMEx project manager Eric Hamonou for his logistical help.
This study received financial support from the MISTRALS/ChArMEx programme funded by CNRS/INSU, CEA, and Météo-France, from ADEME (grant nos. 1262c0009, 1362c0031, and 1462c0031), the French Environmental Ministry, the CaPPA projects, and the Communauté Territoriale de Corse (CORSiCA project). This research has also been supported by the CaPPA project (Chemical and Physical Properties of the Atmosphere) funded by the French National Research Agency (ANR) through the PIA (Programme d'Investissement d'Avenir, grant no. ANR-11-LABX-0005-01) and by the Regional Council Nord-Pas de Calais and the European Funds for Regional Economic Development (FEDER). This research was also funded by the European Union Seventh Framework Programme under the DEFI-VOC project (grant agreement no. 293897) and by CARBO-SOR/Primequal (grant no. 10-MRES-PRIMEQUAL-5-CVS-007-no. 2100082986) and SAF-MED (grant no. ANR-12-BS06-0013-02).
This paper was edited by Xavier Querol and reviewed by two anonymous referees.
Ait-Helal, W., Borbon, A., Sauvage, S., de Gouw, J. A., Colomb, A., Gros, V., Freutel, F., Crippa, M., Afif, C., Baltensperger, U., Beekmann, M., Doussin, J.-F., Durand-Jolibois, R., Fronval, I., Grand, N., Leonardis, T., Lopez, M., Michoud, V., Miet, K., Perrier, S., Prévôt, A. S. H., Schneider, J., Siour, G., Zapf, P., and Locoge, N.: Volatile and intermediate volatility organic compounds in suburban Paris: variability, origin and importance for SOA formation, Atmos. Chem. Phys., 14, 10439–10464, https://doi.org/10.5194/acp-14-10439-2014, 2014.
Atkinson, R.: Gas-phase tropospheric chemistry of organic compounds: a review, Atmos. Environ., 24A, 1–41, https://doi.org/10.1016/j.atmosenv.2007.10.068, 1990.
Atkinson, R.: Atmospheric chemistry of VOCs, and NO(x), Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/S1352-2310(99)00460-4, 2000.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Badol, C., Locoge, N., and Galloo, J.: Using a source-receptor approach to characterise VOC behaviour in a French urban area influenced by industrial emissions Part II: Source contribution assessment using the Chemical Mass Balance (CMB) model, Sci. Total Environ., 9, 429–440, https://doi.org/10.1016/j.scitotenv.2007.09.002, 2008.
Borbon, A., Coddeville, P., Locoge, N., and Galloo, J. C.: Characterising sources, and sinks of rural VOC in eastern France, Chemosphere, 57, 931–942, https://doi.org/10.1016/j.chemosphere.2004.07.034, 2004.
Bressi, M., Sciare, J., Ghersi, V., Mihalopoulos, N., Petit, J.-E., Nicolas, J. B., Moukhtar, S., Rosso, A., Féron, A., Bonnaire, N., Poulakis, E., and Theodosi, C.: Sources and geographical origins of fine aerosols in Paris (France), Atmos. Chem. Phys., 14, 8813–8839, https://doi.org/10.5194/acp-14-8813-2014, 2014.
Buzcu, B. and Fraser, M. P.: Source identification, and apportionment of volatile organic compounds in Houston, TX, Atmos. Environ., 40, 2385–2400, https://doi.org/10.1016/j.atmosenv.2005.12.020, 2006.
Cachier, H., Aulagnier, F., Sarda, R., Gautier, F., Masclet, P., Besombes, J. L., Marchand, N., Despiau, S., Croci, D., Mallet, M., Laj, P., Marinoni, A., Deveau, P. A., Roger, J. C., Putaud, J. P., Van Dingenen, R., Dell'Acqua, A., Viidanoja, J., Martins-Dos Santos, S., Liousse, C., Cousin, F., Rosset, R., Gardrat, E., and Galy-Lacaux, C.: Aerosol studies during the ESCOMPTE experiment: An overview, Atmos. Res., 74, 547–563, https://doi.org/10.1016/j.atmosres.2004.06.013, 2005.
Charron, A., Plaisance, H., Sauvage, S., Coddeville, P., Galloo, J. C., and Guillermo, R.: A study of the source-receptor relationships influencing the acidity of precipitation collected at a rural site in France, Atmos. Environ., 34, 3665–3674, https://doi.org/10.1016/S1352-2310(00)00096-0, 2000.
Cholakian, A., Beekmann, M., Colette, A., Coll, I., Siour, G., Sciare, J., Marchand, N., Couvidat, F., Pey, J., Gros, V., Sauvage, S., Michoud, V., Sellegri, K., Colomb, A., Sartelet, K., Langley DeWitt, H., Elser, M., Prévot, A. S. H., Szidat, S., and Dulac, F.: Simulation of fine organic aerosols in the western Mediterranean area during the ChArMEx 2013 summer campaign, Atmos. Chem. Phys., 18, 7287–7312, https://doi.org/10.5194/acp-18-7287-2018, 2018.
Colette, A., Granier, C., Hodnebrog, Ø., Jakobs, H., Maurizi, A., Nyiri, A., Rao, S., Amann, M., Bessagnet, B., D'Angiola, A., Gauss, M., Heyes, C., Klimont, Z., Meleux, F., Memmesheimer, M., Mieville, A., Rouïl, L., Russo, F., Schucht, S., Simpson, D., Stordal, F., Tampieri, F., and Vrac, M.: Future air quality in Europe: a multi-model assessment of projected exposure to ozone, Atmos. Chem. Phys., 12, 10613–10630, https://doi.org/10.5194/acp-12-10613-2012, 2012.
Colette, A., Bessagnet, B., Vautard, R., Szopa, S., Rao, S., Schucht, S., Klimont, Z., Menut, L., Clain, G., Meleux, F., Curci, G., and Rouïl, L.: European atmosphere in 2050, a regional air quality and climate perspective under CMIP5 scenarios, Atmos. Chem. Phys., 13, 7451–7471, https://doi.org/10.5194/acp-13-7451-2013, 2013.
Coy, L. and Pawson, S.: The major stratospheric sudden warming of January 2013: Analyses, and forecasts in the GEOS-5 data assimilation system, Mon. Weather Rev., 143, 491–510, https://doi.org/10.1175/MWR-D-14-00023.1, 2015.
Debevec, C., Sauvage, S., Gros, V., Sciare, J., Pikridas, M., Stavroulas, I., Salameh, T., Leonardis, T., Gaudion, V., Depelchin, L., Fronval, I., Sarda-Esteve, R., Baisnée, D., Bonsang, B., Savvides, C., Vrekoussis, M., and Locoge, N.: Origin and variability in volatile organic compounds observed at an Eastern Mediterranean background site (Cyprus), Atmos. Chem. Phys., 17, 11355–11388, https://doi.org/10.5194/acp-17-11355-2017, 2017.
Debevec, C., Sauvage, S., Gros, V., Sellegri, K., Sciare, J., Pikridas, M., Stavroulas, I., Leonardis, T., Gaudion, V., Depelchin, L., Fronval, I., Sarda-Esteve, R., Baisnée, D., Bonsang, B., Savvides, C., Vrekoussis, M., and Locoge, N.: Driving parameters of biogenic volatile organic compounds and consequences on new particle formation observed at an eastern Mediterranean background site, Atmos. Chem. Phys., 18, 14297–14325, https://doi.org/10.5194/acp-18-14297-2018, 2018.
de Gouw, J. A., Middlebrook, A. M., Warneke, C., Goldan, P. D., Kuster, W. C., Roberts, J. M., Fehsenfeld, F. C., Worsnop, D. R., Canagaratna, M. R., Pszenny, A. A. P., Keene, W. C., Marchewka, M., Bertman, S. B., and Bates, T. S.: Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002, J. Geophys. Res.-Atmos., 110, D16305, https://doi.org/10.1029/2004JD005623, 2005.
Detournay, A.: Etude de COV oxygénés et biogéniques en milieu rural: du développement métrologique à l'évaluation de l'impact sur la chimie atmosphérique, PhD thesis, Université Lille 1, available at: http://www.theses.fr/2011LIL10139 (last access: 14 June 2020), 2011.
Detournay, A., Sauvage, S., Locoge, N., Gaudion, V., Leonardis, T., Fronval, I., Kaluzny, P., and Galloo, J. C.: Development of a sampling method for the simultaneous monitoring of straight-chain alkanes, straight-chain saturated carbonyl compounds, and monoterpenes in remote areas, J. Environ. Monit., 13, 983–990, https://doi.org/10.1039/c0em00354a, 2011.
Detournay, A., Sauvage, S., Riffault, V., Wroblewski, A., and Locoge, N.: Source, and behavior of isoprenoid compounds at a southern France remote site, Atmos. Environ., 77, 272–282, https://doi.org/10.1016/j.atmosenv.2013.03.041, 2013.
Doche, C., Dufour, G., Foret, G., Eremenko, M., Cuesta, J., Beekmann, M., and Kalabokas, P.: Summertime tropospheric-ozone variability over the Mediterranean basin observed with IASI, Atmos. Chem. Phys., 14, 10589–10600, https://doi.org/10.5194/acp-14-10589-2014, 2014.
Dollard, G. J., Dumitrean, P., Telling, S., Dixon, J., and Derwent, R. G.: Observed trends in ambient concentrations of C2-C8 hydrocarbons in the United Kingdom over the period from 1993 to 2004, Atmos. Environ., 41, 2559–2569, https://doi.org/10.1016/j.atmosenv.2006.11.020, 2007.
Draxler, R. R. and Hess, G. D.: An overview of the HYSPLIT_4 modelling system for trajectories, dispersion, and deposition, Aust. Meteorol. Mag., 47, 295–308, 1998.
Dulac, F.: An overview of the Chemistry-Aerosol Mediterranean Experiment (ChArMEx), EGU General Assembly 2014, 27 April–2 May 2014, Vienna, Austria, EGU2014-11441, 2014.
Dusanter, S.: PTRtoFMS – EMD – Ersa, Cape Corsica, available at: https://mistrals.sedoo.fr/?editDatsId=1261&datsId=1261&project_name=ChArMEx, last access: 31 January 2021.
EEA: Global and European temperatures, available at: https://www.eea.europa.eu/data-and-maps/indicators/global-and-european-temperature-1/assessment (last access: 11 October 2020), 2015.
Elbir, T., Cetin, B., Cetin, E., Bayram, A., and Odabasi, M.: Characterization of volatile organic compounds (VOCs), and their sources in the air of Izmir, Turkey, Environ. Monit. Assess., 133, 149–160, https://doi.org/10.1007/s10661-006-9568-z, 2007.
Encyclopædia Britannica: Corsica, https://www.britannica.com/place/Corsica (last access: 11 October 2020), 2018.
Eyring, V., Köhler, H. W., Van Aardenne, J., and Lauer, A.: Emissions from international shipping: 1. The last 50 years, J. Geophys. Res.-Atmos., 110, D17305, https://doi.org/10.1029/2004JD005619, 2005.
Ferracci, V., Bolas, C. G., Freshwater, R. A., Staniaszek, Z., King, T., Jaars, K., Otu-Larbi, F., Beale, J, Malhi, Y., William Waine, T., Jones, R. L., Ashworth, K., and Harris, N. R. P.: Continuous isoprene measurements in a UK temperate forest for a whole growing season: Effects of drought stress during the 2018 heatwave, Geophys. Res. Lett., 47, e2020GL088885, https://doi.org/10.1029/2020GL088885, 2020.
Finlayson-Pitts, B. J. and Pitts Jr, J. N.: Overview of the chemistry of polluted and remote atmospheres, in: Chemistry of the Lower and Upper Atmosphere – Theory, Experiments and Applications, Academic Press, San Diego, CA, USA, 1–14, 2000.
Friedrich, R. and Obermeier, A.: Anthropogenic emissions of volatile organic compounds, in: Reactive Hydrocarbons in the Atmosphere, Academic Press, San Diego, CA, USA, 1-39, https://doi.org/10.1016/B978-012346240-4/50002-3, 1999.
Fu, T.-M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal , and implications for formation of secondary organic aerosols, J. of Geophys. Res., 13, D15303, https://doi.org/10.1029/2007JD009505, 2008.
Geron, C., Rasmussen, R., Arnts, R. R., and Guenther, A.: A review and synthesis of monoterpene speciation from forests in the United States, Atmos. Environ., 34, 1761–1781, https://doi.org/10.1016/S1352-2310(99)00364-7, 2000a.
Geron, C., Guenther, A., Sharkey, T. O. M., and Arnts, R. R.: Temporal variability in basal isoprene emission factor, Tree Physiol., 20, 799–805, https://doi.org/10.1093/treephys/20.12.799, 2000b.
Giorgi, F.: Climate change hot-spots, Geophys. Res. Lett., 33, L08707, https://doi.org/10.1029/2006GL025734, 2006.
Goldstein, A. H. and Schade, G. W.: Quantifying biogenic, and anthropogenic contributions to acetone mixing ratios in a rural environment, Atmos. Environ., 34, 4997–5006, https://doi.org/10.1016/S1352-2310(00)00321-6, 2000.
Grant, A., Yates, E. L., Simmonds, P. G., Derwent, R. G., Manning, A. J., Young, D., Shallcross, D. E., and O'Doherty, S.: A five year record of high-frequency in situ measurements of non-methane hydrocarbons at Mace Head, Ireland, Atmos. Meas. Tech., 4, 955–964, https://doi.org/10.5194/amt-4-955-2011, 2011.
Guenther, A., Geron, C., Pierce, T., Lamb, B., Harley, P., and Fall, R.: Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America, Atmos. Environ., 34, 2205–2230, https://doi.org/10.1016/S1352-2310(99)00465-3, 2000.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Hakola, H., Hellén, H., and Laurila, T.: Ten years of light hydrocarbons (C2-C6) concentration measurements in background air in Finland, Atmos. Environ., 40, 3621–3630, https://doi.org/10.1016/j.atmosenv.2005.08.019, 2006.
Hellén, H., Hakola, H., and Laurila, T.: Determination of source contributions of NMHCs in Helsinki (60∘ N, 25∘ E) using chemical mass balance, and the Unmix multivariate receptor models, Atmos. Environ., 37, 1413–1424, https://doi.org/10.1016/S1352-2310(02)01049-X, 2003.
Hellén, H., Kouznetsov, R., Anttila, P., and Hakola, H.: Increasing influence of easterly air masses on NMHC concentrations at the Pallas-Sodankylä GAW station, Boreal Environ. Res., 20, 542–552, 2015.
Helmig, D., Tanner, D. M., Honrath, R. E., Owen, R. C., and Parrish, D. D.: Non methane hydrocarbons at Pico Mountain, Azores: 1. Oxidation chemistry in the North Atlantic region, J. Geophys. Res.-Atmos., 113, D20S91, https://doi.org/10.1029/2007JD008930, 2008.
Jacob, D. J., Field, B. D., Jin, E. M., Bey, I., Li, Q., Logan, J. A., Yantosca, R. M., and Singh, H. B.: Atmospheric budget of acetone, J. Geophys. Res.-Atmos., 107, ACH 5-1–ACH5-17, https://doi.org/10.1029/2001JD000694, 2002.
Jaidan, N., El Amraoui, L., Attié, J.-L., Ricaud, P., and Dulac, F.: Future changes in surface ozone over the Mediterranean Basin in the framework of the Chemistry-Aerosol Mediterranean Experiment (ChArMEx), Atmos. Chem. Phys., 18, 9351–9373, https://doi.org/10.5194/acp-18-9351-2018, 2018.
Jardine, K., Harley, P., Karl, T., Guenther, A., Lerdau, M., and Mak, J. E.: Plant physiological and environmental controls over the exchange of acetaldehyde between forest canopies and the atmosphere, Biogeosciences, 5, 1559–1572, https://doi.org/10.5194/bg-5-1559-2008, 2008.
Kalogridis, A.: Caractérisation des composés organiques volatils en région méditerranéenne, PhD thesis, Université Paris Sud – Paris XI, available at: https://tel.archives-ouvertes.fr/tel-01165005 (last access: 14 June 2020), 2014.
Kesselmeier, J. and Staudt, M.: Biogenic Volatile Organic Compounds (VOC): An Overview on Emission, Physiology and Ecology, J. Atmos. Chem., 33, 23–88, https://doi.org/10.1023/A:1006127516791, 1999.
Kopf, S.: World Development Report 2010: Development, and Climate Change, edited by: Bank, T. W., available at: https://openknowledge.worldbank.org/handle/10986/4387 (last access: 31 January 2021), 2010.
Lambert, D., Mallet, M., Ducrocq, V., Dulac, F., Gheusi, F., and Kalthoff, N.: CORSiCA: a Mediterranean atmospheric and oceanographic observatory in Corsica within the framework of HyMeX and ChArMEx, Adv. Geosci., 26, 125–131, https://doi.org/10.5194/adgeo-26-125-2011, 2011.
Lanz, V. A., Henne, S., Staehelin, J., Hueglin, C., Vollmer, M. K., Steinbacher, M., Buchmann, B., and Reimann, S.: Statistical analysis of anthropogenic non-methane VOC variability at a European background location (Jungfraujoch, Switzerland), Atmos. Chem. Phys., 9, 3445–3459, https://doi.org/10.5194/acp-9-3445-2009, 2009.
Laothawornkitkul, J., Taylor, J. E., Paul, N. D., and Hewitt, C. N.: Biogenic volatile organic compounds in the Earth system, New Phytol., 183, 27–51, https://doi.org/10.1111/j.1469-8137.2009.02859.x, 2009.
Legreid, G., Lööv, J. B., Staehelin, J., Hueglin, C., Hill, M., Buchmann, B., Prevot, A. S. H., and Reimann, S.: Oxygenated volatile organic compounds (OVOCs) at an urban background site in Zürich (Europe): Seasonal variation, and source allocation, Atmos. Environ., 41, 8409–8423, https://doi.org/10.1016/j.atmosenv.2007.07.026, 2007.
Legreid, G., Folini, D., Staehelin, J., Lööv, J. B., Steinbacher, M., and Reimann, S.: Measurements of organic trace gases including oxygenated volatile organic compounds at the high alpine site Jungfraujoch (Switzerland): Seasonal variation, and source allocations, J. Geophys. Res.-Atmos., 113, D05307, https://doi.org/10.1029/2007JD008653, 2008.
Lelieveld, J., Hadjinicolaou, P., Kostopoulou, E., Giannakopoulos, C., Pozzer, A., Tanarhte, M., and Tyrlis, E.: Model projected heat extremes, and air pollution in the eastern Mediterranean, and Middle East in the twenty-first century, Reg. Environ. Chang., 14, 1937–1949, https://doi.org/10.1007/s10113-013-0444-4, 2014.
Leuchner, M., Gubo, S., Schunk, C., Wastl, C., Kirchner, M., Menzel, A., and Plass-Dülmer, C.: Can positive matrix factorization help to understand patterns of organic trace gases at the continental Global Atmosphere Watch site Hohenpeissenberg?, Atmos. Chem. Phys., 15, 1221–1236, https://doi.org/10.5194/acp-15-1221-2015, 2015.
Loreto, F. and Schnitzler, J.-P.: Abiotic stresses and induced BVOCs, Trends Plant Sci., 15, 154–166, https://doi.org/10.1016/j.tplants.2009.12.006, 2010.
Lo Vullo, E., Furlani, F., Arduini, J., Giostra, U., Cristofanelli, P., Williams, M. L., and Maione, M.: Non-methane volatile organic compounds in the background atmospheres of a Southern European mountain site (Mt. Cimone, Italy): Annual, and seasonal variability, Aerosol Air Qual. Res., 16, 581–592, https://doi.org/10.4209/aaqr.2015.05.0364, 2016.
Malley, C. S., Braban, C. F., Dumitrean, P., Cape, J. N., and Heal, M. R.: The impact of speciated VOCs on regional ozone increment derived from measurements at the UK EMEP supersites between 1999 and 2012, Atmos. Chem. Phys., 15, 8361–8380, https://doi.org/10.5194/acp-15-8361-2015, 2015.
Matsunaga, S., Mochida, M., and Kawamura, K.: Growth of organic aerosols by biogenic semi-volatile carbonyls in the forestal atmosphere, Atmos. Env., 37, 2045–2050, 2003.
Michoud, V., Sciare, J., Sauvage, S., Dusanter, S., Léonardis, T., Gros, V., Kalogridis, C., Zannoni, N., Féron, A., Petit, J.-E., Crenn, V., Baisnée, D., Sarda-Estève, R., Bonnaire, N., Marchand, N., DeWitt, H. L., Pey, J., Colomb, A., Gheusi, F., Szidat, S., Stavroulas, I., Borbon, A., and Locoge, N.: Organic carbon at a remote site of the western Mediterranean Basin: sources and chemistry during the ChArMEx SOP2 field experiment, Atmos. Chem. Phys., 17, 8837–8865, https://doi.org/10.5194/acp-17-8837-2017, 2017.
Nabat, P., Somot, S., Mallet, M., Chiapello, I., Morcrette, J. J., Solmon, F., Szopa, S., Dulac, F., Collins, W., Ghan, S., Horowitz, L. W., Lamarque, J. F., Lee, Y. H., Naik, V., Nagashima, T., Shindell, D., and Skeie, R.: A 4-D climatology (1979–2009) of the monthly tropospheric aerosol optical depth distribution over the Mediterranean region from a comparative evaluation and blending of remote sensing and model products, Atmos. Meas. Tech., 6, 1287–1314, https://doi.org/10.5194/amt-6-1287-2013, 2013.
Navazo, M., Durana, N., Alonso, L., Gómez, M. C., García, J. A., Ilardia, J. L., Gangoiti, G., and Iza, J.: High temporal resolution measurements of ozone precursors in a rural background station. A two-year study, Environ. Monit. Assess., 136, 53–68, https://doi.org/10.1007/s10661-007-9720-4, 2008.
Niinemets, Ü., Loreto, F., and Reichstein, M.: Physiological and physicochemical controls on foliar volatile organic compound emissions, Trends Plant Sci., 9, 180–186, https://doi.org/10.1016/j.tplants.2004.02.006, 2004.
Norris, G., Duvall, R., Brown, S., and Bai, S.: EPA Positive Matrix Factorization (PMF) 5.0 Fundamentals, and User Guide Prepared for the US Environmental Protection Agency Office of Research, and Development, Washington, DC, available at: https://www.epa.gov/sites/production/files/2015-02/documents/pmf_5.0_user_guide.pdf (last access: 11 October 2020), 2014.
ORT Corse: Les trafics passagers de la Corse au cours de la saison 2013, available at: http://www.corse.developpement-durable.gouv.fr/IMG/pdf/Ete2013.pdf (last access: 11 October 2020), 2013.
Owen, S. M., Boissard, C., and Hewitt, C. N.: Volatile organic compounds (VOCs) emitted from 40 Mediterranean plant species: VOC speciation, and extrapolation to habitat scale, Atmos. Environ., 35, 5393–5409, 2001.
Owen, S. M., Harley, P., Guenther, A., and Hewitt, C. N.: Light dependency of VOC emissions from selected Mediterranean plant species, Atmos. Environ., 36, 3147–3159, https://doi.org/10.1016/S1352-2310(02)00235-2, 2002.
Paatero, P.: Least squares formulation of robust non-negative factor analysis, Chemom. Intell. Lab. Syst., 37, 23–35, https://doi.org/10.1016/S0169-7439(96)00044-5, 1997.
Paatero, P.: The Multilinear Engine–A Table-Driven, Least Squares Program for Solving Multilinear Problems, Including the n-Way Parallel Factor Analysis Model, J. Comput. Graph. Stat., 8, 854–888, https://doi.org/10.1080/10618600.1999.10474853, 1999.
Paatero, P. and Tapper, U.: Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values, Environmetrics, 5, 111–126, https://doi.org/10.1002/env.3170050203, 1994.
Pang, Y., Fuentes, M., and Rieger, P.: Trends in the emissions of Volatile Organic Compounds (VOCs) from light-duty gasoline vehicles tested on chassis dynamometers in Southern California, Atmos. Environ., 83, 127–135, https://doi.org/10.1016/j.atmosenv.2013.11.002, 2014.
Petit, J. E., Favez, O., Albinet, A., and Canonaco, F.: A user-friendly tool for comprehensive evaluation of the geographical origins of atmospheric pollution: Wind, and trajectory analyses, Environ. Model. Softw., 88, 183–187, https://doi.org/10.1016/j.envsoft.2016.11.022, 2017.
Plass-Dülmer, C., Michl, K., Ruf, R., and Berresheim, H.: C2-C8 Hydrocarbon measurement, and quality control procedures at the Global Atmosphere Watch Observatory Hohenpeissenberg, J. Chromatogr. A, 953, 175–197, https://doi.org/10.1016/S0021-9673(02)00128-0, 2002.
Rasmijn, L. M., van der Schrier, G., Barkmeijer, J., Sterl, A., and Hazeleger, W.: Simulating the extreme 2013/2014 winter in a future climate, J. Geophys. Res., 121, 5680–5698, https://doi.org/10.1002/2015JD024492, 2016.
Reimann, S., Wegener, R., Claude, A., and Sauvage, S.: ACTRIS-2 WP3 – Deliverable 3.17. Updated Measurement Guideline for NOx, and VOCs, available at: https://www.actris.eu/Portals/46/Documentation/actris2/Deliverables/public/WP3_D3.17_M42.pdf?ver=2018-11-12-143115-077 (last access: 14 June 2020), 2018.
Rottenberger, S., Kuhn, U., Wolf, A., Schebeske, G., Oliva, S. T., Tavares, T. M., and Kesselmeier, J.: Exchange of short-chain aldehydes between Amazonian vegetation, and the atmosphere, Ecol. Appl., 14, 247–262, 2004.
Rottenberger, S., Kleiss, B., Kuhn, U., Wolf, A., Piedade, M. T. F., Junk, W., and Kesselmeier, J.: The effect of flooding on the exchange of the volatile C2-compounds ethanol, acetaldehyde and acetic acid between leaves of Amazonian floodplain tree species and the atmosphere, Biogeosciences, 5, 1085–1100, https://doi.org/10.5194/bg-5-1085-2008, 2008.
Royer, P., Raut, J.-C., Ajello, G., Berthier, S., and Chazette, P.: Synergy between CALIOP and MODIS instruments for aerosol monitoring: application to the Po Valley, Atmos. Meas. Tech., 3, 893–907, https://doi.org/10.5194/amt-3-893-2010, 2010.
Safieddine, S., Boynard, A., Coheur, P.-F., Hurtmans, D., Pfister, G., Quennehen, B., Thomas, J. L., Raut, J.-C., Law, K. S., Klimont, Z., Hadji-Lazaro, J., George, M., and Clerbaux, C.: Summertime tropospheric ozone assessment over the Mediterranean region using the thermal infrared IASI/MetOp sounder and the WRF-Chem model, Atmos. Chem. Phys., 14, 10119–10131, https://doi.org/10.5194/acp-14-10119-2014, 2014.
Sauvage, S.: NMHC – EMD – EOP – Ersa, Cape Corsica, available at: https://mistrals.sedoo.fr/?editDatsId=723&datsId=723&project_name=ChArMEx, last access: 31 January 2021a.
Sauvage, S.: Light carbonyls – EMD – EOP – Ersa, Cape Corsica, available at: https://mistrals.sedoo.fr/?editDatsId=813&datsId=813&project_name=ChArMEx, last access: 31 January 2021b.
Sauvage, S.: VOC Offline SolidAdsorbant – EMD – EOP – Ersa, Cape Corsica, available at: https://mistrals.sedoo.fr/?editDatsId=1272&datsId=1272&project_name=ChArMEx, last access: 31 January 2021c.
Sauvage, S.: VOC OnlineGC – EMD – Ersa, Cape Corsica, available at: https://mistrals.sedoo.fr/?editDatsId=1262&datsId=1262&project_name=ChArMEx, last access: 31 January 2021d.
Sauvage, S.: OVOC Online – EMD – Ersa, Cape Corsica, available at: https://mistrals.sedoo.fr/?editDatsId=1263&datsId=1263&project_name=ChArMEx, last access: 31 January 2021e.
Sauvage, S.: OVOC Offline DNPH – EMD – Ersa, Cape Corsica, available at: https://mistrals.sedoo.fr/?editDatsId=1265&datsId=1265&project_name=ChArMEx, last access: 31 January 2021f.
Sauvage, S., Plaisance, H., Locoge, N., Wroblewski, A., Coddeville, P., and Galloo, J. C.: Long term measurement, and source apportionment of non-methane hydrocarbons in three French rural areas, Atmos. Environ., 43, 2430–2441, https://doi.org/10.1016/j.atmosenv.2009.02.001, 2009.
Schade, G. W. and Goldstein, A. H.: Fluxes of oxygenated volatile organic compounds from a ponderosa pine plantation, J. Geophys. Res., 106, 3111–3123, 2001.
Schade, G. W. and Goldstein, A. H.: Seasonal measurements of acetone, and methanol: Abundances, and implications for atmospheric budgets, Glob. Biogeochem. Cy., 20, GB1011, https://doi.org/10.1029/2005GB002566, 2006.
Seco, R., Peñuelas, J., and Filella, I.: Short-chain oxygenated VOCs: Emission, and uptake by plants, and atmospheric sources, sinks, and concentrations, Atmos. Environ., 41, 2477–2499, https://doi.org/10.1016/j.atmosenv.2006.11.029, 2007.
Seco, R., Peñuelas, J., Filella, I., Llusià, J., Molowny-Horas, R., Schallhart, S., Metzger, A., Müller, M., and Hansel, A.: Contrasting winter and summer VOC mixing ratios at a forest site in the Western Mediterranean Basin: the effect of local biogenic emissions, Atmos. Chem. Phys., 11, 13161–13179, https://doi.org/10.5194/acp-11-13161-2011, 2011.
Seibert, P., Kromp-Kolb, H., Baltensperger, U., Jost, D. T., and Schwikowski, M.: Trajectory analysis of high-alpine air pollution data, in: Air Pollution Modeling, and Its Application X, edited by Gryning, S.-E. and Millán, M. M., Springer, Boston, USA 595–596, 1994.
Singh, H. B., Salas, L., Chatfield, R. B., Czech, E., Fried, A., Walega, J., Evans, M. J., Field, B. D., Jacob, D. J., Blake, D., Heikes, B., Talbot, R., Sachse, G., Crawford, J. H., Avery, M. A., Sandholm, S., and Fuelberg, H.: Analysis of the atmospheric distribution, sources, and sinks of oxygenated volatile organic chemicals based on measurements over the Pacific during TRACE-P, J. Geophys. Res.-Atmos., 109, D15S07, https://doi.org/10.1029/2003JD003883, 2004.
Solberg, S., Dye, C., Schmidbauer, N., Herzog, A., and Gehrig, R.: Carbonyls, and Nonmethane Hydrocarbons at Rural European Sites from the Mediterranean to the Arctic, J. Atmos. Chem., 25, 33–66, https://doi.org/10.1007/BF00053285, 1996.
Solberg, S., Dye, C., Walker, S. E., and Simpson, D.: Long-term measurements, and model calculations of formaldehyde at rural European monitoring sites, Atmos. Environ., 35, 195–207, https://doi.org/10.1016/S1352-2310(00)00256-9, 2001.
Spivakovsky, C. M., Logan, J. A., Montzka, S. A., Balkanski, Y. J., Foreman-Fowler, M., Jones, D. B. A., Horowitz, L. W., Fusco, A. C., Brenninkmeijer, C. A. M., Prather, M. J., Wofsy, S. C., and McElroy, M. B.: Three-dimensional climatological distribution of tropospheric OH: Update, and evaluation, J. Geophys. Res.-Atmos., 105, 8931–8980, https://doi.org/10.1029/1999JD901006, 2000.
Stein, A. F., Draxler, R. R., Rolph, G. D., Stunder, B. J. B., Cohen, M. D., and Ngan, F.: Noaa's hysplit atmospheric transport, and dispersion modeling system, B. Am. Meteorol. Soc., 96, 2059–2077, https://doi.org/10.1175/BAMS-D-14-00110.1, 2015.
Tørseth, K., Aas, W., Breivik, K., Fjæraa, A. M., Fiebig, M., Hjellbrekke, A. G., Lund Myhre, C., Solberg, S., and Yttri, K. E.: Introduction to the European Monitoring and Evaluation Programme (EMEP) and observed atmospheric composition change during 1972–2009, Atmos. Chem. Phys., 12, 5447–5481, https://doi.org/10.5194/acp-12-5447-2012, 2012.
von Engeln, A. and Teixeira, J.: A planetary boundary layer height climatology derived from ECMWF reanalysis data, J. Climate, 26, 6575–6590, https://doi.org/10.1175/JCLI-D-12-00385.1, 2013.
Van Oldenborgh, G. J., Haarsma, R., De Vries, H., and Allen, M. R.: Cold extremes in North America vs. mild weather in Europe, B. Am. Meteorol. Soc., 96, 707–714, https://doi.org/10.1175/BAMS-D-14-00036.1, 2015.
Waked, A., Favez, O., Alleman, L. Y., Piot, C., Petit, J.-E., Delaunay, T., Verlinden, E., Golly, B., Besombes, J.-L., Jaffrezo, J.-L., and Leoz-Garziandia, E.: Source apportionment of PM10 in a north-western Europe regional urban background site (Lens, France) using positive matrix factorization and including primary biogenic emissions, Atmos. Chem. Phys., 14, 3325–3346, https://doi.org/10.5194/acp-14-3325-2014, 2014.
Waked, A., Sauvage, S., Borbon, A., Gauduin, J., Pallares, C., Vagnot, M.-P., Léonardis, T., and Locoge, N.: Multi-year levels, and trends of non-methane hydrocarbon concentrations observed in ambient air in France, Atmos. Environ., 141, 263–275, https://doi.org/10.1016/j.atmosenv.2016.06.059, 2016.
Waked, A., Bourin, A., Michoud, V., Perdrix, E., Alleman, L. Y., Sauvage, S., Delaunay, T., Vermeesch, S., Petit, J.-E., and Riffault, V.: Investigation of the geographical origins of PM10 based on long, medium and short-range air mass back-trajectories impacting Northern France during the period 2009–2013, Atmos. Environ., 193, 143–152, doi :10.1016/j.atmosenv.2018.08.015, 2018.
Watson, P. A. G., Weisheimer, A., Knight, J. R., and Palmer, T. N.: The role of the tropical West Pacific in the extreme Northern Hemisphere winter of 2013/2014, J. Geophys. Res.-Atmos., 121, 1698–1714, https://doi.org/10.1002/2015JD024048, 2016.
Wildt, J., Kobel, K., Schuh-Thomas, G., and Heiden, A. C.: Emissions of Oxygenated Volatile Organic Compounds from Plants Part II: Emissions of Saturated Aldehydes, J. Atmos. Chem., 45, 173–196, https://doi.org/10.1023/A:1024030821349, 2003.
Winters, A. J., Adams, M. A., Bleby, T. M., Rennenberg, H., Steigner, D., Steinbrecher, R., and Kreuzwieser, J.: Emissions of isoprene, monoterpene, and short-chained carbonyl compounds from Eucalyptus spp. in southern Australia, Atmos. Environ., 43, 3035–3043, https://doi.org/10.1016/j.atmosenv.2009.03.026, 2009.
Wolfe, G. M., Kaiser, J., Hanisco, T. F., Keutsch, F. N., de Gouw, J. A., Gilman, J. B., Graus, M., Hatch, C. D., Holloway, J., Horowitz, L. W., Lee, B. H., Lerner, B. M., Lopez-Hilifiker, F., Mao, J., Marvin, M. R., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., Thornton, J. A., Veres, P. R., and Warneke, C.: Formaldehyde production from isoprene oxidation across NOx regimes, Atmos. Chem. Phys., 16, 2597–2610, https://doi.org/10.5194/acp-16-2597-2016, 2016.
Yáñez-Serrano, A. M., Nölscher, A. C., Bourtsoukidis, E., Derstroff, B., Zannoni, N., Gros, V., Lanza, M., Brito, J., Noe, S. M., House, E., Hewitt, C. N., Langford, B., Nemitz, E., Behrendt, T., Williams, J., Artaxo, P., Andreae, M. O., and Kesselmeier, J.: Atmospheric mixing ratios of methyl ethyl ketone (2-butanone) in tropical, boreal, temperate and marine environments, Atmos. Chem. Phys., 16, 10965–10984, https://doi.org/10.5194/acp-16-10965-2016, 2016.
Zhang, J., Sun, Y., Wu, F., Sun, J., and Wang, Y.: The characteristics, seasonal variation, and source apportionment of VOCs at Gongga Mountain, China, Atmos. Environ., 88, 297–305, https://doi.org/10.1016/j.atmosenv.2013.03.036, 2014.
- Abstract
- Introduction
- Material and methods
- Results
- Discussions on the seasonal variability of VOC concentrations
- VOC concentration variations in continental Europe
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Material and methods
- Results
- Discussions on the seasonal variability of VOC concentrations
- VOC concentration variations in continental Europe
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement