the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Aug 2021
| 19 Aug 2021
Observation and modelling of ozone-destructive halogen chemistry in a passively degassing volcanic plume
Tjarda Roberts
Slimane Bekki
Volcanoes emit halogens into the atmosphere that undergo complex chemical cycling in plumes and cause destruction of ozone. We present a case study of the Mount Etna plume in the summer of 2012, when the volcano was passively degassing, using aircraft observations and numerical simulations with a new 3D model “WRF-Chem Volcano” (WCV), incorporating volcanic emissions and multi-phase halogen chemistry.
Measurements of SO2 – an indicator of plume intensity – and ozone were made in the plume a few tens of kilometres from Etna, revealing a strong negative correlation between ozone and SO2 levels. From these observations, using SO2 as a tracer species, we estimate a mean in-plume ozone loss rate of 1.3×10−5 molecules of O3 per second per molecule of SO2. This value is similar to observation-based estimates reported very close to Etna's vents, indicating continual ozone loss in the plume up to at least tens of kilometres downwind.
The WCV model is run with nested grids to simulate the plume close to the volcano at 1 km resolution. The focus is on the early evolution of passively degassing plumes aged less than 1 h and up to tens of kilometres downwind. The model is able to reproduce the so-called “bromine explosion”: the daytime conversion of HBr into bromine radicals that continuously cycle in the plume. These forms include the radical BrO, a species whose ratio with SO2 is commonly measured in volcanic plumes as an indicator of halogen ozone-destroying chemistry.
The species BrO is produced in the ambient-temperature chemistry, with in-plume BrO SO2 ratios on the order of 10−4 mol/mol, similar to those observed previously in Etna plumes. Wind speed and time of day are identified as non-linear controls on this ratio. Sensitivity simulations confirm the importance of near-vent radical products from high-temperature chemistry in initiating the ambient-temperature plume halogen cycling. Heterogeneous reactions that activate bromine also activate a small fraction of the emitted chlorine; the resulting production of chlorine radical Cl strongly enhances the methane oxidation and hence the formation of formaldehyde (HCHO) in the plume.
Modelled rates of ozone depletion are found to be similar to those derived from aircraft observations. Ozone destruction in the model is controlled by the processes that recycle bromine, with about three-quarters of this recycling occurring via reactions between halogen oxide radicals. Through sensitivity simulations, a relationship between the magnitude of halogen emissions and ozone loss is established.
Volcanic halogen cycling profoundly impacts the overall plume chemistry in the model, notably hydrogen oxide radicals (HOx), nitrogen oxides (NOx), sulfur, and mercury chemistry. In the model, it depletes HOx within the plume, increasing the lifetime of SO2 and hence slowing sulfate aerosol formation. Halogen chemistry also promotes the conversion of NOx into nitric acid (HNO3). This, along with the displacement of nitrate out of background aerosols in the plume, results in enhanced HNO3 levels and an almost total depletion of NOx in the plume. The halogen–mercury model scheme is simple but includes newly identified photo-reductions of mercury halides. With this set-up, the mercury oxidation is found to be slow and in near-balance with the photo-reduction of the plume. Overall, the model findings demonstrate that halogen chemistry has to be considered for a complete understanding of sulfur, HOx, reactive nitrogen, and mercury chemistry and of the formation of sulfate particles in volcanic plumes.
- Article
(3102 KB) - Full-text XML
-
Supplement
(1463 KB) - BibTeX
- EndNote





1.1 Background
Volcanoes emit mixtures of various gases and particulates into the atmosphere. Of the gaseous emissions, H2O, CO2, and SO2 are typically the species with the greatest fluxes. Most studies on the atmospheric impact of volcanic emissions have focused on sulfur because of its well-known effects on atmospheric composition, notably aerosol loading and climate (e.g. Oppenheimer et al., 2011). Several other species are emitted from volcanoes, including other sulfur species (such as H2S, which can, in some cases, be dominant) and the primary focus of this study: halogens. Chlorine, bromine, and fluorine are routinely detected in various proportions within volcanic plumes and are emitted primarily as hydrogen halides. Gerlach (2004) reported that gaseous emissions from arc volcanoes are, on average, 0.84 % HCl, 0.061 % HF, and 0.0025 % HBr. Iodine has also been detected in volcanic plumes (e.g Aiuppa et al., 2005; Bobrowski et al., 2017), but its emission fluxes are typically about 2 orders of magnitude below those of bromine.
The quantities of halogen species emitted from a volcano and their temporal variability appear to provide information on sub-surface processes (Pyle and Mather, 2009). Correlations between the bromine-to-sulfur ratio and volcanic activity have been found in long-term observations (e.g. Bobrowski and Giuffrida, 2012; Dinger et al., 2018; Warnach et al., 2019; Dinger et al., 2021), suggesting that this ratio could potentially be used for monitoring and forecasting of volcanic activity.
However, volcanic halogen emissions are not just potential indicators of sub-surface processes. Importantly, they impact the chemical composition of the atmosphere and hence possibly the climate. For instance ozone, the precursor of the most important atmospheric oxidant (the hydrogen oxide radical OH), is found to be greatly depleted in volcanic plumes containing halogens (Rose et al., 2006; Vance et al., 2010; Kelly et al., 2013; Surl et al., 2015). Ozone is predominately destroyed by bromine chemistry cycles (Bobrowski et al., 2007; von Glasow, 2010; Roberts, 2018). Halogen chemistry also leads to the depletion of nitrogen oxides (NOx) and hydrogen oxide radicals (HOx) (Bobrowski et al., 2007; Roberts et al., 2009; Jourdain et al., 2016) and may oxidize mercury to more soluble, and therefore more easily deposited, forms (von Glasow, 2010). The effects are not limited to the troposphere because volcanic plumes can also reach the tropopause region, sometimes injecting halogens directly into the stratosphere (Rose et al., 2006; Millard et al., 2006). In the case of bromine- or chlorine-rich, large-scale eruptions, this could result in massive stratospheric ozone depletion (Kutterolf et al., 2013; Cadoux et al., 2015; Brenna et al., 2020), dependent upon the fraction of emitted halogens that avoid dry- and wet-removal processes within the tropospheric plume and are therefore able to reach the stratosphere. Estimates for this fraction vary substantially (between 0 and 1), and the fraction depends on both volcanic and atmospheric conditions (see Mather, 2015, and references therein). In recent decades, satellite and aircraft observations have identified the presence of elevated halogens at high altitudes following some (but not all) volcanic eruptions, in particular chlorine as HCl (e.g. Rose et al., 2006; Prata et al., 2007; Carn et al., 2016) and occasionally bromine as BrO (Theys et al., 2009, 2014). Co-injection of volcanic halogens alongside SO2 into the stratosphere modifies the chemistry–climate impacts of the eruptions (Brenna et al., 2020; Ming et al., 2020; Wade et al., 2020). This occurs through feedbacks on SO2 processing to sulfate particles (Lurton et al., 2018) and depletion of ozone and other climate gases such as water vapour and methane, with feedbacks on aerosol microphysics and transport, thereby changing the radiative impacts (Staunton-Sykes et al., 2021). The chemistry–climate impacts of volcanic eruptions also depend on background halogen loading that is currently elevated due to historic CFC emissions. As the stratospheric halogen loading approaches pre-industrial levels in future, volcanic sulfur injections are expected to increase total column ozone, whereas halogen-rich injections would deplete ozone (Klobas et al., 2017).
All these impacts of volcanic halogens depend critically on the extent of the conversion of emitted volcanic hydrogen halides into halogen radicals, a process called halogen activation. Indeed, hydrogen halides are weakly reactive and very soluble. As a result, their direct impact on atmospheric chemistry is very limited and short-lived because they are rapidly removed from the atmosphere. In contrast, halogen radicals are much more reactive chemically, in particular with respect to ozone, and less soluble. Therefore, assessing the atmospheric impacts of volcanic halogens requires a quantitative understanding of the physico-chemical plume processes driving the partitioning of volcanic halogen species, especially bromine species, between radicals and hydrogen halides. This is the primary focus of the present study. Note that only a small fraction of chlorine emissions undertakes reactive chemistry in plumes, and this small fraction is mostly activated as a result of bromine chemistry (Rüdiger et al., 2021, and references therein). As in most studies on volcanic plume halogen chemistry, fluorine and iodine are ignored. The solubility and stability of HF, the main emitted fluorine species, are such that no significant fluorine chemistry occurs within the plume (von Glasow et al., 2009). Iodine, while very reactive, is of substantially lower in-plume abundance than bromine and chlorine (Aiuppa et al., 2005).
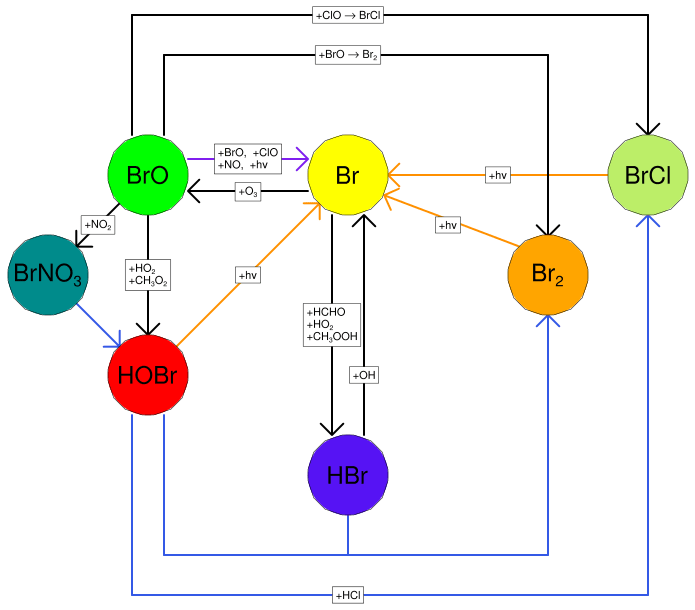
Figure 1The major reactions of the bromine cycle. Orange lines indicate photolysis reactions, blue lines heterogeneous reactions, and black lines other gas-phase reactions. BrO → Br includes one photolysis reaction and three other reactions.
Once emitted at high temperatures in the atmosphere, volcanic volatiles are cooled very quickly by the fast mixing of the plume with the surrounding air. Whilst some halogen radicals as well as HOx and NOx may be formed by high-temperature reactions immediately after emission from the vent (in the so-called “effective source region”; see Bobrowski et al., 2007, and Roberts et al., 2019, and references therein), it is the atmospheric chemistry in the cooled and expanding plume that causes a sustained halogen cycling that impacts tropospheric ozone. This halogen chemistry continues to occur as the plume disperses into the background atmosphere. This chemistry is complex and non-linear. The major bromine cycling driving bromine activation and ozone loss is shown in Fig. 1. Heterogeneous processes involving acidic aerosol are the main pathway by which HBr is converted to reactive forms. The complete cycle requires photolysis reactions and therefore only occurs within the daytime. This bromine cycle is sometimes referred to as the “bromine explosion” (Wennberg, 1999; Bobrowski et al., 2007) because of its autocatalytic nature; bromine extracted from HBr can continue to cycle and generate further reactive bromine from this source. The proportion of emitted bromine that exists as BrO impacts the in-plume BrO SO2 ratio. The observations of Dinger et al. (2021) show that this quantity can vary with meteorological conditions. Although no equivalent “chlorine explosion” exists, heterogeneous reactions can also generate reactive chlorine radicals (via BrCl), which can react with ozone to form chlorine oxide species.
Reactive halogen chemistry in tropospheric volcanic plumes, as evidenced by enhanced BrO, has been observed for many volcanoes worldwide (Gutmann et al., 2018), with satellite observations greatly expanding the quantity and geographic scope of observations (e.g. Hörmann et al., 2013).
A fast interchange exists between Br and BrO. The Br + O3 → BrO + O2 reaction consumes an O3 molecule, while BrO photolysis effectively reverses this as the O radical produced quickly reacts with O2 to form O3. When BrO is reduced to Br via any other pathway (directly or indirectly), the net result is the loss of an O3 molecule.
As well as depleting ozone, halogen chemistry has influences on other chemical systems, such as through reactions with NOx and HOx, as shown in Fig. 1. Volcanic halogens deplete HOx, increasing the lifetime of SO2 with respect to OH oxidation, and oxidation of CH4 by Cl radicals can significantly reduce the in-plume lifetime of CH4. Jourdain et al. (2016) modelled both of these phenomena occurring in an Ambrym plume, finding that halogens' depletive effect on HOx further increased the lifetime of SO2 with respect to oxidation by OH by 36 %. Modelling by Roberts et al. (2009) indicates that volcanic halogen chemistry can result in conversion of in-plume NOx to HNO3.
Finally, volcanoes are also sources of mercury to the atmosphere (Pyle and Mather, 2003) mainly in the inert form Hg(0) (Witt et al., 2008; Bagnato et al., 2007). A 1D model study by von Glasow (2010) suggested that this mercury could be rapidly oxidized by halogen chemistry in a volcanic plume to more soluble forms easily removed from the atmosphere (Seigneur and Lohman, 2008). Significant advances in understanding of the kinetics of halogen–mercury chemistry have been made in the last decade (e.g. Saiz-Lopez et al., 2018, 2019), and these are included in the modelling part of this study.
1.2 Observation and modelling studies
The current understanding of halogen chemistry within volcanic plumes is based upon a body of observations that have used a variety of techniques, coupled with numerical modelling results, most of which have used zero- or one-dimensional chemical box models.
1.2.1 Observations
There are two main methods for measuring halogens in volcanic plumes: remote sensing and in situ sampling.
Remote sensing accounts for most observations of reactive halogens in volcanic plumes. Since the first reported detection by Bobrowski et al. (2003), bromine monoxide (BrO) has been observed within the plume of dozens of volcanoes by differential optical absorption spectroscopy (DOAS) (see Gutmann et al., 2018, for a recent catalogue of such observations). A smaller number of measurements of in-plume ClO and OClO have also been reported. These halogen molecules have spectroscopic signatures within the ultraviolet range, meaning they can be identified from the same data that are used to monitor SO2, including data collected from long-term DOAS monitoring networks at volcanoes (Dinger et al., 2018; Warnach et al., 2019). As well as ground and airborne observations, BrO has been observed in the plumes of some larger volcanic eruptions by satellite-based instruments (e.g. Hörmann et al., 2013; Seo et al., 2019), though such large eruptions are the focus of a future study rather than this one.
In situ sampling of halogens provides the most direct approach to quantifying total halogen emissions: time-averaged sampling has for decades been used to quantify total volcanic halogen emission contents for F, Cl, Br, and I (e.g. Aiuppa et al., 2004; Wittmer et al., 2014). Modern techniques now allow for a degree of speciation in the bromine observed through these methods (Rüdiger et al., 2017, 2021). For most reactive halogen species, these methods required samples to be collected in situ and then subsequently analysed in-lab. Consequently, there are fewer in situ observations of reactive halogens than by remote sensing.
As well as these direct approaches, ozone measurements can provide indirect evidence for halogen chemistry. Ozone destruction in tropospheric volcanic plumes, caused by volcanic halogen cycling, has been measured in a limited number of cases (Vance et al., 2010; Oppenheimer et al., 2010; Kelly et al., 2013; Surl et al., 2015). In ash-rich explosive eruptions, it is possible that uptake of ozone on ash particles may also contribute to some ozone loss (Maters et al., 2017). Measuring ozone in volcanic plumes in the troposphere region downwind from volcanoes is challenging and typically only achieved using instrumented aircraft. Observations suggest a direct relation between bromine content and ozone depletion (Roberts, 2018), although this is based on only three available volcanic datasets: Mount Etna, Mount Redoubt, and Kīlauea. Observations at Mount Redoubt volcano suggest that ozone losses, as a ratio to SO2, increase in magnitude with respect to the distance from the source (Kelly et al., 2013).
For the above methods, the observed halogen gas quantities or ozone depletions are often ratioed to simultaneous sulfur or SO2 measurements. This allows for comparison between plumes of different “strengths” (i.e. density or dilution) and, for example, the tracing of how halogen chemistry changes as a plume disperses as it travels downwind. This use of SO2 as a plume tracer presupposes that it has a long atmospheric lifetime relative to the timescale of the given study.
1.2.2 Numerical modelling
Another tool for studying the chemistry of volcanic plumes is numerical modelling. Most volcanic plume halogen chemistry modelling studies to date have originated from implementations of a few models, in particular MISTRA (Aiuppa et al., 2007; Bobrowski et al., 2007; von Glasow, 2010; Bobrowski et al., 2015; Surl et al., 2015) and PlumeChem (Roberts et al., 2009, 2014, 2018; Kelly et al., 2013). A more recent study by Rüdiger et al. (2021) used the CABBA/MECCA box model. These Lagrangian models are either zero- or one-dimensional and simulate the chemical evolution of the cooled plume by calculating the in-plume rates of chemical reactions and include the continual dilution of the plume with background air. Such models are found to better reproduce observations if the initial halogen emissions include a fraction of halogen radicals. This represents the radicals generated by high-temperature chemistry in the effective source region.
To our best knowledge, Jourdain et al. (2016) is the only prior 3D Eulerian-type mesoscale chemistry-transport modelling study published to date dealing with halogen chemistry in a tropospheric volcanic plume. Volcanic emissions and halogen chemistry were implemented into the CCATT-BRAMS model to simulate the chemistry within the plume of Ambrym during an intense passive-degassing episode in 2005. Their model is similar to the one used in our study. However, their gas emission flux for the Ambrym event is about 6 times greater than the Mount Etna passive-degassing event studied here. Mechanistically, their results showed the formation of BrO as well as ozone depletion occurring within the plume's core that impacts bromine speciation. The study also simulated in-plume depletion of HOx and NOx as well as lengthening of SO2 and methane lifetimes due to halogen chemistry. The model successfully reproduced observed BrO SO2 spatial patterns; however the magnitude was somewhat underestimated, and there were no measurements of ozone to provide constraints on the predictions of ozone depletion, a key feature of reactive halogen chemistry. Finally, Jourdain et al. (2016) focuses on the wider-scale impact of volcanic emissions, whereas this study focuses more on the detailed mechanisms of halogen cycling in the early plume with cross-validation against ozone and halogen radical observations.
WRF-Chem has been used in several studies to model tropospheric volcanic plumes, generally showing good agreement with observations (Stuefer et al., 2013; Burton et al., 2020; Egan et al., 2020; Rizza et al., 2020; Hirtl et al., 2019, 2020). Such studies have predominantly focused on ash and SO2 distribution, and there have been none, to our knowledge, that incorporate halogen chemistry.
1.2.3 This study
The present study is devoted to a plume from Etna during July/August 2012, a period when this volcano was passively degassing. We present new airborne ozone and SO2 measurements which were made during traverses of plumes at distances 7–21 km from the vents. Several other previously published plume measurements were also made around this time. Near-simultaneous near-vent (<500 m) ozone measurements and DOAS observations of BrO SO2 ratios around 10 km downwind were reported by Surl et al. (2015). Additionally in situ sampling of halogen emissions was undertaken that summer at the crater rim by Wittmer et al. (2014), and further DOAS measurements were also made of Etna's plume by Gliß et al. (2015). Consequently, this period of Etna's activity may have the richest overall dataset to date for determining halogen activity in relation to ozone loss in the plume of a volcano.
The observational dataset is analysed using a 3D regional chemistry-transport model (WRF-Chem) modified with respect to its handling of volcanic emissions and with halogen chemistry added to a chemical mechanism. The goal of the modelling is to assess the ability of the 3D model to adequately reproduce the key chemistry features (ozone loss, BrO SO2 ratios) of the Etna plume given reasonable input parameters, such as the typical halogen emission fluxes for the volcano in a passive-degassing state. We then diagnose in-plume chemical processes in the model, exploiting the fact that a model can be interrogated in far greater detail than an observational dataset. The focus is on the chemical processes in the near-downwind plume, up to tens of kilometres from emissions sources for plume ages of up to tens of minutes. As well as halogen chemistry and the associated ozone destruction, the simulated impacts of plume chemistry on the various interconnected chemical systems discussed above (HOx, SO2 lifetime and its oxidation to particles, methane lifetime, reactive nitrogen, and mercury chemistry) are also investigated.
2.1 Aircraft measurements of SO2 and ozone in the plume
The aircraft campaign presented in this study was conducted as part of the Global Mercury Observation System project (http://www.gmos.eu, last access: 13 August 2021) with the same aircraft and instrumentation discussed in Weigelt et al. (2016a) and Weigelt et al. (2016b). We refer the reader to these works for a full description of the campaign set-up and instruments; in this section we highlight the most relevant aspects.
The measurements of this study were performed with a CASA 212 two-engine turboprop aircraft fitted with a specially designed gas inlet system. SO2 was measured with a Thermo Environmental Instruments Model 43C pulsed fluorescence gas analyser with root mean squared (RMS) noise of 1 ppbv and precision of 1 ppbv or 1 % of reading (http://www.thermo.com.cn/Resources/200802/productPDF_12267.pdf, last access: 13 August 2021). Estimates of the interferences from H2O and NO are reported as < 3 ppbv and < 2 % of reading. Ozone was measured with a Teledyne API 400E, with RMS noise and precision < 0.5 % of reading. This measurement is not subject to interference from SO2 or H2O as these interferences are efficiently removed by the comparison of measurements between the ambient air channel and the ozone-scrubbed reference channel (http://www.teledyne-api.com/products/oxygen-compound-instruments/t400, last access: 13 August 2021). Mercury vapour is listed as a potential source of interference; however mercury-detecting instruments were also active during this campaign, and the level of gaseous mercury emission from the volcano was determined to be nil or low (Weigelt et al., 2016a). Both the SO2 and ozone instruments have a temporal resolution of 10 s (averaging time), and their response times are 80 and < 30 s, respectively.
Three flights were conducted, one each on the mornings of 30 July 2012, 31 July, and 1 August, during daylight hours. These flights started and ended at Reggio Calabria Airport and attempted several transects of the plume. The flight paths are shown in Fig. 2.
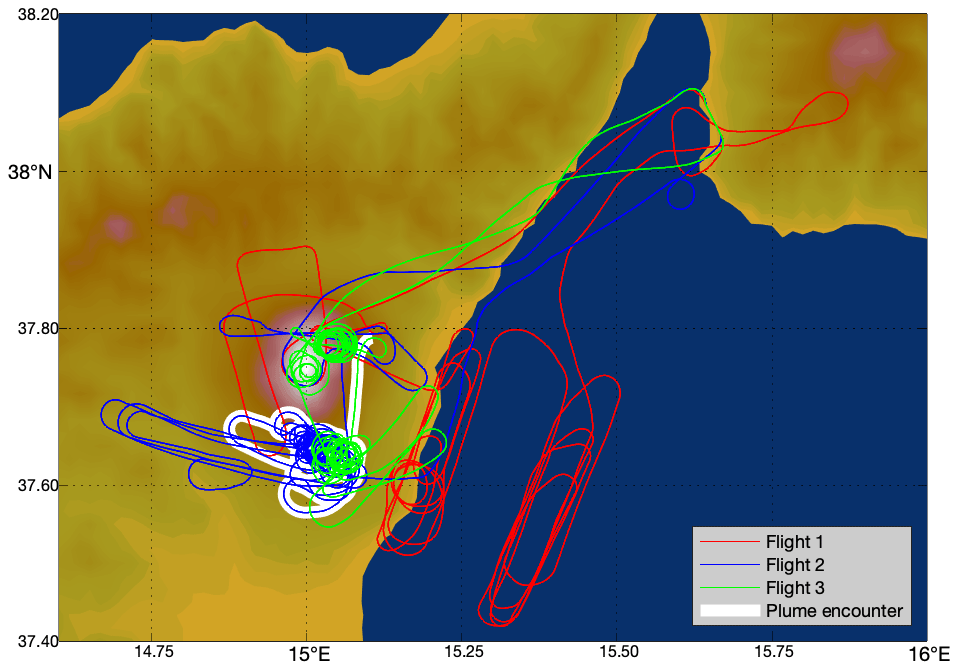
Figure 2Flight paths of the aircraft on the 3 measurement days: red – 30 July 2012, blue – 31 July 2012, green – 1 August 2012. Major plume encounter locations as identified by our algorithm are highlighted in white.
Since ambient concentrations of ozone vary both spatially and temporally, rather than assessing all of the observation data together, we undertook a systematic approach to identify and isolate separate “major plume encounters” from the dataset and separately evaluate the ozone variations within these. This approach was designed such that the majority of the variation in ozone within each major plume encounter could be ascribed to plume chemistry rather than variations in the background. Our approach also fixed a maximum range of distances from the vent that could be considered part of a single major plume encounter so as to minimize any internal variation in ozone losses within a plume encounter due to plume chemistry varying with distance from the source. Inspection of the data showed that, outside of the plume, rapid changes in altitude corresponded with substantial changes in ozone mixing ratio. In order to avoid mistaking such background ozone variation as a plume signal, we fixed a maximum range of altitudes that could be considered part of one major plume encounter. Lastly, encounters that are too short or do not reach a sufficiently high plume intensity so as to allow for an identification of signal above background variation are dismissed.
Our algorithm is therefore as follows:
-
A plume encounter is considered to begin when the SO2 measurement exceeds 10 ppbv and ends when SO2 drops below 10 ppbv.
-
If a data point's altitude is more than 300 m higher or lower than that of any previous data point in the current encounter, the encounter ends, and another immediately begins.
-
If a data point's distance from the source is more than 5 km greater or smaller than that of any previous data point in the current encounter, the encounter ends, and another immediately begins.
-
If a plume encounter has maximum SO2 less than 100 ppbv or lasts for less than 2 min, it is considered a “minor plume encounter” and is discarded.
This process is presented in full as a flowchart in Fig. S1.
2.2 Modelling
We use version 4.1.5 of WRF-Chem (Grell et al., 2005), which is a fully coupled three-dimensional regional model for atmospheric physics, meteorology, and chemistry, including cloud and aerosol radiative feedback processes. We have made several modifications to the code, in particular volcanic gas emissions and chlorine, bromine, and mercury chemistry. We name this new model “WRF-Chem Volcano” (WCV). Our WCV developments build on the WRF-Chem version developed by the Roland von Glasow group at the University of East Anglia (Surl, 2016), and our WCV developments were made with reference to the model code of Badia et al. (2019), another development on the University of East Anglia version with a focus on marine chemistry.
2.2.1 Mechanism
WCV extends the CBMZ-MOSIAC chemistry scheme with eight aerosol size bins (Zaveri and Peters, 1999; Zaveri et al., 2008) to include bromine, chlorine, and mercury chemical mechanisms with gas-phase, photolytic, and heterogeneous reactions involving the following species: HBr, Br, BrO, HOBr, BrNO3 (a.k.a. BrONO2), Br2, HCl, Cl, ClO, OClO, HOCl, ClNO3, Cl2, BrCl, Hg, HgBr, HgCl, HgBr2, HgCl2, and HgBrCl. We exclude BrNO2 as previous studies have found it to be a negligible component (Roberts et al., 2014; Rüdiger et al., 2021). These species are also incorporated into the dry- and wet-deposition schemes and the FastJ photolysis scheme (Wild et al., 2000). The rates of heterogeneous reactions involving HOBr, BrNO3, and ClNO3 on volcanic aerosols are calculated online accounting for the wet surface area of aerosol and gas-phase diffusion limitations as described by Marelle et al. (2021). The products of HOBr reactive uptake are partitioned between Br2 and BrCl (i.e. net overall reaction with HBr or HCl) by a parameterization that assumes fast aqueous-phase equilibria between Br2, Br2Cl−, and BrCl as described by Jourdain et al. (2016). Reactions added to the scheme are listed in the Supplement (Tables S1, S2, S4) along with their rate equations and references for these. Parameters controlling the heterogenous reactions are tabulated in Table S3.
The only SO2 oxidation pathway included in CBMZ-MOSAIC is oxidation by OH, and we did not add any further pathways. Galeazzo et al. (2018) discusses how other pathways could potentially occur in the aqueous phase in volcanic plumes: oxidation by O3aq, oxidation by H2O2, and transition-metal-catalysed oxidation by O2.
Our volcanic emissions pre-processor, a modified version of the PREP-CHEM-SRC utility (Freitas et al., 2011), provides as inputs to the model fluxes of sulfur, bromine, chlorine, and mercury species as well as an “at-source” sulfate particle flux and fluxes of radicals resulting from high-temperature chemistry within the vent (e.g. OH, NO). This emission is time-invariant and is into a single grid cell of the model (cf. the more spatially and temporally sophisticated pre-processor of Hirtl et al., 2020).
We have also introduced artificial tracer species to WCV that do not influence the chemistry but are useful for analysis; tracer1 is a wholly inert species that is emitted with the same flux rate as SO2. It is used to compute mean (weighted average) in-plume values of parameters; here an “in-plume average” of a value (e.g. of ozone mixing ratio) refers to the average of this value across all grid boxes where tracer1 exceeds 3 ppbv, weighted by the tracer1 content of the boxes.
The species tracer2 is similar to tracer1 but undergoes a first-order exponential decay with specified rate. The ratio of tracer1 and tracer2 can therefore be used to derive the mean time since emission of any part of the plume. This approach allows us to accurately calculate plume age in any model grid cell and enables us to monitor how plume parameters (e.g. ozone mixing ratio, BrO SO2 ratio) vary with the plume time evolution since emission.
We have also added to the model output monitoring of several chemistry diagnostics, such as rates of relevant reactions, in order to carry out species' chemical budgets and therefore facilitate the analysis of the underlying halogen and ozone-destructive chemistry.
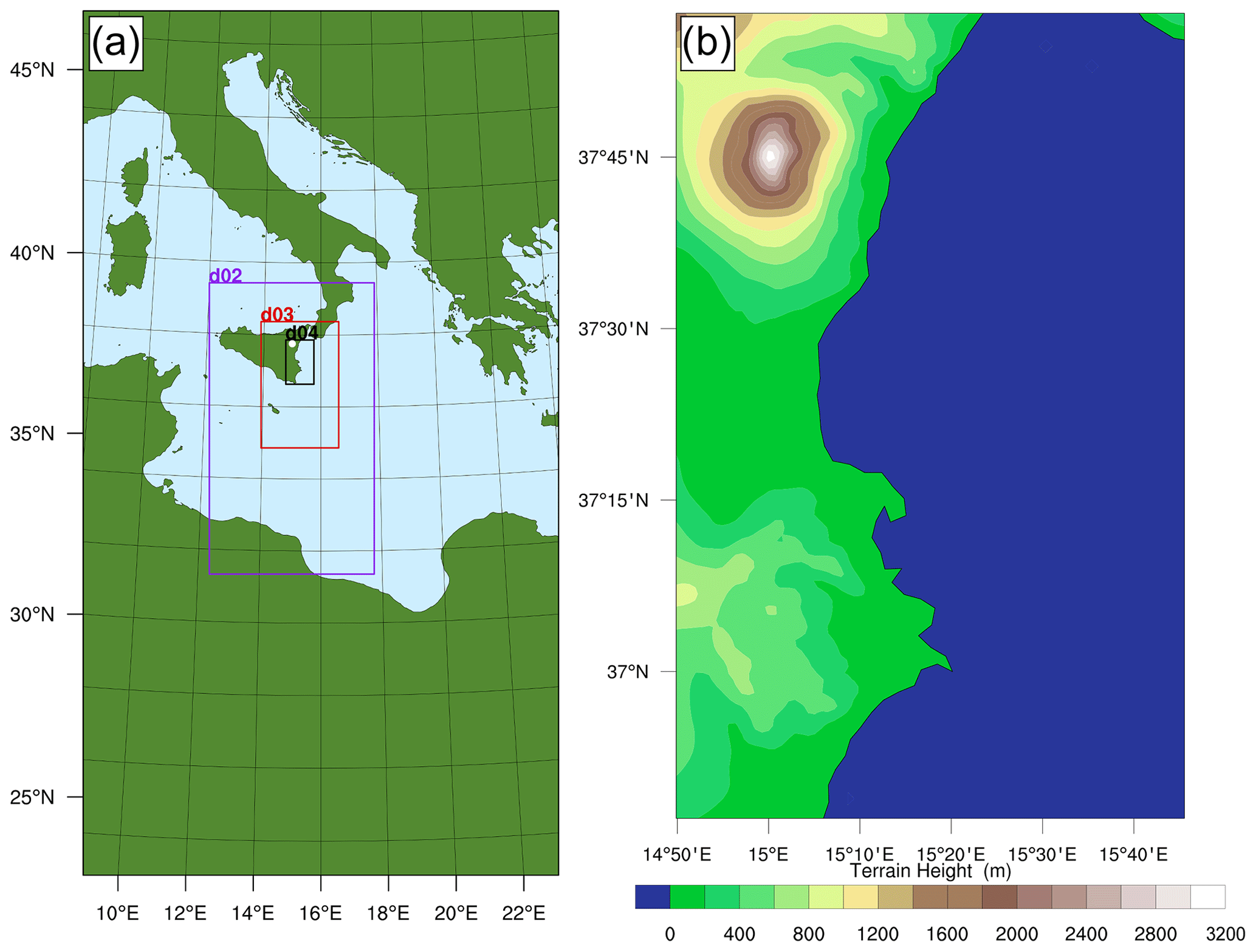
Figure 3(a) The WCV model area; d02, d03, and d04 are progressively nested domains with two-way nesting. (b) Terrain elevation in d04.
2.2.2 Model settings
The total area modelled, at 30 km horizontal resolution, is depicted in Fig. 3a. Also shown are the extents of the progressively nested domains, each modelled with grid dimension 3 times smaller than their parent. These domains are two-way nested in exchanging meteorological, chemical, and physical information between them. Here we focus on the near-downwind plume processing (up to about 90 min). Therefore all the figures and results presented in this study are from the d04 nest (Fig. 3b), which models an 88 km × 134 km area around the east coast of Sicily with a horizontal spatial resolution of 1.1 km. The model has 50 vertical layers, extending up to 50 hPa.
The model was initialized on 29 July 2012 at 00:00 UTC and ran for 4 d, therefore covering all of the days of the aircraft measurements. The first 24 h is considered spin-up; results are presented from > 24 h onwards.
The volcano was considered to be a point source of gas and aerosol, emitting at a constant flux rate throughout the simulation period. Although Mount Etna has several active vents (Northeast Crater, Bocca Nova, Voragine, Southeast Crater) at the volcano summit, the maximum horizontal resolution of the model is not sufficient to distinguish these. Emissions are released into a single grid cell at 37.751∘ N, 14.995∘ S, and 3300 m a.s.l., the location of the summit peak of Etna. This altitude does not correspond to the lowest model level at this location but rather a few levels above it because, even at the maximum model resolution, the sub-grid topography of the Etna volcano is smoothed out in the model grid box. For example, at the 1.1 km resolution, the ground altitude of the Etna grid box is 3088 m. The lower the model resolution is, the more potent the smoothing of sub-grid terrain features is and hence the greater this discrepancy is.
Modelled emissions of SO2 were set to 40 kg s−1. This flux was estimated by adjusting initial estimates according to comparisons between outputs from preliminary runs and observed SO2 mixing ratios. A 40 kg s−1 flux results in SO2 mixing ratios within the centre of the modelled plume being similar to the maximum SO2 mixing ratios observed from the aircraft at the same distance from the source. We assume these observed maxima correspond to transects which cross or come close to the core of the plume. A total of 40 kg s−1 also falls mid-way within the normal range of measured SO2 fluxes 41±30 kg s−1 for Etna's activity during non-eruptive (passive degassing) periods (Salerno et al., 2009). A volcanic H2O flux is set to be 15 times greater (in terms of number of molecules) than the SO2 flux following Aiuppa et al. (2008). Being effectively inert chemically, CO2 and its emissions are ignored in the model.
Mount Etna's plume also contains sulfate-rich particles whose presence in the young plume at the volcano summit (Martin et al., 2012; Roberts et al., 2018) indicates that they are formed early on, possibly in the vent, well below the grid resolution of the model. These “at-source” aerosols are treated as primary aerosols. They are included in the model by setting a volcanic aerosol emission flux which is derived from the reported near-summit sulfate SO2 mass ratio of 0.03 (Roberts et al., 2018). All of this aerosol is taken to be sulfate with a size distribution across the eight MOSAIC size bins following that of Roberts et al. (2018), with an extrapolation made for the smallest bins. During the in situ measurements of Surl et al. (2015) made on 30 June and 1 August the plume was observed to be ash-free. Therefore no ash was included in this modelling.
Chlorine and bromine emission fluxes are specified based on the observed summit chlorine-to-bromine ratio, HCl-to-SO2 ratio, and the SO2 flux specified above. We rely on a comprehensive compositional analysis undertaken between June 2010–June 2012 by Wittmer et al. (2014). The chlorine-to-bromine ratio was fixed to 300 by mass (683 by mole), which is an average calculated for the compositions of Bocca Nova and Northeast Crater reported by Wittmer et al. (2014). The HCl-to-SO2 ratio was set to 0.4 mol/mol, which is about mid-way in the range of ratios (0.29 to 0.56) for these craters.
As stated in the introduction, volcanic gases are believed to react at high temperatures immediately following their release in the vent and into the atmosphere, generating radicals, notably HOx and some halogen radicals. These radicals – as well as the primary aerosols – subsequently initiate the onset of the bromine cycling in the cooled plume (Fig. 1). A representation of the high-temperature radicals is therefore needed for the WCV volcanic input. Thermodynamic models have been used previously to represent this high-temperature “effective source region” (Bobrowski et al., 2007), but their assumption of chemical equilibrium is not considered reliable, whereas recently developed kinetic models do not yet include halogens (see Roberts et al., 2019). Here we choose a simpler approach by, in the main run, partitioning the bromine emission flux into hydrogen halide and radicals, with 75 % of bromine emitted as HBr and 25 % as Br radicals, and by including an HOx emission. This bromine partitioning follows previous thermodynamic modelling estimates (Roberts et al., 2014). Emissions of volcanic HOx are highly uncertain; there exist order-of-magnitude differences between kinetic and thermodynamic model predictions and in the speciation between OH and HO2 (Roberts et al., 2019). Here, we define the volcanic HOx emission by setting an OH SO2 molar ratio of 0.001 (between reported thermodynamic and kinetic model ranges). The immediate reaction of OH with SO2 in WCV will generate HO2 and some additional sulfate. Whilst all volcanic chlorine is emitted as HCl in the model, the reaction with volcanic OH will also quickly generate some Cl radicals.
Although there are open questions regarding the kinetics of high-temperature NO generation in the first few seconds of plume evolution (Martin et al., 2012), we chose to include these emissions to assess their possible effect. We use an NO SO2 molar emission ratio of , which is of the order typically produced by high-temperature thermodynamic modelling of the early plume–air mix (cf. used in Roberts et al., 2014).
Although the aircraft campaign did not find a detectable mercury signal for the plume (Weigelt et al., 2016a), we include a small mercury emission so as to investigate this mechanism. We use a general volcanic emission ratio of mol Hg per mol SO2 from Bagnato et al. (2014), a quantity too small to significantly interfere with other chemical systems. All of this mercury is emitted as Hg(0).
We use external data for model forcing at the boundaries of the outermost domain and for the initial conditions of the model. Meteorological information is sourced from the NCEP FNL Operational Model Global Tropospheric Analyses (National Centers for Environmental Prediction et al., 2000). Chemical information is sourced from CAM-CHEM (Buchholz et al., 2019; Emmons et al., 2020) and applied using the MOZBC utility. Since mercury, bromine, and chlorine species other than HCl were absent from this CAM-CHEM data, we set their initial and boundary values to zero.
As well as the main model run detailed above, several other model runs were made with varying emissions (Table 2). These include a model run with no volcanic emissions (novolc). Differences between novolc and the other runs are used to quantify volcanic impacts. Sensitivity runs were identical to the main run except for the perturbations to the volcanic emissions listed in Table 2. The noHighT run excludes all species expected to be generated in the high-temperature volatile-air mix of the first few seconds after volcanic emission and therefore includes only H2O, SO2, HCl, and HBr (all bromine as HBr) as well as primary sulfate.
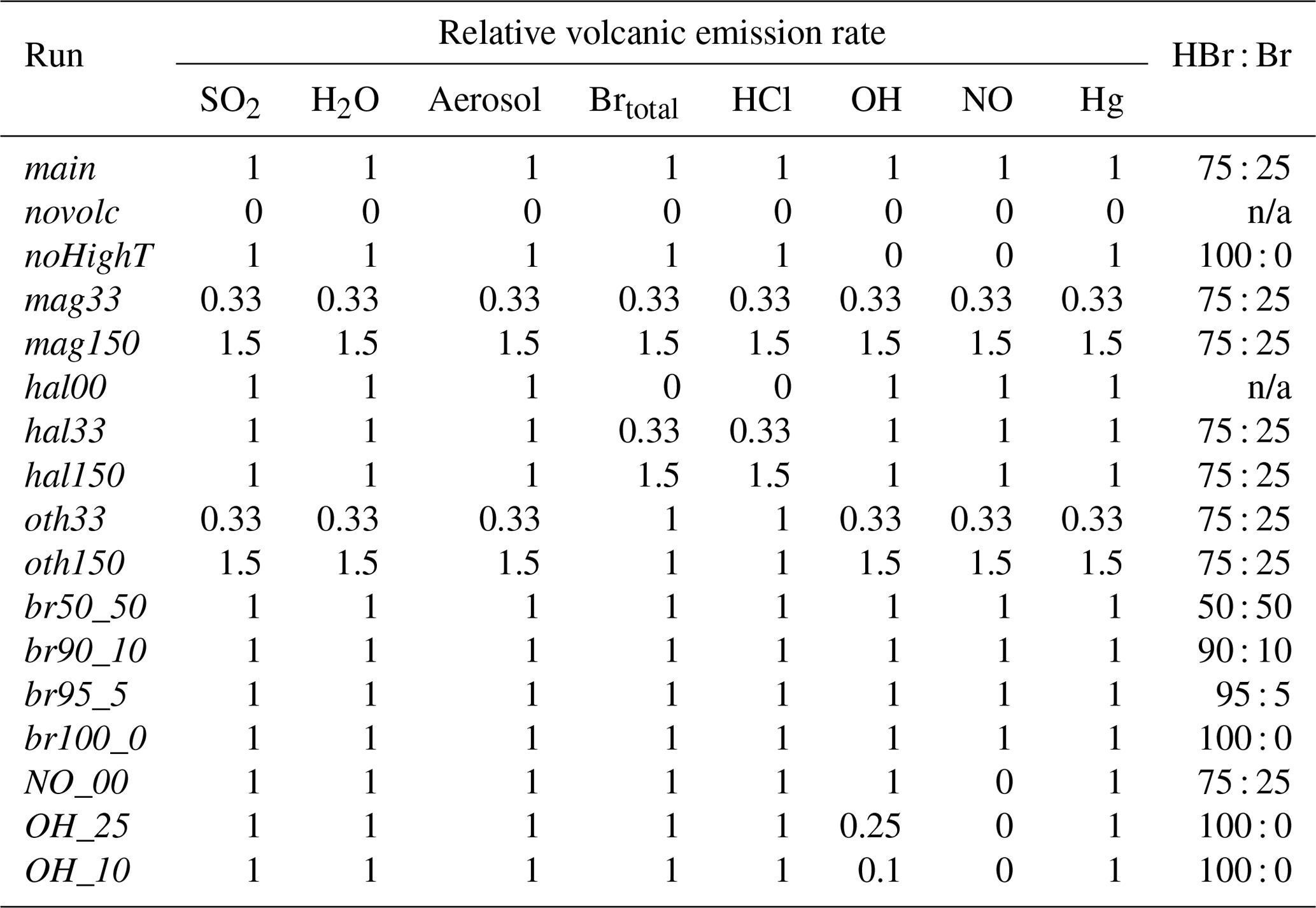
Table 2Volcanic emission rates of species in the model runs of this study relative to those of main and speciation of bromine at emission; n/a stands for not applicable.
The encounter-finding algorithm described in Sect. 2.1 yields 19 major plume encounters, 11 on 31 July and 8 on 1 August 2012. Only minor plume encounters occurred on 30 July. The locations of these major encounters are highlighted in Fig. 2. Plots of O3 vs. SO2 for these encounters are shown in Fig. 4 for 31 July and Fig. 5 for 1 August. These data are also summarized in Table 5.
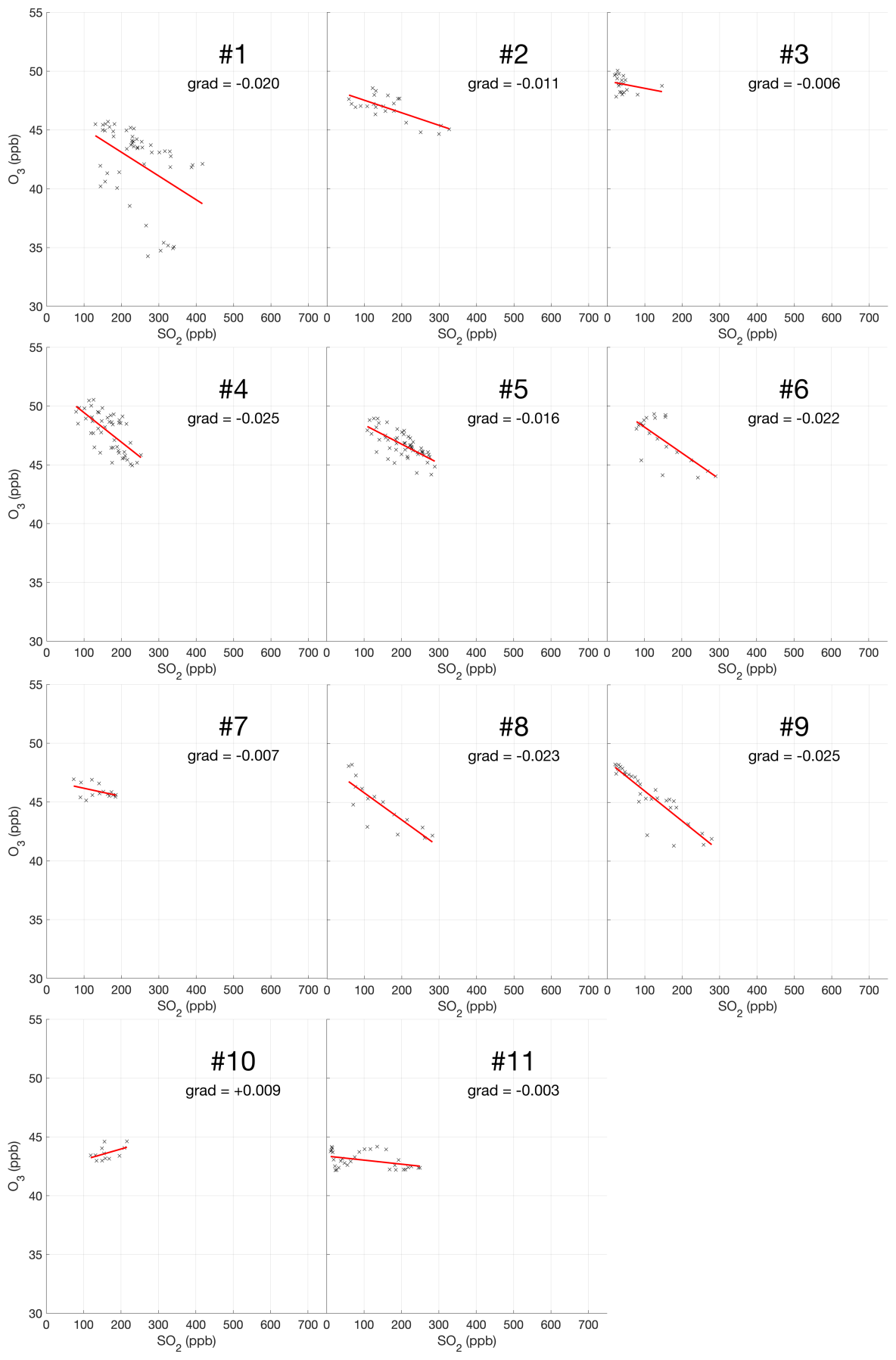
Figure 4Measurements of SO2 and O3 mixing ratios for the 11 major plume encounters on 31 July 2012.
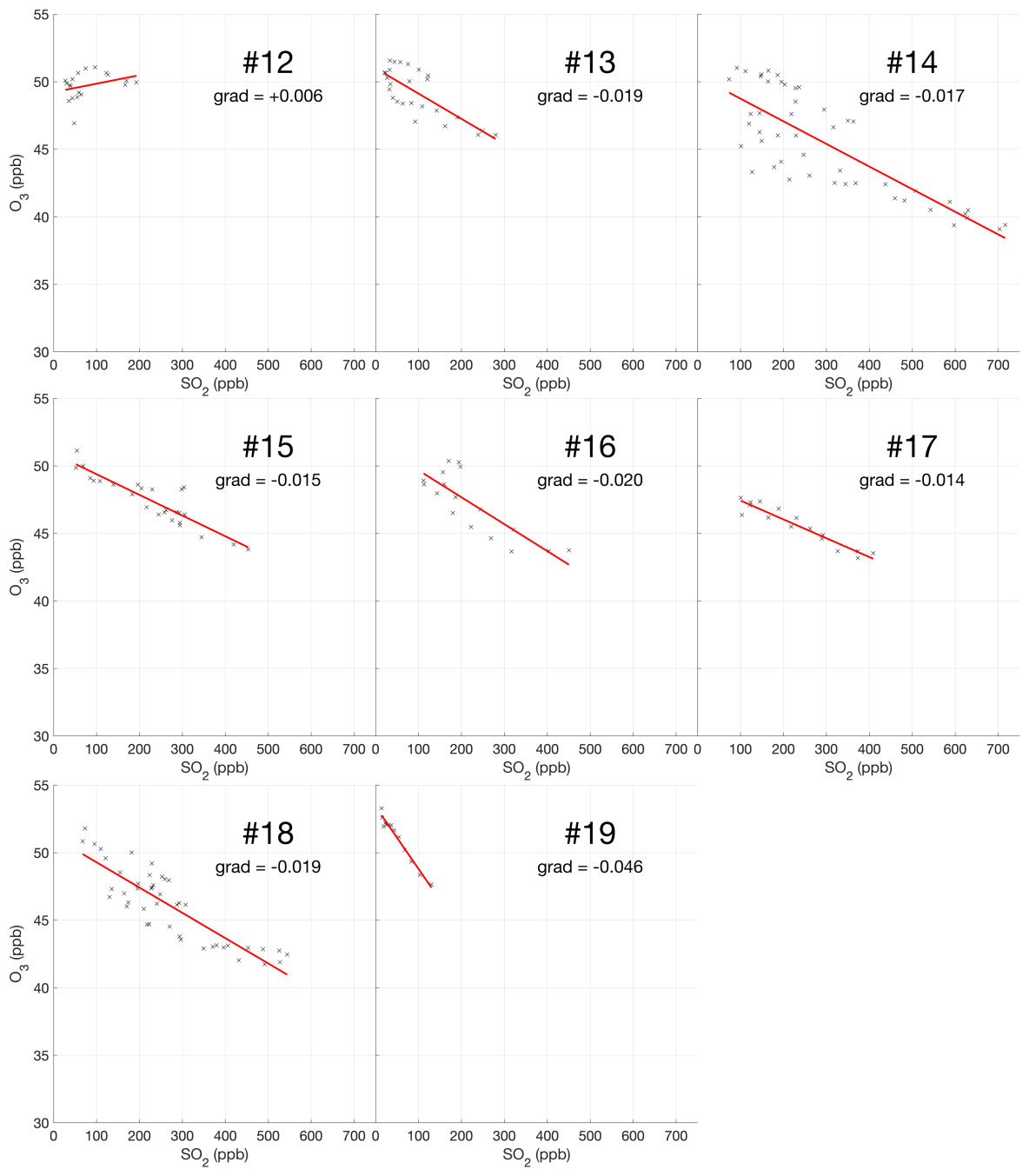
Figure 5Measurements of SO2 and O3 mixing ratios for the eight major plume encounters on 1 August 2012.
For the majority of encounters, there is a clear anti-correlation between O3 and SO2, with linear fits yielding negative gradients for most encounters, which is consistent with continuous O3 destruction during the plume evolution. In several plots, the anti-correlation between SO2 and O3 variations is evident from aligned data points corresponding to consecutive observations. All encounters span ranges of several kilometres, in which the background O3 is liable to vary. We believe this is a significant secondary source of variations in O3 in these encounters. Mixing of the plume with external air with varying levels of O3 would generate random fluctuations in the data and alter the O3 SO2 gradient and its correlation with the plume density. This is the most likely cause for the apparent curvatures when plotting some series of consecutive observations. In the case of encounters 10 and 12, this secondary source of variation obscures the primary effect producing slightly positive gradients.
These observational data show that ozone is depleted within the plume, and this depletion is proportional to the intensity of the plume as quantified by SO2 measurements.
Weighting by the duration of each encounter and their R2 values, the average O3 vs. SO2 gradient for the plume encounters is −0.018 molec. molec.−1, and the average distance from the source is 14 km. Assuming that ozone destruction is a continuous process and that, at distance zero, ozone destruction is zero, these values can be used to quantify the rate of ozone destruction as a ratio of SO2 per kilometre travelled; the resulting value is 0.0015 molec. molec.−1 km−1. This could be converted to a destruction-per-second value by dividing by wind speed. No wind speed data were collected during the flights, so to do this we inspect the meteorological output from the model, which yields for both days a wind speed for the plume of approximately 9 m s−1 at the time of the flights. Using this value yields a rate with respect to time of molec. molec.−1 s−1. Interestingly, this rate is very close to a value of molec. molec.−1 s−1 derived from in situ measurements made within 500 m of the vents on 27–30 July 2012 (Surl et al., 2015), supporting the theory of a continuous process beginning from no ozone depletion at the source.
We note that an analysis to evaluate the trend in ozone depletion with respect to distance within the dataset yielded a null result: ozone-depletion-to-SO2 ratios were calculated for each in-encounter data point by using the y-axis intersect of each plume encounter as an estimate for background ozone for all data points within that encounter. The output from this analysis across the whole dataset was too noisy to discern an overall trend.
The observed depletion of ozone in the Mount Etna plume is consistent with ozone-destructive halogen chemistry in volcanic plumes (Gutmann et al., 2018, and references therein). Halogens were not measured by the aircraft, but ground-based remote sensing confirms the presence of volcanic BrO in the plume during July–August 2012 (Surl et al., 2015).
The following section presents the results of 3D high-resolution WCV model simulations, focusing on the ozone-destructive halogen chemistry in Mount Etna plumes during the 2012 aircraft campaign. The model is used to quantify the ozone depletion and attribute it to specific halogen reactions and to investigate additional impacts of volcanic plume halogen chemistry on atmospheric HOx, NOxy, and mercury. Observational data and the model outputs are compared in Section 4.4.2, where parameters relating to the observed plume encounters and the linear regressions applied to them are also tabulated (Table 5) along with model data.
All results discussed in this section are from the main model run unless otherwise stated. We have paid particular attention to the results relating to 08:00 UTC on 30 July 2012, 31 July, and 1 August 2012 as the aircraft was sampling the plume at approximately this time.
In several cases we use plume age as a variable. This is determined from the tracers, as discussed in the “Methods” section.
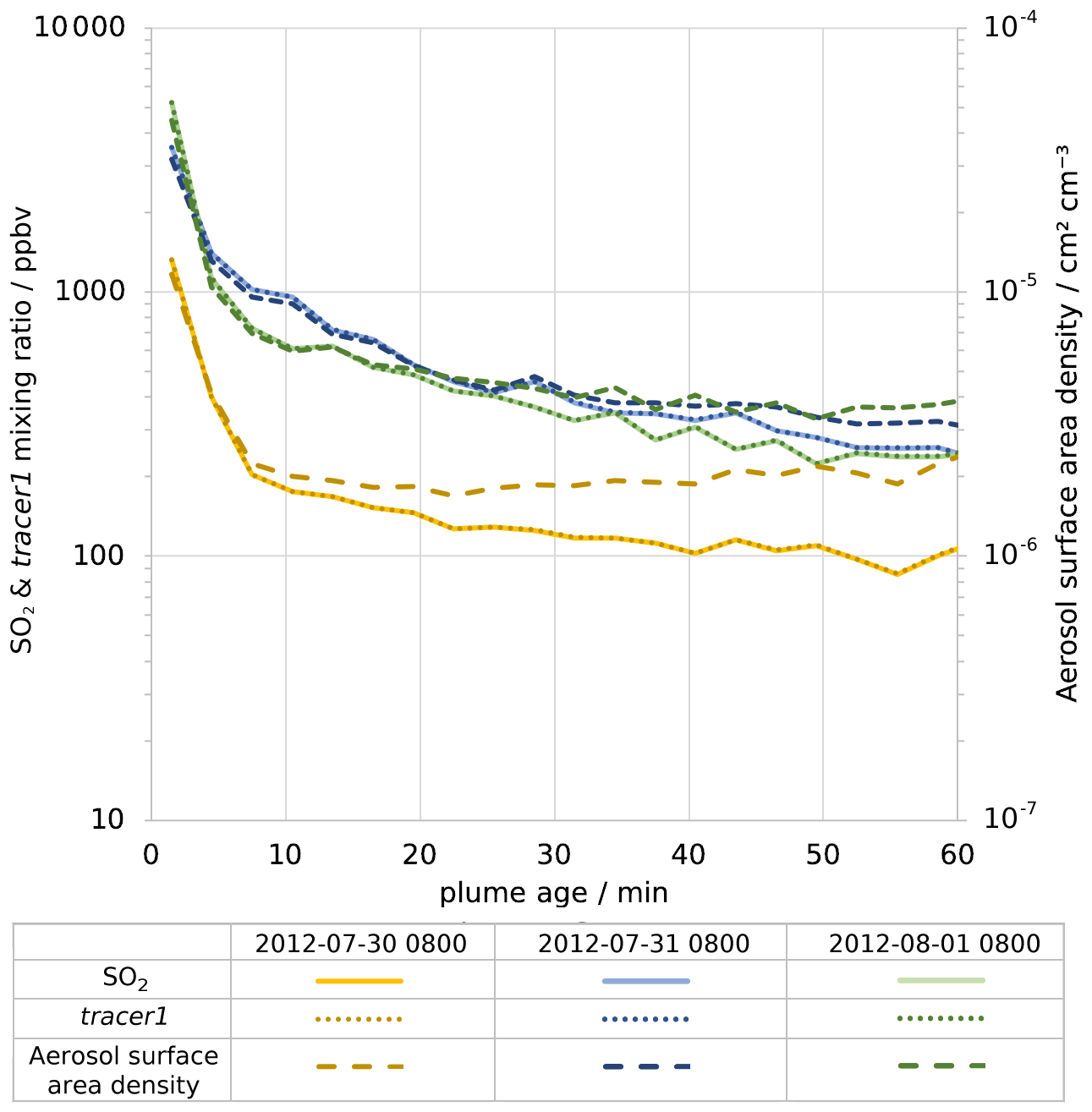
Figure 6Modelled average in-plume mixing ratios of, on left axis, SO2 (solid lines) and tracer1 (dotted lines) and, on right axis, density of aerosol surface area (dashed lines). Values are plotted against plume age for 08:00 UTC for 30 July 2012 (yellow), 31 July 2012 (blue), and 1 August 2012 (green). Note that both y axes are logarithmic.
4.1 SO2, aerosol, and HOx
Before investigating the halogen chemistry of the volcano plume, we first look at SO2. The volcano emits 40 kg of SO2 per second throughout the simulation. This produces a plume that travels downwind, dispersing (i.e. diluting) during transport; SO2 mixing ratios decrease with time and distance from the source (Figs. 6, 14). The CBMZ-MOSAIC reaction scheme includes gas-phase oxidation of SO2 by OH, generating secondary sulfate aerosol. We note that the model does not contain other potentially significant SO2 loss mechanisms which occur in the liquid phase (Galeazzo et al., 2018). The importance of these liquid-phase pathways is likely diminished in a passively degassing plume that does not form a water cloud, which is the case studied here. Nevertheless, the results of this section concerning SO2 oxidation should be interpreted with caution.
For plumes aged less than an hour, the modelled mixing ratios of SO2 and tracer1 in the plume are nearly identical (Fig. 6), indicating that SO2 losses via OH oxidation in this period are negligible. This gives confidence to the use of SO2 as a plume tracer.
The average mixing ratio of SO2 in the plume of 30 July is, for plume ages less than an hour, typically around a third of the equivalent parts of the plume on the other two dates (Fig. 6), although the declining trend of SO2 mixing ratios with age is similar. This difference is due to the fact that the wind speed on 30 July is much higher than on the other dates: the average wind speeds in the < 60 min old plume at 08:00 UTC are 19, 9, and 9 m s−1 for 30 July, 31 July, and 1 August, respectively. Therefore, volcanic emissions are released into a greater volume of air on 30 July, yielding lower concentrations of volcanic volatiles and volcanic aerosol within the plume. As a consequence, the aerosol surface area density within the plume on 30 July is lower, though secondary aerosol production reduces this difference as the plume evolves (Fig. 6).
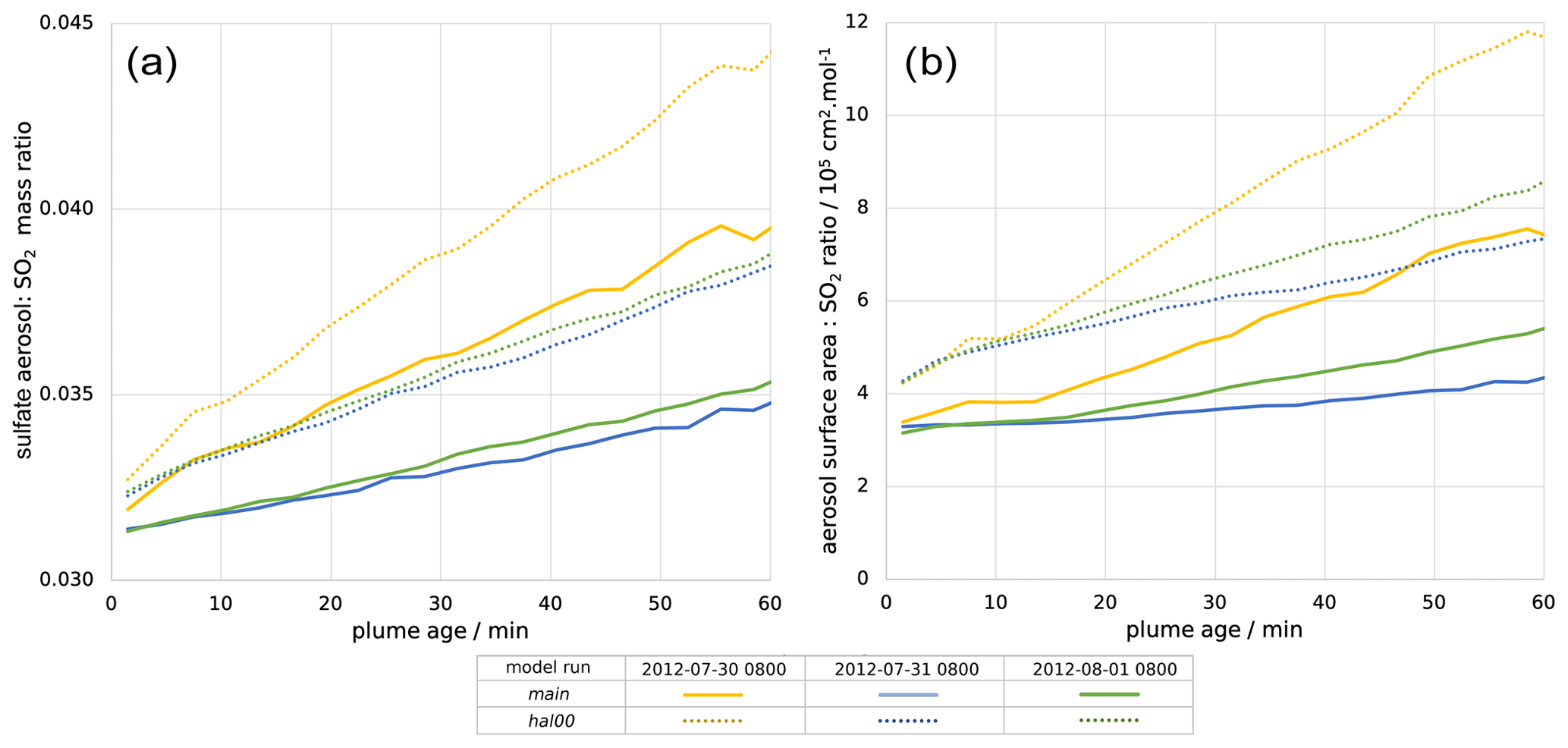
Figure 7(a) Mass ratio of sulfate aerosol to SO2 within the plume and (b) ratio of the total in-plume aerosol surface area to SO2 concentration, both plotted against plume age for 08:00 UTC for 30 July 2012 (yellow), 31 July 2012 (blue), and 1 August 2012 (green). Solid lines are for the main model run; dotted lines are for the hal00 run; see Table 2.
The volcano is a direct source of aerosols, with a flux of 1.2 kg of sulfate per second in the model. This is important for the halogen chemistry as it provides a surface for HOBr uptake, enabling heterogeneous reactions. Figure 7a shows that, shortly after emission, the in-plume ratio of sulfate to SO2 is slightly above the emission ratio of 0.03 because of the early oxidation of SO2 by volcanogenic OH (released in the emission grid box to account for high-temperature radical production), which produces additional sulfate. This ratio continues to increase with plume age due to ambient-temperature oxidation by OH mixing in from background air.
There are similar trends for the ratio of aerosol surface area to SO2 (Fig. 7b). The increase in this ratio shows that the secondary aerosol formation notably increases the surface area available for HOBr uptake. When considered in absolute terms, this secondary aerosol formation partly offsets the decline in aerosol surface area density caused by plume dispersion. This can be seen in Fig. 6, where the aerosol surface area density declines at a slower rate than SO2 mixing ratio. For 30 July the aerosol surface area density is approximately constant after 10 min, indicating that the secondary formation compensates for the dispersion in this regard.
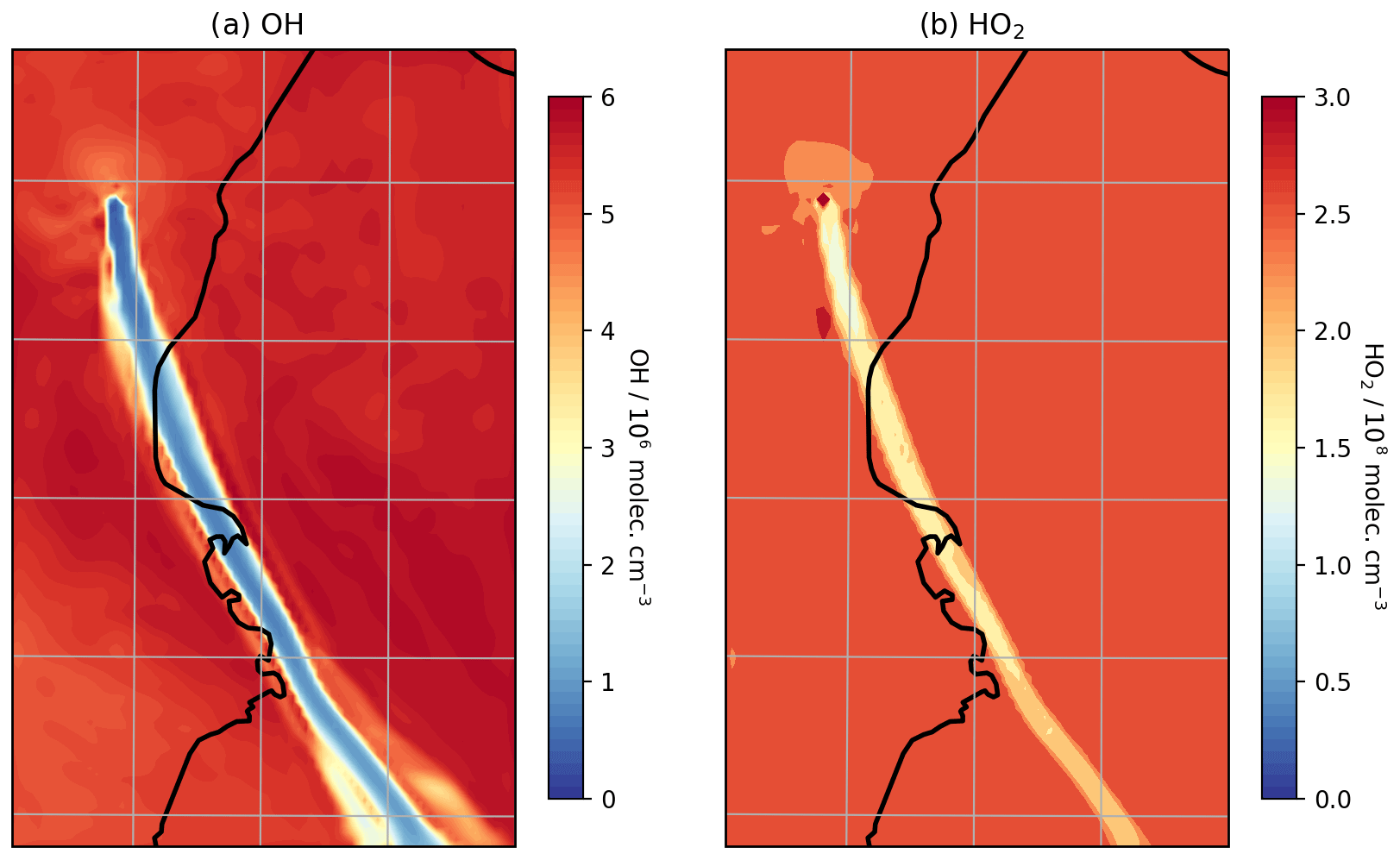
Figure 8Modelled mixing ratios of (a) OH and (b) HO2 at 3300 m a.s.l. at 08:00 UTC on 1 August 2012.
Although the oxidation of SO2 is not significant over these timescales with regard to SO2 mixing ratios, the oxidation that does occur is significant for in-plume aerosol and HOx levels. There is a substantial depletion of OH within the plume and a moderate depletion of HO2 (Fig. 8); for plumes aged around 30 min, OH and HO2 levels are depleted by around 85 % and 40 %, respectively (Table 3). This occurs despite the modelled volcano being a source of OH. This emitted OH is consumed very quickly. This result is consistent with the model findings for the Ambrym plume of Jourdain et al. (2016), who modelled total OH loss occurring in the core of that plume.
Volcanic halogens and SO2 compete for reaction with the available OH in the model. The abundance of SO2 in the plume results in substantial conversion of OH to HO2 via the SO2 + OH reaction. This starts a chain of reactions with short-lived intermediate species that is simplified to a single SO2 + OH → HO2 + H2SO4 reaction in the model (Bekki, 1995; Galeazzo et al., 2018). Volcanic HCl is also abundant and removes OH by the HCl + OH → H2O + Cl reaction. A similar reaction of HBr also occurs, but HBr is much less abundant. HO2 is consumed as part of the bromine cycle in the BrO + HO2 → HOBr reaction.
Table 3 compares the weighted average mixing ratios of HOx species and the instantaneous SO2 lifetime within the plume for the main run, the halogen-free hal00 run, and the equivalent model cells in the plume-free novolc run. Comparing these, it can be seen that the non-halogen components of the plume are sufficient to cause substantial OH depletion, whilst halogens are the cause of HO2 depletion; the hal00 plume actually has greater HO2 compared to the novolc case.
Compared to the novolc case, the instantaneous lifetime of SO2 with respect to oxidation by OH is substantially increased in the halogen-free plume (hal00) due to the reduced OH concentrations, which are caused by high SO2 levels. The addition of halogens to the hal00 scenario, i.e. moving to the main case run, further suppresses OH and hence reduces even more SO2 oxidation, further increasing its lifetime. For the plume part tabulated in Table 3, the instantaneous lifetimes of SO2 for the hal00 and main model runs are, respectively, 4.5 and 7.4 times that of the hal00 run. Slower SO2 oxidation results in slower secondary aerosol production, as seen in terms of both mass and surface area (Fig. 7). This result for a tropospheric volcanic plume mirrors findings from a recent study of a stratospheric volcanic cloud (Lurton et al., 2018).
Table 3In-plume concentrations of HO2 and OH and instantaneous lifetime of SO2 with respect to oxidation by OH for parts of the plume aged 30±5 min at 08:00 UTC on 1 August 2012 for three model runs. Figures for the novolc use the same grid cell weighting as the main model run.

Whilst a detailed analysis of the aerosol microphysics and climate impacts of volcanic aerosols lies beyond the scope of this study, our simulations show substantial differences in the plume sulfate particle surface area density for WCV model simulations with and without volcanic halogen emissions. As plume halogen chemistry exerts an important influence on the oxidation rate of volcanic SO2 and associated formation of secondary aerosol, our results suggest that models simulating chemistry–climate impacts of volcanic sulfur should not ignore the chemistry of volcanic halogens.
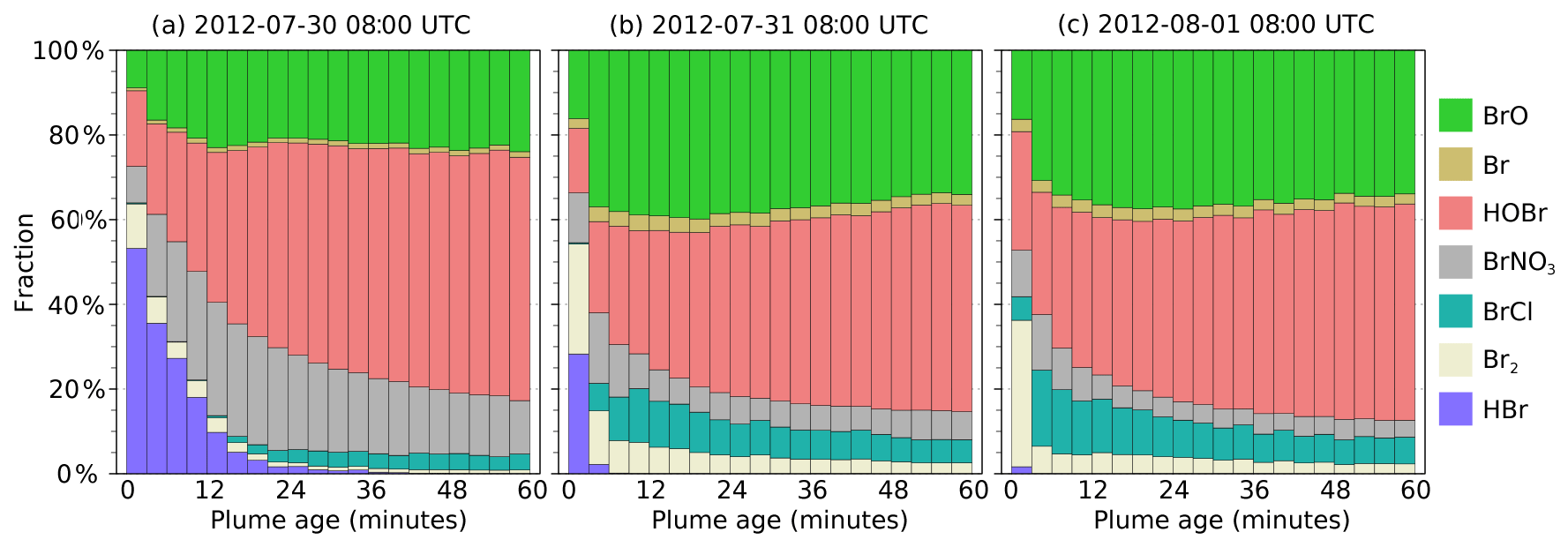
Figure 9Differentiation of bromine between species for parts of the plume at different ages ranging from 0–60 min for plumes at 08:00 UTC on (a) 30 July 2012, (b) 31 July 2012, and (c) 1 August 2012.
4.2 Bromine speciation and BrO SO2 ratio
In the main model run, bromine is emitted from the volcano as HBr and Br in a 3:1 ratio. During daylight this is rapidly converted to other forms, including BrO. Figure 9 shows how the forms which this bromine takes vary between parts of the plume at different ages at 08:00 UTC on 30 July, 31 July, and 1 August. In this model output, HOBr becomes the dominant form of bromine within the plume, followed by BrO. The fraction of BrO increases over approximately the first 20 min before reaching an approximately stable fraction. A significant amount of Br2 is formed shortly after emission, but this fraction declines, with BrCl being the larger fraction of the two halogen dimers. A significant reservoir of BrNO3 that forms shortly after emission declines slowly over time.
There are significant quantitative differences between the bromine evolution on these three dates, although the trends are similar. These differences occur despite the emission parameters being the same for all days. Compared to the other two dates, HBr and BrNO3 persist in the plume for much longer on 30 July. The balance between HOBr and BrO is more greatly tilted towards the former on that day because the in-plume aerosol surface area density is lower on 30 July (see Sect. 4.1 and Fig. 6), reducing the rate of heterogeneous reactions that consume HBr, BrNO3, and HOBr. Additionally, the reaction of BrO with background HO2 to form HOBr is suppressed under more concentrated plume conditions due to the depletion of HO2 discussed in Sect. 4.1.
Although the bromine speciation appears roughly stable after approximately 30 min of evolution, this does not indicate that no further chemistry is occurring; bromine is instead continually cycled between forms. This is shown in Fig. S3, which depicts the rates of transfers between bromine species. “Stability” indicates a state where the chemical formation and loss of each species are approximately balanced, i.e. a steady state.
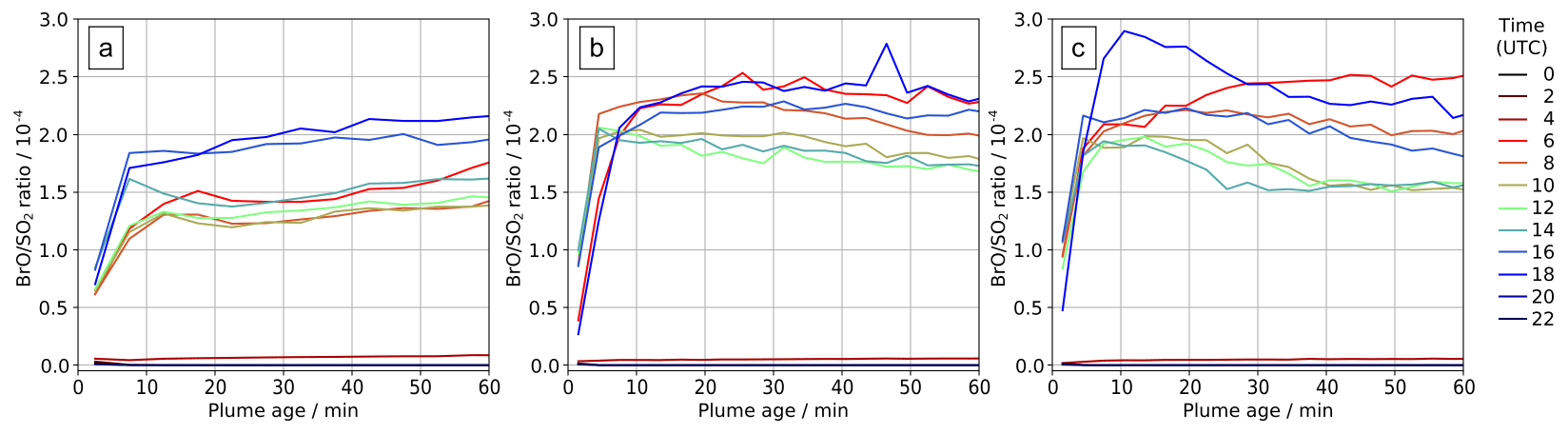
Figure 10In-plume average BrO SO2 ratios for parts of the plume at different ages ranging from 0–60 min at several times on (a) 30 July 2012, (b) 31 July 2012, and (c) 1 August 2012.
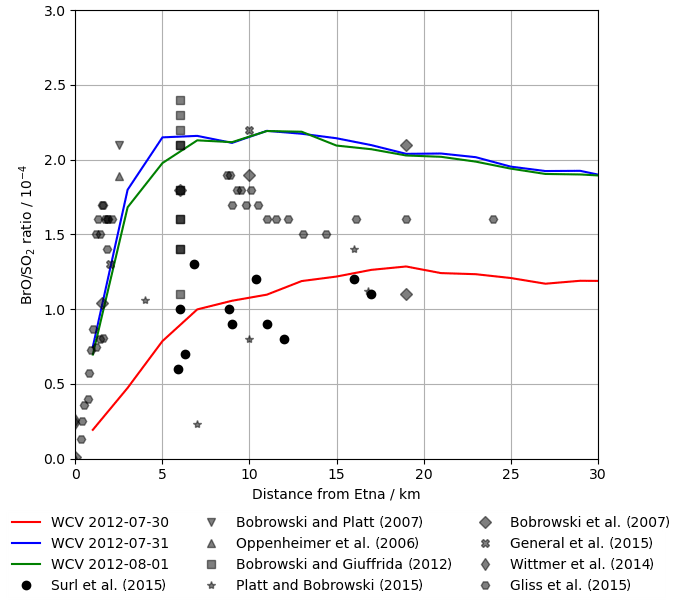
Figure 11Modelled BrO SO2 ratio at 12:00 UTC on 30 July 2012, 31 July 2012, and 1 August 2012 (lines) and observed BrO SO2 ratios in Etna plumes from several studies (markers), plotted against distance from the volcano. We caution that these observations, taken on several days with varying wind speeds, should not be interpreted as representing temporal evolution.
The ratio of total bromine and SO2 is mostly invariant in the plume; therefore the variations in bromine speciation with plume age yield variations in in-plume BrO SO2 ratios. There is an initial rise in BrO SO2, followed by a small decrease in some cases, and then a plateau. This pattern varies with time of day, as shown in Fig. 10. Sunrise is shortly after 04:00 UTC and sunset shortly after 18:00 UTC. Negligible BrO is formed in the model plume at night. We find on all three dates that BrO SO2 ratios are generally greater in the morning and evening than during the middle of the day. This occurs because BrO is more rapidly converted to HOBr in the middle of the day, when atmospheric HO2 is at a maximum. Although moderate in magnitude, this phenomenon may be significant when comparing spectroscopic columns at different times, including datasets from low-earth-orbit satellites with overpasses at different local times.
Our model study identifies how variations in time of day and plume intensity (which is controlled by wind speed in this constant-emission case) impact BrO SO2. These may present additional complications for the interpretation of BrO SO2 ratios observed in passively degassing volcanic plumes. Variations in BrO SO2 are often evaluated with respect to distance from a volcano. We note that the wind speed has a “double effect”. First, faster winds give the impression that the chemistry evolves more slowly as a function of distance downwind because the plume moves further from the source in a given time. Second, all other factors being equal, the along-plume dilution is expected to be stronger at higher wind velocities, affecting the in-plume chemistry and bromine speciation through the influx of background ozone-rich air.
4.2.1 Comparison to observed BrO SO2 ratios
No measurements of in-plume BrO SO2 column ratio were made as part of this study. However the DOAS-measured BrO SO2 columns reported in Surl et al. (2015) span from 24 July 2012 to 2 August 2012, which includes our time period. These observations were made between 5 and 17 km from the volcano at approximately the middle of the day. Figure 11 plots these observations alongside the modelling data for 12:00 UTC each day. These observed ratios, ranging from 0.6–, are at about half of the equivalent modelled ratios for 31 July and 1 August and are comparable, although still slightly lower on average, to those modelled for 30 July, where the initial rise in BrO SO2 ratios occurs over the first 15–20 km.
Although not simultaneous with the time span of this study, BrO SO2 measurements made close to the vents the following month (11 September 2012–26 September 2012) reported by Gliß et al. (2015) are also a relevant comparison. BrO SO2 ratios were found rising rapidly with travel time from the vent, reaching about at about 150 s travel time and then remaining at this level. This rapid rise and higher ratio have better agreement with the observations of 31 July and 1 August.
More generally, the shape of the modelled BrO SO2 versus distance–time trend seen in Figs. 10, 11, and 13a – of an initial rise followed by a steady value – is in agreement with the general trend observed for Etna (Fig. 11) and other volcanoes (Gutmann et al., 2018).
4.2.2 Importance of high-temperature volcanic products
In the main model run, Br and OH radicals are included in the model volcano emission – representing their high-temperature generation in the “effective source region” (see Sect. 2.2.2). The importance of these in the autocatalytic processes of the bromine explosion can be seen by inspecting the output from runs in which the emission strengths of different radicals vary (see Table 2). Figure 12 compares in-plume BrO SO2 ratios for the main case with the results from several other such runs.
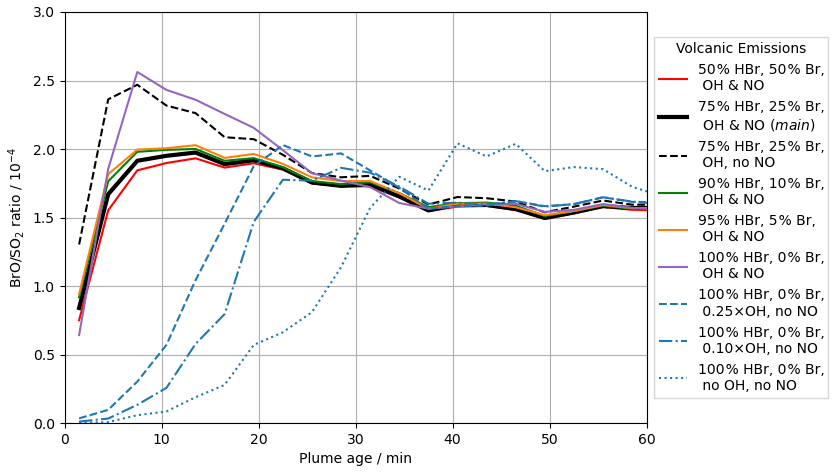
Figure 12Modelled in-plume BrO SO2 ratio at 12:00 UTC on 2012-08-01 versus plume age for the following model runs: br50_50 (red, solid), main (black, solid), NO_00 (black, dashed), br90_10 (green, solid), br95_5 (orange, solid), br100_0 (purple, solid), OH_25 (blue, dashed), OH_10 (blue, dash-dotted), and noHighT (blue, dotted).
The modelled conversion of the emitted bromine into BrO and other forms is much slower when there are no radicals in the volcanic emission (noHighT). The bromine explosion takes tens of minutes to generate in-plume BrO SO2 ratios on the order of 10−4 in the noHighT run. The bromine explosion initiates more quickly in most of the other runs with radical emissions, including the br100_0 case with no volcanogenic Br radical emissions and only OH and NO radicals. These results highlight the importance of high-temperature-generated radicals to initiate the bromine explosion and generate modelled in-plume BrO SO2 ratios of the order observed. We also find low sensitivity to the initial speciation of bromine emissions in the model. We attribute this to halogen radicals (Br and Cl) being formed by reaction of volcanogenic OH with halides. These are sufficient to begin the autocatalytic chemistry even if no Br radicals are directly emitted at the source. The output from the OH_10 and OH_25 runs appears to occupy an intermediate space between the quickly initiating runs and the slow noHighT case, suggesting that the radical emissions in these cases lie around the lower limit of radical emissions required for a quick initiation of the bromine explosion. Note however that the model does not have any background bromine in any runs; in the absence of any volcanogenic radicals, such background bromine mixing into the plume could potentially contribute to initiating the autocatalytic processes. The exact abundance of radicals (e.g. OH, Br, Cl, NO) that may form by high-temperature reactions shortly after emission is not well known (Roberts et al., 2019).
Removing volcanic NO emissions from the model (run NO_00) results in moderately higher in-plume BrO in the early stages of plume evolution as less bromine is held in the BrNO3 reservoir. This difference dissipates with time, with the BrO SO2 ratios for the NO_00 and main runs converging after a few tens of minutes.
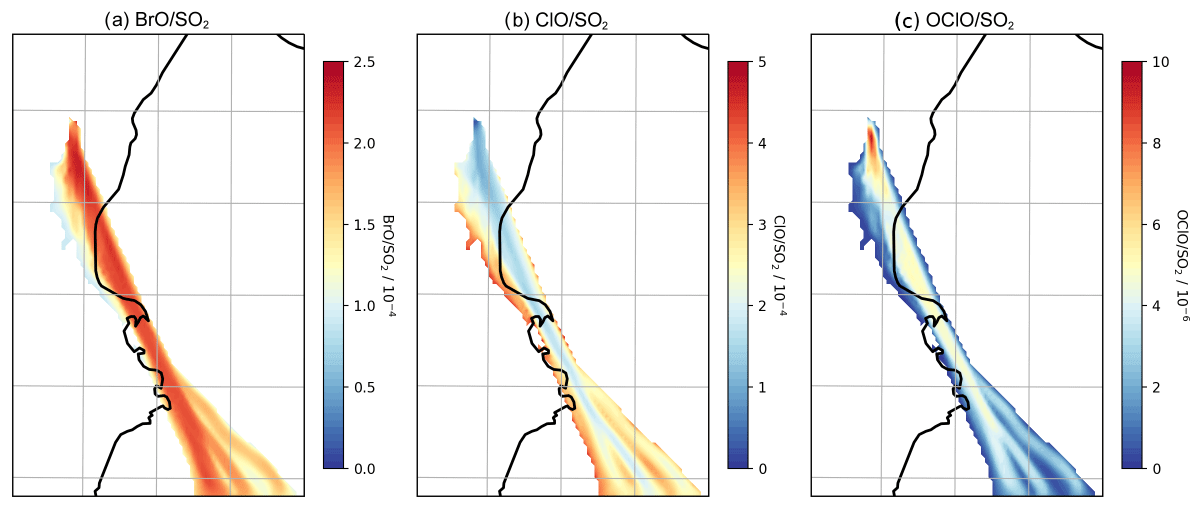
Figure 13Model column ratios at 08:00 UTC on 1 August 2012: (a) BrO SO2, (b) ClO SO2, (c) OClO SO2. Only columns with a modelled SO2 density greater than 2×1016 are plotted.
4.3 Chlorine species
The uptake to particles and subsequent reaction of HOBr with hydrogen halides (HBr, HCl) have the effect of transferring halogen from halides to reactive forms. When HBr and HCl are both present in the plume the product of this is almost exclusively Br2. However after a short time HBr is almost totally depleted in the plume and is consumed as fast as it is produced, whereas HCl remains abundant; the in-plume ratio of HCl to the inert tracer does not significantly change from the emission ratio in the study domain. The subsequent photolysis of BrCl produces reactive chlorine. This reactive chlorine forms the spectroscopically detectable species ClO and OClO within the plume; the reaction of Cl with ozone produces ClO, and the reaction of ClO with BrO produces OClO. Figure 13 shows the vertical columns of these species within the plume as ratios of the SO2 column. ClO SO2 ratios are of similar magnitudes (on the order of 10−4) to BrO SO2. OClO SO2 ratios are about 2 orders of magnitude lower, except for very close to the emission point, where they approach .
Spatially, lower ClO SO2 column ratios are found in the more concentrated parts of the plume (and higher ClO SO2 at the plume edges). Conversely, OClO SO2 shows the opposite spatial pattern and is highest shortly after emission because of the initial burst in OClO production from ClO and BrO in the concentrated early plume.
OClO and ClO are rarely observed above instrumental detection limits in volcanic plumes. Our model results are broadly consistent in magnitude with the few reported OClO observations in Etna plumes reaching (Gliß et al., 2015), whilst General et al. (2015) report OClO SO2 up to 10−4 mol/mol. Some observational studies (Bobrowski et al., 2007; General et al., 2015) report greater BrO SO2 and OClO SO2 at the plume edges compared to the centre, which is not seen in our model. This might be due to the horizontal spatial resolution of WCV, or it might reflect a real difference in the chemistry of the plume. Note that box-modelling findings indicate that the magnitude of such “edge effects” depends on volcanic conditions such as the emitted HBr SO2 (Roberts et al., 2018).
High levels of CH4-oxidizing chlorine radicals (Cl) in the plume reduce the instantaneous lifetime of CH4 in the plume, which, in the early plume considered here, more than compensates for the decrease in CH4 oxidation from the reduced levels of OH. However, at the edges of the plume, the lifetime-extending effect is greater, leading to the spatial pattern seen in Fig. S4. Oxidation of CH4 produces HCHO, and therefore the plume has elevated mixing ratios of this species (Fig. S5).
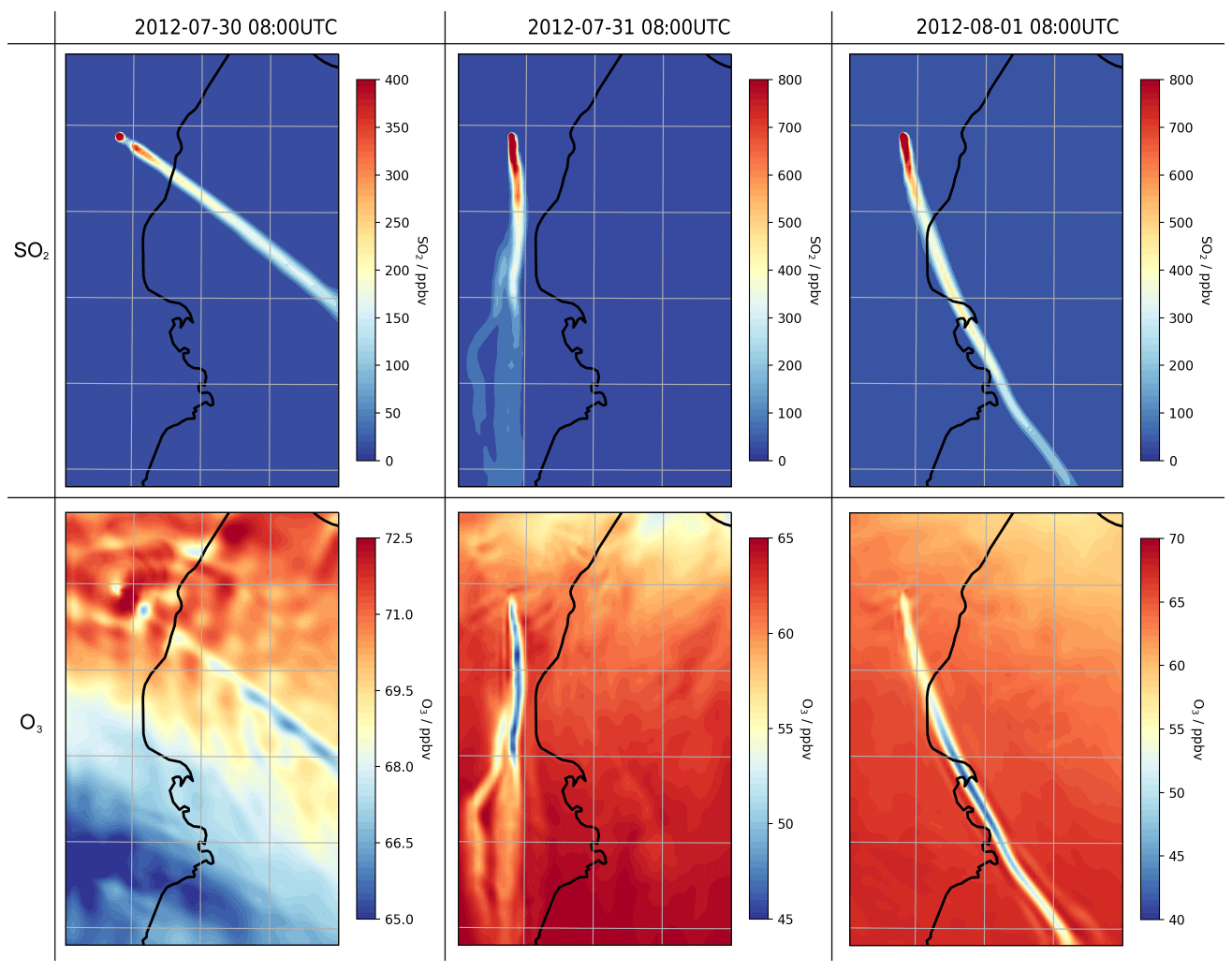
Figure 14Modelled mixing ratios of SO2 (first row) and O3 (second row) at 3300 m a.s.l. (volcano height) in the model at 08:00 UTC on 30 July 2012 (first column), 31 July 2012 (second column), and 1 August 2012 (third column). Different colour scales are used for each subplot.
4.4 Ozone depletion
During the day, ozone levels are lower in the plume than in the surrounding air (Fig. 14). On 30 July, the in-plume ozone loss is only a few parts per billion by volume, whereas it reaches around a few tens of parts per billion by volume on 31 July and 1 August. In terms of ozone columns, the maximum depletions modelled are on the order of 1016 molec. cm−2.
Comparing output from the main and novolc model runs allows for a precise calculation of ΔO3, the change in ozone due to the volcano. Following the approach used in Kelly et al. (2013) and Surl et al. (2015), we compute ΔO3 SO2 to isolate the chemical signal from physical dispersion effects. Figure 15 shows the variation in this ratio with plume age at different times of day. Because this ozone destruction is slower on 30 July compared to the other two dates and because the plume is travelling faster due to the greater wind speed, the smaller ozone loss on 30 compared to the other days is even more apparent if ΔO3 SO2 is plotted against distance (Fig. 16). The absence of halogen chemistry means that, at night, ΔO3 SO2 is close to zero. During the day the ratio is negative and increases in magnitude with plume age. This indicates that chemical ozone destruction in the plume is a continuous, ongoing process. Although the data depicted in Fig. 15 are Eulerian snapshots rather than Lagrangian traces of the plume, these lines' gradients are an indication of the rate of the ozone loss process. For 1 August 2012 at 08:00 UTC, the gradient is about molecules of O3 per molecule SO2 per second.
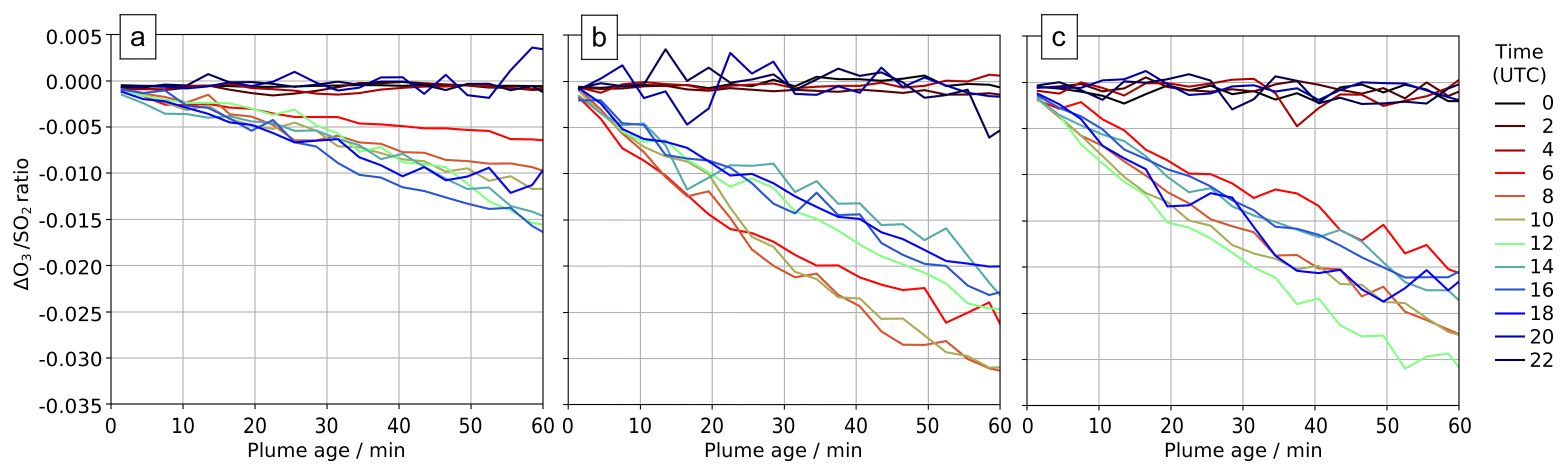
Figure 15In-plume average ΔO3 SO2 ratios for plumes of different ages ranging from 0–60 min at several times on (a) 30 July 2012, (b) 31 July 2012, and (c) 1 August 2012. Only ratios where the corresponding SO2 column exceeds 2.0×1016 molec. cm−2 are shown.
Secondly, by inspecting the rates of reaction for the main model run as shown in Fig. S3 and computing the differences between the rates of ozone-destructive and ozone-forming reactions, we find that these halogen reactions result in an average net ozone loss rate of 3.1×107 molec. cm−3 s−1 within this part of the plume. Dividing this by the weighted average plume SO2 mixing ratio yields an instantaneous loss rate of molecules of O3 per molecule SO2 per second, very close to the value determined by the prior method.
4.4.1 Attribution of ozone loss to halogen reactions
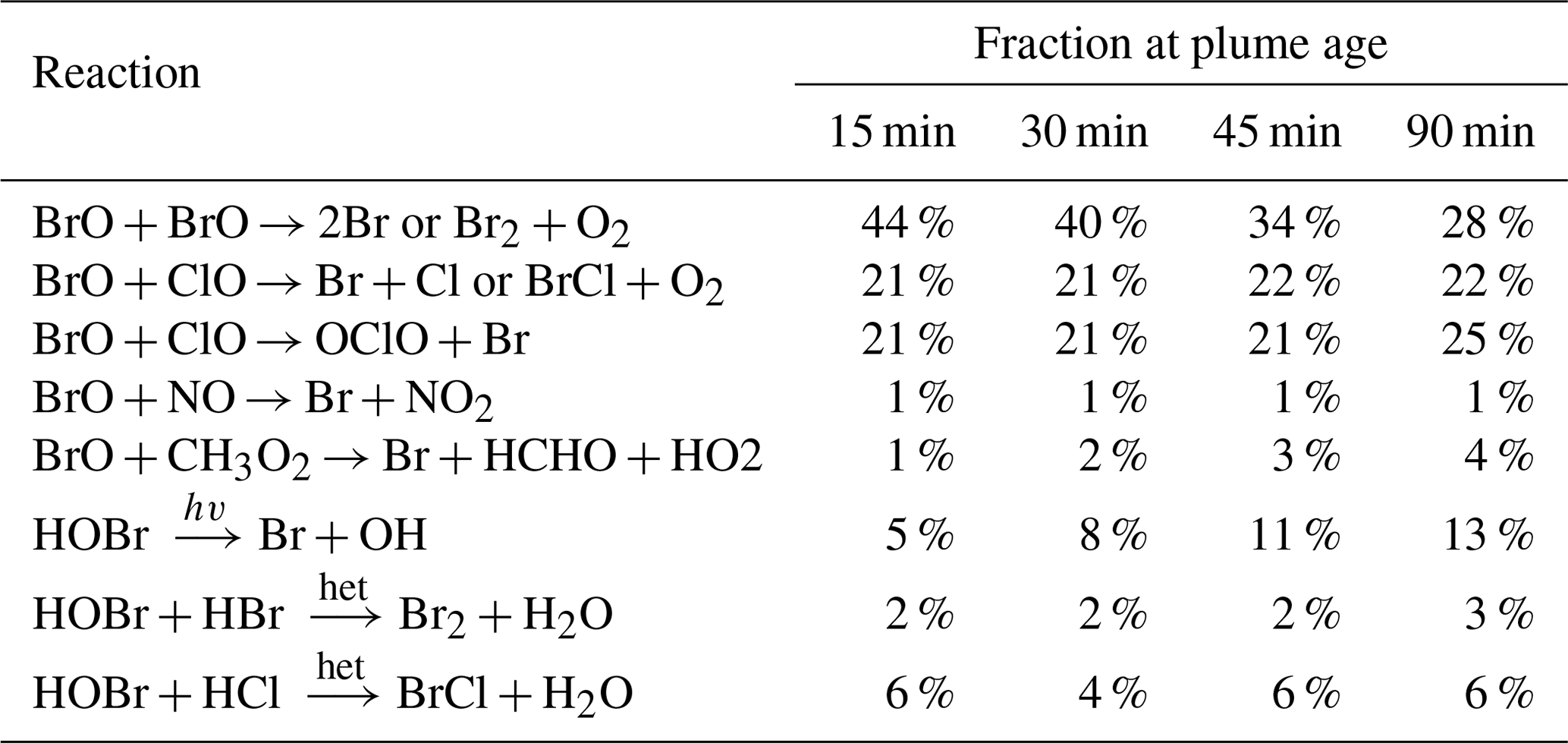
A detailed analysis of the model outputs allows us to attribute ozone loss to specific bromine reaction cycles. Ozone is destroyed by its reaction with Br to form BrO, but the net ozone loss depends on the subsequent fates of BrO and HOBr (the product of BrO and HO2). If BrO undergoes a reduction chemistry that reforms ozone there is no net impact, while if a reduction path does not reform ozone there is a net ozone loss. Table 4 tabulates the relative rates of the BrO and HOBr reduction reactions, which yield Br, Br2, or BrCl without reforming ozone in the plume and therefore can be “credited” with ozone destruction. We find that for this young plume, the most important of these bromine reduction reactions are the reactions of BrO with itself and of BrO with ClO, which together account for about three-quarters of the bromine recycling.
The relative importance of the BrO self-reaction decreases slightly as the plume dissipates and evolves, whilst the two reactions of BrO with ClO maintain approximately the same level of importance. The importance of HOBr photolysis increases over time but remains minor. Although the reactions of HOBr are responsible for only a minor fraction of the bromine reduction in this case, the heterogenous reactions of HOBr are important for transferring bromine from HBr and HOBr to the more potent ozone-destructive forms and for generating the reactive chlorine involved in the BrO + ClO reactions.
Table 4Modelled relative fractions of bromine reduction by the various non-O3-forming reactions for parts of the plume at different ages (±5 min listed age) on 1 August 2012 at 08:00 UTC.
The overall rate of ozone destruction within the plume is dependent upon the quantity of bromine cycling. As shown in Fig. 9, compared to 30 July, bromine is transferred faster out of HBr and BrNO3 on 31 July and 1 August as these are denser (more concentrated) plumes with higher surface area density. Additionally, because several of the reactions listed in Table 4 are between halogen species originating from the volcanic emissions, these are faster in denser plumes. As discussed in Sect. 4.1, the plume is less dense on 30 July due to the higher wind causing volcanic emissions to be injected in larger volumes of air, leading to greater dilution of volcanic emissions. These factors result in a slower ozone destruction for 30, as shown in Fig. 15a. Because this ozone destruction is slower, but the plume travels faster, this difference is magnified if ΔO3 SO2 is plotted against distance (as is done in Fig. 16).
4.4.2 Comparison of model and aircraft data on ozone loss
Here we compare the aircraft observations to model outputs. Because the model plume does not precisely trace the same path as the observed plume, using the exact coordinates of each in-plume observation to identify the plume in the model domain would certainly often result in missing the modelled plume. Instead, for each of the plume encounters discussed in Sect. 3, we identify the equivalent of the observations in the model by the following method. Model data are considered “equivalent” for a plume encounter if they satisfy the following criteria:
-
Time is closest to the median time of the observed encounter.
-
Grid box is wholly or in part within the altitude range of the observed plume encounter.
-
Grid box centre is within the range of distances from Etna for the observed plume encounter.
-
SO2 mixing ratio is in excess of 10 ppbv.
This effectively delimits a 3D space within the model. This 3D space is more likely to include the most concentrated part of the plume than the 1D transect made by the aircraft, and as such, the model data tend to include grid boxes with SO2 greater than the maximum values from the observed plume encounter. For this reason our observation–model comparisons are based on O3 vs. SO2 trends rather than absolute values.
The model output is hourly, and the looping flight path (Fig. 2) means that many of the plume encounters were made minutes apart at nearly the same points in space. As a result, several of these model-equivalent sets share many model data points and are nearly identical. Model equivalents of Figs. 4 and 5 are shown, respectively, in Figs. S8 and S9. The coefficients from simple linear regression of these data are tabulated in Table 5.
Table 5Data relating to the major plume encounters and the equivalent model data, depicted in Figs. 4 and 5 (observations) and Figs. S8 and S9 (model); dur: duration of plume encounter; alt: range of altitudes of plume encounter; dist: distance from Etna; grad: gradient of line of best fit of O3 vs. SO2 slope; int: y intercept of this line.

The gradients of ΔO3 SO2 versus plume age from applying a linear regression to the model and observation data are very similar. Weighting by the quantity and R2 of the fit of the observational data, the root mean square difference in the two gradients is 0.005, with a weighted mean bias of less than 0.001.
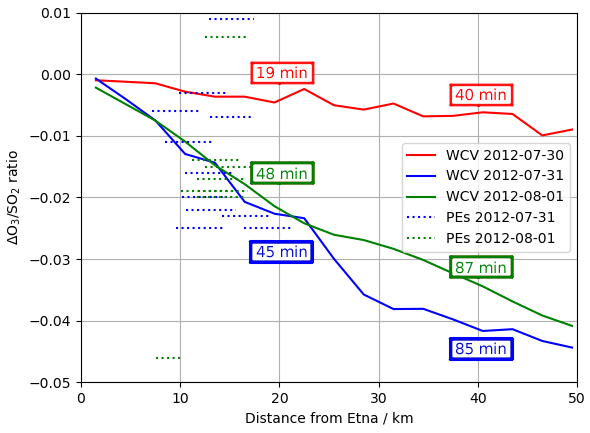
Figure 16Modelled ΔO3 SO2 ratios at 08:00 UTC on 30 July 2012, 1 August 2012, and 31 July 2012 (solid lines) and observed O3 vs. SO2 gradient from each of the major plume encounters (dashed lines), plotted against distance from the volcano. Observations are plotted as horizontal lines spanning the range of distances from the volcano at the plume encounter. Annotations give the average age of the modelled plumes at 20 and 40 km distances.
Figure 16 plots modelled ΔO3 SO2 and these plume encounter gradients against distance from the volcano. The range of distances of the plume encounters is insufficient to determine any trend from the observations beyond those made in Sect. 3; however there is a reasonable match between observed and modelled ΔO3 SO2 values for distances of around 15 km from the volcano. This gives confidence that the model has skill with regards to the ozone chemistry of the plume.
The y intercept of the lines of best fit in the model are consistently 15–20 ppbv lower than those of the observations, reflecting a somewhat higher background ozone. The cause of this is most likely bias in the initial and boundary conditions used in the model. We do not expect this offset to have a significant impact on the main results of this study, which are based on changes in ozone (ΔO3) rather than its absolute magnitude.
4.4.3 Relationship of ozone losses to the magnitude of halogen emissions
As tabulated in Table 2, we ran the model with different volcanic fluxes in order to assess how volcanic impacts on ozone could vary for different passive-degassing emission compositions. In Table 6 we report for the main, mag, hal, and oth runs the average ΔO3 values (compared to the novolc run) in parts per billion by volume for modelled plume aged 60±5 min on 1 August 2012 at 08:00 UTC as a metric for the volcanic impact on ozone. Collectively, these runs explore a two-dimensional parameter space of variations in halogen and other emissions. In all these runs the chlorine emission is scaled to the bromine emission (Cl Br = 300 by mass), the bromine is emitted in the same 3 : 1 fixed proportions of HBr and Br, and all other species (H2O, Hg, OH, NO, at-source aerosol) are scaled to the SO2 emission. In the absence of both SO2 and bromine emissions, the ozone loss relative to the novolc run is, by definition, zero. For the volcanic case without halogens (i.e. the hal00 case), there is a slight ozone production in the plume, as shown in Table 6; model cells for 1 h old plumes have, on average, about half a part per billion by volume more O3 than the equivalent cells in the totally plume-free case. We ascribe this phenomenon to the impact of the volcanic NO emissions, which results in tropospheric ozone production during NOx cycling. Table 6 confirms that it is the halogen emission that causes the ozone loss.
Table 6ΔO3 values (compared to novolc run) in parts per billion by volume for plumes aged 60±5 min on 1 August 2012 at 08:00 UTC for various model runs with varying emissions. For all runs the Cl Br, Br HBr, OH SO2, NO SO2, H2O SO2, Hg SO2, and aerosol SO2 emission ratios remain the same as in main. The 40 kg s−1 row and 30 g s−1 column indicate the emission fluxes used in main.

Increasing the modelled flux of all species other than the halogens above that of the main case does not significantly change the depletion amounts. However, decreasing this flux by two-thirds reduces ozone depletion by around 20 %. As was the case for 30 July in the main model run, the surface area density is insufficient to quickly move bromine from HBr to the ozone-destructive cycle – though in this case this is due to weaker aerosol emissions rather than faster wind speeds.
The four data points for different bromine fluxes at 40 kg SO2 s−1 can be fitted to a second-order polynomial, (ΔO3 ppbv, where x is the volcanic bromine flux in g s−1. We interpret this combination of second- and first-order terms to be the product of the complexities of the chemistry. The reactions that recycle BrO through reaction with other halogen oxide species would be expected to have rates approximately proportional to the square of the quantity of halogens in the plume, while the rates of those that recycle HOBr, including those that generate reactive halogens from hydrogen halides, would vary approximately linearly with this quantity.
We caution against scaling these results to the plumes of large eruptions. Such plumes could have near-total depletion of ozone and/or HOx and produce non-linear effects beyond the scope of this study. The halogen chemistry of large eruptions is being investigated using the WCV model and will be the focus of a future study.

Figure 17(a) Model mixing ratio of NOx, (b) model mixing ratio of HNO3, (c) equivalent mixing ratio of aerosol-phase nitrate at 3300 m a.s.l. in the model at 08:00 UTC on 1 August 2012.
4.5 NOx, NOy, and nitrate aerosol
To conclude this study on passively degassing plumes, we analyse the WCV model outputs to highlight how volcanic halogens also impact nitrogen and mercury species. Although the volcano degassing is a source of NO in the model, the core of the plume is nearly totally depleted in NOx, with concentrations of less than 108 molec. cm−3 compared to background concentrations of around 109 molec. cm−3 (Fig. 17a). The reason for plume NOx being below background levels is the reaction sequence BrO + NO2 → BrNO3 followed by the heterogeneous reaction of BrNO3 with hydrogen halide that has the net effect of converting NO2 into HNO3, a phenomenon discussed by Roberts et al. (2014). As a consequence, as shown in Fig. 17b, the plume is elevated in HNO3 compared to the background; the average in-plume HNO3 concentration is around 13–16×109 molec. cm−3 compared to 7–8×109 molec. cm−3 in the background. Elevated HNO3 has been observed in a number of volcanic plumes (Mather et al., 2004a, b; Martin et al., 2012; Voigt et al., 2014).
As well as the conversion of the volcanogenic and background NOx, displacement of nitrate from background aerosol can also contribute to the in-plume HNO3. The acidic plume, rich in sulfuric and hydrochloric acid, displaces nitrate from background aerosol into the gas phase as HNO3. As shown in Fig. 17c, the aerosol-phase nitrate content within the plume is much lower than the background in the model. The contributions of background NOx (via BrNO3) and background nitrate (via acid displacement) to the plume HNO3 enhancement are of similar magnitude. Because of the conversion of background species, the volcano is not required to be a source of NOx for in-plume HNO3 elevation to occur. In the NO_00 model run, the plume is still elevated in HNO3 due to conversion of background N (Fig. S7); the magnitude of this is about half that of the main case.
As discussed by Martin et al. (2012) it is unclear from reaction kinetics if volcanoes are sources of reactive nitrogen. The levels of background NOx and nitrate aerosol in the free-tropospheric environment modelled in this study (July–August 2012) would be too low to yield, by themselves, HNO3 concentrations of the order measured by Mather et al. (2004a) at Mount Etna summit in May 2002 or those measured in the plume of Etna in a September 2011 aircraft campaign (Voigt et al., 2014). However background NOx and nitrate may be significant contributors to volcanic HNO3 in more nitrogen-polluted environments. Voigt et al. (2014) state that typical conversion times of atmospheric NOx to HNO3 are days in summer mid-latitudes and so cannot explain formation of HNO3 in volcanic plumes. Our modelling results show in-plume gas-phase HNO3 being generated quickly by the mechanisms of acid displacement of background nitrate aerosol and volcanic plume halogen chemistry that converts background NOx as well as volcanic NOx (if present) into HNO3 via BrNO3. These results suggest that analyses of HNO3 measurements within plumes used to assess volcanogenic NOx or NOy need to account for background reactive nitrogen in both the gas and particulate phases.
Due to the formation of the BrNO3 reservoir, a volcanic NOx emission (or, potentially, background NOx of similar magnitude to the plume bromine) could impact the bromine chemistry and speciation and limit the amount of reactive bromine available to form BrO. The quantity of BrNO3 that accumulates depends upon the abundance of NOx available to react with BrO and the rate at which BrNO3 decays via heterogeneous chemistry. Of the three dates modelled, only on 30 July, where the in-plume heterogeneous rates are low (due to a higher wind speed causing greater along-plume dilution), is a substantial fraction of plume bromine held in the BrNO3 reservoir for several minutes.
4.6 Halogen impacts on mercury
The volcano is modelled to emit, per mole of SO2, mol Hg in an unoxidized state. A simplified Hg chemistry scheme has been implemented in WCV to evaluate the extent to which this Hg can become oxidized by interaction with the volcanic halogen chemistry. Importantly, our mechanism includes the recently identified photo-reduction pathways of Hg(I) halides (Saiz-Lopez et al., 2019). For the purposes of this study focused on halogens only, other possible mercury oxidation pathways are ignored. Due to the relatively low quantity of Hg in the plume, the effect of Hg on the halogen chemistry system itself is negligible.
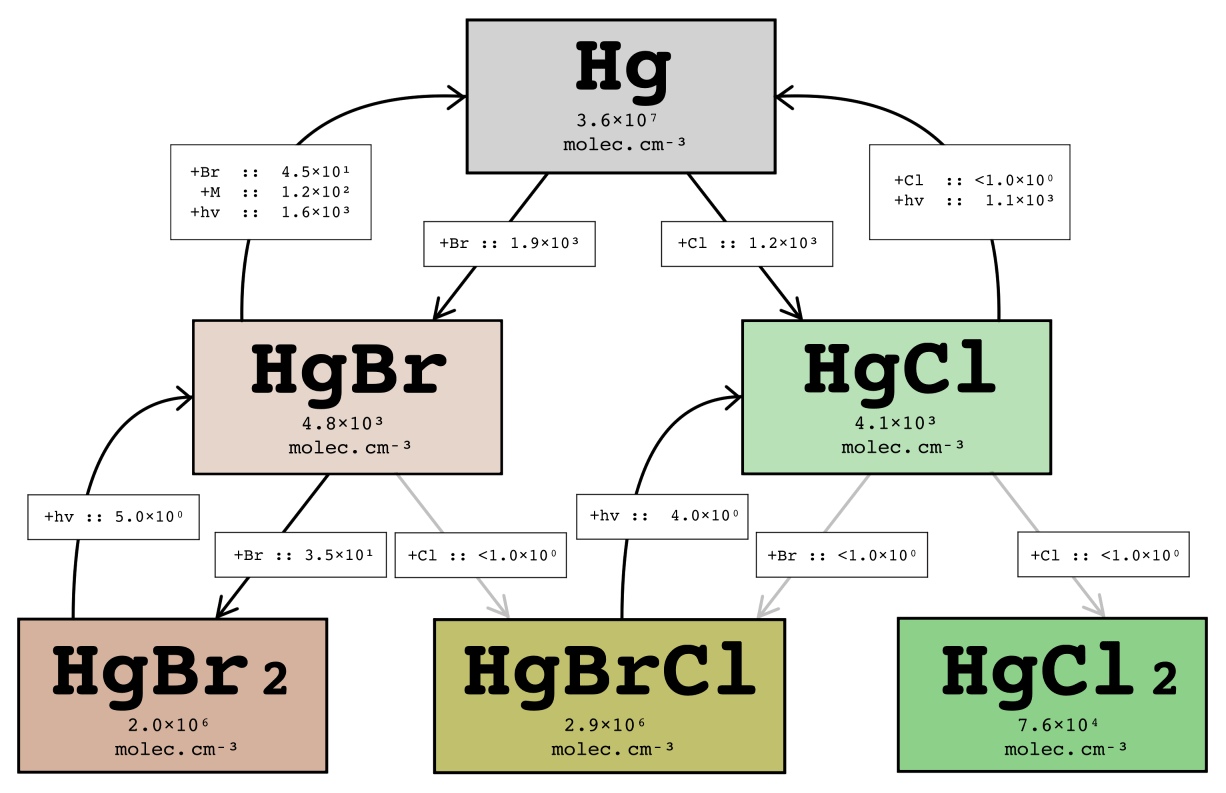
Figure 18Instantaneous average in-plume concentrations and reaction rates (in molec. cm−3 s−1) for the Hg cycle for the part of the plume aged 40–50 min on 1 August 2012 at 12:00 UTC. Rates less than 1 molec. cm−3 s−1 are written as < 1.0×100.
Figure 18 shows the instantaneous average in-plume rates of reactions in the mercury cycle for the part of the plume aged 40–50 min on 1 August 2012 at 12:00 UTC. Although there is a moderately fast oxidation of Hg to HgBr and HgCl, this is mostly offset by reduction. In agreement with global modelling of Saiz-Lopez et al. (2019), photolysis is the dominant Hg(I) reduction process. Overall, there is a small net oxidation of Hg occurring in the plume, but it is slow; for daytime plumes the average in-plume lifetime of Hg(0) is of the order of a few hours, and this oxidation is mostly offset by photo-reduction. Despite the slow rate of oxidation in the plumes aged tens of minutes, we find that in the early plume, modelled levels of mercury oxidation can be several per cent (Fig. S6) from the first few minutes of evolution during both night and day. We attribute this oxidized mercury to oxidation occurring in the very early plume (first few seconds), where volcanogenic radicals are in high concentrations. This near-instantaneous oxidation accounts for the vast majority of oxidized mercury in the modelled plume further downwind. Supporting this interpretation, we find negligible Hg oxidation in the first hour of plume evolution in the output of the noHighT run. Therefore variations in the Hg oxidation with plume age and time of day are due mostly to the conditions at the point of emission rather than any processes occurring within the downwind plume.
We conclude that, for our case study of a Mount Etna passively degassing plume that focused on mercury–halogen interactions only, the net in-plume oxidation rate of mercury by halogen chemistry is near-negligible in the dispersed plume but could be significant very close to the source. Our findings contrast with the model study of von Glasow (2010) that predicted substantial oxidation of mercury to Hg(II) occurring in the dispersed volcanic plume of Mount Etna. However, that study did not include the very fast HgCl and HgBr photo-reduction pathways that critically impact the overall Hg oxidation, although it did include an SO2-mediated reduction in Hg(II) hypothesized by Seigneur et al. (2006). We note that this slow net oxidation rate in the modelled evolved plume occurs despite the absence of this reduction pathway in the mechanism.
We invite caution regarding the interpretation of these results due to the very simple mercury chemical scheme used and the apparent importance of the first few seconds of plume evolution, which would be, spatially, poorly represented even at 1 km grid resolution.
Further investigation of mercury chemistry in volcanic plumes is needed across a range of volcanic and meteorological conditions. This requires both modelling at higher resolution to capture very-near-source processes in the young cooled plume and investigations of hot plume chemistry just after emission and the mercury chemistry in much-larger-eruption plumes that may differ from the passive-degassing case. The mercury oxidation reduction scheme should also be extended to investigate possible roles of other gases (e.g. NOx). More comprehensive observation studies of speciated mercury at Mount Etna and other volcanoes are also needed.
Volcanoes emit halogens that are converted into active chemical radicals in plumes and whose chemistry, notably bromine, causes ozone destruction. However, the plume processes driving this halogen conversion, the so-called halogen activation, and the ensuing ozone depletion within the volcanic plumes are not yet well constrained by the limited observational datasets and numerical model studies. It remains difficult to assess accurately the large-scale impact of volcanic halogens without a quantitative understanding of halogen plume chemistry. This study presents a new dataset of airborne measurements made during the summer 2012 in the degassing plume from the Mount Etna volcano, up to a few tens of kilometres from the source. This chemically reactive plume is simulated using a new numerical 3D model “WRF-Chem Volcano” (WCV), a version of WRF-Chem we have modified to incorporate volcanic emissions (including HBr and HCl) and multi-phase halogen chemistry.
Measurements of SO2 and ozone levels in the plume are found to be strongly anticorrelated. Ozone losses reach up to about 10 ppbv. Accounting for the distance from the source at which these measurements were taken (7–21 km downwind from the summit) and using modelled wind speeds, the ozone destruction rate is estimated at approximately mol O3 per mol SO2 per second. This value is similar to observation-derived estimates reported very close to the Mount Etna vents (< 500 m downwind) (Surl et al., 2015), indicating continual ozone loss in the plume up to tens of kilometres downwind.
The aircraft observations are analysed with the WCV model forced by emission fluxes of volcanic gases, including SO2, mercury, and halogens (HBr, HCl), each set to values within typical ranges observed for Etna in a passive-degassing state. The model initialization also includes a representation of high-temperature radicals (Br, OH, NO) and a volcanic sulfate particle emission. The WCV mechanism includes photolytic, gas-phase, and multi-phase reactions of bromine and chlorine as well as gas-phase oxidation of SO2. WCV was run using two-way nested grids, enabling 1 km resolution for the simulation of plume processes close to the volcano – the focus of this study.
The model simulates the in-plume conversion of emitted HBr into halogen radicals such as BrO and ozone destruction in the volcanic plume. Modelled in-plume BrO SO2 columns are similar to those observed at Mount Etna and other volcanoes, both in terms of magnitude (molar ratio of around 10−4) and their spatial variation (rise and plateau with distance downwind). For the modelled plume corresponding to the observations, the resulting O3 versus SO2 gradients are very similar to those in the observational data. We compute ΔO3, the modelled ozone change due to the plume, by comparing output from model runs with and without volcanic emissions. The variation in ΔO3 SO2 ratios with plume age yields the rate of ozone loss as a ratio of SO2. At the time of the plume measurements on 1 August 2012, this quantity is approximately molec. molec.−1 s−1, with a very similar value from an analysis of the instantaneous rates of reactions. In summary, the WCV model shows apparent skill in reproducing plume halogen chemistry and impacts on tropospheric ozone.
Inspecting the bromine chemical system, we found that HBr, the dominant form of bromine at emission, is converted to other forms within the first few minutes of plume evolution. These forms undergo a continuous cycling in the plume, with BrO and HOBr being the dominant daytime forms. We found that a lower plume density, caused by a greater wind speed, slowed the evolution of the bromine chemical system, with more bromine residing in HBr, HOBr, and BrNO3. The balance between BrO and HOBr varies moderately with time of day due to diurnal variations in HO2, yielding slightly lower BrO SO2 around solar noon. Inspection of the rates of reaction reveals that although the overall proportions of bromine in different forms stabilize after a few tens of minutes of plume evolution, bromine is constantly cycling between forms, and this process includes the ozone-destructive Br + O3 → BrO reaction. Overall the ozone loss depends on several different reactions that reduce oxidized bromine without recreating ozone. We find that, for young plumes (< 1 h old), the most important reactions are those of BrO with halogen monoxides (BrO or ClO).
The conversion of volcanic HBr into reactive bromine forms occurs by the heterogeneous reaction of HOBr. Once plume HBr is depleted, this reaction acts to convert HCl into reactive chlorine, leading to the release of methane-oxidizing chlorine radicals. As a result the lifetime of methane is reduced in the plume core; however at the plume edge methane lifetime increases due to lower OH. The CH4 oxidation initiates organic chemistry processing that results in formaldehyde being elevated in the plume compared to the background. Cl radicals also generate ClO and OClO – species that have also been detected in the plumes of some volcanoes, including Mount Etna. Modelled near-source OClO SO2 ratios are of an approximately similar magnitude (10−5 mol/mol) to those measured by Gliß et al. (2015).
The model plume chemistry is investigated over a range of emission scenarios. If the radical species expected to be produced in the high-temperature volatile-air mix are excluded, the evolution of the halogen chemistry is greatly slowed and delayed. This result highlights the importance of understanding these very early processes in order to have an accurate picture of the overall chemistry. A sensitivity study of the model response to variations in the emissions of both halogens and sulfur finds that ozone loss depends on the bromine emission flux with both linear and quadratic components, reflecting the complexities of the plume chemistry.
Finally, the model outputs are inspected to identify halogen impacts on HOx, sulfur, NOx, and mercury chemistry.
Despite the volcano being an initial source of high-temperature OH radicals, for the early (< 1 h age) plume considered in this study the in-plume instantaneous lifetime of SO2 in the model is substantially increased (from about 2 d to about 2 weeks) due to depletion of OH. These modelling results therefore strengthen the case for using SO2 as a plume tracer on these scales. The substantial depletion of OH is attributed to both SO2 and halogen chemistry, which further reduces OH concentrations within the plume. Halogen chemistry also causes depletion of HO2.
The SO2 oxidation that does occur nevertheless produces sulfate aerosol mass and surface area within the plume. Secondary aerosol is formed more quickly in a simulation that excludes volcanic halogens. This result demonstrates that volcanic halogen chemistry can critically influence sulfur oxidation processes and emphasizes the need to include halogens in studies of volcanic sulfate aerosol impacts.
Despite the volcano being modelled as an initial source of high-T NO, modelled in-plume NOx levels are lower than the surrounding air due to plume chemistry destroying these species. In-plume HNO3 was found to be elevated for two reasons: bromine chemistry converts NOx to HNO3, and background aerosol-phase nitrate is displaced into the gas phase by the acidic plume.
The model includes a very simple mercury scheme which includes photolysis reduction in mercury halides. In this passive-degassing case, WCV predicts some early-stage oxidation of mercury by the initial high-temperature-region Br radicals but – in contrast to previous model studies for Mount Etna passive degassing – also predicts a very slow net oxidation by halogens in the downwind plume. Further model–observation studies of volcanic mercury with more complete schemes are warranted as well as lab experiments assessing the rates of atmospheric mercury reactions.
The rates of the various halogen reactions critical to the main results of this paper are reasonably well known, with the significant exception of the heterogeneous processing of HOBr. The recommended effective uptake coefficient is based on experiments on surfaces that do not necessarily well represent volcanic particles (Ammann et al., 2013). Additionally the surface area available for this reaction is strongly dependant on the quantity and character of volcanic particulate emissions. Such settings used for the modelling of this work are very loosely constrained by observations from other time periods (Roberts et al., 2018), and the summer 2012 emissions of Etna may vary substantially from these. Together with the simplistic assumption of spherical particles, there is overall a large uncertainty regarding this processing. An additional source of uncertainty is the extent to which WRF-Chem overdisperses the plume in the vertical dimension, a known problem with Eulerian models (Lachatre et al., 2020). We note that the emission magnitude in the model was calibrated so as to approximately replicate the aircraft-observed mixing ratios of SO2.
Overall, however, the WCV model appears to show reasonable skill in replicating observed in-plume phenomena of ozone loss specific to this case study and established downwind trends in BrO SO2 for minutes-old passively degassing plumes more generally. Deriving confidence from this, we believe that our model results are at least a reasonable representation of the halogen chemistry occurring within a passively degassing volcanic plume.
This skill gives credence to the assessments of the chemical processes occurring within the plume. WCV operated using nested grids enables 1 km resolution to be reached; however, we suggest caution in using results from the model for processes occurring at the sub-kilometre scale within the very early plume. This study investigated the chemical processes occurring in the passively degassing plume of Mount Etna. We caution against extrapolating these results to stronger-eruption (more dense) plumes as such plumes may experience phenomena out of the scope of this study, such as near-total ozone depletion that perturbs the halogen chemistry. WCV is being applied in a follow-up study to investigate the plume chemical processes in such dense plumes from volcanic eruptions to the troposphere as a contrast to this passive-degassing case. In future, WCV can also be applied to assess the tropospheric impacts of volcanic halogen chemistry in plumes as they disperse and may remain chemically active for up to regional scales.
The code of WCV is available on GitHub (Surl, 2020, https://github.com/LukeSurl/WCV). This repository is being actively maintained. The version of the code used to generate the results of this study is included in this repository as a static branch (etna2012).
The modifications to the PREP-CHEM-SRC utility have been submitted to the maintainers of this software for consideration and are available from the authors on request.
WRF-Chem generates NetCDF files as output. The output relating to the innermost domain (d04 in Fig. 3) is uploaded to a Zenodo online repository (Surl, 2021, https://doi.org/10.5281/zenodo.4415788). This repository also contains the input setting files used for the runs.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-12413-2021-supplement.
LS analysed the observational data, made the model code modifications, ran the model, and analysed and visualized the model output. LS, TR, and SB wrote the manuscript, provided critical feedback, and helped shape the research and analysis. TR supervised the project.
The authors declare that they have no conflict of interest.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
As well as the financial support noted below, the authors would like to acknowledge the following. Computer modelling benefited from access to IDRIS HPC resources (GENCI allocation A007017141) and the IPSL mesoscale computing centre (CICLAD: Calcul Intensif pour le CLimat, l'Atmosphère et la Dynamique). Aircraft measurements were carried out within the Global Mercury Observation System project (GMOS; http://www.gmos.eu, last access: 12 August 2021), and we acknowledge the contribution of the GMOS teams of CNR IIA, Italy, and Helmholtz-Zentrum Geesthacht, Germany. GMOS was financially supported by the European Union within the seventh framework programme (FP-7; project ENV.2010.4.1.3-2). The authors would like to thank Louis Marelle and Jennie Thomas for their assistance with the model development. The model presented in this paper is a development of work undertaken as part of Luke Surl's PhD, which was funded by the UK's Natural Environment Research Council and supervised by Deanna Donohoue and Roland von Glasow. The authors would like to thank the two anonymous referees for suggesting various improvements to this paper.
This paper is a result of a project that has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 800062 as well as ANR Projet de Recherche Collaborative VOLC-HAL-CLIM (Volcanic Halogens: from Deep Earth to Atmospheric Impacts), ANR-18-CE01-0018.
This paper was edited by Marc von Hobe and reviewed by two anonymous referees.
Aiuppa, A., Bellomo, S., D'Alessandro, W., Federico, C., Ferm, M., and Valenza, M.: Volcanic plume monitoring at Mount Etna by diffusive (passive) sampling, J. Geophys. Res.-Atmos., 109, D21308, https://doi.org/10.1029/2003JD004481, 2004. a
Aiuppa, A., Federico, C., Franco, A., Giudice, G., Gurrieri, S., Inguaggiato, S., Liuzzo, M., McGonigle, A. J. S., and Valenza, M.: Emission of bromine and iodine from Mount Etna volcano, Geochem. Geophy. Geosy., 6, Q08008, https://doi.org/10.1029/2005gc000965, 2005. a, b
Aiuppa, A., Franco, A., von Glasow, R., Allen, A. G., D'Alessandro, W., Mather, T. A., Pyle, D. M., and Valenza, M.: The tropospheric processing of acidic gases and hydrogen sulphide in volcanic gas plumes as inferred from field and model investigations, Atmos. Chem. Phys., 7, 1441–1450, https://doi.org/10.5194/acp-7-1441-2007, 2007. a
Aiuppa, A., Giudice, G., Gurrieri, S., Liuzzo, M., Burton, M., Caltabiano, T., McGonigle, A. J. S., Salerno, G., Shinohara, H., and Valenza, M.: Total volatile flux from Mount Etna, Geophys. Res. Lett., 35, L24302, https://doi.org/10.1029/2008GL035871, 2008. a
Ammann, M., Cox, R. A., Crowley, J. N., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates, Atmos. Chem. Phys., 13, 8045–8228, https://doi.org/10.5194/acp-13-8045-2013, 2013. a
Badia, A., Reeves, C. E., Baker, A. R., Saiz-Lopez, A., Volkamer, R., Koenig, T. K., Apel, E. C., Hornbrook, R. S., Carpenter, L. J., Andrews, S. J., Sherwen, T., and von Glasow, R.: Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem, Atmos. Chem. Phys., 19, 3161–3189, https://doi.org/10.5194/acp-19-3161-2019, 2019. a
Bagnato, E., Aiuppa, A., Parello, F., Calabrese, S., D’Alessandro, W., T.A., M., McGonigle, A., Pyle, D., and Wängberg, I.: Degassing of gaseous (elemental and reactive) and particulate mercury from Mount Etna volcano (Southern Italy), Atmos. Environ., 41, 7377–7388, https://doi.org/10.1016/j.atmosenv.2007.05.060, 2007. a
Bagnato, E., Tamburello, G., Avard, G., Martinez Cruz, M., Enrico, M., Fu, X., Sprovieri, M., and Sonke, J.: Mercury fluxes from volcanic and geothermal sources: An update, Geological Society London Special Publications, 410, p. 263, https://doi.org/10.1144/SP410.2, 2014. a
Bekki, S.: Oxidation of volcanic SO2: A sink for stratospheric OH and H2O, Geophys. Res. Lett., 22, 913–916, https://doi.org/10.1029/95gl00534, 1995. a
Bobrowski, N. and Giuffrida, G.: Bromine monoxide / sulphur dioxide ratios in relation to volcanological observations at Mt. Etna 2006–2009, Solid Earth, 3, 433–445, https://doi.org/10.5194/se-3-433-2012, 2012. a
Bobrowski, N., Hönninger, G., Galle, B., and Platt, U.: Detection of bromine monoxide in a volcanic plume, Nature, 423, 273–276, https://doi.org/10.1038/nature01625, 2003. a
Bobrowski, N., von Glasow, R., Aiuppa, A., Inguaggiato, S., Louban, I., Ibrahim, O. W., and Platt, U.: Reactive halogen chemistry in volcanic plumes, J. Geophys. Res.-Atmos., 112, D06311, https://doi.org/10.1029/2006jd007206, 2007. a, b, c, d, e, f, g
Bobrowski, N., von Glasow, R., Giuffrida, G. B., Tedesco, D., Aiuppa, A., Yalire, M., Arellano, S., Johansson, M., and Galle, B.: Gas emission strength and evolution of the molar ratio of BrO SO2 in the plume of Nyiragongo in comparison to Etna, J. Geophys. Res.-Atmos., 120, 277–291, https://doi.org/10.1002/2013jd021069, 2015. a
Bobrowski, N., Giuffrida, G. B., Arellano, S., Yalire, M., Liotta, M., Brusca, L., Calabrese, S., Scaglione, S., Rüdiger, J., Castro, J. M., Galle, B., and Tedesco, D.: Plume composition and volatile flux of Nyamulagira volcano, Democratic Republic of Congo, during birth and evolution of the lava lake, 2014–2015, B. Volcanol., 79, 90, https://doi.org/10.1007/s00445-017-1174-0, 2017. a
Brenna, H., Kutterolf, S., Mills, M. J., and Krüger, K.: The potential impacts of a sulfur- and halogen-rich supereruption such as Los Chocoyos on the atmosphere and climate, Atmos. Chem. Phys., 20, 6521–6539, https://doi.org/10.5194/acp-20-6521-2020, 2020. a, b
Buchholz, R. R., Emmons, L. K., Tilmes, S., and The CESM2 Development Team: CESM2.1/CAM-chem Instantaneous Output for Boundary Conditions – Subset used: 20∘ N–45∘ N, 5∘ E–45∘ E 2018-12-21–2018-12-29, https://doi.org/10.5065/NMP7-EP60, 2019. a
Burton, R. R., Woodhouse, M. J., Gadian, A. M., and Mobbs, S. D.: The Use of a Numerical Weather Prediction Model to Simulate Near-Field Volcanic Plumes, Atmosphere, 11, 594, https://doi.org/10.3390/atmos11060594, 2020. a
Cadoux, A., Scaillet, B., Bekki, S., Oppenheimer, C., and Druitt, T. H.: Stratospheric Ozone destruction by the Bronze-Age Minoan eruption (Santorini Volcano, Greece), Sci. Rep.-UK, 5, 12243, https://doi.org/10.1038/srep12243, 2015. a
Carn, S., Clarisse, L., and Prata, A.: Multi-decadal satellite measurements of global volcanic degassing, J. Volcanol. Geoth. Res., 311, 99–134, https://doi.org/10.1016/j.jvolgeores.2016.01.002, 2016. a
Dinger, F., Bobrowski, N., Warnach, S., Bredemeyer, S., Hidalgo, S., Arellano, S., Galle, B., Platt, U., and Wagner, T.: Periodicity in the BrO SO2 molar ratios in the volcanic gas plume of Cotopaxi and its correlation with the Earth tides during the eruption in 2015, Solid Earth, 9, 247–266, https://doi.org/10.5194/se-9-247-2018, 2018. a, b
Dinger, F., Kleinbek, T., Dörner, S., Bobrowski, N., Platt, U., Wagner, T., Ibarra, M., and Espinoza, E.: SO2 and BrO emissions of Masaya volcano from 2014 to 2020, Atmos. Chem. Phys., 21, 9367–9404, https://doi.org/10.5194/acp-21-9367-2021, 2021. a, b
Egan, S. D., Stuefer, M., Webley, P. W., Lopez, T., Cahill, C. F., and Hirtl, M.: Modeling volcanic ash aggregation processes and related impacts on the April–May 2010 eruptions of Eyjafjallajökull volcano with WRF-Chem, Nat. Hazards Earth Syst. Sci., 20, 2721–2737, https://doi.org/10.5194/nhess-20-2721-2020, 2020. a
Emmons, L. K., Schwantes, R. H., Orlando, J. J., Tyndall, G., Kinnison, D., Lamarque, J.-F., Marsh, D., Mills, M. J., Tilmes, S., Bardeen, C., Buchholz, R. R., Conley, A., Gettelman, A., Garcia, R., Simpson, I., Blake, D. R., Meinardi, S., and Pétron, G.: The Chemistry Mechanism in the Community Earth System Model version 2 (CESM2), J. Adv. Model. Earth Sy., 12, e2019MS001882, https://doi.org/10.1029/2019MS001882, 2020. a
Freitas, S. R., Longo, K. M., Alonso, M. F., Pirre, M., Marecal, V., Grell, G., Stockler, R., Mello, R. F., and Sánchez Gácita, M.: PREP-CHEM-SRC – 1.0: a preprocessor of trace gas and aerosol emission fields for regional and global atmospheric chemistry models, Geosci. Model Dev., 4, 419–433, https://doi.org/10.5194/gmd-4-419-2011, 2011. a
Galeazzo, T., Bekki, S., Martin, E., Savarino, J., and Arnold, S. R.: Photochemical box modelling of volcanic SO2 oxidation: isotopic constraints, Atmos. Chem. Phys., 18, 17909–17931, https://doi.org/10.5194/acp-18-17909-2018, 2018. a, b, c
General, S., Bobrowski, N., Pöhler, D., Weber, K., Fischer, C., and Platt, U.: Airborne I-DOAS measurements at Mt. Etna: BrO and OClO evolution in the plume, J. Volcanol. Geoth. Res., 300, 175–186, https://doi.org/10.1016/j.jvolgeores.2014.05.012, 2015. a, b
Gerlach, T. M.: Volcanic sources of tropospheric ozone-depleting trace gases, Geochem. Geophy. Geosy., 5, Q09007, https://doi.org/10.1029/2004gc000747, 2004. a
Gliß, J., Bobrowski, N., Vogel, L., Pöhler, D., and Platt, U.: OClO and BrO observations in the volcanic plume of Mt. Etna – implications on the chemistry of chlorine and bromine species in volcanic plumes, Atmos. Chem. Phys., 15, 5659–5681, https://doi.org/10.5194/acp-15-5659-2015, 2015. a, b, c, d
Grell, G. A., Peckham, S. E., Schmitz, R., McKeen, S. A., Frost, G., Skamarock, W. C., and Eder, B.: Fully coupled “online” chemistry within the WRF model, Atmos. Environ., 39, 6957–6975, https://doi.org/10.1016/j.atmosenv.2005.04.027, 2005. a
Gutmann, A., Bobrowski, N., Roberts, T. J., Rüdiger, J., and Hoffmann, T.: Advances in Bromine Speciation in Volcanic Plumes, Front. Earth Sci., 6, 213, https://doi.org/10.3389/feart.2018.00213, 2018. a, b, c, d
Hirtl, M., Stuefer, M., Arnold, D., Grell, G., Maurer, C., Natali, S., Scherllin-Pirscher, B., and Webley, P.: The effects of simulating volcanic aerosol radiative feedbacks with WRF-Chem during the Eyjafjallajökull eruption, April and May 2010, Atmos. Environ., 198, 194–206, https://doi.org/10.1016/j.atmosenv.2018.10.058, 2019. a
Hirtl, M., Scherllin-Pirscher, B., Stuefer, M., Arnold, D., Baro, R., Maurer, C., and Mulder, M. D.: Extension of the WRF-Chem volcanic emission preprocessor to integrate complex source terms and evaluation for different emission scenarios of the Grimsvötn 2011 eruption, Nat. Hazards Earth Syst. Sci., 20, 3099–3115, https://doi.org/10.5194/nhess-20-3099-2020, 2020. a, b
Hörmann, C., Sihler, H., Bobrowski, N., Beirle, S., Penning de Vries, M., Platt, U., and Wagner, T.: Systematic investigation of bromine monoxide in volcanic plumes from space by using the GOME-2 instrument, Atmos. Chem. Phys., 13, 4749–4781, https://doi.org/10.5194/acp-13-4749-2013, 2013. a, b
Jourdain, L., Roberts, T. J., Pirre, M., and Josse, B.: Modeling the reactive halogen plume from Ambrym and its impact on the troposphere with the CCATT-BRAMS mesoscale model, Atmos. Chem. Phys., 16, 12099–12125, https://doi.org/10.5194/acp-16-12099-2016, 2016. a, b, c, d, e, f
Kelly, P. J., Kern, C., Roberts, T. J., Lopez, T., Werner, C., and Aiuppa, A.: Rapid chemical evolution of tropospheric volcanic emissions from Redoubt Volcano, Alaska, based on observations of ozone and halogen-containing gases, J. Volcanol. Geoth. Res., 259, 317–333, https://doi.org/10.1016/j.jvolgeores.2012.04.023, 2013. a, b, c, d, e
Klobas, J. E., Wilmouth, D. M., Weisenstein, D. K., Anderson, J. G., and Salawitch, R. J.: Ozone depletion following future volcanic eruptions, Geophys. Res. Lett., 44, 7490–7499, https://doi.org/10.1002/2017GL073972, 2017. a
Kutterolf, S., Hansteen, T., Appel, K., Freundt, A., Krg̈er, K., Pérez, W., and Wehrmann, H.: Combined bromine and chlorine release from large explosive volcanic eruptions: A threat to stratospheric ozone?, Geology, 41, 707–710, https://doi.org/10.1130/G34044.1, 2013. a
Lachatre, M., Mailler, S., Menut, L., Turquety, S., Sellitto, P., Guermazi, H., Salerno, G., Caltabiano, T., and Carboni, E.: New strategies for vertical transport in chemistry transport models: application to the case of the Mount Etna eruption on 18 March 2012 with CHIMERE v2017r4, Geosci. Model Dev., 13, 5707–5723, https://doi.org/10.5194/gmd-13-5707-2020, 2020. a
Lurton, T., Jégou, F., Berthet, G., Renard, J.-B., Clarisse, L., Schmidt, A., Brogniez, C., and Roberts, T. J.: Model simulations of the chemical and aerosol microphysical evolution of the Sarychev Peak 2009 eruption cloud compared to in situ and satellite observations, Atmos. Chem. Phys., 18, 3223–3247, https://doi.org/10.5194/acp-18-3223-2018, 2018. a, b
Marelle, L., Thomas, J. L., Ahmed, S., Tuite, K., Stutz, J., Dommergue, A., Simpson, W. R., Frey, M. M., and Baladima, F.: Implementation and impacts of surface and blowing snow sources of Arctic bromine activation within WRF-Chem 4.1.1, J. Adv. Model. Earth Sy., 13, e2020MS002391, https://doi.org/10.1029/2020MS002391, 2021. a
Martin, R. S., Ilyinskaya, E., and Oppenheimer, C.: The enigma of reactive nitrogen in volcanic emissions, Geochim. Cosmochim. Ac., 95, 93–105, https://doi.org/10.1016/j.gca.2012.07.027, 2012. a, b, c, d
Maters, E. C., Delmelle, P., Rossi, M. J., and Ayris, P. M.: Reactive Uptake of Sulfur Dioxide and Ozone on Volcanic Glass and Ash at Ambient Temperature, J. Geophys. Res.-Atmos., 122, 10,077–10,088, https://doi.org/10.1002/2017JD026993, 2017. a
Mather, T., Allen, A., Davison, B., Pyle, D., Oppenheimer, C., and McGonigle, A.: Nitric acid from volcanoes, Earth Planet. Sci. Lett., 218, 17–30, https://doi.org/10.1016/S0012-821X(03)00640-X, 2004a. a, b
Mather, T. A.: Volcanoes and the environment: Lessons for understanding Earth's past and future from studies of present-day volcanic emissions, J. Volcanol. Geoth. Res., 304, 160–179, https://doi.org/10.1016/j.jvolgeores.2015.08.016, 2015. a
Mather, T. A., Pyle, D. M., and Allen, A. G.: Volcanic source for fixed nitrogen in the early Earth's atmosphere, Geology, 32, 905–908, https://doi.org/10.1130/G20679.1, 2004b. a
Millard, G. A., Mather, T. A., Pyle, D. M., Rose, W. I., and Thornton, B.: Halogen emissions from a small volcanic eruption: Modeling the peak concentrations, dispersion, and volcanically induced ozone loss in the stratosphere, Geophys. Res. Lett., 33, L19815, https://doi.org/10.1029/2006GL026959, 2006. a
Ming, A., Winton, V. H. L., Keeble, J., Abraham, N. L., Dalvi, M. C., Griffiths, P., Caillon, N., Jones, A. E., Mulvaney, R., Savarino, J., Frey, M. M., and Yang, X.: Stratospheric Ozone Changes From Explosive Tropical Volcanoes: Modeling and Ice Core Constraints, J. Geophys. Res.-Atmos., 125, e2019JD032290, https://doi.org/10.1029/2019JD032290, 2020. a
National Centers for Environmental Prediction, National Weather Service, NOAA, and U.S. Department of Commerce: NCEP FNL Operational Model Global Tropospheric Analyses, continuing from July 1999, https://doi.org/10.5065/D6M043C6, 2000. a
Oppenheimer, C., Kyle, P., Eisele, F., Crawford, J., Huey, G., Tanner, D., Kim, S., Mauldin, L., Blake, D., Beyersdorf, A., Buhr, M., and Davis, D.: Atmospheric chemistry of an Antarctic volcanic plume, J. Geophys. Res.-Atmos., 115, D04303, https://doi.org/10.1029/2009jd011910, 2010. a
Oppenheimer, C., Scaillet, B., and Martin, R. S.: Sulfur Degassing From Volcanoes: Source Conditions, Surveillance, Plume Chemistry and Earth System Impacts, Reviews in Mineralogy and Geochemistry, 73, 363–421, https://doi.org/10.2138/rmg.2011.73.13, 2011. a
Prata, A. J., Carn, S. A., Stohl, A., and Kerkmann, J.: Long range transport and fate of a stratospheric volcanic cloud from Soufrière Hills volcano, Montserrat, Atmos. Chem. Phys., 7, 5093–5103, https://doi.org/10.5194/acp-7-5093-2007, 2007. a
Pyle, D. and Mather, T.: Halogens in igneous processes and their fluxes to the atmosphere and oceans from volcanic activity: A review, Chem. Geol., 263, 110–121, https://doi.org/10.1016/j.chemgeo.2008.11.013, 2009. a
Pyle, D. M. and Mather, T. A.: The importance of volcanic emissions for the global atmospheric mercury cycle, Atmos. Environ., 37, 5115–5124, https://doi.org/10.1016/j.atmosenv.2003.07.011, 2003. a
Rizza, U., Brega, E., Caccamo, M. T., Castorina, G., Morichetti, M., Munaò, G., Passerini, G., and Magazù, S.: Analysis of the ETNA 2015 Eruption Using WRF–Chem Model and Satellite Observations, Atmosphere, 11, 1168, https://doi.org/10.3390/atmos11111168, 2020. a
Roberts, T.: Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions, Geosciences, 8, 68, https://doi.org/10.3390/geosciences8020068, 2018. a, b
Roberts, T., Vignelles, D., Liuzzo, M., Giudice, G., Aiuppa, A., Coltelli, M., Salerno, G., Chartier, M., Couté, B., Berthet, G., Lurton, T., Dulac, F., and Renard, J.-B.: The primary volcanic aerosol emission from Mt Etna: Size-resolved particles with SO2 and role in plume reactive halogen chemistry, Geochim. Cosmochim. Ac., 222, 74–93, https://doi.org/10.1016/j.gca.2017.09.040, 2018. a, b, c, d, e, f
Roberts, T., Dayma, G., and Oppenheimer, C.: Reaction Rates Control High-Temperature Chemistry of Volcanic Gases in Air, Front. Earth Sci., 7, 154, https://doi.org/10.3389/feart.2019.00154, 2019. a, b, c, d
Roberts, T. J., Braban, C. F., Martin, R. S., Oppenheimer, C., Adams, J. W., Cox, R. A., Jones, R. L., and Griffiths, P. T.: Modelling reactive halogen formation and ozone depletion in volcanic plumes, Chem. Geol., 263, 151–163, https://doi.org/10.1016/j.chemgeo.2008.11.012, 2009. a, b, c
Roberts, T. J., Martin, R. S., and Jourdain, L.: Reactive bromine chemistry in Mount Etna's volcanic plume: the influence of total Br, high-temperature processing, aerosol loading and plume–air mixing, Atmos. Chem. Phys., 14, 11201–11219, https://doi.org/10.5194/acp-14-11201-2014, 2014. a, b, c, d, e
Rose, W. I., Millard, G. A., Mather, T. A., Hunton, D. E., Anderson, B., Oppenheimer, C., Thornton, B. F., Gerlach, T. M., Viggiano, A. A., Kondo, Y., Miller, T. M., and Ballenthin, J. O.: Atmospheric chemistry of a 33–34 hour old volcanic cloud from Hekla Volcano (Iceland): Insights from direct sampling and the application of chemical box modeling, J. Geophys. Res., 111, D20206, https://doi.org/10.1029/2005jd006872, 2006. a, b, c
Rüdiger, J., Bobrowski, N., Liotta, M., and Hoffmann, T.: Development and application of a sampling method for the determination of reactive halogen species in volcanic gas emissions, Anal. Bioanal. Chem., 409, 5975–5985, https://doi.org/10.1007/s00216-017-0525-1, 2017. a
Rüdiger, J., Gutmann, A., Bobrowski, N., Liotta, M., de Moor, J. M., Sander, R., Dinger, F., Tirpitz, J.-L., Ibarra, M., Saballos, A., Martínez, M., Mendoza, E., Ferrufino, A., Stix, J., Valdés, J., Castro, J. M., and Hoffmann, T.: Halogen activation in the plume of Masaya volcano: field observations and box model investigations, Atmos. Chem. Phys., 21, 3371–3393, https://doi.org/10.5194/acp-21-3371-2021, 2021. a, b, c, d
Saiz-Lopez, A., Sitkiewicz, S. P., Roca-Sanjuán, D., Oliva-Enrich, J. M., Dávalos, J. Z., Notario, R., Jiskra, M., Xu, Y., Wang, F., Thackray, C. P., Sunderland, E. M., Jacob, D. J., Travnikov, O., Cuevas, C. A., Acuña, A. U., Rivero, D., Plane, J. M. C., Kinnison, D. E., and Sonke, J. E.: Photoreduction of gaseous oxidized mercury changes global atmospheric mercury speciation, transport and deposition, Nat. Commun., 9, 4796, https://doi.org/10.1038/s41467-018-07075-3, 2018. a
Saiz-Lopez, A., Acuña, A. U., Trabelsi, T., Carmona-García, J., Dávalos, J. Z., Rivero, D., Cuevas, C. A., Kinnison, D. E., Sitkiewicz, S. P., Roca-Sanjuán, D., and Francisco, J. S.: Gas-Phase Photolysis of Hg(I) Radical Species: A New Atmospheric Mercury Reduction Process, J. Am. Chem. Soc., 141, 8698–8702, https://doi.org/10.1021/jacs.9b02890, 2019. a, b, c
Salerno, G., Burton, M., Oppenheimer, C., Caltabiano, T., Randazzo, D., Bruno, N., and Longo, V.: Three-years of SO2 flux measurements of Mt. Etna using an automated UV scanner array: Comparison with conventional traverses and uncertainties in flux retrieval, Journal of Volcanology and Geothermal Research, 183, 76–83, https://doi.org/10.1016/j.jvolgeores.2009.02.013, 2009. a
Seigneur, C. and Lohman, K.: Effect of bromine chemistry on the atmospheric mercury cycle, J. Geophys. Res.-Atmos., 113, D23309, https://doi.org/10.1029/2008JD010262, 2008. a
Seigneur, C., Vijayaraghavan, K., and Lohman, K.: Atmospheric mercury chemistry: Sensitivity of global model simulations to chemical reactions, J. Geophys. Res., 111, D22306, https://doi.org/10.1029/2005jd006780, 2006. a
Seo, S., Richter, A., Blechschmidt, A.-M., Bougoudis, I., and Burrows, J. P.: First high-resolution BrO column retrievals from TROPOMI, Atmos. Meas. Tech., 12, 2913–2932, https://doi.org/10.5194/amt-12-2913-2019, 2019. a
Staunton-Sykes, J., Aubry, T. J., Shin, Y. M., Weber, J., Marshall, L. R., Luke Abraham, N., Archibald, A., and Schmidt, A.: Co-emission of volcanic sulfur and halogens amplifies volcanic effective radiative forcing, Atmos. Chem. Phys., 21, 9009–9029, https://doi.org/10.5194/acp-21-9009-2021, 2021. a
Stuefer, M., Freitas, S. R., Grell, G., Webley, P., Peckham, S., McKeen, S. A., and Egan, S. D.: Inclusion of ash and SO2 emissions from volcanic eruptions in WRF-Chem: development and some applications, Geosci. Model Dev., 6, 457–468, https://doi.org/10.5194/gmd-6-457-2013, 2013. a
Surl, L.: Modelling the atmospheric chemistry of volcanic plumes, PhD thesis, University of East Anglia, available at: https://ueaeprints.uea.ac.uk/id/eprint/59407/ (last access: 12 August 2021), 2016. a
Surl, L.: WRF-Chem Volcano, Github [software], available at: https://github.com/LukeSurl/WCV (last access: 12 August 2021), 2020. a
Surl, L.: WRF-Chem Volcano output – Etna Summer 2012, Zenodo [data set], https://doi.org/10.5281/zenodo.4415788, 2021. a
Surl, L., Donohoue, D., Aiuppa, A., Bobrowski, N., and von Glasow, R.: Quantification of the depletion of ozone in the plume of Mount Etna, Atmos. Chem. Phys., 15, 2613–2628, https://doi.org/10.5194/acp-15-2613-2015, 2015. a, b, c, d, e, f, g, h, i, j
Theys, N., Van Roozendael, M., Dils, B., Hendrick, F., Hao, N., and De Mazière, M.: First satellite detection of volcanic bromine monoxide emission after the Kasatochi eruption, Geophys. Res. Lett., 36, L03809, https://doi.org/10.1029/2008GL036552, 2009. a
Theys, N., De Smedt, I., Van Roozendael, M., Froidevaux, L., Clarisse, L., and Hendrick, F.: First satellite detection of volcanic OClO after the eruption of Puyehue-Cordón Caulle, Geophys. Res. Lett., 41, 667–672, https://doi.org/10.1002/2013GL058416, 2014. a
Vance, A., McGonigle, A. J. S., Aiuppa, A., Stith, J. L., Turnbull, K., and von Glasow, R.: Ozone depletion in tropospheric volcanic plumes, Geophys. Res. Lett., 37, L22802, https://doi.org/10.1029/2010GL044997, 2010. a, b
Voigt, C., Jessberger, P., Jurkat, T., Kaufmann, S., Baumann, R., Schlager, H., Bobrowski, N., Giuffrida, G., and Salerno, G.: Evolution of CO2, SO2, HCl, and HNO3 in the volcanic plumes from Etna, Geophys. Res. Lett., 41, 2196–2203, https://doi.org/10.1002/2013gl058974, 2014. a, b, c
von Glasow, R.: Atmospheric chemistry in volcanic plumes, P. Natl. Acad. Sci. USA, 107, 6594–6599, https://doi.org/10.1073/pnas.0913164107, 2010. a, b, c, d, e
von Glasow, R., Bobrowski, N., and Kern, C.: The effects of volcanic eruptions on atmospheric chemistry, Chem. Geol., 263, 131–142, https://doi.org/10.1016/j.chemgeo.2008.08.020, 2009. a
Wade, D. C., Vidal, C. M., Abraham, N. L., Dhomse, S., Griffiths, P. T., Keeble, J., Mann, G., Marshall, L., Schmidt, A., and Archibald, A. T.: Reconciling the climate and ozone response to the 1257 CE Mount Samalas eruption, P. Natl. Acad. Sci. USA, 117, 26651–26659, https://doi.org/10.1073/pnas.1919807117, 2020. a
Warnach, S., Bobrowski, N., Hidalgo, S., Arellano, S., Sihler, H., Dinger, F., Lübcke, P., Battaglia, J., Steele, A., Galle, B., Platt, U., and Wagner, T.: Variation of the BrO/SO2 Molar Ratio in the Plume of Tungurahua Volcano Between 2007 and 2017 and Its Relationship to Volcanic Activity, Front. Earth Sci., 7, p. 132, https://doi.org/10.3389/feart.2019.00132, 2019. a, b
Weigelt, A., Ebinghaus, R., Pirrone, N., Bieser, J., Bödewadt, J., Esposito, G., Slemr, F., van Velthoven, P. F. J., Zahn, A., and Ziereis, H.: Tropospheric mercury vertical profiles between 500 and 10 000 m in central Europe, Atmos. Chem. Phys., 16, 4135–4146, https://doi.org/10.5194/acp-16-4135-2016, 2016a. a, b, c
Weigelt, A., Slemr, F., Ebinghaus, R., Pirrone, N., Bieser, J., Bödewadt, J., Esposito, G., and van Velthoven, P. F. J.: Mercury emissions of a coal-fired power plant in Germany, Atmos. Chem. Phys., 16, 13653–13668, https://doi.org/10.5194/acp-16-13653-2016, 2016b. a
Wennberg, P.: Atmospheric chemistry – Bromine explosion, Nature, 397, 299, https://doi.org/10.1038/16805, 1999. a
Wild, O., Zhu, X., and Prather, M. J.: Fast-J: Accurate Simulation of In- and Below-Cloud Photolysis in Tropospheric Chemical Models, J. Atmos. Chem., 37, 245–282, https://doi.org/10.1023/a:1006415919030, 2000. a
Witt, M. L. I., Mather, T. A., Pyle, D. M., Aiuppa, A., Bagnato, E., and Tsanev, V. I.: Mercury and halogen emissions from Masaya and Telica volcanoes, Nicaragua, J. Geophys. Res.-Sol. Ea., 113, B06203, https://doi.org/10.1029/2007JB005401, 2008. a
Wittmer, J., Bobrowski, N., Liotta, M., Giuffrida, G., Calabrese, S., and U., P.: Active alkaline traps to determine acidic-gas ratios in volcanic plumes: sampling technique and analytical Methods, Geochem. Geophys. Geosys., 15, 2797–2820, https://doi.org/10.1002/2013GC005133, 2014. a, b, c, d
Zaveri, R. A. and Peters, L. K.: A new lumped structure photochemical mechanism for large-scale applications, J. Geophys. Res.-Atmos., 104, 30387–30415, https://doi.org/10.1029/1999JD900876, 1999. a
Zaveri, R. A., Easter, R. C., Fast, J. D., and Peters, L. K.: Model for Simulating Aerosol Interactions and Chemistry (MOSAIC), J. Geophys. Res.-Atmos., 113, D13204, https://doi.org/10.1029/2007JD008782, 2008. a
- Abstract
- Introduction
- Methods
- Aircraft observations quantifying ozone destruction in the plume
- Modelling of the atmospheric chemistry of Mount Etna's plume
- Conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Methods
- Aircraft observations quantifying ozone destruction in the plume
- Modelling of the atmospheric chemistry of Mount Etna's plume
- Conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement