the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Jul 2021
| 13 Jul 2021
Measurement report: Nitrogen isotopes (δ15N) and first quantification of oxygen isotope anomalies (Δ17O, δ18O) in atmospheric nitrogen dioxide
Sarah Albertin
Joël Savarino
Slimane Bekki
Albane Barbero
Nicolas Caillon
The isotopic composition of nitrogen and oxygen in nitrogen dioxide (NO2) potentially carries a wealth of information about the dynamics of the nitrogen oxides (NOx = nitric oxide (NO) + NO2) chemistry in the atmosphere. While nitrogen isotopes of NO2 are subtle indicators of NOx emissions and chemistry, oxygen isotopes are believed to reflect only the O3 NOx VOC chemical regime in different atmospheric environments. In order to access this potential tracer of the tropospheric chemistry, we have developed an efficient active method to trap atmospheric NO2 on denuder tubes and measured, for the first time, its multi-isotopic composition (δ15N, δ18O, and Δ17O). The Δ17O values of NO2 trapped at our site in Grenoble, France, show a large diurnal cycle peaking in late morning at (39.2 ± 0.3) ‰ and decreasing at night until (20.5 ± 0.3) ‰. On top of this diurnal cycle, Δ17O also exhibits substantial daytime variability (from 29.7 ‰ to 39.2 ‰), certainly driven by changes in the O3 to peroxyl radicals (RO2) ratio. The nighttime decay of Δ17O(NO2) appears to be driven by NO2 slow removal, mostly from conversion into N2O5, and its formation from the reaction between O3 and freshly emitted NO. As expected from a nighttime Δ17O(NO2) expression, our Δ17O(NO2) measured towards the end of the night is quantitatively consistent with typical values of Δ17O(O3). Daytime N isotope fractionation is estimated using a general expression linking it to Δ17O(NO2). An expression is also derived for the nighttime N isotope fractionation. In contrast to Δ17O(NO2), δ15N(NO2) measurements exhibit little diurnal variability (−11.8 ‰ to −4.9 ‰) with negligible isotope fractionations between NO and NO2, mainly due to high NO2 NOx ratios, excepted during the morning rush hours. The main NOx emission sources are estimated using a Bayesian isotope mixing model, indicating the predominance of traffic emissions in this area. These preliminary results are very promising for using the combination of Δ17O and δ15N of NO2 as a probe of the NOx sources and fate and for interpreting nitrate isotopic composition records.
- Article
(1854 KB) - Full-text XML
-
Supplement
(239 KB) - BibTeX
- EndNote





Nitrogen oxides (NOx = NO2 + NO) are at the heart of tropospheric chemistry, as they are involved in key reaction chains governing the production and destruction of compounds of fundamental interest for health, ecosystems, and climate issues (Brown, 2006; Finlayson-Pitts and Pitts, 2000; Jacob, 1999). For example, NO2 photolysis followed by reaction of NO with peroxy radicals (RO2 = HO2 + RO2) is the only significant source of ozone (O3) in the troposphere, where it serves as a severe air pollutant and a greenhouse gas. Tropospheric O3 also plays a major role in the production processes of radicals which are responsible for the oxidation and removal of compounds emitted into the atmosphere (Crutzen, 1996). This “cleaning” ability is referred to as the atmospheric oxidative capacity (AOC; Prinn, 2003). Additionally, NOx species are at the core of the reactive nitrogen cycle as precursors of atmospheric nitrate (particulate NO + gaseous HNO3), which contributes to soil acidification and eutrophication (Galloway et al., 2004), as well as aerosol radiative forcing (Liao and Seinfeld, 2005). In order to better understand the reactive nitrogen (which includes NOx and HNO3) chemistry, the related AOC, and the contributions of precursor emissions to nitrate deposition, it is necessary to better constrain NOx emission sources and individual oxidation processes.
Stable isotope analysis is a powerful tool for tracing emission sources, individual chemical mechanisms, and budgets of atmospheric trace gases (Kaye, 1987). Because physico-chemical and biological processes favour lighter or heavier isotopologues, the isotopic composition of a chemical species will often vary according to its formation pathway. This phenomenon of isotopic fractionation can thus be used to trace different processes involved in the formation of the chemical species being analysed. Isotopic enrichment (δ) of an element X is expressed in per mill (‰) and defined as with nR being the elemental isotope abundance ratio of the heavy isotope over the light isotope (e.g. for oxygen isotopes 18R(18O 16O) ≡ 18R = x(18O) x(16O) or 17R(17O 16O) ≡ 17R = x(17O) x(16O), with x being the isotopic abundance) in a sample (nRspl) and in a reference (nRref). The Vienna Standard Mean Ocean Water (VSMOW; Li et al., 1988) and atmospheric nitrogen (N2; Mariotti, 1984) are the international references for oxygen and nitrogen ratios, respectively. Most natural isotopic fractionations are mass-dependent fractionations (MDFs; Urey, 1947), as is notably the case for terrestrial oxygenated species in which the triple oxygen composition follows δ17O ≈ 0.52 × δ18O (Thiemens, 1999). Yet, laboratory experiments (Thiemens and Heidenreich, 1983) and atmospheric observations (Johnston and Thiemens, 1997; Krankowsky et al., 1995; Vicars and Savarino, 2014) have shown that the isotopic composition of ozone formed in the atmosphere does not follow this canonical MDF relationship and reflects mass-independent fractionation (MIF) processes. The important deviation from the MDF oxygen relationship is called the oxygen-17 anomaly (Δ17O) and is defined here in its approximate linearized form as Δ17O = δ17O − 0.52 × δ18O. Our choice of this linear definition is mainly motivated by its convenience for mass-balance calculations and its validity for our large Δ17O values and variability. Overall, biases related to our choice of the linear definition are marginal in our conditions (Assonov and Brenninkmeijer, 2005). It follows that Δ17O inherited from ozone can be considered conserved during MDF processes.
The multi-isotopic composition of NOx is therefore a very valuable tracer of its emissions and chemistry in the atmosphere. However, so far, Δ17O of atmospheric NO2 (Δ17O(NO2)) has been investigated only using laboratory (Michalski et al., 2014) and modelling (Alexander et al., 2020, 2009; Lyons, 2001; Morin et al., 2011) approaches with theoretical frameworks, and these results need to be tested against atmospheric observations. Walters et al. (2018) have presented a method of sampling and analysing nitrogen and oxygen stable isotopes of NO2 collected separately at daytime and nighttime in an urban area, but they did not report on Δ17O. Dahal and Hastings (2016) have attempted to measure Δ17O of NO2 collected on passive samplers, but the isotopic signal was partly degraded during the sampling and the analytical procedure. Building on their work, we present here an efficient method to collect atmospheric NO2 for isotopic analysis and present the first measurements of triple oxygen isotopes and double nitrogen isotopes of atmospheric NO2. Combined with mass-balance equations, oxygen isotopes are used to investigate the links between the variability of the oxygen isotope anomaly of NO2 and its formation pathways. We also revisit the Morin et al. (2011) NOx isotopic theoretical framework and extend it to urban environments. After estimating the nitrogen isotopic fractionation between NO and NO2, we infer from δ15N of NO2 (δ15N(NO2)) the major emission sources of NOx influencing our sampling site using an isotopic mixing model (Parnell et al., 2010).
2.1 Sampling method
NO2 was sampled on an active (pumped) collection system using denuder tubes. This method is more efficient to collect NO2 than passive methods (Røyset, 1998), allowing for shorter collection times with a breakthrough of the absorption capacity below 1 % (Buttini et al., 1987; Williams and Grosjean, 1990). The sampled air was pumped through a ChemComb® 3500 speciation cartridge (Thermo Scientific®, USA). Initially used for the speciation of gases and aerosols, these advanced sampling platforms consist of a PM2.5 impactor inlet connected to a stainless-steel cylinder that contains two glass honeycomb denuders connected in series for gas collection and a Teflon stage filter pack for aerosols. To collect NO2, glass tubes were coated with an alkaline guaiacol solution. In basic medium, guaiacol (IUPAC name: 2-methoxyphenol) is known to react with NO2 to form stable NO ions (Nash, 1970), preserving the original NO2 isotopic signal due to the basic nature of the medium (pH = 14 after 10 mL extraction). Because NO or peroxyacetyl nitrate (PAN) are not collected by guaiacol, this methodology avoids potential interferences from these compounds in later analyses (Buttini et al., 1987). Although nitrous acid (HONO) can bind as NO, it is unlikely to adversely impact the results as its mixing ratio (i.e. mole fraction) is much lower than NO2 (by a factor of 10 to 20) even in very polluted cities (e.g. Harris et al., 1982; Michoud et al., 2014; Huang et al., 2017).
To evaluate the sampling system performance, a series of experiments were run with artificial gaseous NO2. Using a commercial gas-standard generator (KinTek FlexStream®) feed with zero air, diluted NO2 (Metronics Dynacal®) was sent through a ChemComb cartridge, while NOx mixing ratio was measured upstream and downstream of the cartridge. From 1 to 30 nmol mol−1 of NO2 (representative of rural to urban atmospheric conditions), mixing ratios coming out of the cartridge were never above the noise level of the NOx monitor (2.5 nmol mol−1). To estimate the denuder's trapping efficiency, we passed different mixing ratios of gaseous NO2 through the collection apparatus and measured the amount of NO collected on the two denuders both connected in series. The denuder efficiency E was then calculated according to the following equation (Buttini et al., 1987):
with a and b representing the amount of NO collected on the first and the second denuder, respectively. From 0.3 to 1.3 µmol of generated NO2 (see Fig. 1), the mean E value was about (97 ± 3) %. The amount of NO measured on the second denuder was reproducible and equivalent to blanks, representing on average 3 % of the quantity measured on the first denuder. In light of these results, the denuder in the second position was not subjected to isotopic analysis and allowed for trapping efficiency control.
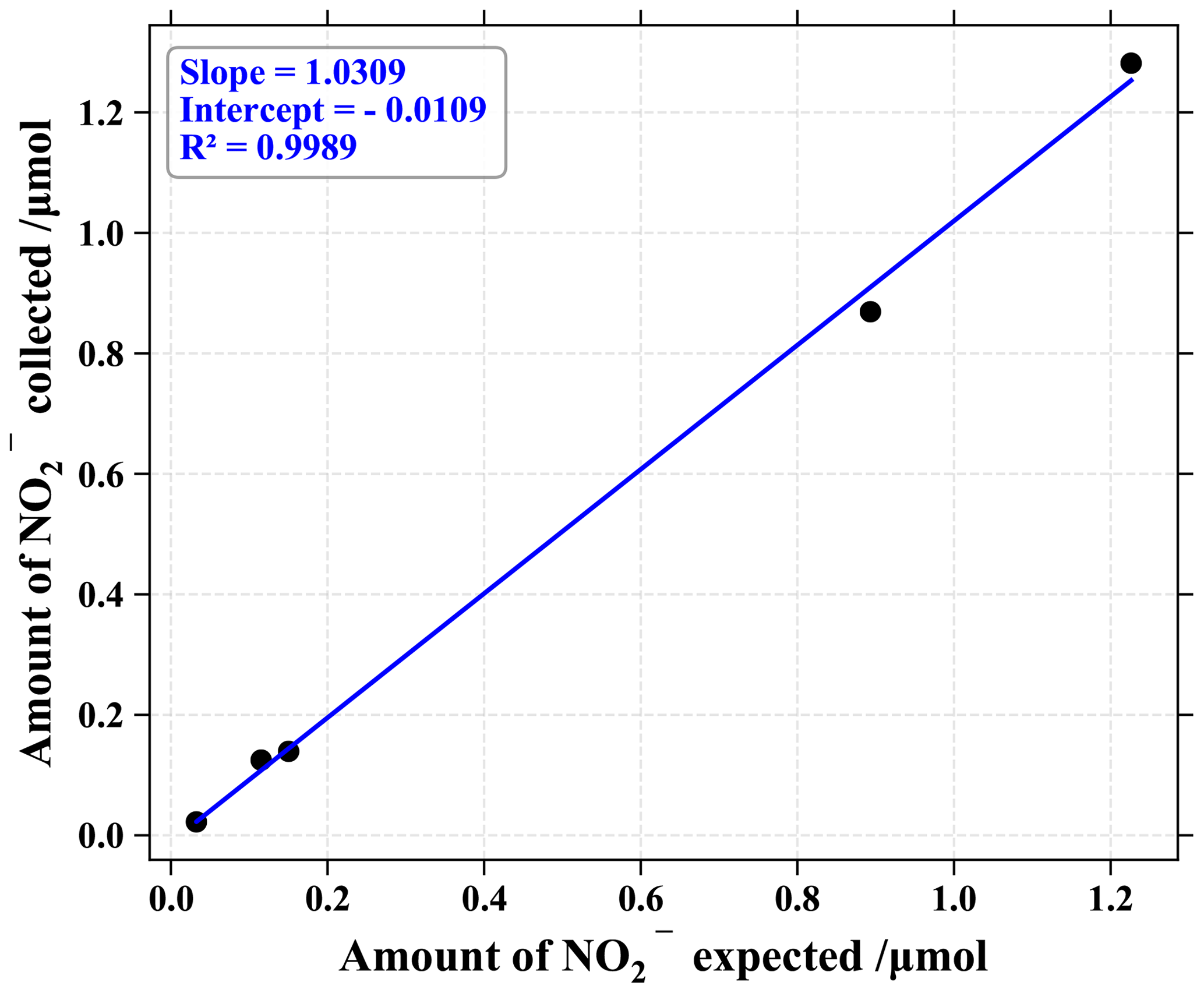
Figure 1Correlation plot of NO2 collected on the first denuder tube of the sampling cartridge vs. NO2 produced by the gas standard generator.
2.2 Isotopic analysis
Simultaneous isotopic analyses of δ15N, δ18O, and δ17O were performed using a Finnigan® MAT253 isotope ratio mass spectrometer (IRMS) following techniques described by Casciotti et al. (2002) and Kaiser et al. (2007). The azide method (McIlvin and Altabet, 2005) was used with ≈ 100 nmol of nitrites converted to N2O using a 50 : 50 by volume mixture of 2 M sodium azide and 100 % acetic acid. This chemical method has the advantage over the bacterial method to be free of nitrate interferences since HNO3 is certainly trapped with NO2 in the basic solution coating of the denuder tube. The principle of identical treatment (Brand, 1996) was strictly respected where the standards and samples possessed the same nitrite amount, water isotopes, total volume and matrix. Three international KNO2 salt standards – RSIL-N7373, RSIL-N10219, and RSIL-N23 with respective δ15N δ18O values of −79.6/4.2 ‰, 2.8/88.5 ‰, and 3.7/11.4 ‰ – were used for normalization of the δ scale. Scale contraction factors were obtained with the linear regression between measured and known values of δ15N and δ18O. Although the three standards cover a wide range of isotopic composition in δ15N and δ18O, they do not have an isotopic anomaly in 17O. For the δ17O scale, MDF fractionation slope (0.52) is assumed for two of these laboratory-prepared nitrite standards (see Appendix A for more details). Accuracy of this analytical method on δ17O, δ18O, and δ15N measurements was estimated as the standard deviation (σ) of the residuals between our measurements of the RSIL standards and their expected values. Additionally, isotopic integrity from denuder's extraction to the analysis by IRMS has been investigated and showed no degradation over several weeks (see Appendix B), confirming that this method is suitable for isotopic analysis of NO2, as first demonstrated by Walters et al. (2018). The uncertainties applied to our measurements of δ15N, δ17O, and δ18O are reported as the propagation error of the measurement uncertainty and the uncertainty resulting from sample storage. Uncertainty on Δ17O is derived from the propagation error of the overall uncertainty on δ17O and δ18O. In our study, average uncertainties on δ15N, δ17O, δ18O, and Δ17O are estimated to be ±0.1 ‰, ±0.8 ‰, ±1.8 ‰, and ±0.3 ‰, respectively (1σ uncertainties).
2.3 Study site and atmospheric NO2 collection
Atmospheric NO2 was collected at the Université Grenoble Alpes campus site. Located in the eastern Grenoble urban area (690 000 inhabitants), the campus stands between a major transportation route and the Isère river. The city is located at the confluence of three valleys surrounded by mountain chains that influence the atmospheric dynamics and the local air quality. During winter, persistent temperature inversions combined with intense domestic heating can lead to severe PM10 pollution events (Largeron and Staquet, 2016) with daily-average mixing ratio above World Health Organization thresholds. In summer, emissions are mainly controlled by road traffic that can result in heightened ozone mixing ratios, especially during stagnant conditions.
Samplings were conducted on a platform 5 m above the ground surface. Ambient air was drawn through the cartridge with a Millipore vacuum pump at a flow rate of 10 L min−1 (room temperature and one atmospheric pressure) adjusted using a Cole-Palmer® flowmeter (accuracy ±3 %). In order to capture the daily variability in NO2 isotopic composition, samples were collected during 24 h with 3 h sampling intervals during the day and 5 h sampling from midnight to 05:00 local time (LT). Ambient NO and NO2 mixing ratios were measured with a 2B Technologies® NO monitor (model 410) paired with a NO2 converter (model 401).
Honeycomb denuders were cleaned and coated the day before sampling. After being generously rinsed (5 min under a stream of deionized water), the denuders were placed in a vacuum chamber (Thermo Scientific® refrigerated vapour trap paired with a SpeedVac concentrator) and dried at 40 ∘C during 1 h. Then, denuder's internal walls were individually coated with 10 mL of a 95 : 5 by volume mixture of 2.5 M KOH (in methanol) and ultrapure guaiacol prepared daily. Denuders were then drawn off, dried in the vacuum chamber at 40 ∘C during 30 min to minimize blanks, hermetically sealed, and stored at ambient temperature in the dark until usage. The different components of the cartridge (impactor, filters, denuders) were cleaned, dried, and fitted together just before use. At the end of the sampling period, both denuders were removed from the ChemComb cartridge and rinsed with 10 mL of deionized water in order to leach trapped NO2 out. A volume of 1 mL of the eluent was rapidly used to determine the nitrite amount using the Griess–Saltzman reaction and UV–Vis spectrometry at 544 nm. Recovered eluent (≈ 7 mL by denuder) was poured into a labelled 15 mL Corning® tube and stored in a freezer until isotopic analysis the following days.
3.1 NOx and O3 atmospheric observations
Figure 2 shows the time evolution of the hourly NO2, NO, and O3 mixing ratios measured during the period covering two nights and 1 d (from 14 May 2019 21:00 LT to 16 May 2019 05:00 LT). Note that most of our NO measurements are found to be within the reported detection limit of the instrument except in the morning (see Table 1) and therefore have to be treated with a lot of caution. NO2 mixing ratios during the sampling period ((6.1 ± 4.2) nmol mol−1; mean ± 1 standard deviation) are in good agreement with the range of values measured at the local air quality site located a kilometre south of the sampling site (https://www.atmo-auvergnerhonealpes.fr/, last access: 18 August 2020).
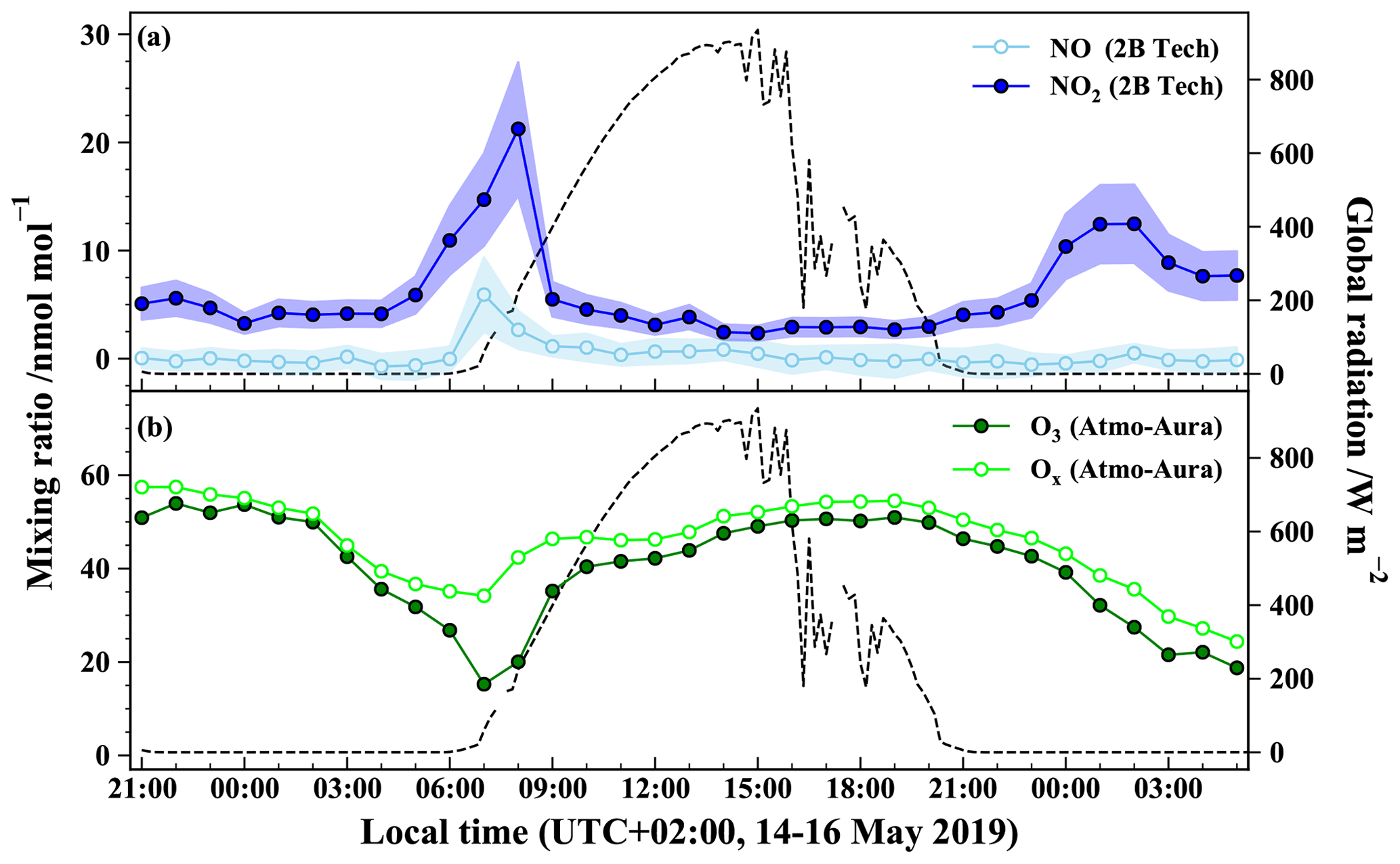
Figure 2Temporal evolution of (a) NO (open circles) and NO2 (closed circles) at the sampling site (the envelops represent ±1σ variations over 1 h) and of (b) O3 (closed circles) and Ox (= O3 + NO2; open circles) at the air quality station during the sampling period. Markers represent for (a) the hourly mean derived from 1 min measurements and for (b) the hourly mean provided by the air quality monitoring station. Global solar radiation flux is represented by dashed lines (measured at 200 m from the sampling site by the IGE weather station with a Skye SP1110 pyranometer).
Table 1Summary table of sampling periods (dates, local times), NO, NO2, and O3 mean mixing ratios over the collection periods, as well as calibrated NO2 isotopic measurements of δ15N, δ18O, and Δ17O. All the sampling periods lasted 3 h except the last one that lasted 5 h. Averaged measurement uncertainties are provided just below the species names.
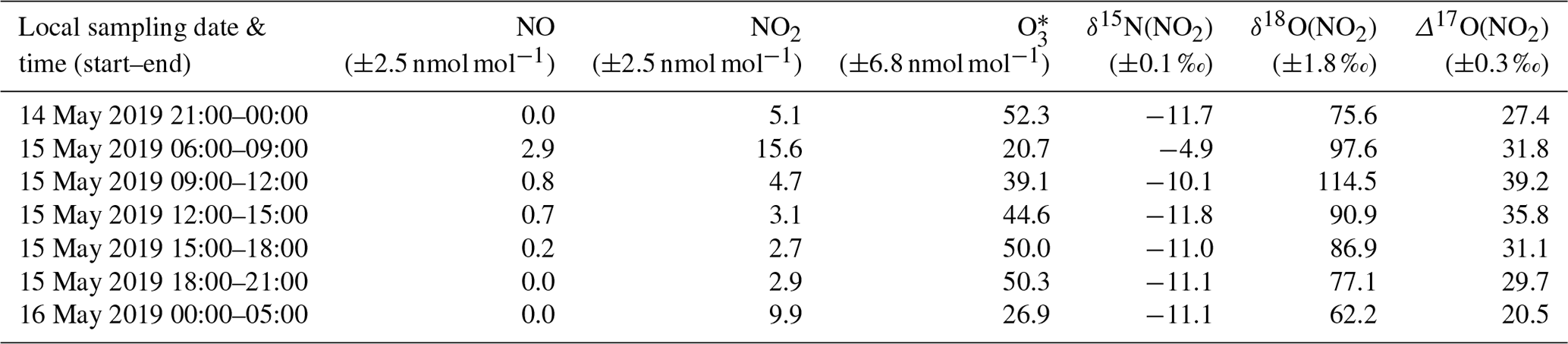
∗ Data monitored at the local air quality monitoring site of Saint-Martin d'Hères located a kilometre south of the sampling site (https://www.atmo-auvergnerhonealpes.fr/).
During both nights, most of the NOx is in the form of NO2. After sunrise, there is a rapid interconversion between NO and NO2, driven by NO2 photolysis and reactions of NO with O3 and peroxy radicals (Jacob, 1999). NO2 levels are at a maximum on 15 May between 04:00 and 10:00 LT with a sharp peak of 21 nmol mol−1 at 08:00 LT. After the morning rise, NO2 decreases to reach a background mixing ratio of about (3.0 ± 0.5) nmol mol−1. This diurnal variation is common in urban/suburban sites characterized by a morning peak caused by important NOx emissions, mainly from road traffic (Mayer, 1999). As morning progresses, the boundary layer height increases rapidly, favouring fast dilution of NOx mixing ratios. Moreover, during the day, NO2 is converted to HNO3, notably by its reaction with OH radicals. Thus, NOx mixing ratio remains low during the day likely because of the combination of atmospheric dilution by vertical mixing and efficient chemical conversion by OH and organic radicals (Tie et al., 2007). In dense urban areas, a second NOx traffic emission peak can occur in late afternoon, but it is not observed at our sampling site for that specific day. This surface pollution peak is usually weaker than the morning peak due to an elevated boundary layer and a longer period of evening commuting. After sunset, NO2 mixing ratio increases gently and reaches a smooth peak with a maximum of 12 nmol mol−1 around 01:00 LT, also recorded at the local air quality site. This NO2 mixing ratio rise may be due to low NO emissions (converted to NO2 by reaction with O3) combined with a decreasing boundary layer height during the night which traps atmospheric species close to the surface (Tie et al., 2007; Villena et al., 2011).
Ozone also exhibits a diurnal variation typical of urban areas (Velasco et al., 2008). O3 peaks around 50 nmol mol−1 at the beginning of both nights and then declines continuously. Indeed, after sunset, O3 production ceases, and its mixing ratio drops due to its dry deposition, reactions with organics, and O3 titration by NO emitted from evening traffic, and industrial activities in the stable nocturnal boundary layer (Klein et al., 2019). O3 reaches a minimum (about 15 nmol mol−1) not at the end of the night but during the morning rush hour peak of NO. The Ox (= O3 + NO2) is a more conservative quantity than O3, because it is less affected by conversion of O3 into NO2 through NO titration which is important in urban environments (Kleinman et al., 2002). For instance, between 06:00 and 08:00 LT, O3 is strongly titrated by freshly emitted NO with its mixing ratio dropping to about 15 nmol mol−1, while Ox reaches a moderate minimum of 34 nmol mol−1. After this morning drop, O3 recovers rapidly to about 46 nmol mol−1 in the late morning, possibly caused by downward O3 flux associated with the formation of the daytime thick boundary layer (Jin and Demerjian, 1993; Klein et al., 2019). During the rest of the day, O3 and Ox keep increasing gently due to photochemical production and reach a close maximum at the end of the afternoon (Geng et al., 2008). After sunset, the important decline of both O3 and Ox highlights the physical losses, notably O3 deposition, and chemical loss of NOx, typical of urban areas.
3.2 Multi-isotopic composition measurements of atmospheric NO2
We present the data for the multi-isotopic composition of seven atmospheric NO2 samples, while two additional samples were rejected as NO amounts were too low to perform a reliable analysis. Table 1 reports ambient mean mixing ratios of NO, NO2, and O3 for the isotopic sampling intervals and corresponding measured NO2 isotopic composition (δ15N(NO2), δ18O(NO2), and Δ17O(NO2)). Figure 3 depicts the time series of measured δ15N, δ18O, and Δ17O of atmospheric NO2. The temporal evolution of NO2 oxygen and nitrogen isotopic composition is interpreted in the following section.
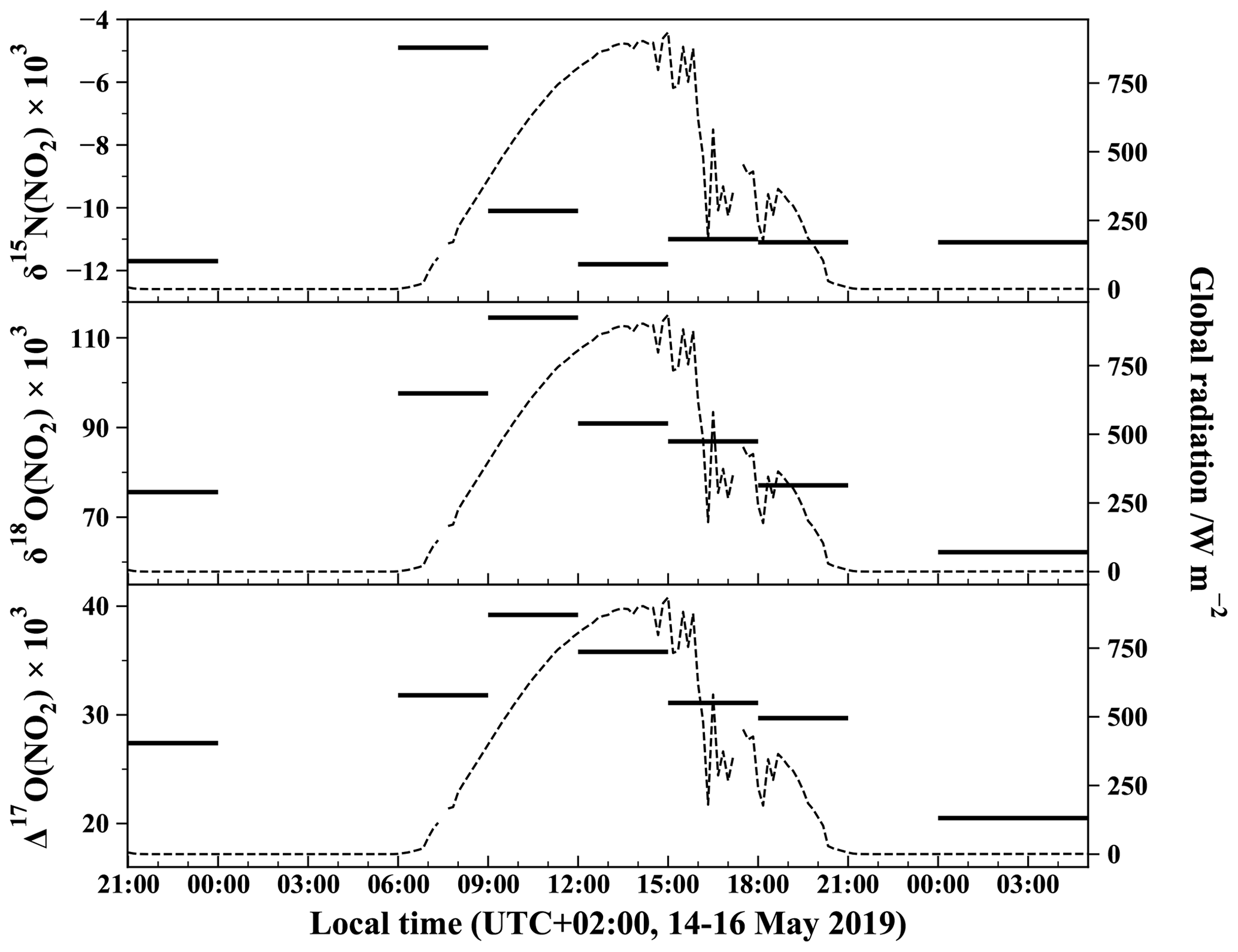
Figure 3Temporal evolution of δ15N, δ18O, and Δ17O of atmospheric NO2 measured with the azide method. Isotopic values for each 3 h slots are from the same NO2 sample collected over 3 h (except for the last period which lasted 5 h). Global solar radiation flux is represented by dashed lines (measured at 200 m from the sampling site by the IGE weather station with a Skye SP1110 pyranometer).
4.1 Oxygen isotope composition
The time evolution of δ18O of atmospheric NO2 (δ18O(NO2)) shown in Fig. 3 exhibits a substantial diurnal variation with a daytime mean of (93.4 ± 13.9) ‰ and a nighttime mean of (68.9 ± 9.5) ‰. A maximum value of 114.5 ‰ is observed in the morning (09:00–12:00 LT interval) and a minimum value of 62.2 ‰ for the late-night interval (00:00–05:00 LT). Using a similar sampling apparatus during summer in the urban/suburban site of West Lafayette, USA, Walters et al. (2018) reported daytime and nighttime mean δ18O(NO2) values of (86.5 ± 14.1) ‰ and (56.3 ± 7.1) ‰, respectively. Although our daytime values are higher than those of Walters et al. (2018), both datasets exhibit the same day–night contrast with a maximum during the day and a minimum at night. As expected from δ18O values, Δ17O(NO2) follows a similar diurnal variation with a maximum value of 39.2 ‰ for the 09:00–12:00 LT interval and a minimum value of 20.5 ‰ for the 00:00–05:00 LT interval. High Δ17O values are expected to reflect the importance of ozone in the oxidation of NO to NO2. Since daytime and nighttime chemistries are radically different, interpretations of our Δ17O measurements and their implications are discussed separately by day and night.
4.1.1 Fundamentals of NOx chemistry and isotopic transfers
NOx is mainly produced under the form of NO by combustion and lighting processes (Dennison et al., 2006; Young, 2002) as well as by the biological activity of soils (Davidson and Kingerlee, 1997). In the daytime, NO and NO2 rapidly interconvert in a timescale of about 1–2 min, establishing a photostationary steady state (PSS; Leighton 1961):
with M=N2 or O2;
This so-called null cycle can be disturbed by RO2 radicals when NOx mixing ratios are relatively high, typically above 30–100 pmol mol−1 (Seinfeld and Pandis, 2006; Monks, 2005):
The reaction between NO and RO2 competes with the NO + O3 reaction, allowing NO2 formation without the consumption of an ozone molecule in the cycle (Monks, 2005). This results in ozone production and can lead to severe ozone build-up in polluted areas. At night, RO2 mixing ratios are strongly reduced making ozone the main NO oxidant following Reaction (R3).
NOx is mainly removed from the atmosphere via the oxidation of NO2 into nitric acid during the day,
and at night,
In this framework, Δ17O(NO2) is driven by the relative importance of the different NO2 production channels, because NO2 loss processes do not fractionate in terms of oxygen mass-independent anomaly. Each NO2 production channel generates a specific mass-independent isotopic anomaly Δ17O on the produced NO2 (Kaiser et al., 2004). Based on the NO2 continuity equation, this can be expressed with the following Δ17O(NO2) mass-balance equation (Morin et al., 2011):
with [NO2] being the atmospheric NO2 mixing ratio, Pi and Lj the NO2 production/emission and loss rates, respectively (i.e. mixing ratio of involved species multiplied by the kinetic constant of the considered chemical reaction), and Δ17Oi(NO2) the specific isotope anomaly transferred to NO2 through the production reaction i.
4.1.2 Δ17Oday(NO2)
By day, the NOx photochemical cycle (Reactions R1 to R4) achieves a steady state in 1–2 min, which is several orders of magnitude faster than NO2 loss reactions (Atkinson et al., 1997) and emission rate (NOx is mainly emitted under the form of NO; Villena et al., 2011). It follows that NO and NO2 short variations can be neglected; i.e. and on short timescales. In addition, fast interconversions between NO and NO2 generate quickly an isotopic equilibrium between NO and NO2 resulting in Δ17O(NO2) ≈ Δ17O(NO) (Michalski et al., 2014; Morin et al., 2007). With these approximations, considering only the main reactions and neglecting halogen chemistry, Eq. (2) yields the following (Morin et al., 2007):
with being the ozone isotopic anomaly transferred to NO during its oxidation to NO2 via Reaction (R3) (also called the transfer function of the isotope anomaly of ozone to NO2; Savarino et al., 2008) and being the RO2 isotopic anomaly transferred to NO during its oxidation to NO2 via Reaction (R4). can be considered to be negligible (Alexander et al., 2020; Michalski et al., 2003), because RO2 is mainly formed by the reactions R + O2 and H + O2 and because the isotopic anomaly of atmospheric O2 is very close to 0 ‰ (Barkan and Luz, 2003). This assumption has been estimated to affect the overall Δ17O of RO2 values by less than 1 ‰ (Röckmann et al., 2001). As a result, Eq. (3) can be simplified, giving a driven by the relative importance of Reaction (R3) (NO + O3) and Reaction (R4) (NO + RO2) in the NO oxidation and by the oxygen isotopic anomaly transferred from O3 to NO2:
with
has been determined experimentally by Savarino et al. (2008). They reported ‰ with Δ17O(O3) being the bulk ozone isotopic anomaly. Δ17O(O3) has been measured in Grenoble in 2012 (Vicars and Savarino, 2014) with a mean value of (26.2 ± 1.3) ‰, corresponding to a value of (37.5 ± 2.8) ‰, which, according to Eq. (4), would give a maximum value of (37.5 ± 2.8) ‰. It is consistent with our maximum measured Δ17O(NO2) value of 39.2 ‰ for the 09:00–12:00 LT interval. In light of the known uncertainties, the small difference is not significant and is much smaller than the diurnal variations of Δ17O(NO2). Note that the Δ17O calibration is not very accurate for the most enriched samples, because nitrite standards with high Δ17O are still not readily available. In a laboratory study, Michalski et al. (2014) measured the Δ17O of NO2 formed by the photochemical NO–NO2–O3 cycle and reported Δ17O(NO2) = (39.3 ± 1.9) ‰. Despite experimental conditions that are not strictly applicable to our atmospheric conditions (e.g. NOx ≫ O3, light source, absence of volatile organic compounds (VOCs)), their value is surprisingly close to our maximum value. Assuming that our maximum Δ17O(NO2) value corresponds to close to unity (Reaction R3 (NO + O3) ≫ Reaction R4 (NO + RO2)), we use a value of 39.2 ‰ for the following calculations. Combining Eqs. (4) and (5), an expression for the RO2 mixing ratio can be derived as
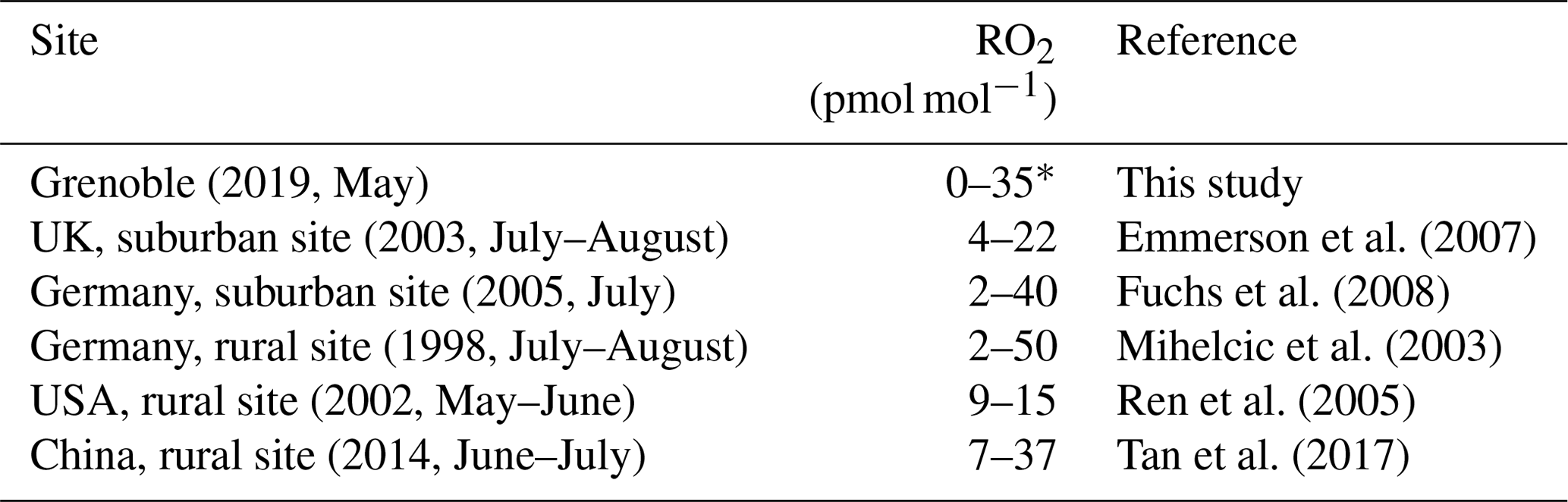
Figure 4 shows the daytime evolution of calculated from Eq. (4) and RO2 calculated from Eq. (6). varies between 0.76 and 1 with a mean daytime value of 0.86 (the measured daytime Δ17O(NO2) mean value is (33.5 ± 3.9) ‰), meaning that 86 % of NO2 is formed via Reaction (R3) (oxidation of NO by O3). The mean estimated RO2 mixing ratio is (13.8 ± 11.2) pmol mol−1. Note that RO2 = 0 pmol mol−1 for the 09:00–12:00 LT interval originates from our assumption of = 1 for our highest Δ17O(NO2) value; in reality, it only means that RO2 is so low that Reaction (R3) (NO + O3) ≫ Reaction (R4) (NO + RO2). Overall, our RO2 values are found to be within the range of values measured at urban/peri-urban sites (see Table 2). However, RO2 diurnal variation at our site does not follow the pattern of previous measurements which usually report a diurnal variation with a maximum varying from noon to early afternoon (Fuchs et al., 2008; Tan et al., 2017), whereas this study shows a maximal mixing ratio in late afternoon. With such a limited dataset (only 1 d of sampling), it is not possible to draw general conclusions on the NOx RO2 chemistry dynamics. An important recommendation for further investigation is to conduct isotopic measurements with accurate measurements of key atmospheric radicals/oxidants, e.g. NO, O3, and possibly RO2, in order to test quantitatively our isotopic approach. Additionally, the use of a chemical box model is also recommended, because it would allow us to account for non-equilibrium effects in isotopic transfers and thus strengthen the interpretation of isotopic measurements in the investigation of the reactive nitrogen cycle in urban atmospheres.
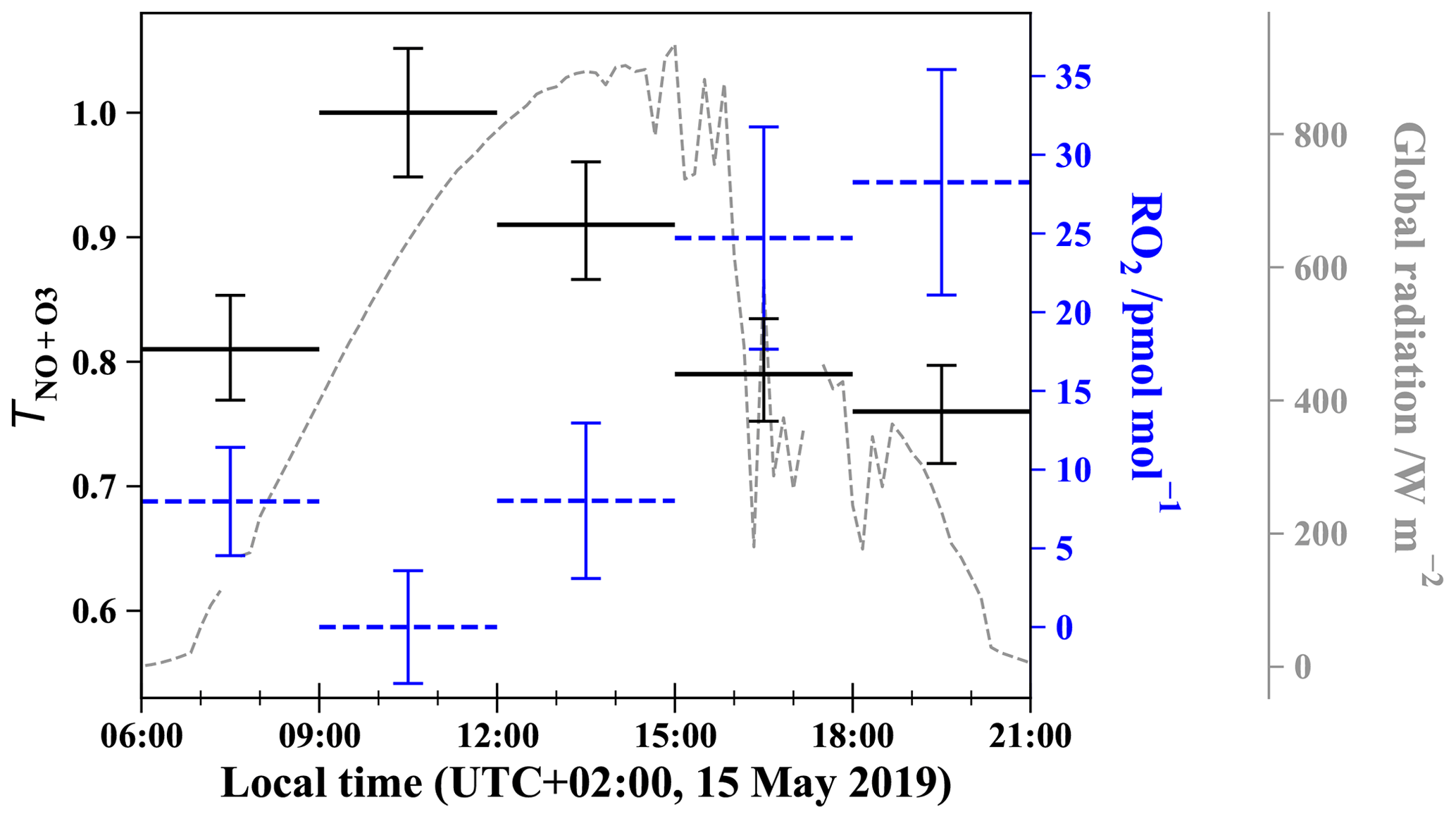
Figure 4Daytime evolution of (solid black lines), estimated from Eq. (4) using measured Δ17O(NO2) in Grenoble, and of RO2 mixing ratios (dashed blue lines), estimated from Eq. (6). Error bars for are derived from standard deviations of Δ17O(NO2) and Δ17O(O) measured in Grenoble (Vicars and Savarino, 2014). RO2 error bars are derived from O3 measurement uncertainties and errors on (by comparison, errors on reaction constants can be neglected). Global solar radiation flux is represented by dashed lines (measured at 200 m from the sampling site by the IGE weather station with a Skye SP1110 pyranometer).
Table 2Mean daytime RO2 mixing ratio ranges measured during field campaigns in various environments and seasons.
∗ Derived from Eq. (6) using Δ17O values of atmospheric NO2 in Grenoble.
Morin et al. (2011) simulated the diurnal variation of Δ17O(NO2) in a remote marine boundary layer without the effect of NOx emissions. They assumed Δ17O(O3) = 30 ‰ ( = 45 ‰) resulting in higher overall Δ17O(NO2) values compared to our study. Their simulated Δ17O(NO2) exhibited large diurnal variations with maximum values at night (close to 41 ‰) and minimum values at noon of 28 ‰. This is consistent with RO2 mixing ratio reaching a maximum around local noon in clean environments. In contrast to their model simulations, our daytime Δ17O(NO2) measurements are higher than our nighttime measurements. We will show in the following section that this difference originates from the absence of NOx emissions in the Morin et al. (2011) photochemical modelling.
4.1.3 Δ17Onight(NO2)
Without photolysis at night and associated RO2 production, ozone is the unique NO oxidant. NO and NO2 are no longer in photochemical equilibrium, because NO2 cannot be converted back into NO. As a result, the oxygen isotopic composition of NO2 formed during the night is determined by the oxygen isotopic composition of O3 and emitted NO. Additionally, in order to estimate the overall isotopic signature of sampled NO2 at night, we need to determine the residuals of NO2 formed during the day that are still present during the night:
with x being the fraction of NO2 formed during the day to the total NO2 measured at night and with (1−x) representing the fraction of NO2 which has been produced during the night to the total NO2 measured at night. NO is mainly emitted by combustion processes in which a nitrogen atom (from atmospheric N2 or N present in fuel) is added to an oxygen atom formed by the thermal decomposition of O2 (Zeldovich, 1946). With Δ17O(O2) being close to 0 ‰ (Barkan and Luz, 2003), NO emissions are very likely to have a Δ17O ≈ 0 ‰ or at least be negligible compared to . Using Eq. (7) and assuming a negligible isotope anomaly for NO, the time evolution of Δ17O(NO2) over the night can be calculated. It is worth pointing out that the x fraction becomes very small at the end of the night, allowing us to further simplify Eq. (7): . Thus, if there are nighttime NO emissions, a measurement of Δ17O(NO2) at the end of the night is also an interesting way of deriving Δ17O(O3) which is difficult to measure directly. The nighttime variation of the x fraction is estimated considering that the nighttime lifetime of NO2 relative to oxidation via ozone and dry deposition is 7.2 h (O3 chemical sink is dominant over deposition by a factor > 104 with cm3 mol−1 s−1; Atkinson et al., 2004; NO2 dry velocity Vd = 0.25 cm s−1; Holland et al., 1999; and assuming a nighttime boundary layer height of 500 m). For the 00:00–05:00 LT interval, we calculate a mean value of Δ17O(NO2) = 19.9 ‰ (with an overall error of about 1.6 ‰), which is very close to our measured Δ17O(NO2) of 20.5 ‰. This first dataset of nighttime Δ17O(NO2) measurements appears to confirm our understanding of nocturnal NO2 formation (Alexander et al., 2020; Michalski et al., 2014). NO emissions in urban areas have a very significant influence on Δ17O(NO2), leading to a behaviour in opposition to the one observed in remote locations. As illustrated by Morin et al. (2011), Δ17O(NO2) is predicted to be maximal at night in remote areas where NO emissions are negligible, reflecting the isotopic signature of NO2 at sunset. In areas where nighttime NO emissions are high, nighttime Δ17O(NO2) can be up to a factor of 2 smaller than in remote areas.
4.2 Nitrogen isotope composition
Measured δ15N(NO2) values range from −11.8 ‰ to −4.9 ‰ with no clear diurnal variation and values clustering around an overall mean of (−10.2 ± 2.2) ‰ (see Fig. 3). Using a similar method, Walters et al. (2018) collected atmospheric NO2 over 1 month in a urban/suburban location during the summer. They reported a mean δ15N value of (−11.4 ± 6.9) ‰, very close to our mean value but with a wider overall range (from −31.4 ‰ to +0.4 ‰). In another urban area but using passive samplers, Dahal and Hastings (2016) reported mean δ15N(NO2) values of (−8.3 ± 0.9) ‰ and (−6.4 ± 1.4) ‰ for summer and winter periods, respectively. All these values are within the δ15N range for NO emitted by industrial combustion and traffic sources which are reported to vary from −19.7 ‰ to −13.7 ‰ and from −9 ‰ to −2 ‰, respectively (Miller et al., 2017; Walters et al., 2015). Interestingly, all the δ15N values measured at our sampling site fall within a narrow range, from about −12 ‰ to −10 ‰, except for the sample collected between 06:00 and 09:00 LT which has a much higher value of −4.9 ‰. This singular value is well correlated with the morning NO traffic emission spike (see Fig. 2). However, once emitted into the atmosphere, NO can undergo isotopic fractionations that modify the nitrogen isotope distribution in NO2 relative to emitted NO (Freyer et al., 1993). In order to use δ15N(NO2) as a tracer of NOx sources, we need to quantify these nitrogen isotopic shifts to correct measured δ15N(NO2). Nitrogen isotopic fractionation, defined as Δ(NO2−NOx) = δ15N(NO2)−δ15N(NOx), is the result of a combination of three effects: (1) an equilibrium isotope effect (EIE) between NO and NO2, (2) a kinetic isotope effect (KIE) during NO oxidation to NO2, and (3) a photochemical isotope fractionation effect (PHIFE) during NO2 photolysis (other NO2 sinks are negligible during the day). The overall daytime nitrogen isotopic shift of NO2 relative to emitted NOx (Δday(NO2−NOx)) can be estimated using the steady-state isotopic mass balance for NO2. Li et al. (2020) derived an expression for Δ(NO2−NOx) by assuming that the conversion of NO to NO2 is solely driven by O3. This could therefore lead to uncertainties on the NO2 shift when other conversion pathways become significant with respect to the NO conversion by O3. A more general expression for Δ(NO2−NOx) can be derived by taking into account the conversion of NO to NO2 via other species, notably RO2 (see Eq. C11 in Appendix C). In our urban environment, we only consider the conversion of NO into NO2 via O3 and RO2 during the day. Assuming (see derivation in Appendix C), Δday(NO2−NOx) can be expressed by
with
and
where , is the fractionation factor of combined KIE and PHIFE (LCIE represents the Leighton cycle isotope effect), and αEIE is the EIE fractionation factor. is defined as the ratio of the NO2 lifetime with respect to isotopic exchanges over the daytime NO2 chemistry lifetime. is the NO2 photolysis rate, is the rate constant of reaction NO + O3, is the rate constant of reaction NO + RO2, and is the rate constant of the isotopic exchange (Reaction CR1) (see Appendix D for rate constants data). Interestingly, we can combine Eqs. (8) and (6) and express as a function of oxygen isotopic variables discussed in the previous section:
Since our NO measurements are not precise, and we do not have direct measurements of or RO2, we use Eq. (9) to estimate the NO2 isotopic fractionation shift. Note that although Li et al. (2020) only consider the NO conversion via O3 in the analysis of their nitrogen isotopic data, they found an excellent agreement between their calculated values and field isotopic measurements at Jülich, Germany (Freyer et al., 1993). Nonetheless, the reason for this accordance remains unclear, as it could be attributable to an equivalent KIE of NO + O3 and NO + RO2 but also to the dominance of the NO oxidation via O3 over RO2.
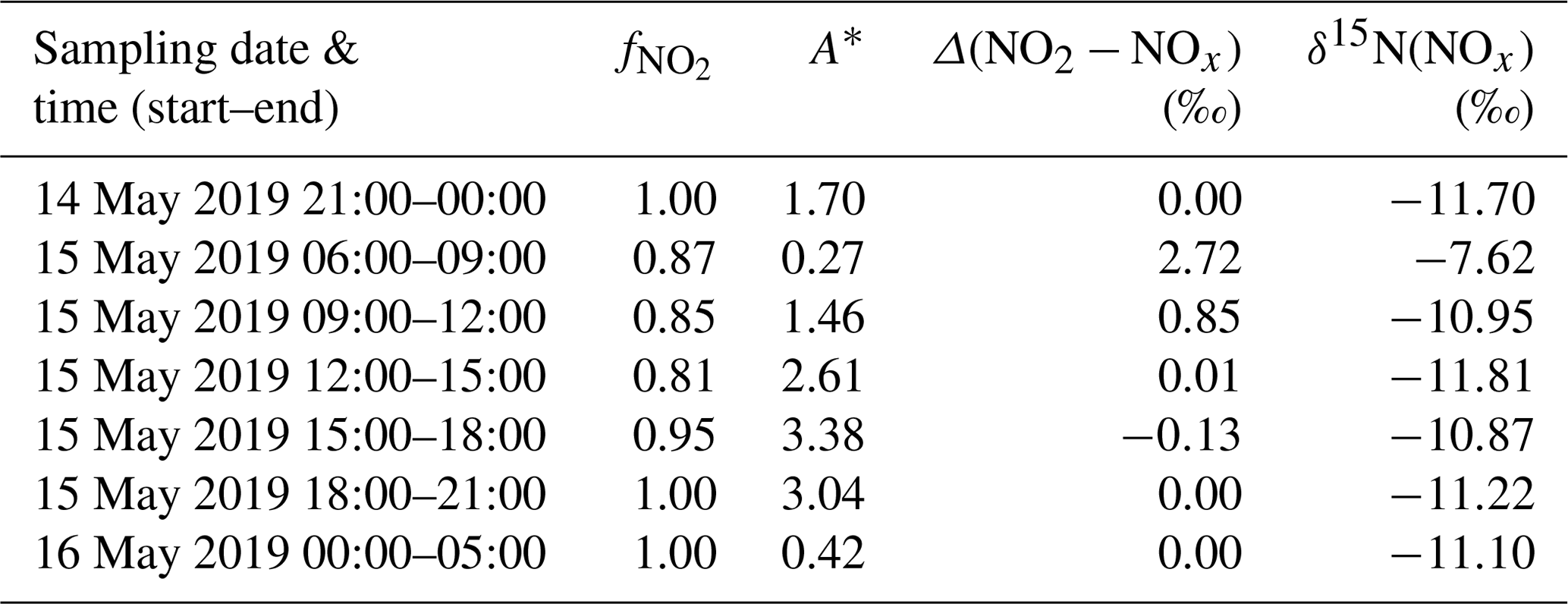
Table 3Summary of measured , calculated A∗ values using Eq. (9) for daytime and Eq. (11) for nighttime, calculated isotopic fractionation between NO2 and NOx (Δ(NO2 − NOx)) using Eq. (8) for daytime and Eq. (10) for nighttime, and δ15N(NOx) estimated with Δ(NO2 − NOx) and measured δ15N(NO2).
At night, the isotopic fractionation shift Δnight(NO2−NOx) is driven by EIE, KIE, the N isotopic composition of NO emissions and , given that is null (see derivation in Appendix C):
where δ15N(NOemis) is the N isotopic composition of NO emissions, and
with E(NO) the NO emission flux. From laboratory experiments, Li et al. (2020) reported fractionation factors of 1.0289 ± 0.0019 and 0.990 ± 0.005 for αEIE and , respectively. Using these experimental values and the ambient mixing ratios of ozone, NO, and NO2 measured at our sampling site, we estimate the time evolution of Δ(NO2−NOx) from Eqs. (8) and (9) for daytime. At night, [NO] ≪ [NO2]; hence, tends towards 1 and (Table 3 provides the calculated values). Δ(NO2−NOx) values are found to be negligible during the entire sampling period except between 06:00 and 09:00 LT with a mean Δ(NO2−NOx) value of 2.7 ‰ due to lower and values. Overall, in our moderately polluted environment, nitrogen oxide isotope effects appear to induce very small nitrogen isotopic shift, reflecting the fact that NOx is overwhelmingly under the form of NO2 (mean = 0.93). Our results are in good agreement with the Δ(NO2−NOx) range (between 1.3 and 2.5 ‰) calculated from isotopic measurements at West Lafayette, USA (Walters et al., 2018). Moreover, Li et al. (2020) calculated a mean Δ(NO2−NOx) of (1.3 ± 3.2) ‰ from isotopic measurements near San Diego, USA (NOx mixing ratios varied from 1 to 9 nmol mol−1).
Using estimated δ15N(NOx), we evaluate the relative contributions of the dominant NOx sources at our site using the Bayesian isotopic mixing model SIAR (Stable Isotope Analysis in R; Parnell et al., 2010). Initially developed for ecological studies (Inger et al., 2006; Samelius et al., 2007), isotopic mixing models have been recently used for atmospheric applications, notably to identify major NOx sources of aerosol nitrate from δ15N (Jin et al., 2021; Zong et al., 2017; Fan et al., 2019). Using as inputs not only isotopic measurements but also their uncertainties, the SIAR model can be used to calculate potential NOx source solutions as probability distributions. A recent emission inventory of NOx in the Grenoble urban area estimated that, in 2016, 52 % of emitted NOx could be attributed to transport, 26 % to industries, 20 % to the residential/tertiary sectors, and 2 % to agriculture (Topin et al., 2019). Looking at the type of energy consumed by each sector, we estimate that at this time of the year, our sampling site was mostly influenced by fossil-fuel combustion NOx sources, mainly gasoline/diesel and natural gas, and by biogenic NOx sources (soils emissions). As shown by previous studies, δ15N of NOx emitted by vehicle exhausts depends on the fuel type, the reduction emission technology, and the vehicle run time with values ranging from −21 ‰ to −2 ‰ (Walters et al., 2015). As 90 % of traffic-related NOx is emitted by diesel-powered engines in the Grenoble urban area (Atmo-Auvergne-Rhônes-Alpes, 2018), we use a value of (−4.7 ± 1.7) ‰, representative of the U.S. vehicle fleet (Miller et al., 2017) for which about 80 % of its traffic-NOx emissions originate from diesel vehicles (Dallmann et al., 2013). For δ15N of NOx emitted by natural gas combustion, we use a value of (−16.5 ± 1.7) ‰ which is the average isotopic signature of natural-gas-burning power plants and residential furnace exhausts (Walters et al., 2015). Despite the large range of δ15N values for biogenic NOx (from −59.8 % to −19.9 %) (Li and Wang, 2008; Yu and Elliott, 2017; Walters et al., 2015), these values are still very distinct from δ15N of fossil-fuel combustion NOx, making possible to roughly estimate the relative contributions of different NOx sources at our sampling site. We use a soil–NOx δ15N value of (−33.8 ± 12.2) ‰ (Zong et al., 2017), which is the average of values taken from several studies on NOx emitted by natural and fertilized soil (Felix and Elliott, 2014; Li and Wang, 2008). Over our sampling period, the SIAR model results indicate traffic as the dominant NOx emission source with a mean relative contribution of (57 ± 8) % (see Fig. 5). Natural gas combustion is found to be the second main NOx emission source ((36 ± 12) %) before soil emissions which account for only (7 ± 5) %. The limited nature of our measurement dataset (only 1 d of sampling) prevents us from drawing any general and robust conclusions on the relative contributions of NOx emissions at our site. Nonetheless, we note that the SIAR overall source apportionment is in close agreement with the Grenoble urban area emission inventory concerning traffic emissions (52 % in 2016), lending some support to the idea that δ15N of NO2 is a reliable tracer of NOx emission sources after correction for LCIE and EIE.
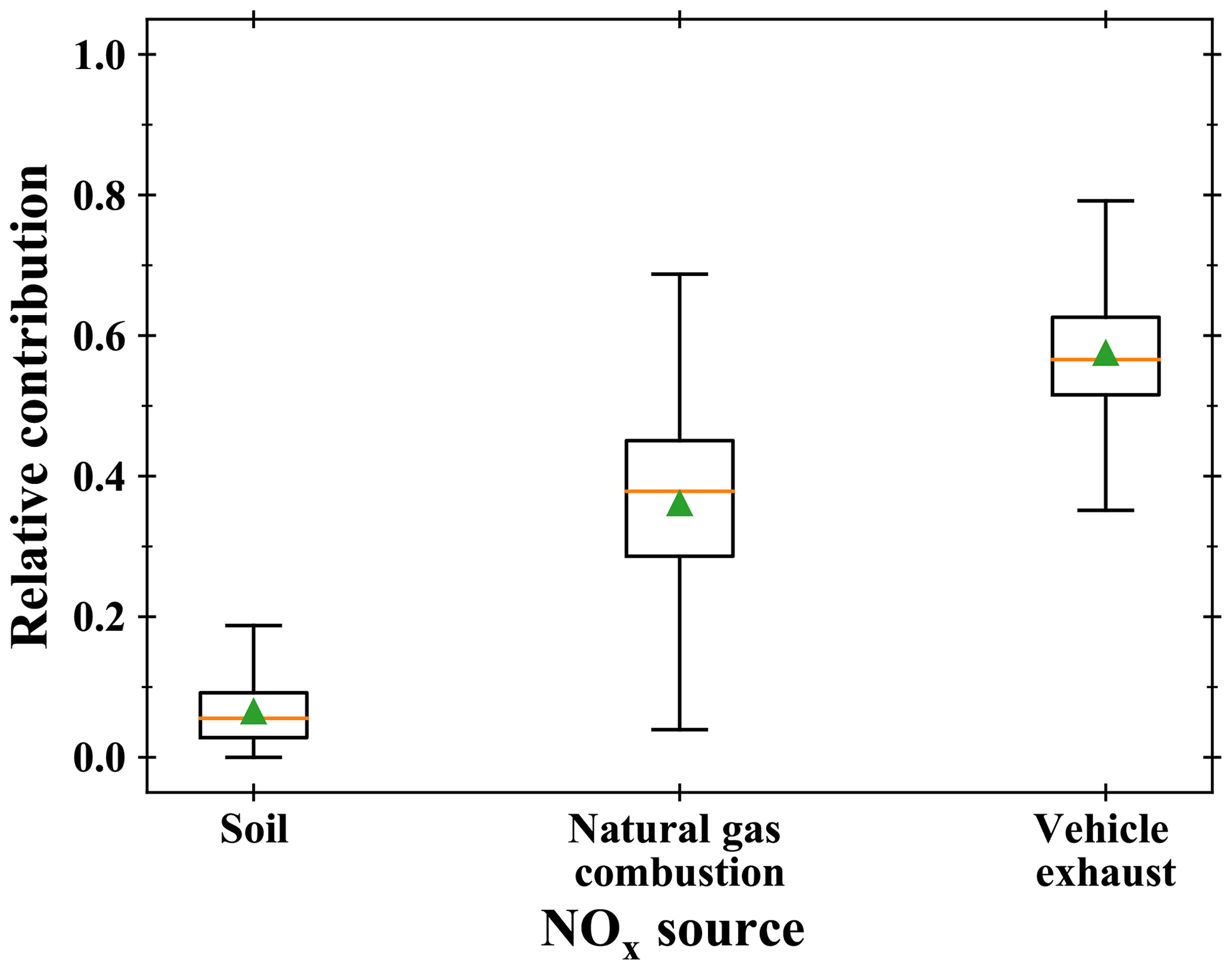
Figure 5Potential NOx emission source partitioning using the SIAR model based on estimated δ15N(NOx). Reference values for each source were taken from Miller et al. (2017), Walters et al. (2015), and Zong et al. (2017).
The primary goal of this preliminary work was to address an efficient and portable sampling system for atmospheric NO2 fitting with accurate isotopic analysis of double nitrogen and triple oxygen isotopes. First simultaneous measurements of the multi-isotopic composition (δ15N, δ18O, and Δ17O) of atmospheric NO2 are reported here, notably at relatively high temporal resolution (3 h). Over the course of more than 1 d in the Grenoble urban/suburban environment, Δ17O(NO2) is found to vary diurnally with a maximum value of (39.2 ± 0.3) ‰ during the day and a minimum value of (20.5 ± 0.3) ‰ at night. At photo-stationary state, high Δ17O(NO2) values result from the ozone predominance in NO oxidation pathways, whereas lower values reflect the influence of peroxy radicals. We estimate from our Δ17O(NO2) measurements that 86 % of NO2 produced by day originates from the oxidation of NO by O3. Moreover, a mean daytime peroxy radical mixing ratio of (13.8 ± 11.2) pmol mol−1 is derived from the oxygen isotopic measurements. At night, NOx photochemistry shuts down; hence, Δ17O(NO2) decreases under the growing influence of the isotopic footprint from NO emitted by night. The Δ17O(NO2) measurement towards the end of the night is found to be quantitatively consistent with typical values of Δ17O(O3). The δ15N(NO2) measurements show little variations, from −11.8 ‰ to −4.9 ‰, with mostly negligible N isotope fractionations between NO and NO2 due to the high NO2 NOx ratios. After correction of nitrogen isotopic fractionations, we use a Bayesian isotope mixing model to estimate the relative contributions of the dominant NOx emissions sources. The results indicate the predominance of traffic NOx emissions in this area at (57 ± 8) %, which is before natural gas combustion and soil emission.
Despite the limited nature of our measurement dataset, our results shed light on the sensitivity of NO2 isotopic signature to the atmospheric chemical regimes and emissions of the local environment. This isotopic approach can be applied to various environments in order to probe further the oxidative chemistry and help to constrain the NOx fate in a more quantitative way. In the future, the interpretation of the isotopic data should be extended with the use of a photochemical box model including isotopic anomaly transfers and local emissions in order to solve persistent issues of atmospheric oxidation mechanisms. Moreover, samplings and multi-isotopic analysis of atmospheric nitrate performed in parallel to those of NO2 would certainly be of interest for the study of the full reactive nitrogen cycle.
This method of analysis induces isotope fractionations during NO N2O conversion and ionization in the spectrometer, as well as isotope exchanges between NO and its medium. Indeed, while isotope exchanges between nitrite and its matrix are minimized due to the basic pH, the chemistry required to convert nitrite to N2O involves a step in an acidic medium that promotes an exchange of oxygen isotopes (Casciotti et al., 2007). In order to eliminate the effects of these isotope splits, the system is calibrated using standards of known isotopic composition, which are subjected to the same treatment as the samples. This is called the identical treatment principle (Brand, 1996). By subjecting compounds of known isotopic composition to the same treatment as the samples, the isotope fractionation induced by the analytical protocol can be estimated, and the sample values can be corrected. Standards are first dissolved in a basic aqueous medium (pH = 12), and then, from this stock solution, five series of each standard are prepared in several amount ranges, namely 40, 80, 100, 120, and 150 nmol, in order to estimate the effects of the amount of a material on its isotopic measurement. The matrix used for their preparation is the same as that of the samples, i.e. a mixture of KOH 2 M/guaiacol in deionized water. Correction factors are obtained by linear regression between the raw and the expected values of δ15N, δ18O, and δ17O of the standards. Three international references of known δ15N and δ18O values are used for this work. These are nitrite salts, named RSIL-N7373, RSIL-N10219, and RSIL-N23 with respective δ15N δ18O values of −79.6/4.2 ‰, 2.8/88.5 ‰, and 3.7/11.4 ‰. Although the three standards cover a wide range of isotopic composition in δ15N and δ18O, they do not have an isotopic anomaly in 17O. As we are not aware of any available international reference nitrite standards with a known 17O anomaly, we are currently in the process of manufacturing our own standards. As this step is still under development and in order to be able to assess the accuracy of our 17O measurements of atmospheric NO2 samples, we have estimated the isotope fractionation that 17O undergoes during the analysis. RSIL-N7373 and RSIL-N23 standards have a Δ17O = 0 ‰, so we estimate their 17O composition such that δ17O = 0.52 × δ18O. For standard RSIL-N10219, we measure a negative Δ17O around −7 ‰. We therefore apply the mass-independent relation such that δ17Ostd(RSIL-N10219) = Δ17Oraw(RSIL-N10219) + 0.5 × δ18Ostd(RSIL-N10219).
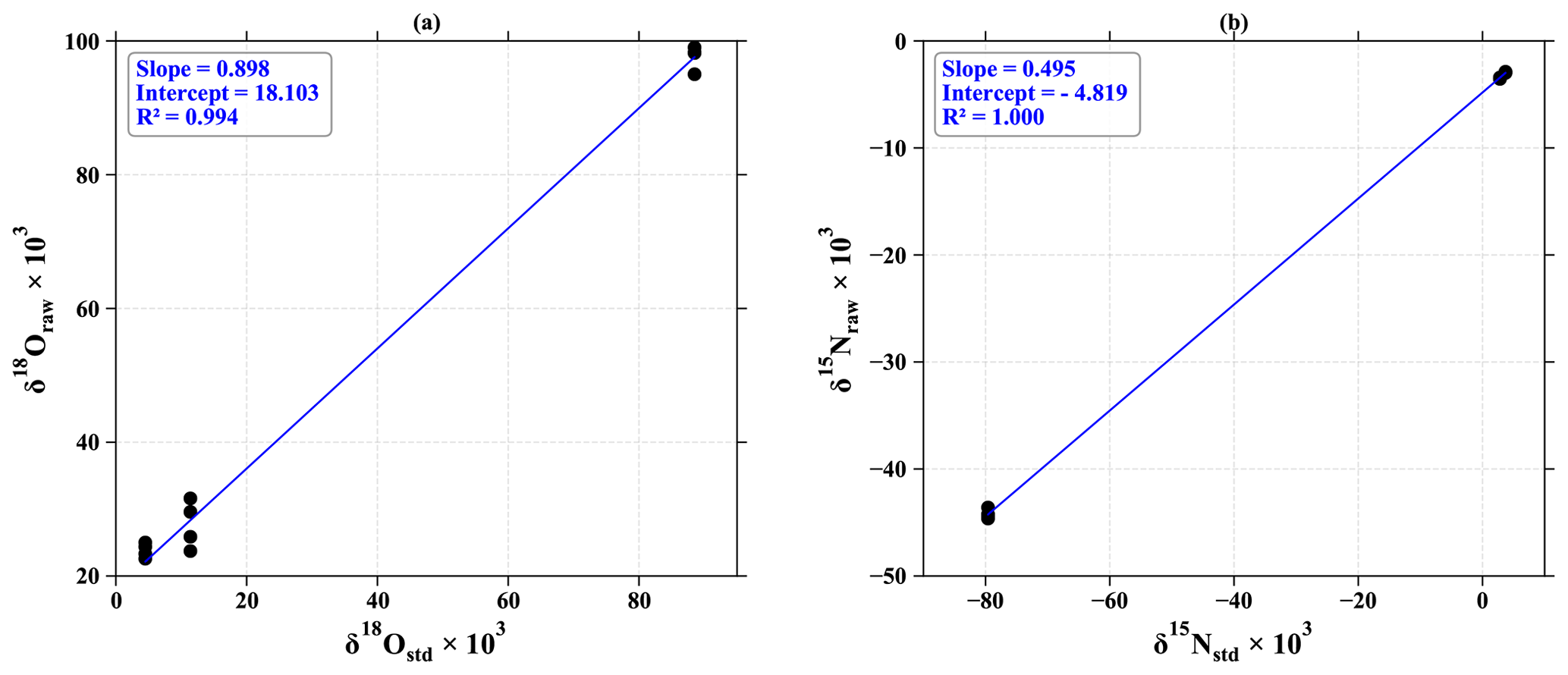
Figure A1Calibration of (a) 18O and (b) 15N with nitrite standards at 100 nmol measured by the chemical azide method. The measured δ18O (δ18Oraw) and δ15N (δ15Nraw) values of NO standards are plotted against their certified reference δ18O (δ18Ostd) and δ15N (δ15Nstd) values.
The isotopic exchange of 18O is estimated at 11 % for standards at 100 nmol (Fig. A1), which is in line with Kobayashi et al. (2021), who have estimated the degree of O isotope exchange in the azide method between H2O and NO to be (10.8 ± 0.3) %. The 15N calibration curve allows us to ensure a good fractionation rate during the analysis. Indeed, given the 1:1 association of the nitrogen atoms of nitrite and azide, the theoretical value of the calibration slope must be 0.5. The slight deviation from our measured value can be attributed to a blank effect, estimated here at 2 % of the size of the standards (6 % for those at 40 nmol).
Oxygen isotopes in nitrites are very labile (Böhlke et al., 2007), but the basic pH of the eluent limits isotopic exchanges. To ensure isotopic integrity from denuder's extraction to analysis by IRMS, we followed the procedure by Walters et al. (2018) to quantify isotopic exchanges that might occur with the eluted matrix during storage. Thus, three solutions each containing 500 nmol of a KNO2 salt (RSIL-N7373, RSIL-N10219 and RSIL-N23) were prepared in the eluted matrix and kept frozen. We monitored the nitrite standard isotopic composition prepared in the eluted guaiacol matrix during 22 d. To do so, 100 nmol were collected from the individual solutions, analysed, and refrozen until the next analysis. The temporal evolution of the δ17O,δ18O, and Δ17O differences between our measurements of RSIL standards (prepared in the KOH/guaiacol eluted matrix) and their certified reference values is plotted in Fig. B1. It represents the temporal drift of the isotopic signal with respect to reference values. If the deviation is constant, it means that the isotopic signal is not degraded with time, and its standard deviation is considered the uncertainty in our δ17O(NO2) and δ18O(NO2) measurements. As shown in Fig. B1, deviation of the three standards was stable over the 22 d experiment with an overall mean of (1.1 ± 0.8) ‰, (2.3 ± 1.8) ‰, and (−0.1 ± 0.3) ‰ for δ17O, δ18O, and Δ17O, respectively. Note that RSIL-N10219 shows higher δ17O and δ18O residuals than the two other standards. The reason for this difference in behaviour is still not fully understood. As residuals remain steady over several weeks, we consider this method suitable for the oxygen analysis of NO2, and the uncertainties applied to our isotopic measurements are reported as the propagation error of the mean measurement uncertainty and the mean uncertainty resulting from NO storage. In our study, average uncertainties on δ17O, δ18O, and Δ17O are estimated to be ±0.8 ‰, ±1.8 ‰, and ± 0.3 ‰, respectively (1σ uncertainties).

Figure B1Temporal evolution of δ17O,δ18O, and Δ17O differences between our measurements of RSIL standards (prepared in the KOH/guaiacol eluted matrix) and their certified reference values. Error bars derived from measurement uncertainties are approximately equivalent to the size of the markers.
We follow the same approach as Li et al. (2020) but take into account all the oxidation pathways of NO into NO2, not only via O3. The reactions considered in deriving the combined isotopic fractionation are the following:
with Xi = RO2, BrO, ClO, …, .
C1 Daytime N fractionation
During the day, NO2 photolysis is the overwhelmingly dominant NO2 sink, and NO oxidation is the main NO2 source (see Fig. C1). The assumption of steady state on 15NO2 for the extended Leighton cycle leads to
where is the rate constant for the nitrogen isotopic exchange between NO and NO2, is the NO2 photolysis rate with αPHIFE being its isotopic fractionation factor, the sum of all the NO oxidation pathways to NO2, Xi the NO oxidant (i.e. O3, RO2, BrO, ClO …), and the rate constant for the reaction of NO + Xi with being its isotopic fractionation factor. Equation (C1) can be rearranged to give
Meanwhile, 14NO2 in steady state leads to
We define as the ratio of the 14NO2 lifetime with respect to isotopic exchange with 14NO () over the daytime 14NO2 chemical lifetime () (Li et al., 2020):
Using Eq. (C3), Eq. (C4) becomes
We also define as the relative importance of the oxidation pathway of NO into NO2 via the oxidant Xi:
with necessarily .
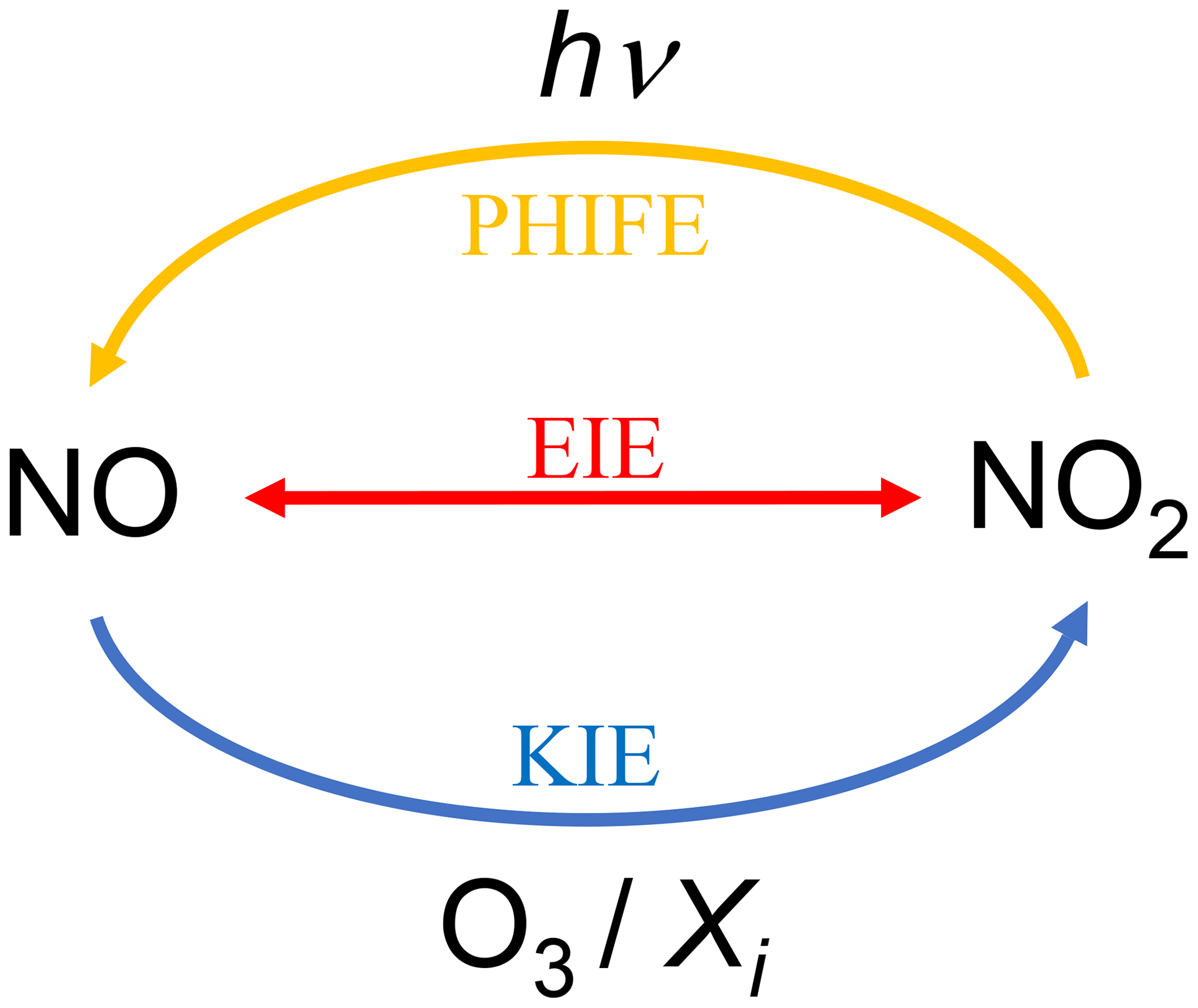
Figure C1Sketch of the nitrogen fractionation processes between NO and NO2. PHIFE represents the photochemical isotope fractionation effect, KIE represents the kinetic isotope effect, and EIE represents the equilibrium isotope effect.
Using the definitions (C5) and (C6), Eq. (C2) becomes
with .
Using (with ) and (with ), Eq. (C7) becomes
As a result, the daytime isotopic shift of NO2 relative to NO, defined as Δ(NO2−NO) = δ15N(NO2)−δ15N(NO), is given by
Using the isotopic balance with = [NO2] [NOx] (Li et al., 2020), the isotopic shift of NO2 relative to NOx, defined as , can be expressed by
Since fractionation factors are close to unity and , Eq. (C11) can be further simplified by keeping only the dominant terms (Li et al., 2020):
with .
Considering the localization of our sampling site (urban mid-latitude area), only NO + RO2 and NO + O3 are thought to be significant as NO2 formation pathways; hence, becomes
and Eq. (C5) becomes
Equations (4) and (5) from Sect. 4.1.2 can be combined to give
Using Eq. (C15), Eq. (C14) becomes
We consider several particular cases. The first case is when . Previous studies found that the NO + O3 reaction falls within the family of “normal kinetic isotope fractionation” with the NO2 produced being depleted in 15N (Walters and Michalski, 2016) compared to residual reactant NO. To our knowledge, no such experiment has been carried out for the NO + RO2 reaction. Nonetheless, considering the very close, and both very low, activation energies for the reaction NO + O3 and NO + RO2, it is quite likely that the fractionation factors of these two reactions are similar. It follows that we obtain the same expression for as for the αLCIE given in Li et al. (2020):
And Eq. (C12) becomes
Equation (C18) with given by Eq. (C16) is the expression that we use to analyse our daytime nitrogen isotopic measurements. Another particular case considered by Li et al. (2020) is ; in that case, is still given by Eq. (C12) but A∗ is simplified:
Equation (C16) with given by Eq. (C19) and given by Eq. (C17) is the same expression as Eq. (8) in Li et al. (2020).
C2 Nighttime N fractionation
An expression similar to Eq. (C1) can be derived for nighttime conditions, when NO2 photolysis is null and hence when there is no recycling between NO and NO2. In addition, the conversion of NO into NO2 occurs only via reaction with O3 because the mixing ratios of other NO oxidants are usually negligible at night. The main source of NOx at night is the NO emissions. The assumption of steady state on short timescales can only hold for 14NO and 15NO, not NO2, leading to an equation equivalent to Eq. (C1):
with E(15NO) being the 15NO emission flux and αKIE is the fractionation factor of NO + O3. Equation (C20) can be rearranged to give
Meanwhile, the steady state on 14NO gives (nitrogen isotopic exchanges are neglected as NO emissions are largely dominated by 14NO)
with E(14NO) being the 14NO emission flux. For nighttime, we define as the ratio of the 14NO lifetime with respect to isotopic exchange with 14NO2 (τexchange−NO) over the nighttime 14NO chemical lifetime (τchem−NO):
Using Eq. (C22), Eq. (C23) gives
NOx is overwhelmingly emitted in the form of NO. The isotopic signature of NO emissions can be characterized with and . Using the definition of , Eqs. (C23), and (C24), Eq. (C21) becomes
And then, using Eq. (C24), Eq. (C25) becomes
Following the approach used in the derivation of daytime isotopic shift, the nighttime isotopic shift of NO2 relative to NO is given by
Using the isotopic balance , the nighttime isotopic shift of NO2 relative to NOx, can be expressed by
where δ15N(NOemis) is the nitrogen isotopic composition of NO emissions.
Keeping the dominant terms, Eq. (C29) can be further simplified following the daytime derivation:
We consider two particular cases. When (), i.e isotopic exchange much faster than NO oxidation, Eq. (C28) becomes
Keeping the dominant terms, Eq. (C31) can be simplified:
As expected, the nighttime isotopic shift of NO2 relative to NO depends only on the isotopic exchange fractionation in that case. In the same way, Eq. (C29) becomes
Keeping the dominant terms, Eq. (C33) can be simplified:
When (), i.e NO oxidation much faster than isotopic exchange, Eq. (C28) yields
leading to
As expected, the nighttime isotopic shift of NO relative to NO emissions depends only on the isotopic fractionation factor of the NO + O3 reaction in that case. It is possible to estimate the nighttime isotopic shift of NO2 relative to NO or NOx on long timescales by assuming crudely that 14NO2 is in steady state:
with representing the equivalent of a first-order rate constant. If the 14NO2 loss is a second-order reaction such as NO2 + O3 loss, then . In the same way, assuming that the NO2 oxidation into nitrate via O3 is not fractionating, 15NO2 in steady state gives
Using Eq. (C38), Eq. (C39) becomes
Using Eq. (C37), Eq. (C40) becomes
or
Under those conditions (negligible isotopic exchange), a measurement of δ15N(NO2) is a measurement of δ15N(NOemis), preferably towards the end of the night in order for 14NO2 to move towards steady state.
In the text, 14NO2 and 14NO are referred to as NO2 and NO for convenience.

The Bayesian mixing model used for this study is available to download from the packages section of the Comprehensive R Archive Network site (CRAN; https://CRAN.R-project.org/package=siar, Parnell and Jackson, 2013).
Data used for this study have been included in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-10477-2021-supplement.
Sampling and analysis protocol were developed by SA under the supervision of JS. NC and AB contributed with technical and knowledge support to SA for isotopic mass spectrometry and more general atmospheric measurements. SB and JS, supervisors of SA's PhD thesis, helped SA in interpreting the results and writing the article.
The authors declare that they have no conflict of interest.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work benefited from the IGE infrastructures and laboratory platforms. This is the publication number 2 of the PANDA platform on which isotope analyses were performed. The authors acknowledge the support of the ALPACA program (Alaskan Layered Pollution and Chemical Analysis). Finally, the authors thank Elsa Gauthier, Sophie Darfeuil, and Pete Akers for help with laboratory work and more general scientific discussions.
This research has been supported by the ANR project ANR-15-IDEX-02, Labex OSUG@2020, Investissements d’avenir – ANR10 LABX56, the French polar institute (IPEV, Institut polaire français Paul-Emile Victor) and INSU-CNRS (National Institute of Sciences of the Universe) via its national LEFE program (Les Enveloppes Fluides et l'Environnement).
This paper was edited by Jan Kaiser and reviewed by two anonymous referees.
Alexander, B., Hastings, M. G., Allman, D. J., Dachs, J., Thornton, J. A., and Kunasek, S. A.: Quantifying atmospheric nitrate formation pathways based on a global model of the oxygen isotopic composition (Δ17O) of atmospheric nitrate, Atmos. Chem. Phys., 9, 5043–5056, https://doi.org/10.5194/acp-9-5043-2009, 2009.
Alexander, B., Sherwen, T., Holmes, C. D., Fisher, J. A., Chen, Q., Evans, M. J., and Kasibhatla, P.: Global inorganic nitrate production mechanisms: comparison of a global model with nitrate isotope observations, Atmos. Chem. Phys., 20, 3859–3877, https://doi.org/10.5194/acp-20-3859-2020, 2020.
Assonov, S. S. and Brenninkmeijer, C. a. M.: Reporting small Δ17O values: existing definitions and concepts, Rapid Commun. Mass Spectrom., 19, 627–636, https://doi.org/10.1002/rcm.1833, 2005.
Atkinson, R., Baulch, D. L., Cox, R. A., Hampson, R. F., Kerr, J. A., Rossi, M. J., and Troe, J.: Evaluated Kinetic, Photochemical and Heterogeneous Data for Atmospheric Chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 26, 521–1011, https://doi.org/10.1063/1.556011, 1997.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Atmo-Auvergne-Rhônes-Alpes: Bilan Qualité de l'Air 2018 – Isère, available at: http://www.atmo-auvergnerhonealpes.fr (last access: 18 August 2020), 2018.
Barkan, E. and Luz, B.: High-precision measurements of 17O 16O and 18O 16O of O2 and O2 Ar ratio in air, Rapid Commun. Mass Spectrom., 17, 2809–2814, https://doi.org/10.1002/rcm.1267, 2003.
Böhlke, J. K., Smith, R. L., and Hannon, J. E.: Isotopic Analysis of N and O in Nitrite and Nitrate by Sequential Selective Bacterial Reduction to N2O, Anal. Chem., 79, 5888–5895, https://doi.org/10.1021/ac070176k, 2007.
Brand, W. A.: High Precision Isotope Ratio Monitoring Techniques in Mass Spectrometry, J. Mass Spectrom., 31, 225–235, https://doi.org/10.1002/(SICI)1096-9888(199603)31:3<225::AID-JMS319>3.0.CO;2-L, 1996.
Brown, S. S.: Variability in Nocturnal Nitrogen Oxide Processing and Its Role in Regional Air Quality, Science, 311, 67–70, https://doi.org/10.1126/science.1120120, 2006.
Buttini, P., Di Palo, V., and Possanzini, M.: Coupling of denuder and ion chromatographic techniques for NO2 trace level determination in air, Sci. Total Environ., 61, 59–72, https://doi.org/10.1016/0048-9697(87)90356-1, 1987.
Casciotti, K. L., Sigman, D. M., Hastings, M. G., Böhlke, J. K., and Hilkert, A.: Measurement of the oxygen isotopic composition of nitrate in seawater and freshwater using the denitrifier method, Anal. Chem., 74, 4905–4912, https://doi.org/10.1021/ac020113w, 2002.
Casciotti, K. L., Böhlke, J. K., McIlvin, M. R., Mroczkowski, S. J., and Hannon, J. E.: Oxygen Isotopes in Nitrite: Analysis, Calibration, and Equilibration, Anal. Chem., 79, 2427–2436, https://doi.org/10.1021/ac061598h, 2007.
Crutzen, P. J.: My life with O3, NOx and other YZOx compounds (Nobel lecture), Angew. Chem. Int. Ed. Engl., 35, 1759–1776, 1996.
Dahal, B. and Hastings, M. G.: Technical considerations for the use of passive samplers to quantify the isotopic composition of NOx and NO2 using the denitrifier method, Atmos. Environ., 143, 60–66, https://doi.org/10.1016/j.atmosenv.2016.08.006, 2016.
Dallmann, T. R., Kirchstetter, T. W., DeMartini, S. J., and Harley, R. A.: Quantifying On-Road Emissions from Gasoline-Powered Motor Vehicles: Accounting for the Presence of Medium- and Heavy-Duty Diesel Trucks, Environ. Sci. Technol., 47, 13873–13881, https://doi.org/10.1021/es402875u, 2013.
Davidson, E. A. and Kingerlee, W.: A global inventory of nitric oxide emissions from soils, Nutr. Cycl. Agroecosystems, 48, 37–50, https://doi.org/10.1023/A:1009738715891, 1997.
Dennison, P., Charoensiri, K., Roberts, D., Peterson, S., and Green, R.: Wildfire temperature and land cover modeling using hyperspectral data, Remote Sens. Environ., 100, 212–222, https://doi.org/10.1016/j.rse.2005.10.007, 2006.
Emmerson, K. M., Carslaw, N., Carslaw, D. C., Lee, J. D., McFiggans, G., Bloss, W. J., Gravestock, T., Heard, D. E., Hopkins, J., Ingham, T., Pilling, M. J., Smith, S. C., Jacob, M., and Monks, P. S.: Free radical modelling studies during the UK TORCH Campaign in Summer 2003, Atmos. Chem. Phys., 7, 167–181, https://doi.org/10.5194/acp-7-167-2007, 2007.
Fan, M.-Y., Zhang, Y.-L., Lin, Y.-C., Chang, Y.-H., Cao, F., Zhang, W.-Q., Hu, Y.-B., Bao, M.-Y., Liu, X.-Y., Zhai, X.-Y., Lin, X., Zhao, Z.-Y., and Song, W.-H.: Isotope-based source apportionment of nitrogen-containing aerosols: A case study in an industrial city in China, Atmos. Environ., 212, 96–105, https://doi.org/10.1016/j.atmosenv.2019.05.020, 2019.
Felix, J. D. and Elliott, E. M.: Isotopic composition of passively collected nitrogen dioxide emissions: Vehicle, soil and livestock source signatures, Atmos. Environ., 92, 359–366, https://doi.org/10.1016/j.atmosenv.2014.04.005, 2014.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the Upper and Lower Atmosphere, Elsevier, https://doi.org/10.1016/B978-012257060-5/50003-4, 2000.
Freyer, H. D., Kley, D., Volz-Thomas, A., and Kobel, K.: On the interaction of isotopic exchange processes with photochemical reactions in atmospheric oxides of nitrogen, J. Geophys. Res.-Atmos., 98, 14791–14796, https://doi.org/10.1029/93JD00874, 1993.
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 79, 084104, https://doi.org/10.1063/1.2968712, 2008.
Galloway, J. N., Dentener, F. J., Capone, D. G., Boyer, E. W., Howarth, R. W., Seitzinger, S. P., Asner, G. P., Cleveland, C. C., Green, P. A., Holland, E. A., Karl, D. M., Michaels, A. F., Porter, J. H., Townsend, A. R., and Vöosmarty, C. J.: Nitrogen Cycles: Past, Present, and Future, Biogeochemistry, 70, 153–226, https://doi.org/10.1007/s10533-004-0370-0, 2004.
Geng, F., Tie, X., Xu, J., Zhou, G., Peng, L., Gao, W., Tang, X., and Zhao, C.: Characterizations of ozone, NOx and VOCs measured in Shanghai, China, Atmos. Environ., 42, 6873–6883, https://doi.org/10.1016/j.atmosenv.2008.05.045, 2008.
Harris, G. W., Carter, W. P. L., Winer, A. M., Pitts, J. N., Platt, U., and Perner, D.: Observations of nitrous acid in the Los Angeles atmosphere and implications for predictions of ozone-precursor relationships, Environ. Sci. Technol., 16, 414–419, https://doi.org/10.1021/es00101a009, 1982.
Holland, E. A., Dentener, F. J., Braswell, B. H., and Sulzman, J. M.: Contemporary and pre-industrial global reactive nitrogen budgets, Biogeochemistry, 46, 7–43, https://doi.org/10.1023/A:1006148011944, 1999.
Huang, R.-J., Yang, L., Cao, J., Wang, Q., Tie, X., Ho, K.-F., Shen, Z., Zhang, R., Li, G., Zhu, C., Zhang, N., Dai, W., Zhou, J., Liu, S., Chen, Y., Chen, J., and O'Dowd, C. D.: Concentration and sources of atmospheric nitrous acid (HONO) at an urban site in Western China, Sci. Total Environ., 593–594, 165–172, https://doi.org/10.1016/j.scitotenv.2017.02.166, 2017.
Inger, R., Ruxton, G. D., Newton, J., Colhoun, K., Robinson, J. A., Jackson, A. L., and Bearhop, S.: Temporal and intrapopulation variation in prey choice of wintering geese determined by stable isotope analysis, J. Anim. Ecol., 75, 1190–1200, https://doi.org/10.1111/j.1365-2656.2006.01142.x, 2006.
Jacob, D. J.: Introduction to Atmospheric Chemistry, Princeton University Press, Princeton, New Jersey, USA, 1999.
Jin, S. and Demerjian, K.: A photochemical box model for urban air quality study, Atmos. Environ. B-Urb., 27, 371–387, https://doi.org/10.1016/0957-1272(93)90015-X, 1993.
Jin, Z., Qian, L., Shi, Y., Fu, G., Li, G., and Li, F.: Quantifying major NOx sources of aerosol nitrate in Hangzhou, China, by using stable isotopes and a Bayesian isotope mixing model, Atmos. Environ., 244, 117979, https://doi.org/10.1016/j.atmosenv.2020.117979, 2021.
Johnston, J. C. and Thiemens, M. H.: The isotopic composition of tropospheric ozone in three environments, J. Geophys. Res.-Atmos., 102, 25395–25404, 1997.
Kaiser, J., Röckmann, T., and Brenninkmeijer, C. A. M.: Contribution of mass-dependent fractionation to the oxygen isotope anomaly of atmospheric nitrous oxide, J. Geophys. Res.-Atmos., 109, D03305, https://doi.org/10.1029/2003JD004088, 2004.
Kaiser, J., Hastings, M. G., Houlton, B. Z., Röckmann, T., and Sigman, D. M.: Triple oxygen isotope analysis of nitrate using the denitrifier method and thermal decomposition of N2O, Anal. Chem., 79, 599–607, https://doi.org/10.1021/ac061022s, 2007.
Kaye, J. A.: Mechanisms and observations for isotope fractionation of molecular species in planetary atmospheres, Rev. Geophys., 25, 1609–1658, https://doi.org/10.1029/RG025i008p01609, 1987.
Klein, A., Ravetta, F., Thomas, J. L., Ancellet, G., Augustin, P., Wilson, R., Dieudonné, E., Fourmentin, M., Delbarre, H., and Pelon, J.: Influence of vertical mixing and nighttime transport on surface ozone variability in the morning in Paris and the surrounding region, Atmos. Environ., 197, 92–102, https://doi.org/10.1016/j.atmosenv.2018.10.009, 2019.
Kleinman, L. I., Daum, P. H., Lee, Y.-N., Nunnermacker, L. J., Springston, S. R., Weinstein-Lloyd, J., and Rudolph, J.: Ozone production efficiency in an urban area, J. Geophys. Res.-Atmos., 107, 4733, https://doi.org/10.1029/2002JD002529, 2002.
Kobayashi, K., Fukushima, K., Onishi, Y., Nishina, K., Makabe, A., Yano, M., Wankel, S. D., Koba, K., and Okabe, S.: Influence of δ18O of water on measurements of δ18O of nitrite and nitrate, Rapid Commun. Mass Spectrom., 35, e8979, https://doi.org/10.1002/rcm.8979, 2021.
Krankowsky, D., Bartecki, F., Klees, G. G., Mauersberger, K., Schellenbach, K., and Stehr, J.: Measurement of heavy isotope enrichment in tropospheric ozone, Geophys. Res. Lett., 22, 1713–1716, 1995.
Largeron, Y. and Staquet, C.: Persistent inversion dynamics and wintertime PM10 air pollution in Alpine valleys, Atmos. Environ., 135, 92–108, https://doi.org/10.1016/j.atmosenv.2016.03.045, 2016.
Leighton, P. A.: Photochemistry of Air Pollution, Academic Press, New York, USA, 1961.
Li, D. and Wang, X.: Nitrogen isotopic signature of soil-released nitric oxide (NO) after fertilizer application, Atmos. Environ., 42, 4747–4754, https://doi.org/10.1016/j.atmosenv.2008.01.042, 2008.
Li, J., Zhang, X., Orlando, J., Tyndall, G., and Michalski, G.: Quantifying the nitrogen isotope effects during photochemical equilibrium between NO and NO2: implications for δ15N in tropospheric reactive nitrogen, Atmos. Chem. Phys., 20, 9805–9819, https://doi.org/10.5194/acp-20-9805-2020, 2020.
Li, W., Ni, B. L., Jin, D. Q., and Zhang, Q. G.: Measurement of the absolute abundance of Oxygen-17 in SMOW, Kexue Tongboa Chin. Sci. Bull., 33, 1610–1613, 1988.
Liao, H. and Seinfeld, J. H.: Global impacts of gas-phase chemistry-aerosol interactions on direct radiative forcing by anthropogenic aerosols and ozone, J. Geophys. Res.-Atmos., 110, D18208, https://doi.org/10.1029/2005JD005907, 2005.
Lyons, J. R.: Transfer of mass-independent fractionation in ozone to other oxygen-containing radicals in the atmosphere, Geophys. Res. Lett., 28, 3231–3234, https://doi.org/10.1029/2000GL012791, 2001.
Mariotti, A.: Natural 15N abundance measurements and atmospheric nitrogen standard calibration, Nature, 311, 251–252, https://doi.org/10.1038/311251a0, 1984.
Mayer, H.: Air pollution in cities, Atmos. Environ., 33, 4029–4037, https://doi.org/10.1016/S1352-2310(99)00144-2, 1999.
McIlvin, M. R. and Altabet, M. A.: Chemical Conversion of Nitrate and Nitrite to Nitrous Oxide for Nitrogen and Oxygen Isotopic Analysis in Freshwater and Seawater, Anal. Chem., 77, 5589–5595, https://doi.org/10.1021/ac050528s, 2005.
Michalski, G., Scott, Z., Kabiling, M., and Thiemens, M. H.: First measurements and modeling of Δ17O in atmospheric nitrate, Geophys. Res. Lett., 30, 1870, https://doi.org/10.1029/2003GL017015, 2003.
Michalski, G., Bhattacharya, S. K., and Girsch, G.: NOx cycle and the tropospheric ozone isotope anomaly: an experimental investigation, Atmos. Chem. Phys., 14, 4935–4953, https://doi.org/10.5194/acp-14-4935-2014, 2014.
Michoud, V., Colomb, A., Borbon, A., Miet, K., Beekmann, M., Camredon, M., Aumont, B., Perrier, S., Zapf, P., Siour, G., Ait-Helal, W., Afif, C., Kukui, A., Furger, M., Dupont, J. C., Haeffelin, M., and Doussin, J. F.: Study of the unknown HONO daytime source at a European suburban site during the MEGAPOLI summer and winter field campaigns, Atmos. Chem. Phys., 14, 2805–2822, https://doi.org/10.5194/acp-14-2805-2014, 2014.
Mihelcic, D., Holland, F., Hofzumahaus, A., Hoppe, L., Konrad, S., Müsgen, P., Pätz, H.-W., Schäfer, H.-J., Schmitz, T., Volz‐Thomas, A., Bächmann, K., Schlomski, S., Platt, U., Geyer, A., Alicke, B., and Moortgat, G. K.: Peroxy radicals during BERLIOZ at Pabstthum: Measurements, radical budgets and ozone production, J. Geophys. Res.-Atmos., 108, 8254, https://doi.org/10.1029/2001JD001014, 2003.
Miller, D. J., Wojtal, P. K., Clark, S. C., and Hastings, M. G.: Vehicle NOx emission plume isotopic signatures: Spatial variability across the eastern United States, J. Geophys. Res.-Atmos., 122, 4698–4717, https://doi.org/10.1002/2016JD025877, 2017.
Monks, P. S.: Gas-phase radical chemistry in the troposphere, Chem. Soc. Rev., 34, 376–395, https://doi.org/10.1039/B307982C, 2005.
Morin, S., Savarino, J., Bekki, S., Gong, S., and Bottenheim, J. W.: Signature of Arctic surface ozone depletion events in the isotope anomaly (Δ17O) of atmospheric nitrate, Atmos. Chem. Phys., 7, 1451–1469, https://doi.org/10.5194/acp-7-1451-2007, 2007.
Morin, S., Sander, R., and Savarino, J.: Simulation of the diurnal variations of the oxygen isotope anomaly (Δ17O) of reactive atmospheric species, Atmos. Chem. Phys., 11, 3653–3671, https://doi.org/10.5194/acp-11-3653-2011, 2011.
Nash, T.: An efficient absorbing reagent for nitrogen dioxide, Atmos. Environ. 1967, 661–665, https://doi.org/10.1016/0004-6981(70)90039-9, 1970.
Parnell, A. and Jackson, A.: siar: Stable Isotope Analysis in R, R package version 4.2, https://CRAN.R-project.org/package=siar (last access: 8 February 2021), 2013.
Parnell, A. C., Inger, R., Bearhop, S., and Jackson, A. L.: Source Partitioning Using Stable Isotopes: Coping with Too Much Variation, PLOS ONE, 5, e9672, https://doi.org/10.1371/journal.pone.0009672, 2010.
Prinn, R. G.: The Cleasing Capacity of the Atmosphere, Annu. Rev. Environ. Resour., 28, 29–57, https://doi.org/10.1146/annurev.energy.28.011503.163425, 2003.
Ren, X., Brune, W. H., Cantrell, C. A., Edwards, G. D., Shirley, T., Metcalf, A. R., and Lesher, R. L.: Hydroxyl and Peroxy Radical Chemistry in a Rural Area of Central Pennsylvania: Observations and Model Comparisons, J. Atmos. Chem., 52, 231–257, https://doi.org/10.1007/s10874-005-3651-7, 2005.
Röckmann, T., Kaiser, J., Crowley, J. N., Brenninkmeijer, C. A. M., and Crutzen, P. J.: The origin of the anomalous or “mass-independent” oxygen isotope fractionation in tropospheric N2O, Geophys. Res. Lett., 28, 503–506, https://doi.org/10.1029/2000GL012295, 2001.
Røyset, O.: Comparison of passive and active sampling methods for the determination of nitrogen dioxide in urban air, Fresenius J. Anal. Chem., 360, 69–73, https://doi.org/10.1007/s002160050644, 1998.
Samelius, G., Alisauskas, R. T., Hobson, K. A., and Larivière, S.: Prolonging the arctic pulse: long-term exploitation of cached eggs by arctic foxes when lemmings are scarce, J. Anim. Ecol., 76, 873–880, https://doi.org/10.1111/j.1365-2656.2007.01278.x, 2007.
Savarino, J., Bhattacharya, S. K., Morin, S., Baroni, M., and Doussin, J.-F.: The NO+O3 reaction: A triple oxygen isotope perspective on the reaction dynamics and atmospheric implications for the transfer of the ozone isotope anomaly, J. Chem. Phys., 128, 194303, https://doi.org/10.1063/1.2917581, 2008.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, 2nd ed., Wiley, Hoboken, NJ, USA, 1203 pp., 2006.
Sharma, H. D., Jervis, R. E., and Wong, K. Y.: Isotopic exchange reactions in nitrogen oxides, J. Phys. Chem., 74, 923–933, https://doi.org/10.1021/j100699a044, 1970.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Thiemens, M. H.: Mass-Independent Isotope Effects in Planetary Atmospheres and the Early Solar System, Science, 283, 341–345, https://doi.org/10.1126/science.283.5400.341, 1999.
Thiemens, M. H. and Heidenreich, J. E.: The Mass-Independent Fractionation of Oxygen: A Novel Isotope Effect and Its Possible Cosmochemical Implications, Science, 219, 1073–1075, https://doi.org/10.1126/science.219.4588.1073, 1983.
Tie, X., Madronich, S., Li, G., Ying, Z., Zhang, R., Garcia, A. R., Lee-Taylor, J., and Liu, Y.: Characterizations of chemical oxidants in Mexico City: A regional chemical dynamical model (WRF-Chem) study, Atmos. Environ., 41, 1989–2008, https://doi.org/10.1016/j.atmosenv.2006.10.053, 2007.
Topin, C., Mouthuy, L., Colomer, C., and Chauvin, P.: Rapport de l'Evaluaton Environnementale Stratégique du PCAET 2020–2030 de Grenoble Alpes Métropole, I Care & Consult, Grenoble Alpes Metrople, 2019.
Urey, H. C.: The thermodynamic properties of isotopic substances, J. Chem. Soc., 562–581, https://doi.org/10.1039/JR9470000562, 1947.
Velasco, E., Márquez, C., Bueno, E., Bernabé, R. M., Sánchez, A., Fentanes, O., Wöhrnschimmel, H., Cárdenas, B., Kamilla, A., Wakamatsu, S., and Molina, L. T.: Vertical distribution of ozone and VOCs in the low boundary layer of Mexico City, Atmos. Chem. Phys., 8, 3061–3079, https://doi.org/10.5194/acp-8-3061-2008, 2008.
Vicars, W. C. and Savarino, J.: Quantitative constraints on the 17O-excess (Δ17O) signature of surface ozone: Ambient measurements from 50∘ N to 50∘ S using the nitrite-coated filter technique, Geochim. Cosmochim. Ac., 135, 270–287, https://doi.org/10.1016/j.gca.2014.03.023, 2014.
Villena, G., Kleffmann, J., Kurtenbach, R., Wiesen, P., Lissi, E., Rubio, M. A., Croxatto, G., and Rappenglück, B.: Vertical gradients of HONO, NOx and O3 in Santiago de Chile, Atmos. Environ., 45, 3867–3873, https://doi.org/10.1016/j.atmosenv.2011.01.073, 2011.
Walters, W. W. and Michalski, G.: Ab initio study of nitrogen and position-specific oxygen kinetic isotope effects in the NO + O3 reaction, J. Chem. Phys., 145, 224311, https://doi.org/10.1063/1.4968562, 2016.
Walters, W. W., Tharp, B. D., Fang, H., Kozak, B. J., and Michalski, G.: Nitrogen isotope composition of thermally produced NOx from various fossil-fuel combustion sources, Environ. Sci. Technol., 49, 11363–11371, https://doi.org/10.1021/acs.est.5b02769, 2015.
Walters, W. W., Fang, H., and Michalski, G.: Summertime diurnal variations in the isotopic composition of atmospheric nitrogen dioxide at a small midwestern United States city, Atmos. Environ., 179, 1–11, https://doi.org/10.1016/j.atmosenv.2018.01.047, 2018.
Williams, E. L. and Grosjean, D.: Removal of atmospheric oxidants with annular denuders, Environ. Sci. Technol., 24, 811–814, https://doi.org/10.1021/es00076a002, 1990.
Young, G. L.: NOx formation in rotary kilns producing cement clinker applicable NOx control techniques and cost effectiveness of these control techniques, in: IEEE-IAS/PCS Cement Industry Technical Conference, 7–9 May 2002, Piscataway, NJ, USA, 239–254, https://doi.org/10.1109/CITCON.2002.1006510, 2002.
Yu, Z. and Elliott, E. M.: Novel Method for Nitrogen Isotopic Analysis of Soil-Emitted Nitric Oxide, Environ. Sci. Technol., 51, 6268–6278, https://doi.org/10.1021/acs.est.7b00592, 2017.
Zeldovich, Y. B.: The Oxidation of Nitrogen in Combustion and Explosions, Acta Physicochim. Acad. Sci. USSR, 21, 577–628, 1946.
Zong, Z., Wang, X., Tian, C., Chen, Y., Fang, Y., Zhang, F., Li, C., Sun, J., Li, J., and Zhang, G.: First Assessment of NOx Sources at a Regional Background Site in North China Using Isotopic Analysis Linked with Modeling, Environ. Sci. Technol., 51, 5923–5931, https://doi.org/10.1021/acs.est.6b06316, 2017.
- Abstract
- Introduction
- Materials and methods
- Atmospheric observations and multi-isotopic measurements
- Discussion of the multi-isotopic composition of atmospheric NO2
- Conclusion
- Appendix A: Isotopic standards and calibration
- Appendix B: Sample's isotopic stability
- Appendix C: Deriving the N isotopic fractionation from isotopic exchange and the extended Leighton cycle
- Appendix D: Kinetic data used
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Materials and methods
- Atmospheric observations and multi-isotopic measurements
- Discussion of the multi-isotopic composition of atmospheric NO2
- Conclusion
- Appendix A: Isotopic standards and calibration
- Appendix B: Sample's isotopic stability
- Appendix C: Deriving the N isotopic fractionation from isotopic exchange and the extended Leighton cycle
- Appendix D: Kinetic data used
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement