the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Aug 2020
| 21 Aug 2020
Evaluation of the chemical composition of gas- and particle-phase products of aromatic oxidation
Archit Mehra
Yuwei Wang
Jordan E. Krechmer
Andrew Lambe
Francesca Majluf
Melissa A. Morris
Michael Priestley
Thomas J. Bannan
Daniel J. Bryant
Kelly L. Pereira
Jacqueline F. Hamilton
Andrew R. Rickard
Mike J. Newland
Harald Stark
Philip Croteau
John T. Jayne
Douglas R. Worsnop
Manjula R. Canagaratna
Aromatic volatile organic compounds (VOCs) are key anthropogenic pollutants emitted to the atmosphere and are important for both ozone and secondary organic aerosol (SOA) formation in urban areas. Recent studies have indicated that aromatic hydrocarbons may follow previously unknown oxidation chemistry pathways, including autoxidation that can lead to the formation of highly oxidised products. In this study we evaluate the gas- and particle-phase ions measured by online mass spectrometry during the hydroxyl radical oxidation of substituted C9-aromatic isomers (1,3,5-trimethylbenzene, 1,2,4-trimethylbenzene, propylbenzene and isopropylbenzene) and a substituted polyaromatic hydrocarbon (1-methylnaphthalene) under low- and medium-NOx conditions.
A time-of-flight chemical ionisation mass spectrometer (ToF-CIMS) with iodide–anion ionisation was used with a filter inlet for gases and aerosols (FIGAERO) for the detection of products in the particle phase, while a Vocus proton-transfer-reaction mass spectrometer (Vocus-PTR-MS) was used for the detection of products in the gas phase. The signal of product ions observed in the mass spectra were compared for the different precursors and experimental conditions. The majority of mass spectral product signal in both the gas and particle phases comes from ions which are common to all precursors, though signal distributions are distinct for different VOCs. Gas- and particle-phase composition are distinct from one another. Ions corresponding to products contained in the near-explicit gas phase Master Chemical Mechanism (MCM version 3.3.1) are utilised as a benchmark of current scientific understanding, and a comparison of these with observations shows that the MCM is missing a range of highly oxidised products from its mechanism.
In the particle phase, the bulk of the product signal from all precursors comes from ring scission ions, a large proportion of which are more oxidised than previously reported and have undergone further oxidation to form highly oxygenated organic molecules (HOMs). Under the perturbation of OH oxidation with increased NOx, the contribution of HOM-ion signals to the particle-phase signal remains elevated for more substituted aromatic precursors. Up to 43 % of product signal comes from ring-retaining ions including HOMs; this is most important for the more substituted aromatics. Unique products are a minor component in these systems, and many of the dominant ions have ion formulae concurrent with other systems, highlighting the challenges in utilising marker ions for SOA.
- Article
(1814 KB) - Full-text XML
-
Supplement
(1145 KB) - BibTeX
- EndNote





Volatile organic compounds (VOCs) are emitted from both natural and anthropogenic sources, and their oxidation in the troposphere is important for reactive chemistry leading to ozone (Atkinson, 2000; Derwent et al., 1998) and secondary organic aerosol (SOA) formation (Ziemann et al., 2012). This can have severe air quality, environmental and health impacts (Hallquist et al., 2009). Globally, VOCs such as isoprene have been estimated to be the largest contributors to SOA formation (Guenther et al., 1995; Simpson et al., 1999). However, in urban and industrialised areas, other sources of VOCs have become increasingly important (Borbon et al., 2013; Karl et al., 2009; Liu et al., 2008b; McDonald et al., 2018). Aromatics are one such class of VOCs which are emitted from fuel use, biomass burning, solvent use and industry (Corrêa and Arbilla, 2006; Liu et al., 2008a).
In the troposphere, aromatic hydrocarbons primarily react with the hydroxyl radical (OH; Calvert, 2002). Detailed mechanisms of aromatic oxidation have been generated previously from experimental work in simulation chambers (Calvert, 2002), and this chemistry has been included in chemical mechanisms such as the Master Chemical Mechanism (MCM: http://mcm.york.ac.uk, last access: 11 August 2020) and the Statewide Air Pollution Research Center (SAPRC; of the University of California, Riverside) mechanism, whose primary goal is to describe ozone formation (Bloss et al., 2005; Carter, 1988; Carter and Heo, 2013; Metzger et al., 2008; Suh et al., 2003; Volkamer et al., 2002). The aromatic oxidation mechanism in the MCM has not been updated since 2005 (Jenkin et al., 2003, Bloss et al., 2005), and this can only be achieved once sufficient evidence of the rate, branching ratios and product distributions has been obtained. Explicit chemical mechanisms often lead to large discrepancies between modelled and measured SOA formation (Johnson et al., 2006; Khan et al., 2017; Volkamer et al., 2006), which may be associated with both uncertainty in the formation pathways of semi-volatile and low-volatility species and a poor representation of aerosol volatility and partitioning.
SOA is made up of thousands of oxidised organic compounds which each exist at very low, often sub-parts per billion levels in the atmosphere (Hallquist et al., 2009). This makes measurement of generated products challenging even under controlled laboratory conditions. However, the increasing availability of high-resolution instrumentation that enables real-time detection of thousands of molecules (Hallquist et al., 2009) has allowed more detailed chemical characterisation of the gas- and particle-phase VOC-oxidation products. Chemical ionisation is extremely powerful in this area due to soft ionisation schemes, such as I−, and H3O+, that form clusters with the types of molecules that sit within atmospherically relevant oxidation ranges (Isaacman-Vanwertz et al., 2017, 2018). These techniques have enabled the identification of many new oxidation products, thereby supporting more detailed mechanistic studies (Molteni et al., 2018; Wang et al., 2015).
Recent mechanistic studies have highlighted the importance of previously unknown pathways, such as autoxidation (Molteni et al., 2018; Wang et al., 2017) and multigenerational OH attack (Garmash et al., 2020; Zaytsev et al., 2019), for the formation of highly oxygenated organic molecules (HOMs) which may contribute to new particle formation (NPF) and rapid SOA formation and growth. Recent work has shown that autoxidation may play a major role in SOA formation at regional and global scales due to the potential for autoxidation of aromatics to continue to be favourable even under higher NO conditions (Crounse et al., 2013; Pye et al., 2019). Furthermore, reductions in NOx in the USA and Europe are enabling autoxidation to play an increasing role under urban conditions (Praske et al., 2018). In order to ascertain the importance of aromatic SOA, detailed laboratory characterisation of SOA composition and aromatic oxidation mechanisms for a broad suite of relevant aromatics is required alongside ambient measurements.
Gas-phase chemistry and SOA formation have been well studied for the most ubiquitous aromatic VOCs, namely benzene and toluene (Bruns et al., 2016; Molteni et al., 2018; Ng et al., 2007; Suh et al., 2003; Volkamer et al., 2002). However, more recently the importance of substituted aromatic hydrocarbons in urban areas has been highlighted for both ozone and SOA formation (Kansal, 2009; Monod et al., 2001). Furthermore, substituted aromatics have high OH reactivities and aerosol mass yields (Odum et al., 1996), so despite existing at generally smaller concentrations than their less substituted homologues, reductions in their emissions may be essential for improving urban air quality (Von Schneidemesser et al., 2010). Uncertainty exists in the emission inventories of C7–C9 aromatics, and this could be important when considering the differences between ozone and SOA formation in developed and developing megacities, particular with a lack of speciation of aromatics in fuels globally (Borbon et al., 2013). Recent work has begun to fill this gap in the chemical knowledge, and it can be seen that though other pollutants have been decreased in newer fuels, an increasing trend is being observed in the levels of aromatics in cities such as Beijing (Peng et al., 2017). In urban environments, the impact of this is not easy to constrain, particularly as these emissions can have complex interactions with biogenic VOCs (Kari et al., 2019), impacting upon SOA composition and yields (McFiggans et al., 2019).
C9 aromatics are of particular interest in urban areas, with major emission sources including unburnt exhaust emissions (Corrêa and Arbilla, 2006; Na et al., 2005), evaporative losses (Miracolo et al., 2012; Rubin et al., 2006) and solvent use (Zhang et al., 2013). Another group of aromatics which are increasingly a focus of research are polyaromatic hydrocarbons (PAHs), which can be emitted from vehicular emissions (Miguel et al., 1998) and biomass burning (Bruns et al., 2016). With < 50 % of SOA products identified (Hamilton et al., 2005; Sato et al., 2007), it is important to build up a detailed chemical insight in order to improve mechanistic understanding of which chemical pathways are leading to SOA formation (Atkinson and Arey, 2007). Previous studies of HOMs have focused on either very different precursors (Mentel et al., 2015) or on a small range with limited comparison between isomers (Molteni et al., 2018).
In this study we focus on the photochemical oxidation of substituted aromatic hydrocarbons which have recently been found to form HOMs through rapid intramolecular autoxidation reactions (Wang et al., 2017). Though VOCs are often arbitrarily grouped in models (Carter, 2000; Yarwood et al., 2005), recent work has found that the location, number and isomeric structure of substituent groups on the benzene ring can have implications for SOA yield, chemical composition and physical properties (Li et al., 2016). In this study we investigate a range of isomers of C9 aromatics to evaluate this effect in more detail. Of the aromatics studied in these experiments, molecular understanding of SOA products is most well developed for the trimethylbenzene isomers (Huang et al., 2014; Li and Wang, 2014; Sato et al., 2012, 2019). Though other aromatics have been studied, these have either lacked molecular-level detail or focused on gas-phase oxidation and lacked detailed characterisation of SOA (Chan et al., 2009; Li et al., 2016; Molteni et al., 2018; Ping, 2013). Here we present a detailed characterisation of the chemical composition of the oxidation products of a range of substituted aromatics (isomers of C3-substituted aromatics and a PAH), namely propylbenzene (PROPBENZ), isopropylbenzene (IPRBENZ), 1,3,5-trimethylbenzene (TMB135), 1,2,4-trimethylbenzene (TMB124) and a substituted PAH, 1-methylnaphthalene (METHNAP). We evaluate the trends in chemistry across different isomers under low- and medium-NOx conditions and discuss the relative importance of different oxidation pathways.
2.1 Oxidation of VOCs
We conduct our experiments in a potential aerosol mass oxidation flow reactor (PAM-OFR; Aerodyne Research Inc.; Lambe et al., 2011), affording short experimental timescales and the ability to generate consistent and reproducible oxidation conditions. Aromatic VOCs were injected into a carrier gas of synthetic air through the use of an automated syringe pump either neat or diluted in carbon tetrachloride (CCl4). Their concentrations (Table 1) were optimised to obtain similar aerosol mass concentrations in the different experiments. In the OFR, OH, HO2 and NO radicals were generated via O2 + H2O + N2O photolysis at 254 and 185 nm via the following reactions:
Table 1VOC concentrations during single experiments for each precursor. Note: ppb – parts per billion.
All experiments were carried out at a temperature of 26 ∘C, relative humidity of 35 % and constant gas flow of 10 SLM through the OFR, including the injection of ∼ 3 % N2O at the inlet to generate NO in a subset of experiments (Lambe et al., 2017; Peng et al., 2018). Under these conditions, the estimated OH exposure in the OFR was in the range of 1.5 × 1012–1.7 × 1012 . These elevated exposures are not attainable in conventional environmental chambers and may prove relevant for understanding photochemistry in urban areas with high oxidation capacities such as those observed in Chinese megacities (Lu et al., 2019; Tan et al., 2019). For example, OH concentrations in Beijing can reach as high as ∼ 1 × 107 molecules cm−3 (Bryant et al., 2019), an order of magnitude higher than the global average of ∼ 1 × 106 molecules cm−3 (Lelieveld et al., 2016), which is used to relate OH exposure in an OFR to days of equivalent atmospheric oxidation (Lambe et al., 2015). This changes what would be 11 d of equivalent atmospheric oxidation in the OFR under global average OH concentrations to just over 1 d of oxidation in Beijing.
In experiments where N2O was added to the OFR, the NO:HO2 concentration ratio was approximately 0.5, as calculated using an adapted version of the OFR photochemical box model described in Li et al. (2015) and Peng et al. (2015). The use of non-tropospheric radiation to generate radicals in the OFR can result in the photolysis of reactants; however, operational conditions can be optimised to ensure OH loss is the dominant removal process of the VOCs (Peng and Jimenez, 2020). In this study, under the conditions used, > 98 % of the loss of VOCs can be attributed to reaction with OH. This is based on calculations under the relevant conditions for 1,3,5-trimethylbenzene, 1,2,4-trimethylbenzene and 1-methylnaphthalene for which photolysis rate data exist. The relevant photolysis data did not exist for propylbenzene and isopropylbenzene, and thus, to the extent that results for trimethylbenzene (TMB) and methylnaphthalene are also applicable to propyl and isopropylbenzene, we anticipate that their loss is also dominated by OH oxidation. The photolysis of products or intermediates are extremely difficult to fully clarify, as discussed in Peng and Jimenez (2020), and thus the practical approach taken here is to optimise the OH oxidation to ensure that OH loss is the dominant pathway for the VOCs themselves.
Between experiments, the OFR was flushed with humidified synthetic air at full lamp power for 12–36 h until the particle mass that was generated was reduced to background concentrations measured by a scanning mobility particle sizer (SMPS) and an aerosol mass spectrometer (AMS). During this time, the walls were cleaned of the semi-volatiles and precursors measured in the gas phase by iodide ToF-CIMS and Vocus-PTR-MS. Backgrounds were determined under lights off or on and with or without precursor injection conditions and spectra recorded with all of the instrumentation. The conditions discussed herein are low NOx ([NO] < 0.1 ppb) and medium NOx ([NO] > 1 ppb, [NO]: [HO2] = 0.5).
2.2 Measurements
2.2.1 FIGAERO–I-CIMS
A time-of-fight chemical ionisation mass spectrometer (Lee et al., 2014) using an iodide ionisation system was coupled with a filter inlet for gases and aerosols (FIGAERO; Lopez-Hilfiker et al., 2014) for the detection of particle-phase composition (herein I-CIMS). Particle mass concentrations were monitored using a TSI scanning mobility particle sizer, and collection time on the FIGAERO filter was varied to ensure a comparable mass of aerosol in each sample for the different precursors. The FIGAERO thermal desorption cycle consisted of a 15 min temperature ramp to 200 ∘C, held at that temperature for 10 min and then cooled down over 15 min.
The gas-phase inlet consisted of a piece of 0.5 m length 1/4” inner diameter perfluoroalkoxy alkene (PFA) tubing from which the I-CIMS subsampled 2 slpm. The aerosol-phase inlet consisted of 0.5 m stainless steel through which 2 slpm was pulled over a Teflon filter. The I− reagent ion was made by flowing N2 over a permeation tube containing methyl iodide (CH3I), followed by ionisation through a polonium-210 ionisation source. This flow enters an ion molecule reaction region (IMR) which was maintained at a pressure of 100 mbar using an SSH-112 pump fitted with a pressure controller. The IMR pressure was automatically maintained at a set value, using an actuated valve connected to the pump line.
2.2.2 Vocus-PTR
A Vocus proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS; Vocus hereafter) was used in this experiment to measure gas-phase organic compounds (Krechmer et al., 2018). Equipped with a newly designed focusing ion–molecule reactor (FIMR), the Vocus was able to measure organics with a wide range of volatilities (Riva et al., 2019). A short-segmented quadrupole (SSQ) pressure of 2.0 mbar and an axial voltage of 420 V were used, corresponding to an E∕N ratio of ∼ 100 Td, which was lower than typical to reduce the fragmentation of labile semi-volatile organic compounds (SVOCs; Krechmer et al., 2018). A total sample flow of 2.2 LPM was maintained by a pump to minimise delay times due to the inlet (Pagonis et al., 2017), of which approximately 125 sccm was sampled into the FIMR through the PEEK tube. To reduce inlet blockages from the high mass loadings of aerosols from the OFR, a filter was connected just before the entrance of the Vocus, which may have reduced the transmission of some SVOCs and low-volatility organic compounds (LVOCs). Background checks by injections of clean air and calibrations by injections from a multicomponent standard cylinder (Apel-Riemer Environmental, Inc.) were performed every 2 h in this experiment. With a resolving power of 12000 Th Th−1 at 200 Th, molecular formula could be assigned to most of detected ions with a mass accuracy better than 3 ppm.
2.2.3 Orbitrap liquid chromatography–mass spectrometry (LC–MS)
Ultra-performance liquid chromatography ultrahigh-resolution mass spectrometry (Dionex 3000; Orbitrap Q Exactive; Thermo Fisher Scientific, USA) was used for the sample analysis. Compound separation was achieved using a reverse-phase C18 column (Accucore; Thermo Fisher Scientific, USA) with the following dimensions: 100 mm (length) × 2.1 mm (width) and 2.6 µm particle size. The column was heated to 40 ∘C during analysis. The solvent composition consisted of water with 0.1 % (v∕v) of formic acid (A) and methanol (B; optima liquid chromatography–mass spectrometry (LC–MS) grade; Thermo Fisher Scientific, USA). Gradient elution was used, starting at 90 % (A) with a 1 min post-injection hold, decreasing to 10 % (A) at 26 min, returning to the starting mobile-phase conditions at 28 min, with a 2 min hold to re-equilibrate the column (total runtime = 30 min). The flow rate was set to 0.3 mL min−1, with a sample injection volume of 2 µL. Samples were stored in a temperature-controlled autosampler tray during analysis, which was set to 4 ∘C. The mass spectrometer was operated in negative and positive ionisation modes with a scan range of m∕z 85 to 750. Heated electrospray ionisation was used with the following parameters: capillary and auxiliary gas temperature of 320 ∘C, sheath gas flow rate of 70 (arbitrary – arb.) and auxiliary gas-flow rate of 3 (arb.). Tandem mass spectrometry was performed using higher energy collision dissociation with a normalised collision energy of 65 and 115. Data were analysed using the Compound Discoverer software (version 2.1; Thermo Fisher Scientific, USA). Full details regarding the data-processing methodology can be found in Pereira et al. (2020). Briefly, molecular formulae assignments were allowed unlimited C, H, and O atoms, up to 2 S atoms, and 5 N atoms plus > 2 Na atoms and 1 K atom in the positive ionisation mode. Only compounds with a mass error < 3 ppm and a signal-to-noise ratio > 3, a hydrogen-to-carbon ratio of 0.5 to 3 and an oxygen-to-carbon ratio of 0.05 to 2 were included in the data set. Instrument artefacts were removed from the sample data if the sample-to-artefact peak area ratio > 3.
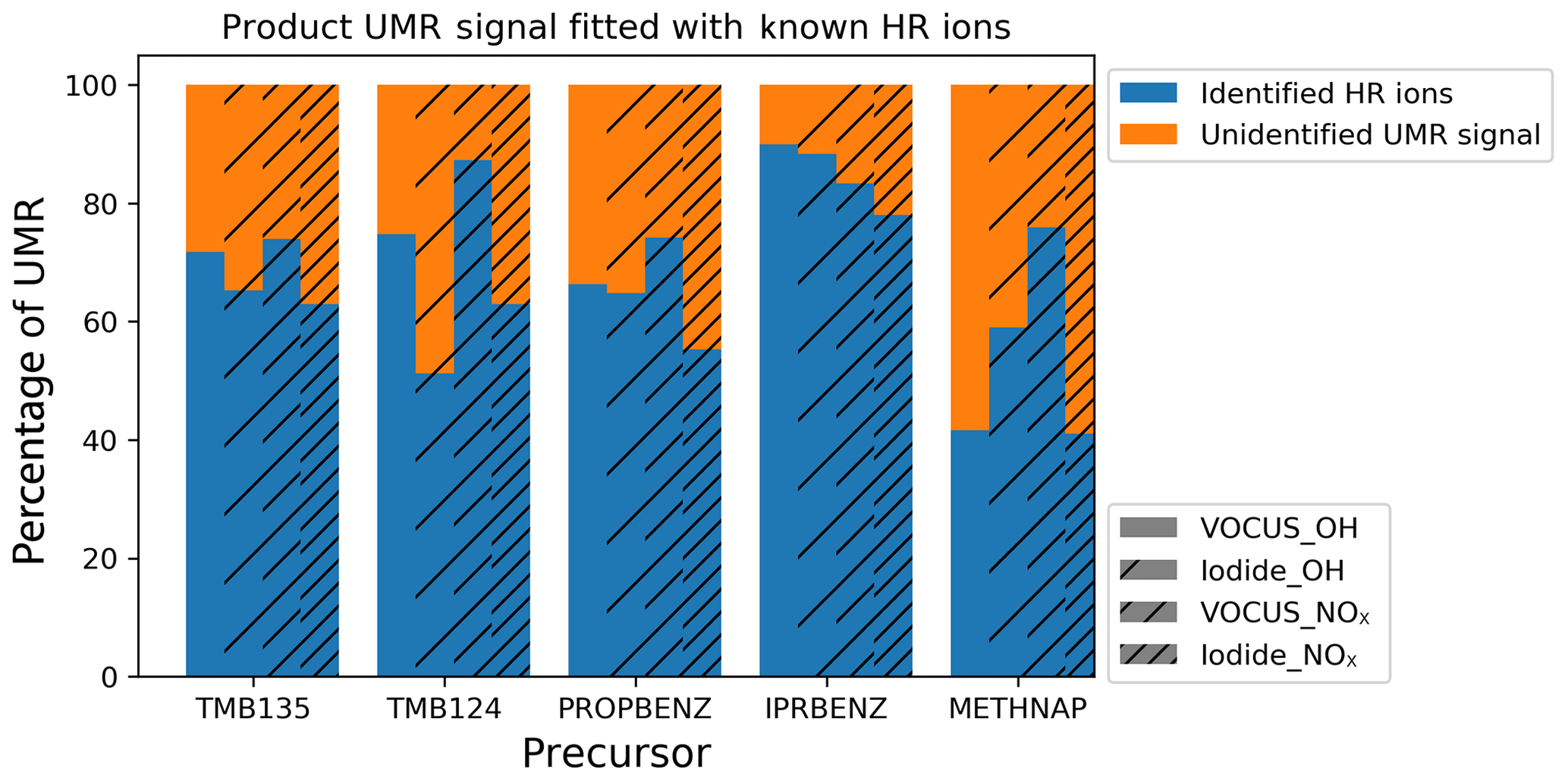
Figure 1Proportion of the unit mass resolution (UMR) signal fitted by identified high-resolution (HR) ions for all precursors and experimental conditions from I-CIMS and Vocus.
2.3 Data analysis
Data analysis of I-CIMS and Vocus was performed using the Tofware package (version 3.1.0) running in the Igor Pro (WaveMetrics, USA) environment (Stark et al., 2015). Time-of-flight values were converted to mass-to-charge ratios in the I-CIMS using I-, and . The instrument was operated at a ∼ 8000 Th Th−1 resolving power. Several impurity ions, ions from ubiquitous VOCs and ions from oxidation of products were used for the mass calibration of Vocus, which are all clear and unique peaks in the mass spectrum, including C2H5O+ (acetaldehyde; 45.033491 Th), (hydrate ion of acetic acid; 79.038971 Th), (common oxidation product of all precursors in our experiments; 129.054621 Th), (common oxidation product of all precursors in our experiments; 139.075356 Th), and (decamethylcyclopentasiloxane – D5; contaminations from personal care products; Coggon et al., 2018; 371.101233 Th). It should be noted that glyoxal, a major product from aromatic oxidation is detected by the Vocus as , which is dominated in these experiments by protonated acetone. The sensitivity of glyoxal in the PTR is almost one-tenth of that of acetone, and furthermore, can also be influenced by fragmentation and is thus not included in the analysis herein (Stönner et al., 2017). Further analysis was carried out in custom Python 3 procedures using the packages of Pandas, Matplotlib and NumPy.
In this work, the chemical composition of the gas-phase species generated in all the experiments is characterised by Vocus. While FIGAERO is capable of providing information about both gas and particle phases, the gas-phase I-CIMS spectra obtained during the medium-NOx experiments was complicated by the presence of high nitric acid formed in the OFR from the N2O precursor (Lambe et al., 2017; Peng et al., 2018). High nitric acid depletes the I− reagent ion to form , which subsequently acts as an additional reagent ion and complicates the interpretation of the observed CIMS spectrum. Thus, the gas-phase FIGAERO measurements were deemed unsuitable for the comparison of medium- and low-NOx conditions, and the high-resolution (HR) analysis of the I-CIMS spectra has been carried out only for the particle-phase I-CIMS–FIGAERO data obtained in all the experiments for which nitric acid levels were lower, resulting in pure iodide reagent ion chemistry. While this setup provides thermal desorption profiles which have previously been used to draw conclusions about the volatility distribution of ions (Lopez-Hilfiker et al., 2016b; Schobesberger et al., 2018; Stark et al., 2017), in this study we instead integrate the desorption profiles in order to compare the overall composition of the different precursors.
The observed mass spectra were first mass calibrated (10 ppm mass accuracy), and then the observed high-resolution peaks were fitted using a multipeak fitting algorithm. The exact mass of the multiple peaks are then matched with the most likely elemental formula. A consistent approach was taken for the high-resolution (HR) peak identification in all experiments. To eliminate bias, separate peak lists were generated for each precursor and experimental condition. Mass ranges were selected in order to focus on the more oxidised products, namely m∕z 200–500 for I-CIMS and m∕z 75–300 for Vocus (except m∕z 79–81 and 117–123 for all precursors, and 143–144 only for methylnaphthalene; the range was dominated by the signals of the precursor, solvent, hydrated ion of acetic acid and their isotopes). It should be noted that this mass range omits methylglyoxal, a dominant product of aromatic oxidation, which was observed by Vocus in all experiments. The mass spectra in this range were fitted with peaks in the order of descending signal contribution until the signal to noise made peak identification impossible. Predicted oxidation products for each aromatic precursor were obtained from the MCM (version 3.3.1) using the “extract” function on the web portal (http://mcm.york.ac.uk/, last access: 11 August 2020). Fitted unknown peaks were firstly compared with this list of MCM products and then assigned through the use of the iterative peak assignment method, which assigns unknown peaks to elemental formulae in a reference list if they are in the correct position (Stark et al., 2015).
Quantification of highly oxidised species measured by I-CIMS is challenging due to the lack of availability of standards for many of the observed products. Previous attempts at quantification have used functional group dependencies, collision limit sensitivities or those derived from ion-adduct declustering scans (Lopez-Hilfiker et al., 2016a). Experimental limitations exist in the use of these techniques, meaning that quantification remains a challenge. For PTR, it has previously been reported that the H3O+ capture rate, and therefore the sensitivity of PTR to all ions lies within a very narrow distribution, unlike iodide sensitivity which can vary by several orders of magnitude. Due to the unusually low E∕N value in the Vocus–FIMR used in these experiments, absolute contributions of certain ions are challenging to ascertain due to the formation of both protonated molecules and hydrate cluster ions of the same molecule alongside fragmentation. Unpicking these is not trivial, though the impact of hydrate formation upon the overall product distribution is not significant to the analysis carried out herein.
Considering these factors, we did not correct for sensitivity and used raw ion signal for both I-CIMS and Vocus measurements. Gas-phase I-CIMS observations were used for cross calibration between I-CIMS and Vocus under low-NOx conditions (Fig. S2 in the Supplement), showing a generally consistent calibration factor for ions containing 1–6 oxygen atoms, indicating that these results are not influenced by vastly different I-CIMS sensitivities. This is further confirmed by similar ion signal distributions observed between both the I-CIMS and Vocus. Comparison of I-CIMS and Vocus data (Fig. S1) shows that 30 %–60 % of the I-CIMS signal is from ions with chemical formulas which are also observed in the Vocus. It should be noted that we have not attempted to relate ion signals to yields, and thus the comparison of ion signals in this study should not be assumed to be related to product yields derived from models such as those implementing mechanisms including the MCM.
The signal fitted by identified HR ions was compared with the total unit mass resolution (UMR) signal, excluding ions known to be unrelated to the experiment using OFR and instrument backgrounds. This comparison shows that between 40 % and 90 % of the observed signal has been assigned elemental formulae, with the residual signal (orange) of unidentified signal either being the result of poor signal to noise or complex overlapping peaks, making peak identification infeasible. This signal may also have some contribution from ion fragments in the Vocus and from the thermal decomposition of desorbed products for the I-CIMS data. Analysis herein has been carried out for the blue proportion of signal for which elemental formulae have been selected.
3.1 Overview of gas- and particle-phase oxidation products
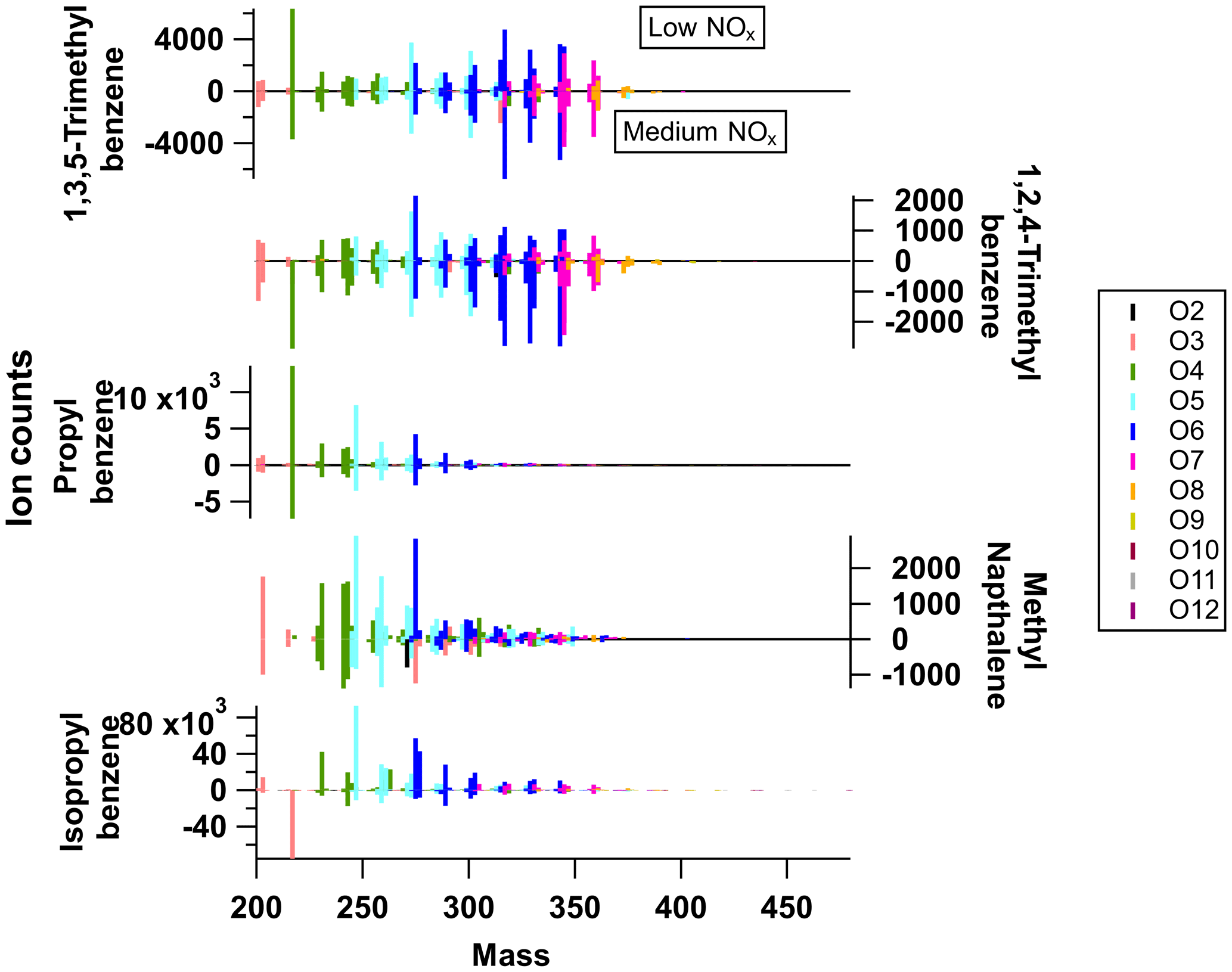
Figure 2Average mass spectra of particle-phase measurements from I-CIMS for all precursors under low-NOx conditions (upwards) and medium-NOx conditions (downwards; negative values correspond to the magnitude of the positive signal).
The gas-phase (low-NOx) mass spectra are dominated by small non-HOM oxidation products, with most of the signal in the C2–C4 range. A similar distribution is seen for the gas-phase (medium-NOx) mass spectra, although 1,3,5-trimethylbenzene and n-propylbenzene show an increase in the contribution of the signal at higher C numbers, which will be discussed below. As expected, the particle-phase mass spectra show a higher fraction of signal at higher C numbers and an increase in the relative proportion of HOM signal. There are marked differences between the distributions of particle-phase products, although there are many common ion formulas observed, which will be discussed in Sect. 4.1.2. The particle-phase mass spectra obtained for each precursor are also included in Fig. 2, with the particle-phase (low-NOx) spectra shown pointing upwards and the particle-phase (medium-NOx) spectra pointing downwards. The ion peaks have been coloured according to the number of O atoms found in the molecular formula. The two TMB isomers have very similar mass spectra. These two highly substituted ring species have a larger proportion of very oxidised ions (O6–O8) than the other precursors, whose spectra are dominated by compounds with O3–O5. N-containing ions do not contribute significantly to product signal under medium-NOx conditions (<10 %; except for 1-methylnaphthalene).
To compare the carbon skeleton of the oxidation products, the ion intensity of all ions with the same carbon number in the average mass spectra have been summed and are presented for each precursor in Fig. 3 for the gas phase and particle phases. These data have been further split into non-HOMs (O≤5) and HOMs (O≥6), based on the definition in Bianchi et al., 2019. Comparison shows clear differences in the distribution of products from different precursors and, in particular, between isomers of the C9 aromatics.
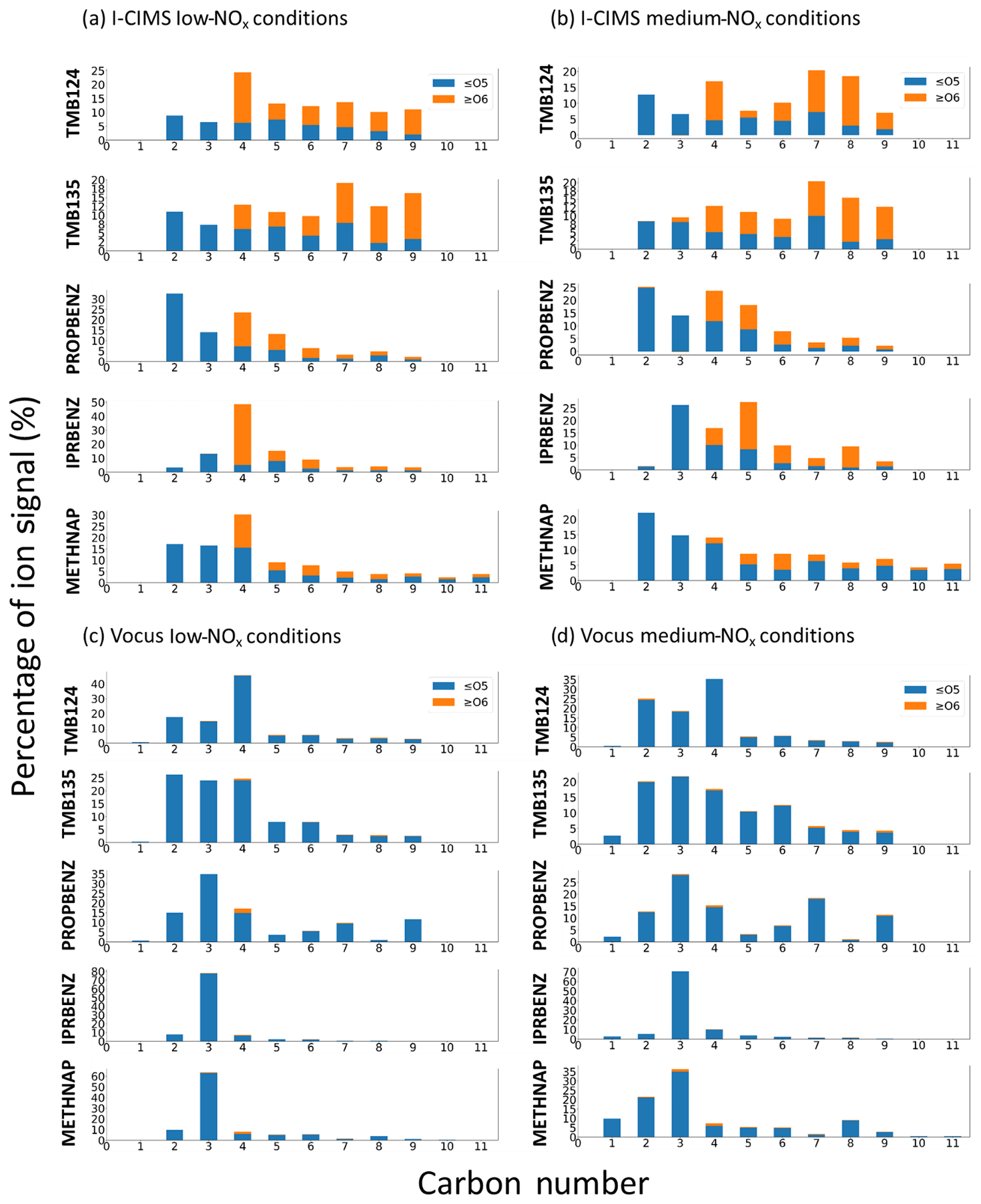
Figure 3Signal distribution across carbon numbers and coloured by the proportion of HOM signals for (a) I-CIMS particle phase under low-NOx conditions, (b) I-CIMS particle phase under medium-NOx conditions, (c) Vocus gas phase under low-NOx conditions and (d) Vocus gas phase under medium-NOx conditions. Only signal contributions up to the precursor carbon numbers are included, namely C9 for TMB124, TMB135, PROPBENZ and IPRBENZ and C11 for METHNAP, to capture both the ring-retaining and ring scission products for mechanistic discussions below. There is very little signal above C9 for the C9-aromatic precursors.
Through comparison of these precursors under similar oxidation conditions, we can see that the lumping of VOC isomers may not be valid in the context of SOA formation. It is possible that lumping VOCs based on single and multi-substituents could prove useful for model parameterisations. Furthermore, the evident HOM formation under elevated NOx conditions is important to consider for effective policy implementation. Detailed trends in gas and particle composition for each precursor will be discussed in the following sections.
3.2 SOA product composition for individual VOCs
3.2.1 1,2,4-Trimethylbenzene
In the gas-phase (low-NOx) mass spectrum (Fig. 3a), over 40 % of the signal comes from C4 ions, with the two largest ions being C4H6O2 and C4H8O3. In the particle-phase (low-NOx) mass spectrum, the signal is distributed more broadly across ions of different carbon numbers. The largest contribution is also from the C4 ions (25 %), with the C5–C9 all contributing ∼ 15 % each. The majority of the C4 product signal comes from C4H4O6, with the other largest single ions being C5H6O5, C7H10O6 and C9H12O6.
Under medium-NOx conditions, the dominant gas-phase ions are consistent with those produced under low-NOx conditions, and the product signal shows a broadly similar distribution to the medium-NOx conditions with a small reduction in the fraction of C4 product ions and an increase in C2 product signal, which can be attributed to an increase in C2H4O3. A small contribution to the signal comes from N-containing ions (3.8 %). In the particle phase, the mass spectrum and C number distribution is fairly similar to the low-NOx conditions, although there is a doubling in the fraction of HOMs with C7–C8. In addition, 9.3 % of the particle product signal is from N-containing ions; specific ions have not been included in further analysis due to potential contributions from thermal decomposition.
3.2.2 1,3,5-Trimethylbenzene
The most abundant contribution to the gas-phase (low-NOx) mass spectrum comes from an almost equal split between C2, C3 and C4 ions (together ∼ 75 % of product signal), with the most dominant ions being C4H6O2 and C3H6O2. The spread of signal in the particle-phase (low-NOx) mass spectrum is across ions with a broader range of carbon numbers, with C7–C9 HOM ions contributing ∼ 40 % of product signal. The most dominant of these ions are C7H10O6 and C9H12O6.
In the gas-phase medium-NOx mass spectrum, there is a shift, with an increased contribution from ions with larger carbon numbers than under the low-NOx conditions. The most dominant gas-phase ions were C4H6O2, C3H6O2, C2H4O3 and C5H6O3. Nitrogen-containing ions contribute only 2.8 % of the observed signal. In the particle-phase (medium-NOx) mass spectrum the most abundant ions are consistent with those under low-NOx conditions. There is a 7.8 % contribution to the product signal from nitrogen-containing ions.
3.2.3 Propylbenzene
Similar to the TMB isomers, the majority of the gas-phase (low-NOx) mass spectrum signal comes from C2–C4 ions, but in this case the C3 ions have the highest intensity (C3H8O2 and C3H6O2). A total of 10 % of the signal contribution is from C9 ions, with the most dominant ion being C9H10O, most likely propiophenone, which is the first-generation ketone from the OH attack on the n-propyl chain (Bloss et al., 2005). In contrast, the bulk of signal in the particle-phase (low-NOx) mass spectrum exists at < C5, with a smaller contribution from HOM ions than the TMB isomers. The majority of ions observed correspond to predicted oxidation products in the MCM. Abundant oxidised ions not included in the MCM include C5H6O6 and C5H6O5.

Figure 4Signal distribution across carbon numbers and coloured by the proportion of HOM signals for (a) Orbitrap negative mode and (b) Orbitrap positive mode. Only signal contributions up to the precursor carbon numbers are included, namely C9 for TMB135 and PROPBENZ and C11 for METHNAP, to capture the relevant products for mechanistic discussions below. Ions which contributed less than 0.5 % of observed signal were assumed to be background and removed.
The gas-phase (medium-NOx) mass spectrum product distribution is largely similar to low-NOx conditions; however there is an enhanced contribution from C7H6O, most likely benzaldehyde. N-containing ions contribute 3.5 % to the gas-phase product signal. In the particle-phase (medium-NOx) mass spectrum, the dominant ions are largely consistent with those observed under low-NOx conditions. N-containing ions contribute 5.8 % of the signal.
3.2.4 Isopropylbenzene
In the gas-phase (low-NOx) mass spectrum ∼ 80 % of the product signal can be attributed to C3 ions potentially formed from the scission of the isopropyl group from the ring, with the two largest ions being C3H8O2 and C3H6O2. In the particle-phase (low-NOx) mass spectrum; however, by far the largest contribution (∼ 50 %) was from highly oxygenated C4 ions, namely C4H4O6 and C4H6O6.
The gas-phase (medium-NOx) mass spectral product distribution was largely similar to low-NOx conditions, with a slightly increased contribution from higher carbon number ions. There is a 3.6 % contribution to signal from N-containing ions. The particle-phase (medium-NOx) mass spectral product distribution was strikingly different compared to low-NOx conditions, with a larger contribution from C3, C5 and higher carbon number ions, and the largest single ions were C3H6O3, C4H4O4 and C5H6O6. In particular, there is a significant contribution from C5–C8 HOM ions. Overall, there was a 2.5 % contribution to signal from N-containing ions.
3.2.5 Methylnaphthalene
In the gas-phase (low-NOx) mass spectrum, ∼ 60 % of gas-phase product signal is attributable to just a few C3 ions (C3H8O2, C3H6O2 and C3H4O3). The particle phase shows a broader spread of product signal across the carbon number range, with a significant contribution of oxidised (> O6) ions ranging from C4–C10, with the largest contribution being from C4H4O6 and C4H4O5.
The gas-phase (medium-NOx) mass spectrum showed an increased contribution from C2 and C8 ions when compared with low-NOx conditions. The most dominant ions observed were largely consistent with low-NOx conditions. Overall, N-containing ions contributed a much larger fraction of the signal in both gas (16.2 %) and particle phases (22.3 %) than for the other precursors. In the particle-phase (medium-NOx) mass spectrum, the product distribution was again similar to low-NOx conditions, with a slight shift in the signal distribution to higher carbon numbers. The most dominant ions in the particle phase were consistent with those observed under low-NOx conditions.
3.3 Comparison with Orbitrap analysis
Offline filters of SOA generated from the low-NOx oxidation of 1-methylnaphthalene, propylbenzene and 1,3,5-trimethylbenzene were analysed by Orbitrap LC–MS. The data presented show all ions above 0.5 % of the total ion signal; this was selected as a threshold to remove contributions from ions which may be contaminants. Though some ions are observed at higher carbon and oxygen numbers in Orbitrap measurements, these are not included in this analysis, and we focus on products up to the precursor carbon number as we can apply current mechanistic understanding in the interpretation of these data.
Results from both negative and positive modes are shown in Fig. 4; the negative mode shows the most similarity to the composition observed by I-CIMS, while the positive mode looks very different. The positive mode is generally more sensitive to peroxide and carbonyl species, and similar ion distributions are observed for the C9 aromatics, with a relatively larger contribution from C5 ions in the case of propylbenzene. 1-Methylnaphthalene shows a major contribution from C8–C9 products, which are minor components in I-CIMS.
The distribution of ions for 1,3,5-trimethylbenzene is similar between I-CIMS and the negative mode, with a larger relative contribution of C5–C6 ions and a smaller relative contribution of C2–C3 ions in the negative mode compared with I-CIMS. Propylbenzene also shows very similar ion distributions to I-CIMS data, with a smaller contribution from C2–C3 ions and a larger relative C7–C9 contribution in both HOM and non-HOM ions. 1-Methylnaphthalene shows significant differences between the I-CIMS and negative mode data for higher carbon numbers (C7–C9 ion contributions are small in I-CIMS but contribute significantly to negative mode), but they are largely similar for a low carbon number, with C4 ions contributing the dominant proportion of signal.
4.1 Composition
4.1.1 Comparison with the MCM
The Master Chemical Mechanism (MCM; http://mcm.york.ac.uk/, last access: 11 August 2020) is a near-explicit chemical mechanism which describes the detailed gas-phase chemical processes involved in the atmospheric degradation of a series of primary emitted VOCs. These include a large number of major-emitted anthropogenic and biogenic species and all of the C9-aromatic isomers studied in these experiments.
The construction protocol developed to allow the building of comprehensive, consistent gas-phase degradation schemes for aromatic VOCs in the MCM is given in Jenkin et al. (2003), which was subsequently updated and optimised using an extensive range of chamber experiments in 2005 by Bloss et al. (2005a, 2005b). The general philosophy behind the MCM is to directly use the most up-to-date literature available on the kinetics and products of elementary reactions relevant to VOC oxidation in order to build near-explicit representations of atmospheric degradation mechanisms. A fundamental assumption in the mechanism-construction process is that the kinetics and products of a large number of unstudied reactions can be defined on the basis of the studied reactions of a smaller subset of similar chemical species, both by analogy and with the use of structure–reactivity correlations (structure–activity relationships – SARs; Vereecken et al., 2018) to estimate the otherwise unknown parameters needed to construct the mechanisms.
Comparisons of the observed ions against the gas-phase oxidation products of aromatic hydrocarbons produced in the aromatic chemical schemes in the MCM can act as benchmarks to determine the relative importance of new pathways for different aromatic precursors. Note that the MCM was not run for any particular conditions for these comparisons, and instead, the potential products included in the MCM were simply compared with the ions observed in this study. It also provides a basis for understanding where gas-phase mechanisms could be improved in order to describe SOA formation. For the four average spectra of each precursor (1-methylnaphthalene is not within the MCM), the percentage of the mass spectral-signal-associated ion formulas also predicted by the MCM was calculated and is shown in Fig. 5.
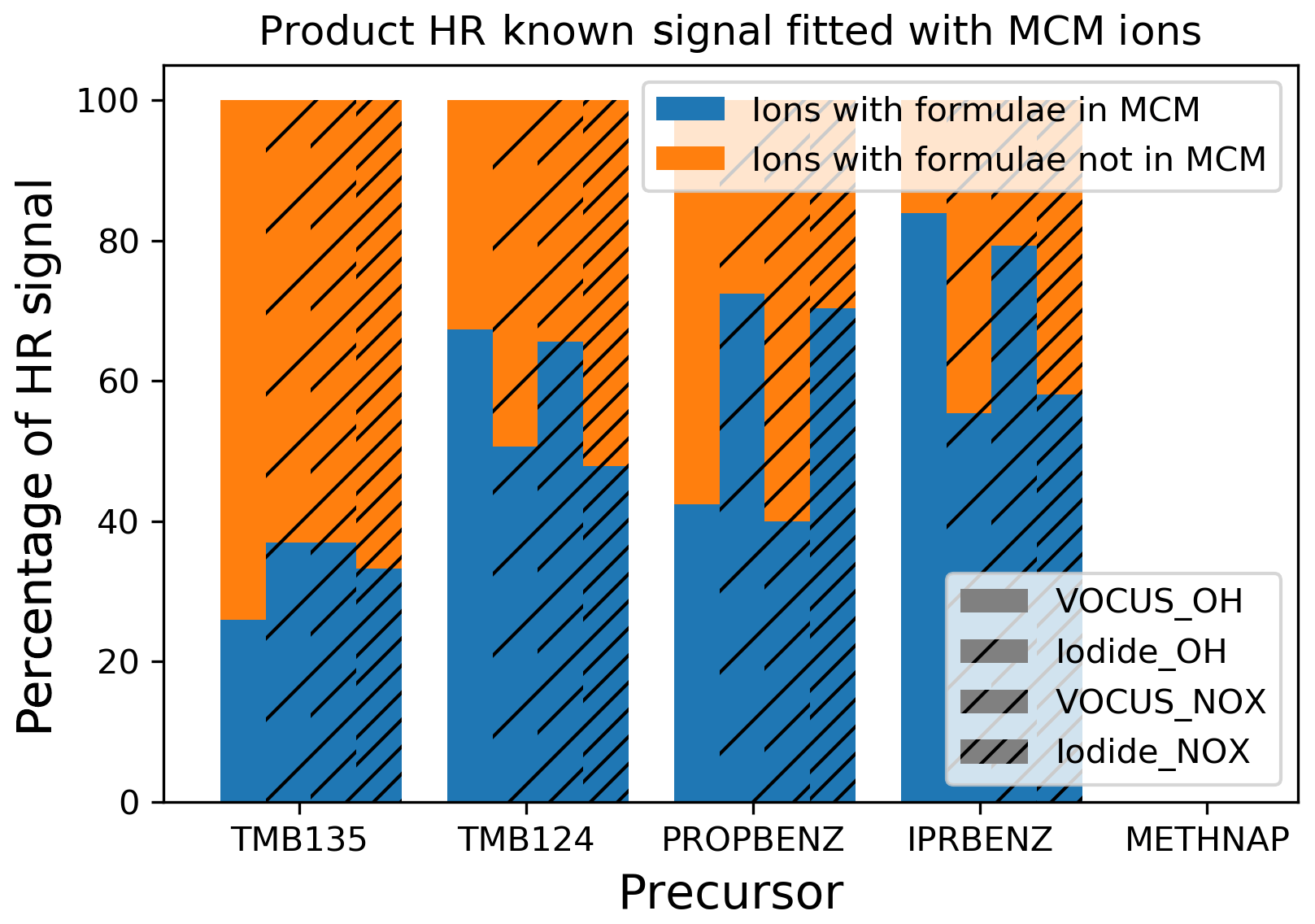
Figure 5Comparison of the signal contribution of ions predicted to form by the MCM and those observed.
The proportion of the mass spectral signal associated with ions consistent with MCM products was variable between precursors and techniques, ranging from 25 %, gas phase (low NOx) for 1,3,5-trimethylbenzene, to 80 %, gas phase (low NOx) for isopropylbenzene. The mechanisms contained within the MCM do not capture the same product profile for the trimethylbenzene isomers, though it should be noted that this agreement would not be expected in the particle phase as the MCM is a gas-phase mechanism. Previous chamber experiments have shown good agreement between gas-phase measurements and MCM results, suggesting that its gas-phase oxidation is well represented (Metzger et al., 2008; Rickard et al., 2010; Wyche et al., 2009). It may be that conditions in the OFR are dissimilar to these previous chamber experiments or that the measurements herein are more sensitive to previously undetected species. These are signal comparisons and, though in I-CIMS these may be affected by differences in sensitivity, the fact that the Vocus and I-CIMS see similar trends suggests that this does not affect our conclusion that the common ions contribute the majority of product signal, as discussed in Sect. 2.3.
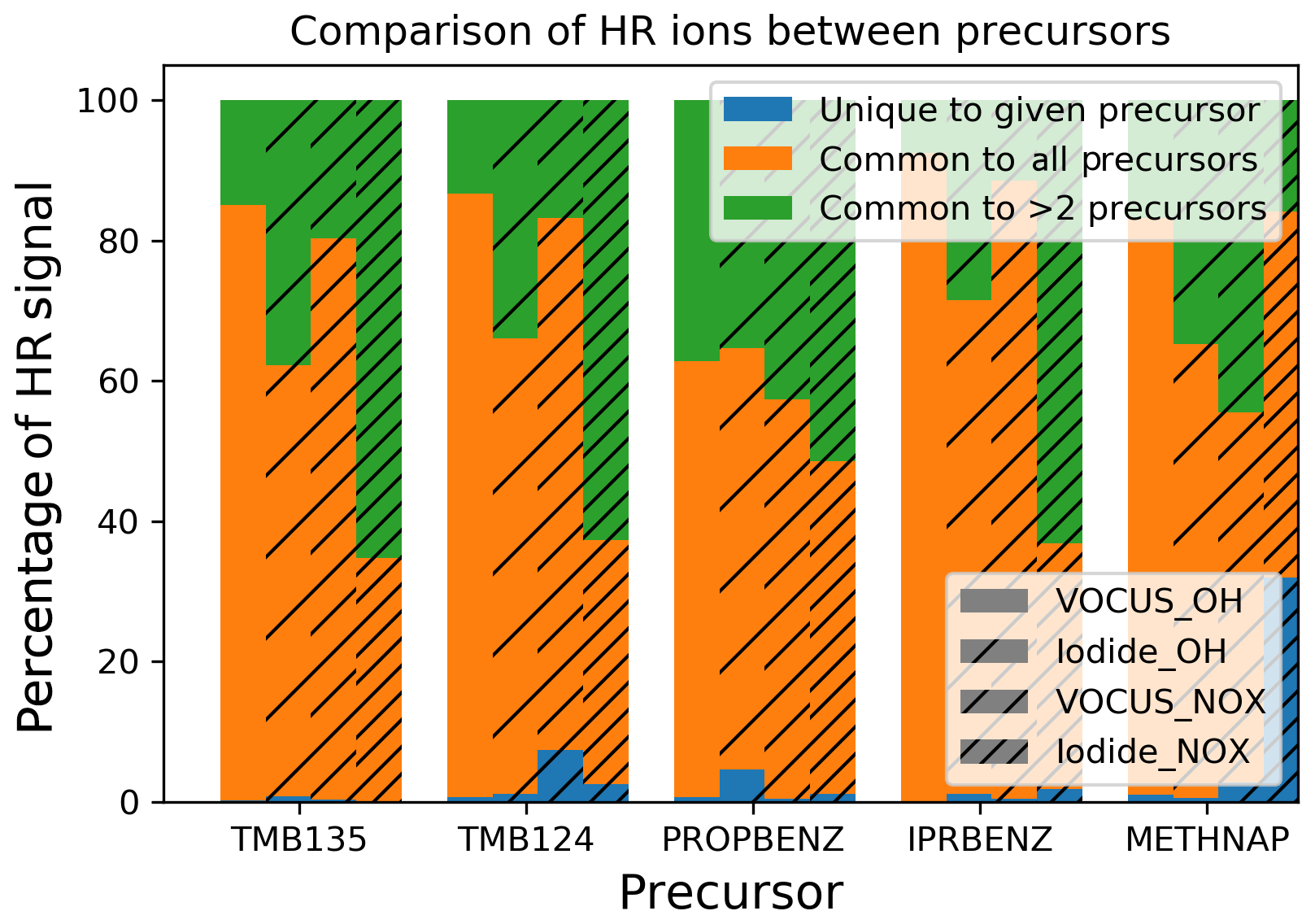
Figure 6Comparison of signal contributions of ions which were commonly observed from all precursors (orange), ions common to two or more precursors (green) and ions unique to the given precursor (blue) for all precursors and experiments.
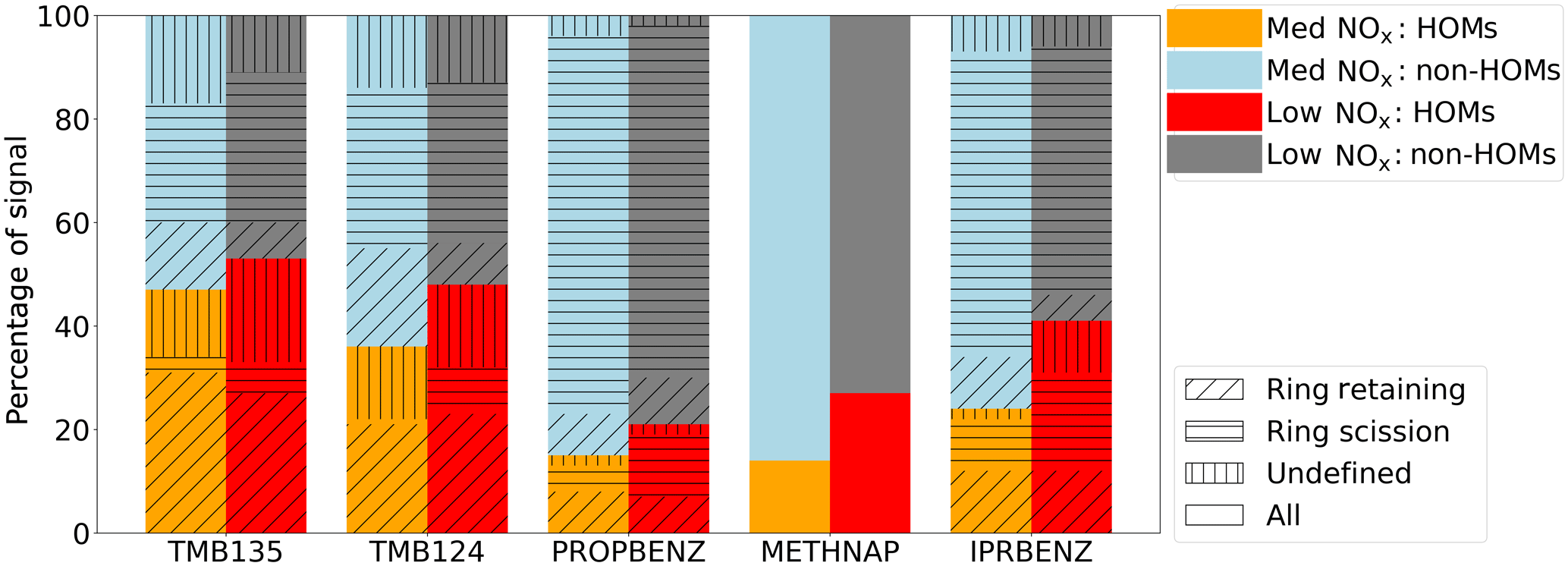
Figure 7Contribution to I-CIMS product signal from HOM and non-HOM ions under low- and medium-NOx conditions. The hatched and shaded areas are grouped by ring retaining, scission and those for which we have no definition; the derivation of these classifications will be discussed in Sect. 4.2. Methylnaphthalene is split by HOM and non-HOM classifications only.
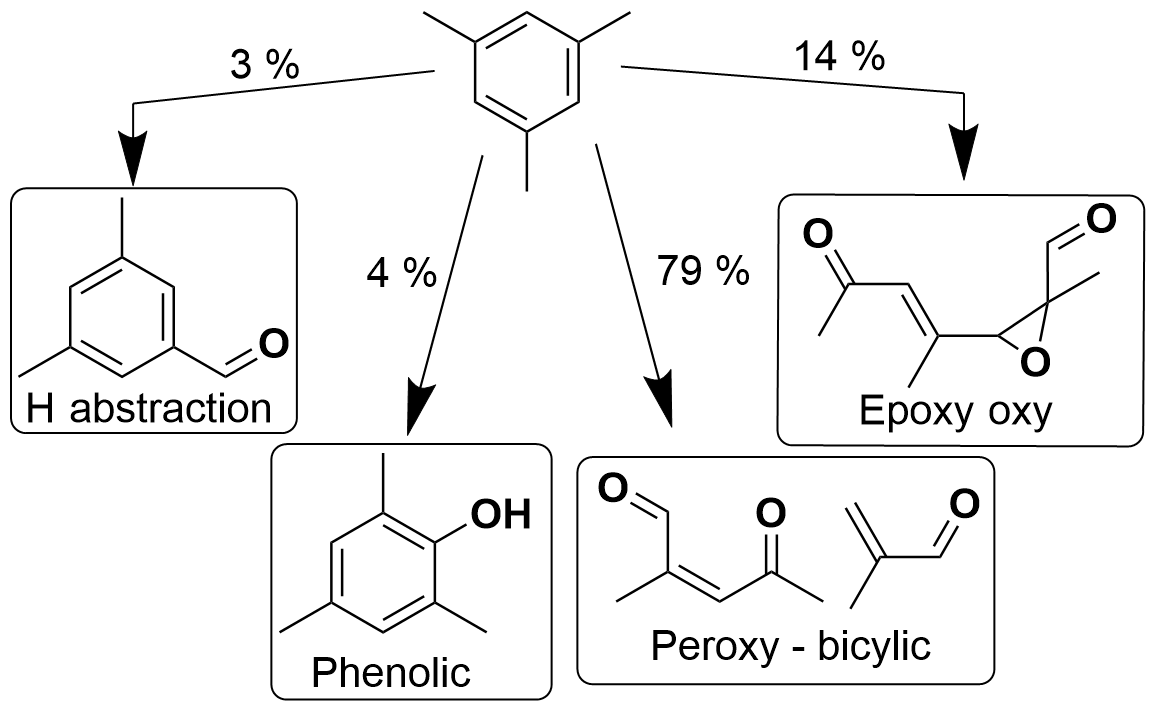
Figure 8Schematic of OH-initiated oxidation pathways of 1,3,5-trimethylbenzene as included in MCM version 3.1, adapted from Metzger et al. (2008).
4.1.2 Comparison between precursors
Here we investigate the similarities and differences in the observed ions across precursors. Products within the MCM are expected to be common between the different aromatic precursors as the decomposition pathways built into the mechanism for the aromatics follow a specific mechanism building protocol (Bloss et al., 2005). The majority of product signal (average of 94 % for I-CIMS; average of 98 % for Vocus) comes from ions with molecular formulae that are common between either multiple (green) or all (orange) of the precursors investigated, leaving an incredibly small contribution from ions which are unique (blue) to any given precursor. As discussed previously, most commonalities are between the C9 precursors (Fig. S4) and within this subset, namely between the trimethylbenzenes. In all cases, unique oxidation products from these different precursors contribute only a tiny amount to the overall product signal, even in cases where they may contribute significantly to the number of ions (Fig. S3). These patterns between observed products are consistent under both low- and medium-NOx conditions for the gas and the particle phase. The majority of the common products are ring scission products, while a large proportion are also HOMs.
HOMs have varying contributions to the SOA of different aromatic precursors; the proportions of the particle phase (low- and medium-NOx) mass spectra (medium NOx) that can be attributed to HOMs are shown in Fig. 7. The fraction of the particle mass spectra assigned as HOMs is within a difference of few percentage points under the low- and medium-NOx conditions for all precursors, except 1-methylnaphthalene where the proportion drops by half when NOx is added (27 % vs. 14 %). The precursor with the highest fraction of HOMs is 1,3,5-trimethylbenzene (44 %–47 %), and the lowest fraction is for propylbenzene (12 %–15 %).
4.2 Mechanisms
Aromatic oxidation can proceed via hydrogen abstraction or OH addition to the ring, with OH addition giving the highest branching ratio (Bloss et al., 2005). Upon OH addition, further oxidation proceeds via the phenolic, epoxy-oxy or bicyclic peroxy radical (BPR) pathways, which have varying relative yields for the different aromatics. These different pathways can result in a variety of ring-retaining or ring scission products, both of which are expected to contribute to SOA (Calvert, 2002; Schwantes et al., 2017).
Benzaldehyde and phenolic channels are predicted in the MCM to be less important for more substituted aromatics. Recent work has identified a potential source of ring-retaining products from pathways of ipso addition followed by dealkylation (Loison et al., 2012; Noda et al., 2009); however, this has been reported as negligible and thus is not included in the new MCM SARs (Jenkin et al., 2018). Yields from the epoxy-oxy pathway have been measured as significantly smaller than predicted by the MCM (Zaytsev et al., 2019), which are consistent with theoretical work (Li and Wang, 2014; Wu et al., 2014). The relative yields of different pathways are not known for the propylbenzene isomers and are estimated based on toluene (Bloss et al., 2005). The epoxy-oxy radical route is included in the MCM to represent the balance of chemistry not accounted for by other routes (Jenkin et al., 2003). This highlights the importance of more detailed mechanistic work to evaluate the relative importance of different pathways for different isomers.
Decomposition via the BPR intermediate or its stabilised isomer is the dominant pathway for all precursors (Jenkin et al., 2018), and recent work has shown that this pathway, which has previously been expected to yield only ring scission products, can also contribute to the formation of ring-retaining HOMs via oxygen addition (Jenkin et al., 2018) and subsequent autoxidation (Molteni et al., 2018; Wang et al., 2017). It has been suggested that H migration in BPRs of substituted aromatics may be faster and compete with HO2/NO reactions, thereby resulting in increased HOM formation (Wang et al., 2017). Due to the formation of both ring-retaining and ring scission products from the BPR intermediate, it is challenging to predict which precursors would be expected to form more ring scission or ring-retaining products.
Ring scission products, mainly formed from the decomposition of BPR, have been commonly observed from different aromatic precursors (Arey et al., 2009; Wang et al., 2007; Yu et al., 1997). Theoretical studies expect the decomposition of these intermediates into 1,2-dicarbonyls and co-products (Li and Wang, 2014; Wu et al., 2014). The larger co-products have been found at systematically lower yields than the corresponding 1,2-dicarbonyl products (Arey et al., 2009), suggesting that further photochemical processing is taking place. Though most studies of these dicarbonyls have been in the gas phase, they have recently been observed in the particle phase from the oxidation of aromatics (Zaytsev et al., 2019).
In this study, we provide a comparison of the composition of the aromatic precursors and compare the relative importance of different oxidation pathways for different C9 aromatics. This comparison is important for understanding differences in SOA formation and composition from these often grouped precursors. A companion paper provides detailed discussion of the mechanisms responsible for formation of the dominant oxidised products from these precursors (Wang et al., 2020).
In order to evaluate the ions in a mechanistic context, they have been classified as ring scission or ring retaining. Ring scission products can be unambiguously defined as ions ≤ C5. This leaves a subset of larger carbon number ions which could be ring retaining or ring scission products. To obtain a more accurate representation of ring-retaining products, the double bond equivalency (DBE) was calculated with ring-retaining ions having a DBE of at least 4. Further classification was attempted through use of an aromaticity index (AI); however, this proved unsuitable for HOMs which have a high oxygen content. The discussion herein is based on ring scission products having ≤ C5 and ring-retaining products being ≥ C6 with a DBE of 4. However, there may be some species, for instance, from the epoxy-oxy route that will be mislabelled using this approach, but their contribution is expected to be minor.
4.2.1 Ring scission products
Ring scission from different aromatics is known to yield a range of alpha-dicarbonyl products and alpha, beta-unsaturated-gamma dicarbonyl, furanone and small acid co-products (Arey et al., 2009; Calvert, 2002; Smith et al., 1998, 1999), many of which are included within the MCM (Bloss et al., 2005). We compare our data to scission products common to a range of aromatics identified by Arey et al. (2009), and all of these were observed in our experiments by I-CIMS (I), Vocus (V) or both (V + I; Table 2). Ring scission products are largely dominant in the gas phase, though they play a role in the particle-phase composition, particularly so for the less substituted aromatics. In some cases, such as 1,3,5-trimethylbenzene, known ring scission ions, given in Arey et al., are abundant products; for example, C5H6O3 is the 10th largest signal in the particle phase under low-NOx conditions. In the MCM, BPR is near-exclusively decomposed into ring scission products which proves suitable for describing the major observed particle-phase products for propyl and isopropylbenzene (C5H6O6, C5H6O5 and C4H4O6) and explains the larger proportion of product signal observed (Fig. 5).
A large proportion of the ring scission products observed in the particle phase are more oxidised than those previously reported (Arey et al., 2009), and many fit the definition of HOMs by Bianchi et al. (2019). These products appear to have undergone oxygen addition to the same carbon backbone as many of the small gas-phase dicarbonyls, suggesting that further gas-phase oxidation and partitioning of dicarbonyl co-products may be an important contributor to SOA. Further processing of these ring-opened products may explain the discrepancy between 1,2-dicarbonyls and their co-products observed in many gas-phase studies of aromatic oxidation (Arey et al., 2009). This has been shown previously for SOA formed from 1,3,5-trimethyl benzene where 3-methyl maleic anhydride (C5H4O3), 2-methyl-4-oxo-2-pentenal (C6H8O2) and 3,5-dimethyl-5(2H)-2-furanone (C6H8O2) contribute to SOA formation and growth through further oxidative processing (Johnson et al., 2005; Rickard et al., 2010). Further examples of this are exhibited in our results, including C7H10O2, which is observed from both trimethyl benzene isomers in Vocus gas phase mass spectra, and C7H10O(4−7) formulas that are some of the most dominant product ions observed in SOA from both trimethyl benzene isomers. In the case of 1,2,4-trimethylbenzene, C5H6O2 is observed in the gas phase while C5H6O(3−7) are dominant ions in the particle phase. This is also the case for propylbenzene, where C5H6O5 and C5H6O6 are the two largest contributors to the particle phase. Other observed products can be explained by further oxidation of scission products within the MCM such as C4H6O6, which is a major ion from isopropylbenzene oxidation.
Table 2Dicarbonyl products observed in the gas and particle phase from Arey (2009) under low-NOx conditions (V – Vocus; I – I-CIMS). Note: PRBZ – PROPBENZ; MN – METHNAP; IPRBZ – IPRBENZ.
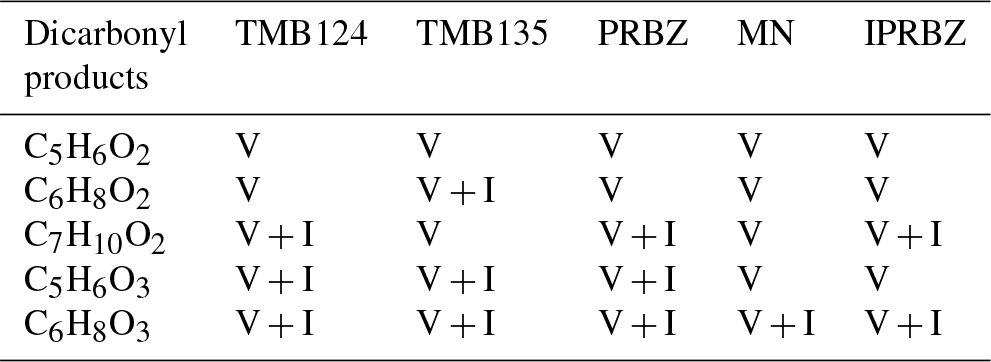
The most dominant of the scission products we observed in the gas and particle phases have a lower carbon number than those within the MCM, with C3 products being most dominant in the gas phase for PROPBENZ, IPRBENZ and METHNAP, and C4 products being more important in the TMBs. In the particle phase, C4 scission products have the largest contribution for TMB124 and METHNAP. Scission products we observe have previously been detected from oxidation of 1,2,4-trimethyl benzene including C5H8O3, C4H6O3 and C3H4O2 (Zaytsev et al., 2019). Ring scission remains a dominant pathway under the NOx conditions, though the spread across carbon numbers increases in the gas and particle phases, as shown in the carbon number distributions. Ring scission HOMs are most commonly observed in the particle phase for the C4 and C5 products and are particularly important in the case of the C9 aromatics. Newly proposed epoxy-dicarbonyl products (Li and Wang, 2014) have also been observed in this study (C4H4O3 – epoxy-butanedial; C5H6O3 – methyl-epoxy-butanedial; and C6H8O3 – dimethyl-epoxy-butanedial) from all aromatic precursors in the particle phase.
4.2.2 Ring-retaining products
Ring-retaining products can form from two different pathways, namely hydrogen abstraction (from the attached alkyl groups), which can result in fragmented ring-retaining products, or via the BPR pathway, which results in non-fragmented ring-retaining products.
Ring-retaining HOMs from 1,3,5-trimethylbenzene oxidation have been identified from experiments carried out in a range of different chambers and conditions (Molteni et al., 2018; Sato et al., 2012; Tsiligiannis et al., 2019). Our observations show good agreement with ion formulas observed in these experiments, with divergence at significantly higher NOx conditions (Tsiligiannis et al., 2019). This includes observations of ions such as C9H12O(2−8), which are observed by Molteni et al. (2018), and are consistent with closed-shell products expected by the mechanisms outlined in Wang et al. (2017) alongside ions observed by LC-ToF-MS (Sato et al., 2012, 2019), which were all important in our experiments (Table S1). Ring retention was previously found to contribute ∼ 25 % of SOA mass from a study of 1,2,4-trimethylbenzene under elevated NOx conditions, which were attributed to BPR, phenolic and benzaldehyde channels despite low ring-retaining concentrations in the gas phase, consistent with our observations. These results identified ions such as C9H12O(4−6), which are also dominant in our observations for both 1,2,4- and 1,3,5-trimethylbenzenes, though we also observed a significant contribution from more oxidised ions such as C9H12O6 and C9H14O6 (Zaytsev et al., 2019).
Molteni et al. (2018) suggested that ring-retaining products with ion formulas containing 4–6 more hydrogen atoms more than their parent hydrocarbon could be attributed to multiple OH attacks. More recently, Tsiligiannis et al. (2019) have shown that C9H14Oz products from oxidation of 1,3,5-trimethylbenzene have characteristics of second-generation products due to their enhanced contributions in experiments with higher OH exposures. Thus, in order to obtain a lower-bound estimate of the contribution of the multigenerational OH attack, ions with greater than 14 hydrogen atoms were classed as multigenerational products. Overall, their contribution to signal is < 10 % in all experiments (Figs. S5–S6).
Previous results have suggested that C8 and C7 compounds, such as C8H10O(4−5) and C7H8O(4−5) from 1,2,4-trimethylbenzene oxidation, can be formed from the dealkylation pathway, though they may also be formed from multiple pathways (Noda et al., 2009; Zaytsev et al., 2019). However, other studies have shown that dealkylation is not a significant pathway (Aschmann et al., 2010; Loison et al., 2012). In our results, C8 ions are only important for the trimethylbenzenes, indicating that this pathway may be important for these precursors but not for others. Though we observe the less oxidised C8 products, the major C7−8 products we observe are C8H10O6 for TMB124 and C8H10O7 for TMB135.
To gain a broader overview of the importance of ring-retaining products across the various precursors, we have estimated the contribution of ring-retaining ions to total product signal. Our results show that the ring-retained HOM formation is most important for TMB135, where these ions contribute 27 % of total observed product signal in the particle phase measured by I-CIMS, with a smaller contribution of 23 % to TMB124 and a minor contribution to IPRBENZ (12 %) and PROPBENZ (7 %) under low-NOx conditions (Fig. 7).
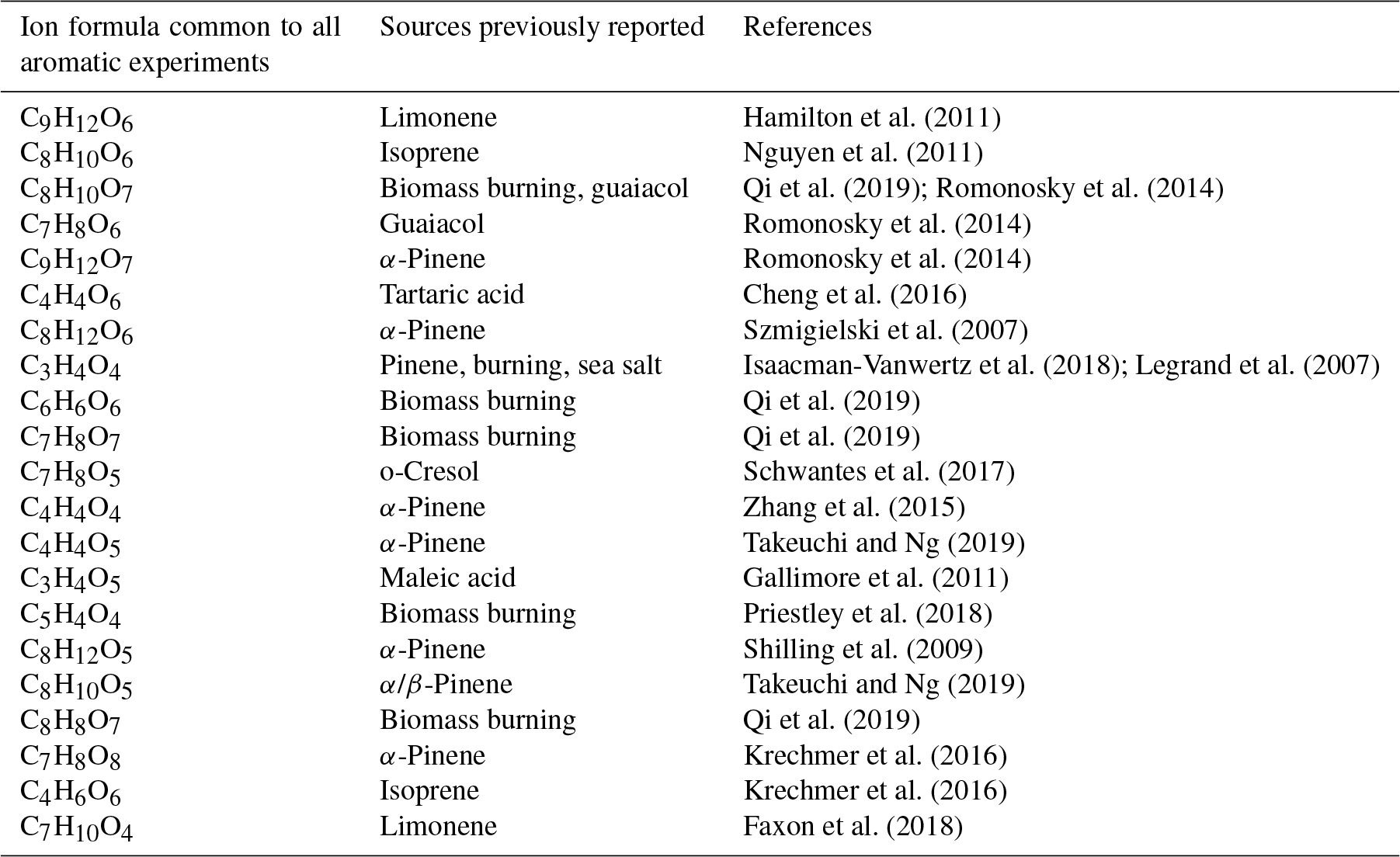
Table 3Comparison of ions observed commonly from all aromatic precursors with these ions reported in literature.
This indicates that ring-retaining-product formation is important for the aromatic hydrocarbons with more substituent groups on the ring rather than a single alkyl side chain, suggesting that the substitution plays a role in favouring the retention of the ring during both H abstraction and OH addition. This may be due to the stabilisation of the substituent ring or due to the steric hindrance of the methyl groups, which make OH attack less favourable, and result in a larger-than-estimated branching ratio of H abstraction from the methyl group. It is apparent in our results that ring-retaining HOMs persist upon perturbation by NO, suggesting that favourable transition-state geometry of the more substituted aromatics may play a role in ensuring the autoxidation of BPR remains competitive at higher NO conditions. This is not observed for the less substituted aromatics, which generally produce fewer HOMs in the presence of NOx.
Under low-NOx conditions, the relative importance of autoxidation for the formation of ring-retaining HOMs from BPR intermediates varies for the different precursors and, as such, impacts upon the relative yields of ring-retaining vs. ring scission products. In the cases of propyl and isopropylbenzene, ring-retaining HOM formation has a minor contribution to products, highlighting the role that substituent groups on the aromatic ring play in favouring autoxidation leading to ring-retaining HOM formation. Isopropylbenzene SOA has a larger contribution from ring-opened HOMs in the particle phase, despite a larger contribution from ring-retaining products in the gas phase, suggesting that, though their formation occurs, they undergo further oxidation and scission before partitioning.
Though we lack knowledge of specific pathways for methylnaphthalene, our observations of products which are assumed to contain a single ring (≤C10) are consistent with those of the C9 aromatics, suggesting that beyond the opening of the first aromatic ring, the oxidation proceeds via similar pathways to those of the C9 aromatics. Overlap in observations between C9 aromatics and the PAHs could prove useful as a basic description of the major products formed in the photooxidation of methylnaphthalene.
4.3 Implications for ambient observations
Aromatic oxidation leads to the formation of oxidised products, which are largely common to all aromatic precursors. This makes identifying their contribution to ambient SOA challenging when using online techniques. Furthermore, unique product ions contribute a tiny proportion of the signal, and thus it is challenging to differentiate between different aromatics. However, markers are widely used for the interpretation of ambient measurements, and many of the ions we observe correspond to formulas which have been reported as markers of other precursors or oxidation pathways. This is shown in Table 3, where we compare the major ions observed from all aromatics with previous literature, and suggests that some of these source attributions may not be valid when aromatics are present.
A rapid increase in the development and application of novel mass spectrometric techniques has given a wealth of near-molecular level detail which is unprecedented in atmospheric chemistry (Isaacman-VanWertz et al., 2017; Laskin et al., 2018). Such high-resolution observational data sets are extremely useful for the development and evaluation of detailed chemical mechanisms used to understand and simulate gas- and aerosol-phase composition in a wide variety of science and policy applications related to air quality and climate. Comparing our results with other laboratory and field experimental observations shows that some major ions from aromatic oxidation are concurrent with other sources or VOC systems, which have implications for marker identification in ambient data sets; for example, C9H12O6, observed in all aromatic oxidation experiments which have previously been identified in cases of biomass burning (Kourtchev et al., 2016) and also as a product of limonene oxidation (Hamilton et al., 2011). C7H8O6 and C9H12O7 have been observed from aromatic autoxidation (Molteni et al., 2018); however, C7H8O6 is also as a product of alpha phellandrene ozonolysis (Mackenzie-Rae et al., 2018). Another product observed from all aromatic precursors is C8H12O6, which is widely reported as 3‐methyl‐1,2,3‐butanetricarboxylic acid (MBTCA), a marker of monoterpene SOA (Hu et al., 2013; Kourtchev et al., 2016). The majority of these observations are from CIMS approaches which lack the separation of liquid chromatography (LC) or gas chromatography (GC).
Though we observe some unique ion signal, this signal is distributed amongst many ions so no single ion has a large enough signal intensity to be used as a marker, and these low-signal-intensity ions are thus likely to be absent in complex ambient spectra. Even in single-component experiments they are not ideal unambiguous markers of SOA; thus, in more complex and ambient SOA samples, it is hard to see how they could be used effectively. Furthermore, due to these ions being minor components, they are difficult to assign formulas to due to a relatively poor signal-to-noise ratio, and as many of them are small carbon numbers, they could be the result of low signal intensity. Thus, comparison of precursors suggests that identifying tracers from aromatic precursors using online techniques with no pre-separation is not possible. Most striking is the fact that methylnaphthalene, which is a C11 molecule has > 68 % of its signal in common with the other precursors in all cases, despite the fact that its oxidation will proceed differently. Tracer-based detection of SOA is thus not likely to be useful in ambient measurements, and though potentially isomeric information may provide unique markers, ion formulas themselves are unlikely to uniquely identify sources between different aromatic precursors.
Oxidation of different C9-aromatic isomers results in the formation of similar products; however, the signal distribution of these products in the gas phase and particle phases are distinct. Gas- and particle-phase compositions are distinct from one another, with many HOMs being observed almost exclusively in the particle-phase I-CIMS measurements and many small scission products exclusively in the Vocus measurements.
Comparison of the observations with the gas-phase MCM mechanism highlights that the MCM does not capture the full extent of observed ions, particularly the more oxidised HOM products formed from autoxidation pathways which have not been included in this mechanism. Comparing the online I-CIMS and offline Orbitrap LC–MS observations shows good agreement in the negative mode, with a large overlap in ring-retaining HOM ions observed by both techniques.
Evaluating products in terms of their formation mechanisms shows that the oxidation of the more substituted aromatic precursors leads to a larger contribution of ring-retaining products, while the single-substituent aromatics yield large proportions of scission products. Overall, ring-retaining HOM products are a minor contributor to SOA product signal (<40 %), while known scission products appear to undergo further oxidation and are major contributors to SOA from all aromatic precursors.
Perturbation of the oxidation system with NOx shows that HOM formation from aromatics proceeds at higher NO, and this effect is most important for the more substituted trimethylbenzene isomers which continue to form ring-retaining HOMs at higher NOx. The variation in composition between the C9-aromatic isomers suggests that grouping VOCs by carbon number for SOA formation may not be valid, and the number of substituent groups may be a more valid grouping to capture their different SOA product distributions. Comparison with 1-methylnaphthalene shows that the products from a different starting precursor are largely similar, with the distributions being the key to differentiation of SOA from different precursors.
Comparison of the ions observed from aromatic oxidation in the literature shows an overlap with those from other sources, suggesting that some source attributions may not be valid when aromatics are present. Correspondences between laboratory and ambient spectra have been observed for oxidation products of biogenic VOCs (Yan et al., 2016); however, this approach has not been taken for anthropogenic VOCs. Through the co-measurement of many ions, these results are expected to support studies evaluating the importance of aromatics for urban SOA formation. This study has focused on single components; however, future work is required to identify the impact that mixing aromatics with biogenics has upon SOA composition (McFiggans et al., 2019).
Data are available upon request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-9783-2020-supplement.
AM and HC designed the experiments. Instrument deployment and operation were carried out by AM, YW, JEK, AL, FM, PC, MAM and MRC. I-CIMS data analysis was carried out by AM, and Vocus data analysis was done by YW. DJB prepared the aerosol samples for LC–MS analysis. KLP designed the non-targeted LC–MS analytical and data-processing methodology, analysed the samples, and provided the processed data. AM, YW and JFH wrote the paper. All co-authors discussed the results and comments on the paper.
The authors declare that they have no conflict of interest.
Archit Mehra has been fully funded by the Natural Environment Research Council (NERC) and acknowledges the funding from the NERC EAO Doctoral Training Partnership (grant no: NE/L002469/1) and CASE partnership support from Aerodyne Research, Inc. Lin Wang acknowledges the funding from the Newton Advanced Fellowships (grant no. NA140106). Hugh Coe and Archit Mehra acknowledge funding from Airpro (grant no. NE/N00695X/1).
This research has been supported by the Natural Environment Research Council (grant nos. NE/L002469/1 and NE/N00695X/1) and the Newton Fund (grant no. NA140106).
This paper was edited by Min Shao and reviewed by two anonymous referees.
Arey, J., Obermeyer, G., Aschmann, S. M., Chattopadhyay, S., Cusick, R. D., and Atkinson, R.: Dicarbonyl Products of the OH Radical-Initiated Reaction of a Series of Aromatic Hydrocarbons, Environ. Sci. Technol., 43, 683–689, https://doi.org/10.1021/es8019098, 2009.
Aschmann, S. M., Arey, J., and Atkinson, R.: Extent of H-atom abstraction from OH + p-cymene and upper limits to the formation of cresols from OH + m-xylene and OH + p-cymene, Atmos. Environ., 44, 3970–3975, https://doi.org/10.1016/j.atmosenv.2010.06.059, 2010.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, 2000.
Atkinson, R. and Arey, J.: Mechanisms of the Gas-Phase Reactions of Aromatic Hydrocarbons and PAHs with OH and NO3 Radicals, Polycycl. Aromat. Comp., 27, 15–40, https://doi.org/10.1080/10406630601134243, 2007.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bloss, C., Wagner, V., Bonzanini, A., Jenkin, M. E., Wirtz, K., Martin-Reviejo, M., and Pilling, M. J.: Evaluation of detailed aromatic mechanisms (MCMv3 and MCMv3.1) against environmental chamber data, Atmos. Chem. Phys., 5, 623–639, https://doi.org/10.5194/acp-5-623-2005, 2005.
Borbon, A., Gilman, J. B., Kuster, W. C., Grand, N., Chevaillier, S., Colomb, A., Dolgorouky, C., Gros, V., Lopez, M., Sarda-Esteve, R., Holloway, J., Stutz, J., Petetin, H., Mckeen, S., Beekmann, M., Warneke, C., Parrish, D. D., and De Gouw, J. A.: Emission ratios of anthropogenic volatile organic compounds in northern mid-latitude megacities: Observations versus emission inventories in Los Angeles and Paris, J. Geophys. Res.-Atmos., 118, 2041–2057, https://doi.org/10.1002/jgrd.50059, 2013.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6, 27881, https://doi.org/10.1038/srep27881, 2016.
Bryant, D. J., Dixon, W. J., Hopkins, J. R., Dunmore, R. E., Pereira, K. L., Shaw, M., Squires, F. A., Bannan, T. J., Mehra, A., Worrall, S. D., Bacak, A., Coe, H., Percival, C. J., Whalley, L. K., Heard, D. E., Slater, E. J., Ouyang, B., Cui, T., Surratt, J. D., Liu, D., Shi, Z., Harrison, R., Sun, Y., Xu, W., Lewis, A. C., Lee, J. D., Rickard, A. R., and Hamilton, J. F.: Strong anthropogenic control of secondary organic aerosol formation from isoprene in Beijing, Atmos. Chem. Phys., 20, 7531–7552, https://doi.org/10.5194/acp-20-7531-2020, 2020.
Calvert, J. (Ed.): The mechanisms of atmospheric oxidation of aromatic hydrocarbons, Oxford University Press, available at: https://global.oup.com/academic/product/the-mechanisms-of-atmospheric-oxidation-of-the-aromatic-hydrocarbons-9780195146288?cc=gb&lang=en&# (last access: 17 August 2020), 2002.
Carter, W.: Documentation for the SAPRC atmospheric photochemical mechanism preparation and emissions processing programs for implementation in airshed models, available at: http://www.engr.ucr.edu/~carter/pubs/arbprep.pdf (last access: 20 April 2020), 1988.
Carter, W. P. L.: Implementation of the Saprc-99 Chemical Mechanism Into the Models-3 Framework, Report to the United States Environmental Protection Agency, available at: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.25.293&rep=rep1&type=pdf (last access: 17 August 2020), 2000.
Carter, W. P. L. and Heo, G.: Development of revised SAPRC aromatics mechanisms, Atmos. Environ., 77, 404–414, https://doi.org/10.1016/j.atmosenv.2013.05.021, 2013.
Chan, A. W. H., Kautzman, K. E., Chhabra, P. S., Surratt, J. D., Chan, M. N., Crounse, J. D., Kürten, A., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: implications for oxidation of intermediate volatility organic compounds (IVOCs), Atmos. Chem. Phys., 9, 3049–3060, https://doi.org/10.5194/acp-9-3049-2009, 2009.
Cheng, C. T., Chan, M. N., and Wilson, K. R.: Importance of Unimolecular HO2 Elimination in the Heterogeneous OH Reaction of Highly Oxygenated Tartaric Acid Aerosol, J. Phys. Chem. A, 120, 5887–5896, https://doi.org/10.1021/acs.jpca.6b05289, 2016.
Coggon, M. M., McDonald, B. C., Vlasenko, A., Veres, P. R., Bernard, F., Koss, A. R., Yuan, B., Gilman, J. B., Peischl, J., Aikin, K. C., Durant, J., Warneke, C., Li, S. M., and De Gouw, J. A.: Diurnal Variability and Emission Pattern of Decamethylcyclopentasiloxane (D5) from the Application of Personal Care Products in Two North American Cities, Environ. Sci. Technol., 52, 5610–5618, https://doi.org/10.1021/acs.est.8b00506, 2018.
Corrêa, S. M. and Arbilla, G.: Aromatic hydrocarbons emissions in diesel and biodiesel exhaust, Atmos. Environ., 40, 6821–6826, https://doi.org/10.1016/j.atmosenv.2006.05.068, 2006.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Derwent, R. G., Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: Photochemical ozone creation potentials for organic compounds in northwest Europe calculated with a master chemical mechanism, Atmos. Environ., 32, 2429–2441, https://doi.org/10.1016/S1352-2310(98)00053-3, 1998.
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018.
Gallimore, P. J., Achakulwisut, P., Pope, F. D., Davies, J. F., Spring, D. R., and Kalberer, M.: Importance of relative humidity in the oxidative ageing of organic aerosols: case study of the ozonolysis of maleic acid aerosol, Atmos. Chem. Phys., 11, 12181–12195, https://doi.org/10.5194/acp-11-12181-2011, 2011.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Guenther, A., Hewitt, C. N., Erickson, D., Guenther, A., Hewitt, N. C., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hamilton, J. F., Webb, P. J., Lewis, A. C., and Reviejo, M. M.: Quantifying small molecules in secondary organic aerosol formed during the photo-oxidation of toluene with hydroxyl radicals, Atmos. Environ., 39, 7263–7275, https://doi.org/10.1016/j.atmosenv.2005.09.006, 2005.
Hamilton, J. F., Rami Alfarra, M., Wyche, K. P., Ward, M. W., Lewis, A. C., McFiggans, G. B., Good, N., Monks, P. S., Carr, T., White, I. R., and Purvis, R. M.: Investigating the use of secondary organic aerosol as seed particles in simulation chamber experiments, Atmos. Chem. Phys., 11, 5917–5929, https://doi.org/10.5194/acp-11-5917-2011, 2011.
Hu, Q.-H., Xie, Z.-Q., Wang, X.-M., Kang, H., He, Q.-F., and Zhang, P.: Secondary organic aerosols over oceans via oxidation of isoprene and monoterpenes from Arctic to Antarctic, Sci. Rep., 3, 2280, https://doi.org/10.1038/srep02280, 2013.
Huang, M., Hu, C., Guo, X., Gu, X., Zhao, W., Wang, Z., Fang, L., and Zhang, W.: Chemical composition of gas and particle–phase products of OH-initiated oxidation of 1,3,5-trimethylbenzene, Atmos. Pollut. Res., 5, 73–78, https://doi.org/10.5094/APR.2014.009, 2014.
Isaacman-VanWertz, G., Massoli, P., O'Brien, R. E., Nowak, J. B., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., Su, L., Knopf, D. A., Misztal, P. K., Arata, C., Goldstein, A. H., and Kroll, J. H.: Using advanced mass spectrometry techniques to fully characterize atmospheric organic carbon: current capabilities and remaining gaps, Faraday Discuss., 200, 579–598, https://doi.org/10.1039/C7FD00021A, 2017.
Isaacman-Vanwertz, G., Massoli, P., OBrien, R., Lim, C., Franklin, J. P., Moss, J. A., Hunter, J. F., Nowak, J. B., Canagaratna, M. R., Misztal, P. K., Arata, C., Roscioli, J. R., Herndon, S. T., Onasch, T. B., Lambe, A. T., Jayne, J. T., Su, L., Knopf, D. A., Goldstein, A. H., Worsnop, D. R., and Kroll, J. H.: Chemical evolution of atmospheric organic carbon over multiple generations of oxidation, Nat. Chem., 10, 462–468, https://doi.org/10.1038/s41557-018-0002-2, 2018.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9329–9349, https://doi.org/10.5194/acp-18-9329-2018, 2018.
Johnson, D., Jenkin, M. E., Wirtz, K., and Martin-Reviejo, M.: Simulating the Formation of Secondary Organic Aerosol from the Photooxidation of Aromatic Hydrocarbons, Environ. Chem., 2, 35–48, 2005.
Johnson, D., Utembe, S. R., Jenkin, M. E., Derwent, R. G., Hayman, G. D., Alfarra, M. R., Coe, H., and McFiggans, G.: Simulating regional scale secondary organic aerosol formation during the TORCH 2003 campaign in the southern UK, Atmos. Chem. Phys., 6, 403–418, https://doi.org/10.5194/acp-6-403-2006, 2006.
Kansal, A.: Sources and reactivity of NMHCs and VOCs in the atmosphere: A review, J. Hazard. Mater., 166, 17–26, https://doi.org/10.1016/j.jhazmat.2008.11.048, 2009.
Kari, E., Hao, L., Ylisirniö, A., Buchholz, A., Leskinen, A., Yli-Pirilä, P., Nuutinen, I., Kuuspalo, K., Jokiniemi, J., Faiola, C. L., Schobesberger, S., and Virtanen, A.: Potential dual effect of anthropogenic emissions on the formation of biogenic secondary organic aerosol (BSOA), Atmos. Chem. Phys., 19, 15651–15671, https://doi.org/10.5194/acp-19-15651-2019, 2019.
Karl, T., Apel, E., Hodzic, A., Riemer, D. D., Blake, D. R., and Wiedinmyer, C.: Emissions of volatile organic compounds inferred from airborne flux measurements over a megacity, Atmos. Chem. Phys., 9, 271–285, https://doi.org/10.5194/acp-9-271-2009, 2009.
Khan, M. A. H., Jenkin, M. E., Foulds, A., Derwent, R. G., Percival, C. J., and Shallcross, D. E.: A modeling study of secondary organic aerosol formation from sesquiterpenes using the STOCHEM global chemistry and transport model, J. Geophys. Res.-Atmos., 122, 4426–4439, https://doi.org/10.1002/2016JD026415, 2017.
Kourtchev, I., Godoi, R. H. M., Connors, S., Levine, J. G., Archibald, A. T., Godoi, A. F. L., Paralovo, S. L., Barbosa, C. G. G., Souza, R. A. F., Manzi, A. O., Seco, R., Sjostedt, S., Park, J.-H., Guenther, A., Kim, S., Smith, J., Martin, S. T., and Kalberer, M.: Molecular composition of organic aerosols in central Amazonia: an ultra-high-resolution mass spectrometry study, Atmos. Chem. Phys., 16, 11899–11913, https://doi.org/10.5194/acp-16-11899-2016, 2016.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and De Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion- Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Krechmer, J. E., Groessl, M., Zhang, X., Junninen, H., Massoli, P., Lambe, A. T., Kimmel, J. R., Cubison, M. J., Graf, S., Lin, Y.-H., Budisulistiorini, S. H., Zhang, H., Surratt, J. D., Knochenmuss, R., Jayne, J. T., Worsnop, D. R., Jimenez, J.-L., and Canagaratna, M. R.: Ion mobility spectrometry–mass spectrometry (IMS–MS) for on- and offline analysis of atmospheric gas and aerosol species, Atmos. Meas. Tech., 9, 3245–3262, https://doi.org/10.5194/amt-9-3245-2016, 2016.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D) + N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Lambe, A. T., Ahern, A. T., Williams, L. R., Slowik, J. G., Wong, J. P. S., Abbatt, J. P. D., Brune, W. H., Ng, N. L., Wright, J. P., Croasdale, D. R., Worsnop, D. R., Davidovits, P., and Onasch, T. B.: Characterization of aerosol photooxidation flow reactors: heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements, Atmos. Meas. Tech., 4, 445–461, https://doi.org/10.5194/amt-4-445-2011, 2011.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Laskin, J., Laskin, A., and Nizkorodov, S. A.: Mass Spectrometry Analysis in Atmospheric Chemistry, Anal. Chem., 90, 166–189, https://doi.org/10.1021/acs.analchem.7b04249, 2018.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., Thornton, J. A., Kurte, T., Worsnop, D. R., Thornton, J. A., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Legrand, M., Preunkert, S., Oliveira, T., Pio, C. A., Hammer, S., Gelencsér, A., Kasper-Giebl, A., and Laj, P.: Origin of C2–C5 dicarboxylic acids in the European atmosphere inferred from year-round aerosol study conducted at a west-east transect, J. Geophys. Res., 112, https://doi.org/10.1029/2006JD008019, 2007.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Li, L., Tang, P., Nakao, S., and Cocker III, D. R.: Impact of molecular structure on secondary organic aerosol formation from aromatic hydrocarbon photooxidation under low-NOx conditions, Atmos. Chem. Phys., 16, 10793–10808, https://doi.org/10.5194/acp-16-10793-2016, 2016.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., De Gouw, J. A., and Jimenez, J. L.: Modeling the Radical Chemistry in an Oxidation Flow Reactor: Radical Formation and Recycling, Sensitivities, and the OH Exposure Estimation Equation, J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Li, Y. and Wang, L.: The atmospheric oxidation mechanism of 1,2,4-trimethylbenzene initiated by OH radicals, Phys. Chem. Chem. Phys., 16, 17908–17917, https://doi.org/10.1039/C4CP02027H, 2014.
Liu, Y., Shao, M., Fu, L., Lu, S., Zeng, L., and Tang, D.: Source profiles of volatile organic compounds (VOCs) measured in China: Part I, Atmos. Environ., 42, 6247–6260, https://doi.org/10.1016/j.atmosenv.2008.01.070, 2008a.
Liu, Y., Shao, M., Lu, S., Chang, C.-C., Wang, J.-L., and Chen, G.: Volatile Organic Compound (VOC) measurements in the Pearl River Delta (PRD) region, China, Atmos. Chem. Phys., 8, 1531–1545, https://doi.org/10.5194/acp-8-1531-2008, 2008b.
Loison, J. C., Rayez, M. T., Rayez, J. C., Gratien, A., Morajkar, P., Fittschen, C., and Villenave, E.: Gas-phase reaction of hydroxyl radical with hexamethylbenzene, J. Phys. Chem. A, 116, 12189–12197, https://doi.org/10.1021/jp307568c, 2012.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016a.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016b.
Lu, K., Guo, S., Tan, Z., Wang, H., Shang, D., Liu, Y., Li, X., Wu, Z., Hu, M., and Zhang, Y.: Exploring atmospheric free-radical chemistry in China: the self-cleansing capacity and the formation of secondary air pollution, Natl. Sci. Rev., 6, 579–594, https://doi.org/10.1093/nsr/nwy073, 2019.
Mackenzie-Rae, F. A., Wallis, H. J., Rickard, A. R., Pereira, K. L., Saunders, S. M., Wang, X., and Hamilton, J. F.: Ozonolysis of α-phellandrene – Part 2: Compositional analysis of secondary organic aerosol highlights the role of stabilised Criegee intermediates, Atmos. Chem. Phys., 18, 4673–4693, https://doi.org/10.5194/acp-18-4673-2018, 2018.
McDonald, B. C., De Gouw, J. A., Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L., Lee-Taylor, J., Hayes, P. L., McKeen, S. A., Cui, Y. Y., Kim, S. W., Gentner, D. R., Isaacman-VanWertz, G., Goldstein, A. H., Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Metzger, A., Dommen, J., Gaeggeler, K., Duplissy, J., Prevot, A. S. H., Kleffmann, J., Elshorbany, Y., Wisthaler, A., and Baltensperger, U.: Evaluation of 1,3,5 trimethylbenzene degradation in the detailed tropospheric chemistry mechanism, MCMv3.1, using environmental chamber data, Atmos. Chem. Phys., 8, 6453–6468, https://doi.org/10.5194/acp-8-6453-2008, 2008.
Miguel, A. H., Kirchstetter, T. M., and Harley, R. A.: On-Road Emissions of Particulate Polycyclic Aromatic Hydrocarbons and Black Carbon from Gasoline and Diesel Vehicles, Environ. Sci. Technol, 32, 450–455, 1998.
Miracolo, M. A., Drozd, G. T., Jathar, S. H., Presto, A. A., Lipsky, E. M., Corporan, E., and Robinson, A. L.: Fuel Composition and Secondary Organic Aerosol Formation: Gas- Turbine Exhaust and Alternative Aviation Fuels, Environ. Sci. Technol., 46, 8493–8501, https://doi.org/10.1021/es300350c, 2012.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Monod, A., Monod, A., Sive, B. C., Avino, P., Chen, T., Blake, D. R., and Rowland, F. S.: Monoaromatic compounds in ambient air of various cities: a focus on correlations between the xylenes and ethylbenzene, Atmos. Environ., 35, 135–149, 2001.
Na, K., Moon, K.-C., and Kim, Y. P.: Source contribution to aromatic VOC concentration and ozone formation potential in the atmosphere of Seoul, Atmos. Environ., 39, 5517–5524, https://doi.org/10.1016/j.atmosenv.2005.06.005, 2005.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
Nguyen, T. B., Roach, P. J., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 11, 6931–6944, https://doi.org/10.5194/acp-11-6931-2011, 2011.
Noda, J., Volkamer, R., and Molina, M. J.: Dealkylation of Alkylbenzenes: A Significant Pathway in the Toluene, o-, m-, p-Xylene + OH Reaction, J. Phys. Chem. A, 113, 9658–9666, https://doi.org/10.1021/jp901529k, 2009.
Odum, J. ., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943, 1996.
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017.
Peng, J., Hu, M., Du, Z., Wang, Y., Zheng, J., Zhang, W., Yang, Y., Qin, Y., Zheng, R., Xiao, Y., Wu, Y., Lu, S., Wu, Z., Guo, S., Mao, H., and Shuai, S.: Gasoline aromatics: a critical determinant of urban secondary organic aerosol formation, Atmos. Chem. Phys., 17, 10743–10752, https://doi.org/10.5194/acp-17-10743-2017, 2017.
Peng, Z. and Jimenez, J. L.: Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev, 49, 2570, https://doi.org/10.1039/c9cs00766k, 2020.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Palm, B. B., Day, D. A., Talukdar, R. K., Hu, W., Lambe, A. T., Brune, W. H., and Jimenez, J. L.: Model Evaluation of New Techniques for Maintaining High-NO Conditions in Oxidation Flow Reactors for the Study of OH-Initiated Atmospheric Chemistry, ACS Earth Sp. Chem., 2, 72–86, https://doi.org/10.1021/acsearthspacechem.7b00070, 2018.
Pereira, K. L., Ward, M. W., Wilkinson, J. L., Sallach, J. B., Bryant, D. J., Dixon, W. J., Hamilton, J. F., and Lewis, A. C.: An automated methodology for non-targeted compositional analysis of small molecules in high complexity environmental matrices, Environ. Sci. Technol., in review, 2020.
Ping, T.: Secondary Organic Aerosol Formation From Aromatic Hydrocarbon, University of California Riverside, available at: https://escholarship.org/uc/item/1zc132bj (last access: 20 March 2019), 2013.
Praske, E., Otkjaer, R. V, Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., Wennberg, P. O., and Lester, M. I.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Priestley, M., Le Breton, M., Bannan, T. J., Leather, K. E., Bacak, A., Reyes-Villegas, E., De Vocht, F., Shallcross, B. M. A., Brazier, T., Anwar Khan, M., Allan, J., Shallcross, D. E., Coe, H., and Percival, C. J.: Observations of Isocyanate, Amide, Nitrate, and Nitro Compounds From an Anthropogenic Biomass Burning Event Using a ToF-CIMS, J. Geophys. Res.-Atmos., 123, 7687–7704, https://doi.org/10.1002/2017JD027316, 2018.
Pye, H. O. T., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Qi, L., Chen, M., Stefenelli, G., Pospisilova, V., Tong, Y., Bertrand, A., Hueglin, C., Ge, X., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 2: Biomass burning influences in winter, Atmos. Chem. Phys., 19, 8037–8062, https://doi.org/10.5194/acp-19-8037-2019, 2019.
Rickard, A. R., Wyche, K. P., Metzger, A., Monks, P. S., Ellis, A. M., Dommen, J., Baltensperger, U., Jenkin, M. E., and Pilling, M. J.: Gas phase precursors to anthropogenic secondary organic aerosol: Using the Master Chemical Mechanism to probe detailed observations of 1,3,5-trimethylbenzene photo-oxidation, Atmos. Environ., 44, 5423–5433, https://doi.org/10.1016/j.atmosenv.2009.09.043, 2010.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Romonosky, D. E., Laskin, A., Laskin, J., and Nizkorodov, S. A.: High-Resolution Mass Spectrometry and Molecular Characterization of Aqueous Photochemistry Products of Common Types of Secondary Organic Aerosols, J. Phys. Chem. A, 119, 2594–2606, https://doi.org/10.1021/jp509476r, 2014.
Rubin, J. I., Kean, A. J., Harley, R. A., Millet, D. B., Goldstein, A. H., Kean, A. J., Harley, R. A., Millet, D. B., and Goldstein, A. H.: Temperature dependence of volatile organic compound evaporative emissions from motor vehicles, J. Geophys. Res, 111, D03305, https://doi.org/10.1029/2005JD006458, 2006.
Sato, K., Hatakeyama, S., and Imamura, T.: Secondary Organic Aerosol Formation during the Photooxidation of Toluene: NOx Dependence of Chemical Composition, J. Phys. Chem. A, 111, 9796–9808, https://doi.org/10.1021/jp071419f, 2007.
Sato, K., Takami, A., Kato, Y., Seta, T., Fujitani, Y., Hikida, T., Shimono, A., and Imamura, T.: AMS and LC/MS analyses of SOA from the photooxidation of benzene and 1,3,5-trimethylbenzene in the presence of NOx: effects of chemical structure on SOA aging, Atmos. Chem. Phys., 12, 4667–4682, https://doi.org/10.5194/acp-12-4667-2012, 2012.
Sato, K., Fujitani, Y., Inomata, S., Morino, Y., Tanabe, K., Hikida, T., Shimono, A., Takami, A., Fushimi, A., Kondo, Y., Imamura, T., Tanimoto, H., and Sugata, S.: A study of volatility by composition, heating, and dilution measurements of secondary organic aerosol from 1,3,5-trimethylbenzene, Atmos. Chem. Phys., 19, 14901–14915, https://doi.org/10.5194/acp-19-14901-2019, 2019.
Schobesberger, S., D'Ambro, E. L., Lopez-Hilfiker, F. D., Mohr, C., and Thornton, J. A.: A model framework to retrieve thermodynamic and kinetic properties of organic aerosol from composition-resolved thermal desorption measurements, Atmos. Chem. Phys., 18, 14757–14785, https://doi.org/10.5194/acp-18-14757-2018, 2018.
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017.
Shilling, J. E., Chen, Q., King, S. M., Rosenoern, T., Kroll, J. H., Worsnop, D. R., DeCarlo, P. F., Aiken, A. C., Sueper, D., Jimenez, J. L., and Martin, S. T.: Loading-dependent elemental composition of α-pinene SOA particles, Atmos. Chem. Phys., 9, 771–782, https://doi.org/10.5194/acp-9-771-2009, 2009.
Simpson, D., Winiwarter, W., Cinderby, S., Ferreiro, A., Guenther, A., Nicholas Hewitt, C., Janson, R., Aslam Khalil, M. K., Owen, S., Pierce, T. E., Puxbaum, H., Shearer, M., Skiba, U., Steinbrecher, R., Tarras, L., and Öquist, M. G.: Inventorying emissions from nature in Europe Publication Date, J. Geophys. Res., 104, 8113–8152, https://doi.org/10.1029/98JD02747, 1999.
Smith, D. F., McIver, C. D., and Kleindienst, T. E.: Primary product distribution from the reaction of hydroxyl radicals with toluene at ppb NO(X) mixing ratios, J. Atmos. Chem., 30, 209–228, https://doi.org/10.1023/A:1005980301720, 1998.
Smith, D. F., Kleindienst, T. E., and McIver, C. D.: Primary product distributions from the reaction of OH with m-, p-xylene, 1,2,4- and 1,3,5-trimethylbenzene, J. Atmos. Chem., 34, 339–364, https://doi.org/10.1023/A:1006277328628, 1999.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kimmel, J. R., Cubison, M. J., Chhabra, P. S., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Methods to extract molecular and bulk chemical information from series of complex mass spectra with limited mass resolution, Int. J. Mass Spectrom., 389, 26–38, https://doi.org/10.1016/j.ijms.2015.08.011, 2015.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Stönner, C., Derstroff, B., Klüpfel, T., Crowley, J. N., and Williams, J.: Glyoxal measurement with a proton transfer reaction time of flight mass spectrometer (PTR-TOF-MS): characterization and calibration, J. Mass Spectrom., 52, 30–35, https://doi.org/10.1002/jms.3893, 2017.
Suh, I., Zhang, R., Molina, L. T., and Molina, M. J.: Oxidation Mechanism of Aromatic Peroxy and Bicyclic Radicals from OH-Toluene Reactions, J. Am. Chem. Soc., 125, 12655–12665, https://doi.org/10.1021/ja0350280, 2003.
Szmigielski, R., Surratt, J. D., Gómez-González, Y., Van der Veken, P., Kourtchev, I., Vermeylen, R., Blockhuys, F., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Seinfeld, J. H., Maenhaut, W., and Claeys, M.: 3-methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Lett., 34, L24811, https://doi.org/10.1029/2007GL031338, 2007.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Tan, Z., Lu, K., Jiang, M., Su, R., Wang, H., Lou, S., Fu, Q., Zhai, C., Tan, Q., Yue, D., Chen, D., Wang, Z., Xie, S., Zeng, L., and Zhang, Y.: Daytime atmospheric oxidation capacity in four Chinese megacities during the photochemically polluted season: a case study based on box model simulation, Atmos. Chem. Phys., 19, 3493–3513, https://doi.org/10.5194/acp-19-3493-2019, 2019.
Tsiligiannis, E., Hammes, J., Salvador, C. M., Mentel, T. F., and Hallquist, M.: Effect of NOx on 1,3,5-trimethylbenzene (TMB) oxidation product distribution and particle formation, Atmos. Chem. Phys., 19, 15073–15086, https://doi.org/10.5194/acp-19-15073-2019, 2019.
Vereecken, L., Aumont, B., Barnes, I., Bozzelli, J. W., Goldman, M. J., Green, W. H., Madronich, S., Mcgillen, M. R., Mellouki, A., Orlando, J. J., Picquet-Varrault, B., Rickard, A. R., Stockwell, W. R., Wallington, T. J., and Carter, W. P. L.: Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry, Int. J. Chem. Kinet., 50, 435–469, https://doi.org/10.1002/kin.21172, 2018.
Volkamer, R., Klotz, B., Barnes, I., Imamura, T., Wirtz, K., Washida, N., Becker, K. H., and Platt, U.: OH-initiated oxidation of benzene Part I. Phenol formation under atmospheric conditions, Phys. Chem. Chem. Phys., 4, 1598–1610, https://doi.org/10.1039/b108747a, 2002.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006GL026899, 2006.
von Schneidemesser, E., Monks, P. S., and Plass-Duelmer, C.: Global comparison of VOC and CO observations in urban areas, Atmos. Environ., 44, 5053–5064, https://doi.org/10.1016/j.atmosenv.2010.09.010, 2010.
Wang, L., Atkinson, R., and Arey, J.: Dicarbonyl Products of the OH Radical-Initiated Reactions of Naphthalene and the C1-and C2-Alkylnaphthalenes, Environ. Sci. Technol., 41, 2803–2810, https://doi.org/10.1021/es0628102, 2007.
Wang, S., Nan, J., Shi, C., Fu, Q., Gao, S., Wang, D., Cui, H., Saiz-Lopez, A., and Zhou, B.: Atmospheric ammonia and its impacts on regional air quality over the megacity of Shanghai, China, Nat. Sci. Reports, 5, 15842, https://doi.org/10.1038/srep15842, 2015.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: Autoxidation and accretion, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2020-165, in review, 2020.
Wu, R., Pan, S., Li, Y., and Wang, L.: Atmospheric Oxidation Mechanism of Toluene, J. Phys. Chem. A, 118, 4533–4547, https://doi.org/10.1021/jp500077f, 2014.
Wyche, K. P., Monks, P. S., Ellis, A. M., Cordell, R. L., Parker, A. E., Whyte, C., Metzger, A., Dommen, J., Duplissy, J., Prevot, A. S. H., Baltensperger, U., Rickard, A. R., and Wulfert, F.: Gas phase precursors to anthropogenic secondary organic aerosol: detailed observations of 1,3,5-trimethylbenzene photooxidation, Atmos. Chem. Phys., 9, 635–665, https://doi.org/10.5194/acp-9-635-2009, 2009.
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016.
Yarwood, G., Whitten, G., and Rao, S.: Updates to the Carbon Bond 4 photochemical mechanism, available at: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.616.4133&rep=rep1&type=pdf (last access: 17 August 2020), 2005.
Yu, J., Jeffries, H. E., and Sexton, K. G.: Atmospheric photooxidation of alkylbenzenes – I. Carbonyl product analyses, Atmos. Environ., 31, 2261–2280, https://doi.org/10.1016/S1352-2310(97)00011-3, 1997.
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019.
Zhang, X., Mcvay, R. C., Huang, D. D., Dalleska, N. F., Aumont, B., Flagan, R. C., and Seinfeld, J. H.: Formation and evolution of molecular products in α-pinene secondary organic aerosol, P. Natl. Acad. Sci. USA, 112, 14168–14173, https://doi.org/10.1073/pnas.1517742112, 2015.
Zhang, Y., Wang, X., Barletta, B., Simpson, I. J., Blake, D. R., Fu, X., Zhang, Z., He, Q., Liu, T., Zhao, X., and Ding, X.: Source attributions of hazardous aromatic hydrocarbons in urban, suburban and rural areas in the Pearl River Delta (PRD) region, J. Hazard. Mater., 250–251, 403–411, https://doi.org/10.1016/j.jhazmat.2013.02.023, 2013.
Ziemann, P. J., Atkinsonz, R., and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.