the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Aug 2020
| 05 Aug 2020
OH and HO2 radical chemistry in a midlatitude forest: measurements and model comparisons
Michelle M. Lew
Pamela S. Rickly
Brandon P. Bottorff
Emily Reidy
Sofia Sklaveniti
Thierry Léonardis
Nadine Locoge
Sebastien Dusanter
Shuvashish Kundu
Ezra Wood
Reactions of the hydroxyl (OH) and peroxy (HO2 and RO2) radicals play a central role in the chemistry of the atmosphere. In addition to controlling the lifetimes of many trace gases important to issues of global climate change, OH radical reactions initiate the oxidation of volatile organic compounds (VOCs) which can lead to the production of ozone and secondary organic aerosols in the atmosphere. Previous measurements of these radicals in forest environments characterized by high mixing ratios of isoprene and low mixing ratios of nitrogen oxides (NOx) (typically less than 1–2 ppb) have shown serious discrepancies with modeled concentrations. These results bring into question our understanding of the atmospheric chemistry of isoprene and other biogenic VOCs under low NOx conditions.
During the summer of 2015, OH and HO2 radical concentrations, as well as total OH reactivity, were measured using laser-induced fluorescence–fluorescence assay by gas expansion (LIF-FAGE) techniques as part of the Indiana Radical Reactivity and Ozone productioN InterComparison (IRRONIC). This campaign took place in a forested area near Indiana University's Bloomington campus which is characterized by high mixing ratios of isoprene (average daily maximum of approximately 4 ppb at 28 ∘C) and low mixing ratios of NO (diurnal average of approximately 170 ppt). Supporting measurements of photolysis rates, VOCs, NOx, and other species were used to constrain a zero-dimensional box model based on the Regional Atmospheric Chemistry Mechanism (RACM2) and the Master Chemical Mechanism (MCM 3.2), including versions of the Leuven isoprene mechanism (LIM1) for HOx regeneration (RACM2-LIM1 and MCM 3.3.1). Using an OH chemical scavenger technique, the study revealed the presence of an interference with the LIF-FAGE measurements of OH that increased with both ambient concentrations of ozone and temperature with an average daytime maximum equivalent OH concentration of approximately 5×106 cm−3. Subtraction of the interference resulted in measured OH concentrations of approximately 4×106 cm−3 (average daytime maximum) that were in better agreement with model predictions although the models underestimated the measurements in the evening. The addition of versions of the LIM1 mechanism increased the base RACM2 and MCM 3.2 modeled OH concentrations by approximately 20 % and 13 %, respectively, with the RACM2-LIM1 mechanism providing the best agreement with the measured concentrations, predicting maximum daily OH concentrations to within 30 % of the measured concentrations. Measurements of HO2 concentrations during the campaign (approximately a 1×109 cm−3 average daytime maximum) included a fraction of isoprene-based peroxy radicals () and were found to agree with model predictions to within 10 %–30 %. On average, the measured reactivity was consistent with that calculated from measured OH sinks to within 20 %, with modeled oxidation products accounting for the missing reactivity, however significant missing reactivity (approximately 40 % of the total measured reactivity) was observed on some days.
- Article
(6382 KB) - Full-text XML
-
Supplement
(1178 KB) - BibTeX
- EndNote





The hydroxyl radical (OH) is one of the primary oxidants in the atmosphere (Levy, 1972). The OH radical initiates the oxidation of volatile organic compounds (VOCs) that leads to the production of hydroperoxy radicals (HO2) and organic peroxy radicals (RO2). In the presence of nitrogen oxides (), reactions of these radicals can lead to the production of ozone and secondary organic aerosols in the atmosphere, the primary components of photochemical smog. Because of their short atmospheric lifetimes, measurements of OH and HO2 (together HOx) and total OH reactivity can provide a robust test of our understanding of this complex chemistry (Heard and Pilling, 2003).
Multiple field campaigns have been conducted over the years measuring OH and HO2 radicals in both urban and forested environments. Measurements of OH in urban areas characterized by high mixing ratios of NOx and anthropogenic VOCs have been generally consistent with model predictions (Ren et al., 2003; Shirley et al., 2006; Kanaya et al., 2007a; Dusanter et al., 2009b; Hofzumahaus et al., 2009; Griffith et al., 2016; Tan et al., 2017, 2018, 2019), while measurements in remote forested environments characterized by low mixing ratios of NOx and high mixing ratios of biogenic VOCs have often been greater than model predictions (Tan et al., 2001; Lelieveld et al., 2008; Whalley et al., 2011; Rohrer et al., 2014).
However, recent measurements by Mao et al. (2012) in a northern Californian forest using a new chemical scavenging technique that removes ambient OH before air enters the detection cell revealed a significant interference associated with their laser-induced fluorescence (LIF) measurements of OH. The unknown interference was a factor of 2 to 3 times higher than ambient OH concentrations (Mao et al., 2012). Similar results were observed in a boreal forest by Novelli et al. (2014), who observed an interference using a similar chemical scrubbing technique that was a factor of 3 to 4 times higher than ambient OH concentrations. One possible source of this observed interference may be the decomposition of Criegee intermediates produced from the ozonolysis of biogenic emissions in the low-pressure detection cells used by LIF instruments, however the ambient concentration of these intermediates in the atmosphere may be too low to explain all of the observed interference (Novelli et al., 2017; Rickly and Stevens, 2018). Another proposed source of the interference is the decomposition of ROOOH molecules inside the fluorescence assay by gas expansion (FAGE) detection cell formed from the reaction of OH with RO2 radicals (Fittschen et al., 2019). Nevertheless, interferences associated with measurements of OH could explain part of the discrepancies between measured and modeled OH concentrations in forested environments. Monitoring potential interferences associated with OH measurements using LIF techniques may be crucial for understanding the discrepancies between measurements and models.
In contrast to measurements of OH, the agreement between measured and modeled HO2 concentrations has been highly variable. In urban environments, measured HO2 concentrations were sometimes found to agree with model predictions (Shirley et al., 2006; Emmerson et al., 2007; Dusanter et al., 2009b; Michoud et al., 2012; Lu et al., 2013; Ren et al., 2013; Griffith et al., 2016; Tan et al., 2017), while other times the measurements were found to be both lower (George et al., 1999; Konrad et al., 2003) and higher than model predictions (Martinez et al., 2003; Ren et al., 2003; Emmerson et al., 2005; Kanaya et al., 2007a; Chen et al., 2010; Sheehy et al., 2010; Czader et al., 2013; Griffith et al., 2016; Tan et al., 2018). In forested environments, measured HO2 concentrations were sometimes found to agree with model predictions (Tan et al., 2001; Ren et al., 2005, 2006) but were often found to be either lower (Carslaw et al., 2001; Kanaya et al., 2007b; Whalley et al., 2011; Kanaya et al., 2012; Mao et al., 2012; Griffith et al., 2013; Mallik et al., 2018) or higher than model predictions (Carslaw et al., 2001; Kubistin et al., 2010; Kim et al., 2013; Hens et al., 2014). Part of this variability may be due to interferences from alkene- and aromatic-based RO2 radicals converting to HO2 in systems that detect HO2 through the conversion to OH by the addition of NO in the sample cell. The degree to which the RO2 species can interfere with HO2 measurements has been quantified through several laboratory experiments (Fuchs et al., 2011; Whalley et al., 2013; Lew et al., 2018) and estimated in some field studies (Hens et al., 2014; Crowley et al., 2018; Mallik et al., 2018). However, the extent of RO2 radical contributions during HO2 measurements in many of the campaigns mentioned above is unclear.
Total OH reactivity measurements can complement HOx measurements by providing a constraint on the total loss of OH that can be compared to that calculated from colocated measurements of OH sinks. Several recent studies have identified discrepancies between measured and calculated OH reactivity in which the measured values are significantly greater than the calculated values (Di Carlo et al., 2004; Hansen et al., 2014; Nölscher et al., 2016; Zannoni et al., 2016; Bsaibes et al., 2020). This difference has been attributed to OH loss from unmeasured VOCs and their oxidation products. In general, significant missing OH reactivity has not been observed as often in urban environments as it has in forested areas, bringing into question our understanding of the chemistry of biogenic emissions and their oxidation products (Dusanter and Stevens, 2017).
This study reports measurements and model simulations of HOx radical chemistry and OH reactivity for a forested site located in Bloomington, Indiana, USA, during the 2015 IRRONIC (Indiana Radical Reactivity and Ozone productioN InterComparison) field campaign. This work compares the measured HOx radical concentrations to model predictions incorporating the Regional Atmospheric Chemistry Mechanism 2 (RACM2) in addition to a version updated to include the Leuven isoprene mechanism (RACM2-LIM1), as well as the Master Chemical Mechanism (MCM) versions 3.2 and 3.3.1, in order to test the ability of each model to reproduce the observed radical concentrations and total OH reactivity.
2.1 IRRONIC location and supporting measurements
The IRRONIC campaign site was located within a mixed deciduous forest (sugar maple, sycamore, tulip poplar, ash, and hickory trees) at the Indiana University Research and Teaching Preserve (IU-RTP) field lab (39.1908∘ N, 86.502∘ W) located approximately 2.5 km northeast of the center of the Indiana University campus and 1 km from the IN 45∕46 bypass at the northern perimeter. The goals of the campaign included an informal intercomparison of peroxy radical measurements by two different techniques (Kundu et al., 2019), an analysis of ozone production sensitivity at this site (Sklaveniti et al., 2018), a comparison of measured OH radical reactivity with that calculated from measured VOCs, and a comparison of measured OH, HO2, and RO2 radicals with model predictions. The main biogenic emission within this area was isoprene with an average daytime maximum mixing ratio of approximately 4 ppb during the campaign. This area exhibited low anthropogenic influences from the campus area with an average daytime maximum mixing ratio of NO of approximately 315 ppt and an average daytime maximum NO2 mixing ratio of approximately 2 ppb. Measurements were conducted on top of two scaffolding platforms adjacent to the field lab approximately 1.8 m from the ground. At this site, solar noon occurred at approximately 13:52 EDT. Additional information regarding the field site and the IRRONIC campaign can be found in Sklaveniti et al. (2018) and Kundu et al. (2019).
Table 1Measurements conducted during the IRRONIC field campaign.
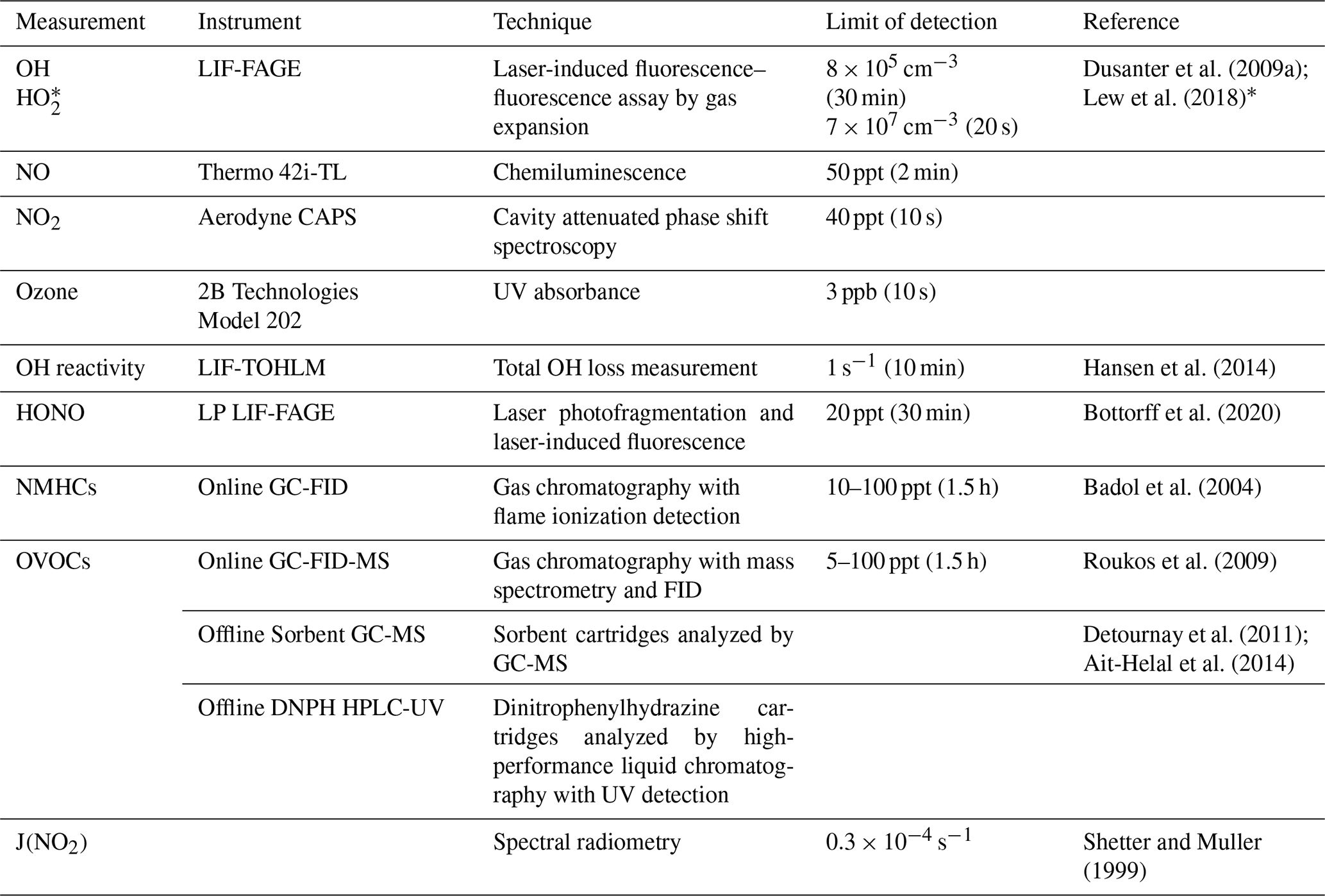
* signifies HO2 plus a fraction of certain RO2 radicals (see text).
Table 1 summarizes the major instrumentation employed during the campaign. NO was measured every 10 s using a chemiluminescence instrument (Thermo model 42i-TL; detection limit 50 ppt, 2 min time resolution). Periodic problems with the sensor's high voltage power supply that required an eventual replacement limited the coverage of the measurements. NO2 was measured every 1 s by a cavity attenuated phase shift (CAPS) instrument (detection limit 40 ppt, 10 s), and ozone was measured every 10 s using a 2B Technologies model 202 UV absorbance instrument (detection limit 3 ppb, 10 s). Further details on the calibration and baseline measurements for the NO, NO2, and O3 measurements are described in Kundu et al. (2019). Non-methane hydrocarbons, including C2–C10 alkanes and alkenes, butadiene, C6–C9 aromatic compounds, isoprene, α-pinene, and β-pinene, were measured using a thermal desorption gas chromatography with flame ionization detection (GC-FID) instrument with a 1.5 h time resolution. Oxygenated VOCs (OVOCs), including C2–C10 aldehydes, C2–C6 ketones, and C2–C4 alcohols, were measured by thermal desorption GC-FID with mass spectrometry (GC-FID-MS) with a 1.5 h time resolution. Offline sampling focused on measurements of oxygenated VOCs, including formaldehyde and C2–C6 aldehydes, acetone, methyl ethyl ketone (MEK), glyoxal, and methylglyoxal, using dinitrophenylhydrazine (DNPH) cartridges, and high-performance liquid chromatography with UV detection (HPLC-UV) analysis. C6–C16 VOCs, including α-pinene, β-pinene, limonene, camphene, heptane-hexadecane, and methylpentene-pentadecene, were measured using Sorbent cartridges and GC-MS analysis. Measurements of J(NO2) were made by spectral radiometry courtesy of the University of Houston, USA. HONO was measured using a newly developed laser photofragmentation–laser-induced fluorescence instrument (Bottorff et al., 2015, 2020).
2.2 HOx radical measurements
The Indiana University laser-induced fluorescence–fluorescence assay by gas expansion (LIF-FAGE) instrument (IU-FAGE) has been described in detail previously and consists of a single axis for alternating measurements of OH and HO2 or (Dusanter et al., 2009a; Griffith et al., 2013, 2016). In the LIF-FAGE technique, OH radicals are detected by laser-induced fluorescence after the expansion of ambient air to low pressure. This extends the OH fluorescence lifetime, allowing temporal filtering of the fluorescence from laser scatter (Heard and Pilling, 2003). Ambient air is expanded through a 0.64 mm diameter orifice located at the top of a cylindrical nozzle (5 cm in diameter and 20 cm long), resulting in a flow rate of approximately 3 slpm (standard liters per minute) through the sampling nozzle. Two scroll pumps (Edwards XDS35i) connected in parallel maintain a pressure inside the cell of 7.3 hPa.
The laser system used in this study consisted of a Spectra-Physics Navigator II YHP40-532Q that produces approximately 8 W of radiation at 532 nm at a repetition rate of 10 kHz which is used to pump a Sirah Credo Dye laser (255 mg L−1 of Rhodamine 610 and 80 mg L−1 of Rhodamine 101 in ethanol), resulting in 40 to 100 mW of radiation at 308 nm. After exiting the dye laser, a fraction of the radiation is focused onto the entrance of a 12 m optical fiber to transmit the radiation to the sampling cell which was placed on top of the 1.8 m platform adjacent to the field lab. In the detection cell, the laser crosses the expanded air perpendicular to the flow in a White cell configuration with 24 passes. For this campaign, the laser power entering the sampling cell ranged from 0.5 to 4.4 mW and was monitored using a photodiode at the exit of the White cell. This does not reflect the laser power density inside the detection cell due to overlap of the beams in the multipass configuration.
OH radicals are excited and detected using the A2Σ+ transition near 308 nm (Stevens et al., 1994). The net signal is measured by spectral modulation by tuning the wavelength on and off resonance in successive modulation cycles. A reference cell where OH is produced by the thermal dissociation of water vapor is used to ensure that the laser is tuned on and off of the OH transition. The OH fluorescence is detected using a microchannel plate photomultiplier tube (MCP-PMT) detector (Hamamatsu R5946U-50), a preamplifier (Stanford Research Systems SR445), and a gated photon counter (Stanford Research Systems SR400). The MCP-PMT is switched off during the laser pulse through the use of electronic gating, allowing the OH fluorescence to be temporally filtered from laser scattered light. A Teflon injector located approximately 2.5 cm below the inlet and 17.5 cm above the detection axis allowed for the addition of NO (approximately 2 sccm, standard cubic centimeters per minute, 1.4×1013 cm−3, 10 % in N2; Matheson Gas) to convert ambient HO2 to OH through the fast reaction, allowing for indirect measurements of HO2.
The IU-FAGE instrument is calibrated by producing known quantities of OH and HO2 from the photolysis of water vapor in air using a mercury pen lamp within the calibration source, as described previously (Dusanter et al., 2008). For these calibrations, zero air was sent through a humidifier and delivered at a flow rate of 38–50 L min−1 to the calibration source. Uncertainties associated with the UV water photolysis calibration technique have been described previously (Dusanter et al., 2008) and are estimated to be 18 % (1σ) for both OH and HO2.
2.2.1 Measurement of OH interferences
The LIF-FAGE measurements are subject to potential interferences when OH radicals are generated inside the detection cell. For example, ozone can be photolyzed by the laser and, in the presence of water vapor, can produce hydroxyl radicals (Davis et al., 1981a, b) (Reactions R1 and R2):
This interference in the IU-FAGE instrument is monitored through laboratory calibrations utilizing various concentrations of ozone, water vapor, and laser power. To characterize this and any other interference during ambient measurements, a chemical scrubbing technique is used to remove ambient OH prior to entering the detection cell (Griffith et al., 2016; Rickly and Stevens, 2018). This chemical modulation technique is used to monitor levels of the laser-generated ozone–water interference and any other processes that may produce OH radicals within the excitation axis.
Hexafluoropropylene (C3F6, 95.5 % in N2; Matheson Gas) is added through a circular injector 1 cm above the nozzle with a flow rate of approximately 3.5 sccm to remove 95 % of externally generated OH (Rickly and Stevens, 2018). During ambient measurements, the chemical addition of C3F6 is modulated in between ambient OH measurements every 15 min for a duration of 10 min. The differences between the measured OH during C3F6 addition and OH measurements including the interference represent the net ambient OH concentration in the atmosphere. Taking the measurement of potential interferences into account results in a limit of detection for OH for this campaign of approximately 7.9×105 cm−3 for a 30 min average ().
2.2.2 Contribution of RO2 interferences during HO2 measurements
As discussed above, HO2 radicals are measured indirectly after sampling ambient air at low pressure through chemical conversion to OH by the addition of NO and the subsequent detection of OH by LIF:
It was previously believed that the detection of HO2 radicals using this technique was free from interferences from the reaction of RO2 radicals with NO as model simulations and measurements suggested that the rate of conversion of RO2 radicals to HO2 by Reactions (R4) and (R5) and subsequent conversion to OH through Reaction (R3) was negligible. This was due to the slow rate of Reaction (R5) under the reduced oxygen concentration in the low pressure LIF-FAGE cell and the short reaction time between the injection of NO and the detection of OH (Heard and Pilling, 2003):
For example, RO2 radicals produced from the OH-initiated oxidation of small alkanes were found to produce a negligible yield of HO2 (Stevens et al., 1994; Kanaya et al., 2001; Tan, et al., 2001; Creasey et al., 2002; Holland et al., 2003). However, recent laboratory studies have shown that there are interferences associated with measurements of HO2 from the conversion of RO2 radicals derived from the OH-initiated oxidation of alkenes and aromatics to HO2 (and subsequently OH) through the reaction with NO. The high conversion efficiency of alkene-based peroxy radicals to HO2 is due to the ability of the β-hydroxyalkoxy radicals produced from OH + VOC reactions to rapidly decompose and form a hydroxyalkyl radical which then reacts rapidly with O2 leading to the production of a carbonyl compound and HO2 (Fuchs et al., 2011; Whalley et al., 2013; Lew et al., 2018). Because of this interference, measurements of peroxy radicals that are sensitive to this interference are denoted as (; ). The conversion efficiency depends on the instrumental characteristics and configurations employed, as well as the amount of NO added. The RO2-to-HO2 conversion efficiencies for a number of different peroxy radicals have been characterized for current and past configurations of the IU-FAGE instrument (Lew et al., 2018). For the configuration of the IU-FAGE instrument used in this study, the conversion efficiency of isoprene-based peroxy radicals was found to be approximately 83 %, while the conversion efficiency of propane peroxy radicals was found to be approximately 15 %. A high concentration of NO leading to a high conversion efficiency of isoprene-based peroxy radicals to HO2 was used throughout the study to provide a useful intercomparison of the IU-FAGE measurements with the RO2+HO2 measurements by the Drexel University's Ethane–Nitric Oxide Chemical Amplifier (ECHAMP) instrument (Kundu et al., 2019) as HO2- and isoprene-based peroxy radicals accounted for approximately 70 % of the total peroxy radicals at this site (see below). To maximize measurement overlap for the intercomparison, low concentrations of added NO were not used, and, as a result, no measurements of HO2 with minimal interference from RO2 radicals were obtained. The instrumental precision for the measurement based on the variability of the background signal due to laser scatter and detector noise results in a limit of detection for during this campaign of 7×107 cm−3 for a 30 s average ().
2.3 OH reactivity measurements
The IU total OH loss rate measurement (TOHLM) instrument is based on the method of Kovacs and Brune (2001) and is described in detail elsewhere (Hansen et al., 2014). Briefly, the instrument is comprised of a flow tube reactor measuring 5 cm in diameter and 75 cm in length. Ambient air is introduced through an 8 cm diameter perfluoroalkoxy polymer film hose attached to the flow tube at a flow rate of approximately 180 slpm using a regenerative blower (Spencer VB001) to establish turbulent flow conditions. Previous measurements have demonstrated that different lengths of this inlet tubing do not significantly impact the measured OH reactivity (Hansen et al., 2014). A pitot-static tube (Dwyer Instruments) is positioned just before the exit of the flow tube facing the turbulent core of the flow approximately 1 cm from the flow tube wall. The pitot-static tube is connected to a differential pressure gauge (MKS Instruments) to measure the total flow tube velocity.
OH radicals are produced in a movable injector that houses a mercury pen lamp (Pen-Ray UV) the top of which was positioned at the end of the injector just before a spiral Teflon spray nozzle used to promote mixing within the flow tube (McMaster-Carr). In addition, a turbulizer is attached to the injector tube 24 cm before the spray nozzle consisting of four 1 cm wide fins to promote turbulent flow conditions, as well as to provide support for the injector throughout the flow tube. The injector is inserted along the main axis and is configured for automated movement to acquire continuous measurements in the forward and backward directions. A nitrogen flow of 10 slpm is bubbled through high-purity water (EMD Chemicals) producing water vapor which is directed through the injector and photolyzed by the pen lamp to produce OH with typical concentrations on the order of 109 cm−3. This method is known to also produce HO2 radicals, which can lead to a regeneration of OH at NO mixing ratios greater than 1 ppbv (Kovacs and Brune, 2001). However, because the average NO mixing ratio measured over the course of the campaign was below this value, no correction to the measured reactivity was applied (Hansen et al., 2014).
OH radicals were measured using a similar FAGE detection cell described above. Ambient air was expanded through a 1 mm diameter orifice to a total pressure of approximately 8 hPa. OH radicals were excited by a portion of the 308 nm output of the dye laser, and the resulting fluorescence was detected by a gated channel photomultiplier tube detector (Excelitas MP 1300) and monitored by a photon counter (Stanford Research SRS400). A 2 m long optical fiber was used to transmit the 308 nm laser beam to the OH reactivity detection cell which was located inside the field lab. The laser power was measured at the exit of the detection cell and monitored with a photodiode.
As ambient air entered the flow tube, the automated OH source injector allowed for varying reaction times with the ambient air over a distance of approximately 15 cm for a period of 2.5 min. This produced an OH decay over a reaction time of 0–0.15 s from which the OH reactivity was determined. Losses of OH on the walls of the flow tube were measured by flowing high-purity nitrogen (Indiana Oxygen) at 180 slpm through the flow tube in addition to the OH production through the injector to measure the decay of OH in the absence of any VOCs. Several measurements of this wall loss (kb) resulted in an average value of 10±2 s−1 (1σ).
The calculated OH reactivity for a measured compound X (kX) can be determined from the product of the concentration of X and its second-order rate constant with OH:
The summation of this value for each reacting species gives the total OH reactivity (kOH):
Under pseudo-first-order conditions ([OH]≪[X]), the OH concentration within the flow tube can be expressed as a first-order exponential decay:
Solving for kOH, the OH reactivity gives
Measurements of the change in the concentration of OH over the reaction time produces the measured OH reactivity value. These measurements can be compared to the calculated total reactivity from measured OH sinks (Eq. 2) to determine whether the measured total OH reactivity can be accounted for by the measured sinks. The difference between the measured and calculated total OH reactivity is referred to as the “missing” OH reactivity.
Laboratory measurements of the reactivity of several VOCs with well-known rate constants, including butane, isoprene, and propane, showed that the OH reactivity measurements for these compounds were on average 30 % lower than calculated when the measured velocity of the turbulent core is used to determine the reaction time. This consistent underestimation of the OH reactivity is likely due to either incomplete mixing of the reactants or a systematic underestimation of the reaction time and is similar to that measured previously by Hansen et al. (2014). As a result, the measured ambient OH reactivity values were scaled by a factor of 1.41. Measurements performed over a range of OH reactivity values suggest that the IU-TOHLM instrument can measure OH reactivity up to 63 s−1 with a precision (1σ) of of the measured value for a 10 min average (Hansen et al., 2014).
2.4 Modeling HOx concentrations and OH reactivity
Ambient measurements of OH, , and total OH reactivity were modeled with the Regional Atmospheric Chemistry Mechanism (RACM2) (Goliff et al., 2013) and the Master Chemical Mechanism (MCM) version 3.2 (Jenkin et al., 1997; Saunders et al., 2003). While the MCM model provides a near-explicit chemical mechanism and is expected to better represent complex chemical atmospheres, the lumped RACM mechanism is easier to use in radical budget calculations. The isoprene oxidation mechanism in RACM2 was updated, as described in Tan et al. (2017), to include the Leuven isoprene mechanism (LIM1) originally proposed by Peeters et al. (2009) which involves peroxy radical isomerization reactions leading to additional HOx radical production. This is a condensed version of the LIM1 mechanism and includes the updated bulk reaction rate constants for the isoprene peroxy radical 1,6-H-shift isomerization reactions as parameterized in Peeters et al. (2014). These isomerization reactions lead to the formation of HO2 and hydroxyperoxy aldehydes (HPALDs) (Crounse et al., 2011; Teng et al., 2017; Berndt et al., 2019), which can subsequently photolyze leading to OH production, as well as di-hydroperoxy carbonyl peroxy radicals (di-HPCARP-RO2) which can rapidly decompose to produce additional OH radicals (Teng et al., 2017; Wennberg et al., 2018). The addition also includes a revision of the chemistry of first-generation isoprene oxidation products, including methyl vinyl ketone (MVK), methacrolein (MACR), and isoprene hydroperoxides (ISHPs) (Tan et al., 2017).
In addition, the ambient measurements were also modeled with version 3.3.1 of the Master Chemical Mechanism. In comparison to version 3.2, MCM 3.3.1 incorporates the explicit LIM1 mechanism, including the equilibrium between different isoprene peroxy radical isomers and H-shift isomerization reactions of specific isomers, resulting in HOx radical recycling through the production of HPALDs, as well as di-HPCARP-RO2 radicals (Jenkin et al., 2015). Based on the recommendation of Peeters (2015), the equilibrium rate coefficients between different peroxy radical isomers were increased, and the 1,6 H-shift isomerization rate constants were decreased in order to match early experimental results of Crounse et al. (2014). These changes resulted in effective bulk 1,6-H-shift peroxy radical isomerization rate constants in MCM 3.3.1 that are approximately a factor of 5 lower than the original LIM1 recommended rates (Novelli et al., 2020).
The Framework for 0-D Atmospheric Modeling (F0AM) was used to calculate the radical concentrations and OH reactivity observed at the IRRONIC site (Wolfe et al., 2016). The model was constrained by the 30 min average measured mixing ratios of ozone, NOx, and VOCs and processed through a 5 d spin-up to generate unmeasured secondary oxidation products. Table S1 in the Supplement summarizes the measured compounds and includes their grouping into the condensed RACM2 model inputs. Because the VOC measurements occurred every 90 min, the measurements were interpolated into 30 min bins before input to the model. Due to the minimal overlap of the NO measurements with the HOx measurements, the model was only run for the days when there were measurements of NO, limiting the number of days for comparison with the radical measurements. Zero-dimensional models cannot explicitly account for emissions, and NO is emitted both by vehicles on the nearby highway 1 km to the southwest and by soil. Such local perturbations to the NOx and O3 radical chemistry necessitate using constrained measurements of NO, NO2, and O3. The measured J(NO2) was used to scale the model-calculated J(NO2) and other photolysis rates. The model uncertainty is approximately 30 % (1σ), estimated from uncertainties associated with the input parameters and the rate constants for each reaction (Griffith et al., 2013; Wolfe et al., 2016).
Campaign diurnal average measurements of J(NO2), temperature, isoprene, O3, NO2, and NO are summarized in Fig. 1. The maximum average mixing ratio of NO of approximately 315 ppt was observed at approximately 08:00 EDT, while the average mixing ratio of NO2 reached a maximum of 2 ppb around 10:00 EDT. Average mixing ratios of isoprene ranged from 0.4 to 4.4 ppb, reaching a maximum around 18:00 EDT. The relatively high nighttime mixing ratios often observed at this site are likely due to the fact that the measurements were made below the forest canopy and relatively close to the surface. As a result, vertical stratification likely resulted in higher concentrations of isoprene near the surface during several nights, which is similar to other measurements of biogenic VOCs below the forest canopy (Bsaibes et al., 2020). Anthropogenic VOCs were relatively low at this site with maximum mixing ratios of benzene less than 80 ppt. Day-to-day profiles (10–25 July) are illustrated in Fig. 2 which show measurements of O3, temperature, isoprene, NOx, , and OH. Unfortunately, instrumental problems limited the NO measurements prior to 19 July.
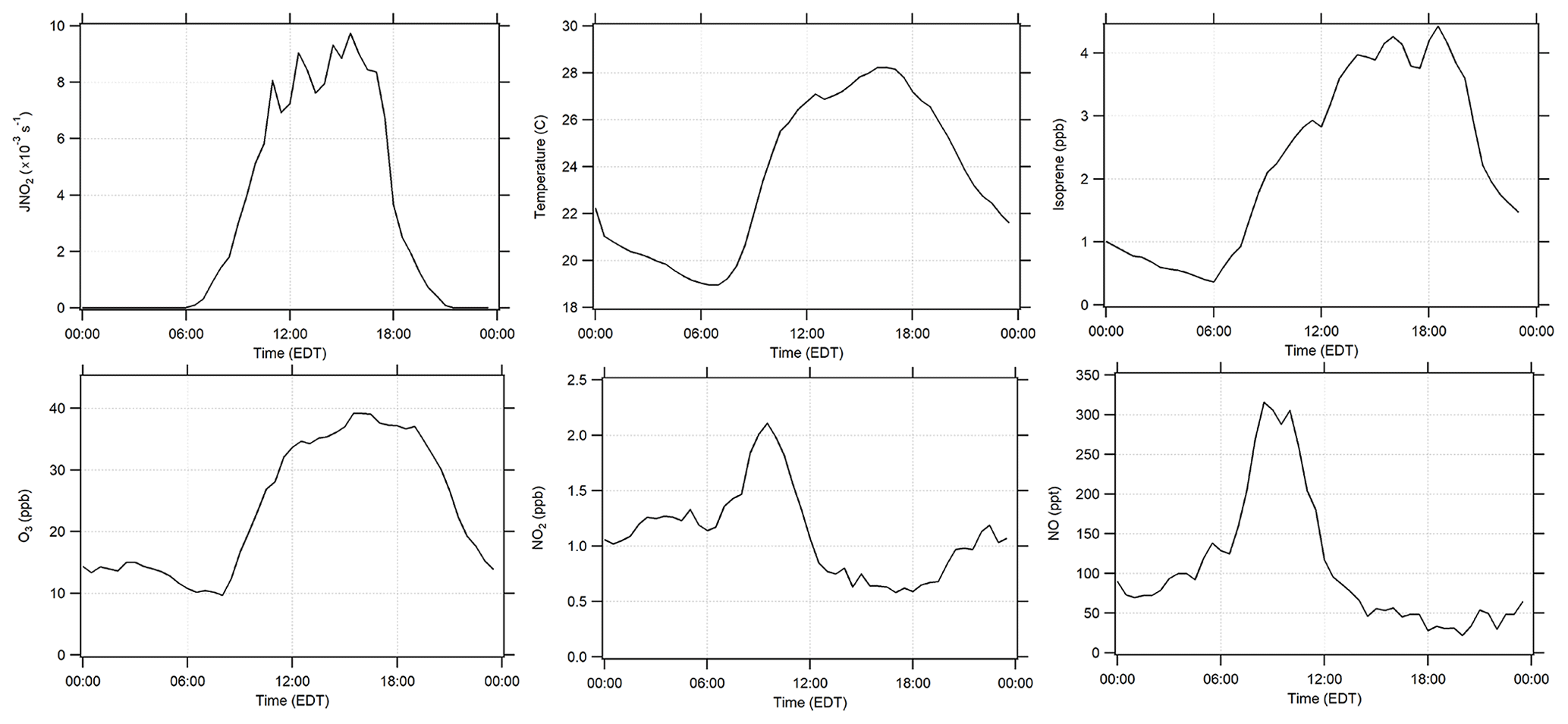
Figure 1Diurnal campaign average profiles of J(NO2), temperature, isoprene, O3, NO2, and NO.
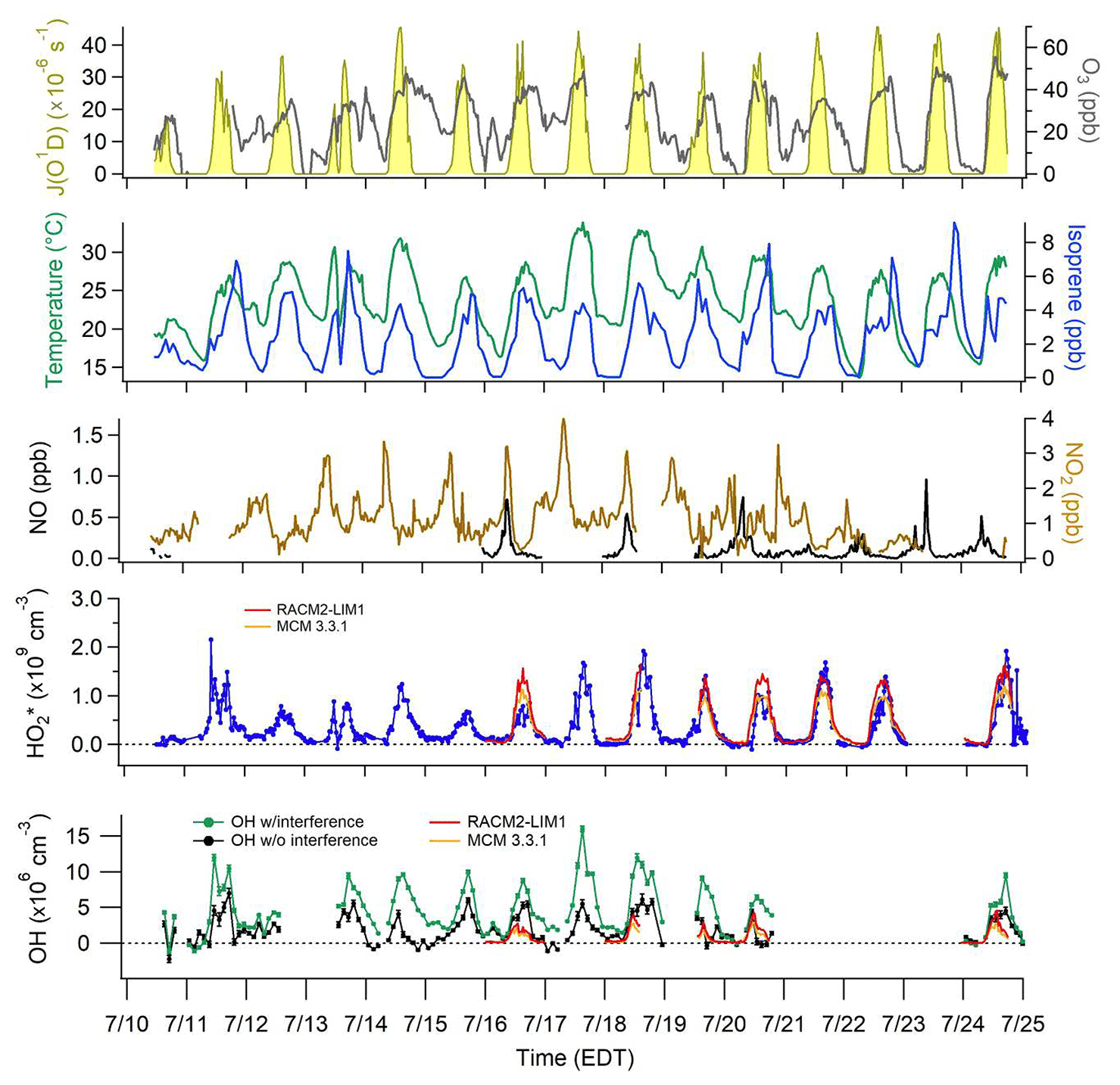
Figure 2Time series of OH and from 10 to 25 July with model-calculated J(O1D) scaled to the measured J(NO2), and measured ozone, temperature, isoprene, and NOx. OH measurements with interference (±1σ) are represented by the green line and measurements without interference (±1σ) by the black line. For clarity, OH data shown are 2 h averages. data are 30 s averages every 30 min. The daily RACM2-LIM1 and MCM 3.3.1 model results for the periods when NO was measured simultaneously are also shown.
3.1 OH measurements and model comparison
OH concentrations were determined using the chemical modulation technique described above utilizing external C3F6 addition to scavenge ambient OH and measure interferences producing OH inside the IU-FAGE detection cell and laser generated OH. The measured interferences were subtracted from the total OH signal determined from spectral modulation, resulting in net ambient OH concentrations (Fig. 2). As can be seen from this figure, the measured interference was a significant fraction of the total OH signal on many days.
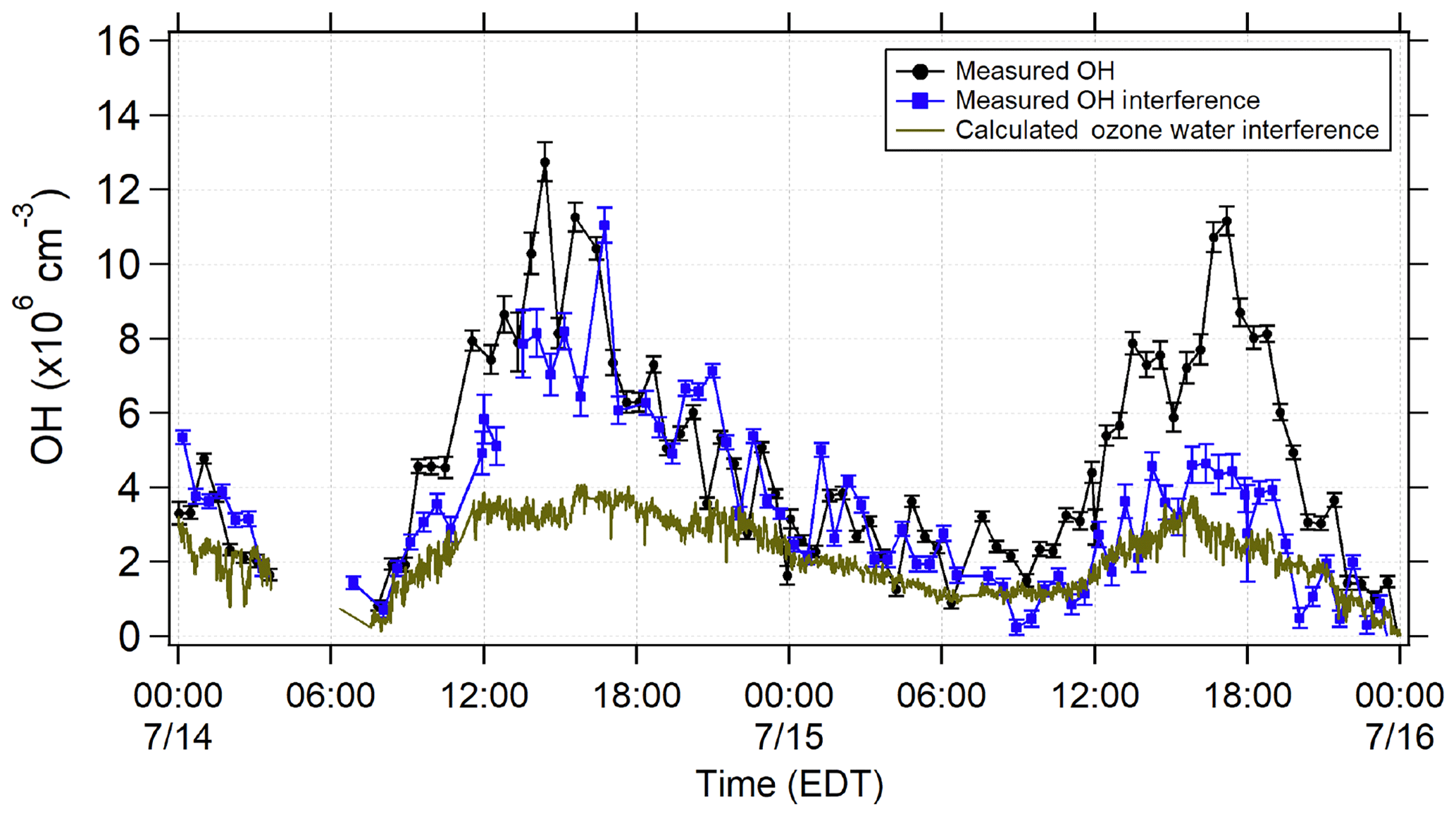
Figure 3Averaged measured total OH signal using spectral modulation (black) and the measured interference using chemical modulation (blue) during 14 and 15 July. The calculated laser-generated interference from ozone photolysis for these days (Reactions R1 and R2; green points) is also shown.
Figure 3 illustrates the total measured OH radical signal by spectral modulation (black circles), the measured interference (blue squares), and the expected laser-generated interference from Reactions (R3) and (R4) calculated from laboratory calibrations (Griffith et al., 2016) (green points) during 14 and 15 July. On 15 July, the measured interference was similar to the calculated interference, suggesting that the majority of the measured interference was laser-generated. However, on 14 July, the measured interference was much larger than the calculated interference, suggesting that the majority of the measured interference was due to an unknown source. Subtraction of the calculated laser-generated interference from the measured interference on all days resulted in a measurement of the unknown interference that increased with both ozone and temperature during the campaign (Fig. 4).
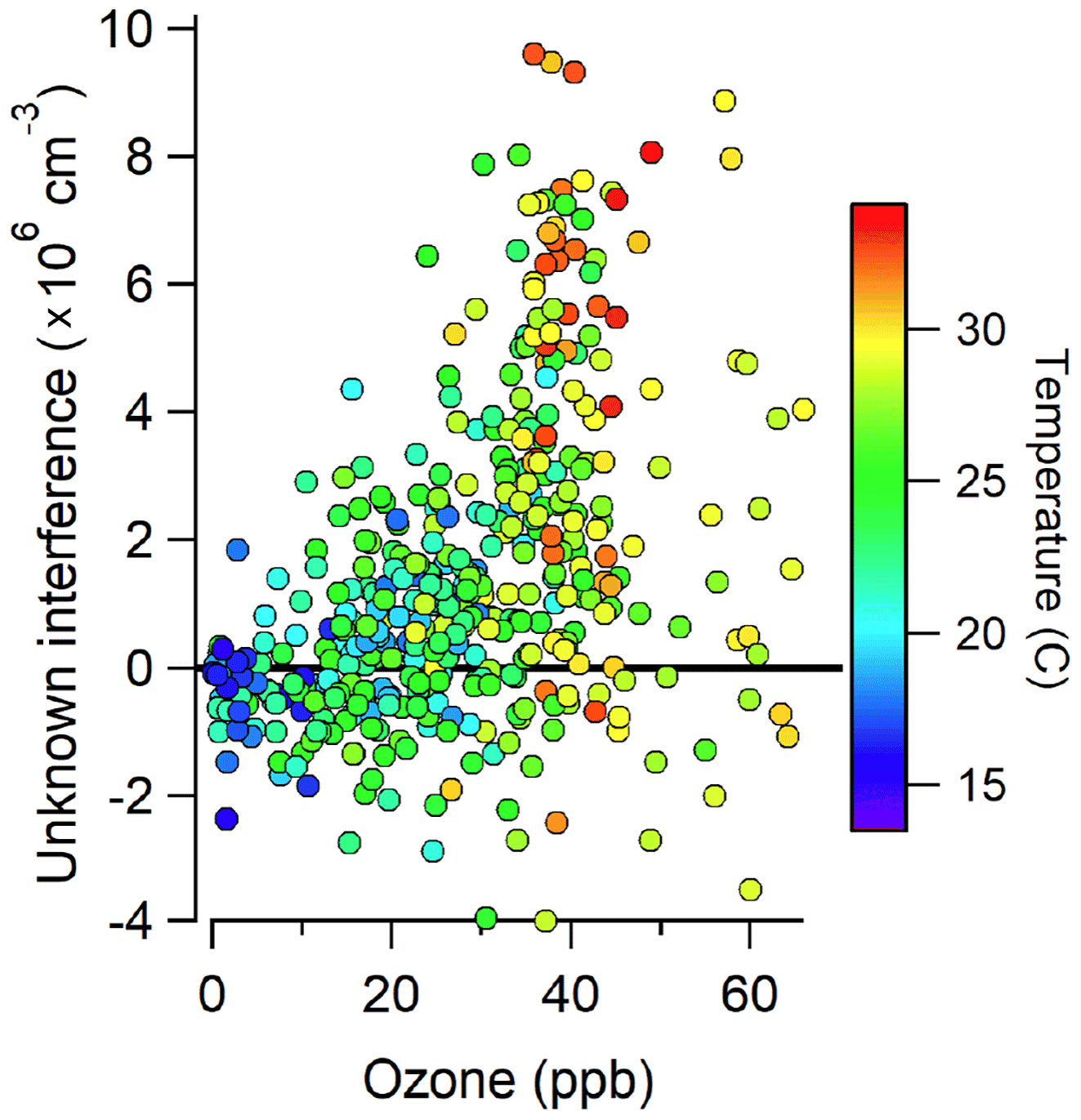
Figure 4Measurements of the unknown interference as a function of ozone and temperature during the campaign.
This result is consistent with the observations from Mao et al. (2012) who found that the interference measured in their LIF-FAGE instrument using a similar chemical modulation technique increased with ozone and total OH reactivity. The observed increase in the magnitude of the unknown interference with ozone and temperature suggests that the interference may be related to the ozonolysis of biogenic VOCs, whose emissions increase with temperature. This result is also consistent with the measurements of Novelli et al. (2017) who found that their observed interference correlated with the product of ozone and biogenic VOC concentrations, although the correlation in the present study was weak (R2=0.15). Previous measurements have shown that some LIF-FAGE instruments, including the IU-FAGE instrument, are susceptible to an interference under high concentrations of ozone and biogenic VOCs perhaps due to the decomposition of Criegee intermediates inside the FAGE detection cell (Novelli et al., 2014, 2017; Fuchs et al., 2016; Rickly and Stevens, 2018). However, estimated concentrations of Criegee intermediates in similar environments on the order of 5×104 cm−3 (Novelli et al., 2017) are too low to explain the observed interference during the IRRONIC campaign.
The observation of a significant interference during this campaign is in contrast to previous measurements of OH by the IU-FAGE instrument in a forested environment during the CABINEX 2009 campaign (Griffth et al., 2013). During this campaign, several tests were conducted where C3F6 or CO was added to remove ambient OH. These tests did not reveal any significant interference, and measurements of OH were found to be in good agreement with model predictions (Griffith et al., 2013). One possible explanation for this discrepancy with the measurements during IRRONIC is the lower levels of ozone and temperatures observed during CABINEX compared to IRRONIC. Average mixing ratios of ozone during CABINEX were near 30 ppb, and average temperatures were near 20 ∘C during the day with average mixing ratios of isoprene less than 2 ppb in the afternoon. These levels of ozone and temperature are lower than those where the interference was observed during IRRONIC (Fig. 4), suggesting that a similar interference was likely undetectable during CABINEX.
Recent measurements have found that NO3 radicals can lead to an interference in FAGE instruments (Fuchs et al., 2016) although the mechanism for the production of this interference is not known. Such an interference in the IU-FAGE instrument could explain the observed interference during some nights (Fig. 3) but is unlikely the source of the interference during the daytime. Another possible source of the interference is the decomposition of ROOOH molecules inside the FAGE detection cell formed from the reaction of OH with RO2 radicals (Fittschen et al., 2019). However, assuming a rate constant of for the OH+RO2 reaction, it is unlikely that a significant fraction of RO2 radicals will react to form ROOOH under the mixing ratios of NO observed at this site as the estimated lifetime of RO2 radicals with respect to reactions with NO was an-order-of-magnitude shorter than that for reactions with OH. Additional measurements and laboratory tests will be needed to identify and minimize interferences associated with LIF-FAGE measurements of OH.
The day-to-day measurements of OH after the interference has been subtracted for 10–20 July and 24–25 July are also illustrated in Fig. 2. Because the interference was not measured simultaneously as the ambient OH measurements, the subtraction of the measured interference often resulted in both apparent negative concentrations and large positive concentrations at night. These large positive and negative values reflect the fact that the nighttime measurements of the interference were much greater than the ambient OH signals and were highly variable between measurement cycles (Fig. 3), resulting in an ambient OH measurement uncertainty that was sometimes larger than the precision calculated from a quadratic propagation of the errors associated with the individual measurements of the ambient OH plus the interference and the interference alone. Measurements on 21–22 July focused on measurements of as part of the peroxy radical informal instrumental intercomparison (Kundu et al., 2019), with NO added continuously to the detection cell to provide measurements with a higher time resolution. Thus OH measurements were not conducted on these days. This figure further illustrates the day-to-day model results for OH and for the days when NO was also measured simultaneously and shows the MCM 3.3.1 and the RACM2-LIM1 models for simplicity.
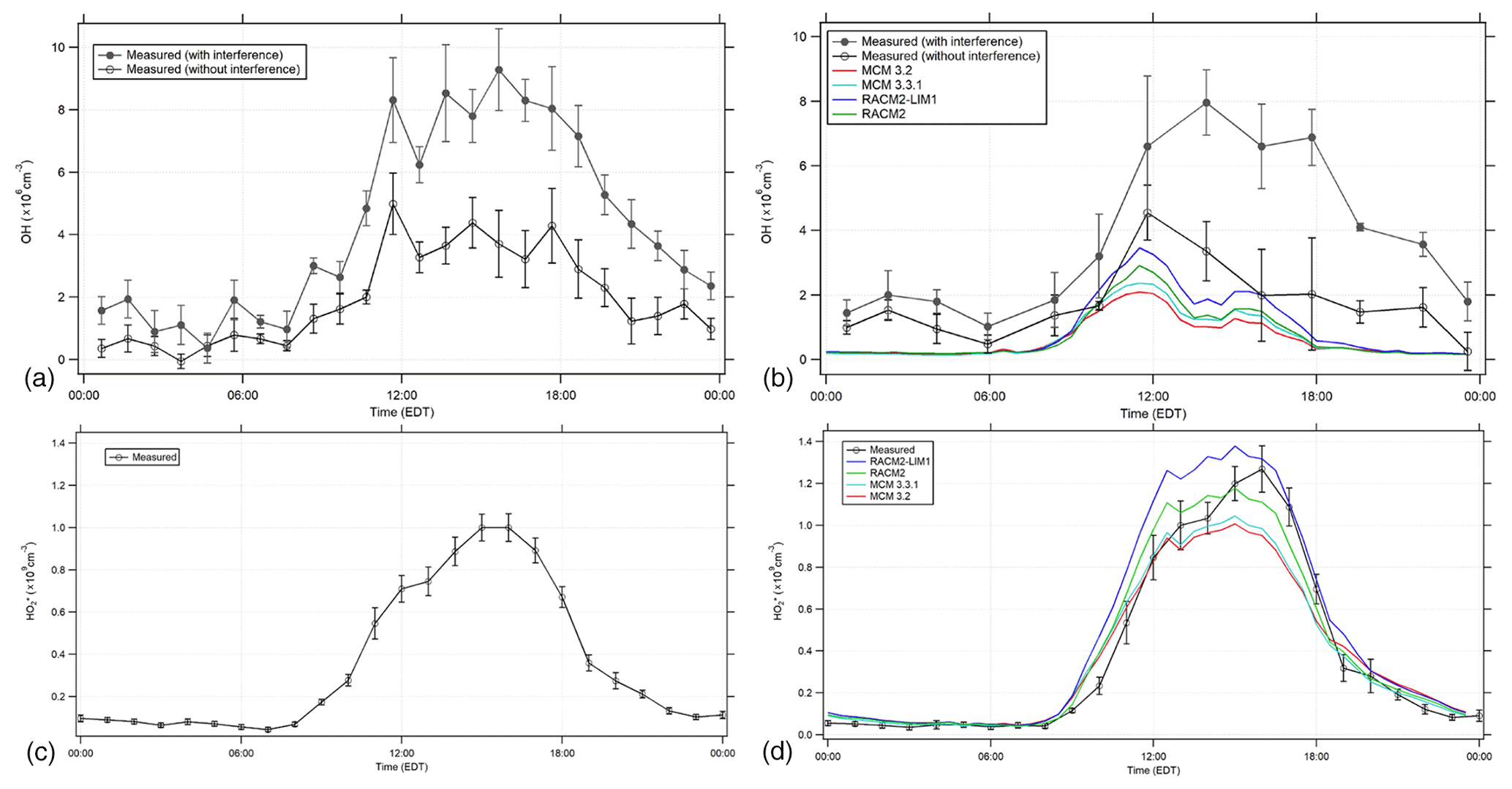
Figure 5Diurnal profiles of OH (a, b) and (c, d) with the RACM2, RACM2-LIM1, MCM 3.2, and MCM 3.3.1 model results. (a, c) illustrate the average of all measurements during the campaign, while (b, d) illustrate the average measurements when NO was measured simultaneously (see text). The open circles represent the 1 or 2 h mean ±1σ standard error of OH and measurements. The filled circles represent the 1 or 2 h mean ±1σ standard error of the OH measurements with the interference.
Figure 5a and b show the average diurnal profile of the 15 min OH measurements, both with and without the measured interference, binned into 1 h time periods for all the days illustrated in Fig. 2 (Fig. 5a) and binned into 2 h time periods along with the MCM and RACM model results for the days when NO was measured simultaneously (16, 18–20, 24 July; Fig. 5b). The average ambient diurnal OH radical concentration measured during the entire campaign reached a maximum of approximately 4–5×106 cm−3 after the measured interference was subtracted (Fig. 5a) and was slightly lower during the period when NO measurements were available (Fig. 5b). If the measured interference was not subtracted from the total OH signal determined by spectral modulation, the resulting OH radical concentrations would be as high as 9×106 cm−3 (Fig. 5), much greater than the MCM and RACM2 modeled diurnal average maximum concentrations of approximately 2–3×106 cm−3. The daytime OH radical concentration measurements after the interference has been subtracted are in better agreement with the model results. Including versions of the LIM1 mechanism in both the MCM (3.3.1) and RACM2 (RACM2-LIM1) models increases the predicted daytime concentrations of OH by approximately 13 % and 20 %, respectively, compared to the base mechanisms during the day (9:00–17:00 EDT), when mixing ratios of NO on these days decreased from an average morning maximum near 500 ppt to approximately 50 ppt in the afternoon. The RACM2-LIM1 results are generally within 30 % of the measured concentrations during the day (Fig. 5b) with the difference at 14:00 EDT within the combined measurement precision, the accuracy of the calibration (18 %, 1σ), and the model uncertainty (30 %). In contrast the MCM 3.3.1 model underpredicted the measurements during this period by approximately a factor of 2. These results are similar to those of Novelli et al. (2020) who found that the MCM 3.3.1 underpredicted measurements of OH by a factor of approximately 1.4 during isoprene oxidation experiments in the SAPHIR (Simulation of Atmospheric PHotochemistry In a large Reaction) chamber when mixing ratios of NO were less than 0.2 ppb, which are similar to the mixing ratios of NO measured in this study. The measured OH concentrations could be reproduced using a model that incorporated the larger equilibrium rate coefficients between the different peroxy radical isomers in the MCM 3.3.1 mechanism with the larger 1,6-H-shift peroxy radical rate constants from the Caltech isoprene mechanism (Teng et al., 2017; Wennberg et al., 2018). This combination increased production of HPALDs and di-HPCARP-RO2 radicals in the oxidation mechanism, resulting in an effective bulk isoprene peroxy radical isomerization rate similar to that in the original LIM1 mechanism (Novelli et al., 2020). These larger bulk peroxy radical isomerization rates are similar to those incorporated into the RACM2-LIM1 mechanism used in the present study (Tan et al., 2017), leading to the higher modeled radical concentrations compared to the MCM 3.3.1 model results shown in Fig. 5.
Although the RACM2-LIM1 mechanism appears to be able to reproduce the daytime OH radical measurements compared to the MCM 3.3.1 mechanism, all the models underestimate the measurements in the early evening and night. While there is uncertainty associated with these nighttime measurements due to the large interference that was subtracted, similar concentrations of OH were observed during the evening by both the IU-FAGE instrument and the University of Colorado chemical ionization mass spectrometry (CIMS) instrument at this site in 2017 during an informal instrument intercomparison (Rosales et al., 2018; Reidy et al., 2018). These results suggest that there may be a missing radical source during this period, such as the ozonolysis of unmeasured biogenic VOCs, and additional measurements will be required to resolve this discrepancy.
3.2 measurements and model comparison
The day-to-day measurements of are also illustrated in Fig. 2 with the MCM 3.3.1 and RACM2-LIM1 model results for the days when NO was also measured simultaneously. Figure 5 shows the average diurnal profile of the measurements for all the days illustrated in Fig. 2 (Fig. 5c), which were similar to the measured concentrations for the days when NO was measured simultaneously along with the MCM and RACM model results (Fig. 5d). The contribution of modeled RO2 radicals to the modeled is based on laboratory calibrations of the RO2-to-HO2 conversion efficiencies for the sampling conditions used in this study (Lew et al., 2018) and is incorporated into both versions of the RACM2 and MCM peroxy radical categories. Under the instrumental conditions during the campaign, the conversion efficiency of isoprene-based peroxy radicals to HO2 was determined to be approximately 83±7 %, while the conversion efficiency of methyl peroxy radicals was estimated to be approximately 5 % (Lew et al., 2018). These two peroxy radicals accounted for the majority of RO2 radicals predicted by the models (see below). The maximum measured concentration each day during the campaign was generally between approximately 2×108 and 2×109 cm−3 (Fig. 2) with an average daily maximum value of approximately 1×109 cm−3 (Fig. 5). The RACM2-LIM1 and MCM 3.3.1 modeled diurnal averaged values reached a maximum of approximately 1.4×109 cm−3 and 1.0×109 cm−3, respectively, compared to values of 1.2×109 and 1.0×109 cm−3, respectively, for the RACM2 and MCM 3.2 modeled (Fig. 5).
The predicted concentrations by the base RACM2 model are in good agreement with the measured concentrations when NO was measured simultaneously with the model results within approximately ±10 % of the measurements on average and within the calibration uncertainty of the measurements (38 %, 2σ). Including the LIM1 mechanism in the RACM2 mechanism increases the modeled by approximately 15 %–20 % due to the modeled increase in HOx radical production from the isomerization of isoprene-based peroxy radicals, overpredicting the measurements by approximately 10 %–30 %. These results are in contrast to those observed during the CABINEX campaign, where a RACM-based model overpredicted the measured by as much as a factor of 2 (Griffith et al., 2013), which is likely related to the higher concentrations of NO observed during IRRONIC compared to CABINEX increasing the importance of the HO2+NO and RO2+NO reactions in determining the fate of these radicals. The MCM-based model results are also in good agreement with the measured (Figs. 2 and 5). The concentrations predicted by the MCM 3.3.1 and 3.2 mechanisms are approximately 5 %–30 % less than the measurements between 12:00 and 18:00 EDT but within the calibration uncertainty of the measurements, with the MCM 3.3.1 mechanism predicting slightly greater concentrations due to the inclusion of HOx production from the isomerization of isoprene-based peroxy radicals.
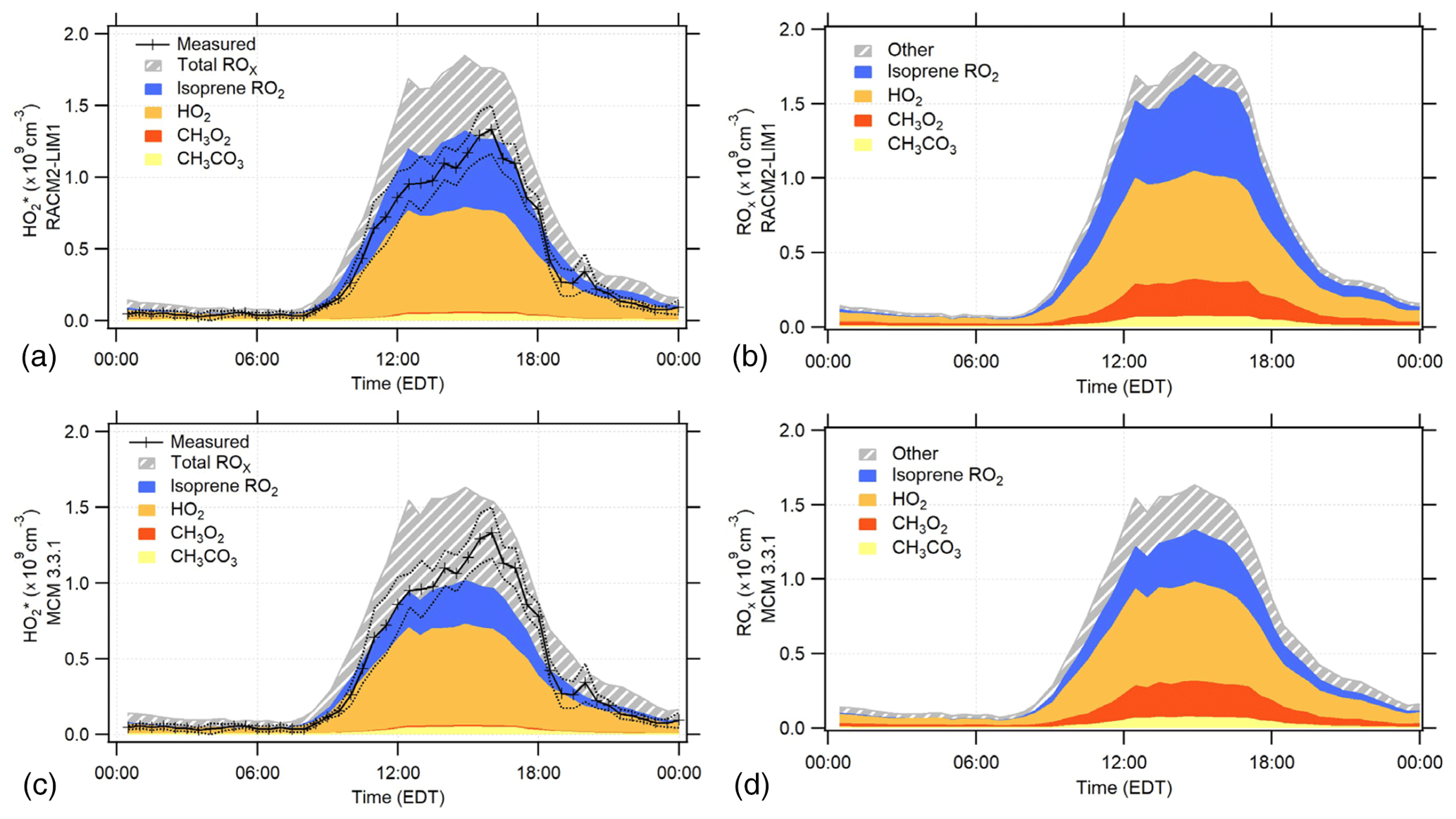
Figure 6The RACM2-LIM1 and MCM 3.3.1 diurnal average modeled peroxy radical concentration and composition for the days when NO was measured simultaneously (see text). (a, c) The modeled contribution to the concentrations. The measured 30 min mean concentrations are shown by the black line with ±1σ standard error of the measurements shown by the dotted lines. The calibration uncertainty of the measurements (not shown) is 38 % (2σ). (b, d) The total ROx (RO2+HO2) composition predicted by each model.
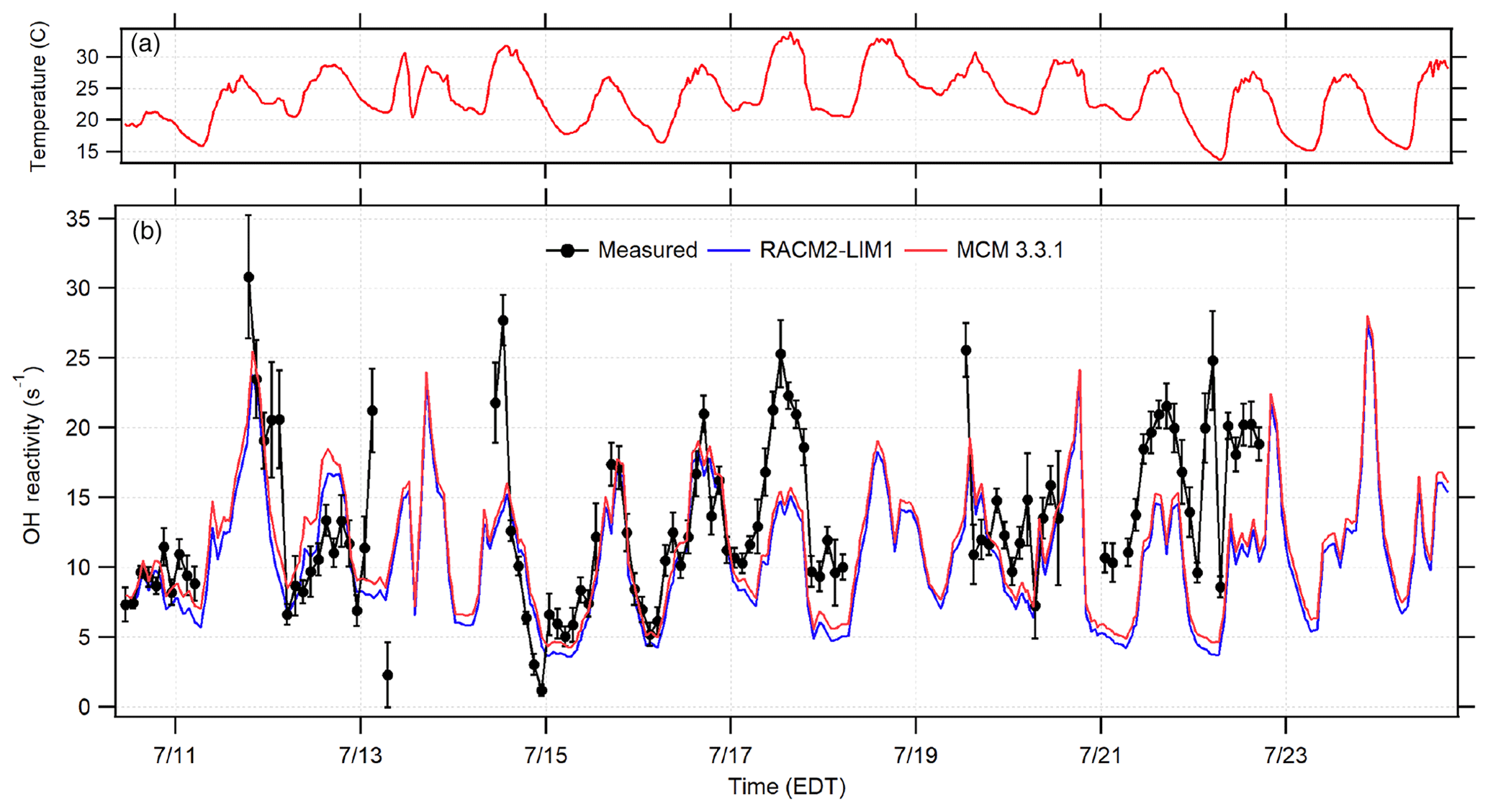
Figure 7Time series of the 2 h averaged OH reactivity measurements (black circles) in comparison to the calculated RACM2-LIM1 and MCM 3.3.1 OH reactivity based on measured OH sinks along with ambient temperature (a). Error bars represent the standard error of the average measurement.
The RACM2-LIM1 and MCM 3.3.1 diurnal average modeled concentrations and the model contribution of peroxy radicals to are shown in Fig. 6 for the days when NO was measured simultaneously (panels a and c). The diurnal profile of the radical concentration predicted by these models includes contributions primarily from isoprene peroxy radicals and HO2 radicals with smaller contributions from methyl peroxy and acetyl peroxy radicals (Fig. 6). The RACM2 and MCM 3.2 models produced similar results, with HO2 and isoprene peroxy radicals contributing to the majority of the modeled concentrations (Fig. S1 in the Supplement). The total modeled ROx (RO2+HO2) concentrations by the different mechanisms for these days are also shown in Fig. 6 (panels b and d). The RACM2-LIM1 model predicted that the diurnal average total ROx concentration consisted primarily of HO2 (42 %), isoprene peroxy radicals (30 %), methyl peroxy (CH3O2, 14 %), and acetyl peroxy (CH3CO3, 4 %) with daytime (08:00–20:00 EDT) contributions of 42 %, 32 %, 13 %, and 4 % for HO2, isoprene peroxy, CH3O2, and CH3CO3, respectively. The MCM 3.3.1 model predicted that HO2 (43 %), isoprene peroxy (18 %), methyl peroxy (14 %), and acetyl peroxy (4 %) were the major contributors to the modeled diurnal average total ROx concentration with daytime contributions of 43 %, 20 %, 13 %, and 4 % (Fig. 6). Similar results were obtained from the MCM 3.2 and RACM2 models (Fig. S1 in the Supplement). As discussed above, the configuration of the IU-FAGE instrument used in this study converted approximately 83 % of isoprene peroxy radicals to HO2 upon the addition of NO and minimally converts methyl peroxy radicals to HO2 (<5 %) (Lew et al., 2018). Thus, the majority of the contributing species to the measured is HO2 and isoprene peroxy radicals which together account for approximately 70 % of the total peroxy radical concentration predicted by these models. Measurements of the total HO2+RO2 radical concentrations using an Ethane–Nitric Oxide Chemical Amplifier (ECHAMP) were found to be in good agreement with the measurements reported here and are summarized in Kundu et al. (2019).
3.3 Total OH reactivity measurements and model comparison
The measured total OH reactivity and that calculated from measured OH sinks using both the RACM and MCM mechanisms are shown in Fig. 7, in which the measured OH reactivity is averaged into 2 h bins. As illustrated in this figure, the calculated OH reactivity was in relatively good agreement with the measured OH reactivity on some days and nights, specifically 15–16 July, with missing reactivity observed later in the campaign. Overall, the averaged measured OH reactivity varied between the instrumental limit of detection of 1 s−1 to a maximum of approximately 31 s−1 with an overall diurnal average value of approximately 13 s−1.
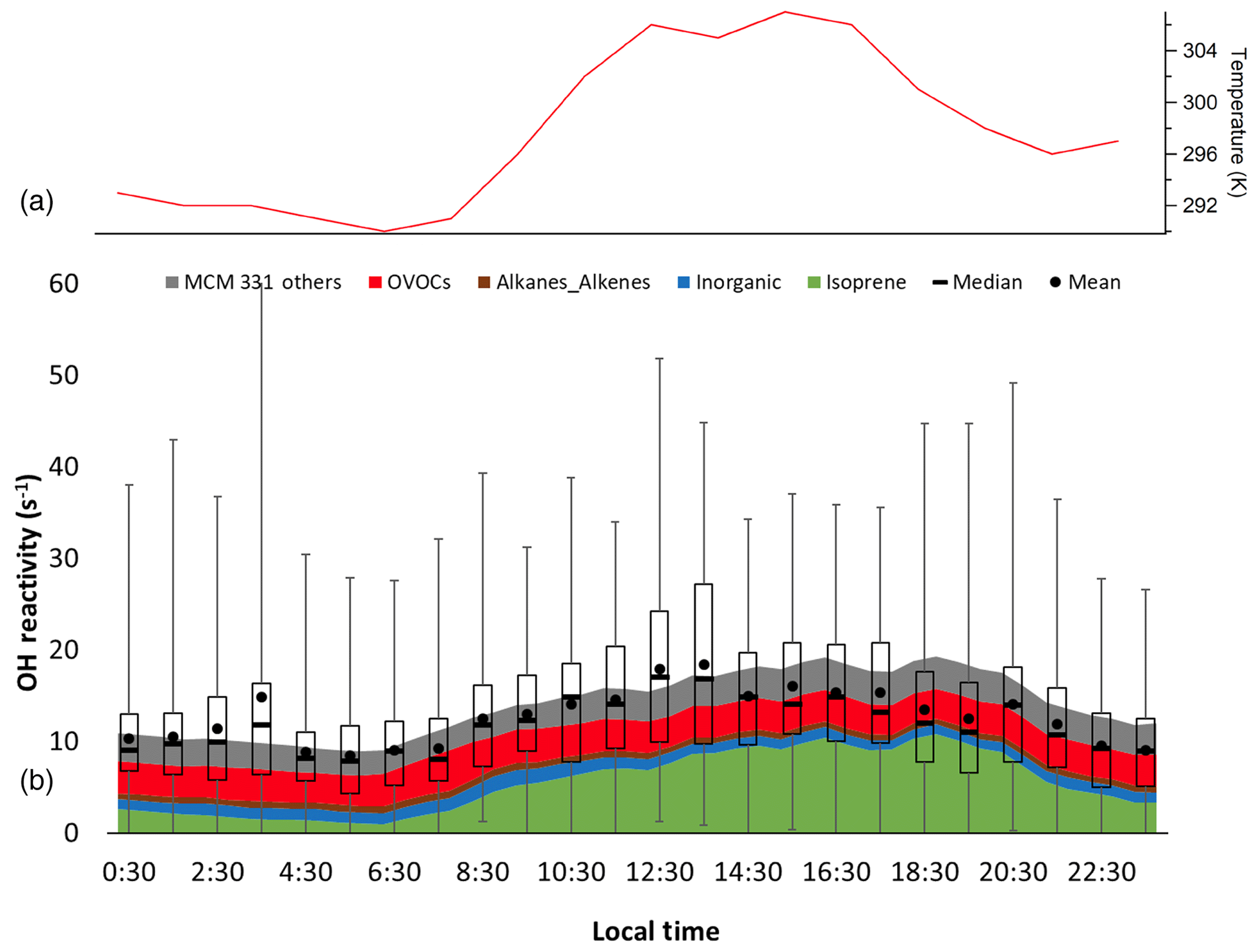
Figure 8Diurnal temperature (a) and box and whiskers plot of observed total OH reactivity measured during the entire campaign showing the mean and median values for each hour with the mean-calculated values from the measured OH sinks, as well as the unmeasured oxidation products from the MCM 3.3.1 model results (b). Error bars show the range of individual 5 min measurements, and bars show Q1 and Q3 for the measured OH reactivity.
The campaign diurnal averaged measured OH reactivity is shown in Fig. 8 along with the calculated total OH reactivity from the measured OH sinks. As expected for this deciduous forest environment, isoprene was the dominant contributor making up 37 % of the diurnally averaged total reactivity, followed by OVOCs (28 %), inorganics (10 %), alkanes and alkenes (5 %), anthropogenic non-methane hydrocarbons (NMHC) (1 %), and monoterpenes (<1 %) with missing reactivity accounting for the remaining 18 % (Fig. S2 in the Supplement). During the daytime (08:00 and 20:00 EDT), the contributions are similar, with isoprene being the largest contributor at 47 % followed by OVOCs (24 %), inorganics (8 %), alkanes and alkenes (4 %), anthropogenic NMHC (1 %), and monoterpenes (<1 %) with missing reactivity accounting for the remaining 14 %. During the nighttime (20:00 to 08:00 EDT), OVOCs were the dominant contributor to the modeled OH reactivity at 32 % followed by isoprene (24 %), inorganics (11 %), alkanes and alkenes (6 %), anthropogenic NMHC (2 %), and monoterpenes (<1 %) with missing reactivity of 24 % (Fig. S2 in the Supplement).
The campaign diurnal average (Fig. 8) shows a correlation with temperature, with the maximum average OH reactivity of approximately 20 s−1 occurring around 13:30 EDT. The calculated reactivity was consistent with the measured reactivity for temperatures less than 294 K, while the observed reactivity is greater than that calculated from the measured sinks for higher temperatures, although at temperatures above 302 K the measured reactivity appears to be less than that calculated (Fig. S3 in the Supplement). These results are similar to several previous studies in which the measured missing reactivity appeared to increase with temperature (Di Carlo et al., 2004; Hansen et al., 2014; Bsaibes, et al., 2020).
Figure 8 also shows the campaign average OH reactivity including the reactivity of unmeasured oxidation products predicted by the MCM 3.3.1 model. On average, including the contribution of unmeasured oxidation products can account for the majority of the missing reactivity. While the model tends to overpredict the average measured reactivity in the afternoon and evening, the model results agree to within the combined uncertainty of the model and the precision of the measurement (Hansen et al., 2014). Similar results were obtained by the RACM2 models, although the predicted reactivity of unmeasured oxidation products by the RACM2 models is approximately a factor of 2 smaller than that predicted by the MCM models (Fig. S4 in the Supplement). These results suggest that the models are generally able to reproduce the measured OH reactivity at this site and that the missing reactivity observed during IRRONIC may be due to unmeasured oxidation products, with isoprene nitrates and isoprene epoxides within the RACM2 and MCM mechanisms being the primary contributors to the missing reactivity.
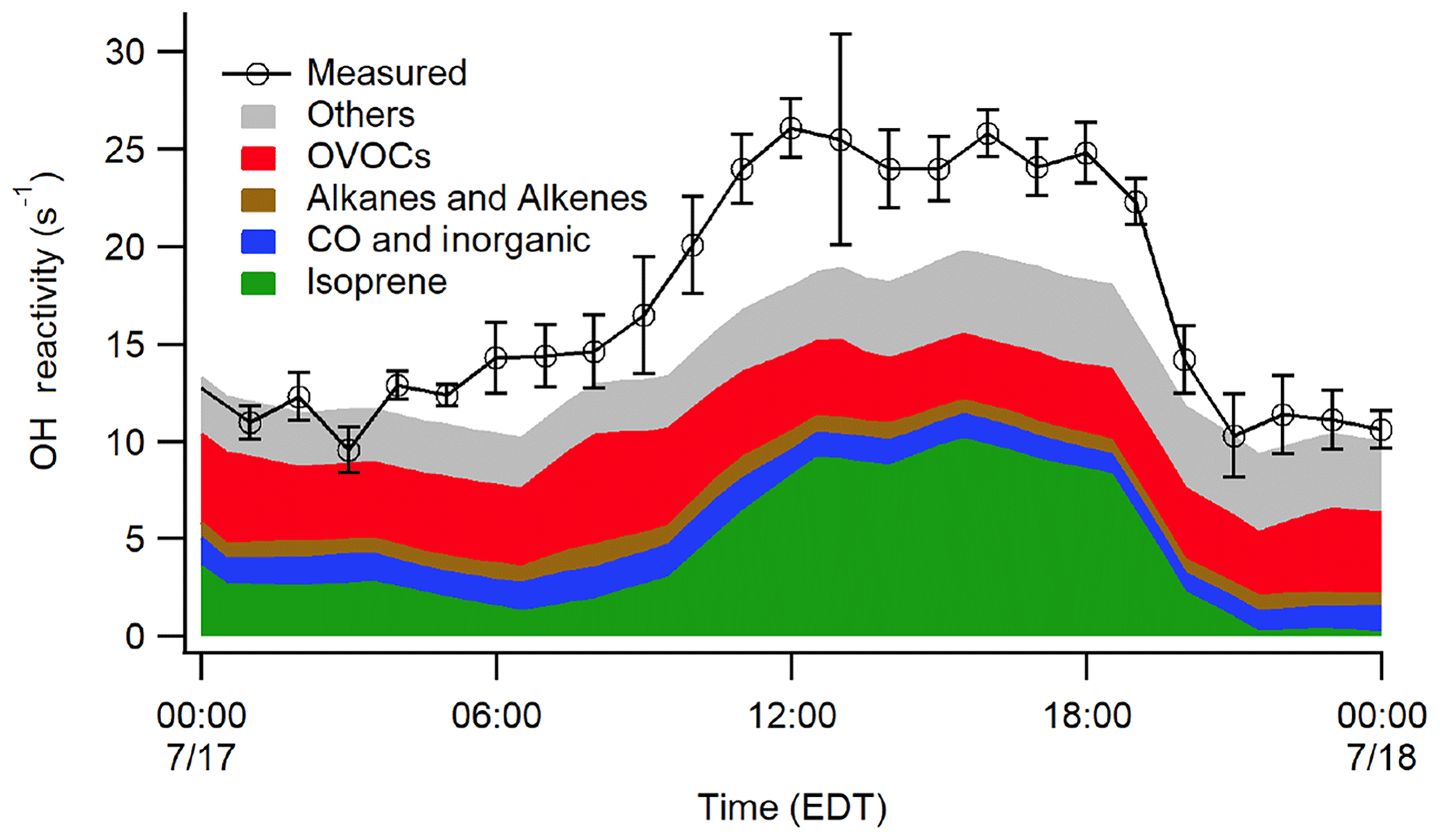
Figure 9Median OH reactivity measurements from 17 July in comparison to modeled reactivity on this day from the MCM 3.3.1 mechanism.
While the campaign averaged OH reactivity measurements appear to be in reasonable agreement with the calculated reactivity based on measured compounds, there were several days that displayed large missing reactivity similar to that observed by Hansen et al. (2014). The MCM 3.3.1 model results for a day with the largest missing reactivity (17 July) are shown in Fig. 9, which indicate that the modeled reactivity including unmeasured oxidation products cannot explain the observed reactivity on this day. The reason for this discrepancy is unclear as the missing reactivity on this day did not appear to correlate with changes in wind speed, direction, trajectory, or meteorological conditions but may indicate the presence of additional unmeasured emissions or oxidation products not accounted for by the model. Additional measurements and analyses will be necessary to determine the source of the missing reactivity.
3.4 Radical budgets
The analysis of the rates of radical initiation, propagation, and termination can provide insight into the importance of individual radical sources and sinks. For the IRRONIC campaign, the OH radical budget is illustrated in Fig. 10, in which OH radical production reactions are represented in shades of blue and loss reactions are represented in shades of red. Daytime production includes reactions with both initiation and propagation that produce OH radicals (positive rates), while daytime OH loss reactions are represented by propagation and termination reactions that remove OH (negative rates). For simplicity, only the RACM2 and RACM2-LIM1 radical budgets are shown.
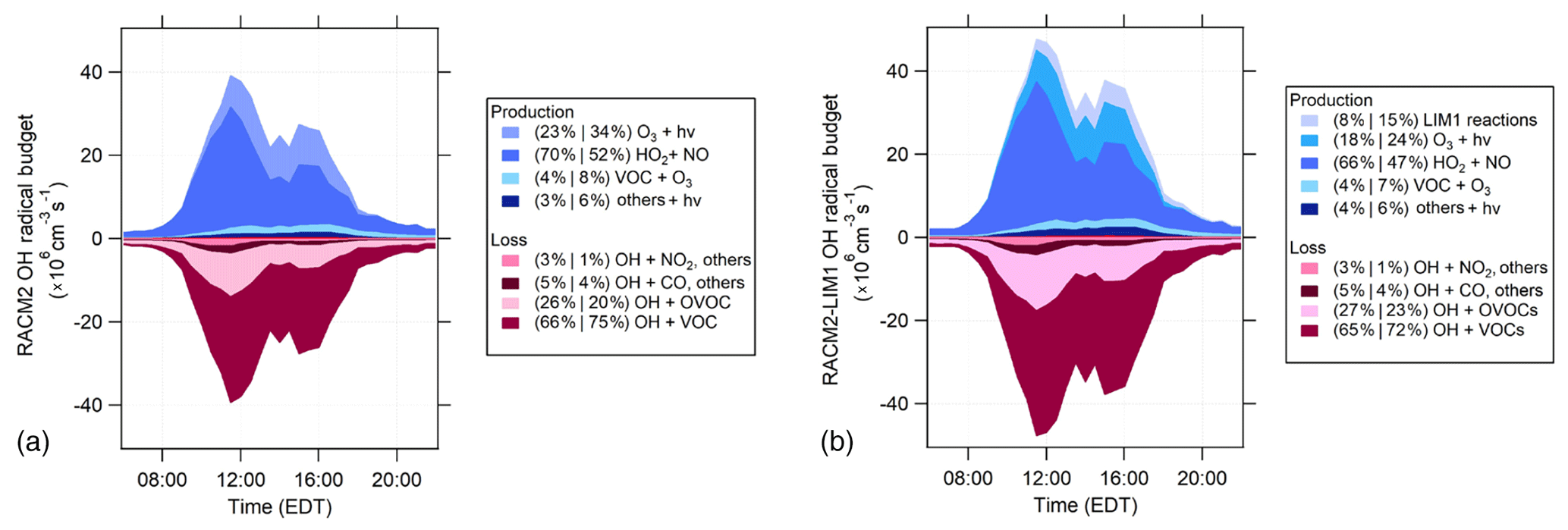
Figure 10RACM2 (a) and RACM2-LIM1 (b) OH radical budgets for the days with NO measurements. Shades of blue represent production reactions, and the shades of red represent loss rates. The percent contribution of each reaction to total production or loss is divided into two periods (10:00 to 14:00 EDT and 14:00 to 18:00 EDT). The “Others+hν” category includes the photolysis of HONO, H2O2, organic peroxides, and HNO3 (see text).
The maximum rates for the OH radical budget of approximately 5×107 from the RACM2-LIM1 model were higher than the maximum value of approximately 4×107 in RACM2. The addition of the LIM1 mechanism increases the OH radical production rate mostly from the photolysis of hydroxyperoxy aldehydes (HPALDs) produced from the isomerization of isoprene-based peroxy radicals and their subsequent chemistry (Peeters et al., 2014; Tan et al., 2017). In the RACM2-LIM1 model, the daytime OH radical production is dominated by the HO2+NO reaction from 10:00 to 14:00 EDT (66 %) and drops to 47 % from 14:00 to 18:00 EDT. Ozone photolysis and the LIM1 mechanism contribute up to 18 %–24 % and 8 %–15 % of the total OH radical production during the morning and afternoon, with ozonolysis (VOC+O3) and photolysis of HONO, H2O2, organic peroxides (OP1, OP2), and HNO3 all contributing 4 %–7 % and 4 %–6 % of the total OH radical production (Fig. 10). A majority of the OH radical loss is due to OH reactions with VOCs (65 %–72 %) and OVOCs (27 %–23 %) during the morning and afternoon. As described above, the measured total OH reactivity was in reasonable agreement with the modeled OH reactivity; therefore, it is likely that the total OH loss is well represented in the model. An experimental radical budget for the period when the measurements were complete suggests that the total measured OH production rate during the day is nearly balanced by the total OH loss rate based on the measured total OH reactivity (Fig. S5 in the Supplement), which is consistent with the agreement between the measured and modeled OH on these days as discussed above and within the combined uncertainties of all the measurements (38 %, 2σ, for OH and for , for example) and similar to that observed previously (Tan et al., 2019). However, the experimental budget is not balanced in the evening, suggesting that there may be a missing radical source during this period, which is consistent with the model underprediction of the OH measurements discussed above.
The total radical (ROx) budget from the RACM2 mechanisms of OH, HO2, and RO2 radicals is illustrated in Fig. 11. Overall, total radical initiation in the RACM2-LIM1 mechanism was larger with a maximum value approaching 3×107 compared to RACM2's maximum value of approximately 2×107 . The increase in total radical initiation in the RACM2-LIM1 model is due to both the added radical initiation from the photolysis of HPALDs and increased radical initiation from other aldehydes produced in the LIM1 mechanism. Overall, radical initiation from the photolysis of HPALDs and the subsequent chemistry from the LIM1 mechanism contributed 16 %–22 % of total radical initiation during the morning and afternoon, while the photolysis of formaldehyde and others (HONO, H2O2, aldehydes, organic peroxides, HNO3) together contributed approximately 42 % of total radical initiation, with ozone photolysis contributing 29 %–33 % of radical initiation (Fig. 11). In contrast, ozone photolysis contributes approximately 44 %–46 % of radical initiation in the RACM2 mechanism, with formaldehyde and other aldehydes together contributing 43 % (Fig. 11). Radical termination for both mechanisms is dominated by peroxy radical self-reactions, such as the HO2+HO2 reaction, as well as the reaction of HO2 with isoprene-based peroxy (ISOP) radicals and other peroxy radicals (RO2). These reactions account for approximately 67 %–95 % of radical termination due to the low levels of NOx used in the models, with reactions of OH+NO2 and other NOx radical reactions accounting for approximately 5 %–33 % of radical termination in these models during the day (Fig. 11).
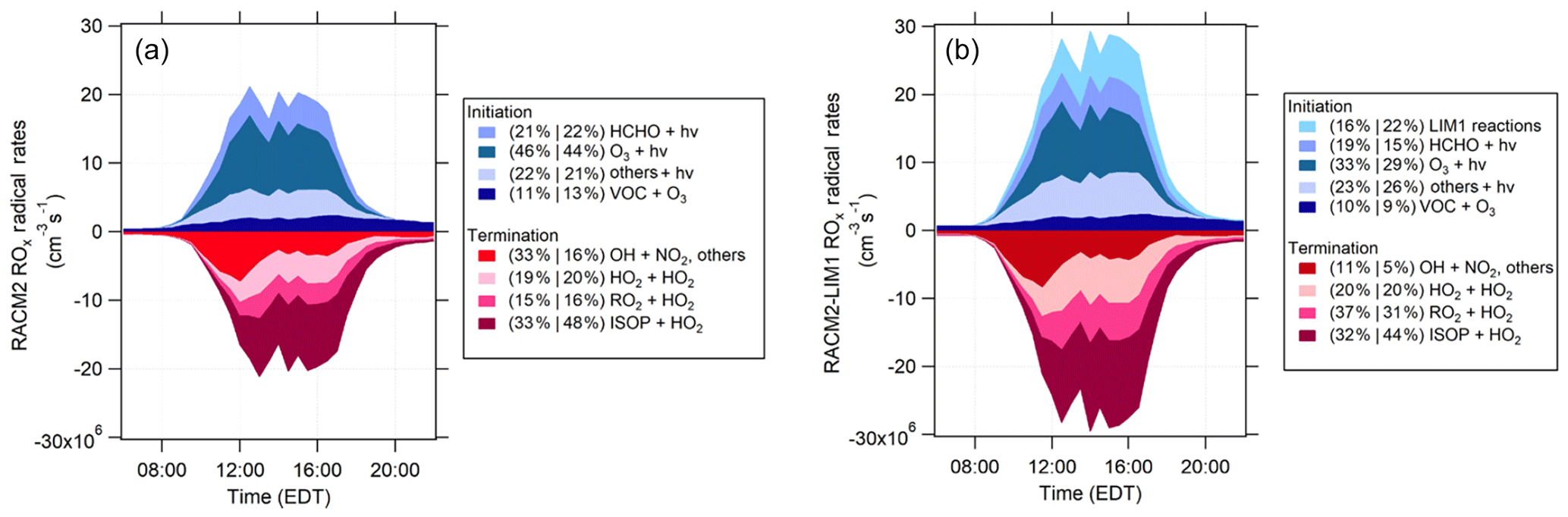
Figure 11RACM2 (a) and RACM2-LIM1 (b) total ROx radical budgets for the days with NO measurements. Shades of blue represent initiation rates and the shades of red represent termination rates. The percent contribution of each reaction to total initiation/termination are divided into two periods (10:00 to 14:00 EDT and 14:00 to 18:00 EDT). The “Others+hν” category includes the photolysis of HONO, H2O2, aldehydes, organic peroxides, and HNO3 (see text).
The partitioning of the total radical budget production for IRRONIC is similar to the modeled budget observed during PROPHET 2008 and CABINEX 2009 (Griffith et al., 2013). The updated RACM model used during these campaigns predicted that radical termination was dominated by HO2+RO2 reactions (including the HO2+ISOP reaction), contributing to approximately 80 % of total radical termination, similar to the 67 %–84 % for the HO2+ISOP, HO2+RO2, and HO2+HO2 reactions predicted here by the RACM2 model. The photolysis of ozone accounted for approximately 20 %–30 % of total radical initiation during these campaigns, based on an updated version of the RACM model (Griffith et al., 2013), compared to approximately 45 % predicted by the RACM2 mechanism during IRRONIC due to higher concentrations observed during this campaign. Ozonolysis reactions contributed approximately 20 %–30 % of total radical initiation during PROPHET and CABINEX compared to approximately 12 % during IRRONIC. Photolysis of HCHO contributed approximately 22 %–24 % of the total rate of radical initiation during IRRONIC compared to 23 % and 5 % during PROPHET 2008 and CABINEX 2009, respectively, with the low contribution during CABINEX primarily due to the lower mixing ratios of HCHO observed during this campaign (Griffith et al., 2013). In contrast, photolysis of HONO was a significant radical source during PROPHET and CABINEX, contributing 14 %–17 % of radical initiation compared to approximately 3 % of total radical production during IRRONIC due to the lower mixing ratios of HONO observed during IRRONIC. On average, mixing ratios of HONO during IRRONIC were approximately 40 ppt at night, decreasing to approximately 10 ppt during the day (Fig. S6 in the Supplement) which can be compared to daytime mixing ratios between 50 and 75 ppt during PROPHET and CABINEX (Griffith et al., 2013). The reason for the difference in the measured HONO values between these two sites is unclear but may be related to increased production from the photolysis of nitric acid on the forest canopy surfaces at the PROPHET site (Zhou et al., 2011).
Measurements of OH radical concentrations using the IU-FAGE instrument during the IRRONIC campaign revealed a significant unknown interference that appeared to correlate with both temperature and ozone. The average measured OH radical concentration, after the interference was subtracted, reached an average daytime maximum of approximately 4–5×106 cm−3. This is in contrast to the measurements including the interference which reached an average daytime maximum of approximately 9×106 cm−3. Similar concentrations of OH were observed after any measured interference was subtracted at this site in 2017 during an informal intercomparison between the IU-FAGE instrument and the University of Colorado chemical ionization mass spectrometry (CIMS) instrument (Rosales et al., 2018; Reidy et al., 2018).
After subtracting the interference, the OH measurements were in better agreement with model simulations utilizing the Regional Atmospheric Chemical Mechanism 2 (RACM2) with an updated Leuven isoprene mechanism (LIM1), as well as the Master Chemical Mechanism versions 3.2 and 3.3.1. Both the RACM2-LIM1 and MCM 3.3.1 mechanisms add radical recycling reactions for isoprene oxidation that increase the modeled OH and peroxy radical concentrations. Similar to the results of Novelli et al. (2020), the RACM2-LIM1 model results were in better agreement compared to the MCM 3.3.1 mechanism, predicting maximum daytime OH concentrations that were within 30 % of the measurements likely due to a larger bulk isoprene peroxy radical isomerization rate leading to a greater rate of radical production. However, the models tend to underpredict the measured concentrations during the evening, suggesting that a significant radical source may be missing from the models. Additional measurements are needed in order to resolve this discrepancy. Nevertheless, it is clear that if the measured interference was not taken into account, the apparent OH concentrations would have been a factor of 2–4 greater than predicted by the model mechanisms, which is comparable to previous measurements under low NOx and high isoprene conditions (Rohrer et al., 2014). These results are similar to those reported by Mao et al. (2012) and Mallik et al. (2018) who found good agreement between their OH measurements and model predictions when measured interferences were taken into account. Because of differences in instrument design (geometry, cell pressure, flow, etc.), these interferences may not significantly impact other LIF-FAGE instruments. However, future OH measurements using the LIF-FAGE technique should include methods to quantify potential instrumental artifacts, even if they are insignificant, to demonstrate that the measurements are free from interferences.
Measurements of total OH reactivity were in reasonable agreement with those calculated from measured OH sinks, with isoprene contributing approximately 37 % and OVOCs 28 % of the diurnally averaged measured reactivity and with 18 % of the measured reactivity missing. However, on average the missing reactivity fraction can be explained by unmeasured oxidation products specifically from isoprene nitrates and isoprene epoxides within the RACM2 and MCM mechanisms. This indicates that these mechanisms are accurately representing the total OH loss at this site.
Measurements of HO2 radicals by the IU-FAGE instrument using chemical conversion to OH by the addition of NO have been shown to be sensitive to alkene-based peroxy radicals (Lew et al., 2018). As a result, the measurements represent the sum of HO2 and a fraction of RO2 radicals in the atmosphere (). During the IRRONIC campaign, the measured concentration primarily reflected the sum of HO2- and isoprene-based peroxy radicals, which contributed approximately 70 % of the total modeled peroxy radicals. The average daytime ambient measurements reached maximum concentrations of approximately 1×109 cm−3. Both MCM models predicted concentrations that were within 10 %–30 % but generally lower than the measurements, while the RACM mechanisms resulted in predicted concentrations that were within 10 %–30 % but generally greater than the measurements. However, all models predicted concentrations that were within the combined uncertainty of both the model and the measurement. These results are in contrast to some previous measurements in forest environments in which model predictions were found to be significantly greater than measured concentrations (Griffith et al., 2013) perhaps as a result of the lower mixing ratios of NO observed at these sites. Additional measurements are needed in order to resolve this discrepancy, which may be related to a gap in our understanding of peroxy radical chemistry under low NO conditions.
Data are available upon request from the corresponding author (pstevens@indiana.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-9209-2020-supplement.
PS, SD, and EW designed the research project. ML, PR, BB, and PS were responsible for the LIF-FAGE OH, , and OH reactivity and HONO measurements. SK and EW were responsible for the supporting measurements of NO, NO2, and O3. SD, SS, TL, and NL were responsible for the measurements of VOCs and OVOCs. ML, PR, ER, and PS conducted the analysis and photochemical modeling and wrote the paper with feedback from all coauthors. ML and PR contributed equally to the paper.
The authors declare that they have no conflict of interest.
We would like to thank James Flynn (University of Houston) for the spectroradiometer used to obtain the J(NO2) measurements.
This study was supported by the National Science Foundation, Directorate for Geosciences (grant nos. AGS-1440834 to Indiana University, AGS-1443842 to the University of Massachusetts, and AGS-1719918 to Drexel University). This work was also supported by the French National Research Agency (grant no. ANR-11-LABX-0005-01) and the European Funds for Regional Economic Development (FEDER) through the CaPPA (Chemical and Physical Properties of the Atmosphere) project.
This paper was edited by Andreas Hofzumahaus and reviewed by three anonymous referees.
Ait-Helal, W., Borbon, A., Sauvage, S., de Gouw, J. A., Colomb, A., Gros, V., Freutel, F., Crippa, M., Afif, C., Baltensperger, U., Beekmann, M., Doussin, J.-F., Durand-Jolibois, R., Fronval, I., Grand, N., Leonardis, T., Lopez, M., Michoud, V., Miet, K., Perrier, S., Prévôt, A. S. H., Schneider, J., Siour, G., Zapf, P., and Locoge, N.: Volatile and intermediate volatility organic compounds in suburban Paris: variability, origin and importance for SOA formation, Atmos. Chem. Phys., 14, 10439–10464, https://doi.org/10.5194/acp-14-10439-2014, 2014.
Badol, C., Borbon, A., Locoge, N., Leonardis, T., and Galloo, J. C.: An automated monitoring system for VOC ozone precursors in ambient air: development, implementation and data analysis, Anal. Bioanal. Chem., 378, 7, 1815–1827, 2004.
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Commun. Chem., 2, 21, https://doi.org/10.1038/s42004-019-0120-9, 2019.
Bottorff, B., Stevens, P. S., Lew, M., Rickly, P., and Dusanter, S.: Measurements of Nitrous Acid (HONO) in an Indiana Forest by Laser Photofragmentation/Laser-Induced Flourescence (LP/LIF), Abstract A21B-0116 presented at 2015 Fall Meeting, AGU, San Francisco, CA, 14–18 December, 2015.
Bottorff, B., Reidy, E., Mielke, L., Dusanter, S., and Stevens, P. S.: Development of Laser Photo-fragmentation Laser-Induced Fluorescence instrument for the measurement of nitrous acid in the atmosphere, Atmos. Meas. Tech., in preparation, 2020.
Bsaibes, S., Al Ajami, M., Mermet, K., Truong, F., Batut, S., Hecquet, C., Dusanter, S., Léornadis, T., Sauvage, S., Kammer, J., Flaud, P.-M., Perraudin, E., Villenave, E., Locoge, N., Gros, V., and Schoemaecker, C.: Variability of hydroxyl radical (OH) reactivity in the Landes maritime pine forest: results from the LANDEX campaign 2017, Atmos. Chem. Phys., 20, 1277–1300, https://doi.org/10.5194/acp-20-1277-2020, 2020.
Carslaw, N., Creasey, D. J., Harrison, D., Heard, D. E., Hunter, M. C., Jacobs, P. J., Jenkin, M. E., Lee, J. D., Lewis, A. C., Pilling, M. J., Saunders, S. M., and Seakins, P. W.: OH and HO2 Radical Chemistry in a Forested Region of North-Western Greece, Atmos. Environ., 35, 4725–4737, 2001.
Chen, S., Ren, X., Mao, J., Chen, Z., Brune, W. H., Lefer, B., Rappenglück, B., Flynn, J., Olson, J., and Crawford, J. H.: A Comparison of Chemical Mechanisms Based on Tramp-2006 Field Data, Atmos. Environ., 44, 4116–4125, 2010.
Creasey, D. J., Heard, D. E., and Lee, J. D.: Eastern Atlantic Spring Experiment 1997 (Ease97) 1. Measurements of OH and HO2 Concentrations at Mace Head, Ireland, J. Geophys. Res., 107, 4091, https://doi.org/10.1029/2001jd000892, 2002.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, 2011.
Crounse, J. D., Teng, A., and Wennberg, P. O.: Experimental constrains on the distribution and fate of peroxy radicals formed in the reactions of isoprene + OH + O2 presented at the Atmospheric Chemical Mechanisms: Simple Models – Real world Complexities, Universtiy of California, Davis, USA, 10–12 December 2014.
Crowley, J. N., Pouvesle, N., Phillips, G. J., Axinte, R., Fischer, H., Petäjä, T., Nölscher, A., Williams, J., Hens, K., Harder, H., Martinez-Harder, M., Novelli, A., Kubistin, D., Bohn, B., and Lelieveld, J.: Insights into HOx and ROx chemistry in the boreal forest via measurement of peroxyacetic acid, peroxyacetic nitric anhydride (PAN) and hydrogen peroxide, Atmos. Chem. Phys., 18, 13457–13479, https://doi.org/10.5194/acp-18-13457-2018, 2018.
Czader, B. H., Li, X., and Rappenglueck, B.: Cmaq Modeling and Analysis of Radicals, Radical Precursors, and Chemical Transformations, J. Geophys. Res., 118, 11376–11387, https://doi.org/10.1002/jgrd.50807, 2013.
Davis, D. D., Rodgers, M. O., Fischer, S. D., and Asai, K.: An Experimental Assessment of the O3/H2O Interference Problem in the Detection of Natural Levels of OH Via Laser Induced Fluorescence, Geophys. Res. Lett., 8, 69–72, https://doi.org/10.1029/GL008i001p00069, 1981a.
Davis, D. D., Rodgers, M. O., Fischer, S. D., and Heaps, W. S.: A Theoretical Assessment of the O3∕H2O Interference Problem in the Detection of Natural Levels of OH Via Laser Induced Fluorescence, Geophys. Res. Lett., 8, 73–76, https://doi.org/10.1029/GL008i001p00073, 1981b.
Detournay, A., Sauvage, S., Locoge, N., Gaudion, V., Leonardis, T., Fronval, I., Kaluzny, P., and Galloo, J.-C.: Development of a sampling method for the simultaneous monitoring of straightchain alkanes, straight-chain saturated carbonyl compounds and monoterpenes in remote areas, J. Environ. Monit., 13, 983–990, 2011.
Di Carlo, P., Brune, W. H., Martinez, M., Harder, H., Lesher, R., Ren, X., Thornberrry, T., Carroll, M. A., Young, V., Shepson, P. B., Riemer, D., Apel, E., and Campbell, C.: Missing OH Reactivity in a Forest: Evidence for Unknown Reactive Biogenic VOCs, Science, 304, 722–725, https://doi.org/10.1126/science.1094392, 2004.
Dusanter, S. and Stevens, P. S.: Recent Advances in the Chemistry of OH and HO2 Radicals in the Atmosphere: Field and Laboratory Measurements, in: Advances in Atmospheric Chemistry, Vol. 1, edited by: Barker, J. R., Steiner, A. L., Wallington, T. J., World Scientific Publishing Co. Pte. Ltd, New Jersey, pp. 493–579, 2017.
Dusanter, S., Vimal, D., and Stevens, P. S.: Technical note: Measuring tropospheric OH and HO2 by laser-induced fluorescence at low pressure. A comparison of calibration techniques, Atmos. Chem. Phys., 8, 321–340, https://doi.org/10.5194/acp-8-321-2008, 2008.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., and Molina, L. T.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 1: Deployment of the Indiana University laser-induced fluorescence instrument, Atmos. Chem. Phys., 9, 1665–1685, https://doi.org/10.5194/acp-9-1665-2009, 2009a.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., Molina, L. T., Baker, A., Meinardi, S., Blake, D., Sheehy, P., Merten, A., Zhang, R., Zheng, J., Fortner, E. C., Junkermann, W., Dubey, M., Rahn, T., Eichinger, B., Lewandowski, P., Prueger, J., and Holder, H.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 9, 6655–6675, https://doi.org/10.5194/acp-9-6655-2009, 2009b.
Emmerson, K. M., Carslaw, N., Carpenter, L. J., Heard, D. E., Lee, J. D., and Pilling, M. J.: Urban Atmospheric Chemistry During the Puma Campaign 1: Comparison of Modelled OH and HO2 Concentrations with Measurements, J. Atmos. Chem., 52, 143–164, 2005.
Emmerson, K. M., Carslaw, N., Carslaw, D. C., Lee, J. D., McFiggans, G., Bloss, W. J., Gravestock, T., Heard, D. E., Hopkins, J., Ingham, T., Pilling, M. J., Smith, S. C., Jacob, M., and Monks, P. S.: Free radical modelling studies during the UK TORCH Campaign in Summer 2003, Atmos. Chem. Phys., 7, 167–181, https://doi.org/10.5194/acp-7-167-2007, 2007.
Fittschen, C., Al Ajami, M., Batut, S., Ferracci, V., Archer-Nicholls, S., Archibald, A. T., and Schoemaecker, C.: ROOOH: a missing piece of the puzzle for OH measurements in low-NO environments?, Atmos. Chem. Phys., 19, 349–362, https://doi.org/10.5194/acp-19-349-2019, 2019.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Fuchs, H., Tan, Z., Hofzumahaus, A., Broch, S., Dorn, H.-P., Holland, F., Künstler, C., Gomm, S., Rohrer, F., Schrade, S., Tillmann, R., and Wahner, A.: Investigation of potential interferences in the detection of atmospheric ROx radicals by laser-induced fluorescence under dark conditions, Atmos. Meas. Tech., 9, 1431–1447, https://doi.org/10.5194/amt-9-1431-2016, 2016.
George, L. A., Hard, T. M., and O'Brien, R. J.: Measurement of Free Radicals OH and HO2 in Los Angeles Smog, J. Geophys. Res., 104, 11643–11655, https://doi.org/10.1029/1998jd100113, 1999.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The Regional Atmospheric Chemistry Mechanism, Version 2, Atmos. Environ., 68, 174–185, 2013.
Griffith, S. M., Hansen, R. F., Dusanter, S., Stevens, P. S., Alaghmand, M., Bertman, S. B., Carroll, M. A., Erickson, M., Galloway, M., Grossberg, N., Hottle, J., Hou, J., Jobson, B. T., Kammrath, A., Keutsch, F. N., Lefer, B. L., Mielke, L. H., O'Brien, A., Shepson, P. B., Thurlow, M., Wallace, W., Zhang, N., and Zhou, X. L.: OH and HO2 radical chemistry during PROPHET 2008 and CABINEX 2009 – Part 1: Measurements and model comparison, Atmos. Chem. Phys., 13, 5403–5423, https://doi.org/10.5194/acp-13-5403-2013, 2013.
Griffith, S. M., Hansen, R. F., Dusanter, S., Michoud, V., Gilman, J. B., Kuster, W. C., Veres, P. R., Graus, M., de Gouw, J. A., Roberts, J., Young, C., Washenfelder, R., Brown, S. S., Thalman, R., Waxman, E., Volkamer, R., Tsai, C., Stutz, J., Flynn, J. H., Grossberg, N., Lefer, B., Alvarez, S. L., Rappenglueck, B., Mielke, L. H., Osthoff, H. D., and Stevens, P. S.: Measurements of Hydroxyl and Hydroperoxy Radicals During CalNex-LA: Model Comparisons and Radical Budgets, J. Geophys. Res., 121, 4211–4232, https://doi.org/10.1002/2015JD024358, 2016.
Hansen, R. F., Griffith, S. M., Dusanter, S., Rickly, P. S., Stevens, P. S., Bertman, S. B., Carroll, M. A., Erickson, M. H., Flynn, J. H., Grossberg, N., Jobson, B. T., Lefer, B. L., and Wallace, H. W.: Measurements of total hydroxyl radical reactivity during CABINEX 2009 – Part 1: field measurements, Atmos. Chem. Phys., 14, 2923–2937, https://doi.org/10.5194/acp-14-2923-2014, 2014.
Heard, D. E. and Pilling, M. J.: Measurement of OH and HO2 in the Troposphere, Chem. Rev., 103, 5163–5198, 2003.
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified Trace Gas Removal in the Troposphere, Science, 324, 1702–1704, 2009.
Holland, F., Hofzumahaus, A., Schäfer, J., Kraus, A., and Pätz, H.-W.: Measurements of OH and HO2 Radical Concentrations and Photolysis Frequencies During Berlioz, J. Geophys. Res., 108, 8246, https://doi.org/10.1029/2001JD001393, 2003.
Jenkin, M. E., Saunders, S. M., Pilling, M. J. The Tropospheric Degradation of Volatile Organic Compounds: A Protocol for Mechanism Development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Kanaya, Y., Sadanaga, Y., Hirokawa, J., Kajii, Y., and Akimoto, H.: Development of a Ground-Based LIF Instrument for Measuring HOx Radicals: Instrumentation and Calibrations, J. Atmos. Chem., 38, 73–110, 2001.
Kanaya, Y., Cao, R., Akimoto, H., Fukuda, M., Komazaki, Y., Yokouchi, Y., Koike, M., Tanimoto, H., Takegawa, N., and Kondo, Y.: Urban Photochemistry in Central Tokyo: 1. Observed and Modeled OH and HO2 Radical Concentrations During the Winter and Summer of 2004, J. Geophys. Res., 112, C11007, https://doi.org/10.1029/2007jd008670, 2007a.
Kanaya, Y., Cao, R., Kato, S., Miyakawa, Y., Kajii, Y., Tanimoto, H., Yokouchi, Y., Mochida, M., Kawamura, K., and Akimoto, H.: Chemistry of OH and HO2 Radicals Observed at Rishiri Island, Japan, in September 2003: Missing Daytime Sink of HO2 and Positive Nighttime Correlations with Monoterpenes, J. Geophys. Res., 112, D11308, https://doi.org/10.1029/2006JD007987, 2007b.
Kanaya, Y., Hofzumahaus, A., Dorn, H.-P., Brauers, T., Fuchs, H., Holland, F., Rohrer, F., Bohn, B., Tillmann, R., Wegener, R., Wahner, A., Kajii, Y., Miyamoto, K., Nishida, S., Watanabe, K., Yoshino, A., Kubistin, D., Martinez, M., Rudolf, M., Harder, H., Berresheim, H., Elste, T., Plass-Dülmer, C., Stange, G., Kleffmann, J., Elshorbany, Y., and Schurath, U.: Comparisons of observed and modeled OH and HO2 concentrations during the ambient measurement period of the HOxComp field campaign, Atmos. Chem. Phys., 12, 2567–2585, https://doi.org/10.5194/acp-12-2567-2012, 2012.
Kim, S., Wolfe, G. M., Mauldin, L., Cantrell, C., Guenther, A., Karl, T., Turnipseed, A., Greenberg, J., Hall, S. R., Ullmann, K., Apel, E., Hornbrook, R., Kajii, Y., Nakashima, Y., Keutsch, F. N., DiGangi, J. P., Henry, S. B., Kaser, L., Schnitzhofer, R., Graus, M., Hansel, A., Zheng, W., and Flocke, F. F.: Evaluation of HOx sources and cycling using measurement-constrained model calculations in a 2-methyl-3-butene-2-ol (MBO) and monoterpene (MT) dominated ecosystem, Atmos. Chem. Phys., 13, 2031–2044, https://doi.org/10.5194/acp-13-2031-2013, 2013.
Konrad, S., Schmitz, T., Buers, H. J., Houben, N., Mannschreck, K., Mihelcic, D., Müsgen, P., Pätz, H. W., Holland, F., Hofzumahaus, A., Schäfer, H. J., Schröder, S., Volz-Thomas, A., Bächmann, K., Schlomski, S., Moortgat, G., and Großmann, D.: Hydrocarbon Measurements at Pabstthum During the Berlioz Campaign and Modeling of Free Radicals, J. Geophys. Res., 108, 8251, https://doi.org/10.1029/2001jd000866, 2003.
Kovacs, T. A. and Brune, W. H.: Total OH Loss Rate Measurement, J. Atmos. Chem., 39, 105–122, 2001.
Kubistin, D., Harder, H., Martinez, M., Rudolf, M., Sander, R., Bozem, H., Eerdekens, G., Fischer, H., Gurk, C., Klüpfel, T., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Taraborrelli, D., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 10, 9705–9728, https://doi.org/10.5194/acp-10-9705-2010, 2010.
Kundu, S., Deming, B. L., Lew, M. M., Bottorff, B. P., Rickly, P., Stevens, P. S., Dusanter, S., Sklaveniti, S., Leonardis, T., Locoge, N., and Wood, E. C.: Peroxy radical measurements by ethane – nitric oxide chemical amplification and laser-induced fluorescence during the IRRONIC field campaign in a forest in Indiana, Atmos. Chem. Phys., 19, 9563–9579, https://doi.org/10.5194/acp-19-9563-2019, 2019.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric Oxidation Capacity Sustained by a Tropical Forest, Nature, 452, 737–740, 2008.
Levy, H.: Photochemistry of the Lower Troposphere, Planet. Space Sci., 20, 919–935, 1972.
Lew, M. M., Dusanter, S., and Stevens, P. S.: Measurement of interferences associated with the detection of the hydroperoxy radical in the atmosphere using laser-induced fluorescence, Atmos. Meas. Tech., 11, 95–109, https://doi.org/10.5194/amt-11-95-2018, 2018.
Lu, K. D., Hofzumahaus, A., Holland, F., Bohn, B., Brauers, T., Fuchs, H., Hu, M., Häseler, R., Kita, K., Kondo, Y., Li, X., Lou, S. R., Oebel, A., Shao, M., Zeng, L. M., Wahner, A., Zhu, T., Zhang, Y. H., and Rohrer, F.: Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006, Atmos. Chem. Phys., 13, 1057–1080, https://doi.org/10.5194/acp-13-1057-2013, 2013.
Mallik, C., Tomsche, L., Bourtsoukidis, E., Crowley, J. N., Derstroff, B., Fischer, H., Hafermann, S., Hüser, I., Javed, U., Keßel, S., Lelieveld, J., Martinez, M., Meusel, H., Novelli, A., Phillips, G. J., Pozzer, A., Reiffs, A., Sander, R., Taraborrelli, D., Sauvage, C., Schuladen, J., Su, H., Williams, J., and Harder, H.: Oxidation processes in the eastern Mediterranean atmosphere: evidence from the modelling of HOx measurements over Cyprus, Atmos. Chem. Phys., 18, 10825–10847, https://doi.org/10.5194/acp-18-10825-2018, 2018.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J.-H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Martinez, M., Harder, H., Kovacs, T. A., Simpas, J. B., Bassis, J., Lesher, R., Brune, W. H., Frost, G. J., Williams, E. J., Stroud, C. A., Jobson, B. T., Roberts, J. M., Hall, S. R., Shetter, R. E., Wert, B., Fried, A., Alicke, B., Stutz, J., Young, V. L., White, A. B., and Zamora, R. J.: OH and HO2 Concentrations, Sources, and Loss Rates During the Southern Oxidants Study in Nashville, Tennessee, Summer 1999, J. Geophys. Res., 108, 4011, https://doi.org/10.1029/2003jd003551, 2003.
Michoud, V., Kukui, A., Camredon, M., Colomb, A., Borbon, A., Miet, K., Aumont, B., Beekmann, M., Durand-Jolibois, R., Perrier, S., Zapf, P., Siour, G., Ait-Helal, W., Locoge, N., Sauvage, S., Afif, C., Gros, V., Furger, M., Ancellet, G., and Doussin, J. F.: Radical budget analysis in a suburban European site during the MEGAPOLI summer field campaign, Atmos. Chem. Phys., 12, 11951–11974, https://doi.org/10.5194/acp-12-11951-2012, 2012.
Nölscher, A. C., Yañez-Serrano, A. M., Wolff, S., de Araujo, A. C., Lavrič, J. V., Kesselmeier, J., and Williams, J.: Unexpected seasonality in quantity and composition of Amazon rainforest air reactivity, Nat. Commun., 7, 10383, https://doi.org/10.1038/ncomms10383, 2016.
Novelli, A., Hens, K., Tatum Ernest, C., Kubistin, D., Regelin, E., Elste, T., Plass-Dülmer, C., Martinez, M., Lelieveld, J., and Harder, H.: Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals, Atmos. Meas. Tech., 7, 3413–3430, https://doi.org/10.5194/amt-7-3413-2014, 2014.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Novelli, A., Vereecken, L., Bohn, B., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Reimer, D., Rohrer, F., Rosanka, S., Taraborrelli, D., Tillmann, R., Wegener, R., Yu, Z., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 3333–3355, https://doi.org/10.5194/acp-20-3333-2020, 2020.
Peeters, J.: Interactive comment on “The MCM v3.3. degradation scheme for isoprene” by M. E. Jenkin et al., Atmos. Chem. Phys. Discuss., 15, C2486–C2486, available at: https://www.atmos-chem-phys-discuss.net/15/C2486/2015/acpd-15-C2486-2015.pdf, 2015.
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx Radical Regeneration in the Oxidation of Isoprene, Phys. Chem. Chem. Phys., 11, , 5935–5939, https://doi.org/10.1039/B908511D, 2009.
Peeters, J., Müller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl Radical Recycling in Isoprene Oxidation Driven by Hydrogen Bonding and Hydrogen Tunneling: The Upgraded LIM1 Mechanism, J. Phys. Chem. A, 118, 8625–8643, 2014.
Reidy, E., Rosales, C., Bottorff, B., Stevens, P. S., Cantrell, C. A., Mauldin, L. Anderson, D. C., and Wood, E. C. D. An Informal Intercomparison of Ambient OH, HO2, and RO2 Measurements in an Indiana Forest Part 2: Comparison with Model Predictions, Abstract A43M-3281 presented at 2018 Fall Meeting, AGU, Washington, D.C., 10–14 December, 2018.
Ren, X., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Simpas, J. B., Brune, W. H., Schwab, J. J., Demerjian, K. L., He, Y., Zhou, X., and Gao, H.: OH and HO2 Chemistry in the Urban Atmosphere of New York City, Atmos. Environ., 37, 3639–3651, 2003.
Ren, X., Brune, W., Cantrell, C., Edwards, G., Shirley, T., Metcalf, A., and Lesher, R.: Hydroxyl and Peroxy Radical Chemistry in a Rural Area of Central Pennsylvania: Observations and Model Comparisons, J. Atmos. Chem., 52, 231–257, 2005.
Ren, X., Brune, W. H., Oliger, A., Metcalf, A. R., Simpas, J. B., Shirley, T., Schwab, J. J., Bai, C., Roychowdhury, U., Li, Y., Cai, C., Demerjian, K. L., He, Y., Zhou, X., Gao, H., and Hou, J.: OH, HO2, and OH Reactivity During the PMTACS-NY Whiteface Mountain 2002 Campaign: Observations and Model Comparison, J. Geophys. Res., 111, D06102, https://doi.org/10.1029/2005JD006126, 2006.
Ren, X., van Duin, D., Cazorla, M., Chen, S., Mao, J., Zhang, L., Brune, W. H., Flynn, J. H., Grossberg, N., Lefer, B. L., Rappenglück, B., Wong, K. W., Tsai, C., Stutz, J., Dibb, J. E., Thomas Jobson, B., Luke,W. T., and Kelley, P.: Atmospheric oxidation chemistry and ozone production: Results from SHARP 2009 in Houston, Texas, J. Geophys. Res., 118, 5770–5780, 2013.
Rickly, P. and Stevens, P. S.: Measurements of a potential interference with laser-induced fluorescence measurements of ambient OH from the ozonolysis of biogenic alkenes, Atmos. Meas. Tech., 11, 1–16, https://doi.org/10.5194/amt-11-1-2018, 2018.
Rohrer, F., Lu, K., Hofzumahaus, A., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Haseler, R., Holland, F., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S., Oebel, A., Shao, M., Zeng, L., Zhu, T., Zhang, Y., and Wahner, A.: Maximum Efficiency in the Hydroxyl-Radical-Based Self-Cleansing of the Troposphere, Nat. Geosci., 7, 559–563, 2014.
Rosales, C., Reidy, E., Bottorff, B., Stevens, P. S., Cantrell, C. A., Mauldin, L. Anderson, D. C., and Wood, E. C. D., An Informal Intercomparison of Ambient Measurements of OH, HO2, and RO2 Radicals in an Indiana Forest, Part 1: Comparison of Instrumental Measurements, Abstract A43M-3280 presented at 2018 Fall Meeting, AGU, Washington, D.C., 10–14 December, 2018.
Roukos, J., Plaisance, H., Leonardis, T., Bates, M., and Locoge, N.: Development and validation of an automated monitoring system for oxygenated volatile organic compounds and nitrile compounds in ambient air, J. Chromatogr. A, 1216, 8642–8651, 2009.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Sheehy, P. M., Volkamer, R., Molina, L. T., and Molina, M. J.: Oxidative capacity of the Mexico City atmosphere – Part 2: A ROx radical cycling perspective, Atmos. Chem. Phys., 10, 6993–7008, https://doi.org/10.5194/acp-10-6993-2010, 2010.
Shetter, R. E., and M. Muller, Photolysis frequency measurements using actinic flux spectroradiometry during the PEM-Tropics mission: Instrumentation description and some results, J. Geophys. Res., 104, 5647–5661, https://doi.org/10.1029/98JD01381, 1999.
Shirley, T. R., Brune, W. H., Ren, X., Mao, J., Lesher, R., Cardenas, B., Volkamer, R., Molina, L. T., Molina, M. J., Lamb, B., Velasco, E., Jobson, T., and Alexander, M.: Atmospheric oxidation in the Mexico City Metropolitan Area (MCMA) during April 2003, Atmos. Chem. Phys., 6, 2753–2765, https://doi.org/10.5194/acp-6-2753-2006, 2006.
Sklaveniti, S., Locoge, N., Stevens, P. S., Wood, E., Kundu, S., and Dusanter, S.: Development of an instrument for direct ozone production rate measurements: measurement reliability and current limitations, Atmos. Meas. Tech., 11, 741–761, https://doi.org/10.5194/amt-11-741-2018, 2018.
Stevens, P. S., Mather, J. H., and Brune, W. H.: Measurement of Tropospheric OH and HO2 by Laser-Induced Fluorescence at Low Pressure, J. Geophys. Res., 99, 3543–3557, https://doi.org/10.1029/93jd03342, 1994.
Tan, D., Faloona, I., Simpas, J. B., Brune, W., Shepson, P. B., Couch, T. L., Sumner, A. L., Carroll, M. A., Thornberry, T., Apel, E., Riemer, D., and Stockwell, W.: HOx Budgets in a Deciduous Forest: Results from the PROPHET Summer 1998 Campaign, J. Geophys. Res., 106, 24407–24427, https://doi.org/10.1029/2001jd900016, 2001.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tan, Z., Lu, K., Hofzumahaus, A., Fuchs, H., Bohn, B., Holland, F., Liu, Y., Rohrer, F., Shao, M., Sun, K., Wu, Y., Zeng, L., Zhang, Y., Zou, Q., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 19, 7129–7150, https://doi.org/10.5194/acp-19-7129-2019, 2019.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 139, 5367–5377, 2017.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St. Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, 2018.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Whalley, L. K., Blitz, M. A., Desservettaz, M., Seakins, P. W., and Heard, D. E.: Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 6, 3425–3440, https://doi.org/10.5194/amt-6-3425-2013, 2013.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Zannoni, N., Gros, V., Lanza, M., Sarda, R., Bonsang, B., Kalogridis, C., Preunkert, S., Legrand, M., Jambert, C., Boissard, C., and Lathiere, J.: OH reactivity and concentrations of biogenic volatile organic compounds in a Mediterranean forest of downy oak trees, Atmos. Chem. Phys., 16, 1619–1636, https://doi.org/10.5194/acp-16-1619-2016, 2016.
Zhou, X., Zhang, N., TerAvest, M., Tang, D., Hou, J., Bertman, S., Alaghmand, M., Shepson, P. B., Carroll, M. A., Griffith, S., Dusanter, S., and Stevens, P. S.: Nitric acid photolysis on forest canopy surface as a source for tropospheric nitrous acid, Nat. Geosci., 4, 440–443, 2011.