the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Jul 2020
| 17 Jul 2020
Chemical characterization of secondary organic aerosol at a rural site in the southeastern US: insights from simultaneous high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) and FIGAERO chemical ionization mass spectrometer (CIMS) measurements
Yunle Chen
Masayuki Takeuchi
Theodora Nah
Manjula R. Canagaratna
Harald Stark
Karsten Baumann
Francesco Canonaco
André S. H. Prévôt
L. Gregory Huey
Rodney J. Weber
The formation and evolution of secondary organic aerosol (SOA) were investigated at Yorkville, GA, in late summer (mid-August to mid-October 2016). The organic aerosol (OA) composition was measured using two online mass spectrometry instruments, the high-resolution time-of-flight aerosol mass spectrometer (AMS) and the Filter Inlet for Gases and AEROsols coupled to a high-resolution time-of-flight iodide-adduct chemical ionization mass spectrometer (FIGAERO-CIMS). Through analysis of speciated organics data from FIGAERO-CIMS and factorization analysis of data obtained from both instruments, we observed notable SOA formation from isoprene and monoterpenes during both day and night. Specifically, in addition to isoprene epoxydiol (IEPOX) uptake, we identified isoprene SOA formation from non-IEPOX pathways and isoprene organic nitrate formation via photooxidation in the presence of NOx and nitrate radical oxidation. Monoterpenes were found to be the most important SOA precursors at night. We observed significant contributions from highly oxidized acid-like compounds to the aged OA factor from FIGAERO-CIMS. Taken together, our results showed that FIGAERO-CIMS measurements are highly complementary to the extensively used AMS factorization analysis, and together they provide more comprehensive insights into OA sources and composition.
- Article
(1699 KB) - Full-text XML
-
Supplement
(1551 KB) - BibTeX
- EndNote





Organic aerosol (OA), known for its complexity, represents a substantial fraction of tropospheric submicron aerosol (Kanakidou et al., 2005; Zhang et al., 2007; Kroll and Seinfeld, 2008; Jimenez et al., 2009). Global and regional measurements have revealed that the majority of OA can be secondary in nature (Lim and Turpin, 2002; Zhang et al., 2007; Weber et al., 2007; Lanz et al., 2007; Huang et al., 2014). The southeastern United States (US) is known for its large biogenic volatile organic compound (VOC) emissions from both conifer and deciduous forests under the influence of intensive anthropogenic activities (Weber et al., 2007; Xu et al., 2015a). Isoprene and monoterpenes (α-pinene, β-pinene, and limonene) are the most dominant biogenic VOC and secondary OA (SOA) precursors in the southeastern US, and there is substantial interest in these compounds. For isoprene-derived SOA, isoprene epoxydiol (IEPOX) uptake, followed by subsequent condensed-phase reactions (Surratt et al., 2010; Lin et al., 2012; Paulot et al., 2009), is known to be the major pathway in the southeastern US, approximately contributing 18 %–36 % to total OA in warm seasons (Budisulistiorini et al., 2013; Hu et al., 2015; Xu et al., 2015a, b). Isoprene organic nitrates formed from both photooxidation and nitrate radical oxidation have been characterized in ambient measurements and included in models (Lee et al., 2016; Bates and Jacob, 2019), as has non-IEPOX SOA formed from isoprene hydroxy hydroperoxide (ISOPOOH) oxidation (Krechmer et al., 2015; Nagori et al., 2019). Monoterpene nocturnal reactions have been shown to be an important source of particulate organic nitrates in the southeastern US (Xu et al., 2015a, b; Pye et al., 2015), while more recent studies have demonstrated that monoterpenes are also the prominent source of total OA in the southeastern US given the large fraction of non-nitrogen-containing monoterpene-derived species (Zhang et al., 2018; Xu et al., 2018).
A better understanding of OA composition has been aided by advances in state-of-the-art real-time aerosol instrumentation in the past 2 decades. Each instrument, with its unique capabilities, provides one piece of information to solve the SOA puzzle. The high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne; henceforth referred to as AMS) (DeCarlo et al., 2006; Canagaratna et al., 2007), for example, has been widely used in both laboratory experiments and field measurements. Designed to quantitatively characterize the chemical composition of submicron non-refractory (NR-PM1) aerosol, the AMS produces ensemble average mass spectra for organic and inorganic species. Different methods have been used to deconvolve AMS OA mass spectra, e.g., multiple component analysis (Zhang et al., 2007) and positive matrix factorization (PMF) (Ulbrich et al., 2009; Canonaco et al., 2013). Oxygenated OA (OOA) is a subgroup, or factor, that has been ubiquitously resolved by AMS factorization analysis and normally used as a surrogate for SOA, while other OA factors can be more regional and seasonal, e.g., isoprene-derived OA (isoprene-OA) and biomass burning OA (BBOA) (Jimenez et al., 2009; Ng et al., 2010; Hu et al., 2015; Xu et al., 2015a; Cubison et al., 2011). OOA can be further divided into more oxidized OOA (MO-OOA, characterized by a higher O:C ratio) and less oxidized OOA (LO-OOA, characterized by a lower O:C ratio) (Setyan et al., 2012; Xu et al., 2015a), which have also been called low-volatility OOA (LV-OOA) and semi-volatile OOA (SV-OOA), respectively, in some studies (Ng et al., 2010; Jimenez et al., 2009). In general, LO-OOA corresponds to fresh SOA and MO-OOA corresponds to aged SOA (Zhang et al., 2005, 2007; Jimenez et al., 2009; Ng et al., 2010). The two OOA factors account for a large fraction of submicron OA worldwide (Jimenez et al., 2009), but the sources of LO-OOA and MO-OOA at different locations are still largely unknown. A chemical ionization mass spectrometer (henceforth referred to as CIMS) is a well-established piece of equipment for online measurements of gaseous species (Huey, 2007), and the recent combination of a Filter Inlet for Gases and AEROsols (henceforth referred to as FIGAERO) with the CIMS (henceforth referred to as FIGAERO-CIMS) allowed for the application of CIMS to aerosol molecular composition characterization (Lopez-Hilfiker et al., 2014). Source apportionment analysis has been performed on CIMS gas- and particle-phase measurements in previous studies in a similar manner to that of AMS measurements (Yan et al., 2016; Massoli et al., 2018; Lee et al., 2018). Compared to traditional AMS source apportionment, FIGAERO-CIMS can provide more information on the identity of each factor, e.g., chemical formulae of tracer molecules and the location of the maximum desorption signal in temperature space (Tmax), by which the enthalpy of sublimation and compound vapor pressure can be evaluated (Lopez-Hilfiker et al., 2014). The FIGAERO-CIMS is highly complementary to the AMS and could substantially expand our knowledge of the AMS OA factors that have been known for over a decade.
Here, we present results from 2-month measurements at Yorkville, GA, a rural site in the southeastern US, during a transitional season from summer to fall. Along with a suite of additional instrumentation (see Nah et al., 2018a, b), AMS and FIGAERO-CIMS were deployed, and factorization analysis was applied to measurements from both instruments in an effort to gain new insights into established AMS OA factors. By combining AMS and FIGAERO-CIMS measurements, we show that isoprene and monoterpenes were dominant OA precursors during both day and night. We also identify notable isoprene oxidation pathways, besides IEPOX uptake, and their contribution to particulate organic nitrates, which was less recognized by previous AMS measurements.
2.1 Site description
The ambient measurements took place from mid-August to mid-October 2016 at the SouthEastern Aerosol Research and Characterization (SEARCH) field site at Yorkville, Georgia (33.92833∘ N, 85.04555∘ W; 394 m a.s.l.). The instruments were housed in an air-conditioned trailer. The Yorkville site was a long-term field site located in a rural environment approximately 55 km northwest of Atlanta, immediately surrounded by forests and open pastures for cattle grazing. Compared to previous measurements at this site (Xu et al., 2015a, b), the sampling period of this study was characterized by a transition from a warmer to colder season, which had a direct influence on biogenic VOC emissions; e.g., the isoprene mixing ratio decreased from more than 2 ppb at the beginning of the campaign to below 1 ppb at the end (daily average). More details of this 2016 Yorkville campaign have been presented in recent publications by Nah et al. (2018a, b).
2.2 Instrumentation
An AMS (DeCarlo et al., 2006; Canagaratna et al., 2007) was used to characterize the composition of NR-PM1. Ambient air was sampled through a PM1 cyclone (URG Corp.) at 16.7 L min−1 to remove coarse particles. A Nafion dryer was placed upstream of the AMS to dry the particles (RH < 20 %) in order to eliminate the influence of relative humidity (RH) on particle collection efficiency (CE) in the AMS (Matthew et al., 2008; Middlebrook et al., 2012). Measurements were taken every minute and post-averaged to a 5 min time interval. Gas-phase interference was eliminated by subtracting the signals when the AMS sampled through a HEPA filter. Ionization efficiency (IE) calibrations were performed with 300 nm ammonium nitrate particles, and sulfate relative ionization efficiency (RIE) calibrations were performed with 300 nm ammonium sulfate particles. Both calibrations were conducted on a weekly basis. AMS data were analyzed using the data analysis toolkits SQUIRREL (v1.57) and PIKA (v1.16G) within the Igor Pro software (v6.37, Wavemetrics, Portland, OR). The organics data matrix and error matrix for source apportionment analysis were also generated from PIKA v1.16G. Elemental ratios, including the oxygen-to-carbon ratio (O : C), hydrogen-to-carbon ratio (H : C), and nitrogen-to-carbon ratio (N : C), were obtained using the method outlined by Canagaratna et al. (2015). By comparing AMS with a parallel particle-into-liquid sampler (PILS) coupled to ion chromatograph (IC) and filter measurements, a constant CE of 0.9 was applied to AMS measurements (Nah et al., 2018a).
An iodide-adduct FIGAERO-CIMS was used to characterize particle-phase multifunctional organic species, given the advantage of its high selectivity towards highly polarizable species, such as carboxylic acids and polyols. A detailed description of FIGAERO-CIMS can be found in Lopez-Hilfiker et al. (2014), while a detailed description of the iodide ionization mechanisms can be found in Huey et al. (1995) and Lee et al. (2014). In brief, ambient air was sampled through a URG PM1 cyclone, and PM1 particles were collected on a perfluorotetrafluoroethylene (PTFE) filter (2 µm pore size Zefluor™, Pall Corporation) in the FIGAERO unit for 25 min at a flow rate of 16.7 L min−1. To prevent potential positive artifacts arising from gases sticking onto the filter during sampling, a 30 cm long parallel-plate activated-carbon denuder (Eatough et al., 1993) was installed upstream of the FIGAERO inlet. After collection, particles were immediately desorbed off the PTFE filter by heated N2 flowing through the filter. The thermal desorption process took 35 min, during which the temperature was increased from room temperature (∼25 ∘C) to ∼200 ∘C in 15 min, held at ∼200 ∘C for another 15 min, and cooled for 5 min. One filter background measurement was taken for every five cycles by keeping the filter on the desorption line. Raw data were saved every second and were pre-averaged to a 10 s time interval before data processing. The data were analyzed using the data analysis toolkit Tofware (v2.5.11, Tofwerk, Thun, Switzerland and Aerodyne, Billerica, MA) within the Igor Pro software (v6.37, Wavemetrics, Portland, OR). The FIGAERO-CIMS particle data matrix was also generated from Tofware v2.5.11. The signals reported for particles in the later discussion were integrations over the thermal desorption process, with background subtracted. The signals reported are in counts per second (Hz) if not specified in the following discussion. As no further sensitivity conversion is applied to the data, reporting the data in hertz implicitly assumes a uniform sensitivity for FIGAERO-CIMS measurements. Due to the nature of iodide reagent ion, which has a higher sensitivity towards oxygenated organic compounds (Lee et al., 2014), the importance of more oxidized compounds will be overemphasized, while less oxidized compounds are underemphasized. Nevertheless, a good correlation (R=0.84) between total OA measured by AMS and FIGAERO-CIMS (Fig. S1 in the Supplement) suggests that the assumption of uniform sensitivity to some extent could be reasonable in this study. When we compared the FIGAERO-CIMS measurements with AMS measurements, the FIGAERO-CIMS signals were converted to mass concentrations by multiplying ion signals in Hz with the molecular weight (MW) of each ion, and the new unit is grams per mole per second (g mol−1 s−1). This conversion allows for an easier cross-instrument comparison between AMS and FIGAERO-CIMS. It is noted that the unit (g mol−1 s−1) is a scalar of the ion signal based on MW and not an actual mass concentration.
This study focuses on AMS and FIGAERO-CIMS measurements. Other co-located instruments included PILS-ICs to measure water-soluble inorganic and organic acid species, CIMSs to measure gaseous species, PILS and mist chambers coupled to a total organic carbon (TOC) analyzer to measure particle- and gas-phase water-soluble organic carbon, a gas chromatography–flame ionization detector (GC-FID) with a focusing trap to measure hourly resolved VOC, and a chemiluminescence monitor to measure NO and NO2.
2.3 Source apportionment methods
As organic measurements from the AMS and FIGAERO-CIMS are comprised of hundreds of species, source apportionment methods were applied to both measurements for a better understanding of OA sources and composition. Two widely used source apportionment methods, positive matrix factorization (PMF) and the multilinear engine (ME-2) algorithm, were used here. PMF is the most commonly used source apportionment method for AMS data (Lanz et al., 2007; Ulbrich et al., 2009; Jimenez et al., 2009; Ng et al., 2010; Zhang et al., 2011). It is a least-squares approach based on a receptor-only multivariate factor analytic model to solve bilinear unmixing problems. PMF deconvolves the observed data matrix as a linear combination of various factors with constant mass spectra but varying concentrations across the dataset. The model solution of PMF is not unique due to rotational ambiguity. The ME-2 solver works in a similar manner to PMF. The difference between PMF and ME-2 is that ME-2 allows users to introduce a priori information in the form of a known factor time series and/or a factor profile as inputs to the model to constrain the solution (Canonaco et al., 2013). In the following discussion, we applied PMF analysis to both AMS and FIGAERO-CIMS datasets. For the AMS dataset, we found that unconstrained PMF runs failed to identify reasonable solutions, so we performed ME-2 analysis on the AMS dataset and constrained it with a fixed isoprene-OA factor profile. The constraining method was known as the a-value approach (Canonaco et al., 2013; Crippa et al., 2014), whereby the a value (ranging from 0 to 1) determines how much a factor profile is allowed to vary from the input source profile. The isoprene-OA factor profile (anchor profile) we used to constrain the ME-2 analysis was previously resolved by PMF from Centreville, Alabama, during the SOAS campaign (Xu et al., 2015a, b). A description of our unconstrained PMF and ME-2 analyses is provided in Sect. 3.3.
2.4 Estimating mass concentration of organic nitrate functionality from AMS measurements
The mass concentration of organic nitrate functionality (NO3,org) was calculated based on from AMS measurements (Farmer et al., 2010) by Eqs. (1)–(2).
Here, Rmeas is the ratio from field measurements; RAN is the ratio of pure ammonium nitrate; and RON is the ratio of pure organic nitrates. Note that NO3,org refers to the mass concentration of nitrate functionality only (-ONO2). In this study, an RAN of 3 (average value from three IE calibrations of ammonium nitrate throughout the field measurements) was adopted for NO3,org calculation. For RON, two values, an upper bound of 10 and a lower bound of 5, derived from β-pinene + NO3• and isoprene + NO3• systems, respectively, were adopted to acquire an NO3,org range for field measurements (Bruns et al., 2010; Boyd et al., 2015; Xu et al., 2015b).
3.1 Campaign overview and OA bulk properties
The meteorological data from the campaign have already been discussed in detail in Nah et al. (2018a). Briefly, the 2-month measurements were characterized by moderate temperature (24.0±4.0 ∘C, average ± SE if not specified hereafter) and high RH (68.9±17.9 %). Isoprene was the most abundant VOC (1.21±1.08 ppb), followed by propane (0.84±0.39 ppb), α-pinene (0.37±0.40 ppb), and β-pinene (0.32±0.29 ppb), making biogenic VOCs the predominant OA precursors at Yorkville. A clear decreasing trend was observed for the isoprene concentration as temperature decreased throughout the campaign, which is consistent with the seasonal variation of isoprene emissions (Seinfeld and Pandis, 2016). The Yorkville site is located in a rural environment with a low but non-negligible NOx level, with average NO and NO2 concentrations of 0.15±0.35 and 2.2±1.8 ppb, respectively. NO was probably transported from roadways, peaking at around 09:00 EDT.
Organic species were the dominant component of NR-PM1 (5.0±2.3 µg m−3), contributing 75 % to the total NR-PM1 aerosol mass measured by AMS. The study mean diurnal trends of OA elemental ratios measured by both the AMS and FIGAERO-CIMS are shown in Fig. 1. Since the nitrate functionality of organic nitrates largely fragments into NO+ and in the AMS (Farmer et al., 2010) and will result in underestimated O : C and N : C values for OA, the nitrogen mass and oxygen mass from NO3,org have been added back in the AMS O : C and N : C analysis. Compared to the OA measured by AMS, the OA measured by FIGAERO-CIMS was more oxidized, with a lower H : C (by 0.08 compared to AMS H : C) and a higher O : C (by 0.17 compared to original AMS O : C and by 0.10 compared to the upper bound of AMS O : C after including oxygen atoms from NO3,org). This difference can be explained by the selective sensitivity of the iodide reagent ion, which has a higher sensitivity towards oxygenated organic compounds (Lee et al., 2014). After including NO3,org in the AMS N : C calculation, the AMS N : C measurements fell into the range of the FIGAERO N : C measurements (average of 0.017 from FIGAERO; average of 0.006 to 0.025 from AMS). Both AMS and FIGAERO-CIMS measurements consistently showed that O : C peaked in the afternoon, while N : C peaked at night, suggesting that OA at Yorkville was more oxidized in the afternoon and organic nitrates accounted for a larger OA fraction at night.
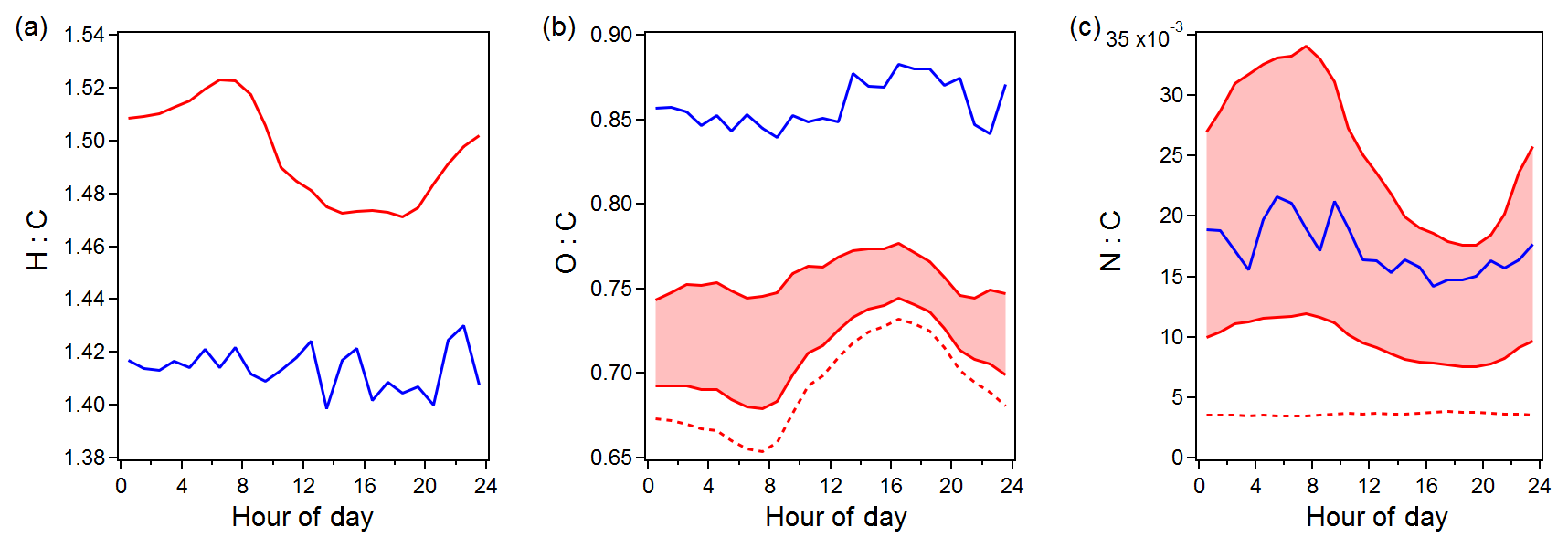
Figure 1Study mean diurnal trends of elemental ratios measured by AMS (red) and FIGAERO-CIMS (blue). The AMS O:C and N:C with and without including NO3,org are shown by the shaded area (with an ratio of 5 and 10) and the dashed line, respectively.
3.2 Overview of organic compounds detected by FIGAERO-CIMS
Figure 2a shows the normalized spectra (signals in mixing ratio) of FIGAERO-CIMS measurements. In total, 769 multifunctional organic compounds possessing 1–18 carbons have been identified in this study, 423 of which were non-nitrogen-containing organic species (pOC, containing at least one carbon atom, at least one oxygen atom, and an even number of hydrogen atoms), and 346 were nitrogen-containing organic species that match the formula of a particulate organic nitrate (pON, containing one nitrogen atom, at least one carbon atom, three or more oxygen atoms, and an odd number of hydrogen atoms). Compounds not attached to an iodide ion were excluded, as their ionization mechanisms were uncertain. Organic nitrates containing two or more nitrogen atoms were not included in the discussion given that they are much less abundant compared to organic mononitrates. Since FIGAERO-CIMS cannot distinguish compounds of the same molecular formula but with different molecular structures, the detected organic nitrate compounds can be peroxy nitrates or multifunctional alkyl nitrates.
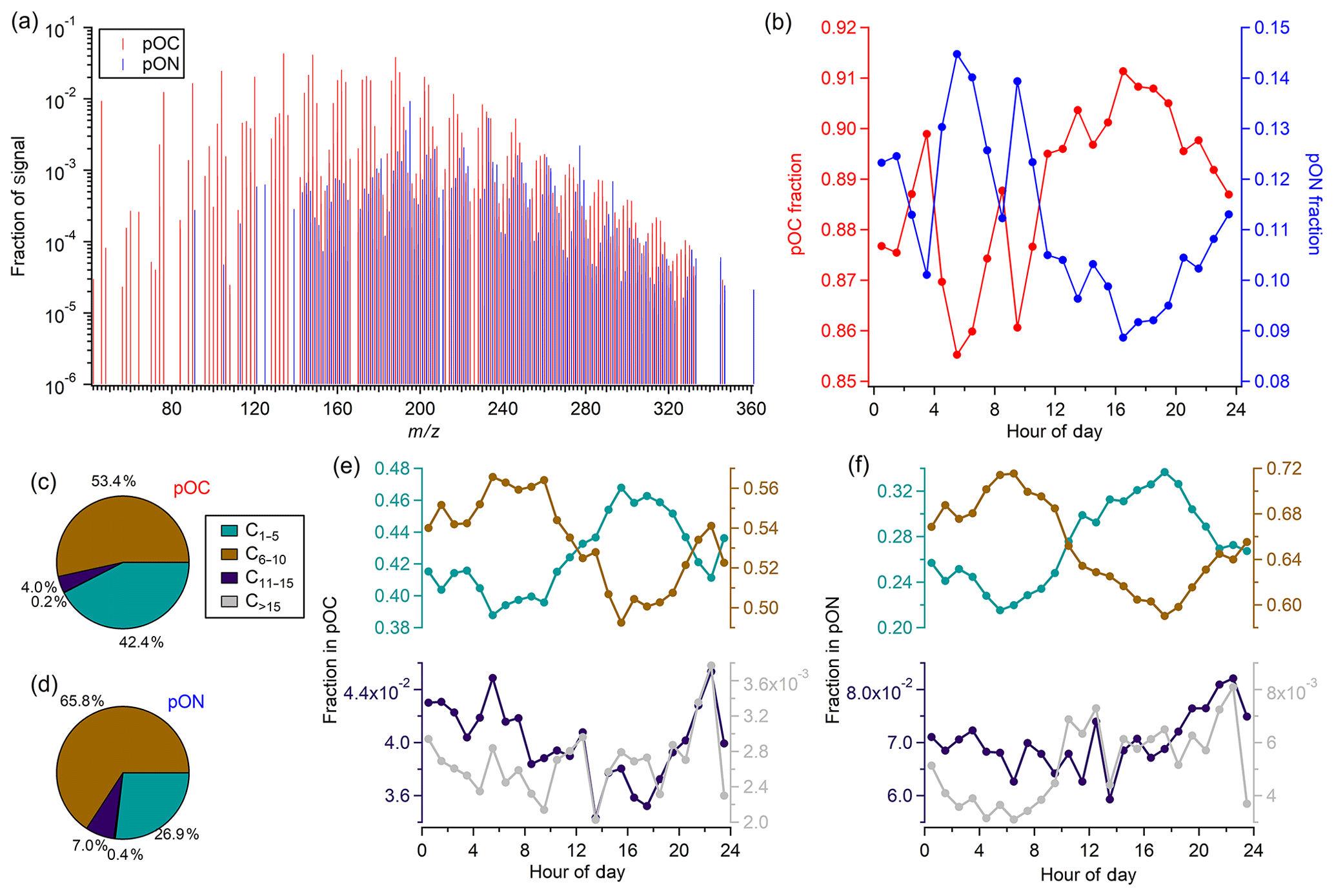
Figure 2Study mean (a) FIGAERO mass spectra (CxHyOz ions in red and CzHyOzN1 ions in blue), (b) fraction of pOC and pON compounds plotted as a function of time of a day, (c, d) fraction of ions of different carbon numbers (grouped as C1–5, C6–10, C11–15, and C>15) in pOC and pON, and (e, f) fraction of C1–5, C6–10, C11–15, and C>15 compounds in pOC and pON plotted as a function of time of day.
On average, pOC and pON contributed 87.7±10.8 % and 12.3±10.8 %, respectively, to the total FIGAERO-CIMS signals, while pOC and pON showed distinct diurnal patterns. pON had a higher contribution at night (Fig. 2b), consistent with our observations of higher N : C at night, which was reported by previous FIGAERO-CIMS studies at other sites (Lee et al., 2016; Huang et al., 2019). A 10:00 EDT peak was also observed for the pON fraction, following the NO peak that happened around 09:00 EDT, likely due to enhanced organic nitrate formation as the NO level increased. The pON fraction was also estimated using AMS nitrate measurements, whereby we calculated lower and upper bounds of NO3,org using an ratio of 10 and 5, respectively, and then applied an average MW of 220 g mol−1 (effective MW of all pON measured by FIGAERO-CIMS) to convert AMS NO3,org to the mass concentration of organic nitrates (sum of the mass of both organic and nitrate functionalities of the organic nitrates). The resulting pON fraction (), 5 %–18 %) was comparable to FIGAERO-CIMS measurements and also agreed with previous studies in the southeastern US (Xu et al., 2015b; Ng et al., 2017). For a group of pON or pOC with the same carbon atom number, a bell-shaped distribution was observed as a function of oxygen atom number (Figs. S2 and S3), similar to observations from previous field measurements (Lee et al., 2016, 2018; Huang et al., 2019).
The average effective formulae of pOC and pON are C6.4H9.0O5.3N0 and C7.5H11.6O6.5N1, respectively. A series of small organic compounds (MW < 80 g mol−1) were detected by FIGAERO-CIMS in this study, some of which were in high abundance, e.g., CH2O2 and C2H4O3. These ions should not be detected in the particle phase due to expected high volatility and were likely thermal decomposition products of less volatile molecules, not uncommon in FIGAERO thermograms (Stark et al., 2017; Schobesberger et al., 2018). The presence of these ions biased effective formulae and MW calculations, and thus the values reported in Table 1 could be smaller than the actual molecules. Meanwhile, these small but highly oxidized fragments may also have a higher carbon oxidation state () and bias the FIGAERO-CIMS elemental ratio calculation. pON molecules on average had around one more carbon than pOC molecules, meaning pON was composed of larger molecules compared to pOC. In Fig. 2, to better illustrate the difference between the pOC and pON composition, we grouped pOC and pON species into four subgroups based on the carbon atom number, C1–5, C6–10, C11–15, and C>15. For both pOC and pON, compounds with fewer than 15 carbon atoms accounted for the majority of total signals (99.8±0.1 % for pOC and 99.6±0.2 % for pON), with C6–10 being the most dominant subgroup (53.4±33.3 % in pOC and 65.8±5.4 % in pON), followed by C1–5 (42.4±33.8 % in pOC and 26.9±5.3 % in pON) and C11–15 (4.0±0.7 % in pOC and 7.0±1.1 % in pON) (Fig. 2c and d). pON contained a higher fraction from C6–10, while pOC contained a higher fraction from C1–5, explaining the difference in their average formulae. Each subgroup showed distinct diurnal patterns, while the same subgroup exhibited similar trends in pOC and pON (Fig. 2e and f). Specifically, C1–5 species had a larger contribution during the daytime, while C6–10 species were more dominant during the night. This is consistent with the emission of their potential precursors; C1–5 species were more likely to arise from isoprene oxidation, while C6–10 species were more likely to arise from monoterpenes, though contributions from other sources, the fragmentation of monoterpene products, and dimer formation in isoprene oxidation are also possible. There was a lack of a clear day–night contrast for C11–15 species, likely due to their low concentrations, low instrument sensitivity, and/or formation from various sources.
3.3 AMS OA factors
We started our analysis with unconstrained PMF runs using the Solution Finder (SoFi 6.4) software. Three factors can be resolved by unconstrained runs, which are isoprene-OA, LO-OOA, and MO-OOA. This three-factor solution was consistent with previous AMS measurements conducted in summer at Yorkville (Xu et al., 2015a, b), in which no primary OA factor was resolved. However, the contribution from isoprene-OA appeared to be largely overestimated in our unconstrained PMF runs. The campaign-average isoprene-OA fraction was 45±15 % (Fig. S4), and the fraction was as high as 90 % at the beginning of the campaign when the emission of isoprene was higher. However, previous measurements at the same site showed that isoprene-OA only accounted for 33 % of total OA in July (Xu et al., 2015a, b). Meanwhile, the (, a tracer for isoprene-derived SOA; Hu et al., 2015) of the resolved isoprene-OA was 7.0 ‰ (Fig. S4c), while in previous studies isoprene-OA had an of around 20 ‰ (Hu et al., 2015; Xu et al., 2015b). These discrepancies indicated that the isoprene-OA factor resolved by unconstrained PMF likely included interferences from other types of OA, as measurements were conducted during a transition in seasons (isoprene emissions), and that unconstrained PMF alone was not sufficient to identify the correct solution for this dataset. Therefore, we applied constraints in the form of an isoprene-OA profile. In previous studies, only the POA profile, rather than SOA, has been fixed in ME-2 analysis (Crippa et al., 2014; Elser et al., 2016). However, as isoprene-OA is a commonly resolved biogenic SOA in the southeastern US during summertime (Xu et al., 2015a, b; Hu et al., 2015; Budisulistiorini et al., 2016; Rattanavaraha et al., 2016) and its profile shows consistency in different studies (Hu et al., 2015), we constrained the isoprene-OA profile with a “clean” isoprene-OA profile resolved in the southeastern US during summer 2013 SOAS measurements at Centreville (Xu et al., 2015a, b). The rotations were explored using the a-value approach (Lanz et al., 2008; Canonaco et al., 2013; Crippa et al., 2014). We tested five a values for the isoprene-OA profile, from 0 to 0.8, with an increment of 0.2. The determination of a final solution was guided by three criteria: the mass fraction of each factor (Fig. S5b), the correlation between factor time series with tracers, and the of resolved isoprene-OA (Fig. S5c). Different tracers were also used for identifying OA factors. 2-methyltetrol is the ring-opening product of IEPOX and can be measured by I−-CIMS (Surratt et al., 2010; Lin et al., 2012; Hu et al., 2015). Lopez-Hilfiker et al. (2016) showed that the 2-methyltetrol signal detected in FIGAERO-CIMS may be derived from the thermal decomposition of accretion products or other organics of lower volatility, but IEPOX uptake is still the major source for this fragment. Here, we still used the 2-methyltetrol (C5H12O4) signal measured by FIGAERO-CIMS as a tracer species for isoprene-OA. Xu et al. (2015a) showed that organic nitrates made up a substantial portion of LO-OOA in the southeastern US, correlating well with LO-OOA. Thus, in this work we used organic nitrate functionality as a tracer for LO-OOA.
Based on the above criteria, a three-factor solution with an a value of 0 was chosen for the AMS dataset. The chosen three-factor solution gave the best correlations between isoprene-OA and the C5H12O4 signal (R=0.85), LO-OOA and NO3,org (R=0.84), and the highest (23 ‰) (Fig. S5). The mass spectra and time series for the factors are shown in Fig. 3. With ME-2 analysis, the fraction of isoprene-OA was lower compared to unconstrained PMF. On average, isoprene-OA, LO-OOA, and MO-OOA contributed 17±5 %, 33±15 %, and 50±13 % to total OA, respectively. Over the course of the campaign, the fraction of isoprene-OA in total OA decreased from 26 % to 8 % (daily averages), consistent with the decreasing temperature during season transition (Fig. S6). Similar to previous measurements at the same site (Xu et al., 2015a, b), MO-OOA was characterized by a wide afternoon peak, likely related to strong daytime photochemistry, while LO-OOA had a nighttime enhancement, which can arise from changes in boundary layer height, temperature-driven partitioning, and nocturnal OA formation such as the nitrate radical oxidation of biogenic VOCs. The diurnal trend of isoprene-OA also showed an afternoon enhancement, but the day–night contrast was less pronounced compared to MO-OOA. MO-OOA had the highest O : C (0.91), followed by isoprene-OA (0.63) and LO-OOA (0.49).
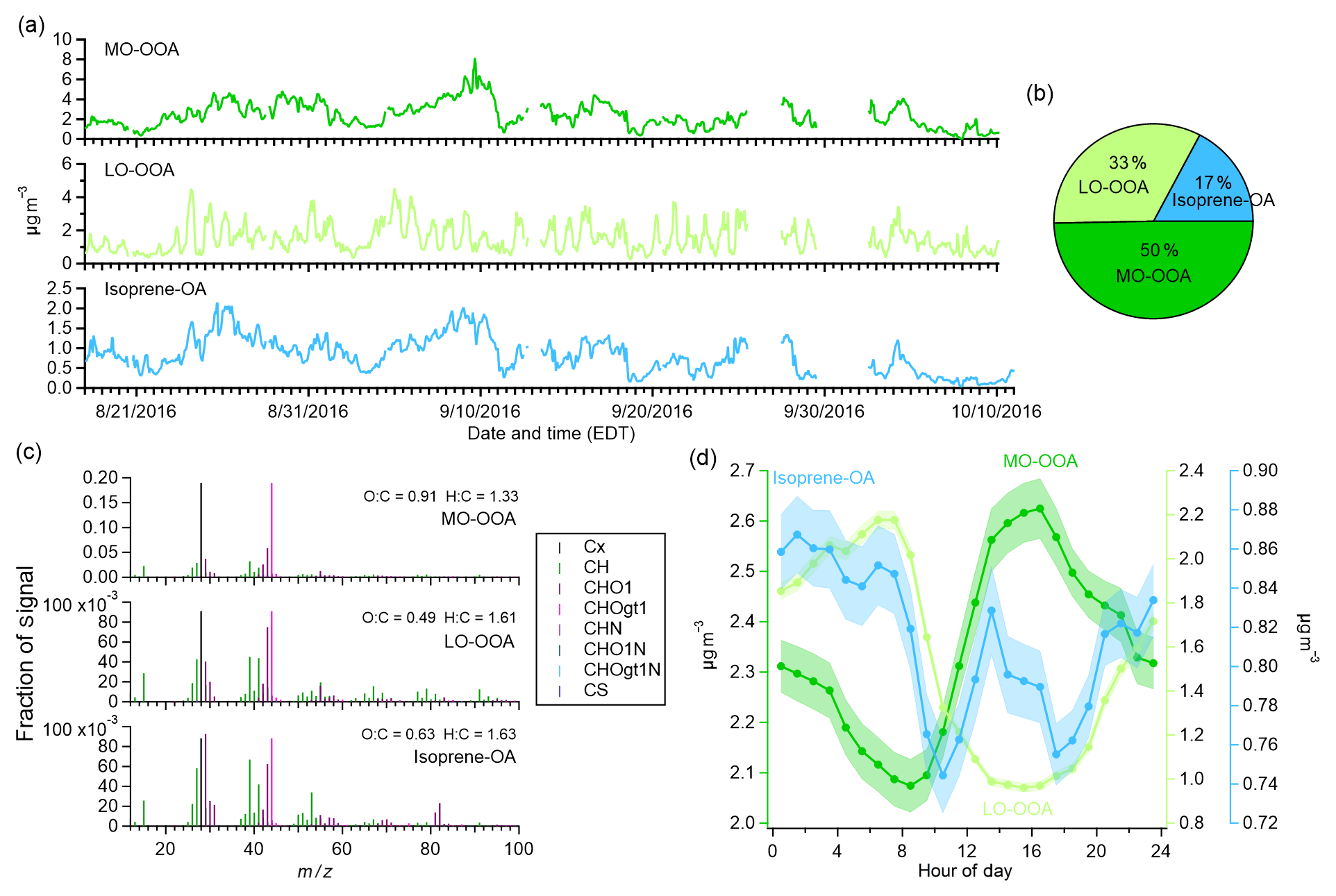
Figure 3(a) Time series and study mean (b) mass fraction, (c) normalized mass spectra, and (d) diurnal profiles (standard deviations in shaded areas) of AMS OA factors resolved by ME-2.
3.4 FIGAERO-CIMS OA factors
The integration of each thermogram, with background subtracted, was taken as the total particle-phase signal (255 desorption cycles were measured in total). The factorization analysis was performed on the integrated total particle-phase signals in the Igor Pro-based PMF Evaluation Tool (version 2.06). Initially, the errors of integrated signals were estimated using Poisson statistics as follows:
where I is the integrated ion signal in the unit of ions. However, we noticed that the σ values estimated by Poisson statistics only provide a lower limit for the real noise, probably due to unaccounted for variabilities introduced by thermogram integration, which can be subjected to overlapping peaks and fragmented ions. As a consequence, the Q∕Qexp from the PMF analyses is ≫1 (Fig. S7), indicating that the estimated errors were underrepresented (Ulbrich et al., 2009). Given the complexity of uncertainties associated with the thermal desorption processes and a lack of well-developed methods to estimate these uncertainties, we developed an empirical scaling factor by comparing the time series of several pairs of highly correlated ions (Fig. S8). Figure S8a, for example, shows a scatter plot of two ions that are highly correlated as a function of time. The Poisson uncertainties for each data point, calculated according to Eq. (3), are also shown. The measured scatter does not have any clear trend with time and is clearly much larger than the calculated Poisson uncertainties. Thus, the uncertainties input into the PMF analysis were empirically increased by a factor of 10 to better account for the observed scatter. This empirical scaling factor of 10 was applied to all errors, which gives more reasonable Q∕Qexp values (Fig. S9) and now only requires one factor to explain the highly correlated ions. As discussed above, thermal decomposition processes could result in the production of a series of small organic compounds (MW < 80 g mol−1). We included these small ions in the PMF analysis, since their time variations reflected those of their parent compounds, but including them will likely result in the overestimation of the and the underestimation of the effective MWs of the factors in a later discussion.
The of each FIGAERO-CIMS factor was calculated using a formula modified from that in Kroll et al. (2011) to include organic nitrate contributions. A group oxidation state of −1 was applied to -ONO2 functionality:
which can be rewritten as
As mentioned above, the iodide reagent ion has a higher sensitivity towards oxygenated organic compounds. Meanwhile, the small and highly oxidized organic compounds formed in potential thermal decomposition may have a higher than their parent molecules. Thus, the average calculated for FIGAERO-CIMS factors could be higher than the actual values.
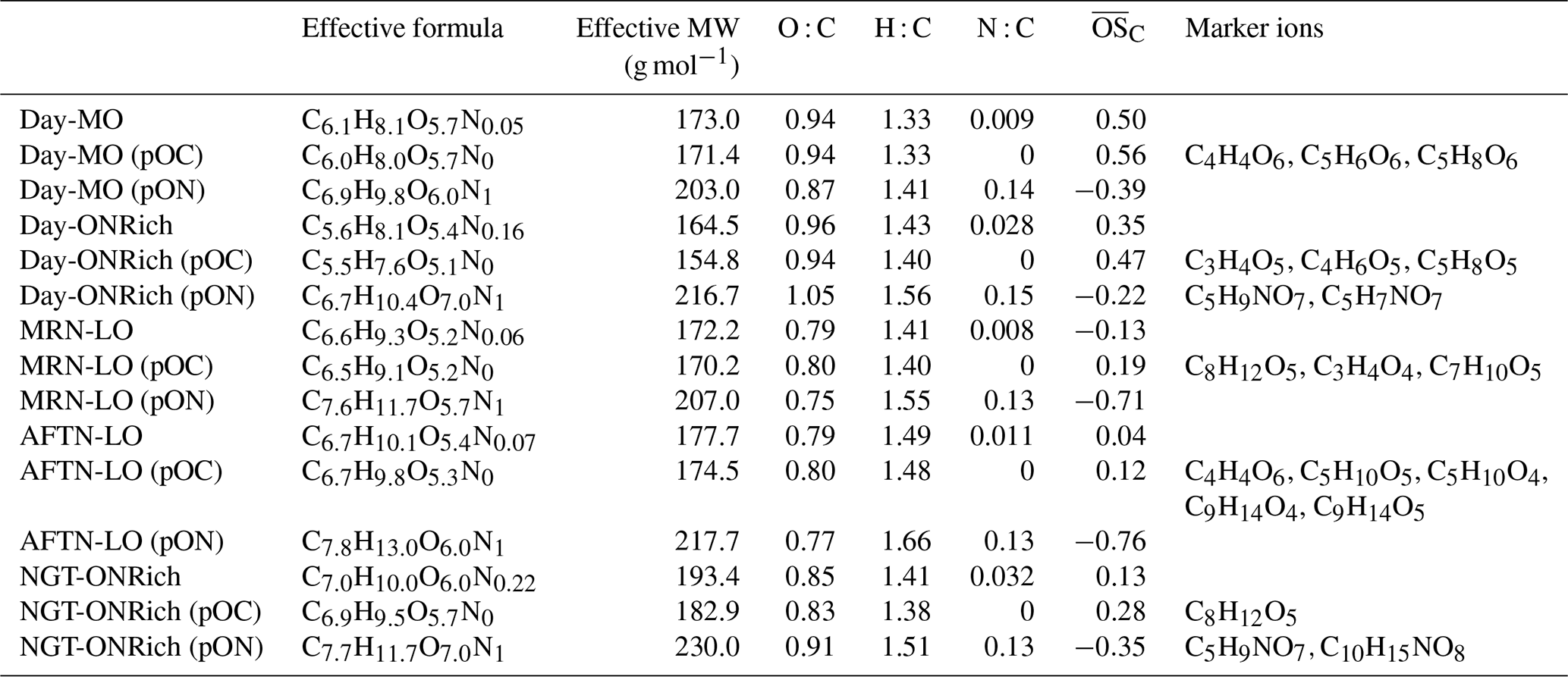
Five FIGAERO-CIMS OA factors were resolved (Figs. 4 and S10). Two factors showing clearly higher N : C (0.028 and 0.032) were distinguished by their diurnal trends and thus denoted as the Day-ONRich (daytime ON-rich) factor and NGT-ONRich (nighttime ON-rich) factor. For the remaining three daytime factors with lower N : C (0.008, 0.009, and 0.011), one showed a significantly higher and was denoted as the Day-MO (daytime more oxidized, ) factor, while the other two were distinguished by their diurnal trends and thus denoted as the MRN-LO (morning less oxidized) factor and AFTN-LO (afternoon less oxidized) factor. The Day-MO, Day-ONRich, MRN-LO, AFTN-LO, and NGT-ONRich factors accounted for 25±15 %, 12±10 %, 21±13 %, 23±16 %, and 18±13 % of total signals measured by FIGAERO-CIMS, respectively. The average effective formulae and MWs were calculated for each factor, as well as for their pOC and pON components, and are shown in Table 1. Similar to the discussion in Sect. 3.2, the pOC and pON species of each factor were grouped into and discussed as C1–5, C6–10, C11–15, and C>15 subgroups (Figs. 5 and S11). The concentration of the C>15 subgroup was negligible, so we excluded them from the following discussion. Below, we evaluate and discuss tracer ions for each FIGAERO-CIMS OA factor based on both their absolute abundance (i.e., ions of the highest signal in the mass spectrum of each factor) and their fractional abundance (i.e., ions dominantly present in a certain factor).
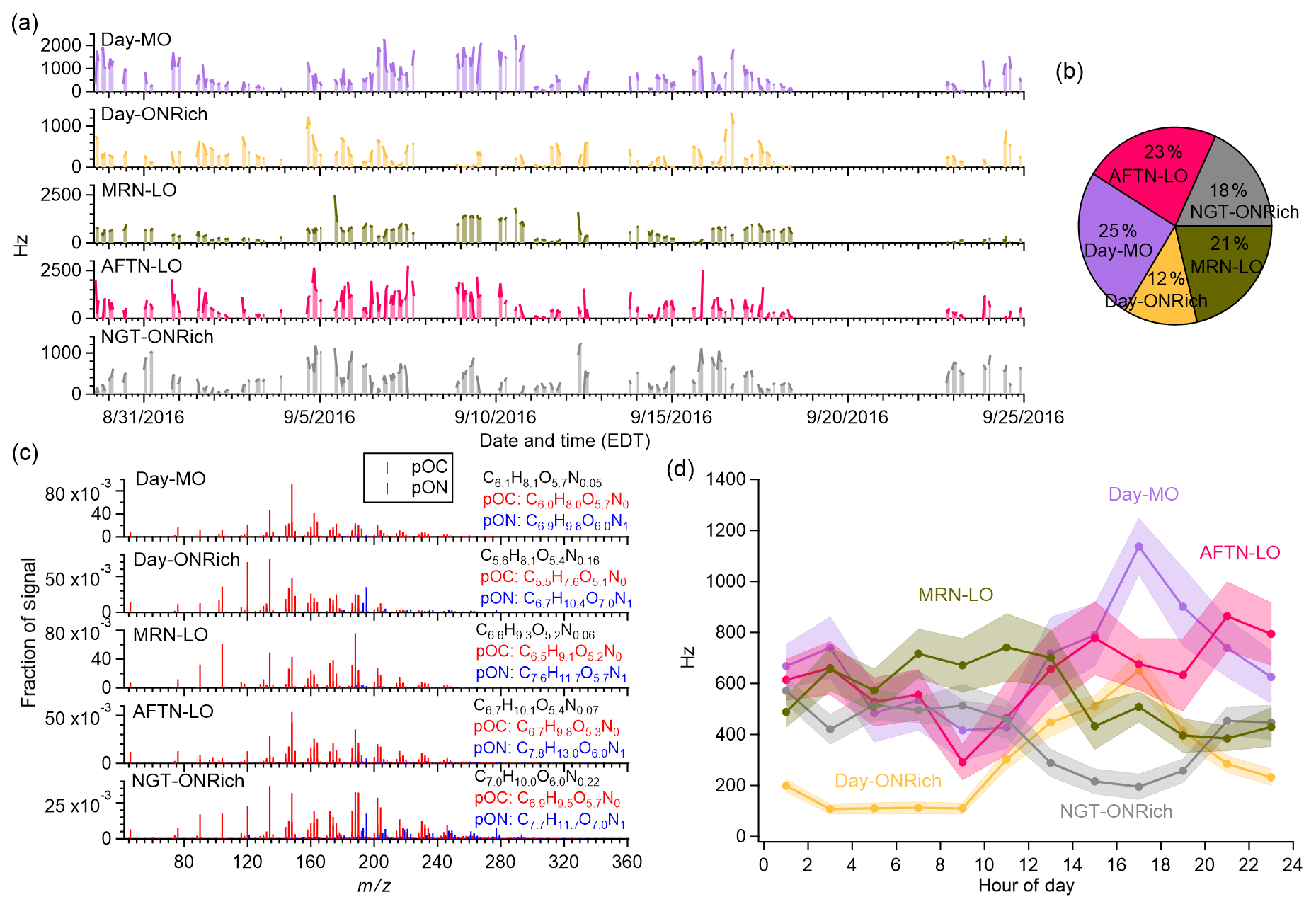
Figure 4(a) Time series and study mean (b) fraction, (c) normalized mass spectra, and (d) diurnal profiles (standard deviations in shaded areas) of FIGAERO OA factors resolved by PMF. Note that the time series in (a) are shaded to guide the eyes.
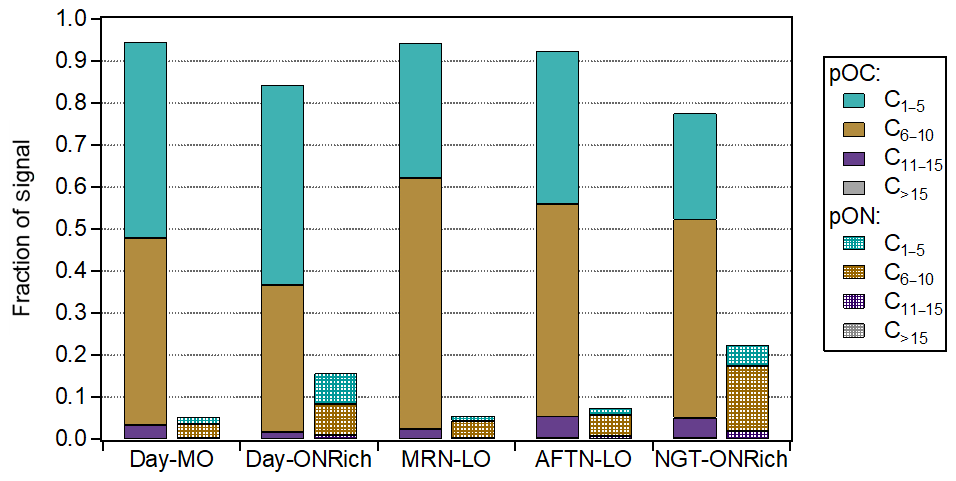
Figure 5Fraction of pOC and pON ions of different carbon numbers (grouped as C1–5, C6–10, C11–15, and C>15) in each FIGAERO OA factor.
NGT-ONRich had the largest MW (193.4 g mol−1), highest effective carbon atom number (7.0), and lowest (0.13), meaning this factor was composed of larger and less oxidized molecules. This feature can be seen more clearly in Fig. 5. Compared to the other four factors, both pOC and pON of NGT-ONRich had a larger fraction from the C6–10 and C11–15 subgroups, as well as a smaller fraction from the C1–5 subgroup. NGT-ONRich also had the highest effective nitrogen atom number (0.22), meaning one in every five molecules was an organic nitrate. The most abundant pON species in NGT-ONRich were C5H9NO7 and C10H15NO8, accounting for 7.8 % and 3.5 % of pON signals in this factor, respectively. C10H15NO8 has been characterized in multiple chamber studies as a major product of α-/β-pinene ∕ limonene + NO3• and α-/β-pinene photooxidation with the presence of NOx (Nah et al., 2016; Lee et al., 2016; Faxon et al., 2018; Takeuchi and Ng, 2019). At Yorkville, the majority of C10H15NO8 was present in NGT-ONRich, implying that nocturnal chemistry is its most important source. Besides C10H15NO8, a series of C9,10 pON ( and ) was also dominantly present in NGT-ONRich, which was similar to fingerprint ions reported by Massoli et al. (2018) for the gaseous terpene nitrate factor at Centreville during the SOAS campaign. The NGT-ONRich we resolved here is likely the particle-phase counterpart of that gaseous terpene nitrate factor. C5H9NO7 was not solely present in NGT-ONRich. Instead, it contributed an even higher fraction to Day-ONRich, suggesting that both daytime and nighttime pathways were critical for C5H9NO7 at Yorkville. This is consistent with C5H9NO7 being detected in previous laboratory studies on isoprene + NO3• and isoprene photooxidation in the presence of NOx (Ng et al., 2008; Lee et al., 2016). Both C5H9NO7 and C10H15NO8 were also identified at Centreville in rural Alabama, US, during SOAS, among the top 10 most abundant pON species (Lee et al., 2016). In another field study at the boreal forest research station SMEAR II located in Hyytiälä, southern Finland, C10H15NO8 has been suggested to be a fingerprint molecule for a daytime factor measured with -based CI-APi-TOF (Yan et al., 2016), but in this study it was more abundant at night. The pOC tracer of NGT-ONRich was C8H12O5, likely corresponding to 2-hydroxyterpenylic acid, which was proposed to be an α-pinene SOA tracer formed from the further oxidation of terpenylic acid (Eddingsaas et al., 2012a; Kahnt et al., 2014a, b; Sato et al., 2016). Taken together, the high contribution from the C6–10 subgroup and the presence of quite a few monoterpene SOA tracers in NGT-ONRich strongly related this factor to monoterpene chemistry, with a non-negligible contribution from isoprene organic nitrates. NGT-ONRich also contained the highest fraction of the C11–15 group. While most signals were from C11 ions, we also observed some C14 and C15 compounds, e.g., pOC C14H18–22O5–7 and C15H20–24O5–7, as well as pON C14H21–25NO7 and C15H23–27NO7, which possibly originated from sesquiterpene oxidation, though more fundamental laboratory studies are needed to further constrain this.
Day-ONRich had an effective nitrogen atom number of 0.16, which is lower compared to NGT-ONRich but still significantly higher than other daytime factors. A total of 23 % of the Day-ONRich pON signal was from C5H9NO7, implying isoprene as the crucial precursor of Day-ONRich, even considering that half of the C5H9NO7 signal may arise from the fragmentation of other larger molecules (Fig. S12a). The second-highest pON ion, C5H7NO7, was also likely from isoprene. The high signals from C5H7NO7 and C5H9NO7 made the C1–5 ON subgroup as prevalent as the C6–10 ON subgroup, which was a distinctive feature for Day-ONRich (Fig. 5). Meanwhile, the pOC of Day-ONRich also contained noticeably more C1–5 ions than other factors, probably due to fragmentation process being a favored pathway under high-NO conditions (Kroll and Seinfeld, 2008). As a result, Day-ONRich had the lowest effective MW (164.5 g mol−1) and the lowest effective carbon number (5.6). The most abundant pOC species of Day-ONRich were C3H4O5, C4H6O5, and C5H8O5. The formula of C3H4O5 implied dicarboxylic acid, and it has been reported in aqueous processes (Lim et al., 2010). However, the average thermogram of C3H4O5 showed two dominant peaks (Fig. S12b): the first peak ( ∘C) roughly matched the volatility of C3 dicarboxylic acids, and the second peak ( ∘C) likely came from the thermal decomposition of molecules of lower volatility. Similar multiple-peak behavior was observed for C3H4O4, a tracer compound for Day-MO (Fig. S12c). C4H6O5, possibly malic acid, has been reported as a higher-generation product of unsaturated fatty acid photochemistry (Kawamura et al., 1996) but has also been found in isoprene SOA in several studies, including particle-phase reactions in isoprene photooxidation in the presence of NOx, the non-IEPOX pathway via the ISOPOOH + OH• reaction (ISOPOOH-SOA), and isoprene ozonolysis (Nguyen et al., 2010; Xu et al., 2014; Krechmer et al., 2015). One isomer of C5H8O5, 3-hydroxyglutaric acid, has been used as a tracer for α-/β-pinene photooxidation SOA (Claeys et al., 2007), while other studies have identified C5H8O5 in isoprene SOA when the IEPOX pathway was suppressed (Nguyen et al., 2011; Krechmer et al., 2015; Liu et al., 2016). C5H8O5 was also found in the oxidation of 1,3,5-trimethylbenzene (Praplan et al., 2014), toluene (Kleindienst et al., 2007), and levoglucosan (Zhao et al., 2014). There was no sign of prevalent anthropogenic emissions or biomass burning events during the measurements, so the presence of C5H8O5 was more likely linked to monoterpene photooxidation and/or non-IEPOX isoprene chemistry.
Day-MO was dominated by pOC signals (accounting for 95 % of signals) and characterized by the highest (0.50) of all factors. The tracer ions of Day-MO were C4H4O6, C5H6O6, and C5H8O6. Given their lower degree of saturation and considerably high O : C, these compounds were likely carboxylic acids, particularly di- or even tri-carboxylic acids. For instance, C4H4O6, likely 2-hydroxy-3-oxosuccinic acid, was identified in the OH•-initiated oxidation of aqueous succinic and tartaric acids (Chan et al., 2014; Cheng et al., 2016). C5H8O6 was likely 2,3-dihydroxy-2-methylsuccinic acid, a product of the aqueous cross-photoreaction of glycolic and pyruvic acids (Xia et al., 2018), or methyltartaric acid (MTA), which is a tracer of aged isoprene SOA (Jaoui et al., 2019). However, we cannot rule out the possibility that they were fragments from the thermal decomposition of larger molecules. Techniques without thermal desorption processes will be beneficial in understanding the nature of highly oxidized OA molecules in future studies.
Similar to Day-MO, pOC accounted for more than 90 % of total signals in MRN-LO and AFTN-LO. These two factors had similar fractions from each subgroup (Fig. 5), though they were dominated by different ions. For MRN-LO, the dominating ions were C8H12O5 and C3H4O4, while C7H10O5 also stood out. C8H12O5, as discussed above, was related to α-/β-pinene SOA, and C7H10O5 also likely corresponded to an α-pinene SOA tracer, i.e., 3-acetylpentanedioic acid (Kleindienst et al., 2007). C3H4O4 could correspond to malonic acid or its isomers, but given its high desorption temperature (Fig. S12c), C3H4O4 was more likely composed of fragments of larger molecules. For AFTN-LO, the most prominent ions were C4H4O6, C5H10O4,5, and C9H14O4,5. C4H4O6. C4H4O6 was likely related to aqueous processing as discussed above. C9H14O4, likely pinic acid (Seinfeld and Pandis, 2016), was a well-established fresh α-pinene SOA tracer, and C9H14O5 was probably related to α-/β-pinene SOA (Kahnt et al., 2014a, b; Sato et al., 2016). C5H10O5 has been shown to be a dominant product of ISOPOOH-SOA (Krechmer et al., 2015; D'Ambro et al., 2017) but has also been detected in isoprene ozonolysis and isoprene photooxidation under high NO (Jaoui et al., 2019). It is interesting that a non-IEPOX isoprene SOA product was found to be one of the prominent tracers for an afternoon low-NO fresh SOA factor in our study. Previous factorization analysis of AMS measurements alone suggested that ISOPOOH-SOA accounted for only ∼2 % of ambient OA at Centreville during summer 2013 SOAS measurements (Krechmer et al., 2015). If the C5H10O5 we observed in AFTN-LO was dominantly from the ISOPOOH + OH• reaction via the non-IEPOX pathway, ISOPOOH-SOA may account for a more considerable fraction of fresh isoprene SOA in our study compared to that reported in Centreville. Thus, the initial difficulty we encountered when resolving isoprene-OA, which is believed to form mainly via the IEPOX pathway, from PMF analysis of AMS data may be explained to some extent. Taken together, although both MRN-LO and AFTN-LO were relatively fresh SOA, MRN-LO had more contribution from monoterpenes, while AFTN-LO was more dominated by isoprene SOA.
3.5 Tracer species detected by FIGAERO-CIMS and their implications
As discussed in Sect. 3.4, a series of biogenic SOA tracers, mostly from isoprene and monoterpenes, has established their importance in more than one FIGAERO-CIMS OA factor. To better understand the OA formation mechanisms, we selected six isoprene and monoterpene SOA tracers to represent different oxidation pathways and examined their distributions in the five FIGAERO-CIMS OA factors (Fig. 6).

Figure 6Diurnal data on selected tracer species for isoprene and monoterpene SOA. (a) C5H9NO7 (isoprene + NO3•, isoprene + OH• + NOx); (b) C5H12O4 (isoprene + OH•, IEPOX uptake); (c) C5H10O5 (isoprene + OH•, non-IEPOX pathway); (d) C10H15NO8 (α-/β-pinene + NO3•, α-/β-pinene + OH• + NOx); (e) C9H14O4 (fresh monoterpene SOA); (f) C8H12O6 (aged monoterpene SOA).
For isoprene SOA, C5H9NO7 was chosen here as a pON tracer, C5H12O4 as an IEPOX uptake tracer, and C5H10O5 as a non-IEPOX tracer. Note that C5H10O5 can form from isoprene oxidation under various conditions: while C5H10O5 is a major product in ISOPOOH + OH• when the IEPOX uptake pathway is suppressed (Krechmer et al., 2015; D'Ambro et al., 2017), it also forms in isoprene + O3 and isoprene + OH• + NOx (Jaoui et al., 2019). Most of the C5H9NO7 signals were found in Day-ONRich (39 %) and NGT-ONRich (32 %), suggesting a non-negligible isoprene ON formation during both day and night. The efficient nocturnal isoprene oxidation is possibly via the reaction with nitrate radicals rather than with ozone (Ng et al., 2008; Brown et al., 2009; Schwantes et al., 2015; Fry et al., 2018). In addition, the recent work by Fry et al. (2018) suggested a substantially longer nighttime peroxy radical lifetime in ambient air versus under chamber conditions, which allows for the formation of lower-volatility products and thus higher SOA yields from isoprene nocturnal chemistry. C5H12O4 was only noticeable in daytime non-ON-rich factors, consistent with its low-NO photochemistry origin. C5H10O5 was also only present in daytime factors. However, different from C5H12O4, a noticeable fraction of its signal was in Day-ONRich, implying that C5H10O5 can also be formed under high-NO conditions. One interesting observation was that while C5H12O4 is an early-generation product of isoprene oxidation, it had a larger fraction in Day-MO (expected to be aged SOA) than in AFTN-LO (expected to be fresh SOA). Here, we hypothesize that the Day-MO factor was closely related to particle-phase aqueous processes, and the presence of C5H12O4 in Day-MO can be explained by the fact that IEPOX uptake to the particle phase requires aerosol water. Aqueous chemistry can also explain the acid-like ions observed in large abundance in Day-MO.
For monoterpene SOA, C10H15NO8 was used here as a pON tracer, C9H14O4, likely pinic acid (Seinfeld and Pandis, 2016), as a fresh SOA tracer, and C8H12O6, likely 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) (Szmigielski et al., 2007; Zhang et al., 2010; Müller et al., 2012; Eddingsaas et al., 2012b), as an aged SOA tracer. C10H15NO8 was prominently present in the nighttime factor NGT-ONRich, implying that nocturnal oxidation, likely by nitrate radicals, was its major source. The majority of the C9H14O4 signal was found in MRN-LO and AFTN-LO as expected, consolidating MRN-LO and AFTN-LO as daytime fresh SOA factors. C8H12O6 was suggested to form from the OH-initiated oxidation of pinonic acid (Müller et al., 2012; Szmigielski et al., 2007). At Yorkville it was present in comparable abundance in MRN-LO, AFTN-LO, Day-MO, and NGT-ONRich, suggesting that complex aging pathways of fresh monoterpene SOA took place both day and night.
3.6 Correlations between AMS OA factors and FIGAERO-CIMS OA factors
To compare AMS OA factors with FIGAERO-CIMS OA factors, we first converted FIGAERO-CIMS signals (Hz) to mass concentrations (Hz g mol−1) by simply applying the effective MW to the time series of each factor, while still assuming uniform sensitivity for all compounds. The hourly averages were used for cross-instrument comparison, and results are shown in Fig. 7.
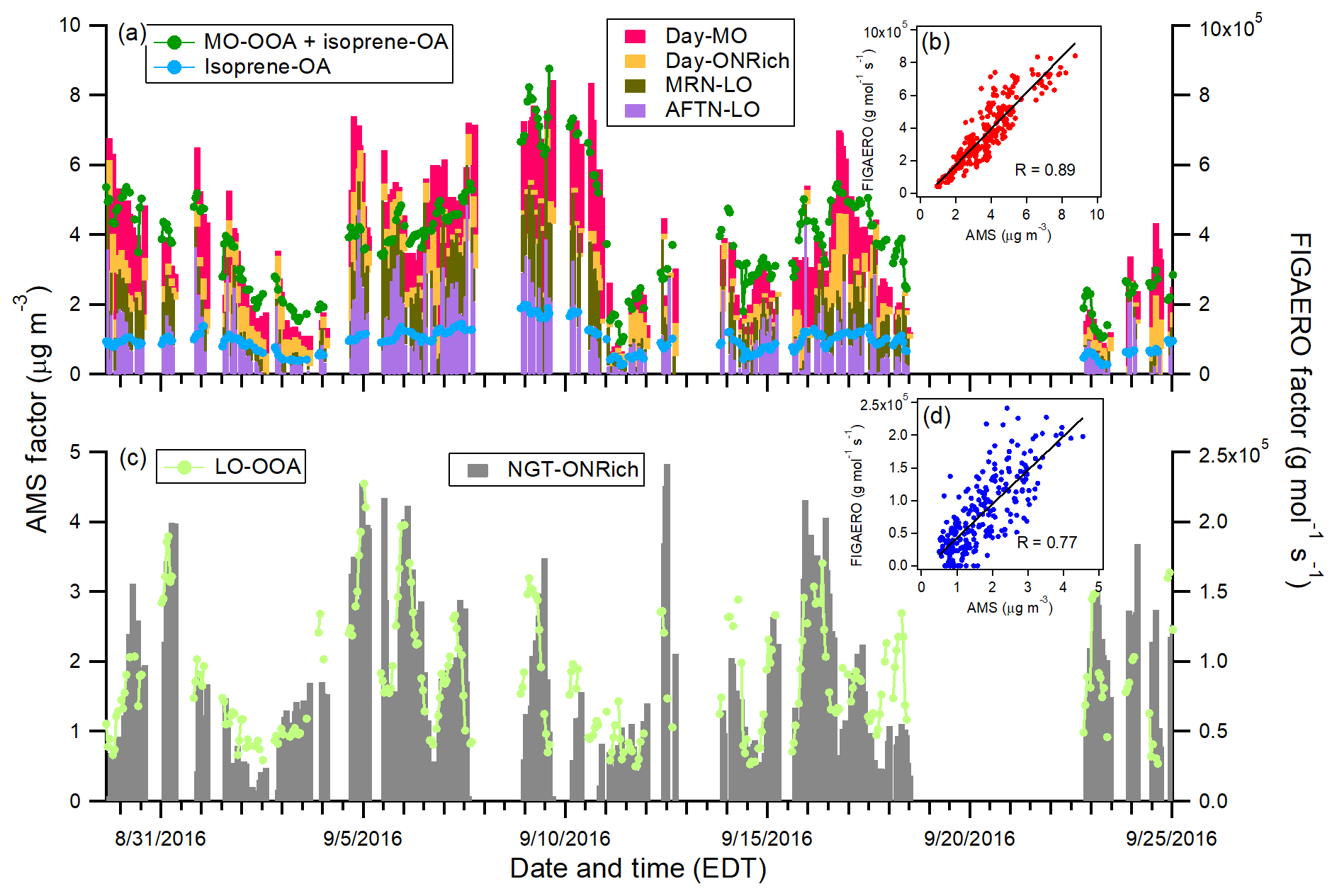
Figure 7Comparison between AMS daytime factors and FIGAERO-CIMS daytime factors (a, b), as well as the AMS nighttime factor and FIGAERO-CIMS nighttime factor (c, d).
For both AMS and FIGAERO-CIMS measurements, only one nighttime factor was resolved, LO-OOA from AMS and NGT-ONRich from FIGAERO-CIMS. A good correlation (R=0.77) in time series was observed between them (Fig. 7c and d). As discussed above, the FIGAERO-CIMS measurements strongly related this factor to monoterpene chemistry, which was consistent with previous AMS measurements in the southeastern US (Xu et al., 2015a, b). NGT-ONRich also showed a prevalent contribution from organic nitrates, with one-fourth of the molecules being pON species. However, FIGAERO-CIMS also identified a non-negligible presence of isoprene-derived pON species in this factor, which the AMS was unable to resolve, implying the potential contribution from isoprene nocturnal organic nitrate formation. In a recent study, Xu et al. (2018) showed that the major source of LO-OOA in the southeastern US is from monoterpenes, but it also includes contributions from sesquiterpene oxidation pathways. Our observation of a series of C14 and C15 species in NGT-ONRich is consistent with the presence of sesquiterpene SOA, though it cannot provide a further quantitative constraint.
Two daytime factors were resolved for AMS measurements, while four were resolved for FIGAERO-CIMS measurements. A strong correlation was observed for the summation of the AMS daytime factors (isoprene-OA + MO-OOA) and the summation of the FIGAERO-CIMS daytime factors (Day-MO + Day-ONRich + MRN-LO + AFTN-LO), with R=0.89 (Fig. 7a and b). For daytime factors, the Day-ONRich factor was unique to FIGAERO-CIMS. In the AMS, the nitrate functionalities of pON fragmented into NO+ and ions, which were not included in source apportionment analysis, and may explain the difficulty of resolving daytime ON-rich factors for the AMS dataset. Both AMS and FIGAERO-CIMS resolved one daytime aged SOA factor, i.e., the AMS MO-OOA factor and FIGAERO-CIMS Day-MO factor, and these two factors were mildly correlated (R=0.71). For AMS MO-OOA, different theories regarding its sources and formation pathways have been proposed (which are not mutually exclusive), including photochemical aging of fresh OA (Jimenez et al., 2009; Ng et al., 2010; Bougiatioti et al., 2014), aqueous processes (Xu et al., 2017), the formation of highly oxygenated molecules (HOMs) (Ehn et al., 2014), long-range transport (Hayes et al., 2013), and the entrainment of aged SOA from the residual layer (Nagori et al., 2019). In our previous discussion, we tentatively related FIGAERO-CIMS Day-MO, which correlated with AMS MO-OOA, to aqueous processes but cannot rule out other processes. AMS resolved only one daytime fresh SOA factor, isoprene-OA. Isoprene-OA was largely, but not entirely, attributed to IEPOX uptake (Xu et al., 2015a; Schwantes et al., 2015), and the enhanced signal at m∕z 82 (C5H6O+) may arise from methylfuran-like structures (Robinson et al., 2011; Budisulistiorini et al., 2013; Hu et al., 2015). FIGAERO-CIMS resolved two daytime fresh SOA factors, MRN-LO and AFTN-LO. The summation of MRN-LO and AFTN-LO showed good correlation with the AMS isoprene-OA factor (R=0.76). We observed various ions with a high abundance in MRN-LO and AFTN-LO that were likely associated with isoprene organic nitrates, isoprene oxidation via non-IEPOX pathways, and monoterpene oxidation. Previous studies have shown that IEPOX-SOA was enhanced even under high-NO conditions (Jacobs et al., 2014; Schwantes et al., 2019) and that α-pinene SOA could interfere with AMS isoprene-OA apportionment (Xu et al., 2018). All these observations may suggest a more complex origin for the AMS isoprene-OA factor (i.e., not just IEPOX uptake).
3.7 Change in the abundance of biogenic VOC and AMS OA factors in a transitional period
This field campaign took place during the transition in seasons from summer to fall, when decreasing temperature led to changes in the abundances of SOA precursors. Figure 8 shows the mixing ratios of major VOCs (isoprene, α-pinene, and β-pinene) and mass concentrations of AMS OA factors as a function of temperature. The FIGAERO-CIMS factors were not discussed here because fewer data points were measured by FIGAERO-CIMS and were not sufficient to provide statistically reliable results. To eliminate the influence of daily meteorological variations, two sampling periods with relatively stable meteorological conditions were chosen to represent daytime (12:00–16:00 EDT, high temperature and boundary layer height, peak solar radiation) and nighttime (00:00–04:00 EDT, low temperature and boundary layer height, zero solar radiation). The isoprene mixing ratio showed a strong dependence on temperature in both day and night. The mixing ratios of α-pinene and β-pinene were moderately dependent on temperature when the temperature was lower than 25 ∘C and remained relatively constant when the temperature was higher than 25 ∘C, at which most daytime data points resided. For AMS factors, isoprene-OA increased with temperature and followed the trend of isoprene, as expected. Meanwhile, different from isoprene, for the same temperature bin, the nighttime isoprene-OA concentration was always higher than the daytime concentration. This can be explained by the longer lifetime of aerosol compared to gas species, and as a result the high concentration of nighttime isoprene-OA was residue from daytime formation. The strong dependence of isoprene-OA on temperature suggested isoprene as the dominant precursor of this factor, implying that isoprene-OA resolved from AMS measurements is still a good surrogate of isoprene-derived SOA even with the potential interference from monoterpene SOA as discussed above. LO-OOA showed similar trends to monoterpenes, consistent with our discussion above and previous literature in that monoterpenes are the dominant precursors to LO-OOA in this region. For MO-OOA, a mild dependence on temperature was observed, suggesting that at least some of its sources were affected by temperature, e.g., through aging of isoprene-derived SOA (the emission of isoprene is temperature-dependent).
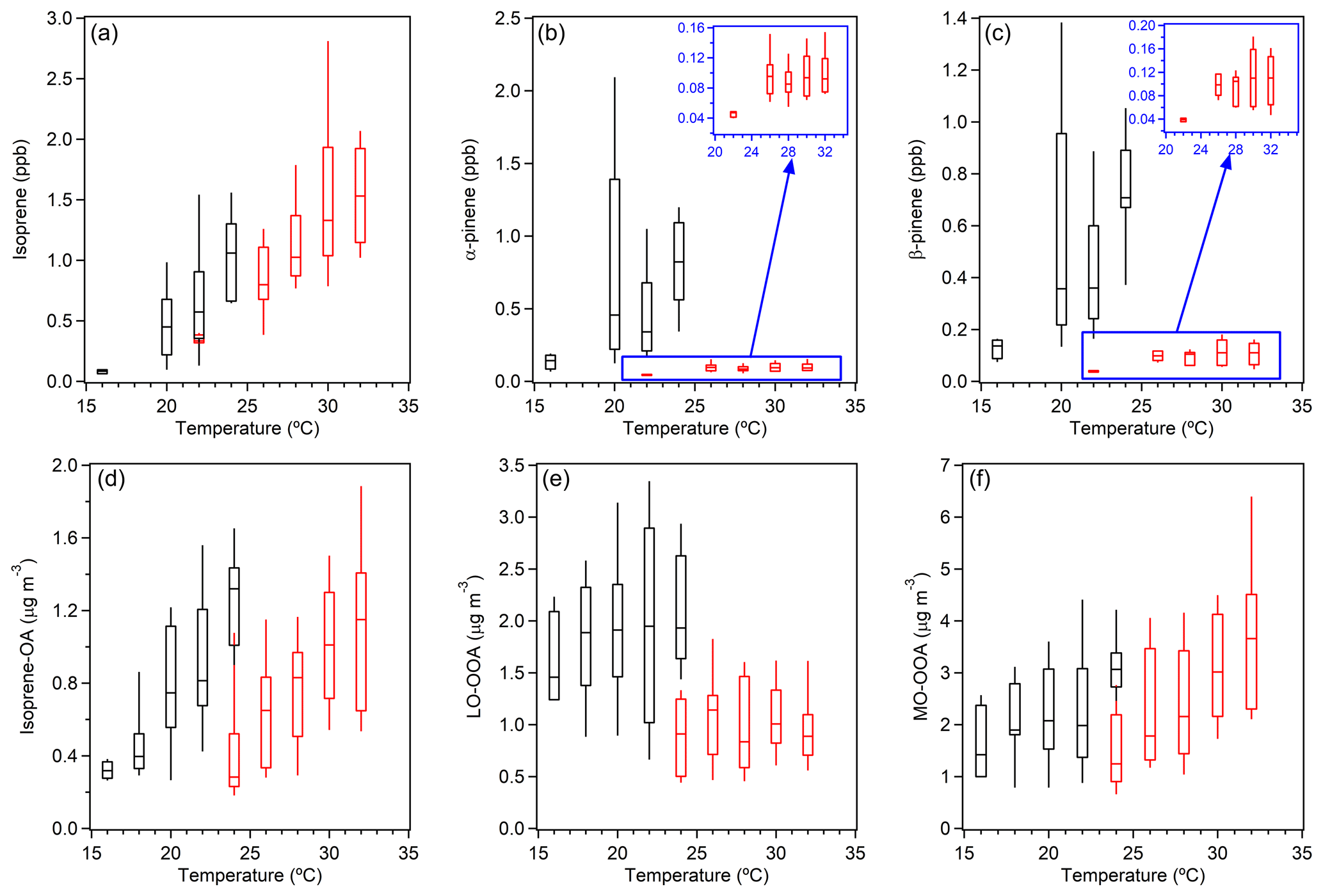
Figure 8Main biogenic VOC mixing ratios (a, b, c) and AMS OA factor mass concentrations (d, e, f) as a function of temperature. The data points are grouped into different temperature bins with a 2 ∘C increment and colored by time of day; afternoon (12:00–16:00 EDT) measurements are in red, and night (00:00–04:00 EDT) measurements are in black. The midpoint line, lower and upper boxes, and lower and upper whiskers represent the median and the 25th, 75th, 10th, and 90th percentiles, respectively.
A total of 2 months of measurements were performed at a rural site in the southeastern US during a transition in seasons. AMS and FIGAERO-CIMS measurements were combined to provide a better understanding of OA sources, composition, and properties. Both instruments consistently identified more oxidized OA in the afternoon and enhanced pON formation during the night, although the OA measured by FIGAERO-CIMS was more oxidized than that by AMS due to the nature of the iodide reagent ion that was used in FIGAERO-CIMS. Similar AMS OA factors were resolved compared to previous summer measurements at the same site, which were isoprene-OA, LO-OOA, and MO-OOA (and no primary OA factors). The fraction of AMS isoprene-OA in total OA decreased from 26 % to 8 % over the campaign, concurrent with a decreasing isoprene mixing ratio, which was strongly dependent on temperature. For FIGAERO-CIMS, three daytime fresh OA factors with low N : C (MRN-LO, AFTN-LO, and Day-MO) each accounted for about one-fourth of the total signals measured by FIGAERO-CIMS, and two factors with high N : C (Day-ONRich and NGT-ONRich) together accounted for the rest. MRN-LO and AFTN-LO were likely fresh biogenic SOA, with MRN-LO more dominated by monoterpene SOA and AFTN-LO more dominated by isoprene SOA. Day-MO was hypothesized to be a mixture of aged and fresh SOA whose formation was possibly aided by aerosol water. NGT-ONRich was mostly from nocturnal monoterpene chemistry, while daytime isoprene oxidation under the effects of NOx was more important to Day-ONRich. Lastly, a series of C14 and C15 compounds was identified by FIGAERO-CIMS, possibly originating from sesquiterpene oxidation pathways. In this study, a uniform sensitivity was assumed for all species measured by FIGAERO-CIMS, resulting in some uncertainties in the overall elemental ratios and carbon numbers. Future studies are warranted to continue to characterize and optimize instrument sensitivity for further quantitative analysis.
Previous studies (Qi et al., 2019; Stefenelli et al., 2019) have shown that combinations of AMS and molecular-based mass spectrometric information is a way forward to provide more insights into the nature of SOA in general. In this study, factor analysis of FIGAERO-CIMS data provided new insights into the sources and composition of the typical AMS OA factors observed in the southeastern US. Specifically, while the AMS isoprene-OA factor has been largely attributed to IEPOX uptake in previous studies, we identified more pathways of isoprene oxidation that contributed to isoprene SOA formation in addition to IEPOX uptake. Notable isoprene pON formation was observed, likely from photooxidation in the presence of NOx and nitrate radical oxidation, as well as notable ISOPOOH-SOA (ISOPOOH oxidation products via non-IEPOX pathways); both pathways have not been resolved by AMS analysis before. The AMS LO-OOA factor correlated well with the NGT-ONRich factor resolved by FIGAERO-CIMS, which contained a series of monoterpene SOA tracers, consolidating the idea that LO-OOA was mostly attributed to monoterpene SOA in the southeastern US. Nonetheless, the non-negligible isoprene-derived pON in the NGT-ONRich factor also related it to nocturnal isoprene chemistry, which was not identified by previous AMS factorization analysis.
Data are available upon request from the corresponding author (ng@chbe.gatech.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-8421-2020-supplement.
YC, RJW, and NLN designed the research. YC, TN, and KB performed the research. YC and MT processed AMS and FIGAERO-CIMS data. LX, MRC, HS, FC, and ASHP provided insights for source apportionment analysis. YC and NLN analyzed the data and wrote the paper.
The authors declare that they have no conflict of interest.
This work has not been formally reviewed by the EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the EPA. The EPA does not endorse any products or commercial services mentioned in this publication.
This work was supported by U.S. Environmental Protection Agency STAR grant R835882. Masayuki Takeuchi and Lu Xu acknowledge support from NSF CAREER AGS-1555034. The FIGAERO HR-ToF-CIMS was purchased through NSF Major Research Instrumentation (MRI) grant 1428738. The authors want to thank Eric S. Edgerton for providing SEARCH network measurements and meteorological data, as well as Qian Zhang, James Rowe, and Linghan Zeng for their help during the campaign.
This research has been supported by the U.S. EPA (STAR grant R835882) and the NSF (grant nos. CAREER AGS-1555034 and Major Research Instrumentation (MRI) 1428738).
This paper was edited by Stefania Gilardoni and reviewed by two anonymous referees.
Bates, K. H. and Jacob, D. J.: A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 19, 9613–9640, https://doi.org/10.5194/acp-19-9613-2019, 2019.
Bougiatioti, A., Stavroulas, I., Kostenidou, E., Zarmpas, P., Theodosi, C., Kouvarakis, G., Canonaco, F., Prévôt, A. S. H., Nenes, A., Pandis, S. N., and Mihalopoulos, N.: Processing of biomass-burning aerosol in the eastern Mediterranean during summertime, Atmos. Chem. Phys., 14, 4793–4807, https://doi.org/10.5194/acp-14-4793-2014, 2014.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Brown, S. S., deGouw, J. A., Warneke, C., Ryerson, T. B., Dubé, W. P., Atlas, E., Weber, R. J., Peltier, R. E., Neuman, J. A., Roberts, J. M., Swanson, A., Flocke, F., McKeen, S. A., Brioude, J., Sommariva, R., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Nocturnal isoprene oxidation over the Northeast United States in summer and its impact on reactive nitrogen partitioning and secondary organic aerosol, Atmos. Chem. Phys., 9, 3027–3042, https://doi.org/10.5194/acp-9-3027-2009, 2009.
Bruns, E. A., Perraud, V., Zelenyuk, A., Ezell, M. J., Johnson, S. N., Yu, Y., Imre, D., Finlayson-Pitts, B. J., and Alexander, M. L.: Comparison of FTIR and Particle Mass Spectrometry for the Measurement of Particulate Organic Nitrates, Environ. Sci. Technol., 44, 1056–1061, https://doi.org/10.1021/es9029864, 2010.
Budisulistiorini, S. H., Canagaratna, M. R., Croteau, P. L., Marth, W. J., Baumann, K., Edgerton, E. S., Shaw, S. L., Knipping, E. M., Worsnop, D. R., Jayne, J. T., Gold, A., and Surratt, J. D.: Real-Time Continuous Characterization of Secondary Organic Aerosol Derived from Isoprene Epoxydiols in Downtown Atlanta, Georgia, Using the Aerodyne Aerosol Chemical Speciation Monitor, Environ. Sci. Technol., 47, 5686–5694, https://doi.org/10.1021/es400023n, 2013.
Budisulistiorini, S. H., Baumann, K., Edgerton, E. S., Bairai, S. T., Mueller, S., Shaw, S. L., Knipping, E. M., Gold, A., and Surratt, J. D.: Seasonal characterization of submicron aerosol chemical composition and organic aerosol sources in the southeastern United States: Atlanta, Georgia,and Look Rock, Tennessee, Atmos. Chem. Phys., 16, 5171–5189, https://doi.org/10.5194/acp-16-5171-2016, 2016.
Canagaratna, M., Jayne, J., Jimenez, J., Allan, J., Alfarra, M., Zhang, Q., Onasch, T., Drewnick, F., Coe, H., and Middlebrook, A.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, 2007.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Canonaco, F., Crippa, M., Slowik, J. G., Baltensperger, U., and Prévôt, A. S. H.: SoFi, an IGOR-based interface for the efficient use of the generalized multilinear engine (ME-2) for the source apportionment: ME-2 application to aerosol mass spectrometer data, Atmos. Meas. Tech., 6, 3649–3661, https://doi.org/10.5194/amt-6-3649-2013, 2013.
Chan, M. N., Zhang, H., Goldstein, A. H., and Wilson, K. R.: Role of Water and Phase in the Heterogeneous Oxidation of Solid and Aqueous Succinic Acid Aerosol by Hydroxyl Radicals, J. Phys. Chem. C, 118, 28978–28992, https://doi.org/10.1021/jp5012022, 2014.
Cheng, C. T., Chan, M. N., and Wilson, K. R.: Importance of Unimolecular HO2 Elimination in the Heterogeneous OH Reaction of Highly Oxygenated Tartaric Acid Aerosol, J. Phys. Chem. A, 120, 5887–5896, https://doi.org/10.1021/acs.jpca.6b05289, 2016.
Claeys, M., Szmigielski, R., Kourtchev, I., Van der Veken, P., Vermeylen, R., Maenhaut, W., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., and Edney, E. O.: Hydroxydicarboxylic Acids: Markers for Secondary Organic Aerosol from the Photooxidation of α-Pinene, Environ. Sci. Technol., 41, 1628–1634, https://doi.org/10.1021/es0620181, 2007.
Crippa, M., Canonaco, F., Lanz, V. A., Äijälä, M., Allan, J. D., Carbone, S., Capes, G., Ceburnis, D., Dall'Osto, M., Day, D. A., DeCarlo, P. F., Ehn, M., Eriksson, A., Freney, E., Hildebrandt Ruiz, L., Hillamo, R., Jimenez, J. L., Junninen, H., Kiendler-Scharr, A., Kortelainen, A.-M., Kulmala, M., Laaksonen, A., Mensah, A. A., Mohr, C., Nemitz, E., O'Dowd, C., Ovadnevaite, J., Pandis, S. N., Petäjä, T., Poulain, L., Saarikoski, S., Sellegri, K., Swietlicki, E., Tiitta, P., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H.: Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach, Atmos. Chem. Phys., 14, 6159–6176, https://doi.org/10.5194/acp-14-6159-2014, 2014.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
D'Ambro, E. L., Møller, K. H., Lopez-Hilfiker, F. D., Schobesberger, S., Liu, J., Shilling, J. E., Lee, B. H., Kjaergaard, H. G., and Thornton, J. A.: Isomerization of Second-Generation Isoprene Peroxy Radicals: Epoxide Formation and Implications for Secondary Organic Aerosol Yields, Environ. Sci. Technol., 51, 4978–4987, https://doi.org/10.1021/acs.est.7b00460, 2017.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., and Docherty, K. S.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, 2006.
Eatough, D. J., Wadsworth, A., Eatough, D. A., Crawford, J. W., Hansen, L. D., and Lewis, E. A.: A multiple-system, multi-channel diffusion denuder sampler for the determination of fine-particulate organic material in the atmosphere, Atmos. Environ. A-Gen., 27, 1213–1219, https://doi.org/10.1016/0960-1686(93)90247-V, 1993.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012a.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 1: Gas-phase composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 6489–6504, https://doi.org/10.5194/acp-12-6489-2012, 2012b.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Elser, M., Huang, R.-J., Wolf, R., Slowik, J. G., Wang, Q., Canonaco, F., Li, G., Bozzetti, C., Daellenbach, K. R., Huang, Y., Zhang, R., Li, Z., Cao, J., Baltensperger, U., El-Haddad, I., and Prévôt, A. S. H.: New insights into PM2.5 chemical composition and sources in two major cities in China during extreme haze events using aerosol mass spectrometry, Atmos. Chem. Phys., 16, 3207–3225, https://doi.org/10.5194/acp-16-3207-2016, 2016.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018.
Fry, J. L., Brown, S. S., Middlebrook, A. M., Edwards, P. M., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Allen, H. M., Ryerson, T. B., Pollack, I., Graus, M., Warneke, C., de Gouw, J. A., Brock, C. A., Gilman, J., Lerner, B. M., Dubé, W. P., Liao, J., and Welti, A.: Secondary organic aerosol (SOA) yields from NO3 radical + isoprene based on nighttime aircraft power plant plume transects, Atmos. Chem. Phys., 18, 11663–11682, https://doi.org/10.5194/acp-18-11663-2018, 2018.
Hayes, P. L., Ortega, A. M., Cubison, M. J., Froyd, K. D., Zhao, Y., Cliff, S. S., Hu, W. W., Toohey, D. W., Flynn, J. H., Lefer, B. L., Grossberg, N., Alvarez, S., Rappenglueck, B., Taylor, J. W., Allan, J. D., Holloway, J. S., Gilman, J. B., Kuster, W. C., De Gouw, J. A., Massoli, P., Zhang, X., Liu, J., Weber, R. J., Corrigan, A. L., Russell, L. M., Isaacman, G., Worton, D. R., Kreisberg, N. M., Goldstein, A. H., Thalman, R., Waxman, E. M., Volkamer, R., Lin, Y. H., Surratt, J. D., Kleindienst, T. E., Offenberg, J. H., Dusanter, S., Griffith, S., Stevens, P. S., Brioude, J., Angevine, W. M., and Jimenez, J. L.: Organic aerosol composition and sources in Pasadena, California, during the 2010 CalNex campaign, J. Geophys. Res.-Atmos., 118, 9233–9257, https://doi.org/10.1002/jgrd.50530, 2013.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Huang, W., Saathoff, H., Shen, X. L., Ramisetty, R., Leisner, T., and Mohr, C.: Chemical Characterization of Highly Functionalized Organonitrates Contributing to Night-Time Organic Aerosol Mass Loadings and Particle Growth, Environ. Sci. Technol., 53, 1165–1174, https://doi.org/10.1021/acs.est.8b05826, 2019.
Huey, L. G.: Measurement of trace atmospheric species by chemical ionization mass spectrometry: Speciation of reactive nitrogen and future directions, Mass Spectrom. Rev., 26, 166–184, https://doi.org/10.1002/mas.20118, 2007.
Huey, L. G., Hanson, D. R., and Howard, C. J.: Reactions of SF6- and I- with Atmospheric Trace Gases, J. Phys. Chem., 99, 5001–5008, https://doi.org/10.1021/j100014a021, 1995.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jaoui, M., Szmigielski, R., Nestorowicz, K., Kolodziejczyk, A., Sarang, K., Rudzinski, K. J., Konopka, A., Bulska, E., Lewandowski, M., and Kleindienst, T. E.: Organic Hydroxy Acids as Highly Oxygenated Molecular (HOM) Tracers for Aged Isoprene Aerosol, Environ. Sci. Technol., 53, 14516–14527, https://doi.org/10.1021/acs.est.9b05075, 2019.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kahnt, A., Iinuma, Y., Blockhuys, F., Mutzel, A., Vermeylen, R., Kleindienst, T. E., Jaoui, M., Offenberg, J. H., Lewandowski, M., Böge, O., Herrmann, H., Maenhaut, W., and Claeys, M.: 2-Hydroxyterpenylic Acid: An Oxygenated Marker Compound for α-Pinene Secondary Organic Aerosol in Ambient Fine Aerosol, Environ. Sci. Technol., 48, 4901–4908, https://doi.org/10.1021/es500377d, 2014a.
Kahnt, A., Iinuma, Y., Mutzel, A., Böge, O., Claeys, M., and Herrmann, H.: Campholenic aldehyde ozonolysis: a mechanism leading to specific biogenic secondary organic aerosol constituents, Atmos. Chem. Phys., 14, 719–736, https://doi.org/10.5194/acp-14-719-2014, 2014b.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kawamura, K., Seméré, R., Imai, Y., Fujii, Y., and Hayashi, M.: Water soluble dicarboxylic acids and related compounds in Antarctic aerosols, J. Geophys. Res.-Atmos., 101, 18721–18728, https://doi.org/10.1029/96jd01541, 1996.
Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Lewis, C. W., Bhave, P. V., and Edney, E. O.: Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location, Atmos. Environ., 41, 8288–8300, https://doi.org/10.1016/j.atmosenv.2007.06.045, 2007.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., Clair, J. M. S., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, 2008.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, 2011.
Lanz, V. A., Alfarra, M. R., Baltensperger, U., Buchmann, B., Hueglin, C., and Prévôt, A. S. H.: Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra, Atmos. Chem. Phys., 7, 1503–1522, https://doi.org/10.5194/acp-7-1503-2007, 2007.
Lanz, V. A., Alfarra, M. R., Baltensperger, U., Buchmann, B., Hueglin, C., Szidat, S., Wehrli, M. N., Wacker, L., Weimer, S., Caseiro, A., Puxbaum, H., and Prevot, A. S. H.: Source Attribution of Submicron Organic Aerosols during Wintertime Inversions by Advanced Factor Analysis of Aerosol Mass Spectra, Environ. Sci. Technol., 42, 214–220, https://doi.org/10.1021/es0707207, 2008.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Lee, B. H., Lopez-Hilfiker, F. D., D'Ambro, E. L., Zhou, P., Boy, M., Petäjä, T., Hao, L., Virtanen, A., and Thornton, J. A.: Semi-volatile and highly oxygenated gaseous and particulate organic compounds observed above a boreal forest canopy, Atmos. Chem. Phys., 18, 11547–11562, https://doi.org/10.5194/acp-18-11547-2018, 2018.
Lim, H.-J. and Turpin, B. J.: Origins of Primary and Secondary Organic Aerosol in Atlanta: Results of Time-Resolved Measurements during the Atlanta Supersite Experiment, Environ. Sci. Technol., 36, 4489–4496, https://doi.org/10.1021/es0206487, 2002.
Lim, Y. B., Tan, Y., Perri, M. J., Seitzinger, S. P., and Turpin, B. J.: Aqueous chemistry and its role in secondary organic aerosol (SOA) formation, Atmos. Chem. Phys., 10, 10521–10539, https://doi.org/10.5194/acp-10-10521-2010, 2010.
Lin, Y.-H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene Epoxydiols as Precursors to Secondary Organic Aerosol Formation: Acid-Catalyzed Reactive Uptake Studies with Authentic Compounds, Environ. Sci. Technol., 46, 250–258, https://doi.org/10.1021/es202554c, 2012.
Liu, J., D'Ambro, E. L., Lee, B. H., Lopez-Hilfiker, F. D., Zaveri, R. A., Rivera-Rios, J. C., Keutsch, F. N., Iyer, S., Kurten, T., Zhang, Z., Gold, A., Surratt, J. D., Shilling, J. E., and Thornton, J. A.: Efficient Isoprene Secondary Organic Aerosol Formation from a Non-IEPOX Pathway, Environ. Sci. Technol., 50, 9872–9880, https://doi.org/10.1021/acs.est.6b01872, 2016.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
Massoli, P., Stark, H., Canagaratna, M. R., Krechmer, J. E., Xu, L., Ng, N. L., Mauldin, R. L., Yan, C., Kimmel, J., Misztal, P. K., Jimenez, J. L., Jayne, J. T., and Worsnop, D. R.: Ambient Measurements of Highly Oxidized Gas-Phase Molecules during the Southern Oxidant and Aerosol Study (SOAS) 2013, ACS Earth Space Chem., 2, 653–672, https://doi.org/10.1021/acsearthspacechem.8b00028, 2018.
Matthew, B. M., Middlebrook, A. M., and Onasch, T. B.: Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols, Aerosol Sci. Tech., 42, 884–898, https://doi.org/10.1080/02786820802356797, 2008.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Müller, L., Reinnig, M.-C., Naumann, K. H., Saathoff, H., Mentel, T. F., Donahue, N. M., and Hoffmann, T.: Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging, Atmos. Chem. Phys., 12, 1483–1496, https://doi.org/10.5194/acp-12-1483-2012, 2012.
Nagori, J., Janssen, R. H. H., Fry, J. L., Krol, M., Jimenez, J. L., Hu, W., and Vilà-Guerau de Arellano, J.: Biogenic emissions and land–atmosphere interactions as drivers of the daytime evolution of secondary organic aerosol in the southeastern US, Atmos. Chem. Phys., 19, 701–729, https://doi.org/10.5194/acp-19-701-2019, 2019.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Nah, T., Guo, H., Sullivan, A. P., Chen, Y., Tanner, D. J., Nenes, A., Russell, A., Ng, N. L., Huey, L. G., and Weber, R. J.: Characterization of aerosol composition, aerosol acidity, and organic acid partitioning at an agriculturally intensive rural southeastern US site, Atmos. Chem. Phys., 18, 11471–11491, https://doi.org/10.5194/acp-18-11471-2018, 2018a.
Nah, T., Ji, Y., Tanner, D. J., Guo, H., Sullivan, A. P., Ng, N. L., Weber, R. J., and Huey, L. G.: Real-time measurements of gas-phase organic acids using SF chemical ionization mass spectrometry, Atmos. Meas. Tech., 11, 5087–5104, https://doi.org/10.5194/amt-11-5087-2018, 2018.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Bateman, A. P., Bones, D. L., Nizkorodov, S. A., Laskin, J., and Laskin, A.: High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene, Atmos. Environ., 44, 1032–1042, https://doi.org/10.1016/j.atmosenv.2009.12.019, 2010.
Nguyen, T. B., Roach, P. J., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 11, 6931–6944, https://doi.org/10.5194/acp-11-6931-2011, 2011.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009.
Praplan, A. P., Hegyi-Gaeggeler, K., Barmet, P., Pfaffenberger, L., Dommen, J., and Baltensperger, U.: Online measurements of water-soluble organic acids in the gas and aerosol phase from the photooxidation of 1,3,5-trimethylbenzene, Atmos. Chem. Phys., 14, 8665–8677, https://doi.org/10.5194/acp-14-8665-2014, 2014.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 49, 14195–14203, https://doi.org/10.1021/acs.est.5b03738, 2015.
Qi, L., Chen, M., Stefenelli, G., Pospisilova, V., Tong, Y., Bertrand, A., Hueglin, C., Ge, X., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 2: Biomass burning influences in winter, Atmos. Chem. Phys., 19, 8037–8062, https://doi.org/10.5194/acp-19-8037-2019, 2019.
Rattanavaraha, W., Chu, K., Budisulistiorini, S. H., Riva, M., Lin, Y.-H., Edgerton, E. S., Baumann, K., Shaw, S. L., Guo, H., King, L., Weber, R. J., Neff, M. E., Stone, E. A., Offenberg, J. H., Zhang, Z., Gold, A., and Surratt, J. D.: Assessing the impact of anthropogenic pollution on isoprene-derived secondary organic aerosol formation in PM2.5 collected from the Birmingham, Alabama, ground site during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 16, 4897–4914, https://doi.org/10.5194/acp-16-4897-2016, 2016.
Robinson, N. H., Hamilton, J. F., Allan, J. D., Langford, B., Oram, D. E., Chen, Q., Docherty, K., Farmer, D. K., Jimenez, J. L., Ward, M. W., Hewitt, C. N., Barley, M. H., Jenkin, M. E., Rickard, A. R., Martin, S. T., McFiggans, G., and Coe, H.: Evidence for a significant proportion of Secondary Organic Aerosol from isoprene above a maritime tropical forest, Atmos. Chem. Phys., 11, 1039–1050, https://doi.org/10.5194/acp-11-1039-2011, 2011.
Sato, K., Jia, T., Tanabe, K., Morino, Y., Kajii, Y., and Imamura, T.: Terpenylic acid and nine-carbon multifunctional compounds formed during the aging of β-pinene ozonolysis secondary organic aerosol, Atmos. Environ., 130, 127–135, https://doi.org/10.1016/j.atmosenv.2015.08.047, 2016.
Schobesberger, S., D'Ambro, E. L., Lopez-Hilfiker, F. D., Mohr, C., and Thornton, J. A.: A model framework to retrieve thermodynamic and kinetic properties of organic aerosol from composition-resolved thermal desorption measurements, Atmos. Chem. Phys., 18, 14757–14785, https://doi.org/10.5194/acp-18-14757-2018, 2018.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St. Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2+HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, 10.1021/acs.jpca.5b06355, 2015.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics : from air pollution to climate change, 3rd Edn., John Wiley & Sons, Inc., Hoboken, New Jersey, 2016.
Setyan, A., Zhang, Q., Merkel, M., Knighton, W. B., Sun, Y., Song, C., Shilling, J. E., Onasch, T. B., Herndon, S. C., Worsnop, D. R., Fast, J. D., Zaveri, R. A., Berg, L. K., Wiedensohler, A., Flowers, B. A., Dubey, M. K., and Subramanian, R.: Characterization of submicron particles influenced by mixed biogenic and anthropogenic emissions using high-resolution aerosol mass spectrometry: results from CARES, Atmos. Chem. Phys., 12, 8131–8156, https://doi.org/10.5194/acp-12-8131-2012, 2012.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W. W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Stefenelli, G., Pospisilova, V., Lopez-Hilfiker, F. D., Daellenbach, K. R., Hüglin, C., Tong, Y., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 1: Biogenic influences and day–night chemistry in summer , Atmos. Chem. Phys., 19, 14825–14848, https://doi.org/10.5194/acp-19-14825-2019, 2019.
Surratt, J. D., Chan, A. W., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, 2010.
Szmigielski, R., Surratt, J. D., Gómez-González, Y., Van der Veken, P., Kourtchev, I., Vermeylen, R., Blockhuys, F., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Seinfeld, J. H., Maenhaut, W., and Claeys, M.: 3-methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Lett., 34, L24811, https://doi.org/10.1029/2007gl031338, 2007.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Weber, R. J., Sullivan, A. P., Peltier, R. E., Russell, A., Yan, B., Zheng, M., de Gouw, J., Warneke, C., Brock, C., Holloway, J. S., Atlas, E. L., and Edgerton, E.: A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States, J. Geophys. Res.-Atmos., 112, D13302, https://doi.org/10.1029/2007JD008408, 2007.
Xia, S.-S., Eugene, A. J., and Guzman, M. I.: Cross Photoreaction of Glyoxylic and Pyruvic Acids in Model Aqueous Aerosol, J. Phys. Chem. A, 122, 6457–6466, https://doi.org/10.1021/acs.jpca.8b05724, 2018.
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the Volatility of Secondary Organic Aerosol from Isoprene Photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S. H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015a.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015b.
Xu, L., Pye, H. O. T., He, J., Chen, Y., Murphy, B. N., and Ng, N. L.: Experimental and model estimates of the contributions from biogenic monoterpenes and sesquiterpenes to secondary organic aerosol in the southeastern United States, Atmos. Chem. Phys., 18, 12613–12637, https://doi.org/10.5194/acp-18-12613-2018, 2018.
Xu, W., Han, T., Du, W., Wang, Q., Chen, C., Zhao, J., Zhang, Y., Li, J., Fu, P., Wang, Z., Worsnop, D. R., and Sun, Y.: Effects of Aqueous-Phase and Photochemical Processing on Secondary Organic Aerosol Formation and Evolution in Beijing, China, Environ. Sci. Technol., 51, 762–770, https://doi.org/10.1021/acs.est.6b04498, 2017.
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhang, Q., Worsnop, D. R., Canagaratna, M. R., and Jimenez, J. L.: Hydrocarbon-like and oxygenated organic aerosols in Pittsburgh: insights into sources and processes of organic aerosols, Atmos. Chem. Phys., 5, 3289–3311, https://doi.org/10.5194/acp-5-3289-2005, 2005.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, 2011.
Zhang, Y. Y., Müller, L., Winterhalter, R., Moortgat, G. K., Hoffmann, T., and Pöschl, U.: Seasonal cycle and temperature dependence of pinene oxidation products, dicarboxylic acids and nitrophenols in fine and coarse air particulate matter, Atmos. Chem. Phys., 10, 7859–7873, https://doi.org/10.5194/acp-10-7859-2010, 2010.
Zhao, R., Mungall, E. L., Lee, A. K. Y., Aljawhary, D., and Abbatt, J. P. D.: Aqueous-phase photooxidation of levoglucosan – a mechanistic study using aerosol time-of-flight chemical ionization mass spectrometry (Aerosol ToF-CIMS), Atmos. Chem. Phys., 14, 9695–9706, https://doi.org/10.5194/acp-14-9695-2014, 2014.