the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Jul 2020
| 16 Jul 2020
Polar semivolatile organic compounds in biomass-burning emissions and their chemical transformations during aging in an oxidation flow reactor
Deep Sengupta
Chiranjivi Bhattarai
Adam C. Watts
Hans Moosmüller
Andrey Y. Khlystov
Semivolatile organic compounds (SVOCs) emitted from open biomass burning (BB) can contribute to chemical and physical properties of atmospheric aerosols and also may cause adverse health effects. The polar fraction of SVOCs is a prominent part of BB organic aerosols, and thus it is important to characterize the chemical composition and reactivity of this fraction. In this study, globally and regionally important representative fuels (Alaskan peat, Moscow peat, Pskov peat, eucalyptus, Malaysian peat, and Malaysian agricultural peat) were burned under controlled conditions using the combustion chamber facility at the Desert Research Institute (DRI). Gas- and particle-phase biomass-burning emissions were aged in an oxidation flow reactor (OFR) to mimic 5–7 d of atmospheric aging. Fresh and OFR-aged biomass-burning aerosols were collected on Teflon-impregnated glass fiber filters (TIGF) in tandem with XAD resin media for organic carbon speciation. The polar fraction extracted with dichloromethane and acetone was analyzed with gas chromatography mass spectrometry (GC-MS) for 84 polar organic compounds – including mono- and dicarboxylic acids, methoxylated phenols, aromatic acids, anhydrosugars, resin acids, and sterols. For all these compounds, fuel-based emission factors (EFs) were calculated for fresh and OFR-aged samples. The carbon mass of the quantified polar compounds was found to constitute 5 % to 7 % of the total organic compound mass. A high abundance of methoxyphenols (239 mg kg−1 for Pskov peat; 22.6 % of total GC-MS characterized mass) and resin acids (118 mg kg−1 for Alaskan peat; 14.5 % of total GC-MS characterized mass) was found in peat-burning emissions (smoldering combustion). The concentration of some organic compounds (e.g., tetracosanoic acid) with a molecular weight (MW) above 350 g mol−1 decreased after OFR aging, while abundances of low-MW compounds (e.g., hexanoic acid) increased. This indicated a significant extent of fragmentation reactions in the OFR. Methoxyphenols decreased after OFR aging, while a significant increase (3.7 to 8.6 times) in the abundance of dicarboxylic acids emission factors (EFs), especially maleic acid (10 to 60 times), was observed. EFs for fresh and ratios from fresh-to-aged BB samples reported in this study can be used to perform source apportionment and predict processes occurring during atmospheric transport.
- Article
(9469 KB) - Full-text XML
-
Supplement
(1174 KB) - BibTeX
- EndNote





Biomass burning (BB), including both wildfires and prescribed burns, is a major source of carbonaceous aerosols in the atmosphere (Penner et al., 1991) and can contribute up to 75 % of total atmospheric aerosol mass loading (Andreae and Merlet, 2001; Park et al., 2007). These carbonaceous aerosols have a significant impact on both regional and global radiative forcing (Ramanathan and Carmichael, 2008). BB emissions also can cause adverse health effects (Arbex et al., 2007; Regalado et al., 2006) because of the toxicological properties of particle-bound organic compounds (Chen et al., 2017; Pardo et al., 2020; Pavagadhi et al., 2013; Sigsgaard et al., 2015; Yang et al., 2010). Therefore, the comprehensive, molecular-level characterization of BB emissions is essential for understanding health effects. Such a molecular characterization of BB carbonaceous aerosols in the atmosphere, however, is challenging, as these aerosols are composed of tens of thousands of compounds (Goldstein and Galbally, 2007).
The simulation of natural fires in a laboratory environment using a BB chamber is one way to characterize the chemical composition of BB emissions (Yokelson et al., 2003). A number of studies characterizing the molecular composition of combustion emissions from fuels that represent different geographical regions have been completed: temperate conifers (Oros and Simoneit, 2001a), deciduous trees (Oros and Simoneit, 2001b), grasses (Oros et al., 2006), and peats (Samburova et al., 2016; Iinuma et al., 2007). Akagi et al. (2011) compiled fuel-based emission factors (EFs) from different fuels from throughout the world, including the peatlands of southern Asia, and found that burning condition (flaming and smoldering) can influence the EFs of individual compounds. These data have been used for modeling work in predicting ozone-forming potential and other air quality impacts (Alvarado et al., 2015). Very few studies (e.g., Samburova et al., 2016) have focused on peat emissions. However, the importance of investigating the combustion products from burning peat soils is multifaceted. Peat soils, comprised predominantly of partially decomposed organic material, represent one fourth to one third of global terrestrial carbon and are under threat of increased fire activity in both boreal and tropical latitudes, areas of widespread peatland occurrence (Turetsky et al., 2015). In addition to the implications of peat fires for the global C cycle, local impacts from the burning of peatlands include problems of public health and safety from degraded air quality and the reduction of surface albedo upon peat smoke deposition on snow surfaces (Beres et al., 2020), as well as ecological changes due to altered surface hydrology in low-relief areas (Watts et al., 2015). Most source apportionment studies, however, focused on the characterization of fresh emissions and emissions of either particle-phase or gas-phase compounds.
Significant changes in organic aerosol composition during atmospheric transport have been reported (Liu et al., 2017; Decker et al., 2019). These changes can impact local and regional air quality. Also, the role of Siberian peat burning in haze formation in the Korean Peninsula (Jung et al., 2016) demonstrates the global impact of BB emissions and their atmospheric transport on regional air quality. Some laboratory studies found an increase in the mass of organic aerosol (OA) after photochemical aging (Ortega et al., 2013; Grieshop et al., 2009), while others observed a modest decrease (Bhattarai et al., 2018). There are still limited data on the evolution of the chemical composition of primary organic aerosols (POAs) during atmospheric aging. Some laboratory experiments demonstrated a degradation of levoglucosan (Hennigan et al., 2010; Kessler et al., 2010) and an oxidation of methoxyphenols in the gas phase (Yee et al., 2013) and aqueous phase (Net et al., 2011). These studies have more mechanistic implications than quantifying gross change after atmospheric oxidation. The reactivity of a pool of organic compounds in a complex mixture such as BB emissions is expected to be different mechanistically from individual compounds. This necessitates the need for studies of the evolution of organic compounds through the bulk molecular-level characterization of BB emissions. Recently, Fortenberry et al. (2018) characterized the chemical fingerprints of aged biomass-burning aerosols (leaf and hardwood of white oak) by performing oxidation in a potential-aerosol-mass oxidation flow reactor (PAM-OFR) and chemical analysis with a thermal desorption aerosol gas chromatograph aerosol mass spectrometer (TAG-AMS). In this study, denuders were used to remove gases, and particles were introduced to the OFR to evaluate only the changes in particulate BB emissions during OFR oxidation. However, the presence of both gas- and particle-phase emissions in real BB emissions and the partitioning of organic compounds in such a complex mixture can affect the reactivity inside the OFR and hence the fate of organic compounds during OFR aging. Bertrand et al. (2018) analyzed 71 organic compounds in BB emissions, sampled from a smog chamber, with high-resolution time of flight mass spectrometry (HR-ToF-AMS). This study confirms that nitroaromatic compounds are formed after OFR oxidation and that they can be used as a tracer of secondary organic aerosol (SOA). However, this study was focused on controlled wood (pellet) burning that is substantially different from wildland BB emissions. There is still a lack of understanding, however, regarding (1) major organic compounds emitted from BB (especially from peat fuels), (2) their roles in atmospheric photochemical reactions, and (3) what compounds are responsible for the light absorption of fresh and aged BB emissions.
In this study, emissions from the laboratory combustion of six globally important fuels (Alaskan peat, Moscow peat, Pskov peat, eucalyptus, Malaysian peat, and Malaysian agricultural peat; Watts et al., 2020) were quantitatively analyzed for more than 250 individual organic species, and the analysis of 84 polar organic species is presented in this paper. BB emissions generated in a combustion chamber were run though the OFR, mimicking approximately 5 to 7 d of atmospheric oxidation (Bhattarai et al., 2018), and the OFR output was analyzed to characterize aged BB emissions. BB emissions were collected on filter and XAD media to identify the distribution of organic species between the gas and particle phases. For the polar fraction of collected organic compounds, we quantitatively analyzed a total of 84 compounds (methoxyphenol derivatives, dicarboxylic acids, monocarboxylic acids, aromatic acids, resin acids, and anhydrosugars). In the analyzed anhydrosugars, we paid special attention to levoglucosan, a derivative of cellulose (Simoneit et al., 1999), since levoglucosan has been widely used as a molecular tracer of BB emissions (Bonvalot et al., 2016; Maenhaut et al., 2016). Methoxyphenols also have been used in source apportionment studies (Schauer et al., 2001; Schmidl et al., 2008, b). These source apportionment studies, however, have not combined such a wide range of different groups in a single investigation. Here we provide a detailed targeted chemical analysis of both gas- and particle-phase BB emissions from the combustion of individual biomass fuels from diverse geographical locations for both fresh and aged emissions. The goal of this research was to characterize the EFs of gas- and particle-phase individual polar organic species for six compound groups (methoxyphenols, dicarboxylic acids, monocarboxylic acids, aromatic acids, anhydrosugars, and resin acids) and to analyze their fate after OFR oxidation. In separate sections we discussed EFs obtained for fresh and OFR-aged BB samples. The fresh-to-aged ratio and top contributing organic species also are discussed. The comparison between fresh and OFR-aged BB emissions helps one to understand the chemical evolution of BB plumes in the atmosphere, and the obtained data can be used in future source apportionment and atmospheric-modeling studies.
2.1 Fuel description
We selected six globally and regionally important BB fuels: Alaskan peat, Moscow peat, Pskov peat, eucalyptus, Malaysian peat, and Malaysian agricultural peat (Watts et al., 2020). Five of these were peat fuels selected from different geographical locations, with four representing smoldering combustion and one (eucalyptus) representing flaming combustion.
Peatland ecosystems, generally wetland or mesic ecosystems underlain by soils composed primarily of partially decomposed biomass, contain mostly organic carbon and more than 20 % of mineral content, represent a vast terrestrial carbon pool, and are potentially vast sources of carbon flux to the atmosphere during wildfires that consume peat (Harden et al., 2000). Peatlands in high-latitude temperate and boreal regions are particularly vulnerable to increased fire-related carbon emissions resulting from climatic warming and increases in the fire season length, while peatlands in low-latitude and tropical regions are threatened by factors such as deforestation for agriculture, urbanization, and drainage (Turetsky et al., 2015). We collected Alaskan peat samples from the upper 10 cm of soils within black spruce (Picea mariana) near crown forests (Chakrabarty et al., 2016). High-latitude and Eurasian samples here are from communities dominated by Sphagnum and cotton grass (Eriophorum spp.), collected from the Moscow (Odintsovo and Shatura districts) and Pskov regions of Russia. These regions are representative of oligotrophic peat bogs found widely across Siberia as well. Tropical peat in this study includes samples from two areas in Malaysian Borneo. One set of samples is from a Dipterocarp-dominated lowland forest with largely intact native land cover, while the second set is from a cleared agricultural area in the Kota Samarahan region. All peat soils were extracted from the top 15 cm of the soil profile. The mass of the dry peat fuels varied from 70 to 100 g. The peat fuels were burned inside a metal pot which contains thermally insulating materials on its bottom and sides.
We selected eucalyptus as a test fuel because of its prevalence across Australia and its important contribution to Australian wildland fires. In addition, economic losses and risk to life and property from fires in eucalypt forests are magnified by their proximity to both fire-prone ecosystems and large urban areas; often eucalypt-dominated stands form boundaries between these two land-use types. There are nearly 900 species of the genera Eucalyptus, Corymbia, and Angophora, which collectively comprise woody plants known as eucalypts. Native to Australia, eucalypt-dominated forests cover nearly 92×106 ha (Hills and Brown, 1978). In addition, the fast and hardy growth characteristics of eucalypts have made them popular in warm ecoregions of Europe as well as North America and South America, where they readily escape cultivation and become established, dominant community types near urban areas where they were originally introduced. Because of their high oil content, rapid and dense growth, and vegetative structure, eucalypts are highly flammable and contribute to high fire risk in areas where they occur (Goodrick and Stanturf, 2012). The mass of the eucalyptus fuel burned (i.e., ∼650 g) was higher than that of peat fuels, and this fuel was arranged similar to a common arrangement for campfires.
2.2 Reagents and materials
We obtained high-performance liquid chromatography (HPLC) grade methanol and hexane from Fisher Scientific (Fair Lawn, NJ, USA) and used the following filters for sampling and further chemical analyses: pre-fired (900∘ C for 4 h) 47 mm diameter quartz-fiber filter (2500 Pallflex QAT-UP, Pall Life Sciences, Ann Arbor, MI, USA) for thermo-optical organic-carbon–elemental-carbon (OC∕EC) analysis, Teflon filters (2500 Pallflex QAT-UP, Pall Life Science, Ann Arbor, MI, USA) for gravimetric particulate-matter (PM) mass analysis, and Teflon-impregnated glass fiber (TIGF) 47 mm diameter filters (Fiber FilmT60A20, Pall Life Sciences, Ann Arbor, MI, USA) for organic analysis. We purchased the following deuterated internal standards from Cambridge Isotope Laboratories (Tewksbury, MA, USA) and CDN Isotopes (Pointe-Claire, Quebec, Canada): hexanoic-d11 acid, succinic-d4 acid, decanoic-d19 acid, adipic-d10 acid, suberic-d12 acid, homovanillic-2,2-d2 acid, myristic-d27 acid, heptadecanoicd33 acid, oleic-9,10-d2 acid, and tetradecanedioic-d24 acid (CDN Isotopes, Quebec, Canada) and benzoic-d5 acid, levoglucosan-d7, and cholesterol-2,2,3,4,4,6-d6 (Cambridge Isotope Laboratories, Inc., MA, USA)
2.3 Biomass-burning (BB) experiments
BB experiments were conducted using the Desert Research Institute (DRI) BB facility for the combustion of the selected fuels under controlled conditions. A close replicate of this facility was described previously (Tian et al., 2015), and a detailed description of the experimental setup was presented elsewhere (Bhattarai et al., 2018; Sengupta et al., 2018).
We mixed laboratory-generated BB emissions with humidified zero air (Airgas Inc., Sparks, NV, USA) using 4 m long spiral copper tubing (12.7 mm outer diameter – OD). Before it was mixed with the BB emissions, the zero air was humidified by bubbling through Nanopure water in a glass 500 mL volume impinger. The flow rate was controlled with a mass flow controller (810C-CE-RFQ-1821, Sierra Instruments, Monterey, CA, USA). An oxidation flow reactor (OFR; Aerodyne Research Inc., Billerica, MA, USA) was used to mimic approximately 7 d of equivalent atmospheric aging (Bhattarai et al., 2018). The OFR consisted of an alodine-coated aluminum cylinder (46 cm length and 22 cm diameter) with an internal volume of 13.3 L. Two sets of lamps emitted UV radiation at wavelengths of 185 and 254 nm (Atlantic Ultraviolet Corporation, Hauppauge, NY, USA) in the OFR to produce ozone and OH radicals (Li et al., 2015). UV irradiance in the OFR was quantified using a photodiode detector with a wavelength range of 225 to 287 nm (TOCON_C6, sglux GmbH, Berlin, Germany). Ultra-high-purity nitrogen (Airgas Inc., Reno, NV, USA) was used to purge the UV lamp compartments to prevent the lamps from overheating. A probe that monitored relative humidity and temperature inside the OFR (from Aerodyne Inc., MA, USA) was mounted toward the outlet side of the OFR. A detailed characterization of the OFR – such as particle loss, OH production rate, and time scales of various processes – can be found in Bhattarai et al. (2018).
The duration of smoldering-combustion experiments ranged from 69 to 255 min, whereas the average duration of flaming-combustion experiments was 50 min. During all experiments, both fresh (directly from the chamber) and aged (oxidized in the OFR) emissions were continuously collected on a TIGF filter (for the particle phase) followed by an XAD cartridge (for the gas phase) for detailed chemical speciation. We used several online instruments to characterize gas- and particle-phase pollutants (see Fig. 1). Simultaneous collection of samples for thermo-optical carbon analysis on quartz-fiber filters (Pall Gelman, 47 mm diameter, pre-heated) was conducted but only for eucalyptus and Malaysian peat. The online instruments alternated every 10 min between sampling fresh and aged emissions using a computer-controlled valve system. A description of all instruments and the detailed experimental setup can be found elsewhere (Bhattarai et al., 2018).
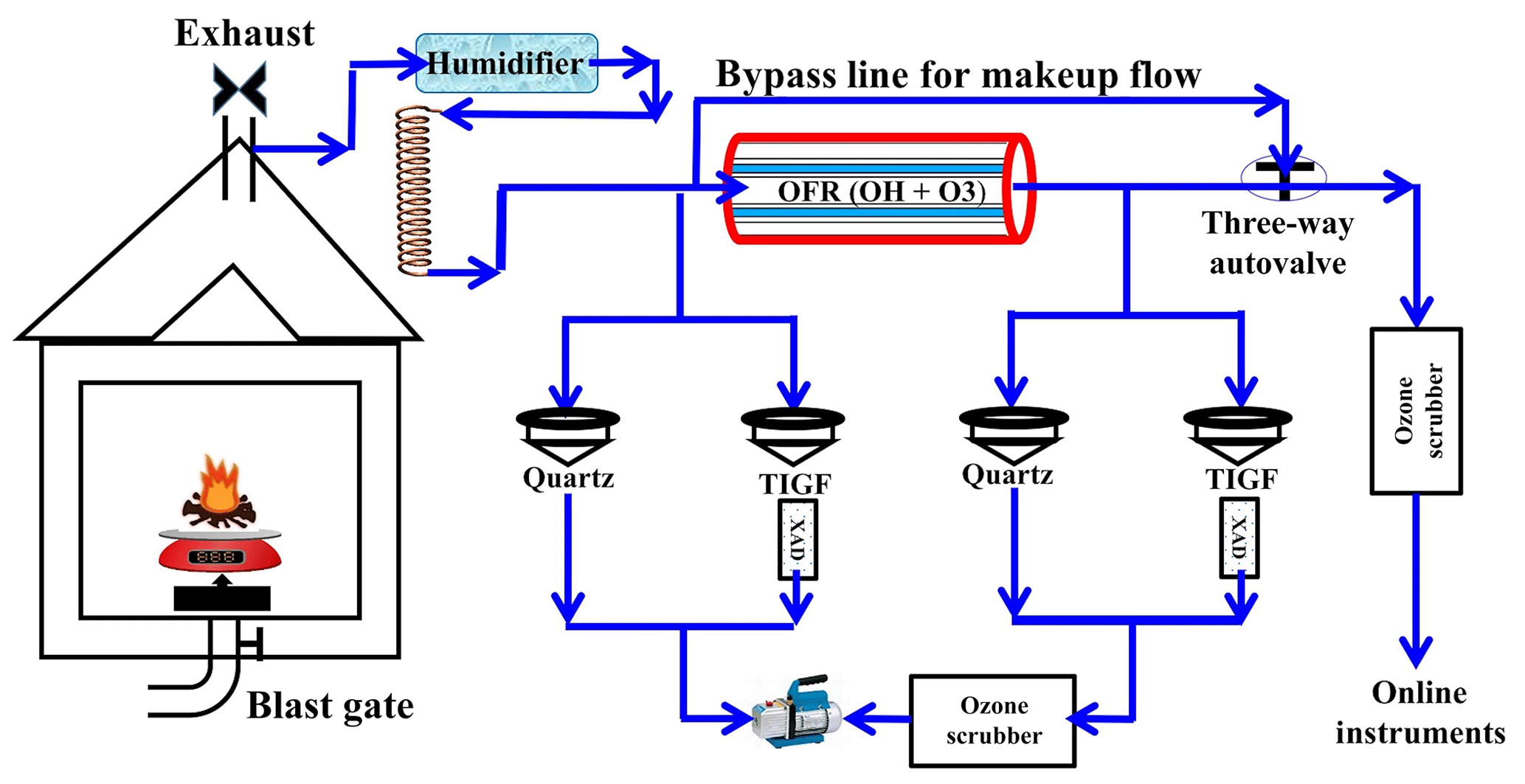
Figure 1Desert Research Institute (DRI) biomass-burning (BB) facility with oxidative flow reactor (OFR) and flow setup.
We employed a bypass flow to keep the flow from the BB chamber and through the OFR constant when online instruments switched between sampling fresh and aged emissions. To protect online instruments from high ozone concentrations produced in the OFR, ozone scrubbers were installed in front of the instruments' inlets. The ozone scrubbers were loaded with charcoal followed by a CARULITE 200 catalyst (Carus Corp., Peru, IL, USA). There were no ozone scrubbers before the filter–XAD setup, which could cause the further oxidation of organic compounds on filter surfaces during sampling. The reaction rates between organics and ozone, however, are orders of magnitude lower than OH oxidation reactions (Finlayson-Pitts and Pitts, 1999). Therefore, we assumed that reactions with OH radicals were primarily responsible for changes in organic compounds associated with fresh-gas and particulate emissions. We also acknowledge that the ozone oxidation on the filters during the sampling can affect the chemistry of the collected BB aerosols (Sect. 3.4).
2.4 Organic-carbon–elemental-carbon (OC∕EC) analysis
Emissions from the combustion of two fuels (eucalyptus and Malaysian peat) were sampled with quartz-fiber filters, collected simultaneously with TIGF filters, for both fresh and aged BB aerosols (Fig. S1 in the Supplement). Punches (area of 1.5 cm2) from these quartz filters were analyzed with a thermo-optical carbon analyzer (Atmoslytic Inc., Calabasas, CA, USA) following the IMPROVE (US Interagency Monitoring of PROtected Visual Environments; Chow et al., 1993, 2004) protocol for the mass of total organic carbon (OCTotal) and elemental carbon (EC).
2.5 Analytical methodology for gas chromatography mass spectrometry (GC-MS)
We extracted filter and XAD samples for gas chromatography mass spectrometry (GC-MS) analysis (Table S1 in the Supplement) yielding concentrations of 84 polar organic compounds. In addition, levoglucosan concentrations were determined using ion chromatography coupled with a pulsed amperometric detector (IC-PAD). Prior to the extraction, sampled TIGF filters and XAD resin cartridges were spiked with deuterated internal standards (see Sect. 2.2). The TIGF filters and XAD cartridges were extracted separately with an accelerated solvent extractor (Dionex ASE 300, Sunnyvale, CA, USA) at the following conditions: 80 ∘C temperature, 250 mL extraction volume, and subsequent extraction with dichloromethane and acetone. The XAD and filters were treated separately to evaluate the speciation of gas- and particle-phase semivolatile polar compounds. The extracts were concentrated with a rotary evaporator (Buchi R-124, Switzerland), filtered using 0.2 µm pore size syringe filters (Thermo Scientific, Redwood, TN, USA), and pre-concentrated with nitrogen to a volume of 4 mL. Then we split the extracts into two fractions. One fraction was transferred to 2.0 mL volume deactivated glass maximum recovery vials (Waters Corporation, Milford, MA, USA), pre-concentrated to 50 µL volume under ultra-high-purity nitrogen (Airgas, Reno, NV, USA), and derivatized with N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA with 1 % of trimethylchlorosilane; Thermo-Scientific, Bellefonte, PA, USA) and pyridine as described elsewhere (Rinehart et al., 2006). Derivatized samples were analyzed by electron impact ionization using a Varian CP-3400 gas chromatograph with a CP-8400 autosampler and interfaced to a Varian 4000 ion trap mass spectrometer (Varian Inc., Palo Alto, CA, USA). The second fraction of non-derivatized extracts was kept for further analysis of nonpolar organic species (e.g., alkanes and polycyclic aromatic hydrocarbons), and those results will be presented in future publications.
Since, we have not performed any replicate burning experiments in this study, we used uncertainty values from our previous work, Yatavelli et al. (2017), as follows. We took mean (μc) and standard-deviation (σc) values for all 84 compounds from Alaskan peat (three replicates) and cheatgrass (three replicates) combustion and calculated fractional uncertainties (fc) using Eq. (1).
These fractional uncertainty values are multiplied by the emission factors for each compound (both fresh and aged) from the current study to obtain individual uncertainties or standard-deviation (SD) values as demonstrated in Eq. (2).
The group uncertainties for each different subclass were computed from individual uncertainties by applying the standard propagation of error method (root sum of squares).
Considering the different nature of combustion, the fractional uncertainties derived from the combustion of Alaskan peat were applied to emissions from all peat fuels, and the uncertainties obtained from cheatgrass combustion were applied to emissions from eucalyptus combustion.
2.6 Levoglucosan analysis
Portions of quartz filters collected for OC∕EC analysis also were used for the quantitative analysis of levoglucosan concentration with IC-PAD. Prior to the analysis, quartz filters were extracted with 15 mL of deionized water (18.2 MΩ), sonicated for 1 h, and refrigerated overnight. The column temperature for IC was 25 ∘C. Analytes along with a mixture of two eluents (48 % hydroxide solution and 52 % deionized water) were passed through the IC column with a 0.4 mL min−1 eluent flow rate and detected using an electrochemical detector. See Chow and Watson (2017) for details. Uncertainties or standard deviations associated with levoglucosan results indicate only analytical uncertainties of the IC-PAD method.
3.1 Gas- and particle-phase emission factors
Organic compounds (84 in total) in fresh emissions identified and quantified in this study were assigned to six major groups (Table S1): methoxyphenol derivatives, dicarboxylic acids, monocarboxylic acids, aromatic acids, resin acids, and levoglucosan. First, we report individual emission factors (EFs) belonging to a particular group calculated by the summation of gas- and particle-phase EFs of individual compounds. The relative abundance of these compounds is reported next followed by a comparison of the contributions of each group (EFgroup) among fuels and a comparison with previously reported results.
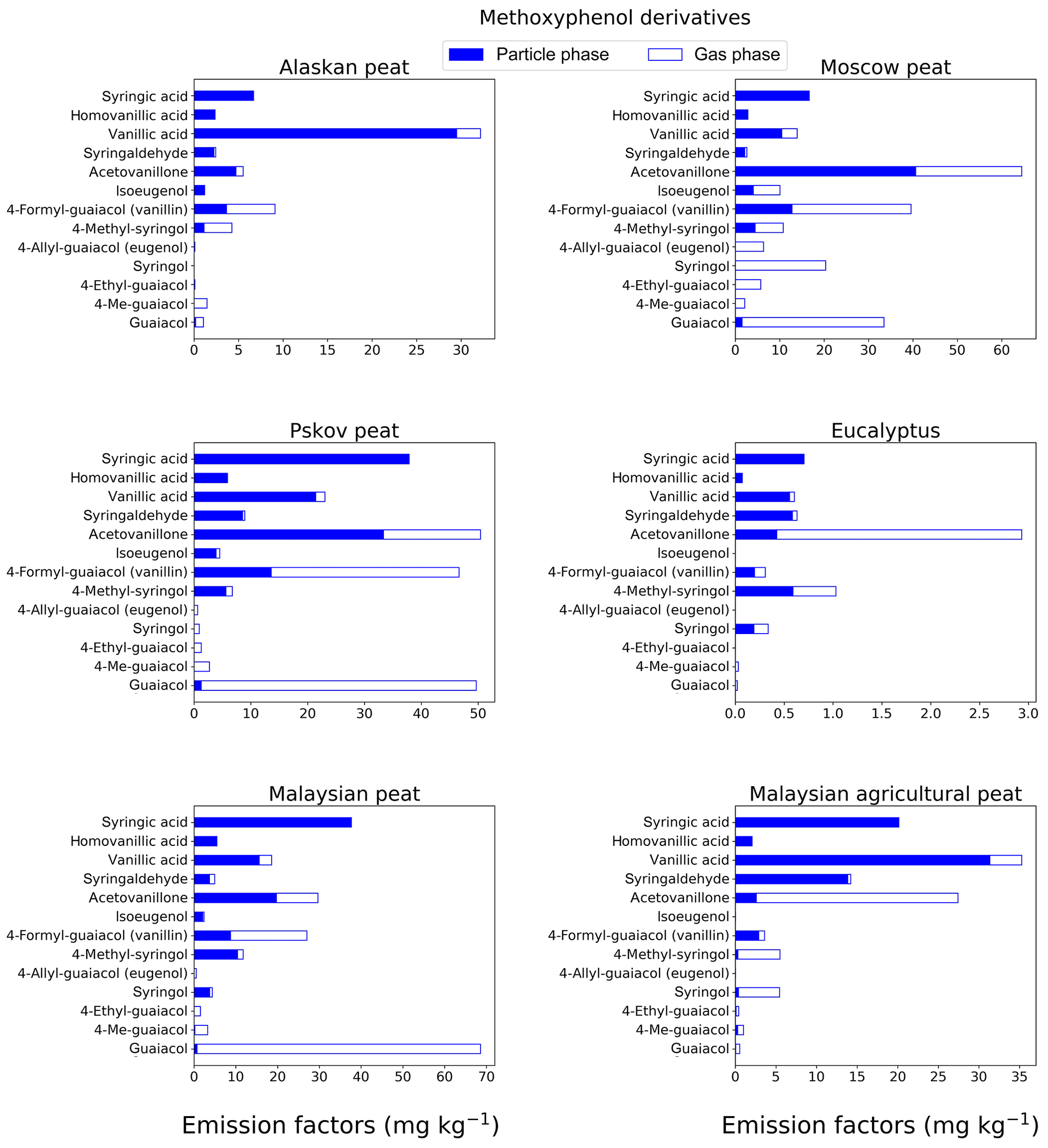
Figure 2EFs for methoxyphenols in both the particle phase (solid bars, filter samples) and gas phase (open bars, XAD samples) from fresh biomass-burning emissions for six different fuel types. We did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on the replicate analysis of emissions from similar fuels (with identical experimental conditions) during our previous combustion campaigns (Yatavelli et al., 2017), where SD ranged between 9.7 % and 22 % for methoxyphenol derivatives.
3.1.1 Methoxyphenol derivatives
Methoxyphenols are key compounds in BB smoke, since they constitute from 20 % to 40 % of the total identified organic aerosol mass (Hawthorne et al., 1989; Yee et al., 2013). For this reason, these compounds are considered potential markers for wood combustion (Schauer et al., 2001) and have been used as probable biomarkers to determine human exposure to BB emissions (Simpson and Naeher, 2010; Dills et al., 2006). Our analysis of 13 methoxyphenols (Fig. 2; Table S1) showed that guaiacol (molecular weight – MW – of 124 g mol−1) was the major contributor to EFs of the measured methoxyphenols in Moscow peat (33.5±3.3 mg kg−1), Pskov peat (49.7±4.8 mg kg−1), and Malaysian peat (68.6±6.7 mg kg−1). Syringol, another methoxyphenol commonly found in BB emissions (Schauer et al., 2001), had the highest EF for Moscow peat fresh emissions (20.4±2.7 mg kg−1), while for the other fuels, the EF was much lower (0–5.5 mg kg−1). EFs for syringic acids (MW of 198 g mol−1) were in the range of 0.06–37.9 mg kg−1 for all fresh emissions. Syringols are generally not formed during the pyrolysis of coniferous lignin but during the pyrolysis of deciduous lignin, where both guaiacols and syringols are formed (Mazzoleni et al., 2007). Presence of both guaiacol and syringol moieties in fresh emissions indicates that the part of the plant material that was responsible for peat formation was probably from deciduous trees, and this signature of deciduous trees from peat-burning emission is irrespective of the geographical origin of those peats (also shown by Schauer et al., 2001). Acetovanillone, vanillin, and vanillic acid also were observed in fresh emissions with a high abundance (5–50 mg kg−1). For example, vanillin is an abundant methoxyphenol in the fresh emissions from Pskov peat (46.7±5.4 mg kg−1), which contributed 4.4 % of the total mass of the 84 analyzed compounds.
Low-MW methoxyphenols (e.g., guaiacol) are expected to be found in the gas phase (Yatavelli et al., 2017), in close agreement with our results. For example, guaiacol and substituted guaiacols were mostly present in the gas phase (82 %–100 %) for emissions from the combustion of different fuels (Fig. 2; Table S1). With the addition of more oxygenated functional groups to a molecule, and thus with an MW increase, the equilibrium gas–particle partitioning of the compound tends to shift toward the particle phase, which also was confirmed by our results (e.g., for acetovanillone, a keto form of lignin derivative, from Malaysian peat combustion, 33.5 % of its mass was found in the gas phase; for the more oxygenated syringic acid, 99 % of its mass was found in particle-phase emissions from the same fuel).
The highest methoxyphenol EFgroup from the combustion of all fuels was observed in the fresh Pskov peat (Fig. 7a) emissions (239±11 mg kg−1). For Moscow peat, which was sampled close to the geographical region of Pskov peat, the EFgroup of methoxyphenols was 229±10 mg kg−1 (Fig. 7a), very similar to that for Pskov peat. The methoxyphenol EFgroup values for peat samples were in the range of 66 to 239 mg kg−1 (Fig. 7a) for our 13 analyzed compounds. A previous study analyzed for 30 different compounds (Schauer et al., 2001) and consequently found a larger value of EFgroup of up to 1330 mg kg−1, at least partially a result of the larger number of compounds analyzed. The formation of methoxyphenols during biomass combustion is mainly because of the pyrolysis of lignin (e.g., Simoneit et al., 1993). Lignin, an essential biopolymer of wood tissue, is primarily derived from three aromatic alcohols: p-coumaryl, coniferyl, and sinapyl alcohols (Hedges and Ertel, 1982). The lignins of hardwoods (angiosperms) are enriched with products from sinapyl alcohol; softwoods (gymnosperms) instead have a high proportion of products from coniferyl alcohol with a minor contribution from sinapyl alcohol; grasses have mainly products from p-coumaryl alcohol. The relative proportions of these biomonomers vary considerably among the major plant classes (Sarkanen and Ludwig, 1971), reflected in our total emission factors estimate for 13 methoxyphenols.
3.1.2 Dicarboxylic acids
Dicarboxylic acids play a significant role in the atmospheric organic aerosols budget (Samburova et al., 2013; Yatavelli et al., 2017) via secondary-organic-aerosol formation that either changes radiative forcing directly or indirectly by acting as cloud condensation nuclei (Kawamura and Bikkina, 2016). The EFgroup for dicarboxylic acids (Fig. 7b) varied among the fuels with the highest EF for fresh Pskov peat samples (123±10 mg kg−1) and with the lowest for eucalyptus (1.5±0.1 mg kg−1). This range in EFs can be attributed to the difference in fuel type and burning conditions (smoldering and flaming). We also observed, however, a difference in the EFgroup of dicarboxylic acid between two tropical peats from the same geographical area (EF of Malaysian peat of 35.33±2.9 mg kg−1 and of Malaysian agricultural peat of 12.29±1.02 mg kg−1). The highest EF for individual dicarboxylic acids was observed for azelaic acid. For example, for Pskov peat the EF was 32.1±4.1 mg kg−1, and for Moscow peat it, was 17.6±2.6 mg kg−1. Azelaic acids were mostly found in the particle phase (Fig. 3; Table S1) and their relative abundances in the gas phase varied between 0.77 % (for Pskov peat) and 2.85 % (for eucalyptus). Maleic acid was mostly found in the gas phase (73 %–83 %), since it is a lower-MW compound (MW of 116.0 g mol−1) compared to azelaic (MW of 188.22 g mol−1) and adipic (146.14 g mol−1) acids. Succinic and methylsuccinic acids are found in both the gas and particle phases (Table S1), and their abundance in the particle phase was 19 %–59 % and 53 %–100 %, respectively. For Malaysian peat BB emissions, succinic acid was present only in the particle phase. A distinguishable increase in dicarboxylic-acid mass concentrations was observed for ambient aerosols followed by a biomass-burning event (Cao et al., 2017) compared to normal ambient concentrations. The formation of saturated dicarboxylic acids (e.g., succinic acid) and unsaturated dicarboxylic acids (e.g., maleic acid) also was reported for ambient aerosols collected near a biomass-burning event (Graham et al., 2002; Kundu et al., 2010; Zhu et al., 2018) and in ice core records historically affected by biomass burning (Müller-Tautges et al., 2016).
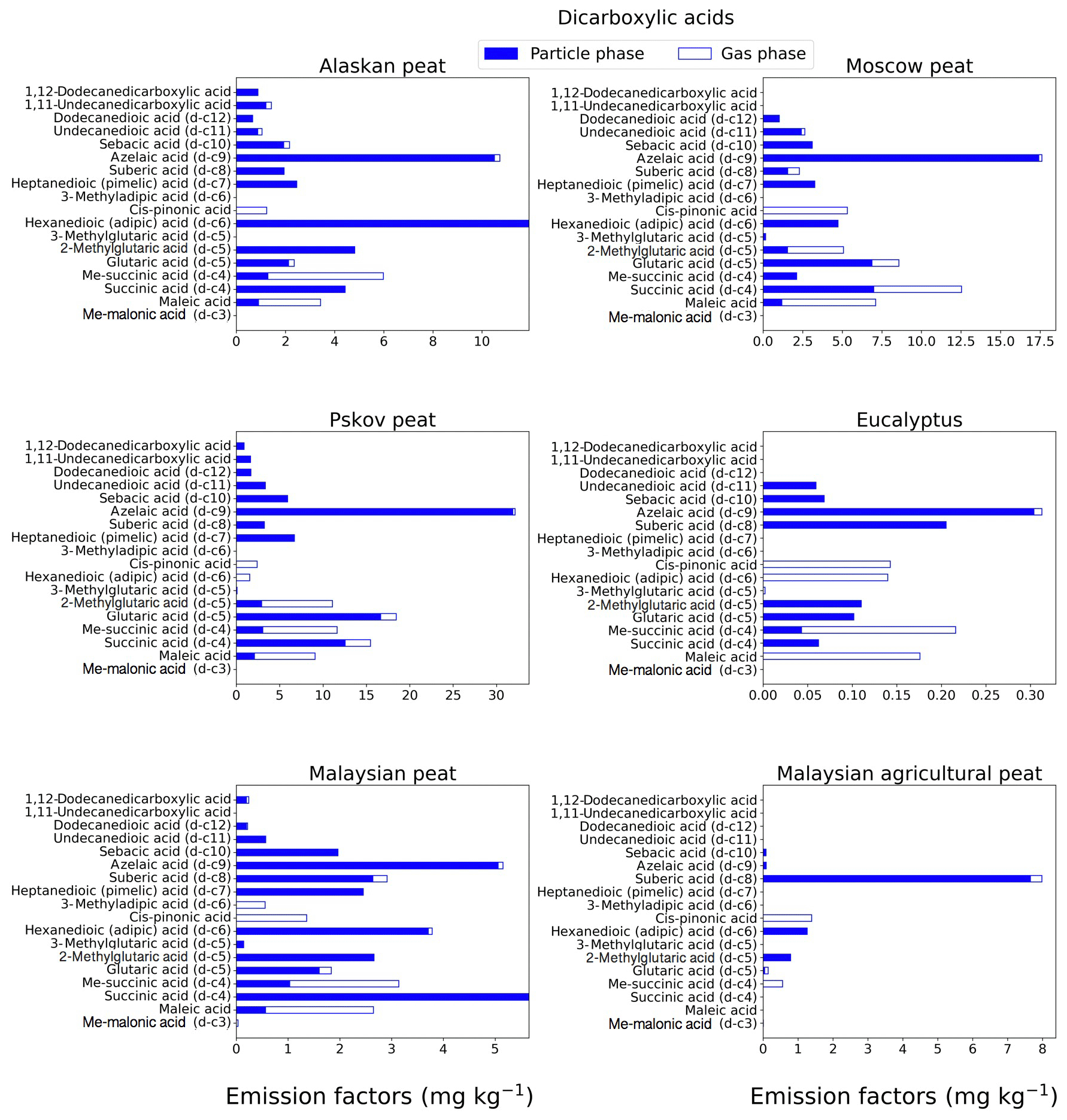
Figure 3EFs for dicarboxylic acids in both the particle phase (solid bars, filter samples) and gas phase (open bars, XAD samples) from fresh biomass-burning emissions for six different fuel types. We did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on replicate analysis of emissions from similar fuels (with identical experimental conditions) during our previous combustion campaigns (Yatavelli et al., 2017), where SD ranged between 10 % and 17 % for dicarboxylic acids.
3.1.3 Monocarboxylic acids
Monocarboxylic acids can constitute up to 30%–40 % of total identified organic aerosol mass from BB emissions (Oros et al., 2006). In our study, we characterized the range from C6 to C24, where some unsaturated monocarboxylic acids (e.g., oleic acid) also are included. For Alaskan and Malaysian peat fresh emissions (Fig. 4), the highest EF (gas + particle) among all analyzed monocarboxylic acids was for hexadecanoic acid (C16) with EFs of 55.7±6.6 and 51.8±6.2 mg kg−1, respectively. The dominance of hexadecanoic acid among other monocarboxylic acids in combustion emissions also was observed in ambient measurements during biomass-burning events in southeastern Asia (Fang et al., 1999). For Moscow (41.5±6.5 mg kg−1) and Pskov (49.8±7.8 mg kg−1) peats, tetradecanoic acid (C14) had the highest EFs in fresh samples (Fig. 4). For eucalyptus and Malaysian agricultural peat fresh samples, the largest contributor to monocarboxylic acids was tetracosanoic acid (C24; Fig. 4) with EFs of 3.81±0.5 and 42.0±5.1 mg kg−1, respectively. As we expected, low-MW monocarboxylic acids like hexanoic acid (MW of 116 g mol−1) was mostly present in the gas phase, and the gas phase mass fraction varied between 72 % (for Malaysian agricultural peat) and 98 % (for Moscow peat). Similar trends were observed for other low-MW monocarboxylic acids. For example, the relative abundance of octanoic acid (C8) in the gas phase was 93.4 % for Alaskan peat. High-MW monocarboxylic acids (C16>) abundance in the gas phase was <2 % for all analyzed fuels.
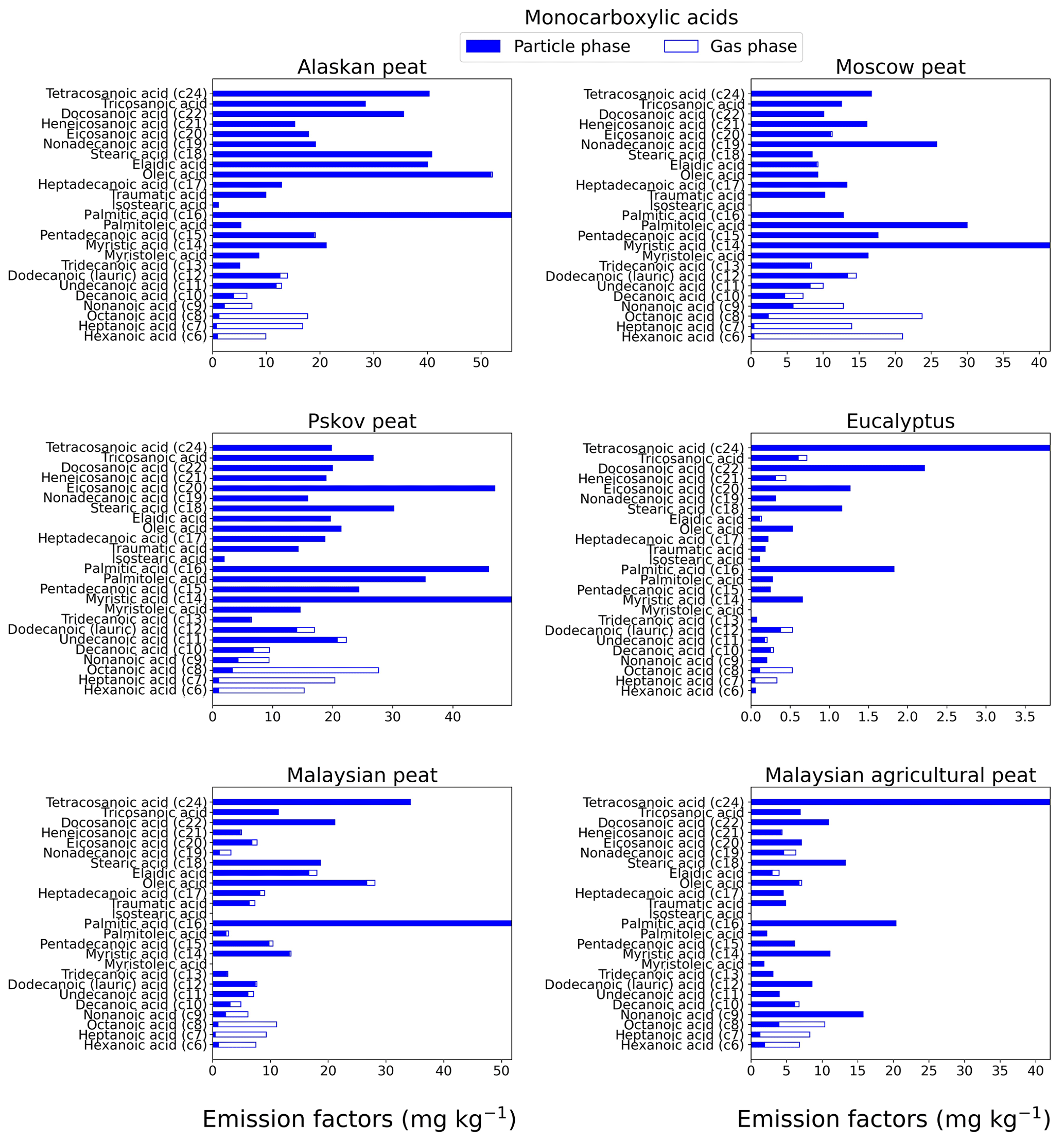
Figure 4EFs for monocarboxylic acids in both the particle phase (solid bars, filter samples) and gas phase (open bars, XAD samples) from fresh biomass-burning emissions for six different fuel types. We did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on replicate analysis of emissions from similar fuels (with identical experimental conditions) during our previous combustion campaigns (Yatavelli et al., 2017), where SD ranged between 9.4 % and 12 % for monocarboxylic acids.
The carbon preference index (CPI) is generally used for the source apportionment of organic aerosols (Fang et al., 1999). We also computed the carbon preference index for monocarboxylic acids from all analyzed fuel combustion emissions by taking the ratio of even carbon numbers to odd carbon numbers on the EFs of monocarboxylic-acid ranges from C6 to C24 (Fig. S1). For fresh emission samples, the CPI values ranged from 1.28 (for Moscow peat) to 4.53 (for eucalyptus). The CPI values are higher for fresh emissions of tropical peats (for example, 2.78 for Malaysian peat) than for emissions from peats from high latitudes (for example, 1.74 for Pskov peat). An average CPI index of 3.7 for monocarboxylic acids was reported for combustion emissions from sedimentary bogs (Freimuth et al., 2019), in the range of our reported values (CPI of 1.3–4.5).
The sums of the EFs for 25 monocarboxylic acids are shown in Fig. 7c. The EFgroup was in the range of 5 to 515 mg kg−1 for all fuels. This range is comparable to the EFgroup reported previously for grass (tundra, cotton, Pampas, and ryegrass) combustion (32–250 mg kg−1; Oros et al., 2006). Overall, the trend of low EFs associated with the flaming combustion of eucalyptus (16±0.7 mg kg−1) compared to smoldering-peat combustion is also evident for this compound category. The combustion of peat fuels from tropical regions (e.g., Malaysian agricultural peat) resulted in monocarboxylic-acid EFs of 212±9.6 mg kg−1 compared to higher EFs from Alaskan peat combustion (505±23 mg kg−1). The origin of monocarboxylic acid is mostly plant wax and oils (Simoneit, 2002). The relative proportion of plant wax and oils can vary widely among vegetation taxa, and also their concentrations in peat depend on biogeochemical processes involved in peat formation. The differences in the relative abundance of waxes and plant oils in living vegetation and the differences between biogeochemical processes involved in peat formation for arctic and tropical regions may be responsible for diverse EFs for monocarboxylic acids.
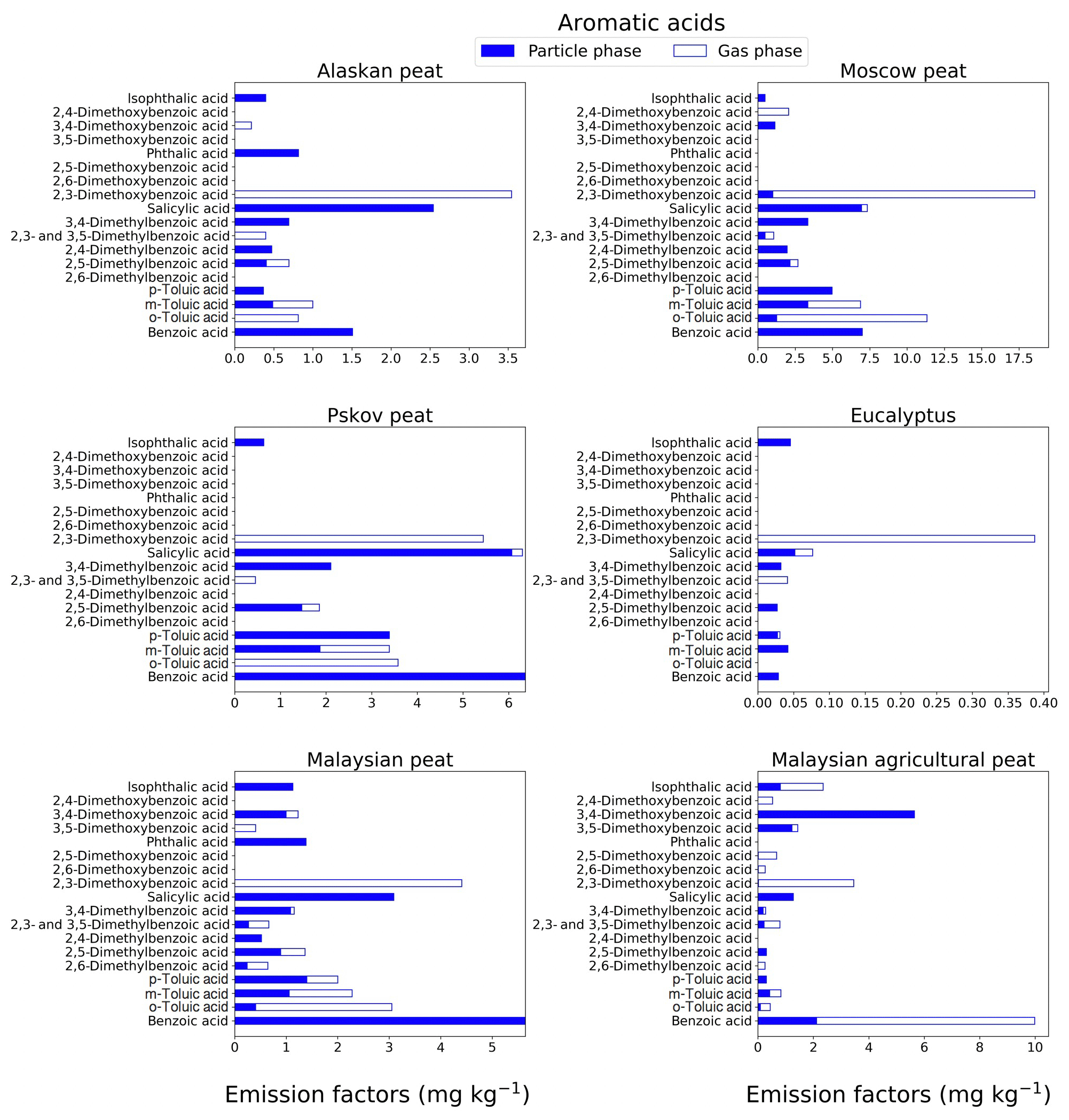
Figure 5EFs for aromatic acids in both the particle phase (solid bars, filter samples) and gas phase (open bars, XAD samples) from fresh biomass-burning emissions for six different fuel types. We did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on replicate analysis of emissions from similar fuels (with identical experimental conditions) during our previous combustion campaigns (Yatavelli et al., 2017), where SD ranged between 9.5 % and 15 % for aromatic acids. Please note that benzoic acid was found in high concentrations in the XAD blanks that introduced a substantial uncertainty to the quantification of this compound.
3.1.4 Aromatic acids
Aromatic acids from BB emissions can contribute up to 20 %–35 % of total identified organic mass (Wan et al., 2019). In our study, the aromatic acids (e.g., p-hydroxy benzoic acid) excluded methoxyphenol derivatives and resin acids. For most of the fuels, low-MW aromatic acids (MW<150 g mol−1; e.g., benzoic acid and o-, m-, and p-toluic acids) contributed more (almost 40 % of total aromatic-acid emissions) toward total fresh emissions, compared to high-MW aromatic acids (MW>150 g mol−1). For example, the benzoic-acid EF for Malaysian agricultural peat fresh emission (Fig. 5) is 9.98±1 mg kg−1, and the EF for the same benzoic acid is 6.36±0.6 mg kg−1 for Pskov peat (Fig. 5). Although, o-toluic and p-toluic acids are found in the gas phase with 50 %–100 % abundance in Alaskan and Moscow peat, benzoic acid is found only in the particle phase. Benzoic acid was found in high concentrations in the XAD blanks that introduced a substantial uncertainty to the quantification of this compound. One of the most abundant aromatic acids in fresh peat emissions was 2,3-dimethoxybenzoic acid. For example, for Moscow peat the EF was 18.6±4.7 mg kg−1. The acid was mostly found in the gas phase (91 %–100 %) for all fuels (Fig. 5; Table S1). 2,3-dimethoxybenzoic acid is potentially derived from the combustion of lignin moieties of biomass, and the emission of this compound is more than an order of magnitude lower in emissions from flaming-combustion samples (EF of 0.38±0.09 mg kg−1) than in emissions from smoldering-combustion samples (EF of 5.44±1.36 mg kg−1). Emissions of 3,4 dimethoxybenzoic acid were only observed for peats from tropical regions (EF of 5.64±0.8 mg kg−1) with 80 %–100 % abundance in the particle phase. This compound can be used for the source apportionment of aerosols emitted from the burning of tropical peat and also can potentially help one to distinguish between emissions from tropical and high-latitude peatland fires.
The EFgroup for aromatic acids in fresh-combustion emissions from eucalyptus fuel is extremely low (0.71±0.05 mg kg−1) compared to that for peat fuels (13–69 mg kg−1). Among all peat samples, the Alaskan peat fresh EF was the lowest EF (13.5±0.9 mg kg−1), whereas Moscow peat fresh emissions yielded the highest EF (69±4.4 mg kg−1). The difference in total aromatic-acid emissions can be attributed to the variation in the lignin content of the fuels and burning conditions (Simoneit, 2002).
3.1.5 Resin acids
We quantitatively analyzed combustion emissions for isomers in six resin acids (Table S1). The most abundant resin acid (78 % of total resin acid emission) is dehydroabietic acid (C20) that does not have isomers. The preponderance of this acid over other resin acids in emissions from oak and pine biomass burning was reported by Simoneit et al. (1993). We found that dehydroabietic acid (C20) content in fresh emissions is 15–30 times higher in fuels from high-latitude peatlands than in those of tropical origin. For example, the EF for dehydroabietic acid in fresh Alaskan peat emissions is 92.2±14 mg kg−1 (Fig. 6), whereas the same in fresh Malaysian peat emissions is 3.44±0.5 mg kg−1 (Fig. 6). Resin acids are supposed to be found mostly in the particle phase based on their MW and functional groups (Asher et al., 2002; Karlberg et al., 1988; Pankow and Asher, 2008), confirmed by our results (80 %–100 % in the particle phase) with the exception of Malaysian peat emissions, where 56.6 % abundance of dehydroabietic acid was found in the gas phase. Although a distinct peak of dehydroabietic acid was observed at the desired retention time for this sample during GC-MS analysis, we believe this result can be attributed to some unknown interference from our analysis procedure.
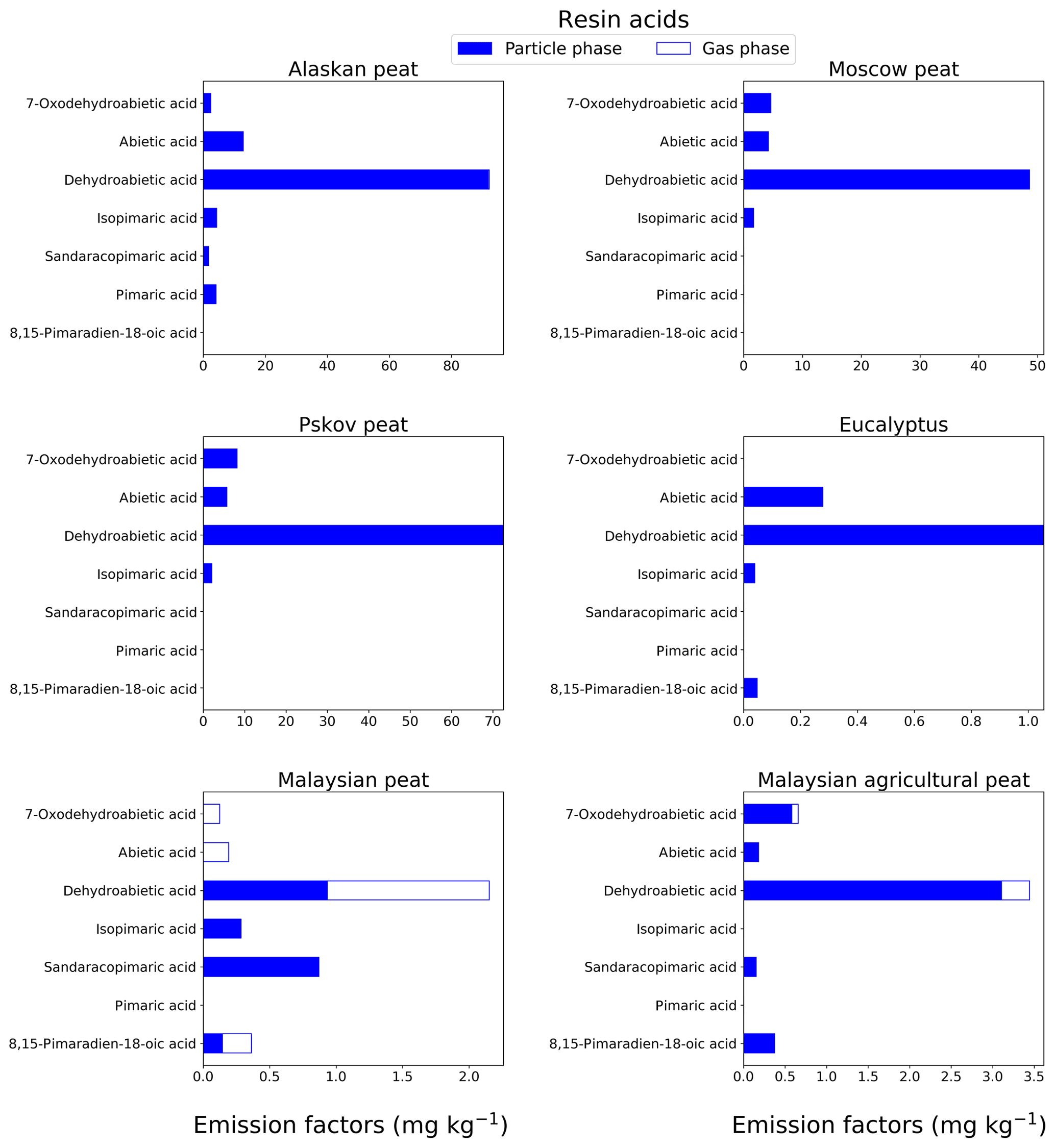
Figure 6EFs of resin acids in both the particle phase (solid bars, filter samples) and gas phase (open bars, XAD samples) from fresh biomass-burning emissions for six different biomass types. As in prior cases, we did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on replicate analysis of similar fuels (with same experimental conditions) from our previous campaigns (Yatavelli et al., 2017), where SD varies between 9.7 % and 15 % for resin acids.
The EFgroup for seven resin acids are presented in Fig. 7e. A high EFgroup was observed for Alaskan (117±15 mg kg−1), Pskov (89±12 mg kg−1), and Moscow (59±7.7 mg kg−1) peats representing midlatitude and arctic peats. Resin acids (e.g., pimaric acid) are biosynthesized mainly by conifers (gymnosperms) in temperate regions. In previous work, Iinuma et al. (2007) gave a range of resin-acid EFs from 0 to 110 mg kg−1, in agreement with our results. Very low resin-acid EFs were found for peat from tropical regions (e.g., 4 mg kg−1 for Malaysian peat fresh samples). As deciduous trees in tropical zones are not prolific resin and mucilage (gum) producers, compositional data on smoke from such sources should not be expected to show moderate concentrations of resin acids. This is supported by earlier work by Iinuma et al. (2007), where resin acids were not even detectable for emissions from Indonesian peat combustion.
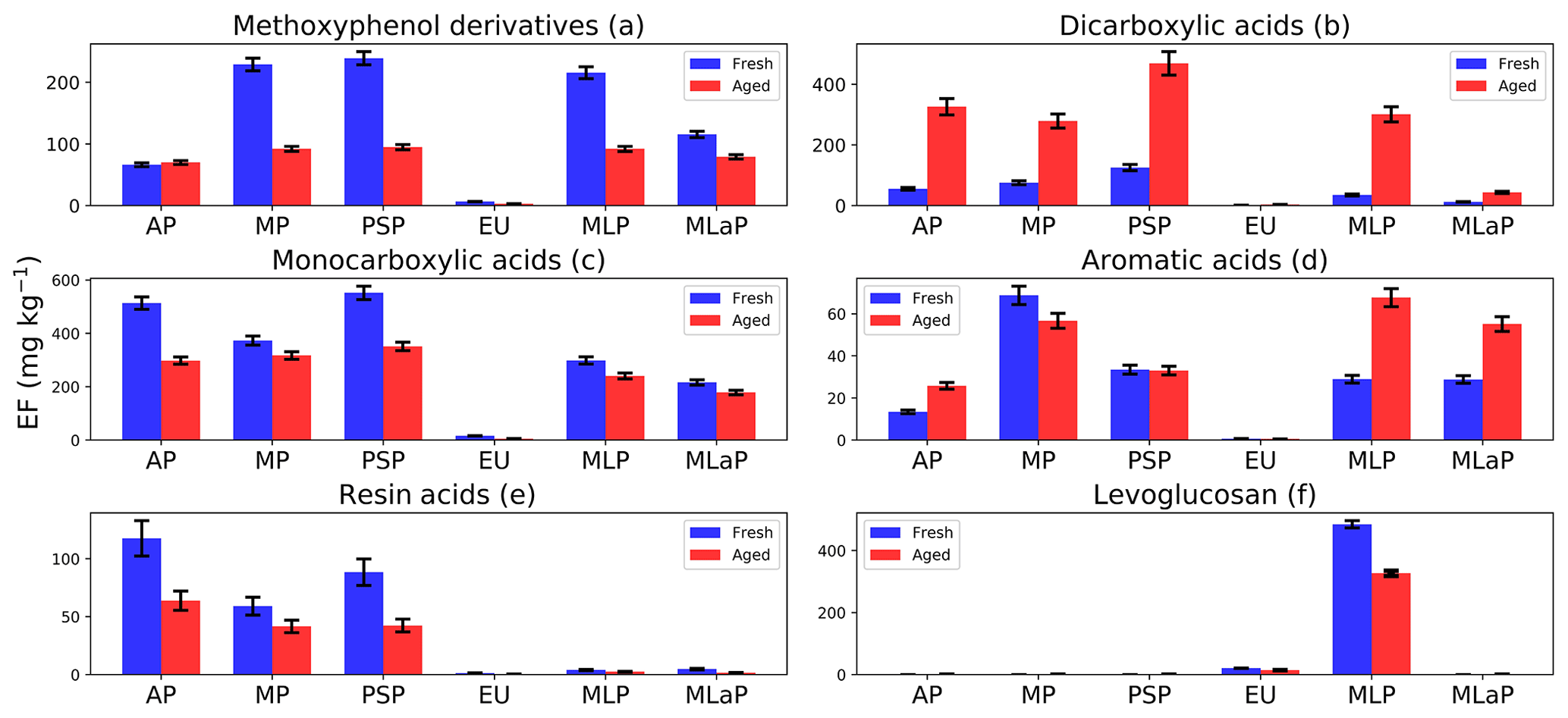
Figure 7Fuel-based emission factors (EFs) of organic compounds assigned to six chemical groups for six different fuels: Alaskan peat (AP), Moscow peat (MP), Pskov peat (PSP), eucalyptus (EU), Malaysian peat (MLP), and Malaysian agricultural peat (MLaP). EFs are presented as a sum of gas- and particle-phase species mass measured in fresh and OFR-aged BB emissions (in units of mg kg−1; mass of emissions per fuel mass combusted). We did not burn fuels in replicates, and the standard-deviation (SD) values of all chemical groups were calculated based on the replicate analysis of similar fuels (with the same experimental conditions) from our previous campaign based on the data reported by Yatavelli et al. (2017).
3.1.6 Levoglucosan
Levoglucosan can be found mostly in the particle phase (Simoneit, 2002). We report levoglucosan EFs from our analysis of the quartz filter using the IC-PAD technique (no gas-phase EFs reported). The EFs of levoglucosan (Fig. 7f) were found to be 20.9±0.68 and 485±11.8 mg kg−1 for eucalyptus and Malaysian peat, respectively, and their carbon content is approximately 1.8 % and 2.5 % of the total organic carbon mass characterized by the thermo-optical technique. Fine et al. (2002) reported 9 % to 16 % contribution of levoglucosan to total OC from residential wood combustion, a relatively higher percentage than values obtained in our study. Anhydrosugars (e.g., levoglucosan and its isomers) are found in great abundance and have been widely used as a BB tracer because of their atmospheric stability, as summarized by Bhattarai et al. (2019). We found that levoglucosan constituted 36 % and 51 % of GC-MS characterized polar (listed in our method) organic aerosol mass for eucalyptus and Malaysian peat, respectively, which also is consistent with the previous BB literature assembled in the recent review article by Bhattarai et al. (2019).
3.2 Emission factors of total (gas + particle) organic compounds of six chemical groups and their changes upon OFR oxidations
Here we describe changes in the EFgroup followed by OFR oxidation for all six chemical groups. Levoglucosan and the most abundant resin acid, dehydroabietic acid, also are reported in this section.
3.2.1 Methoxyphenol derivatives after OFR oxidation (Fig. 7a)
Methoxyphenols can undergo gas-phase oxidation reactions via either aromatic-ring fragmentation or opening to form short-chain ketones, acids, esters, and double bonds in conjugations with all functional groups or the further hydroxylation of aromatic rings to form multiple substituted aromatic compounds (Yee et al., 2013). In either case, a decrease in methoxyphenols after oxidation was expected. In our study, a decrease in methoxyphenol's EFgroup with OFR oxidations was observed for all fuels (e.g., for Pskov peat from 239±11 to 95±4 mg kg−1) except for Alaskan peat, where an insignificant increase from 66±3 to 70±3 mg kg−1 after OFR oxidation was observed.
3.2.2 Dicarboxylic-acid group after OFR oxidation (Fig. 7b)
A significant increase (2.5–8.5 times) in the EFgroup of dicarboxylic acids was observed for OFR-aged samples. For example, the EFgroup of dicarboxylic acids increased from 35±3 to 301±25 mg kg−1 for Malaysian peat and from 56±5 to 326±27 mg kg−1 for Alaskan peat. The oxidation of aerosols potentially produces more oxygenated functional groups (Jimenez et al., 2009), demonstrated by an increase in O:C ratios (from 0.45 to 0.65) in the recent laboratory oxidation of BB emissions by Bertrand et al. (2018), where TAG-AMS was used to identify the fate of organic compounds. In this work, however, the number of identifiable compounds with highly functional groups is constrained by the elution technique used in the TAG method. Our results on the fate of BB organic aerosols with 18 dicarboxylic acids can provide a better mechanistic understanding about the processes inside the OFR.
3.2.3 Monocarboxylic-acid group after OFR oxidation (Fig. 7c)
We observed a decrease in the monocarboxylic-acid EFgroup from OFR aging for all fuels. For example, the EFgroup for monocarboxylic acids from Alaskan peat combustion decreased from 514±23 mg kg−1 (fresh) to 298±14 mg kg−1 (aged). A relatively small decrease compared to Alaskan peat was observed for Malaysian agricultural peat (from 216±10 mg kg−1 – fresh – to 179±8 mg kg−1 – aged) and Malaysian peat (from 298±14 mg kg−1 – fresh – to 240±11 mg kg−1 – aged) too. This is probably because of the formation of low-MW monocarboxylic acids (e.g., hexanoic acids; MW of 116 g mol−1) after OFR oxidation demonstrated in Fig. 10 and Table S2c and will be discussed further in Sect. 3.3. Monocarboxylic acids can be oxidized in the atmosphere (Charbouillot et al., 2012), leading to the formation of dicarboxylic acids from C2 to C6 (Ervens et al., 2004). This is consistent with our results (Fig. 7b, c; Table S2b). Moreover, monocarboxylic acids, during their atmospheric transformations, can produce a potential precursor for the formation of high-MW compounds, such as a humic-like substance (HULIS; Carlton et al., 2007; Tan et al., 2012).
3.2.4 Aromatic-acid group after OFR oxidation (Fig. 7d)
Levels of aromatic acids from peat burning increased for Alaskan peat, Malaysian peat, and Malaysian agricultural peat (e.g., from 29±2 to 68±4 mg kg−1 for Malaysian peat) by OFR aging (Fig. 7e; Table S2e). This increase could be from the oxidation of phenols and methoxyphenols in the OFR chamber (Akagi et al., 2011; Legrand et al., 2016). For eucalyptus and Moscow and Pskov peats, it was an insignificantly small decrease (e.g., from 69±4 to 57±3 mg kg−1 for Moscow peat and from 34±2 to 33±2 mg kg−1 for Pskov peat). This small decrease in the EFgroup of monocarboxylic acids is insignificant. The oxidation processes occurring in the OFR are complex, especially in the case of multicomponent BB emissions. The decrease observed for aromatic acids after the OFR may be attributed, however, to multiple generations of oxidation leading to the breaking of aromatic rings and formation of low-MW organic compounds via fragmentation (Jimenez et al., 2009).
3.2.5 Resin acids after OFR oxidation (Figs. 7e)
Resin acids can be oxidized to corresponding oxo acids (e.g., 7-oxodehydroabietic acid; Karlberg et al., 1988), and they are considered to be stronger contact allergens than the resin acids themselves (Sadhra et al., 1998). Our data showed a small decrease in 7-oxodehydroabietic-acid levels after the OFR (e.g., from 8.2±0.8 to 4.8±0.5 mg kg−1 for Pskov peat). We noted a significant decrease in the EFgroup of resin acids from 117±15 mg kg−1 (fresh) and 63±8 mg kg−1 (aged) for Alaskan peat after OFR oxidation, mostly because of some individual compounds like dehydroabietic acid. Although resin acids are considered to be stable atmospheric tracers for biomass burning (Simoneit et al., 1993), we observed a decrease in the dehydroabietic-acid (most abundant) EF after OFR oxidation of emissions from all fuels (Fig. S2b). For example, for Alaskan peat (Fig. S2b), the decrease was from 92±14 to 57±8 mg kg−1 after OFR oxidation. The fate of resin acids during OFR aging, however, was beyond the scope of this work and may be the subject of future investigations.
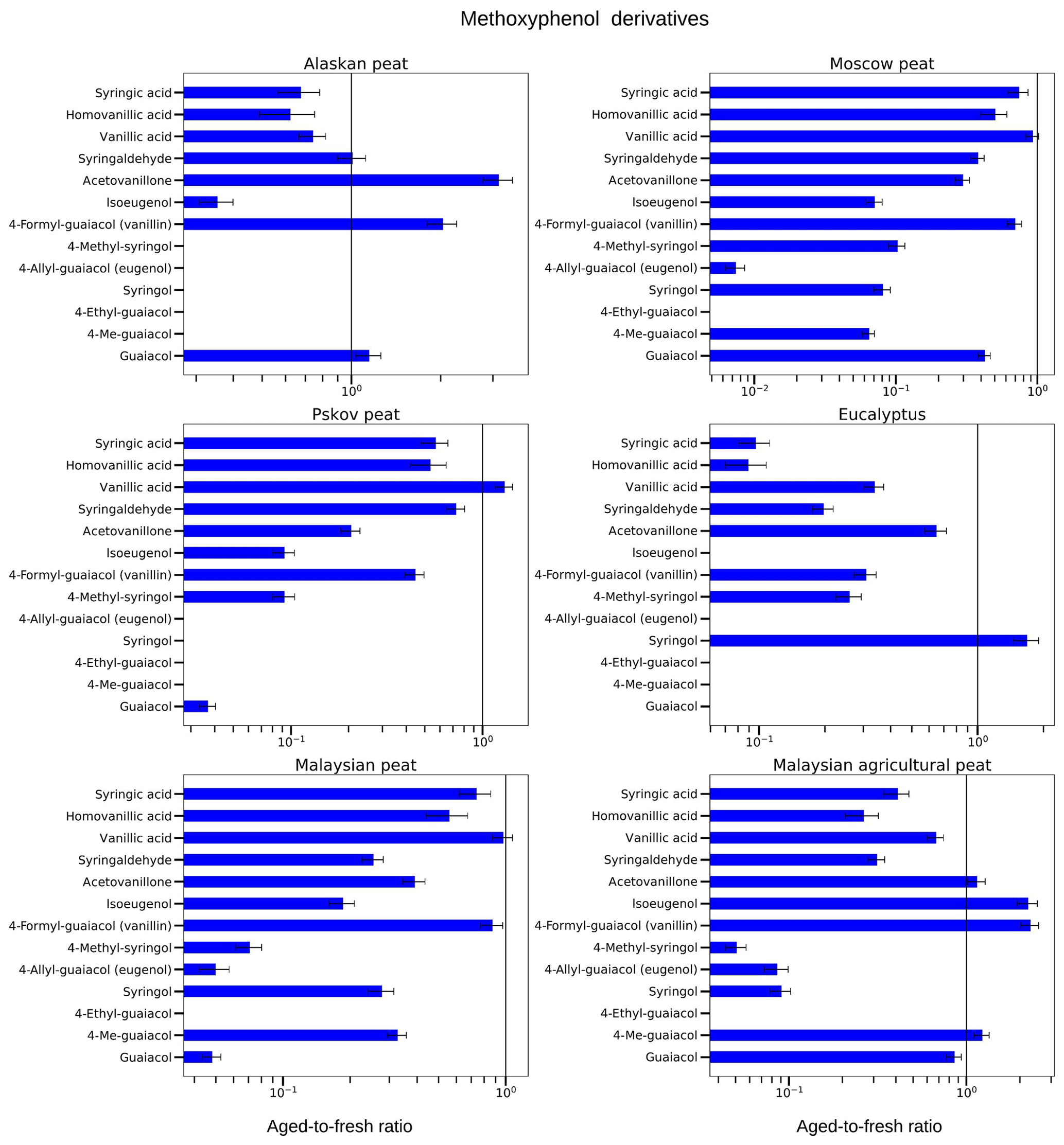
Figure 8Aged-to-fresh ratios of total (gas + particle) EFs for methoxyphenols from biomass-burning emissions for six different biomass types presented in log scale. We did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on the replicate analysis of similar fuels (with the same experimental conditions) from our previous campaigns. SD values derived from EFs were scaled to ratio.
3.2.6 Levoglucosan after OFR oxidation (Fig. 7f)
Levoglucosan is one of the most popular tracers of BB emissions, since it has been considered a stable compound in the atmosphere (Oros et al., 2006; Simoneit, 2002; Simoneit et al., 1999). Several laboratory studies, however, have demonstrated the degradation of levoglucosan in the presence of OH radicals (Hennigan et al., 2010). Here we observed a decrease of 30 % in levoglucosan levels following OFR oxidation. For example, Malaysian peat decreased from 485±12 to 327±8 mg kg−1. For eucalyptus, the decrease was from 20±0.7 to 14±0.6 mg kg−1. This decrease also can be attributed to the degradation process during OH oxidation (Hoffmann et al., 2010). Levoglucosan oxidation should be studied more so that it can be adequately used as a tracer of BB emissions.
3.3 Aged-to-fresh ratios of the total (gas + particle) emission factors of individual organic compounds assigned to six chemical groups and their changes upon OFR oxidation
We computed aged-to-fresh ratios of individual compounds for all fuels. If the aged-to-fresh ratio of one compound is greater than one, this implies that the compound is formed during OFR oxidation; if the ratio is less than one, then the compound must have decomposed inside the OFR.
3.3.1 Methoxyphenol derivatives
Overall, we found that abundances for methoxyphenol derivatives rapidly decreased upon OFR oxidation (Fig. 8; Table S2a). Some compounds – vanillic acid, acetovanillone, and syringic acids – demonstrated both increasing and decreasing trends. For example, for Pskov peat, the aged-to-fresh ratio of guaiacol was 0.04±0.01 reflecting a significant decrease during OFR oxidation. For Pskov peat, we also observed a ratio of less than one for vanillin (0.44±0.05), indicating that vanillin also decreased during OFR oxidation for the same fuel but not to the extent of guaiacol. At the same time and for the same fuel, a slight increase (aged-to-fresh ratio >1) in vanillic acid was observed (1.30±0.13) in the OFR-oxidized sample. This increase in vanillic-acid concentration can be attributed to the oxidation of vanillin, one of the abundant methoxyphenol compounds in the fresh emissions from Pskov peat (Fig. 8; Table S2a). For the combustion of other peats, vanillic-acid concentrations also decreased (e.g., aged-to-fresh ratios were 0.74±0.08 and 0.67±0.07 for Alaskan peat and Malaysian agricultural peat, respectively). Acetovanillone increased by a factor of 3 during OFR oxidation for Alaskan peat and around 15 % for Malaysian agricultural peat (aged-to-fresh ratio of 1.15±0.13), but the increase for Malaysian agricultural peat was not significant. For other fuels, acetovanillone decreased during OFR oxidation. For example, for Moscow peat, the aged-to-fresh ratio for acetovanillone was 0.30±0.03. We still need to investigate the reason why both acetovanillone and vanillic acid increased for some fuels and decreased for others. The reduction of acetovanillone and vanillic acid was because of a photochemical decomposition process in the OFR with the formation of lower MW products, such as succinic acid and maleic acid (Schnitzler and Abbatt, 2018).
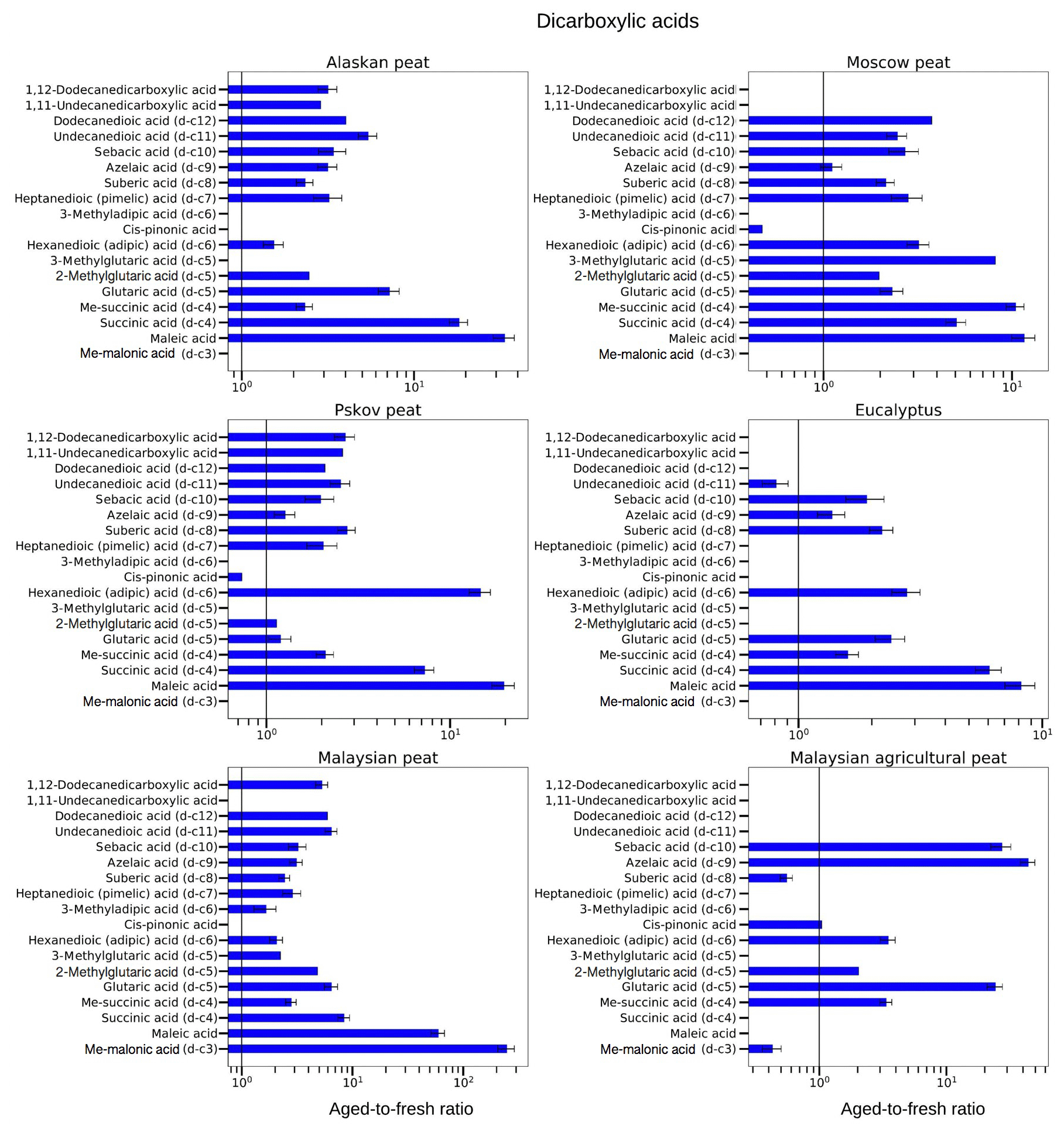
Figure 9Aged-to-fresh ratios of total (gas + particle) EFs for dicarboxylic acids from biomass-burning emissions for six different biomass types presented in log scale. We did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on the replicate analysis of similar fuels (with the same experimental conditions) from our previous campaigns. SD values derived from EFs were scaled to ratio.
3.3.2 Dicarboxylic acids
In the case of dicarboxylic acids, we observed an increase of 2–20 times in EFs, but the degree of enhancement of low-MW dicarboxylic-acid EFs was higher than for high-MW dicarboxylic-acid EFs. For example, a 20-fold increase in maleic acid, a low-MW dicarboxylic acid (MW of 116.07 g mol−1), was observed during OFR oxidation of Pskov peat emissions (aged-to-fresh ratio of 9.6±2.8), whereas for 1,11-undecanedicarboxylic acid, a high-MW dicarboxylic acid (MW of 244.33 g mol−1), the EF increased 2.6 times (aged-to-fresh ratio of 2.60±0.01) for the same fuel. Similarly, the concentration of succinic acid, a low-MW dicarboxylic acid (MW of 118.09 g mol−1), increased almost by 5 times after OFR oxidation (aged-to-fresh ratio of 5.07±0.62), whereas that of undecanedioic acid, a high-MW dicarboxylic acid (MW of 230.30 g mol−1), increased 2.5 times (aged-to-fresh ratio of 2.46±0.30) for Moscow peat. This trend was in accordance with results from ambient observations after BB events (Cao et al., 2017; Kawamura and Bikkina, 2016).
3.3.3 Monocarboxylic acids
Our analysis of OFR-aged samples showed that concentrations of monocarboxylic acids with different MWs changed during OFR oxidation, but the changes varied from one fuel to another. For example, the EF of hexanoic acid (C6) was reduced for eucalyptus (aged-to-fresh ratio of 0.5±0.05) and fuels from other high-latitude peatlands like Alaskan (aged-to-fresh ratio of 0.89±0.09), Moscow (aged-to-fresh ratio of 0.88±0.09) and Pskov peat (aged-to-fresh ratio of 0.33±0.03). The reduction of hexadecenoic acid was insignificant for Alaskan and Moscow peat. The peats from tropical regions showed exactly the opposite change. Hexanoic acid increased for both Malaysian (aged-to-fresh ratio of 1.47±0.14) and Malaysian agricultural peat (aged-to-fresh ratio of 1.40±0.14). We observed a similar trend for heptanoic acid (C7) during OFR oxidation. The tropical peats clearly demonstrated increases (for example, aged-to-fresh ratio of 1.92±0.22 for Malaysian peat) in heptanoic-acid concentration. For Moscow peat, even though hexanoic-acid concentrations were insignificantly decreased, heptanoic-acid concentrations increased significantly (aged-to-fresh ratio of 2.37±0.27). This contrast between changes in hexanoic and heptanoic acid can be explained by a decrease in CPI indices during OFR oxidations (for example, from 2.78 to 1.7 for Malaysian peat). The reduction of CPI indices indicated that during oxidation more monocarboxylic acids with odd carbon numbers were formed than monocarboxylic acids with even carbon numbers. The abundance of hexadecenoic acid (C16) was reduced during OFR oxidation for all fuels (for example, aged-to-fresh ratio of 0.63±0.08 for Alaskan peat) except for the Moscow and Pskov peat aged-to-fresh ratio of 1.05±0.13 for peat), and we believe this small increase is insignificant. Similarly, tetracosanoic acid (C20) was reduced for all fuels (for example, aged-to-fresh ratio of 0.61±0.07 for Malaysian peat) except for Pskov peat (aged-to-fresh ratio of 1.24±0.15). The small increase in tetracosanoic acid (C20) concentration was again insignificant. Even though our results indicated the possibility of the fragmentation of high-MW monocarboxylic acids and formation of low-MW monocarboxylic acids, this was because of the complexity of the OFR oxidation environment. We are not able to hypothesize what is the main reactive mechanism.
3.4 Top contributing compounds
We have identified the top contributing compounds for both fresh and aged BB emissions of each fuel to understand how emissions vary from one fuel to another. The top 10 compounds for fresh and aged emission were different, and we merged the top 10 compounds from fresh and aged emissions resulting in different numbers of the total top compounds for different fuels. Here we discuss Alaskan peat emissions in their particle phase (with solid bars) and gas phase (with open bars) as an example, while the remaining results are given in the Supplement (Figs. S3 and S4). It is clear that the top compounds vary between fuels, likely because of both the different chemical nature of these fuels and nature of combustion.
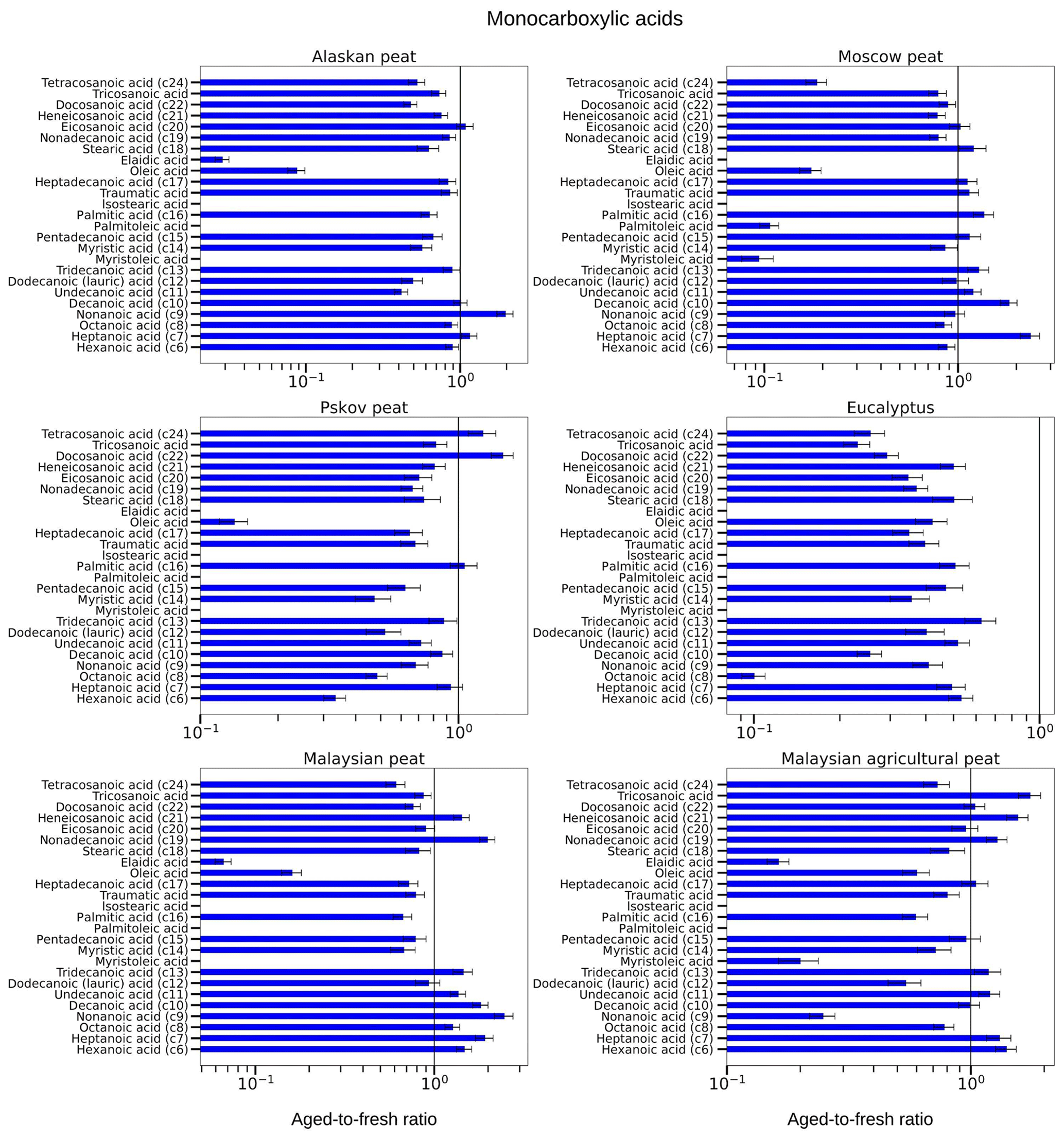
Figure 10Aged-to-fresh ratios of total (gas + particle) EFs for monocarboxylic acids from biomass-burning emissions for six different biomass types presented in log scale. We did not burn fuels in replicates, and the standard-deviation (SD) values were calculated based on replicate analysis of similar fuels (with same experimental conditions) from our previous campaigns. SD values derived from EFs were scaled to ratio.
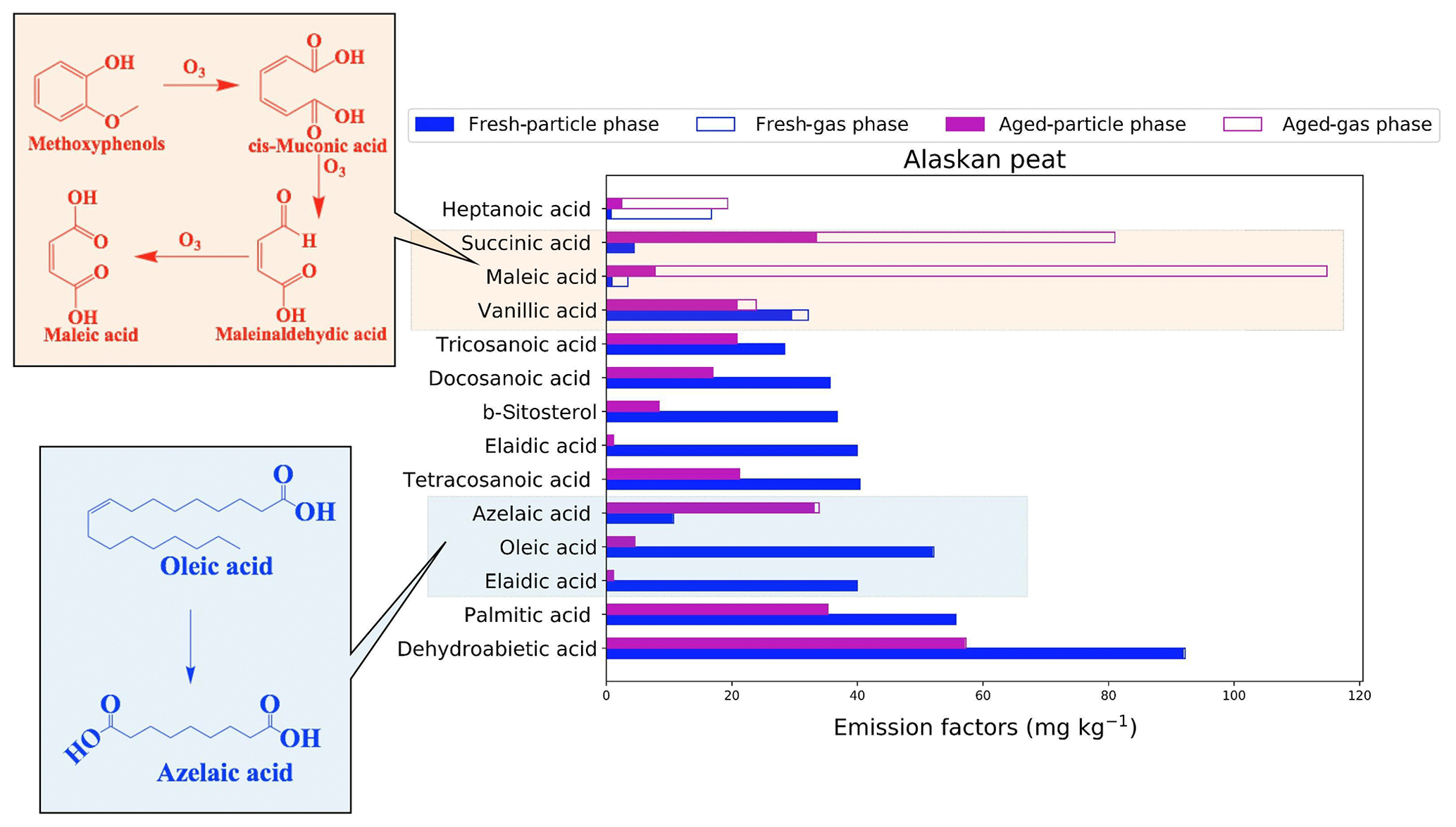
Figure 11Emission factors (EFs) of the top contributing organic compounds assigned to Alaskan peat. The top 10 contributing compounds were selected from both fresh and aged emissions based on EF values. Since fresh and aged samples do not have the same set of compounds after the selection, we included the top 10 compounds for both fresh and aged emissions. Hence, the number of the top contributing compounds varies from one fuel to another. Solid bars of each type represent a chemical group from the particulate emission of BB fuels, and open bars of the same color represent gas-phase BB emissions. The formation of maleic acid is described in Pillar et al. (2014).
Dehydroabietic acid, a resin acid, is the compound with the highest EF (92.2 mg kg−1) for fresh-combustion emissions from Alaskan peat. Monocarboxylic acids including palmitic acid (EF of 55.7 mg kg−1), tetracosanoic acid (EF of 40.35 mg kg−1), and docosanoic acid (EF of 35.38 mg kg−1) were also found in high abundance in fresh emissions from the combustion of this fuel. The high contributions of β-sitosterol (EF of 36.84 mg kg−1) and alkenoic acids (e.g., oleic-acid EF of 52.1 mg kg−1) to emissions are unique to Alaskan peat. All the compounds described above are found in the particle phase. After OFR oxidation, both dehydroabietic acid and β-sitosterol, considered to be potential markers for biomass-burning emissions (Simoneit et al., 1993b), decreased from 91.9 to 57.2 and 36.8 to 8.38 mg kg−1 in the particle phase, respectively. This reduction in EF because of OFR oxidation for both dehydroabietic acid and β-sitosterol must be considered when using these compounds as biomass-burning markers. We observed the formation of low-MW organic compounds, particularly in the gas phase, from OFR oxidation. For example, the EF of heptanoic acid increased from 2.42 to 16.9 mg kg−1 and that of maleic acid increased from 7.8 to 107 mg kg−1 in the gas phase because of OFR oxidation. Such a significant increase in the EF of maleic acid can be explained by the aqueous-phase oxidation of methoxyphenols (El Zein et al., 2015) in the presence of ozone. We found that the oxidation inside the chamber was happening under dry conditions and understand that the reactions of organic compounds with OH radicals inside the OFR chamber will prevail over reactions with ozone. As we had our ozone scrubbers placed after sampling media (Fig. 1) to prevent the pumps and online instruments from ozone-induced damage, we suspect that the maleic acid was not formed inside the OFR chamber but rather by the potential oxidation of organic compounds on filters with a relatively longer exposure of ozone (40–60 min for smoldering combustion). Succinic-acid EFs increased in both the gas phase (from 0.0 to 47.5 mg kg−1) and the particle phase (from 4.43 to 33.5 mg kg−1). Azelaic-acid EFs showed mainly an increase in the particle phase (from 10.5 to 33.1 mg kg−1), and we think that this was because of the oxidation of oleic and eladic acid during OFR oxidation.
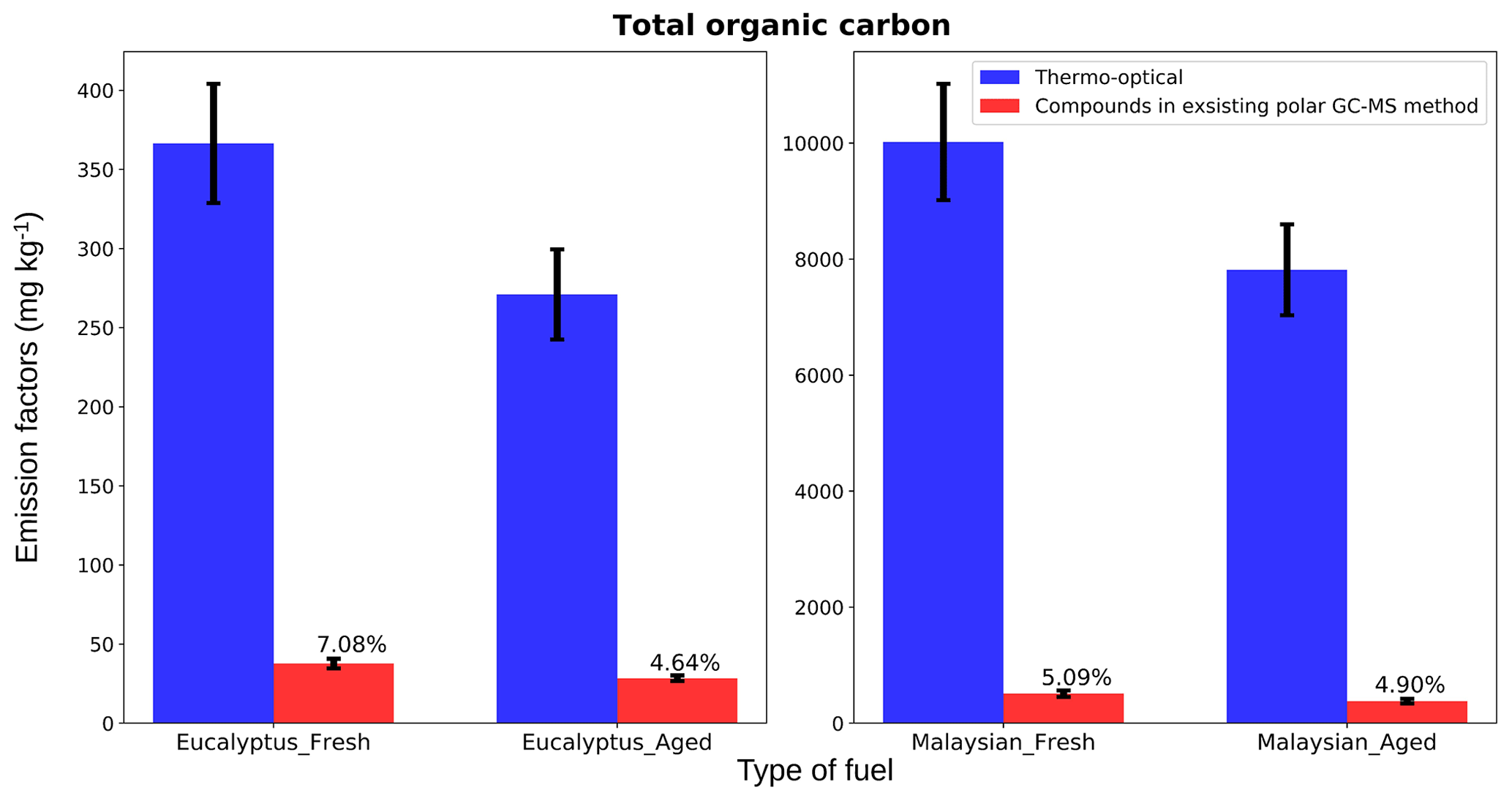
Figure 12Contribution of GC-MS characterized polar compound carbon mass to total thermo-optical organic carbon mass. The y axis shows the total carbon mass with dimensions of mass per mass. The error bars represent the analytical uncertainties of the methods. For the thermo-optical method, uncertainties are the standard deviation of results from multiple punches on the same filter, and for the GC-MS method, uncertainties were computed by taking the square root of the sum of the squares of individual analytical uncertainties of all compounds included.
3.5 Contribution of polar fraction to total organic carbon
For Fig. 12, we calculated the carbon content of total GC-MS characterized mass of identified polar organic compounds and compared results with the total OC mass characterized by the thermo-optical technique to estimate the contribution of polar compounds. The OC emissions were higher for smoldering-combustion (10 209±5 mg kg−1 for Malaysian peat fresh emissions) than for flaming-combustion (366.5±7 mg kg−1 for eucalyptus fresh emissions) samples, similar to the observation of flaming and smoldering combustion by Akagi et al. (2011). Total OC emissions are highly dependent on the type of fuel. For example, the fuel-based OC emission factor for rice crop residue burning is 1960 mg kg−1 (Cao et al., 2008), whereas burning of corn and conifer forest yields emission factors of ∼1457 mg kg−1 (Andreae and Rosenfeld, 2008) and ∼7800 mg kg−1 (Akagi et al., 2011), respectively. Figure 12 shows that the 84 identified polar compounds in our study constituted 4.5 % to 7 % of total OC mass for both fresh and aged emissions. From Indonesian peat combustion emissions, Jayarathne et al. (2018) were able to identify polar compounds that constituted 5.446 % of total organic carbon mass. In recent work, based on both a field campaign with prescribed burning and laboratory investigations, Jen et al. (2019) quantified a fraction (10 %–65 %) of only identified compounds (not a fraction of total mass) by the use of the 2D GC-MS technique. In our work, we identified only up to 7 % (Fig. 12) of the total particle-phase OC, and further analysis of unidentified compounds is needed to improve understanding of atmospheric chemistry of BB emissions.
In this study, we chemically characterized the polar fraction of biomass-burning aerosols from the laboratory combustion of six different globally and regionally important fuels – five of them representing smoldering and one of them representing flaming combustion. Our objective was to understand how emissions of the polar compounds (e.g., methoxyphenols) varied from one fuel to another during these combustion experiments and what the relative distribution of these polar compounds in the gas and particle phases are. We also identified the fates of these polar compounds following laboratory oxidation and aging (OFR aging). Resin acids were found mostly in emissions from the combustion of peats from high-latitude regions but not in emissions of tropical peatlands (e.g., EFgroup of 117±15 mg kg−1 for Alaskan peat and EFgroup of 4.0±0.5 mg kg−1 for Malaysian peat). Similarly, monocarboxylic acids were found in higher abundance in emissions from the combustion of high-latitude peatlands compared to those from tropical peatlands (e.g., EFgroup of 505±36 mg kg−1 for Alaskan peat and EFgroup of 212±15 mg kg−1 for Malaysian agricultural peat). The presence of both guaiacol and syringol moieties in all fuels indicated a part of the biomass, considered as representative of a particular geographical region, is deciduous for all fuels. Low-MW compounds are mostly found in the gas phase (e.g., guaiacol found in the gas phase 82 %–100 %), whereas high-MW (e.g., monocarboxylic acids – >C16 – more than 98 % for all fuels) and highly oxygenated compounds (e.g., syringic acid and acetovanillone 65 %–100 % in the particle phase) are found in the particle phase with high abundance. Monocarboxylic acids (1.2–3 times) and methoxyphenols (1.5–2.5 times) decreased after OFR oxidation, whereas dicarboxylic acids increased by 3–9 times followed by OFR oxidation. Relatively low-MW hexanoic acid increased for both Malaysian (aged-to-fresh ratio of 1.47±0.14) and Malaysian agricultural peat (aged- to-fresh ratio of 1.40±0.14), whereas high-MW tetracosanoic acid (C20) was reduced for all fuel increases (e.g., aged-to-fresh ratio of 0.61±0.07 for Malaysian peat). This indicated fragmentation occurring inside the OFR chamber. With the relative distribution of the top 10–15 compounds from Alaskan peat, we were able to identify transformation of unsaturated fatty acids (e.g., oleic acid) to dicarboxylic acids (e.g., azelaic acid). We identified only up to 7 % of the total particle-phase OC, and further analysis of unidentified compounds with a GC-MS full scan is needed for a better understanding of the atmospheric chemistry of BB emissions.
Data can be provided upon request to andrey.khlystov@dri.edu.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-8227-2020-supplement.
DS, VS, and AK designed experiments. DS and CB performed sample and data collection. DS and CB performed extractions, derivatizations, and the GC-MS analysis. DS summarized data and wrote the paper. AW provided biomass fuels. VS, AK, and HM provided input on the interpretation of the results. VS, HM, and AK revised the paper.
The authors declare that they have no conflict of interest.
The authors would like to thank Anna Tsibar (Lomonosov Moscow State University, Moscow, Russia) for providing peat fuels from Russia. We acknowledge Benjamin Nault (CIRES, UC Boulder), William Stockwell (DRI), and Andrew Lambe (Aerodyne) for their insightful discussion leading to the identification of a potential artifact in our experimental setup associated with high maleic-acid formation. The authors also thank Rodger Kreidberg for revising the paper.
This research has been mainly supported by the National Science Foundation, Division of Atmospheric and Geospace Sciences (grant no. 1544425); the National Science Foundation, Division of Atmospheric and Geospace Sciences (grant no. 1408241); and the National Aeronautics and Space Administration (grant no. NNX15AI48G). This research is also partially supported by the internal funding of Division of Atmospheric Sciences, Desert Research Institute.
This paper was edited by Manish Shrivastava and reviewed by two anonymous referees.
Akagi, S. K., Yokelson, R. J., Wiedinmyer, C., Alvarado, M. J., Reid, J. S., Karl, T., Crounse, J. D., and Wennberg, P. O.: Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 11, 4039–4072, https://doi.org/10.5194/acp-11-4039-2011, 2011.
Alvarado, M. J., Lonsdale, C. R., Yokelson, R. J., Akagi, S. K., Coe, H., Craven, J. S., Fischer, E. V., McMeeking, G. R., Seinfeld, J. H., Soni, T., Taylor, J. W., Weise, D. R., and Wold, C. E.: Investigating the links between ozone and organic aerosol chemistry in a biomass burning plume from a prescribed fire in California chaparral, Atmos. Chem. Phys., 15, 6667–6688, https://doi.org/10.5194/acp-15-6667-2015, 2015.
Andreae, M. O. and and Merlet, P.: Emission of trace gases and aerosols from biomass burning, Global Biogeochem. Cy., 15, 955–966, https://doi.org/10.1029/2000GB001382, 2001.
Andreae, M. O. and Rosenfeld, D.: Aerosol-cloud-precipitation interactions. Part 1. The nature and sources of cloud-active aerosols, Earth-Sci. Rev., 89, 13–41, https://doi.org/10.1016/j.earscirev.2008.03.001, 2008.
Arbex, M. A., Martins, L. C., Carvalho De Oliveira, R., Pereira, A. A., Arbex, F. F., Eduardo, J., Cançado, D., Hilário, P., Saldiva, N., Luís, A., and Braga, F.: Air pollution from biomass burning and asthma hospital admissions in a sugar cane plantation area in Brazil, J. Epidemiol. Commun. H., 61, 395–400, https://doi.org/10.1136/jech.2005.044743, 2007.
Asher, W. E., Pankow, J. F., Erdakos, G. B., and Seinfeld, J. H.: Estimating the vapor pressures of multi-functional oxygen-containing organic compounds using group contribution methods, Atmos. Environ., 36, 1483–1498, https://doi.org/10.1016/S1352-2310(01)00564-7, 2002.
Beres, N. D., Sengupta, D., Samburova, V., Khlystov, A. Y., and Moosmüller, H.: Deposition of brown carbon onto snow: changes in snow optical and radiative properties, Atmos. Chem. Phys., 20, 6095–6114, https://doi.org/10.5194/acp-20-6095-2020, 2020.
Bertrand, A., Stefenelli, G., Jen, C. N., Pieber, S. M., Bruns, E. A., Ni, H., Temime-Roussel, B., Slowik, J. G., Goldstein, A. H., El Haddad, I., Baltensperger, U., Prévôt, A. S. H., Wortham, H., and Marchand, N.: Evolution of the chemical fingerprint of biomass burning organic aerosol during aging, Atmos. Chem. Phys., 18, 7607–7624, https://doi.org/10.5194/acp-18-7607-2018, 2018.
Bhattarai, C., Samburova, V., Sengupta, D., Iaukea-Lum, M., Watts, A. C., Moosmüller, H., and Khlystov, A. Y.: Physical and chemical characterization of aerosol in fresh and aged emissions from open combustion of biomass fuels, Aerosol Sci. Technol., 52, 1266–1282, https://doi.org/10.1080/02786826.2018.1498585, 2018.
Bhattarai, H., Saikawa, E., Wan, X., Zhu, H., Ram, K., Gao, S., Kang, S., Zhang, Q., Zhang, Y., Wu, G., Wang, X., Kawamura, K., Fu, P., and Cong, Z.: Levoglucosan as a tracer of biomass burning: Recent progress and perspectives, Atmos. Res., 220, 20–33, https://doi.org/10.1016/j.atmosres.2019.01.004, 2019.
Bonvalot, L., Tuna, T., Fagault, Y., Jaffrezo, J.-L., Jacob, V., Chevrier, F., and Bard, E.: Estimating contributions from biomass burning, fossil fuel combustion, and biogenic carbon to carbonaceous aerosols in the Valley of Chamonix: a dual approach based on radiocarbon and levoglucosan, Atmos. Chem. Phys., 16, 13753–13772, https://doi.org/10.5194/acp-16-13753-2016, 2016.
Cao, F., Zhang, S. C., Kawamura, K., Liu, X., Yang, C., Xu, Z., Fan, M., Zhang, W., Bao, M., Chang, Y., Song, W., Liu, S., Lee, X., Li, J., Zhang, G., and Zhang, Y. L.: Chemical characteristics of dicarboxylic acids and related organic compounds in PM2.5 during biomass-burning and non-biomass-burning seasons at a rural site of Northeast China, Environ. Pollut., 231, 654–662, https://doi.org/10.1016/j.envpol.2017.08.045, 2017.
Cao, G., Zhang, X., Gong, S., and Zheng, F.: Investigation on emission factors of particulate matter and gaseous pollutants from crop residue burning, J. Environ. Sci., 20, 50–55, https://doi.org/10.1016/S1001-0742(08)60007-8, 2008.
Carlton, A. G., Turpin, B. J., Altieri, K. E., Seitzinger, S., Reff, A., Lim, H. J., and Ervens, B.: Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments, Atmos. Environ., 41, 7588–7602, https://doi.org/10.1016/j.atmosenv.2007.05.035, 2007.
Chakrabarty, R. K., Gyawali, M., Yatavelli, R. L. N., Pandey, A., Watts, A. C., Knue, J., Chen, L.-W. A., Pattison, R. R., Tsibart, A., Samburova, V., and Moosmüller, H.: Brown carbon aerosols from burning of boreal peatlands: microphysical properties, emission factors, and implications for direct radiative forcing, Atmos. Chem. Phys., 16, 3033–3040, https://doi.org/10.5194/acp-16-3033-2016, 2016.
Charbouillot, T., Gorini, S., Voyard, G., Parazols, M., Brigante, M., Deguillaume, L., Delort, A. M., and Mailhot, G.: Mechanism of carboxylic acid photooxidation in atmospheric aqueous phase: Formation, fate and reactivity, Atmos. Environ., 56, 1–8, https://doi.org/10.1016/j.atmosenv.2012.03.079, 2012.
Chen, J., Li, C., Ristovski, Z., Milic, A., Gu, Y., Islam, M. S., Wang, S., Hao, J., Zhang, H., He, C., Guo, H., Fu, H., Miljevic, B., Morawska, L., Thai, P., LAM, Y. F., Pereira, G., Ding, A., Huang, X., and Dumka, U. C.: A review of biomass burning: Emissions and impacts on air quality, health and climate in China, Sci. Total Environ., 579, 1000–1034, https://doi.org/10.1016/j.scitotenv.2016.11.025, 2017.
Chow, J. C. and Watson, J. G.: Enhanced Ion Chromatographic Speciation of Water-Soluble PM2.5 to Improve Aerosol Source Apportionment, Aerosol Sci. Eng., 1, 7–24, https://doi.org/10.1007/s41810-017-0002-4, 2017.
Chow, J. C., Watson, J. G., Pritchett, L. C., Pierson, W. R., Frazier, C. A., and Purcell, R. G.: The DRI thermal optical reflectance carbon analysis system - description, evaluation and applications in United-States air quality studies, Atmos. Environ. A-Gen., 27, 1185–1201, 1993.
Chow, J. C., Watson, J. G., Chen, L. W. A., Arnott, W. P., Moosmüller, H., and Fung, K.: Equivalence of elemental carbon by thermal/optical reflectance and transmittance with different temperature protocols, Environ. Sci. Technol., 38, 4414–4422, https://doi.org/10.1021/es034936u, 2004.
Decker, Z. C. J., Zarzana, K. J., Coggon, M., Min, K. E., Pollack, I., Ryerson, T. B., Peischl, J., Edwards, P., Dubé, W. P., Markovic, M. Z., Roberts, J. M., Veres, P. R., Graus, M., Warneke, C., De Gouw, J., Hatch, L. E., Barsanti, K. C., and Brown, S. S.: Nighttime Chemical Transformation in Biomass Burning Plumes: A Box Model Analysis Initialized with Aircraft Observations, Environ. Sci. Technol., 53, 2529–2538, https://doi.org/10.1021/acs.est.8b05359, 2019.
Dills, R. L., Paulsen, M., Ahmad, J., Kalman, D. A., Elias, F. N., and Simpson, C. D.: Evaluation of urinary methoxyphenols as biomarkers of woodsmoke exposure, Environ. Sci. Technol., 40, 2163–2170, https://doi.org/10.1021/es051886f, 2006.
El Zein, A., Coeur, C., Obeid, E., Lauraguais, A., and Fagniez, T.: Reaction Kinetics of Catechol (1,2-Benzenediol) and Guaiacol (2-Methoxyphenol) with Ozone, J. Phys. Chem. A, 119, 6759–6765, https://doi.org/10.1021/acs.jpca.5b00174, 2015.
Ervens, B., Feingold, G., Frost, G. J., and Kreidenweis, S. M.: A modeling of study of aqueous production of dicarboxylic acids: 1. Chemical pathways and speciated organic mass production, J. Geophys. Res., 109, D15205, https://doi.org/10.1029/2003JD004387, 2004.
Fang, M., Zheng, M., Wang, F., To, K. L., Jaafar, A. B., and Tong, S. L.: The solvent-extractable organic compounds in the Indonesia biomass burning aerosols - Characterization studies, Atmos. Environ., 33, 783–795, https://doi.org/10.1016/S1352-2310(98)00210-6, 1999.
Fine, P. M., Cass, G. R., and Simoneit, B. R. T.: Organic compounds in biomass smoke from residential wood combustion: Emissions characterization at a continental scale, J. Geophys. Res., 107, 8349, https://doi.org/10.1029/2001JD000661, 2002.
Finlayson-Pitts, B. J. and Pitts Jr., J. N.: Chemistry of the upper and lower atmosphere: theory, experiments, and applications, Elsevier, San Diego, California, 1999.
Fortenberry, C. F., Walker, M. J., Zhang, Y., Mitroo, D., Brune, W. H., and Williams, B. J.: Bulk and molecular-level characterization of laboratory-aged biomass burning organic aerosol from oak leaf and heartwood fuels, Atmos. Chem. Phys., 18, 2199–2224, https://doi.org/10.5194/acp-18-2199-2018, 2018.
Freimuth, E. J., Diefendorf, A. F., Lowell, T. V., and Wiles, G. C.: Sedimentary n-alkanes and n-alkanoic acids in a temperate bog are biased toward woody plants, Org. Geochem., 128, 94–107, https://doi.org/10.1016/j.orggeochem.2019.01.006, 2019.
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Goodrick, S. L. and Stanturf, J. A.: Evaluating Potential Changes in Fire Risk from Eucalyptus Plantings in the Southern United States, International Journal of Forestry Research, 2012, 9 pp., https://doi.org/10.1155/2012/680246, 2012.
Graham, B., Mayol-Bracero, O. L., Guyon, P., Roberts, G. C., Decesari, S., Facchini, M. C., Artaxo, P., Maenhaut, W., Köll, P., and Andreae, M. O.: Water-soluble organic compounds in biomass burning aerosols over Amazonia 1. Characterization by NMR and GC-MS, J. Geophys. Res., 107, LBA 14-1–LBA 14-16, https://doi.org/10.1029/2001JD000336, 2002.
Grieshop, A. P., Donahue, N. M., and Robinson, A. L.: Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 2: analysis of aerosol mass spectrometer data, Atmos. Chem. Phys., 9, 2227–2240, https://doi.org/10.5194/acp-9-2227-2009, 2009.
Harden, J., Trumbore, S., Stocks, B., Hirsch, A., Gower, S., O'neill, K., and Kasischke, E.: The role of fire in the boreal carbon budget, Glob. Change Biol., 6, 174–184, https://doi.org/10.1046/j.1365-2486.2000.06019.x, 2000.
Hawthorne, S. B., Krieger, M. S., Miller, D. J., and Mathiason, M. B.: Collection and Quantitation of Methoxylated Phenol Tracers for Atmospheric Pollution from Residential Wood Stoves, Environ. Sci. Technol., 23, 470–475, https://doi.org/10.1021/es00181a013, 1989.
Hedges, J. I. and Ertel, J. R.: Characterization of Lignin by Gas Capillary Chromatography of Cupric Oxide Oxidation Products, Anal. Chem., 54, 174–178, https://doi.org/10.1021/ac00239a007, 1982.
Hennigan, C. J., Sullivan, A. P., Collett, J. L., and Robinson, A. L.: Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals, Geophys. Res. Lett., 37, L09806, https://doi.org/10.1029/2010GL043088, 2010.
Hills, W. E. and Brown, A. G.: Eucalypts for wood production, CSIRO, Canberra, 1978.
Hoffmann, D., Tilgner, A., Iinuma, Y., and Herrmann, H.: Atmospheric stability of levoglucosan: A detailed laboratory and modeling study, Environ. Sci. Technol., 44, 694–699, https://doi.org/10.1021/es902476f, 2010.
Iinuma, Y., Brüggemann, E., Gnauk, T., Müller, K., Andreae, M. O., Helas, G., Parmar, R., and Herrmann, H.: Source characterization of biomass burning particles: The combustion of selected European conifers, African hardwood, savanna grass, and German and Indonesian peat, J. Geophys. Res., 112, D08209, https://doi.org/10.1029/2006JD007120, 2007.
Jayarathne, T., Stockwell, C. E., Gilbert, A. A., Daugherty, K., Cochrane, M. A., Ryan, K. C., Putra, E. I., Saharjo, B. H., Nurhayati, A. D., Albar, I., Yokelson, R. J., and Stone, E. A.: Chemical characterization of fine particulate matter emitted by peat fires in Central Kalimantan, Indonesia, during the 2015 El Niño, Atmos. Chem. Phys., 18, 2585–2600, https://doi.org/10.5194/acp-18-2585-2018, 2018.
Jen, C. N., Hatch, L. E., Selimovic, V., Yokelson, R. J., Weber, R., Fernandez, A. E., Kreisberg, N. M., Barsanti, K. C., and Goldstein, A. H.: Speciated and total emission factors of particulate organics from burning western US wildland fuels and their dependence on combustion efficiency, Atmos. Chem. Phys., 19, 1013–1026, https://doi.org/10.5194/acp-19-1013-2019, 2019.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jung, J., Lyu, Y., Lee, M., Hwang, T., Lee, S., and Oh, S.: Impact of Siberian forest fires on the atmosphere over the Korean Peninsula during summer 2014, Atmos. Chem. Phys., 16, 6757–6770, https://doi.org/10.5194/acp-16-6757-2016, 2016.
Karlberg, A.-T., Boman, A., Hacksell, U., Jacobsson, S., and Nilsson, J. L. G.: Contact allergy to dehydroabietic acid derivatives isolated from Portuguese colophony, Contact Dermatitis, 19, 166–174, 1988.
Kawamura, K. and Bikkina, S.: A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation, Atmos. Res., 170, 140–160, https://doi.org/10.1016/j.atmosres.2015.11.018, 2016.
Kessler, S. H., Smith, J. D., Che, D. L., Worsnop, D. R., Wilson, K. R., and Kroll, J. H.: Chemical Sinks of Organic Aerosol: Kinetics and Products of the Heterogeneous Oxidation of Erythritol and Levoglucosan, Environ. Sci. Technol., 44, 7005–7010, https://doi.org/10.1021/es101465m, 2010.
Kundu, S., Kawamura, K., Andreae, T. W., Hoffer, A., and Andreae, M. O.: Molecular distributions of dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in biomass burning aerosols: implications for photochemical production and degradation in smoke layers, Atmos. Chem. Phys., 10, 2209–2225, https://doi.org/10.5194/acp-10-2209-2010, 2010.
Legrand, M., McConnell, J., Fischer, H., Wolff, E. W., Preunkert, S., Arienzo, M., Chellman, N., Leuenberger, D., Maselli, O., Place, P., Sigl, M., Schüpbach, S., and Flannigan, M.: Boreal fire records in Northern Hemisphere ice cores: a review, Clim. Past, 12, 2033–2059, https://doi.org/10.5194/cp-12-2033-2016, 2016.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., de Gouw, J. A., and Jimenez, J. L.: Modeling the Radical Chemistry in an Oxidation Flow Reactor: Radical Formation and Recycling, Sensitivities, and the OH Exposure Estimation Equation, J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Liu, X., Huey, L. G., Yokelson, R. J., Selimovic, V., Simpson, I. J., Müller, M., Jimenez, J. L., Campuzano-Jost, P., Beyersdorf, A. J., Blake, D. R., Butterfield, Z., Choi, Y., Crounse, J. D., Day, D. A., Diskin, G. S., Dubey, M. K., Fortner, E., Hanisco, T. F., Hu, W., King, L. E., Kleinman, L., Meinardi, S., Mikoviny, T., Onasch, T. B., Palm, B. B., Peischl, J., Pollack, I. B., Ryerson, T. B., Sachse, G. W., Sedlacek, A. J., Shilling, J. E., Springston, S., St. Clair, J. M., Tanner, D. J., Teng, A. P., Wennberg, P. O., Wisthaler, A., and Wolfe, G. M.: Airborne measurements of western U.S. wildfire emissions: Comparison with prescribed burning and air quality implications, J. Geophys. Res., 122, 6108–6129, https://doi.org/10.1002/2016JD026315, 2017.
Maenhaut, W., Vermeylen, R., Claeys, M., Vercauteren, J., and Roekens, E.: Sources of the PM10 aerosol in Flanders, Belgium, and re-assessment of the contribution from wood burning, Sci. Total Environ., 562, 550–560, https://doi.org/10.1016/j.scitotenv.2016.04.074, 2016.
Mazzoleni, L. R., Zielinska, B., and Moosmüller, H.: Emissions of levoglucosan, methoxy phenols, and organic acids from prescribed burns, laboratory combustion of wildland fuels, and residential wood combustion, Environ. Sci. Technol., 41, 2115–2122, https://doi.org/10.1021/es061702c, 2007.
Müller-Tautges, C., Eichler, A., Schwikowski, M., Pezzatti, G. B., Conedera, M., and Hoffmann, T.: Historic records of organic compounds from a high Alpine glacier: influences of biomass burning, anthropogenic emissions, and dust transport, Atmos. Chem. Phys., 16, 1029–1043, https://doi.org/10.5194/acp-16-1029-2016, 2016.
Net, S., Alvarez, E. G., Gligorovski, S., and Wortham, H.: Heterogeneous reactions of ozone with methoxyphenols, in presence and absence of light, Atmos. Environ., 45, 3007–3014, https://doi.org/10.1016/j.atmosenv.2011.03.026, 2011.
Oros, D. R. and Simoneit, B. R. T.: Identification and emission factors of molecular tracers in organic aerosols from biomass burning Part 1. Temperate climate conifers, Appl. Geochem., 16, 1513–1544, https://doi.org/10.1016/S0883-2927(01)00021-X, 2001a.
Oros, D. R. and Simoneit, B. R. T.: Identification and emission factors of molecular tracers in organic aerosols from biomass burning Part 2. Deciduous trees, Appl. Geochem., 16, 1545–1565, https://doi.org/10.1016/S0883-2927(01)00022-1, 2001b.
Oros, D. R., Abas, M. R. B., Omar, N. Y. M. J., Rahman, N. A., and Simoneit, B. R. T.: Identification and emission factors of molecular tracers in organic aerosols from biomass burning: Part 3. Grasses, Appl. Geochem., 21, 919–940, https://doi.org/10.1016/j.apgeochem.2006.01.008, 2006.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Pardo, M., Li, C., He, Q., Levin-Zaidman, S., Tsoory, M., Yu, Q., Wang, X., and Rudich, Y.: Mechanisms of lung toxicity induced by biomass burning aerosols, Part. Fibre Toxicol., 17, 4, https://doi.org/10.1186/s12989-020-0337-x, 2020.
Park, R. J., Jacob, D. J., and Logan, J. A.: Fire and biofuel contributions to annual mean aerosol mass concentrations in the United States, Atmos. Environ., 41, 7389–7400, https://doi.org/10.1016/j.atmosenv.2007.05.061, 2007.
Pavagadhi, S., Betha, R., Venkatesan, S., Balasubramanian, R., and Hande, M. P.: Physicochemical and toxicological characteristics of urban aerosols during a recent Indonesian biomass burning episode, Environ. Sci. Pollut. R., 20, 2569–2578, https://doi.org/10.1007/s11356-012-1157-9, 2013.
Penner, J. E., Ghan, S. J., and Walton, J. J.: The role of biomass burning in the budget and cycle of carbonaceous soot aerosols and their climate impact, available at: https://inis.iaea.org/search/search.aspx?orig_q=RN:23067068 (last access: 8 September 2019), 1991.
Pillar, E. A., Camm, R. C., and Guzman, M. I.: Catechol oxidation by ozone and hydroxyl radicals at the air-water interface, Environ. Sci. Technol., 48(24), 14352–14360, https://doi.org/10.1021/es504094x, 2014.
Ramanathan, V. and Carmichael, G.: Global and regional climate changes due to black carbon, Nat. Geosci., 1, 221–227, https://doi.org/10.1038/ngeo156, 2008.
Regalado, J., Pérez-Padilla, R., Sansores, R., Ramirez, J. I. P., Brauer, M., Paré, P., and Vedal, S.: The effect of biomass burning on respiratory symptoms and lung function in rural Mexican women, Am. J. Resp. Crit. Care, 174, 901–905, https://doi.org/10.1164/rccm.200503-479OC, 2006.
Rinehart, L. R., Fujita, E. M., Chow, J. C., Magliano, K., and Zielinska, B.: Spatial distribution of PM2.5 associated organic compounds in central California, Atmos. Environ., 40, 290–303, https://doi.org/10.1016/j.atmosenv.2005.09.035, 2006.
Sadhra, S., Foulds, I. S., and Gray, C. N.: Oxidation of resin acids in colophony (rosin) and its implications for patch testing, Contact Dermatitis, 39, 58–63, https://doi.org/10.1111/j.1600-0536.1998.tb05833.x, 1998.
Samburova, V., Hallar, A. G., Mazzoleni, L. R., Saranjampour, P., Lowenthal, D., Kohl, S. D., and Zielinska, B.: Composition of water-soluble organic carbon in non-urban atmospheric aerosol collected at the Storm Peak Laboratory, Environ. Chem., 10, 370–380, https://doi.org/10.1071/EN13079, 2013.
Samburova, V., Connolly, J., Gyawali, M., Yatavelli, R. L. N., Watts, A. C., Chakrabarty, R. K., Zielinska, B., Moosmüller, H., and Khlystov, A.: Polycyclic aromatic hydrocarbons in biomass-burning emissions and their contribution to light absorption and aerosol toxicity, Sci. Total Environ., 568, 391–401, https://doi.org/10.1016/j.scitotenv.2016.06.026, 2016.
Sarkanen, K. V. and Ludwig, C. H.: Lignins: occurrence, formation, structure and reactions, Wiley-interscience, New York, 1971.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources. 3. C1–C29 organic compounds from fireplace combustion of wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Schmidl, C., Marr, I. L., Caseiro, A., Kotianová, P., Berner, A., Bauer, H., Kasper-Giebl, A., and Puxbaum, H.: Chemical characterisation of fine particle emissions from wood stove combustion of common woods growing in mid-European Alpine regions, Atmos. Environ., 42, 126–141, https://doi.org/10.1016/j.atmosenv.2007.09.028, 2008a.
Schmidl, C., Bauer, H., Dattler, A., Hitzenberger, R., Weissenboeck, G., Marr, I. L., and Puxbaum, H.: Chemical characterisation of particle emissions from burning leaves, Atmos. Environ., 42, 9070–9079, https://doi.org/10.1016/j.atmosenv.2008.09.010, 2008b.
Schnitzler, E. G. and Abbatt, J. P. D.: Heterogeneous OH oxidation of secondary brown carbon aerosol, Atmos. Chem. Phys., 18, 14539–14553, https://doi.org/10.5194/acp-18-14539-2018, 2018.
Sengupta, D., Samburova, V., Bhattarai, C., Kirillova, E., Mazzoleni, L., Iaukea-Lum, M., Watts, A., Moosmüller, H., and Khlystov, A.: Light absorption by polar and non-polar aerosol compounds from laboratory biomass combustion, Atmos. Chem. Phys., 18, 10849–10867, https://doi.org/10.5194/acp-18-10849-2018, 2018.
Sigsgaard, T., Forsberg, B., Annesi-Maesano, I., Blomberg, A., Bølling, A., Boman, C., Bønløkke, J., Brauer, M., Bruce, N., Héroux, M. E., Hirvonen, M. R., Kelly, F., Künzli, N., Lundbäck, B., Moshammer, H., Noonan, C., Pagels, J., Sallsten, G., Sculier, J. P., and Brunekreef, B.: Health impacts of anthropogenic biomass burning in the developed world, Eur. Respir. J., 46, 1577–1588, https://doi.org/10.1183/13993003.01865-2014, 2015.
Simoneit, B. R. T.: Biomass burning – a review of organic tracers for smoke from incomplete combustion, Appl. Geochem., 17, 129–162, https://doi.org/10.1016/S0883-2927(01)00061-0, 2002.
Simoneit, B. R. T., Rogge, W. F., Mazurek, M. A., Standley, L. J., Hildemann, L. M., and Cass, G. R.: Lignin pyrolysis products, lignans, and resin acids as specific tracers of plant classes in emissions from biomass combustion, Environ. Sci. Technol., 27, 2533–2541, https://doi.org/10.1021/es00048a034, 1993.
Simoneit, B. R. T., Schauer, J. J., Nolte, C. G., Oros, D. R., Elias, V. O., Fraser, M. P., Rogge, W. F., and Cass, G. R.: Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles, Atmos. Environ., 33, 173–182, https://doi.org/10.1016/S1352-2310(98)00145-9, 1999.
Simpson, C. D. and Naeher, L. P.: Biological monitoring of wood-smoke exposure, Inhal. Toxicol., 22, 99–103, https://doi.org/10.3109/08958370903008862, 2010.
Tan, Y., Lim, Y. B., Altieri, K. E., Seitzinger, S. P., and Turpin, B. J.: Mechanisms leading to oligomers and SOA through aqueous photooxidation: insights from OH radical oxidation of acetic acid and methylglyoxal, Atmos. Chem. Phys., 12, 801–813, https://doi.org/10.5194/acp-12-801-2012, 2012.
Tian, J., Chow, J. C., Cao, J., Han, Y., Ni, H., Chen, L. A., Wang, X., Huang, R., Moosmüller, H., and Watson, J. G.: A Biomass Combustion Chamber: Design, Evaluation, and a Case Study of Wheat Straw Combustion Emission Tests, Aerosol Air Qual. Res., 15, 2104–2114, https://doi.org/10.4209/aaqr.2015.03.0167, 2015.
Turetsky, M. R., Benscoter, B., Page, S., Rein, G., Van Der Werf, G. R., and Watts, A.: Global vulnerability of peatlands to fire and carbon loss, Nat. Geosci., 8, 11–14, https://doi.org/10.1038/ngeo2325, 2015.
Wan, X., Kawamura, K., Ram, K., Kang, S., Loewen, M., Gao, S., Wu, G., Fu, P., Zhang, Y., Bhattarai, H., and Cong, Z.: Aromatic acids as biomass-burning tracers in atmospheric aerosols and ice cores: A review, Environ. Pollut., 247, 216–228, https://doi.org/10.1016/j.envpol.2019.01.028, 2019.
Watts, A. C., Schmidt, C. A., McLaughlin, D. L., and Kaplan, D. A.: Hydrologic implications of smoldering fires in wetland landscapes, Freshw. Sci., 34, 1394–1405, https://doi.org/10.1086/683484, 2015.
Watts, A. C., Samburova, V., and H. Moosmüller, H.: Criteria-Based Identification of Important Fuels for Wildland Fire Emission Research, Atmosphere, 11, 640, https://doi.org/10.3390/atmos11060640, 2020.
Yang, X. Y., Igarashi, K., Tang, N., Lin, J. M., Wang, W., Kameda, T., Toriba, A., and Hayakawa, K.: Indirect- and direct-acting mutagenicity of diesel, coal and wood burning-derived particulates and contribution of polycyclic aromatic hydrocarbons and nitropolycyclic aromatic hydrocarbons, Mutat. Res.-Gen. Tox. En., 695, 29–34, https://doi.org/10.1016/j.mrgentox.2009.10.010, 2010.
Yatavelli, R. L. N., Chen, L.-W. A., Knue, J., Samburova, V., Gyawali, M., Watts, A. C., Chakrabarty, R. K., Moosmüller, H., Hodzic, A., Wang, X., Zielinska, B., Chow, J. C., and Watson, J. G.: Emissions and Partitioning of Intermediate-Volatility and Semi-Volatile Polar Organic Compounds (I/SV-POCs) During Laboratory Combustion of Boreal and Sub-Tropical Peat, Aerosol Science and Engineering, 1, 25–32, https://doi.org/10.1007/s41810-017-0001-5, 2017.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Yokelson, R. J., Bertschi, I. T., Christian, T. J., Hobbs, P. V., Ward, D. E., and Hao, W. M.: Trace gas measurements in nascent, aged, and cloud-processed smoke from African savanna fires by airborne Fourier transform infrared spectroscopy (AFTIR), J. Geophys. Res., 108, 8478, https://doi.org/10.1029/2002JD002322, 2003.
Zhu, Y., Yang, L., Chen, J., Kawamura, K., Sato, M., Tilgner, A., van Pinxteren, D., Chen, Y., Xue, L., Wang, X., Simpson, I. J., Herrmann, H., Blake, D. R., and Wang, W.: Molecular distributions of dicarboxylic acids, oxocarboxylic acids and α-dicarbonyls in PM2.5 collected at the top of Mt. Tai, North China, during the wheat burning season of 2014, Atmos. Chem. Phys., 18, 10741–10758, https://doi.org/10.5194/acp-18-10741-2018, 2018.