the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Jun 2020
| 12 Jun 2020
MAX-DOAS measurements of NO2, SO2, HCHO, and BrO at the Mt. Waliguan WMO GAW global baseline station in the Tibetan Plateau
Steffen Dörner
Sebastian Donner
Junli Jin
Siyang Cheng
Junrang Guo
Zhanfeng Zhang
Jianqiong Wang
Peng Liu
Guoqing Zhang
Janis Pukite
Johannes Lampel
Mt. Waliguan Observatory (WLG) is a World Meteorological Organization (WMO) Global Atmosphere Watch (GAW) global baseline station in China. WLG is located at the northeastern part of the Tibetan Plateau (36∘17′ N, 100∘54′ E, 3816 m a.s.l.) and is representative of the pristine atmosphere over the Eurasian continent. We made long-term ground-based multi-axis differential optical absorption spectroscopy (MAX-DOAS) measurements at WLG during the period 2012–2015. In this study, we retrieve the differential slant column densities (dSCDs) and estimate the tropospheric background mixing ratios of different trace gases, including NO2, SO2, HCHO, and BrO, using the measured spectra at WLG. Averaging of 10 original spectra is found to be an “optimum option” for reducing both the statistical error of the spectral retrieval and systematic errors in the analysis. The dSCDs of NO2, SO2, HCHO, and BrO under clear-sky and low-aerosol-load conditions are extracted from measured spectra at different elevation angles at WLG. By performing radiative transfer simulations with the model TRACY-2, we establish approximate relationships between the trace gas dSCDs at 1∘ elevation angle and the corresponding average tropospheric background volume mixing ratios. Mixing ratios of these trace gases in the lower troposphere over WLG are estimated to be in a range of about 7 ppt (January) to 100 ppt (May) for NO2, below 0.5 ppb for SO2, between 0.4 and 0.9 ppb for HCHO, and lower than 0.3 ppt for BrO. The chemical box model simulations constrained by the NO2 concentration from our MAX-DOAS measurements show that there is a little net ozone loss (−0.8 ppb d−1) for the free-tropospheric conditions and a little net ozone production (0.3 ppb d−1) for the boundary layer conditions over WLG during summertime. Our study provides valuable information and data sets for further investigating tropospheric chemistry in the background atmosphere and its links to anthropogenic activities.
- Article
(5015 KB) - Full-text XML
-
Supplement
(2073 KB) - BibTeX
- EndNote




Nitrogen oxides (), sulfur dioxide (SO2), formaldehyde (HCHO), and bromine monoxide (BrO) are important traces gases in tropospheric chemistry. Both NOx and HCHO participate in the control of the strong oxidant O3, which is an indicator of photochemical smog, and the strongest atmospheric oxidizing agent OH, which determines the lifetimes of many gaseous pollutants and greenhouse gases in the atmosphere (Seinfeld and Pandis, 2006; Ma et al., 2012; Lelieveld et al., 2016). NOx and SO2 are gaseous precursors of nitrate and sulfate aerosols, and large amounts of these aerosols can result in haze pollution and exert a strong negative radiative forcing on climate change (Seinfeld and Pandis, 2006; Forster et al., 2007; Ma et al., 2010). NOx and SO2 are released from various anthropogenic emission sources, e.g., the burning of coal, oil, gas, wood, and straw (Granier et al., 2011; Zhao et al., 2012). NOx is also emitted via natural processes including lightning and microbial activities in soils (Lee et al., 1997). Nitric oxide (NO) dominates NOx released from these sources, but it can be quickly converted to nitrogen dioxide (NO2) by reaction with ozone (O3) in the atmosphere. Natural sources of SO2 in the troposphere include volcanic eruptions and the atmospheric oxidation of dimethyl sulfide (DMS: CH3SCH3) emitted from the ocean (Dentener et al., 2006). HCHO in the remote atmosphere is produced through the oxidation of methane (CH4) and non-methane volatile organic compounds (NMVOCs), and it is also emitted from anthropogenic combustion processes, biomass burning, and natural vegetation (Stavrakou et al., 2009).
Global emissions and atmospheric abundance of gaseous pollutants (e.g., NOx and SO2) and aerosols have changed significantly over the past few decades as revealed predominantly by satellite observations (e.g., De Smedt et al., 2015; Xing et al., 2015; Bauwens et al., 2016; Fioletov et al., 2016; Krotkov et al., 2016; Klimont et al., 2017; Li et al., 2017; Georgoulias et al., 2019; Hammer et al., 2018; Ziemke et al., 2019). Worldwide ground-based monitoring of the concentrations and trends of trace gases and aerosols in the atmosphere is essential for the validation of and filling gaps in satellite observations, in order to quantitatively assess the impacts of atmospheric composition change on global air quality and climate changes (Stohl et al., 2015; De Mazière et al., 2018). The international global measurement networks have been set up sequentially over the past decades to establish long-term databases for detecting changes and trends in the chemical and physical state of the atmosphere. Among the networks are the Global Atmosphere Watch (GAW) program of the World Meteorological Organization (WMO) and the Network for the Detection of Atmospheric Composition Change (NDACC), and in the latter measurements are performed mainly by ground-based remote-sensing techniques (De Mazière et al., 2018). In contrast to Europe and North America, the stations under the networks in Asia, especially in the remote areas, are very sparse.
The China Global Atmosphere Watch Baseline Observatory at Mt. Waliguan (WLG) is an in-land GAW baseline station affiliated with the WMO. The site (3816 m a.s.l.), located at the northeastern part of the Tibetan Plateau, is representative of the pristine atmosphere over the Eurasian continent. Air masses at WLG are highly representative of the remote free troposphere (Ma et al., 2002a). Previous model simulations constrained by measured mixing ratios of ozone and its precursors indicated a net destruction of ozone at WLG in the summertime of 1996 (Ma et al., 2002a). In contrast, new insights from model calculations based on more recent measurements showed that ozone was net produced by in situ photochemistry at WLG in late spring and summer of 2003 (Xue et al., 2013). The level of NOx plays a key role in determining the sign of net ozone production in the remote troposphere. However, it is not determined whether the difference in the estimated net ozone production at WLG between the two studies is caused by increasing NOx from 1996 to 2003 or by the uncertainties in the measurements of NOx. Similar to NO2, other reactive gases (e.g., SO2 and HCHO) were also measured by filter or canister sampling methods at WLG at irregular times (Mu et al., 2007; Meng et al., 2010; Lin et al., 2013). It is necessary to start a new measurement program with advanced techniques at WLG for the purpose of precisely monitoring the levels and trends of atmospheric composition in the global pristine atmosphere.
Multi-axis differential optical absorption spectroscopy (MAX-DOAS) has the potential to retrieve the vertical distributions of trace gases and aerosols in the immediate vicinity of the station from the scattered sunlight measured at multiple elevation angles (Hönninger and Platt, 2002; Bobrowski et al., 2003; Van Roozendael et al., 2003; Hönninger et al., 2004; Wittrock et al., 2004). As relatively simple and cheap ground-based instrumentation, UV–visible MAX-DOAS will be included in the certified NDACC measurement techniques for the observation of lower-tropospheric NO2, HCHO, and O3 (De Mazière et al., 2018). Successful measurement and retrieval of trace gases (e.g., NO2, SO2, and HCHO) depend on various factors, including their molecular absorption features and atmospheric abundances as well as the atmospheric visibility and instrumental signal ∕ noise ratio. In contrast to extensive ground-based measurements of NO2, SO2, and HCHO in rural and urban areas worldwide, including highly polluted areas in eastern China (e.g., Ma et al., 2013; Hendrick et al., 2014; Wang et al., 2014, 2017; Jin et al., 2016), measurements of these trace gases by MAX-DOAS in the remote background areas have been very sparse (Gomez et al., 2014; Gil-Ojeda et al., 2015; Schreier et al., 2016). Gomez et al. (2014) proposed a modified geometrical approach (MGA) to estimate long-path-averaged mixing ratios of trace gases from mountain MAX-DOAS measurements. A NO2 level of 20 ppt, which is below the detection limit of the in situ instrumentation, was observed (Gomez et al., 2014). The MAX-DOAS technique has been applied to monitor the absolute column densities and plumes of SO2 from large volcano eruptions (e.g., Lübcke et al., 2016; Tulet et al., 2017), but measuring SO2 in the background free troposphere still remains challenging.
Bromine oxide (BrO) plays an important role in the catalytic destruction of ozone in the remote troposphere (Platt and Hönninger, 2003; von Glasow and Crutzen, 2007). The earliest MAX-DOAS measurements were focused on the retrieval of the mixing ratio levels and vertical profiles of BrO in the boundary layer of Arctic, salt lake, and marine areas (Hönninger and Platt, 2002; Stutz et al., 2002; Leser et al., 2003; Frieß et al., 2004; Saiz-Lopez et al., 2004). Measurement results showed that the BrO mixing ratio could reach up to 30 ppt in the Arctic and 10 ppt in the marine boundary layer (Platt and Hönninger, 2003; Martin et al., 2009). BrO in the free troposphere at the global scale was estimated to be at a level of 0.5–2 ppt based on spaceborne, ground-based, and sounding measurements with the DOAS technique (Harder et al., 1998; Fitzenberger et al., 2000; Richter et al., 2002; Van Roozendael et al., 2002; Hendrick et al., 2007; Theys et al., 2007; Werner et al., 2017). Model calculations indicated that inorganic bromine can influence the chemical budgets of ozone in the free troposphere to a considerable extent, reducing O3 concentration locally by up to 40 % (von Glasow et al., 2004; Lary, 2005; Yang et al., 2005, 2010). Until now, measurements of BrO and related species by ground-based MAX-DOAS have been frequently carried out in the Arctic (Peterson et al., 2015, 2017; Simpson et al., 2017; Luo et al., 2018), Antarctic (Wagner et al., 2007b; Roscoe et al., 2012; Prados-Roman et al., 2018), and coastal atmosphere (Coburn et al., 2011). To our knowledge, no MAX-DOAS measurements have been reported for BrO on continents other than in polar, salt lake, and coastal areas.
We made long-term ground-based MAX-DOAS measurements at WLG during the period 2012–2015. For this study we analyzed the measured spectra to retrieve the free-tropospheric background mixing ratios of different trace gases, including NO2, SO2, HCHO, and BrO, from MAX-DOAS measurements at WLG. Large effort was spent on the spectral analysis, because in spite of the rather long atmospheric light paths at high altitude the respective trace gas absorptions are close to or below the detection limit. In Sect. 2, we give a description of the WLG measurement site, the meteorological conditions, and the MAX-DOAS instrument used in the study. Section 3 describes the method and settings we used in the spectral retrieval. Section 4 introduces the radiative transfer simulations we performed for in-depth analysis of measurement data. Section 5 describes the methods to filter the measurement data for the clear-sky and low-aerosol-load conditions. In Sect. 6, we provide the differential slant column density values of the investigated trace gases and their corresponding tropospheric background mixing ratios over WLG, and we compare the levels of these trace gases reported by different studies. Conclusions are given in Sect. 7.
2.1 WLG station
The WLG station is sited at the top of Mt. Waliguan (36∘17′ N, 100∘54′ E, 3816 m a.s.l.), located in Qinghai Province of China (Fig. 1a). It is one of the WMO GAW global baseline stations and the only one in the hinterland of the Eurasian continent. Mt. Waliguan is an isolated mountain with an elevation of about 600 m relative to the surrounding landmass; it is surrounded by highland steppes, tundra, deserts, and salt lakes (Fig. 1b). With a low population density of about 6 capitals km−2 and hardly any industry within 30 km, WLG has the advantage of being rather isolated from industry, forest, and population centers. It is relatively dry, windy, and short on precipitation with a typical continental plateau climate (Tang et al., 1995). Xining City (the capital of Qinghai Province, located about 90 km northeast of WLG) and Lanzhou City (the capital of Gansu Province, located about 260 km away to its east) are considered to be the nearest large pollution sources that may have impacts on the WLG site. There are several high mountains (∼4000 m a.s.l.) between Xining and Mt. Waliguan. Total column ozone, surface ozone, solar radiation, precipitation chemistry, greenhouse gases, aerosol optical depth, and aerosol scattering and absorption coefficient together with basic meteorological parameters have become operational measurement items at WLG in succession since the year 1991 (Tang et al., 1995). In addition to routine observations, intensive measurements and model analyses were performed to investigate the regional and global representativeness of WLG and the effects of chemical transformations and physical and transport processes on various atmospheric compositions, e.g., surface ozone, short-lifetime reactive gases (such as NO, NO2, SO2, CO, H2O2, HNO3, HCHO, other carbonyls, and non-methane hydrocarbons – NMHCs), greenhouse gases, persistent organic pollutants, metal and isotopes, and aerosols.
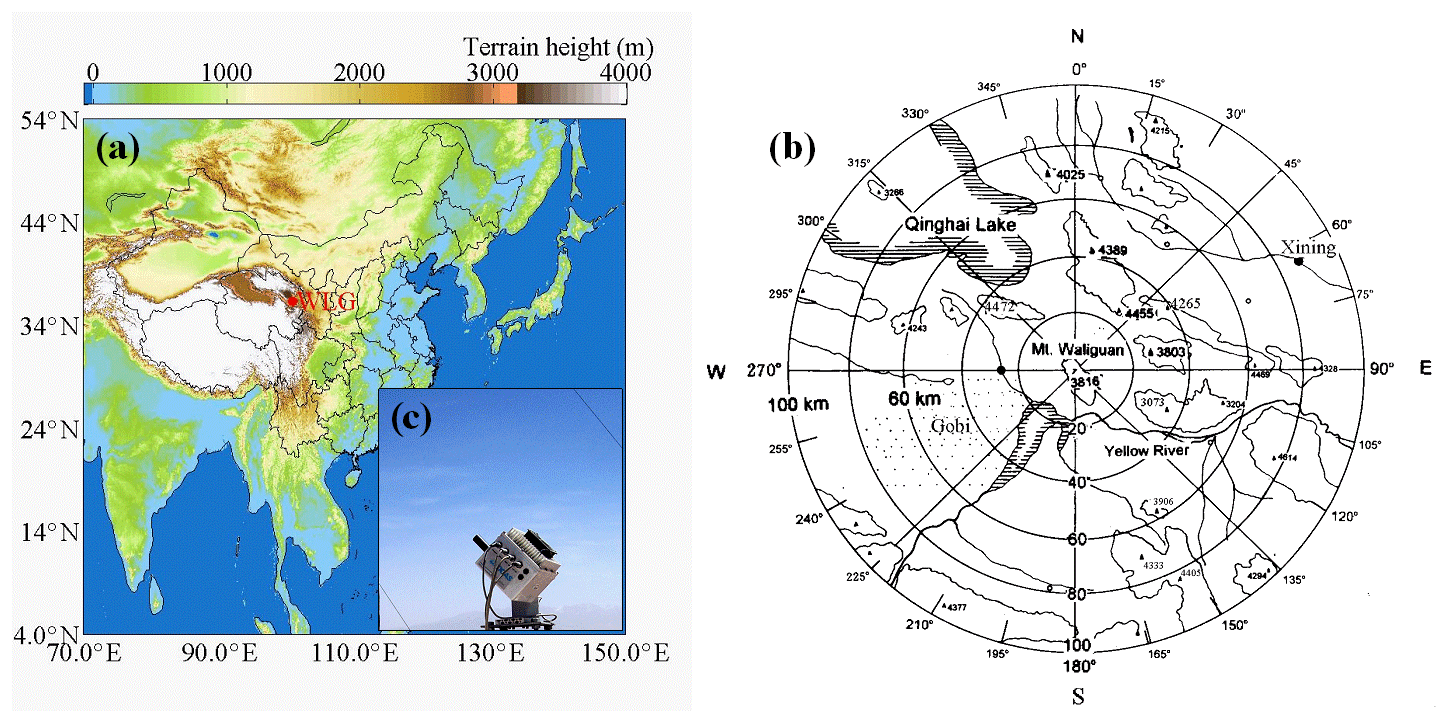
Figure 1(a) Position of Mt. Waliguan in East Asia. (b) Surrounding topography within 100 km of WLG. (c) The MAX-DOAS instrument installed at WLG.
2.2 Meteorological conditions
We used the European Centre for Medium-Range Weather Forecasts (ECMWF) reanalysis data to investigate meteorological conditions over the WLG site, with data for 3–4 km altitude representing the ground level at WLG. As shown in Fig. S1 in the Supplement, temperature is high in summer (around 283 K) and low in winter (around 265 K), pressure is high in summer (around 643 hPa) and low in winter (around 635 hPa), wind speed is low in summer (around 3 m s−1) and high in winter (around 5 m s−1), and wind direction (0 means the wind is blowing from the north) is from the southeast in summer (around 140∘) and from the west in winter (around 260∘). These seasonal variation characteristics are similar to those in earlier years at the station as reported in previous work (Tang et al., 1995). We evaluated the ECMWF reanalysis data for 3–4 km altitude using meteorological data from in situ measurements at the WLG station, and we found that the ECMWF data are in good (temperature, pressure) and reasonable (wind speed and direction) agreement with in situ data (see Fig. S2).
2.3 MAX-DOAS instrument
We started the ground-based MAX-DOAS measurement program at WLG on 26 September 2010. An automated and compact (13 cm × 19 cm × 14 cm) mini MAX-DOAS instrument from Hoffmann Messtechnik GmbH in Germany, which had been used at the Gucheng site in the North China Plain (Jin et al., 2016), was moved to and installed at WLG (Fig. 1c). This instrument is designed for the spectral analysis of scattered sunlight and the application of the MAX-DOAS technique (Hönninger et al., 2004). The same type of instrument was used in previous studies, e.g., in Beijing and the surrounding area (Ma et al., 2013; Jin et al., 2016). The entrance optics, fiber-coupled spectrograph, and controlling electronics are hermetically sealed in a metal box of about 3 L volume. A stepper motor, mounted outside the box, can rotate the whole instrument to control the elevation viewing angle, i.e., the angle between the horizontal and the viewing direction, and thus it can scan vertically at different elevation angles. The spectrograph covers a wavelength range of 290–437 nm and its entrance slit has a width of 50 µm. A Sony ILX511 charged-coupled device (CCD) detects the light in 2048 individual pixels. The whole spectrograph is cooled by a Peltier stage to maintain a stable temperature of the optical setup and to guarantee a small dark current signal. The measurement process and spectra data logging are controlled by a laptop using the MiniMax software package developed by Udo Frieß at the Institute of Environmental Physics, the University of Heidelberg in Germany.
The instrument was mounted on a bracket, fixed on the building roof, at an azimuth viewing direction exactly towards the north. After a winter of pilot run, we added heating elements to the outside of the instrument so that the temperature of the spectrograph could be kept at a stable but not very low value, e.g., −10 ∘C in winter. In other seasons, the temperature of the optical setup was set at a higher value, typically 0 ∘C, below the ambient temperature. Dark current spectra were measured using 10 000 ms and one scan and electronic offset spectra with 3 ms and 1000 scans. Measurements of these signal spectra were made generally month by month or whenever the working temperature of the instrument was changed. Over the pilot run period in the years 2010 and 2011, measurements had been made with the same sequence of elevation angles as used at the Gucheng site in the North China Plain (Jin et al., 2016), with no elevation angles lower than 3∘ available. After the beginning of the year 2012, the elevation angles were set to be −1, 0, 1, 2, 3, 5, 10, 20, 30, and 90∘ in a sequence. Unfortunately, during the data analysis it turned out that the elevation calibration was wrong by −4∘ (see Fig. S3). Thus finally only a few elevation angles from the original selection were found to be above the horizon. After the correction by −4∘ the remaining elevation angles are 1, 6, 16, 26, and 86∘. From the comparison between the measured and simulated elevation angle dependencies of the different quantities (see Fig. S3), we estimate the accuracy of the corrected elevation angles to be within ±0.5∘. The exposure time for each elevation angle was about 1 min. The data from 3 years of measurements over the period April 2012 through April 2015 are used for this study.
At the WLG site the atmospheric trace gas absorptions are usually rather low. Thus the settings of the spectral analysis were optimized for low detection limits. In general this can be achieved by
- a.
co-adding of individual spectra,
- b.
using rather broad spectral ranges, and
- c.
selecting only spectra of high signal-to-noise ratio.
Since at the WLG station different detector temperatures were used for different time periods, the spectral analysis was performed with different spectral calibrations (and corresponding sets of convoluted cross sections) for each of these periods. In addition, the spectral analysis was carried out with consistent settings for the different periods. In order to limit the amount of work, only “long periods” that contained at least 60 measurement days were selected for the data analysis (see Table S1 in the Supplement).
In order to achieve a large reduction of the statistical error, as much as possible individual spectra should be averaged. However, systematic errors tend to increase if an increasing number of spectra are averaged. In this study we performed the analysis of spectra averaged from 10 original spectra, for which a minimum of the fit error was found (see Fig. S4). In this study individual measurements at 26∘ elevation of each elevation sequence are used as Fraunhofer reference spectra, for the reasons explained in Sect. S3.2 in the Supplement.
The spectral ranges for the retrieval of the different trace gases were determined in dedicated sensitivity studies (see Sect. S3.3). Examples of the spectral analyses are shown in Fig. 2. The errors of the retrieved differential slant column densities (dSCDs) of the trace gas were also estimated based on the sensitivity studies described in Sect. S3.3. Table 1 summarizes the spectral fitting ranges and systematic and random errors for the retrieved trace gas dSCDs. For all trace gases, the overall error for individual measurements (averages of 10 original spectra) is dominated by random errors. These errors, however, become much smaller if a large number of measurements are averaged.
Table 1Overview on the different fitting ranges and systematic and random errors for the retrieved trace gas dSCDs (for spectra averaged from 10 original spectra). More details are found in the Supplement.
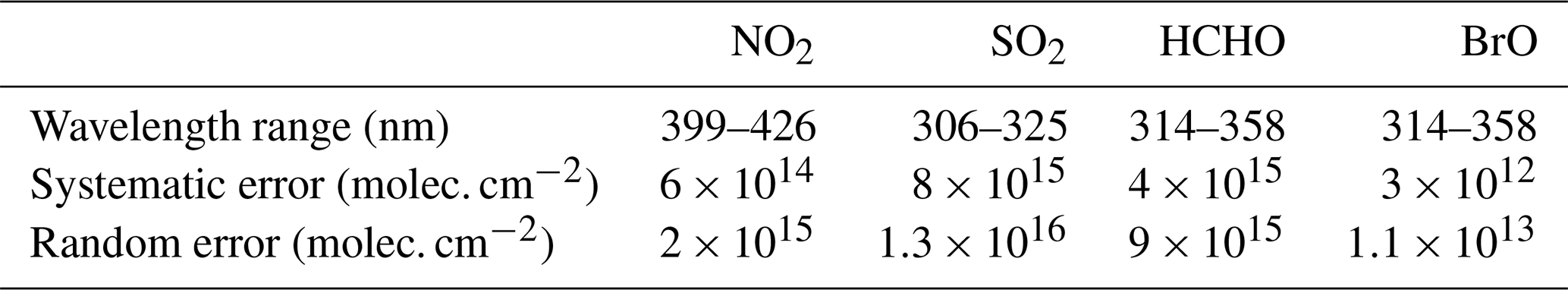
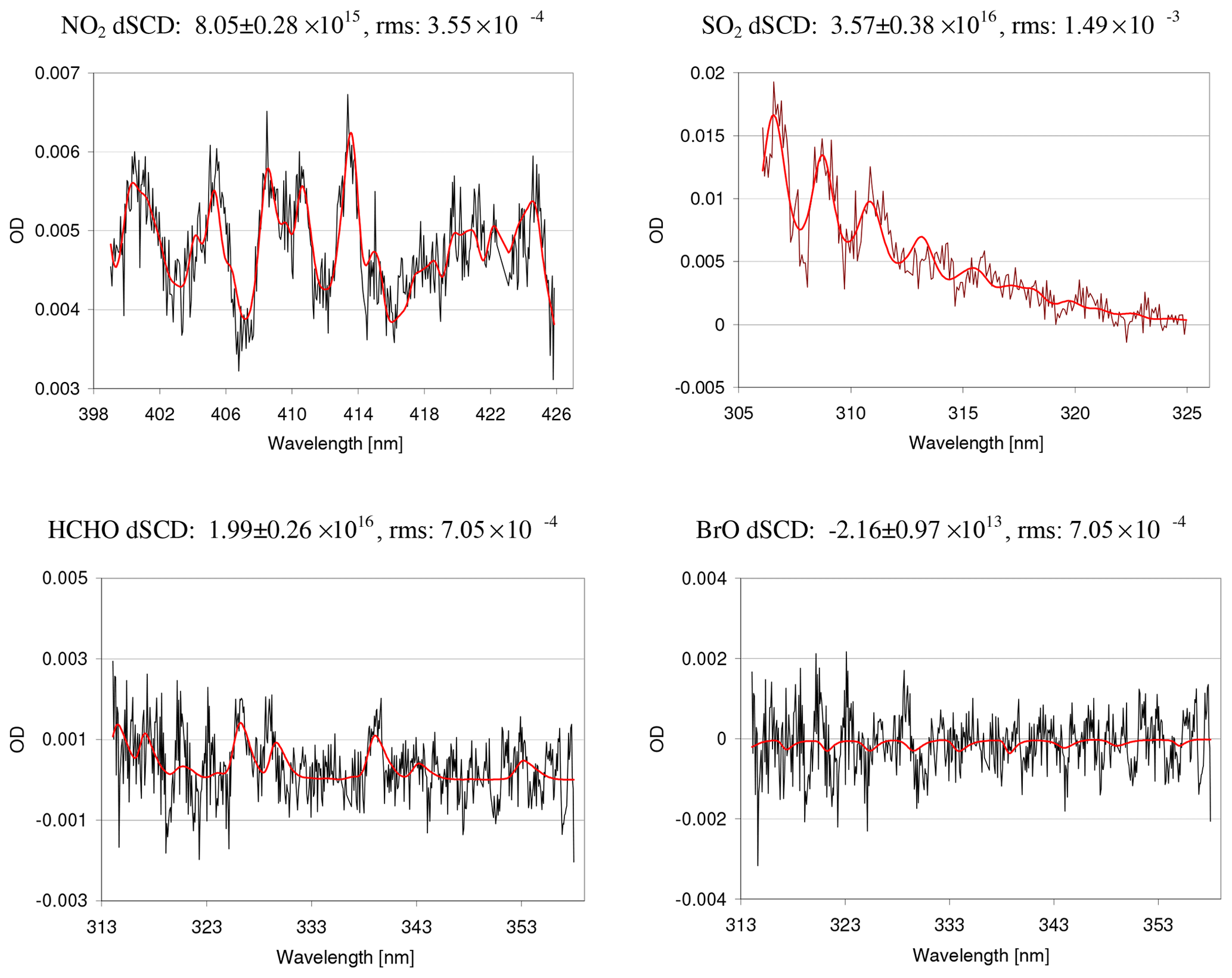
Figure 2Fit result for NO2, SO2, HCHO, and BrO for a spectrum (average of 10 original spectra) taken on 13 May 2013 (00:34–04:19) at 1∘ elevation. On this day, enhanced absorptions of SO2 and NO2 were observed.
Similar to Gomez et al. (2014), we use the dSCDs from MAX-DOAS measurements to estimate the mixing ratios of NO2, SO2, HCHO, and BrO at WLG in this study. The relationships between the dSCDs and mixing ratios of these different trace gases are set up by radiative transfer simulations using the radiative transfer model TRACY-2 (Wagner et al., 2007a). This model allows us to explicitly consider the variation in the topography around the measurement station (for more details on the effect of topography, see Sect. 4.1 below). This option was, however, only used in one dimension (in the viewing direction) in order to minimize the computational effort. The variation in the surface terrain height and the results of the radiative transfer simulations are illustrated in Fig. 3.
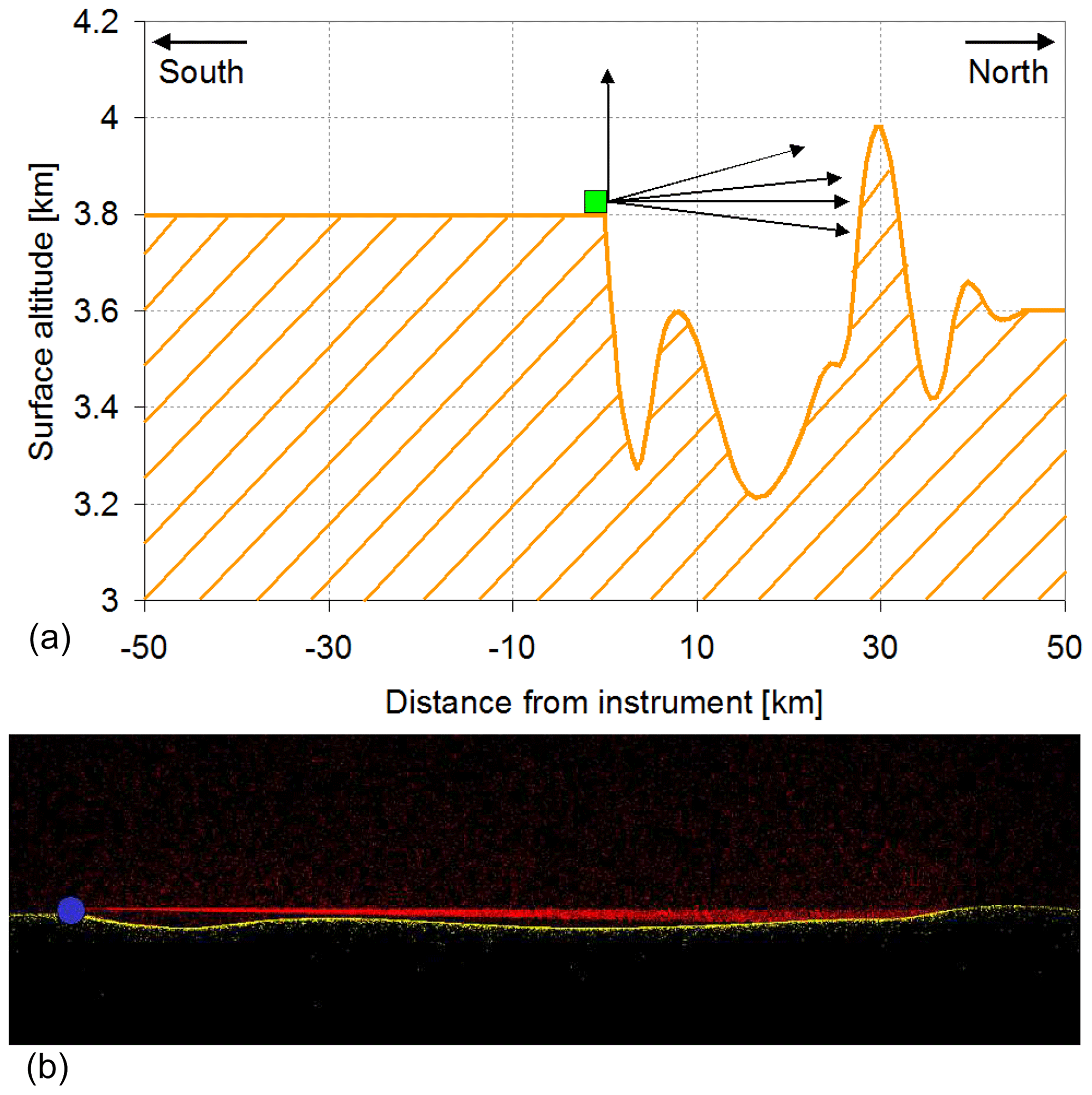
Figure 3(a) Variation in the surface altitude in the viewing direction (towards the north). The variation in the surface altitude across the viewing direction was not explicitly considered to minimize the computational effort. For the same reason, the topography “behind” the instrument towards the south was also assumed to be flat. (b) Illustration of the results of the radiative transfer simulations for the area between the instrument and the high mountain 30 km away. The blue dot indicates the position of the instrument. The small red and yellow dots indicate Rayleigh-scattering events and surface reflection of the simulated solar photons, respectively.
The surface albedo was set to be 7.5 %. Sensitivity studies indicated that the exact choice is not critical: simulations with high surface albedo representative for snow surfaces yielded almost the same results. This finding can be understood by the fact that the measurements and the Fraunhofer reference spectra are affected by changes of the surface albedo in the same way. Thus, for the retrieved dSCDs the effect of the surface albedo cancels out. The aerosol extinction was varied, but it was assumed to be constant between 2600 and 5600 m altitude. Different aerosol loads (aerosol optical depth (AOD) between 0 and 0.5) were assumed. Here it should be noted that only a fraction of 60 % of the total AOD is located above the instrument.
Simulations of trace gas air mass factors (AMFs) were performed for specific viewing geometries for a whole diurnal cycle in January and July. These months were chosen because they represent the most extreme viewing geometries (winter and summer) during the whole year. From the derived diurnal variations in the trace gas AMFs, daily averages for measurements with a solar zenith angle (SZA) below 65∘ were calculated. For these calculations the individual AMFs were weighted by the corresponding simulated intensities. Finally, the simulated AMFs for a 26∘ elevation angle were subtracted from the AMFs for the lower elevation angles, yielding the respective differential AMFs (dAMFs). This procedure was applied in order to calculate trace gas dAMFs which can be directly compared to the trace gas dSCDs derived from the measurements.
In order to relate the measured trace gas dSCDs to atmospheric trace gas mixing ratios, assumptions about the vertical distributions of the trace gases have to be made. The assumed trace gas profiles are described in Sect. S4. Two types of input profiles are used. For the first group of trace gases, the influence of the stratospheric absorptions can be neglected. This is the case for SO2 and HCHO, for which the stratospheric amounts (except for strong volcanic eruptions) are very small and can be neglected. Although for NO2 the contribution from the stratospheric absorption can be rather large, it is found that the stratospheric NO2 absorptions are very similar for the different elevation angles (see Sect. S5.1), and the stratospheric absorptions almost completely cancel out for the derived trace gas dSCDs using sequential Fraunhofer reference spectra. Thus, the tropospheric partial dSCD can be simulated independently from the stratospheric absorptions and can be directly compared to the measured NO2 dSCDs.
For BrO, the situation is different: since the stratospheric BrO profile is located at rather low altitudes, the corresponding absorptions depend substantially on the elevation angle. Thus they do not cancel out in the retrieved BrO dSCDs. In fact, the dSCDs for low elevation angles can even become negative (see Sect. S5.2) due to the stratospheric BrO absorptions. Therefore, for BrO the stratospheric and tropospheric profiles of BrO always have to be considered simultaneously in the radiative transfer simulations.
From the simulation results for a 1∘ elevation angle (and low aerosol load: AOD = 0.1) approximate relationships between the trace gas dSCDs (at 1∘ elevation angle) and the corresponding volume mixing ratios are derived. They are given below for the different trace gases.
-
SO2. A dSCD of 1×1015 molec. cm−2 corresponds to a mixing ratio of 60 ppt.
-
NO2. A dSCD of 1×1015 molec. cm−2 corresponds to a mixing ratio of 33 ppt.
-
HCHO. A dSCD of 1×1015 molec. cm−2 corresponds to a mixing ratio of 55 ppt.
-
BrO. A dSCD of 1×1013 molec. cm−2 corresponds to a mixing ratio of 0.6 ppt.
Here it should be noted that for BrO the relationship is valid for the increase in the BrO dSCD compared to a scenario with no BrO in the troposphere (see Sect. S5.2 and Fig. S24). It should also be noted that while in the simulations a constant mixing ratio throughout the atmosphere was assumed, the measured trace gas dSCDs are mainly sensitive to the trace gas concentrations in the atmospheric layers close to the instrument. Thus the derived trace gas mixing ratios are most representative for the free troposphere in the altitude range between about 4 and 5 km.
4.1 Effects of topography
We investigated the effect of the surface topography in more detail. In addition to the setup with the “true” topography (Fig. 3), we also performed simulations with flat surfaces at sea level or 3700 m altitude, because these options might be used as alternative scenarios for radiative transfer models without the option to consider the true topography. In all three setups the atmospheric properties and the altitude of the detector (3800 m) are kept the same. In Fig. 4 the O4 AMFs for the simulations with flat surfaces are plotted versus the O4 AMFs for the true surface topography. While the results for a flat surface at 3700 m are very similar to those for the true topography, the results for a flat surface at sea level show systematically higher values. With increasing aerosol load these differences even increase. This finding can become very important for the interpretation of measurements in mountainous environments if no radiative transfer simulations considering the true topography are available. However, this finding needs further investigations, especially if an instrument is operated on a more isolated mountain. In such cases, the agreement between simulations with true topography and flat surface at high altitude might be worse. Further studies should also investigate the effects for trace gases with different altitude profiles.
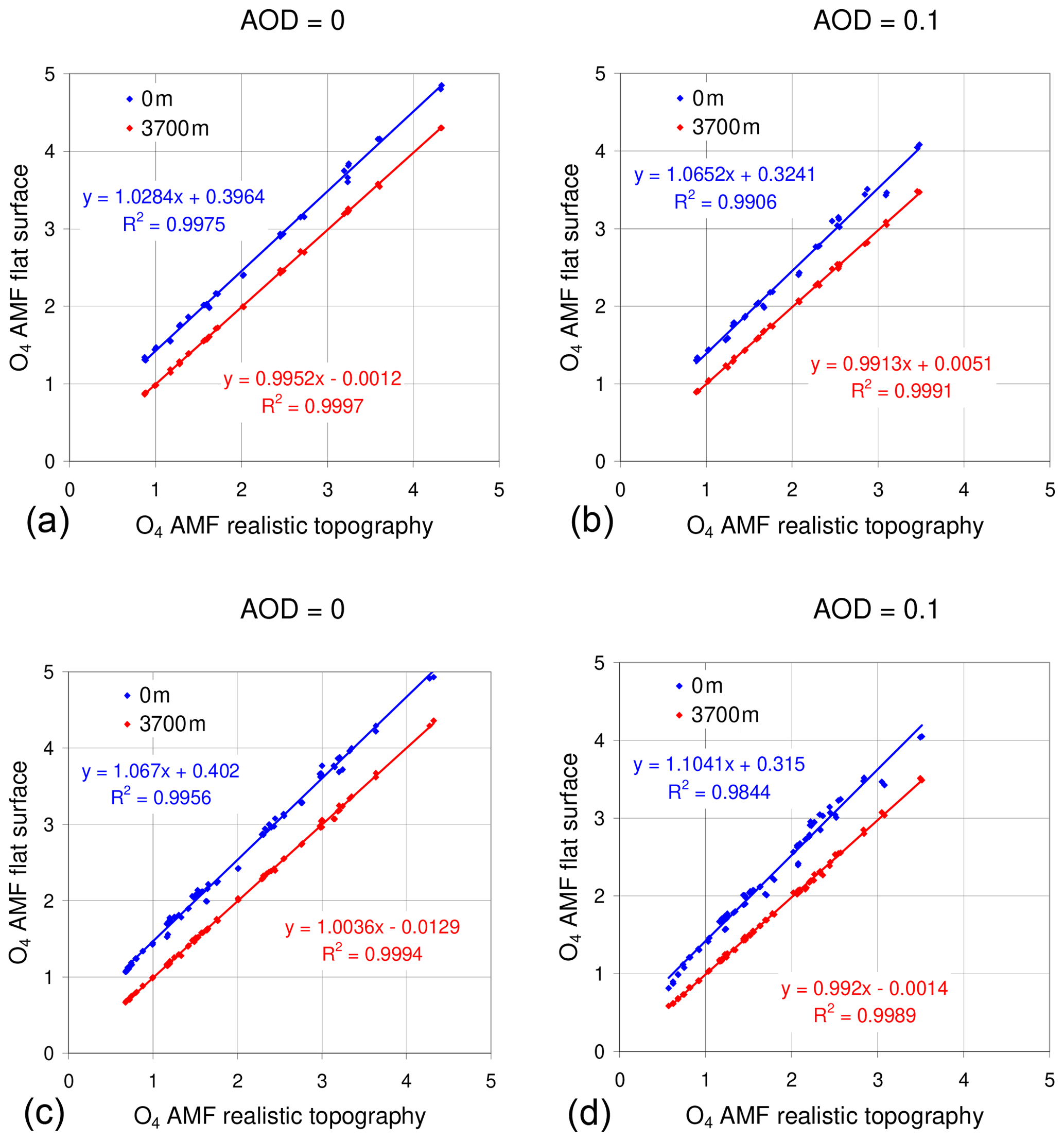
Figure 4Comparison of simulated O4 AMFs for different choices of the surface topography. Results for flat surfaces at sea level (blue) or 3700 m (red) are plotted versus the results for a realistic surface topography (see Fig. 3). (a, b) Results for winter and (c, d) results for summer. (a, c) Results for simulation without aerosols and (b, d) results for an AOD of 0.1.
Besides the simulation of the air mass factors, we also compared the results for other simulated quantities: in Fig. 5 the dependence of the simulated radiance (top) O4 AMF (center) and color index (bottom) is shown as a function of the elevation angle (including negative elevation angles). Again, the results for the true topography and the flat surface at 3700 m altitude are very similar, while the results for a flat surface at sea level are systematically different. The results of these comparisons indicate that for our measurements the assumption of a flat surface at the approximate altitude of the surrounding terrain is a very good approximation for the true surface topography. However, for zero and negative elevation angles, the consideration of the true topography might become important. Here it is interesting to note that these differences will probably increase for measurements in other mountainous scenarios, especially for more measurements at isolated mountains. These effects should also be investigated in more detail in future studies.

Figure 5Simulation results of the radiance (a), O4 AMF (b), and color index (c) for different choices of the surface topography as function of the elevation angle (including negative elevation angles). Simulation results are for summer noontime (see also Fig. S3).
For the quantitative interpretation of the measurements, they are compared to results from radiative transfer simulations. These simulations are performed for well-defined, particularly cloud-free conditions. Thus only measurements for such conditions have to be selected. Moreover, to take advantage of the high sensitivity of the MAX-DOAS measurements, situations with low aerosol load and thus high visibility have to be selected. The following two subsections describe how measurements under cloudy conditions and high aerosol loads are identified.
5.1 Cloud filter
Cloudy-sky conditions can be identified and classified by different quantities (see, e.g., Gielen et al., 2014; Wagner et al., 2014, 2016). In this study, to minimize the computational effort, we only use the color index (CI) measured in the zenith direction (note that the measurements in the zenith direction were not used for trace gas retrievals because of suspected direct sun impact). We chose the wavelength pair 330 and 390 nm:
Moreover, in order to minimize the potential effects of instrument degradation, the absolute value of the CI is not used for the cloud classification. Instead, two derived quantities are calculated.
5.1.1 The temporal smoothness indicator (TSI)
The TSI is derived from the zenith measurements. If the CI between subsequent zenith measurements changes rapidly, this indicates the presence of clouds. The TSI is calculated according to the following formula:
Here i indicates the number of an elevation sequence. For clear sky (and homogenous cloud cover) the TSI is small. For broken clouds the TSI is large.
5.1.2 The spread (SP) of the CI for one elevation sequence
The SP is calculated as the difference between the maximum and minimum of the CI for a selected elevation sequence:
Here max(CIi) and min(CIi) indicate the maximum and minimum CI of the considered elevation sequence. For clear sky, the SP is large; for (homogenous) clouds the SP is low.
Based on the calculated TSI and SP the cloud situation of an individual elevation sequence is classified as clear sky, broken clouds, or continuous clouds according to the following thresholds:
- a.
clear sky – TSI < 0.012 and SP > 0.15,
- b.
broken clouds – TSI > 0.012,
- c.
continuous clouds – TSI < 0.012 and SP < 0.15.
Note that for cases (a) and (c) both TSIs (at the beginning and the end of the elevation sequence) have to be <0.012; for case (b) the condition is fulfilled if one of both TSIs is >0.012.
5.2 Aerosol filter
For the low elevation angles the atmospheric visibility and thus the length of the light path depend strongly on the aerosol load. Thus measurements with high aerosol loads have decreased sensitivity to the trace gas absorptions and thus have to be identified and removed from further processing. For that purpose the retrieved O4 absorption is used (Wagner et al., 2004; Lampel et al., 2018). In the following, a threshold for the retrieved O4 dSCD at a 1∘ elevation angle of 1.4×1043 molec.2 cm−5 is used, which corresponds to an O4 dAMF of 1.2 (see Fig. S27). This threshold represents an AOD of about 0.1 at 360 nm.
In Fig. S28 the seasonal variation in the O4 dSCDs at 1∘ elevation is shown. High values are typically found in winter, indicating low AOD. In other seasons smaller O4 dSCDs are found, indicating higher AOD. This seasonal dependence is in good agreement with measurements of the AOD (Che et al., 2011).
5.3 Summary of sky conditions
In Fig. 6 the seasonal variation in the sky conditions is shown. It is derived by applying the cloud and aerosol classification algorithms described above. The statistics is based on the number of observations (at 1, 6, or 16∘ elevation angle) of spectra averaged from 10 original spectra (April 2012–April 2015). Only measurements with more than 800 scans are considered. The basic colors indicate the cloud properties (clear, broken clouds, continuous clouds). The full or light colors indicate observations with low or high aerosol loads, respectively.
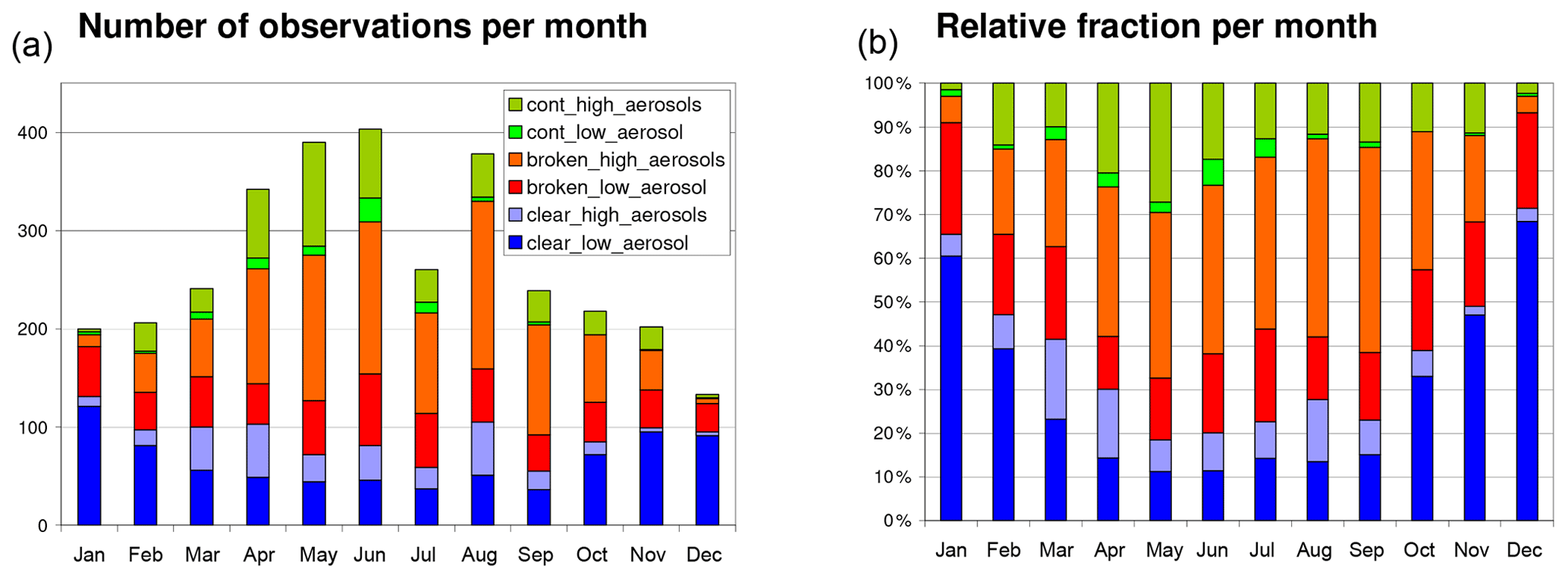
Figure 6Absolute (a) and relative (b) frequency of the different sky conditions. The statistics is based on the number of observations at a 1∘ elevation angle of spectra averaged from 10 original spectra (April 2012–April 2015). Only measurements with more than 800 scans are considered. The basic colors indicate the cloud properties (clear, broken clouds, continuous clouds). The dark or light colors indicate observations with low or high aerosol loads, respectively.
While the absolute frequency of clear-sky observations (low and high aerosol load) stays almost constant over the year, the relative fraction changes strongly with the highest probability of clear-sky observations in winter and the lowest probability in summer. The relative fraction of low aerosol cases is largest in winter. The corresponding seasonal frequency plots for the different trace gas analyses (after application of the individual rms filters) are shown in Fig. S29. While the absolute amount of valid data is different for the different analyses, the seasonal frequency is almost the same.
6.1 Seasonal means of the dSCDs
In Fig. 7 (left) time series of daily averages of the individual trace gas dSCDs for a 1∘ elevation angle are shown. On the right side, the corresponding monthly mean values are shown. In Fig. 8 the seasonal cycles for all elevation angles (1, 6, 16∘) are shown for clear sky. In this figure, the systematic uncertainties of the trace gas dSCDs are also indicated by the blue dotted lines. The systematic uncertainties can be regarded as indicators for the lower bounds of the detection limit (which might be reached if a large number of measurements are averaged). For NO2, SO2, and HCHO, approximate mixing ratios derived for the measurements at 1∘ elevation angles are also indicated by the y axes at the right side (see Sect. 4). The results for broken clouds are similar to those for clear sky (see Fig. S30). This is useful information to confirm the results for clear sky, since for broken clouds, the atmospheric light paths are often similar to those for clear sky.
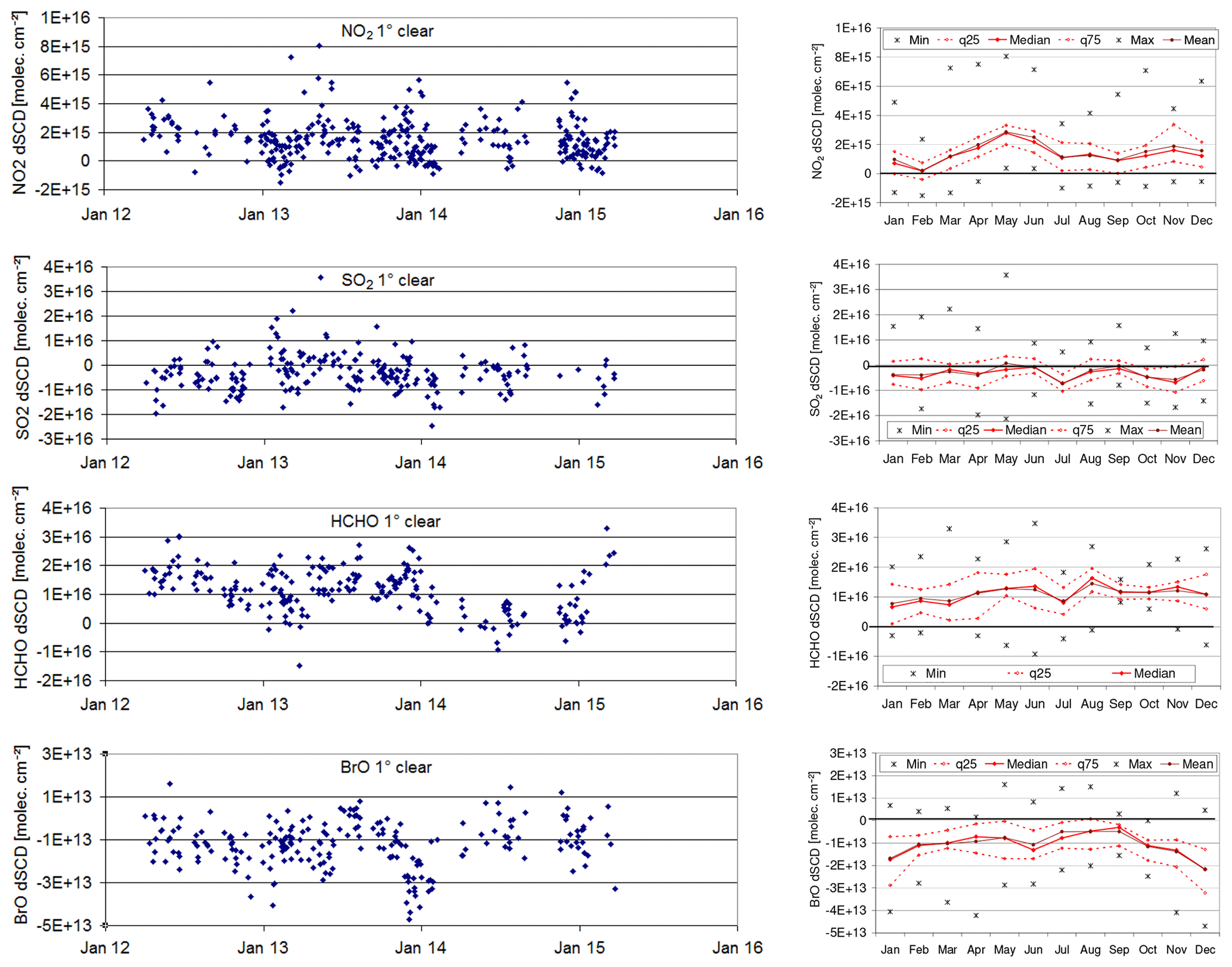
Figure 7Left: time series of daily averaged trace gas dSCDs at 1∘ elevation for clear-sky spectra and low aerosol load from 2012 to 2015, with “Jan 12” referring to “1 Jan 2012”. Right: corresponding seasonal averages.

Figure 8Seasonal means of the trace gas dSCDs for different elevation angles for clear sky and low aerosol load. For NO2, SO2, and HCHO the right axes represent the approximate mixing ratios for measurements at a 1∘ elevation angle. The blue dotted lines indicate the systematic uncertainties, which can be considered the lower bound of the detection limit.
The main findings are as follows.
- i.
For NO2 and HCHO higher dSCDs are found for lower elevation angles (see also Fig. S20), and this indicates enhanced trace gas concentrations in the lower troposphere (at least in the atmospheric layers between about 4 and 5 km a.s.l.) compared to the upper troposphere.
- ii.
The highest NO2 values are found in a period from April to June, most likely due to the influence of long-range transport of NO2 and its reservoir from both human and natural sources (Ma et al., 2002b; Wang et al., 2006).
- iii.
For BrO the opposite dependence is found, and this is mainly caused by the influence of stratospheric BrO (see Sect. S5.2).
- iv.
For SO2 no clear elevation dependence is found (the values are below the detection limit).
6.2 Estimation of a free-tropospheric background mixing ratio from the dSCDs
In this section the lower tropospheric mixing ratios of the NO2, SO2, HCHO, and BrO at WLG are estimated based on the respective dSCDs at 1∘ (see Fig. 8) and the relationships between the dSCDs and the tropospheric mixing ratios (Sect. 4).
Below are our estimates.
-
NO2. The dSCDs at 1∘ between 0.2×1015 molec. cm−2 (January) and 3×1015 molec. cm−2 (May) correspond to mixing ratios between about 7 (January) and 100 ppt (May).
-
SO2. The dSCDs at 1∘ below 8×1015 molec. cm−2 correspond to mixing ratios below 0.5 ppb.
-
HCHO. The dSCDs at 1∘ between 0.7×1015 molec. cm−2 (winter) and 1.7×1015 molec. cm−2 (summer) correspond to mixing ratios between about 0.4 (winter) and 0.9 ppb (summer).
-
BrO. From the dSCDs at 1∘ an upper limit for a troposphere BrO mixing ratio of 0.3 ppt is derived (see Sect. S8).
It should again be noted that while in the simulations a constant mixing ratio throughout the atmosphere was assumed, the measured trace gas dSCDs are mainly sensitive to the trace gas concentrations in the atmospheric layers close to the instrument. Thus the derived trace gas mixing ratios are most representative for the free troposphere in the altitude range between about 4 and 5 km.
6.3 Comparisons of measured results with previous studies
Measurements of the very reactive trace gases are very scarce at remote sites like WLG. Table 2 summarizes the mixing ratios of NO2, SO2, HCHO, and BrO at WLG measured by MAX-DOAS presented in this study in comparison to those recorded in other studies using different methods.
Table 2Comparisons between trace gas mixing ratios at WLG and those recorded in other remote areas at low latitudes.
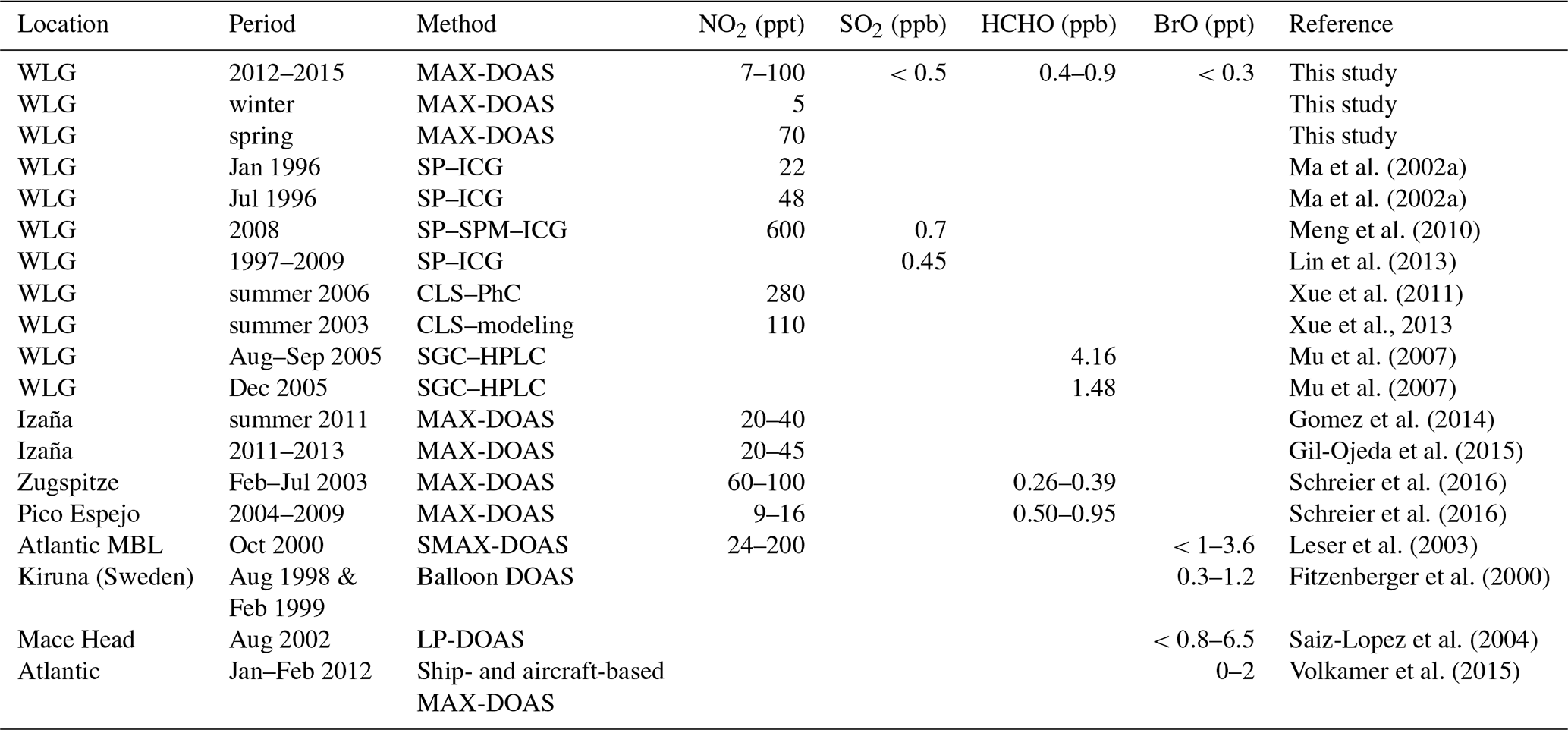
The passive sampling method was used to measure the weekly and monthly mean mixing ratios of NO2 and SO2 at WLG (Ma et al., 2002a; Meng et al., 2010; Lin et al., 2013). The monthly mean mixing ratios and standard deviations of NO2 at WLG were 0.022±0.010 ppb in January and 48±17 ppt in July for the year 1996 (Ma et al., 2002a). For the measurement in 1996, the NO2 was sampled with filter packs (SP) with exposure times of typically 3–5 d and then analyzed by ion chromatography (ICG) equipment (Yu et al., 1997). During the measurement experiment in 2008, the samplers were exposed about 10 d in a month. The extracted NO2 samples (Ogawa passive samplers) were analyzed using a spectrophotometer (SPM) while the extracted SO2 samples were analyzed by ICG (Meng et al., 2010). The average mixing ratios of NO2 and SO2 at WLG in 2008 typically varied in the ranges of 0.6±0.4 and 0.7±0.4 ppb, respectively (Meng et al., 2010). Long-term continuous measurements, by filter sampling with exposure times of 3–5 d followed by ICG chemical analysis, showed that the mixing ratio of SO2 varied typically in a range of 0.45±0.14 ppb at WLG from 1997 to 2009 (Lin et al., 2013). The NO2 levels obtained from this study are in accordance with those recorded by Ma et al. (2002a), but an order of magnitude lower than reported by Meng et al. (2010). The time differences between Meng et al. (2010) and the other two studies seem not to be the main cause. The sampling filters were likely to be polluted by local human activities, such as increasing frequency of occasional cars and vans to the station, which could not be excluded from the recording due to their long exposure time. The level of SO2 might also be overestimated by Meng et al. (2010) considering that there had not been a significant increasing trend for SO2 at WLG during 1997–2009 (Lin et al., 2013). It can be deduced that the SO2 level from this study is reasonable with comparison to the results from Lin et al. (2013).
The daytime NO and NO2 mixing ratios at WLG in the summer of 2006 were 71±45 ppt and 0.28±0.13 ppb, respectively, as measured by chemiluminescence (CLS) analyzer coupled with a photolytic converter (PhC) (Xue et al., 2011). Also measured by CLS, the daytime average NO mixing ratios at WLG were 0.072±0.079 ppb in the late spring and 0.047±0.032 ppb in the summer of 2003 (Wang et al., 2006; Xue et al., 2013). Assuming the same NO2∕NO ratio (i.e., 3.9) as in the summer of 2006, the NO2 mixing ratios at WLG would be 0.28±0.31 ppb in the late spring and 0.18±0.12 ppb in the summer of 2003. The NO2 levels obtained from the CLS method are much higher than those from MAX-DOAS presented in this study. This can be explained by the fact that in situ measurements at WLG are strongly influenced during the daytime by the underlying boundary layer, where NO2 levels are high due to soil emissions and other occasional pollution sources. In contrast, the upwelling air masses have a relatively small influence on the MAX-DOAS measurements, which represent a long optical path.
The ambient concentrations of carbonyl compounds were measured at WLG in 2005, by using the silica gel cartridge–high-performance liquid chromatography (SGC–HPLC) method. The means and standard deviations of HCHO mixing ratios were 4.16±1.89 ppb in August–September and 1.48±0.42 ppb in December (Mu et al., 2007). The HCHO mixing ratios obtained by the SGC–HPLC method are nearly an order of magnitude higher than those by MAX-DOAS presented in this study. Mu et al. (2007) reported that the variability of HCHO concentrations was significant (by a factor of >2) if the air samples were collected at different places at the site. HCHO forms through oxidation reactions associated with methane and various NMVOCs, most of which (e.g., isoprene) have high spatial variability in abundance due to short lifetimes and thus can influence local HCHO concentrations significantly. Therefore, isoprene and other very active NMVOCs emitted from natural vegetation can accumulate near the surface and result in high levels of HCHO by oxidation. While in situ sampling methods as used by Mu et al. (2007) measured HCHO on the ground level within the boundary layer, MAX-DOAS measured the HCHO mixing ratios at a large scale, partly including the free troposphere. The model sensitivity experiment shows that the HCHO mixing ratio is larger in the boundary layer than in the free troposphere over WLG (see Sect. 6.4).
Also listed in Table 2 are the levels of NO2, HCHO, and BrO measured by MAX-DOAS in other remote areas at middle and subtropical latitudes. Note that to our knowledge, no results for SO2 were reported in those studies. Our purpose here is to show what low levels of these gases, BrO in particular, had been detected by MAX-DOAS in these remote regions.
Gomez et al. (2014) retrieved the gas concentrations at a very long horizontal path of over 60 km from high-mountain MAX-DOAS measurements at the Izaña Atmospheric Observatory (28∘18′ N, 16∘29′ W, 2373 m a.s.l.) on the Canary Islands using the modified geometrical approach (MGA). The NO2 mixing ratios were within the range of 20–40 ppt in the summer of 2011 (Gomez et al., 2014). Gil-Ojeda et al. (2015) applied the same technique to a longer data set obtained at the same station and showed that the free-tropospheric NO2 mixing ratios varied in the range of 20–45 ppt between 2011 and 2013, with the lowest values in winter and highest values in summer. MAX-DOAS measurements were also performed at Zugspitze, Germany (47.5∘ N, 11∘ E, 2650 m a.s.l.), during February–July 2003 and Pico Espejo, Venezuela (8.5∘ N, 71∘ W, 4765 m a.s.l.), from March 2004 to February 2009. Based on the MGA method, the monthly mean mixing ratios of free-tropospheric NO2 were estimated to be in the range of 60–100 ppt at Zugspitze and 8.5–15.5 ppt at Pico Espejo. The values for HCHO were in the range of 500–950 ppt at Zugspitze and 255–385 ppt at Pico Espejo (Schreier et al., 2016).
BrO was detected directly in the midlatitude marine environment for the first time, in the region north of the Canary Islands (around 35∘ N, 13∘ W), when a research cruise including ship MAX-DOAS (SMAX-DOAS) measurements was carried out over the Atlantic Ocean in October 2000 (Leser et al., 2003). Typical levels of BrO in the marine lower troposphere were <1 to 3.6 ppt in most cases, and upper limits of NO2 for clean air in the Atlantic west of Africa were observed to be 24 to 100 ppt (Leser et al., 2003). BrO was measured by long-path MAX-DOAS (LP-MAX-DOAS) at the Mace Head observatory on the west coast of Ireland (53∘20′ N, 9∘54′ W) in August 2002. The BrO mixing ratios were shown to vary from below the detection limit (0.8 ppt) at night to a maximum daytime concentration of 6.5 ppt, indicating that significant bromine activation occurs over the open ocean (Saiz-Lopez et al., 2004). Tropospheric BrO profiles over the remote Atlantic were obtained by aircraft- and ship-based MAX-DOAS measurements by Volkamer et al. (2015). In the marine boundary layer (MBL) mixing ratios close to zero were obtained, which increase with altitude in the free troposphere. In the middle troposphere mixing ratios up to 1 ppt were found. Fitzenberger et al. (2000) derived BrO profiles from balloon-borne measurements in Kiruna (Sweden) with mixing ratios up to about 1 ppt in the free troposphere.
It can be seen that the levels of NO2 at WLG derived from this study are comparable to those measured by MAX-DOAS in other remote areas. However, the upper limit for the free-tropospheric mixing ratios of BrO at WLG (about 0.3 ppt) derived from this study is lower than those observed under the MBL conditions at the same latitude band.
6.4 Simulations of chemical ozone production and OH concentrations
We performed chemical box model simulations to evaluate the effects of the variability and trend in NO2 levels on the photochemistry and oxidation capacity of the background atmosphere over WLG. The model, named NCAR's Master Mechanism (MM), was originally developed by Madronich and Calvert (1989) and applied to estimate the chemical budget for ozone at WLG by Ma et al. (2002a). For this study, we simulated the net ozone production and OH in summer for the free-tropospheric conditions, which were characterized by air masses with relatively high ozone and low water vapor and by excluding very-short-lifetime non-methane hydrocarbons (NMHCs) like isoprene, as described by Ma et al. (2002a). All simulations were constrained by the same diurnal cycles of temperature, water vapor, and ozone and fixed NMHC values measured at WLG, as reported in the work of Ma et al. (2002a). Three different NOx levels were considered and fixed to constrain the simulations, with scenarios described below.
-
M02. The daily average of NO2 mixing ratio is 48 ppt, corresponding to the result derived by the SP method from Ma et al. (2002a).
-
M20. The daytime (daily) average of NO2 mixing ratio is 60 ppt (70 ppt), corresponding to the value derived by MAX-DOAS from this study.
-
X13. The daily average of NO2 mixing ratio is 110 ppt, corresponding to the result derived by the CLS method from Xue et al. (2013).
Figure 9 shows the diurnal variations in the net ozone production () and OH concentration (COH) at WLG simulated by the model constrained by the NOx concentrations corresponding to these three NO2 scenarios. Among the three NO2 conditions, both the daytime and COH are the smallest for M02 and the largest for X13. There occurs a net ozone destruction under the M02 condition and net ozone production under the X13 condition, with daily average being −2.2 and 1.8 ppb d−1, respectively. These findings are qualitatively in agreement with the results of Ma et al. (2002a) and Xue et al. (2013), although the latter was derived under different NMHC and water vapor conditions. Under the M20 condition, while net ozone production is found to occur instantaneously in the early morning and late afternoon, ozone is still slightly net destructed over the entire day with a value of −0.8 ppb d−1.
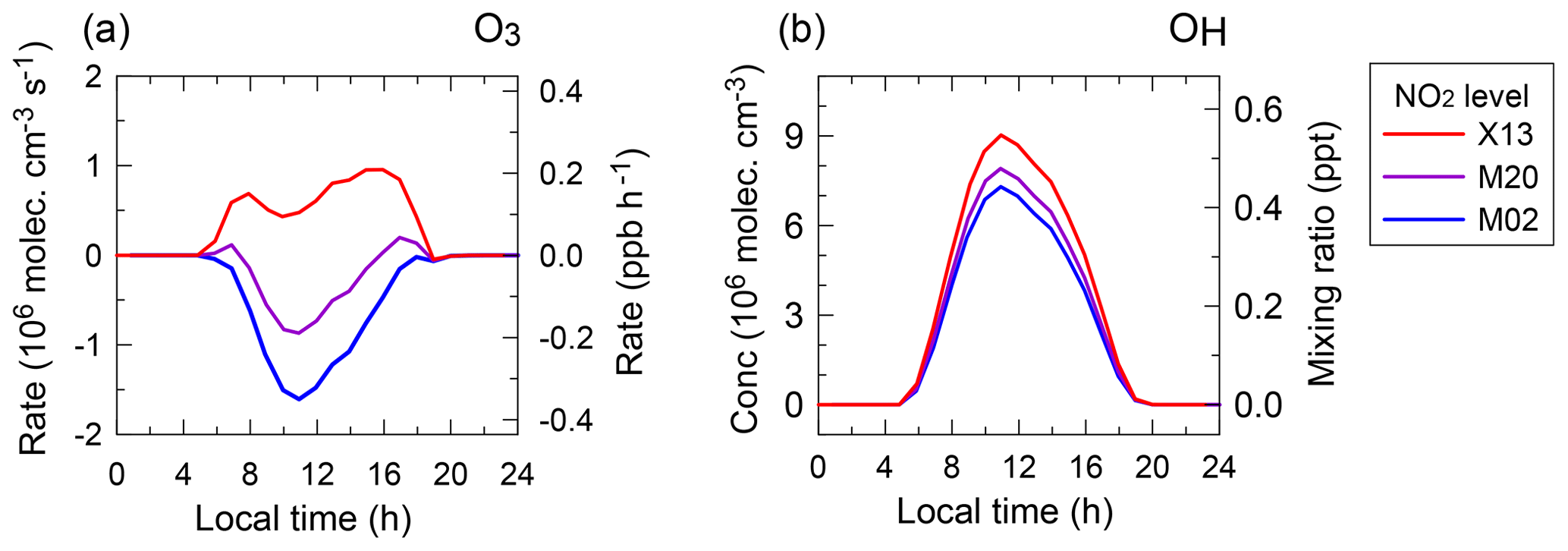
Figure 9(a) Simulated daily variations in the net ozone production rates and (b) in the OH concentrations and mixing ratios for the free-tropospheric conditions over WLG in summer at different NO2 levels.
As mentioned above, the free-tropospheric conditions, which generally have less ozone production with respect to the planetary boundary conditions at WLG as characterized by Ma et al. (2002a), are taken into account. We also made a sensitivity test, for which the short-lived NMHC conditions from Ma et al. (2002a) were added to the M20 conditions (denoted as M20-BL). For the scenario with the added NMHC species, high intermediate oxidation products were predicted for the planetary boundary condition (M20-BL) compared to the free-tropospheric condition (M20). For example, the daily average HCHO mixing ratio was simulated to be 1.9 ppb in M20-BL and 0.4 ppb in M20. Compared to the M20 condition, the instantaneous net ozone production and destruction are more in balance over the course of the day under the M20-BL condition, with a daily average value of 0.3 ppb d−1, indicating that ozone is slightly net produced.
We also performed an additional model simulation (denoted as M20-BrO, not shown in the figure) to investigate the effects of bromine chemistry on and OH in the free troposphere over WLG, by including the gas-phase bromine chemical mechanism provided by the MM. All input variable values were the same as in M20, except that the simulation was constrained with a daily maximum BrO mixing ratio of 0.3 ppt (occurring around noon) for M20-BrO. The result shows a daily net ozone destruction of −1.0 ppb d−1, slightly lower than that for M20. Compared to M20, the OH concentrations in the daily cycle decrease in M20-BrO, with the peak COH value ranging from 7.9×106 molec cm−3 for M20 to 7.4×106 molec cm−3 for M20-BrO. The perturbation of BrO on the net ozone production estimated here (i.e., about −0.2 ppb d−1) may be considered negligible considering that the variability of other environmental conditions, such as the NMHCs and water vapor concentrations, can have larger effects on the simulated value (Ma et al., 2002a).
We made long-term ground-based MAX-DOAS measurements at the WLG WMO GAW global baseline station during the period 2012–2015. For this study we analyzed the measured spectra to estimate the tropospheric background mixing ratios of different trace gases, including NO2, SO2, HCHO, and BrO, from MAX-DOAS measurements at WLG.
For the spectral retrieval, we find that averaging of spectra increases the signal-to-noise ratio and thus reduces the statistical error of the spectral retrieval on the one hand, and systematic errors caused by imperfect correction of the Ring effect tend to increase if an increasing number of spectra are averaged on the other hand. Averages of 10 original spectra have been proven to be an “optimum option” in the spectral analysis for this study. We determined the settings and spectral ranges for the retrieval of the different trace gases by a large number of dedicated sensitivity studies. We performed radiative transfer simulations with the radiative transfer model (RTM) TRACY-2, which allows us to explicitly consider the variation in the topography around the measurement station in the viewing direction. From the simulation results, approximate relationships between the trace gas dSCDs (at the 1∘ elevation angle and low aerosol load: AOD = 0.1) and the corresponding volume mixing ratios are derived. We used the temporal variation and the spread of the color index (CI) derived from our MAX-DOAS measurements to select measurement data for clear sky and low aerosol load, and then we retrieved the corresponding daily averages and seasonal cycles of the trace gas dSCDs at elevation angles of 1, 6, and 16∘.
For NO2 and HCHO, higher dSCDs are found for lower elevation angles than for higher elevation angles, indicating higher trace gas concentrations in the lower troposphere compared to the upper troposphere over WLG. For BrO the opposite dependence is found, reflecting the influence of stratospheric BrO. For SO2 no clear elevation dependence is found. The highest NO2 dSCDs are found in a period from April to June, most likely due to the long-range transport of NO2 and its reservoirs to WLG. From the dSCDs at 1∘ elevation, mixing ratios of NO2 in the lower troposphere over WLG are estimated to be between about 7 ppt (January) and 100 ppt (May), and mixing ratios of SO2 fall below 0.5 ppb. Mixing ratios of HCHO range between about 0.4 (winter) and 0.9 ppb (summer). These mixing ratios are most representative for atmospheric layers between about 4 and 5 km. Mixing ratios of BrO are estimated to be smaller than 0.3 ppt. Retrieving BrO in the continental background troposphere remains a challenge, which should be further addressed in future studies.
The summertime NO2 level at WLG derived by MAX-DOAS from this study (M20) is higher than the result from in situ measurement by the SP method in 1996 (Ma et al., 2002a) (M02) but lower than that by the CLS method in 2003 (Xue et al., 2013) (X13) and 2006 (Xue et al., 2011). The chemical box model simulations show that the daily net ozone production in the free troposphere over WLG is −2.2, 1.8, and −0.8 ppb d−1 under the NO2 levels of M02, X13, and M20, respectively. It is most likely that the ozone production over WLG increased from 1996 to the 2000s and 2010s. The model sensitivity simulation indicates a negligible effect of bromine chemistry on the ozone production in the background troposphere, with a perturbation of −0.2 ppb d−1 over WLG. Within this study, existing retrieval strategies for MAX-DOAS measurements were adapted and improved for measurements at high-altitude stations and in environments with low trace gas abundances. These improvements will be important to similar studies and include the following main aspects.
- a.
In order to achieve low detection limits, spectra were pre-filtered and averaged to minimize the spectral noise. Spectral interferences between different absorbers were investigated and minimized using measured and synthetic spectra. Maximum wide spectral ranges were used to make the best use of the information content.
- b.
Radiative transfer simulations were performed taking into account the surface topography. While it turned out for this study that the effects of surface topography were not very important, this might be different for measurements in other mountainous scenarios, especially for measurements on isolated mountains. These effects should be investigated in more detail in future studies.
- c.
At high-mountain sites the elevation angle dependence of stratospheric absorptions can become important, especially for trace gases like BrO which have their concentration maximum close to the tropopause.
An obvious conclusion from our study is that future measurements should be performed with more elevation angles and with instruments that have a better signal-to-noise ratio.
The spectral analysis and model simulation results here are available upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-6973-2020-supplement.
JM and TW designed the study. JM, JJ, JG, ZZ, JW, PL, and GZ contributed to the measurements. TW, JM, SDö, SDo, JJ, SC, JP, and JL contributed to the data analyses. TW and JM prepared the manuscript with consent by all co-authors.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Study of ozone, aerosols and radiation over the Tibetan Plateau (SOAR-TP) (ACP/AMT inter-journal SI)”. It is not associated with a conference.
We thank all the staff from WLG for assisting measurement work.
This research has been supported by the Ministry of Science and Technology of the People's Republic of China (grant no. 2018YFC1505703), the National Natural Science Foundation of China (grant nos. 41275140 and 41875146), and the China Meteorological Administration (grant no. GYHY201106023).
The article processing charges for this open-access
publication were covered by the Max Planck Society.
This paper was edited by Tao Wang and reviewed by two anonymous referees.
Bauwens, M., Stavrakou, T., Müller, J.-F., De Smedt, I., Van Roozendael, M., van der Werf, G. R., Wiedinmyer, C., Kaiser, J. W., Sindelarova, K., and Guenther, A.: Nine years of global hydrocarbon emissions based on source inversion of OMI formaldehyde observations, Atmos. Chem. Phys., 16, 10133–10158, https://doi.org/10.5194/acp-16-10133-2016, 2016.
Bobrowski, N., Honninger, G., Galle, B., and Platt, U.: Detection of bromine monoxide in a volcanic plume, Nature, 423, 273–276, 2003.
Che, H., Wang, Y., and Sun, J.: Aerosol optical properties at Mt. Waliguan Observatory, China, Atmos. Environ., 45, 6004–6009, https://doi.org/10.1016/j.atmosenv.2011.07.050, 2011.
Coburn, S., Dix, B., Sinreich, R., and Volkamer, R.: The CU ground MAX-DOAS instrument: characterization of RMS noise limitations and first measurements near Pensacola, FL of BrO, IO, and CHOCHO, Atmos. Meas. Tech., 4, 2421–2439, https://doi.org/10.5194/amt-4-2421-2011, 2011.
De Mazière, M., Thompson, A. M., Kurylo, M. J., Wild, J. D., Bernhard, G., Blumenstock, T., Braathen, G. O., Hannigan, J. W., Lambert, J.-C., Leblanc, T., McGee, T. J., Nedoluha, G., Petropavlovskikh, I., Seckmeyer, G., Simon, P. C., Steinbrecht, W., and Strahan, S. E.: The Network for the Detection of Atmospheric Composition Change (NDACC): history, status and perspectives, Atmos. Chem. Phys., 18, 4935–4964, https://doi.org/10.5194/acp-18-4935-2018, 2018.
Dentener, F., Kinne, S., Bond, T., Boucher, O., Cofala, J., Generoso, S., Ginoux, P., Gong, S., Hoelzemann, J. J., Ito, A., Marelli, L., Penner, J. E., Putaud, J.-P., Textor, C., Schulz, M., van der Werf, G. R., and Wilson, J.: Emissions of primary aerosol and precursor gases in the years 2000 and 1750 prescribed data-sets for AeroCom, Atmos. Chem. Phys., 6, 4321–4344, https://doi.org/10.5194/acp-6-4321-2006, 2006.
De Smedt, I., Stavrakou, T., Hendrick, F., Danckaert, T., Vlemmix, T., Pinardi, G., Theys, N., Lerot, C., Gielen, C., Vigouroux, C., Hermans, C., Fayt, C., Veefkind, P., Müller, J.-F., and Van Roozendael, M.: Diurnal, seasonal and long-term variations of global formaldehyde columns inferred from combined OMI and GOME-2 observations, Atmos. Chem. Phys., 15, 12519–12545, https://doi.org/10.5194/acp-15-12519-2015, 2015.
Fioletov, V. E., McLinden, C. A., Krotkov, N., Li, C., Joiner, J., Theys, N., Carn, S., and Moran, M. D.: A global catalogue of large SO2 sources and emissions derived from the Ozone Monitoring Instrument, Atmos. Chem. Phys., 16, 11497–11519, https://doi.org/10.5194/acp-16-11497-2016, 2016.
Fitzenberger, R., Bösch, H., Camy, Peyret, C., Chipperfield, M. P., Harder, H., Platt, U., Sinnhuber, B. M., Wagner, T., and Pfeilsticker, K.: First profile measurements of tropospheric BrO, Geophys. Res. Lett., 27, 2921–2924, https://doi.org/10.1029/2000gl011531, 2000.
Forster, P., Ramasway, V., Artaxo, P., Berntsen, T., Betts, R., Fahey, D. W., Haywood, J., Lean, J., Lowe, D. C., Myhre, G., Nganga, J., Prinn, R., Raga, G., Schulz, M., and Van Dorlando, R.: Changes in atmospheric constituents and in radiative forcing, in: Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Solomon, S., Qin, D., Manning, M., Chen, Z., Marquist, M., Averyt, K. B., Tignor, M., and Miller, H. L., Cambridge Univ. Press, Cambridge, UK, and New York, USA, 129–234, 2007.
Frieß, U., Hollwedel, J., König-Langlo, G., Wagner, T., and Platt, U.: Dynamics and chemistry of tropospheric bromine explosion events in the Antarctic coastal region, J. Geophys. Res., 109, D06305, https://doi.org/10.1029/2003jd004133, 2004.
Georgoulias, A. K., van der A, R. J., Stammes, P., Boersma, K. F., and Eskes, H. J.: Trends and trend reversal detection in 2 decades of tropospheric NO2 satellite observations, Atmos. Chem. Phys., 19, 6269–6294, https://doi.org/10.5194/acp-19-6269-2019, 2019.
Gielen, C., Van Roozendael, M., Hendrick, F., Pinardi, G., Vlemmix, T., De Bock, V., De Backer, H., Fayt, C., Hermans, C., Gillotay, D., and Wang, P.: A simple and versatile cloud-screening method for MAX-DOAS retrievals, Atmos. Meas. Tech., 7, 3509–3527, https://doi.org/10.5194/amt-7-3509-2014, 2014.
Gil-Ojeda, M., Navarro-Comas, M., Gómez-Martín, L., Adame, J. A., Saiz-Lopez, A., Cuevas, C. A., González, Y., Puentedura, O., Cuevas, E., Lamarque, J.-F., Kinninson, D., and Tilmes, S.: NO2 seasonal evolution in the north subtropical free troposphere, Atmos. Chem. Phys., 15, 10567–10579, https://doi.org/10.5194/acp-15-10567-2015, 2015.
Gomez, L., Navarro-Comas, M., Puentedura, O., Gonzalez, Y., Cuevas, E., and Gil-Ojeda, M.: Long-path averaged mixing ratios of O3 and NO2 in the free troposphere from mountain MAX-DOAS, Atmos. Meas. Tech., 7, 3373–3386, https://doi.org/10.5194/amt-7-3373-2014, 2014.
Granier, C., Bessagnet, B., Bond, T., D'Angiola, A., Denier van der Gon, H., Frost, G. J., Heil, A., Kaiser, J. W., Kinne, S., Klimont, Z., Kloster, S., Lamarque, J.-F., Liousse, C., Masui, T., Meleux, F., Mieville, A., Ohara, T., Raut, J.-C., Riahi, K., Schultz, M. G., Smith, S. J., Thompson, A., van Aardenne, J., van der Werf, G. R., and van Vuuren, D. P.: Evolution of anthropogenic and biomass burning emissions of air pollutants at global and regional scales during the 1980–2010 period, Climatic Change, 109, 163, https://doi.org/10.1007/s10584-011-0154-1, 2011.
Hammer, M. S., Martin, R. V., Li, C., Torres, O., Manning, M., and Boys, B. L.: Insight into global trends in aerosol composition from 2005 to 2015 inferred from the OMI Ultraviolet Aerosol Index, Atmos. Chem. Phys., 18, 8097–8112, https://doi.org/10.5194/acp-18-8097-2018, 2018.
Harder, H., Camy, Peyret, C., Ferlemann, F., Fitzenberger, R., Hawat, T., Osterkamp, H., Schneider, M., Perner, D., Platt, U., Vradelis, P., and Pfeilsticker, K.: Stratospheric BrO profiles measured at different latitudes and seasons: Atmospheric observations, Geophys. Res. Lett., 25, 3843–3846, https://doi.org/10.1029/1998gl900026, 1998.
Hendrick, F., Van Roozendael, M., Chipperfield, M. P., Dorf, M., Goutail, F., Yang, X., Fayt, C., Hermans, C., Pfeilsticker, K., Pommereau, J.-P., Pyle, J. A., Theys, N., and De Mazière, M.: Retrieval of stratospheric and tropospheric BrO profiles and columns using ground-based zenith-sky DOAS observations at Harestua, 60∘ N, Atmos. Chem. Phys., 7, 4869–4885, https://doi.org/10.5194/acp-7-4869-2007, 2007.
Hendrick, F., Müller, J.-F., Clémer, K., Wang, P., De Mazière, M., Fayt, C., Gielen, C., Hermans, C., Ma, J. Z., Pinardi, G., Stavrakou, T., Vlemmix, T., and Van Roozendael, M.: Four years of ground-based MAX-DOAS observations of HONO and NO2 in the Beijing area, Atmos. Chem. Phys., 14, 765–781, https://doi.org/10.5194/acp-14-765-2014, 2014.
Hönninger, G. and Platt, U.: Observations of BrO and its vertical distribution during surface ozone depletion at Alert, Atmos. Environ., 36, 2481–2489, 2002.
Hönninger, G., von Friedeburg, C., and Platt, U.: Multi axis differential optical absorption spectroscopy (MAX-DOAS), Atmos. Chem. Phys., 4, 231–254, https://doi.org/10.5194/acp-4-231-2004, 2004.
Jin, J., Ma, J., Lin, W., Zhao, H., Shaiganfar, R., Beirle, S., and Wagner, T.: MAX-DOAS measurements and satellite validation of tropospheric NO2 and SO2 vertical column densities at a rural site of North China, Atmos. Environ., 133, 12–25, https://doi.org/10.1016/j.atmosenv.2016.03.031, 2016.
Klimont, Z., Kupiainen, K., Heyes, C., Purohit, P., Cofala, J., Rafaj, P., Borken-Kleefeld, J., and Schöpp, W.: Global anthropogenic emissions of particulate matter including black carbon, Atmos. Chem. Phys., 17, 8681–8723, https://doi.org/10.5194/acp-17-8681-2017, 2017.
Krotkov, N. A., McLinden, C. A., Li, C., Lamsal, L. N., Celarier, E. A., Marchenko, S. V., Swartz, W. H., Bucsela, E. J., Joiner, J., Duncan, B. N., Boersma, K. F., Veefkind, J. P., Levelt, P. F., Fioletov, V. E., Dickerson, R. R., He, H., Lu, Z., and Streets, D. G.: Aura OMI observations of regional SO2 and NO2 pollution changes from 2005 to 2015, Atmos. Chem. Phys., 16, 4605–4629, https://doi.org/10.5194/acp-16-4605-2016, 2016.
Lampel, J., Zielcke, J., Schmitt, S., Pöhler, D., Frieß, U., Platt, U., and Wagner, T.: Detection of O4 absorption around 328 and 419 nm in measured atmospheric absorption spectra, Atmos. Chem. Phys., 18, 1671–1683, https://doi.org/10.5194/acp-18-1671-2018, 2018.
Lary, D. J.: Halogens and the chemistry of the free troposphere, Atmos. Chem. Phys., 5, 227–237, https://doi.org/10.5194/acp-5-227-2005, 2005.
Lee, D. S., Köhler, I., Grobler, E., Rohrer, F., Sausen, R., Gallardo-Klenner, L., Olivier, J. G. J., Dentener, F. J., and Bouwman, A. F.: Estimations of global no, emissions and their uncertainties, Atmos. Environ., 31, 1735–1749, https://doi.org/10.1016/S1352-2310(96)00327-5, 1997.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Leser, H., Hönninger, G., and Platt, U.: MAX-DOAS measurements of BrO and NO2 in the marine boundary layer, Geophys. Res. Lett., 30, 1537, https://doi.org/10.1029/2002gl015811, 2003.
Li, C., Martin, R. V., van Donkelaar, A., Boys, B. L., Hammer, M. S., Xu, J.-W., Marais, E. A., Reff, A., Strum, M., Ridley, D. A., Crippa, M., Brauer, M., and Zhang, Q.: Trends in Chemical Composition of Global and Regional Population-Weighted Fine Particulate Matter Estimated for 25 Years, Environ. Sci. Technol., 51, 11185–11195, https://doi.org/10.1021/acs.est.7b02530, 2017.
Lin, W., Xu, X., Yu, X., Zhang, X., and Huang, J.: Observed levels and trends of gaseous SO2 and HNO3 at Mt. Waliguan, China: Results from 1997 to 2009, J. Environ. Sci., 25, 726–734, https://doi.org/10.1016/S1001-0742(12)60143-0, 2013.
Lübcke, P., Lampel, J., Arellano, S., Bobrowski, N., Dinger, F., Galle, B., Garzón, G., Hidalgo, S., Chacón Ortiz, Z., Vogel, L., Warnach, S., and Platt, U.: Retrieval of absolute SO2 column amounts from scattered-light spectra: implications for the evaluation of data from automated DOAS networks, Atmos. Meas. Tech., 9, 5677–5698, https://doi.org/10.5194/amt-9-5677-2016, 2016.
Luo, Y., Si, F., Zhou, H., Dou, K., Liu, Y., and Liu, W.: Observations and source investigations of the boundary layer bromine monoxide (BrO) in the Ny-Ålesund Arctic, Atmos. Chem. Phys., 18, 9789–9801, https://doi.org/10.5194/acp-18-9789-2018, 2018.
Ma, J., Tang, J., Zhou, X., and Zhang, X.: Estimates of the chemical budget for ozone at Waliguan Observatory J. Atmos. Chem., 41, 21–48, https://doi.org/10.1023/A:1013892308983, 2002a.
Ma, J., Zhou, X., and Hauglustaine, D.: Summertime tropospheric ozone over China simulated with a regional chemical transport model 2. Source contributions and budget, J. Geophys. Res.-Atmos., 107, 4612, https://doi.org/10.1029/2001jd001355, 2002b.
Ma, J. Z., Chen, Y., Wang, W., Yan, P., Liu, H. J., Yang, S. Y., Hu, Z. J., and Lelieveld, J.: Strong air pollution causes widespread haze-clouds over China, J. Geophys. Res., 115, D18204, https://doi.org/10.1029/2009jd013065, 2010.
Ma, J. Z., Wang, W., Chen, Y., Liu, H. J., Yan, P., Ding, G. A., Wang, M. L., Sun, J., and Lelieveld, J.: The IPAC-NC field campaign: a pollution and oxidization pool in the lower atmosphere over Huabei, China, Atmos. Chem. Phys., 12, 3883–3908, https://doi.org/10.5194/acp-12-3883-2012, 2012.
Ma, J. Z., Beirle, S., Jin, J. L., Shaiganfar, R., Yan, P., and Wagner, T.: Tropospheric NO2 vertical column densities over Beijing: results of the first three years of ground-based MAX-DOAS measurements (2008–2011) and satellite validation, Atmos. Chem. Phys., 13, 1547–1567, https://doi.org/10.5194/acp-13-1547-2013, 2013.
Madronich, S. and Calvert, J. G.: The NCAR master mechanism of the gas-phase chemistry-Version 2.0, The National Center for Atmospheric Research, Boulder, Colorado, Rep. NCAR/TN-333+STR, 1989.
Martin, M., Pöhler, D., Seitz, K., Sinreich, R., and Platt, U.: BrO measurements over the Eastern North-Atlantic, Atmos. Chem. Phys., 9, 9545–9554, https://doi.org/10.5194/acp-9-9545-2009, 2009.
Meng, Z.-Y., Xu, X.-B., Wang, T., Zhang, X.-Y., Yu, X.-L., Wang, S.-F., Lin, W.-L., Chen, Y.-Z., Jiang, Y.-A., and An, X.-Q.: Ambient sulfur dioxide, nitrogen dioxide, and ammonia at ten background and rural sites in China during 2007–2008, Atmos. Environ., 44, 2625–2631, https://doi.org/10.1016/j.atmosenv.2010.04.008, 2010.
Mu, Y., Pang, X., Quan, J., and Zhang, X.: Atmospheric carbonyl compounds in Chinese background area: A remote mountain of the Qinghai-Tibetan Plateau, J. Geophys. Res.-Atmos., 112, D22302, https://doi.org/10.1029/2006jd008211, 2007.
Peterson, P. K., Simpson, W. R., Pratt, K. A., Shepson, P. B., Frieß, U., Zielcke, J., Platt, U., Walsh, S. J., and Nghiem, S. V.: Dependence of the vertical distribution of bromine monoxide in the lower troposphere on meteorological factors such as wind speed and stability, Atmos. Chem. Phys., 15, 2119–2137, https://doi.org/10.5194/acp-15-2119-2015, 2015.
Peterson, P. K., Pöhler, D., Sihler, H., Zielcke, J., General, S., Frieß, U., Platt, U., Simpson, W. R., Nghiem, S. V., Shepson, P. B., Stirm, B. H., Dhaniyala, S., Wagner, T., Caulton, D. R., Fuentes, J. D., and Pratt, K. A.: Observations of bromine monoxide transport in the Arctic sustained on aerosol particles, Atmos. Chem. Phys., 17, 7567–7579, https://doi.org/10.5194/acp-17-7567-2017, 2017.
Platt, U. and Hönninger, G.: The role of halogen species in the troposphere, Chemosphere, 52, 325–338, 2003.
Prados-Roman, C., Gómez-Martín, L., Puentedura, O., Navarro-Comas, M., Iglesias, J., de Mingo, J. R., Pérez, M., Ochoa, H., Barlasina, M. E., Carbajal, G., and Yela, M.: Reactive bromine in the low troposphere of Antarctica: estimations at two research sites, Atmos. Chem. Phys., 18, 8549–8570, https://doi.org/10.5194/acp-18-8549-2018, 2018.
Richter, A., Wittrock, F., Ladstätter-Weissenmayer, A., and Burrows, J. P.: GOME measurements of stratospheric and tropospheric BrO, Adv. Space Res., 29, 1667–1672, 2002.
Roscoe, H. K., Brough, N., Jones, A. E., Wittrock, F., Richter, A., Van Roozendael, M., and Hendrick, F.: Resolution of an important discrepancy between remote and in-situ measurements of tropospheric BrO during Antarctic enhancements, Atmos. Meas. Tech. Discuss., 5, 5419–5448, https://doi.org/10.5194/amtd-5-5419-2012, 2012.
Saiz-Lopez, A., Plane, J. M. C., and Shillito, J. A.: Bromine oxide in the mid-latitude marine boundary layer, Geophys. Res. Lett., 31, L03111, https://doi.org/10.1029/2003gl018956, 2004.
Schreier, S. F., Richter, A., Wittrock, F., and Burrows, J. P.: Estimates of free-tropospheric NO2 and HCHO mixing ratios derived from high-altitude mountain MAX-DOAS observations at midlatitudes and in the tropics, Atmos. Chem. Phys., 16, 2803–2817, https://doi.org/10.5194/acp-16-2803-2016, 2016.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd Edn., Hohn Wiley & Sons Inc., Hoboken, New Jersey, 2006.
Simpson, W. R., Peterson, P. K., Frieß, U., Sihler, H., Lampel, J., Platt, U., Moore, C., Pratt, K., Shepson, P., Halfacre, J., and Nghiem, S. V.: Horizontal and vertical structure of reactive bromine events probed by bromine monoxide MAX-DOAS, Atmos. Chem. Phys., 17, 9291–9309, https://doi.org/10.5194/acp-17-9291-2017, 2017.
Stavrakou, T., Müller, J.-F., De Smedt, I., Van Roozendael, M., van der Werf, G. R., Giglio, L., and Guenther, A.: Evaluating the performance of pyrogenic and biogenic emission inventories against one decade of space-based formaldehyde columns, Atmos. Chem. Phys., 9, 1037–1060, https://doi.org/10.5194/acp-9-1037-2009, 2009.
Stohl, A., Aamaas, B., Amann, M., Baker, L. H., Bellouin, N., Berntsen, T. K., Boucher, O., Cherian, R., Collins, W., Daskalakis, N., Dusinska, M., Eckhardt, S., Fuglestvedt, J. S., Harju, M., Heyes, C., Hodnebrog, Ø., Hao, J., Im, U., Kanakidou, M., Klimont, Z., Kupiainen, K., Law, K. S., Lund, M. T., Maas, R., MacIntosh, C. R., Myhre, G., Myriokefalitakis, S., Olivié, D., Quaas, J., Quennehen, B., Raut, J.-C., Rumbold, S. T., Samset, B. H., Schulz, M., Seland, Ø., Shine, K. P., Skeie, R. B., Wang, S., Yttri, K. E., and Zhu, T.: Evaluating the climate and air quality impacts of short-lived pollutants, Atmos. Chem. Phys., 15, 10529–10566, https://doi.org/10.5194/acp-15-10529-2015, 2015.
Stutz, J., Ackermann, R., Fast, J. D., and Barrie, L.: Atmospheric reactive chlorine and bromine at the Great Salt Lake, Utah, Geophys. Res. Lett., 29, 1380, https://doi.org/10.1029/2002gl014812, 2002.
Tang, J., Wen, Y. P., Xu, X. B., Zheng, X. D., Guo, S., and Zhao, Y. C.: China Global Atmosphere Watch Baseline Observatory and its measurement program, in: CAMS Annual Report 1994–1995, Beijing, 56–65, 1995.
Theys, N., Van Roozendael, M., Hendrick, F., Fayt, C., Hermans, C., Baray, J.-L., Goutail, F., Pommereau, J.-P., and De Mazière, M.: Retrieval of stratospheric and tropospheric BrO columns from multi-axis DOAS measurements at Reunion Island (21∘ S, 56∘ E), Atmos. Chem. Phys., 7, 4733–4749, https://doi.org/10.5194/acp-7-4733-2007, 2007.
Tulet, P., Di Muro, A., Colomb, A., Denjean, C., Duflot, V., Arellano, S., Foucart, B., Brioude, J., Sellegri, K., Peltier, A., Aiuppa, A., Barthe, C., Bhugwant, C., Bielli, S., Boissier, P., Boudoire, G., Bourrianne, T., Brunet, C., Burnet, F., Cammas, J.-P., Gabarrot, F., Galle, B., Giudice, G., Guadagno, C., Jeamblu, F., Kowalski, P., Leclair de Bellevue, J., Marquestaut, N., Mékies, D., Metzger, J.-M., Pianezze, J., Portafaix, T., Sciare, J., Tournigand, A., and Villeneuve, N.: First results of the Piton de la Fournaise STRAP 2015 experiment: multidisciplinary tracking of a volcanic gas and aerosol plume, Atmos. Chem. Phys., 17, 5355–5378, https://doi.org/10.5194/acp-17-5355-2017, 2017.
Van Roozendael, M., Wagner, T., Richter, A., Pundt, I., Arlander, D., Burrows, J. P., Chipperfield, M., Fayt, C., Johnston, P. V., Lambert, J.-C., Kreher, K., Pfeilsticker, K., Platt, U., Pommereau, J.-P., Sinnhuber, B.-M., Tornkvist, K. K., and Wittrock, F.: Intercomparison of BrO measurements from ERS-2 GOME, ground-based and balloon platforms, Adv. Space Res., 29, 1661–1666, 2002.
Van Roozendael, M., Fayt, C., Post, P., Hermans, C., and Lambert, J.-C.: Retrieval of BrO and NO2 from UV-Visible Observations, in: Sounding the troposphere from space: a new Era for Atmospheric Chemistry, The TROPOSAT Final Report, edited by: Borrell, P., Borrell, P. M., Burrows, J. P., and Platt, U., Springer, Dordrecht, 155–166, 2003.
Volkamer, R., Baidar, S., Campos, T. L., Coburn, S., DiGangi, J. P., Dix, B., Eloranta, E. W., Koenig, T. K., Morley, B., Ortega, I., Pierce, B. R., Reeves, M., Sinreich, R., Wang, S., Zondlo, M. A., and Romashkin, P. A.: Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2–O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements, Atmos. Meas. Tech., 8, 2121–2148, https://doi.org/10.5194/amt-8-2121-2015, 2015.
von Glasow, R. and Crutzen, P. J.: Tropospheric Halogen Chemistry, in: Treatise on Geochemistry Update1, edited by: Holland, H. D. and Turekian, K. K., Elsevier-Pergamon, Oxford, UK, 1–67, 2007.
von Glasow, R., von Kuhlmann, R., Lawrence, M. G., Platt, U., and Crutzen, P. J.: Impact of reactive bromine chemistry in the troposphere, Atmos. Chem. Phys., 4, 2481–2497, https://doi.org/10.5194/acp-4-2481-2004, 2004.
Wagner, T., Dix, B., Friedeburg, C. v., Frieß, U., Sanghavi, S., Sinreich, R., and Platt, U.: MAX-DOAS O4 measurements: A new technique to derive information on atmospheric aerosols – Principles and information content, J. Geophys. Res., 109, D22205, https://doi.org/10.1029/2004jd004904, 2004.
Wagner, T., Burrows, J. P., Deutschmann, T., Dix, B., von Friedeburg, C., Frieß, U., Hendrick, F., Heue, K.-P., Irie, H., Iwabuchi, H., Kanaya, Y., Keller, J., McLinden, C. A., Oetjen, H., Palazzi, E., Petritoli, A., Platt, U., Postylyakov, O., Pukite, J., Richter, A., van Roozendael, M., Rozanov, A., Rozanov, V., Sinreich, R., Sanghavi, S., and Wittrock, F.: Comparison of box-air-mass-factors and radiances for Multiple-Axis Differential Optical Absorption Spectroscopy (MAX-DOAS) geometries calculated from different UV/visible radiative transfer models, Atmos. Chem. Phys., 7, 1809–1833, https://doi.org/10.5194/acp-7-1809-2007, 2007a.
Wagner, T., Ibrahim, O., Sinreich, R., Frieß, U., von Glasow, R., and Platt, U.: Enhanced tropospheric BrO over Antarctic sea ice in mid winter observed by MAX-DOAS on board the research vessel Polarstern, Atmos. Chem. Phys., 7, 3129–3142, https://doi.org/10.5194/acp-7-3129-2007, 2007b.
Wagner, T., Apituley, A., Beirle, S., Dörner, S., Friess, U., Remmers, J., and Shaiganfar, R.: Cloud detection and classification based on MAX-DOAS observations, Atmos. Meas. Tech., 7, 1289–1320, https://doi.org/10.5194/amt-7-1289-2014, 2014.
Wagner, T., Beirle, S., Remmers, J., Shaiganfar, R., and Wang, Y.: Absolute calibration of the colour index and O4 absorption derived from Multi AXis (MAX-)DOAS measurements and their application to a standardised cloud classification algorithm, Atmos. Meas. Tech., 9, 4803–4823, https://doi.org/10.5194/amt-9-4803-2016, 2016.
Wang, T., Wong, H. L. A., Tang, J., Ding, A., Wu, W. S., and Zhang, X. C.: On the origin of surface ozone and reactive nitrogen observed at a remote mountain site in the northeastern Qinghai-Tibetan Plateau, western China, J. Geophys. Res.-Atmos., 111, D08303, https://doi.org/10.1029/2005jd006527, 2006.
Wang, T., Hendrick, F., Wang, P., Tang, G., Clémer, K., Yu, H., Fayt, C., Hermans, C., Gielen, C., Müller, J.-F., Pinardi, G., Theys, N., Brenot, H., and Van Roozendael, M.: Evaluation of tropospheric SO2 retrieved from MAX-DOAS measurements in Xianghe, China, Atmos. Chem. Phys., 14, 11149–11164, https://doi.org/10.5194/acp-14-11149-2014, 2014.
Wang, Y., Lampel, J., Xie, P., Beirle, S., Li, A., Wu, D., and Wagner, T.: Ground-based MAX-DOAS observations of tropospheric aerosols, NO2, SO2 and HCHO in Wuxi, China, from 2011 to 2014, Atmos. Chem. Phys., 17, 2189–2215, https://doi.org/10.5194/acp-17-2189-2017, 2017.
Werner, B., Stutz, J., Spolaor, M., Scalone, L., Raecke, R., Festa, J., Colosimo, S. F., Cheung, R., Tsai, C., Hossaini, R., Chipperfield, M. P., Taverna, G. S., Feng, W., Elkins, J. W., Fahey, D. W., Gao, R.-S., Hintsa, E. J., Thornberry, T. D., Moore, F. L., Navarro, M. A., Atlas, E., Daube, B. C., Pittman, J., Wofsy, S., and Pfeilsticker, K.: Probing the subtropical lowermost stratosphere and the tropical upper troposphere and tropopause layer for inorganic bromine, Atmos. Chem. Phys., 17, 1161–1186, https://doi.org/10.5194/acp-17-1161-2017, 2017.
Wittrock, F., Oetjen, H., Richter, A., Fietkau, S., Medeke, T., Rozanov, A., and Burrows, J. P.: MAX-DOAS measurements of atmospheric trace gases in Ny-Ålesund – Radiative transfer studies and their application, Atmos. Chem. Phys., 4, 955–966, https://doi.org/10.5194/acp-4-955-2004, 2004.
Xing, J., Mathur, R., Pleim, J., Hogrefe, C., Gan, C.-M., Wong, D. C., Wei, C., Gilliam, R., and Pouliot, G.: Observations and modeling of air quality trends over 1990–2010 across the Northern Hemisphere: China, the United States and Europe, Atmos. Chem. Phys., 15, 2723–2747, https://doi.org/10.5194/acp-15-2723-2015, 2015.
Xue, L. K., Wang, T., Zhang, J. M., Zhang, X. C., Deliger, Poon, C. N., Ding, A. J., Zhou, X. H., Wu, W. S., Tang, J., Zhang, Q. Z., and Wang, W. X.: Source of surface ozone and reactive nitrogen speciation at Mount Waliguan in western China: New insights from the 2006 summer study, J. Geophys. Res.-Atmos., 116, D07306, https://doi.org/10.1029/2010jd014735, 2011.
Xue, L. K., Wang, T., Guo, H., Blake, D. R., Tang, J., Zhang, X. C., Saunders, S. M., and Wang, W. X.: Sources and photochemistry of volatile organic compounds in the remote atmosphere of western China: results from the Mt. Waliguan Observatory, Atmos. Chem. Phys., 13, 8551–8567, https://doi.org/10.5194/acp-13-8551-2013, 2013.
Yang, X., Cox, R. A., Warwick, N. J., Pyle, J. A., Carver, G. D., O'Connor, F. M., and Savage, N. H.: Tropospheric bromine chemistry and its impacts on ozone: A model study, J. Geophys. Res., 110, D23311, https://doi.org/10.1029/2005jd006244, 2005.
Yang, X., Pyle, J. A., Cox, R. A., Theys, N., and Van Roozendael, M.: Snow-sourced bromine and its implications for polar tropospheric ozone, Atmos. Chem. Phys., 10, 7763–7773, https://doi.org/10.5194/acp-10-7763-2010, 2010.
Yu, X., Tang, J., Li, X. S., and Liang, B. S.: Observation and analysis of SO2 and NO2 in clean air of western China, Q. J. Appl. Meteorol., 8, 62–68, 1997.
Zhao, B., Wang, P., Ma, J. Z., Zhu, S., Pozzer, A., and Li, W.: A high-resolution emission inventory of primary pollutants for the Huabei region, China, Atmos. Chem. Phys., 12, 481–501, https://doi.org/10.5194/acp-12-481-2012, 2012.
Ziemke, J. R., Oman, L. D., Strode, S. A., Douglass, A. R., Olsen, M. A., McPeters, R. D., Bhartia, P. K., Froidevaux, L., Labow, G. J., Witte, J. C., Thompson, A. M., Haffner, D. P., Kramarova, N. A., Frith, S. M., Huang, L.-K., Jaross, G. R., Seftor, C. J., Deland, M. T., and Taylor, S. L.: Trends in global tropospheric ozone inferred from a composite record of TOMS/OMI/MLS/OMPS satellite measurements and the MERRA-2 GMI simulation , Atmos. Chem. Phys., 19, 3257–3269, https://doi.org/10.5194/acp-19-3257-2019, 2019.
- Abstract
- Introduction
- Field experiment
- Spectral retrieval
- Radiative transfer simulations
- Identification of measurements made under cloudy-sky conditions and high aerosol loads
- Results
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Field experiment
- Spectral retrieval
- Radiative transfer simulations
- Identification of measurements made under cloudy-sky conditions and high aerosol loads
- Results
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement