the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Jun 2020
| 03 Jun 2020
Photochemical transformation of residential wood combustion emissions: dependence of organic aerosol composition on OH exposure
Petri Tiitta
Mika Ihalainen
Pasi Yli-Pirilä
Jürgen Orasche
Hendryk Czech
Miika Kortelainen
Heikki Lamberg
Heikki Suhonen
Hanna Koponen
Liqing Hao
Ralf Zimmermann
Jorma Jokiniemi
Jarkko Tissari
Olli Sippula
Residential wood combustion (RWC) emits large amounts of gaseous and particulate organic aerosol (OA). In the atmosphere, the emission is transformed via oxidative reactions, which are under daylight conditions driven mainly by hydroxyl radicals (OH). This continuing oxidative ageing produces secondary OA and may change the health- and climate-related properties of the emission. However, it is not well known how the composition of RWC-originated OA changes as the function of OH exposure. In this work, emissions from two modern residential logwood combustion appliances were photochemically aged in an oxidation flow reactor (OFR) with various OH exposure levels, reaching up to 6×1011 s cm−3 (equivalent to 1 week in the atmosphere). Gaseous organic compounds were analysed by proton transfer reaction time-of-flight mass spectrometry (PTR-ToF-MS), while particulate OA was analysed online by a high-resolution soot particle aerosol mass spectrometer (SP-HR-ToF-AMS) and offline by in situ derivatization thermal desorption–gas chromatography–time-of-flight mass spectrometry (IDTD-GC-ToF-MS). Photochemical reactions increased the mass of particulate organic carbon by a factor of 1.3–3.9. The increase in mass took place during the first atmospheric equivalent day of ageing, after which the enhancement was independent of the extent of photochemical exposure. However, ageing increased the oxidation state of the particulate OA linearly throughout the assessed range, with slopes between −0.17 and −0.49 in van Krevelen space. Ageing led to an increase in acidic fragmentation products in both phases, as measured by the IDTD-GC-ToF-MS for the particulate and PTR-ToF-MS for the gaseous phase. For the gaseous organic compounds, the formation of small carbonylic compounds combined with the rapid degradation of primary volatile organic compounds such as aromatic compounds led to a continuous increase in both the O : C and H : C ratios. Overall, the share of polycyclic aromatic compounds (PACs) in particles degraded rapidly during ageing, although some oxygen-substituted PACs, most notably naphthaldehydic acid, increased, in particular during relatively short exposures. Similarly, the concentrations of particulate nitrophenols rose extensively during the first atmospheric equivalent day. During continuous photochemical ageing, the dominant transformation mechanisms shifted from the initial gas-phase functionalization/condensation to the transformation of the particulate OA by further oxidation reactions and fragmentation. The observed continuous transformation of OA composition throughout a broad range of OH exposures indicates that the entire atmospheric lifetime of the emission needs to be explored to fully assess the potential climate and health effects of RWC emissions.
- Article
(2869 KB) - Full-text XML
-
Supplement
(3416 KB) - BibTeX
- EndNote





Biomass combustion is a major source of atmospheric particulate matter (PM) and is considered the main anthropogenic source of organic matter and the third largest contributor of black carbon (BC) emissions globally (Klimont et al., 2017). The use of wood fuels in small-scale residential settings is a main source for ambient organic aerosol (OA) in many parts of the world. For example, residential wood combustion (RWC) has been identified as a major source of ambient air fine particles in several European cities, where its relative contribution has been estimated to further increase in the future, while PM emissions from other sources, such as industry and traffic, are decreasing (Denier Van Der Gon et al., 2015; Klimont et al., 2017). The amount and contents of the RWC emissions depend greatly on combustion conditions, which are generally affected by the combustion procedure, fuel, and appliance technology (Bhattu et al., 2019; Nuutinen et al., 2014; Orasche et al., 2013; Tissari et al., 2009). In logwood-fired appliances, there is also a strong variation in the emissions during the different combustion phases of batches, with ignition producing the highest organic emissions (Bhattu et al., 2019; Kortelainen et al., 2018). However, the highest black carbon concentrations are emitted during the flaming phase, while the char burnout phase typically emits large amounts of carbon monoxide but low, mainly inorganic particulate emissions (Kortelainen et al., 2018). Combustion conditions also affect the emissions of many toxic compounds, such as polycyclic aromatic compounds (PACs) (Kim et al., 2013; Orasche et al., 2013), and are consequently strongly linked with the adverse health effects of the emissions (Bølling et al., 2009; Kanashova et al., 2018; Kasurinen et al., 2018).
Many of the main organic species in fresh, not atmospherically aged wood smoke are connected to the composition of the wood fuel, such as levoglucosan, a common biomass burning marker, and lignin degradation products such as methoxyphenols and their derivatives (Elsasser et al., 2013; McDonald et al., 2000; Orasche et al., 2013). In addition, the gaseous organic emission from RWC contains hundreds of organic gaseous compounds (OGCs) (Bhattu et al., 2019; Bruns et al., 2017; Hartikainen et al., 2018; Hatch et al., 2017; McDonald et al., 2000), a majority of which can be classified as volatile organic compounds (VOCs) (Hatch et al., 2017), meaning that under ambient conditions they exist purely in gaseous phase. Notably, RWC is an important anthropogenic source for OGCs with high potential for secondary particulate organic aerosol (SOA) formation. The most potent SOA-precursor compounds include aromatic hydrocarbons and oxygenated species, such as phenolic and furanoic compounds (Bruns et al., 2016; Hartikainen et al., 2018), and removal of these compounds via either improved combustion conditions or for example catalytic cleaning has been shown to be efficient in lowering the SOA potential of RWC emissions (Czech et al., 2017; Pieber et al., 2018). Furthermore, the gaseous emissions contain high amounts of carbonyls, such as formaldehyde and acetaldehyde, with adverse health effects (Reda et al., 2015; USEPA, 2019).
OA has an atmospheric lifetime of approximately 1 week (Hodzic et al., 2016), during which its chemical composition and potential environmental and health effects are likely to transform extensively. In daylight conditions, hydroxyl radicals (OH) dominate this ageing process, where the oxidation of OGCs forms a variety of functionalized products. Some of these oxidized secondary organic species partition into the particulate phase, resulting in an enhancement of ambient air particulate organic matter concentrations (Robinson et al., 2007). While aromatic compounds are noted as the main SOA precursors from RWC, the complete pathway for SOA formation and the final SOA yields of complex OGC mixtures under different atmospheric conditions remain unclear (Bruns et al., 2016; Hartikainen et al., 2018; Hatch et al., 2017; McFiggans et al., 2019). Recent experiments on RWC exhaust estimate the mass of particulate OA to increase by a factor of 1.6–5.3 within approximately 1 d of photochemical ageing (Bertrand et al., 2017; Bruns et al., 2015b; Grieshop et al., 2009; Heringa et al., 2011; Tiitta et al., 2016). In addition, heterogeneous oxidation reactions of the particulate matter may be significant during the atmospheric transformation of RWC emissions: it has been reported that only a minority of the primary particulate OA (POA) from biomass burning remains unreacted after a few hours of atmospheric ageing (Hennigan et al., 2011; Tiitta et al., 2016). Photochemical ageing also decomposes hydrocarbonaceous PACs (i.e. polycyclic aromatic hydrocarbons, PAHs), which cause adverse health effects due to their carcinogenic properties. On the other hand, atmospheric ageing may lead to formation of even more toxic, oxygen- or nitrogen-substituted PACs (Finlayson-Pitts and Pitts, 2000; Keyte et al., 2013; Miersch et al., 2019). For instance, these substituted PACs have been reported to cause a substantial part of the particle-induced mutagenicity in Beijing, with a contribution of only 8 % relative to the concentration of hydrocarbon PACs (Wang et al., 2011). Similarly, oxidation in nitrogen oxide (NOx)-rich conditions can produce nitrophenols, which are known to be harmful to plant growth and human health (Harrison et al., 2005) and have been identified as an important constituent of light-absorbing organic matter (“brown carbon”) (Moise et al., 2015; Zhang et al., 2011), thereby affecting atmospheric radiative forcing.
The photochemical ageing of RWC emissions has previously been studied mainly in smog chambers (Bertrand et al., 2017; Bruns et al., 2015a, b; Heringa et al., 2011; Tiitta et al., 2016), where the ageing was monitored as a batch process from fresh emission to up to 1 atmospheric equivalent day of exposure (eqv. d; assuming an ambient average OH concentration of 106 molec cm−3; Prinn et al., 2005). As an alternative, oxidation flow reactors (OFRs) with continuous sample flow have been increasingly utilized in combustion emission studies (Bruns et al., 2015a; Czech et al., 2017; Pieber et al., 2018). To achieve similar to or higher oxidant exposures than chambers in shorter residence times, OFRs have been used with high ozone concentrations together with high-intensity, low-wavelength UV lamps to generate OH-radical concentrations orders of magnitudes higher than those of smog chambers (Kang et al., 2007; Ihalainen et al., 2019; Simonen et al., 2017). Thus, OFRs enable measurements with better temporal resolution, which is a benefit when assessing the ageing of aerosols from dynamic sources, such as batchwise logwood combustion.
In this study, we aim to determine how atmospheric ageing changes the composition of organic aerosol, emitted by logwood-fired stoves, as the function of OH exposure. The atmospheric transformation of emissions from two RWC appliances fired with spruce and beech logwood was simulated using the photochemical emission ageing flow tube reactor (PEAR OFR; Ihalainen et al., 2019) at different exposure levels ranging up to a week of atmospheric age. The transformation of emissions and the formation of related secondary organic emissions were monitored with a comprehensive setup (Fig. 1), including analysis of gas-phase by proton transfer reactor time-of-flight mass spectrometry (PTR-ToF-MS) and investigation of the particulate phase online by aerosol mass spectrometry (SP-HR-ToF-AMS) and offline by targeted gas chromatography mass spectrometry and thermal–optical analyses. Together, these analyses enable the assessment of the OA chemical composition in both bulk and molecular level, from fresh emission to up to exposures representative of long-range transported smoke.
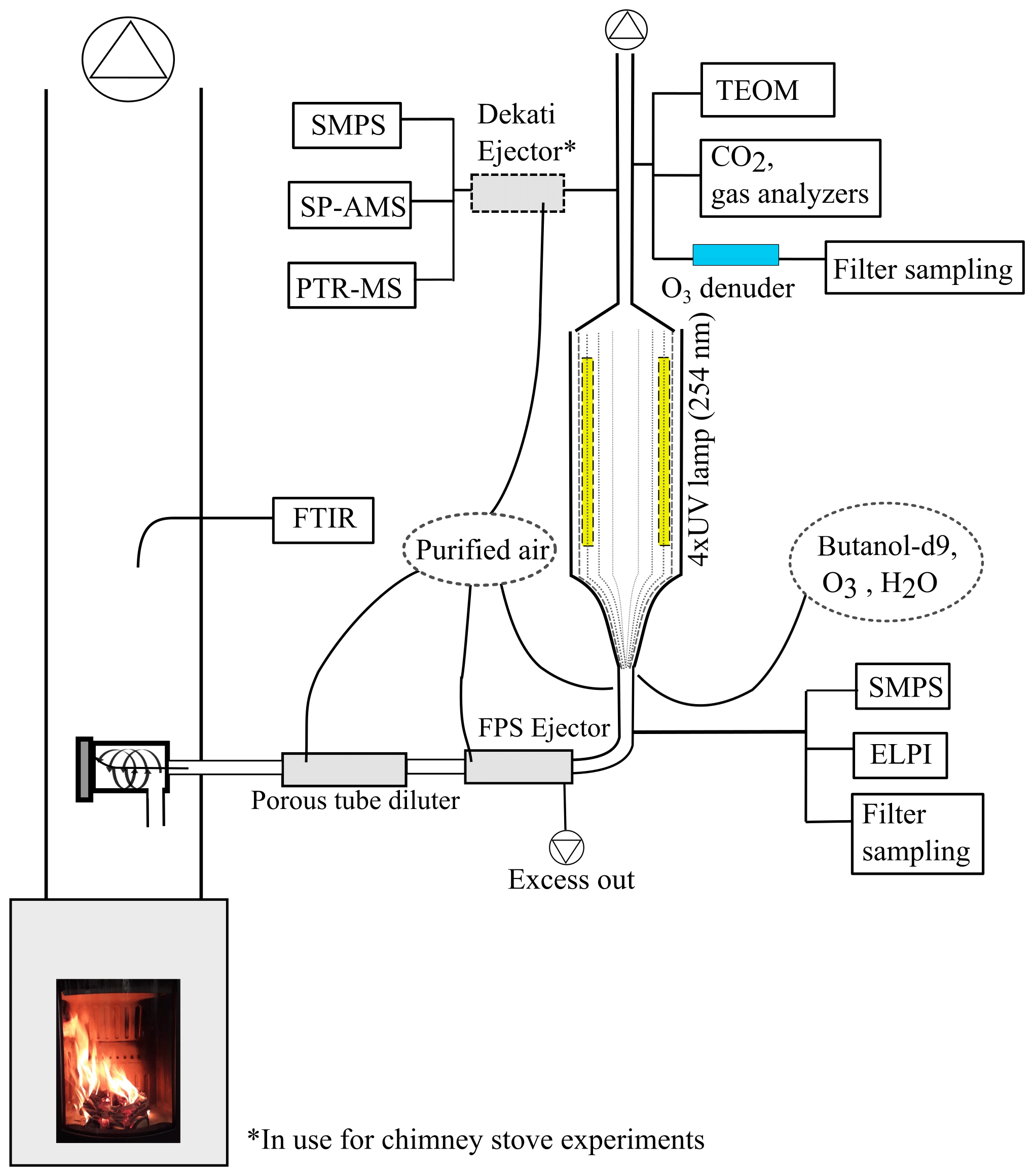

Figure 2CO2 and CO and the OGC in the dry exhaust gas from (a) masonry heater and (b) chimney stove, measured by FTIR. Averages over all experiments are shown with solid lines, whereas different experiments are shown with dotted lines. Average phase lengths are marked on the top panel with the yellow (ignition), green (flaming), and purple (burnout).
Table 1Combustion conditions and experimental conditions of each experiment.
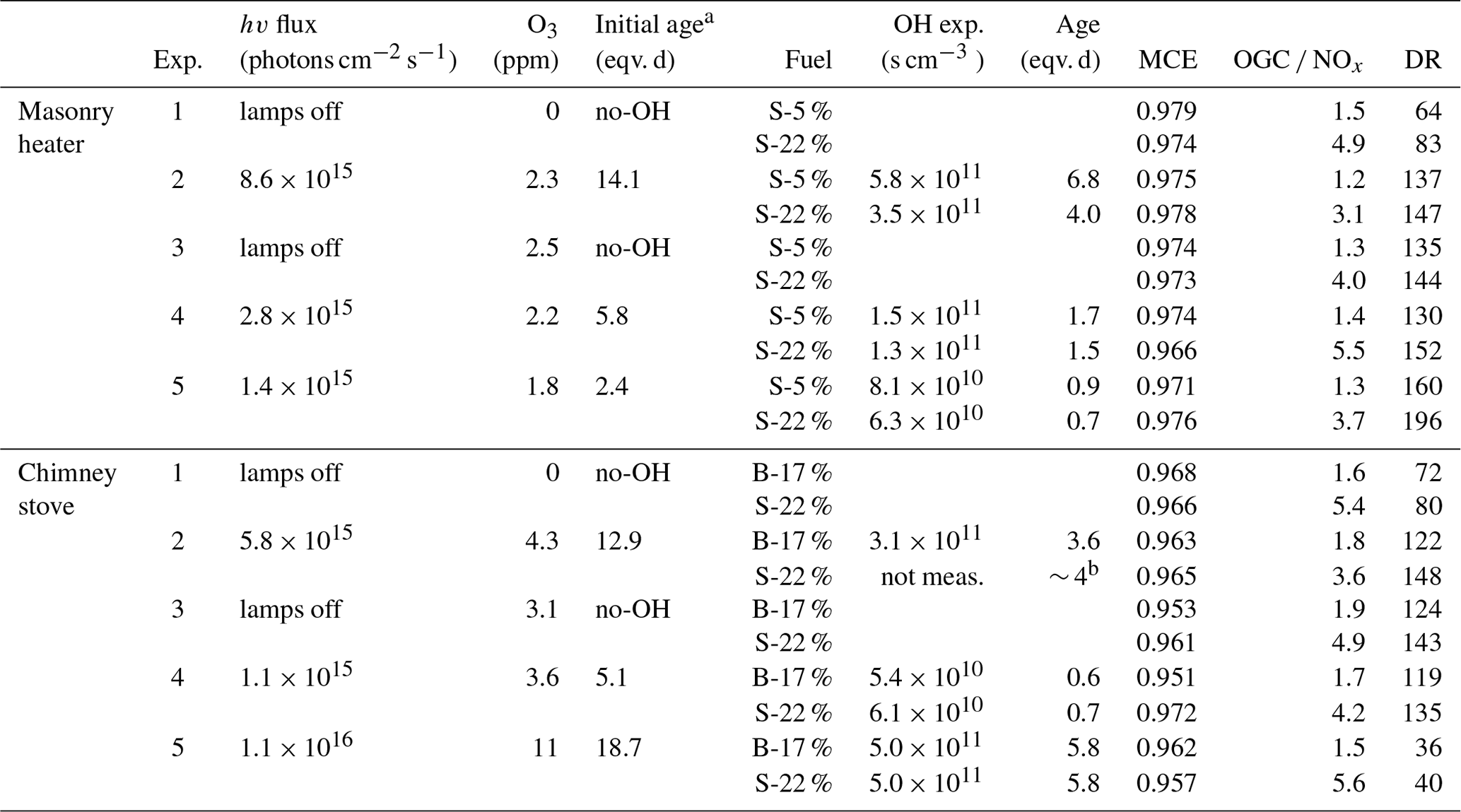
a Age based on the OH exposure of clean air prior to sample input. b Direct OH exposure measurements not available for chimney stove Exp. 2 S-22 %; it is approximately similar to Exp. 2 B-17 %.
2.1 Experimental conditions
Experiments were conducted in the ILMARI laboratory of the University of Eastern Finland (http://www.uef.fi/ilmari, last access: 20 May 2020) with the experimental setup shown in Fig. 1. The two combustion appliances used were modern stoves with improved air intakes. First, a heat-storing masonry heater (Hiisi 4, Tulikivi Ltd., Finland) representing the typical modern logwood combustion technology utilized in northern Europe, was fired with spruce logwood. The combustion procedure in the masonry heater consisted of three 2.5 kg batches (35 min combustion time) of spruce logwood representing kiln-dried fuel (5 % moisture content, S-5 %), after which there was a 25 min char burning period prior to two 45 min batches with moist (22 % moisture content, S-22 %) spruce logwood. Second, a non-heat-retaining chimney stove (Aduro 9.3, Denmark) representing central European modern logwood stoves was fired with both beech and spruce logs. The chimney stove experiments consisted of five 2 kg batches (combustion time 40–55 min) of beech logwood (17 % H2O, B-17 %), followed by two 50 min batches of S-22 %. Beech was used in these experiments because it is the most common firewood used in central Europe, while spruce is used both in northern and central Europe. For ignition, 150 g of dry kindling was placed on the top of the first batch in the cold furnace. Each batch was divided into three parts: ignition, flaming, and burnout phase. The ignition phase was determined to last from the beginning of the batch to the moment of batch maximum flue gas CO2 concentration, and the burnout phase began from the moment when the CO concentration started to elevate and remained at a high level until the end of the batch (Fig. 2). The modified combustion efficiency (MCE) as a function of time was calculated from primary flue-gas CO2 and CO concentrations as .
The exhaust was sampled from the stack with a 10 µm precut cyclone. The sample was diluted with a combination of a porous tube diluter and an ejector diluter (Dekati FPS ejector, Finland) and had a dilution ratio (DR) of 40–150 (Table 1) when fed to the PEAR OFR. In the chimney stove experiments, an additional ejector diluter with a DR of 8 (Dekati DI-1000, Finland) was placed before the secondary online aerosol instruments. Compared to the no-ageing experiments, a higher DR was selected for the ageing experiments in response to the expected increase in particulate matter in the PEAR OFR during photochemical ageing. Measured concentrations were corrected for the dilution and normalized to stoichiometric dry flue-gas by multiplication with the stoichiometric correction factor (SCF) of Eq. (1) based on the background CO2 concentration (CO2,bg), the CO2 concentration in the diluted sample downstream of the PEAR OFR (CO2,sec), and the fact that the dry flue gas of wood combustion with no excess air contains 20.2 % CO2.
In addition, the diluted concentrations were normalized to 13 % flue-gas oxygen content.
2.2 Use of the PEAR OFR
The PEAR OFR (Ihalainen et al., 2019) was used to continuously age the sample stream. In the setup, the extent of photon flux and consequential photochemical ageing were controlled by adjusting the voltage of the 254 nm UV lamps. The total flow through the PEAR OFR was 60 L min−1, and the residence time within the PEAR was 139 s. In addition to the 55 L min−1 sample flow, humified and purified air was introduced to the PEAR OFR to obtain a relative humidity of 45±5 %. In the PEAR OFR, the photolysis of the ozone forms hydroxyl radicals via Reactions (R1) and (R2). Thus, the extent of photochemical ageing in the PEAR OFR depends on the photon flux inside the reactor and the introduction of OH precursors, namely the externally fed O3 and H2O vapours.
In the ageing experiments, ozone and butanol-d9 mixed with the main sample flow before the reactor inlet (Fig. 1) resulted in concentrations of 2–11 ppm of O3 (Table 1) and 80–200 ppb of butanol-d9 in the PEAR OFR before initiation of photochemical ageing. The OH exposure (OHexp) was determined continuously based on the butanol-d9 concentrations downstream of the PEAR according to the method presented by Barmet et al. (2012). The OHexp for clean air in the PEAR prior to the exhaust input ranged from 8.2×1010 to 1.6×1012 molec cm−3 s, which is equivalent to 1–18 d in an ambient atmosphere (eqv. d) with an estimated 24 h average global OH concentration of 1×106 molec cm−3 (Prinn et al., 2005). The alternate reaction pathways in the PEAR OFR, namely exposure to photolysis (flux254 nm,exp), excited (O(1D)exp), atomic oxygen (O(3P)exp), and ozone (O3,exp), were assessed using the OFR exposure estimation equations of Peng et al. (2016) for OFRs with 254 nm lamps. Both the OHexp and the alternate reaction pathways were affected by the external OH reactivity (OHRext) (Li et al., 2015) of the gases in the sample fed to the PEAR OFR. The OHRext was calculated with Eq. (2) from the concentrations (ci) of the primary gaseous compounds (Table S1) and the butanol-d9 in the PEAR OFR, with their OH reaction rate constants ki.
Due to the differences in the emission concentrations during logwood combustion, the OHRext and consequently OHexp vary also within a batch (Fig. S1). Average OHRext was in the range 130–1300 s−1 with the highest contributions from CO, NO, and unsaturated hydrocarbons (Fig. S2).
Particulate wall losses inside the PEAR OFR were minimized by conductive stainless-steel walls, laminar flow, and a relatively small surface-to-volume ratio (2.28 m2 : 139 L) (Ihalainen et al., 2019). The particulate losses were estimated to be approximately 6 % based on the loss of elemental carbon (EC) in the PEAR OFR, determined from the difference in the thermal–optical EC concentrations measured upstream and downstream of the reactor. The fate of the gaseous organic compounds capable of irreversible condensation under the present experimental conditions, that is, the low-volatility organic compounds (LVOCs), was estimated based on Palm et al. (2016) by considering three possible depletion pathways: condensation onto particles, reactions with OH radicals, and condensation onto walls. To estimate the fraction of LVOCs condensing onto particle phase, condensation sinks (Fig. S3) were calculated according to Lehtinen et al. (2003) using an average of the particle size distributions up- and downstream of the PEAR OFR. See Sect. S4.2 in the Supplement for further information on LVOC fate estimation.
2.3 Offline filter sampling and analysis
PM1 filter samples were collected on Teflon (PTFE, Pall Corporation, P/N R2PJO47) and quartz fibre (QF; Pall Corporation, Tissuquartz) filters simultaneously from primary and secondary exhaust at a 10 L min−1 flow rate, following the methodology presented by Sippula et al. (2009). A pre-impactor (Dekati PM-10 impactor) was used to separate the particles with aerodynamic diameters of less than 1 µm (PM1). For the masonry heater, samples were collected separately from the S-5 % combustion (100 min collection time) and S-22 % combustion (90 min collection time). From the chimney stove, two sample pairs were collected from the combustion of beech: full first two batches (85 min collection time) and fourth and fifth batches excluding the last 15 min (85 min collection time). The third chimney stove collection consisted of the combustion of two full batches of moist spruce (100 min collection time). The Teflon filters were weighted before and after sample collection to determine the total PM1 mass of the emission. The amount of organic (OC) and elemental carbon (EC) in PM1 was determined from the QF filters by using a thermal–optical carbon analyser (Sunset Laboratory Inc.) following the protocol NIOSH5040 (NIOSH, 1999). In situ derivatization thermal desorption–gas chromatography–time-of-flight mass spectrometry (IDTD-GC-ToF-MS) (Orasche et al., 2011), was applied for targeted analysis of organic compounds in the particulate phase from the QF filters. Nonpolar and polar compounds were identified and quantified using mixtures of isotopically labelled internal standards and calibration standards; see the Supplement, Sect. S5.1, for further information on the IDTD-GC-ToF-MS analysis procedure. The majority of the targeted compounds can be classified as either semi- or intermediately volatile, meaning that under the present experimental conditions they can be partitioned into both gaseous and particulate phases.
2.4 Online gas-phase measurements
A Fourier transform infrared spectrometer (FTIR DX4000, Gasmet Technologies Inc.) was implemented on the stack to measure the amount of NOx, CO2, CO, and 27 OGCs (Table S1) in the fresh exhaust. The measured primary non-methane OGCs were grouped into four subgroups: alkanes, oxygenated compounds, and unsaturated and aromatic hydrocarbons. The concentrations of CO2, O3, NOx, and SO2 were monitored at the outlet of the PEAR OFR with a trace-level single-gas analysers (ABB CO2 analyser, Siemens), and OGCs were measured by PTR-ToF-MS (PTR-TOF 8000, Ionicon Analytik, Innsbruck, Austria), with H3O+ as the reagent ion and an electric field to gas number density ratio (E ∕ N) of 130. Mass calibration was done with H3O+ (m∕z 21.02) and 1,3-diiodobenzene (m∕z 203.94), which was added as a calibrant for higher m∕z. The processing of the PTR-ToF-MS data was done in a manner similar to that of earlier work (Hartikainen et al., 2018). Reaction rates by Cappellin et al. (2012) were used when available; for other compounds, the reaction rate with H3O+ was assumed to be 2×109 cm3 s−1 (Table S2).
2.5 Online particulate-phase measurements
Particle concentrations and mobility size distributions were monitored with scanning mobility particle sizers before (SMPS 3082, TSI, size range 14.6–661.2 nm) and after the PEAR OFR (SMPS 3080, TSI, size range 15.1–661.2 nm). An electrical low-pressure impactor (ELPI; 10 L min−1, Dekati) measured the particle aerodynamic size distribution and the concentration of primary particles in the size range 18.6–5950 nm. The total particulate mass after PEAR OFR was measured with a tapered element oscillating microbalance monitor (TEOM, Model 1405, Thermo Scientific).
The composition of submicron particulate matter after the PEAR OFR was measured by soot particle aerosol mass spectrometer (SP-HR-ToF-AMS, Aerodyne Research Inc). The SP-HR-ToF-AMS dual vaporizer mode was used, with the combination of the thermal vaporizer (600 ∘C) and the continuous wave laser vaporizer (1064 nm) enabling the study of both nonrefractory (NR-PM) and refractory light-absorbing submicron aerosol particles (R-PM, e.g. refractory black carbon, rBC) (Onasch et al., 2012). Standard mass-based calibrations were performed for the ionization efficiency in the SP-HR-ToF-AMS using ammonium nitrate and Regal Black (Regal 400R Pigment Black, Cabot Corp.) particles (Jayne et al., 2000; Onasch et al., 2012). SP-HR-ToF-AMS was operated in V-mode from 12 to 555 m∕z, and the two vaporizer configurations were alternated every 120 s, including the particle time-of-flight (PTOF) mode (duration 20 s). The SP-HR-ToF-AMS data were analysed using the standard analysis tools SQUIRREL v1.62A and PIKA v1.22D adapted in Igor Pro 8 (Wavemetrics). The data were corrected by time-dependent background gas-phase CO2 subtraction using the online HEPA filter measurement technique. The interactions of inorganic salts with pre-deposited carbon on the tungsten vaporizer were corrected according to Pieber et al. (2016). The CO2-AMS interference calibration value was 0.3 % of the NO3 concentration determined by NO3NH4 calibration and corrected via the fragmentation table according to Pieber et al. (2016). Before correction, the relative effect of the interference on O : C ratio was typically under 1 % and 5 % at maximum. The applied relative ionization efficiency (RIE) was 1.4, which agrees with the OA mass concentrations of biomass-burning emissions within the stated ±38 % uncertainty in AMS (Bahreini et al., 2009; Xu et al., 2018). The elemental analysis of the OA was conducted using the improved-ambient method (Canagaratna et al., 2015), and the average carbon oxidation state (OSC) of the OA was estimated as OS O : C − H : C (Kroll et al., 2011).
The SP-HR-ToF-AMS OA mass spectra was further examined by positive matrix factorization (PMF) using the method described in Lanz et al. (2007) and Ulbrich et al. (2009). Similar methods have been used previously for example for the assessment of RWC generated POA and SOA in a chamber (Bruns et al., 2015a; Tiitta et al., 2016) and in an OFR (Bruns et al., 2015a) or for time-resolved analysis of RWC OA emission constituents (Elsasser et al., 2013; Czech et al., 2016). The PMF Evaluation Tool v.3.05 was applied, and the standard data pretreatment process was completed based on Ulbrich et al. (2009), including the application of minimum error criteria and down-weighting weak variables as well as m∕z 44 () and water-related peaks. The final four-factor PMF solution covered 98 % of the OA spectra (2.2 % residual). Additional factors did not increase the realistic physical meaning of the solution, while fewer factors were insufficient for a meaningful presentation of the data. The factor identification was confirmed by comparing the time series and mass spectra of each factor with external tracers (nitrate, sulfate, ammonium, chloride, PAH, , C2H3O+, , and ). Furthermore, the factors were compared to logwood combustion mass spectra measured by Tiitta et al. (2016) from the ageing of spruce logwood exhaust in a smog chamber. The agreement of spectra was denoted with both a coefficient of determination (R2) and the angle between two mass spectra vectors (Kostenidou et al., 2009), where an angle of less than 15∘ indicates a good agreement between two mass spectra.
The polycyclic aromatic hydrocarbons and other PAC in the exhaust were analysed by using the PAC molecular ions as a proxy, following the P-MIP methodology presented by Herring et al. (2015). The base molecular ions [M]+, their fragments ([M–H]+ and [M–2H]+), and isotopes (2H, 13C, 13C2, 15N, 17O, and 18O) were isolated and quantified using the SP-HR-ToF-AMS high-resolution analysis software tool (version 1.22D). The ions targeted with the high-resolution (HR)-PAH analysis included those previously connected with PACs (Herring et al., 2015) and compounds typically released from RWC (Avagyan et al., 2016; Bertrand et al., 2018; Bruns et al., 2015b; Czech et al., 2018; Miersch et al., 2019). The 61 PACs considered (Table S3) were separated into five subgroups: unsubstituted PAHs (UnSubPAHs), oxygenated PACs (OPAHs), methylated PACs (MPAH), nitrogen-substituted PACs (NPAHs), and amino-PACs (APAHs).
This study comprehensively characterizes the chemical properties of RWC exhaust at different atmospheric ageing times by combining extensive information gathered from gas-phase and particulate-phase chemical analyses. In this section, we first discuss the dynamic combustion conditions and the characteristics of primary emissions from the logwood stoves utilized with different fuels, which define the starting point for the ageing experiments. Next, the ageing conditions in the PEAR OFR are evaluated in order to validate the atmospheric relevance of the results. Finally, we assess the changes in the gaseous and particulate OA during the ageing process under a variety of different oxidant concentrations. The observations of changes both in bulk- and molecular level chemical composition of the aerosol demonstrate that the major transformation pathway of OA changes from initial gas-phase functionalization followed by condensation to the transformation of the particulate OA by heterogeneous oxidation reactions and fragmentation. The study shows a linear dependency between OH exposure and organic aerosol oxidation state. Furthermore, OH-exposure-dependencies of specific OA constituents, such as nitrophenols, carboxylic acids, and PACs, are established.
3.1 Combustion conditions
The average modified combustion efficiency was greater than 0.97 for the masonry heater and greater than 0.95 for the chimney stove, with lower MCE occurring mainly during the char burnout periods (Fig. S4). These values are typical for modern batchwise-operated logwood appliances (Bhattu et al., 2019; Czech et al., 2018; Heringa et al., 2011). In addition to the variation within a combustion batch, there were also differences between the individual batches (Fig. 2). Notably, the combustion conditions during the first batch were distinct from later batches, because of the ignition in a cold firebox. This caused a longer ignition period (determined by the rising CO2 concentrations), which lasted for 24±5 % (masonry heater) and 35±4 % (chimney stove) of the total duration of the batch, whereas in the later batches the firebox temperatures are higher, and the ignition shortened to 9±3 % (masonry heater) or 8±4 % (chimney stove) of the total combustion time. Furthermore, the flaming phases of the first batches were shorter than those of the following batches of dry spruce or beech fuels. The emission profiles were affected by these batchwise differences, with ignition being the period for enhanced organic emissions, whereas the flaming phase was characterized by an increase in particulate emissions consisting mainly of black carbon, as expected based on previous work (Kortelainen et al., 2018). The char burnout phases with these fuels were characterized by high CO concentrations, whereas in moist spruce combustion elevated CO concentrations were measured throughout the batch, thus making the burnout phases less distinguishable.
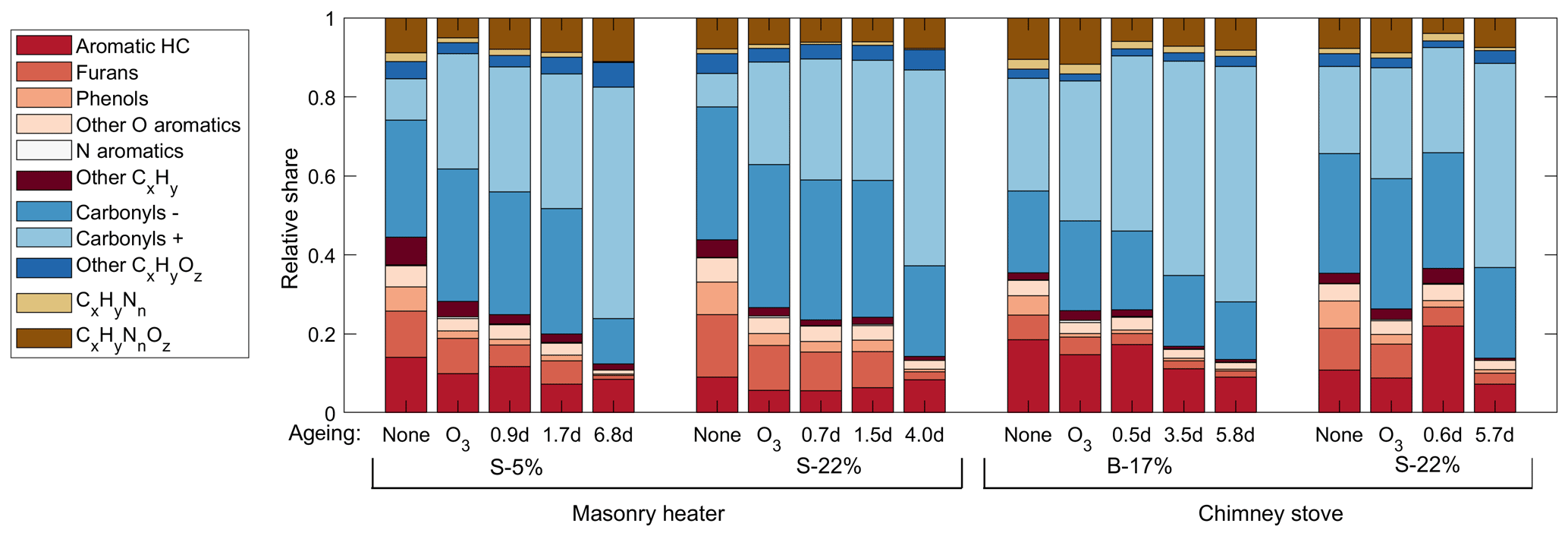
Figure 3Relative shares of the OGC groups in the exhaust downstream of the PEAR OFR as measured by PTR-ToF-MS, averaged over the total experiment time covering 2–5 batches.
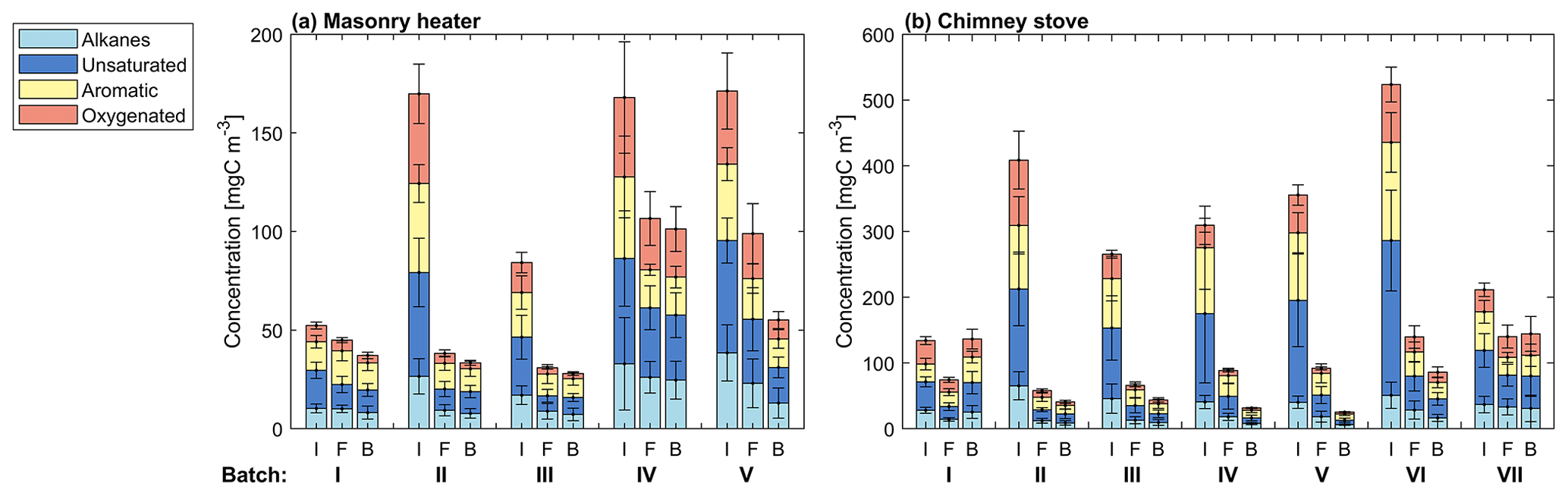
Figure 4Average concentrations of organic gaseous compounds in the dry primary exhaust at the different combustion phases (I: ignition, F: flaming, and B: char burnout phase) for each batch measured by FTIR from the stack. Error bars denote standard deviations between experiments (n=5).
3.2 Primary emissions
3.2.1 Gaseous organic emissions
The primary organic gaseous emissions in the undiluted flue gas were measured continuously by an FTIR multicomponent analyser. Additionally, the PTR-ToF-MS measured diluted, unaged emissions during the no-ageing experiments. These datasets complemented each other because, while FTIR was calibrated for 27 typical combustion-derived compounds (Table S1), a more detailed insight into the composition of the gaseous organic phase was acquired via the PTR-ToF-MS, with which 126 different molecular formulas for OGC in the primary emissions were identified in the m∕z range of 40–180 (Table S2). However, PTR-ToF-MS is unable to detect compounds with proton affinities lower than that of water (691 kJ mol−1), such as alkanes. Furthermore, fragmentation in the PTR-ToF-MS limits the identification of compounds with similar mass-to-charge ratios as common fragment ions, including unsaturated aliphatic compounds, such as propene () at m∕z 41.04 or butene () at m∕z 57.08. However, these compounds were detected by the FTIR.
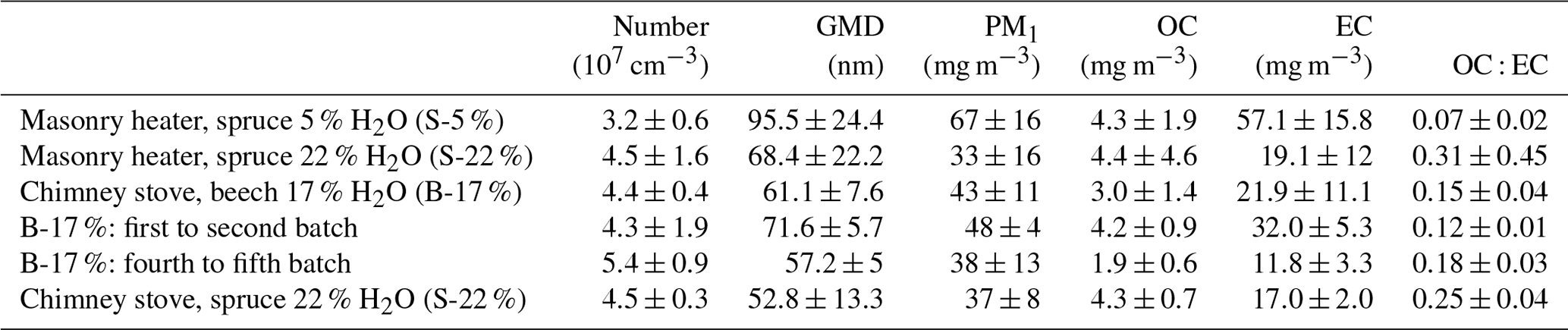
Table 2Primary PM1 concentrations in dry, 13 % O2 flue gas conditions. The number concentration and geometric mean mobility diameter (GMD) were derived from a scanning mobility particle sizer and PM1, organic carbon, and elemental carbon from filter samples.
The major OGC groups measured by PTR-ToF-MS were carbonyls, aromatic hydrocarbons (ArHCs), furans, and phenols (Fig. 3; Table S2). In addition, unsaturated aliphatic compounds constituted a substantial fraction of the total non-methane OGC (NM-OGC) measured using the FTIR (Fig. 4). The NM-OGC emissions based on FTIR were 42.6±9.2 and 101±24.6 mg C m−3 for dry and moist spruce combustion in the masonry heater, respectively, and 89.4±11.3 and 148.3±23.0 mg C m−3 for beech and moist spruce in the chimney stove, respectively. Thus, the lowest NM-OGC concentrations were measured from the dry spruce combustion. Moist spruce combustion produced a factor of 1.7–3.6 higher emission than dry wood, which is well in line with studies by e.g. McDonald et al. (2000), where moist fuel produced 2–4 times more NM-OGC than dry fuel combustion. The difference between concentrations was highest for oxygenated compounds (factor of 4.2±1.7) and for unsaturated compounds (2.7±0.6), but statistically significant for all subgroups (paired t-test p values ≤ 0.02 for all groups for the consecutive dry/moist experiments; Table S4). Differences in NM-OGC emissions were also observed between the masonry heater and the chimney stove. Emissions from moist spruce combusted in the chimney stove were higher by a factor of 1.5 (p value of 0.05) compared to the masonry heater, with a statistically significant increase in unsaturated and aromatic hydrocarbons (factors of 1.6 and 1.7, respectively, p values < 0.01) based on FTIR measurements.
The NM-OGC emissions of the first batches exhibited a distinct time-dependent behaviour in comparison to the following batches: the emitted concentrations always increased both after ignition and at the end of flaming phase (Fig. 2), whereas in the following batches there was a sharp emission peak at ignition, after which the concentrations declined as soon as the flaming phase began. These findings agree with those by Kortelainen et al. (2018) and are influenced by the fact that cold ignition is performed from the top of a fuel batch, while the following batches are ignited from the bottom by the glowing embers. NM-OGC emissions were lowest during the burnout period when most of the fuel was already consumed (Fig. 2). Thus, NM-OGC emissions had a reverse time profile to the CO emissions, which peak during the char burnout phase. Furthermore, the contribution of the different organic compound groups to the total NM-OGC concentration differed in relation to time (Fig. 4). For example, the importance of aromatic species increased for dry fuels (S-5 % and B-17 %) for the flaming and burnout phases, while other species were pronouncedly emitted at ignition. In addition, the composition of aromatic species measured by PTR-ToF-MS in the unaged emission depended on the phase (Fig. S5). In the masonry heater, ArHCs had the highest contribution during the ignition phase, but their relative share decreased during flaming phase, while the share of furanoic and phenolic compounds increased. The relative importance of furanoic and phenolic compounds in the fresh exhaust of the flaming phase from a masonry heater has been also previously established (Czech et al., 2016). Overall, the share of ArHC in the fresh exhaust is higher and less phase dependent for beech combustion in a chimney stove than for other experiments. The share of N-containing aromatic compounds, namely nitrophenol and nitrocresol, also increased after ignition. These findings are important also when considering the potential of SOA formation, as aromatic compounds have been observed to be the major SOA precursors in RWC emissions, and removal of ArHCs from the flue gas either by improving combustion conditions or using e.g. catalytic cleaning has been noted to decrease also the resulting SOA formation (Bruns et al., 2016; Hartikainen et al., 2018; Pieber et al., 2018).
3.2.2 Particulate emissions
The average primary PM1 mass concentrations, 33–67 mg m−3, and the number concentrations, 3.2×107 cm−3–5.4×107 cm−3 (Table 2), were on a similar level with earlier studies reporting emissions from modern logwood stoves (Kortelainen et al., 2018; Nuutinen et al., 2014; Tissari et al., 2009). The combustion of dry spruce in the masonry heater emitted 1.5 and 2 times as much PM1 mass compared to that of moist spruce and beech, respectively, mainly because of the higher elemental carbon emissions. Moist spruce generated similar PM1 emissions with both combustion appliances. The ratio of organic carbon to elemental carbon (OC : EC) of dry spruce combustion was very low (0.07±0.02, Table 2) compared to that of moist spruce combustion (0.31±0.45 for the masonry heater and 0.25±0.04 for the chimney stove) and beech combustion (0.15±0.04). Such low OC : EC ratios have been previously reported for emissions from modern masonry heaters operating with dry logwood (Czech et al., 2018; Miersch et al., 2019; Nuutinen et al., 2014).
The particle size distribution (Fig. S3) from dry spruce combustion was clearly distinguishable from those of other wood fuels and had a geometric mean mobility diameter (GMD) of 95.5 nm, which was considerably larger than those of other fuels (GMD 52.8–68.4 nm). The soot-dominated composition of S-5 % exhaust likely increases the GMD, because soot particles are typically present as larger agglomerates than particles of inorganic origin (ash), which mainly form ultrafine particles (Tissari et al., 2008). The size distribution and number concentration of particles in an exhaust are important not only because of their link to potential health effects but also during ageing of the exhaust, because they affect the condensation sink (CS) (Lehtinen et al., 2003) of condensable vapours during the dilution and ageing process. Thus, the particle number concentrations and size distributions affect the fate of condensable vapours and the overall OA enhancement ratios.
The thermal–optically measured elemental carbon in both the primary and the secondary exhausts correlated well with the refractory black carbon (rBC) measured by SP-HR-ToF-AMS from the secondary exhaust (R2=0.74 and 0.76, respectively; see Fig. S6). Analogous to the elemental carbon results, rBC emission was highest during the combustion of kiln-dried spruce (49.3±13.7 mg m−3). Dry fuel has recently been found to increase the soot and PAC emissions also for combustion of birch logwood in sauna stoves (Tissari et al., 2019). Considering the different combustion phases, rBC emissions were highest during high-temperature flaming combustion, as previously noted also by e.g. Kortelainen et al. (2018). For moist spruce combustion in the masonry heater, the rBC concentration dropped to 24.8±12.3 mg m−3, likely because of the lower temperature and consequentially slower burn rate. Similar rBC concentrations (24.0±6.0 mg m−3) were also measured from spruce combustion in the chimney stove. Unlike the masonry heater where the rBC concentrations were similar throughout the three dry batches, the rBC concentration from the combustion of beech in a chimney stove decreased considerably after the first batch, from 57.7±7.2 to 15.6±5.2 mg m−3. While ageing had no effect on the rBC mass, it plays an important role in the formation of SOA by acting as a seed for condensation during ageing. Furthermore, soot cores composed of elemental carbon are chemically active and may enhance the photooxidation of an OA condensed onto soot agglomerates through electron transfer (Li et al., 2018).
3.3 OFR conditions
During photochemical ageing, OHRext is an important parameter affecting OH-radical consumption and reaction pathways of organic species. For the masonry heater experiments, the average OHRext was 130–150 s−1 for dry spruce samples and 160–220 s−1 for moist spruce. In the chimney stove, the average OHRext was 280–300 s−1 for beech and 320–370 s−1 for moist spruce samples, except for the low-DR experiment (Exp. 5, DR of 30) where the average OHRext reached 820 and 1300 s−1 for the beech and moist spruce samples, respectively. The OFR conditions were divided into good, risky, and bad based on the ratio of the photon flux exposure to OHexp by the definitions of Peng and Jimenez (2017) (Fig. S7). Here, the ageing conditions were defined as mainly “risky” (4×105 cm s−1 < flux254 nm,exp ∕ OHexp < 107 cm s−1) during all the experiments. The OHRext varied considerably during combustion cycles, but the limit for “bad” conditions was exceeded only during ignition phases of chimney stove experiments when the OHRext peaks above 1000 s−1 for brief periods accounting for up to 4 % of total experiment time. In terms of OA emission, the bad conditions accounted for 3 %–7 % of chimney stove OA emissions, excluding the S-22 % combustion for the low-DR experiment where 30 % of OA was emitted under bad conditions.
Evaluation of the OHRext was limited to the externally added butanol-d9 and the compounds measured from the primary exhaust with FTIR, with NO, CO, and unsaturated hydrocarbons as the main OHRext producers (Fig. S2). In other words, the products of later-generation oxidation or from fragmentation from the particulate phase were not considered. The average NOx concentrations input to the PEAR OFR ranged from 150 to 420 ppb, except for the low-DR experiment with concentrations of 1140 and 751 ppb for beech and spruce combustion, respectively. NO is rapidly oxidized to NO2 with the addition of O3 and then partially converted to particulate nitrate. The subsequent low-NO conditions in the PEAR decreased the reactions of organic peroxy radicals (RO2) with NO. Based on the RO2 chemistry model by Peng et al. (2019) for 254 nm OFRs, the high photon fluxes and high concentrations of both OH and HO2 in the PEAR OFR result in lower RO2 lifetime and consequently lower RO2 isomerization rate than in atmosphere, while the importance of RO2+OH reaction is enhanced due to lower-than-ambient HO2-to-OH-ratio (Fig. S8) (Peng et al., 2019).
The average batchwise OH exposure in the PEAR OFR during photochemical ageing experiments ranged from 0.5 to 7 eqv. d depending on the applied photon flux and the OHRext of the sample. During the experiments, the photochemical exposure varied in line with the varying OHRext during batchwise combustion, with the highest exposure occurring during the flaming phase and lower exposure during the ignition period (Fig. S1). The extent of alternative non-OH reaction pathways during photochemical ageing in the PEAR OFR were compared to the tropospheric conditions (Sect. S4.1), and the exposures to O(1D) and O(3P) in the PEAR OFR were estimated to be similar to those in ambient conditions (Fig. S9), excluding the ignition period during the low-DR experiment, where the importance of O(3P)exp briefly exceeded ambient conditions. The exposure to ozone in relation to OH radicals was estimated to be lower in the PEAR OFR than in the troposphere ( < 105). However, our estimations were based on the initial O3 concentrations, whereas O3 is expected to form in the PEAR OFR during photochemical ageing as a product of the OH + OGC reactions (Carter, 1994). This may have led to the being slightly underestimated.
Several of the emitted OGCs were susceptible to photolysis at 254 nm, most importantly the ArHCs, which are among the main SOA precursors from RWC. Photolysis as a degradation pathway can exceed OH reactions also in the atmosphere for compounds with high photolysis rates (Hodzic et al., 2015). Here, the importance of photolysis was notable, e.g. for benzene, of which more than 40 % may have degraded via photolysis (Fig. S10). However, the importance of photolysis during photochemical ageing is inversely proportional to the ratio of the OH reaction rate to the photoabsorption cross section (σabs), and it can be considered a minor pathway for other main OGCs (Table S7), including other aromatic species such as toluene (< 10 % of total degradation during these experiments; Fig. S10).
The detailed results of the LVOC fate calculations are presented in Sect. S4.2. Briefly, the majority of LVOCs were estimated to condense into the particulate phase in all experimental conditions (Fig. S11). Similar to earlier OFR studies (Bruns et al., 2015a; Ihalainen et al., 2019; Simonen et al., 2017), the downstream particle number concentration and condensation sinks were influenced by new particle formation (Fig. S12) with the extent of nucleation depending strongly on the ageing conditions and the concentrations of condensable vapours. However, the distinct formation of ultrafine particles does not necessarily represent the atmospheric fate of condensable vapours because of the faster-than-ambient oxidation, which results in higher saturation ratios and faster gas-to-particle conversion in the PEAR OFR. Gas-to-particle conversion is also affected by the dilution ratio, which was in the PEAR OFR lower than what typically exists in atmosphere. Therefore, it has to be noted that the relative fraction of the measured semivolatile species in the condensed phase are likely lower at ambient dilution ratios than in the conditions of this study.
3.4 Transformation of gaseous phase
The composition of the OGCs changed throughout the studied exposure range, and the abundance of the secondary compounds increased with an increase in photochemical exposure. Namely, the OGCs measured with PTR-ToF-MS from the aged gas phase were governed by small carbonyls and fragmentation products such as acetic acid (C2H4O2, m∕z 61.02). However, while the small oxidized organics became increasingly dominant in the gas phase, photochemical ageing particularly decreased the share of aromatic compounds (Fig. 3). The evolution of the total chemical composition is visualized in the van Krevelen diagram, which is often used for simplified characterization of particulate OA (Heald et al., 2010) and was here extended for the investigation of the gaseous organic phase measured by PTR-ToF-MS (Fig. 5). Photochemical ageing caused several simultaneous and subsequent functionalization reactions of the organic compounds, and the increase in the average H : C ratio together with an increasing O : C ratio led to linear slopes from +0.69 to +1.0 in the van Krevelen diagram for gaseous phase. Similar, positive slopes are derivable also from previous studies of ageing of RWC OGC in smog chambers (Bruns et al., 2017; Hartikainen et al., 2018). Most of the OGCs in primary RWC exhaust have relatively high OH reactivities, and thus their reactions are expected to take place within the first atmosphere equivalent hours of photochemical ageing (Bruns et al., 2017; Hartikainen et al., 2018). Therefore, the secondary OGC products, including the oxidized fragmentation products from the particulate phase, dominate the total OGC in the long-range transported smoke. In addition, the loss of compounds with low H : C and O : C ratios, such as aromatic species, is important for the change in the average composition. Furthermore, the oxidation of compounds undetected by the PTR-ToF-MS, such as alkanes, may introduce secondary products with higher proton affinities and their subsequent appearance in the spectra. Conversely, ageing may also lead to the growth of compounds outside the observed mass range (m∕z 40–180). Compared to previously measured changes in RWC exhaust in a chamber (Hartikainen et al., 2018), ageing in the PEAR OFR led to a slightly higher increase in the H : C ratio. This difference implies an increased fragmentation which is likely the result of faster OH exposure and differences in the RO2 chemistry (Peng et al., 2019) in the PEAR OFR compared to ageing in a smog chamber.
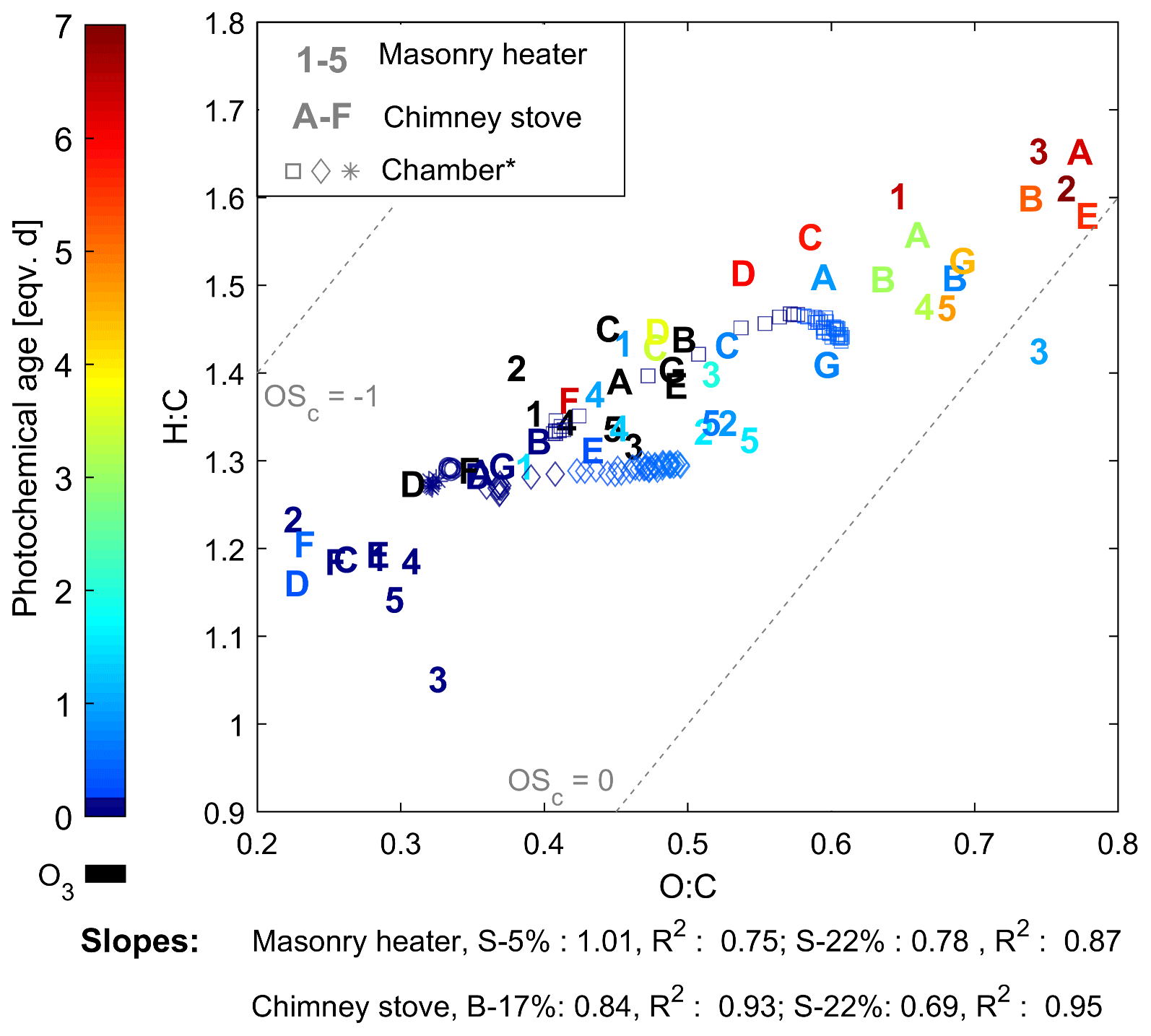
Figure 5Van Krevelen diagram of the OGCs measured by PTR-ToF-MS. Numbers indicate batches combusted in the masonry heater (1–3: dry spruce; 4–5: moist spruce); letters refer to chimney stove batches (A–E: beech; F–G: moist spruce). Ozonolysis experiments (in black) were not considered in the slope calculations. * Behaviour of RWC emission aged in a chamber (Hartikainen et al., 2018).
Aromatic compounds consisting of ArHCs, phenols, furans, N-containing aromatic compounds (N-aromatics), and other oxygenated aromatics were important constituents of the primary organic gas phase and formed 37 %–39 % of the fresh emission from the masonry heater and 33 %–34 % from the chimney stove, as measured by the PTR-ToF-MS (Fig. 3). Similar shares have been measured from fresh RWC exhaust by Bruns et al. (2017) (13 %–33 %) and Hartikainen et al. (2018) (33 %–36 %). However, after 2 eqv. d of exposure, their share decreased to less than 20 % of the identified OGCs, which agrees with previously reported conversions of aromatics during photochemical exposure (Bruns et al., 2017; Hartikainen et al., 2018). Overall, the photochemical reactions of aromatics are an important source of SOA because they form products that efficiently partition into the particulate phase. However, there are large differences between the conversion efficiencies of aromatic compound groups. While ArHCs comprise approximately half of the aromatic compounds in fresh gaseous exhaust, their share grows to over 70 % in 5–7 eqv. d, while the share of oxygenated aromatics decreases with ageing in line with their higher OH reactivity. Similar aromatic behaviour in RWC exhaust was observed earlier in a chamber with spruce exhaust (Hartikainen et al., 2018), where the molar share of ArHCs in the total aromatic content increased from 45 % and 32 % to 63 % and 54 % during ageing of 0.6 and 0.8 eqv. d, respectively, while the share of furanoic and phenolic species decreased. Furthermore, N-aromatics were not detectable here with ages exceeding 1 eqv. d, although they have been observed to form with shorter exposures (Hartikainen et al., 2018). The N-aromatics produced by the first stages of ageing may have partitioned to the particulate phase but are also degraded by subsequent reactions with OH. Simultaneously, the share of aliphatic nitrogen compounds (CHN and CHNO) to the total concentration was not substantially affected by photochemical exposure.
Carbonyls were divided into primary and secondary subgroups based on their behaviour during ageing. The primary carbonyl group, consisting mainly of acetaldehyde and to a smaller extent of compounds such as acrolein and butadiene, was prevalent in the fresh exhaust, but their share of identified compounds decreased from 13 %–27 % in unaged exhaust to 3 %–12 % in the highest exposures. This is the result of the degradation of these compounds and the introduction of high amounts of carbonyls in the secondary carbonyl group. The ratio between the two carbonyl groups increased linearly with age. The secondary carbonyl group was dominated by acetic acid, which was the most prevalent compound after extensive ageing in all experiments and covered over 30 % of the total measured OGC concentration from the highly aged S-5 % and B-17 % exhaust. The mainly small acidic compounds in the secondary carbonyl group were formed from the photochemical reactions of OGCs and from particulate OA, which is a consistent source of oxygenated OGCs such as acetic acid, formic acid, acetaldehyde, and acetone (Malecha and Nizkorodov, 2016). Of these, acetaldehyde is classified as a primary carbonyl because it reacts with OH 2 orders of magnitude faster than the other considered small carbonyls (Atkinson et al., 2006), and thus its concentration remained at a similar level throughout the ageing process.
3.5 Transformation of particulate phase
3.5.1 OA enhancement and composition
The photochemical ageing process increased the mass of particulate organic carbon measured with thermal–optical carbon analysis by factors of 1.3 and 3.9 for dry and moist spruce combustion in the masonry heater, respectively, and by factors of 2.0 and 3.0 for beech and moist spruce combustion in a chimney stove, respectively. This agrees well with the previously observed SP-HR-ToF-AMS-based OA enhancement factors (1.6–5.3) for RWC (Bertrand et al., 2017; Bruns et al., 2015b; Grieshop et al., 2009; Heringa et al., 2011; Tiitta et al., 2016) and with the thermal–optical analysis-based organic carbon enhancements previously measured for the same chimney stove (1.3–1.4 after 1.7–2.5 eqv d) (Miersch et al., 2019). The organic carbon concentrations after the PEAR OFR correlated well with the OA measured with SP-HR-ToF-AMS (R2=0.85, Fig. S6). Ageing led to a linear increase in the SP-HR-ToF-AMS-derived ratio of organic matter to organic carbon (OM : OC, Fig. S13), which rose from the initial average ratio of 1.8–2.2 to 2.7–3.0 during extended ageing. Similarly, the OSC of the OA increased as a function of the photochemical age throughout the tested exposure ranges (Fig. S13), indicating the existence of continuing reactions of the particulate phase after the rapid consumption of the majority of the primary gaseous SOA precursors. As expected, the organic carbon mass enhancement did not increase with continuous ageing because the major SOA precursors were already consumed by relatively short OH exposures. Continuing photochemical exposure may instead reduce the amount of particulate organic carbon (Kroll et al., 2015), which acts as a source for volatile acidic compounds during photochemical ageing (Malecha and Nizkorodov, 2016; Paulot et al., 2011). See Table S8 for experiment-wise average concentrations and chemical compositions of the particulate matter measured by SP-HR-ToF-AMS.
The oxidation states measured after the PEAR OFR without oxidative ageing were highest for the chimney stove OA, with average unaged OSC's of 0.22 and 0.41 for beech and spruce combustion, respectively, while dry and moist spruce in the masonry heater had average unaged OSC's of −0.18 and 0.15, respectively. This indicates the existence of different combustion conditions in the studied appliances, with the masonry heater having lower emissions of highly oxygenated compounds and a higher share of unsaturated hydrocarbons compared to that of the chimney stove emissions. However, as a result of oxidative ageing, OSC surpassed 1.5 after 5 eqv. d regardless of the type of experiment, which exceeds the values typically observed in ambient aerosol (Kroll et al., 2011; Ng et al., 2011). This is likely because highly aged aerosol in ambient air is mixed continuously with fresh and less oxidized sources. However, the chemical evolution of OA in the PEAR OFR followed a similar trend to that observed for typical ambient semivolatile oxygenated aerosol, with a van Krevelen slope of −0.5, which can be interpreted as a result of simultaneous fragmentation and acid-group addition (Ng et al., 2011). Furthermore, the changes in OSC's were similar to those previously observed for ambient aerosol aged extensively in a potential aerosol mass (PAM; Kang et al., 2007) OFR (OSC of up to 2; Ortega et al., 2016). The OSC's of RWC exhaust aged in a PAM OFR have, however, been notably lower than observed here (Fig. 6; Bruns et al., 2015a; Pieber et al., 2018).
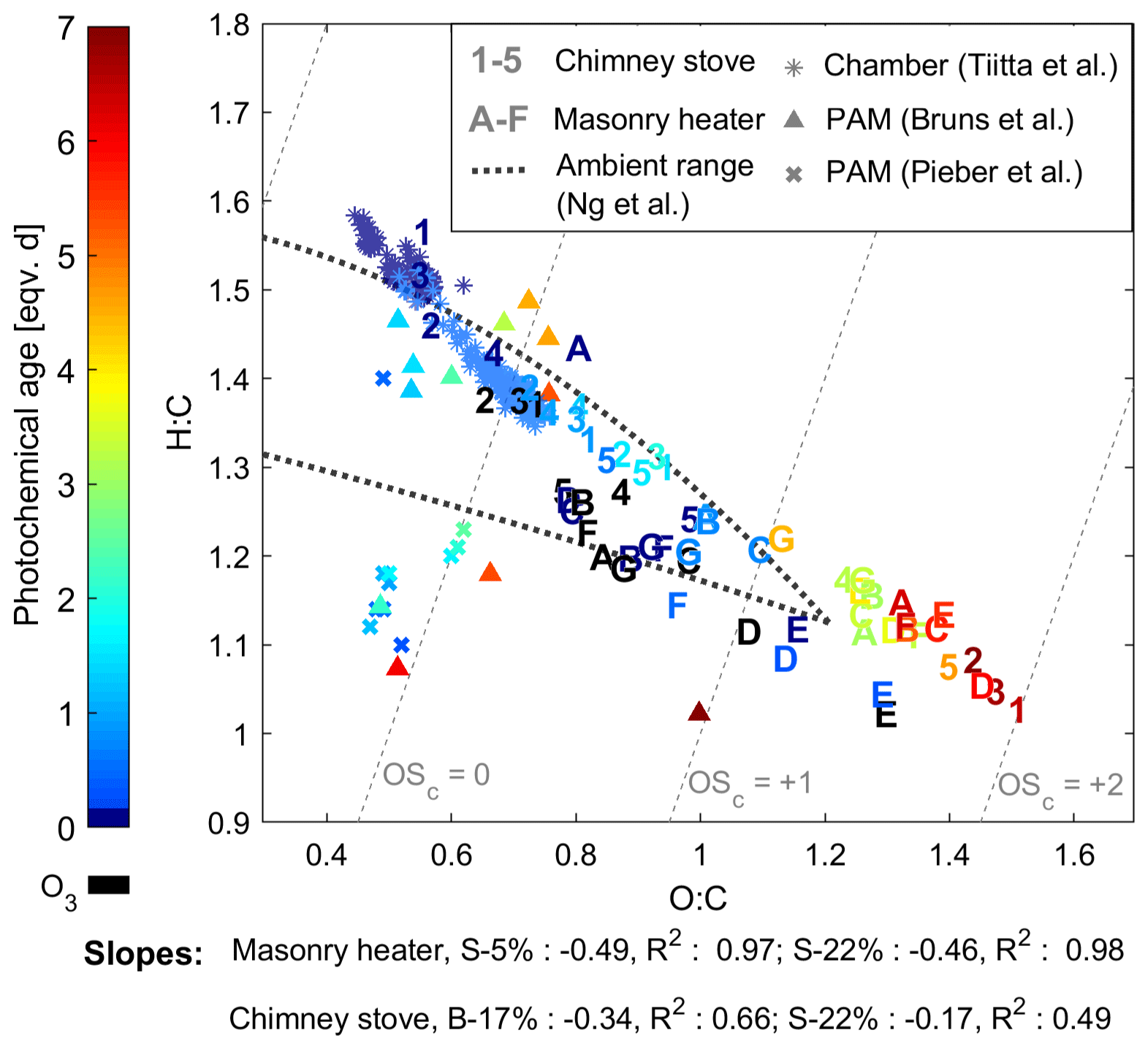
Figure 6Van Krevelen diagram of the particulate organic aerosol measured by SP-HR-ToF-AMS. Numbers indicate batches combusted in the masonry heater (1–3: dry spruce; 4–5: moist spruce), whereas letters refer to chimney stove batches (A–E: beech; F–G: moist spruce) of this study. Ozonolysis experiments are marked with black and were not considered in slope calculations. Results from ageing of RWC exhaust with a PAM OFR by Bruns et al. (2015a) and Pieber et al. (2018), as well as behaviour of RWC OA aged in a chamber (Tiitta et al., 2016) are marked with scatter.

Figure 7Compounds measured by IDTD-GC-ToF-MS in different experiments (ozonolysis, low, and high photochemical exposure) with respect to their carbon number (nC) and oxidation state (OSC). Enhancement ratios (ER) compared to the experiments without oxidative ageing are shown in colour, and the size indicates the dilution-corrected concentrations (normalized dry, 13 % O2 flue gas conditions) in the secondary exhaust.
A higher initial oxidation state in the chimney stove exhaust led to shallower slopes (−0.17 and −0.34) in the van Krevelen diagram than those in the masonry heater exhaust (−0.46 and −0.49, Fig. 6). The slope for the beech combustion aerosol was higher than for spruce, which may result from differences in fuel composition as has been observed also previously for biomass burning OA (BBOA) (Ortega et al., 2013). Previously, the ageing of spruce combustion exhaust from a masonry heater in a chamber has produced similar but slightly steeper van Krevelen slopes of −0.64 to −0.67 (Tiitta et al., 2016). The steepness may be affected by the shorter ageing (< 1 eqv. d) and consequent lower final OSC (maximum of +0.14) in the chamber, as the O : C ratio has been noted to level off at higher oxidation states (Ng et al., 2011). Slopes are also positively affected by fragmentation, which may be enhanced in the PEAR OFR because of the more intensive UV radiation than in smog chambers or in ambient conditions. The van Krevelen slopes agreed well with those previously measured from single precursors aged in the PAM OFR, such as −0.48 for toluene or −0.46 for xylene (Lambe et al., 2011). Similar slopes have been observed also for ageing of OA from pellet combustion (−0.47 to −0.44; Czech et al. 2017) or open biomass combustion (−0.5; Ortega et al. 2013) with a PAM OFR.
The evolution of particulate organic aerosol was assessed also by the IDTD-GC-ToF-MS analysis of filter samples. When comparing the concentrations in the exhaust after the PEAR OFR, the concentrations of compounds with high oxidation states and low carbon number (nC) increased during photochemical ageing (upper-right corner of Fig. 7). The locations of the measured organic compounds in the OSC : nC space are shown in Fig. S14, and their dilution-corrected concentrations downstream of the PEAR OFR in Table S11. In the OSC : nC space, the compounds which exhibited a major increase during photochemical ageing were located in or above the location of the low-volatility oxidized organic aerosol (LV-OOA) classified by Kroll et al. (2011). These compounds are products of the multistep oxidation process including both fragmentation and oxidative reactions.
OA formation is tied to the availability of organic precursors, and thus the formation of SOA was highest at the ignition phase of each batch (Fig. S15). In the masonry heater, the enhancement in particulate OA mass increased considerably with the introduction of moist logs, simultaneously with the increase in gaseous organics in the fresh emissions compared to the preceding dry spruce combustion. In contrast, the low organic emission by dry spruce combustion was reflected as a lower SOA formation. Another aspect related to the primary exhaust was the extent of OH exposure in the PEAR OFR, which was directly connected to the sample concentrations, as discussed in Sect. 3.3. Namely, the photochemical ageing was lower during periods of high emission, leading to lower oxidation states for the OA emitted during ignition.
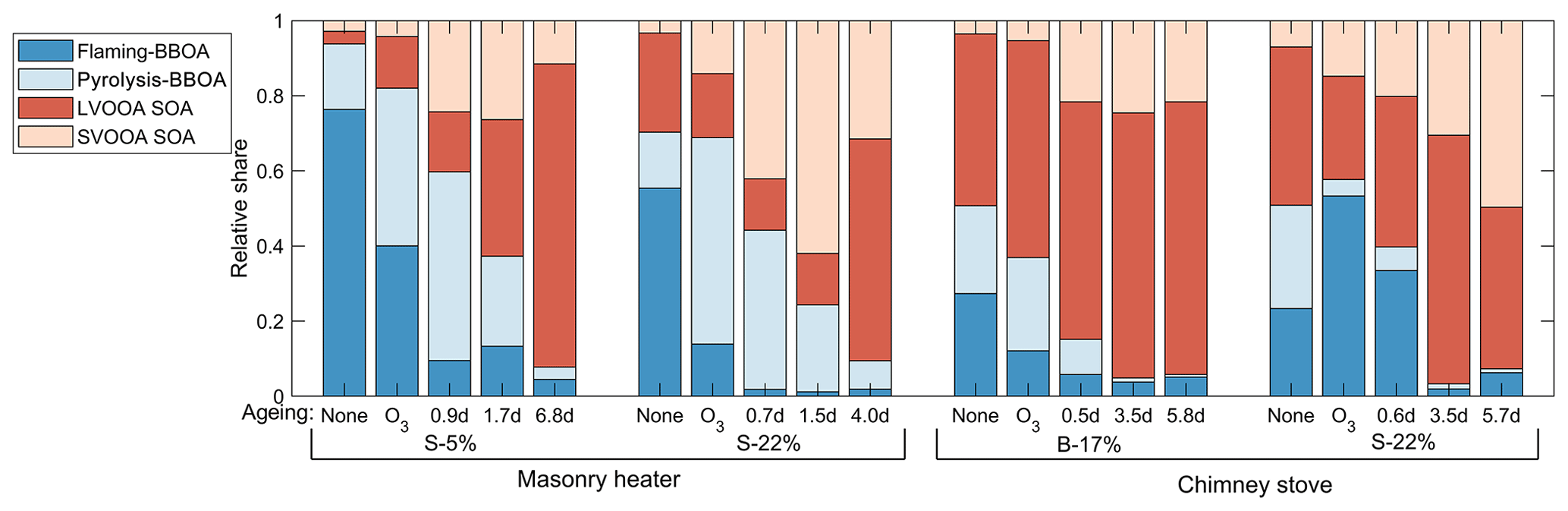
3.5.2 PMF analysis of particulate OA composition
PMF analysis applied to the exhaust produced a four-factor solution for the OA covering 98 % of the spectra. The spectra of the factors are shown in Fig. S16. Two of the factors were associated in particular with the primary OA from biomass combustion: pyrolysis-BBOA, formed especially during ignition, and flaming-BBOA, emitted pronouncedly during the flaming phase. The other two factors, semivolatile oxygenated OA (SV-OOA) and low-volatility oxygenated OA (LV-OOA), represent oxygenated organics with notably higher OSC's than those of the primary OA factors (Table 3). Flaming-BBOA comprised 76 % and 55 % of the unaged OA from dry and moist spruce combustion in the masonry heater, respectively, but only 27 % and 23 % in the beech and moist spruce combustion OA from the chimney stove, respectively (Fig. 8), indicating a less-oxidizing higher-temperature flaming phase in the modern masonry heater. Flaming-BBOA is strongly related to the ion (main peak of m∕z 57; R2=0.90, Fig. S17), which is used as a tracer for hydrocarbon-like compounds (Aiken et al., 2009). However, in contrast to the typical hydrocarbon-like OA factor characterized by a relatively low OSC (−1.7 to −1.6; Kroll et al., 2011), the flaming-BBOA contains more oxygen-containing functional groups and is similar to the primary biomass burning OA factor measured in the RWC exhaust in a chamber (Tiitta et al., 2016; θ=13.6∘; R2=0.95). Pyrolysis-BBOA, on the other hand, consisted of ions typical to the low-temperature pyrolysis products of wood combustion and its time series correlated well with the PACs (R2=0.86, Fig. S17).
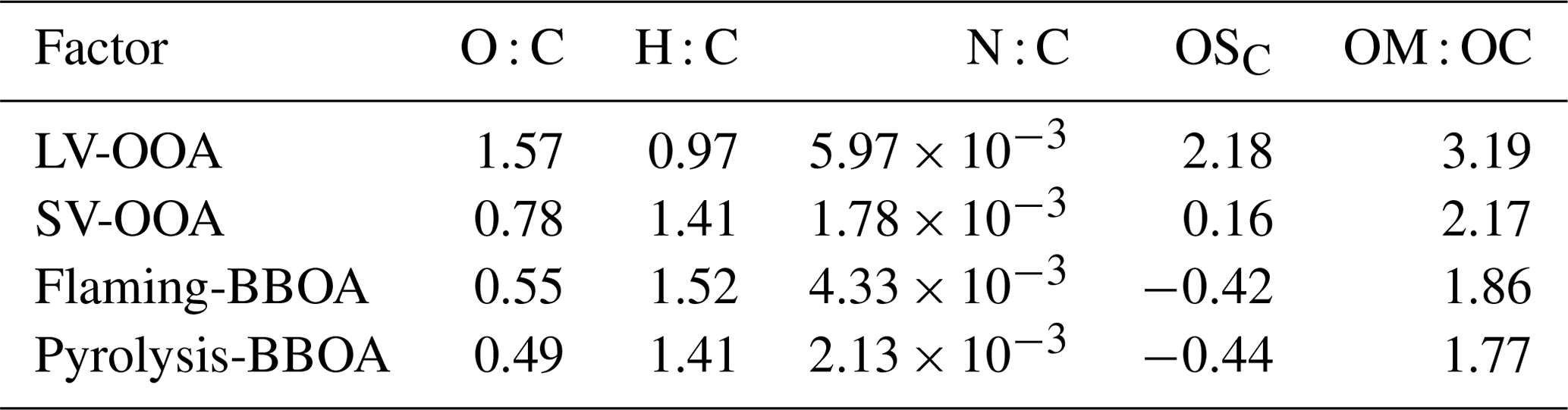
Of the more-oxygenated factors, the LV-OOA was dominated by the CO+ and ions and thus represented highly oxidized OA. The LV-OOA spectra corresponded well with the OH-induced SOA factor identified from the RWC exhaust aged in a smog chamber (Tiitta et al., 2016.; θ=8.4∘; R2=0.98) and was comparable with that of ambient LV-OOA (Aiken et al., 2009) and the LV-OOA spectra of unaged wood combustion exhaust (Kortelainen et al., 2018). LV-OOA was also present in the unaged exhaust of this study, excluding the dry spruce combustion in the masonry heater, which produced the least-oxidized primary exhaust. The SV-OOA, on the other hand, was related to the C2H3O+ ion (R2=0.78), which is indicative of carbonyl formation in the particulate OA (Ng et al., 2010). The SV-OOA was also comparable (; R2=0.96) to a factor of SOA generated in a chamber in the previous work (Tiitta et al., 2016), where this factor was interpreted as ozonolysis-generated organic OA. Interestingly, SV-OOA was formed only during photochemical ageing and increased in line with higher exposures despite semivolatile compounds being products of the initial stages of OA oxidation. This further demonstrates the long-continuing functionalization of OA during ageing alongside with the fragmentation processes.
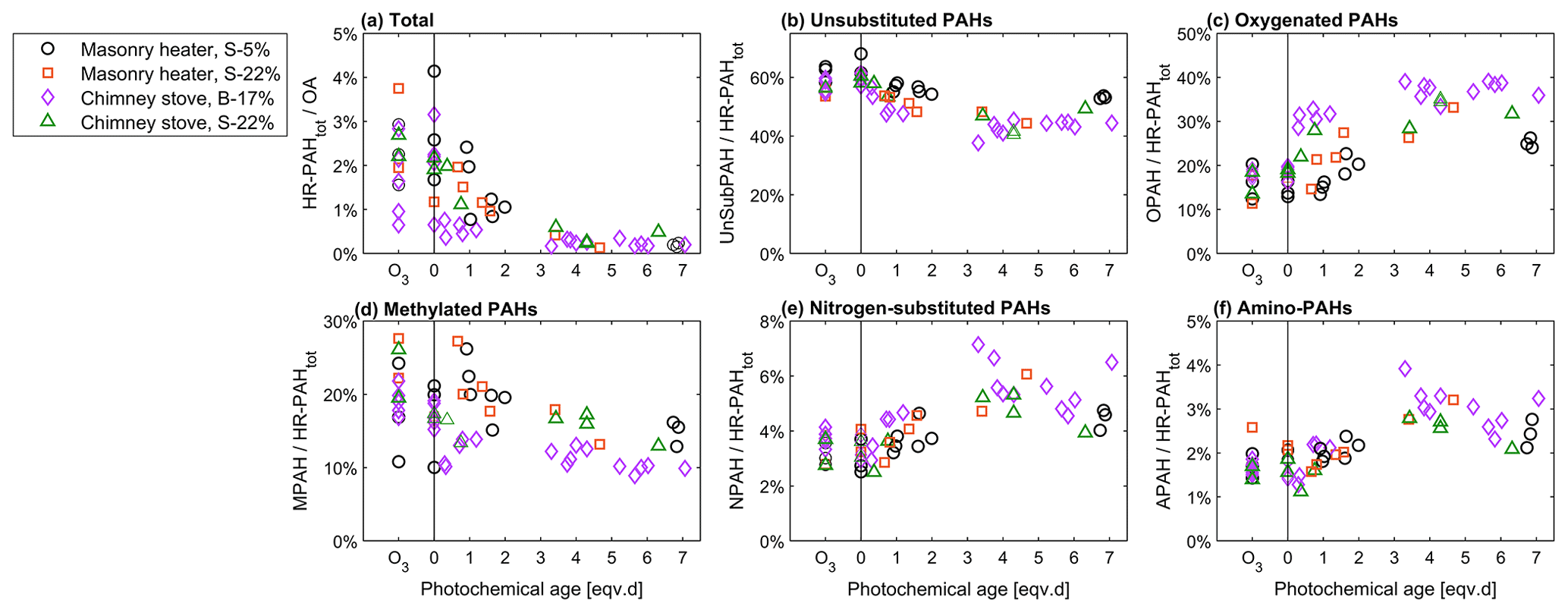
Figure 9Relationship of photochemical ageing to the batchwise average ratios of (a) total HR-PAH concentration to OA concentration and of (b–f) HR-PAH subgroups to the total PAC concentration measured by the SP-HR-ToF-AMS.

Figure 10Dilution-corrected concentrations for selected compounds measured by IDTD-GC-ToF-MS at different exposure levels. Normalized to dry, 13 % O2 flue gas conditions.
3.5.3 Polycyclic aromatic compounds
PACs were overall more prominent in the primary exhaust of dry spruce combustion (3.2 % of total OA, Fig. 9) than from combustion of moist spruce (1.7 % or 2.0 %) or beech (1.7 %). As expected, the total PAC concentrations decreased because of ageing and contributed less than 0.5 % to OA for all cases after 3 eqv. d of ageing. Furthermore, ageing transformed the composition of the PACs assessed with the SP-HR-ToF-AMS HR-PAH analysis (P-MIP analysis, Sect. S5.2; Herring et al., 2015). While UnSubPAHs formed the most prominent PAC group of all the combustion experiments, ageing decreased their share from 60 % in unaged exhaust to 40 %–50 % after 3 eqv. d of ageing. Similarly to the PACs measured with SP-HR-ToF-AMS, ageing decreased the UnSubPAHs analysed with IDTD-GC-ToF-MS by 83 %–85 % in the dry spruce combustion in the masonry heater and by 90 %–98 % in the other situations. Of the most prominent UnSubPAHs, anthracene and fluoranthene degraded within the first eqv. d (Fig. 10). Also benzo[a]pyrene, which is used as the marker for total ambient PAHs (European Council, 2004), degraded by a factor of 5 as a result of photochemical ageing (Table S11).
The change in the PACs is likely to alter the potential health effects of the exhaust; although the total PAC concentration decayed, the simultaneous formation of oxy- and nitro-PAC derivatives known to be detrimental to health was observed. These substituted PACs have lower vapour pressures compared to those of parent PAHs and thus are more likely to condense on the particles (Shen et al., 2012). The share of oxygenated PACs to the total HR-PAH concentration measured by SP-HR-ToF-AMS increased from 15 %–19 % in unaged exhaust to 25 %–38 % in aged exhaust. The concentrations of both the SP-HR-ToF-AMS HR-OPAH and IDTD-GC-ToF-MS-derived OH-PAH also correlated with the SV-OOA PMF factor (Pearson r=0.70 for OPAH and r=0.88 for OH-PAH; see Table S12) pointing towards their continuous formation during ageing. Of the compounds measured by IDTD-GC-ToF-MS, the most notable increase was observed for naphthaldehydic acid, with high concentrations in aged aerosol (up to 100 µg m−3 in dilution-corrected flue gas). Interestingly, its concentration was highest at approximately 1 eqv. d, after which it decreased. Naphthaldehydic acid and other oxygenated PACs have also been previously found to form during photochemical ageing of RWC exhaust (Bruns et al., 2015b; Miersch et al., 2019); however, we found that the photochemically enhanced naphthaldehydic acid concentration degraded after continuous ageing, although remaining considerably higher than in the unaged emissions.
In addition, the share of nitrogen-substituted PACs, including both NPAH and APAH, increased from a combined share of 5 % of HR-PAHtot measured by SP-HR-ToF-AMS in fresh exhaust to a maximum of 9 % in the aged exhaust. In general, particulate nitrogen-substituted PACs are formed in the atmosphere through the oxidation of gaseous PACs or via heterogenous reactions from UnSubPAHs and are also simultaneously degraded by photochemical reactions (Keyte et al., 2013). As UnSubPAHs in the present study were largely consumed after 3 eqv. d, the higher photochemical exposure times consequently led to situation where the precursors for nitrogen-substituted PACs are not available, and therefore no further formation took place.
3.5.4 Organic acids
Photochemical ageing led to a considerable increase in particulate organic acids in exhaust aerosol analysed with IDTD-GC-ToF-MS. The amount of small multifunctional acids such as malic acid (C4H4O4) and tartaric acid (C4H6O6) increased by factors of up to over 200 (Fig. 10, Table S11). Increases in the amounts of also dicarboxylic acids such as succinic (C4H6O4) and glutaric acid (C5H8O4) were evident, although to a lesser factor. The comparison of IDTD-GC-ToF-MS measurements with the SP-HR-ToF-AMS measurements revealed that the concentrations of organic acids were connected to both LV- and SV-OOA factors (r>0.7, Table S12), which also strongly increased during the ageing process. This was expected based on the association of SV-OOA with the C2H3O+ ion indicating carbonyl formation. In the nC–OSC space (Fig. 7), these compounds dominated the upper-right corner with the highest oxidation states and largest enhancement ratios and are close to the LV-OOA region specified for atmospheric OA by Kroll et al. (2011). The measured organic acids were mostly intermediately volatile products of the continuous fragmentation process. Thus, they may partition also to the gaseous phase where they are expected to participate in secondary gas-phase OH reactions. Overall, the concentrations of particulate organic acids were highest at approximately 3 eqv. d, after which they were degraded by further oxidation reactions.
3.5.5 Nitrophenols
An increase in nitrophenols in the particulate phase was evident during photochemical ageing, which increased the concentrations of 4-nitrophenol (4-NP) and 4-nitrocatechol (4-NC) by respective factors of 2–30 and 30–3000 compared to non-aged exhaust (Fig. 10, Table S11) as measured by IDTD-GC-ToF-MS. The highest attained 4-NC concentration corresponded to 2 % of total SP-HR-ToF-AMS-based OA concentration, which is a notable fraction for a single compound in an ageing aerosol. These secondary nitrophenols were products of OH + phenolic-compound reactions and may have originated from both gas-phase and heterogenous reactions (Harrison et al., 2005). Also the ozonolysis experiments led to an extensive formation of 4-NP, whereas the amount of 4-NC did not increase. This discrepancy might be the result of absence of the photochemical production of catechol, which is the precursor for 4-NC formation (Finewax et al., 2018).
Nitrophenol concentrations were highest at relatively low OH exposures (1–2 eqv. d) and decreased with increased ageing. Similar trends with OH exposure were seen in the NPAH and APAH concentrations measured with SP-HR-ToF-AMS. Nitrophenols are reactive towards OH and photolysis in both gaseous and particulate phases, and their amount decreased from both phases after the first equivalent days of ageing. Thus, the measured nitrophenols can be considered to decay efficiently under high-OH exposures and, therefore, are not ideal biomass combustion markers for long-range transported smoke. On the hand, their concentrations still remained higher in the highly aged exhaust than in the fresh exhaust. Furthermore, in the particulate phase, they likely contributed to the formation of organic acids which are formed during the continuous photo-oxidation of the nitrophenols (Hems and Abbatt, 2018).
The photochemical ageing of dynamically changing OA emitted from RWC was evaluated using the PEAR OFR to expose the exhaust to varying photochemical conditions for up to an equivalent of 1 week in the atmosphere. To evaluate typical northern and central European combustion emissions, two different appliances were used with regionally typical logwood fuels. While the primary concentrations of particulate OA were relatively similar for all of the assessed sources, the enhancement of the organic particulate carbon during ageing depended on the type of the fuel. In particular, the fuel moisture content affected the SOA production: dry fuel produced a lower organic mass and OA enhancement during ageing than moist wood, because of the significantly lower emission rates of the organic gases. However, a very low (5 %) logwood moisture content considerably increased the primary PM1 emission because of the extensive soot formation. With current logwood combustion appliances, this presents a conflict in attempts to decrease emissions since the usually preferred dry logwood produces high BC emissions, while high moisture content of the fuel increases the amount of organic emissions, as shown here for spruce logwood. Similar phenomenon has, however, been recently noted also for hardwood fuels (Tissari et al., 2019).
The particulate organic carbon mass in the RWC exhaust increased by a factor of 1.3–3.9 during photochemical ageing. Furthermore, photochemical ageing transformed the overall composition of the OAs. This was observed as a linear increase in the average carbon oxidation state of particulate OA throughout the investigated photochemical exposure range, while the ratio of organic carbon to total organic mass decreased. Photochemical ageing also caused multiform changes in the OA at the molecular level. Notably, small acidic reaction and fragmentation products became increasingly dominant in both particulate and gaseous phases with higher ageing. The concentrations of particulate nitrophenols were at their highest level after 1 eqv. d, after which they began to decay but remained higher than that in the primary exhaust. Similarly, nitrogen-containing aromatics were unobservable in the gaseous phase at longer exposures, although they have been observed to increase during exposures of less than 1 eqv. d (Hartikainen et al., 2018). Ageing also enhanced the share of oxygen- and nitrogen-substituted polycyclic aromatic compounds in the PAC emissions. Of the oxygenated PACs, naphthaldehydic acid in particular increased considerably with concentrations peaking at approximately 1 eqv. d. However, PACs in total degraded almost completely after 3 eqv. d of ageing.
In general, several different oxidation mechanisms of organic aerosol are likely occurring simultaneously both in the atmosphere and in the OFR experiments of this study. However, based on the observed OA chemical compositions under a range of different OH exposures, different major transformation mechanisms for RWC exhaust under photochemical conditions can be roughly outlined. First, the results demonstrate that short OH exposures (∼ 1 eqv. d) are sufficient to functionalize the majority of gaseous SOA precursors of RWC exhaust and lead to their condensation into particulate phase. This mechanism dominates the overall OA transformation until the SOA precursors have been depleted. After this stage, the continuing OH exposure leads to further oxidation of particulate organic aerosol, which is likely explained by heterogeneous reactions between gas-phase oxidants and particles. However, it is also possible that particulate-phase oxidation occurs via evaporation and homogeneous gas-phase oxidation followed by recondensation. It is important to note that partitioning of organic compounds is affected by dilution, which was lower in the PEAR OFR than in ambient air.
While several recent studies (Bertrand et al., 2017; Bruns et al., 2015b; Bruns et al., 2017; Grieshop et al., 2009; Hartikainen et al., 2018; Heringa et al., 2011; Pieber et al., 2018; Tiitta et al., 2016) have assessed the behaviour of RWC emissions in a relatively short timescale of less than 2 eqv. d, this study also highlights the importance of higher exposure levels towards chemical transformation of OA. Due to the potentially long atmospheric lifetimes of OA, long-term ageing is also important to consider in large-scale atmospheric models, which typically estimate SOA formation and characteristics based on short-term ageing experiments. The consideration of only the first stage of gas-phase functionalization and condensation may lead to underestimated oxygenation of the long-transported OA, while specific compound groups, such as nitrophenols or substituted-PACs, can be overestimated. In general, the potential health and climate effects of aerosols are to a large extent determined by their composition, which depends on their sources and the levels of atmospheric ageing. Thus, the characterization of aerosol emissions from different sources and their atmospheric transformation at different exposure levels would be crucial for assessment of the overall environmental effects of ambient air pollution.
The data are available on request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-6357-2020-supplement.
AH, OS, PYP, MI, and HL designed the study. Measurements were performed by AH, PT, MI, PYP, MK, HL, HS, JT, and OS. AH, HC, MI, and OS made the assessments of the PEAR conditions. AH performed analyses of FTIR, SMPS, and PTR-ToF-MS data; PT and LH performed the AMS data analyses; JO performed IDTD-GC-ToF-MS analyses; and HK performed the thermal–optical analyses. OS, JT, RZ, and JJ supervised and acquired funding to the study. The paper was written by AH with contribution from all coauthors.
The authors declare that they have no conflict of interest.
We thank Donna Sueper from Aerodyne Research, Inc., for her support in the SP-HR-ToF-AMS HR-PAH analysis.
This research has been supported by the Academy of Finland, Council for Natural Sciences and Engineering (grants no. 304459 and no. 296645), the German Research Foundation (grant no. ZI 764/14-1), Doctoral School of University of Eastern Finland, the Helmholtz Virtual Institute (https://www.hice-vi.eu/; accessed 20 May 2020), and the aero-HEALTH Helmholtz International Lab (https://www.aerohealth.eu/; accessed 20 May 2020).
This paper was edited by Sergey A. Nizkorodov and reviewed by two anonymous referees.
Aiken, A. C., Salcedo, D., Cubison, M. J., Huffman, J. A., DeCarlo, P. F., Ulbrich, I. M., Docherty, K. S., Sueper, D., Kimmel, J. R., Worsnop, D. R., Trimborn, A., Northway, M., Stone, E. A., Schauer, J. J., Volkamer, R. M., Fortner, E., de Foy, B., Wang, J., Laskin, A., Shutthanandan, V., Zheng, J., Zhang, R., Gaffney, J., Marley, N. A., Paredes-Miranda, G., Arnott, W. P., Molina, L. T., Sosa, G., and Jimenez, J. L.: Mexico City aerosol analysis during MILAGRO using high resolution aerosol mass spectrometry at the urban supersite (T0) – Part 1: Fine particle composition and organic source apportionment, Atmos. Chem. Phys., 9, 6633–6653, https://doi.org/10.5194/acp-9-6633-2009, 2009.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Avagyan, R., Nyström, R., Lindgren, R., Boman, C., and Westerholm, R.: Particulate hydroxy-PAH emissions from a residential wood log stove using different fuels and burning conditions, Atmos. Environ., 140, 1–9, https://doi.org/10.1016/j.atmosenv.2016.05.041, 2016.
Bahreini, R., Ervens, B., Middlebrook, A. M., Warneke, C., de Gouw, J. A., DeCarlo, P. F., Jimenez, J. L., Brock, C. A., Neuman, J. A., Ryerson, T. B., Stark, H., Atlas, E., Brioude, J., Fried, A., Holloway, J. S., Peischl, J., Richter, D., Walega, J., Weibring, P., Wollny, A. G., and Fehsenfeld, F. C.: Organic Aerosol Formation in Urban and Industrial Plumes near Houston and Dallas, Texas, J. Geophys. Res.-Atmos, 114, D00F16, https://doi.org/10.1029/2008JD011493, 2009.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Bertrand, A., Stefenelli, G., Bruns, E. A., Pieber, S. M., Temime-Roussel, B., Slowik, J. G., Prévôt, A. S. H. Wortham, H., El Haddad, I., and Marchand, N.: Primary emissions and secondary aerosol production potential from woodstoves for residential heating: Influence of the stove technology and combustion efficiency, Atmos. Environ., 169, 65–79, https://doi.org/10.1016/j.atmosenv.2017.09.005, 2017.
Bertrand, A., Stefenelli, G., Jen, C. N., Pieber, S. M., Bruns, E. A., Ni, H., Temime-Roussel, B., Slowik, J. G., Goldstein, A. H., El Haddad, I., Baltensperger, U., Prévôt, A. S. H., Wortham, H., and Marchand, N.: Evolution of the chemical fingerprint of biomass burning organic aerosol during aging, Atmos. Chem. Phys., 18, 7607–7624, https://doi.org/10.5194/acp-18-7607-2018, 2018.
Bhattu, D., Zotter, P., Zhou, J., Stefenelli, G., Klein, F., Bertrand, A., Temime-Roussel, B., Marchand, N., Slowik, J. G., Baltensperger, U., Prévôt, A. S. H., Nussbaumer, T., El Haddad, I., and Dommen, J.: Effect of stove technology and combustion conditions on gas and particulate emissions from residential biomass combustion, Environ. Sci. Technol., 53, 2209–2219, https://doi.org/10.1021/acs.est.8b05020, 2019.
Bølling, A. K., Pagels, J., Yttri, K. E., Barregard, L., Sallsten, G., Schwarze, P. E., and Boman, C.: Health effects of residential wood smoke particles: the importance of combustion conditions and physicochemical particle properties, Part. Fibre Toxicol., 6, 29, https://doi.org/10.1186/1743-8977-6-29, 2009.
Bruns, E. A., El Haddad, I., Keller, A., Klein, F., Kumar, N. K., Pieber, S. M., Corbin, J. C., Slowik, J. G., Brune, W. H., Baltensperger, U., and Prévôt, A. S. H.: Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition, Atmos. Meas. Tech., 8, 2315–2332, https://doi.org/10.5194/amt-8-2315-2015, 2015a.
Bruns, E. A., Krapf, M., Orasche, J., Huang, Y., Zimmermann, R., Drinovec, L., Močnik, G., El-Haddad, I., Slowik, J. G., Dommen, J., Baltensperger, U., and Prévôt, A. S. H.: Characterization of primary and secondary wood combustion products generated under different burner loads, Atmos. Chem. Phys., 15, 2825–2841, https://doi.org/10.5194/acp-15-2825-2015, 2015b.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6, 27881, https://doi.org/10.1038/srep27881, 2016.
Bruns, E. A., Slowik, J. G., El Haddad, I., Kilic, D., Klein, F., Dommen, J., Temime-Roussel, B., Marchand, N., Baltensperger, U., and Prévôt, A. S. H.: Characterization of gas-phase organics using proton transfer reaction time-of-flight mass spectrometry: fresh and aged residential wood combustion emissions, Atmos. Chem. Phys., 17, 705–720, https://doi.org/10.5194/acp-17-705-2017, 2017.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Cappellin, L., Karl, T., Probst, M., Ismailova, O., Winkler, P. M., Soukoulis, C., Aprea, E., Märk, T. D., Gasperi, F., and Biasioli, F.: On quantitative determination of volatile organic compound concentrations using proton transfer reaction time-of-flight mass spectrometry, Environ. Sci. Technol., 46, 2283–2290, https://doi.org/10.1021/es203985t, 2012.
Carter, W. P.: Development of ozone reactivity scales for volatile organic compounds, Air Waste, 44, 881–899, 1994.
Czech, H., Sippula, O., Kortelainen, M., Tissari, J., Radischat, C., Passig, J., Streibel, T., Jokiniemi, J., and Zimmermann, R.: On-line analysis of organic emissions from residential wood combustion with single-photon ionisation time-of-flight mass spectrometry (SPI-TOFMS), Fuel, 177, 334–342, https://doi.org/10.1016/j.fuel.2016.03.036, 2016.
Czech, H., Pieber, S. M., Tiitta, P., Sippula, O., Kortelainen, M., Lamberg, H., Grigonyte, J., Streibel, T., Prévôt, A. S., Jokiniemi, J., and Zimmermann, R.: Time-resolved analysis of primary volatile emissions and secondary aerosol formation potential from a small-scale pellet boiler, Atmos. Environ., 158, 236–245, https://doi.org/10.1016/j.atmosenv.2017.03.040, 2017.
Czech, H., Miersch, T., Orasche, J., Abbaszade, G., Sippula, O., Tissari, J., Michalke, B., Schnelle-Kreis, J., Streibel, T., Jokiniemi, J., and Zimmermann, R.: Chemical composition and speciation of particulate organic matter from modern residential small-scale wood combustion appliances, Sci. Total Environ., 612, 636–648, https://doi.org/10.1016/j.scitotenv.2017.08.263, 2018.
Denier van der Gon, H. A. C., Bergström, R., Fountoukis, C., Johansson, C., Pandis, S. N., Simpson, D., and Visschedijk, A. J. H.: Particulate emissions from residential wood combustion in Europe – revised estimates and an evaluation, Atmos. Chem. Phys., 15, 6503–6519, https://doi.org/10.5194/acp-15-6503-2015, 2015.
EC (European Council): Directive on relating to arsenic, cadmium, mercury, nickel and polycyclic aromatic hydrocarbons in ambient air, 2004/107/EC, 2004.
Elsasser, M., Busch, C., Orasche, J., Schön, C., Hartmann, H., Schnelle-Kreis, J., and Zimmermann, R.: Dynamic changes of the aerosol composition and concentration during different burning phases of wood combustion, Energy Fuels, 27, 4959–4968, https://doi.org/10.1021/ef400684f, 2013.
Finewax, Z., de Gouw, J. A., and Ziemann, P. J.: Identification and Quantification of 4-Nitrocatechol Formed from OH and NO3 Radical-Initiated Reactions of Catechol in Air in the Presence of NOx: Implications for Secondary Organic Aerosol Formation from Biomass Burning, Environ. Sci. Technol., 52, 1981–1989, https://doi.org/10.1021/acs.est.7b05864, 2018.
Finlayson-Pitts, B. J. and Pitts Jr., J. N.: Chemistry of the upper and lower atmosphere: theory, experiments, and applications, Academic Press, San Diego, 2000.
Grieshop, A. P., Logue, J. M., Donahue, N. M., and Robinson, A. L.: Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 1: measurement and simulation of organic aerosol evolution, Atmos. Chem. Phys., 9, 1263–1277, https://doi.org/10.5194/acp-9-1263-2009, 2009.
Harrison, M. A., Barra, S., Borghesi, D., Vione, D., Arsene, C., and Olariu, R. I.: Nitrated phenols in the atmosphere: a review, Atmos. Environ., 39, 231–248, https://doi.org/10.1016/j.atmosenv.2004.09.044, 2005.
Hartikainen, A., Yli-Pirilä, P., Tiitta, P., Leskinen, A., Kortelainen, M., Orasche, J., Schnelle-Kreis, J., Lehtinen, K. E., Zimmermann, R., Jokiniemi, J., and Sippula O.: Volatile organic compounds from logwood combustion: emissions and transformation under dark and photochemical aging conditions in a smog chamber, Environ. Sci. Technol., 52, 4979–4988, https://doi.org/10.1021/acs.est.7b06269, 2018.
Hatch, L. E., Yokelson, R. J., Stockwell, C. E., Veres, P. R., Simpson, I. J., Blake, D. R., Orlando, J. J., and Barsanti, K. C.: Multi-instrument comparison and compilation of non-methane organic gas emissions from biomass burning and implications for smoke-derived secondary organic aerosol precursors, Atmos. Chem. Phys., 17, 1471–1489, https://doi.org/10.5194/acp-17-1471-2017, 2017.
Heald, C. L., Kroll, J. H., Jimenez, J. L., Docherty, K. S., DeCarlo, P. F., Aiken, A. C., Chen, Q., Martin, S. T., Farmer, D. K., and Artaxo, P.: A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 37, L08803, https://doi.org/10.1029/2010GL042737, 2010.
Hems, R. F. and Abbatt, J. P.: Aqueous phase photo-oxidation of brown carbon nitrophenols: reaction kinetics, mechanism, and evolution of light absorption, ACS Earth Space Chem., 2, 225–234, https://doi.org/10.1021/acsearthspacechem.7b00123, 2018.
Hennigan, C. J., Miracolo, M. A., Engelhart, G. J., May, A. A., Presto, A. A., Lee, T., Sullivan, A. P., McMeeking, G. R., Coe, H., Wold, C. E., Hao, W.-M., Gilman, J. B., Kuster, W. C., de Gouw, J., Schichtel, B. A., Collett Jr., J. L., Kreidenweis, S. M., and Robinson, A. L.: Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber, Atmos. Chem. Phys., 11, 7669–7686, https://doi.org/10.5194/acp-11-7669-2011, 2011.
Heringa, M. F., DeCarlo, P. F., Chirico, R., Tritscher, T., Dommen, J., Weingartner, E., Richter, R., Wehrle, G., Prévôt, A. S. H., and Baltensperger, U.: Investigations of primary and secondary particulate matter of different wood combustion appliances with a high-resolution time-of-flight aerosol mass spectrometer, Atmos. Chem. Phys., 11, 5945–5957, https://doi.org/10.5194/acp-11-5945-2011, 2011.
Herring, C. L., Faiola, C. L., Massoli, P., Sueper, D., Erickson, M. H., McDonald, J. D., Simpson, C. D., Yost, M. G., Jobson, B. T., and VanReken, T. M.: New methodology for quantifying polycyclic aromatic hydrocarbons (PAHs) using high-resolution aerosol mass spectrometry, Aerosol Sci. Tech., 49, 1131–1148, https://doi.org/10.1080/02786826.2015.1101050, 2015.
Hodzic, A., Madronich, S., Kasibhatla, P. S., Tyndall, G., Aumont, B., Jimenez, J. L., Lee-Taylor, J., and Orlando, J.: Organic photolysis reactions in tropospheric aerosols: effect on secondary organic aerosol formation and lifetime, Atmos. Chem. Phys., 15, 9253–9269, https://doi.org/10.5194/acp-15-9253-2015, 2015.
Hodzic, A., Kasibhatla, P. S., Jo, D. S., Cappa, C. D., Jimenez, J. L., Madronich, S., and Park, R. J.: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 16, 7917–7941, https://doi.org/10.5194/acp-16-7917-2016, 2016.
Ihalainen, M., Tiitta, P., Czech, H., Yli-Pirilä, P., Hartikainen, A., Kortelainen, M., Tissari, J., Stengel, B., Sklorz, M., Suhonen, H., Lamberg, H., Leskinen, A., Kiendler-Scharr, A., Harndorf, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: A novel high-volume Photochemical Emission Aging flow tube Reactor (PEAR), Aerosol Sci. Tech., 53, 276–294, https://doi.org/10.1080/02786826.2018.1559918, 2019.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol. Sci. Technol., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Kanashova, T., Sippula, O., Oeder, S., Streibel, T., Passig, J., Czech, H., Kaoma, T., Sapcariu, S. C., Dilger, M., Paur, H., Schlager, C., Mülhopt, S., Weiss, C., Schmidt-Weber, C., Traidl-Hoffmann, C., Michalke, B., Krebs, T., Karg, E., Jakobi, G., Scholtes, S., Schnelle-Kreis, J., Sklorz, M., Orasche, J., Müller, L., Reda, A., Rüger, C., Neumann, A., Abbaszade, G., Radischat, C., Hiller, K., Grigonyte, J., Kortelainen, M., Kuuspalo, K., Lamberg, H., Leskinen, J., Nuutinen, I., Torvela, T., Tissari, J., Jalava, P., Kasurinen, S., Uski, O., Hirvonen, M.-R., Buters, J., Dittmar, G., Jokiniemi, J. K., and Zimmermann, R.: Emissions from a modern log wood masonry heater and wood pellet boiler: Composition and biological impact on air-liquid interface exposed human lung cancer cells, J. Mol. Clin. Med., 1, 23–35, https://doi.org/10.31083/j.jmcm.2018.01.004, 2018.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Kasurinen, S., Happo, M. S., Rönkkö, T. J., Orasche, J., Jokiniemi, J., Kortelainen, M., Tissari, J., Zimmermann, R., Hirvonen, M., and Jalava, P. I.: Differences between co-cultures and monocultures in testing the toxicity of particulate matter derived from log wood and pellet combustion, PloS one, 13, e0192453, https://doi.org/10.1371/journal.pone.0192453, 2018.
Keyte, I. J., Harrison, R. M., and Lammel, G.: Chemical reactivity and long-range transport potential of polycyclic aromatic hydrocarbons – a review, Chem. Soc. Rev., 42, 9333–9391, https://doi.org/10.1039/c3cs60147a, 2013.
Kim, K., Jahan, S. A., Kabir, E., and Brown, R. J.: A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects, Environ. Int., 60, 71–80, https://doi.org/10.1016/j.envint.2013.07.019, 2013.
Klimont, Z., Kupiainen, K., Heyes, C., Purohit, P., Cofala, J., Rafaj, P., Borken-Kleefeld, J., and Schöpp, W.: Global anthropogenic emissions of particulate matter including black carbon, Atmos. Chem. Phys., 17, 8681–8723, https://doi.org/10.5194/acp-17-8681-2017, 2017.
Kortelainen, M., Jokiniemi, J., Tiitta, P., Tissari, J., Lamberg, H., Leskinen, J., Rodriguez, J. G., Koponen, H., Antikainen, S., Nuutinen, I., Zimmermann, R., and Sippula, O.: Time-resolved chemical composition of small-scale batch combustion emissions from various wood species, Fuel, 233, 224–236, https://doi.org/10.1016/j.fuel.2018.06.056, 2018.
Kostenidou, E., Lee, B., Engelhart, G. J., Pierce, J. R., and Pandis, S. N.: Mass spectra deconvolution of low, medium, and high volatility biogenic secondary organic aerosol, Environ. Sci. Technol., 43, 4884–4889, https://doi.org/10.1021/es803676g, 2009.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kroll, J. H., Lim, C. Y., Kessler, S. H., and Wilson, K. R.: Heterogeneous oxidation of atmospheric organic aerosol: kinetics of changes to the amount and oxidation state of particle-phase organic carbon, J. Phys. Chem. A, 119, 10767–10783, https://doi.org/10.1021/acs.jpca.5b06946, 2015.
Lambe, A. T., Onasch, T. B., Massoli, P., Croasdale, D. R., Wright, J. P., Ahern, A. T., Williams, L. R., Worsnop, D. R., Brune, W. H., and Davidovits, P.: Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA), Atmos. Chem. Phys., 11, 8913–8928, https://doi.org/10.5194/acp-11-8913-2011, 2011.
Lanz, V. A., Alfarra, M. R., Baltensperger, U., Buchmann, B., Hueglin, C., and Prévôt, A. S. H.: Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra, Atmos. Chem. Phys., 7, 1503–1522, https://doi.org/10.5194/acp-7-1503-2007, 2007.
Lehtinen, K. E., Korhonen, H., Maso, M. D., and Kulmala, M.: On the concept of condensation sink diameter, Boreal Environ. Res., 8, 405–412, 2003.
Li, M., Bao, F., Zhang, Y., Song, W., Chen, C., and Zhao, J.: Role of elemental carbon in the photochemical aging of soot, P. Natl. Acad. Sci. USA, 115, 7717–7722, https://doi.org/10.1073/pnas.1804481115, 2018.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., De Gouw, J. A., and Jimenez, J. L.: Modeling the radical chemistry in an oxidation flow reactor: radical formation and recycling, sensitivities, and the OH exposure estimation equation, The J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Malecha, K. T. and Nizkorodov, S. A.: Photodegradation of secondary organic aerosol particles as a source of small, oxygenated volatile organic compounds, Environ. Sci. Technol., 50, 9990–9997, https://doi.org/10.1021/acs.est.6b02313, 2016.
McDonald, J. D., Zielinska, B., Fujita, E. M., Sagebiel, J. C., Chow, J. C., and Watson, J. G.: Fine Particle and Gaseous Emission Rates from Residential Wood Combustion, Environ. Sci. Technol., 34, 2080–2091, https://doi.org/10.1021/es9909632, 2000.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Miersch, T., Czech, H., Hartikainen, A., Ihalainen, M., Orasche, J., Abbaszade, G., Tissari, J., Streibel, T., Jokiniemi, J., Sippula, O., and Zimmermann, R.: Impact of photochemical ageing on Polycyclic Aromatic Hydrocarbons (PAH) and oxygenated PAH (Oxy-PAH/OH-PAH) in logwood stove emissions, Sci. Total Environ., 686, 382–392, https://doi.org/10.1016/j.scitotenv.2019.05.412, 2019.
Moise, T., Flores, J. M., and Rudich, Y.: Optical properties of secondary organic aerosols and their changes by chemical processes, Chem. Rev., 115, 4400–4439, https://doi.org/10.1021/cr5005259, 2015.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011.
Nuutinen, K., Jokiniemi, J., Sippula, O., Lamberg, H., Sutinen, J., Horttanainen, P., and Tissari, J.: Effect of air staging on fine particle, dust and gaseous emissions from masonry heaters, Biomass Bioenergy, 67, 167–178, https://doi.org/10.1016/j.biombioe.2014.04.033, 2014.
Onasch, T. B., Trimborn, A., Fortner, E. C., Jayne, J. T., Kok, G. L., Williams, L. R., Davidovits, P., and Worsnop, D. R.: Soot Particle Aerosol Mass Spectrometer: Development, Validation, and Initial Application, Aerosol Sci. Tech., 46, 804–817, https://doi.org/10.1080/02786826.2012.663948, 2012.
Orasche, J., Schnelle-Kreis, J., Abbaszade, G., and Zimmermann, R.: Technical Note: In-situ derivatization thermal desorption GC-TOFMS for direct analysis of particle-bound non-polar and polar organic species, Atmos. Chem. Phys., 11, 8977–8993, https://doi.org/10.5194/acp-11-8977-2011, 2011.
Orasche, J., Schnelle-Kreis, J., Schön, C., Hartmann, H., Ruppert, H., Arteaga-Salas, J. M., and Zimmermann, R.: Comparison of emissions from wood combustion. Part 2: Impact of combustion conditions on emission factors and characteristics of particle-bound organic species and polycyclic aromatic hydrocarbon (PAH)-related toxicological potential, Energy Fuels, 27, 1482–1491, https://doi.org/10.1021/ef301506h, 2013.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Ortega, A. M., Hayes, P. L., Peng, Z., Palm, B. B., Hu, W., Day, D. A., Li, R., Cubison, M. J., Brune, W. H., Graus, M., Warneke, C., Gilman, J. B., Kuster, W. C., de Gouw, J., Gutiérrez-Montes, C., and Jimenez, J. L.: Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area, Atmos. Chem. Phys., 16, 7411–7433, https://doi.org/10.5194/acp-16-7411-2016, 2016.
Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L.: In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 16, 2943–2970, https://doi.org/10.5194/acp-16-2943-2016, 2016.
Paulot, F., Wunch, D., Crounse, J. D., Toon, G. C., Millet, D. B., DeCarlo, P. F., Vigouroux, C., Deutscher, N. M., González Abad, G., Notholt, J., Warneke, T., Hannigan, J. W., Warneke, C., de Gouw, J. A., Dunlea, E. J., De Mazière, M., Griffith, D. W. T., Bernath, P., Jimenez, J. L., and Wennberg, P. O.: Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 11, 1989–2013, https://doi.org/10.5194/acp-11-1989-2011, 2011.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Peng, Z. and Jimenez, J. L.: Modeling of the chemistry in oxidation flow reactors with high initial NO, Atmos. Chem. Phys., 17, 11991–12010, https://doi.org/10.5194/acp-17-11991-2017, 2017.
Peng, Z., Lee-Taylor, J., Orlando, J. J., Tyndall, G. S., and Jimenez, J. L.: Organic peroxy radical chemistry in oxidation flow reactors and environmental chambers and their atmospheric relevance, Atmos. Chem. Phys., 19, 813–834, https://doi.org/10.5194/acp-19-813-2019, 2019.
Pieber, S. M., El Haddad, I., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. R., Fröhlich, R., Vlachou, A., Klein F., Dommen, J., Miljevic, B., Jiménez J. L., Worsnop D. R., Baltensperger, U., and Prévôt A. S. H.: Inorganic salt interference on CO2 in aerodyne AMS and ACSM organic aerosol composition studies, Environ. Sci. Technol., 50, 10494–10503, https://doi.org/10.1021/acs.est.6b01035, 2016.
Pieber, S. M., Kambolis, A., Ferri, D., Bhattu, D., Bruns, E. A., Elsener, M., Kröcher, O., Prévôt, A. S., and Baltensperger, U.: Mitigation of Secondary Organic Aerosol Formation from Log Wood Burning Emissions by Catalytic Removal of Aromatic Hydrocarbons, Environ. Sci. Technol., 52, 13381–13390, https://doi.org/10.1021/acs.est.8b04124, 2018.
Prinn, R. G., Huang, J., Weiss, R. F., Cunnold, D. M., Fraser, P. J., Simmonds, P. G., McCulloch, A., Harth, C., Reimann, S., Salameh, P., O'Doherty, S., Wang, R. H. J., Porter, L. W., Miller, B. R., and Krummel, P. B.: Evidence for variability of atmospheric hydroxyl radicals over the past quarter century, Geophys. Res. Lett., 32, L07809, https://doi.org/10.1029/2004GL022228, 2005.
Reda, A. A., Czech, H., Schnelle-Kreis, J., Sippula, O., Orasche, J., Weggler, B., Abbaszade, G., Arteaga-Salas, J., Kortelainen, M., Tissari, J., Jokiniemi, J., Streibel, T., and Zimmermann, R.: Analysis of Gas-Phase Carbonyl Compounds in Emissions from Modern Wood Combustion Appliances: Influence of Wood Type and Combustion Appliance, Energy Fuels, 29, 3897–3907, https://doi.org/10.1021/ef502877c, 2015.
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking organic aerosols: semivolatile emissions and photochemical aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007.
Shen, G., Tao, S., Wei, S., Zhang, Y., Wang, R., Wang, B., Li, W., Shen, H., Huang, Y., Chen, Y., Chen, H., Yang, Y., Wang, W., Wang, X., Liu, W., and Simonich, S. L. M.: Emissions of parent, nitro, and oxygenated polycyclic aromatic hydrocarbons from residential wood combustion in rural China, Environ. Sci. Technol., 46, 8123–8130, https://doi.org/10.1021/es301146v, 2012.
Simonen, P., Saukko, E., Karjalainen, P., Timonen, H., Bloss, M., Aakko-Saksa, P., Rönkkö, T., Keskinen, J., and Dal Maso, M.: A new oxidation flow reactor for measuring secondary aerosol formation of rapidly changing emission sources, Atmos. Meas. Tech., 10, 1519–1537, https://doi.org/10.5194/amt-10-1519-2017, 2017.
Sippula, O., Hokkinen, J., Puustinen, H., Yli-Pirilä, P. and Jokiniemi, J.: Particle emissions from small wood-fired district heating units, Energy Fuels, 23, 2974–2982, https://doi.org/10.1021/ef900098v, 2009.
Tiitta, P., Leskinen, A., Hao, L., Yli-Pirilä, P., Kortelainen, M., Grigonyte, J., Tissari, J., Lamberg, H., Hartikainen, A., Kuuspalo, K., Kortelainen, A.-M., Virtanen, A., Lehtinen, K. E. J., Komppula, M., Pieber, S., Prévôt, A. S. H., Onasch, T. B., Worsnop, D. R., Czech, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Transformation of logwood combustion emissions in a smog chamber: formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging, Atmos. Chem. Phys., 16, 13251–13269, https://doi.org/10.5194/acp-16-13251-2016, 2016.
Tissari, J., Lyyränen, J., Hytönen, K., Sippula, O., Tapper, U., Frey, A., Saarnio, K., Pennanen, A. S., Hillamo, R., Salonen, R. O., Hirvonen, M. R., and Jokiniemi, J.: Fine particle and gaseous emissions from normal and smouldering wood combustion in a conventional masonry heater, Atmos. Environ., 42, 7862–7873, https://doi.org/10.1016/j.atmosenv.2008.07.019, 2008.
Tissari, J., Hytönen, K., Sippula, O., and Jokiniemi, J.: The effects of operating conditions on emissions from masonry heaters and sauna stoves, Biomass Bioenergy, 33, 513–520, https://doi.org/10.1016/j.biombioe.2008.08.009, 2009.
Tissari, J., Väätäinen, S., Leskinen, J., Savolahti, M., Lamberg, H., Kortelainen, M., Karvosenoja, N., and Sippula, O.: Fine Particle Emissions from Sauna Stoves: Effects of Combustion Appliance and Fuel, and Implications for the Finnish Emission Inventory, Atmosphere, 10, 775, https://doi.org/10.3390/atmos10120775, 2019.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
United States Environmental Protection Agency (USEPA): Integrated Risk Information System (IRIS); National Center for Environmental Assessment: Washington, DC, available at: http://www.epa.gov/iris, last access: 21 October 2019.
Xu, W., Lambe, A., Silva, P., Hu, W., Onasch, T., Williams, L., Croteau, P., Zhang, X., Renbaum-Wolff, L., Fortner, E., Jimenez, J. L., Jayne, J., Worsnop, D., and Canagaratna, M.: Laboratory evaluation of species-dependent relative ionization efficiencies in the Aerodyne Aerosol Mass Spectrometer, Aerosol Sci. Tech., 52, 626–641, https://doi.org/10.1080/02786826.2018.1439570, 2018.
Zhang, X., Lin, Y., Surratt, J. D., Zotter, P., Prévôt, A. S., and Weber, R. J.: Light-absorbing soluble organic aerosol in Los Angeles and Atlanta: A contrast in secondary organic aerosol, Geophys. Res. Lett., 38, L21810, https://doi.org/10.1029/2011GL049385, 2011.