the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Mar 2020
| 31 Mar 2020
Summertime and wintertime atmospheric processes of secondary aerosol in Beijing
Jing Duan
Ru-Jin Huang
Yongjie Li
Yan Zheng
Yang Chen
Chunshui Lin
Haiyan Ni
Meng Wang
Jurgita Ovadnevaite
Darius Ceburnis
Chunying Chen
Douglas R. Worsnop
Thorsten Hoffmann
Colin O'Dowd
Junji Cao
Secondary aerosol constitutes a large fraction of fine particles in urban air of China. However, its formation mechanisms and atmospheric processes remain largely uncertain despite considerable study in recent years. To elucidate the seasonal variations in fine-particle composition and secondary aerosol formation, an Aerodyne quadrupole aerosol chemical speciation monitor (Q-ACSM), combined with other online instruments, was used to characterize the sub-micrometer particulate matter (diameter < 1 µm, PM1) in Beijing during summer and winter 2015. Our results suggest that photochemical oxidation was the major pathway for sulfate formation during summer, whereas aqueous-phase reaction became an important process for sulfate formation during winter. High concentrations of nitrate (17 % of the PM1 mass) were found during winter, explained by enhanced gas-to-particle partitioning at low temperature, while high nitrate concentrations (19 %) were also observed under the conditions of high relative humidity (RH) during summer, likely due to the hydrophilic property of NH4NO3 and hydrolysis of N2O5. As for organic aerosol (OA) sources, secondary OA (SOA) dominated the OA mass (74 %) during summer, while the SOA contribution decreased to 39 % during winter due to enhanced primary emissions in the heating season. In terms of the SOA formation, photochemical oxidation perhaps played an important role for summertime oxygenated OA (OOA) formation and less-oxidized wintertime OOA (LO-OOA) formation. The wintertime more-oxidized OOA (MO-OOA) showed a good correlation with aerosol liquid water content (ALWC), indicating a more important contribution of aqueous-phase processing over photochemical production to MO-OOA. Meanwhile, the dependence of LO-OOA and the mass ratio of LO-OOA to MO-OOA on atmospheric oxidative tracer (i.e., Ox) both degraded when RH was greater than 60 %, suggesting that RH or aerosol liquid water may also affect LO-OOA formation.
- Article
(2199 KB) - Full-text XML
-
Supplement
(1003 KB) - BibTeX
- EndNote




Haze pollution over the North China Plain (NCP) occurs in all seasons and is characterized by high concentrations of fine particulate matter (PM) (Huang et al., 2014; Yang et al., 2015; An et al., 2019). The haze episodes have become more frequent and severe in recent years, although the annual average concentration of PM2.5 (aerodynamic diameter ≤2.5 µm) in the NCP has decreased due to the implementation of a variety of regulatory measures (Fontes et al., 2017; Sun et al., 2016; Xu et al., 2018). A better understanding of the chemical nature, formation and transformation of fine particles is therefore essential for mitigating the haze pollution.
Compared to primary aerosol that is relatively well constrained in terms of the emission sources, secondary aerosol is still not well understood, likely due to its variable precursors, complex formation and atmospheric transformation processes mediated by meteorological conditions (Ding et al., 2016; Petäjä et al., 2016; Tie et al., 2017; Xu et al., 2017). A number of studies have investigated the formation and aging processes of secondary aerosol (Takegawa et al., 2009; Huang et al., 2014, 2019; Sun et al., 2014, 2016; Cheng et al., 2016; Wang et al., 2016; Duan et al., 2019). For example, field measurements showed that aqueous-phase oxidation of SO2 could be an important formation pathway of sulfate at high RH during haze events (Sun et al., 2014; Elser et al., 2016). Our recent study, however, suggested that the role of photochemical oxidation versus aqueous-phase reactions in sulfate formation largely depends on the meteorological conditions (Duan et al., 2019). Moreover, the aqueous oxidation of SO2 by NO2 has been suggested to be an efficient pathway for sulfate formation (Cheng et al., 2016; Wang et al., 2016), although contribution from this pathway in real air is controversial and is still an open question (Guo et al., 2017; Liu et al., 2017). Model simulation has shown that heterogeneous sulfate formation from SO2 oxidation catalyzed by Fe3+ in aerosol water can significantly improve the sulfate simulation (Li et al., 2017). These studies show that even for sulfate, a simple inorganic component, the formation is complex during haze events. As for secondary organic aerosol (SOA), the formation pathways are much less understood, given the complexity of SOA composition and its precursors. The contribution of SOA to PM2.5 mass was found to be as important as secondary inorganic aerosol (SIA: sulfate, nitrate and ammonium) during severe haze events (Huang et al., 2014). Positive matrix factorization (PMF) analyses have resolved multiple OA factors. In particular, photochemical oxidation has been suggested to be the major pathway of SOA formation during some pollution events in some cities (e.g., Beijing and Xi'an) because SOA correlated tightly with odd oxygen (Ox) and was independent of RH (e.g., Sun et al., 2014; Elser et al., 2016). However, aqueous-phase formation of SOA has also been considered an important pathway during some pollution events in, e.g., Beijing, Shijiazhuang and Baoji (Sun et al., 2016; Wang et al., 2017; Huang et al., 2019). The formation of less-oxidized oxygenated OA (LO-OOA) and more-oxidized oxygenated OA (MO-OOA) in Baoji seemed to be significantly influenced by aqueous-phase chemistry during the period of low atmospheric oxidative capacity (Wang et al., 2017), while in another study Xu et al. (2017) suggested that aqueous-phase processing has a dominant impact on MO-OOA formation but that photochemical oxidation is the dominant pathway for LO-OOA formation in Beijing. It is still unclear whether aqueous-phase processing plays a key role in haze development and what the mechanisms are.
Based on previous studies, it is evident that the formation of secondary aerosol shows large spatial and temporal variation and may change during different events, especially for SOA formation. Further studies are therefore needed to better understand the formation and transformation of secondary aerosol in polluted air. As the pollutant emissions, meteorological conditions and oxidation capacity vary dramatically between summer and winter, in order to further elucidate the atmospheric processes of secondary aerosol under distinctly different atmospheric conditions, here we present results from summertime and wintertime measurements in 2015 in Beijing. The seasonal difference in aerosol chemical composition and in formation mechanisms are discussed.
2.1 Measurement site
The summer (1 July to 19 August 2015) and winter (4 December 2015 to 6 February 2016) campaigns were conducted at an urban site on the roof of a five-story building (∼20 m above the ground) in the National Center for Nanoscience (39.99∘ N, 116.32∘ E) in Beijing, which is close to the 4th Ring Road in Beijing and surrounded by residential, commercial and traffic areas.
2.2 Instrumentation
An Aerodyne quadrupole ACSM (Q-ACSM) with a time resolution of ∼30 min was deployed for the continuous measurement of NR-PM1 species, including organics, sulfate, nitrate, ammonium and chloride. Detailed operation principles of ACSM can be found elsewhere (Ng et al., 2011; Wang et al., 2017; Duan et al., 2019). Briefly, ambient air was sampled through a 0.375 in. stainless steel tube at a flow rate of ∼3 L min−1, and the coarse particles were removed by an University Research Glassware (URG) cyclone (model: URG-2000-30ED) with a 2.5 µm cut in front of the sampling inlet. A Nafion dryer (MD-110-48S, Perma Pure, Inc., Lakewood, NJ, USA) was applied to dry aerosol particles before entering the ACSM, and the sub-micrometer aerosol was subsampled into the ACSM at a flow rate of 85 mL min−1 through a 100 µm diameter critical aperture. The sub-micrometer particles were focused into a narrow beam by an aerodynamic lens and impacted a hot vaporizer (∼600∘). The resulting vapor was ionized via electron impact (70 eV) and chemically characterized with a quadrupole mass spectrometer. Monodispersed 300 nm ammonium nitrate particles, generated by an atomizer (Model 9302, TSI Inc., Shoreview, MN, USA) and selected by a differential mobility analyzer (DMA, TSI model 3080), were used to determine the response factor (RF) and calibrate the ionization efficiency (IE) for the ACSM (Ng et al., 2011).
An aethalometer (Model AE-33, Magee Scientific) was used for the determination of equivalent black carbon (eBC) concentration with a time resolution of 1 min. SO2, CO, NOx and O3 were measured by an Ecotech EC 9850 sulfur dioxide analyzer, a Thermo Scientific Model 48i carbon monoxide analyzer, a Thermo Scientific Model 42i NO–NO2–NOx analyzer and a Thermo Scientific Model 49i ozone analyzer, respectively. Meteorological parameters, including wind speed, wind direction, RH and temperature were measured by an automatic weather station (MAWS201, Vaisala, Vantaa, Finland) and a wind sensor (Vaisala Model QMW101-M2).
2.3 Data analysis
2.3.1 ACSM data analysis
The standard ACSM data analysis software in Igor Pro (WaveMetrics, Inc., Lake Oswego, Oregon USA) was used to analyze the ACSM dataset. IE was determined by comparing the response factors of ACSM to the mass calculated with the known particle size and the number concentration from a condensation particle counter (CPC, TSI model 3772). Standard relative ionization efficiencies (RIEs) were used for organics, nitrate and chloride (i.e., 1.4 for organics, 1.1 for nitrate and 1.3 for chloride) (Canagaratna et al., 2007), and RIEs for ammonium (6.1) and sulfate (1.2) were estimated from the RIE calibrations using NH4NO3 and (NH4)2SO4. The collection efficiency (CE) was introduced to correct for the particle loss due to particle bounce, which is influenced by aerosol acidity, composition and the aerosol liquid water content. Considering that the particles were dried before entering the ACSM and are overall neutralized, the influences of aerosol liquid water and acidity are expected to be negligible. Therefore, CE was determined as CEdry = max (0.45, 0.0833 + 0.9167× ANMF), where ANMF represents the mass fraction of ammonium nitrate in NR-PM1 (Middlebrook et al., 2012).
2.3.2 Source apportionment
PMF was used to perform the source apportionment on the mass spectra of organics as implemented by the multilinear engine (ME-2; Paatero, 1999) via the interface SoFi (Source Finder) coded in Igor Wavemetrics (Canonaco et al., 2013). The OA source apportionment for the summer dataset and the winter dataset was performed separately. First, a range of solutions with different factor numbers from unconstrained runs were examined using conventional PMF. A mixture of two or more factors was found and cannot be separated further for both the summer and winter dataset even after increasing the factor number. Then the ME-2 approach was used, as it can direct the apportionment towards an environmentally meaningful solution by introducing a priori information (profiles) for certain factors (Canonaco et al., 2013; Crippa et al., 2014; Frohlich et al., 2015). The final results are verified based on the rationality of unconstrained factors, distinct mass spectra, time series and good correlations with external tracers for all factors. Details about the PMF and ME-2 source apportionment are provided in the Supplement. In this study, four OA factors, including hydrocarbon-like OA (HOA), cooking OA (COA), isoprene-oxidized OA (ISOOA), and OOA, were resolved during summer and six OA factors, including HOA, COA, biomass burning OA (BBOA), coal combustion OA (CCOA), LO-OOA, and MO-OOA, were resolved during winter. Note that MO-OOA and LO-OOA were defined in winter because of the much higher f44∕43 value for MO-OOA (8.6) than that for LO-OOA (2.6) and the large difference in time series of these two factors. In contrast to the two identified wintertime OOAs, the summertime OOA was mixed and could not be further separated into LO-OOA and MO-OOA, likely due to the limited mass resolution of ACSM (Sun et al., 2012).
2.4 Aerosol liquid water content
Aerosol liquid water content (ALWC) was calculated by the ISORROPIA-II model (Fountoukis and Nenes, 2007) using ACSM aerosol composition and meteorological parameters (temperature and RH) as model inputs. The ISORROPIA-II model then calculated the composition, and the phase state of a –––Cl−–H2O system in thermodynamic equilibrium and the concentration of H+ and ALWC can be resolved.
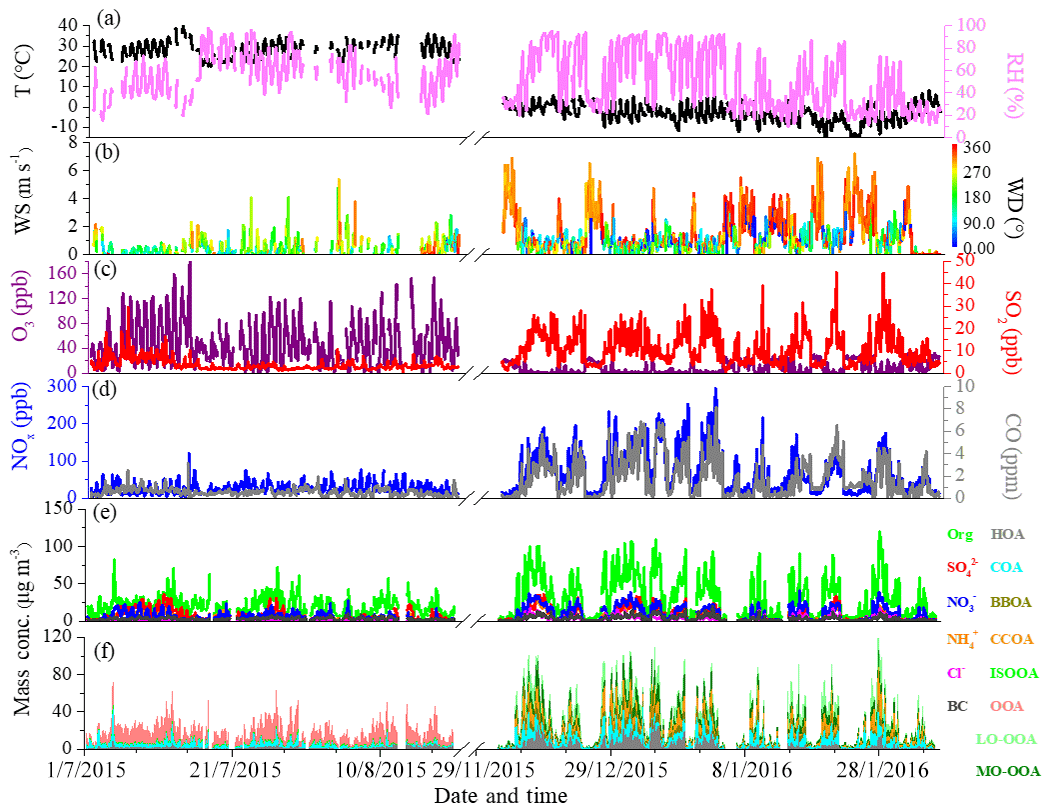
Figure 1Time series of (a) temperature (T) and relative humidity (RH), (b) wind speed (WS) and wind direction (WD), (c) O3 and SO2, (d) CO and NOx, (e) PM1 species, and (f) OA sources between summer and winter measurements.
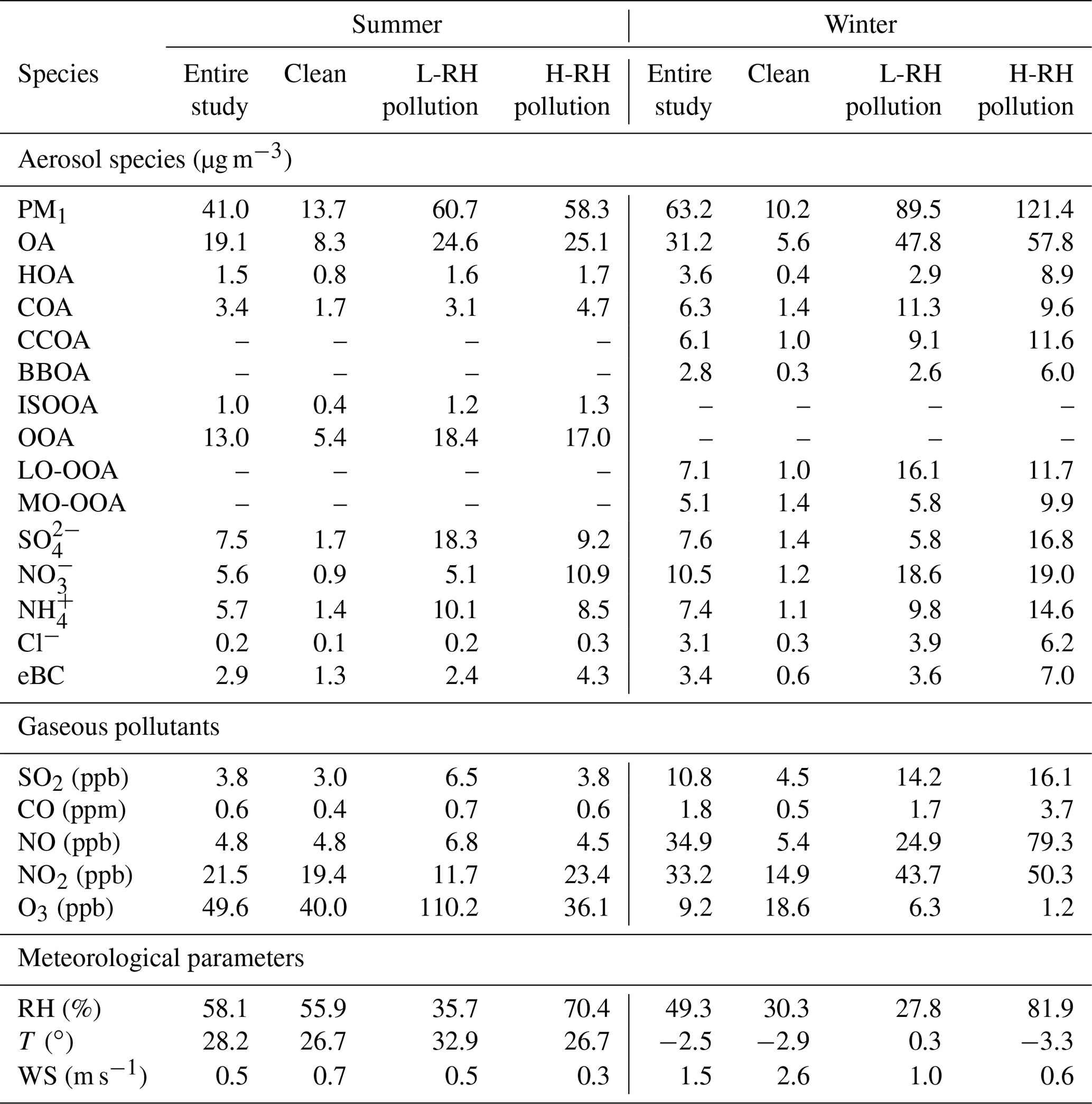
Table 1Summary of mass concentrations of aerosol species, gaseous pollutants and meteorological parameters during different periods between summer and winter in Beijing.
3.1 Chemical composition of PM1 in summer and winter
Figure 1 shows the time series of individual chemical composition of PM1, OA sources, gaseous species and meteorological parameters in summer and winter. The average values of each component in different periods are summarized in Table 1. As shown in Fig. 1, very different time variations in PM1 composition, OA sources and gaseous species were observed between summer and winter. The concentrations of gaseous species such as CO, SO2 and NOx during winter were ∼3 times higher than those during summer, largely due to enhanced emissions and accumulation of pollutants during winter. In contrast, the average O3 concentration during winter (9.2 ppb) was ∼5 times lower than that during summer (49.6 ppb), largely due to titration by enhanced wintertime emissions of NOx. In summer, meteorological conditions were relatively stable, with the wind speeds often being lower than 2 m s−1 during the measurement period. The average mass concentration of PM1 was 41.0±23.4 µg m−3 during summer, and OA constituted a major fraction of PM1 mass (47 %), followed by sulfate (18 %), ammonium (14 %), nitrate (13 %), eBC (7 %) and chloride (1 %). In winter, however, due to frequent changes of meteorological conditions, the time series of PM1 components and gaseous species varied dramatically, such as the rapid build-up of haze pollution under stagnant meteorological conditions with high RH and low wind speed (<2 m s−1) or the sudden occurrence of clean days because of the dilution by clean air masses from the north or northwest at high wind speed (5–10 m s−1). The PM1 mass concentration, with an average value of 63.2±55.1 µg m−3, was observed during winter. OA contributed a mass fraction of 49 %, followed by nitrate (17 %), sulfate (12 %), ammonium (12 %), eBC (5 %) and chloride (5 %). As for secondary inorganic species, sulfate is the largest contributor in summer but is replaced by nitrate in winter largely due to different formation processes between summer and winter (as discussed below). An enhancement of chloride from ∼1 % (0.2 µg m−3 ) in summer to ∼5 % (3.1 µg m−3 ) in winter was also observed, which could be attributed to substantial emissions from coal combustion in winter (Huang et al., 2014; Wang et al., 2015; Elser et al., 2016; Hu et al., 2016).
OA contributed the highest mass fraction to PM1 in both summer and winter, suggesting the important role of OA in PM1 pollution (Zhang et al., 2014; Sun et al., 2015; Hu et al., 2016). As for OA sources, the concentration of HOA increased from 1.5 µg m−3 during summer to 3.6 µg m−3 during winter, and the fractional contribution of HOA increased accordingly from 8 % to 12 %. COA also showed a similar increasing trend from summer to winter, with mass concentration increasing from 3.4 to 6.3 µg m−3 and fractional contribution from 18 % to 20 %. The increase in mass concentration of COA in winter is likely caused by meteorological conditions (e.g., shallower planetary boundary layer heights in winter than in summer) because cooking activities are not expected to have seasonal variation. BBOA and CCOA were only resolved during winter with mass fractions of 9 % and 20 %, respectively. The average wintertime BBOA concentration decreased from 3.6 µg m−3 in 2010 (Hu et al., 2016) to 2.8 µg m−3 in 2015 in our study and further to 2.4 µg m−3 in 2017–2018 (Li et al., 2019), suggesting efficient control of the biomass burning activities in Beijing and surrounding areas in recent years. Two secondary OA sources (OOA and ISOOA) were resolved during summer. OOA was the dominant OA source during summer due to strong atmospheric oxidation capacity, accounting for, on average, 69 % of total OA, with an average mass concentration of 13.0 µg m−3. In addition to OOA, another SOA factor, ISOOA, derived from isoprene oxidation, was resolved in summertime Beijing. ISOOA was generally thought to be formed in environments with low NOx and high biogenic emissions but has recently been observed in urban Nanjing in summer 2013 (Zhang et al., 2017). Similar to the result in summertime Nanjing (4 % of OA), in our study ISOOA is found to contribute ∼5 % of total OA, with an average mass concentration of 1.0 µg m−3 in summertime Beijing. In contrast, ISOOA was not resolved during winter, which is consistent with the low emissions of isoprene. It should be noted that the estimated uncertainty of ISOOA factor is ∼20 %, suggesting large uncertainty in ISOOA source in urban Beijing. In winter, two OOAs, i.e., LO-OOA and MO-OOA, are identified. The fractional contribution of LO-OOA to total OA (23 %, 7.1 µg m−3 ) was larger than that of MO-OOA (16 %, 5.1 µg m−3 ), suggesting more efficient formation of LO-OOA than MO-OOA during winter. The OOA contribution to total OA decreased from 69 % in summer to 39 % in winter, mainly due to enhanced primary emission in winter.
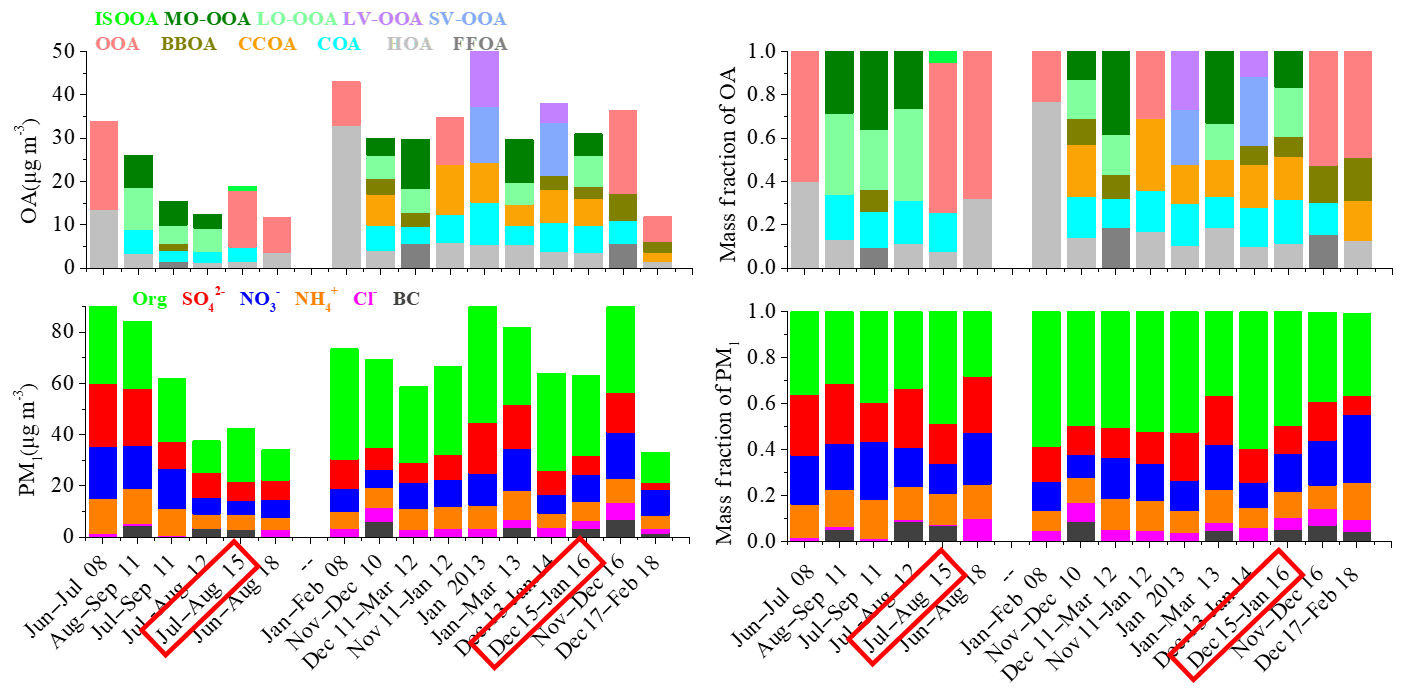
Figure 2Summary of average chemical composition of OA and PM1 in urban Beijing between summer and winter from 2008 to 2018. A more detailed description of the data is presented in Table S1 in the Supplement. As for OA sources, fossil-fuel-related OA (FFOA) is the sum of HOA + CCOA, SV-OOA is semi-volatile OOA and LV-OOA is low-volatility OOA. The date highlighted in red indicate measurements taken during this study in particular.
We also compared our results with previous studies in Beijing. As shown in Fig. 2, the PM1 mass concentration during summer in our study is similar to that observed during summer 2012 (Hu et al., 2017) but ∼30 %–50 % lower than that measured during summer 2008 (Zhang et al., 2013) and summer 2011 (Sun et al., 2015; Hu et al., 2016). From summer 2008 to summer 2015, the fraction contribution of SIA to the PM1 mass decreased from 62 % to 45 % and the OA contribution increased from 36 % to 47 %, suggesting the increasing importance of OA due to the reduction of SO2 and NOx emissions. Meanwhile, we found that the fractional contribution of SOA (i.e., OOAs herein) to total OA also increased from 60 % in summer 2008 to 69 % in summer 2015, indicating the increased importance of SOA formation during summer in recent years. From summer 2015 to summer 2018, the PM1 concentration continued decreasing, while the SIA contribution was again higher than that during summer 2015 and the SOA faction was similar to that in summer 2015 (Zhou et al., 2019). In winter, the average concentration of PM1 observed in our study was similar to that observed in 2014 (Xu et al., 2018) but lower than most of those in 2008–2013 (Zhang et al., 2013, 2014; Sun et al., 2013, 2015, 2016; Hu et al., 2016). In winter 2013, enhanced average PM1 concentrations of 94.0 µg m−3 and 81.7 µg m−3 were observed by Zhang et al. (2014) and Hu et al. (2017), respectively, due to the severe pollution events in January 2013. After 2015, the wintertime PM1 concentration further decreased to 33.2 µg m−3 in 2017–2018 (Li et al., 2019) but showed a peak concentration (92.9 µg m−3) in November–December 2016 because of prevailing air masses from the south and northeast of Beijing and high frequencies of high RH and low wind speeds (Xu et al., 2018). Such changes again suggested that the difference in meteorological conditions is one of the major causes of severe particulate pollution in Beijing. The variations in SIA fractions were not obvious from 2008 to 2015 during wintertime, with SIA contribution ranging from 34 % to 43 %, except a higher SIA contribution of 55 % that was observed by Hu et al. (2017) in winter 2013. However, from winter 2015 to 2018, the SIA fraction increased from 40 % to 54 %, suggesting the increased importance of SIA from 2015 to 2018. As for OA, SOA has a lower contribution during winter than summer in all years from 2008 to 2018. The SOA contributions after 2013 (40 %–54 %) were higher than most of those in 2008–2012 (20 %–30 %) during wintertime, highlighting the increased importance of SOA in winter.
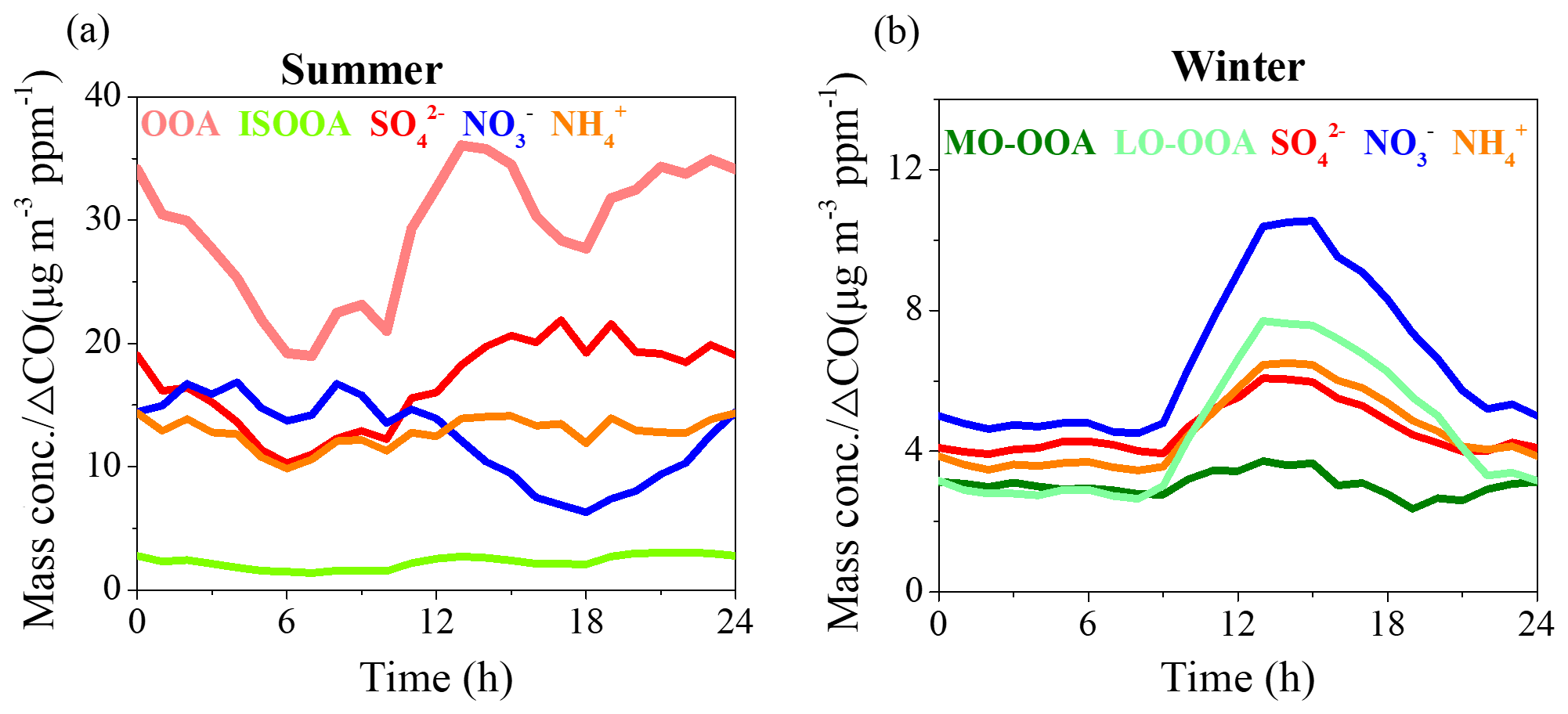
Figure 3Average diurnal variations in the ratios between secondary species and ΔCO during summer (a) and winter (b).
3.2 Daytime evolution of secondary species in summer and winter
The diurnal cycles of PM1 species and OA sources are shown in Fig. S4. The diurnal variations are affected by the evolution of planetary boundary layer (PBL) height, which governs the vertical dispersion of pollutants, and by the diurnal cycle of the emissions and atmospheric processes (Huang et al., 2019). To minimize the effects of PBL height and to investigate the daytime processes of secondary species between summer and winter, data of secondary species were normalized by ΔCO because CO is often used as a tracer to account for dilution on timescales of hours to days. As shown in Fig. 3, after offsetting the PBL dilution effect, OOA displayed the fastest increase from 06:00 to 13:00 (local time, LT), with an average growth rate of 2.4 µg m−3 ppm−1 h−1 during summer. Sulfate also showed a clear increasing trend from 06:00 to 17:00 LT, with an average growth rate of 1.05 µg m−3 ppm−1 h−1, indicating efficient daytime production of OOA and sulfate. However, nitrate displayed a decreasing trend from 11:00 to 18:00 LT, suggesting that evaporation of existing ammonium nitrate was more efficient than production because of its high volatility and thermal instability in the warm afternoon. Associated with sulfate and nitrate, the diurnal cycle of ammonium was affected by both sulfate and nitrate formation mechanisms, which showed a minor increasing trend from 06:00 to 15:00 LT, with an average growth rate of 0.48 µg m−3 ppm−1 h−1. In contrast, all secondary species displayed similar increasing trends from 08:00 to 13:00 LT during winter. Nitrate showed the fastest increasing rate of 1.2 µg m−3 ppm−1 h−1 on average, followed by LO-OOA (1.1 µg m−3 ppm−1 h−1), ammonium (0.6 µg m−3 ppm−1 h−1), sulfate (0.48 µg m−3 ppm−1 h−1) and MO-OOA (0.19 µg m−3 ppm−1 h−1). The high increasing rate of nitrate in winter compared to summer suggested its enhanced daytime production due to lower temperature. LO-OOA displayed a much clearer increasing trend than that of MO-OOA, suggesting that the daytime formation of LO-OOA was much more efficient than that of MO-OOA during winter, which is consistent with the diurnal cycle of MO-OOA concentration (Fig. S4). It should be noted that although the increased rates of sulfate and SOA (LO-OOA + MO-OOA) during winter were lower than those during summer, the enhancement ratios (about 1.5 times higher, from 4.0 to 6.1 µg m−3 ppm−1, for sulfate and about 2.0 times higher, from 5.4 to 11.4 µg m−3 ppm−1, for SOA) during winter were similar to those during summer (about 2.0 times higher, from 10.3 to 21.9 µg m−3 ppm−1, for sulfate and 1.8 times higher, from 20.7 to 38.8 µg m−3 ppm−1, for SOA) indicating that the daytime oxidation formation efficiency of secondary aerosol during winter was comparable to that during summer. We note that wintertime photochemistry was traditionally thought to be low because the low O3 concentrations (e.g., 9.2 ppb in winter versus 49.6 ppb in summer in our study) and reduced solar UV levels resulted in lower OH radical concentrations in wintertime. However, recent measurements of OH radicals at a suburban site in Beijing reported wintertime OH radical concentrations at noontime ranged from 2.4×106 cm−3 during severe pollution events (kOH∼27 s−1) to 3.6×106 cm−3 during relatively clean events (kOH∼5 s−1) (Tan et al., 2018). These OH radical concentrations are nearly an order of magnitude larger than those predicted by global models for northern China in winter (Lelieveld et al., 2016). The higher-than-expected OH concentrations and moderate OH reactivity in Beijing resulted in fast photochemistry in winter (Lu et al., 2019), explaining the high contribution of secondary aerosol during wintertime severe haze events (Huang et al., 2014). Meanwhile, there has been increasing evidence showing that HONO, rather than O3, is the main source of OH radicals in wintertime North China Plain (Hendrick et al., 2014; Xing et al., 2019). Further, Lu et al. (2019) found surprisingly high OH radical oxidation rates of up to 15 ppbv h−1 in Beijing in winter, which is comparable to that in summer and is mainly initiated by the photolysis of HONO and maintained by an extremely efficient radical cycling process driven by nitric oxide.
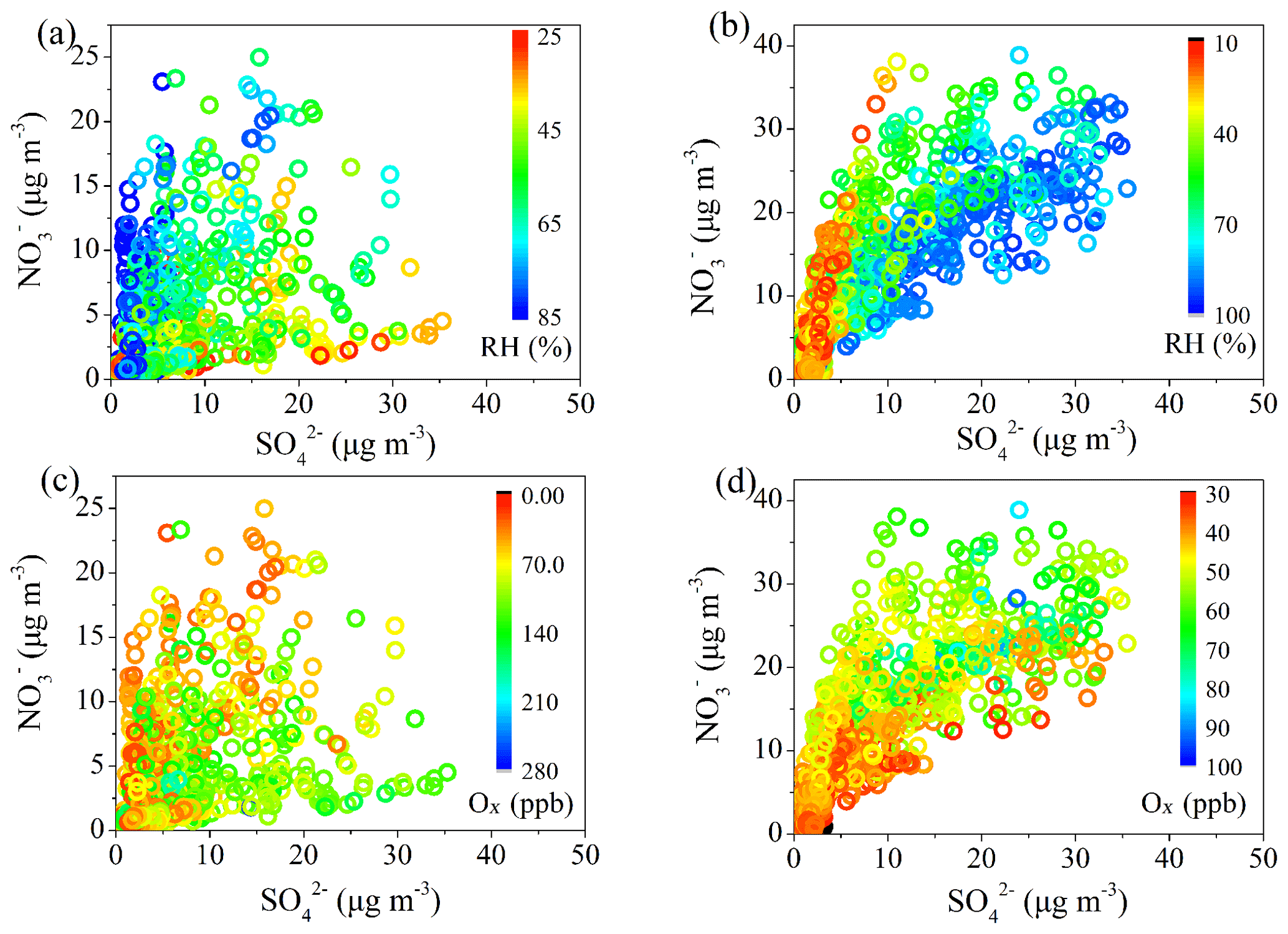
Figure 4Correlations between and , color coded with RH in summer (a) and winter (b), and correlations between and , color coded with Ox concentration in summer (c) and winter (d).
3.3 Sulfate and nitrate formation in summer and winter
To elucidate the formation mechanisms of sulfate and nitrate, the correlations between and colored by RH or Ox are shown in Fig. 4. and correlated well in winter with R2 of 0.6, while there was no good correlation between and in summer. The correlations between and strongly depended on RH and Ox, and this dependence on RH and Ox is reversed between summer and winter. In summer, the correlation between and showed higher slope at higher RH, indicating a faster production rate of than in high RH conditions. The higher concentration of at high RH was likely due to the transformation of HNO3 into particle phase, which could be enhanced in high RH and low-temperature conditions (Sun et al., 2015). In contrast, the slope of correlation between and decreased as Ox concentration increased, indicating the efficient formation of in high Ox concentration due to photochemical oxidation. In contrast, in winter both opposite trends related to RH and Ox were observed. A higher ratio was found at low RH and high Ox conditions, and the ratio decreased dramatically as RH increased due to the fast formation of sulfate in high RH conditions. Such variations between summer and winter suggested that in summer photochemical oxidation was perhaps the major pathway of sulfate formation and that high RH conditions facilitated the nitrate formation due to enhanced gas-to-particle partitioning of NH4NO3, while in winter daytime photochemical process played a dominant role in the formation of nitrate and aqueous-phase processes had significant impacts on the formation of sulfate.
This finding is further supported by the effects of RH and Ox on sulfur oxidation ratio (SOR) and nitrogen oxidation ratio (NOR) (Fig. S5). In summer, there was no obvious dependence between SOR and RH, whereas a positive correlation was found between SOR and Ox. NOR was independent of both RH and Ox, and the higher nitrate concentration in high RH conditions during summer could be attributed to enhanced gas-to-particle partitioning of the highly hydrophilic NH4NO3 and the hydrolysis of N2O5 (Su et al., 2017). In contrast, SOR shows an evident exponential relationship with RH in winter, indicating the important role of aqueous-phase reaction in sulfate formation. NOR shows only a small increasing trend with RH but is strongly correlated with Ox concentration, suggesting the major role of photochemical oxidation in the nitrate formation during winter. It should be noted that SOR also displayed an increasing trend as Ox increased during winter, implying the role of the photochemical process in sulfate formation and that high RH conditions still promoted the gas-to-particle partitioning of nitrate. However, as the aqueous-phase reaction leads to enhanced sulfate formation during winter, the ratio of decreased as RH increased.
It is noted that the diurnal variations in and were similar in winter (Fig. 3). However, the increasing ratio of was much lower than that of . The absolute concentration of wintertime sulfate also showed a decreasing trend in the afternoon, consistent with the diurnal variations in RH and ALWC, while the absolute concentration of wintertime nitrate showed an increasing trend from 08:00 to 18:00 LT (Fig. S4). These results further suggest the difference in dominant formation processes between sulfate and nitrate in winter. The photochemical processes were likely the dominant pathways for daytime nitrate formation but less important for sulfate formation because of large contributions from aqueous-phase processes, as indicated by the exponential relationship between SOR and RH in wintertime Beijing (Fig. S5d).
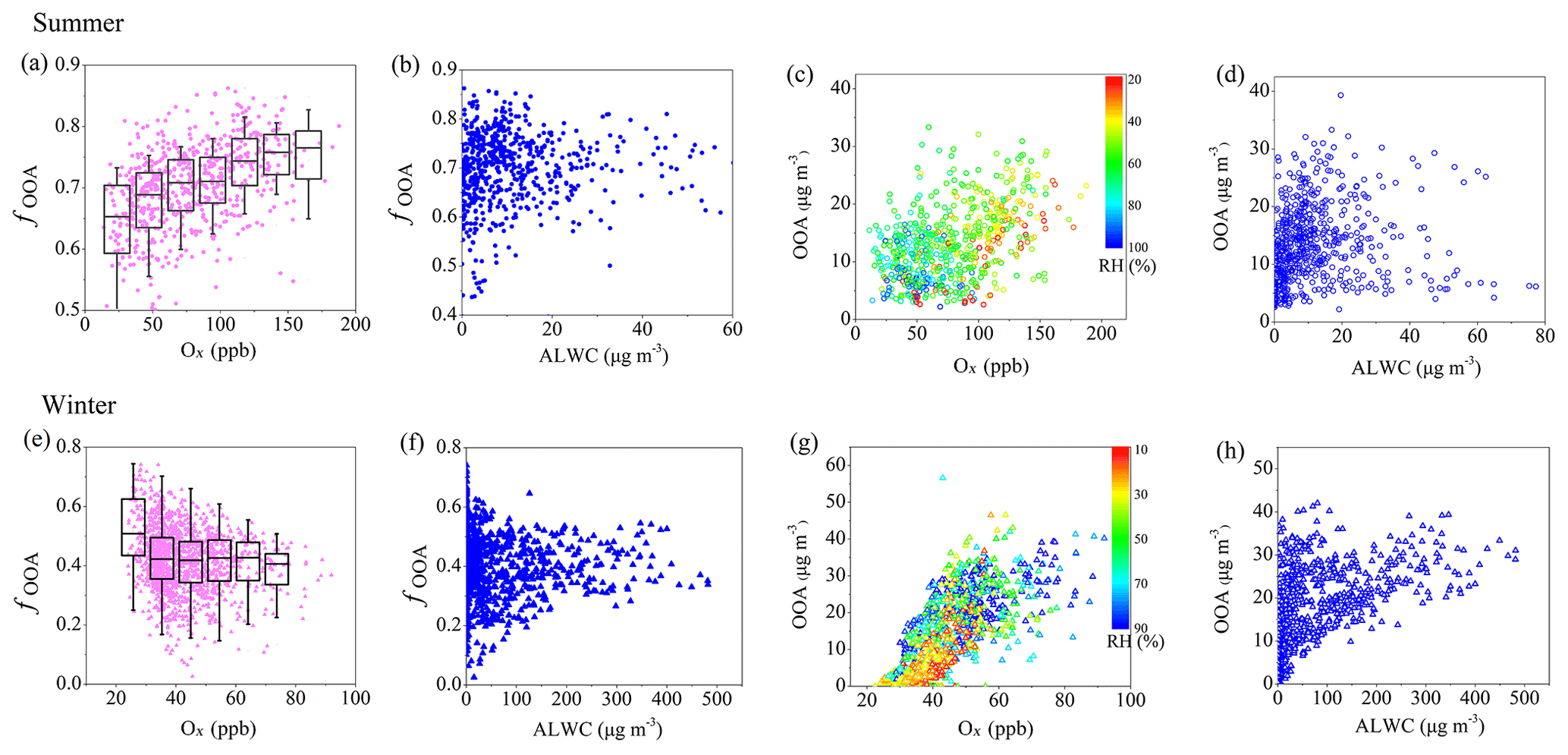
Figure 5Scatterplots of fOOA (mass fraction of OOA (related to OOA in summer and the total of LO-OOA + MO-OOA in winter) to OA) and mass concentration of OOA versus Ox and ALWC in summer (a–d) and winter (e–h).
3.4 SOA formation in summer and winter
The relationships between OOA and Ox or ALWC during summer are shown in Fig. 5. It should be noted that a majority of the data are scattering; therefore, caution should be taken regarding the uncertainties when using Ox and ALWC as indicators of photochemical processing and aqueous processing, respectively. The mass fraction of OOA to OA (fOOA) is positively correlated with Ox, increasing from 0.61 to 0.75 when Ox concentration increases from 20 to 150 ppb, suggesting that photochemical oxidation may play a key role in OOA formation during summer in Beijing. In contrast, fOOA was independent of ALWC during summer. The mass concentration of OOA was also consistently positively correlated with Ox concentration, whereas higher RH was usually located at the bottom-left corner of Fig. 5c, with low Ox concentration and low OOA concentration. This suggests that RH has relatively minor but complex effects on the formation of OOA during summer. During winter, although the fOOA (mass fraction of LO-OOA + MO-OOA to total OA) showed no clear increase or decrease trends with Ox or ALWC (Fig. 5e, f), the correlation between the mass concentration of OOA (total mass of LO-OOA + MO-OOA) and Ox becomes stronger compared with that during summer. Higher OOA concentration was usually related to both higher Ox concentration and higher RH (Fig. 5g). Meanwhile, the increased trend of OOA concentration with ALWC was also more clear in winter than in summer. These suggest that both photochemical oxidation and aqueous-phase reaction play roles in OOA formation during winter in Beijing. It should be noted that temperature could affect both atmospheric oxidative capacity and RH and therefore the photochemistry and aqueous-phase processes. As shown in Fig. S6, both mass concentration and mass fraction of OOA showed positive correlations with temperature, suggesting that high temperature promotes the oxidation and formation of OOA in summer. Further analysis shows a positive correlation between temperature and Ox (R2=0.59) and a strong negative correlation between temperature and RH (R2=0.63). This indicates that high temperature conditions in summer corresponded to high Ox and low RH conditions, further confirming the relatively important role of photochemical oxidation over aqueous-phase processes in OOA formation during summer. In winter, however, both mass concentration and mass fraction of OOA (or LO-OOA and MO-OOA) showed no clear correlation with temperature. Also, there was no clear correlation between temperature and Ox or RH, suggesting a more complex effect of temperature on SOA formation in winter.

Figure 6Relationship between LO-OOA or MO-OOA and Ox (a, e) and relationship between LO-OOA or MO-OOA and ALWC (b, f), as well as mass fractions of LO-OOA and MO-OOA as functions of Ox (c, g) and ALWC (d, h) in winter.
The difference in daytime increasing rate of LO-OOA and MO-OOA suggests a potential difference in their formation during winter (Fig. 3b). The correlations of the two identified OOAs with Ox and ALWC are investigated in Fig. 6. A clear positive correlation between the mass concentration of LO-OOA and Ox is shown (R2=0.51 for the entirety of the data), while the correlation between LO-OOA and ALWC is weaker. Consistently, the correlation coefficient between the mass concentration of LO-OOA and Ox decreased from 0.61 at RH <60 % to 0.29 at RH >60 % (Fig. 6a). The fractional contribution of LO-OOA to total OA increased from 0.17 to 0.35 as Ox concentration increased from 30 to 60 ppb when RH < 60 %, mainly driven by the increased LO-OOA concentrations as Ox increases. The fractional contribution of LO-OOA, however, decreased from 0.25 to 0.18 when ALWC increased from 15 to 300 µg m−3 (Fig. 6d), explained by the more efficient formation of MO-OOA in high RH conditions during winter. Consistently, the mass concentration of MO-OOA showed a strong positive correlation with ALWC during winter with R2 of 0.6, while the correlations between MO-OOA and Ox are weaker than those of LO-OOA both at RH < 60 % and RH > 60 %. The fractional contribution of MO-OOA increased clearly from 0.14 to 0.25 when ALWC increased from 60 to 350 µg m−3 but decreased from 0.3 to 0.18 when Ox concentration increased from 30 to 70 ppb. Similarly, there was a good correlation between the ratio of LO-OOA/MO-OOA and Ox concentration when RH < 60 %, and there was no significant dependence of LO-OOA/MO-OOA on Ox concentration when RH > 60 % (Fig. 7). Our results indicate that Ox likely played a more important role in LO-OOA formation, whereas ALWC was more important for MO-OOA formation in winter. The dependence of LO-OOA and the mass ratio of LO-OOA to MO-OOA on atmospheric oxidative tracer (i.e., Ox) both degraded when RH was greater than 60 %, suggesting that RH or aerosol liquid water may also affect LO-OOA formation.

Figure 7Scatterplots of LO-OOA/MO-OOA (mass ratio) versus Ox at RH > 60 % and RH < 60 % in winter.
3.5 Evolution from clean days to pollution days
In order to better understand aerosol evolution from clean days to pollution days between summer and winter, PM1 composition and OA sources on clean days (daily average PM1 < 20 µg m−3) and pollution days (daily average PM1 > 40 µg m−3 in summer and daily average PM1 > 60 µg m−3 in winter) were analyzed. As shown in Fig. 8, the PM1 composition shows similar evolution trends from clean days to pollution days in both seasons, with an increase in SIA contribution and decrease in OA contribution, suggesting the enhanced formation of SIA during the pollution period. Specifically, the PM1 mass increased from 13.7 µg m−3 during clean days to 59.2 µg m−3 during pollution days in summer, with OA fraction decreasing from 60 % to 44 % and SIA fraction increasing from 29 % to 48 %. In comparison, during winter the average mass concentration of PM1 was 115.5 µg m−3 during polluted days, which was 10 times higher than that during clean days (10.2 µg m−3). The mass fraction of OA decreased from 55 % during clean days to 48 % during pollution days and SIA fraction increased from 36 % to 41 % accordingly. As for OA, the mass concentrations increased substantially during both summer and winter. In summer, the mass fraction of SOA increased from 70 % on clean days to 76 % on polluted days, along with a 3 times greater mass of OA, indicating the importance of SOA formation during pollution events in summer. It should be noted that ISOOA had similar contributions to OA between clean days (5 %) and pollution days (5 %), with the mass concentration increasing from 0.4 to 1.3 µg m−3 and the increase in SOA contribution coming mainly from OOA. In winter, although the mass fraction of SOA (LO-OOA and MO-OOA) decreased from 44 % during clean days to 39 % during pollution days, their mass concentrations increased by 8 times. The mass fraction of POA (HOA, COA, BBOA and CCOA) increased from 56 % to 61 %, indicating that primary contributions are as important as secondary contributions for OA during pollution in winter.
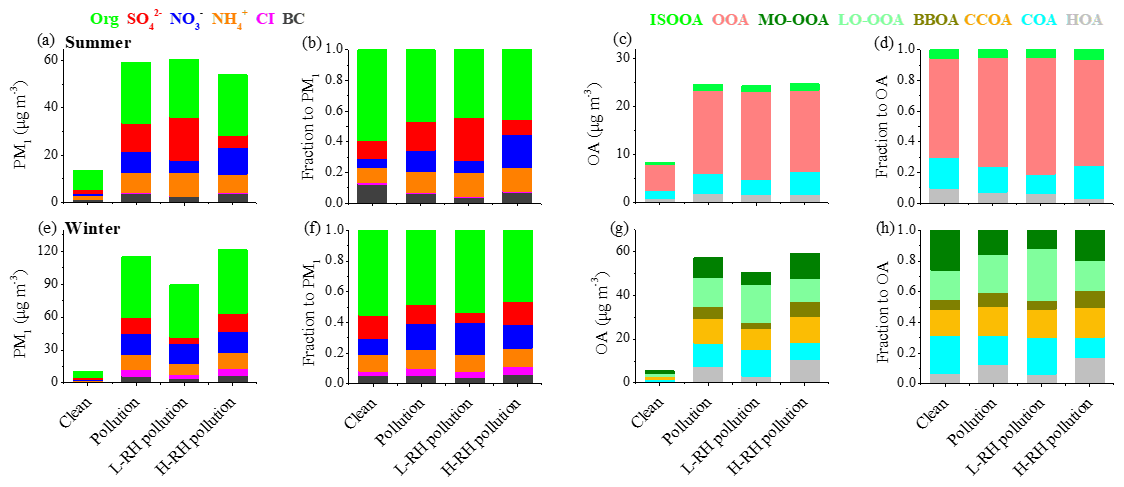
Figure 8Mass concentrations and relative contributions of PM1 species and OA sources during different periods in summer (a–d) and winter (e–h).
To further investigate the effect of RH on aerosol composition, we separated the pollution days into high RH pollution days (H-RH, RH > 60 %) and low RH pollution days (L-RH, RH < 40 %) in summer and winter. As shown in Fig. 8, the PM1 concentration during L-RH pollution days (60.7 µg m−3) was similar to that during H-RH pollution days (58.3 µg m−3) during summer. Interestingly, the mass concentration of sulfate decreased dramatically from 18.3 µg m−3 during L-RH pollution days to 9.2 µg m−3 during H-RH pollution days, and the mass fraction decreased accordingly from 30 % to 16 % in summer. The concentration of nitrate increased from 5.1 µg m−3 during L-RH pollution days to 10.9 µg m−3 during H-RH pollution days, and the mass fraction increased accordingly from 8 % to 19 %. In contrast, during winter the PM1 concentration during H-RH pollution days (121.4 µg m−3) was higher than that during L-RH pollution days (89.5 µg m−3). The mass fraction of sulfate increased from 6 % during L-RH pollution days to 14 % during H-RH pollution days, and the mass concentration increased accordingly from 5.8 to 16.8 µg m−3, while the mass fraction of nitrate decreased from 21 % during L-RH pollution days to 16 % during H-RH pollution days, with similar mass concentrations of 18.6 and 19.0 µg m−3, respectively. The difference between sulfate and nitrate behavior for L-RH and H-RH pollution days in summer and winter suggests that there are factors controlling their production related to aqueous-phase processing other than RH or aerosol liquid water content (e.g., precursors, catalyst and temperature).
As for SOA, the concentration of OOA decreased from 18.4 µg m−3 during L-RH pollution days to 17.0 µg m−3 during H-RH pollution days, with the mass fraction decreasing from 75 % to 68 %. Note that low RH conditions are often related to high temperature and strong oxidation capacity, which facilitates photochemical production during summer. As is consistent with previous understandings of the different importance of photochemical and aqueous production for the two OOAs, LO-OOA and MO-OOA in winter vary differently between L-RH pollution days and H-RH pollution days. The mass fraction of MO-OOA increased from 12 % during L-RH pollution days to 17 % during H-RH pollution days, and the mass concentration increased accordingly from 5.8 to 9.9 µg m−3. In contrast, the mass fraction of LO-OOA decreased from 34 % during L-RH pollution days to 20 % during H-RH pollution days, with mass concentration decreasing from 16.1 to 11.7 µg m−3.
In this study, variations in PM1 composition, OA sources and secondary formation processes between summer and winter in urban Beijing were analyzed. PM1 composition revealed the increase in SIA contribution and decrease in OA contribution during polluted days when compared to clean days, suggesting the important role of SIA during haze pollution. A higher mass ratio of sulfate to nitrate was observed during L-RH pollution days in summer, whereas the same was observed during H-RH pollution days in winter. Analysis of RH and Ox effects indicated that photochemical oxidation was probably the major pathway of sulfate production during summer, while the importance of aqueous-phase reaction increased during winter. In contrast, much higher nitrate (17 %) was found during winter due to preferable gas-to-particle partitioning at low temperature, while higher nitrate (19 %) was also observed under the condition of high relative humidity during summer, likely due to enhanced gas-to-particle partitioning of hydrophilic NH4NO3 and the hydrolysis of N2O5. As for OA sources, SOA-dominated OA mass (74 %) during summer, while the SOA contribution decreased to 39 % during winter due to enhanced primary emission in the heating season. OOA had a higher concentration and fraction during L-RH pollution days than during H-RH pollution days in summer, indicating the possibility of control factors other than RH or aerosol liquid water for aqueous processing. Summertime OOA was likely dominated by gas-phase photochemical processes given the good correlation between OOA and Ox. In comparison, MO-OOA increased during H-RH pollution days in winter, while LO-OOA decreased during H-RH pollution days, indicating that the formation of MO-OOA was perhaps promoted by the aqueous-phase processes, while gas-phase photochemical oxidation facilitates LO-OOA formation during winter. These conclusions were supported by the good correlation between LO-OOA and Ox and tight correlation between MO-OOA and ALWC during winter. Meanwhile, we found that the dependence of LO-OOA and the mass ratio of LO-OOA to MO-OOA on Ox both degraded under conditions of RH > 60 %, suggesting that RH or aerosol liquid water may also affect LO-OOA formation.
Raw data used in this study are archived at the Institute of Earth Environment, Chinese Academy of Sciences, and are available on request by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-3793-2020-supplement.
RJH designed the study. Data analysis and source apportionment were done by JD, RJH and QC. JD and RJH wrote the manuscript. JD and RJH interpreted data and prepared display items. All authors commented on and discussed the manuscript.
Douglas R. Worsnop is an employee of Aerodyne Research, Inc. (ARI), and an ACSM produced by Aerodyne was used in this study.
This work was supported by the National Natural Science Foundation of China (NSFC) under grant nos. 41925015, 91644219, 41877408, and 21661132005; the Chinese Academy of Sciences (no. ZDBS-LY-DQC001); and the National Key Research and Development Program of China (no. 2017YFC0212701). The authors acknowledge financial support from the Cross Innovative Team fund from the State Key Laboratory of Loess and Quaternary Geology (SKLLQG) (no. SKLLQGTD1801). The authors also thank Weikang Ran for assistance with field measurements.
This research has been supported by the National Natural Science Foundation of China (NSFC) (grant nos. 41925015, 91644219, 41877408, and 21661132005), the Chinese Academy of Sciences (grant no. ZDBS-LY-DQC001), the National Key Research and Development Program of China (grant no. 2017YFC0212701), and the Cross Innovative Team fund from the State Key Laboratory of Loess and Quaternary Geology (SKLLQG) (grant no. SKLLQGTD1801).
This paper was edited by Qiang Zhang and reviewed by three anonymous referees.
An, Z., Huang, R.-J., Zhang, R., Tie, X., Li, G., Cao, J., Zhou, W., Shi, Z., Han, Y., Gu, Z., and Ji, Y.: Severe haze in northern China: A synergy of anthropogenic emissions and atmospheric processes, P. Natl. Acad. Sci. USA, 116, 8657–8666, https://doi.org/10.1073/pnas.1900125116, 2019.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the Aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Canonaco, F., Crippa, M., Slowik, J. G., Baltensperger, U., and Prévôt, A. S. H.: SoFi, an IGOR-based interface for the efficient use of the generalized multilinear engine (ME-2) for the source apportionment: ME-2 application to aerosol mass spectrometer data, Atmos. Meas. Tech., 6, 3649–3661, https://doi.org/10.5194/amt-6-3649-2013, 2013.
Cheng, Y. F., Zheng, G. J., Wei, C., Mu, Q., Zheng, B., Wang, Z. B., Gao, M., Zhang, Q., He, K. B., Carmichael, G., Pöschl, U., and Su, H.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Adv., 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Crippa, M., Canonaco, F., Lanz, V. A., Äijälä, M., Allan, J. D., Carbone, S., Capes, G., Ceburnis, D., Dall'Osto, M., Day, D. A., DeCarlo, P. F., Ehn, M., Eriksson, A., Freney, E., Hildebrandt Ruiz, L., Hillamo, R., Jimenez, J. L., Junninen, H., Kiendler-Scharr, A., Kortelainen, A.-M., Kulmala, M., Laaksonen, A., Mensah, A. A., Mohr, C., Nemitz, E., O'Dowd, C., Ovadnevaite, J., Pandis, S. N., Petäjä, T., Poulain, L., Saarikoski, S., Sellegri, K., Swietlicki, E., Tiitta, P., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H.: Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach, Atmos. Chem. Phys., 14, 6159–6176, https://doi.org/10.5194/acp-14-6159-2014, 2014.
Ding, A. J., Huang, X., Nie, W., Sun, J. N., Kerminen, V. M., Petäjä, T., Su, H., Cheng, Y. F., Yang, X. Q., Wang, M. H., Chi, X. G., Wang, J. P., Virkkula, A., Guo, W. D., Yuan, J., Wang, S. Y., Zhang, R. J., Wu, Y. F., Song, Y., Zhu, T., Zilitinkevich, S., Kulmala, M., and Fu, C. B.: Enhanced haze pollution by black carbon in megacities in China, Geophys. Res. Lett., 43, 2873–2879, https://doi.org/10.1002/2016GL067745, 2016.
Duan, J., Huang, R.-J., Lin, C., Dai, W., Wang, M., Gu, Y., Wang, Y., Zhong, H., Zheng, Y., Ni, H., Dusek, U., Chen, Y., Li, Y., Chen, Q., Worsnop, D. R., O'Dowd, C. D., and Cao, J.: Distinctions in source regions and formation mechanisms of secondary aerosol in Beijing from summer to winter, Atmos. Chem. Phys., 19, 10319–10334, https://doi.org/10.5194/acp-19-10319-2019, 2019.
Elser, M., Huang, R.-J., Wolf, R., Slowik, J. G., Wang, Q., Canonaco, F., Li, G., Bozzetti, C., Daellenbach, K. R., Huang, Y., Zhang, R., Li, Z., Cao, J., Baltensperger, U., El-Haddad, I., and Prévôt, A. S. H.: New insights into PM2.5 chemical composition and sources in two major cities in China during extreme haze events using aerosol mass spectrometry, Atmos. Chem. Phys., 16, 3207–3225, https://doi.org/10.5194/acp-16-3207-2016, 2016.
Fontes, T., Li, P. L., Barros, N., and Zhao, P. J.: Trends of PM2.5 concentrations in China: A long term approach, J. Environ. Manage., 196, 719–732, https://doi.org/10.1016/j.jenvman.2017.03.074, 2017.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+––Na+–––Cl−–H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Fröhlich, R., Crenn, V., Setyan, A., Belis, C. A., Canonaco, F., Favez, O., Riffault, V., Slowik, J. G., Aas, W., Aijälä, M., Alastuey, A., Artiñano, B., Bonnaire, N., Bozzetti, C., Bressi, M., Carbone, C., Coz, E., Croteau, P. L., Cubison, M. J., Esser-Gietl, J. K., Green, D. C., Gros, V., Heikkinen, L., Herrmann, H., Jayne, J. T., Lunder, C. R., Minguillón, M. C., Močnik, G., O'Dowd, C. D., Ovadnevaite, J., Petralia, E., Poulain, L., Priestman, M., Ripoll, A., Sarda-Estève, R., Wiedensohler, A., Baltensperger, U., Sciare, J., and Prévôt, A. S. H.: ACTRIS ACSM intercomparison – Part 2: Intercomparison of ME-2 organic source apportionment results from 15 individual, co-located aerosol mass spectrometers, Atmos. Meas. Tech., 8, 2555–2576, https://doi.org/10.5194/amt-8-2555-2015, 2015.
Guo, H., Weber, R. J., and Nenes, A.: High levels of ammonia do not raise fine particle pH sufficiently to yield nitrogen oxide-dominated sulfate production, Sci. Rep., 7, 12109, https://doi.org/10.1038/s41598-017-11704-0, 2017.
Hendrick, F., Müller, J.-F., Clémer, K., Wang, P., De Mazière, M., Fayt, C., Gielen, C., Hermans, C., Ma, J. Z., Pinardi, G., Stavrakou, T., Vlemmix, T., and Van Roozendael, M.: Four years of ground-based MAX-DOAS observations of HONO and NO2 in the Beijing area, Atmos. Chem. Phys., 14, 765–781, https://doi.org/10.5194/acp-14-765-2014, 2014.
Hu, W., Hu, M., Hu, W., Jimenez, J. L., Yuan, B., Chen, W., Wang, M., Wu, Y., Chen, C., Wang, Z., Peng, J., Zeng, L., and Shao, M.: Chemical composition, sources, and aging process of submicron aerosols in Beijing: Contrast between summer and winter, J. Geophys. Res.-Atmos., 121, 1955–1977, https://doi.org/10.1002/2015JD024020, 2016.
Hu, W., Hu, M., Hu, W.-W., Zheng, J., Chen, C., Wu, Y., and Guo, S.: Seasonal variations in high time-resolved chemical compositions, sources, and evolution of atmospheric submicron aerosols in the megacity Beijing, Atmos. Chem. Phys., 17, 9979–10000, https://doi.org/10.5194/acp-17-9979-2017, 2017.
Huang, R. J., Zhang, Y. L., Bozzetti, C., Ho, K. F., Cao, J. J., Han, Y. M., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prevot, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, 2014.
Huang, R.-J., Wang, Y., Cao, J., Lin, C., Duan, J., Chen, Q., Li, Y., Gu, Y., Yan, J., Xu, W., Fröhlich, R., Canonaco, F., Bozzetti, C., Ovadnevaite, J., Ceburnis, D., Canagaratna, M. R., Jayne, J., Worsnop, D. R., El-Haddad, I., Prévôt, A. S. H., and O'Dowd, C. D.: Primary emissions versus secondary formation of fine particulate matter in the most polluted city (Shijiazhuang) in North China, Atmos. Chem. Phys., 19, 2283–2298, https://doi.org/10.5194/acp-19-2283-2019, 2019.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Li, G., Bei, N., Cao, J., Huang, R., Wu, J., Feng, T., Wang, Y., Liu, S., Zhang, Q., Tie, X., and Molina, L. T.: A possible pathway for rapid growth of sulfate during haze days in China, Atmos. Chem. Phys., 17, 3301–3316, https://doi.org/10.5194/acp-17-3301-2017, 2017.
Li, H., Cheng, J., Zhang, Q., Zheng, B., Zhang, Y., Zheng, G., and He, K.: Rapid transition in winter aerosol composition in Beijing from 2014 to 2017: response to clean air actions, Atmos. Chem. Phys., 19, 11485–11499, https://doi.org/10.5194/acp-19-11485-2019, 2019.
Liu, M., Song, Y., Zhou, T., Xu, Z., Yan, C., Zheng, M., Wu, Z., Hu, M., Wu, Y., and Zhu, T.: Fine particle pH during severe haze episodes in northern China, Geophys. Res. Lett., 44, 5213–5221, 2017.
Lu, K., Fuchs, H., Hofzumahaus, A., Tan, Z., Wang, H., Zhang, L., Schmitt, S., Rohrer, F., Bohn, B., Broch, S., Dong, H., Gkatzelis, G., Hohaus, T., Holland, F., Li, X., Liu, Y., Liu, Y. H., Ma, X., Novell, A., Schlag, P., Shao, M., Wu, Y., Wu, Z., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Fast Photochemistry in Wintertime Haze: Consequences for Pollution Mitigation Strategies, Environ. Sci. Technol., 53, 10676–10684, 2019.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of composition-dependent collection efficiencies for the Aerodyne aerosol mass spectrometer using field data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Ng, N. L., Herndon, S. C., Trimborn, A., Canagaratna, M. R., Croteau, P. L., Onasch, T. B., Sueoer, D., Worsnop, D. R., Zhang, Q., Sun, Y. L., and Jayne, J. T.: An Aerosol Chemical Speciation Monitor (ACSM) for routine monitoring of the composition and mass concentrations of ambient aerosol, Aerosol Sci. Technol., 45, 770–784, https://doi.org/10.1080/02786826.2011.560211, 2011.
Paatero, P.: The multilinear engine: a table-driven, least squares program for solving multilinear problems, including the n-way parallel factor analysis model, J. Comput. Graph. Stat., 8, 854–888, https://doi.org/10.1080/10618600.1999.10474853, 1999.
Petäjä, T., Järvi, L., Kerminen, V. M., Ding, A. J., Sun, J. N., Nie, W., Kujansuu, J., Virkkula, A., Yang, X., Fu, C. B., Zilitinkevich, S., and Kulmala, M.: Enhanced air pollution via aerosol-boundary layer feedback in China, Sci. Rep., 6, 18998, https://doi.org/10.1038/srep18998, 2016.
Su, X., Tie, X. X., Li, G. H., Cao, J. J., Huang, R. J., Feng, T., Long, X., and Xu, R. G.: Effect of hydrolysis of N2O5 on nitrate and ammonium formation in Beijing China: WRF-Chem model simulation, Sci. Total. Environ., 579, 221–229, https://doi.org/10.1016/j.scitotenv.2016.11.125, 2017.
Sun, Y. L., Wang, Z., Dong, H., Yang, T., Li, J., Pan, X., Chen, P., and Jayne, J. T.: Characterization of summer organic and inorganic aerosols in Beijing, China with an Aerosol Chemical Speciation Monitor, Atmos. Environ., 51, 250–259, https://doi.org/10.1016/j.atmosenv.2012.01.013, 2012.
Sun, Y. L., Wang, Z. F., Fu, P. Q., Yang, T., Jiang, Q., Dong, H. B., Li, J., and Jia, J. J.: Aerosol composition, sources and processes during wintertime in Beijing, China, Atmos. Chem. Phys., 13, 4577–4592, https://doi.org/10.5194/acp-13-4577-2013, 2013.
Sun, Y., Jiang, Q., Wang, Z., Fu, P., Li, J., Yang, T., and Yin, Y.: Investigation of the sources and evolution processes of severe haze pollution in Beijing in January 2013, J. Geophys. Res. Atmos., 119, 4380–4398, https://doi.org/10.1002/2014JD021641, 2014.
Sun, Y. L., Wang, Z. F., Du, W., Zhang, Q., Wang, Q. Q., Fu, P. Q., Pan, X. L., Li, J., Jayne, J., and Worsnop, D. R.: Long-term real-time measurements of aerosol particle composition in Beijing, China: seasonal variations, meteorological effects, and source analysis, Atmos. Chem. Phys., 15, 10149–10165, https://doi.org/10.5194/acp-15-10149-2015, 2015.
Sun, Y., Du, W., Fu, P., Wang, Q., Li, J., Ge, X., Zhang, Q., Zhu, C., Ren, L., Xu, W., Zhao, J., Han, T., Worsnop, D. R., and Wang, Z.: Primary and secondary aerosols in Beijing in winter: sources, variations and processes, Atmos. Chem. Phys., 16, 8309–8329, https://doi.org/10.5194/acp-16-8309-2016, 2016.
Sun, Y., Xu, W., Zhang, Q., Jiang, Q., Canonaco, F., Prévôt, A. S. H., Fu, P., Li, J., Jayne, J., Worsnop, D. R., and Wang, Z.: Source apportionment of organic aerosol from 2-year highly time-resolved measurements by an aerosol chemical speciation monitor in Beijing, China, Atmos. Chem. Phys., 18, 8469–8489, https://doi.org/10.5194/acp-18-8469-2018, 2018.
Takegawa, N., Miyakawa, T., Kuwata, M., Kondo, Y., Zhao, Y., Han, S., Kita, K., Miyazaki, Y., Deng, Z., Xiao, R., Hu, M., van Pinxteren, D., Herrmann, H., Hofzumahaus, A., Holland, F., Wahner, A., Blake, D. R., Sugimoto, N., and Zhu, T.: Variability of submicron aerosol observed at a rural site in Beijing in the summer of 2006, J. Geophys. Res., 114, D00G05, https://doi.org/10.1029/2008jd010857, 2009.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tie, X., Huang, R. J., Cao, J., Zhang, Q., Cheng, Y., Su, H., Chang, D., Pöschl, U., Hoffmann, T., Dusek, U., Li, G. H., Worsnop, D. R., and O'Dowd, C. D.: Severe pollution in China amplified by atmospheric moisture, Sci. Rep., 7, 15760, https://doi.org/10.1038/s41598-017-15909-1, 2017.
Wang, G. H., Zhang, R. Y., Gomez, M. E., Yang, L. X., Zamora, M. L., Hu, M., Lin, Y., Peng, J. F., Guo, S., Meng, J. J., Li, J. J., Cheng, C. L., Hu, T. F., Ren, Y. Q., Wang, Y. S., Gao, J., Cao, J. J., An, Z. S., Zhou, W. J., Li, G. H., Wang, J. Y., Tian, P. F., Marrero-Ortiz, W., Secrest, J., Du, Z. F., Zheng, J., Shang, D. J., Zeng, L. M., Shao, M., Wang, W. G., Huang, Y., Wang, Y., Zhu, Y. J., Li, Y. X., Hu, J. X., Pan, B., Cai, L., Cheng, Y. T., Ji, Y. M., Zhang, F., Rosenfeld, D., Liss, P. S., Duce, R. A., Kolb, C. E., and Molina, M. J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630–13635, 2016.
Wang, Q., Sun, Y., Jiang, Q., Du, W., Sun, C., Fu, P., and Wang, Z.: Chemical composition of aerosol particles and light extinction apportionment before and during the heating season in Beijing, China, J. Geophys. Res.-Atmos., 120, 12708–12722, https://doi.org/10.1002/2015JD023871, 2015.
Wang, Y. C., Huang, R. J., Ni, H. Y., Chen, Y., Wang, Q. Y., Li, G. H., Tie, X. X., Shen, Z. X., Huang, Y., Liu, S. X., Dong, W. M., Xue, P., Fröhlich, R., Canonaco, F., Elser, M., Daellenbach, K. R., Bozzetti, C., Haddad, E. I., and Cao, J. J.: Chemical composition, sources and secondary processes of aerosols in Baoji city of northwest China, Atmos. Environ., 158, 128–137, https://doi.org/10.1016/j.atmosenv.2017.03.026, 2017.
Xing, L., Wu, J., Elser, M., Tong, S., Liu, S., Li, X., Liu, L., Cao, J., Zhou, J., El-Haddad, I., Huang, R., Ge, M., Tie, X., Prévôt, A. S. H., and Li, G.: Wintertime secondary organic aerosol formation in Beijing-Tianjin-Hebei (BTH): contributions of HONO sources and heterogeneous reactions, Atmos. Chem. Phys., 19, 2343–2359, https://doi.org/10.5194/acp-19-2343-2019, 2019.
Xu, W. Q., Han, T. T., Du, W., Wang, Q. Q., Chen, C., Zhao, J., Zhang, Y. J., Li, J., Fu, P. Q., Wang, Z. F., Worsnop, D. R., and Sun, Y. L.: Effects of Aqueous-Phase and Photochemical Processing on Secondary Organic Aerosol Formation and Evolution in Beijing, China, Environ. Sci. Technol., 51, 762–770, https://doi.org/10.1021/acs.est.6b04498, 2017.
Xu, W. Q., Sun, Y. L., Wang, Q. Q., Zhao, J., Wang, J. F., Ge, X. L., Xie, C. H., Zhou, W., Du, W., Li, J., Fu, P. Q., Wang, Z. F., Worsnop, D. R., and Coe, H.: Changes in aerosol chemistry from 2014 to 2016 in winter in Beijing: insights from high resolution aerosol mass spectrometry, J. Geophys. Res.-Atmos., 124, 1132–1147, 2018.
Yang, Y. R., Liu, X. G., Qu, Y., An, J. L., Jiang, R., Zhang, Y. H., Sun, Y. L., Wu, Z. J., Zhang, F., Xu, W. Q., and Ma, Q. X.: Characteristics and formation mechanism of continuous hazes in China: a case study during the autumn of 2014 in the North China Plain, Atmos. Chem. Phys., 15, 8165–8178, https://doi.org/10.5194/acp-15-8165-2015, 2015.
Zhang, J. K., Sun, Y., Liu, Z. R., Ji, D. S., Hu, B., Liu, Q., and Wang, Y. S.: Characterization of submicron aerosols during a month of serious pollution in Beijing, 2013, Atmos. Chem. Phys., 14, 2887–2903, https://doi.org/10.5194/acp-14-2887-2014, 2014.
Zhang, Y., Sun, J., Zhang, X., Shen, X., Wang, T., and Qin, M.: Seasonal characterization of components and size distributions for submicron aerosols in Beijing, Sci. China Earth Sci., 56, 890–900, https://doi.org/10.1007/s11430-012-4515-z, 2013.
Zhang, Y. J., Tang, L. L., Sun, Y. L., Favez, O., Canonaco, F., Albinet, A., Couvidat, F., Liu, D. T., Jayne, J. T., Wang, Z., Croteau, P. L., Canagaratna, M. R., Zhou, H. C., Prevot, A. S. H., and Worsnop, D. R.: Limited formation of isoprene epoxydiols-derived secondary organic aerosol under NOx-rich environments in Eastern China, Geophys. Res. Lett., 44, 2035–2043, https://doi.org/10.1002/2016GL072368, 2017.
Zhou, W., Gao, M., He, Y., Wang, Q., Xie, C., Xu, W., Zhao, J., Du, W., Qiu, Y., Lei, L., Fu, P., Wang, Z., Worsnop, D.R., Zhang, Q., and Sun, Y.: Response of aerosol chemistry to clean air action in Beijing, China: Insights from two-year ACSM measurements and model simulations, Environ. Pollut., 255, 113345, https://doi.org/10.1016/j.envpol.2019.113345, 2019.